User login
Research could aid platelet production
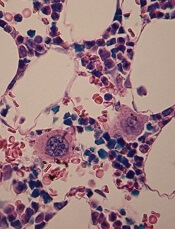
in the bone marrow
Scientists say they’ve shed new light on the mechanism of platelet formation, paving the way to accelerating and enhancing platelet production using stem cells.
The group uncovered their findings by studying the effects of shear stress on megakaryocyte maturation and the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
“Until recently, these microparticles were viewed as inconsequential cell debris,” said Terry Papoutsakis, PhD, of the University of Delaware in Newark.
“We now know that they play a significant biological role in platelet formation. The enhanced generation of preplatelets and platelet-like particles under shear stress correlates with physiological observations—in healthy adults, both acute and prolonged exercise leads to elevated platelet counts.”
“Now, these findings can be used to develop better bioreactor technologies for producing platelets, preplatelets, platelet-like particles, and megakaryocyte microparticles for transfusion medicine, using stem cells as starting material.”
Dr Papoutsakis and his colleagues described these findings in Blood.
The researchers discovered that shear stress accelerated DNA synthesis of immature megakaryocytes, and this was dependent upon exposure time and the shear stress level.
Physiological shear stress increased the formation of preplatelets and platelet-like particles up to 10.8-fold. And it increased megakaryocyte microparticle production up to 47-fold. Platelet-like particles generated under shear flow showed improved function.
Experiments also revealed that phosphatidylserine exposure and caspase-3 activation were enhanced by shear stress. But inhibiting caspase-3 reduced the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
Finally, the researchers found that coculturing megakaryocyte microparticles with hematopoietic stem and progenitor cells promoted differentiation to mature megakaryocytes that synthesized α- and dense-granules, and formed preplatelets without exogenous thrombopoietin.
The team noted that, unlike platelets themselves, these microparticles can be frozen, which will enable them to be stored and used for platelet production on an as-needed basis.
“Knowing that these microparticles have a biological function opens the door to other applications, including genetic therapies,” Dr Papoutsakis said. “We’re hopeful that our discovery can break the vicious cycle of [immune thrombocytopenia] as well as other conditions that cause reduced platelet count and cause life-threatening bleeding.” ![]()
in the bone marrow
Scientists say they’ve shed new light on the mechanism of platelet formation, paving the way to accelerating and enhancing platelet production using stem cells.
The group uncovered their findings by studying the effects of shear stress on megakaryocyte maturation and the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
“Until recently, these microparticles were viewed as inconsequential cell debris,” said Terry Papoutsakis, PhD, of the University of Delaware in Newark.
“We now know that they play a significant biological role in platelet formation. The enhanced generation of preplatelets and platelet-like particles under shear stress correlates with physiological observations—in healthy adults, both acute and prolonged exercise leads to elevated platelet counts.”
“Now, these findings can be used to develop better bioreactor technologies for producing platelets, preplatelets, platelet-like particles, and megakaryocyte microparticles for transfusion medicine, using stem cells as starting material.”
Dr Papoutsakis and his colleagues described these findings in Blood.
The researchers discovered that shear stress accelerated DNA synthesis of immature megakaryocytes, and this was dependent upon exposure time and the shear stress level.
Physiological shear stress increased the formation of preplatelets and platelet-like particles up to 10.8-fold. And it increased megakaryocyte microparticle production up to 47-fold. Platelet-like particles generated under shear flow showed improved function.
Experiments also revealed that phosphatidylserine exposure and caspase-3 activation were enhanced by shear stress. But inhibiting caspase-3 reduced the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
Finally, the researchers found that coculturing megakaryocyte microparticles with hematopoietic stem and progenitor cells promoted differentiation to mature megakaryocytes that synthesized α- and dense-granules, and formed preplatelets without exogenous thrombopoietin.
The team noted that, unlike platelets themselves, these microparticles can be frozen, which will enable them to be stored and used for platelet production on an as-needed basis.
“Knowing that these microparticles have a biological function opens the door to other applications, including genetic therapies,” Dr Papoutsakis said. “We’re hopeful that our discovery can break the vicious cycle of [immune thrombocytopenia] as well as other conditions that cause reduced platelet count and cause life-threatening bleeding.” ![]()
in the bone marrow
Scientists say they’ve shed new light on the mechanism of platelet formation, paving the way to accelerating and enhancing platelet production using stem cells.
The group uncovered their findings by studying the effects of shear stress on megakaryocyte maturation and the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
“Until recently, these microparticles were viewed as inconsequential cell debris,” said Terry Papoutsakis, PhD, of the University of Delaware in Newark.
“We now know that they play a significant biological role in platelet formation. The enhanced generation of preplatelets and platelet-like particles under shear stress correlates with physiological observations—in healthy adults, both acute and prolonged exercise leads to elevated platelet counts.”
“Now, these findings can be used to develop better bioreactor technologies for producing platelets, preplatelets, platelet-like particles, and megakaryocyte microparticles for transfusion medicine, using stem cells as starting material.”
Dr Papoutsakis and his colleagues described these findings in Blood.
The researchers discovered that shear stress accelerated DNA synthesis of immature megakaryocytes, and this was dependent upon exposure time and the shear stress level.
Physiological shear stress increased the formation of preplatelets and platelet-like particles up to 10.8-fold. And it increased megakaryocyte microparticle production up to 47-fold. Platelet-like particles generated under shear flow showed improved function.
Experiments also revealed that phosphatidylserine exposure and caspase-3 activation were enhanced by shear stress. But inhibiting caspase-3 reduced the formation of preplatelets, platelet-like particles, and megakaryocyte microparticles.
Finally, the researchers found that coculturing megakaryocyte microparticles with hematopoietic stem and progenitor cells promoted differentiation to mature megakaryocytes that synthesized α- and dense-granules, and formed preplatelets without exogenous thrombopoietin.
The team noted that, unlike platelets themselves, these microparticles can be frozen, which will enable them to be stored and used for platelet production on an as-needed basis.
“Knowing that these microparticles have a biological function opens the door to other applications, including genetic therapies,” Dr Papoutsakis said. “We’re hopeful that our discovery can break the vicious cycle of [immune thrombocytopenia] as well as other conditions that cause reduced platelet count and cause life-threatening bleeding.” ![]()
Hypercalcemia may be indicator for hematologic cancers

Credit: Graham Colm
Hypercalcemia could be an early indication of cancer, according to a study published in the British Journal of Cancer.
The connection between hypercalcemia and cancer is well known, but this study shows the condition can predate cancer diagnosis in primary care.
The association between hypercalcemia and cancers was particularly strong in men. And myeloma and other hematologic malignancies were among the most common cancers associated with hypercalcemia.
“All previous studies on hypercalcemia and cancer had been carried out with patients who had already been diagnosed with cancer; hypercalcemia was seen as a late effect of the cancer,” said study author Fergus Hamilton, of the University of Bristol in the UK.
“We wanted to look at the issue from a different perspective and find out if high calcium levels in blood could be used as an early indicator of cancer and, therefore, in the diagnosis of cancer.”
So the researchers analyzed the electronic records of 54,267 patients with elevated calcium levels and found that hypercalcemia was strongly associated with cancer, especially in males.
The positive predictive values for cancer in men were 11.5% for calcium levels between 2.60 and 2.79 mmol l-1, 27.9% for 2.8-2.99 mmol l-1, and 50% for >3.0 mmol l-1. In women, the corresponding values were 4.1%, 8.7%, and 16.7%, respectively.
In men, the most common cancers associated with hypercalcemia were lung (34%), prostate (21%), colorectal (8%), myeloma (8%), and other hematologic cancers (8%). There were 12 other cancer types recorded as well (19%).
In women, the most common cancers were myeloma (24%), breast (18%), other hematologic cancers (10%), lung (8%), and metastatic cancer with unknown primary (8%). There were 16 other cancers recorded among women (32%).
The researchers found no difference in calcium levels among the different cancers.
“We were surprised by the gender difference,” Dr Hamilton said. “There are a number of possible explanations for this, but we think it might be because women are much more likely to have hyperparathyroidism, another cause of hypercalcemia. Men rarely get this condition, so their hypercalcemia is more likely to be due to cancer.” ![]()
Credit: Graham Colm
Hypercalcemia could be an early indication of cancer, according to a study published in the British Journal of Cancer.
The connection between hypercalcemia and cancer is well known, but this study shows the condition can predate cancer diagnosis in primary care.
The association between hypercalcemia and cancers was particularly strong in men. And myeloma and other hematologic malignancies were among the most common cancers associated with hypercalcemia.
“All previous studies on hypercalcemia and cancer had been carried out with patients who had already been diagnosed with cancer; hypercalcemia was seen as a late effect of the cancer,” said study author Fergus Hamilton, of the University of Bristol in the UK.
“We wanted to look at the issue from a different perspective and find out if high calcium levels in blood could be used as an early indicator of cancer and, therefore, in the diagnosis of cancer.”
So the researchers analyzed the electronic records of 54,267 patients with elevated calcium levels and found that hypercalcemia was strongly associated with cancer, especially in males.
The positive predictive values for cancer in men were 11.5% for calcium levels between 2.60 and 2.79 mmol l-1, 27.9% for 2.8-2.99 mmol l-1, and 50% for >3.0 mmol l-1. In women, the corresponding values were 4.1%, 8.7%, and 16.7%, respectively.
In men, the most common cancers associated with hypercalcemia were lung (34%), prostate (21%), colorectal (8%), myeloma (8%), and other hematologic cancers (8%). There were 12 other cancer types recorded as well (19%).
In women, the most common cancers were myeloma (24%), breast (18%), other hematologic cancers (10%), lung (8%), and metastatic cancer with unknown primary (8%). There were 16 other cancers recorded among women (32%).
The researchers found no difference in calcium levels among the different cancers.
“We were surprised by the gender difference,” Dr Hamilton said. “There are a number of possible explanations for this, but we think it might be because women are much more likely to have hyperparathyroidism, another cause of hypercalcemia. Men rarely get this condition, so their hypercalcemia is more likely to be due to cancer.” ![]()
Credit: Graham Colm
Hypercalcemia could be an early indication of cancer, according to a study published in the British Journal of Cancer.
The connection between hypercalcemia and cancer is well known, but this study shows the condition can predate cancer diagnosis in primary care.
The association between hypercalcemia and cancers was particularly strong in men. And myeloma and other hematologic malignancies were among the most common cancers associated with hypercalcemia.
“All previous studies on hypercalcemia and cancer had been carried out with patients who had already been diagnosed with cancer; hypercalcemia was seen as a late effect of the cancer,” said study author Fergus Hamilton, of the University of Bristol in the UK.
“We wanted to look at the issue from a different perspective and find out if high calcium levels in blood could be used as an early indicator of cancer and, therefore, in the diagnosis of cancer.”
So the researchers analyzed the electronic records of 54,267 patients with elevated calcium levels and found that hypercalcemia was strongly associated with cancer, especially in males.
The positive predictive values for cancer in men were 11.5% for calcium levels between 2.60 and 2.79 mmol l-1, 27.9% for 2.8-2.99 mmol l-1, and 50% for >3.0 mmol l-1. In women, the corresponding values were 4.1%, 8.7%, and 16.7%, respectively.
In men, the most common cancers associated with hypercalcemia were lung (34%), prostate (21%), colorectal (8%), myeloma (8%), and other hematologic cancers (8%). There were 12 other cancer types recorded as well (19%).
In women, the most common cancers were myeloma (24%), breast (18%), other hematologic cancers (10%), lung (8%), and metastatic cancer with unknown primary (8%). There were 16 other cancers recorded among women (32%).
The researchers found no difference in calcium levels among the different cancers.
“We were surprised by the gender difference,” Dr Hamilton said. “There are a number of possible explanations for this, but we think it might be because women are much more likely to have hyperparathyroidism, another cause of hypercalcemia. Men rarely get this condition, so their hypercalcemia is more likely to be due to cancer.” ![]()
Diabetes Mellitus and Skin Infections
Diabetes mellitus is one of the most common comorbid conditions among patients hospitalized for acute bacterial skin infections.[1, 2, 3, 4, 5, 6] Acute bacterial skin infections in diabetics represent a spectrum of conditions ranging from cellulitis or cutaneous abscess to more complicated infections such as infected ulcers or deep tissue infections. Although most skin infections in diabetics are caused by gram‐positive pathogens (Staphylococcus aureus and streptococci), the risk of gram‐negative pathogens is increased in certain complicated infections such as diabetic foot infections.[7] For such complicated infections, national guidelines therefore recommend broad‐spectrum empiric antibiotic therapy.[7]
The role of gram‐negative pathogens has not been clearly established in diabetics with cellulitis or cutaneous abscess not associated with an infected ulcer or diabetic foot infection. National guidelines for the treatment of cellulitis and abscess recommend antibiotic therapy targeted toward S aureus and streptococcal species irrespective of the presence of diabetes mellitus.[8, 9] However, in a recent multicenter study of patients hospitalized with acute bacterial skin infections in which cases involving infected ulcers or deep tissue infection were excluded, diabetes mellitus was an independent predictor of use of antibiotics with broad gram‐negative activity.[2] This suggests that either gram‐negative pathogens are more common or providers perceive gram‐negative pathogens to be more common among diabetics with otherwise uncomplicated cellulitis or abscess.
A better understanding of the relationship between the microbiology and antibiotic prescribing practices for diabetics with cellulitis or abscess is therefore necessary to promote the most appropriate spectrum of therapy for these patients. We evaluated a large cohort of patients hospitalized with acute bacterial skin infections in order to: (1) compare the microbiology of diabetics and nondiabetics with cellulitis or cutaneous abscess not associated with an ulcer or deep tissue infection; and (2) compare antibiotic prescribing practices among diabetics and nondiabetics. We hypothesized that diabetics would have a similar spectrum of microorganisms as nondiabetics but would be more frequently treated with antibiotics with broad gram‐negative activity.
METHODS
Study Design
This was a secondary analysis of 2 published retrospective studies of patients hospitalized for cellulitis or cutaneous abscess between January 1, 2007 and May 31, 2012.[2, 10] For the purposes of this study, the terms cellulitis and abscess will refer to infections not involving an infected ulcer, osteomyelitis, or other deep tissue infection.
Study Setting and Population
The first of the 2 cohorts analyzed for the present study included patients hospitalized with cellulitis, abscess, or wound infection at 7 academic or community hospitals in Colorado.[2] The second cohort included patients hospitalized with cellulitis or abscess at a single academic medical center (1 of the 7 hospitals above) in Denver, Colorado.[10] The methods of these studies have been reported in detail elsewhere.[2, 10, 11] Briefly, potential cases were identified using International Classification of Diseases, 9th Revision, Clinical Modification codes. The main inclusion and exclusion criteria of the 2 studies were similar. In both studies, cases were excluded that involved infected ulcers or suspected or confirmed deep tissue involvement (eg, osteomyelitis, myositis, fasciitis). Cases were also excluded that involved other infections where empiric antibiotic therapy with gram‐negative activity is standard including infected human or animal bites, periorbital or orbital infections, and perineal infections. The combined cohort in the present study therefore represented a group of patients hospitalized with relatively uncomplicated cellulitis or cutaneous abscess.
Definitions and Study Outcomes
Only 1 of the 2 studies from which the current cohort was derived distinguished between nonpurulent cellulitis, purulent cellulitis, and wound infection.[2] In the other study, cases were more broadly defined as either cellulitis or cutaneous abscess.[10] Infected ulcers and deep tissue infections were excluded from both studies. In combining the data into the current cohort, all nondrainable infections (purulent or nonpurulent cellulitis and wound infection) were categorized generally as cellulitis. All cases with documentation of an abscess in the medical record were categorized as cutaneous abscess. Presence of diabetes mellitus was based on provider documentation of the condition during the hospitalization. Microbiological cultures were obtained at the discretion of treating providers. Exposure to antibiotics with a broad spectrum of gram‐negative activity was defined as receipt of 2 or more calendar days of ‐lactam/‐lactamase inhibitor combinations, second‐ through fifth‐generation cephalosporins, fluoroquinolones, carbapenems, tigecycline, aminoglycosides, or colistin.[2]
The follow‐up periods differed slightly between the 2 studies used to derive the current cohort. In 1 study, all clinical encounters within 30 days of hospital discharge were reviewed to assess clinical outcomes.[10] In the other, clinical encounters within 45 days from the date of hospitalization were reviewed.[2] Clinical failure was defined as any of the following within the 30‐ or 45‐day follow‐up periods, respectively: (1) treatment failure, defined as a change in antibiotic therapy or unplanned drainage procedure due to inadequate clinical response more than 5 days[2] or 7 days[10] after hospital admission; (2) recurrence, defined as reinitiation of antibiotics for skin infection after completion of the initial treatment course; or (3) rehospitalization due to skin infection.[11]
Statistical Analysis
Because the clinical factors, microbiology, and treatment of cellulitis and cutaneous abscesses differ, analyses were performed for the total cohort and stratified by type of infection. Microorganisms cultured, antibiotic selection, and treatment duration were compared between diabetics and nondiabetics using the Wilcoxon rank sum test, 2, or Fisher exact test, as appropriate.
Because we hypothesized that the presence of diabetes mellitus in patients with cellulitis or abscess leads to use of broad gram‐negative therapy, we developed a multivariable logistic regression model to identify factors independently associated with exposure to antibiotics with broad gram‐negative activity. We also developed a linear regression model to explore the relationship between diabetes mellitus and duration of antibiotic therapy after adjusting for covariates. To develop these models, we first performed bivariate analyses and retained variables with a P value 0.25 in the regression models. Variables that did not meet the P value threshold but were considered to be clinically relevant covariates were also included in the model. We assessed for effect modification, multicollinearity, and goodness of fit when developing the models. We used SAS version 9.3 (SAS Institute, Cary, NC) for data analysis.
