User login
FDA approves generic decitabine for MDS

Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Credit: Bill Branson
The US Food and Drug Administration (FDA) has approved decitabine for injection, a generic version of Dacogen, to treat patients with myelodysplastic syndromes (MDS).
Decitabine is indicated for previously treated and untreated patients with de novo and secondary MDS of all French-American-British subtypes—refractory anemia, refractory anemia with ringed sideroblasts, refractory anemia with excess blasts, refractory anemia with excess blasts in transformation, and chronic myelomonocytic leukemia—as well as intermediate-1, intermediate-2, and high-risk International Prognostic Scoring System groups.
Decitabine will be marketed in 20 mL single-dose glass vials containing 50 mg decitabine—the same size and strength as the brand name drug. The dosing regimen is identical as well.
InnoPharma developed the generic formulation of decitabine and entered into an agreement with Sandoz Inc. Sandoz will sell, market, and distribute decitabine in the US. InnoPharma is set to be acquired by Pfizer Inc., but the transaction is subject to US regulatory approval.
The FDA approved another generic form of decitabine for the treatment of MDS in July 2013. That drug is a product of Dr Reddy’s Laboratories Limited.
Dacogen has been FDA-approved to treat MDS since May 2006. Dacogen is a registered trademark used by Eisai Inc. under license from Astex Pharmaceuticals Inc. ![]()
Problems Identified by Advice Line Calls
The period immediately following hospital discharge is particularly hazardous for patients.[1, 2, 3, 4, 5] Problems occurring after discharge may result in high rates of rehospitalization and unscheduled visits to healthcare providers.[6, 7, 8, 9, 10] Numerous investigators have tried to identify patients who are at increased risk for rehospitalizations within 30 days of discharge, and many studies have examined whether various interventions could decrease these adverse events (summarized in Hansen et al.[11]). An increasing fraction of patients discharged by medicine and surgery services have some or all of their care supervised by hospitalists. Thus, hospitals increasingly look to hospitalists for ways to reduce rehospitalizations.
Patients discharged from our hospital are instructed to call an advice line (AL) if and when questions or concerns arise. Accordingly, we examined when these calls were made and what issues were raised, with the idea that the information collected might identify aspects of our discharge processes that needed improvement.
METHODS
Study Design
We conducted a prospective study of a cohort consisting of all unduplicated patients with a matching medical record number in our data warehouse who called our AL between September 1, 2011 and September 1, 2012, and reported being hospitalized or having surgery (inpatient or outpatient) within 30 days preceding their call. We excluded patients who were incarcerated, those who were transferred from other hospitals, those admitted for routine chemotherapy or emergent dialysis, and those discharged to a skilled nursing facility or hospice. The study involved no intervention. It was approved by the Colorado Multiple Institutional Review Board.
Setting
The study was conducted at Denver Health Medical Center, a 525‐bed, university‐affiliated, public safety‐net hospital. At the time of discharge, all patients were given paperwork that listed the telephone number of the AL and written instructions in English or Spanish telling them to call the AL or their primary care physician if they had any of a list of symptoms that was selected by their discharging physician as being relevant to that specific patient's condition(s).
The AL was established in 1997 to provide medical triage to patients of Denver Health. It operates 24 hours a day, 7 days per week, and receives approximately 100,000 calls per year. A language line service is used with nonEnglish‐speaking callers. Calls are handled by a nurse who, with the assistance of a commercial software program (E‐Centaurus; LVM Systems, Phoenix, AZ) containing clinical algorithms (Schmitt‐Thompson Clinical Content, Windsor, CO), makes a triage recommendation. Nurses rarely contact hospital or clinic physicians to assist with triage decisions.
Variables Assessed
We categorized the nature of the callers' reported problem(s) to the AL using the taxonomy summarized in the online appendix (see Supporting Appendix in the online version of this article). We then queried our data warehouse for each patient's demographic information, patient‐level comorbidities, discharging service, discharge date and diagnoses, hospital length of stay, discharge disposition, and whether they had been hospitalized or sought care in our urgent care center or emergency department within 30 days of discharge. The same variables were collected for all unduplicated patients who met the same inclusion and exclusion criteria and were discharged from Denver Health during the same time period but did not call the AL.
Statistics
Data were analyzed using SAS Enterprise Guide 4.1 (SAS Institute, Inc., Cary, NC). Because we made multiple statistical comparisons, we applied the Bonferroni correction when comparing patients calling the AL with those who did not, such that P<0.004 indicated statistical significance. A Student t test or a Wilcoxon rank sum test was used to compare continuous variables depending on results of normality tests. 2 tests were used to compare categorical variables. The intervals between hospital discharge and the call to the AL for patients discharged from medicine versus surgery services were compared using a log‐rank test, with P<0.05 indicating statistical significance.
RESULTS
During the 1‐year study period, 19,303 unique patients were discharged home with instructions regarding the use of the AL. A total of 310 patients called the AL and reported being hospitalized or having surgery within the preceding 30 days. Of these, 2 were excluded (1 who was incarcerated and 1 who was discharged to a skilled nursing facility), leaving 308 patients in the cohort. This represented 1.5% of the total number of unduplicated patients discharged during this same time period (minus the exclusions described above). The large majority of the calls (277/308, 90%) came directly from patients. The remaining 10% came from a proxy, usually a patient's family member. Compared with patients who were discharged during the same time period who did not call the AL, those who called were more likely to speak English, less likely to speak Spanish, more likely to be medically indigent, had slightly longer lengths of stays for their index hospitalization, and were more likely to be discharged from surgery than medicine services (particularly following inpatient surgery) (Table 1).
| Patient Characteristics | Patients Calling Advice Line After Discharge, N=308 | Patients Not Calling Advice Line After Discharge, N=18,995 | P Valuea |
|---|---|---|---|
| |||
| Age, y (meanSD) | 4217 | 3921 | 0.0210 |
| Gender, female, n (%) | 162 (53) | 10,655 (56) | |
| Race/ethnicity, n (%) | 0.1208 | ||
| Hispanic/Latino/Spanish | 129 (42) | 8,896 (47) | |
| African American | 44 (14) | 2,674 (14) | |
| White | 125 (41) | 6,569 (35) | |
| Language, n (%) | <0.0001 | ||
| English | 273 (89) | 14,236 (79) | |
| Spanish | 32 (10) | 3,744 (21) | |
| Payer, n (%) | |||
| Medicare | 45 (15) | 3,013 (16) | |
| Medicaid | 105 (34) | 7,777 (41) | 0.0152 |
| Commercial | 49 (16) | 2,863 (15) | |
| Medically indigentb | 93 (30) | 3,442 (18) | <0.0001 |
| Self‐pay | 5 (1) | 1,070 (5) | |
| Primary care provider, n (%)c | 168 (55) | 10,136 (53) | 0.6794 |
| Psychiatric comorbidity, n (%) | 81 (26) | 4,528 (24) | 0.3149 |
| Alcohol or substance abuse comorbidity, n (%) | 65 (21) | 3,178 (17) | 0.0417 |
| Discharging service, n (%) | <0.0001 | ||
| Surgery | 193 (63) | 7,247 (38) | |
| Inpatient | 123 (40) | 3,425 (18) | |
| Ambulatory | 70 (23) | 3,822 (20) | |
| Medicine | 93 (30) | 6,038 (32) | |
| Pediatric | 4 (1) | 1,315 (7) | |
| Obstetric | 11 (4) | 3,333 (18) | |
| Length of stay, median (IQR) | 2 (04.5) | 1 (03) | 0.0003 |
| Inpatient medicine | 4 (26) | 3 (15) | 0.0020 |
| Inpatient surgery | 3 (16) | 2 (14) | 0.0019 |
| Charlson Comorbidity Index, median (IQR) | |||
| Inpatient medicine | 1 (04) | 1 (02) | 0.0435 |
| Inpatient surgery | 0 (01) | 0 (01) | 0.0240 |
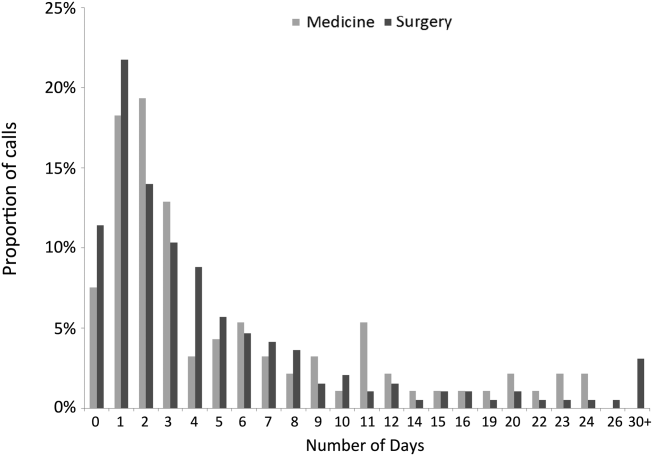
The median time from hospital discharge to the call was 3 days (interquartile range [IQR], 16), but 31% and 47% of calls occurred within 24 or 48 hours of discharge, respectively. Ten percent of patients called the AL the same day of discharge (Figure 1). We found no difference in timing of the calls as a function of discharging service.
The 308 patients reported a total of 612 problems or concerns (meanstandard deviation number of complaints per caller=21), the large majority of which (71%) were symptom‐related (Table 2). The most common symptom was uncontrolled pain, reported by 33% and 40% of patients discharged from medicine and surgery services, respectively. The next most common symptoms related to the gastrointestinal system and to surgical site issues in medicine and surgery patients, respectively (data not shown).
| Total Cohort, n (%) | Patients Discharged From Medicine, n (%) | Patients Discharged From Surgery, n (%) | ||||
|---|---|---|---|---|---|---|
| Patients | Complaints | Patients | Complaints | Patients | Complaints | |
| Symptom related | 280 (91) | 433 (71) | 89 (96) | 166 (77) | 171 (89) | 234 (66) |
| Discharge instructions | 65 (21) | 81 (13) | 18 (19) | 21 (10) | 43 (22) | 56 (16) |
| Medication related | 65 (21) | 87 (14) | 19 (20) | 25 (11) | 39 (20) | 54 (15) |
| Other | 10 (3) | 11 (2) | 4 (4) | 4 (2) | 6 (3) | 7 (2) |
| Total | 612 (100) | 216 (100) | 351 (100) | |||
Sixty‐five patients, representing 21% of the cohort, reported 81 problems understanding or executing discharge instructions. No difference was observed between the fraction of these problems reported by patients from medicine versus surgery (19% and 22%, respectively, P=0.54).
Sixty‐five patients, again representing 21% of the cohort, reported 87 medication‐related problems, 20% from both the medicine and surgery services (P=0.99). Medicine patients more frequently reported difficulties understanding their medication instructions, whereas surgery patients more frequently reported lack of efficacy of medications, particularly with respect to pain control (data not shown).
Thirty percent of patients who called the AL were advised by the nurse to go to the emergency department immediately. Medicine patients were more likely to be triaged to the emergency department compared with surgery patients (45% vs 22%, P<0.0001).
The 30‐day readmission rates and the rates of unscheduled urgent or emergent care visits were higher for patients calling the AL compared with those who did not call (46/308, 15% vs 706/18,995, 4%, and 92/308, 30% vs 1303/18,995, 7%, respectively, both P<0.0001). Similar differences were found for patients discharged from medicine or surgery services who called the AL compared with those who did not (data not shown, both P<0.0001). The median number of days between AL call and rehospitalization was 0 (IQR, 02) and 1 (IQR, 08) for medicine and surgery patients, respectively. Ninety‐three percent of rehospitalizations were related to the index hospitalization, and 78% of patients who were readmitted had no outpatient encounter in the interim between discharge and rehospitalization.
DISCUSSION
We investigated the source and nature of patient telephone calls to an AL following a hospitalization or surgery, and our data revealed the following important findings: (1) nearly one‐half of the calls to the AL occurred within the first 48 hours following discharge; (2) the majority of the calls came from surgery patients, and a greater fraction of patients discharged from surgery services called the AL than patients discharged from medicine services; (3) the most common issues were uncontrolled pain, questions about medications, and problems understanding or executing aftercare instructions (particularly pertaining to the care of surgical wounds); and (4) patients calling the AL had higher rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits.
The utilization of our patient‐initiated call line was only 1.5%, which was on the low end of the 1% to 10% reported in the literature.[7, 12] This can be attributed to a number of issues that are specific to our system. First, the discharge instructions provided to our patients stated that they should call their primary care provider or the AL if they had questions. Accordingly, because approximately 50% of our patients had a primary care provider in our system, some may have preferentially contacted their primary care provider rather than the AL. Second, the instructions stated that the patients should call if they were experiencing the symptoms listed on the instruction sheet, so those with other problems/complaints may not have called. Third, AL personnel identified patients as being in our cohort by asking if they had been discharged or underwent a surgical procedure within 30‐days of their call. This may have resulted in the under‐reporting of patients who were hospitalized or had outpatient surgical procedures. Fourth, there may have been a number of characteristics specific to patients in our system that reduced the frequency with which they utilized the AL (eg, access to telephones or other community providers).
Most previous studies of patient‐initiated call lines have included them as part of multi‐intervention pre‐ and/or postdischarge strategies.[7, 8, 9, 10, 11, 12, 13] One prior small study compared the information reported by 37 patients who called an AL with that elicited by nurse‐initiated patient contact.[12] The most frequently reported problems in this study were medication‐related issues (43%). However, this study only included medicine patients and did not document the proportion of calls occurring at various time intervals.
The problems we identified (in both medicine and surgery patients) have previously been described,[2, 3, 4, 13, 14, 15, 16] but all of the studies reporting these problems utilized calls that were initiated by health care providers to patients at various fixed intervals following discharge (ie, 730 days). Most of these used a scripted approach seeking responses to specific questions or outcomes, and the specific timing at which the problems arose was not addressed. In contrast, we examined unsolicited concerns expressed by patients calling an AL following discharge whenever they felt sufficient urgency to address whatever problems or questions arose. We found that a large fraction of calls occurred on the day of or within the first 48 hours following discharge, much earlier than when provider‐initiated calls in the studies cited above occurred. Accordingly, our results cannot be used to compare the utility of patient‐ versus provider‐initiated calls, or to suggest that other hospitals should create an AL system. Rather, we suggest that our findings might be complementary to those reported in studies of provider‐initiated calls and only propose that by examining calls placed by patients to ALs, problems with hospital discharge processes (some of which may result in increased rates of readmission) may be discovered.
The observation that such a large fraction of calls to our AL occurred within the first 48 hours following discharge, together with the fact that many of the questions asked or concerns raised pertained to issues that should have been discussed during the discharge process (eg, pain control, care of surgical wounds), suggests that suboptimal patient education was occurring prior to discharge as was suggested by Henderson and Zernike.[17] This finding has led us to expand our patient education processes prior to discharge on both medicine and surgery services. Because our hospitalists care for approximately 90% of the patients admitted to medicine services and are increasingly involved in the care of patients on surgery services, they are integrally involved with such quality improvement initiatives.
To our knowledge this is the first study in the literature that describes both medicine and surgery patients who call an AL because of problems or questions following hospital discharge, categorizes these problems, determines when the patients called following their discharge, and identifies those who called as being at increased risk for early rehospitalizations and unscheduled urgent or emergent care visits. Given the financial penalties issued to hospitals with high 30‐day readmission rates, these patients may warrant more attention than is customarily available from telephone call lines or during routine outpatient follow‐up. The majority of patients who called our AL had Medicare, Medicaid, or a commercial insurance, and, accordingly, may have been eligible for additional services such as home visits and/or expedited follow‐up appointments.
Our study has a number of limitations. First, it is a single‐center study, so the results might not generalize to other institutions. Second, because the study was performed in a university‐affiliated, public safety‐net hospital, patient characteristics and the rates and types of postdischarge concerns that we observed might differ from those encountered in different types of hospitals and/or from those in nonteaching institutions. We would suggest, however, that the idea of using concerns raised by patients discharged from any type of hospital in calls to ALs may similarly identify problems with that specific hospital's discharge processes. Third, the information collected from the AL came from summaries provided by nurses answering the calls rather than from actual transcripts. This could have resulted in insufficient or incorrect information pertaining to some of the variables assessed in Table 2. The information presented in Table 1, however, was obtained from our data warehouse after matching medical record numbers. Fourth, we could have underestimated the number of patients who had 30‐day rehospitalizations and/or unplanned for urgent or emergent care visits if patients sought care at other hospitals. Fifth, the number of patients calling the AL was too small to allow us to do any type of robust matching or multivariable analysis. Accordingly, the differences that appeared between patients who called and those who did not (ie, English speakers, being medically indigent, the length of stay for the index hospitalization and the discharging service) could be the result of inadequate matching or interactions among the variables. Although matching or multivariate analysis might have yielded different associations between patients who called the AL versus those who did not, those who called the AL still had an increased risk of readmission and urgent or emergent visits and may still benefit from targeted interventions. Finally, the fact that only 1.5% of unique patients who were discharged called the AL could have biased our results. Because only 55% and 53% of the patients who did or did not call the AL, respectively, saw primary care physicians within our system within the 3 years prior to their index hospitalization (P=0.679), the frequency of calls to the AL that we observed could have underestimated the frequency with which patients had contact with other care providers in the community.
