User login
MEK inhibitors can induce skin eruptions with distinctive duskiness
Case reports of unusual drug hypersensitivity to MEK inhibitors, involving skin eruptions with distinctive central duskiness, have been described online in JAMA Dermatology.
Three patients who were receiving different MEK inhibitors (selumetinib, cobimetinib, and trametinib) developed grade 2 or 3 eruptions, all associated with unique duskiness, reported Dr. Urvi Patel and associates at Washington University, St. Louis.
A 60-year-old man with pancreatic cancer who was receiving selumetinib as part of a clinical trial presented with a grade 2 generalized eruption and pruritus 12 days after initiating therapy. He had diffuse targetoid patches with central duskiness. Selumetinib and other study drugs were withheld, the patient was given topical corticosteroid treatment, and the eruption completely resolved after 4 weeks. The patient did not restart the study drugs because of an elevated alkaline phosphatase level and fatigue.
A woman in her 40s who was receiving cobimetinib and other medication for metastatic melanoma developed grade 2 coalescing urticarial patches with surrounding duskiness on day 28 of treatment. Histopathologic examination showed a superficial perivascular lymphocytic infiltrate with rare eosinophils. After treatment was halted for 7 days and a regimen of oral prednisone was started, cobimetinib therapy was reinstituted at a lower dose. There was no recurrence of the eruption 1 year after cobimetinib therapy was restarted, Dr. Patel and associates reported (JAMA Dermatol. 2015 Jan. 14 [doi:10.1001/jamadermatol.2014.3207]).
The third patient, a woman in her 50s with metastatic melanoma, developed a grade 3 eruption 7 weeks into trametinib treatment together with another drug. The worsening urticarial patches and plaques had surrounding diffuse duskiness. After trametinib treatment was withheld for a week, and a regimen of oral prednisone was begun, trametinib therapy was restarted and the eruption did not return.
“As shown in our patients, successful treatment of this MEK inhibitor–associated cutaneous eruption can include a drug holiday and oral corticosteroid therapy, with reinstitution of the drug at a lower dose without recurrence,” Dr. Patel and his associates wrote.
MEK inhibitors target the mitogen-activated protein kinase pathway. Trametinib has been approved for treating advanced melanoma, and more than a dozen other MEK inhibitors are in clinical trials (including selumetinib and cobimetinib) for treatment of melanoma and other solid-organ malignant neoplasms, including pancreatic, hepatocellular, colorectal, and non–small cell lung cancer, the authors noted.
On Twitter @nikolaideslaura
Case reports of unusual drug hypersensitivity to MEK inhibitors, involving skin eruptions with distinctive central duskiness, have been described online in JAMA Dermatology.
Three patients who were receiving different MEK inhibitors (selumetinib, cobimetinib, and trametinib) developed grade 2 or 3 eruptions, all associated with unique duskiness, reported Dr. Urvi Patel and associates at Washington University, St. Louis.
A 60-year-old man with pancreatic cancer who was receiving selumetinib as part of a clinical trial presented with a grade 2 generalized eruption and pruritus 12 days after initiating therapy. He had diffuse targetoid patches with central duskiness. Selumetinib and other study drugs were withheld, the patient was given topical corticosteroid treatment, and the eruption completely resolved after 4 weeks. The patient did not restart the study drugs because of an elevated alkaline phosphatase level and fatigue.
A woman in her 40s who was receiving cobimetinib and other medication for metastatic melanoma developed grade 2 coalescing urticarial patches with surrounding duskiness on day 28 of treatment. Histopathologic examination showed a superficial perivascular lymphocytic infiltrate with rare eosinophils. After treatment was halted for 7 days and a regimen of oral prednisone was started, cobimetinib therapy was reinstituted at a lower dose. There was no recurrence of the eruption 1 year after cobimetinib therapy was restarted, Dr. Patel and associates reported (JAMA Dermatol. 2015 Jan. 14 [doi:10.1001/jamadermatol.2014.3207]).
The third patient, a woman in her 50s with metastatic melanoma, developed a grade 3 eruption 7 weeks into trametinib treatment together with another drug. The worsening urticarial patches and plaques had surrounding diffuse duskiness. After trametinib treatment was withheld for a week, and a regimen of oral prednisone was begun, trametinib therapy was restarted and the eruption did not return.
“As shown in our patients, successful treatment of this MEK inhibitor–associated cutaneous eruption can include a drug holiday and oral corticosteroid therapy, with reinstitution of the drug at a lower dose without recurrence,” Dr. Patel and his associates wrote.
MEK inhibitors target the mitogen-activated protein kinase pathway. Trametinib has been approved for treating advanced melanoma, and more than a dozen other MEK inhibitors are in clinical trials (including selumetinib and cobimetinib) for treatment of melanoma and other solid-organ malignant neoplasms, including pancreatic, hepatocellular, colorectal, and non–small cell lung cancer, the authors noted.
On Twitter @nikolaideslaura
Case reports of unusual drug hypersensitivity to MEK inhibitors, involving skin eruptions with distinctive central duskiness, have been described online in JAMA Dermatology.
Three patients who were receiving different MEK inhibitors (selumetinib, cobimetinib, and trametinib) developed grade 2 or 3 eruptions, all associated with unique duskiness, reported Dr. Urvi Patel and associates at Washington University, St. Louis.
A 60-year-old man with pancreatic cancer who was receiving selumetinib as part of a clinical trial presented with a grade 2 generalized eruption and pruritus 12 days after initiating therapy. He had diffuse targetoid patches with central duskiness. Selumetinib and other study drugs were withheld, the patient was given topical corticosteroid treatment, and the eruption completely resolved after 4 weeks. The patient did not restart the study drugs because of an elevated alkaline phosphatase level and fatigue.
A woman in her 40s who was receiving cobimetinib and other medication for metastatic melanoma developed grade 2 coalescing urticarial patches with surrounding duskiness on day 28 of treatment. Histopathologic examination showed a superficial perivascular lymphocytic infiltrate with rare eosinophils. After treatment was halted for 7 days and a regimen of oral prednisone was started, cobimetinib therapy was reinstituted at a lower dose. There was no recurrence of the eruption 1 year after cobimetinib therapy was restarted, Dr. Patel and associates reported (JAMA Dermatol. 2015 Jan. 14 [doi:10.1001/jamadermatol.2014.3207]).
The third patient, a woman in her 50s with metastatic melanoma, developed a grade 3 eruption 7 weeks into trametinib treatment together with another drug. The worsening urticarial patches and plaques had surrounding diffuse duskiness. After trametinib treatment was withheld for a week, and a regimen of oral prednisone was begun, trametinib therapy was restarted and the eruption did not return.
“As shown in our patients, successful treatment of this MEK inhibitor–associated cutaneous eruption can include a drug holiday and oral corticosteroid therapy, with reinstitution of the drug at a lower dose without recurrence,” Dr. Patel and his associates wrote.
MEK inhibitors target the mitogen-activated protein kinase pathway. Trametinib has been approved for treating advanced melanoma, and more than a dozen other MEK inhibitors are in clinical trials (including selumetinib and cobimetinib) for treatment of melanoma and other solid-organ malignant neoplasms, including pancreatic, hepatocellular, colorectal, and non–small cell lung cancer, the authors noted.
On Twitter @nikolaideslaura
FROM JAMA DERMATOLOGY
Key clinical point: This MEK inhibitor–associated cutaneous eruption can be treated with a drug holiday and oral corticosteroid treatment, restarting the drug at a lower dose without recurrence.
Major finding: Three patients who were receiving different MEK inhibitors (selumetinib, cobimetinib, and trametinib) developed grade 2 or 3 eruptions, all associated with unique duskiness.
Data source: Three case studies of patients receiving different MEK inhibitors.
Disclosures: Dr. Lynn Cornelius has received a research grant from Genentech and is a clinical subinvestigator for GlaxoSmithKline. Dr. Milan J. Anadkat has received honoraria as a speaker and/or consultant from AstraZeneca, Bristol-Myers Squibb, Eisai, ImClone, and Therakos. No other disclosures were reported.
Real-world data with rivaroxaban similar to trial data

Credit: CDC
A post-marketing study in patients with non-valvular atrial fibrillation (NVAF) suggests that rates and patterns of major bleeding associated with the use of rivaroxaban in routine clinical practice are generally consistent with those observed in a previous phase 3 trial.
The new study, which included more than 27,000 patients, showed a low rate of major bleeding. And gastrointestinal bleeding was more common than intracranial hemorrhage.
While the results are not intended for a direct comparison, the researchers said these data are generally consistent with data from the ROCKET-AF trial.
The results, published in Clinical Cardiology, are the initial findings from an ongoing, 5-year, observational study of patients using rivaroxaban daily over the course of their lives.
“These findings reaffirm the safety profile of Xarelto [rivaroxaban],” said study author W. Frank Peacock, MD, of the Baylor College of Medicine in Houston, Texas.
“We anticipate future findings from this 5-year observational study will continue to provide real-world information about the use of Xarelto in routine clinical practice.”
Dr Peacock and his colleagues analyzed data from January 1, 2013, to March 31, 2014, evaluating major bleeding in 27,467 NVAF patients treated with rivaroxaban in a real-world, clinical setting.
There were a total of 496 major bleeding events in 478 patients. So the incidence of major bleeding was 2.86 per 100 person-years, which was generally consistent with the rate reported in the ROCKET-AF trial.
Most bleeds were gastrointestinal (88.5%), followed by intracranial (7.5%). Fourteen patients each experienced 2 major bleeding events, and 2 patients each experienced 3 events.
Major bleeding was more likely in females and older patients. The incidence of major bleeding was 2.68 for males and 2.99 for females. The mean age of patients with major bleeding was 78.4, compared to 75.7 for those without major bleeding.
Comorbidities tended to be more prevalent among patients who experienced major bleeding. The most common were hypertension (95.6% vs 75.8%), coronary heart disease (64.2% vs 36.7%), heart failure (48.5% vs 23.7%), and renal disease (38.7% vs 16.7%).