RESULTS
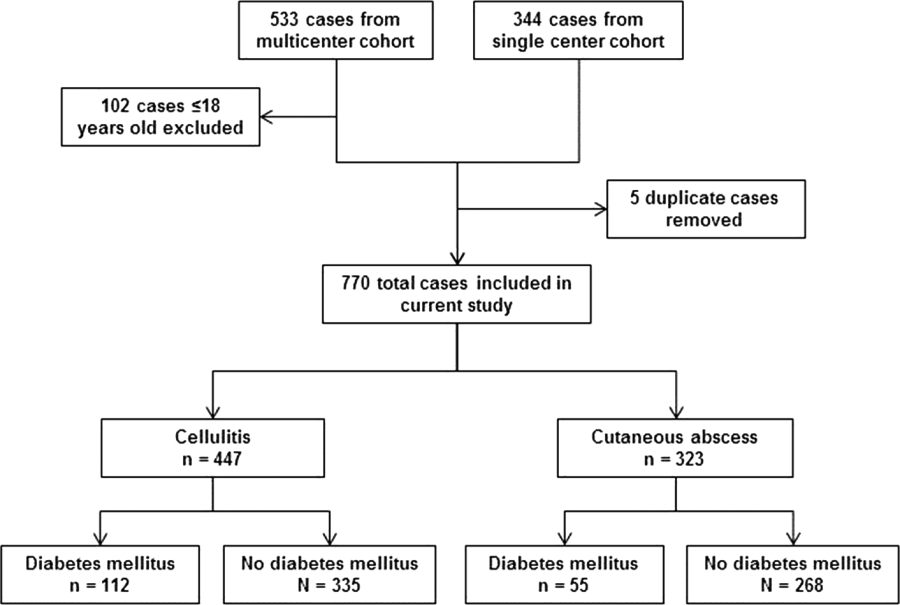
After excluding 102 pediatric cases and removing 5 duplicate cases, 770 total cases were included for analysis: 447 involved cellulitis and 323 involved cutaneous abscess (Figure 1). Overall, 167 (22%) patients had diabetes mellitus. Diabetics were significantly more likely than nondiabetics to have cellulitis as the presenting infection (67% of cases vs 56%, P=0.008) and to have lower extremity involvement (48% vs 33%, P<0.001) (Table 1). Diabetics were also older (median age 55 years vs 48 years, P<0.001), more likely to have cirrhosis or prior skin infection, and less likely to be injection‐drug users or human immunodeficiency virus (HIV) infected. Demographic and clinical characteristics among diabetics and nondiabetics stratified by the categorizations of cellulitis and cutaneous abscess are presented in the Supporting Information, Appendix Table 1, in the online version of this article.
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | |
|---|---|---|
| ||
| Type of infection | ||
| Cellulitis | 112 (67) | 335 (56)a |
| Cutaneous abscess | 55 (33) | 268 (44) |
| Age, y, median (IQR) | 55 (4763) | 48 (3658)a |
| Male | 102 (61) | 405 (67) |
| Injection drug use | 9 (5) | 117 (19)a |
| Alcohol abuse or dependence | 15 (9) | 86 (14) |
| Cirrhosis | 11 (7) | 17 (3)a |
| HIV infection | 0 | 29 (5)a |
| Dialysis dependence | 4 (2) | 5 (1) |
| Peripheral arterial disease | 4 (2) | 5 (1) |
| Saphenous vein harvest | 7 (4) | 11 (2) |
| Prior skin infection | 56 (34) | 125 (21)a |
| Prior MRSA infection or colonization | 20 (12) | 50 (8) |
| Anatomical location | ||
| Lower extremity | 80 (48) | 200 (33)a |
| Upper extremity | 6 (4) | 79 (13)a |
| Head and neck | 14 (8) | 38 (6) |
| Buttock or inguinal | 8 (5) | 35 (6) |
| Chest, abdomen, back, or axilla | 9 (5) | 25 (4) |
| Multiple distinct sites | 7 (4) | 34 (6) |
| Medical primary service | 139 (83) | 395 (66)a |
| Consultation requested | 99 (59) | 294 (49)a |
| Surgery | 58 (35) | 152 (25)a |
| Internal medicine | 18 (11) | 47 (8) |
| Infectious diseases | 41 (25) | 149 (25) |
| Failed initial outpatient antibiotic therapy | 52 (31) | 186 (31) |
| Fever (temperature 38.0C) | 20 (12) | 102 (17) |
| Leukocytosis (WBC >10,000 cells/mm3) | 78 (47) | 311 (52) |
The frequency of use of microbiological cultures was similar among diabetics and nondiabetics (Table 2). In cases of cellulitis, a microorganism was identified in 18% of diabetics and 12% of nondiabetics (P=0.09). In cases of cutaneous abscess, a microorganism was identified more commonly (69% and 74%, respectively, P=0.50). Among cases where a microorganism was identified, aerobic gram‐positive organisms were isolated in 90% of diabetics and 92% of nondiabetics (P=0.59). Aerobic gram‐negative organisms were isolated in 7% of diabetics and 12% of nondiabetics (P=0.28). Specific gram‐negative organisms isolated are shown in the Supporting Information, Appendix Table 2, in the online version of this article; no cases in diabetics involved Pseudomonas aeruginosa. The comparison of microbiological data among diabetics and nondiabetics was similar when stratified by cellulitis versus cutaneous abscess (Table 2).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Any microbiological culture obtaineda | 82 (73) | 234 (70) | 46 (84) | 239 (89) | 128 (77) | 473 (78) | |||
| Wound drainage or swab | 19 (17) | 36 (11) | 1 (2) | 8 (3) | 20 (12) | 44 (7) | |||
| Abscess material | 1 (1) | 3 (1) | 39 (71) | 205 (76) | 40 (24) | 208 (34) | |||
| Tissueb | 2 (2) | 17 (5) | 1 (2) | 8 (3) | 3 (2) | 25 (4) | |||
| Blood | 73 (65) | 212 (63) | 26 (47) | 121 (45) | 99 (59) | 333 (55) | |||
| Any microorganism identifiedc | 20 (18) | 39 (12) | 0.09 | 38 (69) | 197 (74) | 0.50 | 58 (35) | 236 (39) | 0.30 |
| Aerobic gram‐positive | 15 (75) | 36 (92) | 0.11 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 218 (92) | 0.59 |
| Staphylococcus aureus | 11 (55) | 26 (67) | 0.38 | 28 (74) | 132 (67) | 0.42 | 39 (67) | 158 (67) | 0.97 |
| Methicillin‐susceptible | 4 (20) | 15 (38) | 0.15 | 12 (32) | 42 (21) | 0.17 | 16 (28) | 57 (24) | 0.59 |
| Methicillin‐resistant | 5 (25) | 11 (28) | 1.00 | 14 (37) | 85 (43) | 0.47 | 19 (33) | 96 (41) | 0.27 |
| Susceptibility not performed | 2 (10) | 0 | 0.11 | 2 (5) | 5 (3) | 0.32 | 4 (7) | 5 (2) | 0.08 |
| Streptococcal species | 6 (30) | 15 (38) | 0.52 | 12 (32) | 69 (35) | 0.68 | 18 (31) | 84 (36) | 0.51 |
| ‐hemolytic streptococcus | 3 (15) | 13 (33) | 0.13 | 6 (16) | 32 (16) | 0.94 | 9 (16) | 45 (19) | 0.53 |
| Streptococcus anginosus/Streptococcus milleri group | 1 (5) | 0 | 0.34 | 2 (5) | 29 (15) | 0.11 | 3 (5) | 29 (12) | 0.12 |
| Other ‐hemolytic streptococcus | 2 (10) | 2 (5) | 0.60 | 4 (11) | 12 (6) | 0.30 | 6 (10) | 14 (6) | 0.25 |
| Other streptococcus | 0 | 0 | 1 (3) | 3 (2) | 0.51 | 1 (2) | 3 (1) | 0.59 | |
| Staphylococcus aureus or streptococci | 15 (75) | 35 (90) | 0.25 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 217 (92) | 0.60 |
| Enterococcus species | 0 | 2 (5) | 0.54 | 0 | 4 (2) | 1.00 | 0 | 6 (3) | 0.60 |
| Aerobic gram‐negative | 2 (10) | 7 (18) | 0.70 | 2 (5) | 21 (11) | 0.39 | 4 (7) | 28 (12) | 0.28 |
| Anaerobic organism(s) | 2 (10) | 3 (8) | 1.00 | 8 (21) | 30 (15) | 0.37 | 10 (17) | 33 (14) | 0.53 |
| Mixed skin or oral flora | 1 (5) | 1 (3) | 1.00 | 0 | 1 (1) | 1.00 | 1 (2) | 2 (1) | 0.48 |
| Other | 1 (5) | 3 (8) | 1.00 | 2 (5) | 3 (2) | 0.19 | 3 (5) | 6 (3) | 0.39 |
| Polymicrobial | 3 (15) | 17 (45) | 0.03 | 11 (29) | 47 (24) | 0.51 | 14 (24) | 64 (27) | 0.65 |
| Positive blood cultured | 4 (5) | 8 (4) | 0.51 | 2 (8) | 3 (2) | 0.21 | 6 (6) | 11 (3) | 0.24 |
Antibiotic utilization is summarized in Table 3. Among patients who were started on antibiotic therapy in the emergency department or urgent care, the initial regimen included an agent with broad gram‐negative activity in 31% of both diabetics and nondiabetics (P=0.97). During the entire hospital stay (including the emergency department or urgent care), diabetics were significantly more likely to be treated with ‐lactam/‐lactamase inhibitor combinations (42% vs 33%, P=0.04). At the time of hospital discharge, diabetics were more likely to be prescribed fluoroquinolones (11% vs 5%, P=0.01) (Table 3) particularly for cases of cellulitis (13% vs 6%, P=0.008) (see Supporting Information, Appendix Table 3, in the online version of this article). Diabetics were somewhat more likely to be prescribed parenteral antibiotics (10% vs 6%, P=0.07) after discharge. When considering both inpatient and discharge therapy, more diabetics than nondiabetics were exposed to at least 2 calendar days of broad gram‐negative therapy (54% vs 44%, P=0.02) and more were prescribed an antipseudomonal agent (38% vs 25%, P=0.002). In the group of patients who received at least 1 dose of an antibiotic with broad gram‐negative activity, broad gram‐negative agents accounted for 33% of the total days of therapy prescribed for diabetics and 32% for nondiabetics. Overall prescribing patterns were similar when stratified by cellulitis versus cutaneous abscess (see Supporting Information, Appendix Table 3, in the online version of this article).
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
|---|---|---|---|
| |||
| Individual antibiotics prescribed during the inpatient stayab | |||
| Vancomycin | 142 (85) | 504 (84) | 0.65 |
| Clindamycin | 27 (16) | 131 (22) | 0.12 |
| Parenteral ‐lactam/‐lactamase inhibitor | 70 (42) | 200 (33) | 0.04 |
| Second‐generation or higher cephalosporin | 13 (8) | 51 (8) | 0.78 |
| Cefazolin | 17 (10) | 91 (15) | 0.11 |
| Carbapenem | 9 (5) | 34 (6) | 0.90 |
| Fluoroquinolone | 20 (12) | 53 (9) | 0.21 |
| Daptomycin | 8 (5) | 24 (4) | 0.64 |
| Linezolid | 2 (1) | 8 (1) | 1.00 |
| Other ‐lactam | 6 (4) | 30 (5) | 0.45 |
| Trimethoprim‐sulfamethoxazole | 12 (7) | 30 (5) | 0.27 |
| Doxycycline | 15 (9) | 44 (7) | 0.47 |
| Cephalexin | 7 (4) | 22 (4) | 0.74 |
| Amoxicillin‐clavulanate | 11 (7) | 24 (4) | 0.15 |
| Antibiotics prescribed at hospital dischargeb | 163 (98) | 580 (96) | 0.38 |
| Clindamycin | 20 (12) | 95 (16) | 0.23 |
| Trimethoprim‐sulfamethoxazole | 52 (31) | 215 (36) | 0.28 |
| Doxycycline | 32 (19) | 91 (15) | 0.20 |
| Cephalexin | 12 (7) | 46 (8) | 0.85 |
| Amoxicillin‐clavulanate | 24 (14) | 82 (14) | 0.80 |
| Fluoroquinolone | 18 (11) | 32 (5) | 0.01 |
| Linezolid | 8 (5) | 19 (3) | 0.31 |
| Other oral ‐lactam | 3 (2) | 28 (5) | 0.10 |
| Other oral antibiotic | 1 (1) | 2 (0.3) | 0.52 |
| Vancomycin | 8 (5) | 15 (2) | 0.13 |
| Daptomycin | 5 (3) | 10 (2) | 0.34 |
| Other parenteral antibiotic | 4 (2) | 11 (2) | 0.75 |
| Antibiotic with broad gram‐negative activity initiated in emergency department or urgent care | 46/149 (31) | 174/561 (31) | 0.97 |
| Exposed to any antibiotic with broad gram‐negative activityc | 101 (62) | 311 (53) | 0.048 |
| Exposed to any antibiotic with antipseudomonal activity | 62 (38) | 149 (25) | 0.002 |
| Exposed to at least 2 calendar days of antibiotics with broad gram‐negative activityc | 89 (54) | 259 (44) | 0.02 |
| Treatment durationd | |||
| Total duration of therapy, d, median (IQR) | 13 (1015) | 12 (1015) | 0.09 |
| Duration of inpatient therapy, d, median (IQR) | 4 (36) | 4 (35) | 0.03 |
| Duration of therapy after discharge, d, median (IQR) | 8 (710) | 8 (710) | 0.58 |
After adjusting for covariates in the logistic regression model, diabetes mellitus was an independent predictor of exposure to broad gram‐negative therapy (see Supporting Information, Appendix Table 4, in the online version of this article). In addition to diabetes mellitus, culture of an aerobic gram‐negative microorganism, infectious diseases service consultation, presence of fever, and nonmedical admitting services were significantly associated with exposure to broad gram‐negative therapy. Prior methicillin‐resistant S aureus infection or colonization and HIV infection were inversely associated. Compared with nondiabetics, the total duration of antibiotic therapy in diabetics was somewhat longer (median 13 days vs 12 days, P=0.09) (Table 3). After adjusting for covariates in the linear regression model, there was a significant association between diabetes mellitus and treatment duration. On average, diabetics were treated 1 day (95% confidence interval: 0.2‐1.7 days) longer than nondiabetics.
Compared with nondiabetics, diabetics were more likely to have an outpatient follow‐up visit (73% vs 61%, P=0.002) and to be rehospitalized for any reason after discharge (16% vs 9%, P=0.02) (Table 4). Diabetics were overall more likely to be classified as clinical failure (15% vs 9%, P=0.02); this difference was driven by the cellulitis subgroup (19% vs 10%, P=0.01).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Survived to discharge | 111 (99) | 335 (100) | 0.25 | 55 (100) | 268 (100) | 166 (99) | 603 (100) | 0.22 | |
| Outpatient follow‐up documented | 82 (74) | 204 (61) | 0.01 | 40 (73) | 161 (60) | 0.08 | 122 (73) | 365 (61) | 0.002 |
| Rehospitalized | 22 (20) | 34 (10) | 0.008 | 4 (7) | 21 (8) | 1.00 | 26 (16) | 55 (9) | 0.02 |
| Clinical failure | 21 (19) | 34 (10) | 0.01 | 4 (7) | 20 (7) | 1.00 | 25 (15) | 54 (9) | 0.02 |
| Treatment failure | 7 (6) | 17 (5) | 0.62 | 2 (4) | 7 (3) | 0.65 | 9 (5) | 24 (4) | 0.42 |
| Recurrence | 10 (9) | 16 (5) | 0.10 | 1 (2) | 11 (4) | 0.70 | 11 (7) | 27 (4) | 0.26 |
| Rehospitalization due to skin infection | 14 (13) | 17 (5) | 0.01 | 3 (5) | 11 (4) | 0.71 | 17 (10) | 28 (5) | 0.01 |
| Length of stay, d, median (IQR) | 4 (36) | 4 (35) | 0.03 | 4 (36) | 4 (35) | 0.28 | 4 (36) | 4 (35) | 0.02 |
DISCUSSION
Diabetes mellitus is a common comorbidity in patients with acute bacterial skin infections. In this large cohort of patients hospitalized for cellulitis or cutaneous abscess, where those with infected ulcers or deep tissue infections were excluded, microbiological findings in cases associated with positive cultures were similar among diabetics and nondiabetics. Although aerobic gram‐negative microorganisms were not more likely to be identified in diabetics, diabetics were significantly more likely to be exposed to at least 2 calendar days of antibiotics with broad gram‐negative activity. After adjusting for covariates, diabetes mellitus was independently associated with exposure to broad gram‐negative therapy.
To our knowledge, this is the first study to compare the microbiology of cellulitis and cutaneous abscess among diabetics and nondiabetics. Lipsky and colleagues previously described the microbiology of a cohort of diabetic patients hospitalized with a broader range of skin infections including cellulitis, infected ulcers, and surgical site infections.[12] Similar to our findings, gram‐negative pathogens were uncommonly isolated in that study; however, in the absence of a comparator group, whether diabetics were at higher risk for gram‐negative involvement than nondiabetics was not known. Similar to the study by Lipsky and colleagues, most studies of skin infections in diabetics have included a relatively heterogeneous group of infections.[12, 13, 14, 15] The present study therefore contributes to the literature by providing a focused comparison of the microbiology of inpatient cellulitis and abscess in the absence of complicating factors such as an infected ulcer or deep tissue involvement. We found that among cases with a positive culture (13% of cases in the cellulitis group and 73% in the abscess group), the microbiology was similar among diabetics and nondiabetics. Although a microorganism was identified in only a minority of cases of cellulitis, our findings do not support the need for broad gram‐negative therapy in diabetics with cellulitis not associated with an ulcer or deep tissue infection. In diabetics with an abscess, antibiotics with broad gram‐negative activity do not appear to be indicated.
The present study also adds to the literature by providing a detailed comparison of antibiotic utilization patterns among diabetics and nondiabetics. We demonstrated that diabetics were more likely to have significant exposure to antibiotics with broad gram‐negative activity, particularly antipseudomonal agents (the broadest‐spectrum antibiotics). Because initiation of broad gram‐negative therapy in the emergency department or urgent care was not more common among diabetics, the increased use of these agents among diabetics appeared to be driven by inpatient providers. It is also notable that of patients who received any antibiotic with broad gram‐negative activity, these agents accounted for similar proportions of the total days of therapy in both diabetics and nondiabetics. In aggregate, our findings demonstrate that diabetics are more likely to be started on antibiotics with broad gram‐negative activity by inpatient providers, diabetics are not necessarily continued on longer durations of broad gram‐negative therapy once started, and the total amount of exposure to broad gram‐negative agents is substantial.
Overall, our findings suggest that inpatient providers perceive diabetics with cellulitis or abscess to be at increased risk for gram‐negative pathogens. This perhaps reflects an extrapolation of recommendations to use broad‐spectrum empiric therapy in diabetics with certain complicated skin infections.[7] However, for patients with cellulitis or cutaneous abscess, Infectious Diseases Society of America (IDSA) guidelines recommend antibiotic therapy targeted toward S aureus and streptococcal species; there is no suggestion to use a broader spectrum of therapy in diabetics.[8, 9] Our findings therefore highlight an important opportunity to improve antibiotic selection for all patients hospitalized with cellulitis and abscess, but particularly diabetics. It is also noteworthy that by linear regression, diabetes mellitus was independently associated with longer treatment durations. Although the average increase in treatment duration was small (1 day), this finding adds to the evidence that the presence of diabetes mellitus alters providers' treatment approach to cellulitis or abscess.
