User login
The Daily Safety Brief in a Safety Net Hospital: Development and Outcomes
From the MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe the process for the creation and development of the Daily Safety Brief (DSB) in our safety net hospital.
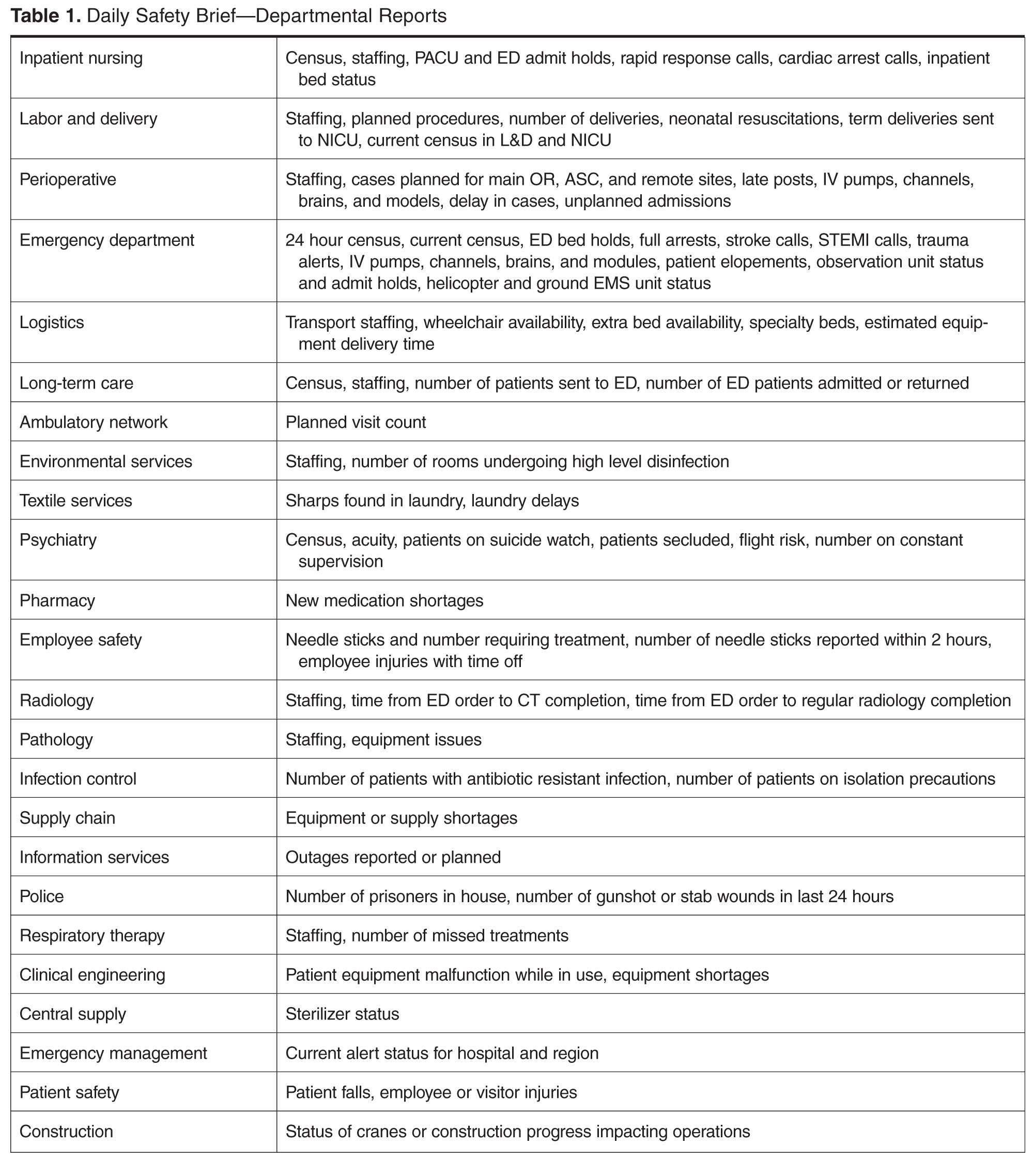
- Methods: We developed the DSB, a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis. Reporting issues while they are small oftentimes makes them easier to manage. The average call length with 25 departments reporting is just 9.5 minutes.
