User login
Dermoscopical screening surpasses app for melanoma diagnosis
A smartphone application was less effective at correctly diagnosing malignant melanoma than clinical diagnosis by dermatologists, based on data from a study evaluating 195 melanocytic lesions.
The app, which used fractal image analysis, was 73% specific and 83% sensitive, whereas the dermatologists’ clinical examinations were 88% sensitive and 97% specific.
Both diagnostic methods’ results were compared to histopathologic analyses of the nevi. The histopathologic analyses found 40 melanomas, 42 dysplastic nevi, and 113 benign nevi.
“The smartphone application ... might be a promising tool in the pre-evaluation of pigmented moles by laypersons,” although the current technology falls short, compared with clinical diagnosis by a dermatologist, according to the study’s researchers.
Find the full study in Journal of the European Academy of Dermatology and Venereology (doi:10.1111/jdv.12648).
A smartphone application was less effective at correctly diagnosing malignant melanoma than clinical diagnosis by dermatologists, based on data from a study evaluating 195 melanocytic lesions.
The app, which used fractal image analysis, was 73% specific and 83% sensitive, whereas the dermatologists’ clinical examinations were 88% sensitive and 97% specific.
Both diagnostic methods’ results were compared to histopathologic analyses of the nevi. The histopathologic analyses found 40 melanomas, 42 dysplastic nevi, and 113 benign nevi.
“The smartphone application ... might be a promising tool in the pre-evaluation of pigmented moles by laypersons,” although the current technology falls short, compared with clinical diagnosis by a dermatologist, according to the study’s researchers.
Find the full study in Journal of the European Academy of Dermatology and Venereology (doi:10.1111/jdv.12648).
A smartphone application was less effective at correctly diagnosing malignant melanoma than clinical diagnosis by dermatologists, based on data from a study evaluating 195 melanocytic lesions.
The app, which used fractal image analysis, was 73% specific and 83% sensitive, whereas the dermatologists’ clinical examinations were 88% sensitive and 97% specific.
Both diagnostic methods’ results were compared to histopathologic analyses of the nevi. The histopathologic analyses found 40 melanomas, 42 dysplastic nevi, and 113 benign nevi.
“The smartphone application ... might be a promising tool in the pre-evaluation of pigmented moles by laypersons,” although the current technology falls short, compared with clinical diagnosis by a dermatologist, according to the study’s researchers.
Find the full study in Journal of the European Academy of Dermatology and Venereology (doi:10.1111/jdv.12648).
Nursing home residents have poor outcomes after lower-extremity revascularization
A substantial number of nursing home residents undergo lower-extremity revascularization each year, but very few of them gain any function and approximately half die within the year, according to a report published online April 6 in JAMA Internal Medicine.
In a population-based analysis of Medicare claims and a database that tracks virtually all U.S. nursing homes, 82% of residents who underwent the procedure during a 3-year period had either died or were unable to walk a year afterward. Most showed a clinically significant decline in function within 3 months of having the procedure, said Dr. Lawrence Oresanya of the department of surgery, University of California, San Francisco, and his associates.
“Our findings can inform conversations between physicians, patients, and families about the risks and expected outcomes of surgery and whether the surgery is likely to be worthwhile. Our findings also highlight the importance of carefully considering a prognosis independent of vascular disease and assessing the goals of care. Ambulatory function … may be impossible to attain,” they wrote.
Lower-extremity revascularization is usually performed to maintain elderly patients’ functional independence by preserving their limbs. But a closer examination of these procedures is warranted in the nursing home population “because nursing home residents, in general, have high levels of functional dependence unrelated to peripheral arterial disease, and higher rates of mortality after most invasive procedures,” the investigators said.
Dr. Oresanya and his colleagues identified 10,784 nursing home residents across the country who underwent lower-extremity revascularization. The procedure was elective in 67% of cases and emergent or urgent in 33%. An endovascular approach was used in 56%, and an open approach in the remainder, with the endovacular approach being more associated with clinical success than open surgery.
The mean patient age was 82 years, and serious comorbidities were very common: 60% had cognitive impairment, 57% had heart failure, and 29% had renal failure. Three-fourths of the patients were nonambulatory at the time of surgery.
The investigators assumed that most patients in this setting had critical limb ischemia rather than claudication. They did not have information about the severity of the lower-extremity ischemia, or about the prevalence or duration of nonhealing wounds or gangrene.
One year after lower-extremity revascularization, mortality was 51% among ambulatory patients and 53% among nonambulatory patients. Only 13% of the entire cohort were able to walk, and only 18% had maintained or improved their presurgical functional status. “Revascularization rarely allowed a nonambulatory resident to become ambulatory,” Dr. Oresanya and his associates wrote (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.0486]).
The researchers were unable to determine whether these poor outcomes resulted from the surgery itself or were due to these patients’ “insufficient physiologic reserve.”
They also cautioned that they confined their study strictly to functional outcomes of lower-extremity revascularization, namely ambulation and mortality. Some patients may have derived other benefits from the procedure, such as relief of pain, healing of wounds, and avoidance of major amputation.
The authors reported having no relevant financial disclosures.
The findings of Oresanya et al. are balanced and valuable, even though the data didn’t give them specific clinical information such as the indications for revascularization and were insensitive to subtle issues such as patient and family wishes for level of care. Such studies point the way to a more rational clinical approach to the care of frail elders with a limited life span but with the prospect of constant pain and discomfort.
But it is important to note that most of the procedures in this study likely were performed to relieve symptoms of ischemic leg pain, nonhealing wounds, or worsening gangrene. In this setting, the surgery should be viewed as a palliative measure rather than as a definitive therapeutic procedure to extend ambulatory function.
William J. Hall, M.D., is at the University of Rochester, New York. He reported having no relevant financial disclosures. Dr. Hall made these remarks in an invited commentary accompanying Dr. Oresanya’s report (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.32]).
The findings of Oresanya et al. are balanced and valuable, even though the data didn’t give them specific clinical information such as the indications for revascularization and were insensitive to subtle issues such as patient and family wishes for level of care. Such studies point the way to a more rational clinical approach to the care of frail elders with a limited life span but with the prospect of constant pain and discomfort.
But it is important to note that most of the procedures in this study likely were performed to relieve symptoms of ischemic leg pain, nonhealing wounds, or worsening gangrene. In this setting, the surgery should be viewed as a palliative measure rather than as a definitive therapeutic procedure to extend ambulatory function.
William J. Hall, M.D., is at the University of Rochester, New York. He reported having no relevant financial disclosures. Dr. Hall made these remarks in an invited commentary accompanying Dr. Oresanya’s report (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.32]).
The findings of Oresanya et al. are balanced and valuable, even though the data didn’t give them specific clinical information such as the indications for revascularization and were insensitive to subtle issues such as patient and family wishes for level of care. Such studies point the way to a more rational clinical approach to the care of frail elders with a limited life span but with the prospect of constant pain and discomfort.
But it is important to note that most of the procedures in this study likely were performed to relieve symptoms of ischemic leg pain, nonhealing wounds, or worsening gangrene. In this setting, the surgery should be viewed as a palliative measure rather than as a definitive therapeutic procedure to extend ambulatory function.
William J. Hall, M.D., is at the University of Rochester, New York. He reported having no relevant financial disclosures. Dr. Hall made these remarks in an invited commentary accompanying Dr. Oresanya’s report (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.32]).
A substantial number of nursing home residents undergo lower-extremity revascularization each year, but very few of them gain any function and approximately half die within the year, according to a report published online April 6 in JAMA Internal Medicine.
In a population-based analysis of Medicare claims and a database that tracks virtually all U.S. nursing homes, 82% of residents who underwent the procedure during a 3-year period had either died or were unable to walk a year afterward. Most showed a clinically significant decline in function within 3 months of having the procedure, said Dr. Lawrence Oresanya of the department of surgery, University of California, San Francisco, and his associates.
“Our findings can inform conversations between physicians, patients, and families about the risks and expected outcomes of surgery and whether the surgery is likely to be worthwhile. Our findings also highlight the importance of carefully considering a prognosis independent of vascular disease and assessing the goals of care. Ambulatory function … may be impossible to attain,” they wrote.
Lower-extremity revascularization is usually performed to maintain elderly patients’ functional independence by preserving their limbs. But a closer examination of these procedures is warranted in the nursing home population “because nursing home residents, in general, have high levels of functional dependence unrelated to peripheral arterial disease, and higher rates of mortality after most invasive procedures,” the investigators said.
Dr. Oresanya and his colleagues identified 10,784 nursing home residents across the country who underwent lower-extremity revascularization. The procedure was elective in 67% of cases and emergent or urgent in 33%. An endovascular approach was used in 56%, and an open approach in the remainder, with the endovacular approach being more associated with clinical success than open surgery.
The mean patient age was 82 years, and serious comorbidities were very common: 60% had cognitive impairment, 57% had heart failure, and 29% had renal failure. Three-fourths of the patients were nonambulatory at the time of surgery.
The investigators assumed that most patients in this setting had critical limb ischemia rather than claudication. They did not have information about the severity of the lower-extremity ischemia, or about the prevalence or duration of nonhealing wounds or gangrene.
One year after lower-extremity revascularization, mortality was 51% among ambulatory patients and 53% among nonambulatory patients. Only 13% of the entire cohort were able to walk, and only 18% had maintained or improved their presurgical functional status. “Revascularization rarely allowed a nonambulatory resident to become ambulatory,” Dr. Oresanya and his associates wrote (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.0486]).
The researchers were unable to determine whether these poor outcomes resulted from the surgery itself or were due to these patients’ “insufficient physiologic reserve.”
They also cautioned that they confined their study strictly to functional outcomes of lower-extremity revascularization, namely ambulation and mortality. Some patients may have derived other benefits from the procedure, such as relief of pain, healing of wounds, and avoidance of major amputation.
The authors reported having no relevant financial disclosures.
A substantial number of nursing home residents undergo lower-extremity revascularization each year, but very few of them gain any function and approximately half die within the year, according to a report published online April 6 in JAMA Internal Medicine.
In a population-based analysis of Medicare claims and a database that tracks virtually all U.S. nursing homes, 82% of residents who underwent the procedure during a 3-year period had either died or were unable to walk a year afterward. Most showed a clinically significant decline in function within 3 months of having the procedure, said Dr. Lawrence Oresanya of the department of surgery, University of California, San Francisco, and his associates.
“Our findings can inform conversations between physicians, patients, and families about the risks and expected outcomes of surgery and whether the surgery is likely to be worthwhile. Our findings also highlight the importance of carefully considering a prognosis independent of vascular disease and assessing the goals of care. Ambulatory function … may be impossible to attain,” they wrote.
Lower-extremity revascularization is usually performed to maintain elderly patients’ functional independence by preserving their limbs. But a closer examination of these procedures is warranted in the nursing home population “because nursing home residents, in general, have high levels of functional dependence unrelated to peripheral arterial disease, and higher rates of mortality after most invasive procedures,” the investigators said.
Dr. Oresanya and his colleagues identified 10,784 nursing home residents across the country who underwent lower-extremity revascularization. The procedure was elective in 67% of cases and emergent or urgent in 33%. An endovascular approach was used in 56%, and an open approach in the remainder, with the endovacular approach being more associated with clinical success than open surgery.
The mean patient age was 82 years, and serious comorbidities were very common: 60% had cognitive impairment, 57% had heart failure, and 29% had renal failure. Three-fourths of the patients were nonambulatory at the time of surgery.
The investigators assumed that most patients in this setting had critical limb ischemia rather than claudication. They did not have information about the severity of the lower-extremity ischemia, or about the prevalence or duration of nonhealing wounds or gangrene.
One year after lower-extremity revascularization, mortality was 51% among ambulatory patients and 53% among nonambulatory patients. Only 13% of the entire cohort were able to walk, and only 18% had maintained or improved their presurgical functional status. “Revascularization rarely allowed a nonambulatory resident to become ambulatory,” Dr. Oresanya and his associates wrote (JAMA Intern. Med. 2015 April 6 [doi:10.1001/jamainternmed.2015.0486]).
The researchers were unable to determine whether these poor outcomes resulted from the surgery itself or were due to these patients’ “insufficient physiologic reserve.”
They also cautioned that they confined their study strictly to functional outcomes of lower-extremity revascularization, namely ambulation and mortality. Some patients may have derived other benefits from the procedure, such as relief of pain, healing of wounds, and avoidance of major amputation.
The authors reported having no relevant financial disclosures.
FROM JAMA INTERNAL MEDICINE
Key clinical point: Many nursing home residents undergo lower-extremity revascularization every year, but few survive and are ambulatory 1 year later.
Major finding: One year after lower-extremity revascularization, mortality was approximately 50%, only 13% of the entire cohort were able to walk, and only 18% had maintained or improved their presurgical functional status.
Data source: A population-based cohort study involving almost all (10,784) U.S. nursing home residents who had lower-extremity revascularization in 2005-2008 and were followed for 1 year.
Disclosures: This study was supported in part by the National Institute on Aging and the University of California, San Francisco, Claude D. Pepper Older Americans Independence Center. Dr. Oresanya and his associates reported having no relevant financial disclosures.
Few PE patients treated with catheter-directed interventions had complications
A review of research suggests that catheter-directed interventions (CDIs) have fewer complications but are not necessarily better at preventing mortality than are standard treatments for pulmonary embolisms, according to Dr. Efthymios D. Avgerinos and Dr. Rabih A. Chaer of the University of Pittsburgh.
Of 594 patients with massive pulmonary embolisms (PEs) who received various forms of CDI, 86.5% survived (range, 40%-100%), according to a systematic review of 35 noncontrolled studies.
“In 95% of these patients, CDIs were initiated without prior intravenous thrombolysis,” while 60%-67% of the patients also received a thrombolytic agent during the procedure, they wrote. The patient survival rate was 91.2% in studies that provided at least 80% of their patients with local thrombolytic therapy during a CDI, compared with 82.8% in studies in which less than 80% of participants received thrombolytic therapy.
Not all findings, however, suggested that it was more favorable for the patients to receive the thrombolytic therapy, Overall, the pooled rates of major and minor complications were 7.9% and 2.4%, respectively. The 25 major complications reported included bleeding complications requiring transfusion, renal failure requiring hemodialysis, cardiopulmonary events, cerebrovascular events, and death.
Other research on CDIs found that right ventricle dilation was reversed in patients with submassive PEs who received fixed-dose, ultrasound-assisted, catheter-directed thrombosis combined with anticoagulation. According to the recently published randomized controlled trial, which compared the effects of fixed-dose, ultrasound-assisted, catheter-directed thrombosis and anticoagulation to anticoagulation alone, the mean right-to-left-ventricle ratio was reduced for patients in the CDI group after 1 day. Such a change did not occur in the control group, but at 90 days, the average ratio “became comparable between the two groups … with a trend in favor of the [CDI],” according to Dr. Avgerinos and Dr. Chaer. None of this study’s participants suffered from major bleeding complications.
“There is increasing evidence that percutaneous CDIs are an essential, effective and safe alternative to systemic thrombolysis or anticoagulation in the contemporary management of massive and submassive PE,” the reviewers noted. More research is needed to confirm the differences in the outcomes between using systemic thrombolysis and catheter-based techniques for treating PEs, as no clinical trial comparing CDIs with systemic thrombolysis for PE has been done, they added.
Read the full review of research in the Journal of Vascular Surgery (doi:10.1016/j.jvs.2014.10.036).
A review of research suggests that catheter-directed interventions (CDIs) have fewer complications but are not necessarily better at preventing mortality than are standard treatments for pulmonary embolisms, according to Dr. Efthymios D. Avgerinos and Dr. Rabih A. Chaer of the University of Pittsburgh.
Of 594 patients with massive pulmonary embolisms (PEs) who received various forms of CDI, 86.5% survived (range, 40%-100%), according to a systematic review of 35 noncontrolled studies.
“In 95% of these patients, CDIs were initiated without prior intravenous thrombolysis,” while 60%-67% of the patients also received a thrombolytic agent during the procedure, they wrote. The patient survival rate was 91.2% in studies that provided at least 80% of their patients with local thrombolytic therapy during a CDI, compared with 82.8% in studies in which less than 80% of participants received thrombolytic therapy.
Not all findings, however, suggested that it was more favorable for the patients to receive the thrombolytic therapy, Overall, the pooled rates of major and minor complications were 7.9% and 2.4%, respectively. The 25 major complications reported included bleeding complications requiring transfusion, renal failure requiring hemodialysis, cardiopulmonary events, cerebrovascular events, and death.
Other research on CDIs found that right ventricle dilation was reversed in patients with submassive PEs who received fixed-dose, ultrasound-assisted, catheter-directed thrombosis combined with anticoagulation. According to the recently published randomized controlled trial, which compared the effects of fixed-dose, ultrasound-assisted, catheter-directed thrombosis and anticoagulation to anticoagulation alone, the mean right-to-left-ventricle ratio was reduced for patients in the CDI group after 1 day. Such a change did not occur in the control group, but at 90 days, the average ratio “became comparable between the two groups … with a trend in favor of the [CDI],” according to Dr. Avgerinos and Dr. Chaer. None of this study’s participants suffered from major bleeding complications.
“There is increasing evidence that percutaneous CDIs are an essential, effective and safe alternative to systemic thrombolysis or anticoagulation in the contemporary management of massive and submassive PE,” the reviewers noted. More research is needed to confirm the differences in the outcomes between using systemic thrombolysis and catheter-based techniques for treating PEs, as no clinical trial comparing CDIs with systemic thrombolysis for PE has been done, they added.
Read the full review of research in the Journal of Vascular Surgery (doi:10.1016/j.jvs.2014.10.036).
A review of research suggests that catheter-directed interventions (CDIs) have fewer complications but are not necessarily better at preventing mortality than are standard treatments for pulmonary embolisms, according to Dr. Efthymios D. Avgerinos and Dr. Rabih A. Chaer of the University of Pittsburgh.
Of 594 patients with massive pulmonary embolisms (PEs) who received various forms of CDI, 86.5% survived (range, 40%-100%), according to a systematic review of 35 noncontrolled studies.
“In 95% of these patients, CDIs were initiated without prior intravenous thrombolysis,” while 60%-67% of the patients also received a thrombolytic agent during the procedure, they wrote. The patient survival rate was 91.2% in studies that provided at least 80% of their patients with local thrombolytic therapy during a CDI, compared with 82.8% in studies in which less than 80% of participants received thrombolytic therapy.
Not all findings, however, suggested that it was more favorable for the patients to receive the thrombolytic therapy, Overall, the pooled rates of major and minor complications were 7.9% and 2.4%, respectively. The 25 major complications reported included bleeding complications requiring transfusion, renal failure requiring hemodialysis, cardiopulmonary events, cerebrovascular events, and death.
Other research on CDIs found that right ventricle dilation was reversed in patients with submassive PEs who received fixed-dose, ultrasound-assisted, catheter-directed thrombosis combined with anticoagulation. According to the recently published randomized controlled trial, which compared the effects of fixed-dose, ultrasound-assisted, catheter-directed thrombosis and anticoagulation to anticoagulation alone, the mean right-to-left-ventricle ratio was reduced for patients in the CDI group after 1 day. Such a change did not occur in the control group, but at 90 days, the average ratio “became comparable between the two groups … with a trend in favor of the [CDI],” according to Dr. Avgerinos and Dr. Chaer. None of this study’s participants suffered from major bleeding complications.
“There is increasing evidence that percutaneous CDIs are an essential, effective and safe alternative to systemic thrombolysis or anticoagulation in the contemporary management of massive and submassive PE,” the reviewers noted. More research is needed to confirm the differences in the outcomes between using systemic thrombolysis and catheter-based techniques for treating PEs, as no clinical trial comparing CDIs with systemic thrombolysis for PE has been done, they added.
Read the full review of research in the Journal of Vascular Surgery (doi:10.1016/j.jvs.2014.10.036).
Low doses of imatinib promote hematopoiesis

Image by Volker Brinkmann
Low doses of the tyrosine kinase inhibitor imatinib can promote hematopoiesis, according to research published in PLOS Pathogens.
Preclinical experiments revealed that the drug can induce differentiation in hematopoietic stem cells (HSCs) and progenitors in the bone marrow,
augment myelopoiesis, and increase the number of myeloid cells in the blood and spleen.
Researchers said these findings suggest imatinib or related drugs could be used to treat infections.
“We think that low doses of imatinib are mimicking ‘emergency hematopoiesis,’ a normal early response to infection,” said study author Daniel Kalman, PhD, of the Emory University School of Medicine in Atlanta, Georgia.
“This was surprising because there are reports that imatinib can be immunosuppressive in some patients. Our data suggest that, at subclinical doses, imatinib can stimulate bone marrow stem cells to produce several types of myeloid cells, such as neutrophils and macrophages, and trigger their exodus from the bone marrow. However, higher doses appear to inhibit this process.”
Dr Kalman and his colleagues observed a 4-fold increase of neutrophils and a 3-fold increase of monocytes in the bone marrow of imatinib-treated mice. However, these mice did not see a significant change in the number of mature B cells, T cells, dendritic cells, eosinophils, or natural killer cells.
Imatinib did not induce the accumulation of HSCs, but it did regulate the accumulation of multipotent progenitors. The drug also reduced the accumulation of HSCs upon infection, which suggests it may increase the flux of HSCs to progenitors.
Imatinib did not increase the number of transplantable HSCs, but it did induce an irreversible commitment of HSCs into progenitors that could differentiate into myeloid cells ex vivo.
Imatinib induced an irreversible differentiation of HSCs or progenitors into myeloid cells in a dose-dependent manner. The drug facilitated the exodus of myeloid cells from the bone marrow only at lower doses. It inhibited this same process at higher doses.
Despite increasing the number of neutrophils, low doses of imatinib did not activate neutrophils. However, the cells retained the capacity to activate upon infection.
In fact, an increase in the number of neutrophils was sufficient to reduce Mycobacterium marinum bacterial load. And imatinib reduced the bacterial load in mice infected with pathogenic Francisella species.
The researchers said these results suggest low doses of imatinib could potentially be used to treat a variety of infections and might prove particularly useful in immunocompromised patients. ![]()
Image by Volker Brinkmann
Low doses of the tyrosine kinase inhibitor imatinib can promote hematopoiesis, according to research published in PLOS Pathogens.
Preclinical experiments revealed that the drug can induce differentiation in hematopoietic stem cells (HSCs) and progenitors in the bone marrow,
augment myelopoiesis, and increase the number of myeloid cells in the blood and spleen.
Researchers said these findings suggest imatinib or related drugs could be used to treat infections.
“We think that low doses of imatinib are mimicking ‘emergency hematopoiesis,’ a normal early response to infection,” said study author Daniel Kalman, PhD, of the Emory University School of Medicine in Atlanta, Georgia.
“This was surprising because there are reports that imatinib can be immunosuppressive in some patients. Our data suggest that, at subclinical doses, imatinib can stimulate bone marrow stem cells to produce several types of myeloid cells, such as neutrophils and macrophages, and trigger their exodus from the bone marrow. However, higher doses appear to inhibit this process.”
Dr Kalman and his colleagues observed a 4-fold increase of neutrophils and a 3-fold increase of monocytes in the bone marrow of imatinib-treated mice. However, these mice did not see a significant change in the number of mature B cells, T cells, dendritic cells, eosinophils, or natural killer cells.
Imatinib did not induce the accumulation of HSCs, but it did regulate the accumulation of multipotent progenitors. The drug also reduced the accumulation of HSCs upon infection, which suggests it may increase the flux of HSCs to progenitors.
Imatinib did not increase the number of transplantable HSCs, but it did induce an irreversible commitment of HSCs into progenitors that could differentiate into myeloid cells ex vivo.
Imatinib induced an irreversible differentiation of HSCs or progenitors into myeloid cells in a dose-dependent manner. The drug facilitated the exodus of myeloid cells from the bone marrow only at lower doses. It inhibited this same process at higher doses.
Despite increasing the number of neutrophils, low doses of imatinib did not activate neutrophils. However, the cells retained the capacity to activate upon infection.
In fact, an increase in the number of neutrophils was sufficient to reduce Mycobacterium marinum bacterial load. And imatinib reduced the bacterial load in mice infected with pathogenic Francisella species.
The researchers said these results suggest low doses of imatinib could potentially be used to treat a variety of infections and might prove particularly useful in immunocompromised patients. ![]()
Image by Volker Brinkmann
Low doses of the tyrosine kinase inhibitor imatinib can promote hematopoiesis, according to research published in PLOS Pathogens.
Preclinical experiments revealed that the drug can induce differentiation in hematopoietic stem cells (HSCs) and progenitors in the bone marrow,
augment myelopoiesis, and increase the number of myeloid cells in the blood and spleen.
Researchers said these findings suggest imatinib or related drugs could be used to treat infections.
“We think that low doses of imatinib are mimicking ‘emergency hematopoiesis,’ a normal early response to infection,” said study author Daniel Kalman, PhD, of the Emory University School of Medicine in Atlanta, Georgia.
“This was surprising because there are reports that imatinib can be immunosuppressive in some patients. Our data suggest that, at subclinical doses, imatinib can stimulate bone marrow stem cells to produce several types of myeloid cells, such as neutrophils and macrophages, and trigger their exodus from the bone marrow. However, higher doses appear to inhibit this process.”
Dr Kalman and his colleagues observed a 4-fold increase of neutrophils and a 3-fold increase of monocytes in the bone marrow of imatinib-treated mice. However, these mice did not see a significant change in the number of mature B cells, T cells, dendritic cells, eosinophils, or natural killer cells.
Imatinib did not induce the accumulation of HSCs, but it did regulate the accumulation of multipotent progenitors. The drug also reduced the accumulation of HSCs upon infection, which suggests it may increase the flux of HSCs to progenitors.
Imatinib did not increase the number of transplantable HSCs, but it did induce an irreversible commitment of HSCs into progenitors that could differentiate into myeloid cells ex vivo.
Imatinib induced an irreversible differentiation of HSCs or progenitors into myeloid cells in a dose-dependent manner. The drug facilitated the exodus of myeloid cells from the bone marrow only at lower doses. It inhibited this same process at higher doses.
Despite increasing the number of neutrophils, low doses of imatinib did not activate neutrophils. However, the cells retained the capacity to activate upon infection.
In fact, an increase in the number of neutrophils was sufficient to reduce Mycobacterium marinum bacterial load. And imatinib reduced the bacterial load in mice infected with pathogenic Francisella species.
The researchers said these results suggest low doses of imatinib could potentially be used to treat a variety of infections and might prove particularly useful in immunocompromised patients. ![]()
Collaboration may help prevent CLABSIs
Collaborative relationships between nurses and physicians may help reduce the rates of healthcare-associated infections in critical care, according to research published in Critical Care Nurse.
Study investigators found lower rates of central line-associated bloodstream infections (CLABSIs) and ventilator-associated pneumonia (VAP) in critical care units in which nurses reported a more favorable perception of nurse-physician collaboration.
“Our findings suggest that raising the quality of collaboration and communication among nurses and physicians has the potential to improve patient safety,” said study author Christine Boev, RN, PhD, CCRN, of Wegmans School of Nursing at St John Fisher College in Rochester, New York.
Dr Boev and her colleagues analyzed 5 years of data from 671 surveys of nurses in 4 specialized intensive care units (ICUs) at a 750-bed New York hospital.
The investigators also collected patient outcome data from those units for the same period, focusing on patients with CLABSIs or VAP. And the team analyzed unit-level variables such as nurses’ skill mix, nursing hours per patient day, and voluntary turnover.
Results revealed a significant association between nurse-physician collaboration and both CLABSIs and VAP. For every 0.5 unit increase in collaboration, the rate of CLABSIs decreased by 2.98 (P=0.005), and the rate of VAP decreased by 1.13 (P=0.005).
In addition, ICUs with a higher proportion of certified nurses had significantly lower incidences of both CLABSIs and VAP—0.43 (P=0.02) and 0.17 (P=0.01), respectively. And ICUs with higher numbers of nursing hours per patient day had significantly lower rates of CLABSIs—0.42 (P=0.05).
However, there was no significant difference in VAP rates according to nursing hours. And there was no significant difference in the rate of either type of infection according to nurses’ skill mix or voluntary turnover.
Dr Boev said these results suggest that efforts to prevent healthcare-associated infections should include interventions to improve nurse-physician collaboration. Such interventions might include multidisciplinary daily patient rounds and interprofessional educational programs, such as shared simulation training. ![]()
Collaborative relationships between nurses and physicians may help reduce the rates of healthcare-associated infections in critical care, according to research published in Critical Care Nurse.
Study investigators found lower rates of central line-associated bloodstream infections (CLABSIs) and ventilator-associated pneumonia (VAP) in critical care units in which nurses reported a more favorable perception of nurse-physician collaboration.
“Our findings suggest that raising the quality of collaboration and communication among nurses and physicians has the potential to improve patient safety,” said study author Christine Boev, RN, PhD, CCRN, of Wegmans School of Nursing at St John Fisher College in Rochester, New York.
Dr Boev and her colleagues analyzed 5 years of data from 671 surveys of nurses in 4 specialized intensive care units (ICUs) at a 750-bed New York hospital.
The investigators also collected patient outcome data from those units for the same period, focusing on patients with CLABSIs or VAP. And the team analyzed unit-level variables such as nurses’ skill mix, nursing hours per patient day, and voluntary turnover.
Results revealed a significant association between nurse-physician collaboration and both CLABSIs and VAP. For every 0.5 unit increase in collaboration, the rate of CLABSIs decreased by 2.98 (P=0.005), and the rate of VAP decreased by 1.13 (P=0.005).
In addition, ICUs with a higher proportion of certified nurses had significantly lower incidences of both CLABSIs and VAP—0.43 (P=0.02) and 0.17 (P=0.01), respectively. And ICUs with higher numbers of nursing hours per patient day had significantly lower rates of CLABSIs—0.42 (P=0.05).
However, there was no significant difference in VAP rates according to nursing hours. And there was no significant difference in the rate of either type of infection according to nurses’ skill mix or voluntary turnover.
Dr Boev said these results suggest that efforts to prevent healthcare-associated infections should include interventions to improve nurse-physician collaboration. Such interventions might include multidisciplinary daily patient rounds and interprofessional educational programs, such as shared simulation training. ![]()
Collaborative relationships between nurses and physicians may help reduce the rates of healthcare-associated infections in critical care, according to research published in Critical Care Nurse.
Study investigators found lower rates of central line-associated bloodstream infections (CLABSIs) and ventilator-associated pneumonia (VAP) in critical care units in which nurses reported a more favorable perception of nurse-physician collaboration.
“Our findings suggest that raising the quality of collaboration and communication among nurses and physicians has the potential to improve patient safety,” said study author Christine Boev, RN, PhD, CCRN, of Wegmans School of Nursing at St John Fisher College in Rochester, New York.
Dr Boev and her colleagues analyzed 5 years of data from 671 surveys of nurses in 4 specialized intensive care units (ICUs) at a 750-bed New York hospital.
The investigators also collected patient outcome data from those units for the same period, focusing on patients with CLABSIs or VAP. And the team analyzed unit-level variables such as nurses’ skill mix, nursing hours per patient day, and voluntary turnover.
Results revealed a significant association between nurse-physician collaboration and both CLABSIs and VAP. For every 0.5 unit increase in collaboration, the rate of CLABSIs decreased by 2.98 (P=0.005), and the rate of VAP decreased by 1.13 (P=0.005).
In addition, ICUs with a higher proportion of certified nurses had significantly lower incidences of both CLABSIs and VAP—0.43 (P=0.02) and 0.17 (P=0.01), respectively. And ICUs with higher numbers of nursing hours per patient day had significantly lower rates of CLABSIs—0.42 (P=0.05).
However, there was no significant difference in VAP rates according to nursing hours. And there was no significant difference in the rate of either type of infection according to nurses’ skill mix or voluntary turnover.
Dr Boev said these results suggest that efforts to prevent healthcare-associated infections should include interventions to improve nurse-physician collaboration. Such interventions might include multidisciplinary daily patient rounds and interprofessional educational programs, such as shared simulation training. ![]()
Mindfulness Meditation for Sleep Problems
Study Overview
Objective. To test the treatment effect of a structured mindfulness meditation program versus sleep hygiene education for improving sleep quality.
Study design. Single-site, parallel-group randomized clinical trial.