Patients with major bleeding were less likely to be on medications such as statins, proton pump inhibitors, and amiodarone—29.1% vs 36.6%.
Fourteen patients died while in the hospital for major bleeding, for a fatal bleeding rate of 0.08 per 100 person-years. None of these patients had multiple bleeding events.
Fatal bleeding events included intracranial hemorrhage with intracerebral bleeding (n=7), gastrointestinal hemorrhage not otherwise specified (n=3), blood in the stool (n=2), subdural hemorrhage (n=1), and intracranial hemorrhage not otherwise specified (n=1).
Patients’ mean age at death was 82.4 years, and the mean time from hospitalization for the bleeding event to death was 3.9 days.
This ongoing study is funded by Janssen Scientific Affairs, LLC, and Bayer Healthcare, the companies developing rivaroxaban. ![]()
Credit: CDC
A post-marketing study in patients with non-valvular atrial fibrillation (NVAF) suggests that rates and patterns of major bleeding associated with the use of rivaroxaban in routine clinical practice are generally consistent with those observed in a previous phase 3 trial.
The new study, which included more than 27,000 patients, showed a low rate of major bleeding. And gastrointestinal bleeding was more common than intracranial hemorrhage.
While the results are not intended for a direct comparison, the researchers said these data are generally consistent with data from the ROCKET-AF trial.
The results, published in Clinical Cardiology, are the initial findings from an ongoing, 5-year, observational study of patients using rivaroxaban daily over the course of their lives.
“These findings reaffirm the safety profile of Xarelto [rivaroxaban],” said study author W. Frank Peacock, MD, of the Baylor College of Medicine in Houston, Texas.
“We anticipate future findings from this 5-year observational study will continue to provide real-world information about the use of Xarelto in routine clinical practice.”
Dr Peacock and his colleagues analyzed data from January 1, 2013, to March 31, 2014, evaluating major bleeding in 27,467 NVAF patients treated with rivaroxaban in a real-world, clinical setting.
There were a total of 496 major bleeding events in 478 patients. So the incidence of major bleeding was 2.86 per 100 person-years, which was generally consistent with the rate reported in the ROCKET-AF trial.
Most bleeds were gastrointestinal (88.5%), followed by intracranial (7.5%). Fourteen patients each experienced 2 major bleeding events, and 2 patients each experienced 3 events.
Major bleeding was more likely in females and older patients. The incidence of major bleeding was 2.68 for males and 2.99 for females. The mean age of patients with major bleeding was 78.4, compared to 75.7 for those without major bleeding.
Comorbidities tended to be more prevalent among patients who experienced major bleeding. The most common were hypertension (95.6% vs 75.8%), coronary heart disease (64.2% vs 36.7%), heart failure (48.5% vs 23.7%), and renal disease (38.7% vs 16.7%).
Patients with major bleeding were less likely to be on medications such as statins, proton pump inhibitors, and amiodarone—29.1% vs 36.6%.
Fourteen patients died while in the hospital for major bleeding, for a fatal bleeding rate of 0.08 per 100 person-years. None of these patients had multiple bleeding events.
Fatal bleeding events included intracranial hemorrhage with intracerebral bleeding (n=7), gastrointestinal hemorrhage not otherwise specified (n=3), blood in the stool (n=2), subdural hemorrhage (n=1), and intracranial hemorrhage not otherwise specified (n=1).
Patients’ mean age at death was 82.4 years, and the mean time from hospitalization for the bleeding event to death was 3.9 days.
This ongoing study is funded by Janssen Scientific Affairs, LLC, and Bayer Healthcare, the companies developing rivaroxaban. ![]()
Credit: CDC
A post-marketing study in patients with non-valvular atrial fibrillation (NVAF) suggests that rates and patterns of major bleeding associated with the use of rivaroxaban in routine clinical practice are generally consistent with those observed in a previous phase 3 trial.
The new study, which included more than 27,000 patients, showed a low rate of major bleeding. And gastrointestinal bleeding was more common than intracranial hemorrhage.
While the results are not intended for a direct comparison, the researchers said these data are generally consistent with data from the ROCKET-AF trial.
The results, published in Clinical Cardiology, are the initial findings from an ongoing, 5-year, observational study of patients using rivaroxaban daily over the course of their lives.
“These findings reaffirm the safety profile of Xarelto [rivaroxaban],” said study author W. Frank Peacock, MD, of the Baylor College of Medicine in Houston, Texas.
“We anticipate future findings from this 5-year observational study will continue to provide real-world information about the use of Xarelto in routine clinical practice.”
Dr Peacock and his colleagues analyzed data from January 1, 2013, to March 31, 2014, evaluating major bleeding in 27,467 NVAF patients treated with rivaroxaban in a real-world, clinical setting.
There were a total of 496 major bleeding events in 478 patients. So the incidence of major bleeding was 2.86 per 100 person-years, which was generally consistent with the rate reported in the ROCKET-AF trial.
Most bleeds were gastrointestinal (88.5%), followed by intracranial (7.5%). Fourteen patients each experienced 2 major bleeding events, and 2 patients each experienced 3 events.
Major bleeding was more likely in females and older patients. The incidence of major bleeding was 2.68 for males and 2.99 for females. The mean age of patients with major bleeding was 78.4, compared to 75.7 for those without major bleeding.
Comorbidities tended to be more prevalent among patients who experienced major bleeding. The most common were hypertension (95.6% vs 75.8%), coronary heart disease (64.2% vs 36.7%), heart failure (48.5% vs 23.7%), and renal disease (38.7% vs 16.7%).
Patients with major bleeding were less likely to be on medications such as statins, proton pump inhibitors, and amiodarone—29.1% vs 36.6%.
Fourteen patients died while in the hospital for major bleeding, for a fatal bleeding rate of 0.08 per 100 person-years. None of these patients had multiple bleeding events.
Fatal bleeding events included intracranial hemorrhage with intracerebral bleeding (n=7), gastrointestinal hemorrhage not otherwise specified (n=3), blood in the stool (n=2), subdural hemorrhage (n=1), and intracranial hemorrhage not otherwise specified (n=1).
Patients’ mean age at death was 82.4 years, and the mean time from hospitalization for the bleeding event to death was 3.9 days.
This ongoing study is funded by Janssen Scientific Affairs, LLC, and Bayer Healthcare, the companies developing rivaroxaban. ![]()
CDC report shows big drop in rate of CLABSIs

through a central line
Credit: Rhoda Baer
Healthcare-associated infections (HAIs) are on the decline in the US, according to a report by the Centers for Disease Control and Prevention (CDC).
The data show that most types of HAIs have decreased in recent years, with a particularly large decrease in the rate of central line-associated bloodstream infections (CLABSIs).
The National and State Healthcare-associated Infection Progress Report is a snapshot of how each state and the country are doing in eliminating the infections that hospitals are required to report to the CDC.
The report summarizes data submitted to the CDC’s National Healthcare Safety Network, the nation’s HAI tracking system, which is used by more than 14,500 healthcare facilities across all 50 states, Washington, DC, and Puerto Rico.
“Hospitals have made real progress to reduce some types of healthcare-associated infections; it can be done,” said CDC Director Tom Frieden, MD.
“The key is for every hospital to have rigorous infection control programs to protect patients and healthcare workers, and for health care facilities and others to work together to reduce the many types of infections that haven’t decreased enough.”
On the national level, the report showed a 46% decrease in CLABSIs between 2008 and 2013. It also revealed a 19% decrease in surgical site infections related to the 10 procedures tracked in the report between 2008 and 2013.
There was an 8% decrease in methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infections between 2011 and 2013 and a 10% decrease in Clostridium difficile infections between 2011 and 2013.
However, there was a 6% increase in catheter-associated urinary tract infections since 2009.
Not all states reported or had enough data to calculate valid infection information on every infection in the report. But the CDC compared the number of infections reported to a national baseline.
And they found that 26 states performed better than the nation on at least 2 of the infection types. Sixteen states performed better than the nation on 3 or more infections, including 6 states performing better on 4 infections.
But 19 states performed worse than the nation on 2 infections, with 8 states performing worse on at least 3 infections.
The national baseline for HAIs will be reset at the end of 2015. Starting in 2016, HAI prevention progress from 2016 to 2020 will be measured in comparison to infection data from 2015. ![]()
through a central line
Credit: Rhoda Baer
Healthcare-associated infections (HAIs) are on the decline in the US, according to a report by the Centers for Disease Control and Prevention (CDC).
The data show that most types of HAIs have decreased in recent years, with a particularly large decrease in the rate of central line-associated bloodstream infections (CLABSIs).
The National and State Healthcare-associated Infection Progress Report is a snapshot of how each state and the country are doing in eliminating the infections that hospitals are required to report to the CDC.
The report summarizes data submitted to the CDC’s National Healthcare Safety Network, the nation’s HAI tracking system, which is used by more than 14,500 healthcare facilities across all 50 states, Washington, DC, and Puerto Rico.
“Hospitals have made real progress to reduce some types of healthcare-associated infections; it can be done,” said CDC Director Tom Frieden, MD.
“The key is for every hospital to have rigorous infection control programs to protect patients and healthcare workers, and for health care facilities and others to work together to reduce the many types of infections that haven’t decreased enough.”
On the national level, the report showed a 46% decrease in CLABSIs between 2008 and 2013. It also revealed a 19% decrease in surgical site infections related to the 10 procedures tracked in the report between 2008 and 2013.
There was an 8% decrease in methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infections between 2011 and 2013 and a 10% decrease in Clostridium difficile infections between 2011 and 2013.
However, there was a 6% increase in catheter-associated urinary tract infections since 2009.
Not all states reported or had enough data to calculate valid infection information on every infection in the report. But the CDC compared the number of infections reported to a national baseline.
And they found that 26 states performed better than the nation on at least 2 of the infection types. Sixteen states performed better than the nation on 3 or more infections, including 6 states performing better on 4 infections.