In summary, information collected from patient‐initiated calls to our AL identified several aspects of our discharge processes that needed improvement. We concluded that our predischarge educational processes for both medicine and surgery services needed modification, especially with respect to pain management, which problems to expect after hospitalization or surgery, and how to deal with them. The high rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits among patients calling the AL identifies them as being at increased risk for these outcomes, although the likelihood of these events may be related to factors other than just calling the AL.
- , , , , . Implementation of the care transitions intervention: sustainability and lessons learned. Prof Case Manag. 2009;14(6):282–293.
- , , , et al. Problems after discharge and understanding of communication with their primary care physicians among hospitalized seniors: a mixed methods study. J Hosp Med. 2010;5(7):385–391.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , , . The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161–167.
- , , . Post‐hospitalization transitions: examining the effects of timing of primary care provider follow‐up. J Hosp Med. 2010;5(7):392–397.
- , , , . Telephone follow‐up after discharge from the hospital: does it make a difference? Appl Nurs Res. 1996;9(2) 47–52.
- , , , et al. The effect of real‐time teleconsultations between hospital‐based nurses and patients with severe COPD discharged after an exacerbation. J Telemed Telecare. 2013;19(8):466–474.
- , , , , , . A randomized, controlled trial of an intensive community nurse‐supported discharge program in preventing hospital readmissions of older patients with chronic lung disease. J Am Geriatr Soc. 2004;52(8):1240–1246.
- , , , et al. Effects of education and support on self‐care and resource utilization in patients with heart failure. Eur Heart J. 1999;20(9):673–682.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281(7):613–620.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , . Complementary telephone strategies to improve postdischarge communication. Am J Med. 2012;125(1):28–30.
- , , , , , . Integrated postdischarge transitional care in a hospitalist system to improve discharge outcome: an experimental study. BMC Med. 2011;9:96.
- , , , , , . Patient experiences after hospitalizations for elective surgery. Am J Surg. 2014;207(6):855–862.
- , , . Complications after discharge for surgical patients. ANZ J Surg. 2004;74(3):92–97.
- , , , . Surgeons are overlooking post‐discharge complications: a prospective cohort study. World J Surg. 2014;38(5):1019–1025.
- , . A study of the impact of discharge information for surgical patients. J Adv Nurs. 2001;35(3):435–441.
The period immediately following hospital discharge is particularly hazardous for patients.[1, 2, 3, 4, 5] Problems occurring after discharge may result in high rates of rehospitalization and unscheduled visits to healthcare providers.[6, 7, 8, 9, 10] Numerous investigators have tried to identify patients who are at increased risk for rehospitalizations within 30 days of discharge, and many studies have examined whether various interventions could decrease these adverse events (summarized in Hansen et al.[11]). An increasing fraction of patients discharged by medicine and surgery services have some or all of their care supervised by hospitalists. Thus, hospitals increasingly look to hospitalists for ways to reduce rehospitalizations.
Patients discharged from our hospital are instructed to call an advice line (AL) if and when questions or concerns arise. Accordingly, we examined when these calls were made and what issues were raised, with the idea that the information collected might identify aspects of our discharge processes that needed improvement.
METHODS
Study Design
We conducted a prospective study of a cohort consisting of all unduplicated patients with a matching medical record number in our data warehouse who called our AL between September 1, 2011 and September 1, 2012, and reported being hospitalized or having surgery (inpatient or outpatient) within 30 days preceding their call. We excluded patients who were incarcerated, those who were transferred from other hospitals, those admitted for routine chemotherapy or emergent dialysis, and those discharged to a skilled nursing facility or hospice. The study involved no intervention. It was approved by the Colorado Multiple Institutional Review Board.
Setting
The study was conducted at Denver Health Medical Center, a 525‐bed, university‐affiliated, public safety‐net hospital. At the time of discharge, all patients were given paperwork that listed the telephone number of the AL and written instructions in English or Spanish telling them to call the AL or their primary care physician if they had any of a list of symptoms that was selected by their discharging physician as being relevant to that specific patient's condition(s).
The AL was established in 1997 to provide medical triage to patients of Denver Health. It operates 24 hours a day, 7 days per week, and receives approximately 100,000 calls per year. A language line service is used with nonEnglish‐speaking callers. Calls are handled by a nurse who, with the assistance of a commercial software program (E‐Centaurus; LVM Systems, Phoenix, AZ) containing clinical algorithms (Schmitt‐Thompson Clinical Content, Windsor, CO), makes a triage recommendation. Nurses rarely contact hospital or clinic physicians to assist with triage decisions.
Variables Assessed
We categorized the nature of the callers' reported problem(s) to the AL using the taxonomy summarized in the online appendix (see Supporting Appendix in the online version of this article). We then queried our data warehouse for each patient's demographic information, patient‐level comorbidities, discharging service, discharge date and diagnoses, hospital length of stay, discharge disposition, and whether they had been hospitalized or sought care in our urgent care center or emergency department within 30 days of discharge. The same variables were collected for all unduplicated patients who met the same inclusion and exclusion criteria and were discharged from Denver Health during the same time period but did not call the AL.
Statistics
Data were analyzed using SAS Enterprise Guide 4.1 (SAS Institute, Inc., Cary, NC). Because we made multiple statistical comparisons, we applied the Bonferroni correction when comparing patients calling the AL with those who did not, such that P<0.004 indicated statistical significance. A Student t test or a Wilcoxon rank sum test was used to compare continuous variables depending on results of normality tests. 2 tests were used to compare categorical variables. The intervals between hospital discharge and the call to the AL for patients discharged from medicine versus surgery services were compared using a log‐rank test, with P<0.05 indicating statistical significance.
RESULTS
During the 1‐year study period, 19,303 unique patients were discharged home with instructions regarding the use of the AL. A total of 310 patients called the AL and reported being hospitalized or having surgery within the preceding 30 days. Of these, 2 were excluded (1 who was incarcerated and 1 who was discharged to a skilled nursing facility), leaving 308 patients in the cohort. This represented 1.5% of the total number of unduplicated patients discharged during this same time period (minus the exclusions described above). The large majority of the calls (277/308, 90%) came directly from patients. The remaining 10% came from a proxy, usually a patient's family member. Compared with patients who were discharged during the same time period who did not call the AL, those who called were more likely to speak English, less likely to speak Spanish, more likely to be medically indigent, had slightly longer lengths of stays for their index hospitalization, and were more likely to be discharged from surgery than medicine services (particularly following inpatient surgery) (Table 1).
| Patient Characteristics | Patients Calling Advice Line After Discharge, N=308 | Patients Not Calling Advice Line After Discharge, N=18,995 | P Valuea |
|---|---|---|---|
| |||
| Age, y (meanSD) | 4217 | 3921 | 0.0210 |
| Gender, female, n (%) | 162 (53) | 10,655 (56) | |
| Race/ethnicity, n (%) | 0.1208 | ||
| Hispanic/Latino/Spanish | 129 (42) | 8,896 (47) | |
| African American | 44 (14) | 2,674 (14) | |
| White | 125 (41) | 6,569 (35) | |
| Language, n (%) | <0.0001 | ||
| English | 273 (89) | 14,236 (79) | |
| Spanish | 32 (10) | 3,744 (21) | |
| Payer, n (%) | |||
| Medicare | 45 (15) | 3,013 (16) | |
| Medicaid | 105 (34) | 7,777 (41) | 0.0152 |
| Commercial | 49 (16) | 2,863 (15) | |
| Medically indigentb | 93 (30) | 3,442 (18) | <0.0001 |
| Self‐pay | 5 (1) | 1,070 (5) | |
| Primary care provider, n (%)c | 168 (55) | 10,136 (53) | 0.6794 |
| Psychiatric comorbidity, n (%) | 81 (26) | 4,528 (24) | 0.3149 |
| Alcohol or substance abuse comorbidity, n (%) | 65 (21) | 3,178 (17) | 0.0417 |
| Discharging service, n (%) | <0.0001 | ||
| Surgery | 193 (63) | 7,247 (38) | |
| Inpatient | 123 (40) | 3,425 (18) | |
| Ambulatory | 70 (23) | 3,822 (20) | |
| Medicine | 93 (30) | 6,038 (32) | |
| Pediatric | 4 (1) | 1,315 (7) | |
| Obstetric | 11 (4) | 3,333 (18) | |
| Length of stay, median (IQR) | 2 (04.5) | 1 (03) | 0.0003 |
| Inpatient medicine | 4 (26) | 3 (15) | 0.0020 |
| Inpatient surgery | 3 (16) | 2 (14) | 0.0019 |
| Charlson Comorbidity Index, median (IQR) | |||
| Inpatient medicine | 1 (04) | 1 (02) | 0.0435 |
| Inpatient surgery | 0 (01) | 0 (01) | 0.0240 |
The median time from hospital discharge to the call was 3 days (interquartile range [IQR], 16), but 31% and 47% of calls occurred within 24 or 48 hours of discharge, respectively. Ten percent of patients called the AL the same day of discharge (Figure 1). We found no difference in timing of the calls as a function of discharging service.
The 308 patients reported a total of 612 problems or concerns (meanstandard deviation number of complaints per caller=21), the large majority of which (71%) were symptom‐related (Table 2). The most common symptom was uncontrolled pain, reported by 33% and 40% of patients discharged from medicine and surgery services, respectively. The next most common symptoms related to the gastrointestinal system and to surgical site issues in medicine and surgery patients, respectively (data not shown).
| Total Cohort, n (%) | Patients Discharged From Medicine, n (%) | Patients Discharged From Surgery, n (%) | ||||
|---|---|---|---|---|---|---|
| Patients | Complaints | Patients | Complaints | Patients | Complaints | |
| Symptom related | 280 (91) | 433 (71) | 89 (96) | 166 (77) | 171 (89) | 234 (66) |
| Discharge instructions | 65 (21) | 81 (13) | 18 (19) | 21 (10) | 43 (22) | 56 (16) |
| Medication related | 65 (21) | 87 (14) | 19 (20) | 25 (11) | 39 (20) | 54 (15) |
| Other | 10 (3) | 11 (2) | 4 (4) | 4 (2) | 6 (3) | 7 (2) |
| Total | 612 (100) | 216 (100) | 351 (100) | |||
Sixty‐five patients, representing 21% of the cohort, reported 81 problems understanding or executing discharge instructions. No difference was observed between the fraction of these problems reported by patients from medicine versus surgery (19% and 22%, respectively, P=0.54).
Sixty‐five patients, again representing 21% of the cohort, reported 87 medication‐related problems, 20% from both the medicine and surgery services (P=0.99). Medicine patients more frequently reported difficulties understanding their medication instructions, whereas surgery patients more frequently reported lack of efficacy of medications, particularly with respect to pain control (data not shown).
Thirty percent of patients who called the AL were advised by the nurse to go to the emergency department immediately. Medicine patients were more likely to be triaged to the emergency department compared with surgery patients (45% vs 22%, P<0.0001).
The 30‐day readmission rates and the rates of unscheduled urgent or emergent care visits were higher for patients calling the AL compared with those who did not call (46/308, 15% vs 706/18,995, 4%, and 92/308, 30% vs 1303/18,995, 7%, respectively, both P<0.0001). Similar differences were found for patients discharged from medicine or surgery services who called the AL compared with those who did not (data not shown, both P<0.0001). The median number of days between AL call and rehospitalization was 0 (IQR, 02) and 1 (IQR, 08) for medicine and surgery patients, respectively. Ninety‐three percent of rehospitalizations were related to the index hospitalization, and 78% of patients who were readmitted had no outpatient encounter in the interim between discharge and rehospitalization.
DISCUSSION
We investigated the source and nature of patient telephone calls to an AL following a hospitalization or surgery, and our data revealed the following important findings: (1) nearly one‐half of the calls to the AL occurred within the first 48 hours following discharge; (2) the majority of the calls came from surgery patients, and a greater fraction of patients discharged from surgery services called the AL than patients discharged from medicine services; (3) the most common issues were uncontrolled pain, questions about medications, and problems understanding or executing aftercare instructions (particularly pertaining to the care of surgical wounds); and (4) patients calling the AL had higher rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits.
The utilization of our patient‐initiated call line was only 1.5%, which was on the low end of the 1% to 10% reported in the literature.[7, 12] This can be attributed to a number of issues that are specific to our system. First, the discharge instructions provided to our patients stated that they should call their primary care provider or the AL if they had questions. Accordingly, because approximately 50% of our patients had a primary care provider in our system, some may have preferentially contacted their primary care provider rather than the AL. Second, the instructions stated that the patients should call if they were experiencing the symptoms listed on the instruction sheet, so those with other problems/complaints may not have called. Third, AL personnel identified patients as being in our cohort by asking if they had been discharged or underwent a surgical procedure within 30‐days of their call. This may have resulted in the under‐reporting of patients who were hospitalized or had outpatient surgical procedures. Fourth, there may have been a number of characteristics specific to patients in our system that reduced the frequency with which they utilized the AL (eg, access to telephones or other community providers).
Most previous studies of patient‐initiated call lines have included them as part of multi‐intervention pre‐ and/or postdischarge strategies.[7, 8, 9, 10, 11, 12, 13] One prior small study compared the information reported by 37 patients who called an AL with that elicited by nurse‐initiated patient contact.[12] The most frequently reported problems in this study were medication‐related issues (43%). However, this study only included medicine patients and did not document the proportion of calls occurring at various time intervals.
The problems we identified (in both medicine and surgery patients) have previously been described,[2, 3, 4, 13, 14, 15, 16] but all of the studies reporting these problems utilized calls that were initiated by health care providers to patients at various fixed intervals following discharge (ie, 730 days). Most of these used a scripted approach seeking responses to specific questions or outcomes, and the specific timing at which the problems arose was not addressed. In contrast, we examined unsolicited concerns expressed by patients calling an AL following discharge whenever they felt sufficient urgency to address whatever problems or questions arose. We found that a large fraction of calls occurred on the day of or within the first 48 hours following discharge, much earlier than when provider‐initiated calls in the studies cited above occurred. Accordingly, our results cannot be used to compare the utility of patient‐ versus provider‐initiated calls, or to suggest that other hospitals should create an AL system. Rather, we suggest that our findings might be complementary to those reported in studies of provider‐initiated calls and only propose that by examining calls placed by patients to ALs, problems with hospital discharge processes (some of which may result in increased rates of readmission) may be discovered.
The observation that such a large fraction of calls to our AL occurred within the first 48 hours following discharge, together with the fact that many of the questions asked or concerns raised pertained to issues that should have been discussed during the discharge process (eg, pain control, care of surgical wounds), suggests that suboptimal patient education was occurring prior to discharge as was suggested by Henderson and Zernike.[17] This finding has led us to expand our patient education processes prior to discharge on both medicine and surgery services. Because our hospitalists care for approximately 90% of the patients admitted to medicine services and are increasingly involved in the care of patients on surgery services, they are integrally involved with such quality improvement initiatives.
To our knowledge this is the first study in the literature that describes both medicine and surgery patients who call an AL because of problems or questions following hospital discharge, categorizes these problems, determines when the patients called following their discharge, and identifies those who called as being at increased risk for early rehospitalizations and unscheduled urgent or emergent care visits. Given the financial penalties issued to hospitals with high 30‐day readmission rates, these patients may warrant more attention than is customarily available from telephone call lines or during routine outpatient follow‐up. The majority of patients who called our AL had Medicare, Medicaid, or a commercial insurance, and, accordingly, may have been eligible for additional services such as home visits and/or expedited follow‐up appointments.
Our study has a number of limitations. First, it is a single‐center study, so the results might not generalize to other institutions. Second, because the study was performed in a university‐affiliated, public safety‐net hospital, patient characteristics and the rates and types of postdischarge concerns that we observed might differ from those encountered in different types of hospitals and/or from those in nonteaching institutions. We would suggest, however, that the idea of using concerns raised by patients discharged from any type of hospital in calls to ALs may similarly identify problems with that specific hospital's discharge processes. Third, the information collected from the AL came from summaries provided by nurses answering the calls rather than from actual transcripts. This could have resulted in insufficient or incorrect information pertaining to some of the variables assessed in Table 2. The information presented in Table 1, however, was obtained from our data warehouse after matching medical record numbers. Fourth, we could have underestimated the number of patients who had 30‐day rehospitalizations and/or unplanned for urgent or emergent care visits if patients sought care at other hospitals. Fifth, the number of patients calling the AL was too small to allow us to do any type of robust matching or multivariable analysis. Accordingly, the differences that appeared between patients who called and those who did not (ie, English speakers, being medically indigent, the length of stay for the index hospitalization and the discharging service) could be the result of inadequate matching or interactions among the variables. Although matching or multivariate analysis might have yielded different associations between patients who called the AL versus those who did not, those who called the AL still had an increased risk of readmission and urgent or emergent visits and may still benefit from targeted interventions. Finally, the fact that only 1.5% of unique patients who were discharged called the AL could have biased our results. Because only 55% and 53% of the patients who did or did not call the AL, respectively, saw primary care physicians within our system within the 3 years prior to their index hospitalization (P=0.679), the frequency of calls to the AL that we observed could have underestimated the frequency with which patients had contact with other care providers in the community.
