User login
Patient safety and tort reform
Question: Developments in medical tort reform include:
A. Continued constitutional challenges to caps on damages.
B. An emphasis on patient safety.
C. Hillary Clinton’s Senate bill.
D. Linking medical tort reform to error reduction.
E. All of the above.

Answer: E. Recent years have witnessed a stabilizing environment for medical liability, although insurance premiums continue to vary greatly by specialty and geographic location.
Recent statistics from the American Medical Association show that 2014 ob.gyn. insurance rates range from less than $50,000 in some areas of California to a high of $215,000 in Nassau and Suffolk counties in New York. The highest average expense in 2013, around a quarter of a million dollars, was for those claims that resulted in plaintiff verdicts, while defendant verdicts were substantially lower and averaged $140,000.
As in the past, most claims were dropped, dismissed, or withdrawn. About one-quarter of claims were settled, with only 2% decided by an alternative dispute resolution. Less than 8% were decided by trial verdict, with the vast majority won by the defendant.
The plaintiff bar continues to mount constitutional challenges to caps on damages. The California Supreme Court had previously ruled that reforms under California’s historic Medical Injury Compensation Reform Act (MICRA)1, which limits noneconomic recovery to $250,000, are constitutional, because they are rationally related to the legitimate legislative goal of reducing medical costs.
However, the statute has again come under challenge, only to be reaffirmed by a California state appeals court. In November 2014, California voters rejected Proposition 46, which sought to increase the cap from $250,000 to $1.1 million.
Texas, another pro-reform state, sides with California, and Mississippi also ruled that its damage cap is constitutional. However, Florida and Oklahoma recently joined jurisdictions such as Georgia, Illinois, and Missouri in ruling that damage caps are unconstitutional.
Asserting that the current health care liability system has been an inefficient and sometimes ineffective mechanism for initiating or resolving claims of medical error, medical negligence, or malpractice, then-U.S. senators Hillary Clinton (D-N.Y.) and Barack Obama (D-Ill.) in 2005 jointly sponsored legislation (S. 1784) to establish a National Medical Error Disclosure and Compensation Program (National MEDiC Act). Although the bill was killed in Senate committee, its key provisions were subsequently published in the New England Journal of Medicine (2006;354:2205-8).
The senators noted that the liability system has failed to the extent that only one medical malpractice claim is filed for every eight medical injuries, that it takes 4-8 years to resolve a claim, and that “solutions to the patient safety, litigation, and medical liability insurance problems have been elusive.”
Accordingly, the bill’s purpose was to promote the confidential disclosure to patients of medical errors in an effort to improve patient-safety systems. At the time of disclosure, there would be negotiations for compensation and proposals to prevent a recurrence of the problem that led to the patient’s injury. However, the patient would retain the right to counsel during negotiations, as well as access to the courts if no agreement were reached. The bill was entirely silent on traditional tort reform measures.
Nearly 4 decades earlier, a no-fault proposal by Professor Jeffrey O’Connell made some of these points, but with sharper focus and specificity, especially regarding damages.2
In marked contrast to the Clinton-Obama bill, his proposal gave the medical provider the exclusive option to tender payment, which would completely foreclose future tort action by the victim. Compensation benefits included net economic loss such as 100% of lost wages, replacement service loss, medical treatment expenses, and reasonable attorney’s fees. But noneconomic losses, such as pain and suffering, were not reimbursable, and payment was net of any benefits from collateral sources.
This proposal elegantly combined efficiency and fairness, and would have ameliorated the financial and emotional toll that comes with litigating injuries arising out of health care. Legislation in the House of Representatives, the Alternative Medical Liability Act (H.R. 5400), incorporated many of these features, and came before the 98th U.S. Congress in 1984. It, too, died in committee.
There may be something to the current trend toward pairing tort reform with error reduction. Thoughtful observers point to “disclosure and offer” programs such as the one at the Lexington (Ky.) Veterans Affairs Medical Center, which boasts average settlements of approximately $15,000 per claim – compared with more than $98,000 at other VA institutions. Its policy has also decreased the average duration of cases, previously 2-4 years, to 2-4 months, as well as reduced costs for legal defense.
Likewise, the program at the University of Michigan Health System has reduced both the frequency and severity of claims, duration of cases, and litigation costs. Aware of these developments, some private insurers, such as the COPIC Insurance Company in Colorado, are adopting a similar approach.
In its updated 2014 tort reform position paper, the American College of Physicians continues to endorse caps on noneconomic and punitive damages, as well as other tort reform measures.
However, it now acknowledges that “improving patient safety and preventing errors must be at the fore of the medical liability reform discussion.” The ACP correctly asserts that “emphasizing patient safety, promoting a culture of quality improvement and coordinated care, and training physicians in best practices to avoid errors and reduce risk will prevent harm and reduce the waste associated with defensive medicine.”
This hybrid approach combining traditional tort reforms with a renewed attention to patient safety through medical error reduction may yet yield additional practical benefits.
Here, the experience in anesthesiology bears recounting: Its dramatic progress in risk management has cut patient death rate from 1 in 5,000 to 1 in 200,000 to 300,000 in the space of 20 years, and this has been associated with a concurrent 37% fall in insurance premiums.
References
1. Medical Injury Compensation Reform Act of 1975, Cal. Civ. Proc. Code § 3333.2 (West 1982).
2. O’Connell, J. No-Fault Insurance for Injuries Arising from Medical Treatment: A Proposal for Elective Coverage. Emory L. J. 1975;24:21.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. Currently, he directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
Question: Developments in medical tort reform include:
A. Continued constitutional challenges to caps on damages.
B. An emphasis on patient safety.
C. Hillary Clinton’s Senate bill.
D. Linking medical tort reform to error reduction.
E. All of the above.
Answer: E. Recent years have witnessed a stabilizing environment for medical liability, although insurance premiums continue to vary greatly by specialty and geographic location.
Recent statistics from the American Medical Association show that 2014 ob.gyn. insurance rates range from less than $50,000 in some areas of California to a high of $215,000 in Nassau and Suffolk counties in New York. The highest average expense in 2013, around a quarter of a million dollars, was for those claims that resulted in plaintiff verdicts, while defendant verdicts were substantially lower and averaged $140,000.
As in the past, most claims were dropped, dismissed, or withdrawn. About one-quarter of claims were settled, with only 2% decided by an alternative dispute resolution. Less than 8% were decided by trial verdict, with the vast majority won by the defendant.
The plaintiff bar continues to mount constitutional challenges to caps on damages. The California Supreme Court had previously ruled that reforms under California’s historic Medical Injury Compensation Reform Act (MICRA)1, which limits noneconomic recovery to $250,000, are constitutional, because they are rationally related to the legitimate legislative goal of reducing medical costs.
However, the statute has again come under challenge, only to be reaffirmed by a California state appeals court. In November 2014, California voters rejected Proposition 46, which sought to increase the cap from $250,000 to $1.1 million.
Texas, another pro-reform state, sides with California, and Mississippi also ruled that its damage cap is constitutional. However, Florida and Oklahoma recently joined jurisdictions such as Georgia, Illinois, and Missouri in ruling that damage caps are unconstitutional.
Asserting that the current health care liability system has been an inefficient and sometimes ineffective mechanism for initiating or resolving claims of medical error, medical negligence, or malpractice, then-U.S. senators Hillary Clinton (D-N.Y.) and Barack Obama (D-Ill.) in 2005 jointly sponsored legislation (S. 1784) to establish a National Medical Error Disclosure and Compensation Program (National MEDiC Act). Although the bill was killed in Senate committee, its key provisions were subsequently published in the New England Journal of Medicine (2006;354:2205-8).
The senators noted that the liability system has failed to the extent that only one medical malpractice claim is filed for every eight medical injuries, that it takes 4-8 years to resolve a claim, and that “solutions to the patient safety, litigation, and medical liability insurance problems have been elusive.”
Accordingly, the bill’s purpose was to promote the confidential disclosure to patients of medical errors in an effort to improve patient-safety systems. At the time of disclosure, there would be negotiations for compensation and proposals to prevent a recurrence of the problem that led to the patient’s injury. However, the patient would retain the right to counsel during negotiations, as well as access to the courts if no agreement were reached. The bill was entirely silent on traditional tort reform measures.
Nearly 4 decades earlier, a no-fault proposal by Professor Jeffrey O’Connell made some of these points, but with sharper focus and specificity, especially regarding damages.2
In marked contrast to the Clinton-Obama bill, his proposal gave the medical provider the exclusive option to tender payment, which would completely foreclose future tort action by the victim. Compensation benefits included net economic loss such as 100% of lost wages, replacement service loss, medical treatment expenses, and reasonable attorney’s fees. But noneconomic losses, such as pain and suffering, were not reimbursable, and payment was net of any benefits from collateral sources.
This proposal elegantly combined efficiency and fairness, and would have ameliorated the financial and emotional toll that comes with litigating injuries arising out of health care. Legislation in the House of Representatives, the Alternative Medical Liability Act (H.R. 5400), incorporated many of these features, and came before the 98th U.S. Congress in 1984. It, too, died in committee.
There may be something to the current trend toward pairing tort reform with error reduction. Thoughtful observers point to “disclosure and offer” programs such as the one at the Lexington (Ky.) Veterans Affairs Medical Center, which boasts average settlements of approximately $15,000 per claim – compared with more than $98,000 at other VA institutions. Its policy has also decreased the average duration of cases, previously 2-4 years, to 2-4 months, as well as reduced costs for legal defense.
Likewise, the program at the University of Michigan Health System has reduced both the frequency and severity of claims, duration of cases, and litigation costs. Aware of these developments, some private insurers, such as the COPIC Insurance Company in Colorado, are adopting a similar approach.
In its updated 2014 tort reform position paper, the American College of Physicians continues to endorse caps on noneconomic and punitive damages, as well as other tort reform measures.
However, it now acknowledges that “improving patient safety and preventing errors must be at the fore of the medical liability reform discussion.” The ACP correctly asserts that “emphasizing patient safety, promoting a culture of quality improvement and coordinated care, and training physicians in best practices to avoid errors and reduce risk will prevent harm and reduce the waste associated with defensive medicine.”
This hybrid approach combining traditional tort reforms with a renewed attention to patient safety through medical error reduction may yet yield additional practical benefits.
Here, the experience in anesthesiology bears recounting: Its dramatic progress in risk management has cut patient death rate from 1 in 5,000 to 1 in 200,000 to 300,000 in the space of 20 years, and this has been associated with a concurrent 37% fall in insurance premiums.
References
1. Medical Injury Compensation Reform Act of 1975, Cal. Civ. Proc. Code § 3333.2 (West 1982).
2. O’Connell, J. No-Fault Insurance for Injuries Arising from Medical Treatment: A Proposal for Elective Coverage. Emory L. J. 1975;24:21.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. Currently, he directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].
Question: Developments in medical tort reform include:
A. Continued constitutional challenges to caps on damages.
B. An emphasis on patient safety.
C. Hillary Clinton’s Senate bill.
D. Linking medical tort reform to error reduction.
E. All of the above.
Answer: E. Recent years have witnessed a stabilizing environment for medical liability, although insurance premiums continue to vary greatly by specialty and geographic location.
Recent statistics from the American Medical Association show that 2014 ob.gyn. insurance rates range from less than $50,000 in some areas of California to a high of $215,000 in Nassau and Suffolk counties in New York. The highest average expense in 2013, around a quarter of a million dollars, was for those claims that resulted in plaintiff verdicts, while defendant verdicts were substantially lower and averaged $140,000.
As in the past, most claims were dropped, dismissed, or withdrawn. About one-quarter of claims were settled, with only 2% decided by an alternative dispute resolution. Less than 8% were decided by trial verdict, with the vast majority won by the defendant.
The plaintiff bar continues to mount constitutional challenges to caps on damages. The California Supreme Court had previously ruled that reforms under California’s historic Medical Injury Compensation Reform Act (MICRA)1, which limits noneconomic recovery to $250,000, are constitutional, because they are rationally related to the legitimate legislative goal of reducing medical costs.
However, the statute has again come under challenge, only to be reaffirmed by a California state appeals court. In November 2014, California voters rejected Proposition 46, which sought to increase the cap from $250,000 to $1.1 million.
