User login
Hospitalists Should Brace for Bitter Flu Season
A severe flu season will have hospitalists dealing with increased patient censuses and the struggle of keeping staffers healthy and available, according to two physicians.
The CDC recently announced that all but seven states are dealing with widespread flu activity. Two of the states not yet seriously affected, California and New York, are among the highest population centers, prodding many to speculate that the flu will worsen in those areas and the overall season will rank among the worst in recent memory.
As a result, hospitalists across the country should expect to see more patients admitted for flu, as well as potential boarding of sick patients as EDs deal with higher patient counts, according to Alice Pong, MD, an infectious disease specialist at Rady Children’s Hospital in San Diego.
"[Hospitalists] deal with the increased number of actually otherwise healthy people who get flu, who then are sick enough to need to be in the hospital," Dr. Pong says, "and they get the chronically ill people with underlying disease who get the flu and get sicker."
Dr. Pong suggests tips to deal with flu season:
- Have a surge plan in place to handle larger censuses. Similarly, have staffing plans to cover shifts when a doctor needs to stay home sick or take care of a sick family member;
- Redouble focus on hygiene. Hand washing is even more important to prevent the spread of infectious disease within hospitals; and
- Work with infectious disease professionals. Take their cues and seek them out with questions about flu prevention.
Team Hospitalist member James O'Callaghan, MD, FHM, says that having staff members or their families get sick can be a larger concern in smaller groups. Dr. O'Callaghan, a regional pediatric hospitalist at EvergreenHealth in Kirkland, Wash., and a medical hospitalist at Seattle Children's, says losing one staff member in a small hospitalist group can be troublesome.
"We're only a 4.0 [full-time equivalent] group, and there's only one of us working per day," he says. "I don't have anybody on standby that I can just, with the press of button, activate if somebody gets sick." TH
Visit our website for more information on infectious diseases.
A severe flu season will have hospitalists dealing with increased patient censuses and the struggle of keeping staffers healthy and available, according to two physicians.
The CDC recently announced that all but seven states are dealing with widespread flu activity. Two of the states not yet seriously affected, California and New York, are among the highest population centers, prodding many to speculate that the flu will worsen in those areas and the overall season will rank among the worst in recent memory.
As a result, hospitalists across the country should expect to see more patients admitted for flu, as well as potential boarding of sick patients as EDs deal with higher patient counts, according to Alice Pong, MD, an infectious disease specialist at Rady Children’s Hospital in San Diego.
"[Hospitalists] deal with the increased number of actually otherwise healthy people who get flu, who then are sick enough to need to be in the hospital," Dr. Pong says, "and they get the chronically ill people with underlying disease who get the flu and get sicker."
Dr. Pong suggests tips to deal with flu season:
- Have a surge plan in place to handle larger censuses. Similarly, have staffing plans to cover shifts when a doctor needs to stay home sick or take care of a sick family member;
- Redouble focus on hygiene. Hand washing is even more important to prevent the spread of infectious disease within hospitals; and
- Work with infectious disease professionals. Take their cues and seek them out with questions about flu prevention.
Team Hospitalist member James O'Callaghan, MD, FHM, says that having staff members or their families get sick can be a larger concern in smaller groups. Dr. O'Callaghan, a regional pediatric hospitalist at EvergreenHealth in Kirkland, Wash., and a medical hospitalist at Seattle Children's, says losing one staff member in a small hospitalist group can be troublesome.
"We're only a 4.0 [full-time equivalent] group, and there's only one of us working per day," he says. "I don't have anybody on standby that I can just, with the press of button, activate if somebody gets sick." TH
Visit our website for more information on infectious diseases.
A severe flu season will have hospitalists dealing with increased patient censuses and the struggle of keeping staffers healthy and available, according to two physicians.
The CDC recently announced that all but seven states are dealing with widespread flu activity. Two of the states not yet seriously affected, California and New York, are among the highest population centers, prodding many to speculate that the flu will worsen in those areas and the overall season will rank among the worst in recent memory.
As a result, hospitalists across the country should expect to see more patients admitted for flu, as well as potential boarding of sick patients as EDs deal with higher patient counts, according to Alice Pong, MD, an infectious disease specialist at Rady Children’s Hospital in San Diego.
"[Hospitalists] deal with the increased number of actually otherwise healthy people who get flu, who then are sick enough to need to be in the hospital," Dr. Pong says, "and they get the chronically ill people with underlying disease who get the flu and get sicker."
Dr. Pong suggests tips to deal with flu season:
- Have a surge plan in place to handle larger censuses. Similarly, have staffing plans to cover shifts when a doctor needs to stay home sick or take care of a sick family member;
- Redouble focus on hygiene. Hand washing is even more important to prevent the spread of infectious disease within hospitals; and
- Work with infectious disease professionals. Take their cues and seek them out with questions about flu prevention.
Team Hospitalist member James O'Callaghan, MD, FHM, says that having staff members or their families get sick can be a larger concern in smaller groups. Dr. O'Callaghan, a regional pediatric hospitalist at EvergreenHealth in Kirkland, Wash., and a medical hospitalist at Seattle Children's, says losing one staff member in a small hospitalist group can be troublesome.
"We're only a 4.0 [full-time equivalent] group, and there's only one of us working per day," he says. "I don't have anybody on standby that I can just, with the press of button, activate if somebody gets sick." TH
Visit our website for more information on infectious diseases.
Hepatitis A Vaccine Recommended for Patients with Chronic Liver Disease
Hospitalists caring for patients with chronic liver disease should consider administering the hepatitis A vaccine upon discharge, says an expert from the CDC.
The recommendation is based on a new study published in Hepatology that shows the rate of hospitalizations caused by a hepatitis A virus (HAV) infection declined significantly in the U.S. from 2002 to 2011. It also reports that older patients and those with chronic liver disease are most likely to be hospitalized with HAV, says Melissa Collier, MD, MPH, a medical epidemiologist within the CDC's division of viral hepatitis.
Dr. Collier and colleagues analyzed ICD-9 codes from the National Inpatient Survey discharge data, focusing on U.S. residents hospitalized with a principal hepatitis A diagnosis and accompanying secondary diagnoses. They found that from 2002 to 2011:
- Rates of hospitalization for hepatitis A as a principal diagnosis decreased from 0.72/100,000 to 0.29/100,000 (P
- Mean age of those hospitalized increased from 37.6 years to 45.5 years (P
- Percentage of hepatitis A hospitalizations covered by Medicare increased from 12.4% to 22.7% (P
- Secondary, comorbid discharge diagnoses, including liver disease, hypertension, ischemic heart disease, disorders of lipid metabolism, and chronic liver disease, increased; and
- No changes were reported in patients’ length of stay or in-hospital deaths from hepatitis A, but persons with liver disease were hospitalized longer.
According to Dr. Collier, the drop in hospitalization rates could be explained by lower incidence of hepatitis A since the Advisory Committee on Immunization Practices (ACIP) recommended universal childhood vaccination in 2006.
Dr. Collier, who is a member of the ACIP hepatitis A working group, also points out that the ACIP recommends all patients with chronic liver disease be vaccinated. She says hospitalists should give patients with chronic liver disease the first dose of the vaccine upon discharge and ask those patients' PCPs to administer the second dose.
"Patients don't think about needing to be vaccinated, and if their physicians aren't recommending it, they aren't going to seek it," she says. Dr. Collier noted that data show only about 12% of people age 19 to 49 have received the hepatitis A vaccine. TH
Visit our website for more information on treating liver disease.
Hospitalists caring for patients with chronic liver disease should consider administering the hepatitis A vaccine upon discharge, says an expert from the CDC.
The recommendation is based on a new study published in Hepatology that shows the rate of hospitalizations caused by a hepatitis A virus (HAV) infection declined significantly in the U.S. from 2002 to 2011. It also reports that older patients and those with chronic liver disease are most likely to be hospitalized with HAV, says Melissa Collier, MD, MPH, a medical epidemiologist within the CDC's division of viral hepatitis.
Dr. Collier and colleagues analyzed ICD-9 codes from the National Inpatient Survey discharge data, focusing on U.S. residents hospitalized with a principal hepatitis A diagnosis and accompanying secondary diagnoses. They found that from 2002 to 2011:
- Rates of hospitalization for hepatitis A as a principal diagnosis decreased from 0.72/100,000 to 0.29/100,000 (P
- Mean age of those hospitalized increased from 37.6 years to 45.5 years (P
- Percentage of hepatitis A hospitalizations covered by Medicare increased from 12.4% to 22.7% (P
- Secondary, comorbid discharge diagnoses, including liver disease, hypertension, ischemic heart disease, disorders of lipid metabolism, and chronic liver disease, increased; and
- No changes were reported in patients’ length of stay or in-hospital deaths from hepatitis A, but persons with liver disease were hospitalized longer.