In summary, information collected from patient‐initiated calls to our AL identified several aspects of our discharge processes that needed improvement. We concluded that our predischarge educational processes for both medicine and surgery services needed modification, especially with respect to pain management, which problems to expect after hospitalization or surgery, and how to deal with them. The high rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits among patients calling the AL identifies them as being at increased risk for these outcomes, although the likelihood of these events may be related to factors other than just calling the AL.
The period immediately following hospital discharge is particularly hazardous for patients.[1, 2, 3, 4, 5] Problems occurring after discharge may result in high rates of rehospitalization and unscheduled visits to healthcare providers.[6, 7, 8, 9, 10] Numerous investigators have tried to identify patients who are at increased risk for rehospitalizations within 30 days of discharge, and many studies have examined whether various interventions could decrease these adverse events (summarized in Hansen et al.[11]). An increasing fraction of patients discharged by medicine and surgery services have some or all of their care supervised by hospitalists. Thus, hospitals increasingly look to hospitalists for ways to reduce rehospitalizations.
Patients discharged from our hospital are instructed to call an advice line (AL) if and when questions or concerns arise. Accordingly, we examined when these calls were made and what issues were raised, with the idea that the information collected might identify aspects of our discharge processes that needed improvement.
METHODS
Study Design
We conducted a prospective study of a cohort consisting of all unduplicated patients with a matching medical record number in our data warehouse who called our AL between September 1, 2011 and September 1, 2012, and reported being hospitalized or having surgery (inpatient or outpatient) within 30 days preceding their call. We excluded patients who were incarcerated, those who were transferred from other hospitals, those admitted for routine chemotherapy or emergent dialysis, and those discharged to a skilled nursing facility or hospice. The study involved no intervention. It was approved by the Colorado Multiple Institutional Review Board.
Setting
The study was conducted at Denver Health Medical Center, a 525‐bed, university‐affiliated, public safety‐net hospital. At the time of discharge, all patients were given paperwork that listed the telephone number of the AL and written instructions in English or Spanish telling them to call the AL or their primary care physician if they had any of a list of symptoms that was selected by their discharging physician as being relevant to that specific patient's condition(s).
The AL was established in 1997 to provide medical triage to patients of Denver Health. It operates 24 hours a day, 7 days per week, and receives approximately 100,000 calls per year. A language line service is used with nonEnglish‐speaking callers. Calls are handled by a nurse who, with the assistance of a commercial software program (E‐Centaurus; LVM Systems, Phoenix, AZ) containing clinical algorithms (Schmitt‐Thompson Clinical Content, Windsor, CO), makes a triage recommendation. Nurses rarely contact hospital or clinic physicians to assist with triage decisions.
Variables Assessed
We categorized the nature of the callers' reported problem(s) to the AL using the taxonomy summarized in the online appendix (see Supporting Appendix in the online version of this article). We then queried our data warehouse for each patient's demographic information, patient‐level comorbidities, discharging service, discharge date and diagnoses, hospital length of stay, discharge disposition, and whether they had been hospitalized or sought care in our urgent care center or emergency department within 30 days of discharge. The same variables were collected for all unduplicated patients who met the same inclusion and exclusion criteria and were discharged from Denver Health during the same time period but did not call the AL.
Statistics
Data were analyzed using SAS Enterprise Guide 4.1 (SAS Institute, Inc., Cary, NC). Because we made multiple statistical comparisons, we applied the Bonferroni correction when comparing patients calling the AL with those who did not, such that P<0.004 indicated statistical significance. A Student t test or a Wilcoxon rank sum test was used to compare continuous variables depending on results of normality tests. 2 tests were used to compare categorical variables. The intervals between hospital discharge and the call to the AL for patients discharged from medicine versus surgery services were compared using a log‐rank test, with P<0.05 indicating statistical significance.
RESULTS
During the 1‐year study period, 19,303 unique patients were discharged home with instructions regarding the use of the AL. A total of 310 patients called the AL and reported being hospitalized or having surgery within the preceding 30 days. Of these, 2 were excluded (1 who was incarcerated and 1 who was discharged to a skilled nursing facility), leaving 308 patients in the cohort. This represented 1.5% of the total number of unduplicated patients discharged during this same time period (minus the exclusions described above). The large majority of the calls (277/308, 90%) came directly from patients. The remaining 10% came from a proxy, usually a patient's family member. Compared with patients who were discharged during the same time period who did not call the AL, those who called were more likely to speak English, less likely to speak Spanish, more likely to be medically indigent, had slightly longer lengths of stays for their index hospitalization, and were more likely to be discharged from surgery than medicine services (particularly following inpatient surgery) (Table 1).
| Patient Characteristics | Patients Calling Advice Line After Discharge, N=308 | Patients Not Calling Advice Line After Discharge, N=18,995 | P Valuea |
|---|---|---|---|
| |||
| Age, y (meanSD) | 4217 | 3921 | 0.0210 |
| Gender, female, n (%) | 162 (53) | 10,655 (56) | |
| Race/ethnicity, n (%) | 0.1208 | ||
| Hispanic/Latino/Spanish | 129 (42) | 8,896 (47) | |
| African American | 44 (14) | 2,674 (14) | |
| White | 125 (41) | 6,569 (35) | |
| Language, n (%) | <0.0001 | ||
| English | 273 (89) | 14,236 (79) | |
| Spanish | 32 (10) | 3,744 (21) | |
| Payer, n (%) | |||
| Medicare | 45 (15) | 3,013 (16) | |
| Medicaid | 105 (34) | 7,777 (41) | 0.0152 |
| Commercial | 49 (16) | 2,863 (15) | |
| Medically indigentb | 93 (30) | 3,442 (18) | <0.0001 |
| Self‐pay | 5 (1) | 1,070 (5) | |
| Primary care provider, n (%)c | 168 (55) | 10,136 (53) | 0.6794 |
| Psychiatric comorbidity, n (%) | 81 (26) | 4,528 (24) | 0.3149 |
| Alcohol or substance abuse comorbidity, n (%) | 65 (21) | 3,178 (17) | 0.0417 |
| Discharging service, n (%) | <0.0001 | ||
| Surgery | 193 (63) | 7,247 (38) | |
| Inpatient | 123 (40) | 3,425 (18) | |
| Ambulatory | 70 (23) | 3,822 (20) | |
| Medicine | 93 (30) | 6,038 (32) | |
| Pediatric | 4 (1) | 1,315 (7) | |
| Obstetric | 11 (4) | 3,333 (18) | |
| Length of stay, median (IQR) | 2 (04.5) | 1 (03) | 0.0003 |
| Inpatient medicine | 4 (26) | 3 (15) | 0.0020 |
| Inpatient surgery | 3 (16) | 2 (14) | 0.0019 |
| Charlson Comorbidity Index, median (IQR) | |||
| Inpatient medicine | 1 (04) | 1 (02) | 0.0435 |
| Inpatient surgery | 0 (01) | 0 (01) | 0.0240 |
The median time from hospital discharge to the call was 3 days (interquartile range [IQR], 16), but 31% and 47% of calls occurred within 24 or 48 hours of discharge, respectively. Ten percent of patients called the AL the same day of discharge (Figure 1). We found no difference in timing of the calls as a function of discharging service.
The 308 patients reported a total of 612 problems or concerns (meanstandard deviation number of complaints per caller=21), the large majority of which (71%) were symptom‐related (Table 2). The most common symptom was uncontrolled pain, reported by 33% and 40% of patients discharged from medicine and surgery services, respectively. The next most common symptoms related to the gastrointestinal system and to surgical site issues in medicine and surgery patients, respectively (data not shown).
| Total Cohort, n (%) | Patients Discharged From Medicine, n (%) | Patients Discharged From Surgery, n (%) | ||||
|---|---|---|---|---|---|---|
| Patients | Complaints | Patients | Complaints | Patients | Complaints | |
| Symptom related | 280 (91) | 433 (71) | 89 (96) | 166 (77) | 171 (89) | 234 (66) |
| Discharge instructions | 65 (21) | 81 (13) | 18 (19) | 21 (10) | 43 (22) | 56 (16) |
| Medication related | 65 (21) | 87 (14) | 19 (20) | 25 (11) | 39 (20) | 54 (15) |
| Other | 10 (3) | 11 (2) | 4 (4) | 4 (2) | 6 (3) | 7 (2) |
| Total | 612 (100) | 216 (100) | 351 (100) | |||
Sixty‐five patients, representing 21% of the cohort, reported 81 problems understanding or executing discharge instructions. No difference was observed between the fraction of these problems reported by patients from medicine versus surgery (19% and 22%, respectively, P=0.54).
Sixty‐five patients, again representing 21% of the cohort, reported 87 medication‐related problems, 20% from both the medicine and surgery services (P=0.99). Medicine patients more frequently reported difficulties understanding their medication instructions, whereas surgery patients more frequently reported lack of efficacy of medications, particularly with respect to pain control (data not shown).
Thirty percent of patients who called the AL were advised by the nurse to go to the emergency department immediately. Medicine patients were more likely to be triaged to the emergency department compared with surgery patients (45% vs 22%, P<0.0001).
The 30‐day readmission rates and the rates of unscheduled urgent or emergent care visits were higher for patients calling the AL compared with those who did not call (46/308, 15% vs 706/18,995, 4%, and 92/308, 30% vs 1303/18,995, 7%, respectively, both P<0.0001). Similar differences were found for patients discharged from medicine or surgery services who called the AL compared with those who did not (data not shown, both P<0.0001). The median number of days between AL call and rehospitalization was 0 (IQR, 02) and 1 (IQR, 08) for medicine and surgery patients, respectively. Ninety‐three percent of rehospitalizations were related to the index hospitalization, and 78% of patients who were readmitted had no outpatient encounter in the interim between discharge and rehospitalization.
DISCUSSION
We investigated the source and nature of patient telephone calls to an AL following a hospitalization or surgery, and our data revealed the following important findings: (1) nearly one‐half of the calls to the AL occurred within the first 48 hours following discharge; (2) the majority of the calls came from surgery patients, and a greater fraction of patients discharged from surgery services called the AL than patients discharged from medicine services; (3) the most common issues were uncontrolled pain, questions about medications, and problems understanding or executing aftercare instructions (particularly pertaining to the care of surgical wounds); and (4) patients calling the AL had higher rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits.
The utilization of our patient‐initiated call line was only 1.5%, which was on the low end of the 1% to 10% reported in the literature.[7, 12] This can be attributed to a number of issues that are specific to our system. First, the discharge instructions provided to our patients stated that they should call their primary care provider or the AL if they had questions. Accordingly, because approximately 50% of our patients had a primary care provider in our system, some may have preferentially contacted their primary care provider rather than the AL. Second, the instructions stated that the patients should call if they were experiencing the symptoms listed on the instruction sheet, so those with other problems/complaints may not have called. Third, AL personnel identified patients as being in our cohort by asking if they had been discharged or underwent a surgical procedure within 30‐days of their call. This may have resulted in the under‐reporting of patients who were hospitalized or had outpatient surgical procedures. Fourth, there may have been a number of characteristics specific to patients in our system that reduced the frequency with which they utilized the AL (eg, access to telephones or other community providers).
Most previous studies of patient‐initiated call lines have included them as part of multi‐intervention pre‐ and/or postdischarge strategies.[7, 8, 9, 10, 11, 12, 13] One prior small study compared the information reported by 37 patients who called an AL with that elicited by nurse‐initiated patient contact.[12] The most frequently reported problems in this study were medication‐related issues (43%). However, this study only included medicine patients and did not document the proportion of calls occurring at various time intervals.
The problems we identified (in both medicine and surgery patients) have previously been described,[2, 3, 4, 13, 14, 15, 16] but all of the studies reporting these problems utilized calls that were initiated by health care providers to patients at various fixed intervals following discharge (ie, 730 days). Most of these used a scripted approach seeking responses to specific questions or outcomes, and the specific timing at which the problems arose was not addressed. In contrast, we examined unsolicited concerns expressed by patients calling an AL following discharge whenever they felt sufficient urgency to address whatever problems or questions arose. We found that a large fraction of calls occurred on the day of or within the first 48 hours following discharge, much earlier than when provider‐initiated calls in the studies cited above occurred. Accordingly, our results cannot be used to compare the utility of patient‐ versus provider‐initiated calls, or to suggest that other hospitals should create an AL system. Rather, we suggest that our findings might be complementary to those reported in studies of provider‐initiated calls and only propose that by examining calls placed by patients to ALs, problems with hospital discharge processes (some of which may result in increased rates of readmission) may be discovered.
The observation that such a large fraction of calls to our AL occurred within the first 48 hours following discharge, together with the fact that many of the questions asked or concerns raised pertained to issues that should have been discussed during the discharge process (eg, pain control, care of surgical wounds), suggests that suboptimal patient education was occurring prior to discharge as was suggested by Henderson and Zernike.[17] This finding has led us to expand our patient education processes prior to discharge on both medicine and surgery services. Because our hospitalists care for approximately 90% of the patients admitted to medicine services and are increasingly involved in the care of patients on surgery services, they are integrally involved with such quality improvement initiatives.
To our knowledge this is the first study in the literature that describes both medicine and surgery patients who call an AL because of problems or questions following hospital discharge, categorizes these problems, determines when the patients called following their discharge, and identifies those who called as being at increased risk for early rehospitalizations and unscheduled urgent or emergent care visits. Given the financial penalties issued to hospitals with high 30‐day readmission rates, these patients may warrant more attention than is customarily available from telephone call lines or during routine outpatient follow‐up. The majority of patients who called our AL had Medicare, Medicaid, or a commercial insurance, and, accordingly, may have been eligible for additional services such as home visits and/or expedited follow‐up appointments.
Our study has a number of limitations. First, it is a single‐center study, so the results might not generalize to other institutions. Second, because the study was performed in a university‐affiliated, public safety‐net hospital, patient characteristics and the rates and types of postdischarge concerns that we observed might differ from those encountered in different types of hospitals and/or from those in nonteaching institutions. We would suggest, however, that the idea of using concerns raised by patients discharged from any type of hospital in calls to ALs may similarly identify problems with that specific hospital's discharge processes. Third, the information collected from the AL came from summaries provided by nurses answering the calls rather than from actual transcripts. This could have resulted in insufficient or incorrect information pertaining to some of the variables assessed in Table 2. The information presented in Table 1, however, was obtained from our data warehouse after matching medical record numbers. Fourth, we could have underestimated the number of patients who had 30‐day rehospitalizations and/or unplanned for urgent or emergent care visits if patients sought care at other hospitals. Fifth, the number of patients calling the AL was too small to allow us to do any type of robust matching or multivariable analysis. Accordingly, the differences that appeared between patients who called and those who did not (ie, English speakers, being medically indigent, the length of stay for the index hospitalization and the discharging service) could be the result of inadequate matching or interactions among the variables. Although matching or multivariate analysis might have yielded different associations between patients who called the AL versus those who did not, those who called the AL still had an increased risk of readmission and urgent or emergent visits and may still benefit from targeted interventions. Finally, the fact that only 1.5% of unique patients who were discharged called the AL could have biased our results. Because only 55% and 53% of the patients who did or did not call the AL, respectively, saw primary care physicians within our system within the 3 years prior to their index hospitalization (P=0.679), the frequency of calls to the AL that we observed could have underestimated the frequency with which patients had contact with other care providers in the community.
In summary, information collected from patient‐initiated calls to our AL identified several aspects of our discharge processes that needed improvement. We concluded that our predischarge educational processes for both medicine and surgery services needed modification, especially with respect to pain management, which problems to expect after hospitalization or surgery, and how to deal with them. The high rates of 30‐day rehospitalization and of unscheduled urgent or emergent care visits among patients calling the AL identifies them as being at increased risk for these outcomes, although the likelihood of these events may be related to factors other than just calling the AL.
- , , , , . Implementation of the care transitions intervention: sustainability and lessons learned. Prof Case Manag. 2009;14(6):282–293.
- , , , et al. Problems after discharge and understanding of communication with their primary care physicians among hospitalized seniors: a mixed methods study. J Hosp Med. 2010;5(7):385–391.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , , . The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161–167.
- , , . Post‐hospitalization transitions: examining the effects of timing of primary care provider follow‐up. J Hosp Med. 2010;5(7):392–397.
- , , , . Telephone follow‐up after discharge from the hospital: does it make a difference? Appl Nurs Res. 1996;9(2) 47–52.
- , , , et al. The effect of real‐time teleconsultations between hospital‐based nurses and patients with severe COPD discharged after an exacerbation. J Telemed Telecare. 2013;19(8):466–474.
- , , , , , . A randomized, controlled trial of an intensive community nurse‐supported discharge program in preventing hospital readmissions of older patients with chronic lung disease. J Am Geriatr Soc. 2004;52(8):1240–1246.
- , , , et al. Effects of education and support on self‐care and resource utilization in patients with heart failure. Eur Heart J. 1999;20(9):673–682.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281(7):613–620.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , . Complementary telephone strategies to improve postdischarge communication. Am J Med. 2012;125(1):28–30.
- , , , , , . Integrated postdischarge transitional care in a hospitalist system to improve discharge outcome: an experimental study. BMC Med. 2011;9:96.
