User login
Dilute Betadine Lavage Reduces Implant-Related Bacterial Burden in a Rabbit Knee Prosthetic Infection Model
Surgical site infection after arthroplasty causes substantial morbidity and potential mortality. Prosthetic joint infection (PJI) ranges from simple superficial wound infection and cellulitis to deep subfascial infection that involves the prosthesis. Consistent use of prophylactic antibiotics has reduced postoperative hip and knee arthroplasty infections to rates of 0.25% to 2%.1-4 Treatment of a patient with PJI commonly includes hospitalization, long-term intravenously administered antibiotics, resection arthroplasty, and staged reimplantation. The estimated cost of interventions reaches tens of millions of dollars annually in the United States and does not include the costs of psychosocial effects on patients and their families.5,6
Betadine (povidone-iodine) is a widely used antiseptic for skin and mucous membrane wounds and has been shown to be effective for the prevention of PJI.7 Dilute Betadine solution has been proposed as an aid in treatment of PJI.8 At a minimum concentration of 5%, cytotoxicity has been observed in chicken tibia osteoblasts.9 A balance of the bactericidal and cytotoxic activities of Betadine, while maintaining its efficacy against resistant organisms, such as methicillin-resistant Staphylococcus aureus (MRSA), is optimized at dilutions between 0.5% and 4%.10-14 We hypothesized that a dilute Betadine lavage of 3.5% would achieve a significant decrease in bacterial counts compared with an isolated saline lavage in an in vivo knee PJI model.
Materials and Methods
Animal Protocol
All surgical procedures were conducted according to the protocol approved by our institutional animal care and use committee. Using a power analysis and data obtained at our institution, we determined that 12 was the minimum number of animals needed to reach significance set at P < .05 and assuming a 50% decrease in colony-forming units (CFU) (SigmaStat Version 2.03; Aspire Software International, Ashburn, Virginia). Eight New Zealand White rabbits were used in our study; because significance was reached early, 12 were not needed. The average weight of the rabbits was 3.5 kg (weight range, 3.2-4.1 kg). All rabbits completed 1 week of acclimation before surgery.
Bacteria Preparation
A broth culture of methicillin-sensitive S aureus (MSSA) (ATCC 25923) was prepared 1 day before surgery. The bacteria were suspended in 5 mL of Trypticase Soy Broth (Becton Dickinson & Co, Franklin Lakes, New Jersey) and incubated at 37°C in a shaking incubator for 16 hours. The next day, the culture was centrifuged and irrigated twice with normal saline to remove the broth and prevent further growth. The bacteria were reconstituted in normal saline, and the concentration was standardized using a turbidity meter (LaMotte 2020e; LaMotte Co, Chestertown, Maryland), which correlated with 106 CFU/100 µl plated on trypticase soy agar plates with 10% sheep blood (Fisher Scientific, Pittsburgh, Pennsylvania).
Surgical and Postoperative Procedures
Our procedure was based on the New Zealand White rabbit knee PJI model.15 General anesthesia was induced with ketamine and xylazine, and maintained with isoflurane inhalation via a nose cone mask. Rabbits were positioned supine, and bilateral knees were shaved, prepped, and draped in a sterile fashion.
A 2-cm longitudinal incision was made over the lateral knee, and arthrotomy was performed, exposing the lateral collateral ligament attachment at the lateral femoral condyle. Using a 4-mm drill bit, a defect was drilled obliquely into the lateral femoral condyle, anterior to the lateral collateral ligament attachment. This produced a defect in the non-weight-bearing, nonarticulating portion of the knee. A fully threaded 4×14-mm stainless steel screw (Synthes, West Chester, Pennsylvania) with a U-shaped ultrahigh-molecular-weight polyethylene washer (Synthes) was inserted into the defect. The joint capsule was closed with a running 3-0 Vicryl suture (Ethicon, Somerville, New Jersey). The knee joint was inoculated with 100 µL of the S aureus preparation using a 22-gauge needle. The skin was closed with a 4-0 Biosyn suture (Ethicon). The procedure was repeated on the contralateral knee (Figures 1A, 1B).
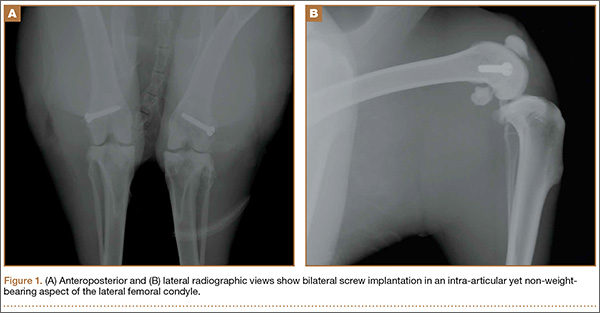
Seven days after the initial surgery, the rabbits were returned to the operating room and were anesthetized, positioned, and prepped for surgery as detailed above. Ceftriaxone (20 mg/kg of body weight) was intravenously administered to all rabbits for the treatment procedure. For each rabbit, a control knee and an experimental knee were randomly assigned. A longitudinal incision was made, exposing the previously placed implants. The screw was loosened slightly to remove the U-shaped polyethylene washer. Each knee then underwent lavage 2 times, for 90 seconds each time, with 3.5% dilute Betadine solution (experimental knee) or with normal saline (control knee). Because Pseudomonas contamination has been reported with povidone-iodine taken from unsterilized bottles,16,17 packets of sterilized povidone-iodine (Aplicare; Clorox, Oakland, California) were used. After the irrigation was complete, a new sterile polyethylene washer was placed and the screw was tightened. The wound closure was repeated as detailed above.
Postoperative analgesia was provided based on a standard institutional animal care and use committee protocol. Rabbits were permitted full cage activity and nutrition ad libitum. Wound healing, body weight, and signs of distress were monitored daily.
Outcome Measures
Seven days after surgery, the rabbits were euthanized with administration of phenobarbital (100 mg/kg of body weight). Arterial blood samples were obtained from the auricular vein to ensure that the rabbits were not systemically infected. Using a sterile technique, the screw, polyethylene washer, lateral femoral condyle bone from the defect, and joint capsule were cultured. Harvested bone and soft tissues were weighed and immediately homogenized (PowerGen Model 35 Handheld Homogenizer; Thermo Fisher Scientific, Inc, Waltham, Massachusetts). Implants were sonicated (UBATH-Y; World Precision Instruments, Inc, Sarasota, Florida) in cold saline to obtain a sensitive culture.18
Bacterial quantification was determined by using trypticase soy agar plates after 24 hours of growth. Final CFU were calculated after serial dilutions and were standardized per gram of biopsied tissues.19 Members of the team were blinded to the treatment type.
Statistical Analysis
Statistical differences in mean bacterial burden were calculated independently for lateral condyle bone, joint capsule, polyethylene, and screws by conducting a Student t test.
Results
Treatment effect was higher than expected, and the study was terminated after 8 animals completed the protocol. All 8 rabbits tolerated the procedures well and were appropriately monitored during the postoperative period. No animals had signs of systemic infection or positive blood culture. All local cultures for screw, polyethylene washer, lateral femoral condyle defect, and joint capsule were positive.
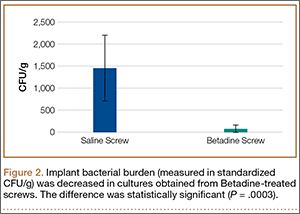
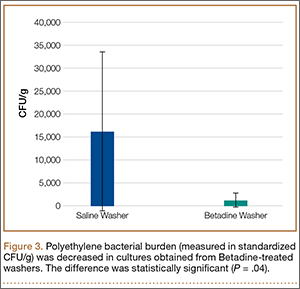
Statistically significant decreases were shown in the bacterial burden of the Betadine-irrigated screws and the Betadine-irrigated polyethylene washers compared with the saline-irrigated controls. Betadine-irrigated screws grew an average of 7.16 × 101 CFU of S aureus/g, whereas screws from control knees grew an average of 1.45 × 103 CFU/g (P = .0003) (Figure 2). Betadine-treated washers grew an average of 1.28 × 103 CFU/g compared with 1.62 × 104 CFU/g for control washers (P =. 04) (Figure 3).
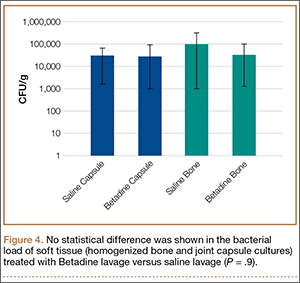
A trend toward decreased bacterial counts was shown in Betadine-treated soft tissues compared with saline-treated soft tissues, but the difference did not reach statistical significance (P = .9). Biopsied joint capsule from knees treated with Betadine grew an average of 2.84 × 104 CFU/g compared with an average of 3.16 × 104 CFU/g in control-rabbit knees (Figure 4). Cultured lateral condyle from Betadine-treated knees had an average bacterial load of 3.22 × 104 CFU/g compared with an average bacterial load of 1 × 105 CFU/g in control knees (Figure 4).
Discussion
Knees irrigated with Betadine showed a significant (P = .0003) decrease in metal implant–related S aureus bacterial counts by 20-fold and a significant (P < .05) decrease in polyethylene implant–related counts by more than 10-fold. This arthroplasty model used Betadine lavage as a treatment adjunct with intravenously administered antibiotics and polyethylene exchange. Our 1-week interval after the index procedure classifies the infection as an acute postoperative arthroplasty infection (occurring less than 4 weeks postoperatively).
The gold standard treatment for these infections is irrigation and débridement with component retention.18 The success rate has been reported to be as high as 71%20 but was closer to 44% in a study by Fridkin and colleagues,21 especially with more virulent bacteria. Staphylococcal species, higher American Society of Anesthesiologists scores, and frank pus around the prosthesis were markers of débridement failure in a recent study by Azzam and colleagues.18
The majority of postoperative joint arthroplasty infections are caused by S aureus, and the incidence of MRSA bacteria continues to rise.22 Community-acquired MRSA is increasing at an alarming rate and is now the predominant organism in skin and soft-tissue infections.23 Organism resistance also occurs at a cellular level by the formation of a glycocalyx layer, or biofilm. This layer assists in changing the phenotypic properties of the organism and decreases the efficacy of antibiotics.24 The self-produced layer of extracellular matrices, deoxyribonucleic acid, and polysaccharides attaches to inert material, preventing phagocytic action by neutrophils. In addition to antibacterial activity, povidone-iodine has antibiofilm activity against Staphylococcal species.25 The active ingredient targets the gene that produces biofilm. This correlates to our study in which the largest decrease in bacterial counts was noted on the implants.
The use of Betadine lavage has shown some promise in vivo as well. A prospective randomized controlled trial26 used 3.5% Betadine irrigation to prevent spine infection. No infections occurred in the Betadine group compared with a deep-infection rate of 2.9% in the control group. Brown and colleagues8 reviewed 1862 hip and knee arthroplasty cases before the use of Betadine lavage and 688 cases after the use of Betadine lavage and found a decrease in infection rate, from 0.97% to 0.15%. S aureus caused 13 of the 18 infections in the control group. These studies8,26 used Betadine lavage for prophylaxis and prevention of deep spine and arthroplasty infection. Betadine lavage as a treatment adjunct for acute arthroplasty infection has not been studied clinically. It has the potential to increase isolated incision and débridement success and to improve component survivorship.
Our arthroplasty model mimics an intra-articular environment and accounts for an implant–polyethylene interface.15 Limitations of our study include the use of MSSA as opposed to MRSA. However, povidone-iodine has the same effects on both MSSA and MRSA.12 We also treated our postoperative infection with 1 dose of antibiotics and not a course, although it should be noted that the single dose of ceftriaxone allowed us to isolate the independent effect of the Betadine lavage. A baseline level of infection severity could have been measured with cultures obtained at the time of irrigation and débridement. Also, a decrease in CFU does not directly correlate to a clinically significant outcome, such as a defined surgical site infection requiring intervention. Nevertheless, it is noteworthy that the decrease in bacterial counts on the stainless steel screws and polyethylene washers were maintained 1 week after the Betadine lavage.
Conclusion
Dilute Betadine lavage is a simple and inexpensive adjunct for the treatment of acute postoperative arthroplasty infection and may increase the rate of component retention. Additionally, the bactericidal and antibiofilm activities of Betadine may improve the effectiveness of systemic antibiotics. Further clinical investigation is warranted.
1. Wilson MG, Kelley K, Thornhill TS. Infection as a complication of total knee-replacement arthroplasty. Risk factors and treatment in sixty-seven cases. J Bone Joint Surg Am. 1990;72(6):878-883.
2. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
3. Mahomed NN, Barrett JA, Katz JN, et al. Rates and outcomes of primary and revision total hip replacement in the United States medicare population. J Bone Joint Surg Am. 2003;85(1):27-32.
4. Mahomed NN, Barrett J, Katz JN, Baron JA, Wright J, Losina E. Epidemiology of total knee replacement in the United States Medicare population. J Bone Joint Surg Am. 2005;87(6):1222-1228.
5. Parvizi J, Zmistowski B, Adeli B. Periprosthetic joint infection: treatment options. Orthopedics. 2010;33(9):659.
6. Poultsides LA, Liaropoulos LL, Malizos KN. The socioeconomic impact of musculoskeletal infections. J Bone Joint Surg Am. 2010;92(11):e13.
7. Chundamala J, Wright JG. The efficacy and risks of using povidone-iodine irrigation to prevent surgical site infection: an evidence-based review. Can J Surg. 2007;50(6):473-481.
8. Brown NM, Cipriano CA, Moric M, Sporer SM, Della Valle CJ. Dilute betadine lavage before closure for the prevention of acute postoperative deep periprosthetic joint infection. J Arthroplasty. 2012;27(1):27-30.
9. Kaysinger KK, Nicholson NC, Ramp WK, Kellam JF. Toxic effects of wound irrigation solutions on cultured tibiae and osteoblasts. J Orthop Trauma. 1995;9(4):303-311.
10. Haley CE, Marling-Cason M, Smith JW, Luby JP, Mackowiak PA. Bactericidal activity of antiseptics against methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 1985;21(6):991-992.
11. Lacey RW, Catto A. Action of povidone-iodine against methicillin-sensitive and -resistant cultures of Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S78-S83.
12. McLure AR, Gordon J. In-vitro evaluation of povidone-iodine and chlorhexidine against methicillin-resistant Staphylococcus aureus. J Hosp Infect. 1992;21(4):291-299.
13. Suzuki J, Komatsuzawa H, Kozai K, Nagasaka N. In vitro susceptibility of Staphylococcus aureus including MRSA to four disinfectants. ASDC J Dent Child. 1997;64(4):260-263.
14. Yasuda T, Yoshimura S, Katsuno Y, et al. Comparison of bactericidal activities of various disinfectants against methicillin-sensitive Staphylococcus aureus and methicillin-resistant Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S66-S69.
15. Craig MR, Poelstra KA, Sherrell JC, Kwon MS, Belzile EL, Brown TE. A novel total knee arthroplasty infection model in rabbits. J Orthop Res. 2005;23(5):1100-1104.
16. Hartman MB, Fehring TK, Jordan L, Norton HJ. Periprosthetic knee sepsis. The role of irrigation and debridement. Clin Orthop. 1991;273:113-118.
17. Mont MA, Waldman B, Banerjee C, Pacheco IH, Hungerford DS. Multiple irrigation, debridement, and retention of components in infected total knee arthroplasty. J Arthroplasty. 1997;12(4):426-433.
18. Azzam KA, Seeley M, Ghanem E, Austin MS, Purtill JJ, Parvizi J. Irrigation and debridement in the management of prosthetic joint infection: traditional indications revisited. J Arthroplasty. 2010;25(7):1022-1027.
19. Stall AC, Becker E, Ludwig SC, Gelb D, Poelstra KA. Reduction of postoperative spinal implant infection using gentamicin microspheres. Spine (Phila Pa 1976). 2009;34(5):479-483.
20. Hota B, Ellenbogen C, Hayden MK, Aroutcheva A, Rice TW, Weinstein RA. Community-associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections at a public hospital: do public housing and incarceration amplify transmission? Arch Intern Med. 2007;167(10):1026-1033.
21. Fridkin SK, Hageman JC, Morrison M, et al, Active Bacterial Core Surveillance Program of the Emerging Infections Program Network. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352(14):1436-1444.
22. Hosman AH, van der Mei HC, Bulstra SK, Busscher HJ, Neut D. Metal-on-metal bearings in total hip arthroplasties: influence of cobalt and chromium ions on bacterial growth and biofilm formation. J Biomed Mater Res A. 2009;88(3):711-716.
23. Oduwole KO, Glynn AA, Molony DC, et al. Anti-biofilm activity of sub-inhibitory povidone-iodine concentrations against Staphylococcus epidermidis and Staphylococcus aureus. J Orthop Res. 2010;28(9):1252-1256.
24. Cheng MT, Chang MC, Wang ST, Yu WK, Liu CL, Chen TH. Efficacy of dilute betadine solution irrigation in the prevention of postoperative infection of spinal surgery. Spine (Phila Pa 1976). 2005;30(15):1689-1693.
25. Anderson RL, Vess RW, Panlilio AL, Favero MS. Prolonged survival of Pseudomonas cepacia in commercially manufactured povidone-iodine. Appl Environ Microbiol. 1990;56(11):3598-3600.
26. Panlilio AL, Beck-Sague CM, Siegel JD, et al. Infections and pseudoinfections due to povidone-iodine solution contaminated with Pseudomonas cepacia. Clin Infect Dis. 1992;14(5):1078-1083.
Surgical site infection after arthroplasty causes substantial morbidity and potential mortality. Prosthetic joint infection (PJI) ranges from simple superficial wound infection and cellulitis to deep subfascial infection that involves the prosthesis. Consistent use of prophylactic antibiotics has reduced postoperative hip and knee arthroplasty infections to rates of 0.25% to 2%.1-4 Treatment of a patient with PJI commonly includes hospitalization, long-term intravenously administered antibiotics, resection arthroplasty, and staged reimplantation. The estimated cost of interventions reaches tens of millions of dollars annually in the United States and does not include the costs of psychosocial effects on patients and their families.5,6
Betadine (povidone-iodine) is a widely used antiseptic for skin and mucous membrane wounds and has been shown to be effective for the prevention of PJI.7 Dilute Betadine solution has been proposed as an aid in treatment of PJI.8 At a minimum concentration of 5%, cytotoxicity has been observed in chicken tibia osteoblasts.9 A balance of the bactericidal and cytotoxic activities of Betadine, while maintaining its efficacy against resistant organisms, such as methicillin-resistant Staphylococcus aureus (MRSA), is optimized at dilutions between 0.5% and 4%.10-14 We hypothesized that a dilute Betadine lavage of 3.5% would achieve a significant decrease in bacterial counts compared with an isolated saline lavage in an in vivo knee PJI model.
Materials and Methods
Animal Protocol
All surgical procedures were conducted according to the protocol approved by our institutional animal care and use committee. Using a power analysis and data obtained at our institution, we determined that 12 was the minimum number of animals needed to reach significance set at P < .05 and assuming a 50% decrease in colony-forming units (CFU) (SigmaStat Version 2.03; Aspire Software International, Ashburn, Virginia). Eight New Zealand White rabbits were used in our study; because significance was reached early, 12 were not needed. The average weight of the rabbits was 3.5 kg (weight range, 3.2-4.1 kg). All rabbits completed 1 week of acclimation before surgery.
Bacteria Preparation
A broth culture of methicillin-sensitive S aureus (MSSA) (ATCC 25923) was prepared 1 day before surgery. The bacteria were suspended in 5 mL of Trypticase Soy Broth (Becton Dickinson & Co, Franklin Lakes, New Jersey) and incubated at 37°C in a shaking incubator for 16 hours. The next day, the culture was centrifuged and irrigated twice with normal saline to remove the broth and prevent further growth. The bacteria were reconstituted in normal saline, and the concentration was standardized using a turbidity meter (LaMotte 2020e; LaMotte Co, Chestertown, Maryland), which correlated with 106 CFU/100 µl plated on trypticase soy agar plates with 10% sheep blood (Fisher Scientific, Pittsburgh, Pennsylvania).
Surgical and Postoperative Procedures
Our procedure was based on the New Zealand White rabbit knee PJI model.15 General anesthesia was induced with ketamine and xylazine, and maintained with isoflurane inhalation via a nose cone mask. Rabbits were positioned supine, and bilateral knees were shaved, prepped, and draped in a sterile fashion.
A 2-cm longitudinal incision was made over the lateral knee, and arthrotomy was performed, exposing the lateral collateral ligament attachment at the lateral femoral condyle. Using a 4-mm drill bit, a defect was drilled obliquely into the lateral femoral condyle, anterior to the lateral collateral ligament attachment. This produced a defect in the non-weight-bearing, nonarticulating portion of the knee. A fully threaded 4×14-mm stainless steel screw (Synthes, West Chester, Pennsylvania) with a U-shaped ultrahigh-molecular-weight polyethylene washer (Synthes) was inserted into the defect. The joint capsule was closed with a running 3-0 Vicryl suture (Ethicon, Somerville, New Jersey). The knee joint was inoculated with 100 µL of the S aureus preparation using a 22-gauge needle. The skin was closed with a 4-0 Biosyn suture (Ethicon). The procedure was repeated on the contralateral knee (Figures 1A, 1B).
Seven days after the initial surgery, the rabbits were returned to the operating room and were anesthetized, positioned, and prepped for surgery as detailed above. Ceftriaxone (20 mg/kg of body weight) was intravenously administered to all rabbits for the treatment procedure. For each rabbit, a control knee and an experimental knee were randomly assigned. A longitudinal incision was made, exposing the previously placed implants. The screw was loosened slightly to remove the U-shaped polyethylene washer. Each knee then underwent lavage 2 times, for 90 seconds each time, with 3.5% dilute Betadine solution (experimental knee) or with normal saline (control knee). Because Pseudomonas contamination has been reported with povidone-iodine taken from unsterilized bottles,16,17 packets of sterilized povidone-iodine (Aplicare; Clorox, Oakland, California) were used. After the irrigation was complete, a new sterile polyethylene washer was placed and the screw was tightened. The wound closure was repeated as detailed above.
Postoperative analgesia was provided based on a standard institutional animal care and use committee protocol. Rabbits were permitted full cage activity and nutrition ad libitum. Wound healing, body weight, and signs of distress were monitored daily.