According to Dr. Collier, the drop in hospitalization rates could be explained by lower incidence of hepatitis A since the Advisory Committee on Immunization Practices (ACIP) recommended universal childhood vaccination in 2006.
Dr. Collier, who is a member of the ACIP hepatitis A working group, also points out that the ACIP recommends all patients with chronic liver disease be vaccinated. She says hospitalists should give patients with chronic liver disease the first dose of the vaccine upon discharge and ask those patients' PCPs to administer the second dose.
"Patients don't think about needing to be vaccinated, and if their physicians aren't recommending it, they aren't going to seek it," she says. Dr. Collier noted that data show only about 12% of people age 19 to 49 have received the hepatitis A vaccine. TH
Visit our website for more information on treating liver disease.
Hospitalists caring for patients with chronic liver disease should consider administering the hepatitis A vaccine upon discharge, says an expert from the CDC.
The recommendation is based on a new study published in Hepatology that shows the rate of hospitalizations caused by a hepatitis A virus (HAV) infection declined significantly in the U.S. from 2002 to 2011. It also reports that older patients and those with chronic liver disease are most likely to be hospitalized with HAV, says Melissa Collier, MD, MPH, a medical epidemiologist within the CDC's division of viral hepatitis.
Dr. Collier and colleagues analyzed ICD-9 codes from the National Inpatient Survey discharge data, focusing on U.S. residents hospitalized with a principal hepatitis A diagnosis and accompanying secondary diagnoses. They found that from 2002 to 2011:
- Rates of hospitalization for hepatitis A as a principal diagnosis decreased from 0.72/100,000 to 0.29/100,000 (P
- Mean age of those hospitalized increased from 37.6 years to 45.5 years (P
- Percentage of hepatitis A hospitalizations covered by Medicare increased from 12.4% to 22.7% (P
- Secondary, comorbid discharge diagnoses, including liver disease, hypertension, ischemic heart disease, disorders of lipid metabolism, and chronic liver disease, increased; and
- No changes were reported in patients’ length of stay or in-hospital deaths from hepatitis A, but persons with liver disease were hospitalized longer.
According to Dr. Collier, the drop in hospitalization rates could be explained by lower incidence of hepatitis A since the Advisory Committee on Immunization Practices (ACIP) recommended universal childhood vaccination in 2006.
Dr. Collier, who is a member of the ACIP hepatitis A working group, also points out that the ACIP recommends all patients with chronic liver disease be vaccinated. She says hospitalists should give patients with chronic liver disease the first dose of the vaccine upon discharge and ask those patients' PCPs to administer the second dose.
"Patients don't think about needing to be vaccinated, and if their physicians aren't recommending it, they aren't going to seek it," she says. Dr. Collier noted that data show only about 12% of people age 19 to 49 have received the hepatitis A vaccine. TH
Visit our website for more information on treating liver disease.
Microneedling
Microneedling, or skin needling, is an aesthetic technique used for decades prior to resurfacing lasers, but it has recently experienced a surge in popularity, particularly for ethnic skin. In 1995, subcision or dermal needling was identified as an effective treatment for scars. Since then, the technique initially referred to as collagen induction therapy has become a staple in the treatment of acne scars, surgical scars, photo aging, and stretch marks.
The skin needling technique involves using fine sterile needles 0.1mm-2.5 mm in length that repeatedly pierce the stratum corneum, producing microscopic “holes” in the dermis. These microscopic wounds lead to the release of growth factors stimulating the formation of new collagen, elastin, and neovascularization in the dermis. There are many brands and manufacturers of microneedling tools on the market, including dermarollers, Dermapen, Dermastamp, Cosmopen, and multiple other in-office and at-home devices. At-home devices usually have shorter needles and provide significantly less penetration and injury, and therefore may be less effective.

Prior to the procedure, patients are often anesthetized with topical anesthesia without vasoconstrictors for 1 hour. The area is cleaned with sterile gauze and alcohol or Hibiclens, and a microneedling device is used to either roll or prick the skin in multiple alternating passes. The depth of penetration, number of passes, and degree of overlap is highly dependent on the underlying condition, the area being treated, the brand of device used, and the length and frequency of the needle insertion. Petechiae and pinpoint bleeding occur during the treatment. Treatments are usually done 4-6 weeks apart. Post procedure, the patient often experiences mild erythema, bruising, and some mild edema.
This technique has been particularly beneficial to patients with skin of color who are not candidates for factional lasers because of the risks of hyperpigmentation and scarring. There is low risk of hyper- or hypopigmentation with microneedling, and multiple treatments can be performed in patients with types III-VI skin and those with a history of melasma.

Contraindications and precautions when considering microneedling include: history of keloid or hypertrophic scarring,recent skin rashes, history of herpes simplex infections if the perioral area is being treated, and the presence of raised moles, warts, or any raised lesions on the targeted area. Absolute contraindications include: scleroderma, collagen vascular diseases clotting problems, active bacterial or fungal infection, and immunosuppression.
Microneedling is a safe, effective, in-office procedure with a range of uses. Many new indications are currently being explored. In my practice, we have used microneedling for atrophic scars, repigmentation of depigmented scars and vitiligo, stimulation of hair regrowth in noninflammatory alopecias, and treatment of burn scars. Patients are generally very happy with the quick treatment time, minimal downtime, and overall long-term results.
References
1. Orentreich DS, Orentreich N. Subcutaneous incisionless (subcision) surgery for the correction of depressed scars and wrinkles. Dermatol. Surg. 1995;21:6543-9.
2. Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast. Surg. 1997;21:48-51.
3. Fernandes D. Minimally invasive percutaneous collagen induction. Oral Maxillofac. Surg. Clin. North Am. 2006;17:51-63.
4. Aust MC, Fernandes D, Kolokythas P, Kaplan HM, Vogt PM. Percutaneous collagen induction therapy: An alternative treatment for scars, wrinkles and skin laxity. Plast. Reconstr. Surg. 2008;21:1421-9.
5. Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin. Dermatol. 2008;26:192-9.
6. Aust MC, Reimers K, Repenning C, Stahl F, Jahn S, Guggenheim M et al. Percutaneous collagen induction: Minimally invasive skin rejuvenation without risk of hyperpigmentation – fact or fiction? Plast. Reconstr. Surg. 2008;122:1553-63.
7. Fabbrocini G, De Vita V, Pastore F, et al. Collagen induction therapy for the treatment of upper lip wrinkles. J. Dermatolog. Treat. 2012;23:144-52. 8. Majid I. Microneedling therapy in atrophic facial scars: an objective assessment. J. Cutan. Aesthet. Surg. 2009;2:26-30.
9. Doddaballapur S. Microneedling with dermaroller. J. Cutan. Aesthet. Surg 2009;2: 110-11.
10. Dogra S, Yadav S. Sarangal R. Microneedling for acne scars in Asian skin type: an effective low cost treatment modality. J. Cosmet. Dermatol. 2014;13:180-7.
Dr. Talakoub and Dr. Wesley are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub.
Microneedling, or skin needling, is an aesthetic technique used for decades prior to resurfacing lasers, but it has recently experienced a surge in popularity, particularly for ethnic skin. In 1995, subcision or dermal needling was identified as an effective treatment for scars. Since then, the technique initially referred to as collagen induction therapy has become a staple in the treatment of acne scars, surgical scars, photo aging, and stretch marks.
The skin needling technique involves using fine sterile needles 0.1mm-2.5 mm in length that repeatedly pierce the stratum corneum, producing microscopic “holes” in the dermis. These microscopic wounds lead to the release of growth factors stimulating the formation of new collagen, elastin, and neovascularization in the dermis. There are many brands and manufacturers of microneedling tools on the market, including dermarollers, Dermapen, Dermastamp, Cosmopen, and multiple other in-office and at-home devices. At-home devices usually have shorter needles and provide significantly less penetration and injury, and therefore may be less effective.