We found that despite more frequent treatment with broad gram‐negative therapy, diabetics were more likely than nondiabetics to be classified as clinical failures. It is important to point out that diabetics were also more likely than nondiabetics to have postdischarge outpatient follow‐up visits, raising the possibility of biased ascertainment of clinical failure events in this group. However, we also demonstrated that diabetics with cellulitis were more likely to be rehospitalized than nondiabetics. This is similar to a finding by Suaya and colleagues who showed that diabetics with skin infections were about twice as likely to be rehospitalized as nondiabetics.[13] One could hypothesize that the increased frequency of clinical failure events among diabetics was due to their older age, hyperglycemia, or vascular insufficiency; however, other factors may have contributed. For example, providers may have mistaken residual erythema for ongoing or recurrent cellulitis, or the diagnosis of cellulitis could have been incorrect to begin with. Additionally, there may have been uncertainty about the microbiology of cellulitis because the infecting pathogen was not usually identified. These factors may have led to alterations in treatment that would have resulted in a classification of clinical failure, and it is possible that providers had a lower threshold to alter treatment in diabetics. It is therefore not clear whether our findings represent a true difference in clinical outcomes between diabetics or nondiabetics. Regardless, in cases associated with a positive culture, our microbiological results do not support that the difference in clinical failure between diabetics and nondiabetics with cellulitis was related to a different spectrum of microorganisms.
In addition to the limitations outlined previously[2, 10] and above, the present study has at least 5 additional limitations. First, this was a secondary analysis of studies that were not designed to evaluate the effect of diabetes mellitus on the microbiology and treatment of skin infections. For example, hemoglobin A1C values were not collected; therefore, we could not examine whether the microbiology and antibiotic prescribing practices differed based on control of diabetes mellitus. Second, there were minor differences in inclusion and exclusion criteria between the 2 cohorts included in this study. Because the proportion of patients with diabetes mellitus was similar among both cohorts, and comparisons were not made between the cohorts, this should not have impacted our results. Third, the broad categorization of cellulitis used when combining the 2 cohorts raised the possibility of differences in infection characteristics between diabetics and nondiabetics (eg, presence of a wound) that could have confounded our findings regarding use of gram‐negative therapy. In the larger of the 2 cohorts from which the combined cohort was derived, only 17 (3%) of 533 patients had wound infections, whereas those with infected ulcers or suspected deep‐tissue infection were excluded from both cohorts. Furthermore, in the combined cohort, the increased frequency of broad gram‐negative therapy among diabetics was also observed in the cutaneous abscess group. It is therefore unlikely that the categorization of cellulitis had a significant impact on our results. Fourth, given the observational nature of the study, the microbiological data were subject to limitations. Importantly, because the infecting pathogen was identified in only 13% of cases of cellulitis, firm conclusions regarding the microbiology of cellulitis cannot be drawn. Finally, the small number of gram‐negative organisms isolated precluded comparisons of specific pathogens among diabetics and nondiabetics. In addition, because a number of gram‐negative organisms were isolated from wound cultures, it is not known whether they were clinically relevant or simply represented colonization.
In conclusion, in cases of cellulitis or abscess associated with a positive culture, gram‐negative microorganisms were not isolated more commonly among diabetics compared with nondiabetics. However, in general, diabetics were more likely to be treated with broad gram‐negative therapy suggesting that, particularly for cutaneous abscesses, this prescribing practice may not be warranted. These findings support current IDSA guidelines that recommend antibiotic therapy targeted toward gram‐positive pathogens for cellulitis or abscess, irrespective of the presence of diabetes mellitus.[8, 9] Because nearly one‐fourth of patients hospitalized with cellulitis or abscess are diabetic, these findings have relevance for national antimicrobial stewardship efforts aimed at curbing antimicrobial resistance through reducing use of antibiotics with broad gram‐negative activity in hospitals.[16]
Disclosures: This work was supported by the National Institute of Allergy and Infectious Diseases, National Institute of Health (TCJ: K23 AI099082). D.M.P. reports potential conflicts of interests with Optimer, Cubist, and Forest Pharmaceuticals. The authors report no other conflicts of interest.
- , , , et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. 2005;41(3):281–288.
- , , , et al. Antibiotic prescribing practices in a multicenter cohort of patients hospitalized for acute bacterial skin and skin structure infection. Infect Control Hosp Epidemiol. 2014;35(10):1241–1250.
- , , , . The role of beta‐hemolytic streptococci in causing diffuse, nonculturable cellulitis: a prospective investigation. Medicine. 2010;89(4):217–226.
- , , , et al. Factors associated with complications and mortality in adult patients hospitalized for infectious cellulitis. Eur J Clin Microbiol Infect Dis. 2003;22(3):151–157.
- , , , et al. Epidemiology and outcomes of complicated skin and soft tissue infections in hospitalized patients. J Clin Microbiol. 2012;50(2):238–245.
- , , , , , . Current management of patients hospitalized with complicated skin and soft tissue infections across Europe (2010–2011): assessment of clinical practice patterns and real‐life effectiveness of antibiotics from the REACH study. Clin Microbiol Infect. 2013;19(9):E377–E385.
- , , , et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132–e173.
- , , , et al. Clinical practice guidelines by the Infectious Diseases Society Of America for the treatment of methicillin‐resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52(3):285–292.
- , , , et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society Of America. Clin Infect Dis. 2014;59(2):e10–e52.
- , , , et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med. 2011;171(12):1072–1079.
- , , , , , . Skin and soft‐tissue infections requiring hospitalization at an academic medical center: opportunities for antimicrobial stewardship. Clin Infect Dis. 2010;51(8):895–903.
- , , , , , . Skin and soft tissue infections in hospitalised patients with diabetes: culture isolates and risk factors associated with mortality, length of stay and cost. Diabetologia. 2010;53(5):914–923.
- , , , . Skin and soft tissue infections and associated complications among commercially insured patients aged 0–64 years with and without diabetes in the U.S. PLoS One. 2013;8(4):e60057.
- , , . A post hoc subgroup analysis of meropenem versus imipenem/cilastatin in a multicenter, double‐blind, randomized study of complicated skin and skin‐structure infections in patients with diabetes mellitus. Clin Ther. 2006;28(8):1164–1174.
- , , , . Treating diabetic foot infections with sequential intravenous to oral moxifloxacin compared with piperacillin‐tazobactam/amoxicillin‐clavulanate. J Antimicr Chemo. 2007;60(2):370–376.
- , , , et al. Vital signs: improving antibiotic use among hospitalized patients. MMWR Morb Mortal Wkly Rep. 2014;63(9):194–200.
Diabetes mellitus is one of the most common comorbid conditions among patients hospitalized for acute bacterial skin infections.[1, 2, 3, 4, 5, 6] Acute bacterial skin infections in diabetics represent a spectrum of conditions ranging from cellulitis or cutaneous abscess to more complicated infections such as infected ulcers or deep tissue infections. Although most skin infections in diabetics are caused by gram‐positive pathogens (Staphylococcus aureus and streptococci), the risk of gram‐negative pathogens is increased in certain complicated infections such as diabetic foot infections.[7] For such complicated infections, national guidelines therefore recommend broad‐spectrum empiric antibiotic therapy.[7]
The role of gram‐negative pathogens has not been clearly established in diabetics with cellulitis or cutaneous abscess not associated with an infected ulcer or diabetic foot infection. National guidelines for the treatment of cellulitis and abscess recommend antibiotic therapy targeted toward S aureus and streptococcal species irrespective of the presence of diabetes mellitus.[8, 9] However, in a recent multicenter study of patients hospitalized with acute bacterial skin infections in which cases involving infected ulcers or deep tissue infection were excluded, diabetes mellitus was an independent predictor of use of antibiotics with broad gram‐negative activity.[2] This suggests that either gram‐negative pathogens are more common or providers perceive gram‐negative pathogens to be more common among diabetics with otherwise uncomplicated cellulitis or abscess.
A better understanding of the relationship between the microbiology and antibiotic prescribing practices for diabetics with cellulitis or abscess is therefore necessary to promote the most appropriate spectrum of therapy for these patients. We evaluated a large cohort of patients hospitalized with acute bacterial skin infections in order to: (1) compare the microbiology of diabetics and nondiabetics with cellulitis or cutaneous abscess not associated with an ulcer or deep tissue infection; and (2) compare antibiotic prescribing practices among diabetics and nondiabetics. We hypothesized that diabetics would have a similar spectrum of microorganisms as nondiabetics but would be more frequently treated with antibiotics with broad gram‐negative activity.
METHODS
Study Design
This was a secondary analysis of 2 published retrospective studies of patients hospitalized for cellulitis or cutaneous abscess between January 1, 2007 and May 31, 2012.[2, 10] For the purposes of this study, the terms cellulitis and abscess will refer to infections not involving an infected ulcer, osteomyelitis, or other deep tissue infection.
Study Setting and Population
The first of the 2 cohorts analyzed for the present study included patients hospitalized with cellulitis, abscess, or wound infection at 7 academic or community hospitals in Colorado.[2] The second cohort included patients hospitalized with cellulitis or abscess at a single academic medical center (1 of the 7 hospitals above) in Denver, Colorado.[10] The methods of these studies have been reported in detail elsewhere.[2, 10, 11] Briefly, potential cases were identified using International Classification of Diseases, 9th Revision, Clinical Modification codes. The main inclusion and exclusion criteria of the 2 studies were similar. In both studies, cases were excluded that involved infected ulcers or suspected or confirmed deep tissue involvement (eg, osteomyelitis, myositis, fasciitis). Cases were also excluded that involved other infections where empiric antibiotic therapy with gram‐negative activity is standard including infected human or animal bites, periorbital or orbital infections, and perineal infections. The combined cohort in the present study therefore represented a group of patients hospitalized with relatively uncomplicated cellulitis or cutaneous abscess.
Definitions and Study Outcomes
Only 1 of the 2 studies from which the current cohort was derived distinguished between nonpurulent cellulitis, purulent cellulitis, and wound infection.[2] In the other study, cases were more broadly defined as either cellulitis or cutaneous abscess.[10] Infected ulcers and deep tissue infections were excluded from both studies. In combining the data into the current cohort, all nondrainable infections (purulent or nonpurulent cellulitis and wound infection) were categorized generally as cellulitis. All cases with documentation of an abscess in the medical record were categorized as cutaneous abscess. Presence of diabetes mellitus was based on provider documentation of the condition during the hospitalization. Microbiological cultures were obtained at the discretion of treating providers. Exposure to antibiotics with a broad spectrum of gram‐negative activity was defined as receipt of 2 or more calendar days of ‐lactam/‐lactamase inhibitor combinations, second‐ through fifth‐generation cephalosporins, fluoroquinolones, carbapenems, tigecycline, aminoglycosides, or colistin.[2]
The follow‐up periods differed slightly between the 2 studies used to derive the current cohort. In 1 study, all clinical encounters within 30 days of hospital discharge were reviewed to assess clinical outcomes.[10] In the other, clinical encounters within 45 days from the date of hospitalization were reviewed.[2] Clinical failure was defined as any of the following within the 30‐ or 45‐day follow‐up periods, respectively: (1) treatment failure, defined as a change in antibiotic therapy or unplanned drainage procedure due to inadequate clinical response more than 5 days[2] or 7 days[10] after hospital admission; (2) recurrence, defined as reinitiation of antibiotics for skin infection after completion of the initial treatment course; or (3) rehospitalization due to skin infection.[11]
Statistical Analysis
Because the clinical factors, microbiology, and treatment of cellulitis and cutaneous abscesses differ, analyses were performed for the total cohort and stratified by type of infection. Microorganisms cultured, antibiotic selection, and treatment duration were compared between diabetics and nondiabetics using the Wilcoxon rank sum test, 2, or Fisher exact test, as appropriate.
Because we hypothesized that the presence of diabetes mellitus in patients with cellulitis or abscess leads to use of broad gram‐negative therapy, we developed a multivariable logistic regression model to identify factors independently associated with exposure to antibiotics with broad gram‐negative activity. We also developed a linear regression model to explore the relationship between diabetes mellitus and duration of antibiotic therapy after adjusting for covariates. To develop these models, we first performed bivariate analyses and retained variables with a P value 0.25 in the regression models. Variables that did not meet the P value threshold but were considered to be clinically relevant covariates were also included in the model. We assessed for effect modification, multicollinearity, and goodness of fit when developing the models. We used SAS version 9.3 (SAS Institute, Cary, NC) for data analysis.
RESULTS
After excluding 102 pediatric cases and removing 5 duplicate cases, 770 total cases were included for analysis: 447 involved cellulitis and 323 involved cutaneous abscess (Figure 1). Overall, 167 (22%) patients had diabetes mellitus. Diabetics were significantly more likely than nondiabetics to have cellulitis as the presenting infection (67% of cases vs 56%, P=0.008) and to have lower extremity involvement (48% vs 33%, P<0.001) (Table 1). Diabetics were also older (median age 55 years vs 48 years, P<0.001), more likely to have cirrhosis or prior skin infection, and less likely to be injection‐drug users or human immunodeficiency virus (HIV) infected. Demographic and clinical characteristics among diabetics and nondiabetics stratified by the categorizations of cellulitis and cutaneous abscess are presented in the Supporting Information, Appendix Table 1, in the online version of this article.
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | |
|---|---|---|
| ||
| Type of infection | ||
| Cellulitis | 112 (67) | 335 (56)a |
| Cutaneous abscess | 55 (33) | 268 (44) |
| Age, y, median (IQR) | 55 (4763) | 48 (3658)a |
| Male | 102 (61) | 405 (67) |
| Injection drug use | 9 (5) | 117 (19)a |
| Alcohol abuse or dependence | 15 (9) | 86 (14) |
| Cirrhosis | 11 (7) | 17 (3)a |
| HIV infection | 0 | 29 (5)a |
| Dialysis dependence | 4 (2) | 5 (1) |
| Peripheral arterial disease | 4 (2) | 5 (1) |
| Saphenous vein harvest | 7 (4) | 11 (2) |
| Prior skin infection | 56 (34) | 125 (21)a |
| Prior MRSA infection or colonization | 20 (12) | 50 (8) |
| Anatomical location | ||
| Lower extremity | 80 (48) | 200 (33)a |
| Upper extremity | 6 (4) | 79 (13)a |
| Head and neck | 14 (8) | 38 (6) |
| Buttock or inguinal | 8 (5) | 35 (6) |
| Chest, abdomen, back, or axilla | 9 (5) | 25 (4) |
| Multiple distinct sites | 7 (4) | 34 (6) |
| Medical primary service | 139 (83) | 395 (66)a |
| Consultation requested | 99 (59) | 294 (49)a |
| Surgery | 58 (35) | 152 (25)a |
| Internal medicine | 18 (11) | 47 (8) |
| Infectious diseases | 41 (25) | 149 (25) |
| Failed initial outpatient antibiotic therapy | 52 (31) | 186 (31) |
| Fever (temperature 38.0C) | 20 (12) | 102 (17) |
| Leukocytosis (WBC >10,000 cells/mm3) | 78 (47) | 311 (52) |
The frequency of use of microbiological cultures was similar among diabetics and nondiabetics (Table 2). In cases of cellulitis, a microorganism was identified in 18% of diabetics and 12% of nondiabetics (P=0.09). In cases of cutaneous abscess, a microorganism was identified more commonly (69% and 74%, respectively, P=0.50). Among cases where a microorganism was identified, aerobic gram‐positive organisms were isolated in 90% of diabetics and 92% of nondiabetics (P=0.59). Aerobic gram‐negative organisms were isolated in 7% of diabetics and 12% of nondiabetics (P=0.28). Specific gram‐negative organisms isolated are shown in the Supporting Information, Appendix Table 2, in the online version of this article; no cases in diabetics involved Pseudomonas aeruginosa. The comparison of microbiological data among diabetics and nondiabetics was similar when stratified by cellulitis versus cutaneous abscess (Table 2).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Any microbiological culture obtaineda | 82 (73) | 234 (70) | 46 (84) | 239 (89) | 128 (77) | 473 (78) | |||
| Wound drainage or swab | 19 (17) | 36 (11) | 1 (2) | 8 (3) | 20 (12) | 44 (7) | |||
| Abscess material | 1 (1) | 3 (1) | 39 (71) | 205 (76) | 40 (24) | 208 (34) | |||
| Tissueb | 2 (2) | 17 (5) | 1 (2) | 8 (3) | 3 (2) | 25 (4) | |||
| Blood | 73 (65) | 212 (63) | 26 (47) | 121 (45) | 99 (59) | 333 (55) | |||
| Any microorganism identifiedc | 20 (18) | 39 (12) | 0.09 | 38 (69) | 197 (74) | 0.50 | 58 (35) | 236 (39) | 0.30 |
| Aerobic gram‐positive | 15 (75) | 36 (92) | 0.11 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 218 (92) | 0.59 |
| Staphylococcus aureus | 11 (55) | 26 (67) | 0.38 | 28 (74) | 132 (67) | 0.42 | 39 (67) | 158 (67) | 0.97 |
| Methicillin‐susceptible | 4 (20) | 15 (38) | 0.15 | 12 (32) | 42 (21) | 0.17 | 16 (28) | 57 (24) | 0.59 |
| Methicillin‐resistant | 5 (25) | 11 (28) | 1.00 | 14 (37) | 85 (43) | 0.47 | 19 (33) | 96 (41) | 0.27 |
| Susceptibility not performed | 2 (10) | 0 | 0.11 | 2 (5) | 5 (3) | 0.32 | 4 (7) | 5 (2) | 0.08 |
| Streptococcal species | 6 (30) | 15 (38) | 0.52 | 12 (32) | 69 (35) | 0.68 | 18 (31) | 84 (36) | 0.51 |
| ‐hemolytic streptococcus | 3 (15) | 13 (33) | 0.13 | 6 (16) | 32 (16) | 0.94 | 9 (16) | 45 (19) | 0.53 |
| Streptococcus anginosus/Streptococcus milleri group | 1 (5) | 0 | 0.34 | 2 (5) | 29 (15) | 0.11 | 3 (5) | 29 (12) | 0.12 |
| Other ‐hemolytic streptococcus | 2 (10) | 2 (5) | 0.60 | 4 (11) | 12 (6) | 0.30 | 6 (10) | 14 (6) | 0.25 |
| Other streptococcus | 0 | 0 | 1 (3) | 3 (2) | 0.51 | 1 (2) | 3 (1) | 0.59 | |
| Staphylococcus aureus or streptococci | 15 (75) | 35 (90) | 0.25 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 217 (92) | 0.60 |
| Enterococcus species | 0 | 2 (5) | 0.54 | 0 | 4 (2) | 1.00 | 0 | 6 (3) | 0.60 |
| Aerobic gram‐negative | 2 (10) | 7 (18) | 0.70 | 2 (5) | 21 (11) | 0.39 | 4 (7) | 28 (12) | 0.28 |
| Anaerobic organism(s) | 2 (10) | 3 (8) | 1.00 | 8 (21) | 30 (15) | 0.37 | 10 (17) | 33 (14) | 0.53 |
| Mixed skin or oral flora | 1 (5) | 1 (3) | 1.00 | 0 | 1 (1) | 1.00 | 1 (2) | 2 (1) | 0.48 |
| Other | 1 (5) | 3 (8) | 1.00 | 2 (5) | 3 (2) | 0.19 | 3 (5) | 6 (3) | 0.39 |
| Polymicrobial | 3 (15) | 17 (45) | 0.03 | 11 (29) | 47 (24) | 0.51 | 14 (24) | 64 (27) | 0.65 |
| Positive blood cultured | 4 (5) | 8 (4) | 0.51 | 2 (8) | 3 (2) | 0.21 | 6 (6) | 11 (3) | 0.24 |
Antibiotic utilization is summarized in Table 3. Among patients who were started on antibiotic therapy in the emergency department or urgent care, the initial regimen included an agent with broad gram‐negative activity in 31% of both diabetics and nondiabetics (P=0.97). During the entire hospital stay (including the emergency department or urgent care), diabetics were significantly more likely to be treated with ‐lactam/‐lactamase inhibitor combinations (42% vs 33%, P=0.04). At the time of hospital discharge, diabetics were more likely to be prescribed fluoroquinolones (11% vs 5%, P=0.01) (Table 3) particularly for cases of cellulitis (13% vs 6%, P=0.008) (see Supporting Information, Appendix Table 3, in the online version of this article). Diabetics were somewhat more likely to be prescribed parenteral antibiotics (10% vs 6%, P=0.07) after discharge. When considering both inpatient and discharge therapy, more diabetics than nondiabetics were exposed to at least 2 calendar days of broad gram‐negative therapy (54% vs 44%, P=0.02) and more were prescribed an antipseudomonal agent (38% vs 25%, P=0.002). In the group of patients who received at least 1 dose of an antibiotic with broad gram‐negative activity, broad gram‐negative agents accounted for 33% of the total days of therapy prescribed for diabetics and 32% for nondiabetics. Overall prescribing patterns were similar when stratified by cellulitis versus cutaneous abscess (see Supporting Information, Appendix Table 3, in the online version of this article).