Texas, another pro-reform state, sides with California, and Mississippi also ruled that its damage cap is constitutional. However, Florida and Oklahoma recently joined jurisdictions such as Georgia, Illinois, and Missouri in ruling that damage caps are unconstitutional.
Asserting that the current health care liability system has been an inefficient and sometimes ineffective mechanism for initiating or resolving claims of medical error, medical negligence, or malpractice, then-U.S. senators Hillary Clinton (D-N.Y.) and Barack Obama (D-Ill.) in 2005 jointly sponsored legislation (S. 1784) to establish a National Medical Error Disclosure and Compensation Program (National MEDiC Act). Although the bill was killed in Senate committee, its key provisions were subsequently published in the New England Journal of Medicine (2006;354:2205-8).
The senators noted that the liability system has failed to the extent that only one medical malpractice claim is filed for every eight medical injuries, that it takes 4-8 years to resolve a claim, and that “solutions to the patient safety, litigation, and medical liability insurance problems have been elusive.”
Accordingly, the bill’s purpose was to promote the confidential disclosure to patients of medical errors in an effort to improve patient-safety systems. At the time of disclosure, there would be negotiations for compensation and proposals to prevent a recurrence of the problem that led to the patient’s injury. However, the patient would retain the right to counsel during negotiations, as well as access to the courts if no agreement were reached. The bill was entirely silent on traditional tort reform measures.
Nearly 4 decades earlier, a no-fault proposal by Professor Jeffrey O’Connell made some of these points, but with sharper focus and specificity, especially regarding damages.2
In marked contrast to the Clinton-Obama bill, his proposal gave the medical provider the exclusive option to tender payment, which would completely foreclose future tort action by the victim. Compensation benefits included net economic loss such as 100% of lost wages, replacement service loss, medical treatment expenses, and reasonable attorney’s fees. But noneconomic losses, such as pain and suffering, were not reimbursable, and payment was net of any benefits from collateral sources.
This proposal elegantly combined efficiency and fairness, and would have ameliorated the financial and emotional toll that comes with litigating injuries arising out of health care. Legislation in the House of Representatives, the Alternative Medical Liability Act (H.R. 5400), incorporated many of these features, and came before the 98th U.S. Congress in 1984. It, too, died in committee.
There may be something to the current trend toward pairing tort reform with error reduction. Thoughtful observers point to “disclosure and offer” programs such as the one at the Lexington (Ky.) Veterans Affairs Medical Center, which boasts average settlements of approximately $15,000 per claim – compared with more than $98,000 at other VA institutions. Its policy has also decreased the average duration of cases, previously 2-4 years, to 2-4 months, as well as reduced costs for legal defense.
Likewise, the program at the University of Michigan Health System has reduced both the frequency and severity of claims, duration of cases, and litigation costs. Aware of these developments, some private insurers, such as the COPIC Insurance Company in Colorado, are adopting a similar approach.
In its updated 2014 tort reform position paper, the American College of Physicians continues to endorse caps on noneconomic and punitive damages, as well as other tort reform measures.
However, it now acknowledges that “improving patient safety and preventing errors must be at the fore of the medical liability reform discussion.” The ACP correctly asserts that “emphasizing patient safety, promoting a culture of quality improvement and coordinated care, and training physicians in best practices to avoid errors and reduce risk will prevent harm and reduce the waste associated with defensive medicine.”
This hybrid approach combining traditional tort reforms with a renewed attention to patient safety through medical error reduction may yet yield additional practical benefits.
Here, the experience in anesthesiology bears recounting: Its dramatic progress in risk management has cut patient death rate from 1 in 5,000 to 1 in 200,000 to 300,000 in the space of 20 years, and this has been associated with a concurrent 37% fall in insurance premiums.
References
1. Medical Injury Compensation Reform Act of 1975, Cal. Civ. Proc. Code § 3333.2 (West 1982).
2. O’Connell, J. No-Fault Insurance for Injuries Arising from Medical Treatment: A Proposal for Elective Coverage. Emory L. J. 1975;24:21.
Dr. Tan is professor emeritus of medicine and former adjunct professor of law at the University of Hawaii. Currently, he directs the St. Francis International Center for Healthcare Ethics in Honolulu. This article is meant to be educational and does not constitute medical, ethical, or legal advice. Some of the articles in this series are adapted from the author’s 2006 book, “Medical Malpractice: Understanding the Law, Managing the Risk,” and his 2012 Halsbury treatise, “Medical Negligence and Professional Misconduct.” For additional information, readers may contact the author at [email protected].

75-Year-Old Woman With Elevated Liver Enzymes
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
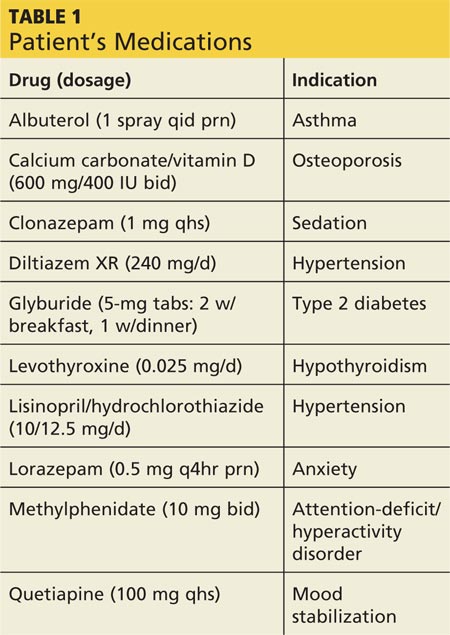
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
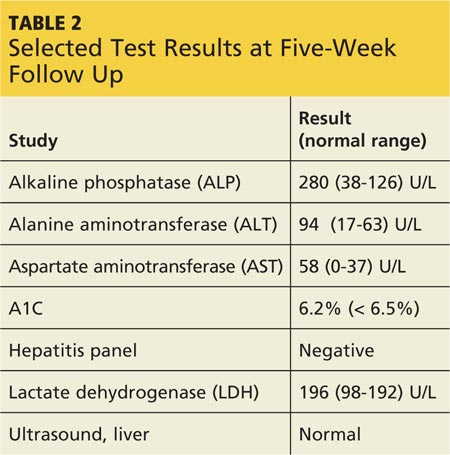
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
A 75-year-old woman, Gladys, was brought to the psychiatric clinic in a manic state by her concerned sister. The patient was disheveled, dehydrated, and having difficulty expressing her thoughts. Vital signs included a blood pressure of 200/94 mm Hg; pulse, 88 beats/min; temperature, 98.4°F; and respiratory rate, 20 breaths/min. Psychiatric history included a diagnosis of schizoaffective disorder with inconsistent adherence to treatment regimens, particularly mood stabilizers; and attention-deficit/hyperactivity disorder, for which she took methylphenidate regularly. Medical history was significant for asthma, osteoporosis, hypertension, type 2 diabetes, and hypothyroidism.
Gladys tended to become involved in personal relationships that adversely affected her mental health. This, in fact, had just happened: A “friend” had taken advantage of her kindness and then abruptly moved away, triggering the patient’s current decompensation. She was referred for admission for psychiatric evaluation and treatment.
During the three-week hospitalization, Gladys was diagnosed with bipolar I disorder. She agreed to take mood-stabilizing medication primarily to alleviate her insomnia during manic episodes. She was discharged on a multidrug regimen for her coexisting conditions (see Table 1). Of note, her blood pressure at discharge was 148/66 mm Hg.
At outpatient follow-up five days later, the patient reported feeling better and stronger. However, five weeks after discharge, Gladys returned with complaints of tiredness during the day (though sleeping well at night), severe dry mouth, aching joints, and poor appetite. Her blood pressure was 100/50 mm Hg. She denied abdominal pain or change in the color of her urine or stool. She also denied use of alcohol, illicit drugs, or OTC medications. Laboratory results revealed elevated levels of several liver enzymes (see Table 2), all of which had been normal when she was admitted to the hospital two months earlier.
Continue for discussion >>
DISCUSSION
Elevations in alanine aminotransferase (ALT) and aspartate aminotransferase (AST) levels may result from a variety of factors. Mild elevations are commonly caused by alcohol consumption, hemochromatosis, medications, nonalcoholic fatty liver disease, and viral hepatitis (with which elevations may range from mild to marked).1 Moderate to marked elevations of ALT and AST are commonly seen with acute biliary obstruction, alcoholic hepatitis, toxic injury, and ischemic injury.2
Abnormal liver enzyme levels are common with use of psychotropic drugs, such as antipsychotics and mood stabilizers.3 In a systematic review that examined the effects of antipsychotics on liver function tests, a median 4% of patients experienced elevated ALT, AST, or gamma-glutamyl transferase (GGT) levels (defined as more than triple the normal level) or alkaline phosphatase (ALP) level (defined as more than twice the normal level).3 Of the studies reviewed, five noted an interval of one to six weeks between initiation of antipsychotic drugs and detection of liver function test abnormalities. None of the included studies reported severe or fatal hepatic injury.
For the atypical antipsychotic quetiapine, elevations in ALT and AST occurred in about 5% and 3% of patients, respectively, in clinical trials of the drug as monotherapy for schizophrenia or bipolar mania.4 These elevations were usually transient, occurring within the first three weeks of treatment initiation and subsiding with continued treatment.
There are rare published reports, however, of serious and even fatal hepatotoxicity induced by quetiapine. One 59-year-old woman developed fulminant hepatic failure (FHF) six weeks after she began taking quetiapine in addition to carbidopa/levodopa for Parkinson disease. She reported nausea, vomiting, poor appetite, and abdominal pain and required a six-week hospitalization, with multidrug treatment that continued after discharge. Liver biopsy identified acute hepatitis with confluent bridging necrosis, a sign that the liver injury was drug-induced. The authors concluded that, because drug-induced hepatotoxicity is the most common cause of FHF in many parts of the world, clinicians should evaluate a patient’s medications for a potential cause.5
In another case report, elevated liver enzymes were identified one month after a 58-year-old woman taking several other medications began treatment with quetiapine (100 mg/d). She developed liver failure and died after a three-week hospitalization. The authors concluded that liver failure was caused by an idiosyncratic reaction to a relatively low dose of quetiapine. This case supports the advisability of close monitoring of liver enzyme levels during quetiapine treatment.6
Naharci et al reported a case of a 77-year-old woman treated with quetiapine (12.5 mg bid for nine days). She developed acute hepatic failure leading to multi-organ system failure and died eight days later. Liver failure was attributed to an idiosyncratic reaction to low-dose quetiapine. The authors concluded that liver function monitoring is essential with quetiapine administration, especially in elderly or fragile patients.7
The initial recommended dosage of quetiapine for elderly patients (defined as age 65 or older) is 50 mg/d, with the dose increased in increments of 50 mg/d, based on clinical response and tolerability. In clinical trials, the mean plasma clearance of quetiapine was reduced by 30% to 50% in the elderly, so dosing adjustments may be necessary in this age-group.4 Gareri et al recommended that atypical antipsychotics be prescribed for elderly patients for the shortest necessary duration and at the lowest effective dose.8
For hepatically impaired patients, recommended initial dosing is 25 mg/d, with increases of 25 to 50 mg/d until an effective and tolerable dose is reached.4 Further, because quetiapine is primarily metabolized via the cytochrome P450 liver enzymes CYP3A4 and CYP2D6,9 when the clinician prescribes a potent CYP3A4 inhibitor (eg, ketoconazole) to a patient taking quetiapine, the quetiapine dosage needs to be reduced. Conversely, when prescribing a CYP3A4 inducer (eg, phenytoin), the quetiapine dosage should be adjusted upward.4
Even when an apparently well-tolerated, effective quetiapine dosage has been reached, clinicians and patients should remain alert to the warning signs of potentially serious events. Adverse effects of atypical antipsychotics, including quetiapine, were summarized by Gareri et al and rated on a scale ranging from no effect to severe effect.8 The most severe adverse effects for quetiapine were hypotension and prolonged QTc interval. Weight gain was identified as a moderate effect, and sedation, gastrointestinal problems (nausea, vomiting, and constipation), and anticholinergic effects as mild. Some effects—tardive dyskinesia, seizures, and hepatic—were deemed “uncertain”; this rating suggests the need for careful monitoring of patients (who should be informed of signs and symptoms that should be reported to the clinician).8
Atasoy et al reviewed the records of 110 patients to assess the effect of atypical antipsychotics on liver function tests. The patients’ records included both baseline liver function tests and repeat testing at six months. Forty-eight patients received quetiapine; 33 patients, olanzapine; and 29 patients, risperidone. Liver enzymes were elevated in 27.1% of the quetiapine group, 30.3% of the olanzapine group, and 27.6% of the risperidone group. In two patients taking olanzapine, liver enzyme levels reached three to four times normal but returned to normal when treatment was stopped. The authors concluded that baseline liver enzyme studies should be done prior to initiation of treatment with atypical antipsychotics, as well as periodically thereafter, especially for patients with preexisting hepatic disorders, those being treated with other potentially hepatotoxic drugs, or those who exhibit signs or symptoms of hepatic impairment.10
Continue for patient outcome >>
PATIENT OUTCOME
Gladys’s ALT and AST levels were mildly elevated. One of the more common causes for this pattern is medication. In addition, her ALP level of more than twice the upper limit of normal further pointed to a viral, alcohol-related, or drug toxicity cause. Since her hepatitis panel was negative and she did not use alcohol, it was determined that elevated liver enzymes were related to the recent addition of quetiapine, which was discontinued. In addition, in light of Gladys’s hypotension (which is also a potential adverse effect of quetiapine8), her dose of lisinopril/hydrochlorothiazide was decreased by half.