Outcome Measures
Seven days after surgery, the rabbits were euthanized with administration of phenobarbital (100 mg/kg of body weight). Arterial blood samples were obtained from the auricular vein to ensure that the rabbits were not systemically infected. Using a sterile technique, the screw, polyethylene washer, lateral femoral condyle bone from the defect, and joint capsule were cultured. Harvested bone and soft tissues were weighed and immediately homogenized (PowerGen Model 35 Handheld Homogenizer; Thermo Fisher Scientific, Inc, Waltham, Massachusetts). Implants were sonicated (UBATH-Y; World Precision Instruments, Inc, Sarasota, Florida) in cold saline to obtain a sensitive culture.18
Bacterial quantification was determined by using trypticase soy agar plates after 24 hours of growth. Final CFU were calculated after serial dilutions and were standardized per gram of biopsied tissues.19 Members of the team were blinded to the treatment type.
Statistical Analysis
Statistical differences in mean bacterial burden were calculated independently for lateral condyle bone, joint capsule, polyethylene, and screws by conducting a Student t test.
Results
Treatment effect was higher than expected, and the study was terminated after 8 animals completed the protocol. All 8 rabbits tolerated the procedures well and were appropriately monitored during the postoperative period. No animals had signs of systemic infection or positive blood culture. All local cultures for screw, polyethylene washer, lateral femoral condyle defect, and joint capsule were positive.
Statistically significant decreases were shown in the bacterial burden of the Betadine-irrigated screws and the Betadine-irrigated polyethylene washers compared with the saline-irrigated controls. Betadine-irrigated screws grew an average of 7.16 × 101 CFU of S aureus/g, whereas screws from control knees grew an average of 1.45 × 103 CFU/g (P = .0003) (Figure 2). Betadine-treated washers grew an average of 1.28 × 103 CFU/g compared with 1.62 × 104 CFU/g for control washers (P =. 04) (Figure 3).
A trend toward decreased bacterial counts was shown in Betadine-treated soft tissues compared with saline-treated soft tissues, but the difference did not reach statistical significance (P = .9). Biopsied joint capsule from knees treated with Betadine grew an average of 2.84 × 104 CFU/g compared with an average of 3.16 × 104 CFU/g in control-rabbit knees (Figure 4). Cultured lateral condyle from Betadine-treated knees had an average bacterial load of 3.22 × 104 CFU/g compared with an average bacterial load of 1 × 105 CFU/g in control knees (Figure 4).
Discussion
Knees irrigated with Betadine showed a significant (P = .0003) decrease in metal implant–related S aureus bacterial counts by 20-fold and a significant (P < .05) decrease in polyethylene implant–related counts by more than 10-fold. This arthroplasty model used Betadine lavage as a treatment adjunct with intravenously administered antibiotics and polyethylene exchange. Our 1-week interval after the index procedure classifies the infection as an acute postoperative arthroplasty infection (occurring less than 4 weeks postoperatively).
The gold standard treatment for these infections is irrigation and débridement with component retention.18 The success rate has been reported to be as high as 71%20 but was closer to 44% in a study by Fridkin and colleagues,21 especially with more virulent bacteria. Staphylococcal species, higher American Society of Anesthesiologists scores, and frank pus around the prosthesis were markers of débridement failure in a recent study by Azzam and colleagues.18
The majority of postoperative joint arthroplasty infections are caused by S aureus, and the incidence of MRSA bacteria continues to rise.22 Community-acquired MRSA is increasing at an alarming rate and is now the predominant organism in skin and soft-tissue infections.23 Organism resistance also occurs at a cellular level by the formation of a glycocalyx layer, or biofilm. This layer assists in changing the phenotypic properties of the organism and decreases the efficacy of antibiotics.24 The self-produced layer of extracellular matrices, deoxyribonucleic acid, and polysaccharides attaches to inert material, preventing phagocytic action by neutrophils. In addition to antibacterial activity, povidone-iodine has antibiofilm activity against Staphylococcal species.25 The active ingredient targets the gene that produces biofilm. This correlates to our study in which the largest decrease in bacterial counts was noted on the implants.
The use of Betadine lavage has shown some promise in vivo as well. A prospective randomized controlled trial26 used 3.5% Betadine irrigation to prevent spine infection. No infections occurred in the Betadine group compared with a deep-infection rate of 2.9% in the control group. Brown and colleagues8 reviewed 1862 hip and knee arthroplasty cases before the use of Betadine lavage and 688 cases after the use of Betadine lavage and found a decrease in infection rate, from 0.97% to 0.15%. S aureus caused 13 of the 18 infections in the control group. These studies8,26 used Betadine lavage for prophylaxis and prevention of deep spine and arthroplasty infection. Betadine lavage as a treatment adjunct for acute arthroplasty infection has not been studied clinically. It has the potential to increase isolated incision and débridement success and to improve component survivorship.
Our arthroplasty model mimics an intra-articular environment and accounts for an implant–polyethylene interface.15 Limitations of our study include the use of MSSA as opposed to MRSA. However, povidone-iodine has the same effects on both MSSA and MRSA.12 We also treated our postoperative infection with 1 dose of antibiotics and not a course, although it should be noted that the single dose of ceftriaxone allowed us to isolate the independent effect of the Betadine lavage. A baseline level of infection severity could have been measured with cultures obtained at the time of irrigation and débridement. Also, a decrease in CFU does not directly correlate to a clinically significant outcome, such as a defined surgical site infection requiring intervention. Nevertheless, it is noteworthy that the decrease in bacterial counts on the stainless steel screws and polyethylene washers were maintained 1 week after the Betadine lavage.
Conclusion
Dilute Betadine lavage is a simple and inexpensive adjunct for the treatment of acute postoperative arthroplasty infection and may increase the rate of component retention. Additionally, the bactericidal and antibiofilm activities of Betadine may improve the effectiveness of systemic antibiotics. Further clinical investigation is warranted.
Surgical site infection after arthroplasty causes substantial morbidity and potential mortality. Prosthetic joint infection (PJI) ranges from simple superficial wound infection and cellulitis to deep subfascial infection that involves the prosthesis. Consistent use of prophylactic antibiotics has reduced postoperative hip and knee arthroplasty infections to rates of 0.25% to 2%.1-4 Treatment of a patient with PJI commonly includes hospitalization, long-term intravenously administered antibiotics, resection arthroplasty, and staged reimplantation. The estimated cost of interventions reaches tens of millions of dollars annually in the United States and does not include the costs of psychosocial effects on patients and their families.5,6
Betadine (povidone-iodine) is a widely used antiseptic for skin and mucous membrane wounds and has been shown to be effective for the prevention of PJI.7 Dilute Betadine solution has been proposed as an aid in treatment of PJI.8 At a minimum concentration of 5%, cytotoxicity has been observed in chicken tibia osteoblasts.9 A balance of the bactericidal and cytotoxic activities of Betadine, while maintaining its efficacy against resistant organisms, such as methicillin-resistant Staphylococcus aureus (MRSA), is optimized at dilutions between 0.5% and 4%.10-14 We hypothesized that a dilute Betadine lavage of 3.5% would achieve a significant decrease in bacterial counts compared with an isolated saline lavage in an in vivo knee PJI model.
Materials and Methods
Animal Protocol
All surgical procedures were conducted according to the protocol approved by our institutional animal care and use committee. Using a power analysis and data obtained at our institution, we determined that 12 was the minimum number of animals needed to reach significance set at P < .05 and assuming a 50% decrease in colony-forming units (CFU) (SigmaStat Version 2.03; Aspire Software International, Ashburn, Virginia). Eight New Zealand White rabbits were used in our study; because significance was reached early, 12 were not needed. The average weight of the rabbits was 3.5 kg (weight range, 3.2-4.1 kg). All rabbits completed 1 week of acclimation before surgery.
Bacteria Preparation
A broth culture of methicillin-sensitive S aureus (MSSA) (ATCC 25923) was prepared 1 day before surgery. The bacteria were suspended in 5 mL of Trypticase Soy Broth (Becton Dickinson & Co, Franklin Lakes, New Jersey) and incubated at 37°C in a shaking incubator for 16 hours. The next day, the culture was centrifuged and irrigated twice with normal saline to remove the broth and prevent further growth. The bacteria were reconstituted in normal saline, and the concentration was standardized using a turbidity meter (LaMotte 2020e; LaMotte Co, Chestertown, Maryland), which correlated with 106 CFU/100 µl plated on trypticase soy agar plates with 10% sheep blood (Fisher Scientific, Pittsburgh, Pennsylvania).
Surgical and Postoperative Procedures
Our procedure was based on the New Zealand White rabbit knee PJI model.15 General anesthesia was induced with ketamine and xylazine, and maintained with isoflurane inhalation via a nose cone mask. Rabbits were positioned supine, and bilateral knees were shaved, prepped, and draped in a sterile fashion.
A 2-cm longitudinal incision was made over the lateral knee, and arthrotomy was performed, exposing the lateral collateral ligament attachment at the lateral femoral condyle. Using a 4-mm drill bit, a defect was drilled obliquely into the lateral femoral condyle, anterior to the lateral collateral ligament attachment. This produced a defect in the non-weight-bearing, nonarticulating portion of the knee. A fully threaded 4×14-mm stainless steel screw (Synthes, West Chester, Pennsylvania) with a U-shaped ultrahigh-molecular-weight polyethylene washer (Synthes) was inserted into the defect. The joint capsule was closed with a running 3-0 Vicryl suture (Ethicon, Somerville, New Jersey). The knee joint was inoculated with 100 µL of the S aureus preparation using a 22-gauge needle. The skin was closed with a 4-0 Biosyn suture (Ethicon). The procedure was repeated on the contralateral knee (Figures 1A, 1B).
Seven days after the initial surgery, the rabbits were returned to the operating room and were anesthetized, positioned, and prepped for surgery as detailed above. Ceftriaxone (20 mg/kg of body weight) was intravenously administered to all rabbits for the treatment procedure. For each rabbit, a control knee and an experimental knee were randomly assigned. A longitudinal incision was made, exposing the previously placed implants. The screw was loosened slightly to remove the U-shaped polyethylene washer. Each knee then underwent lavage 2 times, for 90 seconds each time, with 3.5% dilute Betadine solution (experimental knee) or with normal saline (control knee). Because Pseudomonas contamination has been reported with povidone-iodine taken from unsterilized bottles,16,17 packets of sterilized povidone-iodine (Aplicare; Clorox, Oakland, California) were used. After the irrigation was complete, a new sterile polyethylene washer was placed and the screw was tightened. The wound closure was repeated as detailed above.
Postoperative analgesia was provided based on a standard institutional animal care and use committee protocol. Rabbits were permitted full cage activity and nutrition ad libitum. Wound healing, body weight, and signs of distress were monitored daily.
Outcome Measures
Seven days after surgery, the rabbits were euthanized with administration of phenobarbital (100 mg/kg of body weight). Arterial blood samples were obtained from the auricular vein to ensure that the rabbits were not systemically infected. Using a sterile technique, the screw, polyethylene washer, lateral femoral condyle bone from the defect, and joint capsule were cultured. Harvested bone and soft tissues were weighed and immediately homogenized (PowerGen Model 35 Handheld Homogenizer; Thermo Fisher Scientific, Inc, Waltham, Massachusetts). Implants were sonicated (UBATH-Y; World Precision Instruments, Inc, Sarasota, Florida) in cold saline to obtain a sensitive culture.18
Bacterial quantification was determined by using trypticase soy agar plates after 24 hours of growth. Final CFU were calculated after serial dilutions and were standardized per gram of biopsied tissues.19 Members of the team were blinded to the treatment type.
Statistical Analysis
Statistical differences in mean bacterial burden were calculated independently for lateral condyle bone, joint capsule, polyethylene, and screws by conducting a Student t test.
Results
Treatment effect was higher than expected, and the study was terminated after 8 animals completed the protocol. All 8 rabbits tolerated the procedures well and were appropriately monitored during the postoperative period. No animals had signs of systemic infection or positive blood culture. All local cultures for screw, polyethylene washer, lateral femoral condyle defect, and joint capsule were positive.
Statistically significant decreases were shown in the bacterial burden of the Betadine-irrigated screws and the Betadine-irrigated polyethylene washers compared with the saline-irrigated controls. Betadine-irrigated screws grew an average of 7.16 × 101 CFU of S aureus/g, whereas screws from control knees grew an average of 1.45 × 103 CFU/g (P = .0003) (Figure 2). Betadine-treated washers grew an average of 1.28 × 103 CFU/g compared with 1.62 × 104 CFU/g for control washers (P =. 04) (Figure 3).
A trend toward decreased bacterial counts was shown in Betadine-treated soft tissues compared with saline-treated soft tissues, but the difference did not reach statistical significance (P = .9). Biopsied joint capsule from knees treated with Betadine grew an average of 2.84 × 104 CFU/g compared with an average of 3.16 × 104 CFU/g in control-rabbit knees (Figure 4). Cultured lateral condyle from Betadine-treated knees had an average bacterial load of 3.22 × 104 CFU/g compared with an average bacterial load of 1 × 105 CFU/g in control knees (Figure 4).
Discussion
Knees irrigated with Betadine showed a significant (P = .0003) decrease in metal implant–related S aureus bacterial counts by 20-fold and a significant (P < .05) decrease in polyethylene implant–related counts by more than 10-fold. This arthroplasty model used Betadine lavage as a treatment adjunct with intravenously administered antibiotics and polyethylene exchange. Our 1-week interval after the index procedure classifies the infection as an acute postoperative arthroplasty infection (occurring less than 4 weeks postoperatively).
The gold standard treatment for these infections is irrigation and débridement with component retention.18 The success rate has been reported to be as high as 71%20 but was closer to 44% in a study by Fridkin and colleagues,21 especially with more virulent bacteria. Staphylococcal species, higher American Society of Anesthesiologists scores, and frank pus around the prosthesis were markers of débridement failure in a recent study by Azzam and colleagues.18
The majority of postoperative joint arthroplasty infections are caused by S aureus, and the incidence of MRSA bacteria continues to rise.22 Community-acquired MRSA is increasing at an alarming rate and is now the predominant organism in skin and soft-tissue infections.23 Organism resistance also occurs at a cellular level by the formation of a glycocalyx layer, or biofilm. This layer assists in changing the phenotypic properties of the organism and decreases the efficacy of antibiotics.24 The self-produced layer of extracellular matrices, deoxyribonucleic acid, and polysaccharides attaches to inert material, preventing phagocytic action by neutrophils. In addition to antibacterial activity, povidone-iodine has antibiofilm activity against Staphylococcal species.25 The active ingredient targets the gene that produces biofilm. This correlates to our study in which the largest decrease in bacterial counts was noted on the implants.
The use of Betadine lavage has shown some promise in vivo as well. A prospective randomized controlled trial26 used 3.5% Betadine irrigation to prevent spine infection. No infections occurred in the Betadine group compared with a deep-infection rate of 2.9% in the control group. Brown and colleagues8 reviewed 1862 hip and knee arthroplasty cases before the use of Betadine lavage and 688 cases after the use of Betadine lavage and found a decrease in infection rate, from 0.97% to 0.15%. S aureus caused 13 of the 18 infections in the control group. These studies8,26 used Betadine lavage for prophylaxis and prevention of deep spine and arthroplasty infection. Betadine lavage as a treatment adjunct for acute arthroplasty infection has not been studied clinically. It has the potential to increase isolated incision and débridement success and to improve component survivorship.
Our arthroplasty model mimics an intra-articular environment and accounts for an implant–polyethylene interface.15 Limitations of our study include the use of MSSA as opposed to MRSA. However, povidone-iodine has the same effects on both MSSA and MRSA.12 We also treated our postoperative infection with 1 dose of antibiotics and not a course, although it should be noted that the single dose of ceftriaxone allowed us to isolate the independent effect of the Betadine lavage. A baseline level of infection severity could have been measured with cultures obtained at the time of irrigation and débridement. Also, a decrease in CFU does not directly correlate to a clinically significant outcome, such as a defined surgical site infection requiring intervention. Nevertheless, it is noteworthy that the decrease in bacterial counts on the stainless steel screws and polyethylene washers were maintained 1 week after the Betadine lavage.
Conclusion
Dilute Betadine lavage is a simple and inexpensive adjunct for the treatment of acute postoperative arthroplasty infection and may increase the rate of component retention. Additionally, the bactericidal and antibiofilm activities of Betadine may improve the effectiveness of systemic antibiotics. Further clinical investigation is warranted.
1. Wilson MG, Kelley K, Thornhill TS. Infection as a complication of total knee-replacement arthroplasty. Risk factors and treatment in sixty-seven cases. J Bone Joint Surg Am. 1990;72(6):878-883.
2. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
3. Mahomed NN, Barrett JA, Katz JN, et al. Rates and outcomes of primary and revision total hip replacement in the United States medicare population. J Bone Joint Surg Am. 2003;85(1):27-32.
4. Mahomed NN, Barrett J, Katz JN, Baron JA, Wright J, Losina E. Epidemiology of total knee replacement in the United States Medicare population. J Bone Joint Surg Am. 2005;87(6):1222-1228.
5. Parvizi J, Zmistowski B, Adeli B. Periprosthetic joint infection: treatment options. Orthopedics. 2010;33(9):659.
6. Poultsides LA, Liaropoulos LL, Malizos KN. The socioeconomic impact of musculoskeletal infections. J Bone Joint Surg Am. 2010;92(11):e13.
7. Chundamala J, Wright JG. The efficacy and risks of using povidone-iodine irrigation to prevent surgical site infection: an evidence-based review. Can J Surg. 2007;50(6):473-481.
8. Brown NM, Cipriano CA, Moric M, Sporer SM, Della Valle CJ. Dilute betadine lavage before closure for the prevention of acute postoperative deep periprosthetic joint infection. J Arthroplasty. 2012;27(1):27-30.
9. Kaysinger KK, Nicholson NC, Ramp WK, Kellam JF. Toxic effects of wound irrigation solutions on cultured tibiae and osteoblasts. J Orthop Trauma. 1995;9(4):303-311.
10. Haley CE, Marling-Cason M, Smith JW, Luby JP, Mackowiak PA. Bactericidal activity of antiseptics against methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 1985;21(6):991-992.
11. Lacey RW, Catto A. Action of povidone-iodine against methicillin-sensitive and -resistant cultures of Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S78-S83.
12. McLure AR, Gordon J. In-vitro evaluation of povidone-iodine and chlorhexidine against methicillin-resistant Staphylococcus aureus. J Hosp Infect. 1992;21(4):291-299.
13. Suzuki J, Komatsuzawa H, Kozai K, Nagasaka N. In vitro susceptibility of Staphylococcus aureus including MRSA to four disinfectants. ASDC J Dent Child. 1997;64(4):260-263.
14. Yasuda T, Yoshimura S, Katsuno Y, et al. Comparison of bactericidal activities of various disinfectants against methicillin-sensitive Staphylococcus aureus and methicillin-resistant Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S66-S69.
15. Craig MR, Poelstra KA, Sherrell JC, Kwon MS, Belzile EL, Brown TE. A novel total knee arthroplasty infection model in rabbits. J Orthop Res. 2005;23(5):1100-1104.
16. Hartman MB, Fehring TK, Jordan L, Norton HJ. Periprosthetic knee sepsis. The role of irrigation and debridement. Clin Orthop. 1991;273:113-118.
17. Mont MA, Waldman B, Banerjee C, Pacheco IH, Hungerford DS. Multiple irrigation, debridement, and retention of components in infected total knee arthroplasty. J Arthroplasty. 1997;12(4):426-433.
18. Azzam KA, Seeley M, Ghanem E, Austin MS, Purtill JJ, Parvizi J. Irrigation and debridement in the management of prosthetic joint infection: traditional indications revisited. J Arthroplasty. 2010;25(7):1022-1027.
19. Stall AC, Becker E, Ludwig SC, Gelb D, Poelstra KA. Reduction of postoperative spinal implant infection using gentamicin microspheres. Spine (Phila Pa 1976). 2009;34(5):479-483.
20. Hota B, Ellenbogen C, Hayden MK, Aroutcheva A, Rice TW, Weinstein RA. Community-associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections at a public hospital: do public housing and incarceration amplify transmission? Arch Intern Med. 2007;167(10):1026-1033.
21. Fridkin SK, Hageman JC, Morrison M, et al, Active Bacterial Core Surveillance Program of the Emerging Infections Program Network. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352(14):1436-1444.
22. Hosman AH, van der Mei HC, Bulstra SK, Busscher HJ, Neut D. Metal-on-metal bearings in total hip arthroplasties: influence of cobalt and chromium ions on bacterial growth and biofilm formation. J Biomed Mater Res A. 2009;88(3):711-716.
23. Oduwole KO, Glynn AA, Molony DC, et al. Anti-biofilm activity of sub-inhibitory povidone-iodine concentrations against Staphylococcus epidermidis and Staphylococcus aureus. J Orthop Res. 2010;28(9):1252-1256.
24. Cheng MT, Chang MC, Wang ST, Yu WK, Liu CL, Chen TH. Efficacy of dilute betadine solution irrigation in the prevention of postoperative infection of spinal surgery. Spine (Phila Pa 1976). 2005;30(15):1689-1693.
25. Anderson RL, Vess RW, Panlilio AL, Favero MS. Prolonged survival of Pseudomonas cepacia in commercially manufactured povidone-iodine. Appl Environ Microbiol. 1990;56(11):3598-3600.
26. Panlilio AL, Beck-Sague CM, Siegel JD, et al. Infections and pseudoinfections due to povidone-iodine solution contaminated with Pseudomonas cepacia. Clin Infect Dis. 1992;14(5):1078-1083.
1. Wilson MG, Kelley K, Thornhill TS. Infection as a complication of total knee-replacement arthroplasty. Risk factors and treatment in sixty-seven cases. J Bone Joint Surg Am. 1990;72(6):878-883.
2. Ridgeway S, Wilson J, Charlet A, Kafatos G, Pearson A, Coello R. Infection of the surgical site after arthroplasty of the hip. J Bone Joint Surg Br. 2005;87(6):844-850.
3. Mahomed NN, Barrett JA, Katz JN, et al. Rates and outcomes of primary and revision total hip replacement in the United States medicare population. J Bone Joint Surg Am. 2003;85(1):27-32.
4. Mahomed NN, Barrett J, Katz JN, Baron JA, Wright J, Losina E. Epidemiology of total knee replacement in the United States Medicare population. J Bone Joint Surg Am. 2005;87(6):1222-1228.
5. Parvizi J, Zmistowski B, Adeli B. Periprosthetic joint infection: treatment options. Orthopedics. 2010;33(9):659.
6. Poultsides LA, Liaropoulos LL, Malizos KN. The socioeconomic impact of musculoskeletal infections. J Bone Joint Surg Am. 2010;92(11):e13.
7. Chundamala J, Wright JG. The efficacy and risks of using povidone-iodine irrigation to prevent surgical site infection: an evidence-based review. Can J Surg. 2007;50(6):473-481.
8. Brown NM, Cipriano CA, Moric M, Sporer SM, Della Valle CJ. Dilute betadine lavage before closure for the prevention of acute postoperative deep periprosthetic joint infection. J Arthroplasty. 2012;27(1):27-30.