But 19 states performed worse than the nation on 2 infections, with 8 states performing worse on at least 3 infections.
The national baseline for HAIs will be reset at the end of 2015. Starting in 2016, HAI prevention progress from 2016 to 2020 will be measured in comparison to infection data from 2015. ![]()
through a central line
Credit: Rhoda Baer
Healthcare-associated infections (HAIs) are on the decline in the US, according to a report by the Centers for Disease Control and Prevention (CDC).
The data show that most types of HAIs have decreased in recent years, with a particularly large decrease in the rate of central line-associated bloodstream infections (CLABSIs).
The National and State Healthcare-associated Infection Progress Report is a snapshot of how each state and the country are doing in eliminating the infections that hospitals are required to report to the CDC.
The report summarizes data submitted to the CDC’s National Healthcare Safety Network, the nation’s HAI tracking system, which is used by more than 14,500 healthcare facilities across all 50 states, Washington, DC, and Puerto Rico.
“Hospitals have made real progress to reduce some types of healthcare-associated infections; it can be done,” said CDC Director Tom Frieden, MD.
“The key is for every hospital to have rigorous infection control programs to protect patients and healthcare workers, and for health care facilities and others to work together to reduce the many types of infections that haven’t decreased enough.”
On the national level, the report showed a 46% decrease in CLABSIs between 2008 and 2013. It also revealed a 19% decrease in surgical site infections related to the 10 procedures tracked in the report between 2008 and 2013.
There was an 8% decrease in methicillin-resistant Staphylococcus aureus (MRSA) bloodstream infections between 2011 and 2013 and a 10% decrease in Clostridium difficile infections between 2011 and 2013.
However, there was a 6% increase in catheter-associated urinary tract infections since 2009.
Not all states reported or had enough data to calculate valid infection information on every infection in the report. But the CDC compared the number of infections reported to a national baseline.
And they found that 26 states performed better than the nation on at least 2 of the infection types. Sixteen states performed better than the nation on 3 or more infections, including 6 states performing better on 4 infections.
But 19 states performed worse than the nation on 2 infections, with 8 states performing worse on at least 3 infections.
The national baseline for HAIs will be reset at the end of 2015. Starting in 2016, HAI prevention progress from 2016 to 2020 will be measured in comparison to infection data from 2015. ![]()
Iron overload aids potentially deadly bacteria
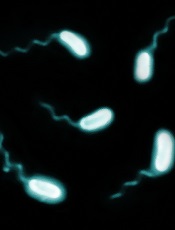
Credit: Paul A. Gulig
Researchers say they’ve determined why patients with hereditary hemochromatosis are so vulnerable to severe illness from Vibrio vulnificus infection.
Patients with hereditary hemochromatosis have a deficiency of the iron-regulating hormone hepcidin and therefore develop excess iron in their blood and tissue, providing prime growth conditions for Vibrio vulnificus.
The researchers also showed that minihepcidin, a medicinal form of the hormone hepcidin that lowers iron levels in blood, could cure the infection by restricting bacterial growth.
The findings appear in Cell Host and Microbe.
“This is the first time that the association of hepcidin deficiency and susceptibility to Vibrio vulnificus infection was tested,” said study author Yonca Bulut, MD, of the David Geffen School of Medicine at UCLA in Los Angeles.
“The dramatic effectiveness of the new treatment, even after the infection was established, was impressive.”
To conduct this study, Dr Bulut and her colleagues compared the fatality of Vibrio vulnificus infection in healthy mice with mice that lacked hepcidin, modeling human hereditary hemochromatosis.
The infection was much more lethal in hepcidin-deficient mice because their bodies could not decrease iron levels in the blood in response to infection, a process mediated by hepcidin in healthy mice.
Giving minihepcidin to hepcidin-deficient mice prevented infection if the hormone was given before Vibrio vulnificus was introduced. And mice given minihepcidin 3 hours after the bacterium was introduced were cured of any infection.
“We found that hepcidin is required for resistance to a Vibrio vulnificus infection,” said study author Joao Arezes, a graduate student from the University of Porto in Portugal.
“The development of the treatment tested in mouse models could reduce the high mortality rate of this disease.”
The next stage of this research is to investigate why Vibrio vulnificus bacteria become so lethal when iron levels are high, and to learn which other microbes respond similarly to excess iron. ![]()
Credit: Paul A. Gulig
Researchers say they’ve determined why patients with hereditary hemochromatosis are so vulnerable to severe illness from Vibrio vulnificus infection.
Patients with hereditary hemochromatosis have a deficiency of the iron-regulating hormone hepcidin and therefore develop excess iron in their blood and tissue, providing prime growth conditions for Vibrio vulnificus.
The researchers also showed that minihepcidin, a medicinal form of the hormone hepcidin that lowers iron levels in blood, could cure the infection by restricting bacterial growth.
The findings appear in Cell Host and Microbe.
“This is the first time that the association of hepcidin deficiency and susceptibility to Vibrio vulnificus infection was tested,” said study author Yonca Bulut, MD, of the David Geffen School of Medicine at UCLA in Los Angeles.
“The dramatic effectiveness of the new treatment, even after the infection was established, was impressive.”
To conduct this study, Dr Bulut and her colleagues compared the fatality of Vibrio vulnificus infection in healthy mice with mice that lacked hepcidin, modeling human hereditary hemochromatosis.
The infection was much more lethal in hepcidin-deficient mice because their bodies could not decrease iron levels in the blood in response to infection, a process mediated by hepcidin in healthy mice.
Giving minihepcidin to hepcidin-deficient mice prevented infection if the hormone was given before Vibrio vulnificus was introduced. And mice given minihepcidin 3 hours after the bacterium was introduced were cured of any infection.
“We found that hepcidin is required for resistance to a Vibrio vulnificus infection,” said study author Joao Arezes, a graduate student from the University of Porto in Portugal.
“The development of the treatment tested in mouse models could reduce the high mortality rate of this disease.”
The next stage of this research is to investigate why Vibrio vulnificus bacteria become so lethal when iron levels are high, and to learn which other microbes respond similarly to excess iron. ![]()
Credit: Paul A. Gulig
Researchers say they’ve determined why patients with hereditary hemochromatosis are so vulnerable to severe illness from Vibrio vulnificus infection.
Patients with hereditary hemochromatosis have a deficiency of the iron-regulating hormone hepcidin and therefore develop excess iron in their blood and tissue, providing prime growth conditions for Vibrio vulnificus.
The researchers also showed that minihepcidin, a medicinal form of the hormone hepcidin that lowers iron levels in blood, could cure the infection by restricting bacterial growth.
The findings appear in Cell Host and Microbe.
“This is the first time that the association of hepcidin deficiency and susceptibility to Vibrio vulnificus infection was tested,” said study author Yonca Bulut, MD, of the David Geffen School of Medicine at UCLA in Los Angeles.
“The dramatic effectiveness of the new treatment, even after the infection was established, was impressive.”
To conduct this study, Dr Bulut and her colleagues compared the fatality of Vibrio vulnificus infection in healthy mice with mice that lacked hepcidin, modeling human hereditary hemochromatosis.
The infection was much more lethal in hepcidin-deficient mice because their bodies could not decrease iron levels in the blood in response to infection, a process mediated by hepcidin in healthy mice.
Giving minihepcidin to hepcidin-deficient mice prevented infection if the hormone was given before Vibrio vulnificus was introduced. And mice given minihepcidin 3 hours after the bacterium was introduced were cured of any infection.
“We found that hepcidin is required for resistance to a Vibrio vulnificus infection,” said study author Joao Arezes, a graduate student from the University of Porto in Portugal.
“The development of the treatment tested in mouse models could reduce the high mortality rate of this disease.”
The next stage of this research is to investigate why Vibrio vulnificus bacteria become so lethal when iron levels are high, and to learn which other microbes respond similarly to excess iron. ![]()
US is leading sponsor of medical research despite slow growth in funding

Credit: Rhoda Baer
An analysis of countries in North America, Europe, and Asia-Oceania showed that the US had the slowest annual growth in medical research funding from 2004 to 2011.
Nevertheless, the US was the leading sponsor of global medical research in 2011, accounting for 44% of the $265 billion spent in all the regions studied.
Hamilton Moses III, MD, of the Alerion Institute and Alerion Advisors LLC, in North Garden, Virginia, and his colleagues reported these discoveries in JAMA.
The researchers examined developments over the past 2 decades in the pattern of who conducts and who supports medical research, as well as resulting patents, publications, and new drug and device approvals.
The group compiled publicly available data from 1994 to 2012, showing trends in US and international research funding, productivity, and disease burden by source and industry type. Patents and publications (1981-2011) were evaluated using citation rates and impact factors.
International research funding
The researchers included data from the major countries of North America (US and Canada), Europe (including the 10 largest European countries in the
Organisation for Economic Co-operation and Development), and Asia-Oceania (Australia, China, India, Japan, Singapore, and South Korea).
Of these regions, the US had the lowest rate of annual growth in funding from 2004 to 2011 (1%). The rate was 4.1% in Europe, 4.5% in Canada, 6.8% in Japan, 9.3% in Australia, 16.9% in China, and 20.8% in the other Asian countries.
Still, in 2011, the US invested $117.2 billion (44%) of the $265 billion spent in all the regions studied. Europe spent $88.6 billion (33%), Japan spent $37.8 billion (14%), China spent $4.9 billion (1.2%), other Asian countries spent $9.7 billion (4%), Australia spent $3.8 billion (1.4%), and Canada spent $3.1 billion (1.2%).
Research outcomes
Dr Moses and his colleagues also compared other aspects of medical research among the regions, such as patent applications, research articles, and drug approvals.
They found that China filed 30% of global life science patent applications in 2011, while the US filed 24%. Japan filed the fewest applications of all the
regions analyzed.