One week later, liver enzyme levels were returning to normal. Gladys, however, began having more difficulty sleeping and more trouble remaining focused and getting things done, despite taking methylphenidate. In place of quetiapine, risperidone (0.5 mg at bedtime) was initiated. At first, Gladys was concerned with her continuing dry mouth symptoms, but when she skipped doses of risperidone, she noticed that she functioned less well. Further, her liver enzyme levels were being closely monitored and were normal. With this reassurance, Gladys remained adherent to risperidone for mood stabilization.
CONCLUSION
Atypical antipsychotic drugs such as quetiapine can cause elevated liver enzyme levels, especially in the elderly, patients with hepatic impairment, or patients on polypharmacotherapy. Rarely, quetiapine has been reported to cause serious hepatotoxicity and even death. Patients taking these drugs should be informed of possible symptoms of liver toxicity, including fatigue, nausea, vomiting, abdominal pain, and change in color of urine or stools. Particularly in more vulnerable patients, liver enzyme levels should be monitored carefully to confirm the continued safety of antipsychotic treatment.
REFERENCES
1. Oh RC, Hustead TR. Causes and evaluation of mildly elevated liver transaminase levels. Am Fam Physician. 2011;84(9):1003-1008.
2. Giannini EG, Testa R, Savarino V. Liver enzyme elevation: a guide for clinicians. CMAJ. 2005;172(3):367-379.
3. Marwick KFM, Taylor M, Walker SW. Antipsychotics and abnormal liver function tests: Systematic review. Clin Neuropharmacol. 2012;35(5):244-253.
4. Seroquel [package insert]. Wilmington, DE: AstraZeneca Pharmaceuticals LP; 2013.
5. Al Mutairi F, Dwivedi G, Al Ameel T. Fulminant hepatic failure in association with quetiapine: A case report. J Med Case Rep. 2012;6:418.
6. El Hajj L, Sharara A, Rockey, DC. Subfulminant liver failure associated with quetiapine. Eur J Gastroenterol Hepatol. 2004;16(12):1415-1418.
7. Naharci MI, Karadurmus N, Demir O, et al. Fatal hepatotoxicity in an elderly patient receiving low-dose quetiapine. Am J Psychiatry. 2011;168(2):212-213.
8. Gareri P, Segura-Garcia C, Manfredi VG, et al. Use of atypical antipsychotics in the elderly: a clinical review. Clin Interv Aging. 2014;16(9):1363-1373.
9. Lin S, Chang Y, Moody DE, Foltz RL. A liquid chromatographic-electrospray-tandem mass spectrometric method for quanititation of quetiapine in human plasma and liver microsomes: application to a study of in vitro metabolism. J Anal Toxicol. 2004;28(6):443-446.
10. Atasoy N, Erdogan A, Yalug I, et al. A review of liver function tests during treatment with atypical antipsychotic drugs: a chart review study. Prog Neuropsychopharmacol Biol Psychiatry. 2007;31(6):1255-1260.
Woman Awakens With Rapid Heart Rate
ANSWER
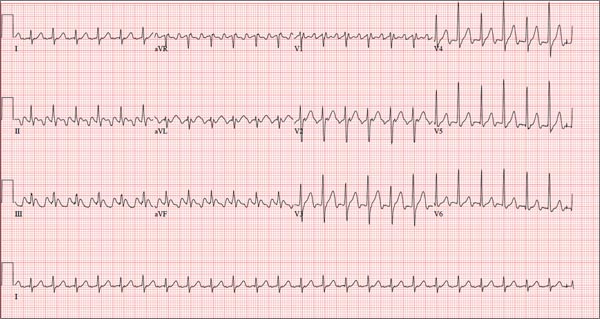
The correct interpretation of this ECG is atrial flutter with a 2:1 block. Careful inspection of lead I reveals a P wave at the terminal portion of the QRS complex, in addition to the P wave seen with a consistent PR interval of 150 ms. This results in two P waves for each QRS complex. Given the presence of the flutter waves, an accurate assessment of the ST segment is not possible.
ANSWER
The correct interpretation of this ECG is atrial flutter with a 2:1 block. Careful inspection of lead I reveals a P wave at the terminal portion of the QRS complex, in addition to the P wave seen with a consistent PR interval of 150 ms. This results in two P waves for each QRS complex. Given the presence of the flutter waves, an accurate assessment of the ST segment is not possible.
ANSWER
The correct interpretation of this ECG is atrial flutter with a 2:1 block. Careful inspection of lead I reveals a P wave at the terminal portion of the QRS complex, in addition to the P wave seen with a consistent PR interval of 150 ms. This results in two P waves for each QRS complex. Given the presence of the flutter waves, an accurate assessment of the ST segment is not possible.
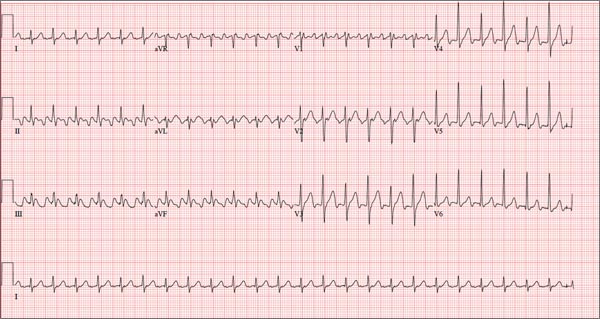
Three nights ago, a 44-year-old woman awoke with a regular, rapid heart rate that lasted about 15 minutes before abruptly terminating. The next morning, at the hospital where she works as an emergency department (ED) nurse, she had a colleague perform an undocumented ECG that, by the patient’s account, was normal. Early this morning, she was again awakened by a similar regular but rapid heart rate. Not wanting anyone at her facility to know about the problem, she presents to your ED instead. She denies chest pain but admits that she is slightly short of breath, adding that her symptoms remind her of how she feels when finishing a 10K run. The patient has been in excellent health with no underlying medical problems and no prior cardiac history. She is an avid runner, averaging three miles a day, and does not smoke. She does report drinking two or three glasses of wine in the evenings and admits she likes to party on the weekends, frequently consuming three or four margaritas with her coworkers on Saturday nights. She experimented with cannabis in college but hasn’t used illicit or recreational drugs since graduating. The patient is divorced, has a steady boyfriend, and has no children or siblings. Her parents are alive and well, with no history of arrhythmias, cardiovascular disease, or diabetes. She has no known drug allergies. Her medications include an oral contraceptive and occasional ibuprofen for soreness following exercise. The review of systems is remarkable for menses, which began yesterday. She denies that she is pregnant. Her vision is corrected with contact lenses. Physical examination reveals a thin, athletic-appearing woman who seems anxious. Her height is 67 in and her weight, 132 lb. Vital signs include a blood pressure of 118/68 mm Hg; pulse, 150 beats/min; respiratory rate, 14 breaths/min-1; and temperature, 37.8°C. The HEENT exam is normal with the presence of contact lenses. There is no thyromegaly. The lungs are clear in all fields. Her cardiac exam reveals a regular, rapid rate of 150 beats/min, without murmurs, rubs, or extra heart sounds. The abdomen is soft and nontender without palpable masses. The peripheral pulses are strong and equal bilaterally. There is no peripheral edema. The neurologic exam is intact. Laboratory tests, including a complete blood count, thyroid panel, and chemistry panel, are performed. All values are within normal limits. An ECG reveals a ventricular rate of 149 beats/min; PR interval, 150 ms; QRS interval, 102 ms; QT/QTc interval, 270/425 ms; P axis, 103°; R axis, 78°; and T axis, –18°. What is your interpretation of this ECG?
Left Arm Pain, Numbness, and Weakness
ANSWER
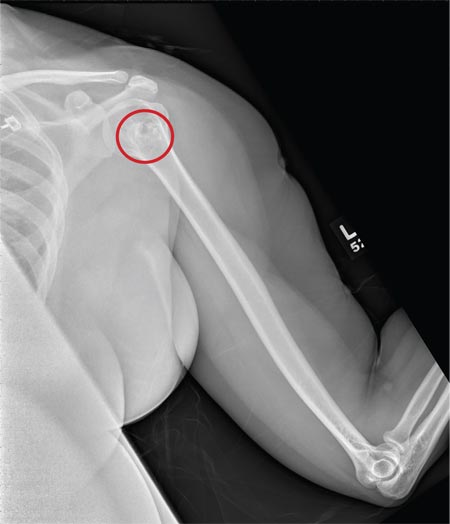
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
ANSWER
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
ANSWER
The radiograph shows no evidence of a fracture. However, there is a 2-cm focal sclerotic area noted within the juncture of the humeral neck and head. This finding could represent an enchondroma, a bone cyst, or a bone infarct. Additional imaging, including MRI and bone scan, is warranted, as is orthopedic evaluation. This finding is likely incidental, as the patient’s clinical exam is suggestive of a cervical radiculitis referable to the herniated disc in her neck.
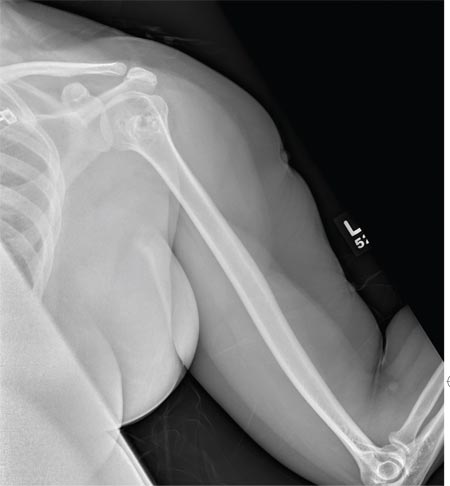
A 40-year-old woman presents to the urgent care clinic complaining of left arm pain with associated numbness and weakness. She denies any injury or trauma, adding that the pain manifested several months ago but has recently progressed. She has already undergone outpatient MRI of her neck; she was told she had some “herniated discs” and would need to see a specialist. Her medical history is significant for hypertension. On physical examination, the patient appears uncomfortable but in no obvious distress. Vital signs are normal. Tenderness is present at the left trapezius and the left shoulder. Mild weakness is present in the left arm; strength is 4/5 and grip strength, 3/5. Pulses are normal, and sensation is intact. Available medical records include a report from her recent MRI of the cervical spine. Findings include a moderate left-sided disc osteophyte at the C6-C7 level and resultant cervical stenosis. A radiograph of the left shoulder is obtained. What is your impression?
Lesions’ Pattern Helps Line Up Diagnosis
ANSWER
The correct answer is lichen nitidus (choice “b”), a harmless, self-limited condition of unknown origin. The lesions’ flat-topped (planar) surfaces and tendency to form in linear configurations along lines of trauma (so-called Koebner phenomenon) are also features seen in lichen planus (choice “d”) lesions; however, the latter are almost always pruritic and purple in color. Ironically, the histologic pattern seen in both is almost identical.
An extremely common condition, molluscum contagiosum (choice “a”) presents with multiple tiny papules. But these are not planar, and most will have an umbilicated center. See the Discussion for ways to distinguish it from lichen nitidus.