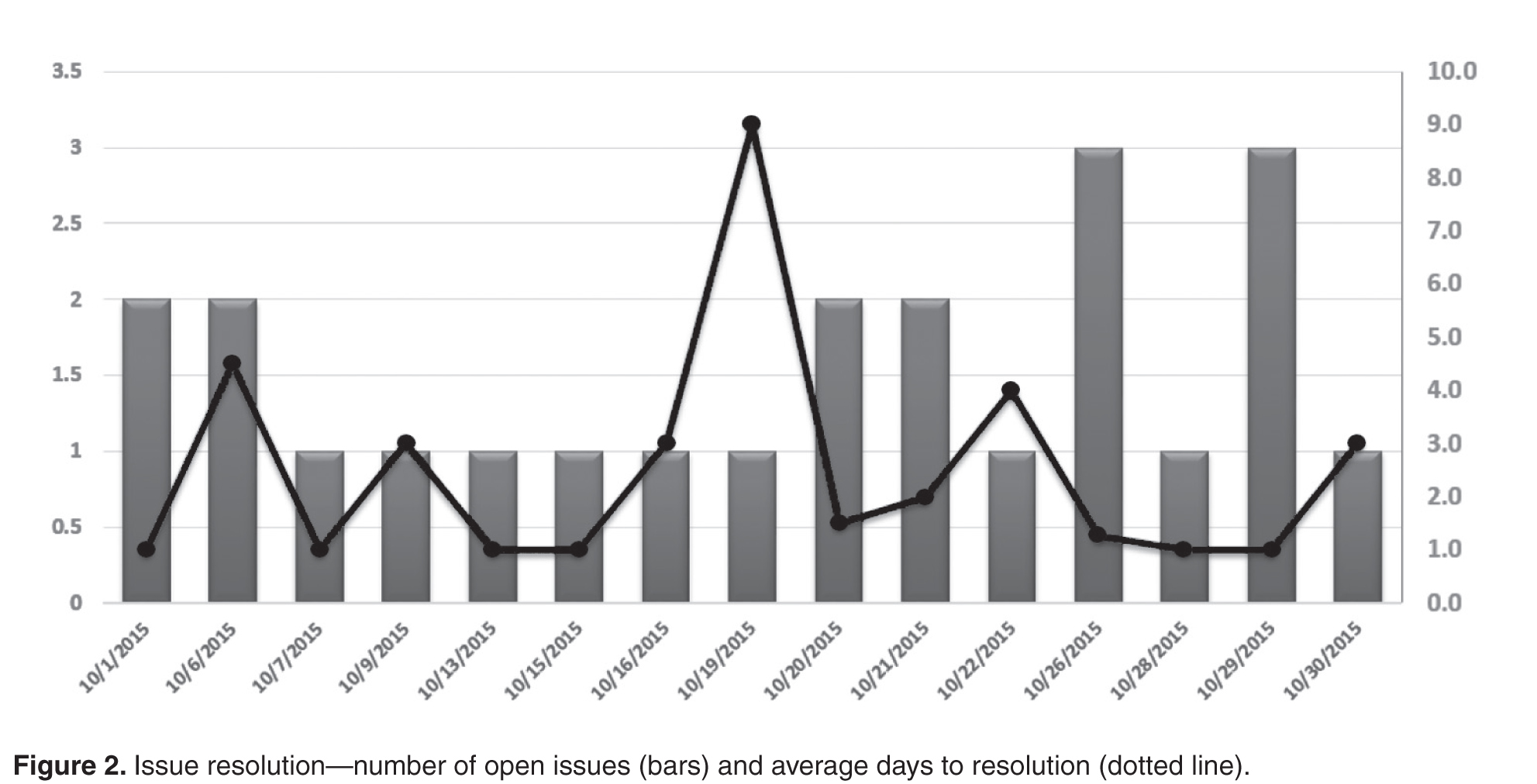
- Results: Survey results reveal an overall average improvement in awareness among DSB participants about hospital safety issues. Average days to issue resolution is currently 2.3 days, with open issues tracked and reported on daily.