The US and the European Union were neck-and-neck with regard to the share of biomedical research articles published in all regions in 2009—33.4% and 32.8%, respectively. China’s share was only 5%, but the country had the greatestgrowth in contribution from 2000 through 2009, at 18.7%.
And the European Medicines Agency (EMA) outstripped the US Food and Drug Administration (FDA) when it came to drug approvals. In 2013, the EMA approved 57 new molecular entities and biologics, compared to the FDA’s 27. From 2003 to2013, the FDA averaged 26 approvals per year, and the EMA averaged 42. ![]()
Credit: Rhoda Baer
An analysis of countries in North America, Europe, and Asia-Oceania showed that the US had the slowest annual growth in medical research funding from 2004 to 2011.
Nevertheless, the US was the leading sponsor of global medical research in 2011, accounting for 44% of the $265 billion spent in all the regions studied.
Hamilton Moses III, MD, of the Alerion Institute and Alerion Advisors LLC, in North Garden, Virginia, and his colleagues reported these discoveries in JAMA.
The researchers examined developments over the past 2 decades in the pattern of who conducts and who supports medical research, as well as resulting patents, publications, and new drug and device approvals.
The group compiled publicly available data from 1994 to 2012, showing trends in US and international research funding, productivity, and disease burden by source and industry type. Patents and publications (1981-2011) were evaluated using citation rates and impact factors.
International research funding
The researchers included data from the major countries of North America (US and Canada), Europe (including the 10 largest European countries in the
Organisation for Economic Co-operation and Development), and Asia-Oceania (Australia, China, India, Japan, Singapore, and South Korea).
Of these regions, the US had the lowest rate of annual growth in funding from 2004 to 2011 (1%). The rate was 4.1% in Europe, 4.5% in Canada, 6.8% in Japan, 9.3% in Australia, 16.9% in China, and 20.8% in the other Asian countries.
Still, in 2011, the US invested $117.2 billion (44%) of the $265 billion spent in all the regions studied. Europe spent $88.6 billion (33%), Japan spent $37.8 billion (14%), China spent $4.9 billion (1.2%), other Asian countries spent $9.7 billion (4%), Australia spent $3.8 billion (1.4%), and Canada spent $3.1 billion (1.2%).
Research outcomes
Dr Moses and his colleagues also compared other aspects of medical research among the regions, such as patent applications, research articles, and drug approvals.
They found that China filed 30% of global life science patent applications in 2011, while the US filed 24%. Japan filed the fewest applications of all the
regions analyzed.
The US and the European Union were neck-and-neck with regard to the share of biomedical research articles published in all regions in 2009—33.4% and 32.8%, respectively. China’s share was only 5%, but the country had the greatestgrowth in contribution from 2000 through 2009, at 18.7%.
And the European Medicines Agency (EMA) outstripped the US Food and Drug Administration (FDA) when it came to drug approvals. In 2013, the EMA approved 57 new molecular entities and biologics, compared to the FDA’s 27. From 2003 to2013, the FDA averaged 26 approvals per year, and the EMA averaged 42. ![]()
Credit: Rhoda Baer
An analysis of countries in North America, Europe, and Asia-Oceania showed that the US had the slowest annual growth in medical research funding from 2004 to 2011.
Nevertheless, the US was the leading sponsor of global medical research in 2011, accounting for 44% of the $265 billion spent in all the regions studied.
Hamilton Moses III, MD, of the Alerion Institute and Alerion Advisors LLC, in North Garden, Virginia, and his colleagues reported these discoveries in JAMA.
The researchers examined developments over the past 2 decades in the pattern of who conducts and who supports medical research, as well as resulting patents, publications, and new drug and device approvals.
The group compiled publicly available data from 1994 to 2012, showing trends in US and international research funding, productivity, and disease burden by source and industry type. Patents and publications (1981-2011) were evaluated using citation rates and impact factors.
International research funding
The researchers included data from the major countries of North America (US and Canada), Europe (including the 10 largest European countries in the
Organisation for Economic Co-operation and Development), and Asia-Oceania (Australia, China, India, Japan, Singapore, and South Korea).
Of these regions, the US had the lowest rate of annual growth in funding from 2004 to 2011 (1%). The rate was 4.1% in Europe, 4.5% in Canada, 6.8% in Japan, 9.3% in Australia, 16.9% in China, and 20.8% in the other Asian countries.
Still, in 2011, the US invested $117.2 billion (44%) of the $265 billion spent in all the regions studied. Europe spent $88.6 billion (33%), Japan spent $37.8 billion (14%), China spent $4.9 billion (1.2%), other Asian countries spent $9.7 billion (4%), Australia spent $3.8 billion (1.4%), and Canada spent $3.1 billion (1.2%).
Research outcomes
Dr Moses and his colleagues also compared other aspects of medical research among the regions, such as patent applications, research articles, and drug approvals.
They found that China filed 30% of global life science patent applications in 2011, while the US filed 24%. Japan filed the fewest applications of all the
regions analyzed.
The US and the European Union were neck-and-neck with regard to the share of biomedical research articles published in all regions in 2009—33.4% and 32.8%, respectively. China’s share was only 5%, but the country had the greatestgrowth in contribution from 2000 through 2009, at 18.7%.
And the European Medicines Agency (EMA) outstripped the US Food and Drug Administration (FDA) when it came to drug approvals. In 2013, the EMA approved 57 new molecular entities and biologics, compared to the FDA’s 27. From 2003 to2013, the FDA averaged 26 approvals per year, and the EMA averaged 42. ![]()
Transsplenic TIPS procedure catching on for portal vein thrombosis
CHICAGO – Portal vein recanalization using a transsplenic approach can be utilized to improve transplant candidacy in patients with cirrhosis and chronic portal vein thrombosis, according to Dr. Bartley Thornburg.
“It’s a safe and effective procedure that allows for end-to-end anastomoses at transplant in patients who otherwise would not be able to have them or would require thrombectomy without transplant, and we know that end-to-end anastomoses are associated with decreased morbidity and mortality,” said Dr. Thornburg of Northwestern University Medical Center in Chicago.
Historically, and at his institution, portal vein thrombosis (PVT) is a relative contraindication to liver transplant. The American Association for the Study of Liver Diseases also recommends that transjugular intrahepatic portosystemic shunt (TIPS) placement be avoided in patients with a Model for End-State Liver Disease (MELD) score >18.

In 2013, colleague Dr. Riad Salem demonstrated the efficacy of portal vein recanalization during TIPS using a transhepatic approach in 44 patients, with only one technical failure and three cases of rethrombosis. Among six patients with a baseline MELD score >18, four went on to successful liver transplant, one is awaiting transplant, and one died as a result of bleeding 45 days post procedure despite an improvement in MELD score.
The last three patients in the series underwent TIPS with a transsplenic approach, and not only were the results equally good, but the approach was technically easier, Dr. Thornburg said at a symposium on vascular surgery sponsored by Northwestern University.
Since then, this increasingly common alternative approach has been assessed in another 11 consecutive patients with cirrhosis, portal hypertension, and chronic PVT. All patients had been denied listing for transplant because of their PVT, and four had a baseline MELD score >18.
At the end of the procedure, thrombus persisted in 45% (5 of 11 patients). On a 1-month follow-up venogram, however, three of the five patients had complete resolution of the thrombus without any added anticoagulation, one had persistent partial thrombus that was smaller than at stent placement, and one went on to transplant, Dr. Thornburg said.
All six of the patients with portal vein (PV) patency post procedure have retained patency after a median follow-up of 6.4 months.
“What we’ve learned from doing these cases is that complete elimination of the portal vein thrombus at the time of TIPS placement isn’t necessary,” he said. “It would be easy to get carried away and do suction or AngioJet [mechanical thrombectomy], but what we’ve found is that because of how much flow there is in the portal vein once it’s recanalized and the TIPS is in place, basically establishing a flow allowed for clot clearance by 1 month in all patients, except one.”
The procedure starts like a typical TIPS, with access achieved by advancing a 21-guage needle into the peripheral splenic vein or hilum under ultrasound guidance. A 5-French sheath is then placed through the parenchyma to the origin of splenic vein or the clot and an intrahepatic venogram performed to confirm occlusion, Dr. Thornburg said.
A 5-French Kumpe catheter and glide wire are used to recanalize the thrombosed portal vein, with a 10-mm gooseneck snare placed through the Kumpe in the peripheral portal vein as a target for the TIPS needle.
“Then, we basically get through-and-through access from the IJ [internal jugular] through this splenic access, and that gives us the workability to get our sheath across the portal vein and place our TIPS,” he said.
The remainder of the procedure is similar to that of the transhepatic approach. Angioplasty of the thrombosed PV is performed with an 8-by-40-mm balloon, followed by deployment of a Viatorr stent graft. The stent and PV are dilated with a 10-by-40-mm balloon and the splenic tract embolized with a couple of 4-by-14-cm Nester coils.
Based on their experience, short TIPS are always placed to maximize the amount of portal vein that is available at transplant for the end-to-end anastomoses, Dr. Thornburg said.
All patients who went on to transplant have received end-to-end anastomoses on what transplant surgeons have described as “totally normal” walled portal veins, including one patient who underwent transplant just 1 week post TIPS, he added.
There have been no major bleeding events with the transsplenic approach and only two adverse events: one case of transient encephalopathy and one low-grade fever.
Dr. Thornburg and Dr. Salem reported having no relevant financial disclosures.
CHICAGO – Portal vein recanalization using a transsplenic approach can be utilized to improve transplant candidacy in patients with cirrhosis and chronic portal vein thrombosis, according to Dr. Bartley Thornburg.
“It’s a safe and effective procedure that allows for end-to-end anastomoses at transplant in patients who otherwise would not be able to have them or would require thrombectomy without transplant, and we know that end-to-end anastomoses are associated with decreased morbidity and mortality,” said Dr. Thornburg of Northwestern University Medical Center in Chicago.