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
|---|---|---|---|
| |||
| Individual antibiotics prescribed during the inpatient stayab | |||
| Vancomycin | 142 (85) | 504 (84) | 0.65 |
| Clindamycin | 27 (16) | 131 (22) | 0.12 |
| Parenteral ‐lactam/‐lactamase inhibitor | 70 (42) | 200 (33) | 0.04 |
| Second‐generation or higher cephalosporin | 13 (8) | 51 (8) | 0.78 |
| Cefazolin | 17 (10) | 91 (15) | 0.11 |
| Carbapenem | 9 (5) | 34 (6) | 0.90 |
| Fluoroquinolone | 20 (12) | 53 (9) | 0.21 |
| Daptomycin | 8 (5) | 24 (4) | 0.64 |
| Linezolid | 2 (1) | 8 (1) | 1.00 |
| Other ‐lactam | 6 (4) | 30 (5) | 0.45 |
| Trimethoprim‐sulfamethoxazole | 12 (7) | 30 (5) | 0.27 |
| Doxycycline | 15 (9) | 44 (7) | 0.47 |
| Cephalexin | 7 (4) | 22 (4) | 0.74 |
| Amoxicillin‐clavulanate | 11 (7) | 24 (4) | 0.15 |
| Antibiotics prescribed at hospital dischargeb | 163 (98) | 580 (96) | 0.38 |
| Clindamycin | 20 (12) | 95 (16) | 0.23 |
| Trimethoprim‐sulfamethoxazole | 52 (31) | 215 (36) | 0.28 |
| Doxycycline | 32 (19) | 91 (15) | 0.20 |
| Cephalexin | 12 (7) | 46 (8) | 0.85 |
| Amoxicillin‐clavulanate | 24 (14) | 82 (14) | 0.80 |
| Fluoroquinolone | 18 (11) | 32 (5) | 0.01 |
| Linezolid | 8 (5) | 19 (3) | 0.31 |
| Other oral ‐lactam | 3 (2) | 28 (5) | 0.10 |
| Other oral antibiotic | 1 (1) | 2 (0.3) | 0.52 |
| Vancomycin | 8 (5) | 15 (2) | 0.13 |
| Daptomycin | 5 (3) | 10 (2) | 0.34 |
| Other parenteral antibiotic | 4 (2) | 11 (2) | 0.75 |
| Antibiotic with broad gram‐negative activity initiated in emergency department or urgent care | 46/149 (31) | 174/561 (31) | 0.97 |
| Exposed to any antibiotic with broad gram‐negative activityc | 101 (62) | 311 (53) | 0.048 |
| Exposed to any antibiotic with antipseudomonal activity | 62 (38) | 149 (25) | 0.002 |
| Exposed to at least 2 calendar days of antibiotics with broad gram‐negative activityc | 89 (54) | 259 (44) | 0.02 |
| Treatment durationd | |||
| Total duration of therapy, d, median (IQR) | 13 (1015) | 12 (1015) | 0.09 |
| Duration of inpatient therapy, d, median (IQR) | 4 (36) | 4 (35) | 0.03 |
| Duration of therapy after discharge, d, median (IQR) | 8 (710) | 8 (710) | 0.58 |
After adjusting for covariates in the logistic regression model, diabetes mellitus was an independent predictor of exposure to broad gram‐negative therapy (see Supporting Information, Appendix Table 4, in the online version of this article). In addition to diabetes mellitus, culture of an aerobic gram‐negative microorganism, infectious diseases service consultation, presence of fever, and nonmedical admitting services were significantly associated with exposure to broad gram‐negative therapy. Prior methicillin‐resistant S aureus infection or colonization and HIV infection were inversely associated. Compared with nondiabetics, the total duration of antibiotic therapy in diabetics was somewhat longer (median 13 days vs 12 days, P=0.09) (Table 3). After adjusting for covariates in the linear regression model, there was a significant association between diabetes mellitus and treatment duration. On average, diabetics were treated 1 day (95% confidence interval: 0.2‐1.7 days) longer than nondiabetics.
Compared with nondiabetics, diabetics were more likely to have an outpatient follow‐up visit (73% vs 61%, P=0.002) and to be rehospitalized for any reason after discharge (16% vs 9%, P=0.02) (Table 4). Diabetics were overall more likely to be classified as clinical failure (15% vs 9%, P=0.02); this difference was driven by the cellulitis subgroup (19% vs 10%, P=0.01).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Survived to discharge | 111 (99) | 335 (100) | 0.25 | 55 (100) | 268 (100) | 166 (99) | 603 (100) | 0.22 | |
| Outpatient follow‐up documented | 82 (74) | 204 (61) | 0.01 | 40 (73) | 161 (60) | 0.08 | 122 (73) | 365 (61) | 0.002 |
| Rehospitalized | 22 (20) | 34 (10) | 0.008 | 4 (7) | 21 (8) | 1.00 | 26 (16) | 55 (9) | 0.02 |
| Clinical failure | 21 (19) | 34 (10) | 0.01 | 4 (7) | 20 (7) | 1.00 | 25 (15) | 54 (9) | 0.02 |
| Treatment failure | 7 (6) | 17 (5) | 0.62 | 2 (4) | 7 (3) | 0.65 | 9 (5) | 24 (4) | 0.42 |
| Recurrence | 10 (9) | 16 (5) | 0.10 | 1 (2) | 11 (4) | 0.70 | 11 (7) | 27 (4) | 0.26 |
| Rehospitalization due to skin infection | 14 (13) | 17 (5) | 0.01 | 3 (5) | 11 (4) | 0.71 | 17 (10) | 28 (5) | 0.01 |
| Length of stay, d, median (IQR) | 4 (36) | 4 (35) | 0.03 | 4 (36) | 4 (35) | 0.28 | 4 (36) | 4 (35) | 0.02 |
DISCUSSION
Diabetes mellitus is a common comorbidity in patients with acute bacterial skin infections. In this large cohort of patients hospitalized for cellulitis or cutaneous abscess, where those with infected ulcers or deep tissue infections were excluded, microbiological findings in cases associated with positive cultures were similar among diabetics and nondiabetics. Although aerobic gram‐negative microorganisms were not more likely to be identified in diabetics, diabetics were significantly more likely to be exposed to at least 2 calendar days of antibiotics with broad gram‐negative activity. After adjusting for covariates, diabetes mellitus was independently associated with exposure to broad gram‐negative therapy.
To our knowledge, this is the first study to compare the microbiology of cellulitis and cutaneous abscess among diabetics and nondiabetics. Lipsky and colleagues previously described the microbiology of a cohort of diabetic patients hospitalized with a broader range of skin infections including cellulitis, infected ulcers, and surgical site infections.[12] Similar to our findings, gram‐negative pathogens were uncommonly isolated in that study; however, in the absence of a comparator group, whether diabetics were at higher risk for gram‐negative involvement than nondiabetics was not known. Similar to the study by Lipsky and colleagues, most studies of skin infections in diabetics have included a relatively heterogeneous group of infections.[12, 13, 14, 15] The present study therefore contributes to the literature by providing a focused comparison of the microbiology of inpatient cellulitis and abscess in the absence of complicating factors such as an infected ulcer or deep tissue involvement. We found that among cases with a positive culture (13% of cases in the cellulitis group and 73% in the abscess group), the microbiology was similar among diabetics and nondiabetics. Although a microorganism was identified in only a minority of cases of cellulitis, our findings do not support the need for broad gram‐negative therapy in diabetics with cellulitis not associated with an ulcer or deep tissue infection. In diabetics with an abscess, antibiotics with broad gram‐negative activity do not appear to be indicated.
The present study also adds to the literature by providing a detailed comparison of antibiotic utilization patterns among diabetics and nondiabetics. We demonstrated that diabetics were more likely to have significant exposure to antibiotics with broad gram‐negative activity, particularly antipseudomonal agents (the broadest‐spectrum antibiotics). Because initiation of broad gram‐negative therapy in the emergency department or urgent care was not more common among diabetics, the increased use of these agents among diabetics appeared to be driven by inpatient providers. It is also notable that of patients who received any antibiotic with broad gram‐negative activity, these agents accounted for similar proportions of the total days of therapy in both diabetics and nondiabetics. In aggregate, our findings demonstrate that diabetics are more likely to be started on antibiotics with broad gram‐negative activity by inpatient providers, diabetics are not necessarily continued on longer durations of broad gram‐negative therapy once started, and the total amount of exposure to broad gram‐negative agents is substantial.
Overall, our findings suggest that inpatient providers perceive diabetics with cellulitis or abscess to be at increased risk for gram‐negative pathogens. This perhaps reflects an extrapolation of recommendations to use broad‐spectrum empiric therapy in diabetics with certain complicated skin infections.[7] However, for patients with cellulitis or cutaneous abscess, Infectious Diseases Society of America (IDSA) guidelines recommend antibiotic therapy targeted toward S aureus and streptococcal species; there is no suggestion to use a broader spectrum of therapy in diabetics.[8, 9] Our findings therefore highlight an important opportunity to improve antibiotic selection for all patients hospitalized with cellulitis and abscess, but particularly diabetics. It is also noteworthy that by linear regression, diabetes mellitus was independently associated with longer treatment durations. Although the average increase in treatment duration was small (1 day), this finding adds to the evidence that the presence of diabetes mellitus alters providers' treatment approach to cellulitis or abscess.
We found that despite more frequent treatment with broad gram‐negative therapy, diabetics were more likely than nondiabetics to be classified as clinical failures. It is important to point out that diabetics were also more likely than nondiabetics to have postdischarge outpatient follow‐up visits, raising the possibility of biased ascertainment of clinical failure events in this group. However, we also demonstrated that diabetics with cellulitis were more likely to be rehospitalized than nondiabetics. This is similar to a finding by Suaya and colleagues who showed that diabetics with skin infections were about twice as likely to be rehospitalized as nondiabetics.[13] One could hypothesize that the increased frequency of clinical failure events among diabetics was due to their older age, hyperglycemia, or vascular insufficiency; however, other factors may have contributed. For example, providers may have mistaken residual erythema for ongoing or recurrent cellulitis, or the diagnosis of cellulitis could have been incorrect to begin with. Additionally, there may have been uncertainty about the microbiology of cellulitis because the infecting pathogen was not usually identified. These factors may have led to alterations in treatment that would have resulted in a classification of clinical failure, and it is possible that providers had a lower threshold to alter treatment in diabetics. It is therefore not clear whether our findings represent a true difference in clinical outcomes between diabetics or nondiabetics. Regardless, in cases associated with a positive culture, our microbiological results do not support that the difference in clinical failure between diabetics and nondiabetics with cellulitis was related to a different spectrum of microorganisms.
In addition to the limitations outlined previously[2, 10] and above, the present study has at least 5 additional limitations. First, this was a secondary analysis of studies that were not designed to evaluate the effect of diabetes mellitus on the microbiology and treatment of skin infections. For example, hemoglobin A1C values were not collected; therefore, we could not examine whether the microbiology and antibiotic prescribing practices differed based on control of diabetes mellitus. Second, there were minor differences in inclusion and exclusion criteria between the 2 cohorts included in this study. Because the proportion of patients with diabetes mellitus was similar among both cohorts, and comparisons were not made between the cohorts, this should not have impacted our results. Third, the broad categorization of cellulitis used when combining the 2 cohorts raised the possibility of differences in infection characteristics between diabetics and nondiabetics (eg, presence of a wound) that could have confounded our findings regarding use of gram‐negative therapy. In the larger of the 2 cohorts from which the combined cohort was derived, only 17 (3%) of 533 patients had wound infections, whereas those with infected ulcers or suspected deep‐tissue infection were excluded from both cohorts. Furthermore, in the combined cohort, the increased frequency of broad gram‐negative therapy among diabetics was also observed in the cutaneous abscess group. It is therefore unlikely that the categorization of cellulitis had a significant impact on our results. Fourth, given the observational nature of the study, the microbiological data were subject to limitations. Importantly, because the infecting pathogen was identified in only 13% of cases of cellulitis, firm conclusions regarding the microbiology of cellulitis cannot be drawn. Finally, the small number of gram‐negative organisms isolated precluded comparisons of specific pathogens among diabetics and nondiabetics. In addition, because a number of gram‐negative organisms were isolated from wound cultures, it is not known whether they were clinically relevant or simply represented colonization.
In conclusion, in cases of cellulitis or abscess associated with a positive culture, gram‐negative microorganisms were not isolated more commonly among diabetics compared with nondiabetics. However, in general, diabetics were more likely to be treated with broad gram‐negative therapy suggesting that, particularly for cutaneous abscesses, this prescribing practice may not be warranted. These findings support current IDSA guidelines that recommend antibiotic therapy targeted toward gram‐positive pathogens for cellulitis or abscess, irrespective of the presence of diabetes mellitus.[8, 9] Because nearly one‐fourth of patients hospitalized with cellulitis or abscess are diabetic, these findings have relevance for national antimicrobial stewardship efforts aimed at curbing antimicrobial resistance through reducing use of antibiotics with broad gram‐negative activity in hospitals.[16]
Disclosures: This work was supported by the National Institute of Allergy and Infectious Diseases, National Institute of Health (TCJ: K23 AI099082). D.M.P. reports potential conflicts of interests with Optimer, Cubist, and Forest Pharmaceuticals. The authors report no other conflicts of interest.
Diabetes mellitus is one of the most common comorbid conditions among patients hospitalized for acute bacterial skin infections.[1, 2, 3, 4, 5, 6] Acute bacterial skin infections in diabetics represent a spectrum of conditions ranging from cellulitis or cutaneous abscess to more complicated infections such as infected ulcers or deep tissue infections. Although most skin infections in diabetics are caused by gram‐positive pathogens (Staphylococcus aureus and streptococci), the risk of gram‐negative pathogens is increased in certain complicated infections such as diabetic foot infections.[7] For such complicated infections, national guidelines therefore recommend broad‐spectrum empiric antibiotic therapy.[7]
The role of gram‐negative pathogens has not been clearly established in diabetics with cellulitis or cutaneous abscess not associated with an infected ulcer or diabetic foot infection. National guidelines for the treatment of cellulitis and abscess recommend antibiotic therapy targeted toward S aureus and streptococcal species irrespective of the presence of diabetes mellitus.[8, 9] However, in a recent multicenter study of patients hospitalized with acute bacterial skin infections in which cases involving infected ulcers or deep tissue infection were excluded, diabetes mellitus was an independent predictor of use of antibiotics with broad gram‐negative activity.[2] This suggests that either gram‐negative pathogens are more common or providers perceive gram‐negative pathogens to be more common among diabetics with otherwise uncomplicated cellulitis or abscess.
A better understanding of the relationship between the microbiology and antibiotic prescribing practices for diabetics with cellulitis or abscess is therefore necessary to promote the most appropriate spectrum of therapy for these patients. We evaluated a large cohort of patients hospitalized with acute bacterial skin infections in order to: (1) compare the microbiology of diabetics and nondiabetics with cellulitis or cutaneous abscess not associated with an ulcer or deep tissue infection; and (2) compare antibiotic prescribing practices among diabetics and nondiabetics. We hypothesized that diabetics would have a similar spectrum of microorganisms as nondiabetics but would be more frequently treated with antibiotics with broad gram‐negative activity.
METHODS
Study Design
This was a secondary analysis of 2 published retrospective studies of patients hospitalized for cellulitis or cutaneous abscess between January 1, 2007 and May 31, 2012.[2, 10] For the purposes of this study, the terms cellulitis and abscess will refer to infections not involving an infected ulcer, osteomyelitis, or other deep tissue infection.