9. Kaysinger KK, Nicholson NC, Ramp WK, Kellam JF. Toxic effects of wound irrigation solutions on cultured tibiae and osteoblasts. J Orthop Trauma. 1995;9(4):303-311.
10. Haley CE, Marling-Cason M, Smith JW, Luby JP, Mackowiak PA. Bactericidal activity of antiseptics against methicillin-resistant Staphylococcus aureus. J Clin Microbiol. 1985;21(6):991-992.
11. Lacey RW, Catto A. Action of povidone-iodine against methicillin-sensitive and -resistant cultures of Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S78-S83.
12. McLure AR, Gordon J. In-vitro evaluation of povidone-iodine and chlorhexidine against methicillin-resistant Staphylococcus aureus. J Hosp Infect. 1992;21(4):291-299.
13. Suzuki J, Komatsuzawa H, Kozai K, Nagasaka N. In vitro susceptibility of Staphylococcus aureus including MRSA to four disinfectants. ASDC J Dent Child. 1997;64(4):260-263.
14. Yasuda T, Yoshimura S, Katsuno Y, et al. Comparison of bactericidal activities of various disinfectants against methicillin-sensitive Staphylococcus aureus and methicillin-resistant Staphylococcus aureus. Postgrad Med J. 1993;69(3 suppl):S66-S69.
15. Craig MR, Poelstra KA, Sherrell JC, Kwon MS, Belzile EL, Brown TE. A novel total knee arthroplasty infection model in rabbits. J Orthop Res. 2005;23(5):1100-1104.
16. Hartman MB, Fehring TK, Jordan L, Norton HJ. Periprosthetic knee sepsis. The role of irrigation and debridement. Clin Orthop. 1991;273:113-118.
17. Mont MA, Waldman B, Banerjee C, Pacheco IH, Hungerford DS. Multiple irrigation, debridement, and retention of components in infected total knee arthroplasty. J Arthroplasty. 1997;12(4):426-433.
18. Azzam KA, Seeley M, Ghanem E, Austin MS, Purtill JJ, Parvizi J. Irrigation and debridement in the management of prosthetic joint infection: traditional indications revisited. J Arthroplasty. 2010;25(7):1022-1027.
19. Stall AC, Becker E, Ludwig SC, Gelb D, Poelstra KA. Reduction of postoperative spinal implant infection using gentamicin microspheres. Spine (Phila Pa 1976). 2009;34(5):479-483.
20. Hota B, Ellenbogen C, Hayden MK, Aroutcheva A, Rice TW, Weinstein RA. Community-associated methicillin-resistant Staphylococcus aureus skin and soft tissue infections at a public hospital: do public housing and incarceration amplify transmission? Arch Intern Med. 2007;167(10):1026-1033.
21. Fridkin SK, Hageman JC, Morrison M, et al, Active Bacterial Core Surveillance Program of the Emerging Infections Program Network. Methicillin-resistant Staphylococcus aureus disease in three communities. N Engl J Med. 2005;352(14):1436-1444.
22. Hosman AH, van der Mei HC, Bulstra SK, Busscher HJ, Neut D. Metal-on-metal bearings in total hip arthroplasties: influence of cobalt and chromium ions on bacterial growth and biofilm formation. J Biomed Mater Res A. 2009;88(3):711-716.
23. Oduwole KO, Glynn AA, Molony DC, et al. Anti-biofilm activity of sub-inhibitory povidone-iodine concentrations against Staphylococcus epidermidis and Staphylococcus aureus. J Orthop Res. 2010;28(9):1252-1256.
24. Cheng MT, Chang MC, Wang ST, Yu WK, Liu CL, Chen TH. Efficacy of dilute betadine solution irrigation in the prevention of postoperative infection of spinal surgery. Spine (Phila Pa 1976). 2005;30(15):1689-1693.
25. Anderson RL, Vess RW, Panlilio AL, Favero MS. Prolonged survival of Pseudomonas cepacia in commercially manufactured povidone-iodine. Appl Environ Microbiol. 1990;56(11):3598-3600.
26. Panlilio AL, Beck-Sague CM, Siegel JD, et al. Infections and pseudoinfections due to povidone-iodine solution contaminated with Pseudomonas cepacia. Clin Infect Dis. 1992;14(5):1078-1083.
Ruxolitinib bests standard treatment for PV
The JAK1/2 inhibitor ruxolitinib can outperform standard therapy in patients with polycythemia vera (PV), results of the RESPONSE trial suggest.
In patients who could not tolerate or were resistant to hydroxyurea, ruxolitinib proved superior to standard therapy for controlling hematocrit levels and reducing spleen volume
“This study indicates that ruxolitinib may represent an important advance for this population of patients with PV,” said Alessandro M. Vannucchi, MD, of the University of Florence in Italy.
Dr Vannucchi and his colleagues reported these findings in NEJM. The trial was funded by Incyte Corporation, the company developing ruxolitinib.
The phase 3 study included 222 patients. They were phlebotomy-dependent, had splenomegaly, and could not tolerate or were resistant to hydroxyurea.
The patients were randomized 1:1 to receive either ruxolitinib (starting dose of 10 mg twice daily) or standard therapy, which was defined as investigator-selected monotherapy or observation only. The ruxolitinib dose was adjusted as needed throughout the study.
The primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
So 21% of patients in the ruxolitinib group met the primary endpoint, achieving both hematocrit control and spleen reduction. But only 1% of patients in the standard-therapy group did the same (P<0.001).
In all, 60% of patients in the ruxolitinib arm achieved hematocrit control, compared to 20% of those receiving standard therapy. And 38% of patients in the ruxolitinib arm had at least a 35% spleen reduction, compared to 1% of patients in the standard-therapy arm.
The rate of complete hematologic remission was significantly higher in the ruxolitinib group than in the standard-therapy group, at 24% and 9%, respectively (P=0.003).
And ruxolitinib-treated patients had a greater reduction in overall symptoms. Forty-nine percent of ruxolitinib-treated patients had at least a 50% reduction in their total symptom score at week 32 (as measured by the MPN-SAF 14-item total symptom score), compared to 5% of patients on standard therapy.
Based on these results, most patients in the standard-therapy arm crossed over to receive ruxolitinib immediately after week 32. So the researchers could only compare rates of adverse events through week 32.
They found that grade 3/4 anemia was more common with ruxolitinib than with standard therapy (2% and 0%, respectively). The same was true of grade 3/4 thrombocytopenia (5% and 4%, respectively) and herpes zoster infections of all grades (6% and 0%, respectively).
However, thromboembolic events were more common with standard therapy. They occurred in 6 patients who received standard therapy and 1 ruxolitinib-treated patient.
The most common non-hematologic adverse events in the ruxolitinib arm were headache (16%), diarrhea (15%), and fatigue (15%), which were mainly grade 1 or 2. The rates of these events in the standard therapy arm were 19%, 7%, and 15%, respectively.
The researchers also noted that nearly 85% of patients randomized to ruxolitinib were still receiving treatment at a median follow-up of 81 weeks. ![]()
The JAK1/2 inhibitor ruxolitinib can outperform standard therapy in patients with polycythemia vera (PV), results of the RESPONSE trial suggest.
In patients who could not tolerate or were resistant to hydroxyurea, ruxolitinib proved superior to standard therapy for controlling hematocrit levels and reducing spleen volume
“This study indicates that ruxolitinib may represent an important advance for this population of patients with PV,” said Alessandro M. Vannucchi, MD, of the University of Florence in Italy.
Dr Vannucchi and his colleagues reported these findings in NEJM. The trial was funded by Incyte Corporation, the company developing ruxolitinib.
The phase 3 study included 222 patients. They were phlebotomy-dependent, had splenomegaly, and could not tolerate or were resistant to hydroxyurea.
The patients were randomized 1:1 to receive either ruxolitinib (starting dose of 10 mg twice daily) or standard therapy, which was defined as investigator-selected monotherapy or observation only. The ruxolitinib dose was adjusted as needed throughout the study.
The primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
So 21% of patients in the ruxolitinib group met the primary endpoint, achieving both hematocrit control and spleen reduction. But only 1% of patients in the standard-therapy group did the same (P<0.001).
In all, 60% of patients in the ruxolitinib arm achieved hematocrit control, compared to 20% of those receiving standard therapy. And 38% of patients in the ruxolitinib arm had at least a 35% spleen reduction, compared to 1% of patients in the standard-therapy arm.
The rate of complete hematologic remission was significantly higher in the ruxolitinib group than in the standard-therapy group, at 24% and 9%, respectively (P=0.003).
And ruxolitinib-treated patients had a greater reduction in overall symptoms. Forty-nine percent of ruxolitinib-treated patients had at least a 50% reduction in their total symptom score at week 32 (as measured by the MPN-SAF 14-item total symptom score), compared to 5% of patients on standard therapy.
Based on these results, most patients in the standard-therapy arm crossed over to receive ruxolitinib immediately after week 32. So the researchers could only compare rates of adverse events through week 32.
They found that grade 3/4 anemia was more common with ruxolitinib than with standard therapy (2% and 0%, respectively). The same was true of grade 3/4 thrombocytopenia (5% and 4%, respectively) and herpes zoster infections of all grades (6% and 0%, respectively).
However, thromboembolic events were more common with standard therapy. They occurred in 6 patients who received standard therapy and 1 ruxolitinib-treated patient.
The most common non-hematologic adverse events in the ruxolitinib arm were headache (16%), diarrhea (15%), and fatigue (15%), which were mainly grade 1 or 2. The rates of these events in the standard therapy arm were 19%, 7%, and 15%, respectively.
The researchers also noted that nearly 85% of patients randomized to ruxolitinib were still receiving treatment at a median follow-up of 81 weeks. ![]()
The JAK1/2 inhibitor ruxolitinib can outperform standard therapy in patients with polycythemia vera (PV), results of the RESPONSE trial suggest.
In patients who could not tolerate or were resistant to hydroxyurea, ruxolitinib proved superior to standard therapy for controlling hematocrit levels and reducing spleen volume
“This study indicates that ruxolitinib may represent an important advance for this population of patients with PV,” said Alessandro M. Vannucchi, MD, of the University of Florence in Italy.
Dr Vannucchi and his colleagues reported these findings in NEJM. The trial was funded by Incyte Corporation, the company developing ruxolitinib.
The phase 3 study included 222 patients. They were phlebotomy-dependent, had splenomegaly, and could not tolerate or were resistant to hydroxyurea.
The patients were randomized 1:1 to receive either ruxolitinib (starting dose of 10 mg twice daily) or standard therapy, which was defined as investigator-selected monotherapy or observation only. The ruxolitinib dose was adjusted as needed throughout the study.
The primary endpoint was a composite of hematocrit control and spleen reduction. To meet the endpoint, patients had to experience a 35% or greater reduction in spleen volume from baseline, as assessed by imaging at week 32.
And a patient’s hematocrit was considered under control if he was not eligible for phlebotomy from week 8 through 32 (and had no more than one instance of phlebotomy eligibility between randomization and week 8). Patients who were deemed eligible for phlebotomy had hematocrit that was greater than 45% or had increased 3 or more percentage points from the time they entered the study.
So 21% of patients in the ruxolitinib group met the primary endpoint, achieving both hematocrit control and spleen reduction. But only 1% of patients in the standard-therapy group did the same (P<0.001).
In all, 60% of patients in the ruxolitinib arm achieved hematocrit control, compared to 20% of those receiving standard therapy. And 38% of patients in the ruxolitinib arm had at least a 35% spleen reduction, compared to 1% of patients in the standard-therapy arm.
The rate of complete hematologic remission was significantly higher in the ruxolitinib group than in the standard-therapy group, at 24% and 9%, respectively (P=0.003).
And ruxolitinib-treated patients had a greater reduction in overall symptoms. Forty-nine percent of ruxolitinib-treated patients had at least a 50% reduction in their total symptom score at week 32 (as measured by the MPN-SAF 14-item total symptom score), compared to 5% of patients on standard therapy.
Based on these results, most patients in the standard-therapy arm crossed over to receive ruxolitinib immediately after week 32. So the researchers could only compare rates of adverse events through week 32.
They found that grade 3/4 anemia was more common with ruxolitinib than with standard therapy (2% and 0%, respectively). The same was true of grade 3/4 thrombocytopenia (5% and 4%, respectively) and herpes zoster infections of all grades (6% and 0%, respectively).
However, thromboembolic events were more common with standard therapy. They occurred in 6 patients who received standard therapy and 1 ruxolitinib-treated patient.
The most common non-hematologic adverse events in the ruxolitinib arm were headache (16%), diarrhea (15%), and fatigue (15%), which were mainly grade 1 or 2. The rates of these events in the standard therapy arm were 19%, 7%, and 15%, respectively.
The researchers also noted that nearly 85% of patients randomized to ruxolitinib were still receiving treatment at a median follow-up of 81 weeks. ![]()
Discovery could help make Ras druggable

Credit: Jes Andersen/
University of Copenhagen
Researchers say they have discovered how Ras proteins find their proper place in cells, a finding that may aid the development of novel approaches to treat cancers.
The team noted that cancers develop if Ras proteins start to trigger misregulation, and Ras misregulates if it misses its correct location on the cell wall—the membrane.
What the researchers discovered is that Ras cannot reach its designated location if the membrane has the wrong shape.
“If the curvature of the cell is right, Ras goes to the right place,” said Dimitrios Stamou, PhD, of the University of Copenhagen in Denmark.
“If the membrane is too straight or too bent, it does not. And Ras is very much like any other worker. If it never finds the way to its workplace, it is not likely to get any work done.”
Dr Stamou and his colleagues described this discovery in Nature Chemical Biology.
Ras proteins are thought to be misregulated in upwards of 30% of all cancers. For 3 decades, researchers have been searching for ways to quell the killer protein.
Their lack of success has given Ras a reputation as the “undruggable cancer target,” but Dr Stamou believes we can change by moving in a new direction.
“If Ras goes off the rails because of changes in the curvature of the cell, perhaps we should target whatever changes the shape of the cell membrane,” he said.
Looking for a correlation between cell shape and Ras misregulation was unusual, even bordering on controversial, said study author Jannik Bruun Larsen, PhD, of the University of Copenhagen.
The researchers were investigating how Ras proteins attach themselves to the cell wall, and Dr Larsen tried to attach Ras to a variety of simulated cell membranes formed into small spheres or vesicles of varying sizes.
He found that Ras would attach more readily to smaller spheres, which were more curved than the large ones, and Dr Larsen started to see a pattern.
“For more than a decade, people thought that the constituents of the cell wall was the thing that controlled where Ras was localized,” Dr Larson said. “We have shown that at least one other aspect—namely, membrane curvature—governs where Ras ends up in the cell and is therefore likely to be a factor in cancer development.”
All of the research so far has been conducted in vitro. Dr Stamou said the next big challenge is to uncover how these effects play out in living systems.
“It will be 10 times more difficult to uncover these effects in living systems, but it needs to happen,” he said. “We have started, and we really hope others will follow. It may prove complicated to develop a drug that changes the shape of cells, but I am certain that the discovery of the shape/misregulation-correlation will at least lead to new ways to diagnose cancers.” ![]()
Credit: Jes Andersen/
University of Copenhagen
Researchers say they have discovered how Ras proteins find their proper place in cells, a finding that may aid the development of novel approaches to treat cancers.
The team noted that cancers develop if Ras proteins start to trigger misregulation, and Ras misregulates if it misses its correct location on the cell wall—the membrane.
What the researchers discovered is that Ras cannot reach its designated location if the membrane has the wrong shape.
“If the curvature of the cell is right, Ras goes to the right place,” said Dimitrios Stamou, PhD, of the University of Copenhagen in Denmark.
“If the membrane is too straight or too bent, it does not. And Ras is very much like any other worker. If it never finds the way to its workplace, it is not likely to get any work done.”
Dr Stamou and his colleagues described this discovery in Nature Chemical Biology.
Ras proteins are thought to be misregulated in upwards of 30% of all cancers. For 3 decades, researchers have been searching for ways to quell the killer protein.
Their lack of success has given Ras a reputation as the “undruggable cancer target,” but Dr Stamou believes we can change by moving in a new direction.
“If Ras goes off the rails because of changes in the curvature of the cell, perhaps we should target whatever changes the shape of the cell membrane,” he said.
Looking for a correlation between cell shape and Ras misregulation was unusual, even bordering on controversial, said study author Jannik Bruun Larsen, PhD, of the University of Copenhagen.
The researchers were investigating how Ras proteins attach themselves to the cell wall, and Dr Larsen tried to attach Ras to a variety of simulated cell membranes formed into small spheres or vesicles of varying sizes.
He found that Ras would attach more readily to smaller spheres, which were more curved than the large ones, and Dr Larsen started to see a pattern.
“For more than a decade, people thought that the constituents of the cell wall was the thing that controlled where Ras was localized,” Dr Larson said. “We have shown that at least one other aspect—namely, membrane curvature—governs where Ras ends up in the cell and is therefore likely to be a factor in cancer development.”
All of the research so far has been conducted in vitro. Dr Stamou said the next big challenge is to uncover how these effects play out in living systems.
“It will be 10 times more difficult to uncover these effects in living systems, but it needs to happen,” he said. “We have started, and we really hope others will follow. It may prove complicated to develop a drug that changes the shape of cells, but I am certain that the discovery of the shape/misregulation-correlation will at least lead to new ways to diagnose cancers.” ![]()
Credit: Jes Andersen/
University of Copenhagen
Researchers say they have discovered how Ras proteins find their proper place in cells, a finding that may aid the development of novel approaches to treat cancers.
The team noted that cancers develop if Ras proteins start to trigger misregulation, and Ras misregulates if it misses its correct location on the cell wall—the membrane.
What the researchers discovered is that Ras cannot reach its designated location if the membrane has the wrong shape.
“If the curvature of the cell is right, Ras goes to the right place,” said Dimitrios Stamou, PhD, of the University of Copenhagen in Denmark.
“If the membrane is too straight or too bent, it does not. And Ras is very much like any other worker. If it never finds the way to its workplace, it is not likely to get any work done.”
Dr Stamou and his colleagues described this discovery in Nature Chemical Biology.
Ras proteins are thought to be misregulated in upwards of 30% of all cancers. For 3 decades, researchers have been searching for ways to quell the killer protein.
Their lack of success has given Ras a reputation as the “undruggable cancer target,” but Dr Stamou believes we can change by moving in a new direction.
“If Ras goes off the rails because of changes in the curvature of the cell, perhaps we should target whatever changes the shape of the cell membrane,” he said.
Looking for a correlation between cell shape and Ras misregulation was unusual, even bordering on controversial, said study author Jannik Bruun Larsen, PhD, of the University of Copenhagen.
The researchers were investigating how Ras proteins attach themselves to the cell wall, and Dr Larsen tried to attach Ras to a variety of simulated cell membranes formed into small spheres or vesicles of varying sizes.
He found that Ras would attach more readily to smaller spheres, which were more curved than the large ones, and Dr Larsen started to see a pattern.
“For more than a decade, people thought that the constituents of the cell wall was the thing that controlled where Ras was localized,” Dr Larson said. “We have shown that at least one other aspect—namely, membrane curvature—governs where Ras ends up in the cell and is therefore likely to be a factor in cancer development.”
All of the research so far has been conducted in vitro. Dr Stamou said the next big challenge is to uncover how these effects play out in living systems.
“It will be 10 times more difficult to uncover these effects in living systems, but it needs to happen,” he said. “We have started, and we really hope others will follow. It may prove complicated to develop a drug that changes the shape of cells, but I am certain that the discovery of the shape/misregulation-correlation will at least lead to new ways to diagnose cancers.” ![]()
Cell imaging gets colorful

Credit: Rhoda Baer
The detection and imaging of protein-protein interactions in live cells just got a lot more colorful, researchers have reported in Nature Methods.
The team created a technique that converts biochemical processes into color changes that are easily visualized.
The group said this provides a new tool scientists can use to answer questions about fundamental mechanisms in cell biology, aid the discovery of novel therapeutics, and more.
Robert E. Campbell, PhD, of the University of Alberta in Edmonton, Alberta, Canada, and his colleagues conducted this research.
They developed the technique, dubbed FPX, that employs genetically encoded fluorescent proteins to image dynamic biochemical events in live cells and tissues. The FPX method converts a change in protein-protein interactions into a dramatic green to red (or vice versa) color change that is immediately visible.
“Strategies for converting fluorescent proteins into active biosensors of intracellular biochemistry are few in number and technically challenging,” Dr Campbell said. “With this development, we can immediately image activity happening at the cellular level, offering an alternative to existing methods for detecting and imaging of protein-protein interactions in live cells.”
The FPX method is based on green and red dimerization-dependent fluorescent proteins (ddFPs) that Dr Campbell and his colleagues first reported in 2012.
Yidan Ding, PhD, a research assistant at the University of Alberta and the primary contributor to this work, found she could combine the use of both green and red ddFPs in single cells, such that the proteins could be green or red, but not both, at the same time.
By introducing modified versions of the proteins into live cells, and taking advantage of the fact that green and red fluorescence are mutually exclusive, Dr Ding was able to construct a wide variety of biosensors that underwent dramatic changes in fluorescence in response to biochemical processes of interest.
By adding this new dimension to fluorescent proteins and engineering them to be biosensors that change their color in response to specific biological events, Drs Ding and Campbell and their colleagues have provided a tool for researchers to immediately pinpoint a major change at the cellular level.
This minimizes the need for extensive biosensor optimization and provides a versatile new approach to building the next generation of biosensors.
“This allows for a wide scope of applications,” Dr Campbell said. “It will be immediately relevant to many areas of fundamental cell biology research and practical applications such as drug discovery. Ultimately, it will help researchers achieve breakthroughs in a wide variety of areas in the life sciences, such as neuroscience, diabetes, and cancer.”
Dr Campbell has a patent pending on the technology. ![]()
Credit: Rhoda Baer
The detection and imaging of protein-protein interactions in live cells just got a lot more colorful, researchers have reported in Nature Methods.
The team created a technique that converts biochemical processes into color changes that are easily visualized.
The group said this provides a new tool scientists can use to answer questions about fundamental mechanisms in cell biology, aid the discovery of novel therapeutics, and more.
Robert E. Campbell, PhD, of the University of Alberta in Edmonton, Alberta, Canada, and his colleagues conducted this research.
They developed the technique, dubbed FPX, that employs genetically encoded fluorescent proteins to image dynamic biochemical events in live cells and tissues. The FPX method converts a change in protein-protein interactions into a dramatic green to red (or vice versa) color change that is immediately visible.
“Strategies for converting fluorescent proteins into active biosensors of intracellular biochemistry are few in number and technically challenging,” Dr Campbell said. “With this development, we can immediately image activity happening at the cellular level, offering an alternative to existing methods for detecting and imaging of protein-protein interactions in live cells.”