Setting and participants. Adults aged 55 years and older were recruited from the urban Los Angeles community through a newspaper advertisement and flyers posted in community centers. Participants had to agree to be randomized and have a Pittsburgh Sleep Quality Index (PSQI) score [1] exceeding 5 at screening. Exclusion criteria were current smoking, substance dependence, inability to speak English, depression, cognitive impairment, current daily meditation, and obesity. Also excluded were those who reported a current inflammatory disorder, sleep apnea, restless legs syndrome, illness, or infection.
Intervention. Participants were randomized into 2 standardized treatment conditions: the Mindful Awareness Practices program (MAPs) and sleep hygiene education (SHE). Each treatment consisted of weekly 2-hour group-based classes over the course of the 6-week intervention. The comparison sleep hygiene program matched the MAPs condition for time, attention, group interaction, and expectancy of benefit effects. Eight visits to the study site were requested, including 1 pretreatment assessment visit, 6 intervention sessions, and 1 posttreatment assessment visit. Participants were compensated up to $50 in gift cards and received parking vouchers for visits.
Main outcome measure. The primary outcome measure was the PSQI, a commonly used and validated 19-item self-rated questionnaire that assesses sleep quality and disturbances over a 1-month time interval. A global score greater than 5 yields a diagnostic sensitivity of 89.6% and specificity of 86.5% in distinguishing good and poor sleepers [1]. Secondary outcomes included scores on instruments that measured depression, anxiety, stress, and fatigue.
Results. After screening for eligibility, 49 adults were randomized, 24 to MAPS and 25 to SHE. Session attendance was similar across the groups. Mean (± SD) age of participants was 66.3 (7.4) years and 67% were female. Mean PSQI was 10.2 at baseline and 7.4 postintervention for MAPs, and 10.2 at baseline and 9.1 postintervention for SHE. In the intention-to-treat analyses, PSQI improved by 2.8 in MAPS vs. 1.1 in SHE (between-group mean difference, 1.8; 95% confidence interval, 0.6–2.9) with an effect size of 0.89. Relative improvements in depression scores and daytime fatigue were also noted.
Conclusion. The program improved sleep quality relative to SHE. Mindfulness meditation appears to have a role in addressing the burden of sleep problems in older adults.
Commentary
Older adults commonly report disturbed sleep, and an expanding literature suggests that poor sleep increases the risk of adverse health outcomes, including frailty and lower cognitive function. Current nonpharmacologic treatments for disturbed sleep include sleep hygiene education and cognitive behavioral therapy (CBT), which have been shown to be effective. However, as the current study’s authors point out, clinical interventions like CBT are intensive, require administration by highly trained therapists, and are intended for patients with insomnia [2].
These researchers investigated an alternative intervention consisting of mindfulness meditation. Mindfulness has been defined as being intentionally aware of internal and external experiences that occur at the present moment, without judgment. Mindfulness-based interventions are increasingly being studied for a wide array of health conditions, and courses in the community and online are frequently available.
The results of the current study, which applied mindfulness meditation to the problem of sleep disturbance in older adults, are compelling. The effect size of 0.89 was large and of clinical relevance: as the authors point out, in a meta-analysis of behavioral interventions for insomnia, the average effect size for improvement in subjective sleep outcomes among older adults was 0.76 [3]. It is noteworthy that the authors of the current study recruited patients on the basis of PSQI score and did not require a diagnosis of insomnia. The use of the PSQI means that the sample consisted of patients with self-rated poor sleep quality, and epidemiologic evidence suggests that a PSQI score greater than 5 identifies older persons at risk for adverse health outcomes [4]. Thus, this is a logical group to target. In addition, the sample may have included those with undiagnosed insomnia and other sleep disturbances; this fact makes the findings even more impressive [4].
The use of validated measures are a strength of the study. Limitations include lack of postintervention assessment data for 12% of participants and a preponderance of female and highly educated participants.
Applications for Clinical Practice
Standardized mindfulness programs are becoming more widely available, both online and in the community, and can be be introduced to older adults to help them with moderate sleep disturbances.
1. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
2. Morin CM, Bootzin RR, Buysse DJ, Edinger JD, Espie CA, Lichstein KL. Psychological and behavioral treatment of insomnia: update of the recent evidence (1998-2004). Sleep 2006;29:1398–414.
3. Irwin MR, Cole JC, Nicassio PM. Comparative meta-analysis of behavioral interventions for insomnia and their efficacy in middle-aged adults and in older adults 55+ years of age. Health Psychol 2006;25:3–14.
4. Spira AP. Being mindful of later-life sleep quality and its potential role in prevention. JAMA Intern Med. Published online 16 Feb 2015.
Study Overview
Objective. To test the treatment effect of a structured mindfulness meditation program versus sleep hygiene education for improving sleep quality.
Study design. Single-site, parallel-group randomized clinical trial.
Setting and participants. Adults aged 55 years and older were recruited from the urban Los Angeles community through a newspaper advertisement and flyers posted in community centers. Participants had to agree to be randomized and have a Pittsburgh Sleep Quality Index (PSQI) score [1] exceeding 5 at screening. Exclusion criteria were current smoking, substance dependence, inability to speak English, depression, cognitive impairment, current daily meditation, and obesity. Also excluded were those who reported a current inflammatory disorder, sleep apnea, restless legs syndrome, illness, or infection.
Intervention. Participants were randomized into 2 standardized treatment conditions: the Mindful Awareness Practices program (MAPs) and sleep hygiene education (SHE). Each treatment consisted of weekly 2-hour group-based classes over the course of the 6-week intervention. The comparison sleep hygiene program matched the MAPs condition for time, attention, group interaction, and expectancy of benefit effects. Eight visits to the study site were requested, including 1 pretreatment assessment visit, 6 intervention sessions, and 1 posttreatment assessment visit. Participants were compensated up to $50 in gift cards and received parking vouchers for visits.
Main outcome measure. The primary outcome measure was the PSQI, a commonly used and validated 19-item self-rated questionnaire that assesses sleep quality and disturbances over a 1-month time interval. A global score greater than 5 yields a diagnostic sensitivity of 89.6% and specificity of 86.5% in distinguishing good and poor sleepers [1]. Secondary outcomes included scores on instruments that measured depression, anxiety, stress, and fatigue.
Results. After screening for eligibility, 49 adults were randomized, 24 to MAPS and 25 to SHE. Session attendance was similar across the groups. Mean (± SD) age of participants was 66.3 (7.4) years and 67% were female. Mean PSQI was 10.2 at baseline and 7.4 postintervention for MAPs, and 10.2 at baseline and 9.1 postintervention for SHE. In the intention-to-treat analyses, PSQI improved by 2.8 in MAPS vs. 1.1 in SHE (between-group mean difference, 1.8; 95% confidence interval, 0.6–2.9) with an effect size of 0.89. Relative improvements in depression scores and daytime fatigue were also noted.
Conclusion. The program improved sleep quality relative to SHE. Mindfulness meditation appears to have a role in addressing the burden of sleep problems in older adults.
Commentary
Older adults commonly report disturbed sleep, and an expanding literature suggests that poor sleep increases the risk of adverse health outcomes, including frailty and lower cognitive function. Current nonpharmacologic treatments for disturbed sleep include sleep hygiene education and cognitive behavioral therapy (CBT), which have been shown to be effective. However, as the current study’s authors point out, clinical interventions like CBT are intensive, require administration by highly trained therapists, and are intended for patients with insomnia [2].
These researchers investigated an alternative intervention consisting of mindfulness meditation. Mindfulness has been defined as being intentionally aware of internal and external experiences that occur at the present moment, without judgment. Mindfulness-based interventions are increasingly being studied for a wide array of health conditions, and courses in the community and online are frequently available.
The results of the current study, which applied mindfulness meditation to the problem of sleep disturbance in older adults, are compelling. The effect size of 0.89 was large and of clinical relevance: as the authors point out, in a meta-analysis of behavioral interventions for insomnia, the average effect size for improvement in subjective sleep outcomes among older adults was 0.76 [3]. It is noteworthy that the authors of the current study recruited patients on the basis of PSQI score and did not require a diagnosis of insomnia. The use of the PSQI means that the sample consisted of patients with self-rated poor sleep quality, and epidemiologic evidence suggests that a PSQI score greater than 5 identifies older persons at risk for adverse health outcomes [4]. Thus, this is a logical group to target. In addition, the sample may have included those with undiagnosed insomnia and other sleep disturbances; this fact makes the findings even more impressive [4].
The use of validated measures are a strength of the study. Limitations include lack of postintervention assessment data for 12% of participants and a preponderance of female and highly educated participants.
Applications for Clinical Practice
Standardized mindfulness programs are becoming more widely available, both online and in the community, and can be be introduced to older adults to help them with moderate sleep disturbances.
Study Overview
Objective. To test the treatment effect of a structured mindfulness meditation program versus sleep hygiene education for improving sleep quality.
Study design. Single-site, parallel-group randomized clinical trial.
Setting and participants. Adults aged 55 years and older were recruited from the urban Los Angeles community through a newspaper advertisement and flyers posted in community centers. Participants had to agree to be randomized and have a Pittsburgh Sleep Quality Index (PSQI) score [1] exceeding 5 at screening. Exclusion criteria were current smoking, substance dependence, inability to speak English, depression, cognitive impairment, current daily meditation, and obesity. Also excluded were those who reported a current inflammatory disorder, sleep apnea, restless legs syndrome, illness, or infection.
Intervention. Participants were randomized into 2 standardized treatment conditions: the Mindful Awareness Practices program (MAPs) and sleep hygiene education (SHE). Each treatment consisted of weekly 2-hour group-based classes over the course of the 6-week intervention. The comparison sleep hygiene program matched the MAPs condition for time, attention, group interaction, and expectancy of benefit effects. Eight visits to the study site were requested, including 1 pretreatment assessment visit, 6 intervention sessions, and 1 posttreatment assessment visit. Participants were compensated up to $50 in gift cards and received parking vouchers for visits.
Main outcome measure. The primary outcome measure was the PSQI, a commonly used and validated 19-item self-rated questionnaire that assesses sleep quality and disturbances over a 1-month time interval. A global score greater than 5 yields a diagnostic sensitivity of 89.6% and specificity of 86.5% in distinguishing good and poor sleepers [1]. Secondary outcomes included scores on instruments that measured depression, anxiety, stress, and fatigue.
Results. After screening for eligibility, 49 adults were randomized, 24 to MAPS and 25 to SHE. Session attendance was similar across the groups. Mean (± SD) age of participants was 66.3 (7.4) years and 67% were female. Mean PSQI was 10.2 at baseline and 7.4 postintervention for MAPs, and 10.2 at baseline and 9.1 postintervention for SHE. In the intention-to-treat analyses, PSQI improved by 2.8 in MAPS vs. 1.1 in SHE (between-group mean difference, 1.8; 95% confidence interval, 0.6–2.9) with an effect size of 0.89. Relative improvements in depression scores and daytime fatigue were also noted.
Conclusion. The program improved sleep quality relative to SHE. Mindfulness meditation appears to have a role in addressing the burden of sleep problems in older adults.
Commentary
Older adults commonly report disturbed sleep, and an expanding literature suggests that poor sleep increases the risk of adverse health outcomes, including frailty and lower cognitive function. Current nonpharmacologic treatments for disturbed sleep include sleep hygiene education and cognitive behavioral therapy (CBT), which have been shown to be effective. However, as the current study’s authors point out, clinical interventions like CBT are intensive, require administration by highly trained therapists, and are intended for patients with insomnia [2].
These researchers investigated an alternative intervention consisting of mindfulness meditation. Mindfulness has been defined as being intentionally aware of internal and external experiences that occur at the present moment, without judgment. Mindfulness-based interventions are increasingly being studied for a wide array of health conditions, and courses in the community and online are frequently available.
The results of the current study, which applied mindfulness meditation to the problem of sleep disturbance in older adults, are compelling. The effect size of 0.89 was large and of clinical relevance: as the authors point out, in a meta-analysis of behavioral interventions for insomnia, the average effect size for improvement in subjective sleep outcomes among older adults was 0.76 [3]. It is noteworthy that the authors of the current study recruited patients on the basis of PSQI score and did not require a diagnosis of insomnia. The use of the PSQI means that the sample consisted of patients with self-rated poor sleep quality, and epidemiologic evidence suggests that a PSQI score greater than 5 identifies older persons at risk for adverse health outcomes [4]. Thus, this is a logical group to target. In addition, the sample may have included those with undiagnosed insomnia and other sleep disturbances; this fact makes the findings even more impressive [4].
The use of validated measures are a strength of the study. Limitations include lack of postintervention assessment data for 12% of participants and a preponderance of female and highly educated participants.
Applications for Clinical Practice
Standardized mindfulness programs are becoming more widely available, both online and in the community, and can be be introduced to older adults to help them with moderate sleep disturbances.
1. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
2. Morin CM, Bootzin RR, Buysse DJ, Edinger JD, Espie CA, Lichstein KL. Psychological and behavioral treatment of insomnia: update of the recent evidence (1998-2004). Sleep 2006;29:1398–414.
3. Irwin MR, Cole JC, Nicassio PM. Comparative meta-analysis of behavioral interventions for insomnia and their efficacy in middle-aged adults and in older adults 55+ years of age. Health Psychol 2006;25:3–14.
4. Spira AP. Being mindful of later-life sleep quality and its potential role in prevention. JAMA Intern Med. Published online 16 Feb 2015.
1. Buysse DJ, Reynolds CF 3rd, Monk TH, et al. The Pittsburgh Sleep Quality Index: a new instrument for psychiatric practice and research. Psychiatry Res 1989;28:193–213.
2. Morin CM, Bootzin RR, Buysse DJ, Edinger JD, Espie CA, Lichstein KL. Psychological and behavioral treatment of insomnia: update of the recent evidence (1998-2004). Sleep 2006;29:1398–414.
3. Irwin MR, Cole JC, Nicassio PM. Comparative meta-analysis of behavioral interventions for insomnia and their efficacy in middle-aged adults and in older adults 55+ years of age. Health Psychol 2006;25:3–14.
4. Spira AP. Being mindful of later-life sleep quality and its potential role in prevention. JAMA Intern Med. Published online 16 Feb 2015.
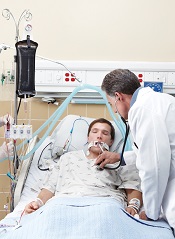
Optimizing Inpatient Pharmacotherapy Using a Single Clinical Policy Streamlining Pharmacy Protocols
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
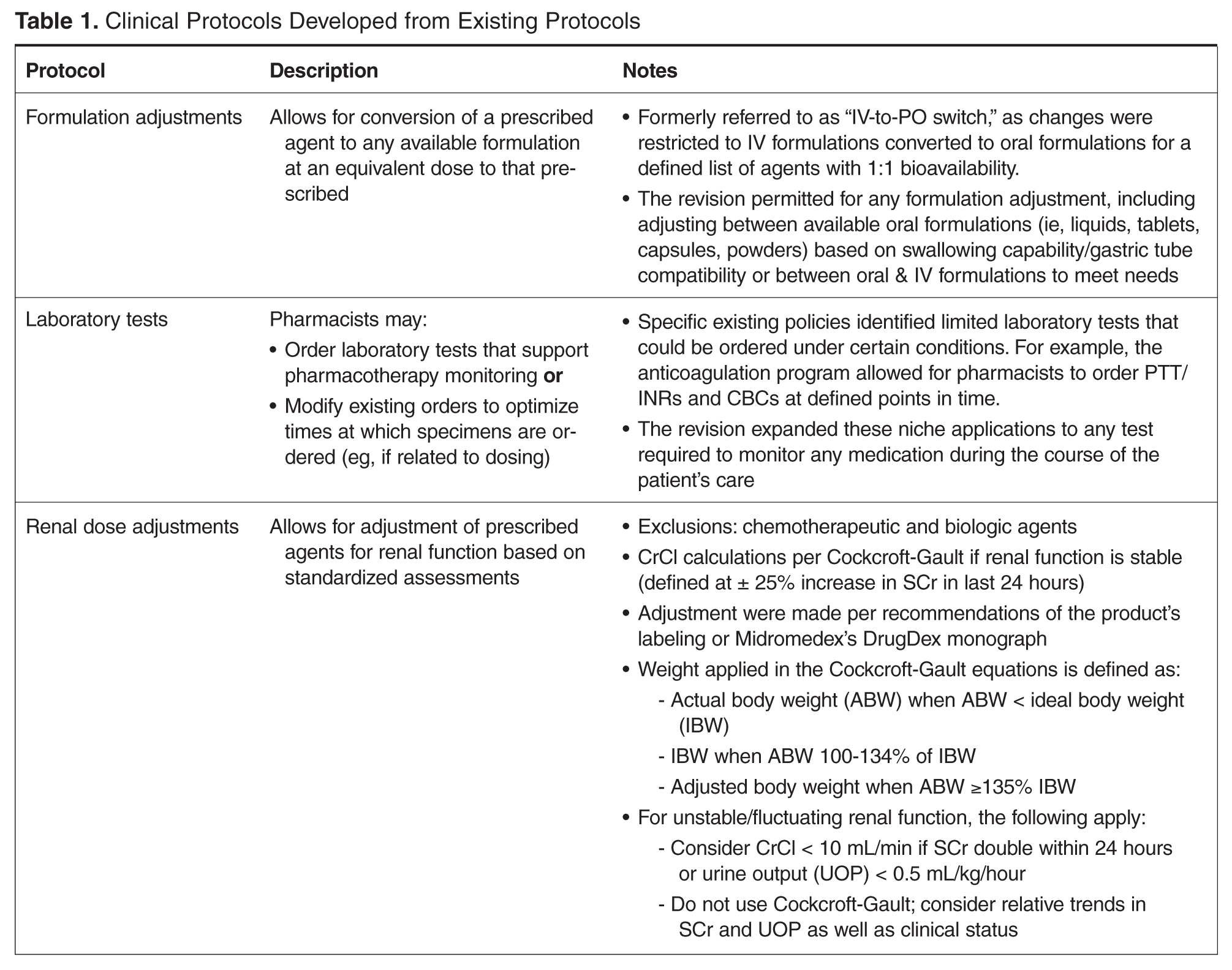
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
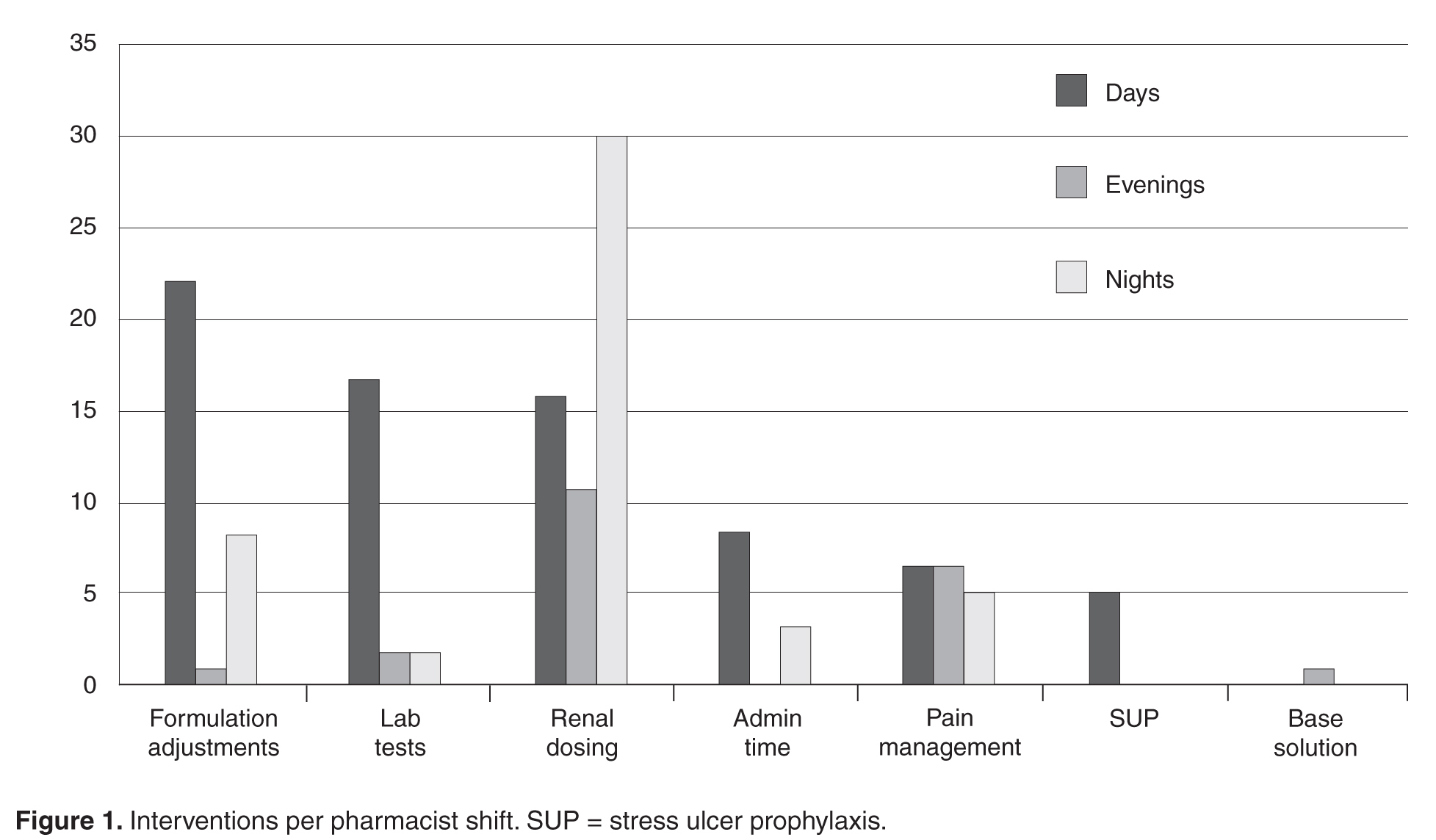
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
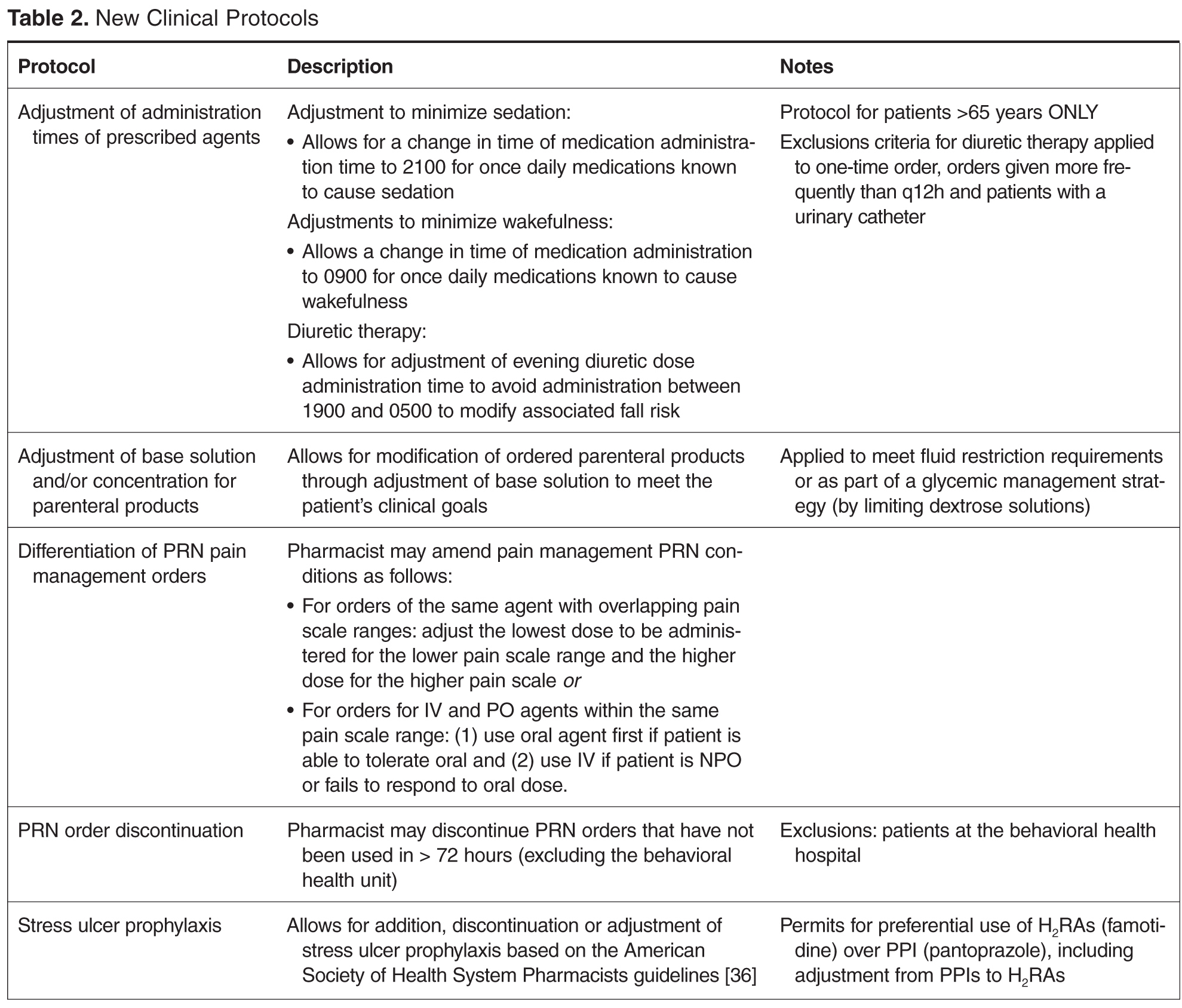
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
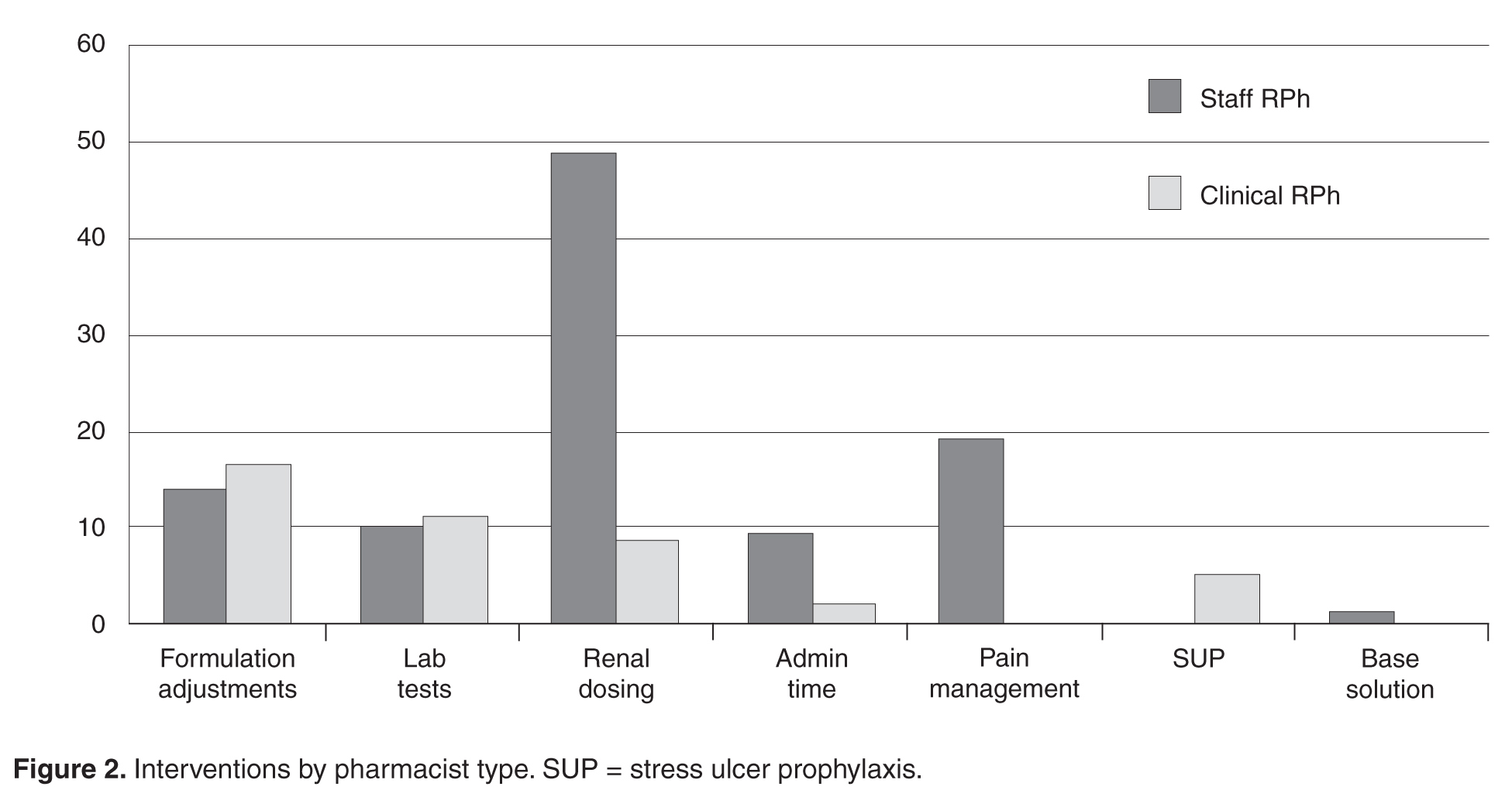
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
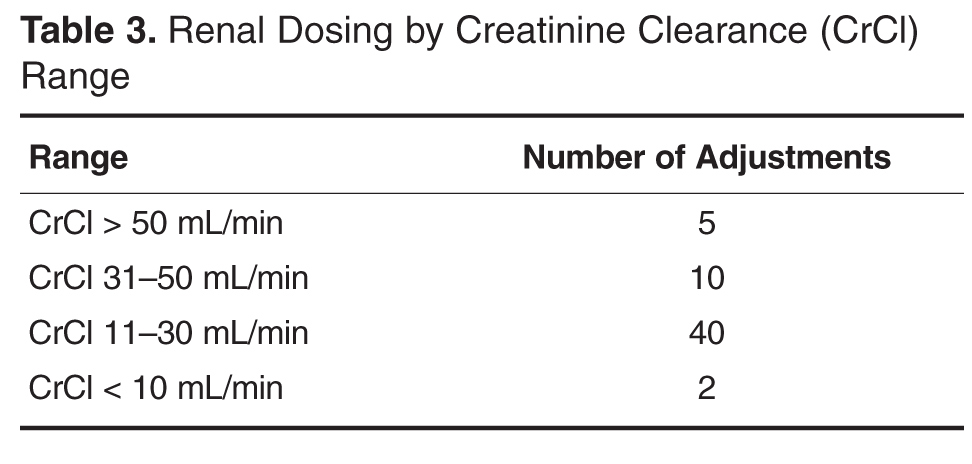
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
From the Ernest Mario School of Pharmacy, Rutgers, The State University of New Jersey, Piscataway, NJ.
Abstract
- Objectives: To describe the implementation of broadly scoped clinical pharmacy protocols positioned as a singular policy in a community hospital. These protocols were designed to expand the established benefits demonstrated using narrower, traditional protocols.
- Methods: A retrospective chart review of protocol interventions in the first year of the policy’s implementation was conducted to evaluate prescriber acceptance of protocol interventions. Interventions were identified from required email notifications. The frequency of use of each protocol was assessed, including evaluation of novel characteristics of specific protocols. Pharmacist utilization patterns were assessed for job classification, shift, and practice setting (ie, centralized or decentralized).
- Results: In the 1-year assessment period, 145 interventions were reported and 144 were accepted by the prescribing physicians. Interventions involved orders from hospitalists and intensivists most frequently, with the renal dosing and dose formulations protocols being the most commonly utilized. Staff pharmacists used the policy more frequently than clinical pharmacists, primarily during day shift from decentralized locations on the patient care units.
- Conclusions: The implementation of broadly scoped clinical pharmacy protocols for items our pharmacists routinely contact physicians about (and our physicians deemed were within the practice of pharmacy) instituted a cultural shift that expanded the elements considered to be part of routine pharmacy practice. As a result, pharmacists more seamlessly applied their expertise as pharmacotherapy specialists to optimize pharmacotherapy, which streamlined workflow for both pharmacists and physicians. This expanded the proven benefits of allowing professionals to work to their fullest extent, as established in the literature.