Flat warts (choice “c”), known as verruca plana, can strongly resemble lichen nitidus, but they are not as flat-topped and do not appear white. They do Koebnerize, however, which occasionally makes the distinction difficult.
DISCUSSION
Lichen nitidus (LN) is an unusual but benign condition primarily affecting children and young adults. Due to the contrast, the white planar papules are easier to see on darker skin. As is the case with many dermatologic diagnoses, LN is easily identified if you’ve heard of it and therefore know what to expect—but much more difficult if you haven’t.
LN’s unique manifestation distinguishes it from other items in the differential. For example, molluscum and LN can easily be confused, especially since both primarily affect children. But the pathognomic central umbilication of molluscum lesions is the key distinguishing feature; the best way to highlight it is with a short blast of liquid nitrogen. (Usually, though, the planar surfaces of LN are sufficient to distinguish it from other conditions.)
In the United States, the term Koebner phenomenon refers to the tendency for lesions to form along areas of trauma, usually in a linear configuration. All four items in our differential can present in this way. However, the term auto-inoculation might be more properly applied to conditions such as warts and molluscum, since the trauma has merely inoculated the organism into the skin. Inflammatory conditions such as LN and lichen planus are not truly “spread” by the trauma.
Linearly configured lesions are sufficiently unusual in dermatology to warrant their own differential. Among those that can present in this manner are psoriasis, lichen sclerosus et atrophicus, and vitiligo.
TREATMENT/PROGNOSIS
Our LN patient did not require any treatment, nor was any possible. The condition is quite likely to clear on its own, leaving little if any evidence in its wake.
I often show affected patients and/or their parents pictures of these types of conditions from our textbooks, for added reassurance. And in this day and age, I direct them to websites where they can do more investigation on their own time.
The effective practice of dermatology (and of all medicine, for that matter) includes more than merely making a correct diagnosis: I believe we’re obliged to “sell” it as well.
ANSWER
The correct answer is lichen nitidus (choice “b”), a harmless, self-limited condition of unknown origin. The lesions’ flat-topped (planar) surfaces and tendency to form in linear configurations along lines of trauma (so-called Koebner phenomenon) are also features seen in lichen planus (choice “d”) lesions; however, the latter are almost always pruritic and purple in color. Ironically, the histologic pattern seen in both is almost identical.
An extremely common condition, molluscum contagiosum (choice “a”) presents with multiple tiny papules. But these are not planar, and most will have an umbilicated center. See the Discussion for ways to distinguish it from lichen nitidus.
Flat warts (choice “c”), known as verruca plana, can strongly resemble lichen nitidus, but they are not as flat-topped and do not appear white. They do Koebnerize, however, which occasionally makes the distinction difficult.
DISCUSSION
Lichen nitidus (LN) is an unusual but benign condition primarily affecting children and young adults. Due to the contrast, the white planar papules are easier to see on darker skin. As is the case with many dermatologic diagnoses, LN is easily identified if you’ve heard of it and therefore know what to expect—but much more difficult if you haven’t.
LN’s unique manifestation distinguishes it from other items in the differential. For example, molluscum and LN can easily be confused, especially since both primarily affect children. But the pathognomic central umbilication of molluscum lesions is the key distinguishing feature; the best way to highlight it is with a short blast of liquid nitrogen. (Usually, though, the planar surfaces of LN are sufficient to distinguish it from other conditions.)
In the United States, the term Koebner phenomenon refers to the tendency for lesions to form along areas of trauma, usually in a linear configuration. All four items in our differential can present in this way. However, the term auto-inoculation might be more properly applied to conditions such as warts and molluscum, since the trauma has merely inoculated the organism into the skin. Inflammatory conditions such as LN and lichen planus are not truly “spread” by the trauma.
Linearly configured lesions are sufficiently unusual in dermatology to warrant their own differential. Among those that can present in this manner are psoriasis, lichen sclerosus et atrophicus, and vitiligo.
TREATMENT/PROGNOSIS
Our LN patient did not require any treatment, nor was any possible. The condition is quite likely to clear on its own, leaving little if any evidence in its wake.
I often show affected patients and/or their parents pictures of these types of conditions from our textbooks, for added reassurance. And in this day and age, I direct them to websites where they can do more investigation on their own time.
The effective practice of dermatology (and of all medicine, for that matter) includes more than merely making a correct diagnosis: I believe we’re obliged to “sell” it as well.
ANSWER
The correct answer is lichen nitidus (choice “b”), a harmless, self-limited condition of unknown origin. The lesions’ flat-topped (planar) surfaces and tendency to form in linear configurations along lines of trauma (so-called Koebner phenomenon) are also features seen in lichen planus (choice “d”) lesions; however, the latter are almost always pruritic and purple in color. Ironically, the histologic pattern seen in both is almost identical.
An extremely common condition, molluscum contagiosum (choice “a”) presents with multiple tiny papules. But these are not planar, and most will have an umbilicated center. See the Discussion for ways to distinguish it from lichen nitidus.
Flat warts (choice “c”), known as verruca plana, can strongly resemble lichen nitidus, but they are not as flat-topped and do not appear white. They do Koebnerize, however, which occasionally makes the distinction difficult.
DISCUSSION
Lichen nitidus (LN) is an unusual but benign condition primarily affecting children and young adults. Due to the contrast, the white planar papules are easier to see on darker skin. As is the case with many dermatologic diagnoses, LN is easily identified if you’ve heard of it and therefore know what to expect—but much more difficult if you haven’t.
LN’s unique manifestation distinguishes it from other items in the differential. For example, molluscum and LN can easily be confused, especially since both primarily affect children. But the pathognomic central umbilication of molluscum lesions is the key distinguishing feature; the best way to highlight it is with a short blast of liquid nitrogen. (Usually, though, the planar surfaces of LN are sufficient to distinguish it from other conditions.)
In the United States, the term Koebner phenomenon refers to the tendency for lesions to form along areas of trauma, usually in a linear configuration. All four items in our differential can present in this way. However, the term auto-inoculation might be more properly applied to conditions such as warts and molluscum, since the trauma has merely inoculated the organism into the skin. Inflammatory conditions such as LN and lichen planus are not truly “spread” by the trauma.
Linearly configured lesions are sufficiently unusual in dermatology to warrant their own differential. Among those that can present in this manner are psoriasis, lichen sclerosus et atrophicus, and vitiligo.
TREATMENT/PROGNOSIS
Our LN patient did not require any treatment, nor was any possible. The condition is quite likely to clear on its own, leaving little if any evidence in its wake.
I often show affected patients and/or their parents pictures of these types of conditions from our textbooks, for added reassurance. And in this day and age, I direct them to websites where they can do more investigation on their own time.
The effective practice of dermatology (and of all medicine, for that matter) includes more than merely making a correct diagnosis: I believe we’re obliged to “sell” it as well.
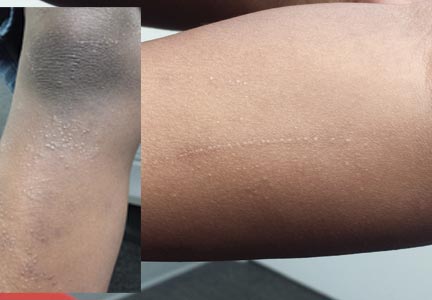
Six months ago, a 6-year-old boy developed asymptomatic lesions on his elbows, then his knees. They slowly spread to other areas, including his forearms. One primary care provider diagnosed probable warts; another, molluscum. The prescribed treatments—liquid nitrogen and tretinoin, respectively—had no effect. The boy’s mother became alarmed when the lesions started to form in long lines on his arms. At that point, she decided to bring him to dermatology for evaluation. Aside from his skin condition, the child is healthy, according to both his mother and the records provided by his primary care provider’s office. The lesions are particularly numerous over the extensor surfaces of the legs—especially the knees—but are also seen on the extensor forearms and elbows. The lesions are exquisitely discrete, identical, tiny white pinpoint papules, all with flat tops. None are umbilicated. In several areas of the arms, linear collections of lesions, some extending as long as 6 cm, are noted. The rest of his exposed type V skin is unremarkable.

Breaking down midurethral sling approaches
It has been nearly 20 years since the first minimally invasive midurethral sling was introduced. This development was followed 5 years later with the introduction of the transobturator midurethral sling. The advent of both ambulatory techniques has essentially changed the landscape in the surgical treatment of stress urinary incontinence; midurethral slings are certainly considered the procedure of choice for many women.
The midurethral sling has continued to evolve. Not only does the surgeon have the choice of placing a retropubic midurethral sling (bottom to top or top to bottom) and the transobturator midurethral sling (inside-out or outside-in), but, as of late, single incision midurethral slings (mini-slings or mini-tape) as well.

In the previous Master Class on urinary incontinence, Dr. Eric Sokol discussed issues of sling selection and the evidence in favor of various types of retropubic and transobturator slings. This month, we’ll discuss the technique behind these two approaches. I have elicited the assistance of Dr. Sokol, as well as Dr. Charles Rardin.
Dr. Sokol is an associate professor of obstetrics and gynecology, associate professor of urology (by courtesy), and cochief of the division of urogynecology and pelvic reconstructive surgery at Stanford (Calif.) University. He has published many articles regarding urogynecology and minimally invasive surgery. Dr. Sokol has been awarded numerous teaching awards, and he is a reviewer for multiple prestigious, peer-reviewed journals.
Dr. Rardin is the director of the robotic surgery program at Women & Infants Hospital of Rhode Island, Providence, a surgeon in the division of urogynecology and reconstructive pelvic surgery, and is the director of the hospital’s fellowship in urogynecology and reconstructive pelvic surgery. He is also an assistant professor at Brown University, also in Providence. He has published numerous articles in peer-reviewed journals.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, immediate past president of the International Society for Gynecologic Endoscopy (ISGE), and a past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville, Ill., and Schaumburg, Ill.; the director of minimally invasive gynecologic surgery and the director of the AAGL/SRS fellowship in minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. Dr. Miller is a consultant and on the speaker’s bureau for Ethicon.
It has been nearly 20 years since the first minimally invasive midurethral sling was introduced. This development was followed 5 years later with the introduction of the transobturator midurethral sling. The advent of both ambulatory techniques has essentially changed the landscape in the surgical treatment of stress urinary incontinence; midurethral slings are certainly considered the procedure of choice for many women.
The midurethral sling has continued to evolve. Not only does the surgeon have the choice of placing a retropubic midurethral sling (bottom to top or top to bottom) and the transobturator midurethral sling (inside-out or outside-in), but, as of late, single incision midurethral slings (mini-slings or mini-tape) as well.
In the previous Master Class on urinary incontinence, Dr. Eric Sokol discussed issues of sling selection and the evidence in favor of various types of retropubic and transobturator slings. This month, we’ll discuss the technique behind these two approaches. I have elicited the assistance of Dr. Sokol, as well as Dr. Charles Rardin.
Dr. Sokol is an associate professor of obstetrics and gynecology, associate professor of urology (by courtesy), and cochief of the division of urogynecology and pelvic reconstructive surgery at Stanford (Calif.) University. He has published many articles regarding urogynecology and minimally invasive surgery. Dr. Sokol has been awarded numerous teaching awards, and he is a reviewer for multiple prestigious, peer-reviewed journals.
Dr. Rardin is the director of the robotic surgery program at Women & Infants Hospital of Rhode Island, Providence, a surgeon in the division of urogynecology and reconstructive pelvic surgery, and is the director of the hospital’s fellowship in urogynecology and reconstructive pelvic surgery. He is also an assistant professor at Brown University, also in Providence. He has published numerous articles in peer-reviewed journals.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, immediate past president of the International Society for Gynecologic Endoscopy (ISGE), and a past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville, Ill., and Schaumburg, Ill.; the director of minimally invasive gynecologic surgery and the director of the AAGL/SRS fellowship in minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. Dr. Miller is a consultant and on the speaker’s bureau for Ethicon.
It has been nearly 20 years since the first minimally invasive midurethral sling was introduced. This development was followed 5 years later with the introduction of the transobturator midurethral sling. The advent of both ambulatory techniques has essentially changed the landscape in the surgical treatment of stress urinary incontinence; midurethral slings are certainly considered the procedure of choice for many women.
The midurethral sling has continued to evolve. Not only does the surgeon have the choice of placing a retropubic midurethral sling (bottom to top or top to bottom) and the transobturator midurethral sling (inside-out or outside-in), but, as of late, single incision midurethral slings (mini-slings or mini-tape) as well.