The FPX method is based on green and red dimerization-dependent fluorescent proteins (ddFPs) that Dr Campbell and his colleagues first reported in 2012.
Yidan Ding, PhD, a research assistant at the University of Alberta and the primary contributor to this work, found she could combine the use of both green and red ddFPs in single cells, such that the proteins could be green or red, but not both, at the same time.
By introducing modified versions of the proteins into live cells, and taking advantage of the fact that green and red fluorescence are mutually exclusive, Dr Ding was able to construct a wide variety of biosensors that underwent dramatic changes in fluorescence in response to biochemical processes of interest.
By adding this new dimension to fluorescent proteins and engineering them to be biosensors that change their color in response to specific biological events, Drs Ding and Campbell and their colleagues have provided a tool for researchers to immediately pinpoint a major change at the cellular level.
This minimizes the need for extensive biosensor optimization and provides a versatile new approach to building the next generation of biosensors.
“This allows for a wide scope of applications,” Dr Campbell said. “It will be immediately relevant to many areas of fundamental cell biology research and practical applications such as drug discovery. Ultimately, it will help researchers achieve breakthroughs in a wide variety of areas in the life sciences, such as neuroscience, diabetes, and cancer.”
Dr Campbell has a patent pending on the technology. ![]()
Credit: Rhoda Baer
The detection and imaging of protein-protein interactions in live cells just got a lot more colorful, researchers have reported in Nature Methods.
The team created a technique that converts biochemical processes into color changes that are easily visualized.
The group said this provides a new tool scientists can use to answer questions about fundamental mechanisms in cell biology, aid the discovery of novel therapeutics, and more.
Robert E. Campbell, PhD, of the University of Alberta in Edmonton, Alberta, Canada, and his colleagues conducted this research.
They developed the technique, dubbed FPX, that employs genetically encoded fluorescent proteins to image dynamic biochemical events in live cells and tissues. The FPX method converts a change in protein-protein interactions into a dramatic green to red (or vice versa) color change that is immediately visible.
“Strategies for converting fluorescent proteins into active biosensors of intracellular biochemistry are few in number and technically challenging,” Dr Campbell said. “With this development, we can immediately image activity happening at the cellular level, offering an alternative to existing methods for detecting and imaging of protein-protein interactions in live cells.”
The FPX method is based on green and red dimerization-dependent fluorescent proteins (ddFPs) that Dr Campbell and his colleagues first reported in 2012.
Yidan Ding, PhD, a research assistant at the University of Alberta and the primary contributor to this work, found she could combine the use of both green and red ddFPs in single cells, such that the proteins could be green or red, but not both, at the same time.
By introducing modified versions of the proteins into live cells, and taking advantage of the fact that green and red fluorescence are mutually exclusive, Dr Ding was able to construct a wide variety of biosensors that underwent dramatic changes in fluorescence in response to biochemical processes of interest.
By adding this new dimension to fluorescent proteins and engineering them to be biosensors that change their color in response to specific biological events, Drs Ding and Campbell and their colleagues have provided a tool for researchers to immediately pinpoint a major change at the cellular level.
This minimizes the need for extensive biosensor optimization and provides a versatile new approach to building the next generation of biosensors.
“This allows for a wide scope of applications,” Dr Campbell said. “It will be immediately relevant to many areas of fundamental cell biology research and practical applications such as drug discovery. Ultimately, it will help researchers achieve breakthroughs in a wide variety of areas in the life sciences, such as neuroscience, diabetes, and cancer.”
Dr Campbell has a patent pending on the technology. ![]()
Broad application of JNC-8 would save lives, reduce costs
Antihypertensive therapy would prevent about 56,000 cardiovascular events annually and 13,000 deaths from strokes, myocardial infarctions, and other causes if it were used by all U.S. adults who qualify for treatment under 2014 Joint National Committee hypertension guidelines, according to computer modeling published online Jan. 28 in the New England Journal of Medicine.
Even though the new Joint Committee guidelines are a bit less stringent than the committee’s prior 2003 advice, blood pressure remains inadequately controlled in 44% of the 64 million U.S. adults with hypertension, according to the investigators, led by Dr. Andrew Moran of Columbia University Medical Center, New York (N. Engl. J. Med. 2015;372:447-55).

The team used data from the National Health and Nutrition Examination Survey, the Framingham Heart Study, and other sources to estimate costs and benefits of expanding treatment to all U.S. adults aged 35-74 years who meet the 2014 benchmarks. They then calculated cost-effectiveness of expanding use in various subpopulations, using $50,000/quality-adjusted life-year (QALY) gained, or less, as their cut-off.
Overall, the investigators found that fuller implementation of the Joint Committee goals would pay for itself in reduced cardiovascular morbidity and mortality. The results were driven primarily by secondary prevention in patients with cardiovascular disease and primary prevention in patients with stage 2 hypertension, meaning systolic BP of 160 mm Hg or higher or diastolic BP of 100 mm Hg or higher.
“There is an enormous potential for improving population health by expanding treatment and improving control. Our findings clearly show that it would be worthwhile to significantly increase spending on office visits, home blood pressure monitoring, and interventions to improve treatment adherence. In fact, we could double treatment and monitoring spending for some patients – namely those with severe hypertension – and still break even,” Dr. Moran said in a statement announcing the results.
Treatment of patients with existing cardiovascular disease or stage 2 hypertension would save lives and costs in all men 35-74 years old and in women aged 45-74 years. The treatment of more modest hypertension – systolic BP of 140-159 mm Hg or a diastolic BP of 90-99 mm Hg – was cost effective for all men and for women also between the ages of 45 and 74 years, but treating women 35-44 years old with moderate hypertension and diabetes or kidney disease had intermediate cost-effectiveness ($125,000 per QALY), and low cost-effectiveness ($181,000 per QALY) if those comorbidities were not present.
“Some people will be alarmed about our conclusion that it may not be cost effective to treat hypertension in young adults, especially young women. It’s worth noting that our analysis didn’t capture the cumulative, lifetime effects of hypertension. It may well turn out to be cost effective to treat this group if we look at data on costs and benefits over several decades,” Dr. Moran said.
The team assumed a medication adherence rate of 75%. The costs of treatment included medications, monitoring, and drug side effects.
They did not analyze the effect of diet and lifestyle interventions for lowering blood pressure, or compare the cost-effectiveness of specific antihypertensive medication classes or combinations.
The work was funded by the National Heart, Lung, and Blood Institute, among others. The authors reported no relevant financial disclosures.
Antihypertensive therapy would prevent about 56,000 cardiovascular events annually and 13,000 deaths from strokes, myocardial infarctions, and other causes if it were used by all U.S. adults who qualify for treatment under 2014 Joint National Committee hypertension guidelines, according to computer modeling published online Jan. 28 in the New England Journal of Medicine.
Even though the new Joint Committee guidelines are a bit less stringent than the committee’s prior 2003 advice, blood pressure remains inadequately controlled in 44% of the 64 million U.S. adults with hypertension, according to the investigators, led by Dr. Andrew Moran of Columbia University Medical Center, New York (N. Engl. J. Med. 2015;372:447-55).
The team used data from the National Health and Nutrition Examination Survey, the Framingham Heart Study, and other sources to estimate costs and benefits of expanding treatment to all U.S. adults aged 35-74 years who meet the 2014 benchmarks. They then calculated cost-effectiveness of expanding use in various subpopulations, using $50,000/quality-adjusted life-year (QALY) gained, or less, as their cut-off.
Overall, the investigators found that fuller implementation of the Joint Committee goals would pay for itself in reduced cardiovascular morbidity and mortality. The results were driven primarily by secondary prevention in patients with cardiovascular disease and primary prevention in patients with stage 2 hypertension, meaning systolic BP of 160 mm Hg or higher or diastolic BP of 100 mm Hg or higher.
“There is an enormous potential for improving population health by expanding treatment and improving control. Our findings clearly show that it would be worthwhile to significantly increase spending on office visits, home blood pressure monitoring, and interventions to improve treatment adherence. In fact, we could double treatment and monitoring spending for some patients – namely those with severe hypertension – and still break even,” Dr. Moran said in a statement announcing the results.
Treatment of patients with existing cardiovascular disease or stage 2 hypertension would save lives and costs in all men 35-74 years old and in women aged 45-74 years. The treatment of more modest hypertension – systolic BP of 140-159 mm Hg or a diastolic BP of 90-99 mm Hg – was cost effective for all men and for women also between the ages of 45 and 74 years, but treating women 35-44 years old with moderate hypertension and diabetes or kidney disease had intermediate cost-effectiveness ($125,000 per QALY), and low cost-effectiveness ($181,000 per QALY) if those comorbidities were not present.
“Some people will be alarmed about our conclusion that it may not be cost effective to treat hypertension in young adults, especially young women. It’s worth noting that our analysis didn’t capture the cumulative, lifetime effects of hypertension. It may well turn out to be cost effective to treat this group if we look at data on costs and benefits over several decades,” Dr. Moran said.
The team assumed a medication adherence rate of 75%. The costs of treatment included medications, monitoring, and drug side effects.
They did not analyze the effect of diet and lifestyle interventions for lowering blood pressure, or compare the cost-effectiveness of specific antihypertensive medication classes or combinations.
The work was funded by the National Heart, Lung, and Blood Institute, among others. The authors reported no relevant financial disclosures.
Antihypertensive therapy would prevent about 56,000 cardiovascular events annually and 13,000 deaths from strokes, myocardial infarctions, and other causes if it were used by all U.S. adults who qualify for treatment under 2014 Joint National Committee hypertension guidelines, according to computer modeling published online Jan. 28 in the New England Journal of Medicine.
Even though the new Joint Committee guidelines are a bit less stringent than the committee’s prior 2003 advice, blood pressure remains inadequately controlled in 44% of the 64 million U.S. adults with hypertension, according to the investigators, led by Dr. Andrew Moran of Columbia University Medical Center, New York (N. Engl. J. Med. 2015;372:447-55).
The team used data from the National Health and Nutrition Examination Survey, the Framingham Heart Study, and other sources to estimate costs and benefits of expanding treatment to all U.S. adults aged 35-74 years who meet the 2014 benchmarks. They then calculated cost-effectiveness of expanding use in various subpopulations, using $50,000/quality-adjusted life-year (QALY) gained, or less, as their cut-off.
Overall, the investigators found that fuller implementation of the Joint Committee goals would pay for itself in reduced cardiovascular morbidity and mortality. The results were driven primarily by secondary prevention in patients with cardiovascular disease and primary prevention in patients with stage 2 hypertension, meaning systolic BP of 160 mm Hg or higher or diastolic BP of 100 mm Hg or higher.
“There is an enormous potential for improving population health by expanding treatment and improving control. Our findings clearly show that it would be worthwhile to significantly increase spending on office visits, home blood pressure monitoring, and interventions to improve treatment adherence. In fact, we could double treatment and monitoring spending for some patients – namely those with severe hypertension – and still break even,” Dr. Moran said in a statement announcing the results.
Treatment of patients with existing cardiovascular disease or stage 2 hypertension would save lives and costs in all men 35-74 years old and in women aged 45-74 years. The treatment of more modest hypertension – systolic BP of 140-159 mm Hg or a diastolic BP of 90-99 mm Hg – was cost effective for all men and for women also between the ages of 45 and 74 years, but treating women 35-44 years old with moderate hypertension and diabetes or kidney disease had intermediate cost-effectiveness ($125,000 per QALY), and low cost-effectiveness ($181,000 per QALY) if those comorbidities were not present.
“Some people will be alarmed about our conclusion that it may not be cost effective to treat hypertension in young adults, especially young women. It’s worth noting that our analysis didn’t capture the cumulative, lifetime effects of hypertension. It may well turn out to be cost effective to treat this group if we look at data on costs and benefits over several decades,” Dr. Moran said.
The team assumed a medication adherence rate of 75%. The costs of treatment included medications, monitoring, and drug side effects.
They did not analyze the effect of diet and lifestyle interventions for lowering blood pressure, or compare the cost-effectiveness of specific antihypertensive medication classes or combinations.
The work was funded by the National Heart, Lung, and Blood Institute, among others. The authors reported no relevant financial disclosures.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Key clinical point: Even young patients, in many cases, can benefit from adherence to theJoint National Committee 2014 hypertension guidelines.
Major finding: The treatment of modest hypertension is cost effective for men 35-74 years old, and women between the ages of 45 and 74 years, meaning that each quality-adjusted life-year gained would cost less than $50,000.
Data source: Computer estimates of the impact of applying JNC-8 to all hypertensive U.S. adults 35-74 years old.
Disclosures: The work was funded by the National Heart, Lung, and Blood Institute, among others. The authors reported no relevant financial disclosures.

Icatibant rapidly resolved ACE inhibitor–induced angioedema
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Angioedema caused by ACE inhibitors resolved 70% more rapidly with icatibant than did standard therapy in a multicenter phase II study in Germany, which was reported online Jan. 29 in the New England Journal of Medicine.
Because of the increasing use of ACE inhibitors, approximately one-third of all cases of angioedema treated in emergency departments now are attributed to these agents. The current standard ED treatment of ACE inhibitor–induced angioedema is glucocorticoids plus antihistamines. However, patients generally don’t respond to this therapy, likely because this form of angioedema isn’t a histamine-mediated reaction. Instead, it is thought by some to be a bradykinin-mediated reaction, said Dr. Murat Bas of the department of otorhinolaryngology, Technische Universität München (Germany), and his associates.
Icatibant injections (Firazyr) are approved by the Food and Drug Administration for the treatment of acute attacks of hereditary angioedema in adults 18 years of age and older. The drug also is being studied in the United States for the treatment of ACE-inhibitor–induced angioedema.
Since ACE inhibitors interfere with the breakdown of bradykinin, and bradykinin-mediated hereditary angioedema is usually treated with bradykinin-receptor antagonists such as icatibant, the investigators performed a double-blind randomized trial comparing subcutaneous icatibant against standard treatment in 27 adults who presented to four German EDs during a 1.5-year period.
The primary endpoint – the time to complete resolution of ACE inhibitor–induced angioedema – was 8 hours with icatibant and 27 hours with standard therapy. Angioedema resolved within 4 hours in five patients (38%) given icatibant; none of the patients given standard therapy responded that quickly. The onset of symptom relief was 2 hours with icatibant and 12 hours with standard glucocorticoids plus antihistamines, a significant difference as judged by the study participants and the researchers. Also, the physician-assessed severity of angioedema began to abate within 1 hour of icatibant administration and within 8 hours for standard treatment (N. Engl. J. Med. 2015 Jan. 29 [doi:10.1056/NEJMoa1312524]).
“Although the sample size in this trial was too small to allow for a robust evaluation of safety, no patient discontinued participation in the study owing to adverse events,” Dr. Bas and his associates added.
Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Key clinical point: Icatibant may prove to be a more effective treatment than glucocorticoids and antihistamines for ACE inhibitor–induced angioedema.
Major finding: The time to complete resolution of ACE inhibitor–induced angioedema was 8 hours with icatibant and 27 hours with standard therapy.
Data source: A multicenter double-blind randomized phase II clinical trial involving 27 adults hospitalized in Germany for ACE inhibitor–induced angioedema during a 1.5-year period.
Disclosures: This study was supported by an educational grant from Shire and by the Federal Ministry of Education and Research of Germany. Dr. Bas reported receiving grants and personal fees from Shire, the maker of icatibant, as did some of his associates.
Fluid Resuscitation in Sepsis: A Systematic Review and Network Meta-Analysis
Clinical question: Is there any difference between different resuscitative fluids (crystalloids or colloids) on mortality in critically ill sepsis patients?
Background: Fluid resuscitation, in addition to antibiotics and source control, is a cornerstone of initial management of sepsis. Resuscitation with crystalloids compared with colloids for critically ill patients has been evaluated in large randomized controlled trials and meta-analyses; however, whether any of these fluid properties translates into a survival advantage, particularly regarding the optimal fluid for resuscitation in critically ill sepsis patients, remains unclear.
Study design: Systematic review, network meta-analysis (NMA).
Setting: Database search in MEDLINE, Embase, ACP Journal Club, Cumulative Index to Nursing and Allied Health Literature (CINAHL), HealthSTAR, the Allied and Complementary Medicine Database (AMED), and the Cochrane Central Register of Controlled Trials (CENTRAL).
Synopsis: Of 9,875 records that involved adult (age =16 years) critically ill patients with severe sepsis or septic shock who required fluid resuscitation, with no restrictions on language or publication date, 14 randomized controlled trials were considered eligible. Interventions studied included any fluid or fluid strategy used for resuscitation compared with another fluid or fluid strategy. The endpoint was 90-day mortality or, if not available, 30-day intensive care unit or hospital mortality, whichever was longest.
The analysis classified fluids as crystalloids or colloids. The relevant analyses were a four-node NMA (crystalloids versus albumin versus hydroxyethyl starch [HES] versus gelatin) and a six-node NMA (crystalloids versus albumin versus HES versus gelatin, with crystalloids divided into balanced or unbalanced and HES divided into low or high molecular weight), and a conventional fixed-effects meta-analytic comparison of crystalloids versus colloids.
In the four-node analysis, the results suggested higher mortality with starches (versus crystalloids) and lower mortality with albumin (versus crystalloids and starches). In the six-node analysis, the results suggested that albumin is superior to saline and low-molecular-weight starch and that balanced crystalloids are superior to saline and starch (both high and low molecular weight).
These results highlight potentially important differences in mortality among solutions; they suggest an advantage of balanced crystalloids versus saline and low- or high-molecular-weight starch, with similar mortality results for balanced crystalloids and albumin. These differences were not detectable using a standard meta-analytic approach directly comparing “any crystalloids versus any colloids.” Biological rationale is consistent with the findings of lower mortality with balanced crystalloid solutions than with saline as it mimics the homeostatic composition of body fluids to a greater extent than unbalanced fluids. These results raise concerns about using unbalanced crystalloids in the acute resuscitation of patients with sepsis.
Bottom line: Among patients with sepsis, resuscitation with balanced crystalloids or albumin is associated with reduced mortality compared to other fluids.
Clinical question: Is there any difference between different resuscitative fluids (crystalloids or colloids) on mortality in critically ill sepsis patients?
Background: Fluid resuscitation, in addition to antibiotics and source control, is a cornerstone of initial management of sepsis. Resuscitation with crystalloids compared with colloids for critically ill patients has been evaluated in large randomized controlled trials and meta-analyses; however, whether any of these fluid properties translates into a survival advantage, particularly regarding the optimal fluid for resuscitation in critically ill sepsis patients, remains unclear.
Study design: Systematic review, network meta-analysis (NMA).
Setting: Database search in MEDLINE, Embase, ACP Journal Club, Cumulative Index to Nursing and Allied Health Literature (CINAHL), HealthSTAR, the Allied and Complementary Medicine Database (AMED), and the Cochrane Central Register of Controlled Trials (CENTRAL).
Synopsis: Of 9,875 records that involved adult (age =16 years) critically ill patients with severe sepsis or septic shock who required fluid resuscitation, with no restrictions on language or publication date, 14 randomized controlled trials were considered eligible. Interventions studied included any fluid or fluid strategy used for resuscitation compared with another fluid or fluid strategy. The endpoint was 90-day mortality or, if not available, 30-day intensive care unit or hospital mortality, whichever was longest.
The analysis classified fluids as crystalloids or colloids. The relevant analyses were a four-node NMA (crystalloids versus albumin versus hydroxyethyl starch [HES] versus gelatin) and a six-node NMA (crystalloids versus albumin versus HES versus gelatin, with crystalloids divided into balanced or unbalanced and HES divided into low or high molecular weight), and a conventional fixed-effects meta-analytic comparison of crystalloids versus colloids.
In the four-node analysis, the results suggested higher mortality with starches (versus crystalloids) and lower mortality with albumin (versus crystalloids and starches). In the six-node analysis, the results suggested that albumin is superior to saline and low-molecular-weight starch and that balanced crystalloids are superior to saline and starch (both high and low molecular weight).
These results highlight potentially important differences in mortality among solutions; they suggest an advantage of balanced crystalloids versus saline and low- or high-molecular-weight starch, with similar mortality results for balanced crystalloids and albumin. These differences were not detectable using a standard meta-analytic approach directly comparing “any crystalloids versus any colloids.” Biological rationale is consistent with the findings of lower mortality with balanced crystalloid solutions than with saline as it mimics the homeostatic composition of body fluids to a greater extent than unbalanced fluids. These results raise concerns about using unbalanced crystalloids in the acute resuscitation of patients with sepsis.
Bottom line: Among patients with sepsis, resuscitation with balanced crystalloids or albumin is associated with reduced mortality compared to other fluids.
Clinical question: Is there any difference between different resuscitative fluids (crystalloids or colloids) on mortality in critically ill sepsis patients?
Background: Fluid resuscitation, in addition to antibiotics and source control, is a cornerstone of initial management of sepsis. Resuscitation with crystalloids compared with colloids for critically ill patients has been evaluated in large randomized controlled trials and meta-analyses; however, whether any of these fluid properties translates into a survival advantage, particularly regarding the optimal fluid for resuscitation in critically ill sepsis patients, remains unclear.
Study design: Systematic review, network meta-analysis (NMA).
Setting: Database search in MEDLINE, Embase, ACP Journal Club, Cumulative Index to Nursing and Allied Health Literature (CINAHL), HealthSTAR, the Allied and Complementary Medicine Database (AMED), and the Cochrane Central Register of Controlled Trials (CENTRAL).
Synopsis: Of 9,875 records that involved adult (age =16 years) critically ill patients with severe sepsis or septic shock who required fluid resuscitation, with no restrictions on language or publication date, 14 randomized controlled trials were considered eligible. Interventions studied included any fluid or fluid strategy used for resuscitation compared with another fluid or fluid strategy. The endpoint was 90-day mortality or, if not available, 30-day intensive care unit or hospital mortality, whichever was longest.
The analysis classified fluids as crystalloids or colloids. The relevant analyses were a four-node NMA (crystalloids versus albumin versus hydroxyethyl starch [HES] versus gelatin) and a six-node NMA (crystalloids versus albumin versus HES versus gelatin, with crystalloids divided into balanced or unbalanced and HES divided into low or high molecular weight), and a conventional fixed-effects meta-analytic comparison of crystalloids versus colloids.
In the four-node analysis, the results suggested higher mortality with starches (versus crystalloids) and lower mortality with albumin (versus crystalloids and starches). In the six-node analysis, the results suggested that albumin is superior to saline and low-molecular-weight starch and that balanced crystalloids are superior to saline and starch (both high and low molecular weight).