Allowing pharmacists to apply their expertise has been associated with improved outcomes in both pharmacotherapy quality (eg, reduction in mortality and length of stay [1]) and savings in health care dollars. Studies of focused protocols, including intravenous-to-oral (IV-to-PO) switch [2–20], renal dosing [21], stress ulcer prophylaxis [22] and anticoagulation management [1,23,24] demonstrate these benefits in a multitude of practice areas. While such protocols have become commonplace in the acute care setting [25–28], most continue to be singularly focused and impose patient population restrictions that preclude comprehensive patient evaluation. Many are administered as a task within the pharmacist workflow using a patient list generated by the limited protocol criteria, which are often restricted to agent or patient characteristics.
Better outcomes are associated with permitting professionals such as pharmacists to work to the fullest extent of their scope and expertise [29–31]. In specific cases, studies evaluating pharmacists’ impact within a multi-disciplinary health care team have demonstrated improved outcomes in regard to both patient care and cost [29–31]. Recognizing this, accountable care organizations (ACOs) have developed practice models that are based on this benefit. Each team member is expected to robustly apply their training and expertise to achieve the best outcomes [32,33]. As health care moves toward a more integrative approach, it is paramount that pharmacists utilize the full scope of the skills in which they are trained.
This report describes the development, implementation, and outcomes of a singular policy outlining comprehensively scoped protocols allowing acute care hospital pharmacists within Princeton HealthCare System to optimize pharmacotherapy during the course of their usual clinical practice.
Methods
Setting
The University Medical Center of Princeton at Plainsboro (UMCPP), part of the Princeton HealthCare System, is a 230-bed community acute care hospital located in central New Jersey. The hospital facility relocated in May 2012 from its previous location in Princeton to a new state-of-the-art facility in Plainsboro. As an affiliate of the Robert Wood Johnson Medical School and the Ernest Mario School of Pharmacy at Rutgers, The State University of New Jersey (ie, Rutgers), it is an academic teaching hospital with a mixed model for providing patient care. UMCPP employs both faculty physicians leading academic teams alongside hospitalists and private attendings.
Pharmacy services are provided on facility 24 hours a day, 365 days a year. The department of pharmacy services provides a full scope medication services from a centralized location with 3 full-time day pharmacists and 1 oncology satellite pharmacist. During weekdays, decentralized pharmacists provide medication review, patient education, and medication reconciliation on 2 to 3 inpatient care units. Centralized support decreases to 2 pharmacists in the evening and 1 overnight. Clinical pharmacists, both hospital-based and Rutgers faculty, work in conjunction with the staff pharmacists to ensure appropriate management of patients throughout different levels of care.
Program Overview and Implementation
To enhance protocols allowing pharmacists to more holistically and robustly optimize pharmacotherapy, UMCPP implemented the Clinical Pharmacy Services policy in February 2012. The policy outlined 8 protocols through which registered pharmacists within the acute care hospital could implement outlined medication order adjustments for adults of inpatient status. Pediatric patients or those treated outside of the acute care hospital (eg, in the psychiatric hospital, surgical center or outpatient facilities) were excluded. While the hospital had existing traditional programs such as IV-to-PO conversions, the programs were restricted to specific agents or conditions. As such, pharmacists were assigned to review queues in the clinical computer system to which orders for the agents outlined by the specific program would flow. Review would occur at set intervals and focus on that detail of the patient’s care as opposed to broadly encompassing an evaluation of the patient’s comprehensive pharmacotherapy. The goal of the new policy was to better utilize the pharmacists’ expertise by broadening these assessments to all applicable agents, refine workflow (by allowing protocol management instead of requiring individual prescriber calls for each issue) and integrate holistic refinement of pharmacotherapy regimens during the usual course of the pharmacist’s clinical care.
In the state of New Jersey, the Pharmacy Practice Act (updated on 14 January 2004) formally recognizes pharmacists as health care professionals and permits for collaborative practice in the community setting [34]. However, pharmacist management by protocol in the acute care hospital setting is defined separately, requiring only medical approvals within the system [35]. In accordance, the policy and associated protocols were approved by the institution’s multidisciplinary pharmacy and therapeutics (P&T) and medical executive committee processes.
To ensure appropriate oversight, the policy required that the pharmacist making changes submit notification of protocol intervention to the patient’s attending physician, the physician who generated the original order (if other than the attending) and a designated clinical pharmacist (for auditing purposes). All notifications were made via email within the clinical computer system in “interrupt” status to ensure active recognition by the prescriber(s).
Program Evaluation
An evaluation of the first year’s interventions was conducted to validate the program, describe its utility, and provide a basis for re-evaluation and continued evolution. The aim was to evaluate the institution’s experience with the program, focusing on both specific physician and pharmacist elements. One of the primary goals was to evaluate which physician’s orders were associated with interventions as well as the rate of physician acceptance of protocol interventions, as their acceptance clearly validates the pharmacist’s ability to appropriately apply the protocols in patient-specific contexts.
To evaluate the pharmacist’s experience, trends in pharmacist utilization were captured, including which pharmacist by job classification (ie, staff or clinical pharmacist) implemented interventions, during which shift, and in what operational capacity (ie, centralized or decentralized) the pharmacist was practicing. Lastly, the study sought to characterize the frequency to which each protocol was applied. Based on the existing experiences described in the literature as well as with consideration of institutional culture and operation, we hypothesized that all pharmacists would apply protocols with equal efficacy with more interventions likely generated by staff pharmacists due to their role in primary order review and that the types of interventions would vary based on shift and location.
A retrospective review of cases throughout the first year of the policy’s implementation was conducted, including interventions made between 1 February 2012 and 31 January 2013. Cases were identified through the required email notification of the auditing clinical pharmacist. The patient’s electronic medical record for that defined visit was reviewed. To assess pharmacist utilization patterns, data captured included the agent involved in the intervention, date, day of week and shift, whether the pharmacist was centralized or decentralized, and whether that pharmacist was classified as staff or clinical. Decentralized pharmacists were defined as a pharmacist working on the patient care unit with direct access to other practitioners and patients, rather than those performing their functions from within the confines of the pharmacy department.
Prescribers were described both by status (ie, attending or resident/training) and specialty. Physician acceptance was assessed through evaluation of order trends as the electronic medical record allows for all changes to an order to be audited and tracked; a review of progress notes to capture any commentary or rationale regarding interventions or the surrounding circumstances; as well as a review of any associated laboratory or diagnostic reports and nursing notes. If the order was not altered by the physician within 24 hours (ie, the time frame in which orders must be reviewed by the prescriber per institutional standards) of the pharmacist’s protocol change it was deemed accepted by the physician. Changes made within 24 hours for clinical reasons unrelated to the protocol change as verified by documentation in the progress notes were considered as accepted. These included, for example, the discontinuation of empiric antibiotics that had been dose adjusted by the pharmacist for patients in whom infection had been ruled out or a change from the adjusted agent to one of another class (such as might occur during de-escalation of antibiotic therapy). Interventions were excluded if there were insufficient patient and/or intervention details to allow complete assessment.
For protocol evaluation, details concerning the nature of the adjustment were collected. For formulation changes, agents were classified by their bioavailability. Renal dose adjustments were classified by the patient’s estimated creatinine clearance range since interventions were not restricted to ranges or agents. Stress ulcer prophylaxis adjustments were classified as those involving initiation, changes or discontinuation of therapy. For parenteral product adjustments, the initial and final base solution and/or the change in concentration was captured. Pain management order adjustments were classified as those involving the same agent with overlapping indications or those with oral and intravenous orders for the same pain scale range. When laboratory tests were ordered, the type of test was captured.
The study was approved by the institutional review boards of Princeton HealthCare System and Rutgers.
Results
There were 145 interventions occurring between 1 February 2012 and 31 January 2013, with 144 (99.3%) of those being accepted by the prescriber. The 1 intervention that was not accepted involved an IV to oral conversion of levothyroxine. The pharmacist performed the conversion appropriately as the patient was tolerating other oral medications. However, on the day of the change, the patient refused all oral medications despite having the ability to accept them and, as a result, all medications were converted back to parenteral formulations.
Pharmacist Evaluation
Prescriber Evaluation
An evaluation of prescribers revealed that the primary physician groups (ie, order generators) involved were hospitalists (n = 32) and critical care attendings (n = 24) at 22% and 17% of all orders, respectively. The remaining 89 interventions were distributed across other attending types (including general medicine physicians, specialty physicians and surgeons) and trainees (residents and fellows) with no more than eight orders for any individual physician category.
Protocol Evaluation
The total number of laboratory tests ordered accounted for 14% (n = 21) of all interventions. Studies related to the management of anti-infective agents and blood formation, coagulation, and thrombosis agents consisted of the majority of the lab tests ordered; INR/PTT and vancomycin levels were the most commonly ordered. Thirteen percent (n = 19) of all interventions include pain management adjustments with an even distribution between pain medications.
Several protocols were less frequently used, specifically the stress ulcer prophylaxis protocol (representing 3% of all interventions or n = 5), base solution changes (< 1% of all interventions or n = 1), and adjustment of administration time (7.6% of all interventions, n = 11). Of the time adjustments, more than 50% (n = 6) involved furosemide.
Discussion
While the literature has many studies describing pharmacists improving outcomes through successful provision of clinical programs by protocol in the acute care hospital setting, the majority of studies are limited to single or focused protocols [2–24,27,37,38]. This approach fails to recognize or limits application of a pharmacist’s expertise in pharmacotherapy, as intervention is permitted only on defined agents under specific circumstances. This is the only report we are aware of that addresses a broader approach in permitting pharmacists to optimize pharmaco-therapy during the course of their usual practice through a single policy. As better outcomes are associated with allowing professionals to work to the fullest extent of their expertise, a broad range of protocols identified as pharmacy clinical services were selected and integrated into a singular policy that would be the foundation for instituting cultural change in regard to the elements considered to be routine pharmacy practice. Thus, the protocols applied here did not specify agents that could be adjusted for renal function or classes for which formulation conversion were permissible. This is also the case for dose formulation adjustments, where the protocol allowed for the pharmacist to apply their expertise beyond 1:1 conversions using standardized drug information references (Table 1 and Table 2). As such, the protocols allowed for the full application of the pharmacist’s expertise as a pharmacotherapy consultant within these intervention categories to assure that therapies are optimized. Additionally, eliminating phone calls streamlined the workflow for both the pharmacist and physicians, thus minimizing interruptions that distract from the other functions in which they are engaged.
During the approval process, physicians inquired whether all pharmacists were equally capable of making the clinical judgments involved with the protocols as described and, thusly, whether protocol management should be limited to clinical pharmacists who have less traditional dispensing roles and more experience and time at the bedside. During those discussions we contended that the nature of these protocols were fundamental and applicable to all practicing pharmacists and, if limited, would result in missed opportunities as the clinical pharmacists are focused in specialized areas during weekdays only at UMCPP. For example, a single, centralized night-shift pharmacist could make routine dose or formulation adjustments without the need to awaken a physician as the UMCPP electronic medical record makes available all progress notes, laboratory results, and diagnostics crucial to clinical decision making. All pharmacists, regardless of job title, meet the same requirements for licensure. Post-doctoral residency or fellowship training and advanced certifications in specialty areas of practice exist among both groups as well. The study results support the validity of this argument. The majority of interventions were successfully performed by staff pharmacists with involvement from all shifts, including a third that occurred overnight. This is important because, like at most hospitals, the UMCPP staffing ratio decreases throughout the course of the day presenting changing workflow challenges throughout different shifts.
Several limitations of this study should be noted. Due to its retrospective nature, it is likely that not all interventions were captured. Some decentralized pharmacists reported not emailing interventions as they had verbally communicated the adjustments prior to having the opportunity to send the email. Four interventions could not be assessed as the email notification did not contain all the required patient identifiers or intervention information to permit for appropriate evaluation. The hospital also moved to a newly built facility in the fourth month of protocol implementation, which required significant changes in drug distribution methods, and this could have contributed to the small sample size of interventions. The move temporarily shifted departmental resources to support operational needs.
Another important factor is the voluntary nature of the policy; while it was within the pharmacist’s professional judgment to apply the protocols, pharmacists were encouraged to contact prescribers if there was any ambiguity. Therefore, while one might have expected more resident physicians to be involved with orders that were adjusted, the UMCPP practice philosophy supports contacting training physicians about changes so that they may learn from the discussion to support developing stronger prescribing habits. Future development should therefore support more universal protocol application to all eligible patients to optimize the benefits described here. Lastly, data measuring the clinical outcomes and time savings or increased productivity secondary to the elimination of physician phone calls was not directly measured. We thus sought to first demonstrate to the physician base that pharmacists could successfully apply a variety of protocols that were broader than those formally studied with equal accuracy. With that effectiveness established, future studies should explore if broader protocol application produces a greater optimization of outcomes.
After the study was completed, a survey was conducted of the pharmacists to assess perceptions and guide further policy development. We received a 63.6% response rate (14 of 22 possible respondents) with a strong majority of the respondents expressing a favorable perception of the protocols. A few respondents indicated some protocols were infrequently utilized and there was limited familiarity with others. We anticipate this is largely based on various shift and unit assignments that would make some protocols more applicable than others to the populations serviced. One of the survey questions polled the respondents on the necessity of the email notification to the prescriber given that this practice is of a higher level of notification than other established hospital protocols which only requires a notation of the change within the medication order. Seventy-one percent (n = 10) of respondents favored removing the email notification, citing primarily that it would be consistent with physician comments regarding the existing notifications. Pharmacists also identified further areas of protocol development including electrocardiogram ordering for QTc monitoring, implementation of a standardized vancomycin dosing protocol, discontinuation of duplicate orders, product substitution for nonformulary items and addition of a protocol for pharmacists to order over-the-counter or nonprescription products as they would in a community setting. This input will shape the revision of the policy and its protocols.
Conclusion
Consistent with the published literature, pharmacists effectively performed pharmacotherapy interventions in a multitude of practice categories for adult inpatients of an acute care community-teaching hospital using a single, comprehensive clinical policy. Providing these broadly scoped protocols in a singular policy allowed pharmacists to increase the autonomy with which they applied their pharmacotherapy expertise during the course of their routine, prospective care and expanded the established benefit of allowing professionals to work to their fullest extent. Pharmacist protocol intervention was met with a high physician acceptance rate.
Acknowledgment: We thank all the pharmacists at UMCPP for supporting our efforts to refine pharmacy practice for our patients.
Corresponding author: Liza Barbarello Andrews, PharmD, BSPharm, BCPS, Rutgers, The State University of New Jersey, 160 Frelinghuysen Rd, Piscataway, NJ 08854, [email protected].
Financial disclosures: None.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
1. Bond CA, Raehl CL. Pharmacist provided anticoagulation management in United States hospitals: death rates, length of stay, Medicare charges, bleeding complications and transfusions. Pharmacother 2004;24:953–63.
2. Yen YH, Chen HY, Wuan-Jin L, et al. Clinical and economic impact of a pharmacist-managed iv-to-po conversion service for levofloxacin in Taiwan. Int J Clin Pharmacol Ther 2012;50:136–41.
3. Buyle F, Vogelaers D, PelemanR, et al. Implementation of guidelines for sequential therapy with fluoroquinolones in a Belgian hospital. Pharm World Sci 2010;32:404–10.
4. Davis SL, Delgado G, McKinnon PS. Pharmacoeconomic consideration associated with the use of intravenous-to-oral moxifloxacin for community-acquired pneumonia Clin Infect Dis 2005;41Supp2;5:136–43.
5. Ho BP, Lau TT, Balen RM, et al. The impact of a pharmacist-managed dosage form conversion service on ciprofloxacin usage at a major Canadian teaching hospital: a pre- and post-intervention study. BMC Health Serv Res 2005;5:48.
6. Kuti JL, Le TN, Nightingale CH, et al. Pharmacoeconomics of a pharmacist-managed program for automatically converting levofloxacin route from iv to oral. Am J Health Sys Pharm 2002;59:2209–15.
7. Cohen SM, Lipsett PA, Buchman TG, et al. Comparison of intravenous/oral ciprofloxacin plus metronidazole versus piperacillin/tazobactam in the treatment of complicated intraabdominal infections. Ann Surg 2000;232:254–62.
8. Plouffe J, Schwartz DB, Kolokathis A, et al. Clinical efficacy of intravenous followed by oral azithromycin monotherapy in hospitalized patients with community-acquired pneumonia. Antimicrob Agents Chemother 2000;44:1796–802.
9. Wetzstein GA. Intravenous to oral (IV:PO) anti-infective combination therapy. Cancer Control 2000;7:170–6.
10. Paladino JA. Pharmacoeconomics of antimicrobial therapy. Am J Healthsys Pharm 1999;56(Supp3):S25–8.
11. Ahkee S, Smith S, et al. Early switch from intravenous to oral antibiotics in hospitalized patient with infections: a six-month prospective study. Pharmacotherapy 1997;17:569–75.
12. Przybylski KG, Rybak MJ, Martin PR, et al. A pharmacist-initiated program of intravenous to oral antibiotic conversion. Pharmacotherapy 1997;17:271–6.
13. Jewesson P. Cost-effectiveness and value of an IV switch. Pharmacoeconomics 1994;5(Supp2):20–6.
14. Ramirez J. Advances in antibiotic use: switch therapy. Curr Ther Res 1994;55(suppA):30–3.
15. Shepard MF. Making the switch from IV to PO. Am J Healthsys Pharm 1994;50:2510.
16. Chessin LN. When to switch from IV to oral antibiotics. Patient Care 1993;15:113–25.
17. Cohen MR. Important news! IV route not needed to justify hospitalization for antibiotics. Hospital Pharmacy 1993;28:946.
18. Schentag JJ. Changes in antimicrobial agent usage resulting from interactions among clinical pharmacy, infectious disease division and the microbiology laboratory. Diagnos Microbiol Infect Dis 1993;16:255–64.
19. Fighetto L. Intravenous to oral stepdown program: four years’ experience in a large teaching hospital. Ann Pharmacother 1992;26:1447–51.
20. Powers T. Clinical and economic effect of ciprofloxacin as an alternative to injectable antimicrobial therapy. Am J Healthsys Pharm 1990;47:1781–4.
21. Ament PW, McGuire WM. Setting up an automatic pharmacist-initiated pharmacokinetic dosing service. Hosp Formul 1993;28:589–92.
22. Mousavi M, Dashti-Khavidaki S, Khalili H, et al. Impact of clinical pharmacy services on stress ulcer prophylaxis prescribing and related cost in patients with renal insufficiency. Int J Pharm Pract 2012;Nov 9.
23. Damaske DL, Baird RW. Development and implementation of a pharmacist managed inpatient warfarin protocol. Proc (Baylor Univ Med Cent) 2005;18:397–400.
24. Radley AS, Hall J, Farrow M, et al. Evaluation of anticoagulation control in a pharmacist operated anticoagulation clinic. J Clin Pathol 1995;48:545–47.
25. Pharmacy and Therapeutics Committee. Medication policies and protocols of Nebraska Methodist Hospital, Methodist Women’s Hospital.
26. University of Kentucky Pharmacy Services. Therapeutic interchanges [Internet]. Available at www.hosp.uky.edu/pharmacy/interchange.asp
27. Bayshore Community Hospital Department of Pharmacy. Conversion of intravenous azithromycin (Zithromax), ceftriaxone (Rocephin), ciprofloxacin (Cipro), fluconazole (Diflucan), lansoprazole (Prevacid), levofloxacin (Levaquin), linezolid (Zyvox), metronidazole (Flagyl), moxifloxacin (Avelox), potassium chloride (KC), or ranitidine (Zantac) to oral medication [Internet]. [cited 2013 May 9]. Available at www.ashp.org/s_ashp/docs/files/R-IVtoPOConvPol-2.pdf.
28. Medication Education Safety Approval Committee, Massachusetts General Hospital. Automatic intravenous to oral protocol [Internet]. MESAC memo; 2005 July [cited 2013 March 9]. Available at www2.massgeneral.org/pharmacy/mesac/mesac_memo3.pdf.
29. Hanson RL, Habibi M, Khamo N, et al. Integrated clinical and specialty pharmacy practice model for management of patients with multiple sclerosis. Am J Health Syst Pharm 2014;71:463–9.
30. Making pharmacists part of the multidisciplinary team. Hosp Case Manag 2014;22:13–6.
31. Preslaski CR, Lat I, MacLaren R, et al. Pharmacist contributions as members of the multidisciplinary ICU team. Chest 2013;144:1687–95.
32. Ripley TL, Adamson PB, Hennebry TA, et al. Collaborative practice model between cardiologists and clinical pharmacist for management of patients with cardiovascular disease in an outpatient clinic. Ann Pharmacother 2014;48:412–9.
33. Smith M, Bates DW, Bodenheimer TS. Pharmacists belong in accountable care organizations and integrated care teams. Health Aff (Millwood) 2013;32:1963–70.
34. Ukens C. New Jersey rewrites its state pharmacy practice act. Drug Topics 2004;148:41.
35. New Jersey Board of Pharmacy Laws. Statute 45:14-64. Inapplicability relative to collaborative drug therapy management in hospital. [Internet] July 2011 [cited 11 September 14]. Available from: www.njconsumeraffairs.gov/laws/pharmlaws.pdf.
36. ASHP Commission on Therapeutics. ASHP therapeutic guidelines on stress ulcer prophylaxis. Am J Health Syst Pharm 1999;56:347–79.
37. Bond CA, Raehl CL. Clinical pharmacy services, pharmacy staffing, and hospital mortality rates. Pharmacotherapy 2007;27:481–93.
38. Bond CA, Raehl CL, Franke T. Interrelationships among mortality rates, drug costs, total cost of care, and length of stay in United States hospitals: summary and recommendations for clinical pharmacy services and staffing. Pharmacotherapy 2001;21:129–41.
Management of Acute Decompensated Heart Failure in Hospitalized Patients
From Ohio Health, Riverside Methodist Hospital, Columbus, OH.
Abstract
- Objective: To review the current in-hospital management of patients with acute decompensated heart failure (ADHF).
- Methods: Review of the literature.
- Results: Heart failure is a leading cause of hospitalization in the elderly, and morbidity, mortality, and hospital readmission rates for ADHF remain high. The patient’s hemodynamic status along with the use of prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms, and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion; however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy.
- Conclusion: Patients with ADHF are at increased risk for readmission to the hospital as well as at increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Heart failure is a major public health problem in the United States and the leading cause of hospitalization in patients 65 years of age and older [1]. Patients hospitalized with acute decompensated heart failure (ADHF) have a readmission rate as high as 50% within 6 months and 25% within 30 days [2]. It is estimated that $32 billion is spent on heart failure care each year, the majority of which is directly related to inpatient care. Projections show that by 2030 the total cost of heart failure will increase to $70 billion per year [1]. Despite the growing burden, advances in treatment have been limited [2,3] and management continues to be a challenge. In this article, we review the current in-hospital management of patients with ADHF.
Case Study
Initial Presentation
A 64-year-old woman with a nonischemic dilated cardiomyopathy presents to the emergency department (ED) with a 4-day history of progressive dyspnea on exertion. She can not ambulate more than 50 feet without having to stop due to dyspnea and reports increased lower extremity edema. She is found to have a heart rate of 105 bpm, a respiratory rate of 30 breaths/min, and a blood pressure of 90/51 mm Hg. Physical examination is remarkable for distended neck vein, S3 gallop, end expiratory wheezing in the bases, and lower extremity edema. Blood tests, including a B-type natriuretic peptide level, are pending. Electrocardiogram and chest radiograph are ordered. The physician suspects that the patient has ADHF and admits her for further management.
What are aspects of initial management in the ED?
Most patients that present for evaluation and management of ADHF are first evaluated in the ED. Initial management includes an assessment of oxygenation, hemodynamic status, and adequacy of tissue perfusion, as well as for possibility of an acute coronary syndrome. A complete history, physical examination, chest radiography, 12-lead electrocardiogram, cardiac troponin T or I, electrolytes, and complete blood count should be obtained to allow rapid diagnosis and triage followed by prompt, aggressive treatment in the ED or observation unit. This should alleviate the patient’s symptoms sooner, and it is intuitive that this would lessen morbidity and length of hospital stay [4].
How are patients with ADHF classified?
What risk assessment tools are available?
B-type natriuretic peptide (BNP) and N-terminal fragment proBNP (NT-proBNP) were recently validated as diagnostic aids for the differentiation of etiologies of dypnea in patients in the ED with possible symptoms of ADHF. Use of these biomarkers can help reduce diagnostic uncertainty and associated mismanagement of patients presenting with nonspecific symptoms of dysp-nea [4,5,7]. Low or normal levels (BNP < 100 pg/ml or NT-proBNP < 500 pg/ml) have a high negative predictive value for excluding heart failure.
Elevated BNP or NT-proBNP levels may also yield prognostic information, identifying patients at increased risk of mortality or rehospitalization when value does not fall after aggressive heart failure management [8,9].In a recent study by Fonarow et al, the levels of BNP on hospital admission correlated directly with the risk of in-hospital mortality in patients admitted with ADHF independent of left ventricular ejection fraction. When the levels of BNP were below 430 pg/ml, the in-hospital mortality was 1.9%, and when the levels were above 1730 pg/ml, the mortality went up to 6% (P < 0.001) [8]. Additionally, elevated pre-discharge BNP levels (BNP > 350 ng/l; P < 0.001) in patients with ADHF seem to identify those at increased risk of death or readmission after in-patient management [9]. Elevated cardiac troponin T or I in hospitalized patients with ADHF also are associated with increased mortality, including in those without acute coronary syndrome or underlying coronary artery disease [10,11].
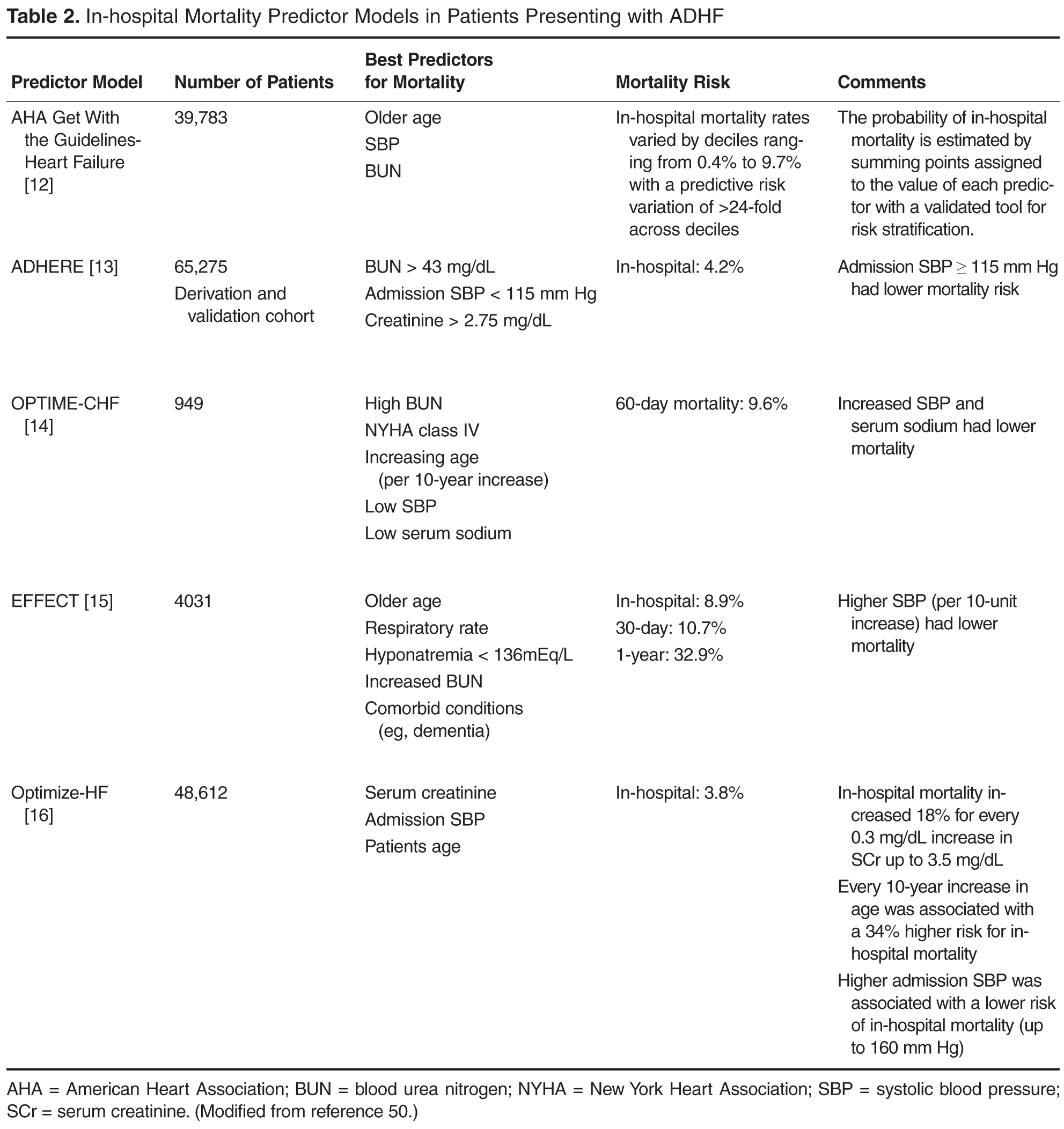
Case Continued
Upon further evaluation by a cardiologist, the patient is cool and clammy with elevated neck veins and prominent S3 confirmed. She continues to report severe shortness of breath after 1 dose of intravenous (IV) furosemide in the ED. Repeat vital signs shows a blood pressure of 83/49 mm Hg and respiratory rate of 33. Her electrocardiogram shows sinus tachycardia. The cardiologist determines that the patient’s clinical profile is “cold and wet” and admits the patient to the cardiac care unit (CCU) with a diagnosis of ADHF.
Initial blood tests show a BNP level of 1830 pg/ml, troponin I is 0.63 and stable after 2 measurements, serum creatinine is 1.6 mg/dL, BUN is 44 mg/dL, and serum sodium is 132 mg/dL. The GWTG-HF risk score for in-hospital mortality was calculated based on admission data and the probability of death was estimated at > 5% to 10% [12]. Prompt aggressive medical therapy was instituted in the CCU consisting of furosemide infusion to reduce congestion and IV dobutamine to improve systemic perfusion. Enoxoparin 40 mg subcutaneously once daily was initiated for venous thromboembolism prophylaxis.
What are important aspects of therapy for ADHF?
Several days to weeks prior to the appearance of signs and symptoms of volume overload, patients may develop hemodynamic congestion, defined as an elevation of ventricular filling pressure/pulmonary capillary wedge pressure independent of clinical evidence of fluid overload [17]. Elevated filling pressure is the culprit in the development of most of the signs and symptoms of ADHF and is the target for treatment.

Although use of vasoactive medications such as nitroglycerin or nitroprusside are not routinely recommended for use in all ADHF patients admitted to the hospital, retrospective analysis of the ADHEREdatabase suggests that there is a significant reduction of mortality, hospital length of stay, admission to intensive care unit, invasive procedures, and prolonged hospitalizations when IV diuretics, vasodilators (nitroglycerin, nitroprusside, nesiritide,) and/or positive inotropes (milrinone, dobutamine) are initiated in the ED within 6 hours of an ADHF presentation [18,19].However, whether prompt ED intervention impacts intermediate- to long-term outcomes is unknown [4].
Hospitalized patients with ADHF are at increased risk of venous thromboembolism mainly due to reduced cardiac output, increased systemic venous pressure, and reduced activity levels. Therefore, it is recommended that during the hospitalization ADHF patients receive prophylaxis against venous thromboembolism with low-dose unfractionated heparin or low-molecular-weight heparin if there is no contraindication [5].Individual therapeutic choices for ADHF are reviewed in detail below.
What treatments are used to relieve congestion?
Diuresis
In patients admitted to the hospital with ADHF, initial effective diuresis is vital to lowering cardiac filling pressures and relieving symptoms of congestion. Intravenous loop diuretics represent the first line of treatment and have long been the mainstay of therapy for decompensated heart failure with preserved or reduced ejection fraction, reducing fluid overload, and relieving symptoms.