In the previous Master Class on urinary incontinence, Dr. Eric Sokol discussed issues of sling selection and the evidence in favor of various types of retropubic and transobturator slings. This month, we’ll discuss the technique behind these two approaches. I have elicited the assistance of Dr. Sokol, as well as Dr. Charles Rardin.
Dr. Sokol is an associate professor of obstetrics and gynecology, associate professor of urology (by courtesy), and cochief of the division of urogynecology and pelvic reconstructive surgery at Stanford (Calif.) University. He has published many articles regarding urogynecology and minimally invasive surgery. Dr. Sokol has been awarded numerous teaching awards, and he is a reviewer for multiple prestigious, peer-reviewed journals.
Dr. Rardin is the director of the robotic surgery program at Women & Infants Hospital of Rhode Island, Providence, a surgeon in the division of urogynecology and reconstructive pelvic surgery, and is the director of the hospital’s fellowship in urogynecology and reconstructive pelvic surgery. He is also an assistant professor at Brown University, also in Providence. He has published numerous articles in peer-reviewed journals.
Dr. Miller is clinical associate professor at the University of Illinois at Chicago, immediate past president of the International Society for Gynecologic Endoscopy (ISGE), and a past president of the AAGL. He is a reproductive endocrinologist and minimally invasive gynecologic surgeon in private practice in Naperville, Ill., and Schaumburg, Ill.; the director of minimally invasive gynecologic surgery and the director of the AAGL/SRS fellowship in minimally invasive gynecologic surgery at Advocate Lutheran General Hospital, Park Ridge, Ill.; and the medical editor of this column, Master Class. Dr. Miller is a consultant and on the speaker’s bureau for Ethicon.

Expert tips on retropubic vs. transobturator sling approaches
Midurethral slings – both retropubic and transobturator – have been extensively studied and have evolved to become standard therapies for the treatment of stress urinary incontinence. The two approaches utilize different routes for sling delivery, but in many other respects, they are similar. Improvements in technique are continually being developed. In this column, Dr. Sokol and Dr. Rardin share key parts of their technique and give their pearls of advice for midurethral sling surgery.
Dr. Sokol’s retropubic approach
I use newer retropubic midurethral slings with smaller trocars that have evolved from first-generation tension-free vaginal tape (TVT) slings. The slings I prefer are placed in a bottom-up fashion, with curved needles passed from a small vaginal incision up through the retropubic space to exit through two suprapubic incisions.
I find it helpful to place patients in a high lithotomy position with the legs supported in candy cane stirrups rather than Allen-type stirrups; sling placement is a short procedure with minimal to no risk of neuropathy.

To precisely identify the midurethral point, I use a 16-FR Foley catheter. When the catheter balloon is filled with 10 mm of fluid and gently pulled back, the urethrovesical junction can be identified. Then, by looking at the urethral meatus relative to the bladder neck, I can mark the midpoint between the two.
The suprapubic exit points are marked at two finger-widths lateral to the midline, just above the pubic symphysis. For precise identification of these points, the Foley catheter may be pulled up exactly midline (with the collection bag detached), and the two finger-widths measured on either side. I also aim for the ipsilateral shoulder, imagining a straight line from the urethral meatus to the ipsilateral shoulder on each side. Together, these measurements and visual cues serve as a good safety check.
With two Allis clamps, the vaginal wall on either side of the midline is grasped transversely at the level of the midurethral mark. The clamps will sit a couple of centimeters apart so that the midurethral point can be visualized. This helps to stabilize and elevate the midurethra.
To safely and efficiently develop a paraurethral passage, I perform a hydrodissection and hemostatic injection at the level of the midurethra using a control top 10-cc syringe with a 22-gauge needle. I find that dilute vasopressin saline solution affords better hemostasis than does a dilute lidocaine epinephrine solution, though I use dilute lidocaine if the sling is being done under local anesthesia.
The needle is inserted into a full thickness of skin, to a point shy of the urethra, and 10 cc is rapidly injected. The vaginal epithelium will appear blanched and will balloon out, like a white marble. The process basically lifts the vaginal skin away from the urethra itself, not only creating hemostasis but also providing a zone of safety to help avoid a urethral injury.
With a second syringe identical to the first, I inject 5 cc on each side of the midurethral point, aiming precisely at the underside of the pubic bone toward the ipsilateral shoulder. This creates a hydrodissected tunnel around each side of the midurethra. With a final syringe, I then inject 5 cc on each side suprapubically at my marked exit points.
With a #15 blade scalpel, I make two very small “poke” incisions transversely at the suprapubic sites. The suburethral incision is larger – just over a centimeter – and is made through a full thickness of skin under the area of hydrodissection, but not so deep as to injure the urethra. To finish development of the paraurethral passage, I pass standard Metzenbaum scissors through each hydrodissected tunnel until I feel the underside of the pubic bone, but no further.
For sling placement (after ensuring the bladder is completely empty), I lower the table such that my arm will be at a right angle to pass the sling while standing.
With my index finger underneath the pubic bone, the trocar tip, with the attached sling, is advanced with my thumb directly toward the ipsilateral shoulder just until it pops through the retropubic space. The depth of the trocar tip can be palpated with the index finger of the same hand, which is positioned just below the pelvic bone.
After the sling “pops” into the retropubic space, I remove my hand from the vagina and place it on the abdominal wall at the ipsilateral suprapubic poke site. In one smooth pass, I hug the pubic bone and advance the sling, again aiming directly and consistently at the shoulder. The trocar handle stays steady, never deviating in any direction. Cystoscopy is performed after the sling is placed on both sides to ensure bladder and urethral integrity.
For tensioning, I raise the table back up and, after reinserting the Foley catheter and a Sims retractor, I place my finger in the middle of the sling and pull the suprapubic ends of the sling up until my finger rests right under the urethra.
I then remove the vaginal clamps and use Metzenbaum scissors as a spacer between the sling and the urethra. With the scissors parallel to and right under the Foley catheter, at the same angle as the urethra, I tighten the sling and remove the plastic sheaths.
Dr. Sokol’s transobturator (TOT) approach
I most often use an outside-in sling. I utilize the same patient positioning and identify the midurethral point in the same way as with the retropubic approach.
On the thigh, I identify the adductor longus tendon as well as a little soft spot or depression just beneath the tendon and lateral to the descending ischial pubic ramus. With my thumb on the soft spot, I can actually grasp the adductor longus tendon between my thumb and index finger. This spot, which is also approximately at the level of the clitoris, marks the entry point for sling placement. It is the thinnest point between the groin and the vagina at the level of the midurethra.
I perform a similar hydrodissection under the urethra as I do in a retropubic procedure, though instead of injecting 5 cc’s to the underside of the pubic symphysis on each side, I instead inject toward the obturator internus muscles. I then inject my final syringe of dilute vasopressin saline solution at the groin poke incision sites, directed toward the projected trocar path, as opposed to suprapubically.
After the full-thickness vaginal incision is made with the scalpel, the dissection is performed sharply with Metzenbaum scissors and is more like the dissection done for cystocele repair than for a retropubic sling. Rather than a poke, the midurethral incision is long enough – about 1.5 cm – for me to reach a finger behind the obturator internus muscle after having sharply dissected the suburethral tissue and fascia. The angle of the dissection is more lateral than for a retropubic sling, toward the underside of the descending ischiopubic ramus and obturator internus muscle.
To place the sling, I have one hand with an index finger in the midurethral tunnel under the obturator internus muscle to protect the urethra. The thumb of that same hand is used to push the helical trocar straight through the thigh poke incision with the handle starting at a 35-degree angle from vertical. The trocar tip is pushed until it can no longer go straight and is ready to be tightly turned around the descending ischiopubic ramus with the opposite hand. A distinct pop can be felt as the trocar tip advances through the obturator membrane and muscles. As the tip is advanced, the angle of the trocar is rotated from 35 degrees to vertical, almost perpendicular to the floor. At this point, the tip of the trocar should be guided out of the midurethral tunnel against the opposite index finger.
I utilize the same technique for tensioning a TOT sling as I do the retropubic sling.
Dr. Rardin’s retropubic approach
I continue to use the original TVT sling with a 5-mm stainless steel, mechanically cut trocar and reusable handle. The newer slender needles may advance with less pressure, but I worry about them bending during passage. I feel more assured and comfortable using the older trocars.
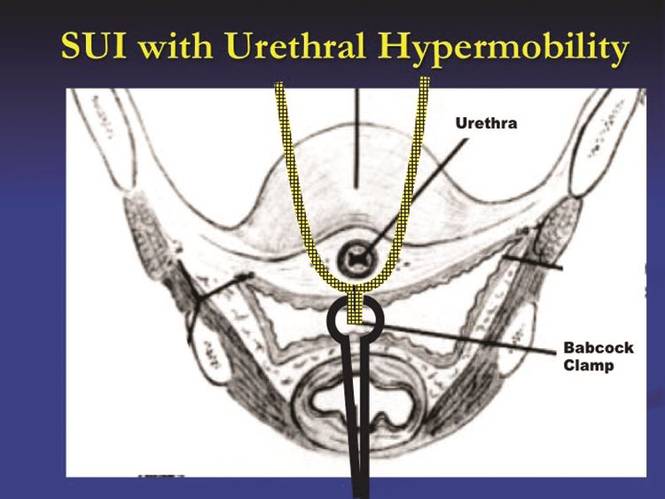
I perform retropubic hydrodissection with a spinal needle using a top-down approach. With 40 cc of a very dilute solution of bupivacaine (Marcaine) with epinephrine on each side of the urethra, I create columns of hydrodissected space. Studies are inconsistent about the benefits of hydrodissection, but theoretically, it decreases the risk of bladder injury by pushing the bladder away from the pubic bone, creates effective hemostasis, and can provide analgesia that will be on board when the patient wakes up.
I bring the spinal needle down behind the pubic bone to the location of the urethral incision site, with my finger in the vagina, so I can feel the tip of the needle next to the urethra/Foley catheter. For each side, I will inject 20 cc of the solution in this location, and the other 20 cc as I withdraw the needle upward.
For trocar passage, some surgeons are taught to advance the trocar until a pop is felt, then drop the handle and push upward. I view the maneuver as a consistent, smooth arch; for every degree that I advance the trocar, I drop the handle slightly in order to maintain contact with the back of the pubic bone throughout the pass. I continuously and simultaneously drop the handle and advance the trocar. Contact with the back of the pubic bone is maintained with a slight pulling on the back end of the trocar handle, while the forward hand pushes the trocar upward.
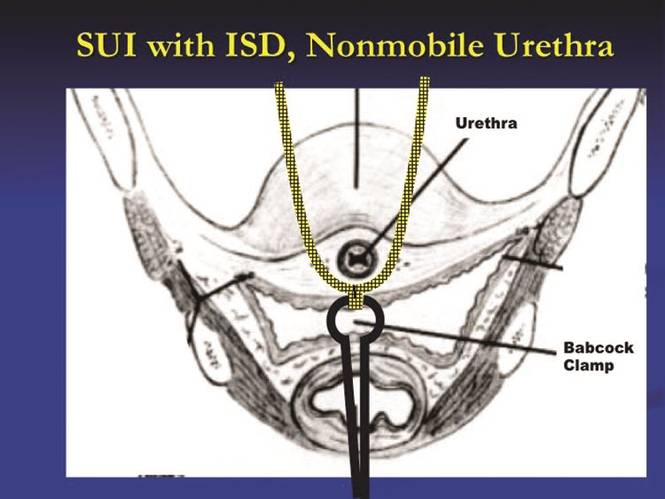
The target that I visualize for a retropubic pass is the patient’s ipsilateral shoulder. A cadaver study showed that if you aim as far lateral as the patient’s outstretched elbow, you can enter the iliac vasculature (Obstet. Gynecol. 2003;101:933-6).
If the patient is draped such that I cannot see the ipsilateral shoulder, I ask the anesthesia team to show me. I have also identified and marked the suprapubic points about 2-3 cm from each other on either side of the midline just about the pubic symphysis, but I consider the broader anatomic picture and purposeful visualization toward the ipsilateral shoulder to be an essential part of safe technique. In general, it is safer to be more medial than more lateral for the needle passage.
I continue to use a rigid catheter guide to deflect the bladder neck while passing the needles.