Study Setting and Population
The first of the 2 cohorts analyzed for the present study included patients hospitalized with cellulitis, abscess, or wound infection at 7 academic or community hospitals in Colorado.[2] The second cohort included patients hospitalized with cellulitis or abscess at a single academic medical center (1 of the 7 hospitals above) in Denver, Colorado.[10] The methods of these studies have been reported in detail elsewhere.[2, 10, 11] Briefly, potential cases were identified using International Classification of Diseases, 9th Revision, Clinical Modification codes. The main inclusion and exclusion criteria of the 2 studies were similar. In both studies, cases were excluded that involved infected ulcers or suspected or confirmed deep tissue involvement (eg, osteomyelitis, myositis, fasciitis). Cases were also excluded that involved other infections where empiric antibiotic therapy with gram‐negative activity is standard including infected human or animal bites, periorbital or orbital infections, and perineal infections. The combined cohort in the present study therefore represented a group of patients hospitalized with relatively uncomplicated cellulitis or cutaneous abscess.
Definitions and Study Outcomes
Only 1 of the 2 studies from which the current cohort was derived distinguished between nonpurulent cellulitis, purulent cellulitis, and wound infection.[2] In the other study, cases were more broadly defined as either cellulitis or cutaneous abscess.[10] Infected ulcers and deep tissue infections were excluded from both studies. In combining the data into the current cohort, all nondrainable infections (purulent or nonpurulent cellulitis and wound infection) were categorized generally as cellulitis. All cases with documentation of an abscess in the medical record were categorized as cutaneous abscess. Presence of diabetes mellitus was based on provider documentation of the condition during the hospitalization. Microbiological cultures were obtained at the discretion of treating providers. Exposure to antibiotics with a broad spectrum of gram‐negative activity was defined as receipt of 2 or more calendar days of ‐lactam/‐lactamase inhibitor combinations, second‐ through fifth‐generation cephalosporins, fluoroquinolones, carbapenems, tigecycline, aminoglycosides, or colistin.[2]
The follow‐up periods differed slightly between the 2 studies used to derive the current cohort. In 1 study, all clinical encounters within 30 days of hospital discharge were reviewed to assess clinical outcomes.[10] In the other, clinical encounters within 45 days from the date of hospitalization were reviewed.[2] Clinical failure was defined as any of the following within the 30‐ or 45‐day follow‐up periods, respectively: (1) treatment failure, defined as a change in antibiotic therapy or unplanned drainage procedure due to inadequate clinical response more than 5 days[2] or 7 days[10] after hospital admission; (2) recurrence, defined as reinitiation of antibiotics for skin infection after completion of the initial treatment course; or (3) rehospitalization due to skin infection.[11]
Statistical Analysis
Because the clinical factors, microbiology, and treatment of cellulitis and cutaneous abscesses differ, analyses were performed for the total cohort and stratified by type of infection. Microorganisms cultured, antibiotic selection, and treatment duration were compared between diabetics and nondiabetics using the Wilcoxon rank sum test, 2, or Fisher exact test, as appropriate.
Because we hypothesized that the presence of diabetes mellitus in patients with cellulitis or abscess leads to use of broad gram‐negative therapy, we developed a multivariable logistic regression model to identify factors independently associated with exposure to antibiotics with broad gram‐negative activity. We also developed a linear regression model to explore the relationship between diabetes mellitus and duration of antibiotic therapy after adjusting for covariates. To develop these models, we first performed bivariate analyses and retained variables with a P value 0.25 in the regression models. Variables that did not meet the P value threshold but were considered to be clinically relevant covariates were also included in the model. We assessed for effect modification, multicollinearity, and goodness of fit when developing the models. We used SAS version 9.3 (SAS Institute, Cary, NC) for data analysis.
RESULTS
After excluding 102 pediatric cases and removing 5 duplicate cases, 770 total cases were included for analysis: 447 involved cellulitis and 323 involved cutaneous abscess (Figure 1). Overall, 167 (22%) patients had diabetes mellitus. Diabetics were significantly more likely than nondiabetics to have cellulitis as the presenting infection (67% of cases vs 56%, P=0.008) and to have lower extremity involvement (48% vs 33%, P<0.001) (Table 1). Diabetics were also older (median age 55 years vs 48 years, P<0.001), more likely to have cirrhosis or prior skin infection, and less likely to be injection‐drug users or human immunodeficiency virus (HIV) infected. Demographic and clinical characteristics among diabetics and nondiabetics stratified by the categorizations of cellulitis and cutaneous abscess are presented in the Supporting Information, Appendix Table 1, in the online version of this article.
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | |
|---|---|---|
| ||
| Type of infection | ||
| Cellulitis | 112 (67) | 335 (56)a |
| Cutaneous abscess | 55 (33) | 268 (44) |
| Age, y, median (IQR) | 55 (4763) | 48 (3658)a |
| Male | 102 (61) | 405 (67) |
| Injection drug use | 9 (5) | 117 (19)a |
| Alcohol abuse or dependence | 15 (9) | 86 (14) |
| Cirrhosis | 11 (7) | 17 (3)a |
| HIV infection | 0 | 29 (5)a |
| Dialysis dependence | 4 (2) | 5 (1) |
| Peripheral arterial disease | 4 (2) | 5 (1) |
| Saphenous vein harvest | 7 (4) | 11 (2) |
| Prior skin infection | 56 (34) | 125 (21)a |
| Prior MRSA infection or colonization | 20 (12) | 50 (8) |
| Anatomical location | ||
| Lower extremity | 80 (48) | 200 (33)a |
| Upper extremity | 6 (4) | 79 (13)a |
| Head and neck | 14 (8) | 38 (6) |
| Buttock or inguinal | 8 (5) | 35 (6) |
| Chest, abdomen, back, or axilla | 9 (5) | 25 (4) |
| Multiple distinct sites | 7 (4) | 34 (6) |
| Medical primary service | 139 (83) | 395 (66)a |
| Consultation requested | 99 (59) | 294 (49)a |
| Surgery | 58 (35) | 152 (25)a |
| Internal medicine | 18 (11) | 47 (8) |
| Infectious diseases | 41 (25) | 149 (25) |
| Failed initial outpatient antibiotic therapy | 52 (31) | 186 (31) |
| Fever (temperature 38.0C) | 20 (12) | 102 (17) |
| Leukocytosis (WBC >10,000 cells/mm3) | 78 (47) | 311 (52) |
The frequency of use of microbiological cultures was similar among diabetics and nondiabetics (Table 2). In cases of cellulitis, a microorganism was identified in 18% of diabetics and 12% of nondiabetics (P=0.09). In cases of cutaneous abscess, a microorganism was identified more commonly (69% and 74%, respectively, P=0.50). Among cases where a microorganism was identified, aerobic gram‐positive organisms were isolated in 90% of diabetics and 92% of nondiabetics (P=0.59). Aerobic gram‐negative organisms were isolated in 7% of diabetics and 12% of nondiabetics (P=0.28). Specific gram‐negative organisms isolated are shown in the Supporting Information, Appendix Table 2, in the online version of this article; no cases in diabetics involved Pseudomonas aeruginosa. The comparison of microbiological data among diabetics and nondiabetics was similar when stratified by cellulitis versus cutaneous abscess (Table 2).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Any microbiological culture obtaineda | 82 (73) | 234 (70) | 46 (84) | 239 (89) | 128 (77) | 473 (78) | |||
| Wound drainage or swab | 19 (17) | 36 (11) | 1 (2) | 8 (3) | 20 (12) | 44 (7) | |||
| Abscess material | 1 (1) | 3 (1) | 39 (71) | 205 (76) | 40 (24) | 208 (34) | |||
| Tissueb | 2 (2) | 17 (5) | 1 (2) | 8 (3) | 3 (2) | 25 (4) | |||
| Blood | 73 (65) | 212 (63) | 26 (47) | 121 (45) | 99 (59) | 333 (55) | |||
| Any microorganism identifiedc | 20 (18) | 39 (12) | 0.09 | 38 (69) | 197 (74) | 0.50 | 58 (35) | 236 (39) | 0.30 |
| Aerobic gram‐positive | 15 (75) | 36 (92) | 0.11 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 218 (92) | 0.59 |
| Staphylococcus aureus | 11 (55) | 26 (67) | 0.38 | 28 (74) | 132 (67) | 0.42 | 39 (67) | 158 (67) | 0.97 |
| Methicillin‐susceptible | 4 (20) | 15 (38) | 0.15 | 12 (32) | 42 (21) | 0.17 | 16 (28) | 57 (24) | 0.59 |
| Methicillin‐resistant | 5 (25) | 11 (28) | 1.00 | 14 (37) | 85 (43) | 0.47 | 19 (33) | 96 (41) | 0.27 |
| Susceptibility not performed | 2 (10) | 0 | 0.11 | 2 (5) | 5 (3) | 0.32 | 4 (7) | 5 (2) | 0.08 |
| Streptococcal species | 6 (30) | 15 (38) | 0.52 | 12 (32) | 69 (35) | 0.68 | 18 (31) | 84 (36) | 0.51 |
| ‐hemolytic streptococcus | 3 (15) | 13 (33) | 0.13 | 6 (16) | 32 (16) | 0.94 | 9 (16) | 45 (19) | 0.53 |
| Streptococcus anginosus/Streptococcus milleri group | 1 (5) | 0 | 0.34 | 2 (5) | 29 (15) | 0.11 | 3 (5) | 29 (12) | 0.12 |
| Other ‐hemolytic streptococcus | 2 (10) | 2 (5) | 0.60 | 4 (11) | 12 (6) | 0.30 | 6 (10) | 14 (6) | 0.25 |
| Other streptococcus | 0 | 0 | 1 (3) | 3 (2) | 0.51 | 1 (2) | 3 (1) | 0.59 | |
| Staphylococcus aureus or streptococci | 15 (75) | 35 (90) | 0.25 | 37 (97) | 182 (92) | 0.48 | 52 (90) | 217 (92) | 0.60 |
| Enterococcus species | 0 | 2 (5) | 0.54 | 0 | 4 (2) | 1.00 | 0 | 6 (3) | 0.60 |
| Aerobic gram‐negative | 2 (10) | 7 (18) | 0.70 | 2 (5) | 21 (11) | 0.39 | 4 (7) | 28 (12) | 0.28 |
| Anaerobic organism(s) | 2 (10) | 3 (8) | 1.00 | 8 (21) | 30 (15) | 0.37 | 10 (17) | 33 (14) | 0.53 |
| Mixed skin or oral flora | 1 (5) | 1 (3) | 1.00 | 0 | 1 (1) | 1.00 | 1 (2) | 2 (1) | 0.48 |
| Other | 1 (5) | 3 (8) | 1.00 | 2 (5) | 3 (2) | 0.19 | 3 (5) | 6 (3) | 0.39 |
| Polymicrobial | 3 (15) | 17 (45) | 0.03 | 11 (29) | 47 (24) | 0.51 | 14 (24) | 64 (27) | 0.65 |
| Positive blood cultured | 4 (5) | 8 (4) | 0.51 | 2 (8) | 3 (2) | 0.21 | 6 (6) | 11 (3) | 0.24 |
Antibiotic utilization is summarized in Table 3. Among patients who were started on antibiotic therapy in the emergency department or urgent care, the initial regimen included an agent with broad gram‐negative activity in 31% of both diabetics and nondiabetics (P=0.97). During the entire hospital stay (including the emergency department or urgent care), diabetics were significantly more likely to be treated with ‐lactam/‐lactamase inhibitor combinations (42% vs 33%, P=0.04). At the time of hospital discharge, diabetics were more likely to be prescribed fluoroquinolones (11% vs 5%, P=0.01) (Table 3) particularly for cases of cellulitis (13% vs 6%, P=0.008) (see Supporting Information, Appendix Table 3, in the online version of this article). Diabetics were somewhat more likely to be prescribed parenteral antibiotics (10% vs 6%, P=0.07) after discharge. When considering both inpatient and discharge therapy, more diabetics than nondiabetics were exposed to at least 2 calendar days of broad gram‐negative therapy (54% vs 44%, P=0.02) and more were prescribed an antipseudomonal agent (38% vs 25%, P=0.002). In the group of patients who received at least 1 dose of an antibiotic with broad gram‐negative activity, broad gram‐negative agents accounted for 33% of the total days of therapy prescribed for diabetics and 32% for nondiabetics. Overall prescribing patterns were similar when stratified by cellulitis versus cutaneous abscess (see Supporting Information, Appendix Table 3, in the online version of this article).
| Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
|---|---|---|---|
| |||
| Individual antibiotics prescribed during the inpatient stayab | |||
| Vancomycin | 142 (85) | 504 (84) | 0.65 |
| Clindamycin | 27 (16) | 131 (22) | 0.12 |
| Parenteral ‐lactam/‐lactamase inhibitor | 70 (42) | 200 (33) | 0.04 |
| Second‐generation or higher cephalosporin | 13 (8) | 51 (8) | 0.78 |
| Cefazolin | 17 (10) | 91 (15) | 0.11 |
| Carbapenem | 9 (5) | 34 (6) | 0.90 |
| Fluoroquinolone | 20 (12) | 53 (9) | 0.21 |
| Daptomycin | 8 (5) | 24 (4) | 0.64 |
| Linezolid | 2 (1) | 8 (1) | 1.00 |
| Other ‐lactam | 6 (4) | 30 (5) | 0.45 |
| Trimethoprim‐sulfamethoxazole | 12 (7) | 30 (5) | 0.27 |
| Doxycycline | 15 (9) | 44 (7) | 0.47 |
| Cephalexin | 7 (4) | 22 (4) | 0.74 |
| Amoxicillin‐clavulanate | 11 (7) | 24 (4) | 0.15 |
| Antibiotics prescribed at hospital dischargeb | 163 (98) | 580 (96) | 0.38 |
| Clindamycin | 20 (12) | 95 (16) | 0.23 |
| Trimethoprim‐sulfamethoxazole | 52 (31) | 215 (36) | 0.28 |
| Doxycycline | 32 (19) | 91 (15) | 0.20 |
| Cephalexin | 12 (7) | 46 (8) | 0.85 |
| Amoxicillin‐clavulanate | 24 (14) | 82 (14) | 0.80 |
| Fluoroquinolone | 18 (11) | 32 (5) | 0.01 |
| Linezolid | 8 (5) | 19 (3) | 0.31 |
| Other oral ‐lactam | 3 (2) | 28 (5) | 0.10 |
| Other oral antibiotic | 1 (1) | 2 (0.3) | 0.52 |
| Vancomycin | 8 (5) | 15 (2) | 0.13 |
| Daptomycin | 5 (3) | 10 (2) | 0.34 |
| Other parenteral antibiotic | 4 (2) | 11 (2) | 0.75 |
| Antibiotic with broad gram‐negative activity initiated in emergency department or urgent care | 46/149 (31) | 174/561 (31) | 0.97 |
| Exposed to any antibiotic with broad gram‐negative activityc | 101 (62) | 311 (53) | 0.048 |
| Exposed to any antibiotic with antipseudomonal activity | 62 (38) | 149 (25) | 0.002 |
| Exposed to at least 2 calendar days of antibiotics with broad gram‐negative activityc | 89 (54) | 259 (44) | 0.02 |
| Treatment durationd | |||
| Total duration of therapy, d, median (IQR) | 13 (1015) | 12 (1015) | 0.09 |
| Duration of inpatient therapy, d, median (IQR) | 4 (36) | 4 (35) | 0.03 |
| Duration of therapy after discharge, d, median (IQR) | 8 (710) | 8 (710) | 0.58 |
After adjusting for covariates in the logistic regression model, diabetes mellitus was an independent predictor of exposure to broad gram‐negative therapy (see Supporting Information, Appendix Table 4, in the online version of this article). In addition to diabetes mellitus, culture of an aerobic gram‐negative microorganism, infectious diseases service consultation, presence of fever, and nonmedical admitting services were significantly associated with exposure to broad gram‐negative therapy. Prior methicillin‐resistant S aureus infection or colonization and HIV infection were inversely associated. Compared with nondiabetics, the total duration of antibiotic therapy in diabetics was somewhat longer (median 13 days vs 12 days, P=0.09) (Table 3). After adjusting for covariates in the linear regression model, there was a significant association between diabetes mellitus and treatment duration. On average, diabetics were treated 1 day (95% confidence interval: 0.2‐1.7 days) longer than nondiabetics.
Compared with nondiabetics, diabetics were more likely to have an outpatient follow‐up visit (73% vs 61%, P=0.002) and to be rehospitalized for any reason after discharge (16% vs 9%, P=0.02) (Table 4). Diabetics were overall more likely to be classified as clinical failure (15% vs 9%, P=0.02); this difference was driven by the cellulitis subgroup (19% vs 10%, P=0.01).
| Cellulitis | Cutaneous Abscess | All Cases | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Diabetes Mellitus, N=112 | No Diabetes Mellitus, N=335 | P | Diabetes Mellitus, N=55 | No Diabetes Mellitus, N=268 | P | Diabetes Mellitus, N=167 | No Diabetes Mellitus, N=603 | P | |
| |||||||||
| Survived to discharge | 111 (99) | 335 (100) | 0.25 | 55 (100) | 268 (100) | 166 (99) | 603 (100) | 0.22 | |
| Outpatient follow‐up documented | 82 (74) | 204 (61) | 0.01 | 40 (73) | 161 (60) | 0.08 | 122 (73) | 365 (61) | 0.002 |
| Rehospitalized | 22 (20) | 34 (10) | 0.008 | 4 (7) | 21 (8) | 1.00 | 26 (16) | 55 (9) | 0.02 |
| Clinical failure | 21 (19) | 34 (10) | 0.01 | 4 (7) | 20 (7) | 1.00 | 25 (15) | 54 (9) | 0.02 |
| Treatment failure | 7 (6) | 17 (5) | 0.62 | 2 (4) | 7 (3) | 0.65 | 9 (5) | 24 (4) | 0.42 |
| Recurrence | 10 (9) | 16 (5) | 0.10 | 1 (2) | 11 (4) | 0.70 | 11 (7) | 27 (4) | 0.26 |
| Rehospitalization due to skin infection | 14 (13) | 17 (5) | 0.01 | 3 (5) | 11 (4) | 0.71 | 17 (10) | 28 (5) | 0.01 |
| Length of stay, d, median (IQR) | 4 (36) | 4 (35) | 0.03 | 4 (36) | 4 (35) | 0.28 | 4 (36) | 4 (35) | 0.02 |
DISCUSSION
Diabetes mellitus is a common comorbidity in patients with acute bacterial skin infections. In this large cohort of patients hospitalized for cellulitis or cutaneous abscess, where those with infected ulcers or deep tissue infections were excluded, microbiological findings in cases associated with positive cultures were similar among diabetics and nondiabetics. Although aerobic gram‐negative microorganisms were not more likely to be identified in diabetics, diabetics were significantly more likely to be exposed to at least 2 calendar days of antibiotics with broad gram‐negative activity. After adjusting for covariates, diabetes mellitus was independently associated with exposure to broad gram‐negative therapy.