- Conclusion: The DSB has improved real-time communication and awareness about safety issues in our organization.
As health care organizations strive to ensure a culture of safety for patients and staff, they must also be able to demonstrate reliability in that culture. The concept of highly reliable organizations originated in aviation and military fields due to the high-stakes environment and need for rapid and effective communication across departments. High reliability in health care organizations is described by the Joint Commission as consistent excellence in quality and safety for every patient, every time [1].
Highly reliable organizations put systems in place that makes them resilient with methods that lead to consistent accomplishment of goals and strategies to avoid potentially catastrophic errors [2]. An integral component to success in all high reliability organizations is a method of “Plan-of-the-Day” meetings to keep staff apprised of critical updates throughout the health system impacting care delivery [3]. Leaders at MetroHealth Medical Center believed that a daily safety briefing would help support the hospital’s journey to high reliability. We developed the Daily Safety Brief (DSB), a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis [4]. Reporting issues while they are small oftentimes makes them easier to manage. This article will describe the development and implementation of the DSB in our hospital.
Setting
MetroHealth Medical Center is an academic medical center in Cleveland, OH, affiliated with Case Western Reserve University. Metrohealth is a public safety net hospital with 731 licensed beds and a total of 1,160,773 patient visits in 2014, with 27,933 inpatient stays and 106,000 emergency department (ED) visits. The staff includes 507 physicians, 374 resident physicians, and 1222 nurses.
Program Development
As Metrohealth was contemplating the DSB, a group of senior leaders, including the chief medical officer, visited the Cincinnati Children’s Hospital, which had a DSB process in place. Following that visit, a larger group of physicians and administrators from intake points, procedural areas, and ancillary departments were invited to listen in live to Cincinnati’s DSB. This turned out to be a pivotal step in gaining buy-in. The initial concerns from participants were that this would be another scheduled meeting in an already busy day. What we learned from listening in was that the DSB was conducted in a manner that was succinct and professional. Issues were identified without accusations or unrelated agendas. Following the call, participants discussed how impressed they were and clearly saw the value of the information that was shared. They began to brainstorm about what they could report that would be relevant to the audience.
It was determined that a leader and 2 facilitators would be assigned to each call. The role of the DSB leader is to trigger individual department report outs and to ensure follow-up on unresolved safety issues from the previous DSB. Leaders are recruited by senior leadership and need to be familiar with the effects that issues can have across the health care system. Leaders need to be able to ask pertinent questions, have the credibility to raise concerns, and have access to senior administration when they need to bypass usual administrative channels.
The role of the facilitators, who are all members of the Center for Quality, is to connect to the conference bridge line, to keep the DSB leader on task, and to record all departmental data and pertinent details of the DSB. The facilitators maintain the daily DSB document, which outlines the order in which departments are called to report and identifies for the leader any open items identified in the previous day’s DSB.