Cystoscopy is performed after both needles are placed but not yet pulled through. I fill the bladder until the ureteral orifices appear flattened, which confirms that the bladder is under enough distension to preclude any mucosal wrinkles.
The technique I utilize for adjusting the tension of the TVT sling was taught to me by Dr. Peter L. Rosenblatt of Mt. Auburn Hospital in Cambridge, Mass. At the midline of the sling, I advance the sheaths just enough so that I can grasp a 2-3 mm “knuckle” of the midportion of the sling with a Babcock clamp. I then pull the sling ends until the Babcock comes into gentle contact with the suburethral tissue. The sheaths encasing the sling are then removed and the Babcock clamp is released to assure a tension-free deployment.
The amount of tape that is pinched with the Babcock – the size of the “knuckle” – determines the tension. For a patient with a more profound problem, such as intrinsic sphincter deficiency or a lack or urethral hypermobility, I will grasp a smaller knuckle.
These steps ensure that the midportion of the tape will not tighten or become deformed under tension. Rather than use a spacer, I like to protect the midportion of the tape and prevent it from being stretched. I find that the approach is reproducible and results in a reliable amount of space between the urethra and sling when the procedure is completed.
Dr. Rardin’s TOT approach
I employ an inside-out technique to the TOT procedure, and I utilize devices with segment of mesh that is shorter – only about 13 cm in length – than the original full-length mesh used in many TOT procedures. Once placed, the ends of the mesh penetrate the obturator membrane and obturator externus but not the adductor compartment of the thigh and groin.
In a study we presented last year at the annual meeting of the Society of Gynecologic Surgeons meeting, the shortened tape reduced postoperative groin pain, compared with full-length TOT tape without any reduction in subjective benefit. It appears that with shortened tape, we are anchoring the sling in tissues that provide critical support while avoiding the muscles that relate to the inner thigh/groin pain experienced by some patients. Effectiveness was not reduced, compared with full-length TOT slings.
These shortened slings are distinct from a single-incision sling, which is basically pushed into place. We still pass the needle all the way through a vaginal incision and out through the obturator foramen, and we pull the sling into place as we would any other TOT sling. The difference is that we’re not leaving any mesh in the groin.
I prefer an inside-out approach for two reasons: I always feel that I have more control over where a needle enters than where it exits, and precision is important with suburethral slings. Secondly, the dissection tunnel created for an inside-out pass is much smaller than the tunnel that must be dissected for an outside-in approach. In theory, less dissection means less devascularization, less denervation, and less opportunity for erosion.
Hydrodissection for TOT slings is more minimal and involves less fluid than does hydrodissection for retropubic slings, mainly because we do not want to anesthetize the obturator nerve. I pass the spinal needle from the outside in. At the start, prior to making any incisions, it is important to identify the arcus tendineus, a linear thickening of the superior fascia that is sometimes called the “white line.” This is where the sulcus is affixed to the sidewall. I will be sure to penetrate the sidewall at or above the level of the arcus.
With the TOT approach, the likelihood of bladder injury is so low that I usually drive the trocars all the way through prior to cystoscopy, as opposed to leaving the needles in place as I do with the retropubic approach.
Tensioning is achieved in the same manner, by using the Babcock clamp to avoid distortion of the critical part of the mesh while creating the space needed given the patient’s clinical scenario. It is worth remembering, at this point, that overall risks for retention appear to be lower for obturator slings, compared with retropubic slings.
I place most of my patients receiving retropubic slings in dorsal lithotomy position; but for obturator sling placement, I favor a few degrees into higher lithotomy because this pulls the obturator neurovascular bundle a little further out of the path of the needle.
Dr. Sokol reported that he owns stock in Pelvalon and is a clinical adviser to that company. He also is a national principal investigator for American Medical Systems and the recipient of research grants from Acell and several other companies. Dr. Rardin reported that he has no relevant financial disclosures. To view a video related to this article, go to SurgeryU at aagl.org/obgyn-news.
Midurethral slings – both retropubic and transobturator – have been extensively studied and have evolved to become standard therapies for the treatment of stress urinary incontinence. The two approaches utilize different routes for sling delivery, but in many other respects, they are similar. Improvements in technique are continually being developed. In this column, Dr. Sokol and Dr. Rardin share key parts of their technique and give their pearls of advice for midurethral sling surgery.
Dr. Sokol’s retropubic approach
I use newer retropubic midurethral slings with smaller trocars that have evolved from first-generation tension-free vaginal tape (TVT) slings. The slings I prefer are placed in a bottom-up fashion, with curved needles passed from a small vaginal incision up through the retropubic space to exit through two suprapubic incisions.
I find it helpful to place patients in a high lithotomy position with the legs supported in candy cane stirrups rather than Allen-type stirrups; sling placement is a short procedure with minimal to no risk of neuropathy.
To precisely identify the midurethral point, I use a 16-FR Foley catheter. When the catheter balloon is filled with 10 mm of fluid and gently pulled back, the urethrovesical junction can be identified. Then, by looking at the urethral meatus relative to the bladder neck, I can mark the midpoint between the two.
The suprapubic exit points are marked at two finger-widths lateral to the midline, just above the pubic symphysis. For precise identification of these points, the Foley catheter may be pulled up exactly midline (with the collection bag detached), and the two finger-widths measured on either side. I also aim for the ipsilateral shoulder, imagining a straight line from the urethral meatus to the ipsilateral shoulder on each side. Together, these measurements and visual cues serve as a good safety check.
With two Allis clamps, the vaginal wall on either side of the midline is grasped transversely at the level of the midurethral mark. The clamps will sit a couple of centimeters apart so that the midurethral point can be visualized. This helps to stabilize and elevate the midurethra.
To safely and efficiently develop a paraurethral passage, I perform a hydrodissection and hemostatic injection at the level of the midurethra using a control top 10-cc syringe with a 22-gauge needle. I find that dilute vasopressin saline solution affords better hemostasis than does a dilute lidocaine epinephrine solution, though I use dilute lidocaine if the sling is being done under local anesthesia.
The needle is inserted into a full thickness of skin, to a point shy of the urethra, and 10 cc is rapidly injected. The vaginal epithelium will appear blanched and will balloon out, like a white marble. The process basically lifts the vaginal skin away from the urethra itself, not only creating hemostasis but also providing a zone of safety to help avoid a urethral injury.
With a second syringe identical to the first, I inject 5 cc on each side of the midurethral point, aiming precisely at the underside of the pubic bone toward the ipsilateral shoulder. This creates a hydrodissected tunnel around each side of the midurethra. With a final syringe, I then inject 5 cc on each side suprapubically at my marked exit points.
With a #15 blade scalpel, I make two very small “poke” incisions transversely at the suprapubic sites. The suburethral incision is larger – just over a centimeter – and is made through a full thickness of skin under the area of hydrodissection, but not so deep as to injure the urethra. To finish development of the paraurethral passage, I pass standard Metzenbaum scissors through each hydrodissected tunnel until I feel the underside of the pubic bone, but no further.
For sling placement (after ensuring the bladder is completely empty), I lower the table such that my arm will be at a right angle to pass the sling while standing.
With my index finger underneath the pubic bone, the trocar tip, with the attached sling, is advanced with my thumb directly toward the ipsilateral shoulder just until it pops through the retropubic space. The depth of the trocar tip can be palpated with the index finger of the same hand, which is positioned just below the pelvic bone.
After the sling “pops” into the retropubic space, I remove my hand from the vagina and place it on the abdominal wall at the ipsilateral suprapubic poke site. In one smooth pass, I hug the pubic bone and advance the sling, again aiming directly and consistently at the shoulder. The trocar handle stays steady, never deviating in any direction. Cystoscopy is performed after the sling is placed on both sides to ensure bladder and urethral integrity.
For tensioning, I raise the table back up and, after reinserting the Foley catheter and a Sims retractor, I place my finger in the middle of the sling and pull the suprapubic ends of the sling up until my finger rests right under the urethra.
I then remove the vaginal clamps and use Metzenbaum scissors as a spacer between the sling and the urethra. With the scissors parallel to and right under the Foley catheter, at the same angle as the urethra, I tighten the sling and remove the plastic sheaths.
Dr. Sokol’s transobturator (TOT) approach
I most often use an outside-in sling. I utilize the same patient positioning and identify the midurethral point in the same way as with the retropubic approach.
On the thigh, I identify the adductor longus tendon as well as a little soft spot or depression just beneath the tendon and lateral to the descending ischial pubic ramus. With my thumb on the soft spot, I can actually grasp the adductor longus tendon between my thumb and index finger. This spot, which is also approximately at the level of the clitoris, marks the entry point for sling placement. It is the thinnest point between the groin and the vagina at the level of the midurethra.
I perform a similar hydrodissection under the urethra as I do in a retropubic procedure, though instead of injecting 5 cc’s to the underside of the pubic symphysis on each side, I instead inject toward the obturator internus muscles. I then inject my final syringe of dilute vasopressin saline solution at the groin poke incision sites, directed toward the projected trocar path, as opposed to suprapubically.
After the full-thickness vaginal incision is made with the scalpel, the dissection is performed sharply with Metzenbaum scissors and is more like the dissection done for cystocele repair than for a retropubic sling. Rather than a poke, the midurethral incision is long enough – about 1.5 cm – for me to reach a finger behind the obturator internus muscle after having sharply dissected the suburethral tissue and fascia. The angle of the dissection is more lateral than for a retropubic sling, toward the underside of the descending ischiopubic ramus and obturator internus muscle.
To place the sling, I have one hand with an index finger in the midurethral tunnel under the obturator internus muscle to protect the urethra. The thumb of that same hand is used to push the helical trocar straight through the thigh poke incision with the handle starting at a 35-degree angle from vertical. The trocar tip is pushed until it can no longer go straight and is ready to be tightly turned around the descending ischiopubic ramus with the opposite hand. A distinct pop can be felt as the trocar tip advances through the obturator membrane and muscles. As the tip is advanced, the angle of the trocar is rotated from 35 degrees to vertical, almost perpendicular to the floor. At this point, the tip of the trocar should be guided out of the midurethral tunnel against the opposite index finger.
I utilize the same technique for tensioning a TOT sling as I do the retropubic sling.
Dr. Rardin’s retropubic approach
I continue to use the original TVT sling with a 5-mm stainless steel, mechanically cut trocar and reusable handle. The newer slender needles may advance with less pressure, but I worry about them bending during passage. I feel more assured and comfortable using the older trocars.
I perform retropubic hydrodissection with a spinal needle using a top-down approach. With 40 cc of a very dilute solution of bupivacaine (Marcaine) with epinephrine on each side of the urethra, I create columns of hydrodissected space. Studies are inconsistent about the benefits of hydrodissection, but theoretically, it decreases the risk of bladder injury by pushing the bladder away from the pubic bone, creates effective hemostasis, and can provide analgesia that will be on board when the patient wakes up.
I bring the spinal needle down behind the pubic bone to the location of the urethral incision site, with my finger in the vagina, so I can feel the tip of the needle next to the urethra/Foley catheter. For each side, I will inject 20 cc of the solution in this location, and the other 20 cc as I withdraw the needle upward.
For trocar passage, some surgeons are taught to advance the trocar until a pop is felt, then drop the handle and push upward. I view the maneuver as a consistent, smooth arch; for every degree that I advance the trocar, I drop the handle slightly in order to maintain contact with the back of the pubic bone throughout the pass. I continuously and simultaneously drop the handle and advance the trocar. Contact with the back of the pubic bone is maintained with a slight pulling on the back end of the trocar handle, while the forward hand pushes the trocar upward.
The target that I visualize for a retropubic pass is the patient’s ipsilateral shoulder. A cadaver study showed that if you aim as far lateral as the patient’s outstretched elbow, you can enter the iliac vasculature (Obstet. Gynecol. 2003;101:933-6).
If the patient is draped such that I cannot see the ipsilateral shoulder, I ask the anesthesia team to show me. I have also identified and marked the suprapubic points about 2-3 cm from each other on either side of the midline just about the pubic symphysis, but I consider the broader anatomic picture and purposeful visualization toward the ipsilateral shoulder to be an essential part of safe technique. In general, it is safer to be more medial than more lateral for the needle passage.
I continue to use a rigid catheter guide to deflect the bladder neck while passing the needles.