- , , , , , . Patient experiences after hospitalizations for elective surgery. Am J Surg. 2014;207(6):855–862.
- , , . Complications after discharge for surgical patients. ANZ J Surg. 2004;74(3):92–97.
- , , , . Surgeons are overlooking post‐discharge complications: a prospective cohort study. World J Surg. 2014;38(5):1019–1025.
- , . A study of the impact of discharge information for surgical patients. J Adv Nurs. 2001;35(3):435–441.
- , , , , . Implementation of the care transitions intervention: sustainability and lessons learned. Prof Case Manag. 2009;14(6):282–293.
- , , , et al. Problems after discharge and understanding of communication with their primary care physicians among hospitalized seniors: a mixed methods study. J Hosp Med. 2010;5(7):385–391.
- , , , et al. Adverse events among medical patients after discharge from hospital. CMAJ. 2004;170(3):345–349.
- , , , , . The incidence and severity of adverse events affecting patients after discharge from the hospital. Ann Intern Med. 2003;138(3):161–167.
- , , . Post‐hospitalization transitions: examining the effects of timing of primary care provider follow‐up. J Hosp Med. 2010;5(7):392–397.
- , , , . Telephone follow‐up after discharge from the hospital: does it make a difference? Appl Nurs Res. 1996;9(2) 47–52.
- , , , et al. The effect of real‐time teleconsultations between hospital‐based nurses and patients with severe COPD discharged after an exacerbation. J Telemed Telecare. 2013;19(8):466–474.
- , , , , , . A randomized, controlled trial of an intensive community nurse‐supported discharge program in preventing hospital readmissions of older patients with chronic lung disease. J Am Geriatr Soc. 2004;52(8):1240–1246.
- , , , et al. Effects of education and support on self‐care and resource utilization in patients with heart failure. Eur Heart J. 1999;20(9):673–682.
- , , , et al. Comprehensive discharge planning and home follow‐up of hospitalized elders: a randomized clinical trial. JAMA. 1999;281(7):613–620.
- , , , , . Interventions to reduce 30‐day rehospitalization: a systematic review. Ann Intern Med. 2011;155(8):520–528.
- , , , . Complementary telephone strategies to improve postdischarge communication. Am J Med. 2012;125(1):28–30.
- , , , , , . Integrated postdischarge transitional care in a hospitalist system to improve discharge outcome: an experimental study. BMC Med. 2011;9:96.
- , , , , , . Patient experiences after hospitalizations for elective surgery. Am J Surg. 2014;207(6):855–862.
- , , . Complications after discharge for surgical patients. ANZ J Surg. 2004;74(3):92–97.
- , , , . Surgeons are overlooking post‐discharge complications: a prospective cohort study. World J Surg. 2014;38(5):1019–1025.
- , . A study of the impact of discharge information for surgical patients. J Adv Nurs. 2001;35(3):435–441.
© 2014 Society of Hospital Medicine
Aggression and angry outbursts
Introduction
Aggressive behavior is one of the most common child psychiatric symptoms for which parents seek help. The difficulty with managing aggressive behavior is determining whether it is out of the ordinary from typical child development and then assessing the causes of the behavior before tackling the tough job of intervening. The following case is typical of what might present to the pediatrics office and provides a few ideas for the assessment and management of aggressive behavior.
Case summary

Dakota is a 6-year-old boy who presents for a well-child check with his father, Joe. Dakota is just finishing his kindergarten year, and the teachers have expressed concerns about his behavior in the classroom and on the playground. They note that he is often irritable and touchy, and that he will frequently have aggressive outbursts, particularly when asked to do something that he doesn’t like. He will often interrupt other children’s games, and will force children to play by his rules with a threat of, or occasional use of, hitting. In the classroom, he has been removed multiple times to the principal’s office, where he will go only with marked reluctance. He has been noted by his teachers to have difficulty attending to the classroom instructions, and frequently removes himself during circle time. They allow him to do this to avoid a power struggle. Similarly, Joe notes that the entire family is "walking on eggshells" because they never know what might set him off. They’ve tried "everything," including time out, sticker charts, and spanking, but with little effect. Joe says that he was "just like Dakota" when he was a child, and that he was "straightened out" in the Army. He wonders if some kind of boot camp or "scared straight" program would help Dakota learn his lesson.
Discussion
Diagnosis. Irritability and aggression are common manifestations of multiple child psychiatric conditions. While it’s easy to jump to the conclusion that the patient has oppositional-defiant disorder (ODD) or conduct disorder (CD) and move straight to treatment, care must be taken to evaluate common causes and co-occurring disorders that might change the treatment plan.
The differential diagnosis includes a primary mood disorder like depression, other disruptive behavior disorders such as attention-deficit/hyperactivity disorder (ADHD), a primary anxiety disorder, posttraumatic stress disorder, a learning or language disorder, and/or intellectual disability. One also must determine whether the aggression exhibited is greater than that shown by other boys his age. For this reason, the use of a scale that has normative values by age and sex makes sense. Having a standardized instrument filled out by parents and by the teachers also will help give an indication of how he is performing in multiple settings. Using a broad-based instrument that also covers mood, anxiety, and attention problems can be a quick and useful way to examine what type of co-occurring symptoms are present.
Aggression, while a heritable trait, also has a significant component from the environment. It is important to see how much of the aggression is being "caught, not taught" in the family setting. Querying as to the general level of negative, coercive parenting can be performed quickly by asking for a description of how the last outburst was managed – what the precipitant, the course, and the outcome were. Frequently, with ODD in particular, you will find a cycle of escalating threats and illogical consequences that serve to reinforce, rather than to reduce, aggressive and oppositional behavior. Practically, while the busy pediatrician may be able to manage some of this screening in a well-child check, it is likely that a separate appointment will be needed to go over the results of the screening instruments and to more fully assess the parenting environment.
At the scheduled visit designed to specifically assess the aggression:
• Make sure that both the parents and the child see this as a family-based problem. A treatment alliance with both parties is necessary to get the buy-in for any type of intervention that will occur.
• Assess the level of impairment. Are these outbursts severe only at home? In the school setting? With other people such as coaches or health care providers?
• Review the broadband screening instruments from multiple settings to make sure that this is a primary disruptive behavior disorder and not something else, particularly ADHD or a mood disorder, which will need to be managed differently.
• Determine if the aggressive behavior is impulsive/reactive or if it is planned/predatory. Is there remorse afterward (about the action, as opposed to remorse about being caught)? Lack of remorse could be an indicator of callous-unemotional traits, which have a worse prognosis.
Pearl: When asking about aggressive outbursts, make sure to concentrate not just on the outburst, but on the behavior and mood between outbursts. If the mood between outbursts is chronically irritable or sad, this might indicate a mood disorder rather than a primary disruptive behavior disorder.
Treatment. Treatment for aggressive behavior really calls for an "all hands on deck" family-based intervention. Parenting interventions will work best when the parents themselves are as healthy as they can be. Working with them to ensure that aggressive behavior, substance abuse, or anxiety is adequately treated through referral is an important step.
Next, the parenting interventions should involve those best informed by evidence-based practice, which typically include components of reducing the cycle of reinforcing aggressive behavior, noticing and rewarding prosocial behavior, and ceasing corporal punishment and replacing it with predictable, logical consequences for aggressive behavior. There are several excellent programs that therapists can use with parents, and referring to a therapist working with an evidence-based treatment program makes sense. There is a table listing parent management training packages that can be found in the American Academy of Child and Adolescent Psychiatry (AACAP) Practice Parameters for ODD (J. Am. Acad. Child. Adolesc. Psychiatry 2007;46:126-41).
Wellness interventions such as ensuring hydration and adequate caloric intake can make a difference in the management of aggression. It’s harder to maintain control when you are concentrating on the grumbling of your stomach. Further, using exercise and sports as an intervention allows children to channel some of their negative aggressive impulses into positive, prosocial activities.
Pharmacotherapy is not indicated for ODD or CD, except to target co-occurring symptoms. For example, treatment of ADHD or anxiety can quite successfully reduce impulsive or reactive aggression, and can make it easier to treat the ODD or CD through parent management techniques. In very severe cases of aggression, treatment with other agents such as mood stabilizers or antipsychotics might be indicated, but this would likely be implemented only in consultation with a child and adolescent psychiatrist.
Finally, there is little to no evidence for a mock incarceration or boot camp approach with children who exhibit oppositional behavior. In fact, it’s very possible that these kinds of programs can make the behaviors worse (J. Am. Acad. Child Adolesc. Psychiatry 1999;38:1320-1; "Aggression and Antisocial Behavior in Children and Adolescents: Research and Treatment" [New York: The Guilford Press, 2002]).
When to consult? Uncomplicated aggressive behavior can be managed by the primary care team with consultation from a therapist using evidence-based approaches. If there is poor treatment response, or if the aggression is severe enough to cause serious physical injury, or if there is concern for a cycling mood disorder (such as bipolar disorder – a topic for a later column), then consultation with a child psychiatrist is likely appropriate.
Dr. Althoff is an associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Dr. Althoff has received grants/research support from the National Institute of Mental Health, the National Institute of General Medical Sciences, and the Klingenstein Third Generation Foundation, and honoraria from the Oakstone General Publishing for CME presentations.
Introduction
Aggressive behavior is one of the most common child psychiatric symptoms for which parents seek help. The difficulty with managing aggressive behavior is determining whether it is out of the ordinary from typical child development and then assessing the causes of the behavior before tackling the tough job of intervening. The following case is typical of what might present to the pediatrics office and provides a few ideas for the assessment and management of aggressive behavior.
Case summary
Dakota is a 6-year-old boy who presents for a well-child check with his father, Joe. Dakota is just finishing his kindergarten year, and the teachers have expressed concerns about his behavior in the classroom and on the playground. They note that he is often irritable and touchy, and that he will frequently have aggressive outbursts, particularly when asked to do something that he doesn’t like. He will often interrupt other children’s games, and will force children to play by his rules with a threat of, or occasional use of, hitting. In the classroom, he has been removed multiple times to the principal’s office, where he will go only with marked reluctance. He has been noted by his teachers to have difficulty attending to the classroom instructions, and frequently removes himself during circle time. They allow him to do this to avoid a power struggle. Similarly, Joe notes that the entire family is "walking on eggshells" because they never know what might set him off. They’ve tried "everything," including time out, sticker charts, and spanking, but with little effect. Joe says that he was "just like Dakota" when he was a child, and that he was "straightened out" in the Army. He wonders if some kind of boot camp or "scared straight" program would help Dakota learn his lesson.
Discussion
Diagnosis. Irritability and aggression are common manifestations of multiple child psychiatric conditions. While it’s easy to jump to the conclusion that the patient has oppositional-defiant disorder (ODD) or conduct disorder (CD) and move straight to treatment, care must be taken to evaluate common causes and co-occurring disorders that might change the treatment plan.
The differential diagnosis includes a primary mood disorder like depression, other disruptive behavior disorders such as attention-deficit/hyperactivity disorder (ADHD), a primary anxiety disorder, posttraumatic stress disorder, a learning or language disorder, and/or intellectual disability. One also must determine whether the aggression exhibited is greater than that shown by other boys his age. For this reason, the use of a scale that has normative values by age and sex makes sense. Having a standardized instrument filled out by parents and by the teachers also will help give an indication of how he is performing in multiple settings. Using a broad-based instrument that also covers mood, anxiety, and attention problems can be a quick and useful way to examine what type of co-occurring symptoms are present.
Aggression, while a heritable trait, also has a significant component from the environment. It is important to see how much of the aggression is being "caught, not taught" in the family setting. Querying as to the general level of negative, coercive parenting can be performed quickly by asking for a description of how the last outburst was managed – what the precipitant, the course, and the outcome were. Frequently, with ODD in particular, you will find a cycle of escalating threats and illogical consequences that serve to reinforce, rather than to reduce, aggressive and oppositional behavior. Practically, while the busy pediatrician may be able to manage some of this screening in a well-child check, it is likely that a separate appointment will be needed to go over the results of the screening instruments and to more fully assess the parenting environment.
At the scheduled visit designed to specifically assess the aggression:
• Make sure that both the parents and the child see this as a family-based problem. A treatment alliance with both parties is necessary to get the buy-in for any type of intervention that will occur.
• Assess the level of impairment. Are these outbursts severe only at home? In the school setting? With other people such as coaches or health care providers?
• Review the broadband screening instruments from multiple settings to make sure that this is a primary disruptive behavior disorder and not something else, particularly ADHD or a mood disorder, which will need to be managed differently.
• Determine if the aggressive behavior is impulsive/reactive or if it is planned/predatory. Is there remorse afterward (about the action, as opposed to remorse about being caught)? Lack of remorse could be an indicator of callous-unemotional traits, which have a worse prognosis.
Pearl: When asking about aggressive outbursts, make sure to concentrate not just on the outburst, but on the behavior and mood between outbursts. If the mood between outbursts is chronically irritable or sad, this might indicate a mood disorder rather than a primary disruptive behavior disorder.
Treatment. Treatment for aggressive behavior really calls for an "all hands on deck" family-based intervention. Parenting interventions will work best when the parents themselves are as healthy as they can be. Working with them to ensure that aggressive behavior, substance abuse, or anxiety is adequately treated through referral is an important step.
Next, the parenting interventions should involve those best informed by evidence-based practice, which typically include components of reducing the cycle of reinforcing aggressive behavior, noticing and rewarding prosocial behavior, and ceasing corporal punishment and replacing it with predictable, logical consequences for aggressive behavior. There are several excellent programs that therapists can use with parents, and referring to a therapist working with an evidence-based treatment program makes sense. There is a table listing parent management training packages that can be found in the American Academy of Child and Adolescent Psychiatry (AACAP) Practice Parameters for ODD (J. Am. Acad. Child. Adolesc. Psychiatry 2007;46:126-41).
Wellness interventions such as ensuring hydration and adequate caloric intake can make a difference in the management of aggression. It’s harder to maintain control when you are concentrating on the grumbling of your stomach. Further, using exercise and sports as an intervention allows children to channel some of their negative aggressive impulses into positive, prosocial activities.
Pharmacotherapy is not indicated for ODD or CD, except to target co-occurring symptoms. For example, treatment of ADHD or anxiety can quite successfully reduce impulsive or reactive aggression, and can make it easier to treat the ODD or CD through parent management techniques. In very severe cases of aggression, treatment with other agents such as mood stabilizers or antipsychotics might be indicated, but this would likely be implemented only in consultation with a child and adolescent psychiatrist.
Finally, there is little to no evidence for a mock incarceration or boot camp approach with children who exhibit oppositional behavior. In fact, it’s very possible that these kinds of programs can make the behaviors worse (J. Am. Acad. Child Adolesc. Psychiatry 1999;38:1320-1; "Aggression and Antisocial Behavior in Children and Adolescents: Research and Treatment" [New York: The Guilford Press, 2002]).
When to consult? Uncomplicated aggressive behavior can be managed by the primary care team with consultation from a therapist using evidence-based approaches. If there is poor treatment response, or if the aggression is severe enough to cause serious physical injury, or if there is concern for a cycling mood disorder (such as bipolar disorder – a topic for a later column), then consultation with a child psychiatrist is likely appropriate.
Dr. Althoff is an associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Dr. Althoff has received grants/research support from the National Institute of Mental Health, the National Institute of General Medical Sciences, and the Klingenstein Third Generation Foundation, and honoraria from the Oakstone General Publishing for CME presentations.
Introduction
Aggressive behavior is one of the most common child psychiatric symptoms for which parents seek help. The difficulty with managing aggressive behavior is determining whether it is out of the ordinary from typical child development and then assessing the causes of the behavior before tackling the tough job of intervening. The following case is typical of what might present to the pediatrics office and provides a few ideas for the assessment and management of aggressive behavior.
Case summary
Dakota is a 6-year-old boy who presents for a well-child check with his father, Joe. Dakota is just finishing his kindergarten year, and the teachers have expressed concerns about his behavior in the classroom and on the playground. They note that he is often irritable and touchy, and that he will frequently have aggressive outbursts, particularly when asked to do something that he doesn’t like. He will often interrupt other children’s games, and will force children to play by his rules with a threat of, or occasional use of, hitting. In the classroom, he has been removed multiple times to the principal’s office, where he will go only with marked reluctance. He has been noted by his teachers to have difficulty attending to the classroom instructions, and frequently removes himself during circle time. They allow him to do this to avoid a power struggle. Similarly, Joe notes that the entire family is "walking on eggshells" because they never know what might set him off. They’ve tried "everything," including time out, sticker charts, and spanking, but with little effect. Joe says that he was "just like Dakota" when he was a child, and that he was "straightened out" in the Army. He wonders if some kind of boot camp or "scared straight" program would help Dakota learn his lesson.
Discussion
Diagnosis. Irritability and aggression are common manifestations of multiple child psychiatric conditions. While it’s easy to jump to the conclusion that the patient has oppositional-defiant disorder (ODD) or conduct disorder (CD) and move straight to treatment, care must be taken to evaluate common causes and co-occurring disorders that might change the treatment plan.