Historically, and at his institution, portal vein thrombosis (PVT) is a relative contraindication to liver transplant. The American Association for the Study of Liver Diseases also recommends that transjugular intrahepatic portosystemic shunt (TIPS) placement be avoided in patients with a Model for End-State Liver Disease (MELD) score >18.
In 2013, colleague Dr. Riad Salem demonstrated the efficacy of portal vein recanalization during TIPS using a transhepatic approach in 44 patients, with only one technical failure and three cases of rethrombosis. Among six patients with a baseline MELD score >18, four went on to successful liver transplant, one is awaiting transplant, and one died as a result of bleeding 45 days post procedure despite an improvement in MELD score.
The last three patients in the series underwent TIPS with a transsplenic approach, and not only were the results equally good, but the approach was technically easier, Dr. Thornburg said at a symposium on vascular surgery sponsored by Northwestern University.
Since then, this increasingly common alternative approach has been assessed in another 11 consecutive patients with cirrhosis, portal hypertension, and chronic PVT. All patients had been denied listing for transplant because of their PVT, and four had a baseline MELD score >18.
At the end of the procedure, thrombus persisted in 45% (5 of 11 patients). On a 1-month follow-up venogram, however, three of the five patients had complete resolution of the thrombus without any added anticoagulation, one had persistent partial thrombus that was smaller than at stent placement, and one went on to transplant, Dr. Thornburg said.
All six of the patients with portal vein (PV) patency post procedure have retained patency after a median follow-up of 6.4 months.
“What we’ve learned from doing these cases is that complete elimination of the portal vein thrombus at the time of TIPS placement isn’t necessary,” he said. “It would be easy to get carried away and do suction or AngioJet [mechanical thrombectomy], but what we’ve found is that because of how much flow there is in the portal vein once it’s recanalized and the TIPS is in place, basically establishing a flow allowed for clot clearance by 1 month in all patients, except one.”
The procedure starts like a typical TIPS, with access achieved by advancing a 21-guage needle into the peripheral splenic vein or hilum under ultrasound guidance. A 5-French sheath is then placed through the parenchyma to the origin of splenic vein or the clot and an intrahepatic venogram performed to confirm occlusion, Dr. Thornburg said.
A 5-French Kumpe catheter and glide wire are used to recanalize the thrombosed portal vein, with a 10-mm gooseneck snare placed through the Kumpe in the peripheral portal vein as a target for the TIPS needle.
“Then, we basically get through-and-through access from the IJ [internal jugular] through this splenic access, and that gives us the workability to get our sheath across the portal vein and place our TIPS,” he said.
The remainder of the procedure is similar to that of the transhepatic approach. Angioplasty of the thrombosed PV is performed with an 8-by-40-mm balloon, followed by deployment of a Viatorr stent graft. The stent and PV are dilated with a 10-by-40-mm balloon and the splenic tract embolized with a couple of 4-by-14-cm Nester coils.
Based on their experience, short TIPS are always placed to maximize the amount of portal vein that is available at transplant for the end-to-end anastomoses, Dr. Thornburg said.
All patients who went on to transplant have received end-to-end anastomoses on what transplant surgeons have described as “totally normal” walled portal veins, including one patient who underwent transplant just 1 week post TIPS, he added.
There have been no major bleeding events with the transsplenic approach and only two adverse events: one case of transient encephalopathy and one low-grade fever.
Dr. Thornburg and Dr. Salem reported having no relevant financial disclosures.
CHICAGO – Portal vein recanalization using a transsplenic approach can be utilized to improve transplant candidacy in patients with cirrhosis and chronic portal vein thrombosis, according to Dr. Bartley Thornburg.
“It’s a safe and effective procedure that allows for end-to-end anastomoses at transplant in patients who otherwise would not be able to have them or would require thrombectomy without transplant, and we know that end-to-end anastomoses are associated with decreased morbidity and mortality,” said Dr. Thornburg of Northwestern University Medical Center in Chicago.
Historically, and at his institution, portal vein thrombosis (PVT) is a relative contraindication to liver transplant. The American Association for the Study of Liver Diseases also recommends that transjugular intrahepatic portosystemic shunt (TIPS) placement be avoided in patients with a Model for End-State Liver Disease (MELD) score >18.
In 2013, colleague Dr. Riad Salem demonstrated the efficacy of portal vein recanalization during TIPS using a transhepatic approach in 44 patients, with only one technical failure and three cases of rethrombosis. Among six patients with a baseline MELD score >18, four went on to successful liver transplant, one is awaiting transplant, and one died as a result of bleeding 45 days post procedure despite an improvement in MELD score.
The last three patients in the series underwent TIPS with a transsplenic approach, and not only were the results equally good, but the approach was technically easier, Dr. Thornburg said at a symposium on vascular surgery sponsored by Northwestern University.
Since then, this increasingly common alternative approach has been assessed in another 11 consecutive patients with cirrhosis, portal hypertension, and chronic PVT. All patients had been denied listing for transplant because of their PVT, and four had a baseline MELD score >18.
At the end of the procedure, thrombus persisted in 45% (5 of 11 patients). On a 1-month follow-up venogram, however, three of the five patients had complete resolution of the thrombus without any added anticoagulation, one had persistent partial thrombus that was smaller than at stent placement, and one went on to transplant, Dr. Thornburg said.
All six of the patients with portal vein (PV) patency post procedure have retained patency after a median follow-up of 6.4 months.
“What we’ve learned from doing these cases is that complete elimination of the portal vein thrombus at the time of TIPS placement isn’t necessary,” he said. “It would be easy to get carried away and do suction or AngioJet [mechanical thrombectomy], but what we’ve found is that because of how much flow there is in the portal vein once it’s recanalized and the TIPS is in place, basically establishing a flow allowed for clot clearance by 1 month in all patients, except one.”
The procedure starts like a typical TIPS, with access achieved by advancing a 21-guage needle into the peripheral splenic vein or hilum under ultrasound guidance. A 5-French sheath is then placed through the parenchyma to the origin of splenic vein or the clot and an intrahepatic venogram performed to confirm occlusion, Dr. Thornburg said.
A 5-French Kumpe catheter and glide wire are used to recanalize the thrombosed portal vein, with a 10-mm gooseneck snare placed through the Kumpe in the peripheral portal vein as a target for the TIPS needle.
“Then, we basically get through-and-through access from the IJ [internal jugular] through this splenic access, and that gives us the workability to get our sheath across the portal vein and place our TIPS,” he said.
The remainder of the procedure is similar to that of the transhepatic approach. Angioplasty of the thrombosed PV is performed with an 8-by-40-mm balloon, followed by deployment of a Viatorr stent graft. The stent and PV are dilated with a 10-by-40-mm balloon and the splenic tract embolized with a couple of 4-by-14-cm Nester coils.
Based on their experience, short TIPS are always placed to maximize the amount of portal vein that is available at transplant for the end-to-end anastomoses, Dr. Thornburg said.
All patients who went on to transplant have received end-to-end anastomoses on what transplant surgeons have described as “totally normal” walled portal veins, including one patient who underwent transplant just 1 week post TIPS, he added.
There have been no major bleeding events with the transsplenic approach and only two adverse events: one case of transient encephalopathy and one low-grade fever.
Dr. Thornburg and Dr. Salem reported having no relevant financial disclosures.
AT THE NORTHWESTERN VASCULAR SYMPOSIUM

Mango
Mangifera indica (mango) is a member of the Anacardiaceae family with a tradition of use as a medicinal plant. Mango extracts have been characterized as exhibiting antioxidant, anti-inflammatory, analgesic, and immunomodulatory activities (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9; Drug Chem. Toxicol. 2009;32:53-8). Mango is grown in more than 100 countries, primarily in Asia, in tropical as well as subtropical regions (Molecules 2014;19:17107-29). Mango stem bark and leaves have been used in traditional medicine to treat anemia, cutaneous infections, diabetes, diarrhea, scabies, syphilis, and malignant tumors (Pharmacol. Res. 2007;55:351-8). Polyphenols and carotenoids are among the phytonutrients identified as responsible for the biologic activity of mango (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).

Various biologic activities and traditional uses
Ojewole investigated the anti-inflammatory, analgesic, and antidiabetic activity of M. indica stem bark aqueous extract in rats and mice in 2005. In mice, mango extract dose-dependently delivered significant analgesic effects against thermally and chemically-generated pain. The investigators attributed the observed salutary effects of the plant to its constituent polyphenolics, flavonoids, triterpenoids, and mangiferin. They also noted that their findings support the folkloric uses of the plant for treating arthritic and other inflammatory conditions, as well as type 2 diabetes (Methods Find. Exp. Clin. Pharmacol. 2005;27:547-54).
Another important constituent of mango (also found in olive, strawberry, fig, and various medicinal herbs) is the triterpene lupeol, which has been characterized as exhibiting potent antioxidant, antimutagenic, anti-inflammatory, and antiarthritic activity (Oncogene 2004;23:5203-14). A 2014 study by Sahu et al. also showed that M. indica leaves display some antityrosinase activity, though not as strongly as other medicinal plants, such as Emblica officinalis (Pak. J. Biol. Sci. 2014;17:146-50).
Anticancer, antioxidant, and antiphotoaging activity
In 2004, Saleem et al. demonstrated that topically applied lupeol exhibited anti–tumor-promoting effects in a CD-1 mouse skin tumorigenesis model. Pretreatment with the mango constituent time- and dose-dependently inhibited multiple 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-mediated increases in edema, hyperplasia, epidermal ornithine decarboxylase (ODC) activity, as well as protein expression of ODC, cyclooxygenase 2 (COX-2) and nitric oxide synthase. Pretreated animals also experienced significantly lower tumor incidence and tumor body burden as well as a significant delay in tumor latency period. The researchers concluded that lupeol exerts anti–skin tumor promoting effects on CD-1 mice (Oncogene 2004;23:5203-14).