Cystoscopy is performed after both needles are placed but not yet pulled through. I fill the bladder until the ureteral orifices appear flattened, which confirms that the bladder is under enough distension to preclude any mucosal wrinkles.
The technique I utilize for adjusting the tension of the TVT sling was taught to me by Dr. Peter L. Rosenblatt of Mt. Auburn Hospital in Cambridge, Mass. At the midline of the sling, I advance the sheaths just enough so that I can grasp a 2-3 mm “knuckle” of the midportion of the sling with a Babcock clamp. I then pull the sling ends until the Babcock comes into gentle contact with the suburethral tissue. The sheaths encasing the sling are then removed and the Babcock clamp is released to assure a tension-free deployment.
The amount of tape that is pinched with the Babcock – the size of the “knuckle” – determines the tension. For a patient with a more profound problem, such as intrinsic sphincter deficiency or a lack or urethral hypermobility, I will grasp a smaller knuckle.
These steps ensure that the midportion of the tape will not tighten or become deformed under tension. Rather than use a spacer, I like to protect the midportion of the tape and prevent it from being stretched. I find that the approach is reproducible and results in a reliable amount of space between the urethra and sling when the procedure is completed.
Dr. Rardin’s TOT approach
I employ an inside-out technique to the TOT procedure, and I utilize devices with segment of mesh that is shorter – only about 13 cm in length – than the original full-length mesh used in many TOT procedures. Once placed, the ends of the mesh penetrate the obturator membrane and obturator externus but not the adductor compartment of the thigh and groin.
In a study we presented last year at the annual meeting of the Society of Gynecologic Surgeons meeting, the shortened tape reduced postoperative groin pain, compared with full-length TOT tape without any reduction in subjective benefit. It appears that with shortened tape, we are anchoring the sling in tissues that provide critical support while avoiding the muscles that relate to the inner thigh/groin pain experienced by some patients. Effectiveness was not reduced, compared with full-length TOT slings.
These shortened slings are distinct from a single-incision sling, which is basically pushed into place. We still pass the needle all the way through a vaginal incision and out through the obturator foramen, and we pull the sling into place as we would any other TOT sling. The difference is that we’re not leaving any mesh in the groin.
I prefer an inside-out approach for two reasons: I always feel that I have more control over where a needle enters than where it exits, and precision is important with suburethral slings. Secondly, the dissection tunnel created for an inside-out pass is much smaller than the tunnel that must be dissected for an outside-in approach. In theory, less dissection means less devascularization, less denervation, and less opportunity for erosion.
Hydrodissection for TOT slings is more minimal and involves less fluid than does hydrodissection for retropubic slings, mainly because we do not want to anesthetize the obturator nerve. I pass the spinal needle from the outside in. At the start, prior to making any incisions, it is important to identify the arcus tendineus, a linear thickening of the superior fascia that is sometimes called the “white line.” This is where the sulcus is affixed to the sidewall. I will be sure to penetrate the sidewall at or above the level of the arcus.
With the TOT approach, the likelihood of bladder injury is so low that I usually drive the trocars all the way through prior to cystoscopy, as opposed to leaving the needles in place as I do with the retropubic approach.
Tensioning is achieved in the same manner, by using the Babcock clamp to avoid distortion of the critical part of the mesh while creating the space needed given the patient’s clinical scenario. It is worth remembering, at this point, that overall risks for retention appear to be lower for obturator slings, compared with retropubic slings.
I place most of my patients receiving retropubic slings in dorsal lithotomy position; but for obturator sling placement, I favor a few degrees into higher lithotomy because this pulls the obturator neurovascular bundle a little further out of the path of the needle.
Dr. Sokol reported that he owns stock in Pelvalon and is a clinical adviser to that company. He also is a national principal investigator for American Medical Systems and the recipient of research grants from Acell and several other companies. Dr. Rardin reported that he has no relevant financial disclosures. To view a video related to this article, go to SurgeryU at aagl.org/obgyn-news.
Midurethral slings – both retropubic and transobturator – have been extensively studied and have evolved to become standard therapies for the treatment of stress urinary incontinence. The two approaches utilize different routes for sling delivery, but in many other respects, they are similar. Improvements in technique are continually being developed. In this column, Dr. Sokol and Dr. Rardin share key parts of their technique and give their pearls of advice for midurethral sling surgery.
Dr. Sokol’s retropubic approach
I use newer retropubic midurethral slings with smaller trocars that have evolved from first-generation tension-free vaginal tape (TVT) slings. The slings I prefer are placed in a bottom-up fashion, with curved needles passed from a small vaginal incision up through the retropubic space to exit through two suprapubic incisions.
I find it helpful to place patients in a high lithotomy position with the legs supported in candy cane stirrups rather than Allen-type stirrups; sling placement is a short procedure with minimal to no risk of neuropathy.
To precisely identify the midurethral point, I use a 16-FR Foley catheter. When the catheter balloon is filled with 10 mm of fluid and gently pulled back, the urethrovesical junction can be identified. Then, by looking at the urethral meatus relative to the bladder neck, I can mark the midpoint between the two.
The suprapubic exit points are marked at two finger-widths lateral to the midline, just above the pubic symphysis. For precise identification of these points, the Foley catheter may be pulled up exactly midline (with the collection bag detached), and the two finger-widths measured on either side. I also aim for the ipsilateral shoulder, imagining a straight line from the urethral meatus to the ipsilateral shoulder on each side. Together, these measurements and visual cues serve as a good safety check.
With two Allis clamps, the vaginal wall on either side of the midline is grasped transversely at the level of the midurethral mark. The clamps will sit a couple of centimeters apart so that the midurethral point can be visualized. This helps to stabilize and elevate the midurethra.
To safely and efficiently develop a paraurethral passage, I perform a hydrodissection and hemostatic injection at the level of the midurethra using a control top 10-cc syringe with a 22-gauge needle. I find that dilute vasopressin saline solution affords better hemostasis than does a dilute lidocaine epinephrine solution, though I use dilute lidocaine if the sling is being done under local anesthesia.
The needle is inserted into a full thickness of skin, to a point shy of the urethra, and 10 cc is rapidly injected. The vaginal epithelium will appear blanched and will balloon out, like a white marble. The process basically lifts the vaginal skin away from the urethra itself, not only creating hemostasis but also providing a zone of safety to help avoid a urethral injury.
With a second syringe identical to the first, I inject 5 cc on each side of the midurethral point, aiming precisely at the underside of the pubic bone toward the ipsilateral shoulder. This creates a hydrodissected tunnel around each side of the midurethra. With a final syringe, I then inject 5 cc on each side suprapubically at my marked exit points.
With a #15 blade scalpel, I make two very small “poke” incisions transversely at the suprapubic sites. The suburethral incision is larger – just over a centimeter – and is made through a full thickness of skin under the area of hydrodissection, but not so deep as to injure the urethra. To finish development of the paraurethral passage, I pass standard Metzenbaum scissors through each hydrodissected tunnel until I feel the underside of the pubic bone, but no further.
For sling placement (after ensuring the bladder is completely empty), I lower the table such that my arm will be at a right angle to pass the sling while standing.
With my index finger underneath the pubic bone, the trocar tip, with the attached sling, is advanced with my thumb directly toward the ipsilateral shoulder just until it pops through the retropubic space. The depth of the trocar tip can be palpated with the index finger of the same hand, which is positioned just below the pelvic bone.
After the sling “pops” into the retropubic space, I remove my hand from the vagina and place it on the abdominal wall at the ipsilateral suprapubic poke site. In one smooth pass, I hug the pubic bone and advance the sling, again aiming directly and consistently at the shoulder. The trocar handle stays steady, never deviating in any direction. Cystoscopy is performed after the sling is placed on both sides to ensure bladder and urethral integrity.
For tensioning, I raise the table back up and, after reinserting the Foley catheter and a Sims retractor, I place my finger in the middle of the sling and pull the suprapubic ends of the sling up until my finger rests right under the urethra.
I then remove the vaginal clamps and use Metzenbaum scissors as a spacer between the sling and the urethra. With the scissors parallel to and right under the Foley catheter, at the same angle as the urethra, I tighten the sling and remove the plastic sheaths.
Dr. Sokol’s transobturator (TOT) approach
I most often use an outside-in sling. I utilize the same patient positioning and identify the midurethral point in the same way as with the retropubic approach.
On the thigh, I identify the adductor longus tendon as well as a little soft spot or depression just beneath the tendon and lateral to the descending ischial pubic ramus. With my thumb on the soft spot, I can actually grasp the adductor longus tendon between my thumb and index finger. This spot, which is also approximately at the level of the clitoris, marks the entry point for sling placement. It is the thinnest point between the groin and the vagina at the level of the midurethra.
I perform a similar hydrodissection under the urethra as I do in a retropubic procedure, though instead of injecting 5 cc’s to the underside of the pubic symphysis on each side, I instead inject toward the obturator internus muscles. I then inject my final syringe of dilute vasopressin saline solution at the groin poke incision sites, directed toward the projected trocar path, as opposed to suprapubically.
After the full-thickness vaginal incision is made with the scalpel, the dissection is performed sharply with Metzenbaum scissors and is more like the dissection done for cystocele repair than for a retropubic sling. Rather than a poke, the midurethral incision is long enough – about 1.5 cm – for me to reach a finger behind the obturator internus muscle after having sharply dissected the suburethral tissue and fascia. The angle of the dissection is more lateral than for a retropubic sling, toward the underside of the descending ischiopubic ramus and obturator internus muscle.
To place the sling, I have one hand with an index finger in the midurethral tunnel under the obturator internus muscle to protect the urethra. The thumb of that same hand is used to push the helical trocar straight through the thigh poke incision with the handle starting at a 35-degree angle from vertical. The trocar tip is pushed until it can no longer go straight and is ready to be tightly turned around the descending ischiopubic ramus with the opposite hand. A distinct pop can be felt as the trocar tip advances through the obturator membrane and muscles. As the tip is advanced, the angle of the trocar is rotated from 35 degrees to vertical, almost perpendicular to the floor. At this point, the tip of the trocar should be guided out of the midurethral tunnel against the opposite index finger.
I utilize the same technique for tensioning a TOT sling as I do the retropubic sling.
Dr. Rardin’s retropubic approach
I continue to use the original TVT sling with a 5-mm stainless steel, mechanically cut trocar and reusable handle. The newer slender needles may advance with less pressure, but I worry about them bending during passage. I feel more assured and comfortable using the older trocars.
I perform retropubic hydrodissection with a spinal needle using a top-down approach. With 40 cc of a very dilute solution of bupivacaine (Marcaine) with epinephrine on each side of the urethra, I create columns of hydrodissected space. Studies are inconsistent about the benefits of hydrodissection, but theoretically, it decreases the risk of bladder injury by pushing the bladder away from the pubic bone, creates effective hemostasis, and can provide analgesia that will be on board when the patient wakes up.
I bring the spinal needle down behind the pubic bone to the location of the urethral incision site, with my finger in the vagina, so I can feel the tip of the needle next to the urethra/Foley catheter. For each side, I will inject 20 cc of the solution in this location, and the other 20 cc as I withdraw the needle upward.
For trocar passage, some surgeons are taught to advance the trocar until a pop is felt, then drop the handle and push upward. I view the maneuver as a consistent, smooth arch; for every degree that I advance the trocar, I drop the handle slightly in order to maintain contact with the back of the pubic bone throughout the pass. I continuously and simultaneously drop the handle and advance the trocar. Contact with the back of the pubic bone is maintained with a slight pulling on the back end of the trocar handle, while the forward hand pushes the trocar upward.
The target that I visualize for a retropubic pass is the patient’s ipsilateral shoulder. A cadaver study showed that if you aim as far lateral as the patient’s outstretched elbow, you can enter the iliac vasculature (Obstet. Gynecol. 2003;101:933-6).
If the patient is draped such that I cannot see the ipsilateral shoulder, I ask the anesthesia team to show me. I have also identified and marked the suprapubic points about 2-3 cm from each other on either side of the midline just about the pubic symphysis, but I consider the broader anatomic picture and purposeful visualization toward the ipsilateral shoulder to be an essential part of safe technique. In general, it is safer to be more medial than more lateral for the needle passage.
I continue to use a rigid catheter guide to deflect the bladder neck while passing the needles.
Cystoscopy is performed after both needles are placed but not yet pulled through. I fill the bladder until the ureteral orifices appear flattened, which confirms that the bladder is under enough distension to preclude any mucosal wrinkles.