The differential diagnosis includes a primary mood disorder like depression, other disruptive behavior disorders such as attention-deficit/hyperactivity disorder (ADHD), a primary anxiety disorder, posttraumatic stress disorder, a learning or language disorder, and/or intellectual disability. One also must determine whether the aggression exhibited is greater than that shown by other boys his age. For this reason, the use of a scale that has normative values by age and sex makes sense. Having a standardized instrument filled out by parents and by the teachers also will help give an indication of how he is performing in multiple settings. Using a broad-based instrument that also covers mood, anxiety, and attention problems can be a quick and useful way to examine what type of co-occurring symptoms are present.
Aggression, while a heritable trait, also has a significant component from the environment. It is important to see how much of the aggression is being "caught, not taught" in the family setting. Querying as to the general level of negative, coercive parenting can be performed quickly by asking for a description of how the last outburst was managed – what the precipitant, the course, and the outcome were. Frequently, with ODD in particular, you will find a cycle of escalating threats and illogical consequences that serve to reinforce, rather than to reduce, aggressive and oppositional behavior. Practically, while the busy pediatrician may be able to manage some of this screening in a well-child check, it is likely that a separate appointment will be needed to go over the results of the screening instruments and to more fully assess the parenting environment.
At the scheduled visit designed to specifically assess the aggression:
• Make sure that both the parents and the child see this as a family-based problem. A treatment alliance with both parties is necessary to get the buy-in for any type of intervention that will occur.
• Assess the level of impairment. Are these outbursts severe only at home? In the school setting? With other people such as coaches or health care providers?
• Review the broadband screening instruments from multiple settings to make sure that this is a primary disruptive behavior disorder and not something else, particularly ADHD or a mood disorder, which will need to be managed differently.
• Determine if the aggressive behavior is impulsive/reactive or if it is planned/predatory. Is there remorse afterward (about the action, as opposed to remorse about being caught)? Lack of remorse could be an indicator of callous-unemotional traits, which have a worse prognosis.
Pearl: When asking about aggressive outbursts, make sure to concentrate not just on the outburst, but on the behavior and mood between outbursts. If the mood between outbursts is chronically irritable or sad, this might indicate a mood disorder rather than a primary disruptive behavior disorder.
Treatment. Treatment for aggressive behavior really calls for an "all hands on deck" family-based intervention. Parenting interventions will work best when the parents themselves are as healthy as they can be. Working with them to ensure that aggressive behavior, substance abuse, or anxiety is adequately treated through referral is an important step.
Next, the parenting interventions should involve those best informed by evidence-based practice, which typically include components of reducing the cycle of reinforcing aggressive behavior, noticing and rewarding prosocial behavior, and ceasing corporal punishment and replacing it with predictable, logical consequences for aggressive behavior. There are several excellent programs that therapists can use with parents, and referring to a therapist working with an evidence-based treatment program makes sense. There is a table listing parent management training packages that can be found in the American Academy of Child and Adolescent Psychiatry (AACAP) Practice Parameters for ODD (J. Am. Acad. Child. Adolesc. Psychiatry 2007;46:126-41).
Wellness interventions such as ensuring hydration and adequate caloric intake can make a difference in the management of aggression. It’s harder to maintain control when you are concentrating on the grumbling of your stomach. Further, using exercise and sports as an intervention allows children to channel some of their negative aggressive impulses into positive, prosocial activities.
Pharmacotherapy is not indicated for ODD or CD, except to target co-occurring symptoms. For example, treatment of ADHD or anxiety can quite successfully reduce impulsive or reactive aggression, and can make it easier to treat the ODD or CD through parent management techniques. In very severe cases of aggression, treatment with other agents such as mood stabilizers or antipsychotics might be indicated, but this would likely be implemented only in consultation with a child and adolescent psychiatrist.
Finally, there is little to no evidence for a mock incarceration or boot camp approach with children who exhibit oppositional behavior. In fact, it’s very possible that these kinds of programs can make the behaviors worse (J. Am. Acad. Child Adolesc. Psychiatry 1999;38:1320-1; "Aggression and Antisocial Behavior in Children and Adolescents: Research and Treatment" [New York: The Guilford Press, 2002]).
When to consult? Uncomplicated aggressive behavior can be managed by the primary care team with consultation from a therapist using evidence-based approaches. If there is poor treatment response, or if the aggression is severe enough to cause serious physical injury, or if there is concern for a cycling mood disorder (such as bipolar disorder – a topic for a later column), then consultation with a child psychiatrist is likely appropriate.
Dr. Althoff is an associate professor of psychiatry, psychology, and pediatrics at the University of Vermont, Burlington. He is director of the division of behavioral genetics and conducts research on the development of self-regulation in children. Dr. Althoff has received grants/research support from the National Institute of Mental Health, the National Institute of General Medical Sciences, and the Klingenstein Third Generation Foundation, and honoraria from the Oakstone General Publishing for CME presentations.

Group exploits autophagy to fight myeloma
Credit: Sarah Pfau
Researchers have devised a strategy that leverages autophagy to work against multiple myeloma (MM).
Their method involves targeting the p62 protein, which plays a role in disposing of unwanted cellular proteins during autophagy.
The team used anticancer agents to induce autophagy in MM cells and found that simultaneously blocking p62 expression, either pharmacologically or with shRNA, could prompt apoptosis in the cells, both in vitro and in vivo.
Steven Grant, MD, of Virginia Commonwealth University’s Massey Cancer Center, and his colleagues described this work in Molecular and Cellular Biology.
“Therapies that are designed to block the early stages of autophagy do not offer the possibility of exploiting its potentially lethal effects,” Dr Grant said. “Our strategy turns autophagy from a protective process into a toxic one, and these results suggest it could increase the effectiveness of a variety of cancer therapies that induce autophagy.”
The researchers conducted several experiments in MM cell lines and mouse models of the disease. They used an anticancer agent—obatoclax or bortezomib—to induce autophagy and a cyclin-dependent kinase (CDK) inhibitor—flavopiridol or dinaciclib—or shRNA to target p62.
And they discovered that blocking p62 disrupted autophagy and killed far more MM cells than administering anticancer agents alone.
Critical to the success of this strategy is Bik, a protein that plays a significant role in governing cell death and survival. With anticancer treatment, Bik accumulates in MM cells until it triggers apoptosis.
Normally, the MM cells would initiate autophagy to survive, and p62 would rid the cells of Bik by loading the proteins into autophagosomes for disposal.
However, the researchers found that blocking p62 production results in an inefficient form of autophagy, and the accumulation of Bik eventually causes the MM cells to undergo apoptosis.
This research builds upon more than a decade of work by Dr Grant’s lab, in which the team investigated novel treatment strategies and combination therapies that selectively kill MM cells and other blood cancer cells.
“We are now working to identify additional CDK inhibitors that can be used to disrupt autophagy,” Dr Grant said. “The ultimate goal will be to translate these findings into a clinical trial to test the therapy in patients with various blood cancers.”
The technology in his study has been made available for licensing through the Virginia Commonwealth University Office of Research. ![]()
Credit: Sarah Pfau
Researchers have devised a strategy that leverages autophagy to work against multiple myeloma (MM).
Their method involves targeting the p62 protein, which plays a role in disposing of unwanted cellular proteins during autophagy.
The team used anticancer agents to induce autophagy in MM cells and found that simultaneously blocking p62 expression, either pharmacologically or with shRNA, could prompt apoptosis in the cells, both in vitro and in vivo.
Steven Grant, MD, of Virginia Commonwealth University’s Massey Cancer Center, and his colleagues described this work in Molecular and Cellular Biology.
“Therapies that are designed to block the early stages of autophagy do not offer the possibility of exploiting its potentially lethal effects,” Dr Grant said. “Our strategy turns autophagy from a protective process into a toxic one, and these results suggest it could increase the effectiveness of a variety of cancer therapies that induce autophagy.”
The researchers conducted several experiments in MM cell lines and mouse models of the disease. They used an anticancer agent—obatoclax or bortezomib—to induce autophagy and a cyclin-dependent kinase (CDK) inhibitor—flavopiridol or dinaciclib—or shRNA to target p62.
And they discovered that blocking p62 disrupted autophagy and killed far more MM cells than administering anticancer agents alone.
Critical to the success of this strategy is Bik, a protein that plays a significant role in governing cell death and survival. With anticancer treatment, Bik accumulates in MM cells until it triggers apoptosis.
Normally, the MM cells would initiate autophagy to survive, and p62 would rid the cells of Bik by loading the proteins into autophagosomes for disposal.
However, the researchers found that blocking p62 production results in an inefficient form of autophagy, and the accumulation of Bik eventually causes the MM cells to undergo apoptosis.
This research builds upon more than a decade of work by Dr Grant’s lab, in which the team investigated novel treatment strategies and combination therapies that selectively kill MM cells and other blood cancer cells.
“We are now working to identify additional CDK inhibitors that can be used to disrupt autophagy,” Dr Grant said. “The ultimate goal will be to translate these findings into a clinical trial to test the therapy in patients with various blood cancers.”
The technology in his study has been made available for licensing through the Virginia Commonwealth University Office of Research. ![]()
Credit: Sarah Pfau
Researchers have devised a strategy that leverages autophagy to work against multiple myeloma (MM).
Their method involves targeting the p62 protein, which plays a role in disposing of unwanted cellular proteins during autophagy.
The team used anticancer agents to induce autophagy in MM cells and found that simultaneously blocking p62 expression, either pharmacologically or with shRNA, could prompt apoptosis in the cells, both in vitro and in vivo.
Steven Grant, MD, of Virginia Commonwealth University’s Massey Cancer Center, and his colleagues described this work in Molecular and Cellular Biology.
“Therapies that are designed to block the early stages of autophagy do not offer the possibility of exploiting its potentially lethal effects,” Dr Grant said. “Our strategy turns autophagy from a protective process into a toxic one, and these results suggest it could increase the effectiveness of a variety of cancer therapies that induce autophagy.”
The researchers conducted several experiments in MM cell lines and mouse models of the disease. They used an anticancer agent—obatoclax or bortezomib—to induce autophagy and a cyclin-dependent kinase (CDK) inhibitor—flavopiridol or dinaciclib—or shRNA to target p62.
And they discovered that blocking p62 disrupted autophagy and killed far more MM cells than administering anticancer agents alone.
Critical to the success of this strategy is Bik, a protein that plays a significant role in governing cell death and survival. With anticancer treatment, Bik accumulates in MM cells until it triggers apoptosis.
Normally, the MM cells would initiate autophagy to survive, and p62 would rid the cells of Bik by loading the proteins into autophagosomes for disposal.
However, the researchers found that blocking p62 production results in an inefficient form of autophagy, and the accumulation of Bik eventually causes the MM cells to undergo apoptosis.
This research builds upon more than a decade of work by Dr Grant’s lab, in which the team investigated novel treatment strategies and combination therapies that selectively kill MM cells and other blood cancer cells.
“We are now working to identify additional CDK inhibitors that can be used to disrupt autophagy,” Dr Grant said. “The ultimate goal will be to translate these findings into a clinical trial to test the therapy in patients with various blood cancers.”
The technology in his study has been made available for licensing through the Virginia Commonwealth University Office of Research. ![]()
Program improves depression treatment in cancer

Credit: NIH
Results of a large study suggest major depression is common—but largely untreated—among cancer patients in Scotland.
And 2 additional studies of Scottish patients showed that a program specifically designed for individuals with cancer can treat depression and improve quality of life more effectively than current methods of care.
These studies appear in The Lancet, The Lancet Oncology, and The Lancet Psychiatry.
In The Lancet Psychiatry, researchers recounted their analysis of data from 21,151 patients treated at cancer clinics in Scotland. The team found that major depression was substantially more common in cancer patients than in the general population.
Major depression was most common in patients with lung cancer (13%) and lowest in those with genitourinary cancer (6%). Moreover, nearly three-quarters (73%) of depressed cancer patients were not receiving treatment.
To address the problem of inadequate treatment, researchers initiated the SMaRT Oncology-2 trial. They reported the results in The Lancet.
The team evaluated a new treatment program called “Depression Care for People with Cancer” (DCPC). DCPC is delivered by specially trained cancer nurses and psychiatrists, working in collaboration with the patient’s cancer team and general practitioner, and is given as part of cancer care. It is a systematic treatment program that includes both antidepressants and psychological therapy.
The trial included 500 adults with major depression and a cancer with a good prognosis (predicted survival of more than 12 months).
Patients were randomized to receive either DCPC or “usual care,” which was provided by a patient’s general practitioner and might have included prescribing antidepressants or referring the patient to mental health services for assessment or psychological treatment.
Results showed that DCPC was more effective than usual care in reducing depression. At 6 months, 62% of patients who received DCPC responded to treatment (experiencing at least a 50% reduction in the severity of their depression), compared with 17% of those who received the usual care (P<0.0001). This benefit was sustained at 12 months.
In addition, DCPC improved anxiety, pain, fatigue, functioning, and overall quality of life (all P<0.05). The researchers also noted that the cost of providing DCPC was modest (£613 per patient).
“The huge benefit that DCPC delivers for patients with cancer and depression shows what we can achieve for patients if we take as much care with the treatment of their depression as we do with the treatment of their cancer,” said study author Michael Sharpe, MD, of the University of Oxford in the UK.
To see if patients with a poor-prognosis cancer could also benefit from DCPC, researchers initiated the SMaRT Oncology-3 trial. They reported the results in The Lancet Oncology.
The team tested a version of DCPC adapted for cancer patients with a poor prognosis. The trial included 142 patients with lung cancer and major depression.
Patients who received the modified version of DCPC had a significantly greater improvement in depression than those who received the usual care during 32 weeks of follow-up (P=0.0003). DCPC also improved patients’ anxiety (P=0.046), functioning (P=0.0019), and quality of life (P=0.018).
“Patients with lung cancer often have a poor prognosis,” said study author Jane Walker, MBChB, PhD, of the University of Oxford and Sobell House Hospice in Oxford, UK.
“If they also have major depression, that can blight the time they have left to live. This trial shows that we can effectively treat depression in patients with poor-prognosis cancers, like lung cancer, and really improve patients’ lives.” ![]()
Credit: NIH
Results of a large study suggest major depression is common—but largely untreated—among cancer patients in Scotland.
And 2 additional studies of Scottish patients showed that a program specifically designed for individuals with cancer can treat depression and improve quality of life more effectively than current methods of care.
These studies appear in The Lancet, The Lancet Oncology, and The Lancet Psychiatry.
In The Lancet Psychiatry, researchers recounted their analysis of data from 21,151 patients treated at cancer clinics in Scotland. The team found that major depression was substantially more common in cancer patients than in the general population.
Major depression was most common in patients with lung cancer (13%) and lowest in those with genitourinary cancer (6%). Moreover, nearly three-quarters (73%) of depressed cancer patients were not receiving treatment.
To address the problem of inadequate treatment, researchers initiated the SMaRT Oncology-2 trial. They reported the results in The Lancet.
The team evaluated a new treatment program called “Depression Care for People with Cancer” (DCPC). DCPC is delivered by specially trained cancer nurses and psychiatrists, working in collaboration with the patient’s cancer team and general practitioner, and is given as part of cancer care. It is a systematic treatment program that includes both antidepressants and psychological therapy.
The trial included 500 adults with major depression and a cancer with a good prognosis (predicted survival of more than 12 months).
Patients were randomized to receive either DCPC or “usual care,” which was provided by a patient’s general practitioner and might have included prescribing antidepressants or referring the patient to mental health services for assessment or psychological treatment.
Results showed that DCPC was more effective than usual care in reducing depression. At 6 months, 62% of patients who received DCPC responded to treatment (experiencing at least a 50% reduction in the severity of their depression), compared with 17% of those who received the usual care (P<0.0001). This benefit was sustained at 12 months.
In addition, DCPC improved anxiety, pain, fatigue, functioning, and overall quality of life (all P<0.05). The researchers also noted that the cost of providing DCPC was modest (£613 per patient).
“The huge benefit that DCPC delivers for patients with cancer and depression shows what we can achieve for patients if we take as much care with the treatment of their depression as we do with the treatment of their cancer,” said study author Michael Sharpe, MD, of the University of Oxford in the UK.
To see if patients with a poor-prognosis cancer could also benefit from DCPC, researchers initiated the SMaRT Oncology-3 trial. They reported the results in The Lancet Oncology.
The team tested a version of DCPC adapted for cancer patients with a poor prognosis. The trial included 142 patients with lung cancer and major depression.
Patients who received the modified version of DCPC had a significantly greater improvement in depression than those who received the usual care during 32 weeks of follow-up (P=0.0003). DCPC also improved patients’ anxiety (P=0.046), functioning (P=0.0019), and quality of life (P=0.018).
“Patients with lung cancer often have a poor prognosis,” said study author Jane Walker, MBChB, PhD, of the University of Oxford and Sobell House Hospice in Oxford, UK.
“If they also have major depression, that can blight the time they have left to live. This trial shows that we can effectively treat depression in patients with poor-prognosis cancers, like lung cancer, and really improve patients’ lives.” ![]()
Credit: NIH
Results of a large study suggest major depression is common—but largely untreated—among cancer patients in Scotland.
And 2 additional studies of Scottish patients showed that a program specifically designed for individuals with cancer can treat depression and improve quality of life more effectively than current methods of care.
These studies appear in The Lancet, The Lancet Oncology, and The Lancet Psychiatry.