Prior to the procedure, patients are often anesthetized with topical anesthesia without vasoconstrictors for 1 hour. The area is cleaned with sterile gauze and alcohol or Hibiclens, and a microneedling device is used to either roll or prick the skin in multiple alternating passes. The depth of penetration, number of passes, and degree of overlap is highly dependent on the underlying condition, the area being treated, the brand of device used, and the length and frequency of the needle insertion. Petechiae and pinpoint bleeding occur during the treatment. Treatments are usually done 4-6 weeks apart. Post procedure, the patient often experiences mild erythema, bruising, and some mild edema.
This technique has been particularly beneficial to patients with skin of color who are not candidates for factional lasers because of the risks of hyperpigmentation and scarring. There is low risk of hyper- or hypopigmentation with microneedling, and multiple treatments can be performed in patients with types III-VI skin and those with a history of melasma.
Contraindications and precautions when considering microneedling include: history of keloid or hypertrophic scarring,recent skin rashes, history of herpes simplex infections if the perioral area is being treated, and the presence of raised moles, warts, or any raised lesions on the targeted area. Absolute contraindications include: scleroderma, collagen vascular diseases clotting problems, active bacterial or fungal infection, and immunosuppression.
Microneedling is a safe, effective, in-office procedure with a range of uses. Many new indications are currently being explored. In my practice, we have used microneedling for atrophic scars, repigmentation of depigmented scars and vitiligo, stimulation of hair regrowth in noninflammatory alopecias, and treatment of burn scars. Patients are generally very happy with the quick treatment time, minimal downtime, and overall long-term results.
References
1. Orentreich DS, Orentreich N. Subcutaneous incisionless (subcision) surgery for the correction of depressed scars and wrinkles. Dermatol. Surg. 1995;21:6543-9.
2. Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast. Surg. 1997;21:48-51.
3. Fernandes D. Minimally invasive percutaneous collagen induction. Oral Maxillofac. Surg. Clin. North Am. 2006;17:51-63.
4. Aust MC, Fernandes D, Kolokythas P, Kaplan HM, Vogt PM. Percutaneous collagen induction therapy: An alternative treatment for scars, wrinkles and skin laxity. Plast. Reconstr. Surg. 2008;21:1421-9.
5. Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin. Dermatol. 2008;26:192-9.
6. Aust MC, Reimers K, Repenning C, Stahl F, Jahn S, Guggenheim M et al. Percutaneous collagen induction: Minimally invasive skin rejuvenation without risk of hyperpigmentation – fact or fiction? Plast. Reconstr. Surg. 2008;122:1553-63.
7. Fabbrocini G, De Vita V, Pastore F, et al. Collagen induction therapy for the treatment of upper lip wrinkles. J. Dermatolog. Treat. 2012;23:144-52. 8. Majid I. Microneedling therapy in atrophic facial scars: an objective assessment. J. Cutan. Aesthet. Surg. 2009;2:26-30.
9. Doddaballapur S. Microneedling with dermaroller. J. Cutan. Aesthet. Surg 2009;2: 110-11.
10. Dogra S, Yadav S. Sarangal R. Microneedling for acne scars in Asian skin type: an effective low cost treatment modality. J. Cosmet. Dermatol. 2014;13:180-7.
Dr. Talakoub and Dr. Wesley are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub.
Microneedling, or skin needling, is an aesthetic technique used for decades prior to resurfacing lasers, but it has recently experienced a surge in popularity, particularly for ethnic skin. In 1995, subcision or dermal needling was identified as an effective treatment for scars. Since then, the technique initially referred to as collagen induction therapy has become a staple in the treatment of acne scars, surgical scars, photo aging, and stretch marks.
The skin needling technique involves using fine sterile needles 0.1mm-2.5 mm in length that repeatedly pierce the stratum corneum, producing microscopic “holes” in the dermis. These microscopic wounds lead to the release of growth factors stimulating the formation of new collagen, elastin, and neovascularization in the dermis. There are many brands and manufacturers of microneedling tools on the market, including dermarollers, Dermapen, Dermastamp, Cosmopen, and multiple other in-office and at-home devices. At-home devices usually have shorter needles and provide significantly less penetration and injury, and therefore may be less effective.
Prior to the procedure, patients are often anesthetized with topical anesthesia without vasoconstrictors for 1 hour. The area is cleaned with sterile gauze and alcohol or Hibiclens, and a microneedling device is used to either roll or prick the skin in multiple alternating passes. The depth of penetration, number of passes, and degree of overlap is highly dependent on the underlying condition, the area being treated, the brand of device used, and the length and frequency of the needle insertion. Petechiae and pinpoint bleeding occur during the treatment. Treatments are usually done 4-6 weeks apart. Post procedure, the patient often experiences mild erythema, bruising, and some mild edema.
This technique has been particularly beneficial to patients with skin of color who are not candidates for factional lasers because of the risks of hyperpigmentation and scarring. There is low risk of hyper- or hypopigmentation with microneedling, and multiple treatments can be performed in patients with types III-VI skin and those with a history of melasma.
Contraindications and precautions when considering microneedling include: history of keloid or hypertrophic scarring,recent skin rashes, history of herpes simplex infections if the perioral area is being treated, and the presence of raised moles, warts, or any raised lesions on the targeted area. Absolute contraindications include: scleroderma, collagen vascular diseases clotting problems, active bacterial or fungal infection, and immunosuppression.
Microneedling is a safe, effective, in-office procedure with a range of uses. Many new indications are currently being explored. In my practice, we have used microneedling for atrophic scars, repigmentation of depigmented scars and vitiligo, stimulation of hair regrowth in noninflammatory alopecias, and treatment of burn scars. Patients are generally very happy with the quick treatment time, minimal downtime, and overall long-term results.
References
1. Orentreich DS, Orentreich N. Subcutaneous incisionless (subcision) surgery for the correction of depressed scars and wrinkles. Dermatol. Surg. 1995;21:6543-9.
2. Camirand A, Doucet J. Needle dermabrasion. Aesthetic Plast. Surg. 1997;21:48-51.
3. Fernandes D. Minimally invasive percutaneous collagen induction. Oral Maxillofac. Surg. Clin. North Am. 2006;17:51-63.
4. Aust MC, Fernandes D, Kolokythas P, Kaplan HM, Vogt PM. Percutaneous collagen induction therapy: An alternative treatment for scars, wrinkles and skin laxity. Plast. Reconstr. Surg. 2008;21:1421-9.
5. Fernandes D, Signorini M. Combating photoaging with percutaneous collagen induction. Clin. Dermatol. 2008;26:192-9.
6. Aust MC, Reimers K, Repenning C, Stahl F, Jahn S, Guggenheim M et al. Percutaneous collagen induction: Minimally invasive skin rejuvenation without risk of hyperpigmentation – fact or fiction? Plast. Reconstr. Surg. 2008;122:1553-63.
7. Fabbrocini G, De Vita V, Pastore F, et al. Collagen induction therapy for the treatment of upper lip wrinkles. J. Dermatolog. Treat. 2012;23:144-52. 8. Majid I. Microneedling therapy in atrophic facial scars: an objective assessment. J. Cutan. Aesthet. Surg. 2009;2:26-30.
9. Doddaballapur S. Microneedling with dermaroller. J. Cutan. Aesthet. Surg 2009;2: 110-11.
10. Dogra S, Yadav S. Sarangal R. Microneedling for acne scars in Asian skin type: an effective low cost treatment modality. J. Cosmet. Dermatol. 2014;13:180-7.
Dr. Talakoub and Dr. Wesley are cocontributors to a monthly Aesthetic Dermatology column in Dermatology News. Dr. Talakoub is in private practice in McLean, Va. Dr. Wesley practices dermatology in Beverly Hills, Calif. This month’s column is by Dr. Talakoub.

Joining forces, Part 2
The ongoing sea change in medicine has led to a substantial erosion of physician autonomy, and to ever-increasing administrative burdens that hit small practices the hardest. Does this mean that the independent private physician practice model is doomed, as some predict? Absolutely not; but it will force many solo practitioners and small groups to join forces to protect themselves.
Those practices that offer unique services, or fill an unmet niche, may be able to remain small; but most smaller practices will need to consider a larger alternative. In a previous column, I outlined the basics of one such protective strategy – merging two or more small practices into a larger entity – but there are other options to consider.
One attractive and relatively straightforward strategy is the formation of a cooperative group. In most areas, there are very likely several small practices in similar predicaments that might be receptive to discussing a collaboration on billing and purchasing. This allows each participant to maintain independence as a private practice, while pooling resources to ease the administrative burdens of all. Once that arrangement is in place, the group can consider more ambitious projects, such as the joint purchase of an EHR system, sharing of personnel to lower staffing costs, and an integrated scheduling system. The latter will be particularly attractive to participants in later stages of their careers who are considering an intermediate option, somewhere between full-time work and complete retirement.