These results highlight potentially important differences in mortality among solutions; they suggest an advantage of balanced crystalloids versus saline and low- or high-molecular-weight starch, with similar mortality results for balanced crystalloids and albumin. These differences were not detectable using a standard meta-analytic approach directly comparing “any crystalloids versus any colloids.” Biological rationale is consistent with the findings of lower mortality with balanced crystalloid solutions than with saline as it mimics the homeostatic composition of body fluids to a greater extent than unbalanced fluids. These results raise concerns about using unbalanced crystalloids in the acute resuscitation of patients with sepsis.
Bottom line: Among patients with sepsis, resuscitation with balanced crystalloids or albumin is associated with reduced mortality compared to other fluids.
Physician Dashboards, Pay-for-Performance Deliver Better VTE Prophylaxis Rates
The combination of direct feedback from web-based physician dashboards and a pay-for-performance program “significantly improved” hospitalists’ compliance with VTE prophylaxis, according to a recent study in the Journal of Hospital Medicine.
In the report, hospitalist Henry Michtalik, MD, MPH, MHS, and colleagues noted that while physicians’ compliance increased most by using dashboards, the combination of the two methods is recommended to “combine extrinsic and intrinsic motivations.”
“They end up enhancing each other,” says Dr. Michtalik, associate faculty and international consultant at the Armstrong Institute for Patient Safety and Quality at Johns Hopkins Medicine in Baltimore. “I really don’t think you can have one without the other. If you have a pay-for-performance program without a dashboard, that’s the equivalent of driving down an unfamiliar highway without any signs. You’re not really sure where you were, you’re not really sure where you’re going, you think you’re on the right path, but you would sure feel a lot better if you had some markers along the way.”
The study analyzed 3,144 inpatient admissions at a tertiary-care medical center from 2009 to 2012. During the dashboard-only intervention period, providers improved compliance by 4% on average (95% CI, 3–5; P<0.001). With the addition of the pay-for-performance program, providers improved another 4% (95% CI, 3–5; P<0.001). Group compliance improved from 86% (95% CI, 85–88) during the baseline period to 90% (95% CI, 88–93) during the dashboard period (P=0.01) and 94% (95% CI, 93–96) during the pay-for-performance program (P=0.01).
Dr. Michtalik says that in an era of value-based purchasing and broader healthcare reform, most institutions are already collecting data on VTE prophylaxis. The next step, he says, should be bringing data to the provider level and making that an agent for change.
Visit our website for more information on VTE prophylaxis.
The combination of direct feedback from web-based physician dashboards and a pay-for-performance program “significantly improved” hospitalists’ compliance with VTE prophylaxis, according to a recent study in the Journal of Hospital Medicine.
In the report, hospitalist Henry Michtalik, MD, MPH, MHS, and colleagues noted that while physicians’ compliance increased most by using dashboards, the combination of the two methods is recommended to “combine extrinsic and intrinsic motivations.”
“They end up enhancing each other,” says Dr. Michtalik, associate faculty and international consultant at the Armstrong Institute for Patient Safety and Quality at Johns Hopkins Medicine in Baltimore. “I really don’t think you can have one without the other. If you have a pay-for-performance program without a dashboard, that’s the equivalent of driving down an unfamiliar highway without any signs. You’re not really sure where you were, you’re not really sure where you’re going, you think you’re on the right path, but you would sure feel a lot better if you had some markers along the way.”
The study analyzed 3,144 inpatient admissions at a tertiary-care medical center from 2009 to 2012. During the dashboard-only intervention period, providers improved compliance by 4% on average (95% CI, 3–5; P<0.001). With the addition of the pay-for-performance program, providers improved another 4% (95% CI, 3–5; P<0.001). Group compliance improved from 86% (95% CI, 85–88) during the baseline period to 90% (95% CI, 88–93) during the dashboard period (P=0.01) and 94% (95% CI, 93–96) during the pay-for-performance program (P=0.01).
Dr. Michtalik says that in an era of value-based purchasing and broader healthcare reform, most institutions are already collecting data on VTE prophylaxis. The next step, he says, should be bringing data to the provider level and making that an agent for change.
Visit our website for more information on VTE prophylaxis.
The combination of direct feedback from web-based physician dashboards and a pay-for-performance program “significantly improved” hospitalists’ compliance with VTE prophylaxis, according to a recent study in the Journal of Hospital Medicine.
In the report, hospitalist Henry Michtalik, MD, MPH, MHS, and colleagues noted that while physicians’ compliance increased most by using dashboards, the combination of the two methods is recommended to “combine extrinsic and intrinsic motivations.”
“They end up enhancing each other,” says Dr. Michtalik, associate faculty and international consultant at the Armstrong Institute for Patient Safety and Quality at Johns Hopkins Medicine in Baltimore. “I really don’t think you can have one without the other. If you have a pay-for-performance program without a dashboard, that’s the equivalent of driving down an unfamiliar highway without any signs. You’re not really sure where you were, you’re not really sure where you’re going, you think you’re on the right path, but you would sure feel a lot better if you had some markers along the way.”
The study analyzed 3,144 inpatient admissions at a tertiary-care medical center from 2009 to 2012. During the dashboard-only intervention period, providers improved compliance by 4% on average (95% CI, 3–5; P<0.001). With the addition of the pay-for-performance program, providers improved another 4% (95% CI, 3–5; P<0.001). Group compliance improved from 86% (95% CI, 85–88) during the baseline period to 90% (95% CI, 88–93) during the dashboard period (P=0.01) and 94% (95% CI, 93–96) during the pay-for-performance program (P=0.01).
Dr. Michtalik says that in an era of value-based purchasing and broader healthcare reform, most institutions are already collecting data on VTE prophylaxis. The next step, he says, should be bringing data to the provider level and making that an agent for change.
Visit our website for more information on VTE prophylaxis.
Can social media help mental health practitioners prevent suicides?
Suicide is the tenth leading cause of death among Americans and the third leading cause among those age 15 to 24.1 As many as 36% of suicide victims leave a suicide note.2 Researchers have analyzed such notes with the aim of identifying specific content and patterns that might aid in creating more effective strategies for preventing suicide.3-5
One study found that the presence of a suicide note is an indicator of serious intent; that is, when the initial attempt fails, those who had left a suicide note were found to be at increased risk of subsequent completed suicide.4 Researchers also found that 75% of suicide notes contained the theme “apology/shame,” suggesting that many suicide victims might have welcomed an alternative to suicide to solve their personal predicament. Tragically, however, most suicide notes are not discovered until suicide has been attempted or completed.4
That’s where social media comes in. As platforms for self-expression, social networking sites such as Facebook, Twitter, and Tumblr are sources of real-time information that could aid in suicide prevention.6 With that in mind, we:
• present 2 cases in which a patient announced her suicidal ideation on Facebook
• consider the opportunities that social media present for early intervention
• propose high-tech monitoring methods for high-risk patients.
CASE 1 Major depressive disorder (MDD) and nonadherence
Ms. S, age 24, has a 4-year history of MDD and treatment nonadherence. She had no history of suicide attempt or inpatient treatment, but she had briefly engaged in psychotherapy before discontinuing visits. Physically healthy and employed as a security officer, Ms. S recently broke up with her boyfriend who had abused her physically—and against whom she had an order of protection.
On the day in question, Ms. S posted several status updates on Facebook expressing hopelessness, which, over the course of the day, escalated to expression of frank suicidal ideation:
• “I am ugly, no man would ever want to live with me.”
• “I have made no effect on the world and I’m just a waste of space.”
• “It’s sad that I want to die but such is life. We all die one day.”
• “I’m going to kill myself. It was nice knowing you world. Goodbye everyone.”
CASE 2 Substance abuse and previous suicide attempt
Ms. B, age 21, had a remote (approximately age 16) history of a suicide attempt and was actively abusing 3,4-methylenedioxymethamphetamine (MDMA [“Ecstasy,” “Molly”]) and Cannabis. She was not receiving outpatient care. One afternoon, Ms. B walked into the emergency department (ED) and said she had just taken 17 ibuprofen pills with the intent of killing herself.
On initial evaluation, Ms. B was irritable and uncooperative, denying all psychiatric symptoms and refusing to divulge details of her recent behavior. Her mother, who had not accompanied her daughter to the ED, reported that Ms. B had engaged in excessive risk-taking—speeding, driving while intoxicated, having multiple sex partners—for the past 5 years, resulting in several arrests for minor offenses, and she had been depressed and was sleeping and eating poorly in the 2 weeks leading up to the suicide attempt.
Two days ago, her mother added, Ms. B had posted disturbing notes on Facebook: ”Life is useless,” she declared in one post; “I’d be better off dead,” in another.
Suicidal content online
Worldwide, Facebook has 1.35 billion active users each month.7 Thus far, a limited number of posts indicating suicidal intent have been reported in the lay press,8 but evidence suggests that the use of social media for this purpose is an emerging trend.9
A search of the literature yielded only 3 case reports.8,10,11 In one case, a delayed response to a suicide note resulted in a failure to prevent the suicide.8 In another, a clinician’s discovery of a patient’s explicitly suicidal Facebook post led to what the team leader described as a more meaningful therapeutic relationship.10 The clinician’s discovery might have been pivotal in preventing the patient from committing suicide.
The authors of these case reports explored the idea of using Facebook for suicide prevention, raising a number of practical and ethical issues. Among them are the potential for immediate intervention by other Facebook users and the extent to which suicidal posts on social media sites induce copycat suicides.8
Issues associated with clinicians’ use of social media to follow or monitor patients include the ethical concepts of beneficence and nonmaleficence, privacy and confidentiality, clinical judgment, and informed consent,8,10 including potential benefit and harm and the difference between actual and perceived privacy violations. Bennett et al11 recommend developing guidelines for the use of social media to enhance medical care and provide appropriate protections to both patients and providers.
Reporting suicidal content. Although the primary purpose of Facebook is to give users the opportunity to share life events and thoughts with friends and family, the company does address the question of suicidal content in its Help Center (Box 1).12 As our 2 cases illustrate, however, intervention can be significantly delayed. 
CASE 1 CONTINUED Call to 911
Fortunately for Ms. S, a friend who read her Facebook posts called 911; even then, however, 16 hours passed between the initial postings and the patient’s arrival at the ED. When emergency medical services brought Ms. S to the Comprehensive Psychiatry Emergency Program, she acknowledged suicidal ideation without an active plan. Other symptoms included depressed mood, a sense of hopelessness, feelings of worthlessness lasting >2 months, low self-esteem, dissatisfaction with body image, and a recent verbal altercation with a friend.
Ms. S was admitted to the inpatient unit for further observation and stabilization.
CASE 2 CONTINUED No one answered her calls
Ms. B, who did not arrive at the ED until 2 days after her suicidal posts, corroborated the history given by her mother. She also reported that she had attempted to reach out to her friends for support, but no one had answered her phone calls. She felt hurt because of this, Ms. B said, and impulsively ingested the pills.
Ms. B said she regretted the suicide attempt. Nevertheless, in light of her recent attempt and persistent distress, she was admitted to the inpatient unit for observation and stabilization.
Can artificial intelligence help?
There is no effective means of tracking high-risk patients after their first contact with the mental health system, despite the fact that (1) those who attempt suicide are at high risk of subsequent suicide attempts3 and (2) we have the potential to prevent future attempts based on self-expressed online cues. We propose machine learning algorithms—a branch of artificial intelligence—to capture and process suicide notes on Facebook in real time.
Machine learning can be broadly defined as computational methods using experience to improve performance or make accurate predictions. In this context, “experience” refers to past information, typically in the form of electronic data collected and analyzed to design accurate and efficient predictive algorithms. Machine learning, which incorporates fundamental concepts in computer science, as well as statistics, probability, and optimization, already has been established in a variety of applications, such as detecting e-mail spam, natural language processing, and computational biology.13
Affective computing, known as emotion-oriented computing, is a branch of artificial intelligence that involves the design of systems and devices that can recognize, interpret, and process human moods and emotions (Box 2).14
Prediction models, developed by Poulin et al15 to estimate the risk of suicide (based on keywords and multiword phrases from unstructured clinical notes from a national sample of U.S. Veterans Administration medical records), resulted in an inference accuracy of ≥65%. Pestian et al16 created and annotated a collection of suicide notes—a vital resource for scientists to use for machine learning and data mining. Machine learning algorithms based on such notes and clinical data might be used to capture alarming social media posts by high-risk patients and activate crisis management, with potentially life-saving results.
But limitations remain
It is not easy to identify or analyze people’s emotions based on social media posts; emotions can be implicit, based on specific events or situations. To distinguish among different emotions purely on the basis of keywords is to deal in great subtlety. Framing algorithms to include multiple parameters—the duration of suicidal content and the number of suicidal posts, for example—would help mitigate the risk of false alarms.
Another problem is that not all Facebook profiles are public. In fact, only 28% of users share all or most of their posts with anyone other than their friends.17 This limitation could be addressed by urging patients identified as being at high risk of suicide during an initial clinical encounter with a mental health provider to “friend” a generic Web page created by the hospital or clinic to protect patients’ privacy.
As Levahot et al10 wrote in their report of the patient whose clinician discovered a patient’s explicitly suicidal Facebook post, the incident “did not hinder the therapeutic alliance.” Instead, the team leader said, the discovery deepened the therapeutic relationship and helped the patient “better understand his mental illness and need for increased support.”
Bottom Line
Machine learning algorithms offer the possibility of analyzing status updates from patients who express suicidal ideation on social media and alerting clinicians to the need for early intervention. There are steps clinicians can take now, however, to take advantage of Facebook, in particular, to monitor and potentially prevent suicide attempts by those at high risk.
Related Resource
• Ahuja AK, Biesaga K, Sudak DM, et al. Suicide on Facebook. J Psychiatr Pract. 2014;20(2):141-146.
Acknowledgement
Zafar Sharif MD, Associate Clinical Professor of Psychiatry, Columbia University College of Physicians and Surgeons, and Director of Psychiatry, Harlem Hospital Center, New York, New York, and Michael Yogman MD, Assistant Clinical Professor of Pediatrics, Harvard Medical School, Boston Children’s Hospital, Boston, Massachusetts, provided insight into the topic and useful feedback on the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Web-based Injury Statistics Query and Reporting System (WISQARS) 2010. http://www.cdc.gov/injury/wisqars/index.html. Updated July 7, 2014. Accessed January 19, 2015.
2. Shioiri T, Nishimura A, Akazawa K, et al. Incidence of note-leaving remains constant despite increasing suicide rates. Psychiatr Clin Neurosci. 2005;59(2):226-228.
3. Barr W, Leitner M, Thomas J. Self-harm or attempted suicide? Do suicide notes help us decide the level of intent in those who survive? Accid Emerg Nurs. 2007;15(3):122-127.
4. Foster T. Suicide note themes and suicide prevention. Int J Psychiatry Med. 2003;33(4):323-331.
5. Bhatia MS, Verma SK, Murty OP. Suicide notes: psychological and clinical profile. Int J Psychiatry Med. 2006;36(2):163-170.
6. Jashinsky J, Burton SH, Hanson CL, et al. Tracking suicide risk factors through Twitter in the US. Crisis. 2014;35(1):51-59.
7. Facebook news room. Company info. http://newsroom. fb.com/company-info. Accessed January 7, 2015.
8. Ruder TD, Hatch GM, Ampanozi G, et al. Suicide announcement on Facebook. Crisis. 2011;32(5):280-282.
9. Luxton DD, June JD, Fairall JM. Social media and suicide: a public health perspective. Am J Public Health. 2012;102(suppl 2):S195-S200.
10. Lehavot K, Ben-Zeev D, Neville RE. Ethical considerations and social media: a case of suicidal postings on Facebook. Journal of Dual Diagnosis. 2012;8(4):341-346.
11. Bennett A, Pourmand A, Shokoohi H, et al. Impacts of social networking sites on patient care in the emergency department. Telemed J E Health. 2014;20(1):94-96.
12. How to report suicidal content/threats on Facebook. h t tps ://www. facebook.com/notes/amer ican-foundation-for-suicide-prevention/how-to-report-suicidal-contentthreats-on-facebook/10150090259398144. Published February 15, 2011. Accessed January 22, 2015.
13. Mohri M, Rostamizadeh A, Talwalker A. Foundations of machine learning (adaptive computation and machine learning series). Cambridge, MA: MIT Press; 2012:14.
14. Blázquez Gil G, Berlanga de Jesús A, Molina Lopéz JM. Combining machine learning techniques and natural language processing to infer emotions using Spanish Twitter corpus. Communications in Computer and Information Science. 2013;365:149-157.
15. Poulin C, Shiner B, Thompson P, et al. Predicting the risk of suicide by analyzing the text of clinical notes. PLoS One. 2014;9(1):e85733.
16. Pestian JP, Matykiewicz P, Linn-Gust M. What’s in a note: construction of a suicide note corpus. Biomed Inform Insights. 2012;5:1-6.
17. ConsumerReports.org. Facebook & your privacy. http:// www.consumerreports.org/cro/magazine/2012/06/ facebook-your-privacy/index.html. Published June 2012. Accessed January 22, 2015
Suicide is the tenth leading cause of death among Americans and the third leading cause among those age 15 to 24.1 As many as 36% of suicide victims leave a suicide note.2 Researchers have analyzed such notes with the aim of identifying specific content and patterns that might aid in creating more effective strategies for preventing suicide.3-5
One study found that the presence of a suicide note is an indicator of serious intent; that is, when the initial attempt fails, those who had left a suicide note were found to be at increased risk of subsequent completed suicide.4 Researchers also found that 75% of suicide notes contained the theme “apology/shame,” suggesting that many suicide victims might have welcomed an alternative to suicide to solve their personal predicament. Tragically, however, most suicide notes are not discovered until suicide has been attempted or completed.4
That’s where social media comes in. As platforms for self-expression, social networking sites such as Facebook, Twitter, and Tumblr are sources of real-time information that could aid in suicide prevention.6 With that in mind, we:
• present 2 cases in which a patient announced her suicidal ideation on Facebook
• consider the opportunities that social media present for early intervention
• propose high-tech monitoring methods for high-risk patients.
CASE 1 Major depressive disorder (MDD) and nonadherence
Ms. S, age 24, has a 4-year history of MDD and treatment nonadherence. She had no history of suicide attempt or inpatient treatment, but she had briefly engaged in psychotherapy before discontinuing visits. Physically healthy and employed as a security officer, Ms. S recently broke up with her boyfriend who had abused her physically—and against whom she had an order of protection.
On the day in question, Ms. S posted several status updates on Facebook expressing hopelessness, which, over the course of the day, escalated to expression of frank suicidal ideation:
• “I am ugly, no man would ever want to live with me.”
• “I have made no effect on the world and I’m just a waste of space.”
• “It’s sad that I want to die but such is life. We all die one day.”
• “I’m going to kill myself. It was nice knowing you world. Goodbye everyone.”
CASE 2 Substance abuse and previous suicide attempt
Ms. B, age 21, had a remote (approximately age 16) history of a suicide attempt and was actively abusing 3,4-methylenedioxymethamphetamine (MDMA [“Ecstasy,” “Molly”]) and Cannabis. She was not receiving outpatient care. One afternoon, Ms. B walked into the emergency department (ED) and said she had just taken 17 ibuprofen pills with the intent of killing herself.
On initial evaluation, Ms. B was irritable and uncooperative, denying all psychiatric symptoms and refusing to divulge details of her recent behavior. Her mother, who had not accompanied her daughter to the ED, reported that Ms. B had engaged in excessive risk-taking—speeding, driving while intoxicated, having multiple sex partners—for the past 5 years, resulting in several arrests for minor offenses, and she had been depressed and was sleeping and eating poorly in the 2 weeks leading up to the suicide attempt.
Two days ago, her mother added, Ms. B had posted disturbing notes on Facebook: ”Life is useless,” she declared in one post; “I’d be better off dead,” in another.
Suicidal content online
Worldwide, Facebook has 1.35 billion active users each month.7 Thus far, a limited number of posts indicating suicidal intent have been reported in the lay press,8 but evidence suggests that the use of social media for this purpose is an emerging trend.9
A search of the literature yielded only 3 case reports.8,10,11 In one case, a delayed response to a suicide note resulted in a failure to prevent the suicide.8 In another, a clinician’s discovery of a patient’s explicitly suicidal Facebook post led to what the team leader described as a more meaningful therapeutic relationship.10 The clinician’s discovery might have been pivotal in preventing the patient from committing suicide.
The authors of these case reports explored the idea of using Facebook for suicide prevention, raising a number of practical and ethical issues. Among them are the potential for immediate intervention by other Facebook users and the extent to which suicidal posts on social media sites induce copycat suicides.8
Issues associated with clinicians’ use of social media to follow or monitor patients include the ethical concepts of beneficence and nonmaleficence, privacy and confidentiality, clinical judgment, and informed consent,8,10 including potential benefit and harm and the difference between actual and perceived privacy violations. Bennett et al11 recommend developing guidelines for the use of social media to enhance medical care and provide appropriate protections to both patients and providers.
Reporting suicidal content. Although the primary purpose of Facebook is to give users the opportunity to share life events and thoughts with friends and family, the company does address the question of suicidal content in its Help Center (Box 1).12 As our 2 cases illustrate, however, intervention can be significantly delayed.
CASE 1 CONTINUED Call to 911
Fortunately for Ms. S, a friend who read her Facebook posts called 911; even then, however, 16 hours passed between the initial postings and the patient’s arrival at the ED. When emergency medical services brought Ms. S to the Comprehensive Psychiatry Emergency Program, she acknowledged suicidal ideation without an active plan. Other symptoms included depressed mood, a sense of hopelessness, feelings of worthlessness lasting >2 months, low self-esteem, dissatisfaction with body image, and a recent verbal altercation with a friend.
Ms. S was admitted to the inpatient unit for further observation and stabilization.
CASE 2 CONTINUED No one answered her calls
Ms. B, who did not arrive at the ED until 2 days after her suicidal posts, corroborated the history given by her mother. She also reported that she had attempted to reach out to her friends for support, but no one had answered her phone calls. She felt hurt because of this, Ms. B said, and impulsively ingested the pills.
Ms. B said she regretted the suicide attempt. Nevertheless, in light of her recent attempt and persistent distress, she was admitted to the inpatient unit for observation and stabilization.
Can artificial intelligence help?
There is no effective means of tracking high-risk patients after their first contact with the mental health system, despite the fact that (1) those who attempt suicide are at high risk of subsequent suicide attempts3 and (2) we have the potential to prevent future attempts based on self-expressed online cues. We propose machine learning algorithms—a branch of artificial intelligence—to capture and process suicide notes on Facebook in real time.