Despite its long track record, the dose administration of IV diuretics is more of an art than a science. Medication dosage sufficient to produce a rate of diuresis that will optimize volume status and relieve signs and symptoms of congestion without causing kidney injury or hypotension is recommended [5].Due to the relatively short half-life of loop diuretics and concerns about tubular sodium reabsorption in the kidneys, continuous IV diuretic infusion has been suggested to enhance diuresis and avoid sodium and fluid rebound [5,20,21]. However, continuous loop diuretic infusion has not proven superior to intermittent IV bolus dosing in clinical studies. Recent data from the Diuretic Optimization Strategies Evaluation (DOSE) trial comparing bolus versus continuous infusion diuretic strategy in patients with ADHF showed no difference in global symptom relief, diuresis, or any of the clinical secondary endpoints including composite of death, re-hospitalization, or ED visits with either IV bolus versus continuous infusion or low versus high doses of furosemide [22]. Concern has also been previously raised about adverse outcomes utilizing high doses of loop diuretics in the treatment of ADHF [20,23,24]. However, the DOSE trial also evaluated the safety of 2 strategies for furosemide dosing in patients with ADHF. The study randomized ADHF patients with a prior diagnosis of chronic heart failure to 4 different treatment groups, either a high dose (2.5x their daily chronic oral furosemide dose) or low dose (1x their daily chronic oral furosemide dose), which was given either twice daily via IV bolus or via continuous infusion. The study showed no difference in change in renal function from baseline to 72 hours with either IV bolus versus continuous infusion or low versus high doses of furosemide [22].
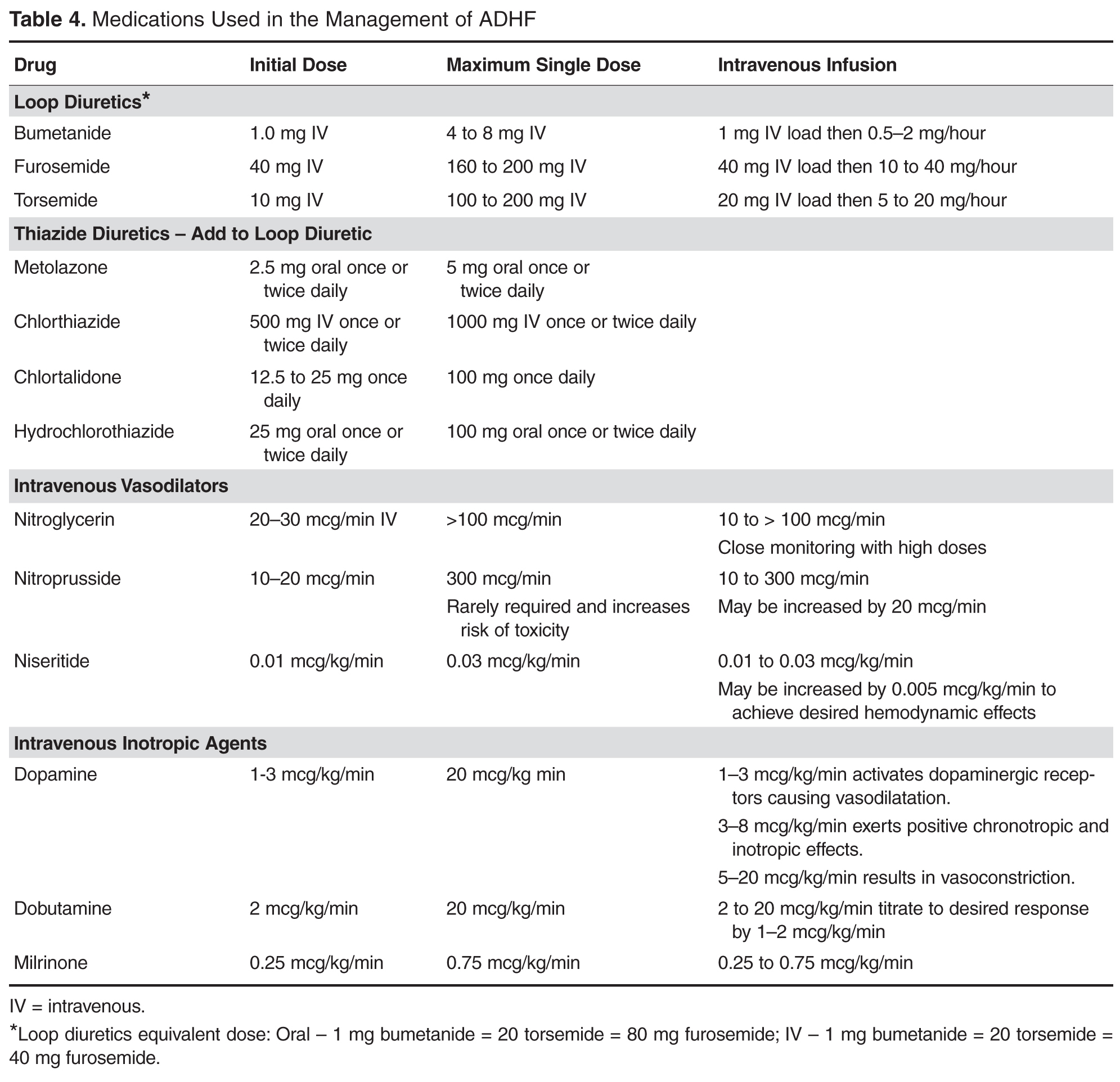
Ultrafiltration
For patients with marked fluid overload who are unresponsive to diuretic therapy, peripheral ultrafiltration may be considered. Initial data demonstrated that early ultrafiltration effectively and safely reduced congestion in patients with ADHF with diuretic resistance and renal insufficiency. Length of stay was reduced, with 60% of discharges in 3 days or less and 1 readmission at 30 days. Neurohormonal activation, indicated by reduction in BNP level, was reduced without worsening glomerular filtration rate, hypotension or electrolyte abnormalities [28]. The UNLOAD trial confirmed these results and extended their findings to show that patients undergoing peripheral ultrafiltration had greater weight and net fluid loss at 48 hours and reduced rate of rehospitalization at 90 days when compared with IV diuretic therapy alone in ADHF patients. Interestingly, there was no difference in the dyspnea score at 48 hours and there was a trend toward worsening of renal function in the ultrafiltration group. The study was not powered to document a survival benefit [29]. However, the more recent Cardiorenal Rescue Study in ADHF (CARRESS-HF) trial involving patients with ADHF and worsening renal function showed that there was no difference in weight loss between patients randomized to ultrafiltration or a strategy of stepped pharmacologic therapy. Additionally, ultrafiltration was associated with a significant increase in creatinine at 96 hours and a higher rate of adverse events related to the procedure, driven by complications from intravenous catheter insertion. There was no difference between the 2 groups in death or rehospitalization for heart failure [30]. At present, ultrafiltration may be a reasonable option if all diuretic strategies are unsuccessful in relieving congestion [5].
Vasopressin-Receptor Antagonists
The vasopressin-receptor antagonists represent a relatively new class of medications that target the vasopressin receptors V1a and V2. Activation of the vasopressin V2 receptors by arginine vasopressin in heart failure causes inappropriate free water retention contributing to the symptoms of congestion and hyponatremia [31]. Currently, the only 2 vasopressin-receptor antagonists available for clinical use are conivaptan (V1a /V2 receptor antagonist) and tolvaptan (V2 receptor antagonist). The effectiveness of tolvaptan was tested in a randomized study (EVEREST) in patients hospitalized with ADHF [32,33]. At 1 year there was no difference seen in the primary endpoints of all-cause mortality, death from cardiovascular causes, or first hospitalization for heart failure [32,33]. However, hyponatremia, when present, was improved in the tolvaptan group. Conivaptan has a similar hemodynamic profile compared to tolvaptan, but without improving signs and symptoms in hospitalized patients with ADHF [34]. Currently, vasopressin antagonists are recommended in the management of ADHF by professional guidelines as only a class IIb indication in hospitalized patients with volume overload and severe hyponatremia [5].
Case Continued
After 24 hours of medical therapy in the CCU, the patient is no longer clammy and cool but continues to have shortness of breath, and peripheral edema is not improving. She continues to have elevated JVP and S3. Her blood pressure is now 120/79 mm Hg and her heart rate is 110. A Swan-Ganz catheter placed this morning showed a cardiac index of 1.8 L/minute/m2 (reference range, 2.5–4.0 L/min/m2); pulmonary capillary wedge pressure is 28 mm Hg (reference range, 6–12 mm Hg) and systemic vascular resistance is 1932 dyne/second/cm5 (reference range, 800–1200 dynes/sec/cm5). The physician decides to add nitroprusside to lower her filling pressure and systemic vascular resistance.
What is the role of vasoactive medications in treatment?
Vasodilators
Nitroglycerin is a venodilating medication with preload reduction properties at low doses and an arterial dilator at high doses [35]. Preload reduction improves left ventricular filling pressures and pulmonary congestion without increasing the oxygen demand in the heart in patients with ADHF. This leads to an improvement of symptoms, including dyspnea, in as early as 5 minutes [36]. For a highly symptomatic patient, nitroglycerin given sublingually can be useful in an acute situation because it is typically immediately available while preparations are made for administration of IV medications. Limitations of nitroglycerin include rapid tachyphylaxis within several hours of continuous exposure at high doses, resistance to the hemodynamic effects of nitroglycerin in up to 20% of patients, and hypotension, which may occur before significant preload reduction effect can be obtained [37]. When symptomatic hypotension becomes a problem, the highest hemodynamically tolerable dose should be given. Another agent with a potent vasodilator effect used in the treatment of heart failure is sodium nitroprusside (SNP). As opposed to nitroglycerin, this drug has an equally potent preload- and afterload-reducing effect [35]. Afterload reduction through its arteriodilator effect has the benefit of increasing cardiac output and decreasing myocardial oxygen demand with improvement of pulmonary congestion [36]. SNP is used in less than 1% of patients hospitalized with heart failure [38], probably due to the potential for causing marked hypotension, its need for invasive hemodynamic monitoring, and the rare risk for thiocyanate toxicity with high doses and/or longer infusions, especially in patients with reduced hepatic perfusion and renal function, as in the case of low-output heart failure [35]. However, data demonstrating safety and efficacy of SNP infusion in patients with ADHF are limited [39].A single-center, retrospective case-control study suggested that the administration of SNP in carefully selected patients with advanced low-output ADHF was safe and may be associated with favorable long-term clinical outcomes [39]. SNP can be attractive in severely congested patients with hypertension or severe mitral regurgitation complicating left ventricular failure, but prospective trials are needed to clarify the safety and efficacy in this patient population.
Nesiritide is a human recombinant form of BNP that has a direct effect on the vascular endothelium by increasing the bioavailability of nitric oxide through stimulation of cyclic guanosine monophosphate. Its primary mechanism of action is to reduce left ventricular filling pressures by a systemic and pulmonary vasodilator effect. It also promotes diuresis and natriuresis [40].The initial efficacy of nesiritide was demonstrated in the VMAC (Vasodilation in the Management of Acute Congestive Heart Failure) study, a randomized trial of IV nesiritide versus IV nitroglycerin or placebo in decompensated heart failure patients. A significant reduction in pulmonary capillary wedge pressure was demonstrated within 15 minutes in the nesiritide group and maintained at 3 hours compared to either nitroglycerin or placebo, with a similar improvement in dyspnea extending out to 24 hours [41].
The large ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) randomized ADHF patients to nesiritide or placebo and tested the hypothesis that nesiritide would be superior to placebo in improving acute dyspnea, all-cause mortality, and heart failure readmission in patients presenting with ADHF [42]. Nesiritide-treated patients showed only a modest early improvement in self-assessed dyspnea and no difference in the composite endpoint of death or rehospitalization at 30 days in patients admitted with ADHF. Reassuringly, there was no increase in renal failure compared to placebo; however, the incidence of symptomatic hypotension was higher with nesiritide [42]. Although nesiritide remains in the armentarium of vasoactive medications for ADHF, less expensive vasodilators such as nitroglycerin or nitroprusside may be preferred by many clinicians.
Overall, vasodilators represent a good treatment option for patients presenting with ADHF characterized by low cardiac output, high filling pressures, and elevated systemic vascular resistance. There is no clear evidence, however, to suggest that IV vasodilators improve survival in hospitalized patients with ADHF; thus, its use should be restricted to the relief of dyspnea in patients with stable blood pressure [5].
Inotropic Therapy
The most commonly used positive inotropic agents in the management of patients with ADHF in the United States are dobutamine (beta-1, beta-2, and alpha adrenoreceptor agonist) and milrinone (phosphodiesterase-III inhibitor) [38]. Inotropes increase cardiac output by increasing myocardial contractility, reduce left and right ventricular filling pressures, and improve hemodynamic parameters. Despite these hemodynamic effects, inotropic agents have not demonstrated a survival benefit in patients with ADHF. A major limitation regarding these agents is that they increase the risk of cardiac arrhythmias by increasing intracellular calcium in cardiac myocytes. In fact, retrospective analyses suggest that most inotropic agents are associated with an increased risk of death [38,43].
Milrinone inhibits type III isoform of the enzyme phosphodiasterase leading to an increase in intracellular cyclic AMP to exert its positive inotropic effect on the myocardium. Milrinone also exerts systemic and pulmonary vasodilator effects in the circulation decreasing right atrial, pulmonary capillary wedge, and mean arterial pressure. In the OPTIME-CHF trial, patients with chronic heart failure admitted to the hospital with ADHF were randomized to short term infusion of milrinone vs. placebo plus standard therapy. Milrinone resulted in more hypotension, atrial fibrillation and ventricular arrhythmias without any benefit on mortality or re-hospitalization [44].A retrospective analysis from the ADHEREregistry showed that in-hospital mortality was twofold higher with the use of dobutamine or milrinone in patients with ADHF when compared to treatment with vasodilators [38].
Dobutamine is a beta-1, beta-2, and alpha adrenoreceptor agonist that works by increasing myocardial contractility leading to an increase in cardiac output as its primary cardiovascular effect. Currently, routine use of IV positive inotropic agents in the absence of imminent cardiogenic shock or low output ADHF with systemic hypoperfusion is generally not recommended due to concerns of adverse effects [5]. The ACCF/AHA guidelines recommend the use of positive inotropic agents to relieve symptoms, improve systemic perfusion and preserve end-organ function in patients with severe left ventricular systolic failure and low output syndrome with evidence of end-organ dysfunction (such as hypotension, altered mentation, cool extremities, low urine output and serum markers indicative of renal and/or hepatic dysfunction) with or without congestion [5].
Continuous outpatient therapy with inotropes may be a viable option in patients with stage D (end stage) heart failure who are deemed unlikely to survive hospital discharge [45].This is also supported by the ACCF/AHA practice guidelines where IV inotropic support may be considered for the previous reasons only after all alternative therapies to achieve stability have failed (Class IIB indication) [5].
Is there a role for morphine?
For decades morphine has been considered an essential component in the armamentarium for the treatment of ADHF. Its preload-reducing effect, anti-anxiety properties, and breathlessness suppression has made morphine a popular medication in the treatment of ADHF. Despite its common use, there is a lack of prospective randomized trials demonstrating the safety and benefit of this drug. In a retrospective analysis from the ADHERE database, IV morphine used for ADHF was associated with higher rates of adverse events, including increase use of mechanical ventilation, prolonged hospitalization, increased intensive care unit admissions, and higher mortality, bringing into question its safety profile [46]. Until a randomized trial is completed demonstrating safety and benefit, caution is advised regarding the use of morphine in ADHF.
Case Continued
Over the next 72 hours the patient’s symptoms improved. She no longer has dyspnea at rest, she has had a proper urine-output response to therapy, her serum creatinine has returned to normal, and her vital signs have remained stable. The IV vasodilator was discontinued, dobutamine was weaned off, and the patient was transitioned to guideline-directed medical therapy with an angiotensin-converting enzyme (ACE) inhibitor while continuing IV furosemide. Hospitalized patients who are hemodynamically stable should be transitioned to guideline-directed medical therapy with an oral ACE inhibitor unless the patient has a contraindication, such as marked azotemia or hyperkalemia. Low-dose carvedilol was initiated after optimization of volume status was confirmed. In the absence of shock and after optimization of volume status, every effort should be made to initiate low-dose beta blockers prior to hospital discharge.
When is mechanical circulatory support indicated in ADHF patients?
Mechanical circulatory support has emerged as a reasonable option in selected patients with acute and reversible cardiogenic shock (ie, acute coronary syndrome or an acute mechanical problem such as a torn papillary muscle or ventricular septal defect) [5]. Recently, the utility of intraaortic balloon pump (IABP) in the setting of cardiogenic shock resulting from acute coronary syndrome was called into question with the negative results from the Intraaortic Balloon Pump in Cardiogenic Shock II (IABP-SHOCK II) trial [47]. The study compared IABP with best available medical therapy alone among patients with acute myocardial infarction complicated by cardiogenic shock for who early revascularization was planned. Use of IABP did not reduce 30-day mortality compared with medical therapy in this patient population [47]. Whether IABP has a significant role in mechanical complications, such as acute ventricular septal rupture or papillary muscle rupture, is unknown due to the paucity of data in the management of patients with such complications. Therefore, when patients present with severe acute cardiogenic shock refractory to medical therapy, mechanical circulatory support with either ventricular assist devices (VAD) or extracorporeal membrane oxygenation (ECMO) is the preferred means to reverse terminal circulatory collapse. VADs are effective in the short-term as a “bridge-to-recovery” or as a “bridge-to-decision” when recovery, transplant candidacy, or neurologic status are still uncertain [48,49]. There are several options currently available for mechanical circulatory support, including surgically implanted VADs or the percutaneously implanted VADs, such as the Impella 2.5, 3.5 and 5.0 (Abiomed, Danvers, MA) and the TandemHeart pump (Cardiac Assist, Pittsburgh, PA).The ideal device and optimal duration of temporary support are yet to be defined. A detailed description of the function and clinical effects of mechanical support devices is beyond the scope of this article, although thorough reviews are available [48,49].
What elements of care may help optimize the discharge process?
Transition of care in hospitalized patients with ADHF to outpatient care is a critical and vulnerable period for patients given the complexity of the discharge planning for heart failure. A multidisciplinary heart failure disease management program is recommended in both the inpatient and outpatient setting to address the barriers to successful transition of care [5]. Physicians and physician extenders, nurses, pharmacists, and social workers can work together to identify risk factors for readmission and bridge the gap between the inpatient and outpatient setting.
Patients at high risk for hospital readmission should be referred to a heart failure disease management program [5,37]. Patients at high risk for hospital readmission include patients with renal insufficiency, low output state, diabetes mellitus, chronic lung disease, persistent NYHA functional class III, IV symptoms, frequent hospitalizations, multiple comorbidities, history of depression, cognitive impairment, or recurrent problems with noncompliance. There is strong evidence that a heart failure disease management program will reduce rehospitalization rates and costs while improving functional status and quality of life of the patient [37].In addition, a heart failure disease management clinic often can see the patient shortly after discharge, which may allow earlier discharge of the patient and shorter length of stay. Proven therapies such as ACE inhibitors, angiotensin-receptor blockers, beta blockers, and aldosterone antagonists can be titrated frequently in this setting.
It is strongly recommended that comprehensive written discharge instructions be provided at the end of hospitalization with special emphasis on diet, discharge medications, activity level, follow-up appointment, daily weight monitoring, and instructions for recurrence of symptoms [5].
Case Conclusion
The patient tolerated well the initiation of guideline-directed medical therapy and is continued on the ACE inhibitor and beta-blocker medications. After 4 days IV furosemide is discontinued and transitioned to oral furosemide. Precipitant causes of heart failure were addressed throughout hospitalization. It was determined that the patient had been taking high doses of nonsteroidal anti-inflammatory drugs due to knee pain. She was educated on this and other potential precipitant factors. Heart failure education was reinforced, including self-care, emergency plans, and need for medication and diet adherence. She is scheduled an early follow-up visit within 2 weeks of hospital discharge in the multidisciplinary heart failure disease management clinic.
Summary
ADHF is a major public health problem commonly encountered and often initially managed in the ED. Initial history and physical examination are important to estimate the degree of congestion and peripheral perfusion. The patient’s hemodynamic status along with the use prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion, however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy. Patients with ADHF are at increased risk for readmission to the hospital as well as increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Corresponding author: Carlos E. Sanchez, MD, 3705 Olentanfy River Rd., Columbus, OH 43214, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CES; analysis and interpretation of data, CES; drafting of article, CES; critical revision of the article, CES, DRR; collection and assembly of data, CES.
1. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 2013;127: e6–245.
2. Fonarow GC. ADHERE Scientific Advisory Committee. The ADHF National Registry (ADHERE): opportunities to improve care of patients hospitalized with ADHF. Rev Cardiovasc Med 2003;4(suppl 7):S21-S30.
3. Philbin EF, Dec GW, Enkins PL, et al. Socioeconomic status as an independent risk factor for hospital readmission for heart failure. Am J Cardiol 2001;87:1367–71.
4. Weintraub NL, Collins SP, Pang PS, et al.; on behalf of the American Heart Association Council on Clinical Cardiology and Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Acute heart failure syndromes: emergency department presentation, treatment, and disposition: current approaches and future aims: a scientific statement from the American Heart Association. Circulation 2010;122:1975–96.
5. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147-239.
6. Nohria A, Tsang SW, Fang JC, et al. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol 2003;41:1797–1804.
7. Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161–7.
8. Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in ADHF. J Am Coll Cardiol 2007;49:1943–50.
9. Logeart D, Thabut G, Jourdian P, et al. Pre-discharge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004;43:635–41.
10. Peacock WFIV, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008;358:2117–26.
11. Ilva T, Lassus J, Siirila-Waris K, et al. Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail 2008;10:772–9.
12. Peterson PN, Rumsfeld JS, Liang L, et al. A validated risk score for inhospital mortality in patients with heart failure from the American Heart Association Get With The Guidelines program. Circ Cardiovasc Qual Outcomes 2010;3:25–32.
13. Fonarow GC, Adams KF, Abraham WT, et al, for the ADHERE Scientific Advisory Committee, Study Groups and Investigators. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA 2005;293:572–80.
14. Felker GM, Leimberger JD, Califf RM, et al. Risk stratification after hospitalization for decompensated heart failure. J Card Fail 2004;10:460–6.
15. Lee DS, Austin PC, Rouleau JL, et al. Predicting mortality among patients hospitalized for heart failure: derivation and validation of a clinical model. JAMA 2003;290:2581–7.
16. Abraham WT, Fonarow GC, Albert NM, et al. Predictors of in-hospital mortality in patients hospitalized for heart failure. J Am Coll Cardiol 2008; 52:347–56.
17. Gheorghiade M, Shin DD, Thomas TO, et al. Congestion is an important diagnostic and therapeutic target in heart failure. Rev Cardiovasc Med 2006;7(suppl l):S12-S24.
18. Peacock WF, Emerman C, Costanzo MR, et al. Early vasoactive drugs improve heart failure outcomes. Congest Heart Fail 2009;15:256–64.
19. Maisel AS, Peacock WF, McMullin N, et al. Timing of immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure: an ADHERE analysis. J Am Coll Cardiol 2008;52:534–40.
20. Salvador DRK, Rey NR, Ramos GC, et al. Continuous infusion versus bolus injection of loop diuretics in congestive heart failure. Cochrane Database Syst Rev 2005(3):CD003178.
21. Pivac N, Rumboldt Z, Sardelic S, et al. Diuretic effects of furosemide infusion versus bolus injection in congestive heart failure. Int J Clin Pharmacol Res 1998;18:121–8.
22. Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with ADHF. N Engl J Med 2011;364:797–805.
23. Cotter G, Metzkor E, Kaluski E, et al. Randomized trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary edema. Lancet 1998;351:389–93.
24. Butler J, Forman DE, Abraham WT, et al. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J 2004;147:331–8.
25. Giamouzis G, Butler J, Starling RC, et al. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in ADHF (DAD-HF) Trial. J Card Fail 2010;16:922–30.
26. Cotter G, Weissgarten J, Metzkor E, et al. Increased toxicity of high-dose furosemide versus low-dose dopamine in the treatment of refractory congestive heart failure. Clin Pharmacol Ther 1997;62:187–93.
27. Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013;310:2533–43.
28. Costanzo MR, Saltzberg M, O’Sullivan J, et al. Early ultrafiltration in patients with decompensated heart failure and diuretic resistance. J Am Coll Cardiol 2005;46:2047–51.
29. Costanzo MR, Guglin ME, Saltzberg MT, et al; UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for ADHF. J Am Coll Cardiol 2007;49:675–83.
30. Bart BA, Goldsmith SR, Lee KL, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012;367:2296-304.
31. Shrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med 1999;341:577–85.
32. Konstam MA, Gheorghiade M, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007;297:1319–31.
33. Gheorghiade M, Konstam MA, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA 2007;297:1332–43.
34. Goldsmith SR, Elkayam U, Haught WH, et al. Efficacy and safety of the vasopressin V1A/V2-receptor antagonist conivaptan in ADHF: a dose-ranging pilot study. J Card Fail 2008;14:641–-7.
35. Shin DD, Brandimarte F, DeLuca L, et al. Review of current and investigational pharmacologic agents for acute heart failure syndromes. Am J Cardiol 2007;99:4A–23A.
36. Mattu A, Martinez JP, Kelly BS. Modern management of cardiogenic pulmonary edema. Emerg Med Clin North Am 2005;23:1105–25.
37. Lindenfeld J, Albert NM, Boehmer JP, et al. Executive Summary: HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 2010;16:e475-e539
38. Abraham WT, Adams KF, Fonarow GC, et al. ADHERE Scientific Advisory Committee and Investigators; ADHERE Study Group. In-hospital mortality in patients with ADHF requiring intravenous vasoactive medications: an analysis from the ADHF national registry (ADHERE). J Am Coll Cardiol 2005;46:57–64.
39. Mullens W, Abrahams Z, Francis GS, et al. Sodium nitroprusside for advanced low-output heart failure. J Am Coll Cardiol 2008;52:200–7.
40. Bhalla V, Willis S, Maisel AS. B-type natriuretic peptide: the level and the drug-partners in the diagnosis and management of congestive heart failure. Congest Heart Fail 2004;10(1 suppl 1):3–27.
41. Publication Committee for the VMAC investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531–40.
42. O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with ADHF. N Engl J Med 2011;365:32–43.
43. Elkayam U, Tasissa G, Binanay C, et al. Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am Heart J 2007;153:98–104.
44. Cuffe MS, Califf RM, Adams KF Jr, et al. Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized control trial. JAMA 2002;287:1541–7.
45. Hershberger RE, Nauman D, Walker TL, et al. Care processes and clinical outcomes of continuous outpatient support with inotropes (COSI) in patients with refractory endstage heart failure. J Card Fail 2003;9:180–7.
46. Peacock WF, Hollander JE, Diercks DB, et al. Morphine and outcomes in ADHF: an ADHERE analysis. Emerg Med J 2008;25:205–9.
47. Thiele H, Zeymer U, Neumann FJ, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med 2012;367:1287–96.
48. Abu-Omar Y, Tsui S. Mechanical circulatory support for AMI and cardiogenic shock. J Card Surg 2010;25:434–41.
49. Ziemba EA, John R. Mechanical circulatory support for bridge to decision: which device and when to decide. J Card Surg 2010;25:425–33.
50. Sanchez CE, Richards DR. Contemporary in-hospital management strategies for ADHF. Cardiol Rev 2011;19:122–9.
From Ohio Health, Riverside Methodist Hospital, Columbus, OH.
Abstract
- Objective: To review the current in-hospital management of patients with acute decompensated heart failure (ADHF).
- Methods: Review of the literature.
- Results: Heart failure is a leading cause of hospitalization in the elderly, and morbidity, mortality, and hospital readmission rates for ADHF remain high. The patient’s hemodynamic status along with the use of prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms, and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion; however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy.
- Conclusion: Patients with ADHF are at increased risk for readmission to the hospital as well as at increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Heart failure is a major public health problem in the United States and the leading cause of hospitalization in patients 65 years of age and older [1]. Patients hospitalized with acute decompensated heart failure (ADHF) have a readmission rate as high as 50% within 6 months and 25% within 30 days [2]. It is estimated that $32 billion is spent on heart failure care each year, the majority of which is directly related to inpatient care. Projections show that by 2030 the total cost of heart failure will increase to $70 billion per year [1]. Despite the growing burden, advances in treatment have been limited [2,3] and management continues to be a challenge. In this article, we review the current in-hospital management of patients with ADHF.
Case Study
Initial Presentation
A 64-year-old woman with a nonischemic dilated cardiomyopathy presents to the emergency department (ED) with a 4-day history of progressive dyspnea on exertion. She can not ambulate more than 50 feet without having to stop due to dyspnea and reports increased lower extremity edema. She is found to have a heart rate of 105 bpm, a respiratory rate of 30 breaths/min, and a blood pressure of 90/51 mm Hg. Physical examination is remarkable for distended neck vein, S3 gallop, end expiratory wheezing in the bases, and lower extremity edema. Blood tests, including a B-type natriuretic peptide level, are pending. Electrocardiogram and chest radiograph are ordered. The physician suspects that the patient has ADHF and admits her for further management.
What are aspects of initial management in the ED?
Most patients that present for evaluation and management of ADHF are first evaluated in the ED. Initial management includes an assessment of oxygenation, hemodynamic status, and adequacy of tissue perfusion, as well as for possibility of an acute coronary syndrome. A complete history, physical examination, chest radiography, 12-lead electrocardiogram, cardiac troponin T or I, electrolytes, and complete blood count should be obtained to allow rapid diagnosis and triage followed by prompt, aggressive treatment in the ED or observation unit. This should alleviate the patient’s symptoms sooner, and it is intuitive that this would lessen morbidity and length of hospital stay [4].
How are patients with ADHF classified?
What risk assessment tools are available?
B-type natriuretic peptide (BNP) and N-terminal fragment proBNP (NT-proBNP) were recently validated as diagnostic aids for the differentiation of etiologies of dypnea in patients in the ED with possible symptoms of ADHF. Use of these biomarkers can help reduce diagnostic uncertainty and associated mismanagement of patients presenting with nonspecific symptoms of dysp-nea [4,5,7]. Low or normal levels (BNP < 100 pg/ml or NT-proBNP < 500 pg/ml) have a high negative predictive value for excluding heart failure.
Elevated BNP or NT-proBNP levels may also yield prognostic information, identifying patients at increased risk of mortality or rehospitalization when value does not fall after aggressive heart failure management [8,9].In a recent study by Fonarow et al, the levels of BNP on hospital admission correlated directly with the risk of in-hospital mortality in patients admitted with ADHF independent of left ventricular ejection fraction. When the levels of BNP were below 430 pg/ml, the in-hospital mortality was 1.9%, and when the levels were above 1730 pg/ml, the mortality went up to 6% (P < 0.001) [8]. Additionally, elevated pre-discharge BNP levels (BNP > 350 ng/l; P < 0.001) in patients with ADHF seem to identify those at increased risk of death or readmission after in-patient management [9]. Elevated cardiac troponin T or I in hospitalized patients with ADHF also are associated with increased mortality, including in those without acute coronary syndrome or underlying coronary artery disease [10,11].
Case Continued
Upon further evaluation by a cardiologist, the patient is cool and clammy with elevated neck veins and prominent S3 confirmed. She continues to report severe shortness of breath after 1 dose of intravenous (IV) furosemide in the ED. Repeat vital signs shows a blood pressure of 83/49 mm Hg and respiratory rate of 33. Her electrocardiogram shows sinus tachycardia. The cardiologist determines that the patient’s clinical profile is “cold and wet” and admits the patient to the cardiac care unit (CCU) with a diagnosis of ADHF.
Initial blood tests show a BNP level of 1830 pg/ml, troponin I is 0.63 and stable after 2 measurements, serum creatinine is 1.6 mg/dL, BUN is 44 mg/dL, and serum sodium is 132 mg/dL. The GWTG-HF risk score for in-hospital mortality was calculated based on admission data and the probability of death was estimated at > 5% to 10% [12]. Prompt aggressive medical therapy was instituted in the CCU consisting of furosemide infusion to reduce congestion and IV dobutamine to improve systemic perfusion. Enoxoparin 40 mg subcutaneously once daily was initiated for venous thromboembolism prophylaxis.
What are important aspects of therapy for ADHF?