The Daily Safety Brief
Rollout
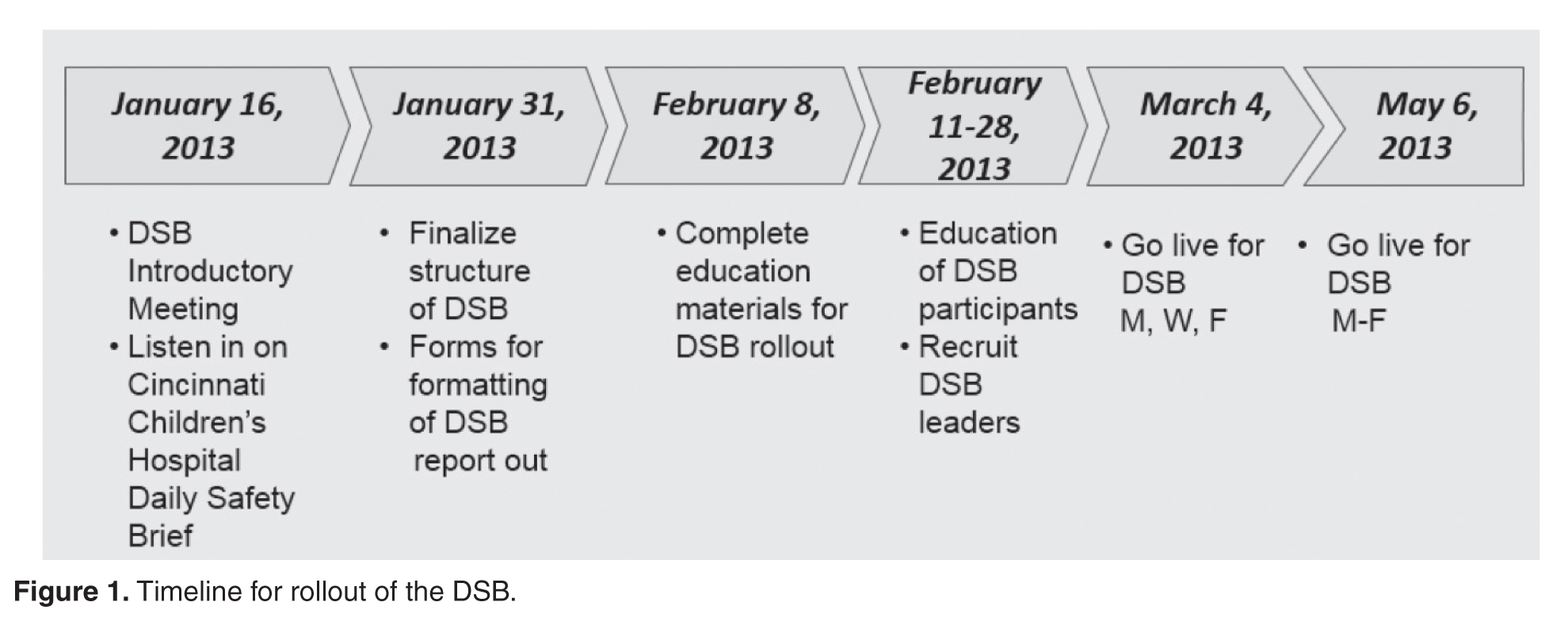
The DSB began 3 days per week on Monday, Wednesday and Friday at 0830. The time was moved to 0800 since participants found the later time difficult as it fell in the middle of an hour, potentially conflicting with other meetings and preparation for the daily bed huddle. We recognized that many meetings began right at the start of the DSB. The CEO requested that all 0800 meetings begin with a call in to listen to the DSB. After 2 months, the frequency was increased to 5 days per week, Monday through Friday. The hospital trialed a weekend DSB, however, feedback from participants found this extremely difficult to attend due to leaner weekend staffing models and found that information shared was not impactful. In particular, items were identified on the weekend daily safety briefs but the staff needed to resolve those items were generally not available until Monday.
Refinements
Coaching occurred to help people be more succinct in sharing information that would impact other areas. Information that was relevant only internally to their department was streamlined. The participants were counseled to identify items that had potential impact on other departments or where other departments had resources that might improve operations.
After a year, participating departments requested the addition of the logistics and construction departments to the DSB. The addition of the logistics department offered the opportunity for clinical departments to communicate what equipment was needed to start the day and created the opportunity for logistics to close the feedback loop by giving an estimate on expected time of arrival of equipment. The addition of the construction department helped communicate issues that may impact the organization, and helps to coordinate care to minimally impact patients and operations.
Examples of Safety Improvements
The DSB keeps the departmental leadership aware of problems developing in all areas of the hospital. Upcoming safety risks are identified early so that plans can be put in place to ameliorate them. The expectation of the DSB leader is that a problem that isn’t readily solved during the DSB must be taken to senior administration for resolution. As an example, an issue involving delays in the purchase of a required neonatal ventilator was taken directly to the CEO by the DSB leader, resulting in completion of the purchase within days. Importantly, the requirement to report at the DSB leads to a preoccupation with risk and reporting and leads to transparency among interdependent departments.