Machine learning can be broadly defined as computational methods using experience to improve performance or make accurate predictions. In this context, “experience” refers to past information, typically in the form of electronic data collected and analyzed to design accurate and efficient predictive algorithms. Machine learning, which incorporates fundamental concepts in computer science, as well as statistics, probability, and optimization, already has been established in a variety of applications, such as detecting e-mail spam, natural language processing, and computational biology.13
Affective computing, known as emotion-oriented computing, is a branch of artificial intelligence that involves the design of systems and devices that can recognize, interpret, and process human moods and emotions (Box 2).14
Prediction models, developed by Poulin et al15 to estimate the risk of suicide (based on keywords and multiword phrases from unstructured clinical notes from a national sample of U.S. Veterans Administration medical records), resulted in an inference accuracy of ≥65%. Pestian et al16 created and annotated a collection of suicide notes—a vital resource for scientists to use for machine learning and data mining. Machine learning algorithms based on such notes and clinical data might be used to capture alarming social media posts by high-risk patients and activate crisis management, with potentially life-saving results.
But limitations remain
It is not easy to identify or analyze people’s emotions based on social media posts; emotions can be implicit, based on specific events or situations. To distinguish among different emotions purely on the basis of keywords is to deal in great subtlety. Framing algorithms to include multiple parameters—the duration of suicidal content and the number of suicidal posts, for example—would help mitigate the risk of false alarms.
Another problem is that not all Facebook profiles are public. In fact, only 28% of users share all or most of their posts with anyone other than their friends.17 This limitation could be addressed by urging patients identified as being at high risk of suicide during an initial clinical encounter with a mental health provider to “friend” a generic Web page created by the hospital or clinic to protect patients’ privacy.
As Levahot et al10 wrote in their report of the patient whose clinician discovered a patient’s explicitly suicidal Facebook post, the incident “did not hinder the therapeutic alliance.” Instead, the team leader said, the discovery deepened the therapeutic relationship and helped the patient “better understand his mental illness and need for increased support.”
Bottom Line
Machine learning algorithms offer the possibility of analyzing status updates from patients who express suicidal ideation on social media and alerting clinicians to the need for early intervention. There are steps clinicians can take now, however, to take advantage of Facebook, in particular, to monitor and potentially prevent suicide attempts by those at high risk.
Related Resource
• Ahuja AK, Biesaga K, Sudak DM, et al. Suicide on Facebook. J Psychiatr Pract. 2014;20(2):141-146.
Acknowledgement
Zafar Sharif MD, Associate Clinical Professor of Psychiatry, Columbia University College of Physicians and Surgeons, and Director of Psychiatry, Harlem Hospital Center, New York, New York, and Michael Yogman MD, Assistant Clinical Professor of Pediatrics, Harvard Medical School, Boston Children’s Hospital, Boston, Massachusetts, provided insight into the topic and useful feedback on the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Suicide is the tenth leading cause of death among Americans and the third leading cause among those age 15 to 24.1 As many as 36% of suicide victims leave a suicide note.2 Researchers have analyzed such notes with the aim of identifying specific content and patterns that might aid in creating more effective strategies for preventing suicide.3-5
One study found that the presence of a suicide note is an indicator of serious intent; that is, when the initial attempt fails, those who had left a suicide note were found to be at increased risk of subsequent completed suicide.4 Researchers also found that 75% of suicide notes contained the theme “apology/shame,” suggesting that many suicide victims might have welcomed an alternative to suicide to solve their personal predicament. Tragically, however, most suicide notes are not discovered until suicide has been attempted or completed.4
That’s where social media comes in. As platforms for self-expression, social networking sites such as Facebook, Twitter, and Tumblr are sources of real-time information that could aid in suicide prevention.6 With that in mind, we:
• present 2 cases in which a patient announced her suicidal ideation on Facebook
• consider the opportunities that social media present for early intervention
• propose high-tech monitoring methods for high-risk patients.
CASE 1 Major depressive disorder (MDD) and nonadherence
Ms. S, age 24, has a 4-year history of MDD and treatment nonadherence. She had no history of suicide attempt or inpatient treatment, but she had briefly engaged in psychotherapy before discontinuing visits. Physically healthy and employed as a security officer, Ms. S recently broke up with her boyfriend who had abused her physically—and against whom she had an order of protection.
On the day in question, Ms. S posted several status updates on Facebook expressing hopelessness, which, over the course of the day, escalated to expression of frank suicidal ideation:
• “I am ugly, no man would ever want to live with me.”
• “I have made no effect on the world and I’m just a waste of space.”
• “It’s sad that I want to die but such is life. We all die one day.”
• “I’m going to kill myself. It was nice knowing you world. Goodbye everyone.”
CASE 2 Substance abuse and previous suicide attempt
Ms. B, age 21, had a remote (approximately age 16) history of a suicide attempt and was actively abusing 3,4-methylenedioxymethamphetamine (MDMA [“Ecstasy,” “Molly”]) and Cannabis. She was not receiving outpatient care. One afternoon, Ms. B walked into the emergency department (ED) and said she had just taken 17 ibuprofen pills with the intent of killing herself.
On initial evaluation, Ms. B was irritable and uncooperative, denying all psychiatric symptoms and refusing to divulge details of her recent behavior. Her mother, who had not accompanied her daughter to the ED, reported that Ms. B had engaged in excessive risk-taking—speeding, driving while intoxicated, having multiple sex partners—for the past 5 years, resulting in several arrests for minor offenses, and she had been depressed and was sleeping and eating poorly in the 2 weeks leading up to the suicide attempt.
Two days ago, her mother added, Ms. B had posted disturbing notes on Facebook: ”Life is useless,” she declared in one post; “I’d be better off dead,” in another.
Suicidal content online
Worldwide, Facebook has 1.35 billion active users each month.7 Thus far, a limited number of posts indicating suicidal intent have been reported in the lay press,8 but evidence suggests that the use of social media for this purpose is an emerging trend.9
A search of the literature yielded only 3 case reports.8,10,11 In one case, a delayed response to a suicide note resulted in a failure to prevent the suicide.8 In another, a clinician’s discovery of a patient’s explicitly suicidal Facebook post led to what the team leader described as a more meaningful therapeutic relationship.10 The clinician’s discovery might have been pivotal in preventing the patient from committing suicide.
The authors of these case reports explored the idea of using Facebook for suicide prevention, raising a number of practical and ethical issues. Among them are the potential for immediate intervention by other Facebook users and the extent to which suicidal posts on social media sites induce copycat suicides.8
Issues associated with clinicians’ use of social media to follow or monitor patients include the ethical concepts of beneficence and nonmaleficence, privacy and confidentiality, clinical judgment, and informed consent,8,10 including potential benefit and harm and the difference between actual and perceived privacy violations. Bennett et al11 recommend developing guidelines for the use of social media to enhance medical care and provide appropriate protections to both patients and providers.
Reporting suicidal content. Although the primary purpose of Facebook is to give users the opportunity to share life events and thoughts with friends and family, the company does address the question of suicidal content in its Help Center (Box 1).12 As our 2 cases illustrate, however, intervention can be significantly delayed.
CASE 1 CONTINUED Call to 911
Fortunately for Ms. S, a friend who read her Facebook posts called 911; even then, however, 16 hours passed between the initial postings and the patient’s arrival at the ED. When emergency medical services brought Ms. S to the Comprehensive Psychiatry Emergency Program, she acknowledged suicidal ideation without an active plan. Other symptoms included depressed mood, a sense of hopelessness, feelings of worthlessness lasting >2 months, low self-esteem, dissatisfaction with body image, and a recent verbal altercation with a friend.
Ms. S was admitted to the inpatient unit for further observation and stabilization.
CASE 2 CONTINUED No one answered her calls
Ms. B, who did not arrive at the ED until 2 days after her suicidal posts, corroborated the history given by her mother. She also reported that she had attempted to reach out to her friends for support, but no one had answered her phone calls. She felt hurt because of this, Ms. B said, and impulsively ingested the pills.
Ms. B said she regretted the suicide attempt. Nevertheless, in light of her recent attempt and persistent distress, she was admitted to the inpatient unit for observation and stabilization.
Can artificial intelligence help?
There is no effective means of tracking high-risk patients after their first contact with the mental health system, despite the fact that (1) those who attempt suicide are at high risk of subsequent suicide attempts3 and (2) we have the potential to prevent future attempts based on self-expressed online cues. We propose machine learning algorithms—a branch of artificial intelligence—to capture and process suicide notes on Facebook in real time.
Machine learning can be broadly defined as computational methods using experience to improve performance or make accurate predictions. In this context, “experience” refers to past information, typically in the form of electronic data collected and analyzed to design accurate and efficient predictive algorithms. Machine learning, which incorporates fundamental concepts in computer science, as well as statistics, probability, and optimization, already has been established in a variety of applications, such as detecting e-mail spam, natural language processing, and computational biology.13
Affective computing, known as emotion-oriented computing, is a branch of artificial intelligence that involves the design of systems and devices that can recognize, interpret, and process human moods and emotions (Box 2).14
Prediction models, developed by Poulin et al15 to estimate the risk of suicide (based on keywords and multiword phrases from unstructured clinical notes from a national sample of U.S. Veterans Administration medical records), resulted in an inference accuracy of ≥65%. Pestian et al16 created and annotated a collection of suicide notes—a vital resource for scientists to use for machine learning and data mining. Machine learning algorithms based on such notes and clinical data might be used to capture alarming social media posts by high-risk patients and activate crisis management, with potentially life-saving results.
But limitations remain
It is not easy to identify or analyze people’s emotions based on social media posts; emotions can be implicit, based on specific events or situations. To distinguish among different emotions purely on the basis of keywords is to deal in great subtlety. Framing algorithms to include multiple parameters—the duration of suicidal content and the number of suicidal posts, for example—would help mitigate the risk of false alarms.
Another problem is that not all Facebook profiles are public. In fact, only 28% of users share all or most of their posts with anyone other than their friends.17 This limitation could be addressed by urging patients identified as being at high risk of suicide during an initial clinical encounter with a mental health provider to “friend” a generic Web page created by the hospital or clinic to protect patients’ privacy.
As Levahot et al10 wrote in their report of the patient whose clinician discovered a patient’s explicitly suicidal Facebook post, the incident “did not hinder the therapeutic alliance.” Instead, the team leader said, the discovery deepened the therapeutic relationship and helped the patient “better understand his mental illness and need for increased support.”
Bottom Line
Machine learning algorithms offer the possibility of analyzing status updates from patients who express suicidal ideation on social media and alerting clinicians to the need for early intervention. There are steps clinicians can take now, however, to take advantage of Facebook, in particular, to monitor and potentially prevent suicide attempts by those at high risk.
Related Resource
• Ahuja AK, Biesaga K, Sudak DM, et al. Suicide on Facebook. J Psychiatr Pract. 2014;20(2):141-146.
Acknowledgement
Zafar Sharif MD, Associate Clinical Professor of Psychiatry, Columbia University College of Physicians and Surgeons, and Director of Psychiatry, Harlem Hospital Center, New York, New York, and Michael Yogman MD, Assistant Clinical Professor of Pediatrics, Harvard Medical School, Boston Children’s Hospital, Boston, Massachusetts, provided insight into the topic and useful feedback on the manuscript of this article.
Disclosures
The authors report no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Centers for Disease Control and Prevention. Web-based Injury Statistics Query and Reporting System (WISQARS) 2010. http://www.cdc.gov/injury/wisqars/index.html. Updated July 7, 2014. Accessed January 19, 2015.
2. Shioiri T, Nishimura A, Akazawa K, et al. Incidence of note-leaving remains constant despite increasing suicide rates. Psychiatr Clin Neurosci. 2005;59(2):226-228.
3. Barr W, Leitner M, Thomas J. Self-harm or attempted suicide? Do suicide notes help us decide the level of intent in those who survive? Accid Emerg Nurs. 2007;15(3):122-127.
4. Foster T. Suicide note themes and suicide prevention. Int J Psychiatry Med. 2003;33(4):323-331.
5. Bhatia MS, Verma SK, Murty OP. Suicide notes: psychological and clinical profile. Int J Psychiatry Med. 2006;36(2):163-170.
6. Jashinsky J, Burton SH, Hanson CL, et al. Tracking suicide risk factors through Twitter in the US. Crisis. 2014;35(1):51-59.
7. Facebook news room. Company info. http://newsroom. fb.com/company-info. Accessed January 7, 2015.
8. Ruder TD, Hatch GM, Ampanozi G, et al. Suicide announcement on Facebook. Crisis. 2011;32(5):280-282.
9. Luxton DD, June JD, Fairall JM. Social media and suicide: a public health perspective. Am J Public Health. 2012;102(suppl 2):S195-S200.
10. Lehavot K, Ben-Zeev D, Neville RE. Ethical considerations and social media: a case of suicidal postings on Facebook. Journal of Dual Diagnosis. 2012;8(4):341-346.
11. Bennett A, Pourmand A, Shokoohi H, et al. Impacts of social networking sites on patient care in the emergency department. Telemed J E Health. 2014;20(1):94-96.
12. How to report suicidal content/threats on Facebook. h t tps ://www. facebook.com/notes/amer ican-foundation-for-suicide-prevention/how-to-report-suicidal-contentthreats-on-facebook/10150090259398144. Published February 15, 2011. Accessed January 22, 2015.
13. Mohri M, Rostamizadeh A, Talwalker A. Foundations of machine learning (adaptive computation and machine learning series). Cambridge, MA: MIT Press; 2012:14.
14. Blázquez Gil G, Berlanga de Jesús A, Molina Lopéz JM. Combining machine learning techniques and natural language processing to infer emotions using Spanish Twitter corpus. Communications in Computer and Information Science. 2013;365:149-157.
15. Poulin C, Shiner B, Thompson P, et al. Predicting the risk of suicide by analyzing the text of clinical notes. PLoS One. 2014;9(1):e85733.
16. Pestian JP, Matykiewicz P, Linn-Gust M. What’s in a note: construction of a suicide note corpus. Biomed Inform Insights. 2012;5:1-6.
17. ConsumerReports.org. Facebook & your privacy. http:// www.consumerreports.org/cro/magazine/2012/06/ facebook-your-privacy/index.html. Published June 2012. Accessed January 22, 2015
1. Centers for Disease Control and Prevention. Web-based Injury Statistics Query and Reporting System (WISQARS) 2010. http://www.cdc.gov/injury/wisqars/index.html. Updated July 7, 2014. Accessed January 19, 2015.
2. Shioiri T, Nishimura A, Akazawa K, et al. Incidence of note-leaving remains constant despite increasing suicide rates. Psychiatr Clin Neurosci. 2005;59(2):226-228.
3. Barr W, Leitner M, Thomas J. Self-harm or attempted suicide? Do suicide notes help us decide the level of intent in those who survive? Accid Emerg Nurs. 2007;15(3):122-127.
4. Foster T. Suicide note themes and suicide prevention. Int J Psychiatry Med. 2003;33(4):323-331.
5. Bhatia MS, Verma SK, Murty OP. Suicide notes: psychological and clinical profile. Int J Psychiatry Med. 2006;36(2):163-170.
6. Jashinsky J, Burton SH, Hanson CL, et al. Tracking suicide risk factors through Twitter in the US. Crisis. 2014;35(1):51-59.
7. Facebook news room. Company info. http://newsroom. fb.com/company-info. Accessed January 7, 2015.
8. Ruder TD, Hatch GM, Ampanozi G, et al. Suicide announcement on Facebook. Crisis. 2011;32(5):280-282.
9. Luxton DD, June JD, Fairall JM. Social media and suicide: a public health perspective. Am J Public Health. 2012;102(suppl 2):S195-S200.
10. Lehavot K, Ben-Zeev D, Neville RE. Ethical considerations and social media: a case of suicidal postings on Facebook. Journal of Dual Diagnosis. 2012;8(4):341-346.
11. Bennett A, Pourmand A, Shokoohi H, et al. Impacts of social networking sites on patient care in the emergency department. Telemed J E Health. 2014;20(1):94-96.
12. How to report suicidal content/threats on Facebook. h t tps ://www. facebook.com/notes/amer ican-foundation-for-suicide-prevention/how-to-report-suicidal-contentthreats-on-facebook/10150090259398144. Published February 15, 2011. Accessed January 22, 2015.
13. Mohri M, Rostamizadeh A, Talwalker A. Foundations of machine learning (adaptive computation and machine learning series). Cambridge, MA: MIT Press; 2012:14.
14. Blázquez Gil G, Berlanga de Jesús A, Molina Lopéz JM. Combining machine learning techniques and natural language processing to infer emotions using Spanish Twitter corpus. Communications in Computer and Information Science. 2013;365:149-157.
15. Poulin C, Shiner B, Thompson P, et al. Predicting the risk of suicide by analyzing the text of clinical notes. PLoS One. 2014;9(1):e85733.
16. Pestian JP, Matykiewicz P, Linn-Gust M. What’s in a note: construction of a suicide note corpus. Biomed Inform Insights. 2012;5:1-6.
17. ConsumerReports.org. Facebook & your privacy. http:// www.consumerreports.org/cro/magazine/2012/06/ facebook-your-privacy/index.html. Published June 2012. Accessed January 22, 2015

Prescriber’s guide to using 3 new antidepressants: Vilazodone, levomilnacipran, vortioxetine
With a prevalence >17%, depression is one of the most common mental disorders in the United States and the second leading cause of disability worldwide.1,2 For decades, primary care and mental health providers have used selective serotonin reuptake inhibitors (SSRIs) as first-line treatment for depression—yet the remission rate after the first trial of an antidepressant is <30%, and continues to decline after a first antidepressant failure.3
That is why clinicians continue to seek effective treatments for depression—ones that will provide quick and sustainable remission—and why scientists and pharmaceutical manufacturers have been competing to develop more effective antidepressant medications.
In the past 4 years, the FDA has approved 3 antidepressants—vilazodone, levomilnacipran, and vortioxetine—with the hope of increasing options for patients who suffer from major depression. These 3 antidepressants differ in their mechanisms of action from other available antidepressants, and all have been shown to have acceptable safety and tolerability profiles.
In this article, we review these novel antidepressants and present some clinical pearls for their use. We also present our observations that each agent appears to show particular advantage in a certain subpopulation of depressed patients who often do not respond, or who do not adequately respond, to other antidepressants.
Vilazodone
Vilazodone was approved by the FDA in 2011 (Table 1). The drug increases serotonin bioavailability in synapses through a strong dual action:
• blocking serotonin reuptake through the serotonin transporter
• partial agonism of the 5-HT1A presynaptic receptor.
Vilazodone also has a moderate effect on the 5-HT4 receptor and on dopamine and norepinephrine uptake inhibition.
The unique presynaptic 5-HT1A partial agonism of vilazodone is similar to that of buspirone, in which both drugs initially inhibit serotonin synthesis and neuronal firing.4 Researchers therefore expected that vilazodone would be more suitable for patients who have depression and a comorbid anxiety disorder; current FDA approval, however, is for depression only.
Adverse effects. The 5-HT4 receptor on which vilazodone acts is present in the gastrointestinal (GI) tract, and contributes to regulating symptoms in patients with irritable bowel syndrome (IBS)5; not surprisingly, the most frequent adverse effects of vilazodone are GI in nature (diarrhea, nausea, vomiting).
Headache is the most common non- GI side effect of vilazodone. Depressed patients who took vilazodone had no significant weight gain and did not report adverse sexual effects, compared with subjects given placebo.6
The following case—a patient with depression, significant anxiety, and IBS— exemplifies the type of patient for whom we find vilazodone most useful.
CASE Ms. A, age 19, is a college student with a history of major depressive disorder, social anxiety, and panic attacks for 2 years and IBS for 3 years. She was taking lubiprostone for IBS, with incomplete relief of GI symptoms. Because the family history included depression in Ms. A’s mother and sister, and both were doing well on escitalopram, we began a trial of that drug, 10 mg/d, that was quickly titrated to 20 mg/d.
Ms. A did not respond to 20 mg of escitalopram combined with psychotherapy.
We then started vilazodone, 10 mg/d after breakfast, for the first week, and reduced escitalopram to 10 mg/d. During Week 2, escitalopram was discontinued and vilazodone was increased to 20 mg/d. During Week 3, vilazodone was titrated to 40 mg/d.
Ms. A tolerated vilazodone well. Her depressive symptoms improved at the end of Week 2.
Unlike her experience with escitalopram, Ms. A’s anxiety symptoms—tenseness, racing thoughts, and panic attacks—all diminished when she switched to vilazodone. Notably, her IBS symptoms also were relieved, and she discontinued lubiprostone.
Ms. A’s depression remained in remission for 2 years, except for a brief period one summer, when she thought she “could do without any medication.” She tapered the vilazodone, week by week, to 10 mg/d, but her anxiety and bowel symptoms resurfaced to a degree that she resumed the 40-mg/d dosage.
Levomilnacipran
This drug is a 2013 addition to the small serotonin–norepinephrine reuptake inhibitor (SNRI) family of venlafaxine, desvenlafaxine, and duloxetine7 (Table 2). Levomilnacipran is the enantiomer of milnacipran, approved in Europe for depression but only for fibromyalgia pain and peripheral neuropathy in the United States.8 (Levomilnacipran is not FDA-approved for treating fibromyalgia pain.)

Levomilnacipran is unique because it is more of an NSRI, so to speak, than an SNRI: That is, the drug’s uptake inhibition of norepinephrine is more potent than its serotonin inhibition. Theoretically, levomilnacipran should help improve cognitive functions linked to the action of norepinephrine, such as concentration and motivation, and in turn, improve social function. The FDA also has approved levomilnacipran for treating functional impairment in depression.9
Adverse effects. The norepinephrine uptake inhibition of levomilnacipran might be responsible for observed increases in heart rate and blood pressure in some patients, and dose-dependent urinary hesitancy and erectile dysfunction in others. The drug has no significant effect on weight in depressed patients, compared with placebo.
Continue to: The benefits of levomilnacipran
The following case illustrates the benefits of levomilnacipran in a depressed patient who suffers from chronic pain and impaired social function.
CASE Mrs. C, age 44, was referred by her outpatient psychologist and her primary care provider for management of refractory depression. She did not respond to an SSRI, an SNRI, or augmentation with bupropion and aripiprazole.
Mrs. C was on disability leave from work because of depression and cervical spine pain that might have been related to repetitive movement as a telephone customer service representative. She complained of loss of motivation, fatigue, and high anxiety about returning to work because of the many unhappy customers she felt she had to soothe.
Levomilnacipran was started at 20 mg/d for 2 days, then titrated to 40 mg/d for 5 days, 80 mg/d for 1 week, and 120 mg/d thereafter. Her previous antidepressants, fluoxetine and bupropion, were discontinued while levomilnacipran was titrated.
Mrs. C continued to receive weekly psychotherapy and physical therapy and to take tizanidine, a muscle relaxant, and over-the-counter medications for pain. Her Patient Health Questionnaire (PHQ-9) score declined from 13 when levomilnacipran was started to 5 at the next visit, 6 weeks later.