Several days to weeks prior to the appearance of signs and symptoms of volume overload, patients may develop hemodynamic congestion, defined as an elevation of ventricular filling pressure/pulmonary capillary wedge pressure independent of clinical evidence of fluid overload [17]. Elevated filling pressure is the culprit in the development of most of the signs and symptoms of ADHF and is the target for treatment.
Although use of vasoactive medications such as nitroglycerin or nitroprusside are not routinely recommended for use in all ADHF patients admitted to the hospital, retrospective analysis of the ADHEREdatabase suggests that there is a significant reduction of mortality, hospital length of stay, admission to intensive care unit, invasive procedures, and prolonged hospitalizations when IV diuretics, vasodilators (nitroglycerin, nitroprusside, nesiritide,) and/or positive inotropes (milrinone, dobutamine) are initiated in the ED within 6 hours of an ADHF presentation [18,19].However, whether prompt ED intervention impacts intermediate- to long-term outcomes is unknown [4].
Hospitalized patients with ADHF are at increased risk of venous thromboembolism mainly due to reduced cardiac output, increased systemic venous pressure, and reduced activity levels. Therefore, it is recommended that during the hospitalization ADHF patients receive prophylaxis against venous thromboembolism with low-dose unfractionated heparin or low-molecular-weight heparin if there is no contraindication [5].Individual therapeutic choices for ADHF are reviewed in detail below.
What treatments are used to relieve congestion?
Diuresis
In patients admitted to the hospital with ADHF, initial effective diuresis is vital to lowering cardiac filling pressures and relieving symptoms of congestion. Intravenous loop diuretics represent the first line of treatment and have long been the mainstay of therapy for decompensated heart failure with preserved or reduced ejection fraction, reducing fluid overload, and relieving symptoms.
Despite its long track record, the dose administration of IV diuretics is more of an art than a science. Medication dosage sufficient to produce a rate of diuresis that will optimize volume status and relieve signs and symptoms of congestion without causing kidney injury or hypotension is recommended [5].Due to the relatively short half-life of loop diuretics and concerns about tubular sodium reabsorption in the kidneys, continuous IV diuretic infusion has been suggested to enhance diuresis and avoid sodium and fluid rebound [5,20,21]. However, continuous loop diuretic infusion has not proven superior to intermittent IV bolus dosing in clinical studies. Recent data from the Diuretic Optimization Strategies Evaluation (DOSE) trial comparing bolus versus continuous infusion diuretic strategy in patients with ADHF showed no difference in global symptom relief, diuresis, or any of the clinical secondary endpoints including composite of death, re-hospitalization, or ED visits with either IV bolus versus continuous infusion or low versus high doses of furosemide [22]. Concern has also been previously raised about adverse outcomes utilizing high doses of loop diuretics in the treatment of ADHF [20,23,24]. However, the DOSE trial also evaluated the safety of 2 strategies for furosemide dosing in patients with ADHF. The study randomized ADHF patients with a prior diagnosis of chronic heart failure to 4 different treatment groups, either a high dose (2.5x their daily chronic oral furosemide dose) or low dose (1x their daily chronic oral furosemide dose), which was given either twice daily via IV bolus or via continuous infusion. The study showed no difference in change in renal function from baseline to 72 hours with either IV bolus versus continuous infusion or low versus high doses of furosemide [22].
Ultrafiltration
For patients with marked fluid overload who are unresponsive to diuretic therapy, peripheral ultrafiltration may be considered. Initial data demonstrated that early ultrafiltration effectively and safely reduced congestion in patients with ADHF with diuretic resistance and renal insufficiency. Length of stay was reduced, with 60% of discharges in 3 days or less and 1 readmission at 30 days. Neurohormonal activation, indicated by reduction in BNP level, was reduced without worsening glomerular filtration rate, hypotension or electrolyte abnormalities [28]. The UNLOAD trial confirmed these results and extended their findings to show that patients undergoing peripheral ultrafiltration had greater weight and net fluid loss at 48 hours and reduced rate of rehospitalization at 90 days when compared with IV diuretic therapy alone in ADHF patients. Interestingly, there was no difference in the dyspnea score at 48 hours and there was a trend toward worsening of renal function in the ultrafiltration group. The study was not powered to document a survival benefit [29]. However, the more recent Cardiorenal Rescue Study in ADHF (CARRESS-HF) trial involving patients with ADHF and worsening renal function showed that there was no difference in weight loss between patients randomized to ultrafiltration or a strategy of stepped pharmacologic therapy. Additionally, ultrafiltration was associated with a significant increase in creatinine at 96 hours and a higher rate of adverse events related to the procedure, driven by complications from intravenous catheter insertion. There was no difference between the 2 groups in death or rehospitalization for heart failure [30]. At present, ultrafiltration may be a reasonable option if all diuretic strategies are unsuccessful in relieving congestion [5].
Vasopressin-Receptor Antagonists
The vasopressin-receptor antagonists represent a relatively new class of medications that target the vasopressin receptors V1a and V2. Activation of the vasopressin V2 receptors by arginine vasopressin in heart failure causes inappropriate free water retention contributing to the symptoms of congestion and hyponatremia [31]. Currently, the only 2 vasopressin-receptor antagonists available for clinical use are conivaptan (V1a /V2 receptor antagonist) and tolvaptan (V2 receptor antagonist). The effectiveness of tolvaptan was tested in a randomized study (EVEREST) in patients hospitalized with ADHF [32,33]. At 1 year there was no difference seen in the primary endpoints of all-cause mortality, death from cardiovascular causes, or first hospitalization for heart failure [32,33]. However, hyponatremia, when present, was improved in the tolvaptan group. Conivaptan has a similar hemodynamic profile compared to tolvaptan, but without improving signs and symptoms in hospitalized patients with ADHF [34]. Currently, vasopressin antagonists are recommended in the management of ADHF by professional guidelines as only a class IIb indication in hospitalized patients with volume overload and severe hyponatremia [5].
Case Continued
After 24 hours of medical therapy in the CCU, the patient is no longer clammy and cool but continues to have shortness of breath, and peripheral edema is not improving. She continues to have elevated JVP and S3. Her blood pressure is now 120/79 mm Hg and her heart rate is 110. A Swan-Ganz catheter placed this morning showed a cardiac index of 1.8 L/minute/m2 (reference range, 2.5–4.0 L/min/m2); pulmonary capillary wedge pressure is 28 mm Hg (reference range, 6–12 mm Hg) and systemic vascular resistance is 1932 dyne/second/cm5 (reference range, 800–1200 dynes/sec/cm5). The physician decides to add nitroprusside to lower her filling pressure and systemic vascular resistance.
What is the role of vasoactive medications in treatment?
Vasodilators
Nitroglycerin is a venodilating medication with preload reduction properties at low doses and an arterial dilator at high doses [35]. Preload reduction improves left ventricular filling pressures and pulmonary congestion without increasing the oxygen demand in the heart in patients with ADHF. This leads to an improvement of symptoms, including dyspnea, in as early as 5 minutes [36]. For a highly symptomatic patient, nitroglycerin given sublingually can be useful in an acute situation because it is typically immediately available while preparations are made for administration of IV medications. Limitations of nitroglycerin include rapid tachyphylaxis within several hours of continuous exposure at high doses, resistance to the hemodynamic effects of nitroglycerin in up to 20% of patients, and hypotension, which may occur before significant preload reduction effect can be obtained [37]. When symptomatic hypotension becomes a problem, the highest hemodynamically tolerable dose should be given. Another agent with a potent vasodilator effect used in the treatment of heart failure is sodium nitroprusside (SNP). As opposed to nitroglycerin, this drug has an equally potent preload- and afterload-reducing effect [35]. Afterload reduction through its arteriodilator effect has the benefit of increasing cardiac output and decreasing myocardial oxygen demand with improvement of pulmonary congestion [36]. SNP is used in less than 1% of patients hospitalized with heart failure [38], probably due to the potential for causing marked hypotension, its need for invasive hemodynamic monitoring, and the rare risk for thiocyanate toxicity with high doses and/or longer infusions, especially in patients with reduced hepatic perfusion and renal function, as in the case of low-output heart failure [35]. However, data demonstrating safety and efficacy of SNP infusion in patients with ADHF are limited [39].A single-center, retrospective case-control study suggested that the administration of SNP in carefully selected patients with advanced low-output ADHF was safe and may be associated with favorable long-term clinical outcomes [39]. SNP can be attractive in severely congested patients with hypertension or severe mitral regurgitation complicating left ventricular failure, but prospective trials are needed to clarify the safety and efficacy in this patient population.
Nesiritide is a human recombinant form of BNP that has a direct effect on the vascular endothelium by increasing the bioavailability of nitric oxide through stimulation of cyclic guanosine monophosphate. Its primary mechanism of action is to reduce left ventricular filling pressures by a systemic and pulmonary vasodilator effect. It also promotes diuresis and natriuresis [40].The initial efficacy of nesiritide was demonstrated in the VMAC (Vasodilation in the Management of Acute Congestive Heart Failure) study, a randomized trial of IV nesiritide versus IV nitroglycerin or placebo in decompensated heart failure patients. A significant reduction in pulmonary capillary wedge pressure was demonstrated within 15 minutes in the nesiritide group and maintained at 3 hours compared to either nitroglycerin or placebo, with a similar improvement in dyspnea extending out to 24 hours [41].
The large ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) randomized ADHF patients to nesiritide or placebo and tested the hypothesis that nesiritide would be superior to placebo in improving acute dyspnea, all-cause mortality, and heart failure readmission in patients presenting with ADHF [42]. Nesiritide-treated patients showed only a modest early improvement in self-assessed dyspnea and no difference in the composite endpoint of death or rehospitalization at 30 days in patients admitted with ADHF. Reassuringly, there was no increase in renal failure compared to placebo; however, the incidence of symptomatic hypotension was higher with nesiritide [42]. Although nesiritide remains in the armentarium of vasoactive medications for ADHF, less expensive vasodilators such as nitroglycerin or nitroprusside may be preferred by many clinicians.
Overall, vasodilators represent a good treatment option for patients presenting with ADHF characterized by low cardiac output, high filling pressures, and elevated systemic vascular resistance. There is no clear evidence, however, to suggest that IV vasodilators improve survival in hospitalized patients with ADHF; thus, its use should be restricted to the relief of dyspnea in patients with stable blood pressure [5].
Inotropic Therapy
The most commonly used positive inotropic agents in the management of patients with ADHF in the United States are dobutamine (beta-1, beta-2, and alpha adrenoreceptor agonist) and milrinone (phosphodiesterase-III inhibitor) [38]. Inotropes increase cardiac output by increasing myocardial contractility, reduce left and right ventricular filling pressures, and improve hemodynamic parameters. Despite these hemodynamic effects, inotropic agents have not demonstrated a survival benefit in patients with ADHF. A major limitation regarding these agents is that they increase the risk of cardiac arrhythmias by increasing intracellular calcium in cardiac myocytes. In fact, retrospective analyses suggest that most inotropic agents are associated with an increased risk of death [38,43].
Milrinone inhibits type III isoform of the enzyme phosphodiasterase leading to an increase in intracellular cyclic AMP to exert its positive inotropic effect on the myocardium. Milrinone also exerts systemic and pulmonary vasodilator effects in the circulation decreasing right atrial, pulmonary capillary wedge, and mean arterial pressure. In the OPTIME-CHF trial, patients with chronic heart failure admitted to the hospital with ADHF were randomized to short term infusion of milrinone vs. placebo plus standard therapy. Milrinone resulted in more hypotension, atrial fibrillation and ventricular arrhythmias without any benefit on mortality or re-hospitalization [44].A retrospective analysis from the ADHEREregistry showed that in-hospital mortality was twofold higher with the use of dobutamine or milrinone in patients with ADHF when compared to treatment with vasodilators [38].
Dobutamine is a beta-1, beta-2, and alpha adrenoreceptor agonist that works by increasing myocardial contractility leading to an increase in cardiac output as its primary cardiovascular effect. Currently, routine use of IV positive inotropic agents in the absence of imminent cardiogenic shock or low output ADHF with systemic hypoperfusion is generally not recommended due to concerns of adverse effects [5]. The ACCF/AHA guidelines recommend the use of positive inotropic agents to relieve symptoms, improve systemic perfusion and preserve end-organ function in patients with severe left ventricular systolic failure and low output syndrome with evidence of end-organ dysfunction (such as hypotension, altered mentation, cool extremities, low urine output and serum markers indicative of renal and/or hepatic dysfunction) with or without congestion [5].
Continuous outpatient therapy with inotropes may be a viable option in patients with stage D (end stage) heart failure who are deemed unlikely to survive hospital discharge [45].This is also supported by the ACCF/AHA practice guidelines where IV inotropic support may be considered for the previous reasons only after all alternative therapies to achieve stability have failed (Class IIB indication) [5].
Is there a role for morphine?
For decades morphine has been considered an essential component in the armamentarium for the treatment of ADHF. Its preload-reducing effect, anti-anxiety properties, and breathlessness suppression has made morphine a popular medication in the treatment of ADHF. Despite its common use, there is a lack of prospective randomized trials demonstrating the safety and benefit of this drug. In a retrospective analysis from the ADHERE database, IV morphine used for ADHF was associated with higher rates of adverse events, including increase use of mechanical ventilation, prolonged hospitalization, increased intensive care unit admissions, and higher mortality, bringing into question its safety profile [46]. Until a randomized trial is completed demonstrating safety and benefit, caution is advised regarding the use of morphine in ADHF.
Case Continued
Over the next 72 hours the patient’s symptoms improved. She no longer has dyspnea at rest, she has had a proper urine-output response to therapy, her serum creatinine has returned to normal, and her vital signs have remained stable. The IV vasodilator was discontinued, dobutamine was weaned off, and the patient was transitioned to guideline-directed medical therapy with an angiotensin-converting enzyme (ACE) inhibitor while continuing IV furosemide. Hospitalized patients who are hemodynamically stable should be transitioned to guideline-directed medical therapy with an oral ACE inhibitor unless the patient has a contraindication, such as marked azotemia or hyperkalemia. Low-dose carvedilol was initiated after optimization of volume status was confirmed. In the absence of shock and after optimization of volume status, every effort should be made to initiate low-dose beta blockers prior to hospital discharge.
When is mechanical circulatory support indicated in ADHF patients?
Mechanical circulatory support has emerged as a reasonable option in selected patients with acute and reversible cardiogenic shock (ie, acute coronary syndrome or an acute mechanical problem such as a torn papillary muscle or ventricular septal defect) [5]. Recently, the utility of intraaortic balloon pump (IABP) in the setting of cardiogenic shock resulting from acute coronary syndrome was called into question with the negative results from the Intraaortic Balloon Pump in Cardiogenic Shock II (IABP-SHOCK II) trial [47]. The study compared IABP with best available medical therapy alone among patients with acute myocardial infarction complicated by cardiogenic shock for who early revascularization was planned. Use of IABP did not reduce 30-day mortality compared with medical therapy in this patient population [47]. Whether IABP has a significant role in mechanical complications, such as acute ventricular septal rupture or papillary muscle rupture, is unknown due to the paucity of data in the management of patients with such complications. Therefore, when patients present with severe acute cardiogenic shock refractory to medical therapy, mechanical circulatory support with either ventricular assist devices (VAD) or extracorporeal membrane oxygenation (ECMO) is the preferred means to reverse terminal circulatory collapse. VADs are effective in the short-term as a “bridge-to-recovery” or as a “bridge-to-decision” when recovery, transplant candidacy, or neurologic status are still uncertain [48,49]. There are several options currently available for mechanical circulatory support, including surgically implanted VADs or the percutaneously implanted VADs, such as the Impella 2.5, 3.5 and 5.0 (Abiomed, Danvers, MA) and the TandemHeart pump (Cardiac Assist, Pittsburgh, PA).The ideal device and optimal duration of temporary support are yet to be defined. A detailed description of the function and clinical effects of mechanical support devices is beyond the scope of this article, although thorough reviews are available [48,49].
What elements of care may help optimize the discharge process?
Transition of care in hospitalized patients with ADHF to outpatient care is a critical and vulnerable period for patients given the complexity of the discharge planning for heart failure. A multidisciplinary heart failure disease management program is recommended in both the inpatient and outpatient setting to address the barriers to successful transition of care [5]. Physicians and physician extenders, nurses, pharmacists, and social workers can work together to identify risk factors for readmission and bridge the gap between the inpatient and outpatient setting.
Patients at high risk for hospital readmission should be referred to a heart failure disease management program [5,37]. Patients at high risk for hospital readmission include patients with renal insufficiency, low output state, diabetes mellitus, chronic lung disease, persistent NYHA functional class III, IV symptoms, frequent hospitalizations, multiple comorbidities, history of depression, cognitive impairment, or recurrent problems with noncompliance. There is strong evidence that a heart failure disease management program will reduce rehospitalization rates and costs while improving functional status and quality of life of the patient [37].In addition, a heart failure disease management clinic often can see the patient shortly after discharge, which may allow earlier discharge of the patient and shorter length of stay. Proven therapies such as ACE inhibitors, angiotensin-receptor blockers, beta blockers, and aldosterone antagonists can be titrated frequently in this setting.
It is strongly recommended that comprehensive written discharge instructions be provided at the end of hospitalization with special emphasis on diet, discharge medications, activity level, follow-up appointment, daily weight monitoring, and instructions for recurrence of symptoms [5].
Case Conclusion
The patient tolerated well the initiation of guideline-directed medical therapy and is continued on the ACE inhibitor and beta-blocker medications. After 4 days IV furosemide is discontinued and transitioned to oral furosemide. Precipitant causes of heart failure were addressed throughout hospitalization. It was determined that the patient had been taking high doses of nonsteroidal anti-inflammatory drugs due to knee pain. She was educated on this and other potential precipitant factors. Heart failure education was reinforced, including self-care, emergency plans, and need for medication and diet adherence. She is scheduled an early follow-up visit within 2 weeks of hospital discharge in the multidisciplinary heart failure disease management clinic.
Summary
ADHF is a major public health problem commonly encountered and often initially managed in the ED. Initial history and physical examination are important to estimate the degree of congestion and peripheral perfusion. The patient’s hemodynamic status along with the use prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion, however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy. Patients with ADHF are at increased risk for readmission to the hospital as well as increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Corresponding author: Carlos E. Sanchez, MD, 3705 Olentanfy River Rd., Columbus, OH 43214, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CES; analysis and interpretation of data, CES; drafting of article, CES; critical revision of the article, CES, DRR; collection and assembly of data, CES.
From Ohio Health, Riverside Methodist Hospital, Columbus, OH.
Abstract
- Objective: To review the current in-hospital management of patients with acute decompensated heart failure (ADHF).
- Methods: Review of the literature.
- Results: Heart failure is a leading cause of hospitalization in the elderly, and morbidity, mortality, and hospital readmission rates for ADHF remain high. The patient’s hemodynamic status along with the use of prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms, and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion; however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy.
- Conclusion: Patients with ADHF are at increased risk for readmission to the hospital as well as at increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Heart failure is a major public health problem in the United States and the leading cause of hospitalization in patients 65 years of age and older [1]. Patients hospitalized with acute decompensated heart failure (ADHF) have a readmission rate as high as 50% within 6 months and 25% within 30 days [2]. It is estimated that $32 billion is spent on heart failure care each year, the majority of which is directly related to inpatient care. Projections show that by 2030 the total cost of heart failure will increase to $70 billion per year [1]. Despite the growing burden, advances in treatment have been limited [2,3] and management continues to be a challenge. In this article, we review the current in-hospital management of patients with ADHF.
Case Study
Initial Presentation
A 64-year-old woman with a nonischemic dilated cardiomyopathy presents to the emergency department (ED) with a 4-day history of progressive dyspnea on exertion. She can not ambulate more than 50 feet without having to stop due to dyspnea and reports increased lower extremity edema. She is found to have a heart rate of 105 bpm, a respiratory rate of 30 breaths/min, and a blood pressure of 90/51 mm Hg. Physical examination is remarkable for distended neck vein, S3 gallop, end expiratory wheezing in the bases, and lower extremity edema. Blood tests, including a B-type natriuretic peptide level, are pending. Electrocardiogram and chest radiograph are ordered. The physician suspects that the patient has ADHF and admits her for further management.
What are aspects of initial management in the ED?
Most patients that present for evaluation and management of ADHF are first evaluated in the ED. Initial management includes an assessment of oxygenation, hemodynamic status, and adequacy of tissue perfusion, as well as for possibility of an acute coronary syndrome. A complete history, physical examination, chest radiography, 12-lead electrocardiogram, cardiac troponin T or I, electrolytes, and complete blood count should be obtained to allow rapid diagnosis and triage followed by prompt, aggressive treatment in the ED or observation unit. This should alleviate the patient’s symptoms sooner, and it is intuitive that this would lessen morbidity and length of hospital stay [4].
How are patients with ADHF classified?
What risk assessment tools are available?
B-type natriuretic peptide (BNP) and N-terminal fragment proBNP (NT-proBNP) were recently validated as diagnostic aids for the differentiation of etiologies of dypnea in patients in the ED with possible symptoms of ADHF. Use of these biomarkers can help reduce diagnostic uncertainty and associated mismanagement of patients presenting with nonspecific symptoms of dysp-nea [4,5,7]. Low or normal levels (BNP < 100 pg/ml or NT-proBNP < 500 pg/ml) have a high negative predictive value for excluding heart failure.
Elevated BNP or NT-proBNP levels may also yield prognostic information, identifying patients at increased risk of mortality or rehospitalization when value does not fall after aggressive heart failure management [8,9].In a recent study by Fonarow et al, the levels of BNP on hospital admission correlated directly with the risk of in-hospital mortality in patients admitted with ADHF independent of left ventricular ejection fraction. When the levels of BNP were below 430 pg/ml, the in-hospital mortality was 1.9%, and when the levels were above 1730 pg/ml, the mortality went up to 6% (P < 0.001) [8]. Additionally, elevated pre-discharge BNP levels (BNP > 350 ng/l; P < 0.001) in patients with ADHF seem to identify those at increased risk of death or readmission after in-patient management [9]. Elevated cardiac troponin T or I in hospitalized patients with ADHF also are associated with increased mortality, including in those without acute coronary syndrome or underlying coronary artery disease [10,11].
Case Continued
Upon further evaluation by a cardiologist, the patient is cool and clammy with elevated neck veins and prominent S3 confirmed. She continues to report severe shortness of breath after 1 dose of intravenous (IV) furosemide in the ED. Repeat vital signs shows a blood pressure of 83/49 mm Hg and respiratory rate of 33. Her electrocardiogram shows sinus tachycardia. The cardiologist determines that the patient’s clinical profile is “cold and wet” and admits the patient to the cardiac care unit (CCU) with a diagnosis of ADHF.
Initial blood tests show a BNP level of 1830 pg/ml, troponin I is 0.63 and stable after 2 measurements, serum creatinine is 1.6 mg/dL, BUN is 44 mg/dL, and serum sodium is 132 mg/dL. The GWTG-HF risk score for in-hospital mortality was calculated based on admission data and the probability of death was estimated at > 5% to 10% [12]. Prompt aggressive medical therapy was instituted in the CCU consisting of furosemide infusion to reduce congestion and IV dobutamine to improve systemic perfusion. Enoxoparin 40 mg subcutaneously once daily was initiated for venous thromboembolism prophylaxis.
What are important aspects of therapy for ADHF?
Several days to weeks prior to the appearance of signs and symptoms of volume overload, patients may develop hemodynamic congestion, defined as an elevation of ventricular filling pressure/pulmonary capillary wedge pressure independent of clinical evidence of fluid overload [17]. Elevated filling pressure is the culprit in the development of most of the signs and symptoms of ADHF and is the target for treatment.
Although use of vasoactive medications such as nitroglycerin or nitroprusside are not routinely recommended for use in all ADHF patients admitted to the hospital, retrospective analysis of the ADHEREdatabase suggests that there is a significant reduction of mortality, hospital length of stay, admission to intensive care unit, invasive procedures, and prolonged hospitalizations when IV diuretics, vasodilators (nitroglycerin, nitroprusside, nesiritide,) and/or positive inotropes (milrinone, dobutamine) are initiated in the ED within 6 hours of an ADHF presentation [18,19].However, whether prompt ED intervention impacts intermediate- to long-term outcomes is unknown [4].
Hospitalized patients with ADHF are at increased risk of venous thromboembolism mainly due to reduced cardiac output, increased systemic venous pressure, and reduced activity levels. Therefore, it is recommended that during the hospitalization ADHF patients receive prophylaxis against venous thromboembolism with low-dose unfractionated heparin or low-molecular-weight heparin if there is no contraindication [5].Individual therapeutic choices for ADHF are reviewed in detail below.
What treatments are used to relieve congestion?
Diuresis
In patients admitted to the hospital with ADHF, initial effective diuresis is vital to lowering cardiac filling pressures and relieving symptoms of congestion. Intravenous loop diuretics represent the first line of treatment and have long been the mainstay of therapy for decompensated heart failure with preserved or reduced ejection fraction, reducing fluid overload, and relieving symptoms.
Despite its long track record, the dose administration of IV diuretics is more of an art than a science. Medication dosage sufficient to produce a rate of diuresis that will optimize volume status and relieve signs and symptoms of congestion without causing kidney injury or hypotension is recommended [5].Due to the relatively short half-life of loop diuretics and concerns about tubular sodium reabsorption in the kidneys, continuous IV diuretic infusion has been suggested to enhance diuresis and avoid sodium and fluid rebound [5,20,21]. However, continuous loop diuretic infusion has not proven superior to intermittent IV bolus dosing in clinical studies. Recent data from the Diuretic Optimization Strategies Evaluation (DOSE) trial comparing bolus versus continuous infusion diuretic strategy in patients with ADHF showed no difference in global symptom relief, diuresis, or any of the clinical secondary endpoints including composite of death, re-hospitalization, or ED visits with either IV bolus versus continuous infusion or low versus high doses of furosemide [22]. Concern has also been previously raised about adverse outcomes utilizing high doses of loop diuretics in the treatment of ADHF [20,23,24]. However, the DOSE trial also evaluated the safety of 2 strategies for furosemide dosing in patients with ADHF. The study randomized ADHF patients with a prior diagnosis of chronic heart failure to 4 different treatment groups, either a high dose (2.5x their daily chronic oral furosemide dose) or low dose (1x their daily chronic oral furosemide dose), which was given either twice daily via IV bolus or via continuous infusion. The study showed no difference in change in renal function from baseline to 72 hours with either IV bolus versus continuous infusion or low versus high doses of furosemide [22].
Ultrafiltration
For patients with marked fluid overload who are unresponsive to diuretic therapy, peripheral ultrafiltration may be considered. Initial data demonstrated that early ultrafiltration effectively and safely reduced congestion in patients with ADHF with diuretic resistance and renal insufficiency. Length of stay was reduced, with 60% of discharges in 3 days or less and 1 readmission at 30 days. Neurohormonal activation, indicated by reduction in BNP level, was reduced without worsening glomerular filtration rate, hypotension or electrolyte abnormalities [28]. The UNLOAD trial confirmed these results and extended their findings to show that patients undergoing peripheral ultrafiltration had greater weight and net fluid loss at 48 hours and reduced rate of rehospitalization at 90 days when compared with IV diuretic therapy alone in ADHF patients. Interestingly, there was no difference in the dyspnea score at 48 hours and there was a trend toward worsening of renal function in the ultrafiltration group. The study was not powered to document a survival benefit [29]. However, the more recent Cardiorenal Rescue Study in ADHF (CARRESS-HF) trial involving patients with ADHF and worsening renal function showed that there was no difference in weight loss between patients randomized to ultrafiltration or a strategy of stepped pharmacologic therapy. Additionally, ultrafiltration was associated with a significant increase in creatinine at 96 hours and a higher rate of adverse events related to the procedure, driven by complications from intravenous catheter insertion. There was no difference between the 2 groups in death or rehospitalization for heart failure [30]. At present, ultrafiltration may be a reasonable option if all diuretic strategies are unsuccessful in relieving congestion [5].
Vasopressin-Receptor Antagonists
The vasopressin-receptor antagonists represent a relatively new class of medications that target the vasopressin receptors V1a and V2. Activation of the vasopressin V2 receptors by arginine vasopressin in heart failure causes inappropriate free water retention contributing to the symptoms of congestion and hyponatremia [31]. Currently, the only 2 vasopressin-receptor antagonists available for clinical use are conivaptan (V1a /V2 receptor antagonist) and tolvaptan (V2 receptor antagonist). The effectiveness of tolvaptan was tested in a randomized study (EVEREST) in patients hospitalized with ADHF [32,33]. At 1 year there was no difference seen in the primary endpoints of all-cause mortality, death from cardiovascular causes, or first hospitalization for heart failure [32,33]. However, hyponatremia, when present, was improved in the tolvaptan group. Conivaptan has a similar hemodynamic profile compared to tolvaptan, but without improving signs and symptoms in hospitalized patients with ADHF [34]. Currently, vasopressin antagonists are recommended in the management of ADHF by professional guidelines as only a class IIb indication in hospitalized patients with volume overload and severe hyponatremia [5].
Case Continued
After 24 hours of medical therapy in the CCU, the patient is no longer clammy and cool but continues to have shortness of breath, and peripheral edema is not improving. She continues to have elevated JVP and S3. Her blood pressure is now 120/79 mm Hg and her heart rate is 110. A Swan-Ganz catheter placed this morning showed a cardiac index of 1.8 L/minute/m2 (reference range, 2.5–4.0 L/min/m2); pulmonary capillary wedge pressure is 28 mm Hg (reference range, 6–12 mm Hg) and systemic vascular resistance is 1932 dyne/second/cm5 (reference range, 800–1200 dynes/sec/cm5). The physician decides to add nitroprusside to lower her filling pressure and systemic vascular resistance.
What is the role of vasoactive medications in treatment?
Vasodilators
Nitroglycerin is a venodilating medication with preload reduction properties at low doses and an arterial dilator at high doses [35]. Preload reduction improves left ventricular filling pressures and pulmonary congestion without increasing the oxygen demand in the heart in patients with ADHF. This leads to an improvement of symptoms, including dyspnea, in as early as 5 minutes [36]. For a highly symptomatic patient, nitroglycerin given sublingually can be useful in an acute situation because it is typically immediately available while preparations are made for administration of IV medications. Limitations of nitroglycerin include rapid tachyphylaxis within several hours of continuous exposure at high doses, resistance to the hemodynamic effects of nitroglycerin in up to 20% of patients, and hypotension, which may occur before significant preload reduction effect can be obtained [37]. When symptomatic hypotension becomes a problem, the highest hemodynamically tolerable dose should be given. Another agent with a potent vasodilator effect used in the treatment of heart failure is sodium nitroprusside (SNP). As opposed to nitroglycerin, this drug has an equally potent preload- and afterload-reducing effect [35]. Afterload reduction through its arteriodilator effect has the benefit of increasing cardiac output and decreasing myocardial oxygen demand with improvement of pulmonary congestion [36]. SNP is used in less than 1% of patients hospitalized with heart failure [38], probably due to the potential for causing marked hypotension, its need for invasive hemodynamic monitoring, and the rare risk for thiocyanate toxicity with high doses and/or longer infusions, especially in patients with reduced hepatic perfusion and renal function, as in the case of low-output heart failure [35]. However, data demonstrating safety and efficacy of SNP infusion in patients with ADHF are limited [39].A single-center, retrospective case-control study suggested that the administration of SNP in carefully selected patients with advanced low-output ADHF was safe and may be associated with favorable long-term clinical outcomes [39]. SNP can be attractive in severely congested patients with hypertension or severe mitral regurgitation complicating left ventricular failure, but prospective trials are needed to clarify the safety and efficacy in this patient population.
Nesiritide is a human recombinant form of BNP that has a direct effect on the vascular endothelium by increasing the bioavailability of nitric oxide through stimulation of cyclic guanosine monophosphate. Its primary mechanism of action is to reduce left ventricular filling pressures by a systemic and pulmonary vasodilator effect. It also promotes diuresis and natriuresis [40].The initial efficacy of nesiritide was demonstrated in the VMAC (Vasodilation in the Management of Acute Congestive Heart Failure) study, a randomized trial of IV nesiritide versus IV nitroglycerin or placebo in decompensated heart failure patients. A significant reduction in pulmonary capillary wedge pressure was demonstrated within 15 minutes in the nesiritide group and maintained at 3 hours compared to either nitroglycerin or placebo, with a similar improvement in dyspnea extending out to 24 hours [41].