After a time, when the structure is stabilized and everyone agrees that his or her individual and shared interests and goals are being met, an outright merger can be contemplated. Projects of this scope require careful planning and implementation, and should not be undertaken without the help of competent legal counsel and an experienced business consultant.
A more complex but increasingly popular option is to join other small practices and providers in an independent practice association (IPA). An IPA is a legal entity organized and directed by physicians for the purpose of negotiating contracts with insurance companies on their behalf. Because of its structure, an IPA is better positioned to enter into such financial arrangements, and to counterbalance the leverage of insurers, but there are legal issues to consider. Many IPAs are vulnerable to antitrust charges because they include competing health care providers. You should check with legal counsel before signing on to an IPA, to make sure that it abides by antitrust and price fixing laws. IPAs have also been known to fail, particularly in states where they are not adequately regulated.
A possible successor to IPAs is the accountable care organization (ACO), an entity born as a component of the Affordable Care Act. While the official definition remains nebulous, an ACO is basically a network of doctors and hospitals that shares financial and medical responsibility for providing coordinated and efficient care to patients. The goal of ACO participants is to limit unnecessary spending, both individually and collectively, according to criteria established by the Centers for Medicare & Medicaid Services, without compromising quality of care in the process. More than 600 ACOs had been approved by the CMS as of the beginning of 2014.
It is important to remember that the ACO model remains very much a work in progress. ACOs make providers jointly accountable for the health of their patients. They offer financial incentives to cooperate, and to save money by avoiding unnecessary tests and procedures. A key component is the sharing of information. Providers who save money while also meeting quality targets are theoretically entitled to a portion of the savings.
As with IPAs, ACO ventures involve a measure of risk. ACOs that fail to meet the CMS performance and savings benchmarks can be stuck with the bill for investments made to improve care, such as equipment and computer purchases and the hiring of mid-level providers and managers, and they may be assessed monetary penalties as well. ACOs sponsored by physicians or rural providers, however, can apply to receive payments in advance to help finance infrastructure investments – a concession the Obama administration made after receiving complaints from rural hospitals.
Clearly, the price of remaining autonomous will be significant, and many private practitioners will be unwilling to pay it: Only 36% of physicians remained in independent practice at the end of the 2013, according to data from the American Medical Association – down from 57% in 2000 – but those of us who remain committed to independence will find ways to preserve it. In medicine, as in life, those most responsive to change will survive and flourish.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.
The ongoing sea change in medicine has led to a substantial erosion of physician autonomy, and to ever-increasing administrative burdens that hit small practices the hardest. Does this mean that the independent private physician practice model is doomed, as some predict? Absolutely not; but it will force many solo practitioners and small groups to join forces to protect themselves.
Those practices that offer unique services, or fill an unmet niche, may be able to remain small; but most smaller practices will need to consider a larger alternative. In a previous column, I outlined the basics of one such protective strategy – merging two or more small practices into a larger entity – but there are other options to consider.
One attractive and relatively straightforward strategy is the formation of a cooperative group. In most areas, there are very likely several small practices in similar predicaments that might be receptive to discussing a collaboration on billing and purchasing. This allows each participant to maintain independence as a private practice, while pooling resources to ease the administrative burdens of all. Once that arrangement is in place, the group can consider more ambitious projects, such as the joint purchase of an EHR system, sharing of personnel to lower staffing costs, and an integrated scheduling system. The latter will be particularly attractive to participants in later stages of their careers who are considering an intermediate option, somewhere between full-time work and complete retirement.
After a time, when the structure is stabilized and everyone agrees that his or her individual and shared interests and goals are being met, an outright merger can be contemplated. Projects of this scope require careful planning and implementation, and should not be undertaken without the help of competent legal counsel and an experienced business consultant.
A more complex but increasingly popular option is to join other small practices and providers in an independent practice association (IPA). An IPA is a legal entity organized and directed by physicians for the purpose of negotiating contracts with insurance companies on their behalf. Because of its structure, an IPA is better positioned to enter into such financial arrangements, and to counterbalance the leverage of insurers, but there are legal issues to consider. Many IPAs are vulnerable to antitrust charges because they include competing health care providers. You should check with legal counsel before signing on to an IPA, to make sure that it abides by antitrust and price fixing laws. IPAs have also been known to fail, particularly in states where they are not adequately regulated.
A possible successor to IPAs is the accountable care organization (ACO), an entity born as a component of the Affordable Care Act. While the official definition remains nebulous, an ACO is basically a network of doctors and hospitals that shares financial and medical responsibility for providing coordinated and efficient care to patients. The goal of ACO participants is to limit unnecessary spending, both individually and collectively, according to criteria established by the Centers for Medicare & Medicaid Services, without compromising quality of care in the process. More than 600 ACOs had been approved by the CMS as of the beginning of 2014.
It is important to remember that the ACO model remains very much a work in progress. ACOs make providers jointly accountable for the health of their patients. They offer financial incentives to cooperate, and to save money by avoiding unnecessary tests and procedures. A key component is the sharing of information. Providers who save money while also meeting quality targets are theoretically entitled to a portion of the savings.
As with IPAs, ACO ventures involve a measure of risk. ACOs that fail to meet the CMS performance and savings benchmarks can be stuck with the bill for investments made to improve care, such as equipment and computer purchases and the hiring of mid-level providers and managers, and they may be assessed monetary penalties as well. ACOs sponsored by physicians or rural providers, however, can apply to receive payments in advance to help finance infrastructure investments – a concession the Obama administration made after receiving complaints from rural hospitals.
Clearly, the price of remaining autonomous will be significant, and many private practitioners will be unwilling to pay it: Only 36% of physicians remained in independent practice at the end of the 2013, according to data from the American Medical Association – down from 57% in 2000 – but those of us who remain committed to independence will find ways to preserve it. In medicine, as in life, those most responsive to change will survive and flourish.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.
The ongoing sea change in medicine has led to a substantial erosion of physician autonomy, and to ever-increasing administrative burdens that hit small practices the hardest. Does this mean that the independent private physician practice model is doomed, as some predict? Absolutely not; but it will force many solo practitioners and small groups to join forces to protect themselves.
Those practices that offer unique services, or fill an unmet niche, may be able to remain small; but most smaller practices will need to consider a larger alternative. In a previous column, I outlined the basics of one such protective strategy – merging two or more small practices into a larger entity – but there are other options to consider.
One attractive and relatively straightforward strategy is the formation of a cooperative group. In most areas, there are very likely several small practices in similar predicaments that might be receptive to discussing a collaboration on billing and purchasing. This allows each participant to maintain independence as a private practice, while pooling resources to ease the administrative burdens of all. Once that arrangement is in place, the group can consider more ambitious projects, such as the joint purchase of an EHR system, sharing of personnel to lower staffing costs, and an integrated scheduling system. The latter will be particularly attractive to participants in later stages of their careers who are considering an intermediate option, somewhere between full-time work and complete retirement.
After a time, when the structure is stabilized and everyone agrees that his or her individual and shared interests and goals are being met, an outright merger can be contemplated. Projects of this scope require careful planning and implementation, and should not be undertaken without the help of competent legal counsel and an experienced business consultant.
A more complex but increasingly popular option is to join other small practices and providers in an independent practice association (IPA). An IPA is a legal entity organized and directed by physicians for the purpose of negotiating contracts with insurance companies on their behalf. Because of its structure, an IPA is better positioned to enter into such financial arrangements, and to counterbalance the leverage of insurers, but there are legal issues to consider. Many IPAs are vulnerable to antitrust charges because they include competing health care providers. You should check with legal counsel before signing on to an IPA, to make sure that it abides by antitrust and price fixing laws. IPAs have also been known to fail, particularly in states where they are not adequately regulated.
A possible successor to IPAs is the accountable care organization (ACO), an entity born as a component of the Affordable Care Act. While the official definition remains nebulous, an ACO is basically a network of doctors and hospitals that shares financial and medical responsibility for providing coordinated and efficient care to patients. The goal of ACO participants is to limit unnecessary spending, both individually and collectively, according to criteria established by the Centers for Medicare & Medicaid Services, without compromising quality of care in the process. More than 600 ACOs had been approved by the CMS as of the beginning of 2014.