In The Lancet Psychiatry, researchers recounted their analysis of data from 21,151 patients treated at cancer clinics in Scotland. The team found that major depression was substantially more common in cancer patients than in the general population.
Major depression was most common in patients with lung cancer (13%) and lowest in those with genitourinary cancer (6%). Moreover, nearly three-quarters (73%) of depressed cancer patients were not receiving treatment.
To address the problem of inadequate treatment, researchers initiated the SMaRT Oncology-2 trial. They reported the results in The Lancet.
The team evaluated a new treatment program called “Depression Care for People with Cancer” (DCPC). DCPC is delivered by specially trained cancer nurses and psychiatrists, working in collaboration with the patient’s cancer team and general practitioner, and is given as part of cancer care. It is a systematic treatment program that includes both antidepressants and psychological therapy.
The trial included 500 adults with major depression and a cancer with a good prognosis (predicted survival of more than 12 months).
Patients were randomized to receive either DCPC or “usual care,” which was provided by a patient’s general practitioner and might have included prescribing antidepressants or referring the patient to mental health services for assessment or psychological treatment.
Results showed that DCPC was more effective than usual care in reducing depression. At 6 months, 62% of patients who received DCPC responded to treatment (experiencing at least a 50% reduction in the severity of their depression), compared with 17% of those who received the usual care (P<0.0001). This benefit was sustained at 12 months.
In addition, DCPC improved anxiety, pain, fatigue, functioning, and overall quality of life (all P<0.05). The researchers also noted that the cost of providing DCPC was modest (£613 per patient).
“The huge benefit that DCPC delivers for patients with cancer and depression shows what we can achieve for patients if we take as much care with the treatment of their depression as we do with the treatment of their cancer,” said study author Michael Sharpe, MD, of the University of Oxford in the UK.
To see if patients with a poor-prognosis cancer could also benefit from DCPC, researchers initiated the SMaRT Oncology-3 trial. They reported the results in The Lancet Oncology.
The team tested a version of DCPC adapted for cancer patients with a poor prognosis. The trial included 142 patients with lung cancer and major depression.
Patients who received the modified version of DCPC had a significantly greater improvement in depression than those who received the usual care during 32 weeks of follow-up (P=0.0003). DCPC also improved patients’ anxiety (P=0.046), functioning (P=0.0019), and quality of life (P=0.018).
“Patients with lung cancer often have a poor prognosis,” said study author Jane Walker, MBChB, PhD, of the University of Oxford and Sobell House Hospice in Oxford, UK.
“If they also have major depression, that can blight the time they have left to live. This trial shows that we can effectively treat depression in patients with poor-prognosis cancers, like lung cancer, and really improve patients’ lives.” ![]()
Drug granted orphan status for PNH in EU
A novel compound has received orphan status in the Europe Union to treat paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening disease that causes severe anemia and confers a high risk of thrombosis.
The compound, AMY-101, works by inhibiting C3, a central component of the complement immune system.
AMY-101 was developed by John Lambris, PhD, of the University of Pennsylvania, and subsequently licensed to Amyndas Pharmaceuticals.
AMY-101’s orphan status provides Amyndas with benefits such as tax incentives, market exclusivity for 10 years, possibilities for additional research funding, and additional guidance from the European Medicines Agency during clinical development.
How AMY-101 works
PNH is caused by the defective expression of regulatory proteins on the surface of blood cells, which leaves them vulnerable to complement attack. This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
The monoclonal antibody eculizumab is often successful in treating PNH, but roughly a third of patients do not respond well to the drug and still require blood transfusions to manage their anemia.
Research has suggested this lack of response is due to fragments of complement C3 proteins on the surface of the patients’ red blood cells, which are eventually attacked by immune cells.
In an attempt to overcome this problem, Dr Lambris and his colleagues developed AMY-101. The drug is designed to inhibit the complement cascade, thereby preventing hemolysis and immune cell recognition.
The researchers have investigated the effects of AMY-101 on self-attack and the resulting hemolysis in human PNH cells and found the drug to be active.
These results have not been published, but the group has published results with a C3 inhibitor known as Cp40, and AMY-101 is based on Cp40.
The researchers reported in Blood that Cp40 and its long-acting form, PEG-Cp40, effectively inhibited hemolysis and efficiently prevented the deposition of C3 fragments on red blood cells from patients with PNH. ![]()
A novel compound has received orphan status in the Europe Union to treat paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening disease that causes severe anemia and confers a high risk of thrombosis.
The compound, AMY-101, works by inhibiting C3, a central component of the complement immune system.
AMY-101 was developed by John Lambris, PhD, of the University of Pennsylvania, and subsequently licensed to Amyndas Pharmaceuticals.
AMY-101’s orphan status provides Amyndas with benefits such as tax incentives, market exclusivity for 10 years, possibilities for additional research funding, and additional guidance from the European Medicines Agency during clinical development.
How AMY-101 works
PNH is caused by the defective expression of regulatory proteins on the surface of blood cells, which leaves them vulnerable to complement attack. This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
The monoclonal antibody eculizumab is often successful in treating PNH, but roughly a third of patients do not respond well to the drug and still require blood transfusions to manage their anemia.
Research has suggested this lack of response is due to fragments of complement C3 proteins on the surface of the patients’ red blood cells, which are eventually attacked by immune cells.
In an attempt to overcome this problem, Dr Lambris and his colleagues developed AMY-101. The drug is designed to inhibit the complement cascade, thereby preventing hemolysis and immune cell recognition.
The researchers have investigated the effects of AMY-101 on self-attack and the resulting hemolysis in human PNH cells and found the drug to be active.
These results have not been published, but the group has published results with a C3 inhibitor known as Cp40, and AMY-101 is based on Cp40.
The researchers reported in Blood that Cp40 and its long-acting form, PEG-Cp40, effectively inhibited hemolysis and efficiently prevented the deposition of C3 fragments on red blood cells from patients with PNH. ![]()
A novel compound has received orphan status in the Europe Union to treat paroxysmal nocturnal hemoglobinuria (PNH), a life-threatening disease that causes severe anemia and confers a high risk of thrombosis.
The compound, AMY-101, works by inhibiting C3, a central component of the complement immune system.
AMY-101 was developed by John Lambris, PhD, of the University of Pennsylvania, and subsequently licensed to Amyndas Pharmaceuticals.
AMY-101’s orphan status provides Amyndas with benefits such as tax incentives, market exclusivity for 10 years, possibilities for additional research funding, and additional guidance from the European Medicines Agency during clinical development.
How AMY-101 works
PNH is caused by the defective expression of regulatory proteins on the surface of blood cells, which leaves them vulnerable to complement attack. This can lead to hemolysis, which results in severe anemia and contributes to a high risk of thrombosis.
The monoclonal antibody eculizumab is often successful in treating PNH, but roughly a third of patients do not respond well to the drug and still require blood transfusions to manage their anemia.
Research has suggested this lack of response is due to fragments of complement C3 proteins on the surface of the patients’ red blood cells, which are eventually attacked by immune cells.
In an attempt to overcome this problem, Dr Lambris and his colleagues developed AMY-101. The drug is designed to inhibit the complement cascade, thereby preventing hemolysis and immune cell recognition.
The researchers have investigated the effects of AMY-101 on self-attack and the resulting hemolysis in human PNH cells and found the drug to be active.
These results have not been published, but the group has published results with a C3 inhibitor known as Cp40, and AMY-101 is based on Cp40.
The researchers reported in Blood that Cp40 and its long-acting form, PEG-Cp40, effectively inhibited hemolysis and efficiently prevented the deposition of C3 fragments on red blood cells from patients with PNH. ![]()
Genes aid classification of polycythemia vera

Credit: AFIP
New research has revealed a molecular method for classifying patients with polycythemia vera (PV).
Investigators identified 102 genes that can be used to distinguish patients with aggressive PV from those with indolent disease.
The 2 patient groups exhibited significant differences with regard to leukemic transformation, disease duration, survival, hemoglobin level, thrombosis, splenomegaly, splenectomy, and chemotherapy exposure.
Jerry L. Spivak, MD, of the Johns Hopkins University School of Medicine in Baltimore, and his colleagues conducted this research and recounted the results in NEJM.
The researchers analyzed gene expression in CD34+ cells from 19 patients with PV and compared the results to healthy control subjects of the same sex.
Males with PV had roughly twice as many differentially regulated genes as females with PV—571 and 253, respectively.
The investigators subtracted the genes with sex-specific expression and were left with 102 genes that were differentially regulated (68 upregulated and 34 downregulated) between PV patients and controls.
And the team found they could use these genes to separate patients with indolent PV from those with aggressive disease, as the expression of the genes differed markedly between the 2 groups.
The 2 groups also differed significantly with regard to a number of clinical characteristics. The median disease duration was 14 years for patients with aggressive disease and 6 years for those with indolent disease (P=0.05).
The number of patients who transformed to acute leukemia was 4 and 1, respectively (P=0.04). And the number of patients who were still alive at the time of analysis was 1 and 11, respectively (P=0.001).
There were also significant differences with regard to hemoglobin level (P=0.007), the incidence of thromboembolic events (P=0.04), the frequency of palpable splenomegaly (P=0.03), the rate of splenectomy (P=0.007), and chemotherapy exposure (P=0.03).
However, there were no significant differences between the 2 groups with regard to age, JAK2 V617F neutrophil allele burden, white cell count, or platelet count. ![]()
Credit: AFIP
New research has revealed a molecular method for classifying patients with polycythemia vera (PV).
Investigators identified 102 genes that can be used to distinguish patients with aggressive PV from those with indolent disease.
The 2 patient groups exhibited significant differences with regard to leukemic transformation, disease duration, survival, hemoglobin level, thrombosis, splenomegaly, splenectomy, and chemotherapy exposure.
Jerry L. Spivak, MD, of the Johns Hopkins University School of Medicine in Baltimore, and his colleagues conducted this research and recounted the results in NEJM.
The researchers analyzed gene expression in CD34+ cells from 19 patients with PV and compared the results to healthy control subjects of the same sex.
Males with PV had roughly twice as many differentially regulated genes as females with PV—571 and 253, respectively.
The investigators subtracted the genes with sex-specific expression and were left with 102 genes that were differentially regulated (68 upregulated and 34 downregulated) between PV patients and controls.
And the team found they could use these genes to separate patients with indolent PV from those with aggressive disease, as the expression of the genes differed markedly between the 2 groups.
The 2 groups also differed significantly with regard to a number of clinical characteristics. The median disease duration was 14 years for patients with aggressive disease and 6 years for those with indolent disease (P=0.05).
The number of patients who transformed to acute leukemia was 4 and 1, respectively (P=0.04). And the number of patients who were still alive at the time of analysis was 1 and 11, respectively (P=0.001).
There were also significant differences with regard to hemoglobin level (P=0.007), the incidence of thromboembolic events (P=0.04), the frequency of palpable splenomegaly (P=0.03), the rate of splenectomy (P=0.007), and chemotherapy exposure (P=0.03).
However, there were no significant differences between the 2 groups with regard to age, JAK2 V617F neutrophil allele burden, white cell count, or platelet count. ![]()
Credit: AFIP
New research has revealed a molecular method for classifying patients with polycythemia vera (PV).
Investigators identified 102 genes that can be used to distinguish patients with aggressive PV from those with indolent disease.
The 2 patient groups exhibited significant differences with regard to leukemic transformation, disease duration, survival, hemoglobin level, thrombosis, splenomegaly, splenectomy, and chemotherapy exposure.
Jerry L. Spivak, MD, of the Johns Hopkins University School of Medicine in Baltimore, and his colleagues conducted this research and recounted the results in NEJM.
The researchers analyzed gene expression in CD34+ cells from 19 patients with PV and compared the results to healthy control subjects of the same sex.
Males with PV had roughly twice as many differentially regulated genes as females with PV—571 and 253, respectively.
The investigators subtracted the genes with sex-specific expression and were left with 102 genes that were differentially regulated (68 upregulated and 34 downregulated) between PV patients and controls.
And the team found they could use these genes to separate patients with indolent PV from those with aggressive disease, as the expression of the genes differed markedly between the 2 groups.
The 2 groups also differed significantly with regard to a number of clinical characteristics. The median disease duration was 14 years for patients with aggressive disease and 6 years for those with indolent disease (P=0.05).
The number of patients who transformed to acute leukemia was 4 and 1, respectively (P=0.04). And the number of patients who were still alive at the time of analysis was 1 and 11, respectively (P=0.001).
There were also significant differences with regard to hemoglobin level (P=0.007), the incidence of thromboembolic events (P=0.04), the frequency of palpable splenomegaly (P=0.03), the rate of splenectomy (P=0.007), and chemotherapy exposure (P=0.03).
However, there were no significant differences between the 2 groups with regard to age, JAK2 V617F neutrophil allele burden, white cell count, or platelet count. ![]()
Was fetus’ wrist injured during cesarean delivery?
Was fetus’ wrist injured during cesarean delivery?
At 34 weeks’ gestation, a 39-year-old woman went to the hospital in preterm labor. Her history included a prior cesarean delivery. Ultrasonography (US) showed that the fetus was in a double-footling breech position. The ObGyn decided to perform a cesarean delivery when the fetal heart-rate monitor indicated distress.
After making a midline incision through the earlier scar, the ObGyn created a low transverse uterine incision with a scalpel. The mother’s uterus was thick because labor had not progressed. When the ObGyn was unable to deliver the baby through the low transverse incision, she performed a T-extension of the incision using bandage scissors while placing her free hand inside the uterus to shield the fetus from injury. After extensive manipulation, the baby was delivered and immediately handed to a neonatologist. After surgery, the neonatologist told the mother that the baby had sustained two lacerations to the ulnar side of the right wrist. The newborn was airlifted to another hospital for treatment of sepsis. There, an orthopedic hand surgeon examined the child and determined that the lacerations were superficial and only required sutures. The orthopedist saw the infant a month later and believed there was no significant wrist injury.
When the child began preschool, she started to experience cold intolerance and difficulty writing with her right hand. The child was referred to a pediatric neurologist, who found no nerve damage and ordered occupational therapy.
The original orthopedic surgeon examined the child when she was 7 years old and determined that the flexor carpi ulnaris tendon had been completely severed with a partial injury to the ulnar nerve. He recommended a return visit at age 14 for full assessment of the wrist injury.
PARENTS’ CLAIM The ObGyn did not properly shield the fetus when performing the T-extension incision during cesarean delivery. The child’s weakness will increase with age, ruling out some occupations.
PHYSICIAN’S DEFENSE The ObGyn was not negligent; she had provided adequate protection of the fetus during both incisions.
VERDICT An Illinois defense verdict was returned.
Woman dies after tubal ligation
After a 42-year-old woman underwent tubal ligation, her surgeon was concerned about a possible bowel perforation and admitted her to the hospital. The next morning, a computed tomography (CT) scan of the abdomen did not reveal bowel injury.
That afternoon, when the patient reported shortness of breath, the surgeon called the hospitalist with concern for pulmonary embolism (PE). The hospitalist immediately ordered a CT scan of the chest, initiated PE protocol, and wrote “r/o PE” on the chart. A radiologist reminded the hospitalist of the earlier CT scan with concern for kidney damage from another dye study. The hospitalist cancelled the CT scan and PE protocol. After waiting 17 hours to run any further tests, a CT scan revealed massive bilateral PE. The patient was transferred to the ICU, but died the next day.
PATIENT’S CLAIM The 17-hour delay was negligent.
PHYSICIAN’S DEFENSE There was no negligence. The patient died of septic shock, not PE.
VERDICT A $4 million Virginia verdict was returned.
Child born without hand and forearm
During prenatal care, a mother underwent US at 20 and 36 weeks; both studies were reported as normal. The child was born missing his left hand and part of his left forearm due to a congenital amputation. The child will require prosthetics for life.
PATIENT’S CLAIM The condition should have been seen during prenatal US; an abortion was still an option at 20 weeks.
DEFENDANTS’ DEFENSE US was properly performed and evaluated. It can be difficult to differentiate the right from left extremities.
VERDICT A California defense verdict was returned.
After starting Yasmin, woman has stroke with permanent paralysis: $16.5M total award
When a 37-year-old woman reported irregular menstruation, her ObGyn prescribed drospirenone/ethinyl estradiol (Yasmin; Bayer). Thirteen days after starting the drug, the patient had a stroke. She is paralyzed on her left side, has limited ability to speak, cannot use her left arm and leg, and requires 24-hour care.
PATIENT’S CLAIM The ObGyn should have recognized that Yasmin was not appropriate for this patient because of the drug’s clotting risks. The patient’s risk factors included her age (over 35), borderline hypertension, overweight, history of smoking, and high cholesterol. The ObGyn should have offered safer alternatives, such as a progesterone-only pill. The US Food and Drug Administration (FDA) issued a safety warning that all drospirenone-containing drugs may be associated with a higher risk of venous thrombosis during the first 6 months of use.
DEFENDANTS’ DEFENSE According to Bayer, Yasmin is safe, and remains on the market. It was an appropriate drug to treat her irregular bleeding.
VERDICT Claims against the medical center that referred the patient to the ObGyn were settled for $2.5 million before trial. A $14 million Illinois verdict was returned against the ObGyn, for a total award of $16.5 million.