Another issue effectively addressed by the DSB was when we received notification of a required mandatory power shutdown for an extended period of time. The local power company informed our facilities management department director that they discovered issues requiring urgent replacement of the transformer within 2 weeks. Facilities management reported this in the morning DSB. The DSB leader requested all stakeholders to stay on the call following completion of the DSB, and plans were set in motion to plan for the shutdown of power. The team agreed to conference call again at noon the same day to continue planning, and the affected building was prepared for the shutdown by the following day.
Another benefit of the DSB is illustrated by our inpatient psychiatry unit, which reports an acuity measure each day on a scale of 1 to 10. The MetroHealth Police Department utilizes the report to adjust their rounding schedule, with increased presence on days with high acuity, which has led to an improvement in morale among psychiatry unit staff.
Challenges and Solutions
Since these reports are available to a wide audience in the organization, it is important to assure the reporters that no repercussions will ensue from any information that they provide. Senior leadership was enlisted to communicate with their departments that no repercussions would occur from reporting. As an example, some managers reported to the DSB development team privately that their supervisors were concerned about reporting of staff shortages on the DSB. As the shortages had patient care implications and affected other clinical departments, the DSB development team met with the involved supervisors to address the need for open reporting. In fact, repeated reporting of shortages in one support department on the DSB resulted in that issue being taken to high levels of administration leading to an increase in their staffing levels.
Scheduling can be a challenge for DSB participants. Holding the DSB at 0800 has led some departments to delegate the reporting or information gathering. For the individual reporting departments, creating a reporting workflow was a challenge. The departments needed to ensure that their DSB report was ready to go by 0800. This timeline forced departments to improve their own interdepartmental communication structure. An unexpected benefit of this requirement is that some departments have created a morning huddle to share information, which has reportedly improved communication and morale. The ambulatory network created a separate shared database for clinics to post concerns meeting DSB reporting criteria. One designated staff member would access this collective information when preparing for the DSB report. While most departments have a senior manager providing their report, this is not a requirement. In many departments, that reporter varies from day to day, although consistently it is someone with some administrative or leadership role in the department.
Conference call technology presented the solution to the problem of acquiring a meeting space for a large group. The DSB is broadcast from one physical location, where the facilitators and leader convene. While this conference room is open to anyone who wants to attend in person, most departments choose to participate through the conference line. The DSB conference call is open to anyone in the organization to access. Typically 35 to 40 phones are accessing the line each DSB. Challenges included callers not muting their phones, creating distracting background noise, and callers placing their phones on hold, which prompted the hospital hold message to play continuously. Multiple repeated reminders via email and at the start of the DSB has rectified this issue for the most part, with occasional reminders made when the issue recurs.
Data Management
Initially, an Excel file was created with columns for each reporting department as well as each item they were asked to report on. This “running” file became cumbersome. Retrieving information on past issues was not automated. Therefore, we enlisted the help of a data analyst to create an Access database. When it was complete, this new database allowed us to save information by individual dates, query number of days to issue resolution, and create reports noting unresolved issues for the leader to reference. Many data points can be queried in the access database. Real-time reports are available at all times and updated with every data entry. The database is able to identify departments not on the daily call and trend information, ie, how many listeners were on the DSB, number of falls, forensic patients in house, number of patients awaiting admission from the ED, number of ambulatory visits scheduled each day, equipment needed, number of cardiac arrest calls, and number of neonatal resuscitations.
At the conclusion of the call, the DSB report is completed and posted to a shared website on the hospital intranet for the entire hospital to access and read. Feedback from participant indicated that they found it cumbersome to access this. The communications department was enlisted to enable easy access and staff can now access the DSB report from the front page of the hospital intranet.