It is important to remember that the ACO model remains very much a work in progress. ACOs make providers jointly accountable for the health of their patients. They offer financial incentives to cooperate, and to save money by avoiding unnecessary tests and procedures. A key component is the sharing of information. Providers who save money while also meeting quality targets are theoretically entitled to a portion of the savings.
As with IPAs, ACO ventures involve a measure of risk. ACOs that fail to meet the CMS performance and savings benchmarks can be stuck with the bill for investments made to improve care, such as equipment and computer purchases and the hiring of mid-level providers and managers, and they may be assessed monetary penalties as well. ACOs sponsored by physicians or rural providers, however, can apply to receive payments in advance to help finance infrastructure investments – a concession the Obama administration made after receiving complaints from rural hospitals.
Clearly, the price of remaining autonomous will be significant, and many private practitioners will be unwilling to pay it: Only 36% of physicians remained in independent practice at the end of the 2013, according to data from the American Medical Association – down from 57% in 2000 – but those of us who remain committed to independence will find ways to preserve it. In medicine, as in life, those most responsive to change will survive and flourish.
Dr. Eastern practices dermatology and dermatologic surgery in Belleville, N.J. He is the author of numerous articles and textbook chapters, and is a longtime monthly columnist for Dermatology News.

RNA sequencing characterized high-risk squamous cell carcinomas
SAN DIEGO – Cutaneous squamous cell carcinomas from organ transplant recipients had a more aggressive molecular profile than did tumor samples from immunocompetent patients, according to an RNA sequencing study presented at the annual meeting of the American Society for Dermatologic Surgery.
Specimens from organ transplant recipients showed greater induction of biologic pathways related to cancer signaling, fibrosis, and extracellular matrix remodeling, said Dr. Cameron Chesnut, a dermatologist in private practice in Spokane, Wash., who carried out the research while he was a dermatologic surgery resident at the University of California, Los Angeles.
Furthermore, the TP53 tumor suppressor gene was inhibited at least five times more in samples from organ transplant recipients, compared with those from immunocompetent patients, Dr. Chesnut said in an interview.
Squamous cell carcinoma (SCC) is the most common cancer to occur after organ transplantation, Dr. Chesnut and his associates noted. The malignancy is 65-250 times more common, is more than 4 times more likely to metastasize, and has a mortality rate of 5% compared with a rate of less than 1% in immunocompetent patients, based on data published online in the journal F1000 Prime Reports, they said.
To characterize these high-risk SCCs and compare them with lower-risk SCCs, the researchers performed RNA sequencing of three normal skin samples and SCC specimens from 15 patients – 7 organ transplant recipients and 8 otherwise healthy individuals. The researchers used an Illumina GAIIx RNA Seq instrument to generate RNA sequencing libraries of the specimens. They also used the web-based Ingenuity Pathway Analysis technique to identify the major biological pathways regulated within the tumors.
In all, 690 highly expressed genes were induced at least fivefold in SCCs from organ transplant recipients compared with those from otherwise healthy patients. These genes encoded pathways related to fibrosis, extracellular remodeling, the cell cycle, and tumor signaling, the investigators said. The COX-2 pathway for prostaglandin synthesis also was induced fivefold or more in the high-risk SCCs compared with those from immunocompetent patients, Dr. Chesnut added.
The researchers also identified 1,290 highly expressed genes that were inhibited at least fivefold in SCCs from organ transplant recipients compared with specimens from immunocompetent patients. The most strongly inhibited pathways were related to sterol biosynthesis and epithelial differentiation, followed by nucleotide excision repair, interleukin-6 and IL-17, and apoptosis, they said.
Based on these findings, novel therapeutics might someday be able to target specific biologic pathways that are highly induced in SCCs from organ transplant recipients, Dr. Chesnut said. “It’s hard to say what the most likely candidates are,” but based on the study findings, “regulating inflammation may be a target,” he added. Dr. Chesnut and his associates reported no external funding sources or conflicts of interest.
SAN DIEGO – Cutaneous squamous cell carcinomas from organ transplant recipients had a more aggressive molecular profile than did tumor samples from immunocompetent patients, according to an RNA sequencing study presented at the annual meeting of the American Society for Dermatologic Surgery.
Specimens from organ transplant recipients showed greater induction of biologic pathways related to cancer signaling, fibrosis, and extracellular matrix remodeling, said Dr. Cameron Chesnut, a dermatologist in private practice in Spokane, Wash., who carried out the research while he was a dermatologic surgery resident at the University of California, Los Angeles.
Furthermore, the TP53 tumor suppressor gene was inhibited at least five times more in samples from organ transplant recipients, compared with those from immunocompetent patients, Dr. Chesnut said in an interview.
Squamous cell carcinoma (SCC) is the most common cancer to occur after organ transplantation, Dr. Chesnut and his associates noted. The malignancy is 65-250 times more common, is more than 4 times more likely to metastasize, and has a mortality rate of 5% compared with a rate of less than 1% in immunocompetent patients, based on data published online in the journal F1000 Prime Reports, they said.
To characterize these high-risk SCCs and compare them with lower-risk SCCs, the researchers performed RNA sequencing of three normal skin samples and SCC specimens from 15 patients – 7 organ transplant recipients and 8 otherwise healthy individuals. The researchers used an Illumina GAIIx RNA Seq instrument to generate RNA sequencing libraries of the specimens. They also used the web-based Ingenuity Pathway Analysis technique to identify the major biological pathways regulated within the tumors.
In all, 690 highly expressed genes were induced at least fivefold in SCCs from organ transplant recipients compared with those from otherwise healthy patients. These genes encoded pathways related to fibrosis, extracellular remodeling, the cell cycle, and tumor signaling, the investigators said. The COX-2 pathway for prostaglandin synthesis also was induced fivefold or more in the high-risk SCCs compared with those from immunocompetent patients, Dr. Chesnut added.
The researchers also identified 1,290 highly expressed genes that were inhibited at least fivefold in SCCs from organ transplant recipients compared with specimens from immunocompetent patients. The most strongly inhibited pathways were related to sterol biosynthesis and epithelial differentiation, followed by nucleotide excision repair, interleukin-6 and IL-17, and apoptosis, they said.
Based on these findings, novel therapeutics might someday be able to target specific biologic pathways that are highly induced in SCCs from organ transplant recipients, Dr. Chesnut said. “It’s hard to say what the most likely candidates are,” but based on the study findings, “regulating inflammation may be a target,” he added. Dr. Chesnut and his associates reported no external funding sources or conflicts of interest.
SAN DIEGO – Cutaneous squamous cell carcinomas from organ transplant recipients had a more aggressive molecular profile than did tumor samples from immunocompetent patients, according to an RNA sequencing study presented at the annual meeting of the American Society for Dermatologic Surgery.
Specimens from organ transplant recipients showed greater induction of biologic pathways related to cancer signaling, fibrosis, and extracellular matrix remodeling, said Dr. Cameron Chesnut, a dermatologist in private practice in Spokane, Wash., who carried out the research while he was a dermatologic surgery resident at the University of California, Los Angeles.
Furthermore, the TP53 tumor suppressor gene was inhibited at least five times more in samples from organ transplant recipients, compared with those from immunocompetent patients, Dr. Chesnut said in an interview.
Squamous cell carcinoma (SCC) is the most common cancer to occur after organ transplantation, Dr. Chesnut and his associates noted. The malignancy is 65-250 times more common, is more than 4 times more likely to metastasize, and has a mortality rate of 5% compared with a rate of less than 1% in immunocompetent patients, based on data published online in the journal F1000 Prime Reports, they said.
To characterize these high-risk SCCs and compare them with lower-risk SCCs, the researchers performed RNA sequencing of three normal skin samples and SCC specimens from 15 patients – 7 organ transplant recipients and 8 otherwise healthy individuals. The researchers used an Illumina GAIIx RNA Seq instrument to generate RNA sequencing libraries of the specimens. They also used the web-based Ingenuity Pathway Analysis technique to identify the major biological pathways regulated within the tumors.
In all, 690 highly expressed genes were induced at least fivefold in SCCs from organ transplant recipients compared with those from otherwise healthy patients. These genes encoded pathways related to fibrosis, extracellular remodeling, the cell cycle, and tumor signaling, the investigators said. The COX-2 pathway for prostaglandin synthesis also was induced fivefold or more in the high-risk SCCs compared with those from immunocompetent patients, Dr. Chesnut added.