Within 4 months of initiating levomilnacipran, Mrs. C returned to work with a series of cue cards to use when speaking with irate or unhappy customers. At that point, her cervical spine pain was barely noticeable and no longer interfered with function.
Vortioxetine
This agent has a novel multimodal mechanism of action (Table 3). It is an SSRI as well as a 5-HT1A full agonist and 5-HT3 receptor antagonist. Vortioxetine also has an inhibitory effect on 5-HT7 and 5-HT1D receptors and partial agonism of 5-HT1B receptors.
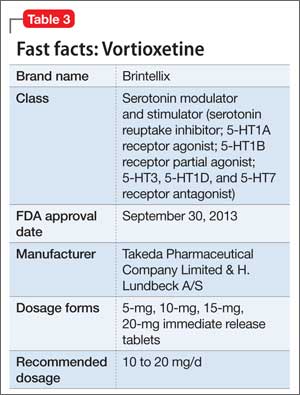
The downstream effect of this multimodal action is an increase in dopamine, norepinephrine, and acetylcholine activity in the prefrontal cortex.10 These downstream effects are thought to help restore some cognitive deficits associated with depression.11
Vortioxetine is the only antidepressant among the 3 discussed in this article that was studied over a long period to ensure that short-term benefits continue beyond the 6- to 8-week acute Phase-III studies. A high remission rate (61%) was observed in patients who were treated on an open-label basis with vortioxetine, 10 mg/d, then randomized to maintenance with vortioxetine or placebo.12
Older patients. Vortioxetine is unique among these 3 antidepressants in that it is the only one studied separately in geriatric patients: In an 8-week Phase-III trial, 452 geriatric patients age 64 to 88 were randomized to 5 mg/d of vortioxetine or placebo.13 Vortioxetine was significantly more effective than placebo at Week 6.
Vortioxetine also is the only antidepressant investigated for an effect on cognitive deficits: In a Phase-III double-blind, placebo-controlled study of 602 patients with major depressive disorder, using duloxetine as active reference, vortioxetine was found to have a significant effect on Digit Symbol Substitution Test scores, compared with placebo, independent of its antidepressant effect (ie, patients who did not show any antidepressant benefit still showed an improvement in attention, speed processing, memory, and executive function).14
We have found, therefore, that vortioxetine is helpful for depressed patients who have cognitive deficits, especially geriatric patients.
CASE Mrs. B, age 84, married, has a 4-year history of depression. She has taken several antidepressants with little consistent relief.
A brief psychiatric hospitalization 2 years ago temporarily reduced the severity of Mrs. B’s depression; gradually, she relapsed. She felt hopeless and resisted another psychiatric evaluation. Mrs. B’s family includes several clinicians, who wondered if she was developing cognitive deficits that were interfering with her recovery.
At initial evaluation, Mrs. B failed to recall 2 of 3 objects but performed the clock drawing test perfectly—qualifying her for a diagnosis of mild cognitive impairment in addition to major depression. Her PHQ-9 score at baseline was 22.
On the assumption that the severity of her depression was contributing to cognitive deficits, vortioxetine, 5 mg/d, was initiated for 2 weeks and then titrated to 10 mg/d.
At 4 weeks’ follow-up, Mrs. B passed the Mini-Cog test; her PHQ-9 score fell to 8. She has remained asymptomatic for 6 months at the 10-mg/d dosage; her lowest PHQ-9 score was 5.
Adverse effects. The most common adverse effects are mild or moderate GI in nature. Weight gain and adverse sexual effects were not significantly different among patients receiving vortioxetine than among patients given placebo.
A note about the safety of these agents
All 3 of these antidepressants carry the standard black-box warning about the elevated risk of suicide in patients taking an antidepressant. None of them are approved for patients age <18.
Continue to: Suicidal ideation was reported
Suicidal ideation was reported in 11.2% of patients taking vortioxetine, compared with 12.5% of those given placebo15; 24% of patients taking levomilnacipran reported suicidal ideation, compared with 22% of those who took placebo.16 In a long-term study of 599 patients taking vilazodone, 4 given placebo exhibited suicidal behavior, compared with 2 who took vilazodone.17
Drug-drug interactions are an important consideration when vilazodone, levomilnacipran, and vortioxetine are prescribed in combination with other medications. See the following discussion.
Vilazodone should be taken with food because it has 72% bioavailability after a meal.18 The drug is metabolized primarily by cytochrome P (CYP) 3A4 and CYP3A5; it does not affect CYP substrates or, it’s likely, produce significant changes to other medications metabolized by the CYP pathway.
Conversely, the dosage of vilazodone should be reduced to 20 mg/d if it is co- administered with a strong CYP3A4 inhibitor (eg, ketoconazole). The dosage should be increased as much as 2-fold when vilazodone is used concomitantly used with a strong CYP3A4 inducer (eg, carbamazepine) for >14 days. The maximum daily dosage should not exceed 80 mg/d.
Levomilnacipran. Unlike vilazodone and vortioxetine, levomilnacipran is affected by renal function.19 Concomitant medications, however, including those that influence CYP renal transporters (particularly CYP3A4, which metabolizes levomilnacipran), do not show an impact on the blood level of levomilnacipran.
No dosage adjustment is needed for patients who have mild renal impairment, but the maintenance dosage of levomilnacipran for patients who have moderate or severe renal impairment should not exceed 80 mg/d in 1 dose, and 60 mg/d in 1 dose, respectively.20
Vortioxetine. Seventy percent of a dose of vortioxetine is absorbed independent of food; the drug has a half-life of 66 hours. Vortioxetine is metabolized primarily by the CYP450 enzyme system, including 2D6, and, to a lesser extent, by CYP3A4, CYP3A5, CYP2C9, and CYP2C19.21
Vortioxetine has minimal effect on P450 substrates in in vitro studies, which was confirmed in 4 other in vivo studies.21-23 In studies of hormonal contraception, bupropion, and omeprazole, vortioxetine did not produce significant changes in the blood level of the other medications. The blood level of vortioxetine increased by 128% when taken with the CYP2D6 inhibitor bupropion,24 but the blood level did not markedly change with other inhibitors because the drug utilizes uses several CYP pathways. Use caution, therefore, when adding bupropion to vortioxetine because the combination elevates the risk of nausea, diarrhea, and headache.
With each agent, specific benefit
Vilazodone, levomilnacipran, and vortioxetine each add distinct benefit to the clinician’s toolbox of treatments for major depressive disorder. Although all antidepressants to some extent alleviate anxiety and pain and reverse cognitive decline associated with depression, our experience suggests using vilazodone for anxious depressed patients; levomilnacipran for depressed patients who experience pain; and vortioxetine for depressed patients who suffer cognitive decline and for geriatric patients.
Bottom Line
The FDA has approved 3 antidepressants in the past 4 years: vilazodone, levomilnacipran, and vortioxetine. The hope is that these agents will bolster treatment options for major depression—perhaps especially so, as we have seen, in the anxious depressed (vilazodone), the depressed in pain (levomilnacipran), and the depressed with cognitive decline, or geriatric patients (vortioxetine).
Related Resources
• Kalia R, Mittal M, Preskorn S. Vilazodone for major depressive disorder. Current Psychiatry. 2011;10(4):84-86,88.
• Lincoln J, Wehler C. Vortioxetine for major depressive disorder. Current Psychiatry. 2014;13(2):67-70.
• Macaluso M, Kazanchi H, Malhotra V. Levomilnacipran for the treatment of major depressive disorder. Current Psychiatry. 2013;12(12):50-52,54,55.
• McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. Int J Neuropsychopharmacol. 2014;17(10):1557-1567.
• Thase ME, Chen D, Edwards J, et al. Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(6):351-356.
Drug Brand Names
Aripiprazole • Abilify Levomilnacipran • Fetzima
Bupropion • Wellbutrin, Zyban Lubiprostone • Amitiza
Buspirone • BuSpar Milnacipran • Savella
Carbamazepine • Tegretol, Equetro Omeprazole • Prilosec
Desvenlafaxine • Pristiq Tizanidine • Zanaflex
Duloxetine • Cymbalta Venlafaxine • Effexor
Escitalopram • Lexapro Vilazodone • Viibryd
Fluoxetine • Prozac Vortioxetine • Brintellix
Ketoconazole • Nizoral
1. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
2. Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547.
3. Warden D, Rush AJ, Trivedi MH, et al. The STAR*D Project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9(6):449-459.
4. Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
5. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
6. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
7. Saraceni MM, Venci JV, Gandhi MA. Levomilnacipran (Fetzima): a new serotonin-norepinephrine reuptake inhibitor for the treatment of major depressive disorder. J Pharm Pract. 2013;27(4):389-395.
8. Deardorff WJ, Grossberg GT. A review of the clinical efficacy, safety and tolerability of the antidepressants vilazodone, levomilnacipran and vortioxetine. Expert Opin Pharmacother. 2014;15(17):2525-2542.
9. Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2013;67(11):1089-1104.
10. Mørk A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666-675.
11. Pehrson AL, Leiser SC, Gulinello M, et al. Treatment of cognitive dysfunction in major depressive disorder-a review of the preclinical evidence for efficacy of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors and the multimodal-acting antidepressant vortioxetine [published online August 5, 2014]. Eur J Pharmacol. doi: 10.1016/j.ejphar.2014.07.044.
12. Baldwin DS, Hansen T, Florea I. Vortioxetine (Lu AA21004) in the long-term open-label treatment of major depressive disorder. Curr Med Res Opin. 2012;28(10):1717-1724.
13. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215-523.
14. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6): 900-909.
15. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
16. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
17. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
18. Boinpally R, Gad N, Gupta S, et al. Influence of CYP3A4 induction/inhibition on the pharmacokinetics of vilazodone in healthy subjects. Clin Ther. 2014; 36(11):1638-1649.
19. Chen L, Boinpally R, Greenberg WM, et al. Effect of hepatic impairment on the pharmacokinetics of levomilnacipran following a single oral dose of a levomilnacipran extended-release capsule in human participants. Clin Drug Investig. 2014;34(5):351-359.
20. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
21. Hvenegaard MG, Bang-Andersen B, Pedersen H, et al. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012; 40(7):1357-1365.
22. Chen G, Lee R, Højer AM, et al. Pharmacokinetic drug interactions involving vortioxetine (Lu AA21004), a multimodal antidepressant. Clin Drug Investig. 2013; 33(10):727-736.
23. Areberg J, Søgaard B, Højer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111(3):198-205.
24. Areberg J, Petersen KB, Chen G, et al. Population pharmacokinetic meta-analysis of vortioxetine in healthy individuals. Basic Clin Pharmacol Toxicol. 2014;115(6):552-559.
With a prevalence >17%, depression is one of the most common mental disorders in the United States and the second leading cause of disability worldwide.1,2 For decades, primary care and mental health providers have used selective serotonin reuptake inhibitors (SSRIs) as first-line treatment for depression—yet the remission rate after the first trial of an antidepressant is <30%, and continues to decline after a first antidepressant failure.3
That is why clinicians continue to seek effective treatments for depression—ones that will provide quick and sustainable remission—and why scientists and pharmaceutical manufacturers have been competing to develop more effective antidepressant medications.
In the past 4 years, the FDA has approved 3 antidepressants—vilazodone, levomilnacipran, and vortioxetine—with the hope of increasing options for patients who suffer from major depression. These 3 antidepressants differ in their mechanisms of action from other available antidepressants, and all have been shown to have acceptable safety and tolerability profiles.
In this article, we review these novel antidepressants and present some clinical pearls for their use. We also present our observations that each agent appears to show particular advantage in a certain subpopulation of depressed patients who often do not respond, or who do not adequately respond, to other antidepressants.
Vilazodone
Vilazodone was approved by the FDA in 2011 (Table 1). The drug increases serotonin bioavailability in synapses through a strong dual action:
• blocking serotonin reuptake through the serotonin transporter
• partial agonism of the 5-HT1A presynaptic receptor.
Vilazodone also has a moderate effect on the 5-HT4 receptor and on dopamine and norepinephrine uptake inhibition.
The unique presynaptic 5-HT1A partial agonism of vilazodone is similar to that of buspirone, in which both drugs initially inhibit serotonin synthesis and neuronal firing.4 Researchers therefore expected that vilazodone would be more suitable for patients who have depression and a comorbid anxiety disorder; current FDA approval, however, is for depression only.
Adverse effects. The 5-HT4 receptor on which vilazodone acts is present in the gastrointestinal (GI) tract, and contributes to regulating symptoms in patients with irritable bowel syndrome (IBS)5; not surprisingly, the most frequent adverse effects of vilazodone are GI in nature (diarrhea, nausea, vomiting).
Headache is the most common non- GI side effect of vilazodone. Depressed patients who took vilazodone had no significant weight gain and did not report adverse sexual effects, compared with subjects given placebo.6
The following case—a patient with depression, significant anxiety, and IBS— exemplifies the type of patient for whom we find vilazodone most useful.
CASE Ms. A, age 19, is a college student with a history of major depressive disorder, social anxiety, and panic attacks for 2 years and IBS for 3 years. She was taking lubiprostone for IBS, with incomplete relief of GI symptoms. Because the family history included depression in Ms. A’s mother and sister, and both were doing well on escitalopram, we began a trial of that drug, 10 mg/d, that was quickly titrated to 20 mg/d.
Ms. A did not respond to 20 mg of escitalopram combined with psychotherapy.
We then started vilazodone, 10 mg/d after breakfast, for the first week, and reduced escitalopram to 10 mg/d. During Week 2, escitalopram was discontinued and vilazodone was increased to 20 mg/d. During Week 3, vilazodone was titrated to 40 mg/d.
Ms. A tolerated vilazodone well. Her depressive symptoms improved at the end of Week 2.
Unlike her experience with escitalopram, Ms. A’s anxiety symptoms—tenseness, racing thoughts, and panic attacks—all diminished when she switched to vilazodone. Notably, her IBS symptoms also were relieved, and she discontinued lubiprostone.
Ms. A’s depression remained in remission for 2 years, except for a brief period one summer, when she thought she “could do without any medication.” She tapered the vilazodone, week by week, to 10 mg/d, but her anxiety and bowel symptoms resurfaced to a degree that she resumed the 40-mg/d dosage.
Levomilnacipran
This drug is a 2013 addition to the small serotonin–norepinephrine reuptake inhibitor (SNRI) family of venlafaxine, desvenlafaxine, and duloxetine7 (Table 2). Levomilnacipran is the enantiomer of milnacipran, approved in Europe for depression but only for fibromyalgia pain and peripheral neuropathy in the United States.8 (Levomilnacipran is not FDA-approved for treating fibromyalgia pain.)
Levomilnacipran is unique because it is more of an NSRI, so to speak, than an SNRI: That is, the drug’s uptake inhibition of norepinephrine is more potent than its serotonin inhibition. Theoretically, levomilnacipran should help improve cognitive functions linked to the action of norepinephrine, such as concentration and motivation, and in turn, improve social function. The FDA also has approved levomilnacipran for treating functional impairment in depression.9
Adverse effects. The norepinephrine uptake inhibition of levomilnacipran might be responsible for observed increases in heart rate and blood pressure in some patients, and dose-dependent urinary hesitancy and erectile dysfunction in others. The drug has no significant effect on weight in depressed patients, compared with placebo.
Continue to: The benefits of levomilnacipran
The following case illustrates the benefits of levomilnacipran in a depressed patient who suffers from chronic pain and impaired social function.
CASE Mrs. C, age 44, was referred by her outpatient psychologist and her primary care provider for management of refractory depression. She did not respond to an SSRI, an SNRI, or augmentation with bupropion and aripiprazole.
Mrs. C was on disability leave from work because of depression and cervical spine pain that might have been related to repetitive movement as a telephone customer service representative. She complained of loss of motivation, fatigue, and high anxiety about returning to work because of the many unhappy customers she felt she had to soothe.
Levomilnacipran was started at 20 mg/d for 2 days, then titrated to 40 mg/d for 5 days, 80 mg/d for 1 week, and 120 mg/d thereafter. Her previous antidepressants, fluoxetine and bupropion, were discontinued while levomilnacipran was titrated.
Mrs. C continued to receive weekly psychotherapy and physical therapy and to take tizanidine, a muscle relaxant, and over-the-counter medications for pain. Her Patient Health Questionnaire (PHQ-9) score declined from 13 when levomilnacipran was started to 5 at the next visit, 6 weeks later.
Within 4 months of initiating levomilnacipran, Mrs. C returned to work with a series of cue cards to use when speaking with irate or unhappy customers. At that point, her cervical spine pain was barely noticeable and no longer interfered with function.
Vortioxetine
This agent has a novel multimodal mechanism of action (Table 3). It is an SSRI as well as a 5-HT1A full agonist and 5-HT3 receptor antagonist. Vortioxetine also has an inhibitory effect on 5-HT7 and 5-HT1D receptors and partial agonism of 5-HT1B receptors.
The downstream effect of this multimodal action is an increase in dopamine, norepinephrine, and acetylcholine activity in the prefrontal cortex.10 These downstream effects are thought to help restore some cognitive deficits associated with depression.11
Vortioxetine is the only antidepressant among the 3 discussed in this article that was studied over a long period to ensure that short-term benefits continue beyond the 6- to 8-week acute Phase-III studies. A high remission rate (61%) was observed in patients who were treated on an open-label basis with vortioxetine, 10 mg/d, then randomized to maintenance with vortioxetine or placebo.12
Older patients. Vortioxetine is unique among these 3 antidepressants in that it is the only one studied separately in geriatric patients: In an 8-week Phase-III trial, 452 geriatric patients age 64 to 88 were randomized to 5 mg/d of vortioxetine or placebo.13 Vortioxetine was significantly more effective than placebo at Week 6.
Vortioxetine also is the only antidepressant investigated for an effect on cognitive deficits: In a Phase-III double-blind, placebo-controlled study of 602 patients with major depressive disorder, using duloxetine as active reference, vortioxetine was found to have a significant effect on Digit Symbol Substitution Test scores, compared with placebo, independent of its antidepressant effect (ie, patients who did not show any antidepressant benefit still showed an improvement in attention, speed processing, memory, and executive function).14
We have found, therefore, that vortioxetine is helpful for depressed patients who have cognitive deficits, especially geriatric patients.
CASE Mrs. B, age 84, married, has a 4-year history of depression. She has taken several antidepressants with little consistent relief.
A brief psychiatric hospitalization 2 years ago temporarily reduced the severity of Mrs. B’s depression; gradually, she relapsed. She felt hopeless and resisted another psychiatric evaluation. Mrs. B’s family includes several clinicians, who wondered if she was developing cognitive deficits that were interfering with her recovery.
At initial evaluation, Mrs. B failed to recall 2 of 3 objects but performed the clock drawing test perfectly—qualifying her for a diagnosis of mild cognitive impairment in addition to major depression. Her PHQ-9 score at baseline was 22.
On the assumption that the severity of her depression was contributing to cognitive deficits, vortioxetine, 5 mg/d, was initiated for 2 weeks and then titrated to 10 mg/d.
At 4 weeks’ follow-up, Mrs. B passed the Mini-Cog test; her PHQ-9 score fell to 8. She has remained asymptomatic for 6 months at the 10-mg/d dosage; her lowest PHQ-9 score was 5.
Adverse effects. The most common adverse effects are mild or moderate GI in nature. Weight gain and adverse sexual effects were not significantly different among patients receiving vortioxetine than among patients given placebo.
A note about the safety of these agents
All 3 of these antidepressants carry the standard black-box warning about the elevated risk of suicide in patients taking an antidepressant. None of them are approved for patients age <18.
Continue to: Suicidal ideation was reported
Suicidal ideation was reported in 11.2% of patients taking vortioxetine, compared with 12.5% of those given placebo15; 24% of patients taking levomilnacipran reported suicidal ideation, compared with 22% of those who took placebo.16 In a long-term study of 599 patients taking vilazodone, 4 given placebo exhibited suicidal behavior, compared with 2 who took vilazodone.17
Drug-drug interactions are an important consideration when vilazodone, levomilnacipran, and vortioxetine are prescribed in combination with other medications. See the following discussion.
Vilazodone should be taken with food because it has 72% bioavailability after a meal.18 The drug is metabolized primarily by cytochrome P (CYP) 3A4 and CYP3A5; it does not affect CYP substrates or, it’s likely, produce significant changes to other medications metabolized by the CYP pathway.
Conversely, the dosage of vilazodone should be reduced to 20 mg/d if it is co- administered with a strong CYP3A4 inhibitor (eg, ketoconazole). The dosage should be increased as much as 2-fold when vilazodone is used concomitantly used with a strong CYP3A4 inducer (eg, carbamazepine) for >14 days. The maximum daily dosage should not exceed 80 mg/d.
Levomilnacipran. Unlike vilazodone and vortioxetine, levomilnacipran is affected by renal function.19 Concomitant medications, however, including those that influence CYP renal transporters (particularly CYP3A4, which metabolizes levomilnacipran), do not show an impact on the blood level of levomilnacipran.
No dosage adjustment is needed for patients who have mild renal impairment, but the maintenance dosage of levomilnacipran for patients who have moderate or severe renal impairment should not exceed 80 mg/d in 1 dose, and 60 mg/d in 1 dose, respectively.20
Vortioxetine. Seventy percent of a dose of vortioxetine is absorbed independent of food; the drug has a half-life of 66 hours. Vortioxetine is metabolized primarily by the CYP450 enzyme system, including 2D6, and, to a lesser extent, by CYP3A4, CYP3A5, CYP2C9, and CYP2C19.21
Vortioxetine has minimal effect on P450 substrates in in vitro studies, which was confirmed in 4 other in vivo studies.21-23 In studies of hormonal contraception, bupropion, and omeprazole, vortioxetine did not produce significant changes in the blood level of the other medications. The blood level of vortioxetine increased by 128% when taken with the CYP2D6 inhibitor bupropion,24 but the blood level did not markedly change with other inhibitors because the drug utilizes uses several CYP pathways. Use caution, therefore, when adding bupropion to vortioxetine because the combination elevates the risk of nausea, diarrhea, and headache.
With each agent, specific benefit
Vilazodone, levomilnacipran, and vortioxetine each add distinct benefit to the clinician’s toolbox of treatments for major depressive disorder. Although all antidepressants to some extent alleviate anxiety and pain and reverse cognitive decline associated with depression, our experience suggests using vilazodone for anxious depressed patients; levomilnacipran for depressed patients who experience pain; and vortioxetine for depressed patients who suffer cognitive decline and for geriatric patients.
Bottom Line
The FDA has approved 3 antidepressants in the past 4 years: vilazodone, levomilnacipran, and vortioxetine. The hope is that these agents will bolster treatment options for major depression—perhaps especially so, as we have seen, in the anxious depressed (vilazodone), the depressed in pain (levomilnacipran), and the depressed with cognitive decline, or geriatric patients (vortioxetine).