To our knowledge, this is the first study to compare the microbiology of cellulitis and cutaneous abscess among diabetics and nondiabetics. Lipsky and colleagues previously described the microbiology of a cohort of diabetic patients hospitalized with a broader range of skin infections including cellulitis, infected ulcers, and surgical site infections.[12] Similar to our findings, gram‐negative pathogens were uncommonly isolated in that study; however, in the absence of a comparator group, whether diabetics were at higher risk for gram‐negative involvement than nondiabetics was not known. Similar to the study by Lipsky and colleagues, most studies of skin infections in diabetics have included a relatively heterogeneous group of infections.[12, 13, 14, 15] The present study therefore contributes to the literature by providing a focused comparison of the microbiology of inpatient cellulitis and abscess in the absence of complicating factors such as an infected ulcer or deep tissue involvement. We found that among cases with a positive culture (13% of cases in the cellulitis group and 73% in the abscess group), the microbiology was similar among diabetics and nondiabetics. Although a microorganism was identified in only a minority of cases of cellulitis, our findings do not support the need for broad gram‐negative therapy in diabetics with cellulitis not associated with an ulcer or deep tissue infection. In diabetics with an abscess, antibiotics with broad gram‐negative activity do not appear to be indicated.
The present study also adds to the literature by providing a detailed comparison of antibiotic utilization patterns among diabetics and nondiabetics. We demonstrated that diabetics were more likely to have significant exposure to antibiotics with broad gram‐negative activity, particularly antipseudomonal agents (the broadest‐spectrum antibiotics). Because initiation of broad gram‐negative therapy in the emergency department or urgent care was not more common among diabetics, the increased use of these agents among diabetics appeared to be driven by inpatient providers. It is also notable that of patients who received any antibiotic with broad gram‐negative activity, these agents accounted for similar proportions of the total days of therapy in both diabetics and nondiabetics. In aggregate, our findings demonstrate that diabetics are more likely to be started on antibiotics with broad gram‐negative activity by inpatient providers, diabetics are not necessarily continued on longer durations of broad gram‐negative therapy once started, and the total amount of exposure to broad gram‐negative agents is substantial.
Overall, our findings suggest that inpatient providers perceive diabetics with cellulitis or abscess to be at increased risk for gram‐negative pathogens. This perhaps reflects an extrapolation of recommendations to use broad‐spectrum empiric therapy in diabetics with certain complicated skin infections.[7] However, for patients with cellulitis or cutaneous abscess, Infectious Diseases Society of America (IDSA) guidelines recommend antibiotic therapy targeted toward S aureus and streptococcal species; there is no suggestion to use a broader spectrum of therapy in diabetics.[8, 9] Our findings therefore highlight an important opportunity to improve antibiotic selection for all patients hospitalized with cellulitis and abscess, but particularly diabetics. It is also noteworthy that by linear regression, diabetes mellitus was independently associated with longer treatment durations. Although the average increase in treatment duration was small (1 day), this finding adds to the evidence that the presence of diabetes mellitus alters providers' treatment approach to cellulitis or abscess.
We found that despite more frequent treatment with broad gram‐negative therapy, diabetics were more likely than nondiabetics to be classified as clinical failures. It is important to point out that diabetics were also more likely than nondiabetics to have postdischarge outpatient follow‐up visits, raising the possibility of biased ascertainment of clinical failure events in this group. However, we also demonstrated that diabetics with cellulitis were more likely to be rehospitalized than nondiabetics. This is similar to a finding by Suaya and colleagues who showed that diabetics with skin infections were about twice as likely to be rehospitalized as nondiabetics.[13] One could hypothesize that the increased frequency of clinical failure events among diabetics was due to their older age, hyperglycemia, or vascular insufficiency; however, other factors may have contributed. For example, providers may have mistaken residual erythema for ongoing or recurrent cellulitis, or the diagnosis of cellulitis could have been incorrect to begin with. Additionally, there may have been uncertainty about the microbiology of cellulitis because the infecting pathogen was not usually identified. These factors may have led to alterations in treatment that would have resulted in a classification of clinical failure, and it is possible that providers had a lower threshold to alter treatment in diabetics. It is therefore not clear whether our findings represent a true difference in clinical outcomes between diabetics or nondiabetics. Regardless, in cases associated with a positive culture, our microbiological results do not support that the difference in clinical failure between diabetics and nondiabetics with cellulitis was related to a different spectrum of microorganisms.
In addition to the limitations outlined previously[2, 10] and above, the present study has at least 5 additional limitations. First, this was a secondary analysis of studies that were not designed to evaluate the effect of diabetes mellitus on the microbiology and treatment of skin infections. For example, hemoglobin A1C values were not collected; therefore, we could not examine whether the microbiology and antibiotic prescribing practices differed based on control of diabetes mellitus. Second, there were minor differences in inclusion and exclusion criteria between the 2 cohorts included in this study. Because the proportion of patients with diabetes mellitus was similar among both cohorts, and comparisons were not made between the cohorts, this should not have impacted our results. Third, the broad categorization of cellulitis used when combining the 2 cohorts raised the possibility of differences in infection characteristics between diabetics and nondiabetics (eg, presence of a wound) that could have confounded our findings regarding use of gram‐negative therapy. In the larger of the 2 cohorts from which the combined cohort was derived, only 17 (3%) of 533 patients had wound infections, whereas those with infected ulcers or suspected deep‐tissue infection were excluded from both cohorts. Furthermore, in the combined cohort, the increased frequency of broad gram‐negative therapy among diabetics was also observed in the cutaneous abscess group. It is therefore unlikely that the categorization of cellulitis had a significant impact on our results. Fourth, given the observational nature of the study, the microbiological data were subject to limitations. Importantly, because the infecting pathogen was identified in only 13% of cases of cellulitis, firm conclusions regarding the microbiology of cellulitis cannot be drawn. Finally, the small number of gram‐negative organisms isolated precluded comparisons of specific pathogens among diabetics and nondiabetics. In addition, because a number of gram‐negative organisms were isolated from wound cultures, it is not known whether they were clinically relevant or simply represented colonization.
In conclusion, in cases of cellulitis or abscess associated with a positive culture, gram‐negative microorganisms were not isolated more commonly among diabetics compared with nondiabetics. However, in general, diabetics were more likely to be treated with broad gram‐negative therapy suggesting that, particularly for cutaneous abscesses, this prescribing practice may not be warranted. These findings support current IDSA guidelines that recommend antibiotic therapy targeted toward gram‐positive pathogens for cellulitis or abscess, irrespective of the presence of diabetes mellitus.[8, 9] Because nearly one‐fourth of patients hospitalized with cellulitis or abscess are diabetic, these findings have relevance for national antimicrobial stewardship efforts aimed at curbing antimicrobial resistance through reducing use of antibiotics with broad gram‐negative activity in hospitals.[16]
Disclosures: This work was supported by the National Institute of Allergy and Infectious Diseases, National Institute of Health (TCJ: K23 AI099082). D.M.P. reports potential conflicts of interests with Optimer, Cubist, and Forest Pharmaceuticals. The authors report no other conflicts of interest.
- , , , et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. 2005;41(3):281–288.
- , , , et al. Antibiotic prescribing practices in a multicenter cohort of patients hospitalized for acute bacterial skin and skin structure infection. Infect Control Hosp Epidemiol. 2014;35(10):1241–1250.
- , , , . The role of beta‐hemolytic streptococci in causing diffuse, nonculturable cellulitis: a prospective investigation. Medicine. 2010;89(4):217–226.
- , , , et al. Factors associated with complications and mortality in adult patients hospitalized for infectious cellulitis. Eur J Clin Microbiol Infect Dis. 2003;22(3):151–157.
- , , , et al. Epidemiology and outcomes of complicated skin and soft tissue infections in hospitalized patients. J Clin Microbiol. 2012;50(2):238–245.
- , , , , , . Current management of patients hospitalized with complicated skin and soft tissue infections across Europe (2010–2011): assessment of clinical practice patterns and real‐life effectiveness of antibiotics from the REACH study. Clin Microbiol Infect. 2013;19(9):E377–E385.
- , , , et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132–e173.
- , , , et al. Clinical practice guidelines by the Infectious Diseases Society Of America for the treatment of methicillin‐resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52(3):285–292.
- , , , et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society Of America. Clin Infect Dis. 2014;59(2):e10–e52.
- , , , et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med. 2011;171(12):1072–1079.
- , , , , , . Skin and soft‐tissue infections requiring hospitalization at an academic medical center: opportunities for antimicrobial stewardship. Clin Infect Dis. 2010;51(8):895–903.
- , , , , , . Skin and soft tissue infections in hospitalised patients with diabetes: culture isolates and risk factors associated with mortality, length of stay and cost. Diabetologia. 2010;53(5):914–923.
- , , , . Skin and soft tissue infections and associated complications among commercially insured patients aged 0–64 years with and without diabetes in the U.S. PLoS One. 2013;8(4):e60057.
- , , . A post hoc subgroup analysis of meropenem versus imipenem/cilastatin in a multicenter, double‐blind, randomized study of complicated skin and skin‐structure infections in patients with diabetes mellitus. Clin Ther. 2006;28(8):1164–1174.
- , , , . Treating diabetic foot infections with sequential intravenous to oral moxifloxacin compared with piperacillin‐tazobactam/amoxicillin‐clavulanate. J Antimicr Chemo. 2007;60(2):370–376.
- , , , et al. Vital signs: improving antibiotic use among hospitalized patients. MMWR Morb Mortal Wkly Rep. 2014;63(9):194–200.
- , , , et al. Increased risk of common infections in patients with type 1 and type 2 diabetes mellitus. Clin Infect Dis. 2005;41(3):281–288.
- , , , et al. Antibiotic prescribing practices in a multicenter cohort of patients hospitalized for acute bacterial skin and skin structure infection. Infect Control Hosp Epidemiol. 2014;35(10):1241–1250.
- , , , . The role of beta‐hemolytic streptococci in causing diffuse, nonculturable cellulitis: a prospective investigation. Medicine. 2010;89(4):217–226.
- , , , et al. Factors associated with complications and mortality in adult patients hospitalized for infectious cellulitis. Eur J Clin Microbiol Infect Dis. 2003;22(3):151–157.
- , , , et al. Epidemiology and outcomes of complicated skin and soft tissue infections in hospitalized patients. J Clin Microbiol. 2012;50(2):238–245.
- , , , , , . Current management of patients hospitalized with complicated skin and soft tissue infections across Europe (2010–2011): assessment of clinical practice patterns and real‐life effectiveness of antibiotics from the REACH study. Clin Microbiol Infect. 2013;19(9):E377–E385.
- , , , et al. 2012 Infectious Diseases Society of America clinical practice guideline for the diagnosis and treatment of diabetic foot infections. Clin Infect Dis. 2012;54(12):e132–e173.
- , , , et al. Clinical practice guidelines by the Infectious Diseases Society Of America for the treatment of methicillin‐resistant Staphylococcus aureus infections in adults and children: executive summary. Clin Infect Dis. 2011;52(3):285–292.
- , , , et al. Practice guidelines for the diagnosis and management of skin and soft tissue infections: 2014 update by the Infectious Diseases Society Of America. Clin Infect Dis. 2014;59(2):e10–e52.
- , , , et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med. 2011;171(12):1072–1079.
- , , , , , . Skin and soft‐tissue infections requiring hospitalization at an academic medical center: opportunities for antimicrobial stewardship. Clin Infect Dis. 2010;51(8):895–903.
- , , , , , . Skin and soft tissue infections in hospitalised patients with diabetes: culture isolates and risk factors associated with mortality, length of stay and cost. Diabetologia. 2010;53(5):914–923.
- , , , . Skin and soft tissue infections and associated complications among commercially insured patients aged 0–64 years with and without diabetes in the U.S. PLoS One. 2013;8(4):e60057.
- , , . A post hoc subgroup analysis of meropenem versus imipenem/cilastatin in a multicenter, double‐blind, randomized study of complicated skin and skin‐structure infections in patients with diabetes mellitus. Clin Ther. 2006;28(8):1164–1174.
- , , , . Treating diabetic foot infections with sequential intravenous to oral moxifloxacin compared with piperacillin‐tazobactam/amoxicillin‐clavulanate. J Antimicr Chemo. 2007;60(2):370–376.
- , , , et al. Vital signs: improving antibiotic use among hospitalized patients. MMWR Morb Mortal Wkly Rep. 2014;63(9):194–200.
© 2014 Society of Hospital Medicine
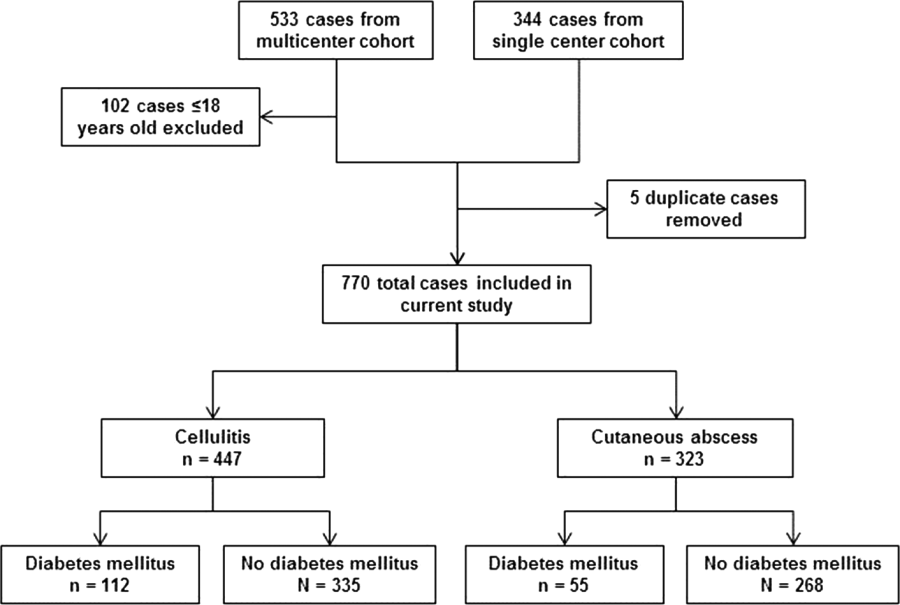
Advancing Orthopedic Postsurgical Pain Management & Multimodal Care Pathways: Improving Clinical & Economic Outcomes


Commentary to "5 Points on Total Ankle Arthroplasty"
There are considerable differences in the design and implantation technique of the current total ankle implants available in the United States, eg, mobile vs. fixed bearing, intramedullary vs. extramedullary guidance, anterior vs. lateral surgical approach, flat vs. curved bone cuts, natural articular design with minimal bone resection (Zimmer Trabecular Metal Total Ankle; Zimmer, Warsaw, Indiana) vs. larger implant construct with more bone resection (Inbone II; Figure 2). There is no evidence that one implant design is superior, and, as the authors conclude, “Direct comparisons between TAA [total ankle arthroplasty] implant systems are needed to determine what clinical benefits are achieved with each design and what contributes to these differences.”
Hsu AR, Anderson RB, Cohen BE. Total Ankle Arthroplasty. Am J Orthop. 2014;43(10):451-457.
There are considerable differences in the design and implantation technique of the current total ankle implants available in the United States, eg, mobile vs. fixed bearing, intramedullary vs. extramedullary guidance, anterior vs. lateral surgical approach, flat vs. curved bone cuts, natural articular design with minimal bone resection (Zimmer Trabecular Metal Total Ankle; Zimmer, Warsaw, Indiana) vs. larger implant construct with more bone resection (Inbone II; Figure 2). There is no evidence that one implant design is superior, and, as the authors conclude, “Direct comparisons between TAA [total ankle arthroplasty] implant systems are needed to determine what clinical benefits are achieved with each design and what contributes to these differences.”
Hsu AR, Anderson RB, Cohen BE. Total Ankle Arthroplasty. Am J Orthop. 2014;43(10):451-457.
There are considerable differences in the design and implantation technique of the current total ankle implants available in the United States, eg, mobile vs. fixed bearing, intramedullary vs. extramedullary guidance, anterior vs. lateral surgical approach, flat vs. curved bone cuts, natural articular design with minimal bone resection (Zimmer Trabecular Metal Total Ankle; Zimmer, Warsaw, Indiana) vs. larger implant construct with more bone resection (Inbone II; Figure 2). There is no evidence that one implant design is superior, and, as the authors conclude, “Direct comparisons between TAA [total ankle arthroplasty] implant systems are needed to determine what clinical benefits are achieved with each design and what contributes to these differences.”
Hsu AR, Anderson RB, Cohen BE. Total Ankle Arthroplasty. Am J Orthop. 2014;43(10):451-457.
5 Points on Total Ankle Arthroplasty
Rivaroxaban rivals standard for VTE in cancer

Credit: CDC
MADRID—A pooled analysis of 2 studies suggests rivaroxaban prevents recurrent venous thromboembolism (VTE) in cancer patients as effectively as standard therapy, while conferring a lower risk of major bleeding.
The analysis included data from the EINSTEIN-DVT and EINSTEIN-PE trials, which were funded by the companies developing rivaroxaban (Xarelto), Bayer HealthCare Pharmaceuticals and Janssen Research & Development, LLC.
The results were published in Lancet Haematology and presented at the ESMO 2014 Congress (abstract LBA48).
The EINSTEIN-PE study included 3449 subjects with acute symptomatic pulmonary embolism (PE), with or without symptomatic deep vein thrombosis (DVT), who received anticoagulant therapy for 6 or 12 months.
The EINSTEIN-DVT study included 4833 patients who had acute symptomatic DVT, but no symptoms of PE, and received treatment for 3, 6, or 12 months.
In both studies, patients received either oral rivaroxaban (15 mg twice daily for 3 weeks, followed by 20 mg once daily) or standard therapy—enoxaparin (1.0 mg/kg twice daily) followed by a vitamin K antagonist (warfarin or acenocoumarol).
A total of 8281 patients were randomized in these studies—4150 to rivaroxaban and 4131 to standard therapy. Of the 655 patients (7.9%) with active cancer, 462 (5.6%) had cancer at baseline, and 193 (2.3%) were diagnosed during the study.
For this analysis, Martin H. Prins, MD, PhD, of Maastricht University Medical Center in The Netherlands, and his colleagues compared the 2 treatments in patients with active cancer.
VTE recurred in 4.5% (16/354) of the patients who were randomized to rivaroxaban and 6.6% (20/301) of patients randomized to standard therapy (hazard ratio [HR]=0.67).