Who is at fault when pelvic mesh erodes?
In January 2011, an ObGyn implanted the Gynecare TVT Obturator System (TVT‑O; Ethicon) during a midurethral sling procedure to treat stress urinary incontinence (SUI) in a woman in her 60s. Shortly thereafter, the ObGyn left practice because of early-onset Alzheimer’s disease, and the patient’s care was taken over by a gynecologist.
At the 2-month postoperative visit, the gynecologist found that the mesh had eroded into the patient’s vagina. The gynecologist simply cut the mesh with a scissor, charted that a small erosion was present, and prescribed estrogen cream.
The patient continued to report pain, discomfort, pressure, difficulty voiding urine, continued incontinence, vaginal discharge, scarring, infection, odor, and bleeding.
PATIENT’S CLAIM The polypropylene mesh used during the midurethral sling procedure has been shown to be incompatible with human tissue. It promotes an immune response, which stimulates degradation of the pelvic tissue and can contribute to the development of severe adverse reactions to the mesh. Ethicon negligently designed, manufactured, marketed, labeled, and packaged the pelvic mesh products.
DEFENDANTS’ DEFENSE Proper warnings were provided about the health risks associated with polypropylene mesh products. The medical device was not properly sized.
VERDICT A Texas jury rejected the patient’s claims that Ethicon did not provide proper warnings about the sling’s health risks and declined to award punitive damages.
However, the jury decided that the mesh implant was defectively designed, and returned a $1.2 million verdict against Ethicon.
Was suspected bowel injury treated properly?
A 40-year-old woman was referred to an ObGyn after reporting abnormal uterine bleeding to her primary care physician. The patient had very light menses every few weeks. The ObGyn performed an ablation procedure, without relief. A month later, the ObGyn performed robot-assisted laparoscopic hysterectomy. The next day, the patient reported abdominal pain. Suspecting a bowel injury, the ObGyn ordered a CT scan; the bowel appeared normal, so the ObGyn referred the patient to a surgeon. During exploratory laparotomy, the surgeon found and repaired a bowel injury. The patient developed significant complications from a necrotizing infection that included respiratory distress and ongoing wound care.
PATIENT’S CLAIM Conservative treatment should have been offered before surgery. The ObGyn should have waited longer after the ablation procedure before doing the hysterectomy. The ObGyn should have checked for a possible bowel injury before closing the hysterectomy.
PHYSICIAN’S DEFENSE The bowel injury is a known complication of the procedure and was recognized and repaired in a timely manner.
VERDICT A Kentucky defense verdict was returned.

Pap smear improperly interpreted: Woman dies from cervical cancer
A 37-year-old woman underwent a pap smear in 2008 that was read by a cytotechnologist as normal. Two years later, the patient was found to have a golf-ball–sized cancerous tumor. She died from cervical cancer in 2011.
ESTATE’S CLAIM The cytotechnologist was negligent in misreading the 2008 Pap smear. If treatment had been started in 2008, the cancer could have been resolved with a simple conization biopsy.
DEFENDANTS’ DEFENSE The Pap smear interpretation was reasonable. The cancer could not have been diagnosed in 2008. The patient was at fault for failing to follow-up Pap smears during the next 2 years.
VERDICT After assigning 75% fault to the cytotechnologist and 25% fault to the patient, a Florida jury returned a $20,870,200 verdict, which was reduced to $15,816,699.
Disastrous off-label use of anticoagulation
When a pelvic abscess was found, a 50-year-old woman was admitted to the hospital for treatment. She was taking warfarin due to a history of venous thromboembolism.
Before the procedure, her physicians attempted to temporarily reverse her anticoagulation by administering Factor IX Complex (Profilnine SD, Grifols Biologicals). The dose ordered for the patient was nearly double the maximum recommended weight-based dose. Almost immediately after receiving the infusion, the patient went into cardiopulmonary arrest and died. An autopsy found the cause of death to be pulmonary emboli (PE).
ESTATE’S CLAIM An excessive dose of Profilnine caused PE. At the time of the incident, Profilnine was not FDA approved for warfarin reversal, although some off-label uses were recognized in emergent situations, such as intracranial bleeds.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $1.25 million Virginia settlement was reached.
Vesicovaginal fistula from ureteral injury
At a women’s health clinic, a patient reported continuous, heavy vaginal bleeding; pain; and shortness of breath when walking. She had a history of endometritis and multiple abdominal surgeries. Examination disclosed a profuse vaginal discharge, a normal cervix, and an enlarged uterus. The patient consented to abdominal hysterectomy and bilateral salpingo-oophorectomy performed by an ObGyn assisted by a resident.
During surgery, the ObGyn found that the patient’s uterus was at 16 to 20 weeks’ gestation size, with multiple serosal uterine fibroids and frank pus and necrosed fibroid tumors within the uterine cavity. The procedure took longer than planned because of extensive adhesions. After surgery, the patient was anemic and was given a beta-blocker for tachycardia. She was discharged 3 days later with 48 hours’ worth of intravenous antibiotics.
A month later, the patient reported urinary incontinence. She saw a urologist, who found a vesicovaginal fistula. The patient underwent nephrostomy-tube placement. Right ureterolysis and a right ureteral reimplant was performed 4 months later.
PATIENT’S CLAIM The ObGyn injured the right ureter during surgery.
DEFENDANTS’ DEFENSE The ureter injury is a known risk of the procedure. The injury was due to an infection or delayed effects of ischemia. The patient had a good recovery with no residual injury.
VERDICT A Michigan defense verdict was returned.
Why did mother die after delivering twins?
After a 35-year-old woman gave birth to twins by cesarean delivery, she died. At autopsy, 4 liters of blood were found in her abdomen.
ESTATE’S CLAIM The ObGyn failed to recognize and treat an arterial or venous bleed during surgery.
DEFENDANTS’ DEFENSE The patient died from amniotic fluid embolism. Autopsy results showed right ventricular heart failure, respiratory failure, and disseminated intravascular coagulation.
VERDICT A Florida defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Was fetus’ wrist injured during cesarean delivery?
At 34 weeks’ gestation, a 39-year-old woman went to the hospital in preterm labor. Her history included a prior cesarean delivery. Ultrasonography (US) showed that the fetus was in a double-footling breech position. The ObGyn decided to perform a cesarean delivery when the fetal heart-rate monitor indicated distress.
After making a midline incision through the earlier scar, the ObGyn created a low transverse uterine incision with a scalpel. The mother’s uterus was thick because labor had not progressed. When the ObGyn was unable to deliver the baby through the low transverse incision, she performed a T-extension of the incision using bandage scissors while placing her free hand inside the uterus to shield the fetus from injury. After extensive manipulation, the baby was delivered and immediately handed to a neonatologist. After surgery, the neonatologist told the mother that the baby had sustained two lacerations to the ulnar side of the right wrist. The newborn was airlifted to another hospital for treatment of sepsis. There, an orthopedic hand surgeon examined the child and determined that the lacerations were superficial and only required sutures. The orthopedist saw the infant a month later and believed there was no significant wrist injury.
When the child began preschool, she started to experience cold intolerance and difficulty writing with her right hand. The child was referred to a pediatric neurologist, who found no nerve damage and ordered occupational therapy.
The original orthopedic surgeon examined the child when she was 7 years old and determined that the flexor carpi ulnaris tendon had been completely severed with a partial injury to the ulnar nerve. He recommended a return visit at age 14 for full assessment of the wrist injury.
PARENTS’ CLAIM The ObGyn did not properly shield the fetus when performing the T-extension incision during cesarean delivery. The child’s weakness will increase with age, ruling out some occupations.
PHYSICIAN’S DEFENSE The ObGyn was not negligent; she had provided adequate protection of the fetus during both incisions.
VERDICT An Illinois defense verdict was returned.
Woman dies after tubal ligation
After a 42-year-old woman underwent tubal ligation, her surgeon was concerned about a possible bowel perforation and admitted her to the hospital. The next morning, a computed tomography (CT) scan of the abdomen did not reveal bowel injury.
That afternoon, when the patient reported shortness of breath, the surgeon called the hospitalist with concern for pulmonary embolism (PE). The hospitalist immediately ordered a CT scan of the chest, initiated PE protocol, and wrote “r/o PE” on the chart. A radiologist reminded the hospitalist of the earlier CT scan with concern for kidney damage from another dye study. The hospitalist cancelled the CT scan and PE protocol. After waiting 17 hours to run any further tests, a CT scan revealed massive bilateral PE. The patient was transferred to the ICU, but died the next day.
PATIENT’S CLAIM The 17-hour delay was negligent.
PHYSICIAN’S DEFENSE There was no negligence. The patient died of septic shock, not PE.
VERDICT A $4 million Virginia verdict was returned.
Child born without hand and forearm
During prenatal care, a mother underwent US at 20 and 36 weeks; both studies were reported as normal. The child was born missing his left hand and part of his left forearm due to a congenital amputation. The child will require prosthetics for life.
PATIENT’S CLAIM The condition should have been seen during prenatal US; an abortion was still an option at 20 weeks.
DEFENDANTS’ DEFENSE US was properly performed and evaluated. It can be difficult to differentiate the right from left extremities.
VERDICT A California defense verdict was returned.
After starting Yasmin, woman has stroke with permanent paralysis: $16.5M total award
When a 37-year-old woman reported irregular menstruation, her ObGyn prescribed drospirenone/ethinyl estradiol (Yasmin; Bayer). Thirteen days after starting the drug, the patient had a stroke. She is paralyzed on her left side, has limited ability to speak, cannot use her left arm and leg, and requires 24-hour care.
PATIENT’S CLAIM The ObGyn should have recognized that Yasmin was not appropriate for this patient because of the drug’s clotting risks. The patient’s risk factors included her age (over 35), borderline hypertension, overweight, history of smoking, and high cholesterol. The ObGyn should have offered safer alternatives, such as a progesterone-only pill. The US Food and Drug Administration (FDA) issued a safety warning that all drospirenone-containing drugs may be associated with a higher risk of venous thrombosis during the first 6 months of use.
DEFENDANTS’ DEFENSE According to Bayer, Yasmin is safe, and remains on the market. It was an appropriate drug to treat her irregular bleeding.
VERDICT Claims against the medical center that referred the patient to the ObGyn were settled for $2.5 million before trial. A $14 million Illinois verdict was returned against the ObGyn, for a total award of $16.5 million.
Who is at fault when pelvic mesh erodes?
In January 2011, an ObGyn implanted the Gynecare TVT Obturator System (TVT‑O; Ethicon) during a midurethral sling procedure to treat stress urinary incontinence (SUI) in a woman in her 60s. Shortly thereafter, the ObGyn left practice because of early-onset Alzheimer’s disease, and the patient’s care was taken over by a gynecologist.
At the 2-month postoperative visit, the gynecologist found that the mesh had eroded into the patient’s vagina. The gynecologist simply cut the mesh with a scissor, charted that a small erosion was present, and prescribed estrogen cream.
The patient continued to report pain, discomfort, pressure, difficulty voiding urine, continued incontinence, vaginal discharge, scarring, infection, odor, and bleeding.
PATIENT’S CLAIM The polypropylene mesh used during the midurethral sling procedure has been shown to be incompatible with human tissue. It promotes an immune response, which stimulates degradation of the pelvic tissue and can contribute to the development of severe adverse reactions to the mesh. Ethicon negligently designed, manufactured, marketed, labeled, and packaged the pelvic mesh products.
DEFENDANTS’ DEFENSE Proper warnings were provided about the health risks associated with polypropylene mesh products. The medical device was not properly sized.
VERDICT A Texas jury rejected the patient’s claims that Ethicon did not provide proper warnings about the sling’s health risks and declined to award punitive damages.
However, the jury decided that the mesh implant was defectively designed, and returned a $1.2 million verdict against Ethicon.
Was suspected bowel injury treated properly?
A 40-year-old woman was referred to an ObGyn after reporting abnormal uterine bleeding to her primary care physician. The patient had very light menses every few weeks. The ObGyn performed an ablation procedure, without relief. A month later, the ObGyn performed robot-assisted laparoscopic hysterectomy. The next day, the patient reported abdominal pain. Suspecting a bowel injury, the ObGyn ordered a CT scan; the bowel appeared normal, so the ObGyn referred the patient to a surgeon. During exploratory laparotomy, the surgeon found and repaired a bowel injury. The patient developed significant complications from a necrotizing infection that included respiratory distress and ongoing wound care.
PATIENT’S CLAIM Conservative treatment should have been offered before surgery. The ObGyn should have waited longer after the ablation procedure before doing the hysterectomy. The ObGyn should have checked for a possible bowel injury before closing the hysterectomy.
PHYSICIAN’S DEFENSE The bowel injury is a known complication of the procedure and was recognized and repaired in a timely manner.
VERDICT A Kentucky defense verdict was returned.
Pap smear improperly interpreted: Woman dies from cervical cancer
A 37-year-old woman underwent a pap smear in 2008 that was read by a cytotechnologist as normal. Two years later, the patient was found to have a golf-ball–sized cancerous tumor. She died from cervical cancer in 2011.
ESTATE’S CLAIM The cytotechnologist was negligent in misreading the 2008 Pap smear. If treatment had been started in 2008, the cancer could have been resolved with a simple conization biopsy.
DEFENDANTS’ DEFENSE The Pap smear interpretation was reasonable. The cancer could not have been diagnosed in 2008. The patient was at fault for failing to follow-up Pap smears during the next 2 years.
VERDICT After assigning 75% fault to the cytotechnologist and 25% fault to the patient, a Florida jury returned a $20,870,200 verdict, which was reduced to $15,816,699.
Disastrous off-label use of anticoagulation
When a pelvic abscess was found, a 50-year-old woman was admitted to the hospital for treatment. She was taking warfarin due to a history of venous thromboembolism.
Before the procedure, her physicians attempted to temporarily reverse her anticoagulation by administering Factor IX Complex (Profilnine SD, Grifols Biologicals). The dose ordered for the patient was nearly double the maximum recommended weight-based dose. Almost immediately after receiving the infusion, the patient went into cardiopulmonary arrest and died. An autopsy found the cause of death to be pulmonary emboli (PE).
ESTATE’S CLAIM An excessive dose of Profilnine caused PE. At the time of the incident, Profilnine was not FDA approved for warfarin reversal, although some off-label uses were recognized in emergent situations, such as intracranial bleeds.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $1.25 million Virginia settlement was reached.
Vesicovaginal fistula from ureteral injury
At a women’s health clinic, a patient reported continuous, heavy vaginal bleeding; pain; and shortness of breath when walking. She had a history of endometritis and multiple abdominal surgeries. Examination disclosed a profuse vaginal discharge, a normal cervix, and an enlarged uterus. The patient consented to abdominal hysterectomy and bilateral salpingo-oophorectomy performed by an ObGyn assisted by a resident.
During surgery, the ObGyn found that the patient’s uterus was at 16 to 20 weeks’ gestation size, with multiple serosal uterine fibroids and frank pus and necrosed fibroid tumors within the uterine cavity. The procedure took longer than planned because of extensive adhesions. After surgery, the patient was anemic and was given a beta-blocker for tachycardia. She was discharged 3 days later with 48 hours’ worth of intravenous antibiotics.
A month later, the patient reported urinary incontinence. She saw a urologist, who found a vesicovaginal fistula. The patient underwent nephrostomy-tube placement. Right ureterolysis and a right ureteral reimplant was performed 4 months later.
PATIENT’S CLAIM The ObGyn injured the right ureter during surgery.
DEFENDANTS’ DEFENSE The ureter injury is a known risk of the procedure. The injury was due to an infection or delayed effects of ischemia. The patient had a good recovery with no residual injury.
VERDICT A Michigan defense verdict was returned.
Why did mother die after delivering twins?
After a 35-year-old woman gave birth to twins by cesarean delivery, she died. At autopsy, 4 liters of blood were found in her abdomen.
ESTATE’S CLAIM The ObGyn failed to recognize and treat an arterial or venous bleed during surgery.
DEFENDANTS’ DEFENSE The patient died from amniotic fluid embolism. Autopsy results showed right ventricular heart failure, respiratory failure, and disseminated intravascular coagulation.
VERDICT A Florida defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Was fetus’ wrist injured during cesarean delivery?
At 34 weeks’ gestation, a 39-year-old woman went to the hospital in preterm labor. Her history included a prior cesarean delivery. Ultrasonography (US) showed that the fetus was in a double-footling breech position. The ObGyn decided to perform a cesarean delivery when the fetal heart-rate monitor indicated distress.
After making a midline incision through the earlier scar, the ObGyn created a low transverse uterine incision with a scalpel. The mother’s uterus was thick because labor had not progressed. When the ObGyn was unable to deliver the baby through the low transverse incision, she performed a T-extension of the incision using bandage scissors while placing her free hand inside the uterus to shield the fetus from injury. After extensive manipulation, the baby was delivered and immediately handed to a neonatologist. After surgery, the neonatologist told the mother that the baby had sustained two lacerations to the ulnar side of the right wrist. The newborn was airlifted to another hospital for treatment of sepsis. There, an orthopedic hand surgeon examined the child and determined that the lacerations were superficial and only required sutures. The orthopedist saw the infant a month later and believed there was no significant wrist injury.