The researchers also identified 1,290 highly expressed genes that were inhibited at least fivefold in SCCs from organ transplant recipients compared with specimens from immunocompetent patients. The most strongly inhibited pathways were related to sterol biosynthesis and epithelial differentiation, followed by nucleotide excision repair, interleukin-6 and IL-17, and apoptosis, they said.
Based on these findings, novel therapeutics might someday be able to target specific biologic pathways that are highly induced in SCCs from organ transplant recipients, Dr. Chesnut said. “It’s hard to say what the most likely candidates are,” but based on the study findings, “regulating inflammation may be a target,” he added. Dr. Chesnut and his associates reported no external funding sources or conflicts of interest.
Key clinical point: Squamous cell carcinomas from organ transplant recipients showed a more aggressive molecular profile than did those from immunocompetent individuals.
Major finding: The high-risk tumors showed greater induction of biologic pathways related to cancer signaling, fibrosis, and extracellular matrix remodeling, and inhibition of the tp53 tumor suppressor gene.
Data source: RNA sequencing of 15 squamous cell carcinomas, including seven from organ transplant recipients.
Disclosures: The investigators reported no external funding sources or conflicts of interest.
Make the Diagnosis - January 2015
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
Diagnosis: Roseola
Roseola, also known as exanthema subitum or sixth disease, is a common viral infection that affects children by the age of 2 years, most commonly between ages 6 and 15 months. Two strains of viruses, human herpes virus 6 and 7, are the most common causes. Roseola spreads from person to person through respiratory secretions. Symptoms can vary in severity and can start with a sudden high fever, often greater than 103° F, that can last several days and may be associated with a febrile seizure. Associated symptoms may include sore throat, runny nose, cough, and lymphadenopathy.
After the fever resolves, erythematous macules and papules develop, starting on the chest, back, or abdomen and subsequently spreading to the neck and arms. The rash is not typically pruritic, is not contagious, and can last anywhere from several hours to several days. It is important to note that a person with roseola is contagious even if no rash is present.
Roseola can be difficult to diagnose given the nonspecific symptoms that are very similar to those of other childhood illnesses. Diagnosis is often clinical, based on history and physical, but can be confirmed by the characteristic rash or by detecting antibodies to roseola.
Treatment of roseola is supportive, with most children recovering within a week. Over-the-counter medications to treat fever, such as acetaminophen and ibuprofen, can be used, but aspirin should be avoided to treat fever in children younger than 2 years given the risk of Reye's syndrome. Antibodies developed against roseola are protective against future infections. Measures should be taken to avoid contact with those infected and hands should be washed frequently to avoid spreading the virus. Adults can contract roseola if they did not have it as a child; the disease is milder in adults but they can spread it to children.
Another common viral childhood illness is hand, foot, and mouth disease, caused by an enterovirus (coxsackievirus) that leads to sores and blisters on the hands, feet, and buttocks/legs that last for a week. The virus is spread through coughing and sneezing and through fecal-to-oral transmission. Symptoms include sore throat and fever with the rash. Diagnosis is clinical, based on history and physical exam, and the condition is treated supportively similar to roseola.
Another common illness is erythema infectiosum, also known as fifth disease, and is caused by parvovirus B19. The virus is common among school-aged children. The rash typically starts as erythematous patches on the cheeks giving the characteristic "slapped cheek" appearance. By the time the rash appears, the child is no longer contagious. The virus is spread through respiratory secretions.
Adults whose occupations put them in close contact with children can become infected with symptoms, including joint pain, rather than the rash. Infection in pregnant women and patients with sickle cell disease carries an increased risk of miscarriage and severe anemia, respectively. Similar to roseola and hand, foot, and mouth disease, diagnosis of fifth disease is clinical, and treatment is largely supportive.
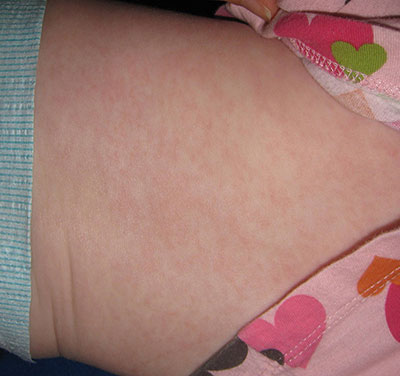
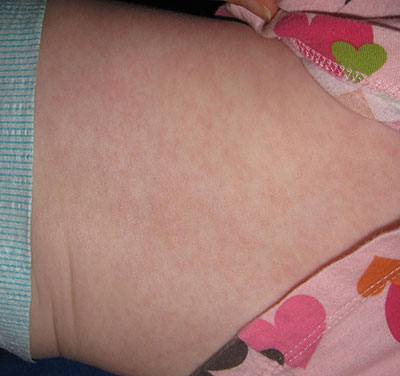
This case was submitted by Parteek Singla, B.S., and Dr. Donna Bilu Martin of Premier Dermatology, MD. An otherwise healthy 10-month-old female developed a high fever and runny nose that lasted for 4 days. On the fifth day, she developed a rash on her trunk that spread to her extremities and lasted for 1 day. Her illness resolved with no sequelae.
Protein may hold key to treating resistant lymphomas

Stephanie Grabow, Liz Valente,
and Andreas Strasser
Photo courtesy of the
Walter and Eliza Hall Insitute
Targeting a cell survival protein could overcome treatment resistance in T-cell lymphomas, according to preclinical research published in Blood.
Investigators found that removing the pro-survival protein MCL-1 prompted the death of lymphoma cells that had become resistant to conventional treatments.
The team noted that half of all cancers become resistant to chemotherapy and radiotherapy by acquiring mutations in the tumor-suppressing p53 protein.
And their research showed that MCL-1 helps these cancer cells survive by subverting the normal process of apoptosis.
“There are several pro-survival proteins that promote the sustained survival of cancer cells; the challenge is to identify which one is the most important in keeping each type of cancer cell alive,” said Stephanie Grabow, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“When we removed MCL-1 in models of T-cell lymphoma that had ‘lost’ the tumor-suppressing protein p53, cancers could not develop, demonstrating that MCL-1 is critical for the development of T-cell lymphomas.”
“Previous work from our colleagues at the institute has shown that MCL-1 is also critical for the survival and therapy-resistance of other blood cancers, including B-cell lymphoma and acute myeloid leukemia, indicating that is a very important target for potential new anticancer treatments.”
So the new finding reinforces the need to develop compounds that specifically target MCL-1, said Andreas Strasser, PhD, also of the Walter and Eliza Hall Institute.
“Investigating the role of MCL-1 and other proteins involved in controlling apoptosis has shown that MCL-1 is a critical protein in the survival of many types of cancer cells,” he said. “Targeting MCL-1 could therefore allow us to develop new, urgently needed therapies to treat cancers that have stopped responding to other anticancer drugs.”
Dr Grabow said the researchers will continue to investigate the role of MCL-1 in the development and progression of other cancers.
“Finding new treatment targets is crucial if we are to reduce the impact of these diseases,” she concluded. ![]()
Stephanie Grabow, Liz Valente,
and Andreas Strasser
Photo courtesy of the
Walter and Eliza Hall Insitute
Targeting a cell survival protein could overcome treatment resistance in T-cell lymphomas, according to preclinical research published in Blood.
Investigators found that removing the pro-survival protein MCL-1 prompted the death of lymphoma cells that had become resistant to conventional treatments.
The team noted that half of all cancers become resistant to chemotherapy and radiotherapy by acquiring mutations in the tumor-suppressing p53 protein.
And their research showed that MCL-1 helps these cancer cells survive by subverting the normal process of apoptosis.
“There are several pro-survival proteins that promote the sustained survival of cancer cells; the challenge is to identify which one is the most important in keeping each type of cancer cell alive,” said Stephanie Grabow, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“When we removed MCL-1 in models of T-cell lymphoma that had ‘lost’ the tumor-suppressing protein p53, cancers could not develop, demonstrating that MCL-1 is critical for the development of T-cell lymphomas.”
“Previous work from our colleagues at the institute has shown that MCL-1 is also critical for the survival and therapy-resistance of other blood cancers, including B-cell lymphoma and acute myeloid leukemia, indicating that is a very important target for potential new anticancer treatments.”
So the new finding reinforces the need to develop compounds that specifically target MCL-1, said Andreas Strasser, PhD, also of the Walter and Eliza Hall Institute.
“Investigating the role of MCL-1 and other proteins involved in controlling apoptosis has shown that MCL-1 is a critical protein in the survival of many types of cancer cells,” he said. “Targeting MCL-1 could therefore allow us to develop new, urgently needed therapies to treat cancers that have stopped responding to other anticancer drugs.”