Outcomes
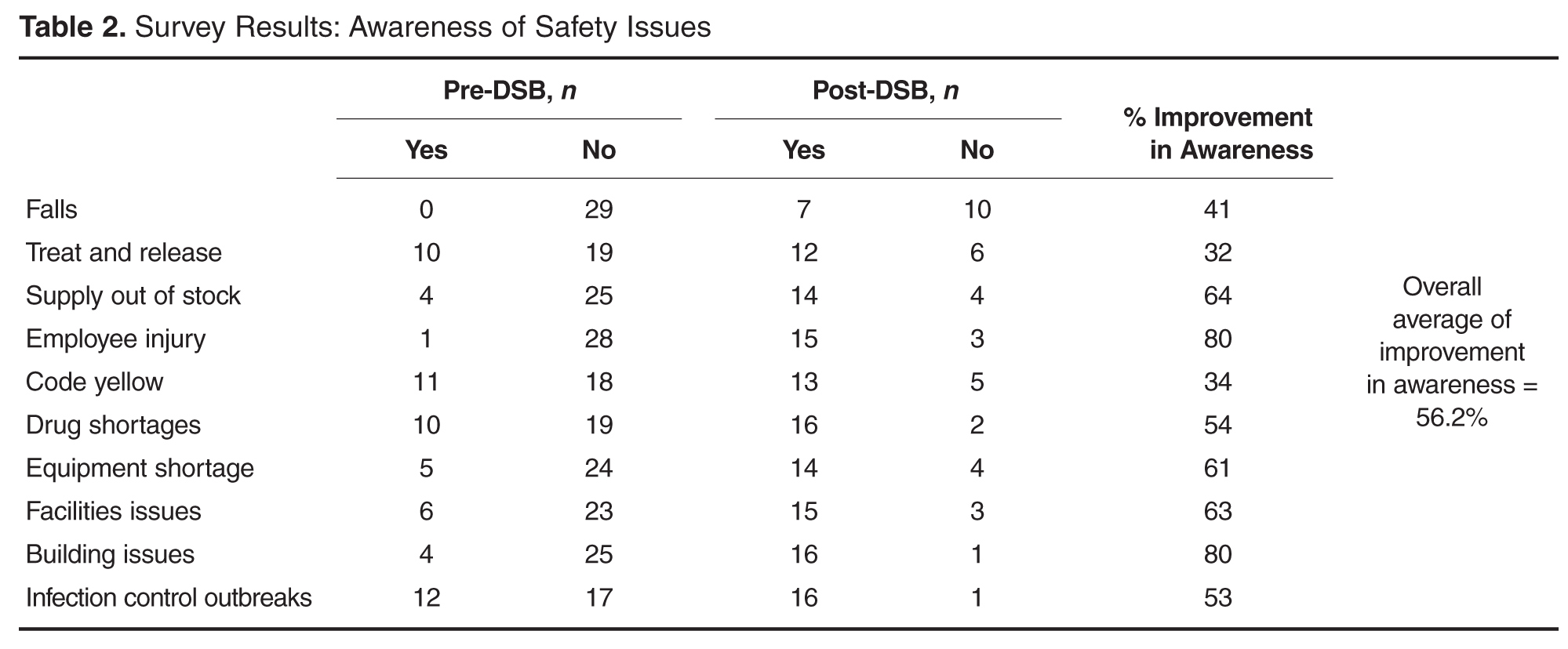
Since initiation of our DSB, we have tracked the average number of minutes spent on each call. When calls began, the average time on the call was 12.4 minutes. With the evolution of the DSB and coaching managers in various departments, the average time on the call is now 9.5 minutes in 2015, despite additional reporting departments joining the DSB.
Summary
The DSB has become an important tool in creating and moving towards a culture of safety and high reliability within the MetroHealth System. Over time, processes have become organized and engrained in all departments. This format has allowed issues to be brought forward timely where immediate attention can be given to achieve resolution in a nonthreatening manner, improving transparency. The fluidity of the DSB allows it to be enhanced and modified as improvements and opportunities are identified in the organization. The DSB has provided opportunities to create situational awareness which allows a look forward to prevention and creates a proactive environment. The results of these efforts has made MetroHealth a safer place for patients, visitors, and employees.
Corresponding author: Anne M. Aulisio, MSN, [email protected].
Financial disclosures: None.
1. Joint Commission Center for Transforming Healthcare. Available at www.centerfortransforminghealthcare.org.
2. Gamble M. 5 traits of high reliability organizations: how to hardwire each in your organization. Becker’s Hospital Review 29 Apr 2013. Accessed at www.beckershospitalreview.com/hospital-management-administration/5-traits-of-high-reliability-organizations-how-to-hardwire-each-in-your-organization.html.