The mortality rate was 16.4% (58/354) among patients randomized to rivaroxaban and 17.6% (53/301) among those randomized to standard therapy (HR=0.93).
Major bleeding occurred in 2.3% (8/353) of patients who received rivaroxaban and 5% (15/298) who received standard therapy (HR=0.42). And clinically relevant bleeding occurred in 13.6% (48/353) and 16.4% (49/298), respectively (HR=0.80).
Given these results, Dr Prins and his colleagues concluded that rivaroxaban can be considered an alternative to standard therapy in patients with cancer-associated VTE.
The team also said there is a need for a head-to-head comparison of rivaroxaban and long-term low-molecular-weight heparin in this patient population. ![]()
Credit: CDC
MADRID—A pooled analysis of 2 studies suggests rivaroxaban prevents recurrent venous thromboembolism (VTE) in cancer patients as effectively as standard therapy, while conferring a lower risk of major bleeding.
The analysis included data from the EINSTEIN-DVT and EINSTEIN-PE trials, which were funded by the companies developing rivaroxaban (Xarelto), Bayer HealthCare Pharmaceuticals and Janssen Research & Development, LLC.
The results were published in Lancet Haematology and presented at the ESMO 2014 Congress (abstract LBA48).
The EINSTEIN-PE study included 3449 subjects with acute symptomatic pulmonary embolism (PE), with or without symptomatic deep vein thrombosis (DVT), who received anticoagulant therapy for 6 or 12 months.
The EINSTEIN-DVT study included 4833 patients who had acute symptomatic DVT, but no symptoms of PE, and received treatment for 3, 6, or 12 months.
In both studies, patients received either oral rivaroxaban (15 mg twice daily for 3 weeks, followed by 20 mg once daily) or standard therapy—enoxaparin (1.0 mg/kg twice daily) followed by a vitamin K antagonist (warfarin or acenocoumarol).
A total of 8281 patients were randomized in these studies—4150 to rivaroxaban and 4131 to standard therapy. Of the 655 patients (7.9%) with active cancer, 462 (5.6%) had cancer at baseline, and 193 (2.3%) were diagnosed during the study.
For this analysis, Martin H. Prins, MD, PhD, of Maastricht University Medical Center in The Netherlands, and his colleagues compared the 2 treatments in patients with active cancer.
VTE recurred in 4.5% (16/354) of the patients who were randomized to rivaroxaban and 6.6% (20/301) of patients randomized to standard therapy (hazard ratio [HR]=0.67).
The mortality rate was 16.4% (58/354) among patients randomized to rivaroxaban and 17.6% (53/301) among those randomized to standard therapy (HR=0.93).
Major bleeding occurred in 2.3% (8/353) of patients who received rivaroxaban and 5% (15/298) who received standard therapy (HR=0.42). And clinically relevant bleeding occurred in 13.6% (48/353) and 16.4% (49/298), respectively (HR=0.80).
Given these results, Dr Prins and his colleagues concluded that rivaroxaban can be considered an alternative to standard therapy in patients with cancer-associated VTE.
The team also said there is a need for a head-to-head comparison of rivaroxaban and long-term low-molecular-weight heparin in this patient population. ![]()
Credit: CDC
MADRID—A pooled analysis of 2 studies suggests rivaroxaban prevents recurrent venous thromboembolism (VTE) in cancer patients as effectively as standard therapy, while conferring a lower risk of major bleeding.
The analysis included data from the EINSTEIN-DVT and EINSTEIN-PE trials, which were funded by the companies developing rivaroxaban (Xarelto), Bayer HealthCare Pharmaceuticals and Janssen Research & Development, LLC.
The results were published in Lancet Haematology and presented at the ESMO 2014 Congress (abstract LBA48).
The EINSTEIN-PE study included 3449 subjects with acute symptomatic pulmonary embolism (PE), with or without symptomatic deep vein thrombosis (DVT), who received anticoagulant therapy for 6 or 12 months.
The EINSTEIN-DVT study included 4833 patients who had acute symptomatic DVT, but no symptoms of PE, and received treatment for 3, 6, or 12 months.
In both studies, patients received either oral rivaroxaban (15 mg twice daily for 3 weeks, followed by 20 mg once daily) or standard therapy—enoxaparin (1.0 mg/kg twice daily) followed by a vitamin K antagonist (warfarin or acenocoumarol).
A total of 8281 patients were randomized in these studies—4150 to rivaroxaban and 4131 to standard therapy. Of the 655 patients (7.9%) with active cancer, 462 (5.6%) had cancer at baseline, and 193 (2.3%) were diagnosed during the study.
For this analysis, Martin H. Prins, MD, PhD, of Maastricht University Medical Center in The Netherlands, and his colleagues compared the 2 treatments in patients with active cancer.
VTE recurred in 4.5% (16/354) of the patients who were randomized to rivaroxaban and 6.6% (20/301) of patients randomized to standard therapy (hazard ratio [HR]=0.67).
The mortality rate was 16.4% (58/354) among patients randomized to rivaroxaban and 17.6% (53/301) among those randomized to standard therapy (HR=0.93).
Major bleeding occurred in 2.3% (8/353) of patients who received rivaroxaban and 5% (15/298) who received standard therapy (HR=0.42). And clinically relevant bleeding occurred in 13.6% (48/353) and 16.4% (49/298), respectively (HR=0.80).
Given these results, Dr Prins and his colleagues concluded that rivaroxaban can be considered an alternative to standard therapy in patients with cancer-associated VTE.
The team also said there is a need for a head-to-head comparison of rivaroxaban and long-term low-molecular-weight heparin in this patient population. ![]()
FDA approves cancer drugs faster, study shows

MADRID—The US Food and Drug Administration (FDA) tends to approve cancer drugs faster than Health Canada and the European Medicines Agency (EMA),
according to a study presented at the ESMO 2014 Congress.
On average, the FDA approved antineoplastic agents about 6 to 8 months faster than the EMA and Health Canada, researchers found.
One of the drugs studied had been FDA-approved for more than 4.5 years before the EMA and Health Canada authorized its use.
The researchers said these results suggest a need for a coordinated international approach to reduce the disparity in approval times.
“There needs to be a dialogue amongst industry, regulatory agencies, patient bodies, [the] research community, and oncology professionals on how best we can reduce the time to approval while ensuring safety for approved drugs,” said study investigator Sunil Verma, MD, of Sunnybrook Odette Cancer Center in Toronto, Canada.
A previous study, published in NEJM in 2012, showed that, between 2001 and 2010, the FDA tended to approve all types of drugs faster than the EMA and Health Canada.
Dr Verma and Nardin Samuel, an MD/PhD student at the University of Toronto, focused their study on cancer drugs and presented their findings at ESMO as abstract 1036O_PR.
The pair analyzed approval data for 41 antineoplastic agents and found the average time to FDA approval for these drugs was 6 months shorter than for the EMA and 7.6 months shorter than for Health Canada.
Azacitidine, which is approved to treat hematologic malignancies, had the greatest delay between FDA and Health Canada approval, at 66.1 months. The EMA approved azacitidine 10.3 months earlier than Health Canada but 55.8 months after the FDA.
The fastest approval time among the drugs studied was for cabazitaxel, which was approved for metastatic prostate cancer by the FDA just 17 days after the drug’s manufacturer filed for approval. In Canada and the European Union, the times to approval for cabazitaxel were 11.63 months and 11.03 months, respectively.
“It is not clear why there were these differences, but they are of some concern . . . ,” said David Cameron, MD, of the Edinburgh Cancer Research Centre in the UK, who was not involved in this research.
“[T]hey suggest that, in the absence of data to the contrary, there may be bureaucratic rather than medical/scientific reasons for differential geographical approval timelines, which, of course, will lead to differential geographical benefits from new agents.”
Dr Cameron added that more work is needed to understand the reasons for these differences, as well as assess any potential impact on patients. ![]()
MADRID—The US Food and Drug Administration (FDA) tends to approve cancer drugs faster than Health Canada and the European Medicines Agency (EMA),
according to a study presented at the ESMO 2014 Congress.
On average, the FDA approved antineoplastic agents about 6 to 8 months faster than the EMA and Health Canada, researchers found.
One of the drugs studied had been FDA-approved for more than 4.5 years before the EMA and Health Canada authorized its use.
The researchers said these results suggest a need for a coordinated international approach to reduce the disparity in approval times.
“There needs to be a dialogue amongst industry, regulatory agencies, patient bodies, [the] research community, and oncology professionals on how best we can reduce the time to approval while ensuring safety for approved drugs,” said study investigator Sunil Verma, MD, of Sunnybrook Odette Cancer Center in Toronto, Canada.
A previous study, published in NEJM in 2012, showed that, between 2001 and 2010, the FDA tended to approve all types of drugs faster than the EMA and Health Canada.
Dr Verma and Nardin Samuel, an MD/PhD student at the University of Toronto, focused their study on cancer drugs and presented their findings at ESMO as abstract 1036O_PR.
The pair analyzed approval data for 41 antineoplastic agents and found the average time to FDA approval for these drugs was 6 months shorter than for the EMA and 7.6 months shorter than for Health Canada.
Azacitidine, which is approved to treat hematologic malignancies, had the greatest delay between FDA and Health Canada approval, at 66.1 months. The EMA approved azacitidine 10.3 months earlier than Health Canada but 55.8 months after the FDA.
The fastest approval time among the drugs studied was for cabazitaxel, which was approved for metastatic prostate cancer by the FDA just 17 days after the drug’s manufacturer filed for approval. In Canada and the European Union, the times to approval for cabazitaxel were 11.63 months and 11.03 months, respectively.
“It is not clear why there were these differences, but they are of some concern . . . ,” said David Cameron, MD, of the Edinburgh Cancer Research Centre in the UK, who was not involved in this research.
“[T]hey suggest that, in the absence of data to the contrary, there may be bureaucratic rather than medical/scientific reasons for differential geographical approval timelines, which, of course, will lead to differential geographical benefits from new agents.”
Dr Cameron added that more work is needed to understand the reasons for these differences, as well as assess any potential impact on patients. ![]()
MADRID—The US Food and Drug Administration (FDA) tends to approve cancer drugs faster than Health Canada and the European Medicines Agency (EMA),
according to a study presented at the ESMO 2014 Congress.
On average, the FDA approved antineoplastic agents about 6 to 8 months faster than the EMA and Health Canada, researchers found.
One of the drugs studied had been FDA-approved for more than 4.5 years before the EMA and Health Canada authorized its use.
The researchers said these results suggest a need for a coordinated international approach to reduce the disparity in approval times.
“There needs to be a dialogue amongst industry, regulatory agencies, patient bodies, [the] research community, and oncology professionals on how best we can reduce the time to approval while ensuring safety for approved drugs,” said study investigator Sunil Verma, MD, of Sunnybrook Odette Cancer Center in Toronto, Canada.
A previous study, published in NEJM in 2012, showed that, between 2001 and 2010, the FDA tended to approve all types of drugs faster than the EMA and Health Canada.
Dr Verma and Nardin Samuel, an MD/PhD student at the University of Toronto, focused their study on cancer drugs and presented their findings at ESMO as abstract 1036O_PR.
The pair analyzed approval data for 41 antineoplastic agents and found the average time to FDA approval for these drugs was 6 months shorter than for the EMA and 7.6 months shorter than for Health Canada.
Azacitidine, which is approved to treat hematologic malignancies, had the greatest delay between FDA and Health Canada approval, at 66.1 months. The EMA approved azacitidine 10.3 months earlier than Health Canada but 55.8 months after the FDA.
The fastest approval time among the drugs studied was for cabazitaxel, which was approved for metastatic prostate cancer by the FDA just 17 days after the drug’s manufacturer filed for approval. In Canada and the European Union, the times to approval for cabazitaxel were 11.63 months and 11.03 months, respectively.
“It is not clear why there were these differences, but they are of some concern . . . ,” said David Cameron, MD, of the Edinburgh Cancer Research Centre in the UK, who was not involved in this research.
“[T]hey suggest that, in the absence of data to the contrary, there may be bureaucratic rather than medical/scientific reasons for differential geographical approval timelines, which, of course, will lead to differential geographical benefits from new agents.”
Dr Cameron added that more work is needed to understand the reasons for these differences, as well as assess any potential impact on patients. ![]()
Telemetry Order Duration Reductions
The Society of Hospital Medicine's Adult Choosing Wisely measures include not ordering continuous telemetry monitoring outside of the ICU [intensive care unit] without using a protocol that governs continuation.[1] Current guidelines for cardiac monitoring use recommend minimum durations for all adult class I and most class II indications.[2] However, telemetry ordering often fails to include timing or criteria for discontinuation. We determined the impact of a reduction in telemetry order duration within our hospital, hypothesizing this reduction would lead to earlier reassessment of telemetry need and therefore decrease overall utilization.
METHODS
Setting
Durham Veterans Affairs Medical Center (DVAMC) is a 151‐bed tertiary care hospital within Veterans Affairs (VA) Integrated Services Network Region 6 (VISN 6) serving as the primary VA hospital for >54,000 patients and a referral hospital for VISN 6. Twenty‐five telemetry units are available for use on 2 wards with 48 potential telemetry beds. All nonintensive care wards contain general medical and surgical patients, without a primary inpatient cardiology service. Most orders are written by housestaff supervised by attending physicians.
Intervention
Prior to our intervention, the maximum allowable duration of telemetry orders was 72 hours. The duration was enforced by nursing staff automatically discontinuing telemetry not renewed within 72 hours. For our intervention, we reduced the duration of telemetry within our electronic ordering system in November 2013 so that orders had to be renewed within 48 hours or they were discontinued. No education regarding appropriate telemetry use was provided. This intervention was created as a quality‐improvement (QI) project affecting all telemetry use within DVAMC and was exempt from institutional review board review.
Outcomes
Outcomes included the mean number of telemetry orders per week, mean duration of telemetry orders, mean duration of telemetry per episode, and the ratio of time on telemetry relative to the total length of stay. As a balancing measure, we examined rates of rapid response and code blue events. All measures were compared for 12 weeks before and 16 weeks after the intervention. Telemetry orders and durations were obtained using the Corporate Data Warehouse.
Analysis
All outcome measurements were continuous variables and compared using the Student t test in Stata version 9.2 (StataCorp, College Station, TX).
RESULTS
Following the intervention, overall order duration decreased by 33% from 66.68.3 hours to 44.52.3 hours per order (P0.01), mirroring the reduction in the maximum telemetry order duration from 72 to 48 hours (Table 1). However, an increase in telemetry order frequency after the intervention resulted in no significant change in telemetry duration per episode or the proportion of the hospitalization on telemetry (59.3 vs 56.3 hours per patient, P=0.43; and 66.4% vs 66.2% of hospitalization, P=0.58). Rapid response and code blue events did not differ significantly relative to the intervention (2.8 events per week before and 3.1 events per week after, P=0.63).
| Before Intervention | After Intervention | P Value | |
|---|---|---|---|
| |||
| No. of hospitalizations with telemetry ordered | 557 | 684 | NA |
| No. of telemetry orders | 952 | 1515 | NA |
| Average no. of orders per week (SD) | 79.3 (9.2) | 94.7 (25.9) | 0.06 |
| Hours of telemetry per order (SD) | 66.6 (8.3) | 44.5 (2.3) | 0.01 |
| Duration of telemetry per patient, h | 59.3 | 56.3 | 0.43 |
| % of hospitalizations receiving telemetry per patient | 66.4% | 66.2% | 0.90 |
| RRT/code blue events per week | 2.8 | 3.1 | 0.63 |
DISCUSSION
Overall, telemetry utilization was unchanged in spite of an intervention successfully reducing telemetry order duration. Providers responded to this decreased order duration by increasing renewal orders, leaving the amount of time patients spent on telemetry unchanged.
Little primary evidence underlies the American Heart Association recommendations for duration of telemetry in general ward patients.[2] The existing literature documents the timing in which arrhythmias occur after cardiac surgery or myocardial infarction, and therefore is limited in guiding patient care outside intensive care unit settings.[3, 4] As such, hospitalists and inpatient providers have little data directing additional telemetry decisions for these patients, and none for patients requiring telemetry for other indications.
As interventions focusing solely on telemetry duration may not lead to changes in usage patterns, reducing telemetry utilization may require active stewardship. For example, explicit justification may be needed for renewal of telemetry orders. Similarly, education on appropriate telemetry indications in tandem with electronic ordering changes may be more likely to change behavior. Alternatively, incorporating data identifying chest pain patients at very low risk of developing arrhythmias or cardiac complications, based on published risk scores at the time of ordering, may lead to better decision making in initiating telemetry.[5, 6]
This QI project had several limitations. First, the intervention occurred in a facility with a previous telemetry order duration limit. In hospitals without a current duration limitation, some reduction in overall telemetry utilization may be possible. Second, this project was a nonrandom before/after study and potentially subject to bias due to confounding. However, our limited number of telemetry resources, the relatively low number of inpatient teams at our facility, and the inability to target geographic locations for team admissions would have made a cluster‐randomized trial impractical. Third, rationales for telemetry ordering were unknown, as well as drivers for increased orders after the intervention. Better understanding these factors could lead to targeted interventions in some settings.
CONCLUSION
In conclusion, a QI initiative reducing telemetry order duration did not reduce overall telemetry utilization but increased the number of telemetry orders written. Interventions incorporating appropriate telemetry indications or event risks may be required to change ordering behaviors.
Disclosure: Nothing to report.
- Society of Hospital Medicine. Society of Hospital Medicine–adult hospital medicine: five things physicians and patients should question. Available at: http://www.choosingwisely.org/doctor‐patient‐lists/society‐of‐hospital‐medicine‐adult‐hospital‐medicine. Accessed June 4, 2014.
- , , , et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical‐Care Nurses. Circulation. 2004;110(17):2721–2746.
- , , , . Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539–549.
- , , , et al. Time‐based risk assessment after myocardial infarction. Implications for timing of discharge and applications to medical decision‐making. Eur Heart J. 2003;24(2):182–189.
- , , , et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med. 2001;110(1):7–11.
- , , , . Lack of utility of telemetry monitoring for identification of cardiac death and life‐threatening ventricular dysrhythmias in low‐risk patients with chest pain. Ann Emerg Med. 2004;43(1):71–76.
The Society of Hospital Medicine's Adult Choosing Wisely measures include not ordering continuous telemetry monitoring outside of the ICU [intensive care unit] without using a protocol that governs continuation.[1] Current guidelines for cardiac monitoring use recommend minimum durations for all adult class I and most class II indications.[2] However, telemetry ordering often fails to include timing or criteria for discontinuation. We determined the impact of a reduction in telemetry order duration within our hospital, hypothesizing this reduction would lead to earlier reassessment of telemetry need and therefore decrease overall utilization.