When the child began preschool, she started to experience cold intolerance and difficulty writing with her right hand. The child was referred to a pediatric neurologist, who found no nerve damage and ordered occupational therapy.
The original orthopedic surgeon examined the child when she was 7 years old and determined that the flexor carpi ulnaris tendon had been completely severed with a partial injury to the ulnar nerve. He recommended a return visit at age 14 for full assessment of the wrist injury.
PARENTS’ CLAIM The ObGyn did not properly shield the fetus when performing the T-extension incision during cesarean delivery. The child’s weakness will increase with age, ruling out some occupations.
PHYSICIAN’S DEFENSE The ObGyn was not negligent; she had provided adequate protection of the fetus during both incisions.
VERDICT An Illinois defense verdict was returned.
Woman dies after tubal ligation
After a 42-year-old woman underwent tubal ligation, her surgeon was concerned about a possible bowel perforation and admitted her to the hospital. The next morning, a computed tomography (CT) scan of the abdomen did not reveal bowel injury.
That afternoon, when the patient reported shortness of breath, the surgeon called the hospitalist with concern for pulmonary embolism (PE). The hospitalist immediately ordered a CT scan of the chest, initiated PE protocol, and wrote “r/o PE” on the chart. A radiologist reminded the hospitalist of the earlier CT scan with concern for kidney damage from another dye study. The hospitalist cancelled the CT scan and PE protocol. After waiting 17 hours to run any further tests, a CT scan revealed massive bilateral PE. The patient was transferred to the ICU, but died the next day.
PATIENT’S CLAIM The 17-hour delay was negligent.
PHYSICIAN’S DEFENSE There was no negligence. The patient died of septic shock, not PE.
VERDICT A $4 million Virginia verdict was returned.
Child born without hand and forearm
During prenatal care, a mother underwent US at 20 and 36 weeks; both studies were reported as normal. The child was born missing his left hand and part of his left forearm due to a congenital amputation. The child will require prosthetics for life.
PATIENT’S CLAIM The condition should have been seen during prenatal US; an abortion was still an option at 20 weeks.
DEFENDANTS’ DEFENSE US was properly performed and evaluated. It can be difficult to differentiate the right from left extremities.
VERDICT A California defense verdict was returned.
After starting Yasmin, woman has stroke with permanent paralysis: $16.5M total award
When a 37-year-old woman reported irregular menstruation, her ObGyn prescribed drospirenone/ethinyl estradiol (Yasmin; Bayer). Thirteen days after starting the drug, the patient had a stroke. She is paralyzed on her left side, has limited ability to speak, cannot use her left arm and leg, and requires 24-hour care.
PATIENT’S CLAIM The ObGyn should have recognized that Yasmin was not appropriate for this patient because of the drug’s clotting risks. The patient’s risk factors included her age (over 35), borderline hypertension, overweight, history of smoking, and high cholesterol. The ObGyn should have offered safer alternatives, such as a progesterone-only pill. The US Food and Drug Administration (FDA) issued a safety warning that all drospirenone-containing drugs may be associated with a higher risk of venous thrombosis during the first 6 months of use.
DEFENDANTS’ DEFENSE According to Bayer, Yasmin is safe, and remains on the market. It was an appropriate drug to treat her irregular bleeding.
VERDICT Claims against the medical center that referred the patient to the ObGyn were settled for $2.5 million before trial. A $14 million Illinois verdict was returned against the ObGyn, for a total award of $16.5 million.
Who is at fault when pelvic mesh erodes?
In January 2011, an ObGyn implanted the Gynecare TVT Obturator System (TVT‑O; Ethicon) during a midurethral sling procedure to treat stress urinary incontinence (SUI) in a woman in her 60s. Shortly thereafter, the ObGyn left practice because of early-onset Alzheimer’s disease, and the patient’s care was taken over by a gynecologist.
At the 2-month postoperative visit, the gynecologist found that the mesh had eroded into the patient’s vagina. The gynecologist simply cut the mesh with a scissor, charted that a small erosion was present, and prescribed estrogen cream.
The patient continued to report pain, discomfort, pressure, difficulty voiding urine, continued incontinence, vaginal discharge, scarring, infection, odor, and bleeding.
PATIENT’S CLAIM The polypropylene mesh used during the midurethral sling procedure has been shown to be incompatible with human tissue. It promotes an immune response, which stimulates degradation of the pelvic tissue and can contribute to the development of severe adverse reactions to the mesh. Ethicon negligently designed, manufactured, marketed, labeled, and packaged the pelvic mesh products.
DEFENDANTS’ DEFENSE Proper warnings were provided about the health risks associated with polypropylene mesh products. The medical device was not properly sized.
VERDICT A Texas jury rejected the patient’s claims that Ethicon did not provide proper warnings about the sling’s health risks and declined to award punitive damages.
However, the jury decided that the mesh implant was defectively designed, and returned a $1.2 million verdict against Ethicon.
Was suspected bowel injury treated properly?
A 40-year-old woman was referred to an ObGyn after reporting abnormal uterine bleeding to her primary care physician. The patient had very light menses every few weeks. The ObGyn performed an ablation procedure, without relief. A month later, the ObGyn performed robot-assisted laparoscopic hysterectomy. The next day, the patient reported abdominal pain. Suspecting a bowel injury, the ObGyn ordered a CT scan; the bowel appeared normal, so the ObGyn referred the patient to a surgeon. During exploratory laparotomy, the surgeon found and repaired a bowel injury. The patient developed significant complications from a necrotizing infection that included respiratory distress and ongoing wound care.
PATIENT’S CLAIM Conservative treatment should have been offered before surgery. The ObGyn should have waited longer after the ablation procedure before doing the hysterectomy. The ObGyn should have checked for a possible bowel injury before closing the hysterectomy.
PHYSICIAN’S DEFENSE The bowel injury is a known complication of the procedure and was recognized and repaired in a timely manner.
VERDICT A Kentucky defense verdict was returned.
Pap smear improperly interpreted: Woman dies from cervical cancer
A 37-year-old woman underwent a pap smear in 2008 that was read by a cytotechnologist as normal. Two years later, the patient was found to have a golf-ball–sized cancerous tumor. She died from cervical cancer in 2011.
ESTATE’S CLAIM The cytotechnologist was negligent in misreading the 2008 Pap smear. If treatment had been started in 2008, the cancer could have been resolved with a simple conization biopsy.
DEFENDANTS’ DEFENSE The Pap smear interpretation was reasonable. The cancer could not have been diagnosed in 2008. The patient was at fault for failing to follow-up Pap smears during the next 2 years.
VERDICT After assigning 75% fault to the cytotechnologist and 25% fault to the patient, a Florida jury returned a $20,870,200 verdict, which was reduced to $15,816,699.
Disastrous off-label use of anticoagulation
When a pelvic abscess was found, a 50-year-old woman was admitted to the hospital for treatment. She was taking warfarin due to a history of venous thromboembolism.
Before the procedure, her physicians attempted to temporarily reverse her anticoagulation by administering Factor IX Complex (Profilnine SD, Grifols Biologicals). The dose ordered for the patient was nearly double the maximum recommended weight-based dose. Almost immediately after receiving the infusion, the patient went into cardiopulmonary arrest and died. An autopsy found the cause of death to be pulmonary emboli (PE).
ESTATE’S CLAIM An excessive dose of Profilnine caused PE. At the time of the incident, Profilnine was not FDA approved for warfarin reversal, although some off-label uses were recognized in emergent situations, such as intracranial bleeds.
DEFENDANTS’ DEFENSE The case was settled during the trial.
VERDICT A $1.25 million Virginia settlement was reached.
Vesicovaginal fistula from ureteral injury
At a women’s health clinic, a patient reported continuous, heavy vaginal bleeding; pain; and shortness of breath when walking. She had a history of endometritis and multiple abdominal surgeries. Examination disclosed a profuse vaginal discharge, a normal cervix, and an enlarged uterus. The patient consented to abdominal hysterectomy and bilateral salpingo-oophorectomy performed by an ObGyn assisted by a resident.
During surgery, the ObGyn found that the patient’s uterus was at 16 to 20 weeks’ gestation size, with multiple serosal uterine fibroids and frank pus and necrosed fibroid tumors within the uterine cavity. The procedure took longer than planned because of extensive adhesions. After surgery, the patient was anemic and was given a beta-blocker for tachycardia. She was discharged 3 days later with 48 hours’ worth of intravenous antibiotics.
A month later, the patient reported urinary incontinence. She saw a urologist, who found a vesicovaginal fistula. The patient underwent nephrostomy-tube placement. Right ureterolysis and a right ureteral reimplant was performed 4 months later.
PATIENT’S CLAIM The ObGyn injured the right ureter during surgery.
DEFENDANTS’ DEFENSE The ureter injury is a known risk of the procedure. The injury was due to an infection or delayed effects of ischemia. The patient had a good recovery with no residual injury.
VERDICT A Michigan defense verdict was returned.
Why did mother die after delivering twins?
After a 35-year-old woman gave birth to twins by cesarean delivery, she died. At autopsy, 4 liters of blood were found in her abdomen.
ESTATE’S CLAIM The ObGyn failed to recognize and treat an arterial or venous bleed during surgery.
DEFENDANTS’ DEFENSE The patient died from amniotic fluid embolism. Autopsy results showed right ventricular heart failure, respiratory failure, and disseminated intravascular coagulation.
VERDICT A Florida defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
Share your thoughts on this article! Send your Letter to the Editor to [email protected]. Please include your name and the city and state in which you practice.
Overcorrection of Hyponatremia
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) A clinic patient of mine was recently admitted to the hospital with hyponatremia (serum sodium, 115 mEq/L). She was treated with 2 L of normal saline and discharged home 48 hours later, at her baseline mental status with a serum sodium level of 132 mEq/L. Two days later, she was readmitted for mental status changes, and MRI showed brain swelling. The neurologist stated this was a result of the initial treatment for her hyponatremia. How is this possible?
The cause-and-effect relationship between rapid correction of chronic hyponatremia and subsequent development of neurologic problems was discovered in the late 1970s. Central pontine and extrapontine myelinolysis (known as osmotic demyelination syndrome or ODS) is a neurologic condition that can occur from rapid sodium correction. It is diagnosed by MRI, which shows hyperintense lesions on T2-weighted images. Clinical signs include upper motor neuron signs, pseudobulbar palsy, spastic quadriparesis, and mental status changes ranging from mild confusion to coma.2
Treatment for hyponatremia should be guided by symptom management.2,3 If a patient is asymptomatic, a simple and effective strategy is to keep NPO for 24 hours, except for medications. Simple food and fluid restriction will likely increase the serum sodium level because of obligate solute losses and urinary electrolyte free water loss.2,4 While the first instinct is to feed these patients, as they often appear malnourished, this can cause a solute load leading to a too-rapid sodium correction. After 24 hours, if intake restriction is not effective, use 0.5% normal saline but with limited dosing orders, as usual saline dosing can cause too rapid a correction.2
For symptomatic patients (confusion, seizures, coma), the goal is to initially elevate sodium by 1 to 2 mEq/L per hour for the first two to three hours. Do not exceed 10 mEq/L in 24 hours or 18 mEq/L in 48 hours. Exceeding these limits puts patients at high risk for ODS. In fact, even when staying within these parameters, there is some risk for overcorrection. It is always better to go slowly.2,3
In the patient with hyponatremia due to low solute intake (eg, beer potomania), diuresis can start spontaneously after a period of food and fluid restriction. It can also be initiated with just a small amount of solute. For example, administering an IV antibiotic with a base solution of 100 mL of normal saline or a “banana bag” (an IV solution containing 0.5 to 1 L of normal saline with multivitamins/minerals that cause the fluid to be yellow) can produce several liters of diuresis.2 Once you open the floodgate, you can unintentionally cause too-rapid correction that could lead to ODS.
In chronic hyponatremic patients, low antidiuretic hormone (ADH) levels are often found; thus when a solute is introduced, there is little ADH in the system to protect against excessive water loss and electrolyte imbalance. At the same time, excessive water loss can translate to higher sodium levels and increase the risk for cerebral edema. If rapid diuresis occurs, an infusion of D5W (5% dextrose in water) to match the rate of urine output may prevent a rapid serum sodium level rise. Frequent monitoring of serum sodium levels is often necessary. In instances where ODS is already present, there are case studies of improved neurologic outcomes with reduction of serum sodium levels.2,3
While the treatment of hyponatremia at first glance seems straightforward—replace that which is lost—it can actually transform a seemingly simple problem into a complicated clinical course requiring intensive care, due to the need for frequent monitoring and intervention.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Hyponatremia: Beer Potomania
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
| Clinician Reviews in partnership with |
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, who is a physician assistant with Metropolitan Nephrology in Alexandria, Virginia, and Clinton, Maryland; she is also past chair of the NKF-CAP. This month’s responses were authored by Kristina Unterseher, MSN, FNP, CNN-NP, who practices at Peacehealth St. John Medical Center in Longview, Washington.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.
Q) Recently, we had a patient admitted for hyponatremia with
a serum sodium level of 117 mEq/L. One of the hospitalists mentioned “beer potomania” in the differential. Not wanting to look dumb, I just agreed. What is beer potomania, and how is it related to low serum sodium?
Potomania is the excessive consumption of alcoholic beverages; beer potomania is used to refer to a dilutional hyponatremia caused by excessive consumption of beer.1 First recognized in 1971, this cause of hyponatremia is not the most common but should be in the differential if the patient is a heavy alcohol imbiber who presents with encephalopathy and low serum sodium.
When considering this diagnosis, keep in mind that hyponatremia is common among chronic alcoholics and can be due to conditions such as cirrhosis, congestive heart failure, syndrome of inappropriate antidiuretic hormone (SIADH) secretion, and hypovolemia. Less common but still belonging in the differential are pseudohyponatremia secondary to alcohol-induced severe hypertriglyceridemia and cerebral salt wasting syndrome.2,3
Beer potomania usually manifests as altered mental status, weakness, and gait disturbance with an average serum sodium concentration of 108 mEq/L.3 Other abnormal lab results consistent with this diagnosis include hypokalemia (mean potassium, 3 mEq/L) and low blood urea nitrogen and urine sodium levels.2,3 Another fairly consistent finding is a recent personal history of binge drinking (more than about 5 L, or 14 cans of beer, in 24 hours) and/or history of illness (vomiting, diarrhea) that predisposed the patient to a rapid drop in serum sodium levels.2
Based on the information presented thus far, you may ask, “Why haven’t I seen this diagnosed more often? There are a lot of beer bingers out there!” Good question. Let’s review the pathophysiology of beer potomania. When patients have poor protein and solute (food, electrolytes) intake, they can experience water intoxication with smaller-than-usual volumes of fluid. The kidneys need a certain amount of solute to facilitate free water clearance (the ability to clear excess fluid from the body). A lack of adequate solute results in a buildup of free water in the vascular system, leading to a dilutional hyponatremia.3
Free water clearance is dependent on both solute excretion and the ability to dilute urine. Someone consuming an average diet will excrete 600 to 900 mOsm/d of solute. This osmolar load in-cludes urea generated from protein (10 g of protein produces about 50 mOsm of urea), along with dietary sodium and potassium. The maximum capacity for urinary dilution is 50 mOsm/L. In a nutritionally sound person, a lot of fluid—about 20 L—would be required to overwhelm the body’s capacity for urinary dilution.2
However, when you don’t eat, the body starts to break down tissue to create energy to survive. This catabolism creates 100 to 150 mOsm/d of urea, allowing you to continue to appropriately excrete a moderate amount of fluid in spite of poor solute intake ... as long as you are not drinking excessive amounts of water.5
Alcoholics get a moderate amount of their calories via beer consumption and do not experience this endogenous protein breakdown or its resultant low urea/solute level. With low solute intake, dramatically lower fluid intake (about 14 cans of beer) will overwhelm the kidneys’ ability to clear excess free water in the body.2 Fortunately, most heavy beer drinkers continue to eat at least modestly, which is sufficient to avoid this rare type of hyponatremia. Chronic alcoholics who go on a drinking binge beyond their normal baseline alcohol consumption, or who develop a flulike illness that causes electrolyte depletion (via diarrhea or vomiting), are at higher risk for beer potomania.
Kristina Unterseher, MSN, FNP, CNN-NP
Peacehealth St. John
Medical Center
Longview, WA
REFERENCES
1. Hilden T, Swensen TL. Electrolyte disturbances in beer drinkers: a specific “hypo-osmolaity syndrome.” Lancet. 1975;2(7928):245-246.
2. Sanghvi SR, Kellerman PS, Nanovic L. Beer potomania: an unusual cause of hyponatremia at high risk of complications from rapid correction. Am J Kidney Dis. 2007;50(4):673-680.
3. Bhattarai N, Poonam K, Panda M. Beer potomania: a case report. BMJ Case Rep. 2010; 2010: bcr10.2009.2414.
4. Campbell M. Hyponatremia and central pontine myelinolysis as a result of beer potomania: a case report. Prim Care Companion J Clin Psychiatry. 2010;12(4):PCC.09100936.
5. Thaler SM, Teitelbaum I, Beri T. “Beer potomania” in non-beer drinkers: effect of low dietary solute intake. Am J Kidney Dis. 1998;31(6):1028-1031.