The large ASCEND-HF (Acute Study of Clinical Effectiveness of Nesiritide in Decompensated Heart Failure) randomized ADHF patients to nesiritide or placebo and tested the hypothesis that nesiritide would be superior to placebo in improving acute dyspnea, all-cause mortality, and heart failure readmission in patients presenting with ADHF [42]. Nesiritide-treated patients showed only a modest early improvement in self-assessed dyspnea and no difference in the composite endpoint of death or rehospitalization at 30 days in patients admitted with ADHF. Reassuringly, there was no increase in renal failure compared to placebo; however, the incidence of symptomatic hypotension was higher with nesiritide [42]. Although nesiritide remains in the armentarium of vasoactive medications for ADHF, less expensive vasodilators such as nitroglycerin or nitroprusside may be preferred by many clinicians.
Overall, vasodilators represent a good treatment option for patients presenting with ADHF characterized by low cardiac output, high filling pressures, and elevated systemic vascular resistance. There is no clear evidence, however, to suggest that IV vasodilators improve survival in hospitalized patients with ADHF; thus, its use should be restricted to the relief of dyspnea in patients with stable blood pressure [5].
Inotropic Therapy
The most commonly used positive inotropic agents in the management of patients with ADHF in the United States are dobutamine (beta-1, beta-2, and alpha adrenoreceptor agonist) and milrinone (phosphodiesterase-III inhibitor) [38]. Inotropes increase cardiac output by increasing myocardial contractility, reduce left and right ventricular filling pressures, and improve hemodynamic parameters. Despite these hemodynamic effects, inotropic agents have not demonstrated a survival benefit in patients with ADHF. A major limitation regarding these agents is that they increase the risk of cardiac arrhythmias by increasing intracellular calcium in cardiac myocytes. In fact, retrospective analyses suggest that most inotropic agents are associated with an increased risk of death [38,43].
Milrinone inhibits type III isoform of the enzyme phosphodiasterase leading to an increase in intracellular cyclic AMP to exert its positive inotropic effect on the myocardium. Milrinone also exerts systemic and pulmonary vasodilator effects in the circulation decreasing right atrial, pulmonary capillary wedge, and mean arterial pressure. In the OPTIME-CHF trial, patients with chronic heart failure admitted to the hospital with ADHF were randomized to short term infusion of milrinone vs. placebo plus standard therapy. Milrinone resulted in more hypotension, atrial fibrillation and ventricular arrhythmias without any benefit on mortality or re-hospitalization [44].A retrospective analysis from the ADHEREregistry showed that in-hospital mortality was twofold higher with the use of dobutamine or milrinone in patients with ADHF when compared to treatment with vasodilators [38].
Dobutamine is a beta-1, beta-2, and alpha adrenoreceptor agonist that works by increasing myocardial contractility leading to an increase in cardiac output as its primary cardiovascular effect. Currently, routine use of IV positive inotropic agents in the absence of imminent cardiogenic shock or low output ADHF with systemic hypoperfusion is generally not recommended due to concerns of adverse effects [5]. The ACCF/AHA guidelines recommend the use of positive inotropic agents to relieve symptoms, improve systemic perfusion and preserve end-organ function in patients with severe left ventricular systolic failure and low output syndrome with evidence of end-organ dysfunction (such as hypotension, altered mentation, cool extremities, low urine output and serum markers indicative of renal and/or hepatic dysfunction) with or without congestion [5].
Continuous outpatient therapy with inotropes may be a viable option in patients with stage D (end stage) heart failure who are deemed unlikely to survive hospital discharge [45].This is also supported by the ACCF/AHA practice guidelines where IV inotropic support may be considered for the previous reasons only after all alternative therapies to achieve stability have failed (Class IIB indication) [5].
Is there a role for morphine?
For decades morphine has been considered an essential component in the armamentarium for the treatment of ADHF. Its preload-reducing effect, anti-anxiety properties, and breathlessness suppression has made morphine a popular medication in the treatment of ADHF. Despite its common use, there is a lack of prospective randomized trials demonstrating the safety and benefit of this drug. In a retrospective analysis from the ADHERE database, IV morphine used for ADHF was associated with higher rates of adverse events, including increase use of mechanical ventilation, prolonged hospitalization, increased intensive care unit admissions, and higher mortality, bringing into question its safety profile [46]. Until a randomized trial is completed demonstrating safety and benefit, caution is advised regarding the use of morphine in ADHF.
Case Continued
Over the next 72 hours the patient’s symptoms improved. She no longer has dyspnea at rest, she has had a proper urine-output response to therapy, her serum creatinine has returned to normal, and her vital signs have remained stable. The IV vasodilator was discontinued, dobutamine was weaned off, and the patient was transitioned to guideline-directed medical therapy with an angiotensin-converting enzyme (ACE) inhibitor while continuing IV furosemide. Hospitalized patients who are hemodynamically stable should be transitioned to guideline-directed medical therapy with an oral ACE inhibitor unless the patient has a contraindication, such as marked azotemia or hyperkalemia. Low-dose carvedilol was initiated after optimization of volume status was confirmed. In the absence of shock and after optimization of volume status, every effort should be made to initiate low-dose beta blockers prior to hospital discharge.
When is mechanical circulatory support indicated in ADHF patients?
Mechanical circulatory support has emerged as a reasonable option in selected patients with acute and reversible cardiogenic shock (ie, acute coronary syndrome or an acute mechanical problem such as a torn papillary muscle or ventricular septal defect) [5]. Recently, the utility of intraaortic balloon pump (IABP) in the setting of cardiogenic shock resulting from acute coronary syndrome was called into question with the negative results from the Intraaortic Balloon Pump in Cardiogenic Shock II (IABP-SHOCK II) trial [47]. The study compared IABP with best available medical therapy alone among patients with acute myocardial infarction complicated by cardiogenic shock for who early revascularization was planned. Use of IABP did not reduce 30-day mortality compared with medical therapy in this patient population [47]. Whether IABP has a significant role in mechanical complications, such as acute ventricular septal rupture or papillary muscle rupture, is unknown due to the paucity of data in the management of patients with such complications. Therefore, when patients present with severe acute cardiogenic shock refractory to medical therapy, mechanical circulatory support with either ventricular assist devices (VAD) or extracorporeal membrane oxygenation (ECMO) is the preferred means to reverse terminal circulatory collapse. VADs are effective in the short-term as a “bridge-to-recovery” or as a “bridge-to-decision” when recovery, transplant candidacy, or neurologic status are still uncertain [48,49]. There are several options currently available for mechanical circulatory support, including surgically implanted VADs or the percutaneously implanted VADs, such as the Impella 2.5, 3.5 and 5.0 (Abiomed, Danvers, MA) and the TandemHeart pump (Cardiac Assist, Pittsburgh, PA).The ideal device and optimal duration of temporary support are yet to be defined. A detailed description of the function and clinical effects of mechanical support devices is beyond the scope of this article, although thorough reviews are available [48,49].
What elements of care may help optimize the discharge process?
Transition of care in hospitalized patients with ADHF to outpatient care is a critical and vulnerable period for patients given the complexity of the discharge planning for heart failure. A multidisciplinary heart failure disease management program is recommended in both the inpatient and outpatient setting to address the barriers to successful transition of care [5]. Physicians and physician extenders, nurses, pharmacists, and social workers can work together to identify risk factors for readmission and bridge the gap between the inpatient and outpatient setting.
Patients at high risk for hospital readmission should be referred to a heart failure disease management program [5,37]. Patients at high risk for hospital readmission include patients with renal insufficiency, low output state, diabetes mellitus, chronic lung disease, persistent NYHA functional class III, IV symptoms, frequent hospitalizations, multiple comorbidities, history of depression, cognitive impairment, or recurrent problems with noncompliance. There is strong evidence that a heart failure disease management program will reduce rehospitalization rates and costs while improving functional status and quality of life of the patient [37].In addition, a heart failure disease management clinic often can see the patient shortly after discharge, which may allow earlier discharge of the patient and shorter length of stay. Proven therapies such as ACE inhibitors, angiotensin-receptor blockers, beta blockers, and aldosterone antagonists can be titrated frequently in this setting.
It is strongly recommended that comprehensive written discharge instructions be provided at the end of hospitalization with special emphasis on diet, discharge medications, activity level, follow-up appointment, daily weight monitoring, and instructions for recurrence of symptoms [5].
Case Conclusion
The patient tolerated well the initiation of guideline-directed medical therapy and is continued on the ACE inhibitor and beta-blocker medications. After 4 days IV furosemide is discontinued and transitioned to oral furosemide. Precipitant causes of heart failure were addressed throughout hospitalization. It was determined that the patient had been taking high doses of nonsteroidal anti-inflammatory drugs due to knee pain. She was educated on this and other potential precipitant factors. Heart failure education was reinforced, including self-care, emergency plans, and need for medication and diet adherence. She is scheduled an early follow-up visit within 2 weeks of hospital discharge in the multidisciplinary heart failure disease management clinic.
Summary
ADHF is a major public health problem commonly encountered and often initially managed in the ED. Initial history and physical examination are important to estimate the degree of congestion and peripheral perfusion. The patient’s hemodynamic status along with the use prognostic models for short-term mortality may facilitate patient triage and encourage the use of evidence-based therapy, especially in high-risk patients. Initial treatment should target the relief of congestive symptoms and intravenous loop diuretics are the mainstay of therapy. The preferred IV vasoactive medication has yet to be determined in a large prospective randomized trial. Positive inotropic agents should be reserved for patients with signs of low cardiac output and tissue hypoperfusion, however, the risk/benefit equation should be evaluated judiciously with each treatment option before initiating therapy. For patients with refractory hemodynamic collapse, ventricular assist devices can allow stabilization until recovery or decision regarding transplantation versus destination therapy. Patients with ADHF are at increased risk for readmission to the hospital as well as increased risk for death. Risk factors need to be identified and referral to a heart disease management program should be considered for those patients deemed at increased risk for rehospitalization.
Corresponding author: Carlos E. Sanchez, MD, 3705 Olentanfy River Rd., Columbus, OH 43214, [email protected].
Financial disclosures: None.
Author contributions: conception and design, CES; analysis and interpretation of data, CES; drafting of article, CES; critical revision of the article, CES, DRR; collection and assembly of data, CES.
1. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 2013;127: e6–245.
2. Fonarow GC. ADHERE Scientific Advisory Committee. The ADHF National Registry (ADHERE): opportunities to improve care of patients hospitalized with ADHF. Rev Cardiovasc Med 2003;4(suppl 7):S21-S30.
3. Philbin EF, Dec GW, Enkins PL, et al. Socioeconomic status as an independent risk factor for hospital readmission for heart failure. Am J Cardiol 2001;87:1367–71.
4. Weintraub NL, Collins SP, Pang PS, et al.; on behalf of the American Heart Association Council on Clinical Cardiology and Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Acute heart failure syndromes: emergency department presentation, treatment, and disposition: current approaches and future aims: a scientific statement from the American Heart Association. Circulation 2010;122:1975–96.
5. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147-239.
6. Nohria A, Tsang SW, Fang JC, et al. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol 2003;41:1797–1804.
7. Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161–7.
8. Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in ADHF. J Am Coll Cardiol 2007;49:1943–50.
9. Logeart D, Thabut G, Jourdian P, et al. Pre-discharge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004;43:635–41.
10. Peacock WFIV, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008;358:2117–26.
11. Ilva T, Lassus J, Siirila-Waris K, et al. Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail 2008;10:772–9.
12. Peterson PN, Rumsfeld JS, Liang L, et al. A validated risk score for inhospital mortality in patients with heart failure from the American Heart Association Get With The Guidelines program. Circ Cardiovasc Qual Outcomes 2010;3:25–32.
13. Fonarow GC, Adams KF, Abraham WT, et al, for the ADHERE Scientific Advisory Committee, Study Groups and Investigators. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA 2005;293:572–80.
14. Felker GM, Leimberger JD, Califf RM, et al. Risk stratification after hospitalization for decompensated heart failure. J Card Fail 2004;10:460–6.
15. Lee DS, Austin PC, Rouleau JL, et al. Predicting mortality among patients hospitalized for heart failure: derivation and validation of a clinical model. JAMA 2003;290:2581–7.
16. Abraham WT, Fonarow GC, Albert NM, et al. Predictors of in-hospital mortality in patients hospitalized for heart failure. J Am Coll Cardiol 2008; 52:347–56.
17. Gheorghiade M, Shin DD, Thomas TO, et al. Congestion is an important diagnostic and therapeutic target in heart failure. Rev Cardiovasc Med 2006;7(suppl l):S12-S24.
18. Peacock WF, Emerman C, Costanzo MR, et al. Early vasoactive drugs improve heart failure outcomes. Congest Heart Fail 2009;15:256–64.
19. Maisel AS, Peacock WF, McMullin N, et al. Timing of immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure: an ADHERE analysis. J Am Coll Cardiol 2008;52:534–40.
20. Salvador DRK, Rey NR, Ramos GC, et al. Continuous infusion versus bolus injection of loop diuretics in congestive heart failure. Cochrane Database Syst Rev 2005(3):CD003178.
21. Pivac N, Rumboldt Z, Sardelic S, et al. Diuretic effects of furosemide infusion versus bolus injection in congestive heart failure. Int J Clin Pharmacol Res 1998;18:121–8.
22. Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with ADHF. N Engl J Med 2011;364:797–805.
23. Cotter G, Metzkor E, Kaluski E, et al. Randomized trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary edema. Lancet 1998;351:389–93.
24. Butler J, Forman DE, Abraham WT, et al. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J 2004;147:331–8.
25. Giamouzis G, Butler J, Starling RC, et al. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in ADHF (DAD-HF) Trial. J Card Fail 2010;16:922–30.
26. Cotter G, Weissgarten J, Metzkor E, et al. Increased toxicity of high-dose furosemide versus low-dose dopamine in the treatment of refractory congestive heart failure. Clin Pharmacol Ther 1997;62:187–93.
27. Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013;310:2533–43.
28. Costanzo MR, Saltzberg M, O’Sullivan J, et al. Early ultrafiltration in patients with decompensated heart failure and diuretic resistance. J Am Coll Cardiol 2005;46:2047–51.
29. Costanzo MR, Guglin ME, Saltzberg MT, et al; UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for ADHF. J Am Coll Cardiol 2007;49:675–83.
30. Bart BA, Goldsmith SR, Lee KL, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012;367:2296-304.
31. Shrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med 1999;341:577–85.
32. Konstam MA, Gheorghiade M, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007;297:1319–31.
33. Gheorghiade M, Konstam MA, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA 2007;297:1332–43.
34. Goldsmith SR, Elkayam U, Haught WH, et al. Efficacy and safety of the vasopressin V1A/V2-receptor antagonist conivaptan in ADHF: a dose-ranging pilot study. J Card Fail 2008;14:641–-7.
35. Shin DD, Brandimarte F, DeLuca L, et al. Review of current and investigational pharmacologic agents for acute heart failure syndromes. Am J Cardiol 2007;99:4A–23A.
36. Mattu A, Martinez JP, Kelly BS. Modern management of cardiogenic pulmonary edema. Emerg Med Clin North Am 2005;23:1105–25.
37. Lindenfeld J, Albert NM, Boehmer JP, et al. Executive Summary: HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 2010;16:e475-e539
38. Abraham WT, Adams KF, Fonarow GC, et al. ADHERE Scientific Advisory Committee and Investigators; ADHERE Study Group. In-hospital mortality in patients with ADHF requiring intravenous vasoactive medications: an analysis from the ADHF national registry (ADHERE). J Am Coll Cardiol 2005;46:57–64.
39. Mullens W, Abrahams Z, Francis GS, et al. Sodium nitroprusside for advanced low-output heart failure. J Am Coll Cardiol 2008;52:200–7.
40. Bhalla V, Willis S, Maisel AS. B-type natriuretic peptide: the level and the drug-partners in the diagnosis and management of congestive heart failure. Congest Heart Fail 2004;10(1 suppl 1):3–27.
41. Publication Committee for the VMAC investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531–40.
42. O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with ADHF. N Engl J Med 2011;365:32–43.
43. Elkayam U, Tasissa G, Binanay C, et al. Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am Heart J 2007;153:98–104.
44. Cuffe MS, Califf RM, Adams KF Jr, et al. Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized control trial. JAMA 2002;287:1541–7.
45. Hershberger RE, Nauman D, Walker TL, et al. Care processes and clinical outcomes of continuous outpatient support with inotropes (COSI) in patients with refractory endstage heart failure. J Card Fail 2003;9:180–7.
46. Peacock WF, Hollander JE, Diercks DB, et al. Morphine and outcomes in ADHF: an ADHERE analysis. Emerg Med J 2008;25:205–9.
47. Thiele H, Zeymer U, Neumann FJ, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med 2012;367:1287–96.
48. Abu-Omar Y, Tsui S. Mechanical circulatory support for AMI and cardiogenic shock. J Card Surg 2010;25:434–41.
49. Ziemba EA, John R. Mechanical circulatory support for bridge to decision: which device and when to decide. J Card Surg 2010;25:425–33.
50. Sanchez CE, Richards DR. Contemporary in-hospital management strategies for ADHF. Cardiol Rev 2011;19:122–9.
1. Go AS, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics–2013 update: a report from the American Heart Association. Circulation 2013;127: e6–245.
2. Fonarow GC. ADHERE Scientific Advisory Committee. The ADHF National Registry (ADHERE): opportunities to improve care of patients hospitalized with ADHF. Rev Cardiovasc Med 2003;4(suppl 7):S21-S30.
3. Philbin EF, Dec GW, Enkins PL, et al. Socioeconomic status as an independent risk factor for hospital readmission for heart failure. Am J Cardiol 2001;87:1367–71.
4. Weintraub NL, Collins SP, Pang PS, et al.; on behalf of the American Heart Association Council on Clinical Cardiology and Council on Cardiopulmonary, Critical Care, Perioperative and Resuscitation. Acute heart failure syndromes: emergency department presentation, treatment, and disposition: current approaches and future aims: a scientific statement from the American Heart Association. Circulation 2010;122:1975–96.
5. Yancy CW, Jessup M, Bozkurt B, et al. 2013 ACCF/AHA guideline for the management of heart failure: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. J Am Coll Cardiol. 2013;62:e147-239.
6. Nohria A, Tsang SW, Fang JC, et al. Clinical assessment identifies hemodynamic profiles that predict outcomes in patients admitted with heart failure. J Am Coll Cardiol 2003;41:1797–1804.
7. Maisel AS, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. N Engl J Med 2002;347:161–7.
8. Fonarow GC, Peacock WF, Phillips CO, et al. ADHERE Scientific Advisory Committee and Investigators. Admission B-type natriuretic peptide levels and in-hospital mortality in ADHF. J Am Coll Cardiol 2007;49:1943–50.
9. Logeart D, Thabut G, Jourdian P, et al. Pre-discharge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. J Am Coll Cardiol 2004;43:635–41.
10. Peacock WFIV, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med 2008;358:2117–26.
11. Ilva T, Lassus J, Siirila-Waris K, et al. Clinical significance of cardiac troponins I and T in acute heart failure. Eur J Heart Fail 2008;10:772–9.
12. Peterson PN, Rumsfeld JS, Liang L, et al. A validated risk score for inhospital mortality in patients with heart failure from the American Heart Association Get With The Guidelines program. Circ Cardiovasc Qual Outcomes 2010;3:25–32.
13. Fonarow GC, Adams KF, Abraham WT, et al, for the ADHERE Scientific Advisory Committee, Study Groups and Investigators. Risk stratification for in-hospital mortality in acutely decompensated heart failure: classification and regression tree analysis. JAMA 2005;293:572–80.
14. Felker GM, Leimberger JD, Califf RM, et al. Risk stratification after hospitalization for decompensated heart failure. J Card Fail 2004;10:460–6.
15. Lee DS, Austin PC, Rouleau JL, et al. Predicting mortality among patients hospitalized for heart failure: derivation and validation of a clinical model. JAMA 2003;290:2581–7.
16. Abraham WT, Fonarow GC, Albert NM, et al. Predictors of in-hospital mortality in patients hospitalized for heart failure. J Am Coll Cardiol 2008; 52:347–56.
17. Gheorghiade M, Shin DD, Thomas TO, et al. Congestion is an important diagnostic and therapeutic target in heart failure. Rev Cardiovasc Med 2006;7(suppl l):S12-S24.
18. Peacock WF, Emerman C, Costanzo MR, et al. Early vasoactive drugs improve heart failure outcomes. Congest Heart Fail 2009;15:256–64.
19. Maisel AS, Peacock WF, McMullin N, et al. Timing of immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure: an ADHERE analysis. J Am Coll Cardiol 2008;52:534–40.
20. Salvador DRK, Rey NR, Ramos GC, et al. Continuous infusion versus bolus injection of loop diuretics in congestive heart failure. Cochrane Database Syst Rev 2005(3):CD003178.
21. Pivac N, Rumboldt Z, Sardelic S, et al. Diuretic effects of furosemide infusion versus bolus injection in congestive heart failure. Int J Clin Pharmacol Res 1998;18:121–8.
22. Felker GM, Lee KL, Bull DA, et al. Diuretic strategies in patients with ADHF. N Engl J Med 2011;364:797–805.
23. Cotter G, Metzkor E, Kaluski E, et al. Randomized trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary edema. Lancet 1998;351:389–93.
24. Butler J, Forman DE, Abraham WT, et al. Relationship between heart failure treatment and development of worsening renal function among hospitalized patients. Am Heart J 2004;147:331–8.
25. Giamouzis G, Butler J, Starling RC, et al. Impact of dopamine infusion on renal function in hospitalized heart failure patients: results of the Dopamine in ADHF (DAD-HF) Trial. J Card Fail 2010;16:922–30.
26. Cotter G, Weissgarten J, Metzkor E, et al. Increased toxicity of high-dose furosemide versus low-dose dopamine in the treatment of refractory congestive heart failure. Clin Pharmacol Ther 1997;62:187–93.
27. Chen HH, Anstrom KJ, Givertz MM, et al. Low-dose dopamine or low-dose nesiritide in acute heart failure with renal dysfunction: the ROSE acute heart failure randomized trial. JAMA 2013;310:2533–43.
28. Costanzo MR, Saltzberg M, O’Sullivan J, et al. Early ultrafiltration in patients with decompensated heart failure and diuretic resistance. J Am Coll Cardiol 2005;46:2047–51.
29. Costanzo MR, Guglin ME, Saltzberg MT, et al; UNLOAD Trial Investigators. Ultrafiltration versus intravenous diuretics for patients hospitalized for ADHF. J Am Coll Cardiol 2007;49:675–83.
30. Bart BA, Goldsmith SR, Lee KL, et al. Ultrafiltration in decompensated heart failure with cardiorenal syndrome. N Engl J Med 2012;367:2296-304.
31. Shrier RW, Abraham WT. Hormones and hemodynamics in heart failure. N Engl J Med 1999;341:577–85.
32. Konstam MA, Gheorghiade M, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Effects of oral tolvaptan in patients hospitalized for worsening heart failure: the EVEREST Outcome Trial. JAMA 2007;297:1319–31.
33. Gheorghiade M, Konstam MA, Burnett Jr JC, et al. Efficacy of Vasopressin Antagonism in Heart Failure Outcome Study With Tolvaptan (EVEREST) Investigators. Short-term clinical effects of tolvaptan, an oral vasopressin antagonist, in patients hospitalized for heart failure: the EVEREST Clinical Status Trials. JAMA 2007;297:1332–43.
34. Goldsmith SR, Elkayam U, Haught WH, et al. Efficacy and safety of the vasopressin V1A/V2-receptor antagonist conivaptan in ADHF: a dose-ranging pilot study. J Card Fail 2008;14:641–-7.
35. Shin DD, Brandimarte F, DeLuca L, et al. Review of current and investigational pharmacologic agents for acute heart failure syndromes. Am J Cardiol 2007;99:4A–23A.
36. Mattu A, Martinez JP, Kelly BS. Modern management of cardiogenic pulmonary edema. Emerg Med Clin North Am 2005;23:1105–25.
37. Lindenfeld J, Albert NM, Boehmer JP, et al. Executive Summary: HFSA 2010 Comprehensive Heart Failure Practice Guideline. J Card Fail 2010;16:e475-e539
38. Abraham WT, Adams KF, Fonarow GC, et al. ADHERE Scientific Advisory Committee and Investigators; ADHERE Study Group. In-hospital mortality in patients with ADHF requiring intravenous vasoactive medications: an analysis from the ADHF national registry (ADHERE). J Am Coll Cardiol 2005;46:57–64.
39. Mullens W, Abrahams Z, Francis GS, et al. Sodium nitroprusside for advanced low-output heart failure. J Am Coll Cardiol 2008;52:200–7.
40. Bhalla V, Willis S, Maisel AS. B-type natriuretic peptide: the level and the drug-partners in the diagnosis and management of congestive heart failure. Congest Heart Fail 2004;10(1 suppl 1):3–27.
41. Publication Committee for the VMAC investigators (Vasodilatation in the Management of Acute CHF). Intravenous nesiritide vs nitroglycerin for treatment of decompensated congestive heart failure: a randomized controlled trial. JAMA 2002;287:1531–40.
42. O’Connor CM, Starling RC, Hernandez AF, et al. Effect of nesiritide in patients with ADHF. N Engl J Med 2011;365:32–43.
43. Elkayam U, Tasissa G, Binanay C, et al. Use and impact of inotropes and vasodilator therapy in hospitalized patients with severe heart failure. Am Heart J 2007;153:98–104.
44. Cuffe MS, Califf RM, Adams KF Jr, et al. Outcomes of a Prospective Trial of Intravenous Milrinone for Exacerbations of Chronic Heart Failure (OPTIME-CHF) Investigators. Short-term intravenous milrinone for acute exacerbation of chronic heart failure: a randomized control trial. JAMA 2002;287:1541–7.
45. Hershberger RE, Nauman D, Walker TL, et al. Care processes and clinical outcomes of continuous outpatient support with inotropes (COSI) in patients with refractory endstage heart failure. J Card Fail 2003;9:180–7.
46. Peacock WF, Hollander JE, Diercks DB, et al. Morphine and outcomes in ADHF: an ADHERE analysis. Emerg Med J 2008;25:205–9.
47. Thiele H, Zeymer U, Neumann FJ, et al. Intraaortic balloon support for myocardial infarction with cardiogenic shock. N Engl J Med 2012;367:1287–96.
48. Abu-Omar Y, Tsui S. Mechanical circulatory support for AMI and cardiogenic shock. J Card Surg 2010;25:434–41.
49. Ziemba EA, John R. Mechanical circulatory support for bridge to decision: which device and when to decide. J Card Surg 2010;25:425–33.
50. Sanchez CE, Richards DR. Contemporary in-hospital management strategies for ADHF. Cardiol Rev 2011;19:122–9.
Predictors of Suboptimal Glycemic Control for Hospitalized Patients with Diabetes: Targets for Clinical Action
From Sharp HealthCare, San Diego, CA (Ms. Thompson, Mr. Koucheki, Dr. Holdy), National University, San Diego, CA (Dr. Smith), and University of California, Irvine (Dr. Bender).
Abstract
- Objective: Suboptimal glycemic control (SGC) puts hospitalized patients with diabetes at risk for poor outcomes. The purpose of this study was to quantify factors with predictive capacity to identify patients at risk for SGC during hospitalization.
- Methods: 32 baseline and demographic variables were extracted from the electronic records of 23,100 patients with diabetes hospitalized between 2009 and 2012. The rate of blood glucose values between 70 and 180 mg/dL was calculated for each patient. A predictive model for SGC was developed using regression modeling, standardized coefficients, and classification tree analysis. Odds ratios (ORs) were calculated to isolate adjusted odds of SGC for top predictors.
- Results: The final predictive model included 13 variables (C statistic = 0.88). HbA1c (OR, 0.60 [95% confidence interval {CI}, 0.58–0.61]), admission blood glucose (OR, 0.91 [CI, 0.91–0.92]), and steroid use (OR, 0.06 [CI 0.04–0.08]) were the highest-ranking predictors of SGC. HbA1c and SGC had a strong linear relationship (R2 = 0.99), with increasing odds for SGC as HbA1c increased. Admission blood glucose and SGC had a polynomial relationship (R2 = 0.95); increasing odds for SGC until 240 mg/dL; then odds started decreasing. Steroid use showed a steady threefold increase in odds for SGC across all rates of use.
- Conclusions: Poor preadmission diabetes control and inpatient steroid use strongly predict SGC. A range of thresholds for these predictors was empirically determined, providing a basis for targeted therapies on admission. Guidelines incorporating empirically derived thresholds should enhance the ability to achieve optimal glycemic control for hospitalized patients with diabetes.
Current recommendations for glycemic control of hospitalized patients include use of multidisciplinary diabetic care teams and standardized insulin order sets [1,2], yet there is still uncertainty how best to target such protocols for patients with diabetes at risk for suboptimal glycemic control [2,3]. Many factors are theorized to hinder optimal inpatient glycemic control, such as steroid use [4,5],comorbid states [6],severity of illness [7,8], and preadmission glycemic control [9,10]. However, there currently exists little evidence of these factors’ ability to predict suboptimal glycemic control (SGC). Identifying straightforward predictive factors of SGC would provide a clinically meaningful basis for targeted therapy. The purpose of this study was to describe the prevalence of a range of potential risk factors in a diverse hospitalized patient population with a secondary diagnosis of diabetes (types 1 and 2), and to determine which factors were predictive of SGC.
Methods
A retrospective cohort study design was used to identify factors predictive of inpatient SGC for patients admitted to any of 3 hospitals aligned with Sharp HealthCare (“Sharp”), a community-based, nonprofit integrated health system headquartered in San Diego, California, that serves more than 27% of the county’s 3 million-plus residents each year.
Inclusion Criteria
We extracted data for 23,100 patients hospitalized between January 2009 and December 2012 with a secondary diagnosis of diabetes (types 1 and 2), a length of stay (LOS) ≥ 3 days, and a minimum of 2 point-of-care (POC) blood glucose tests per day. The LOS and blood glucose minimum are standard criteria for Sharp glycemic monitoring to ensure a minimum quantity of blood glucose monitoring for glycemic management.
Glycemic Control (Independent Variable)
Glycemic control was defined as POC blood glucose values within the target range of 70 to 180 mg/dL during hospitalization. POC tests outside that range were defined as SGC. This range was determined based on current Sharp benchmark targets and was not adjusted for total number of blood glucose values.