Dr Grabow said the researchers will continue to investigate the role of MCL-1 in the development and progression of other cancers.
“Finding new treatment targets is crucial if we are to reduce the impact of these diseases,” she concluded. ![]()
Stephanie Grabow, Liz Valente,
and Andreas Strasser
Photo courtesy of the
Walter and Eliza Hall Insitute
Targeting a cell survival protein could overcome treatment resistance in T-cell lymphomas, according to preclinical research published in Blood.
Investigators found that removing the pro-survival protein MCL-1 prompted the death of lymphoma cells that had become resistant to conventional treatments.
The team noted that half of all cancers become resistant to chemotherapy and radiotherapy by acquiring mutations in the tumor-suppressing p53 protein.
And their research showed that MCL-1 helps these cancer cells survive by subverting the normal process of apoptosis.
“There are several pro-survival proteins that promote the sustained survival of cancer cells; the challenge is to identify which one is the most important in keeping each type of cancer cell alive,” said Stephanie Grabow, PhD, of the Walter and Eliza Hall Institute of Medical Research in Victoria, Australia.
“When we removed MCL-1 in models of T-cell lymphoma that had ‘lost’ the tumor-suppressing protein p53, cancers could not develop, demonstrating that MCL-1 is critical for the development of T-cell lymphomas.”
“Previous work from our colleagues at the institute has shown that MCL-1 is also critical for the survival and therapy-resistance of other blood cancers, including B-cell lymphoma and acute myeloid leukemia, indicating that is a very important target for potential new anticancer treatments.”
So the new finding reinforces the need to develop compounds that specifically target MCL-1, said Andreas Strasser, PhD, also of the Walter and Eliza Hall Institute.
“Investigating the role of MCL-1 and other proteins involved in controlling apoptosis has shown that MCL-1 is a critical protein in the survival of many types of cancer cells,” he said. “Targeting MCL-1 could therefore allow us to develop new, urgently needed therapies to treat cancers that have stopped responding to other anticancer drugs.”
Dr Grabow said the researchers will continue to investigate the role of MCL-1 in the development and progression of other cancers.
“Finding new treatment targets is crucial if we are to reduce the impact of these diseases,” she concluded. ![]()
T-cell receptor ensures Treg functionality

Regulatory T cells (Tregs) need T-cell receptors to fulfill their protective functions, according to research published in Immunity.
The researchers knew that Tregs need T-cell receptors to develop properly, but they were unsure of the receptors’ role after that.
To find out, the team deactivated T-cell receptors on mature Tregs in genetically modified mice.
They found these defective Tregs were not able to carry out their protective function without the T-cell receptors.
Furthermore, the Treg pool fell significantly, as these cells were no longer multiplying.
However, the researchers also discovered that two of Tregs’ most well-known central molecular properties—the production of Foxp3 protein and specific chemical changes to DNA—were still present in the defective T cells.
“Without their receptor, the Tregs are still clearly identifiable as Tregs,” said study author Christoph Vahl, PhD, of the Max Planck Institute of Biochemistry in Martinsried, Germany.
“However, they lose a large part of their cellular identity. They also lose their special ability to suppress excessive immune reactions.”
“The Tregs obviously need continuous contact with their environment to function correctly. This is presumably the reason why they need a receptor that recognizes endogenous substances and continuously sends signals.”
“During the course of our research, we uncovered a very important mechanism for suppressing excessive responses and responses targeted against the human body,” added Marc Schmidt-Supprian, PhD, also of the Max Planck Institute.
“These findings could be relevant for situations where it would be beneficial to weaken the control of Tregs over immune responses—for example, in the treatment of cancer.” ![]()
Regulatory T cells (Tregs) need T-cell receptors to fulfill their protective functions, according to research published in Immunity.
The researchers knew that Tregs need T-cell receptors to develop properly, but they were unsure of the receptors’ role after that.
To find out, the team deactivated T-cell receptors on mature Tregs in genetically modified mice.
They found these defective Tregs were not able to carry out their protective function without the T-cell receptors.
Furthermore, the Treg pool fell significantly, as these cells were no longer multiplying.
However, the researchers also discovered that two of Tregs’ most well-known central molecular properties—the production of Foxp3 protein and specific chemical changes to DNA—were still present in the defective T cells.
“Without their receptor, the Tregs are still clearly identifiable as Tregs,” said study author Christoph Vahl, PhD, of the Max Planck Institute of Biochemistry in Martinsried, Germany.
“However, they lose a large part of their cellular identity. They also lose their special ability to suppress excessive immune reactions.”
“The Tregs obviously need continuous contact with their environment to function correctly. This is presumably the reason why they need a receptor that recognizes endogenous substances and continuously sends signals.”
“During the course of our research, we uncovered a very important mechanism for suppressing excessive responses and responses targeted against the human body,” added Marc Schmidt-Supprian, PhD, also of the Max Planck Institute.
“These findings could be relevant for situations where it would be beneficial to weaken the control of Tregs over immune responses—for example, in the treatment of cancer.” ![]()
Regulatory T cells (Tregs) need T-cell receptors to fulfill their protective functions, according to research published in Immunity.
The researchers knew that Tregs need T-cell receptors to develop properly, but they were unsure of the receptors’ role after that.
To find out, the team deactivated T-cell receptors on mature Tregs in genetically modified mice.
They found these defective Tregs were not able to carry out their protective function without the T-cell receptors.
Furthermore, the Treg pool fell significantly, as these cells were no longer multiplying.
However, the researchers also discovered that two of Tregs’ most well-known central molecular properties—the production of Foxp3 protein and specific chemical changes to DNA—were still present in the defective T cells.
“Without their receptor, the Tregs are still clearly identifiable as Tregs,” said study author Christoph Vahl, PhD, of the Max Planck Institute of Biochemistry in Martinsried, Germany.
“However, they lose a large part of their cellular identity. They also lose their special ability to suppress excessive immune reactions.”
“The Tregs obviously need continuous contact with their environment to function correctly. This is presumably the reason why they need a receptor that recognizes endogenous substances and continuously sends signals.”
“During the course of our research, we uncovered a very important mechanism for suppressing excessive responses and responses targeted against the human body,” added Marc Schmidt-Supprian, PhD, also of the Max Planck Institute.
“These findings could be relevant for situations where it would be beneficial to weaken the control of Tregs over immune responses—for example, in the treatment of cancer.” ![]()
Study reveals potential strategy to treat AML

Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy. ![]()
Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy. ![]()
Credit: Lance Liotta
Researchers have discovered that interactions between two molecules—STAT3 and PRL-3—may provide a therapeutic target for acute myeloid leukemia (AML).
The team found evidence to suggest that the STAT3-PRL-3 regulatory loop contributes to the development of AML.
Chng Wee Joo, MB ChB, PhD, of the National University Cancer Institute in Singapore, and his colleagues reported these findings in Experimental Hematology.
The researchers discovered that STAT3, a transcription factor, binds and promotes the production of PRL-3 in cells. A decrease in STAT3 levels led to a corresponding decrease in the levels of PRL-3 and diminished the malignant properties of leukemic cells.
The team therefore concluded that a disruption of this regulatory loop may offer an attractive anti-AML therapeutic strategy. Furthermore, PRL-3 has the potential to be used as a biomarker in personalized therapy for AML patients.
The group was the first to report that the PRL-3 protein is overexpressed in 47% of bone marrow samples from AML patients. In addition, cellular levels of STAT3 were found to be elevated in about 50% of AML cases.
The researchers created a core STAT3 signature by analyzing datasets in the scientific literature. And they found that STAT3 core signature was significantly enriched in AML cases with high PRL-3 expression.
“Earlier studies on PRL-3 have been conducted in other cancers, but only in recent years has attention been turned to the significance of PRL-3 in blood cancer,” Dr Chng said.
“Previously, the mechanism by which PRL-3 is regulated in AML has also not been fully elucidated. This study reveals a novel connection between these two important oncogenes for the first time and also shows that the STAT3-PRL-3 regulatory loop contributes to the pathogenesis of AML.”
The researchers are now looking into methods to target the STAT3-PRL-3 pathway in AML, which could open up new avenues to treat AML patients with high expression of PRL-3 and offer an attractive anti-leukemia therapeutic strategy. ![]()
Study aims to determine prognostic factors for subset of thyroid cancer patients
CORONADO, CALIF. – In patients with radioactive iodine–refractory differentiated thyroid cancer, those with target lesions less than 1.5 cm in size appeared to derive less benefit from sorafenib in terms of progression-free survival, results from an international study showed.