Three years later, Núñez-Sellés et al. reported that a mango stem bark extract (Vimang) developed in Cuba exhibited antioxidant, analgesic, anti-inflammatory, and immunomodulating activity in basic, preclinical, and clinical studies (Pharmacol. Res. 2007;55:351-8).
A 2009 toxicological analysis of Vimang, which has been formulated into tablets, creams, capsules, syrup, vaginal oval, and suppositories for various applications, revealed via irritant tests conducted on rabbits that the topical formulation was not irritating to the skin, generally, with minimal irritancy noted after vaginal application. No adverse effects were reported (Drug Chem. Toxicol. 2009;32:53-8).
In 2012, Li et al. discovered norathyriol (1,3,6,7-tetrahydroxy-9H-xanthen-9-one), a plant-derived chemopreventive metabolite of mangiferin, found in mango, Hypericum elegans, and Tripterospermum lanceolatum. They found that norathyriol significantly inhibited solar UV-induced skin carcinogenesis in mouse models. In vitro investigations revealed that the compound suppressed cell growth in mouse skin epidermal JB6 P+ cells at the level of G2-M phase arrest. The investigators concluded that this newly identified substance appears to act as a safe chemopreventive agent against UV-induced skin cancer (Cancer Res. 2012;72:260-70).
A year later, Song et al. assessed the protective effects of orally administered mango extract against UVB-induced cutaneous aging in HR-1 hairless male mice. The animals were divided into control, UVB-treated vehicle, and UVB-treated mango extract groups. The researchers found that mango extract significantly suppressed the increase in epidermal thickness and hypertrophy indicative of UVB treatment, with mean length of wrinkles significantly lower in the mango group compared with the UVB-treated vehicle group. Treatment with mango extract also led to a significant increase in collagen bundles in animals treated with UVB. The authors concluded that mango extract displayed antiphotoaging properties in hairless mice exposed to UVB (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).
Further, a 2014 in vitro study revealed that extracts of Helicanthus elastica growing on M. indica exhibited antioxidant activity. H. elastica is a hemiparasite that often grows on mango trees in India and is known to be a rich source of phenolic substances (J. Tradit. Complement. Med. 2014;4:285-8).
Topical delivery
Mandawgade and Patravale developed a mango butter skin care formulation in 2008 that was used to test skin repair in rat excision and incision wound models. A healing response was noted in both animal models. The formulation also was found to be effective in achieving complete repair of worn and cracked skin on the feet of all human volunteers in the study. The investigators concluded that the mango butter preparation delivers superlative emolliency and warrants consideration as an excipient agent in cosmeceutical products (Indian J. Pharm. Sci. 2008;70:539-42).
It is worth noting that cases of “mango dermatitis” (allergic contact dermatitis to the sap or skin of M. indica), manifesting in urticaria and eczematous rashes, have been reported (Australas. J. Dermatol. 1996;37:59-60; Int. J. Dermatol. 2004;43:195-6).
In 2014, Leanpolchareanchai et al. developed a microemulsion system containing Thai mango seed kernel extract that displayed strong skin enhancement results in ex vivo skin permeation studies (penetrating skin layers up to 60-fold higher than controls) and physicochemical stability over 6 months (Drug Chem. Toxicol. 2009;32:53-8). Thai mango seed kernel extract had previously been shown to exhibit anti–methicillin-resistant Staphylococcus aureus and antityrosinase characteristics, as well as strong free radical scavenging, antioxidant, anti-inflammatory, and hepatoprotective activities.(Molecules 2014;19:17107-29).
Conclusion
Evidence on the cutaneous applications of mango is emerging, but does not have a significant track record. That said, this fruit has long been used in traditional medicine for a range of indications, including skin disorders. Much more research is necessary, however, to ascertain how beneficial this fruit and its extracts may be. At the very least, there are few reports of adverse events associated with topical application.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.
Mangifera indica (mango) is a member of the Anacardiaceae family with a tradition of use as a medicinal plant. Mango extracts have been characterized as exhibiting antioxidant, anti-inflammatory, analgesic, and immunomodulatory activities (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9; Drug Chem. Toxicol. 2009;32:53-8). Mango is grown in more than 100 countries, primarily in Asia, in tropical as well as subtropical regions (Molecules 2014;19:17107-29). Mango stem bark and leaves have been used in traditional medicine to treat anemia, cutaneous infections, diabetes, diarrhea, scabies, syphilis, and malignant tumors (Pharmacol. Res. 2007;55:351-8). Polyphenols and carotenoids are among the phytonutrients identified as responsible for the biologic activity of mango (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).
Various biologic activities and traditional uses
Ojewole investigated the anti-inflammatory, analgesic, and antidiabetic activity of M. indica stem bark aqueous extract in rats and mice in 2005. In mice, mango extract dose-dependently delivered significant analgesic effects against thermally and chemically-generated pain. The investigators attributed the observed salutary effects of the plant to its constituent polyphenolics, flavonoids, triterpenoids, and mangiferin. They also noted that their findings support the folkloric uses of the plant for treating arthritic and other inflammatory conditions, as well as type 2 diabetes (Methods Find. Exp. Clin. Pharmacol. 2005;27:547-54).
Another important constituent of mango (also found in olive, strawberry, fig, and various medicinal herbs) is the triterpene lupeol, which has been characterized as exhibiting potent antioxidant, antimutagenic, anti-inflammatory, and antiarthritic activity (Oncogene 2004;23:5203-14). A 2014 study by Sahu et al. also showed that M. indica leaves display some antityrosinase activity, though not as strongly as other medicinal plants, such as Emblica officinalis (Pak. J. Biol. Sci. 2014;17:146-50).
Anticancer, antioxidant, and antiphotoaging activity
In 2004, Saleem et al. demonstrated that topically applied lupeol exhibited anti–tumor-promoting effects in a CD-1 mouse skin tumorigenesis model. Pretreatment with the mango constituent time- and dose-dependently inhibited multiple 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-mediated increases in edema, hyperplasia, epidermal ornithine decarboxylase (ODC) activity, as well as protein expression of ODC, cyclooxygenase 2 (COX-2) and nitric oxide synthase. Pretreated animals also experienced significantly lower tumor incidence and tumor body burden as well as a significant delay in tumor latency period. The researchers concluded that lupeol exerts anti–skin tumor promoting effects on CD-1 mice (Oncogene 2004;23:5203-14).
Three years later, Núñez-Sellés et al. reported that a mango stem bark extract (Vimang) developed in Cuba exhibited antioxidant, analgesic, anti-inflammatory, and immunomodulating activity in basic, preclinical, and clinical studies (Pharmacol. Res. 2007;55:351-8).
A 2009 toxicological analysis of Vimang, which has been formulated into tablets, creams, capsules, syrup, vaginal oval, and suppositories for various applications, revealed via irritant tests conducted on rabbits that the topical formulation was not irritating to the skin, generally, with minimal irritancy noted after vaginal application. No adverse effects were reported (Drug Chem. Toxicol. 2009;32:53-8).
In 2012, Li et al. discovered norathyriol (1,3,6,7-tetrahydroxy-9H-xanthen-9-one), a plant-derived chemopreventive metabolite of mangiferin, found in mango, Hypericum elegans, and Tripterospermum lanceolatum. They found that norathyriol significantly inhibited solar UV-induced skin carcinogenesis in mouse models. In vitro investigations revealed that the compound suppressed cell growth in mouse skin epidermal JB6 P+ cells at the level of G2-M phase arrest. The investigators concluded that this newly identified substance appears to act as a safe chemopreventive agent against UV-induced skin cancer (Cancer Res. 2012;72:260-70).
A year later, Song et al. assessed the protective effects of orally administered mango extract against UVB-induced cutaneous aging in HR-1 hairless male mice. The animals were divided into control, UVB-treated vehicle, and UVB-treated mango extract groups. The researchers found that mango extract significantly suppressed the increase in epidermal thickness and hypertrophy indicative of UVB treatment, with mean length of wrinkles significantly lower in the mango group compared with the UVB-treated vehicle group. Treatment with mango extract also led to a significant increase in collagen bundles in animals treated with UVB. The authors concluded that mango extract displayed antiphotoaging properties in hairless mice exposed to UVB (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).
Further, a 2014 in vitro study revealed that extracts of Helicanthus elastica growing on M. indica exhibited antioxidant activity. H. elastica is a hemiparasite that often grows on mango trees in India and is known to be a rich source of phenolic substances (J. Tradit. Complement. Med. 2014;4:285-8).
Topical delivery
Mandawgade and Patravale developed a mango butter skin care formulation in 2008 that was used to test skin repair in rat excision and incision wound models. A healing response was noted in both animal models. The formulation also was found to be effective in achieving complete repair of worn and cracked skin on the feet of all human volunteers in the study. The investigators concluded that the mango butter preparation delivers superlative emolliency and warrants consideration as an excipient agent in cosmeceutical products (Indian J. Pharm. Sci. 2008;70:539-42).
It is worth noting that cases of “mango dermatitis” (allergic contact dermatitis to the sap or skin of M. indica), manifesting in urticaria and eczematous rashes, have been reported (Australas. J. Dermatol. 1996;37:59-60; Int. J. Dermatol. 2004;43:195-6).
In 2014, Leanpolchareanchai et al. developed a microemulsion system containing Thai mango seed kernel extract that displayed strong skin enhancement results in ex vivo skin permeation studies (penetrating skin layers up to 60-fold higher than controls) and physicochemical stability over 6 months (Drug Chem. Toxicol. 2009;32:53-8). Thai mango seed kernel extract had previously been shown to exhibit anti–methicillin-resistant Staphylococcus aureus and antityrosinase characteristics, as well as strong free radical scavenging, antioxidant, anti-inflammatory, and hepatoprotective activities.(Molecules 2014;19:17107-29).
Conclusion
Evidence on the cutaneous applications of mango is emerging, but does not have a significant track record. That said, this fruit has long been used in traditional medicine for a range of indications, including skin disorders. Much more research is necessary, however, to ascertain how beneficial this fruit and its extracts may be. At the very least, there are few reports of adverse events associated with topical application.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.