The technique I utilize for adjusting the tension of the TVT sling was taught to me by Dr. Peter L. Rosenblatt of Mt. Auburn Hospital in Cambridge, Mass. At the midline of the sling, I advance the sheaths just enough so that I can grasp a 2-3 mm “knuckle” of the midportion of the sling with a Babcock clamp. I then pull the sling ends until the Babcock comes into gentle contact with the suburethral tissue. The sheaths encasing the sling are then removed and the Babcock clamp is released to assure a tension-free deployment.
The amount of tape that is pinched with the Babcock – the size of the “knuckle” – determines the tension. For a patient with a more profound problem, such as intrinsic sphincter deficiency or a lack or urethral hypermobility, I will grasp a smaller knuckle.
These steps ensure that the midportion of the tape will not tighten or become deformed under tension. Rather than use a spacer, I like to protect the midportion of the tape and prevent it from being stretched. I find that the approach is reproducible and results in a reliable amount of space between the urethra and sling when the procedure is completed.
Dr. Rardin’s TOT approach
I employ an inside-out technique to the TOT procedure, and I utilize devices with segment of mesh that is shorter – only about 13 cm in length – than the original full-length mesh used in many TOT procedures. Once placed, the ends of the mesh penetrate the obturator membrane and obturator externus but not the adductor compartment of the thigh and groin.
In a study we presented last year at the annual meeting of the Society of Gynecologic Surgeons meeting, the shortened tape reduced postoperative groin pain, compared with full-length TOT tape without any reduction in subjective benefit. It appears that with shortened tape, we are anchoring the sling in tissues that provide critical support while avoiding the muscles that relate to the inner thigh/groin pain experienced by some patients. Effectiveness was not reduced, compared with full-length TOT slings.
These shortened slings are distinct from a single-incision sling, which is basically pushed into place. We still pass the needle all the way through a vaginal incision and out through the obturator foramen, and we pull the sling into place as we would any other TOT sling. The difference is that we’re not leaving any mesh in the groin.
I prefer an inside-out approach for two reasons: I always feel that I have more control over where a needle enters than where it exits, and precision is important with suburethral slings. Secondly, the dissection tunnel created for an inside-out pass is much smaller than the tunnel that must be dissected for an outside-in approach. In theory, less dissection means less devascularization, less denervation, and less opportunity for erosion.
Hydrodissection for TOT slings is more minimal and involves less fluid than does hydrodissection for retropubic slings, mainly because we do not want to anesthetize the obturator nerve. I pass the spinal needle from the outside in. At the start, prior to making any incisions, it is important to identify the arcus tendineus, a linear thickening of the superior fascia that is sometimes called the “white line.” This is where the sulcus is affixed to the sidewall. I will be sure to penetrate the sidewall at or above the level of the arcus.
With the TOT approach, the likelihood of bladder injury is so low that I usually drive the trocars all the way through prior to cystoscopy, as opposed to leaving the needles in place as I do with the retropubic approach.
Tensioning is achieved in the same manner, by using the Babcock clamp to avoid distortion of the critical part of the mesh while creating the space needed given the patient’s clinical scenario. It is worth remembering, at this point, that overall risks for retention appear to be lower for obturator slings, compared with retropubic slings.
I place most of my patients receiving retropubic slings in dorsal lithotomy position; but for obturator sling placement, I favor a few degrees into higher lithotomy because this pulls the obturator neurovascular bundle a little further out of the path of the needle.
Dr. Sokol reported that he owns stock in Pelvalon and is a clinical adviser to that company. He also is a national principal investigator for American Medical Systems and the recipient of research grants from Acell and several other companies. Dr. Rardin reported that he has no relevant financial disclosures. To view a video related to this article, go to SurgeryU at aagl.org/obgyn-news.

Team discovers new mechanisms of platelet formation
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
in the bone marrow
Researchers have found that megakaryocytes can produce platelets via different polyploidization mechanisms.
Experiments suggested that endomitosis is the major megakaryocyte polyploidization mechanism, but megakaryocytes can generate platelets even in the absence of endomitosis—via endocycles and re-replication.
These findings may have implications for treating cancers as well as thrombocytopenia, the researchers said.
They described their discoveries in Developmental Cell.
Marcos Malumbres, PhD, of Centro Nacional de Investigaciones Oncologicas (CNIO) in Madrid, Spain, and his colleagues performed a genetic analysis of endomitosis, in which megakaryocytes become polyploid by entering mitosis but aborting anaphase.
The team studied mice lacking key proteins involved in mitotic entry (Cdk1 and Cdk2), mitotic progression (Aurora B), or mitotic exit (the anaphase-promoting complex [APC/C] cofactor Cdc20) during megakaryocyte maturation.
They found that Aurora B is not required for megakaryocyte maturation, but Cdc20 is. In mice lacking Cdc20, the researchers observed mitotic arrest and severe thrombocytopenia. The team said this suggests endomitosis is the major megakaryocyte polyploidization mechanism in vivo.
So they were surprised to discover that deleting Cdk1 prevents endomitosis but does not hinder platelet formation.
“Whilst the elimination of the main proteins that regulate megakaryocyte growth generates, as we thought, a reduction in the production of platelets, this didn’t happen when we removed Cdk1,” Dr Malumbres said. “[Even] when Cdk1 was absent, the megakaryocytes were able to grow in size in a similar way to normal cells.”
“[Further] analysis revealed that cells deficient in Cdk1 underwent cellular reprogramming towards a process known as endocycles, which can also be seen in other types of cells, such as certain cells in the placenta,” said Marianna Trakala, also of CNIO.
In endocycles, cells successively replicate their genomes without segregating chromosomes during mitosis.
The researchers uncovered an additional mechanism for megakaryocyte polyploidy when they studied mice lacking both Cdk1 and Cdk2. The deletion of both Cdks resulted in aberrant re-replication, in which the genome is replicated more than once per cell cycle, but platelet levels were not affected.
In fact, the team found that the loss of Cdk1 alone or Cdk1 and Cdk2 together can significantly rescue proplatelet formation and increase platelet levels in Cdc20-null mice.
“We immediately asked ourselves if, by reprogramming the cell cycle towards endocycles, we could correct the thrombocytopenia induced in other models,” Dr Malumbres said.
And when the researchers eliminated Cdk1 in Cdc20-null mice with severe thrombocytopenia, they observed an increase in platelet production. Results were similar when the team ablated Cdk1 and Cdk2 in Cdc20-null mice.
In addition to their implications for treating thrombocytopenia, these results could aid the design of cancer treatments, the researchers said. The findings reveal the different requirements that normal or tumor cells have toward cell-cycle regulators.
Specifically, the research suggests that deleting Cdk1 or other cell-cycle proteins is lethal for tumor cells but does not affect megakaryocytes. So drugs that inhibit these proteins could be used to treat malignancies such as pro-megakaryocytic leukemias. ![]()
Fusion protein fights resistant ALL

In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
In a preclinical study, a fusion protein targeted treatment-resistant leukemia, demonstrating superiority over both chemotherapy and radiation.
The protein, CD19L-sTRAIL, induced apoptosis in radiation-resistant cells from patients with B-precursor acute lymphoblastic leukemia (ALL) and in
mouse models of the disease.
Additionally, mice that received CD19L-sTRAIL had significantly longer event-free survival than mice that received chemotherapy.
An account of this study appears in The Journal of Clinical Investigation.
Study investigators knew that TNF-related apoptosis-inducing ligand (TRAIL) can cause apoptosis in cancer cells by binding to TRAIL-receptor 1 and TRAIL-receptor 2.
“TRAIL is a naturally occurring part of the body’s immune system that kills cancer cells without toxicity to normal cells,” said Faith Uckun, MD, PhD, of Children’s Hospital Los Angeles in California.
“However, earlier clinical trials using TRAIL as a potential anticancer medicine candidate have not been successful, largely because of its propensity to bind, not only to cancer cells, but also to ‘decoy’ receptors.”
But Dr Uckun and her colleagues had discovered CD19-ligand (CD19L), a natural ligand of human CD19 that is expressed by almost all ALL cells. And they hypothesized that fusing CD19L to the portion of TRAIL that can kill cancer cells (known as sTRAIL) would produce a powerful weapon against leukemia cells.
The resulting fusion protein, CD19L-sTRAIL, would seek out, bind, and destroy only leukemia cells carrying CD19 as the target docking site.
In experiments, the investigators showed that their engineering converted sTRAIL into a much more potent “membrane-anchored” form that is capable of triggering apoptosis, even in the most aggressive and therapy-resistant ALL cells.
“Due to its ability to anchor to the surface of cancer cells via CD19, CD19L-sTRAIL was 100,000-fold more potent than sTRAIL and consistently killed more than 99% of aggressive leukemia cells taken directly from children with ALL—not only in the test tube but also in mice,” Dr Uckun said.
At a 2.1 pM concentration, CD19L-sTRAIL caused 84.0±4.7% apoptosis in leukemia cells from patients with B-precursor ALL, whereas radiation with 2 Gy γ-rays caused 45.0±9.0% apoptosis (P<0.0001). Higher concentrations of CD19L-sTRAIL prompted an apoptosis rate of more than 90%.
In cells from mouse models of B-precursor ALL, 2.1 pM of CD19L-sTRAIL caused 91.4±5.4% apoptosis, compared to 16.0±4.6% with 2 Gy γ-rays (P<0.0001).
In addition, administering 2 or 3 doses of CD19L-sTRAIL significantly improved event-free survival in mice challenged with an otherwise fatal dose of leukemia cells.
The median event-free survival was 17 days in control mice; 20 days in mice treated with either vincristine, dexamethasone, and peg-asparaginase or vincristine, doxorubicin, and peg-asparaginase; and 58 days in mice that received 2- to 3-day treatment with CD19L-sTRAIL (P<0.0001 vs controls; P=0.0002 vs chemo).
The investigators said these results support the clinical potential of CD19L-sTRAIL as a new agent against B-precursor ALL.
“We are hopeful that the knowledge gained from this study will open a new range of effective treatment opportunities for children with recurrent leukemia,” Dr Uckun said. ![]()
Product granted orphan designation for GVHD

Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()
Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()
Credit: PLOS ONE
The European Medicines Agency (EMA) has granted orphan drug status to ApoCell for the prevention of graft-vs-host disease (GVHD).
ApoCell consists of matched-donor, mononuclear-enriched, early apoptotic cells. It has been shown to immunomodulate macrophages and dendritic cells, both of which are involved in GVHD pathogenesis.
ApoCell showed early promise in a phase 1/2a study, and a phase 2b/3 trial of the product is set to begin this year.
ApoCell, which is under development by Enlivex Therapeutics, received orphan drug designation in the US in 2013.
In the European Union (EU), orphan designation is granted to products intended for the treatment, prevention, or diagnosis of a life-threatening or chronically debilitating disease with a prevalence of no more than 5 in 10,000, with no satisfactory method of diagnosis, prevention, or treatment authorized by the EMA.
Orphan designation in the EU comes with a number of benefits, including protocol assistance and 10-year market exclusivity following regulatory approval.
Phase 1/2a trial of ApoCell
For this study, researchers tested an infusion of ApoCell, in addition to cyclosporine and methotrexate, as prophylaxis for acute GVHD.
The trial enrolled 13 leukemia patients who had a median age of 37 (range, 20-59). All of the patients received an HLA-matched, myeloablative, allogeneic hematopoietic stem cell transplant (HSCT) from a related donor.
All patients engrafted. The median time to neutrophil recovery was 13 days after HSCT (range, 11 to 19), and the median time to platelet recovery was 15 days (range, 11 to 59).
At 100 days post-HSCT, the cumulative incidence of acute grade 2-4 GVHD was 23.1%, and the incidence of acute grade 3-4 GVHD was 15.4%. None of the patients developed acute GVHD beyond day 100.
Among patients who received the 2 higher doses of ApoCell (n=6), the rate of acute grade 2-4 GVHD was 0%.
Ten patients experienced serious adverse events, but 7 were considered unrelated to ApoCell, and 3 were likely unrelated to the treatment.
The nonrelapse mortality rate was 7.7% at both 100 and 180 days after HSCT. The overall survival rate was 92% at 100 days and 85% at 180 days.
The researchers said these results suggest a single infusion of ApoCell is safe and may effectively prevent acute GVHD. ![]()