Predictive Variables
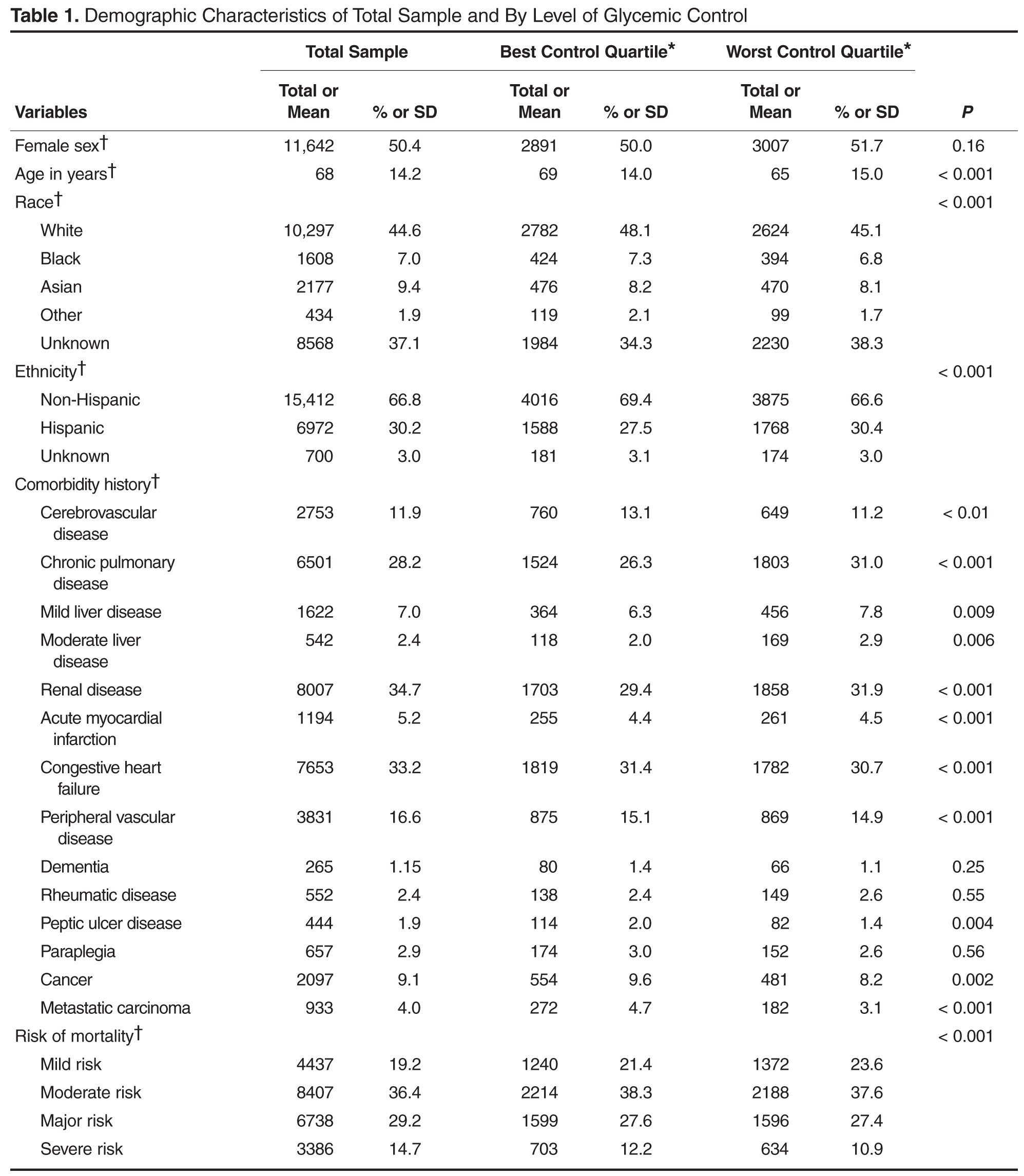
Age, gender, race, ethnicity, payor, facility and LOS were extracted from Sharp’s data warehouse. Medical vs. surgical stay and major diagnostic category was determined from administrative diagnosis coding. Body mass index (BMI) was extracted from Sharp’s electronic health record. Risk of mortality and severity of illness were calculated using 3M APR-DRG proprietary software using administrative diagnosis coding. Comorbidities were determined based on administrative diagnosis codes per published guidelines [11]. Glycosylated hemoglobin (HbA1c) was obtained on admission for patients with a secondary diagnosis of diabetes as part of Sharp's multidisciplinary diabetes care management program and extracted from the electronic health record. Admission blood glucose was defined as the first documented POC blood glucose after admission. ICU stay was calculated as a continuous variable: the percent of LOS spent in the ICU. Steroid use was similarly calculated, and defined as oral or intravenous administration of any quantity or dosage of the following corticosteroids during each day of hospitalization: dexamethasone, hydrocortisone, prednisone, and/or methylprednisone. Adherence to Sharp's multidisciplinary diabetes care management program was measured by use of standardized insulin order sets. Sharp uses evidence-based order sets for continuous infusion and subcutaneous insulin management; subcutaneous orders include basal and rapid-acting insulin. We calculated the total time a person was on an order set during hospitalization by subtracting any time a patient did not have insulin ordered from the total LOS. This was transformed into a variable documenting the percent of LOS the patient was on an insulin order set. Average blood glucose for admission was calculated for all documented POC blood glucoses during admission, omitting the admission blood glucose (the first POC blood glucose of the admission).
Analysis
Univariate analyses including t tests and chi-square tests were conducted to investigate the unadjusted association between variables and glycemic control. Good glycemic control was defined as 90% of all POC blood glucose tests between 70 and 180 mg/dL based on empirical distribution and organization targets. A predictive model of inpatient glycemic control was then developed using a backward stepwise multivariable logistic regression approach. The data were split into a model building and validation set. Variables were included that represented both baseline and transitional state during the hospital stay to account for potentially mediating effects and a sensitivity analysis was conducted with them in and out of the final model to assess impact. Standardized coefficients were calculated to rank order variables in the model allowing indication of the variables with the greatest predictive impact on the outcome. Further investigation of the optimal classification points for the variables was conducted to indicate best differentiation of good glycemic control. Significant variables from the multivariable logistic regression were included in an exploratory classification tree analysis that recursively partitioned data in order to improve the fit, with optimal splitting identified over all variables at all possible split points. Classification tree cut-off points were used to further develop models identifying odds ratios for various thresholds for the top three predictive variables. A series of logistic regression models were then run with differing cut points of top 3 predictors to isolate adjusted odds of good glycemic control. Analytic data set building and statistical analyses were completed using SAS 9.4.
Results
Patient Characteristics
Table 1 shows patient demographic and clinical characteristics for the entire sample and for the top quartile (76% or greater POC blood glucose values within target range) and bottom quartile (25% or less POC blood glucose values within target range). Unadjusted results show a significant difference across quartiles for all factors except age, gender, dementia, rheumatic disease and paraplegia. Patients in the bottom 25th percentile (ie, the poorest control) were more likely than the total population to have a higher admission blood glucose (198 mg/dL vs. 153 mg/dL), higher HbA1c (8.53 [70mmol/mol] vs. 7.35 [57mmol/mol), a medical (74% vs. 66%) and/or respiratory (18% vs. 12%) diagnosis, corticosteroid use (17% vs. 27%), an insulin order set use (80% vs. 70%) and higher mean blood glucose during hospitalization (206.3 vs. 157.1 mg/dL). Patients with poorest control were less likely that the total population to have a high risk of mortality (11% vs. 15%) and severity of illness (13% vs. 18%). They also had less ICU care (8% vs. 13%), and a shorter LOS (5.82 vs. 7.82 days).
Predictive Modeling
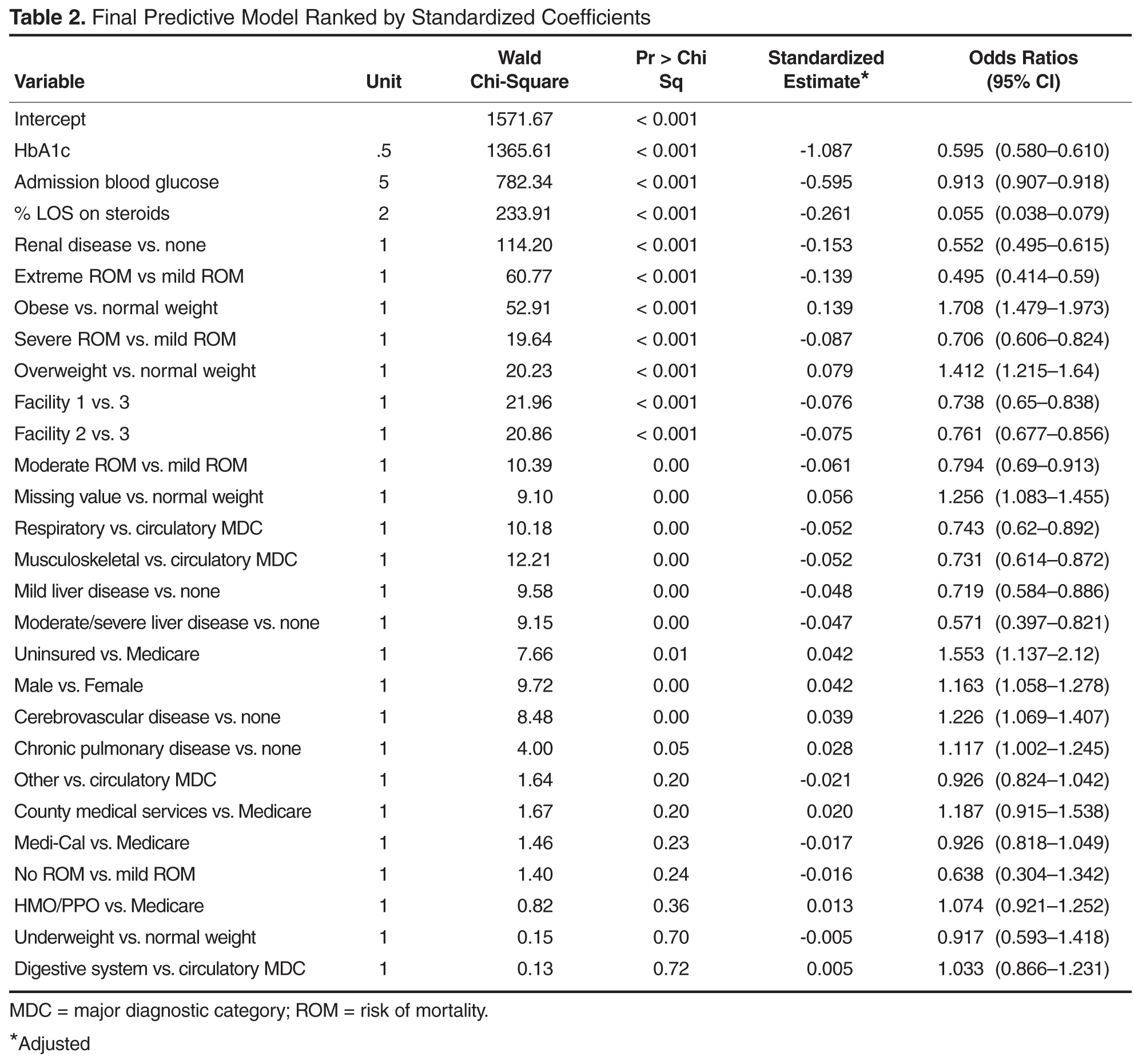
Classification tree analysis resulted in the same top 3 predictors, but in a different order. The analysis also provided cut-off values that predict suboptimal glycemic control. Classification tree analysis showed admission blood glucose was the most influential predictor, with 164.5 mg/dL indicating the optimal cut-point for prediction of SGC, followed by HbA1c with an optimal cut-off point of 6.65% indicating prediction of SGC, followed by treatment with corticosteroids, with an optimal cut-off point of 24% of the LOS on corticosteroids indicating prediction of SGC.
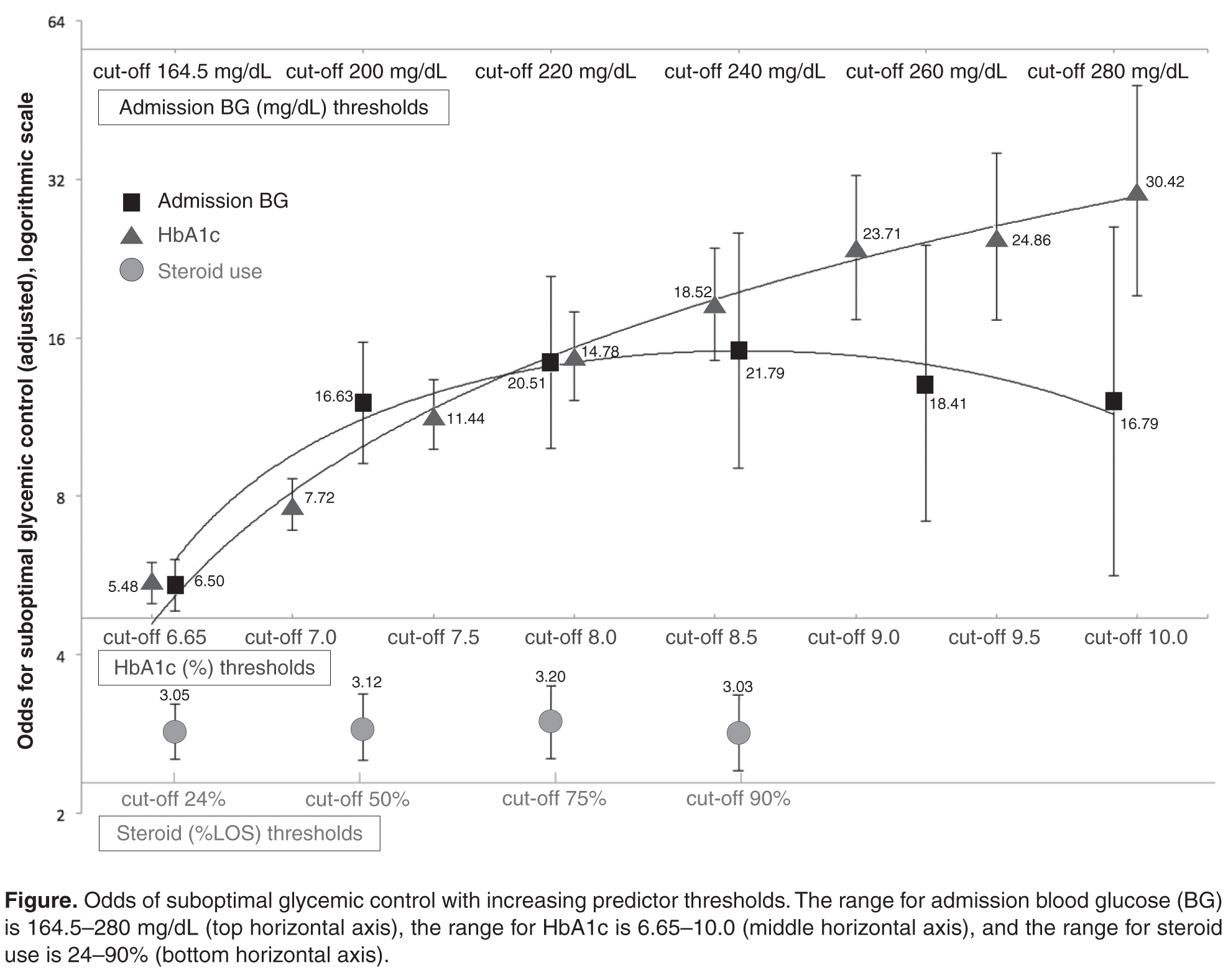
Discussion
Evidence of patient characteristics that consistently predict suboptimal glycemic control during hospitalization is needed to better inform clinical decisions for inpatient glycemic management. Hospitalized patients would greatly benefit from glycemic protocols that incorporate risk stratification tools based evidence-based risk factors for poor glycemic control [12]. The science of inpatient glycemic management is in its infancy, however, and currently there is limited evidence to help identify at-risk populations and guide effective management for at-risk patient populations [13]. This study provides important data that can be used to develop risk stratification tools with implications for improved glycemic management of hospitalized patients with diabetes. Among the 32 factors included in the final multivariate logistic model (Table 2), 10 were statistically significant predictors of SGC. The top 3 predictors of SGC were HbA1c, admission blood glucose, and steroid use. These are straightforward, easily accessed factors that can become the basis for effective risk stratification and targeted clinical therapies.
This study showed that the degree of diabetes control prior to admission, as measured by HbA1c, is one of the strongest predictors of inpatient SGC. Patients with poorly controlled diabetes pre-admission had significantly higher rates of SGC than patients admitted with good diabetes control. Furthermore, there was a strong linear relationship between degree of pre-admission diabetes control and glycemic control during hospitalization: the higher the HbA1c on admission, the higher the odds are for poor glycemic control during hospitalization (Figure). The odds of SGC increased more than fivefold at an HbA1c of just 6.7% (50 mmol/mol) and continued to increase linearly as HbA1c increased. This increase occurred despite the fact that patients with poorly controlled pre-admission diabetes were found to have a significantly greater rate of insulin treatment using standardized order sets (which included basal and rapid-acting insulin) than the total sample in this study. So although patients with poorly controlled diabetes pre-admission were actively managed using evidence-based insulin order sets to control glycemia throughout their hospitalization, this did not translate to better glycemic control. Boord et al [14] and Neubauer et al [15] found similar results in their evaluation of glycemic control in hospitalized patients: while insulin use was high, glycemic control remained suboptimal. Similarly, Schnipper et al [16] found that adherence to insulin orders per se were not associated with better glucose control. They noted that the majority of patients with continued elevated blood glucose values did not have changes made to their insulin orders in response to suboptimal blood glucose values. These observations and this study’s findings suggest that fixed standardized subcutaneous orders may be more effective for well-controlled patients with diabetes than for patients with poorly controlled diabetes on admission. These patients need more frequent modifications to standard order sets based on clinical response during hospitalization to ensure good glycemic control.
This study confirmed the well known, highly correlated relationship between POC blood glucose and HbA1c. Rohlfing et al [17] previously documented a strong relationship (R = 0.82) between blood glucose and HbA1c in patients enrolled in the Diabetes Control and Complications Trial. Nathan et al [18]also documented a strong relationship (R= 0.84) between HbA1c and average blood glucose that was consistent across diverse populations. This study’s findings show odds for SGC based on admission blood glucose followed a very similar trend as HbA1c, and both factors showed much greater odds for SGC than steroid use, independent of other factors.
Admission blood glucose was the second best predictor of SBC using regression modeling (Table 2) and the strongest predictor using classification tree analysis. Odds for SGC were both very high and remarkably similar for a concomitant range of admission blood glucose and HbA1c values (Figure). The overlapping admission blood glucose range was 165 to 240 mg/dL, which corresponded to HbA1c values between 6.65 and 8.5 (49 mmol/mol–69 mmol/mol). The odds for SGC increased from a 6.5-fold increase in odds at 165 mg/dL to 22-fold increased odds at 240 mg/dL. This corresponded to a 5.5-fold increase in odds with HbA1c of 6.65 (49 mmol/mol) to a 19-fold increase in odds with HbA1c of 8.5 (69 mmol/mol).
Notably, an admission blood glucose as low as 164 mg/dL significantly increased the odds of SGC. This relatively mild hyperglycemia is not typically considered a signifier for difficult inpatient glycemic management. Furthermore, the odds of SGC continue to increase until admission blood glucose reached approximately 240 mg/dL, at which point the odds start declining (Figure). This suggests that only exceptionally elevated admission blood glucoses triggered prompt insulin treatment on admission. The data from this study suggests targeted action for an admission blood glucose as low as 164 mg/dL is just as necessary as for those admitted with a much higher blood glucose to ensure optimal glycemic control throughout hospitalization. Implications are that admission blood glucose may be an inexpensive, straightforward, and readily available predictor of SGC and marker for targeted clinical action, especially for hospitals that do not routinely order HbA1c labs during hospitalization.
Steroid treatment was the third strongest predictor of SGC. Additional analysis showed that any proportion of hospital stay with steroid administration resulted in a stable threefold increased odds for SGC, adjusting for other predictive factors (Figure). Developing insulin treatment therapies that are tailored to patients that will be administered steroids at any point during their hospitalization may be a reasonable strategy to reduce SGC in this population. Based on the results of this study and other Sharp data, Sharp is currently piloting a steroid insulin order set that is available in the electronic health record to hospital physicians for use with any patient that is administered steroids. The order set includes eating and non-eating standards, intensified meal dose coverage, a lower blood glucose threshold for starting correction dosing, and a diabetic nurse educator consult. Evaluation will include appropriate order set usage, rate of glycemic control and extreme blood glucose values.
Limitations
A limitation of this study is that it used observational data and was conducted within a single health care system, thus potentially reducing generalizability. Nevertheless, the sample size was large, there were many clinical and demographic characteristics to be leveraged in the analysis, the statistical approach utilized a complementary regression and classification approach to adjust and present the findings, and the sample included patients from three hospitals across San Diego County with diverse patient populations.
Conclusion
While much progress has been made understanding the need for appropriate glycemic management for patient with diabetes to reduce their risk for adverse outcomes, the knowledge base is still quite limited, especially regarding optimal glycemic limits for diverse patient populations. There is a need to identify predictors of SGC if risk stratification tools are to be built that can help target therapies with the potential to reducing the risk of poor glycemic control and adverse patient outcomes. This study identified 3 readily available factors—admission blood glucose, HbA1c and steroid use—that strongly predict SGC, controlling for other patient risk factors. In general, poor pre-admission diabetes control and inpatient steroid use strongly predict SGC, and the data suggests that earlier and frequently calibrated intervention may improve inpatient glycemic control for these patient populations. We identified a range of thresholds for these variables that may provide a basis for targeted treatment on admission. In conclusion, this study has important implications for meaningful use of readily available factors to identify patients at risk for SGC. Clinical therapies and guidelines incorporating empirically derived risk-stratification tools should enhance the ability to achieve the triple aim of better health, better care quality and more efficient care costs for hospitalized patient with diabetes.
Corresponding author: Miriam Bender, PhD, RN, University of California, Irvine, 252 Berk Hall, Irvine, CA 92697, [email protected].
Financial disclosures: None.
Author contributions: Author contributions: conception and design, MB, TCS, JT, KH; analysis and interpretation of data, MB, TCS, JT, KH; drafting of article, MB, TCS; critical revision of the article, MB, TCS, JT, KH; administrative or technical support, JT, KH; collection and assembly of data, MB, TCS, AK.
1. Umpierrez GE, Hellman R, Korytkowski MT, et al; Endocrine Society. Management of hyperglycemia in hospitalized patients in non-critical care setting: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2012;97:16–38.
2. Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care2009;32:1119–31.
3. Coursin DB, Connery LE, Ketzler JT. Perioperative diabetic and hyperglycemic management issues. Crit Care Med 2004;32(4 Suppl):S116–25.
4. Pandit MK, Burke J, Gustafson AB, et al. Drug-induced disorders of glucose tolerance. Ann Intern Med 1993;118:529–39.
5. Donihi AC, Raval D, Saul M, et al. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr Pract 2006;12:358–62.
6. McHugh MD, Shang J, Sloane DM, Aiken LH. Risk factors for hospital-acquired ‘poor glycemic control’: a case-control study. Int J Qual Health Care 2011;23:44–51.
7. McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin 2001;17:107–24.
8. Rady MY, Johnson DJ, Patel BM, et al. Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus. Mayo Clin Proc 2005;80:1558–67.
9. Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010;362:800–11.
10. Kim KS, Kim SK, Lee YK, et al. Diagnostic value of glycated haemoglobin HbA(1c) for the early detection of diabetes in high-risk subjects. Diabet Med 2008;25:997–1000.
11. Quan H, Li B, Couris CM, et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol 2011;173:676–82.
12. Kirk JK, Oldham EC. Hyperglycemia management using insulin in the acute care setting: therapies and strategies for care in the non-critically ill patient. Ann Pharmacother 2010;44:1222–30.
13. Draznin B, Gilden J, Golden SH, et al. Pathways to quality inpatient management of hyperglycemia and diabetes: a call to action. Diabetes Care 2013;36:1807–14.
14. Boord JB, Greevy RA, Braithwaite SS, et al. Evaluation of hospital glycemic control at US academic medical centers. J Hosp Med 2009;4:35–44.
15. Neubauer KM, Schaupp L, Plank J, et al. Failure to control hyperglycemia in noncritically ill diabetes patients despite standard glycemic management in a hospital setting. J Diabetes Sci Technol 2013;7:402–9.
16. Schnipper JL, Barsky EE, Shaykevich S, et al. Inpatient management of diabetes and hyperglycemia among general medicine patients at a large teaching hospital. J Hosp Med 2006;1:145–50.
17. Rohlfing CL, Wiedmeyer HM, Little RR, et al. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275–8.
18. Nathan DM, Kuenen J, Borg R, et al; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008;31:1473–8.
From Sharp HealthCare, San Diego, CA (Ms. Thompson, Mr. Koucheki, Dr. Holdy), National University, San Diego, CA (Dr. Smith), and University of California, Irvine (Dr. Bender).
Abstract
- Objective: Suboptimal glycemic control (SGC) puts hospitalized patients with diabetes at risk for poor outcomes. The purpose of this study was to quantify factors with predictive capacity to identify patients at risk for SGC during hospitalization.
- Methods: 32 baseline and demographic variables were extracted from the electronic records of 23,100 patients with diabetes hospitalized between 2009 and 2012. The rate of blood glucose values between 70 and 180 mg/dL was calculated for each patient. A predictive model for SGC was developed using regression modeling, standardized coefficients, and classification tree analysis. Odds ratios (ORs) were calculated to isolate adjusted odds of SGC for top predictors.
- Results: The final predictive model included 13 variables (C statistic = 0.88). HbA1c (OR, 0.60 [95% confidence interval {CI}, 0.58–0.61]), admission blood glucose (OR, 0.91 [CI, 0.91–0.92]), and steroid use (OR, 0.06 [CI 0.04–0.08]) were the highest-ranking predictors of SGC. HbA1c and SGC had a strong linear relationship (R2 = 0.99), with increasing odds for SGC as HbA1c increased. Admission blood glucose and SGC had a polynomial relationship (R2 = 0.95); increasing odds for SGC until 240 mg/dL; then odds started decreasing. Steroid use showed a steady threefold increase in odds for SGC across all rates of use.
- Conclusions: Poor preadmission diabetes control and inpatient steroid use strongly predict SGC. A range of thresholds for these predictors was empirically determined, providing a basis for targeted therapies on admission. Guidelines incorporating empirically derived thresholds should enhance the ability to achieve optimal glycemic control for hospitalized patients with diabetes.
Current recommendations for glycemic control of hospitalized patients include use of multidisciplinary diabetic care teams and standardized insulin order sets [1,2], yet there is still uncertainty how best to target such protocols for patients with diabetes at risk for suboptimal glycemic control [2,3]. Many factors are theorized to hinder optimal inpatient glycemic control, such as steroid use [4,5],comorbid states [6],severity of illness [7,8], and preadmission glycemic control [9,10]. However, there currently exists little evidence of these factors’ ability to predict suboptimal glycemic control (SGC). Identifying straightforward predictive factors of SGC would provide a clinically meaningful basis for targeted therapy. The purpose of this study was to describe the prevalence of a range of potential risk factors in a diverse hospitalized patient population with a secondary diagnosis of diabetes (types 1 and 2), and to determine which factors were predictive of SGC.
Methods
A retrospective cohort study design was used to identify factors predictive of inpatient SGC for patients admitted to any of 3 hospitals aligned with Sharp HealthCare (“Sharp”), a community-based, nonprofit integrated health system headquartered in San Diego, California, that serves more than 27% of the county’s 3 million-plus residents each year.
Inclusion Criteria
We extracted data for 23,100 patients hospitalized between January 2009 and December 2012 with a secondary diagnosis of diabetes (types 1 and 2), a length of stay (LOS) ≥ 3 days, and a minimum of 2 point-of-care (POC) blood glucose tests per day. The LOS and blood glucose minimum are standard criteria for Sharp glycemic monitoring to ensure a minimum quantity of blood glucose monitoring for glycemic management.
Glycemic Control (Independent Variable)
Glycemic control was defined as POC blood glucose values within the target range of 70 to 180 mg/dL during hospitalization. POC tests outside that range were defined as SGC. This range was determined based on current Sharp benchmark targets and was not adjusted for total number of blood glucose values.
Predictive Variables
Age, gender, race, ethnicity, payor, facility and LOS were extracted from Sharp’s data warehouse. Medical vs. surgical stay and major diagnostic category was determined from administrative diagnosis coding. Body mass index (BMI) was extracted from Sharp’s electronic health record. Risk of mortality and severity of illness were calculated using 3M APR-DRG proprietary software using administrative diagnosis coding. Comorbidities were determined based on administrative diagnosis codes per published guidelines [11]. Glycosylated hemoglobin (HbA1c) was obtained on admission for patients with a secondary diagnosis of diabetes as part of Sharp's multidisciplinary diabetes care management program and extracted from the electronic health record. Admission blood glucose was defined as the first documented POC blood glucose after admission. ICU stay was calculated as a continuous variable: the percent of LOS spent in the ICU. Steroid use was similarly calculated, and defined as oral or intravenous administration of any quantity or dosage of the following corticosteroids during each day of hospitalization: dexamethasone, hydrocortisone, prednisone, and/or methylprednisone. Adherence to Sharp's multidisciplinary diabetes care management program was measured by use of standardized insulin order sets. Sharp uses evidence-based order sets for continuous infusion and subcutaneous insulin management; subcutaneous orders include basal and rapid-acting insulin. We calculated the total time a person was on an order set during hospitalization by subtracting any time a patient did not have insulin ordered from the total LOS. This was transformed into a variable documenting the percent of LOS the patient was on an insulin order set. Average blood glucose for admission was calculated for all documented POC blood glucoses during admission, omitting the admission blood glucose (the first POC blood glucose of the admission).
Analysis
Univariate analyses including t tests and chi-square tests were conducted to investigate the unadjusted association between variables and glycemic control. Good glycemic control was defined as 90% of all POC blood glucose tests between 70 and 180 mg/dL based on empirical distribution and organization targets. A predictive model of inpatient glycemic control was then developed using a backward stepwise multivariable logistic regression approach. The data were split into a model building and validation set. Variables were included that represented both baseline and transitional state during the hospital stay to account for potentially mediating effects and a sensitivity analysis was conducted with them in and out of the final model to assess impact. Standardized coefficients were calculated to rank order variables in the model allowing indication of the variables with the greatest predictive impact on the outcome. Further investigation of the optimal classification points for the variables was conducted to indicate best differentiation of good glycemic control. Significant variables from the multivariable logistic regression were included in an exploratory classification tree analysis that recursively partitioned data in order to improve the fit, with optimal splitting identified over all variables at all possible split points. Classification tree cut-off points were used to further develop models identifying odds ratios for various thresholds for the top three predictive variables. A series of logistic regression models were then run with differing cut points of top 3 predictors to isolate adjusted odds of good glycemic control. Analytic data set building and statistical analyses were completed using SAS 9.4.
Results
Patient Characteristics
Table 1 shows patient demographic and clinical characteristics for the entire sample and for the top quartile (76% or greater POC blood glucose values within target range) and bottom quartile (25% or less POC blood glucose values within target range). Unadjusted results show a significant difference across quartiles for all factors except age, gender, dementia, rheumatic disease and paraplegia. Patients in the bottom 25th percentile (ie, the poorest control) were more likely than the total population to have a higher admission blood glucose (198 mg/dL vs. 153 mg/dL), higher HbA1c (8.53 [70mmol/mol] vs. 7.35 [57mmol/mol), a medical (74% vs. 66%) and/or respiratory (18% vs. 12%) diagnosis, corticosteroid use (17% vs. 27%), an insulin order set use (80% vs. 70%) and higher mean blood glucose during hospitalization (206.3 vs. 157.1 mg/dL). Patients with poorest control were less likely that the total population to have a high risk of mortality (11% vs. 15%) and severity of illness (13% vs. 18%). They also had less ICU care (8% vs. 13%), and a shorter LOS (5.82 vs. 7.82 days).
Predictive Modeling
Classification tree analysis resulted in the same top 3 predictors, but in a different order. The analysis also provided cut-off values that predict suboptimal glycemic control. Classification tree analysis showed admission blood glucose was the most influential predictor, with 164.5 mg/dL indicating the optimal cut-point for prediction of SGC, followed by HbA1c with an optimal cut-off point of 6.65% indicating prediction of SGC, followed by treatment with corticosteroids, with an optimal cut-off point of 24% of the LOS on corticosteroids indicating prediction of SGC.
Discussion
Evidence of patient characteristics that consistently predict suboptimal glycemic control during hospitalization is needed to better inform clinical decisions for inpatient glycemic management. Hospitalized patients would greatly benefit from glycemic protocols that incorporate risk stratification tools based evidence-based risk factors for poor glycemic control [12]. The science of inpatient glycemic management is in its infancy, however, and currently there is limited evidence to help identify at-risk populations and guide effective management for at-risk patient populations [13]. This study provides important data that can be used to develop risk stratification tools with implications for improved glycemic management of hospitalized patients with diabetes. Among the 32 factors included in the final multivariate logistic model (Table 2), 10 were statistically significant predictors of SGC. The top 3 predictors of SGC were HbA1c, admission blood glucose, and steroid use. These are straightforward, easily accessed factors that can become the basis for effective risk stratification and targeted clinical therapies.