METHODS
Setting
Durham Veterans Affairs Medical Center (DVAMC) is a 151‐bed tertiary care hospital within Veterans Affairs (VA) Integrated Services Network Region 6 (VISN 6) serving as the primary VA hospital for >54,000 patients and a referral hospital for VISN 6. Twenty‐five telemetry units are available for use on 2 wards with 48 potential telemetry beds. All nonintensive care wards contain general medical and surgical patients, without a primary inpatient cardiology service. Most orders are written by housestaff supervised by attending physicians.
Intervention
Prior to our intervention, the maximum allowable duration of telemetry orders was 72 hours. The duration was enforced by nursing staff automatically discontinuing telemetry not renewed within 72 hours. For our intervention, we reduced the duration of telemetry within our electronic ordering system in November 2013 so that orders had to be renewed within 48 hours or they were discontinued. No education regarding appropriate telemetry use was provided. This intervention was created as a quality‐improvement (QI) project affecting all telemetry use within DVAMC and was exempt from institutional review board review.
Outcomes
Outcomes included the mean number of telemetry orders per week, mean duration of telemetry orders, mean duration of telemetry per episode, and the ratio of time on telemetry relative to the total length of stay. As a balancing measure, we examined rates of rapid response and code blue events. All measures were compared for 12 weeks before and 16 weeks after the intervention. Telemetry orders and durations were obtained using the Corporate Data Warehouse.
Analysis
All outcome measurements were continuous variables and compared using the Student t test in Stata version 9.2 (StataCorp, College Station, TX).
RESULTS
Following the intervention, overall order duration decreased by 33% from 66.68.3 hours to 44.52.3 hours per order (P0.01), mirroring the reduction in the maximum telemetry order duration from 72 to 48 hours (Table 1). However, an increase in telemetry order frequency after the intervention resulted in no significant change in telemetry duration per episode or the proportion of the hospitalization on telemetry (59.3 vs 56.3 hours per patient, P=0.43; and 66.4% vs 66.2% of hospitalization, P=0.58). Rapid response and code blue events did not differ significantly relative to the intervention (2.8 events per week before and 3.1 events per week after, P=0.63).
| Before Intervention | After Intervention | P Value | |
|---|---|---|---|
| |||
| No. of hospitalizations with telemetry ordered | 557 | 684 | NA |
| No. of telemetry orders | 952 | 1515 | NA |
| Average no. of orders per week (SD) | 79.3 (9.2) | 94.7 (25.9) | 0.06 |
| Hours of telemetry per order (SD) | 66.6 (8.3) | 44.5 (2.3) | 0.01 |
| Duration of telemetry per patient, h | 59.3 | 56.3 | 0.43 |
| % of hospitalizations receiving telemetry per patient | 66.4% | 66.2% | 0.90 |
| RRT/code blue events per week | 2.8 | 3.1 | 0.63 |
DISCUSSION
Overall, telemetry utilization was unchanged in spite of an intervention successfully reducing telemetry order duration. Providers responded to this decreased order duration by increasing renewal orders, leaving the amount of time patients spent on telemetry unchanged.
Little primary evidence underlies the American Heart Association recommendations for duration of telemetry in general ward patients.[2] The existing literature documents the timing in which arrhythmias occur after cardiac surgery or myocardial infarction, and therefore is limited in guiding patient care outside intensive care unit settings.[3, 4] As such, hospitalists and inpatient providers have little data directing additional telemetry decisions for these patients, and none for patients requiring telemetry for other indications.
As interventions focusing solely on telemetry duration may not lead to changes in usage patterns, reducing telemetry utilization may require active stewardship. For example, explicit justification may be needed for renewal of telemetry orders. Similarly, education on appropriate telemetry indications in tandem with electronic ordering changes may be more likely to change behavior. Alternatively, incorporating data identifying chest pain patients at very low risk of developing arrhythmias or cardiac complications, based on published risk scores at the time of ordering, may lead to better decision making in initiating telemetry.[5, 6]
This QI project had several limitations. First, the intervention occurred in a facility with a previous telemetry order duration limit. In hospitals without a current duration limitation, some reduction in overall telemetry utilization may be possible. Second, this project was a nonrandom before/after study and potentially subject to bias due to confounding. However, our limited number of telemetry resources, the relatively low number of inpatient teams at our facility, and the inability to target geographic locations for team admissions would have made a cluster‐randomized trial impractical. Third, rationales for telemetry ordering were unknown, as well as drivers for increased orders after the intervention. Better understanding these factors could lead to targeted interventions in some settings.
CONCLUSION
In conclusion, a QI initiative reducing telemetry order duration did not reduce overall telemetry utilization but increased the number of telemetry orders written. Interventions incorporating appropriate telemetry indications or event risks may be required to change ordering behaviors.
Disclosure: Nothing to report.
The Society of Hospital Medicine's Adult Choosing Wisely measures include not ordering continuous telemetry monitoring outside of the ICU [intensive care unit] without using a protocol that governs continuation.[1] Current guidelines for cardiac monitoring use recommend minimum durations for all adult class I and most class II indications.[2] However, telemetry ordering often fails to include timing or criteria for discontinuation. We determined the impact of a reduction in telemetry order duration within our hospital, hypothesizing this reduction would lead to earlier reassessment of telemetry need and therefore decrease overall utilization.
METHODS
Setting
Durham Veterans Affairs Medical Center (DVAMC) is a 151‐bed tertiary care hospital within Veterans Affairs (VA) Integrated Services Network Region 6 (VISN 6) serving as the primary VA hospital for >54,000 patients and a referral hospital for VISN 6. Twenty‐five telemetry units are available for use on 2 wards with 48 potential telemetry beds. All nonintensive care wards contain general medical and surgical patients, without a primary inpatient cardiology service. Most orders are written by housestaff supervised by attending physicians.
Intervention
Prior to our intervention, the maximum allowable duration of telemetry orders was 72 hours. The duration was enforced by nursing staff automatically discontinuing telemetry not renewed within 72 hours. For our intervention, we reduced the duration of telemetry within our electronic ordering system in November 2013 so that orders had to be renewed within 48 hours or they were discontinued. No education regarding appropriate telemetry use was provided. This intervention was created as a quality‐improvement (QI) project affecting all telemetry use within DVAMC and was exempt from institutional review board review.
Outcomes
Outcomes included the mean number of telemetry orders per week, mean duration of telemetry orders, mean duration of telemetry per episode, and the ratio of time on telemetry relative to the total length of stay. As a balancing measure, we examined rates of rapid response and code blue events. All measures were compared for 12 weeks before and 16 weeks after the intervention. Telemetry orders and durations were obtained using the Corporate Data Warehouse.
Analysis
All outcome measurements were continuous variables and compared using the Student t test in Stata version 9.2 (StataCorp, College Station, TX).
RESULTS
Following the intervention, overall order duration decreased by 33% from 66.68.3 hours to 44.52.3 hours per order (P0.01), mirroring the reduction in the maximum telemetry order duration from 72 to 48 hours (Table 1). However, an increase in telemetry order frequency after the intervention resulted in no significant change in telemetry duration per episode or the proportion of the hospitalization on telemetry (59.3 vs 56.3 hours per patient, P=0.43; and 66.4% vs 66.2% of hospitalization, P=0.58). Rapid response and code blue events did not differ significantly relative to the intervention (2.8 events per week before and 3.1 events per week after, P=0.63).
| Before Intervention | After Intervention | P Value | |
|---|---|---|---|
| |||
| No. of hospitalizations with telemetry ordered | 557 | 684 | NA |
| No. of telemetry orders | 952 | 1515 | NA |
| Average no. of orders per week (SD) | 79.3 (9.2) | 94.7 (25.9) | 0.06 |
| Hours of telemetry per order (SD) | 66.6 (8.3) | 44.5 (2.3) | 0.01 |
| Duration of telemetry per patient, h | 59.3 | 56.3 | 0.43 |
| % of hospitalizations receiving telemetry per patient | 66.4% | 66.2% | 0.90 |
| RRT/code blue events per week | 2.8 | 3.1 | 0.63 |
DISCUSSION
Overall, telemetry utilization was unchanged in spite of an intervention successfully reducing telemetry order duration. Providers responded to this decreased order duration by increasing renewal orders, leaving the amount of time patients spent on telemetry unchanged.
Little primary evidence underlies the American Heart Association recommendations for duration of telemetry in general ward patients.[2] The existing literature documents the timing in which arrhythmias occur after cardiac surgery or myocardial infarction, and therefore is limited in guiding patient care outside intensive care unit settings.[3, 4] As such, hospitalists and inpatient providers have little data directing additional telemetry decisions for these patients, and none for patients requiring telemetry for other indications.
As interventions focusing solely on telemetry duration may not lead to changes in usage patterns, reducing telemetry utilization may require active stewardship. For example, explicit justification may be needed for renewal of telemetry orders. Similarly, education on appropriate telemetry indications in tandem with electronic ordering changes may be more likely to change behavior. Alternatively, incorporating data identifying chest pain patients at very low risk of developing arrhythmias or cardiac complications, based on published risk scores at the time of ordering, may lead to better decision making in initiating telemetry.[5, 6]
This QI project had several limitations. First, the intervention occurred in a facility with a previous telemetry order duration limit. In hospitals without a current duration limitation, some reduction in overall telemetry utilization may be possible. Second, this project was a nonrandom before/after study and potentially subject to bias due to confounding. However, our limited number of telemetry resources, the relatively low number of inpatient teams at our facility, and the inability to target geographic locations for team admissions would have made a cluster‐randomized trial impractical. Third, rationales for telemetry ordering were unknown, as well as drivers for increased orders after the intervention. Better understanding these factors could lead to targeted interventions in some settings.
CONCLUSION
In conclusion, a QI initiative reducing telemetry order duration did not reduce overall telemetry utilization but increased the number of telemetry orders written. Interventions incorporating appropriate telemetry indications or event risks may be required to change ordering behaviors.
Disclosure: Nothing to report.
- Society of Hospital Medicine. Society of Hospital Medicine–adult hospital medicine: five things physicians and patients should question. Available at: http://www.choosingwisely.org/doctor‐patient‐lists/society‐of‐hospital‐medicine‐adult‐hospital‐medicine. Accessed June 4, 2014.
- , , , et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical‐Care Nurses. Circulation. 2004;110(17):2721–2746.
- , , , . Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539–549.
- , , , et al. Time‐based risk assessment after myocardial infarction. Implications for timing of discharge and applications to medical decision‐making. Eur Heart J. 2003;24(2):182–189.
- , , , et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med. 2001;110(1):7–11.
- , , , . Lack of utility of telemetry monitoring for identification of cardiac death and life‐threatening ventricular dysrhythmias in low‐risk patients with chest pain. Ann Emerg Med. 2004;43(1):71–76.
- Society of Hospital Medicine. Society of Hospital Medicine–adult hospital medicine: five things physicians and patients should question. Available at: http://www.choosingwisely.org/doctor‐patient‐lists/society‐of‐hospital‐medicine‐adult‐hospital‐medicine. Accessed June 4, 2014.
- , , , et al. Practice standards for electrocardiographic monitoring in hospital settings: an American Heart Association scientific statement from the Councils on Cardiovascular Nursing, Clinical Cardiology, and Cardiovascular Disease in the Young: endorsed by the International Society of Computerized Electrocardiology and the American Association of Critical‐Care Nurses. Circulation. 2004;110(17):2721–2746.
- , , , . Hazards of postoperative atrial arrhythmias. Ann Thorac Surg. 1993;56(3):539–549.
- , , , et al. Time‐based risk assessment after myocardial infarction. Implications for timing of discharge and applications to medical decision‐making. Eur Heart J. 2003;24(2):182–189.
- , , , et al. Emergency department admissions to inpatient cardiac telemetry beds: a prospective cohort study of risk stratification and outcomes. Am J Med. 2001;110(1):7–11.
- , , , . Lack of utility of telemetry monitoring for identification of cardiac death and life‐threatening ventricular dysrhythmias in low‐risk patients with chest pain. Ann Emerg Med. 2004;43(1):71–76.
Compound can inhibit metastasis in multiple myeloma
Credit: Daniel E. Sabath
A novel compound can prevent metastasis in mouse models of multiple myeloma (MM), according to research published in Cell Reports.
Investigators discovered that this compound, olaptesed pegol, can inhibit stromal cell-derived factor-1 (SDF-1), which attracts certain cells to new locations within the bone marrow.
By blocking the activity of SDF-1, olaptesed pegol renders the bone marrow uninviting to MM cells and prevents metastasis.
“Metastasis remains one of the most formidable complications we face as cancer researchers and physicians,” said study author Irene Ghobrial, MD, of the Dana-Farber Cancer Institute in Boston.
“Improvements in the treatment of metastatic cancers have, for the most part, not been nearly as dramatic as in primary disease.”
Dr Ghobrial and her colleagues studied MM because it is metastatic by nature. Myeloma cells originate in the bone marrow, depart for the bloodstream, and eventually return to the bones, where they form numerous colonies.
The team found that mice with advanced stages of MM had higher levels of SDF-1 at sites in the bones where metastasis had occurred.
“We reasoned that by neutralizing SDF-1, we could change the bone marrow environment to make it less receptive for multiple myeloma cells, reduce myeloma cells’ affinity for the marrow, and thereby inhibit the progression of the disease,” said Aldo Roccaro, MD, PhD, also of Dana-Farber.
Working with the German biotechnology company NOXXON Pharma, the investigators tested olaptesed pegol (a PEGylated mirror-image L-oligonucleotide), which binds to SDF-1.
The team found that olaptesed pegol modulates bone marrow niches and prevents MM cells from homing and engrafting to bone.
This slowed disease progression and prolonged survival in the animals, both compared to control mice and mice treated with AMD3100.
The investigators said it isn’t completely clear what becomes of the blood-borne MM cells that are prevented from metastasizing.
“We know that myeloma cells can’t survive for long if they’re circulating in the blood and can’t adhere to other tissue,” Dr Ghobrial said. “We saw no evidence that they had metastasized and begun to grow in other tissue either.”
“Our findings clearly document a therapeutic effect of olaptesed pegol in a mouse model of advanced myeloma. It is now being tested in a clinical trial of multiple myeloma patients, with more trials to come.” ![]()
Credit: Daniel E. Sabath
A novel compound can prevent metastasis in mouse models of multiple myeloma (MM), according to research published in Cell Reports.
Investigators discovered that this compound, olaptesed pegol, can inhibit stromal cell-derived factor-1 (SDF-1), which attracts certain cells to new locations within the bone marrow.
By blocking the activity of SDF-1, olaptesed pegol renders the bone marrow uninviting to MM cells and prevents metastasis.
“Metastasis remains one of the most formidable complications we face as cancer researchers and physicians,” said study author Irene Ghobrial, MD, of the Dana-Farber Cancer Institute in Boston.
“Improvements in the treatment of metastatic cancers have, for the most part, not been nearly as dramatic as in primary disease.”
Dr Ghobrial and her colleagues studied MM because it is metastatic by nature. Myeloma cells originate in the bone marrow, depart for the bloodstream, and eventually return to the bones, where they form numerous colonies.
The team found that mice with advanced stages of MM had higher levels of SDF-1 at sites in the bones where metastasis had occurred.
“We reasoned that by neutralizing SDF-1, we could change the bone marrow environment to make it less receptive for multiple myeloma cells, reduce myeloma cells’ affinity for the marrow, and thereby inhibit the progression of the disease,” said Aldo Roccaro, MD, PhD, also of Dana-Farber.
Working with the German biotechnology company NOXXON Pharma, the investigators tested olaptesed pegol (a PEGylated mirror-image L-oligonucleotide), which binds to SDF-1.
The team found that olaptesed pegol modulates bone marrow niches and prevents MM cells from homing and engrafting to bone.
This slowed disease progression and prolonged survival in the animals, both compared to control mice and mice treated with AMD3100.
The investigators said it isn’t completely clear what becomes of the blood-borne MM cells that are prevented from metastasizing.
“We know that myeloma cells can’t survive for long if they’re circulating in the blood and can’t adhere to other tissue,” Dr Ghobrial said. “We saw no evidence that they had metastasized and begun to grow in other tissue either.”
“Our findings clearly document a therapeutic effect of olaptesed pegol in a mouse model of advanced myeloma. It is now being tested in a clinical trial of multiple myeloma patients, with more trials to come.” ![]()
Credit: Daniel E. Sabath
A novel compound can prevent metastasis in mouse models of multiple myeloma (MM), according to research published in Cell Reports.
Investigators discovered that this compound, olaptesed pegol, can inhibit stromal cell-derived factor-1 (SDF-1), which attracts certain cells to new locations within the bone marrow.
By blocking the activity of SDF-1, olaptesed pegol renders the bone marrow uninviting to MM cells and prevents metastasis.
“Metastasis remains one of the most formidable complications we face as cancer researchers and physicians,” said study author Irene Ghobrial, MD, of the Dana-Farber Cancer Institute in Boston.
“Improvements in the treatment of metastatic cancers have, for the most part, not been nearly as dramatic as in primary disease.”
Dr Ghobrial and her colleagues studied MM because it is metastatic by nature. Myeloma cells originate in the bone marrow, depart for the bloodstream, and eventually return to the bones, where they form numerous colonies.
The team found that mice with advanced stages of MM had higher levels of SDF-1 at sites in the bones where metastasis had occurred.
“We reasoned that by neutralizing SDF-1, we could change the bone marrow environment to make it less receptive for multiple myeloma cells, reduce myeloma cells’ affinity for the marrow, and thereby inhibit the progression of the disease,” said Aldo Roccaro, MD, PhD, also of Dana-Farber.
Working with the German biotechnology company NOXXON Pharma, the investigators tested olaptesed pegol (a PEGylated mirror-image L-oligonucleotide), which binds to SDF-1.
The team found that olaptesed pegol modulates bone marrow niches and prevents MM cells from homing and engrafting to bone.
This slowed disease progression and prolonged survival in the animals, both compared to control mice and mice treated with AMD3100.
The investigators said it isn’t completely clear what becomes of the blood-borne MM cells that are prevented from metastasizing.
“We know that myeloma cells can’t survive for long if they’re circulating in the blood and can’t adhere to other tissue,” Dr Ghobrial said. “We saw no evidence that they had metastasized and begun to grow in other tissue either.”
“Our findings clearly document a therapeutic effect of olaptesed pegol in a mouse model of advanced myeloma. It is now being tested in a clinical trial of multiple myeloma patients, with more trials to come.” ![]()