Mangifera indica (mango) is a member of the Anacardiaceae family with a tradition of use as a medicinal plant. Mango extracts have been characterized as exhibiting antioxidant, anti-inflammatory, analgesic, and immunomodulatory activities (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9; Drug Chem. Toxicol. 2009;32:53-8). Mango is grown in more than 100 countries, primarily in Asia, in tropical as well as subtropical regions (Molecules 2014;19:17107-29). Mango stem bark and leaves have been used in traditional medicine to treat anemia, cutaneous infections, diabetes, diarrhea, scabies, syphilis, and malignant tumors (Pharmacol. Res. 2007;55:351-8). Polyphenols and carotenoids are among the phytonutrients identified as responsible for the biologic activity of mango (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).
Various biologic activities and traditional uses
Ojewole investigated the anti-inflammatory, analgesic, and antidiabetic activity of M. indica stem bark aqueous extract in rats and mice in 2005. In mice, mango extract dose-dependently delivered significant analgesic effects against thermally and chemically-generated pain. The investigators attributed the observed salutary effects of the plant to its constituent polyphenolics, flavonoids, triterpenoids, and mangiferin. They also noted that their findings support the folkloric uses of the plant for treating arthritic and other inflammatory conditions, as well as type 2 diabetes (Methods Find. Exp. Clin. Pharmacol. 2005;27:547-54).
Another important constituent of mango (also found in olive, strawberry, fig, and various medicinal herbs) is the triterpene lupeol, which has been characterized as exhibiting potent antioxidant, antimutagenic, anti-inflammatory, and antiarthritic activity (Oncogene 2004;23:5203-14). A 2014 study by Sahu et al. also showed that M. indica leaves display some antityrosinase activity, though not as strongly as other medicinal plants, such as Emblica officinalis (Pak. J. Biol. Sci. 2014;17:146-50).
Anticancer, antioxidant, and antiphotoaging activity
In 2004, Saleem et al. demonstrated that topically applied lupeol exhibited anti–tumor-promoting effects in a CD-1 mouse skin tumorigenesis model. Pretreatment with the mango constituent time- and dose-dependently inhibited multiple 12-O-tetradecanoyl-phorbol-13-acetate (TPA)-mediated increases in edema, hyperplasia, epidermal ornithine decarboxylase (ODC) activity, as well as protein expression of ODC, cyclooxygenase 2 (COX-2) and nitric oxide synthase. Pretreated animals also experienced significantly lower tumor incidence and tumor body burden as well as a significant delay in tumor latency period. The researchers concluded that lupeol exerts anti–skin tumor promoting effects on CD-1 mice (Oncogene 2004;23:5203-14).
Three years later, Núñez-Sellés et al. reported that a mango stem bark extract (Vimang) developed in Cuba exhibited antioxidant, analgesic, anti-inflammatory, and immunomodulating activity in basic, preclinical, and clinical studies (Pharmacol. Res. 2007;55:351-8).
A 2009 toxicological analysis of Vimang, which has been formulated into tablets, creams, capsules, syrup, vaginal oval, and suppositories for various applications, revealed via irritant tests conducted on rabbits that the topical formulation was not irritating to the skin, generally, with minimal irritancy noted after vaginal application. No adverse effects were reported (Drug Chem. Toxicol. 2009;32:53-8).
In 2012, Li et al. discovered norathyriol (1,3,6,7-tetrahydroxy-9H-xanthen-9-one), a plant-derived chemopreventive metabolite of mangiferin, found in mango, Hypericum elegans, and Tripterospermum lanceolatum. They found that norathyriol significantly inhibited solar UV-induced skin carcinogenesis in mouse models. In vitro investigations revealed that the compound suppressed cell growth in mouse skin epidermal JB6 P+ cells at the level of G2-M phase arrest. The investigators concluded that this newly identified substance appears to act as a safe chemopreventive agent against UV-induced skin cancer (Cancer Res. 2012;72:260-70).
A year later, Song et al. assessed the protective effects of orally administered mango extract against UVB-induced cutaneous aging in HR-1 hairless male mice. The animals were divided into control, UVB-treated vehicle, and UVB-treated mango extract groups. The researchers found that mango extract significantly suppressed the increase in epidermal thickness and hypertrophy indicative of UVB treatment, with mean length of wrinkles significantly lower in the mango group compared with the UVB-treated vehicle group. Treatment with mango extract also led to a significant increase in collagen bundles in animals treated with UVB. The authors concluded that mango extract displayed antiphotoaging properties in hairless mice exposed to UVB (Photodermatol. Photoimmunol. Photomed. 2013;29:84-9).
Further, a 2014 in vitro study revealed that extracts of Helicanthus elastica growing on M. indica exhibited antioxidant activity. H. elastica is a hemiparasite that often grows on mango trees in India and is known to be a rich source of phenolic substances (J. Tradit. Complement. Med. 2014;4:285-8).
Topical delivery
Mandawgade and Patravale developed a mango butter skin care formulation in 2008 that was used to test skin repair in rat excision and incision wound models. A healing response was noted in both animal models. The formulation also was found to be effective in achieving complete repair of worn and cracked skin on the feet of all human volunteers in the study. The investigators concluded that the mango butter preparation delivers superlative emolliency and warrants consideration as an excipient agent in cosmeceutical products (Indian J. Pharm. Sci. 2008;70:539-42).
It is worth noting that cases of “mango dermatitis” (allergic contact dermatitis to the sap or skin of M. indica), manifesting in urticaria and eczematous rashes, have been reported (Australas. J. Dermatol. 1996;37:59-60; Int. J. Dermatol. 2004;43:195-6).
In 2014, Leanpolchareanchai et al. developed a microemulsion system containing Thai mango seed kernel extract that displayed strong skin enhancement results in ex vivo skin permeation studies (penetrating skin layers up to 60-fold higher than controls) and physicochemical stability over 6 months (Drug Chem. Toxicol. 2009;32:53-8). Thai mango seed kernel extract had previously been shown to exhibit anti–methicillin-resistant Staphylococcus aureus and antityrosinase characteristics, as well as strong free radical scavenging, antioxidant, anti-inflammatory, and hepatoprotective activities.(Molecules 2014;19:17107-29).
Conclusion
Evidence on the cutaneous applications of mango is emerging, but does not have a significant track record. That said, this fruit has long been used in traditional medicine for a range of indications, including skin disorders. Much more research is necessary, however, to ascertain how beneficial this fruit and its extracts may be. At the very least, there are few reports of adverse events associated with topical application.
Dr. Baumann is chief executive officer of the Baumann Cosmetic & Research Institute in the Design District in Miami. She founded the Cosmetic Dermatology Center at the University of Miami in 1997. Dr. Baumann wrote the textbook “Cosmetic Dermatology: Principles and Practice” (New York: McGraw-Hill, 2002), and a book for consumers, “The Skin Type Solution” (New York: Bantam Dell, 2006). She has contributed to the Cosmeceutical Critique column in Dermatology News since January 2001. Her latest book, “Cosmeceuticals and Cosmetic Ingredients,” was published in November 2014. Dr. Baumann has received funding for clinical grants from Allergan, Aveeno, Avon Products, Evolus, Galderma, GlaxoSmithKline, Kythera, Mary Kay, Medicis Pharmaceuticals, Neutrogena, Philosophy, Topix Pharmaceuticals, and Unilever.

New test could improve warfarin monitoring

SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
SAN FRANCISCO—Monitoring warfarin using a modified prothrombin time (PT) test can improve anticoagulation stability and long-term clinical outcomes, result of the Fiix trial suggest.
With Fiix-PT—a test that only measures the activity of coagulation factors II and X—the effect of warfarin fluctuated less than with standard PT.
Fiix-PT also proved superior in reducing long-term, recurrent thromboembolism (TE), and it did not increase bleeding, despite the fact that the test omits factor VII activity.
However, most of these effects occurred only after the 6-month mark.
Pall T. Onundarson, MD, of Landspitali University Hospital and University of Iceland School of Medicine in Reykjavik, presented these results at the 2014 ASH Annual Meeting (abstract 347).
Dr Onundarson is a co-inventor of the Fiix-PT test and has stock in Fiix Diagnostics, a startup company with the two inventors of the test as majority shareholders.
“Why would anyone still be interested in warfarin in 2014,” Dr Onundarson asked the audience at ASH. “Even if warfarin is very efficacious . . ., managing warfarin is trouble. The dose varies between patients, and the main problem is that we have a fluctuating or unstable effect—namely, a fluctuating INR—and this leads to frequent dose adjustments and frequent testing.”
Dr Onundarson said the common wisdom is that the effect of warfarin fluctuates due to food interactions and drug interactions. But he and his colleagues speculated that the fluctuation is partly caused by conventional PT.
They noted that experiments have suggested the anticoagulation effect of warfarin is mainly influenced by a reduction in FII and FX activity, and FVII may have relatively little effect. Due to the short half-life of FVII and its strong influence on the PT, monitoring warfarin using PT may confound dosing.
With that in mind, the Fiix-PT test was designed to measure only the activity of FII and FX. And the researchers conducted the Fiix trial to compare Fiix-PT and conventional PT.
Their study included 1148 patients—573 in the Fiix-PT arm and 575 in the PT arm. Overall, about 69% of patients had atrial fibrillation, and 21% had venous thromboembolism. The median monitoring time was 1.7 years per patient in both arms.
The researchers noted that most outcomes were not significantly different between the 2 arms in the first 6 months, but, after that point, things changed. So they examined outcomes after 6 months in a secondary analysis.
From day 1 to 180, there was no significant difference between the Fiix-PT arm and the PT arm in the number of dose changes per patient year (5.3 and 6.5, respectively, P=0.08). However, from day 181 to 720, there were significantly fewer dose changes in the Fiix-PT arm (4.1 and 5.0, respectively, P=0.01).