In addition, papillary histology was a positive predictive factor and a predictive factor for benefit from sorafenib.
“Patients with radioactive iodine–refractory differentiated thyroid cancer have a poor prognosis, and there is a lack of effective treatments,” Dr. Martin Schlumberger said at the annual meeting of the American Thyroid Association. “The median survival for this subset is estimated to be 2.5-5 years.”

Sorafenib was approved by the Food and Drug Administration in November 2013 for the treatment of radioactive iodine–refractory differentiated thyroid cancer based on results from the randomized, controlled, double-blind phase III DECISION trial (Lancet 2014;384:319-28). Investigators found that the use of sorafenib extended median progression-free survival by 5 months, compared with placebo (10.8 vs 5.8 months; P < .0001). The purpose of the current analysis was to determine which demographic baseline or disease-related characteristics are prognostic for better outcomes in this patient population. To do so, Dr. Schlumberger of the department of nuclear medicine and endocrine oncology at Gustave Roussy, Villejuif, France, and his associates performed multivariate Cox proportional hazards models adjusted for treatment effect.
He reported findings from 417 patients. Of these, 210 were randomized to receive placebo and 207 were randomized to receive sorafenib. Variables found to be prognostic factors for progression-free survival in placebo patients, and in all patients when adjusted for sorafenib treatment, included papillary histology, lower targeted tumor size, baseline thyroglobulin less than 486 ng/mL, lower number of lesions, and residing in Asia vs. Europe and North America. Subgroup analyses of patients in the sorafenib arm revealed that the following baseline or disease-related variables were predictive of progression-free survival: papillary histology, tumor size of at least 1.5 cm, and having only lung metastases.
In a post-hoc exploratory analysis of progression-free survival by thyroid cancer symptoms among all 417 patients at study entry, the researchers found that both symptomatic and asymptomatic patients had improved progression-free survival following treatment with sorafenib.
On the basis of these findings, radioactive iodine–refractory differentiated thyroid cancer patients with no progressive disease and a tumor size of less than 1.5 cm “appear to have a good prognosis and may be candidates for a ‘watch and wait’ approach before initiating treatment with sorafenib,” Dr. Schlumberger concluded.
Dr. Schlumberger is an adviser to AstraZeneca, Bayer, Eisai, Exelixis, and Genzyme. He has also received research support from Genzyme and Bayer.
On Twitter @dougbrunk
CORONADO, CALIF. – In patients with radioactive iodine–refractory differentiated thyroid cancer, those with target lesions less than 1.5 cm in size appeared to derive less benefit from sorafenib in terms of progression-free survival, results from an international study showed.
In addition, papillary histology was a positive predictive factor and a predictive factor for benefit from sorafenib.
“Patients with radioactive iodine–refractory differentiated thyroid cancer have a poor prognosis, and there is a lack of effective treatments,” Dr. Martin Schlumberger said at the annual meeting of the American Thyroid Association. “The median survival for this subset is estimated to be 2.5-5 years.”
Sorafenib was approved by the Food and Drug Administration in November 2013 for the treatment of radioactive iodine–refractory differentiated thyroid cancer based on results from the randomized, controlled, double-blind phase III DECISION trial (Lancet 2014;384:319-28). Investigators found that the use of sorafenib extended median progression-free survival by 5 months, compared with placebo (10.8 vs 5.8 months; P < .0001). The purpose of the current analysis was to determine which demographic baseline or disease-related characteristics are prognostic for better outcomes in this patient population. To do so, Dr. Schlumberger of the department of nuclear medicine and endocrine oncology at Gustave Roussy, Villejuif, France, and his associates performed multivariate Cox proportional hazards models adjusted for treatment effect.
He reported findings from 417 patients. Of these, 210 were randomized to receive placebo and 207 were randomized to receive sorafenib. Variables found to be prognostic factors for progression-free survival in placebo patients, and in all patients when adjusted for sorafenib treatment, included papillary histology, lower targeted tumor size, baseline thyroglobulin less than 486 ng/mL, lower number of lesions, and residing in Asia vs. Europe and North America. Subgroup analyses of patients in the sorafenib arm revealed that the following baseline or disease-related variables were predictive of progression-free survival: papillary histology, tumor size of at least 1.5 cm, and having only lung metastases.
In a post-hoc exploratory analysis of progression-free survival by thyroid cancer symptoms among all 417 patients at study entry, the researchers found that both symptomatic and asymptomatic patients had improved progression-free survival following treatment with sorafenib.
On the basis of these findings, radioactive iodine–refractory differentiated thyroid cancer patients with no progressive disease and a tumor size of less than 1.5 cm “appear to have a good prognosis and may be candidates for a ‘watch and wait’ approach before initiating treatment with sorafenib,” Dr. Schlumberger concluded.
Dr. Schlumberger is an adviser to AstraZeneca, Bayer, Eisai, Exelixis, and Genzyme. He has also received research support from Genzyme and Bayer.
On Twitter @dougbrunk
CORONADO, CALIF. – In patients with radioactive iodine–refractory differentiated thyroid cancer, those with target lesions less than 1.5 cm in size appeared to derive less benefit from sorafenib in terms of progression-free survival, results from an international study showed.
In addition, papillary histology was a positive predictive factor and a predictive factor for benefit from sorafenib.
“Patients with radioactive iodine–refractory differentiated thyroid cancer have a poor prognosis, and there is a lack of effective treatments,” Dr. Martin Schlumberger said at the annual meeting of the American Thyroid Association. “The median survival for this subset is estimated to be 2.5-5 years.”
Sorafenib was approved by the Food and Drug Administration in November 2013 for the treatment of radioactive iodine–refractory differentiated thyroid cancer based on results from the randomized, controlled, double-blind phase III DECISION trial (Lancet 2014;384:319-28). Investigators found that the use of sorafenib extended median progression-free survival by 5 months, compared with placebo (10.8 vs 5.8 months; P < .0001). The purpose of the current analysis was to determine which demographic baseline or disease-related characteristics are prognostic for better outcomes in this patient population. To do so, Dr. Schlumberger of the department of nuclear medicine and endocrine oncology at Gustave Roussy, Villejuif, France, and his associates performed multivariate Cox proportional hazards models adjusted for treatment effect.
He reported findings from 417 patients. Of these, 210 were randomized to receive placebo and 207 were randomized to receive sorafenib. Variables found to be prognostic factors for progression-free survival in placebo patients, and in all patients when adjusted for sorafenib treatment, included papillary histology, lower targeted tumor size, baseline thyroglobulin less than 486 ng/mL, lower number of lesions, and residing in Asia vs. Europe and North America. Subgroup analyses of patients in the sorafenib arm revealed that the following baseline or disease-related variables were predictive of progression-free survival: papillary histology, tumor size of at least 1.5 cm, and having only lung metastases.
In a post-hoc exploratory analysis of progression-free survival by thyroid cancer symptoms among all 417 patients at study entry, the researchers found that both symptomatic and asymptomatic patients had improved progression-free survival following treatment with sorafenib.
On the basis of these findings, radioactive iodine–refractory differentiated thyroid cancer patients with no progressive disease and a tumor size of less than 1.5 cm “appear to have a good prognosis and may be candidates for a ‘watch and wait’ approach before initiating treatment with sorafenib,” Dr. Schlumberger concluded.
Dr. Schlumberger is an adviser to AstraZeneca, Bayer, Eisai, Exelixis, and Genzyme. He has also received research support from Genzyme and Bayer.
On Twitter @dougbrunk
AT THE ATA ANNUAL MEETING
Key clinical point: Radioactive iodine–refractory differentiated thyroid cancer patients with no progressive disease and a tumor size of less than 1.5 cm may be candidates for a “watch and wait” approach before initiating treatment with sorafenib.
Major finding: Baseline or disease-related variables found to be prognostic factors for progression-free survival in placebo patients and in all patients when adjusted for sorafenib treatment included papillary histology, lower targeted tumor size, baseline thyroglobulin less than 486 ng/mL, lower number of lesions, and residing in Asia versus Europe and North America.
Data source: An analysis of 417 patients from the randomized, controlled, double-blind, phase III DECISION trial.
Disclosures: Dr. Schlumberger is an adviser to AstraZeneca, Bayer, Eisai, Exelixis, and Genzyme. He has also received research support from Genzyme and Bayer.