Related Resources
• Kalia R, Mittal M, Preskorn S. Vilazodone for major depressive disorder. Current Psychiatry. 2011;10(4):84-86,88.
• Lincoln J, Wehler C. Vortioxetine for major depressive disorder. Current Psychiatry. 2014;13(2):67-70.
• Macaluso M, Kazanchi H, Malhotra V. Levomilnacipran for the treatment of major depressive disorder. Current Psychiatry. 2013;12(12):50-52,54,55.
• McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. Int J Neuropsychopharmacol. 2014;17(10):1557-1567.
• Thase ME, Chen D, Edwards J, et al. Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(6):351-356.
Drug Brand Names
Aripiprazole • Abilify Levomilnacipran • Fetzima
Bupropion • Wellbutrin, Zyban Lubiprostone • Amitiza
Buspirone • BuSpar Milnacipran • Savella
Carbamazepine • Tegretol, Equetro Omeprazole • Prilosec
Desvenlafaxine • Pristiq Tizanidine • Zanaflex
Duloxetine • Cymbalta Venlafaxine • Effexor
Escitalopram • Lexapro Vilazodone • Viibryd
Fluoxetine • Prozac Vortioxetine • Brintellix
Ketoconazole • Nizoral
With a prevalence >17%, depression is one of the most common mental disorders in the United States and the second leading cause of disability worldwide.1,2 For decades, primary care and mental health providers have used selective serotonin reuptake inhibitors (SSRIs) as first-line treatment for depression—yet the remission rate after the first trial of an antidepressant is <30%, and continues to decline after a first antidepressant failure.3
That is why clinicians continue to seek effective treatments for depression—ones that will provide quick and sustainable remission—and why scientists and pharmaceutical manufacturers have been competing to develop more effective antidepressant medications.
In the past 4 years, the FDA has approved 3 antidepressants—vilazodone, levomilnacipran, and vortioxetine—with the hope of increasing options for patients who suffer from major depression. These 3 antidepressants differ in their mechanisms of action from other available antidepressants, and all have been shown to have acceptable safety and tolerability profiles.
In this article, we review these novel antidepressants and present some clinical pearls for their use. We also present our observations that each agent appears to show particular advantage in a certain subpopulation of depressed patients who often do not respond, or who do not adequately respond, to other antidepressants.
Vilazodone
Vilazodone was approved by the FDA in 2011 (Table 1). The drug increases serotonin bioavailability in synapses through a strong dual action:
• blocking serotonin reuptake through the serotonin transporter
• partial agonism of the 5-HT1A presynaptic receptor.
Vilazodone also has a moderate effect on the 5-HT4 receptor and on dopamine and norepinephrine uptake inhibition.
The unique presynaptic 5-HT1A partial agonism of vilazodone is similar to that of buspirone, in which both drugs initially inhibit serotonin synthesis and neuronal firing.4 Researchers therefore expected that vilazodone would be more suitable for patients who have depression and a comorbid anxiety disorder; current FDA approval, however, is for depression only.
Adverse effects. The 5-HT4 receptor on which vilazodone acts is present in the gastrointestinal (GI) tract, and contributes to regulating symptoms in patients with irritable bowel syndrome (IBS)5; not surprisingly, the most frequent adverse effects of vilazodone are GI in nature (diarrhea, nausea, vomiting).
Headache is the most common non- GI side effect of vilazodone. Depressed patients who took vilazodone had no significant weight gain and did not report adverse sexual effects, compared with subjects given placebo.6
The following case—a patient with depression, significant anxiety, and IBS— exemplifies the type of patient for whom we find vilazodone most useful.
CASE Ms. A, age 19, is a college student with a history of major depressive disorder, social anxiety, and panic attacks for 2 years and IBS for 3 years. She was taking lubiprostone for IBS, with incomplete relief of GI symptoms. Because the family history included depression in Ms. A’s mother and sister, and both were doing well on escitalopram, we began a trial of that drug, 10 mg/d, that was quickly titrated to 20 mg/d.
Ms. A did not respond to 20 mg of escitalopram combined with psychotherapy.
We then started vilazodone, 10 mg/d after breakfast, for the first week, and reduced escitalopram to 10 mg/d. During Week 2, escitalopram was discontinued and vilazodone was increased to 20 mg/d. During Week 3, vilazodone was titrated to 40 mg/d.
Ms. A tolerated vilazodone well. Her depressive symptoms improved at the end of Week 2.
Unlike her experience with escitalopram, Ms. A’s anxiety symptoms—tenseness, racing thoughts, and panic attacks—all diminished when she switched to vilazodone. Notably, her IBS symptoms also were relieved, and she discontinued lubiprostone.
Ms. A’s depression remained in remission for 2 years, except for a brief period one summer, when she thought she “could do without any medication.” She tapered the vilazodone, week by week, to 10 mg/d, but her anxiety and bowel symptoms resurfaced to a degree that she resumed the 40-mg/d dosage.
Levomilnacipran
This drug is a 2013 addition to the small serotonin–norepinephrine reuptake inhibitor (SNRI) family of venlafaxine, desvenlafaxine, and duloxetine7 (Table 2). Levomilnacipran is the enantiomer of milnacipran, approved in Europe for depression but only for fibromyalgia pain and peripheral neuropathy in the United States.8 (Levomilnacipran is not FDA-approved for treating fibromyalgia pain.)
Levomilnacipran is unique because it is more of an NSRI, so to speak, than an SNRI: That is, the drug’s uptake inhibition of norepinephrine is more potent than its serotonin inhibition. Theoretically, levomilnacipran should help improve cognitive functions linked to the action of norepinephrine, such as concentration and motivation, and in turn, improve social function. The FDA also has approved levomilnacipran for treating functional impairment in depression.9
Adverse effects. The norepinephrine uptake inhibition of levomilnacipran might be responsible for observed increases in heart rate and blood pressure in some patients, and dose-dependent urinary hesitancy and erectile dysfunction in others. The drug has no significant effect on weight in depressed patients, compared with placebo.
Continue to: The benefits of levomilnacipran
The following case illustrates the benefits of levomilnacipran in a depressed patient who suffers from chronic pain and impaired social function.
CASE Mrs. C, age 44, was referred by her outpatient psychologist and her primary care provider for management of refractory depression. She did not respond to an SSRI, an SNRI, or augmentation with bupropion and aripiprazole.
Mrs. C was on disability leave from work because of depression and cervical spine pain that might have been related to repetitive movement as a telephone customer service representative. She complained of loss of motivation, fatigue, and high anxiety about returning to work because of the many unhappy customers she felt she had to soothe.
Levomilnacipran was started at 20 mg/d for 2 days, then titrated to 40 mg/d for 5 days, 80 mg/d for 1 week, and 120 mg/d thereafter. Her previous antidepressants, fluoxetine and bupropion, were discontinued while levomilnacipran was titrated.
Mrs. C continued to receive weekly psychotherapy and physical therapy and to take tizanidine, a muscle relaxant, and over-the-counter medications for pain. Her Patient Health Questionnaire (PHQ-9) score declined from 13 when levomilnacipran was started to 5 at the next visit, 6 weeks later.
Within 4 months of initiating levomilnacipran, Mrs. C returned to work with a series of cue cards to use when speaking with irate or unhappy customers. At that point, her cervical spine pain was barely noticeable and no longer interfered with function.
Vortioxetine
This agent has a novel multimodal mechanism of action (Table 3). It is an SSRI as well as a 5-HT1A full agonist and 5-HT3 receptor antagonist. Vortioxetine also has an inhibitory effect on 5-HT7 and 5-HT1D receptors and partial agonism of 5-HT1B receptors.
The downstream effect of this multimodal action is an increase in dopamine, norepinephrine, and acetylcholine activity in the prefrontal cortex.10 These downstream effects are thought to help restore some cognitive deficits associated with depression.11
Vortioxetine is the only antidepressant among the 3 discussed in this article that was studied over a long period to ensure that short-term benefits continue beyond the 6- to 8-week acute Phase-III studies. A high remission rate (61%) was observed in patients who were treated on an open-label basis with vortioxetine, 10 mg/d, then randomized to maintenance with vortioxetine or placebo.12
Older patients. Vortioxetine is unique among these 3 antidepressants in that it is the only one studied separately in geriatric patients: In an 8-week Phase-III trial, 452 geriatric patients age 64 to 88 were randomized to 5 mg/d of vortioxetine or placebo.13 Vortioxetine was significantly more effective than placebo at Week 6.
Vortioxetine also is the only antidepressant investigated for an effect on cognitive deficits: In a Phase-III double-blind, placebo-controlled study of 602 patients with major depressive disorder, using duloxetine as active reference, vortioxetine was found to have a significant effect on Digit Symbol Substitution Test scores, compared with placebo, independent of its antidepressant effect (ie, patients who did not show any antidepressant benefit still showed an improvement in attention, speed processing, memory, and executive function).14
We have found, therefore, that vortioxetine is helpful for depressed patients who have cognitive deficits, especially geriatric patients.
CASE Mrs. B, age 84, married, has a 4-year history of depression. She has taken several antidepressants with little consistent relief.
A brief psychiatric hospitalization 2 years ago temporarily reduced the severity of Mrs. B’s depression; gradually, she relapsed. She felt hopeless and resisted another psychiatric evaluation. Mrs. B’s family includes several clinicians, who wondered if she was developing cognitive deficits that were interfering with her recovery.
At initial evaluation, Mrs. B failed to recall 2 of 3 objects but performed the clock drawing test perfectly—qualifying her for a diagnosis of mild cognitive impairment in addition to major depression. Her PHQ-9 score at baseline was 22.
On the assumption that the severity of her depression was contributing to cognitive deficits, vortioxetine, 5 mg/d, was initiated for 2 weeks and then titrated to 10 mg/d.
At 4 weeks’ follow-up, Mrs. B passed the Mini-Cog test; her PHQ-9 score fell to 8. She has remained asymptomatic for 6 months at the 10-mg/d dosage; her lowest PHQ-9 score was 5.
Adverse effects. The most common adverse effects are mild or moderate GI in nature. Weight gain and adverse sexual effects were not significantly different among patients receiving vortioxetine than among patients given placebo.
A note about the safety of these agents
All 3 of these antidepressants carry the standard black-box warning about the elevated risk of suicide in patients taking an antidepressant. None of them are approved for patients age <18.
Continue to: Suicidal ideation was reported
Suicidal ideation was reported in 11.2% of patients taking vortioxetine, compared with 12.5% of those given placebo15; 24% of patients taking levomilnacipran reported suicidal ideation, compared with 22% of those who took placebo.16 In a long-term study of 599 patients taking vilazodone, 4 given placebo exhibited suicidal behavior, compared with 2 who took vilazodone.17
Drug-drug interactions are an important consideration when vilazodone, levomilnacipran, and vortioxetine are prescribed in combination with other medications. See the following discussion.
Vilazodone should be taken with food because it has 72% bioavailability after a meal.18 The drug is metabolized primarily by cytochrome P (CYP) 3A4 and CYP3A5; it does not affect CYP substrates or, it’s likely, produce significant changes to other medications metabolized by the CYP pathway.
Conversely, the dosage of vilazodone should be reduced to 20 mg/d if it is co- administered with a strong CYP3A4 inhibitor (eg, ketoconazole). The dosage should be increased as much as 2-fold when vilazodone is used concomitantly used with a strong CYP3A4 inducer (eg, carbamazepine) for >14 days. The maximum daily dosage should not exceed 80 mg/d.
Levomilnacipran. Unlike vilazodone and vortioxetine, levomilnacipran is affected by renal function.19 Concomitant medications, however, including those that influence CYP renal transporters (particularly CYP3A4, which metabolizes levomilnacipran), do not show an impact on the blood level of levomilnacipran.
No dosage adjustment is needed for patients who have mild renal impairment, but the maintenance dosage of levomilnacipran for patients who have moderate or severe renal impairment should not exceed 80 mg/d in 1 dose, and 60 mg/d in 1 dose, respectively.20
Vortioxetine. Seventy percent of a dose of vortioxetine is absorbed independent of food; the drug has a half-life of 66 hours. Vortioxetine is metabolized primarily by the CYP450 enzyme system, including 2D6, and, to a lesser extent, by CYP3A4, CYP3A5, CYP2C9, and CYP2C19.21
Vortioxetine has minimal effect on P450 substrates in in vitro studies, which was confirmed in 4 other in vivo studies.21-23 In studies of hormonal contraception, bupropion, and omeprazole, vortioxetine did not produce significant changes in the blood level of the other medications. The blood level of vortioxetine increased by 128% when taken with the CYP2D6 inhibitor bupropion,24 but the blood level did not markedly change with other inhibitors because the drug utilizes uses several CYP pathways. Use caution, therefore, when adding bupropion to vortioxetine because the combination elevates the risk of nausea, diarrhea, and headache.
With each agent, specific benefit
Vilazodone, levomilnacipran, and vortioxetine each add distinct benefit to the clinician’s toolbox of treatments for major depressive disorder. Although all antidepressants to some extent alleviate anxiety and pain and reverse cognitive decline associated with depression, our experience suggests using vilazodone for anxious depressed patients; levomilnacipran for depressed patients who experience pain; and vortioxetine for depressed patients who suffer cognitive decline and for geriatric patients.
Bottom Line
The FDA has approved 3 antidepressants in the past 4 years: vilazodone, levomilnacipran, and vortioxetine. The hope is that these agents will bolster treatment options for major depression—perhaps especially so, as we have seen, in the anxious depressed (vilazodone), the depressed in pain (levomilnacipran), and the depressed with cognitive decline, or geriatric patients (vortioxetine).
Related Resources
• Kalia R, Mittal M, Preskorn S. Vilazodone for major depressive disorder. Current Psychiatry. 2011;10(4):84-86,88.
• Lincoln J, Wehler C. Vortioxetine for major depressive disorder. Current Psychiatry. 2014;13(2):67-70.
• Macaluso M, Kazanchi H, Malhotra V. Levomilnacipran for the treatment of major depressive disorder. Current Psychiatry. 2013;12(12):50-52,54,55.
• McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo-controlled study of vortioxetine on cognitive function in depressed adults. Int J Neuropsychopharmacol. 2014;17(10):1557-1567.
• Thase ME, Chen D, Edwards J, et al. Efficacy of vilazodone on anxiety symptoms in patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(6):351-356.
Drug Brand Names
Aripiprazole • Abilify Levomilnacipran • Fetzima
Bupropion • Wellbutrin, Zyban Lubiprostone • Amitiza
Buspirone • BuSpar Milnacipran • Savella
Carbamazepine • Tegretol, Equetro Omeprazole • Prilosec
Desvenlafaxine • Pristiq Tizanidine • Zanaflex
Duloxetine • Cymbalta Venlafaxine • Effexor
Escitalopram • Lexapro Vilazodone • Viibryd
Fluoxetine • Prozac Vortioxetine • Brintellix
Ketoconazole • Nizoral
1. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
2. Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547.
3. Warden D, Rush AJ, Trivedi MH, et al. The STAR*D Project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9(6):449-459.
4. Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
5. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
6. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
7. Saraceni MM, Venci JV, Gandhi MA. Levomilnacipran (Fetzima): a new serotonin-norepinephrine reuptake inhibitor for the treatment of major depressive disorder. J Pharm Pract. 2013;27(4):389-395.
8. Deardorff WJ, Grossberg GT. A review of the clinical efficacy, safety and tolerability of the antidepressants vilazodone, levomilnacipran and vortioxetine. Expert Opin Pharmacother. 2014;15(17):2525-2542.
9. Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2013;67(11):1089-1104.
10. Mørk A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666-675.
11. Pehrson AL, Leiser SC, Gulinello M, et al. Treatment of cognitive dysfunction in major depressive disorder-a review of the preclinical evidence for efficacy of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors and the multimodal-acting antidepressant vortioxetine [published online August 5, 2014]. Eur J Pharmacol. doi: 10.1016/j.ejphar.2014.07.044.
12. Baldwin DS, Hansen T, Florea I. Vortioxetine (Lu AA21004) in the long-term open-label treatment of major depressive disorder. Curr Med Res Opin. 2012;28(10):1717-1724.
13. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215-523.
14. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6): 900-909.
15. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
16. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
17. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
18. Boinpally R, Gad N, Gupta S, et al. Influence of CYP3A4 induction/inhibition on the pharmacokinetics of vilazodone in healthy subjects. Clin Ther. 2014; 36(11):1638-1649.
19. Chen L, Boinpally R, Greenberg WM, et al. Effect of hepatic impairment on the pharmacokinetics of levomilnacipran following a single oral dose of a levomilnacipran extended-release capsule in human participants. Clin Drug Investig. 2014;34(5):351-359.
20. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
21. Hvenegaard MG, Bang-Andersen B, Pedersen H, et al. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012; 40(7):1357-1365.
22. Chen G, Lee R, Højer AM, et al. Pharmacokinetic drug interactions involving vortioxetine (Lu AA21004), a multimodal antidepressant. Clin Drug Investig. 2013; 33(10):727-736.
23. Areberg J, Søgaard B, Højer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111(3):198-205.
24. Areberg J, Petersen KB, Chen G, et al. Population pharmacokinetic meta-analysis of vortioxetine in healthy individuals. Basic Clin Pharmacol Toxicol. 2014;115(6):552-559.
1. Andrade L, Caraveo-Anduaga JJ, Berglund P, et al. The epidemiology of major depressive episodes: results from the International Consortium of Psychiatric Epidemiology (ICPE) Surveys. Int J Methods Psychiatr Res. 2003;12(1):3-21.
2. Ferrari AJ, Charlson FJ, Norman RE, et al. Burden of depressive disorders by country, sex, age, and year: findings from the global burden of disease study 2010. PLoS Med. 2013;10(11):e1001547.
3. Warden D, Rush AJ, Trivedi MH, et al. The STAR*D Project results: a comprehensive review of findings. Curr Psychiatry Rep. 2007;9(6):449-459.
4. Khan A. Vilazodone, a novel dual-acting serotonergic antidepressant for managing major depression. Expert Opin Investig Drugs. 2009;18(11):1753-1764.
5. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
6. Robinson DS, Kajdasz DK, Gallipoli S, et al. A 1-year, open-label study assessing the safety and tolerability of vilazodone in patients with major depressive disorder. J Clin Psychopharmacol. 2011;31(5):643-646.
7. Saraceni MM, Venci JV, Gandhi MA. Levomilnacipran (Fetzima): a new serotonin-norepinephrine reuptake inhibitor for the treatment of major depressive disorder. J Pharm Pract. 2013;27(4):389-395.
8. Deardorff WJ, Grossberg GT. A review of the clinical efficacy, safety and tolerability of the antidepressants vilazodone, levomilnacipran and vortioxetine. Expert Opin Pharmacother. 2014;15(17):2525-2542.
9. Citrome L. Levomilnacipran for major depressive disorder: a systematic review of the efficacy and safety profile for this newly approved antidepressant—what is the number needed to treat, number needed to harm and likelihood to be helped or harmed? Int J Clin Pract. 2013;67(11):1089-1104.
10. Mørk A, Pehrson A, Brennum LT, et al. Pharmacological effects of Lu AA21004: a novel multimodal compound for the treatment of major depressive disorder. J Pharmacol Exp Ther. 2012;340(3):666-675.
11. Pehrson AL, Leiser SC, Gulinello M, et al. Treatment of cognitive dysfunction in major depressive disorder-a review of the preclinical evidence for efficacy of selective serotonin reuptake inhibitors, serotonin-norepinephrine reuptake inhibitors and the multimodal-acting antidepressant vortioxetine [published online August 5, 2014]. Eur J Pharmacol. doi: 10.1016/j.ejphar.2014.07.044.
12. Baldwin DS, Hansen T, Florea I. Vortioxetine (Lu AA21004) in the long-term open-label treatment of major depressive disorder. Curr Med Res Opin. 2012;28(10):1717-1724.
13. Katona C, Hansen T, Olsen CK. A randomized, double-blind, placebo-controlled, duloxetine-referenced, fixed-dose study comparing the efficacy and safety of Lu AA21004 in elderly patients with major depressive disorder. Int Clin Psychopharmacol. 2012;27(4):215-523.
14. Raskin J, Wiltse CG, Siegal A, et al. Efficacy of duloxetine on cognition, depression, and pain in elderly patients with major depressive disorder: an 8-week, double-blind, placebo-controlled trial. Am J Psychiatry. 2007;164(6): 900-909.
15. Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: a randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. Int Clin Psychopharmacol. 2014;29(3):138-149.
16. Mago R, Forero G, Greenberg WM, et al. Safety and tolerability of levomilnacipran ER in major depressive disorder: results from an open-label, 48-week extension study. Clin Drug Investig. 2013;33(10):761-771.
17. Khan A, Sambunaris A, Edwards J, et al. Vilazodone in the treatment of major depressive disorder: efficacy across symptoms and severity of depression. Int Clin Psychopharmacol. 2014;29(2):86-92.
18. Boinpally R, Gad N, Gupta S, et al. Influence of CYP3A4 induction/inhibition on the pharmacokinetics of vilazodone in healthy subjects. Clin Ther. 2014; 36(11):1638-1649.
19. Chen L, Boinpally R, Greenberg WM, et al. Effect of hepatic impairment on the pharmacokinetics of levomilnacipran following a single oral dose of a levomilnacipran extended-release capsule in human participants. Clin Drug Investig. 2014;34(5):351-359.
20. Asnis GM, Bose A, Gommoll CP, et al. Efficacy and safety of levomilnacipran sustained release 40 mg, 80 mg, or 120 mg in major depressive disorder: a phase 3, randomized, double-blind, placebo-controlled study. J Clin Psychiatry. 2013;74(3):242-248.
21. Hvenegaard MG, Bang-Andersen B, Pedersen H, et al. Identification of the cytochrome P450 and other enzymes involved in the in vitro oxidative metabolism of a novel antidepressant, Lu AA21004. Drug Metab Dispos. 2012; 40(7):1357-1365.
22. Chen G, Lee R, Højer AM, et al. Pharmacokinetic drug interactions involving vortioxetine (Lu AA21004), a multimodal antidepressant. Clin Drug Investig. 2013; 33(10):727-736.
23. Areberg J, Søgaard B, Højer AM. The clinical pharmacokinetics of Lu AA21004 and its major metabolite in healthy young volunteers. Basic Clin Pharmacol Toxicol. 2012;111(3):198-205.
24. Areberg J, Petersen KB, Chen G, et al. Population pharmacokinetic meta-analysis of vortioxetine in healthy individuals. Basic Clin Pharmacol Toxicol. 2014;115(6):552-559.