The researchers assessed the median time within target range (TTR) during 4 periods of the study, and, although results fluctuated, there were significant differences in favor of Fiix-PT.
The median TTR was 85% in the Fiix-PT arm and 81% in the PT arm for days 1 to 180 (P=0.013); 85% and 90%, respectively, for days 181 to 360 (P<0.0003); 80% and 81%, respectively, for days 361 to 540 (P=0.34); and 87% and 79%, respectively, for days 541 to 720 (P<0.005).
Overall, there was no significant difference in the rate of TE or major bleeding between the Fiix-PT and PT arms. However, when the researchers excluded the first 6 months of observation, they noted a significant difference in the rate of TE in favor of Fiix-PT.
In the primary analysis (encompassing days 1 to 720), the TE rate per patient year was 1.2% in the Fiix-PT arm and 2.3% in the PT arm (P=0.09 for superiority, P<0.001 for non-inferiority). The major bleeding rates were 2.2% and 2.5%, respectively (P=0.8 for superiority, P<0.001 for non-inferiority).
In the secondary analysis (from day 181 to 720), Fiix-PT led to a superior long-term reduction in TE compared to conventional PT—1.1% and 2.2%, respectively (P=0.03 for superiority). But there was no significant difference in the rates of major bleeding—1.5% and 2.3%, respectively (P=0.5 for superiority).
“Compared to high-quality PT-INR monitoring, Fiix-PT increased the stability of warfarin anticoagulation,” Dr Onundarson said in closing. “Fiix-PT was clinically at least non-inferior to the INR in the primary analysis, and Fiix-PT led to superior long-term reduction in TE in the secondary analysis. Fiix-PT did not increase major bleeding, despite lowering the long-term thromboembolic rate and despite not being affected by FVII in the test sample.”
“So my overall conclusion is that a fluctuating INR during warfarin treatment is partly a confounding side effect of the PT itself. The data suggests that, if the PT is replaced with a monitoring test that is not affected by FVII, warfarin may become more stable than was previously assumed.” ![]()
NHS cuts 5 blood cancer drugs from CDF, adds 1

Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Credit: Steven Harbour
The National Health Service (NHS) has increased the budget for England’s Cancer Drugs Fund (CDF) and added a new drug to treat 2 hematologic malignancies, but 5 other blood cancer drugs will be removed from the fund in March.
The budget for the CDF will grow from £200 million in 2013/14 to £280 million in 2014/15.
However, 16 drugs (for 25 different indications) will no longer be offered through the fund as of March 12, 2015.
Still, the NHS said it has taken steps to ensure patients can receive appropriate treatment.
Review leads to cuts
A national panel of oncologists, pharmacists, and patient representatives independently reviewed the drug indications currently available through the CDF, plus new applications.
They evaluated the clinical benefit, survival, quality of life, toxicity, and safety associated with each treatment, as well as the level of unmet need and the median cost per patient. In cases where the high cost of a drug would lead to its exclusion from CDF, manufacturers were given an opportunity to reduce prices.
The result of the review is that 59 of the 84 most effective currently approved indications of drugs will rollover into the CDF next year, creating room for new drug indications that will be funded for the first time.
These are panitumumab for bowel cancer, ibrutinib for mantle cell lymphoma, and ibrutinib for chronic lymphocytic leukemia.
However, 16 drugs, including 5 blood cancer drugs—bendamustine, bortezomib, bosutinib, dasatinib, and ofatumumab—will no longer be offered through the CDF.
Following these changes, the NHS will put 4 measures in place to ensure patients can receive appropriate treatment. First, any patient currently receiving a drug through the CDF will continue to receive it, regardless of whether it remains in the CDF.
Second, drugs that are the only therapy for the cancer in question will remain available through the CDF. Third, if the CDF panel removes a drug for a particular indication, some patients may instead be able to receive it in another line of therapy or receive an alternative CDF-approved drug.
And finally, clinicians can apply for their patient to receive a drug not available through the CDF on an exceptional basis.
Cuts to blood cancer drugs
The full list of cuts to the CDF is available on the NHS website, but the following list includes all drugs for hematologic malignancies that will no longer be available. These drugs will still be available for other indications, however.
- Bendamustine for the treatment of low-grade lymphoma that is refractory to rituximab alone or in combination.
- Bortezomib for the treatment of:
- relapsed/refractory mantle cell lymphoma after 1 or more prior chemotherapies or stem cell transplant
- relapsed multiple myeloma patients with a previous partial response or complete response of 6 months or more with bortezomib
- relapsed Waldenstrom’s macroglobulinemia patients who received previous treatment with alkylating agents and purine analogues.
- Bosutinib for the treatment of:
- blast crisis chronic myeloid leukemia (CML) that is refractory to nilotinib or dasatinib if dasatinib was accessed via a clinical trial or via its current approved CDF indication
- blast crisis CML where there is treatment intolerance, specifically, significant intolerance to dasatinib (grade 3 or 4 adverse events) if dasatinib was accessed via its current approved CDF indication.
- Dasatinib for the treatment of lymphoid, blast crisis CML that is refractory to, significantly intolerant of, or resistant to prior therapy including imatinib (grade 3 or 4 adverse events); also when used as the 2nd- or 3rd-line treatment.
- Ofatumumab for the treatment of CML as the 2nd- or 3rd-line indication and if the patient is refractory to treatment with fludarabine in combination and/or alemtuzumab or if treatment with fludarabine in combination and/or alemtuzumab is contraindicated.
More about the CDF and the NHS
The CDF—set up in 2010 and currently due to run until March 2016—is money the government has set aside to pay for cancer drugs that haven’t been approved by the National Institute for Health and Care Excellence (NICE) and aren’t available within the NHS in England. Most cancer drugs are routinely funded outside of the CDF.
NHS England said it is working with cancer charities, the pharmaceutical industry, and NICE to create a sustainable model for the commissioning of chemotherapy. The agency has also updated its procedures for evaluating drugs in the CDF, in an effort to ensure sustainability.
In addition, NHS England has set up an appeals process by which pharmaceutical companies can challenge the decision-making process.
And a newly assembled national taskforce, headed by Harpal Kumar, chief executive of Cancer Research UK, is set to produce a refreshed, 5-year cancer plan for the NHS. ![]()
Drug can increase survival in poor-risk AML
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.” ![]()
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.” ![]()
In a phase 2 trial, an investigational drug conferred a significant improvement in survival when used as salvage therapy in poor-risk patients with acute myeloid leukemia (AML).
On the other hand, the drug did not provide any significant improvements over control treatment (investigator’s choice) in the entire population of AML patients in first relapse.
The drug, CPX-351, is a fixed-ratio combination of cytarabine and daunorubicin inside a lipid vesicle.
Researchers reported these results with CPX-351 in Cancer. The study was funded by Celator Pharmaceuticals, the company developing CPX-351, and the Leukemia and Lymphoma Society.
“Patients with first-relapse AML have generally a poor prognosis, with a limited likelihood of response following salvage treatment,” said study author Jorge Cortes, MD, of the MD Anderson Cancer Center in Houston, Texas.
“This is particularly true for patients classified by the EPI [European Prognostic Index] as poor-risk upon entering the trial.”
For this trial, Dr Cortes and his colleagues evaluated 125 patients, ages 18 to 65, from 35 centers in the US, Canada, and Europe. Patients had AML in first relapse after an initial complete remission lasting a month or longer.
They were stratified per the EPI into favorable-, intermediate-, and poor-risk groups based on the duration of their first complete remission, cytogenetics, age, and transplant history. Most patients (68%) were in the poor-risk group.
Patients were randomized 2:1 to receive CPX-351 (100 units/m2 on days 1, 3, and 5 by 90-minute infusion) or the investigators’ choice of first salvage chemotherapy. The control treatment was usually based on cytarabine and an anthracycline, often with one or more additional agents.
Patient characteristics were largely well-balanced between the treatment arms. However, the CPX-351 group had a younger median age (52 years vs 56 years), more patients with secondary AML (12.3% vs 6.8%), and a higher rate of prior transplant (27.2% vs 15.9%).
Response and survival
Response rates were higher in the CPX-351 arm than in the control arm. The rates of complete response were 37% and 31.8%, respectively. And the rates of complete response with incomplete count recovery were 12.3% and 9.1%, respectively.
However, there was no significant difference in event-free survival (EFS) or overall survival (OS) between the treatment arms.
The median EFS was 4 months in the CPX-351 arm and 1.4 months in the control arm (hazard ratio[HR]=0.66, P=0.08). And the median OS was 8.5 months and 6.3 months, respectively (HR=0.75, P=0.19).
In the poor-risk population, there was no significant improvement in EFS with CPX-351, but there was a significant improvement in OS.
The median EFS was 1.8 months in the CPX-351 arm and 1.2 months in the control arm (HR=0.63, P=0.08). And the median OS was 6.5 months and 4.2 months, respectively (HR=0.55, P=0.02).
Safety and early mortality
The early mortality rate was similar between the treatment arms at 30 days—7.4% in the CPX-351 arm and 4.5% in the control arm—and at 60 days—14.8% and 15.9%, respectively. However, at 90 days, deaths were more frequent in the control arm—21.4% and 37.9%.
Patients in the CPX-351 arm had slower neutrophil recovery than those in the control arm—42 days and 34 days, respectively. The same was true for platelet recovery—45 days and 35 days, respectively.
Delayed hematologic recovery was associated with more infection-related events, such as febrile neutropenia, bacteremia, pneumonia, sepsis, urinary tract infection, pyrexia, and cellulitis.
The researchers believe these results, along with the previously published results from a phase 2 trial of CPX-351 in patients newly diagnosed with AML, support the phase 3 study of CPX-351 as a first-line therapy in older patients with high-risk (secondary) AML.
“We were very pleased to see promising response rates in this difficult-to-treat population,” Dr Cortes said. “And we eagerly await results from Celator’s pivotal phase 3 study of CPX-351, which could fulfill a considerable unmet need in AML.”