This study showed that the degree of diabetes control prior to admission, as measured by HbA1c, is one of the strongest predictors of inpatient SGC. Patients with poorly controlled diabetes pre-admission had significantly higher rates of SGC than patients admitted with good diabetes control. Furthermore, there was a strong linear relationship between degree of pre-admission diabetes control and glycemic control during hospitalization: the higher the HbA1c on admission, the higher the odds are for poor glycemic control during hospitalization (Figure). The odds of SGC increased more than fivefold at an HbA1c of just 6.7% (50 mmol/mol) and continued to increase linearly as HbA1c increased. This increase occurred despite the fact that patients with poorly controlled pre-admission diabetes were found to have a significantly greater rate of insulin treatment using standardized order sets (which included basal and rapid-acting insulin) than the total sample in this study. So although patients with poorly controlled diabetes pre-admission were actively managed using evidence-based insulin order sets to control glycemia throughout their hospitalization, this did not translate to better glycemic control. Boord et al [14] and Neubauer et al [15] found similar results in their evaluation of glycemic control in hospitalized patients: while insulin use was high, glycemic control remained suboptimal. Similarly, Schnipper et al [16] found that adherence to insulin orders per se were not associated with better glucose control. They noted that the majority of patients with continued elevated blood glucose values did not have changes made to their insulin orders in response to suboptimal blood glucose values. These observations and this study’s findings suggest that fixed standardized subcutaneous orders may be more effective for well-controlled patients with diabetes than for patients with poorly controlled diabetes on admission. These patients need more frequent modifications to standard order sets based on clinical response during hospitalization to ensure good glycemic control.
This study confirmed the well known, highly correlated relationship between POC blood glucose and HbA1c. Rohlfing et al [17] previously documented a strong relationship (R = 0.82) between blood glucose and HbA1c in patients enrolled in the Diabetes Control and Complications Trial. Nathan et al [18]also documented a strong relationship (R= 0.84) between HbA1c and average blood glucose that was consistent across diverse populations. This study’s findings show odds for SGC based on admission blood glucose followed a very similar trend as HbA1c, and both factors showed much greater odds for SGC than steroid use, independent of other factors.
Admission blood glucose was the second best predictor of SBC using regression modeling (Table 2) and the strongest predictor using classification tree analysis. Odds for SGC were both very high and remarkably similar for a concomitant range of admission blood glucose and HbA1c values (Figure). The overlapping admission blood glucose range was 165 to 240 mg/dL, which corresponded to HbA1c values between 6.65 and 8.5 (49 mmol/mol–69 mmol/mol). The odds for SGC increased from a 6.5-fold increase in odds at 165 mg/dL to 22-fold increased odds at 240 mg/dL. This corresponded to a 5.5-fold increase in odds with HbA1c of 6.65 (49 mmol/mol) to a 19-fold increase in odds with HbA1c of 8.5 (69 mmol/mol).
Notably, an admission blood glucose as low as 164 mg/dL significantly increased the odds of SGC. This relatively mild hyperglycemia is not typically considered a signifier for difficult inpatient glycemic management. Furthermore, the odds of SGC continue to increase until admission blood glucose reached approximately 240 mg/dL, at which point the odds start declining (Figure). This suggests that only exceptionally elevated admission blood glucoses triggered prompt insulin treatment on admission. The data from this study suggests targeted action for an admission blood glucose as low as 164 mg/dL is just as necessary as for those admitted with a much higher blood glucose to ensure optimal glycemic control throughout hospitalization. Implications are that admission blood glucose may be an inexpensive, straightforward, and readily available predictor of SGC and marker for targeted clinical action, especially for hospitals that do not routinely order HbA1c labs during hospitalization.
Steroid treatment was the third strongest predictor of SGC. Additional analysis showed that any proportion of hospital stay with steroid administration resulted in a stable threefold increased odds for SGC, adjusting for other predictive factors (Figure). Developing insulin treatment therapies that are tailored to patients that will be administered steroids at any point during their hospitalization may be a reasonable strategy to reduce SGC in this population. Based on the results of this study and other Sharp data, Sharp is currently piloting a steroid insulin order set that is available in the electronic health record to hospital physicians for use with any patient that is administered steroids. The order set includes eating and non-eating standards, intensified meal dose coverage, a lower blood glucose threshold for starting correction dosing, and a diabetic nurse educator consult. Evaluation will include appropriate order set usage, rate of glycemic control and extreme blood glucose values.
Limitations
A limitation of this study is that it used observational data and was conducted within a single health care system, thus potentially reducing generalizability. Nevertheless, the sample size was large, there were many clinical and demographic characteristics to be leveraged in the analysis, the statistical approach utilized a complementary regression and classification approach to adjust and present the findings, and the sample included patients from three hospitals across San Diego County with diverse patient populations.
Conclusion
While much progress has been made understanding the need for appropriate glycemic management for patient with diabetes to reduce their risk for adverse outcomes, the knowledge base is still quite limited, especially regarding optimal glycemic limits for diverse patient populations. There is a need to identify predictors of SGC if risk stratification tools are to be built that can help target therapies with the potential to reducing the risk of poor glycemic control and adverse patient outcomes. This study identified 3 readily available factors—admission blood glucose, HbA1c and steroid use—that strongly predict SGC, controlling for other patient risk factors. In general, poor pre-admission diabetes control and inpatient steroid use strongly predict SGC, and the data suggests that earlier and frequently calibrated intervention may improve inpatient glycemic control for these patient populations. We identified a range of thresholds for these variables that may provide a basis for targeted treatment on admission. In conclusion, this study has important implications for meaningful use of readily available factors to identify patients at risk for SGC. Clinical therapies and guidelines incorporating empirically derived risk-stratification tools should enhance the ability to achieve the triple aim of better health, better care quality and more efficient care costs for hospitalized patient with diabetes.
Corresponding author: Miriam Bender, PhD, RN, University of California, Irvine, 252 Berk Hall, Irvine, CA 92697, [email protected].
Financial disclosures: None.
Author contributions: Author contributions: conception and design, MB, TCS, JT, KH; analysis and interpretation of data, MB, TCS, JT, KH; drafting of article, MB, TCS; critical revision of the article, MB, TCS, JT, KH; administrative or technical support, JT, KH; collection and assembly of data, MB, TCS, AK.
From Sharp HealthCare, San Diego, CA (Ms. Thompson, Mr. Koucheki, Dr. Holdy), National University, San Diego, CA (Dr. Smith), and University of California, Irvine (Dr. Bender).
Abstract
- Objective: Suboptimal glycemic control (SGC) puts hospitalized patients with diabetes at risk for poor outcomes. The purpose of this study was to quantify factors with predictive capacity to identify patients at risk for SGC during hospitalization.
- Methods: 32 baseline and demographic variables were extracted from the electronic records of 23,100 patients with diabetes hospitalized between 2009 and 2012. The rate of blood glucose values between 70 and 180 mg/dL was calculated for each patient. A predictive model for SGC was developed using regression modeling, standardized coefficients, and classification tree analysis. Odds ratios (ORs) were calculated to isolate adjusted odds of SGC for top predictors.
- Results: The final predictive model included 13 variables (C statistic = 0.88). HbA1c (OR, 0.60 [95% confidence interval {CI}, 0.58–0.61]), admission blood glucose (OR, 0.91 [CI, 0.91–0.92]), and steroid use (OR, 0.06 [CI 0.04–0.08]) were the highest-ranking predictors of SGC. HbA1c and SGC had a strong linear relationship (R2 = 0.99), with increasing odds for SGC as HbA1c increased. Admission blood glucose and SGC had a polynomial relationship (R2 = 0.95); increasing odds for SGC until 240 mg/dL; then odds started decreasing. Steroid use showed a steady threefold increase in odds for SGC across all rates of use.
- Conclusions: Poor preadmission diabetes control and inpatient steroid use strongly predict SGC. A range of thresholds for these predictors was empirically determined, providing a basis for targeted therapies on admission. Guidelines incorporating empirically derived thresholds should enhance the ability to achieve optimal glycemic control for hospitalized patients with diabetes.
Current recommendations for glycemic control of hospitalized patients include use of multidisciplinary diabetic care teams and standardized insulin order sets [1,2], yet there is still uncertainty how best to target such protocols for patients with diabetes at risk for suboptimal glycemic control [2,3]. Many factors are theorized to hinder optimal inpatient glycemic control, such as steroid use [4,5],comorbid states [6],severity of illness [7,8], and preadmission glycemic control [9,10]. However, there currently exists little evidence of these factors’ ability to predict suboptimal glycemic control (SGC). Identifying straightforward predictive factors of SGC would provide a clinically meaningful basis for targeted therapy. The purpose of this study was to describe the prevalence of a range of potential risk factors in a diverse hospitalized patient population with a secondary diagnosis of diabetes (types 1 and 2), and to determine which factors were predictive of SGC.
Methods
A retrospective cohort study design was used to identify factors predictive of inpatient SGC for patients admitted to any of 3 hospitals aligned with Sharp HealthCare (“Sharp”), a community-based, nonprofit integrated health system headquartered in San Diego, California, that serves more than 27% of the county’s 3 million-plus residents each year.
Inclusion Criteria
We extracted data for 23,100 patients hospitalized between January 2009 and December 2012 with a secondary diagnosis of diabetes (types 1 and 2), a length of stay (LOS) ≥ 3 days, and a minimum of 2 point-of-care (POC) blood glucose tests per day. The LOS and blood glucose minimum are standard criteria for Sharp glycemic monitoring to ensure a minimum quantity of blood glucose monitoring for glycemic management.
Glycemic Control (Independent Variable)
Glycemic control was defined as POC blood glucose values within the target range of 70 to 180 mg/dL during hospitalization. POC tests outside that range were defined as SGC. This range was determined based on current Sharp benchmark targets and was not adjusted for total number of blood glucose values.
Predictive Variables
Age, gender, race, ethnicity, payor, facility and LOS were extracted from Sharp’s data warehouse. Medical vs. surgical stay and major diagnostic category was determined from administrative diagnosis coding. Body mass index (BMI) was extracted from Sharp’s electronic health record. Risk of mortality and severity of illness were calculated using 3M APR-DRG proprietary software using administrative diagnosis coding. Comorbidities were determined based on administrative diagnosis codes per published guidelines [11]. Glycosylated hemoglobin (HbA1c) was obtained on admission for patients with a secondary diagnosis of diabetes as part of Sharp's multidisciplinary diabetes care management program and extracted from the electronic health record. Admission blood glucose was defined as the first documented POC blood glucose after admission. ICU stay was calculated as a continuous variable: the percent of LOS spent in the ICU. Steroid use was similarly calculated, and defined as oral or intravenous administration of any quantity or dosage of the following corticosteroids during each day of hospitalization: dexamethasone, hydrocortisone, prednisone, and/or methylprednisone. Adherence to Sharp's multidisciplinary diabetes care management program was measured by use of standardized insulin order sets. Sharp uses evidence-based order sets for continuous infusion and subcutaneous insulin management; subcutaneous orders include basal and rapid-acting insulin. We calculated the total time a person was on an order set during hospitalization by subtracting any time a patient did not have insulin ordered from the total LOS. This was transformed into a variable documenting the percent of LOS the patient was on an insulin order set. Average blood glucose for admission was calculated for all documented POC blood glucoses during admission, omitting the admission blood glucose (the first POC blood glucose of the admission).
Analysis
Univariate analyses including t tests and chi-square tests were conducted to investigate the unadjusted association between variables and glycemic control. Good glycemic control was defined as 90% of all POC blood glucose tests between 70 and 180 mg/dL based on empirical distribution and organization targets. A predictive model of inpatient glycemic control was then developed using a backward stepwise multivariable logistic regression approach. The data were split into a model building and validation set. Variables were included that represented both baseline and transitional state during the hospital stay to account for potentially mediating effects and a sensitivity analysis was conducted with them in and out of the final model to assess impact. Standardized coefficients were calculated to rank order variables in the model allowing indication of the variables with the greatest predictive impact on the outcome. Further investigation of the optimal classification points for the variables was conducted to indicate best differentiation of good glycemic control. Significant variables from the multivariable logistic regression were included in an exploratory classification tree analysis that recursively partitioned data in order to improve the fit, with optimal splitting identified over all variables at all possible split points. Classification tree cut-off points were used to further develop models identifying odds ratios for various thresholds for the top three predictive variables. A series of logistic regression models were then run with differing cut points of top 3 predictors to isolate adjusted odds of good glycemic control. Analytic data set building and statistical analyses were completed using SAS 9.4.
Results
Patient Characteristics
Table 1 shows patient demographic and clinical characteristics for the entire sample and for the top quartile (76% or greater POC blood glucose values within target range) and bottom quartile (25% or less POC blood glucose values within target range). Unadjusted results show a significant difference across quartiles for all factors except age, gender, dementia, rheumatic disease and paraplegia. Patients in the bottom 25th percentile (ie, the poorest control) were more likely than the total population to have a higher admission blood glucose (198 mg/dL vs. 153 mg/dL), higher HbA1c (8.53 [70mmol/mol] vs. 7.35 [57mmol/mol), a medical (74% vs. 66%) and/or respiratory (18% vs. 12%) diagnosis, corticosteroid use (17% vs. 27%), an insulin order set use (80% vs. 70%) and higher mean blood glucose during hospitalization (206.3 vs. 157.1 mg/dL). Patients with poorest control were less likely that the total population to have a high risk of mortality (11% vs. 15%) and severity of illness (13% vs. 18%). They also had less ICU care (8% vs. 13%), and a shorter LOS (5.82 vs. 7.82 days).
Predictive Modeling
Classification tree analysis resulted in the same top 3 predictors, but in a different order. The analysis also provided cut-off values that predict suboptimal glycemic control. Classification tree analysis showed admission blood glucose was the most influential predictor, with 164.5 mg/dL indicating the optimal cut-point for prediction of SGC, followed by HbA1c with an optimal cut-off point of 6.65% indicating prediction of SGC, followed by treatment with corticosteroids, with an optimal cut-off point of 24% of the LOS on corticosteroids indicating prediction of SGC.
Discussion
Evidence of patient characteristics that consistently predict suboptimal glycemic control during hospitalization is needed to better inform clinical decisions for inpatient glycemic management. Hospitalized patients would greatly benefit from glycemic protocols that incorporate risk stratification tools based evidence-based risk factors for poor glycemic control [12]. The science of inpatient glycemic management is in its infancy, however, and currently there is limited evidence to help identify at-risk populations and guide effective management for at-risk patient populations [13]. This study provides important data that can be used to develop risk stratification tools with implications for improved glycemic management of hospitalized patients with diabetes. Among the 32 factors included in the final multivariate logistic model (Table 2), 10 were statistically significant predictors of SGC. The top 3 predictors of SGC were HbA1c, admission blood glucose, and steroid use. These are straightforward, easily accessed factors that can become the basis for effective risk stratification and targeted clinical therapies.
This study showed that the degree of diabetes control prior to admission, as measured by HbA1c, is one of the strongest predictors of inpatient SGC. Patients with poorly controlled diabetes pre-admission had significantly higher rates of SGC than patients admitted with good diabetes control. Furthermore, there was a strong linear relationship between degree of pre-admission diabetes control and glycemic control during hospitalization: the higher the HbA1c on admission, the higher the odds are for poor glycemic control during hospitalization (Figure). The odds of SGC increased more than fivefold at an HbA1c of just 6.7% (50 mmol/mol) and continued to increase linearly as HbA1c increased. This increase occurred despite the fact that patients with poorly controlled pre-admission diabetes were found to have a significantly greater rate of insulin treatment using standardized order sets (which included basal and rapid-acting insulin) than the total sample in this study. So although patients with poorly controlled diabetes pre-admission were actively managed using evidence-based insulin order sets to control glycemia throughout their hospitalization, this did not translate to better glycemic control. Boord et al [14] and Neubauer et al [15] found similar results in their evaluation of glycemic control in hospitalized patients: while insulin use was high, glycemic control remained suboptimal. Similarly, Schnipper et al [16] found that adherence to insulin orders per se were not associated with better glucose control. They noted that the majority of patients with continued elevated blood glucose values did not have changes made to their insulin orders in response to suboptimal blood glucose values. These observations and this study’s findings suggest that fixed standardized subcutaneous orders may be more effective for well-controlled patients with diabetes than for patients with poorly controlled diabetes on admission. These patients need more frequent modifications to standard order sets based on clinical response during hospitalization to ensure good glycemic control.
This study confirmed the well known, highly correlated relationship between POC blood glucose and HbA1c. Rohlfing et al [17] previously documented a strong relationship (R = 0.82) between blood glucose and HbA1c in patients enrolled in the Diabetes Control and Complications Trial. Nathan et al [18]also documented a strong relationship (R= 0.84) between HbA1c and average blood glucose that was consistent across diverse populations. This study’s findings show odds for SGC based on admission blood glucose followed a very similar trend as HbA1c, and both factors showed much greater odds for SGC than steroid use, independent of other factors.
Admission blood glucose was the second best predictor of SBC using regression modeling (Table 2) and the strongest predictor using classification tree analysis. Odds for SGC were both very high and remarkably similar for a concomitant range of admission blood glucose and HbA1c values (Figure). The overlapping admission blood glucose range was 165 to 240 mg/dL, which corresponded to HbA1c values between 6.65 and 8.5 (49 mmol/mol–69 mmol/mol). The odds for SGC increased from a 6.5-fold increase in odds at 165 mg/dL to 22-fold increased odds at 240 mg/dL. This corresponded to a 5.5-fold increase in odds with HbA1c of 6.65 (49 mmol/mol) to a 19-fold increase in odds with HbA1c of 8.5 (69 mmol/mol).
Notably, an admission blood glucose as low as 164 mg/dL significantly increased the odds of SGC. This relatively mild hyperglycemia is not typically considered a signifier for difficult inpatient glycemic management. Furthermore, the odds of SGC continue to increase until admission blood glucose reached approximately 240 mg/dL, at which point the odds start declining (Figure). This suggests that only exceptionally elevated admission blood glucoses triggered prompt insulin treatment on admission. The data from this study suggests targeted action for an admission blood glucose as low as 164 mg/dL is just as necessary as for those admitted with a much higher blood glucose to ensure optimal glycemic control throughout hospitalization. Implications are that admission blood glucose may be an inexpensive, straightforward, and readily available predictor of SGC and marker for targeted clinical action, especially for hospitals that do not routinely order HbA1c labs during hospitalization.
Steroid treatment was the third strongest predictor of SGC. Additional analysis showed that any proportion of hospital stay with steroid administration resulted in a stable threefold increased odds for SGC, adjusting for other predictive factors (Figure). Developing insulin treatment therapies that are tailored to patients that will be administered steroids at any point during their hospitalization may be a reasonable strategy to reduce SGC in this population. Based on the results of this study and other Sharp data, Sharp is currently piloting a steroid insulin order set that is available in the electronic health record to hospital physicians for use with any patient that is administered steroids. The order set includes eating and non-eating standards, intensified meal dose coverage, a lower blood glucose threshold for starting correction dosing, and a diabetic nurse educator consult. Evaluation will include appropriate order set usage, rate of glycemic control and extreme blood glucose values.
Limitations
A limitation of this study is that it used observational data and was conducted within a single health care system, thus potentially reducing generalizability. Nevertheless, the sample size was large, there were many clinical and demographic characteristics to be leveraged in the analysis, the statistical approach utilized a complementary regression and classification approach to adjust and present the findings, and the sample included patients from three hospitals across San Diego County with diverse patient populations.
Conclusion
While much progress has been made understanding the need for appropriate glycemic management for patient with diabetes to reduce their risk for adverse outcomes, the knowledge base is still quite limited, especially regarding optimal glycemic limits for diverse patient populations. There is a need to identify predictors of SGC if risk stratification tools are to be built that can help target therapies with the potential to reducing the risk of poor glycemic control and adverse patient outcomes. This study identified 3 readily available factors—admission blood glucose, HbA1c and steroid use—that strongly predict SGC, controlling for other patient risk factors. In general, poor pre-admission diabetes control and inpatient steroid use strongly predict SGC, and the data suggests that earlier and frequently calibrated intervention may improve inpatient glycemic control for these patient populations. We identified a range of thresholds for these variables that may provide a basis for targeted treatment on admission. In conclusion, this study has important implications for meaningful use of readily available factors to identify patients at risk for SGC. Clinical therapies and guidelines incorporating empirically derived risk-stratification tools should enhance the ability to achieve the triple aim of better health, better care quality and more efficient care costs for hospitalized patient with diabetes.
Corresponding author: Miriam Bender, PhD, RN, University of California, Irvine, 252 Berk Hall, Irvine, CA 92697, [email protected].
Financial disclosures: None.
Author contributions: Author contributions: conception and design, MB, TCS, JT, KH; analysis and interpretation of data, MB, TCS, JT, KH; drafting of article, MB, TCS; critical revision of the article, MB, TCS, JT, KH; administrative or technical support, JT, KH; collection and assembly of data, MB, TCS, AK.
1. Umpierrez GE, Hellman R, Korytkowski MT, et al; Endocrine Society. Management of hyperglycemia in hospitalized patients in non-critical care setting: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2012;97:16–38.
2. Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care2009;32:1119–31.
3. Coursin DB, Connery LE, Ketzler JT. Perioperative diabetic and hyperglycemic management issues. Crit Care Med 2004;32(4 Suppl):S116–25.
4. Pandit MK, Burke J, Gustafson AB, et al. Drug-induced disorders of glucose tolerance. Ann Intern Med 1993;118:529–39.
5. Donihi AC, Raval D, Saul M, et al. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr Pract 2006;12:358–62.
6. McHugh MD, Shang J, Sloane DM, Aiken LH. Risk factors for hospital-acquired ‘poor glycemic control’: a case-control study. Int J Qual Health Care 2011;23:44–51.
7. McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin 2001;17:107–24.
8. Rady MY, Johnson DJ, Patel BM, et al. Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus. Mayo Clin Proc 2005;80:1558–67.
9. Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010;362:800–11.
10. Kim KS, Kim SK, Lee YK, et al. Diagnostic value of glycated haemoglobin HbA(1c) for the early detection of diabetes in high-risk subjects. Diabet Med 2008;25:997–1000.
11. Quan H, Li B, Couris CM, et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol 2011;173:676–82.
12. Kirk JK, Oldham EC. Hyperglycemia management using insulin in the acute care setting: therapies and strategies for care in the non-critically ill patient. Ann Pharmacother 2010;44:1222–30.
13. Draznin B, Gilden J, Golden SH, et al. Pathways to quality inpatient management of hyperglycemia and diabetes: a call to action. Diabetes Care 2013;36:1807–14.
14. Boord JB, Greevy RA, Braithwaite SS, et al. Evaluation of hospital glycemic control at US academic medical centers. J Hosp Med 2009;4:35–44.
15. Neubauer KM, Schaupp L, Plank J, et al. Failure to control hyperglycemia in noncritically ill diabetes patients despite standard glycemic management in a hospital setting. J Diabetes Sci Technol 2013;7:402–9.
16. Schnipper JL, Barsky EE, Shaykevich S, et al. Inpatient management of diabetes and hyperglycemia among general medicine patients at a large teaching hospital. J Hosp Med 2006;1:145–50.
17. Rohlfing CL, Wiedmeyer HM, Little RR, et al. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275–8.
18. Nathan DM, Kuenen J, Borg R, et al; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008;31:1473–8.
1. Umpierrez GE, Hellman R, Korytkowski MT, et al; Endocrine Society. Management of hyperglycemia in hospitalized patients in non-critical care setting: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2012;97:16–38.
2. Moghissi ES, Korytkowski MT, DiNardo M, et al; American Association of Clinical Endocrinologists; American Diabetes Association. American Association of Clinical Endocrinologists and American Diabetes Association consensus statement on inpatient glycemic control. Diabetes Care2009;32:1119–31.
3. Coursin DB, Connery LE, Ketzler JT. Perioperative diabetic and hyperglycemic management issues. Crit Care Med 2004;32(4 Suppl):S116–25.
4. Pandit MK, Burke J, Gustafson AB, et al. Drug-induced disorders of glucose tolerance. Ann Intern Med 1993;118:529–39.
5. Donihi AC, Raval D, Saul M, et al. Prevalence and predictors of corticosteroid-related hyperglycemia in hospitalized patients. Endocr Pract 2006;12:358–62.
6. McHugh MD, Shang J, Sloane DM, Aiken LH. Risk factors for hospital-acquired ‘poor glycemic control’: a case-control study. Int J Qual Health Care 2011;23:44–51.
7. McCowen KC, Malhotra A, Bistrian BR. Stress-induced hyperglycemia. Crit Care Clin 2001;17:107–24.
8. Rady MY, Johnson DJ, Patel BM, et al. Influence of individual characteristics on outcome of glycemic control in intensive care unit patients with or without diabetes mellitus. Mayo Clin Proc 2005;80:1558–67.
9. Selvin E, Steffes MW, Zhu H, et al. Glycated hemoglobin, diabetes, and cardiovascular risk in nondiabetic adults. N Engl J Med 2010;362:800–11.
10. Kim KS, Kim SK, Lee YK, et al. Diagnostic value of glycated haemoglobin HbA(1c) for the early detection of diabetes in high-risk subjects. Diabet Med 2008;25:997–1000.
11. Quan H, Li B, Couris CM, et al. Updating and validating the Charlson comorbidity index and score for risk adjustment in hospital discharge abstracts using data from 6 countries. Am J Epidemiol 2011;173:676–82.
12. Kirk JK, Oldham EC. Hyperglycemia management using insulin in the acute care setting: therapies and strategies for care in the non-critically ill patient. Ann Pharmacother 2010;44:1222–30.
13. Draznin B, Gilden J, Golden SH, et al. Pathways to quality inpatient management of hyperglycemia and diabetes: a call to action. Diabetes Care 2013;36:1807–14.
14. Boord JB, Greevy RA, Braithwaite SS, et al. Evaluation of hospital glycemic control at US academic medical centers. J Hosp Med 2009;4:35–44.
15. Neubauer KM, Schaupp L, Plank J, et al. Failure to control hyperglycemia in noncritically ill diabetes patients despite standard glycemic management in a hospital setting. J Diabetes Sci Technol 2013;7:402–9.
16. Schnipper JL, Barsky EE, Shaykevich S, et al. Inpatient management of diabetes and hyperglycemia among general medicine patients at a large teaching hospital. J Hosp Med 2006;1:145–50.
17. Rohlfing CL, Wiedmeyer HM, Little RR, et al. Defining the relationship between plasma glucose and HbA(1c): analysis of glucose profiles and HbA(1c) in the Diabetes Control and Complications Trial. Diabetes Care 2002;25:275–8.
18. Nathan DM, Kuenen J, Borg R, et al; A1c-Derived Average Glucose Study Group. Translating the A1C assay into estimated average glucose values. Diabetes Care 2008;31:1473–8.
Mouse model could aid study of malaria
Plasmodium vivax infection
Image by Mae Melvin
Researchers say they have developed a human-chimeric mouse model that can advance the study of the malaria parasite Plasmodium vivax.
The model is engineered to grow human livers that can be infected with P vivax and used for investigations into parasite development, dormancy, and activation, as well as the effect certain drugs have on each aspect of the infection.
The researchers described this model in Cell Host & Microbe.
Stefan Kappe, PhD, of Seattle Biomedical Research Institute in Washington, and his colleagues noted that P vivax malaria parasites are more resistant to control and elimination than other malaria parasites because, after infection, they fall dormant within the liver for months to years.
When these dormant parasites eventually become activated, they replicate, infect the bloodstream, and cause relapsing malaria.
The lack of tractable P vivax animal models has made it difficult to examine P vivax liver-stage infection, but Dr Kappe and his colleagues found a solution in a model known as FRG KO huHep.
To create the FRG KO huHep model, the team transplanted human hepatocytes in the severely immunocompromised FRG KO mouse, which has deletions in fumarylacetoacetate hydrolase (FAH), recombinationactivating gene 2 (Rag2), and interleukin-2 receptor subunit gamma (Il2rg).
The researchers found that FRG KO huHep mice support P vivax sporozoite infection, liver-stage development, and the formation and persistence of dormant liver-stage parasites.
In addition, experiments with this model revealed that the antimalarial drug primaquine could prevent and eliminate liver-stage infection.
The researchers therefore concluded that P vivax-infected FRG KO huHep mice are a suitable model for studying liver-stage development and dormancy and may facilitate the discovery of drugs targeting relapsing malaria.
“This model is a real game-changer,” said Ivo Mueller, PhD, a malaria expert at the Walter and Eliza Hall Institute in Melbourne, Victoria, Australia, who was not involved in this research.
“For the first time, we now have a realistic opportunity that not only allows us to directly study this normally hidden P vivax life-stage, but also to test new drugs and vaccines. This new model is an essential resource in our quest to develop the new anti-vivax interventions that we will need to eliminate P vivax malaria.” ![]()
Plasmodium vivax infection
Image by Mae Melvin
Researchers say they have developed a human-chimeric mouse model that can advance the study of the malaria parasite Plasmodium vivax.
The model is engineered to grow human livers that can be infected with P vivax and used for investigations into parasite development, dormancy, and activation, as well as the effect certain drugs have on each aspect of the infection.
The researchers described this model in Cell Host & Microbe.
Stefan Kappe, PhD, of Seattle Biomedical Research Institute in Washington, and his colleagues noted that P vivax malaria parasites are more resistant to control and elimination than other malaria parasites because, after infection, they fall dormant within the liver for months to years.
When these dormant parasites eventually become activated, they replicate, infect the bloodstream, and cause relapsing malaria.
The lack of tractable P vivax animal models has made it difficult to examine P vivax liver-stage infection, but Dr Kappe and his colleagues found a solution in a model known as FRG KO huHep.
To create the FRG KO huHep model, the team transplanted human hepatocytes in the severely immunocompromised FRG KO mouse, which has deletions in fumarylacetoacetate hydrolase (FAH), recombinationactivating gene 2 (Rag2), and interleukin-2 receptor subunit gamma (Il2rg).
The researchers found that FRG KO huHep mice support P vivax sporozoite infection, liver-stage development, and the formation and persistence of dormant liver-stage parasites.
In addition, experiments with this model revealed that the antimalarial drug primaquine could prevent and eliminate liver-stage infection.
The researchers therefore concluded that P vivax-infected FRG KO huHep mice are a suitable model for studying liver-stage development and dormancy and may facilitate the discovery of drugs targeting relapsing malaria.
“This model is a real game-changer,” said Ivo Mueller, PhD, a malaria expert at the Walter and Eliza Hall Institute in Melbourne, Victoria, Australia, who was not involved in this research.
“For the first time, we now have a realistic opportunity that not only allows us to directly study this normally hidden P vivax life-stage, but also to test new drugs and vaccines. This new model is an essential resource in our quest to develop the new anti-vivax interventions that we will need to eliminate P vivax malaria.” ![]()
Plasmodium vivax infection
Image by Mae Melvin
Researchers say they have developed a human-chimeric mouse model that can advance the study of the malaria parasite Plasmodium vivax.
The model is engineered to grow human livers that can be infected with P vivax and used for investigations into parasite development, dormancy, and activation, as well as the effect certain drugs have on each aspect of the infection.
The researchers described this model in Cell Host & Microbe.
Stefan Kappe, PhD, of Seattle Biomedical Research Institute in Washington, and his colleagues noted that P vivax malaria parasites are more resistant to control and elimination than other malaria parasites because, after infection, they fall dormant within the liver for months to years.
When these dormant parasites eventually become activated, they replicate, infect the bloodstream, and cause relapsing malaria.
The lack of tractable P vivax animal models has made it difficult to examine P vivax liver-stage infection, but Dr Kappe and his colleagues found a solution in a model known as FRG KO huHep.
To create the FRG KO huHep model, the team transplanted human hepatocytes in the severely immunocompromised FRG KO mouse, which has deletions in fumarylacetoacetate hydrolase (FAH), recombinationactivating gene 2 (Rag2), and interleukin-2 receptor subunit gamma (Il2rg).
The researchers found that FRG KO huHep mice support P vivax sporozoite infection, liver-stage development, and the formation and persistence of dormant liver-stage parasites.
In addition, experiments with this model revealed that the antimalarial drug primaquine could prevent and eliminate liver-stage infection.
The researchers therefore concluded that P vivax-infected FRG KO huHep mice are a suitable model for studying liver-stage development and dormancy and may facilitate the discovery of drugs targeting relapsing malaria.
“This model is a real game-changer,” said Ivo Mueller, PhD, a malaria expert at the Walter and Eliza Hall Institute in Melbourne, Victoria, Australia, who was not involved in this research.
“For the first time, we now have a realistic opportunity that not only allows us to directly study this normally hidden P vivax life-stage, but also to test new drugs and vaccines. This new model is an essential resource in our quest to develop the new anti-vivax interventions that we will need to eliminate P vivax malaria.” ![]()