3. Stockmeier C, Clapper C. Daily check-in for safety: from best practice to common practice. Patient Safety Qual Healthcare 2011:23. Accessed at psqh.com/daily-check-in-for-safety-from-best-practice-to-common-practice.
4. Creating situational awareness: a systems approach. In: Institute of Medicine (US) Forum on Medical and Public Health Preparedness for Catastrophic Events. Medical surge capacity: workshop summary. Washington, DC: National Academies Press; 2010. Accessed at www.ncbi.nlm.nih.gov/books/NBK32859/.
5. TeamSTEPPS. Available at www.ahrq.gov/professionals/education/curriculum-tools/teamstepps/index.html.
From the MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe the process for the creation and development of the Daily Safety Brief (DSB) in our safety net hospital.
- Methods: We developed the DSB, a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis. Reporting issues while they are small oftentimes makes them easier to manage. The average call length with 25 departments reporting is just 9.5 minutes.
- Results: Survey results reveal an overall average improvement in awareness among DSB participants about hospital safety issues. Average days to issue resolution is currently 2.3 days, with open issues tracked and reported on daily.
- Conclusion: The DSB has improved real-time communication and awareness about safety issues in our organization.
As health care organizations strive to ensure a culture of safety for patients and staff, they must also be able to demonstrate reliability in that culture. The concept of highly reliable organizations originated in aviation and military fields due to the high-stakes environment and need for rapid and effective communication across departments. High reliability in health care organizations is described by the Joint Commission as consistent excellence in quality and safety for every patient, every time [1].
Highly reliable organizations put systems in place that makes them resilient with methods that lead to consistent accomplishment of goals and strategies to avoid potentially catastrophic errors [2]. An integral component to success in all high reliability organizations is a method of “Plan-of-the-Day” meetings to keep staff apprised of critical updates throughout the health system impacting care delivery [3]. Leaders at MetroHealth Medical Center believed that a daily safety briefing would help support the hospital’s journey to high reliability. We developed the Daily Safety Brief (DSB), a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis [4]. Reporting issues while they are small oftentimes makes them easier to manage. This article will describe the development and implementation of the DSB in our hospital.
Setting
MetroHealth Medical Center is an academic medical center in Cleveland, OH, affiliated with Case Western Reserve University. Metrohealth is a public safety net hospital with 731 licensed beds and a total of 1,160,773 patient visits in 2014, with 27,933 inpatient stays and 106,000 emergency department (ED) visits. The staff includes 507 physicians, 374 resident physicians, and 1222 nurses.
Program Development
As Metrohealth was contemplating the DSB, a group of senior leaders, including the chief medical officer, visited the Cincinnati Children’s Hospital, which had a DSB process in place. Following that visit, a larger group of physicians and administrators from intake points, procedural areas, and ancillary departments were invited to listen in live to Cincinnati’s DSB. This turned out to be a pivotal step in gaining buy-in. The initial concerns from participants were that this would be another scheduled meeting in an already busy day. What we learned from listening in was that the DSB was conducted in a manner that was succinct and professional. Issues were identified without accusations or unrelated agendas. Following the call, participants discussed how impressed they were and clearly saw the value of the information that was shared. They began to brainstorm about what they could report that would be relevant to the audience.
It was determined that a leader and 2 facilitators would be assigned to each call. The role of the DSB leader is to trigger individual department report outs and to ensure follow-up on unresolved safety issues from the previous DSB. Leaders are recruited by senior leadership and need to be familiar with the effects that issues can have across the health care system. Leaders need to be able to ask pertinent questions, have the credibility to raise concerns, and have access to senior administration when they need to bypass usual administrative channels.
The role of the facilitators, who are all members of the Center for Quality, is to connect to the conference bridge line, to keep the DSB leader on task, and to record all departmental data and pertinent details of the DSB. The facilitators maintain the daily DSB document, which outlines the order in which departments are called to report and identifies for the leader any open items identified in the previous day’s DSB.
The Daily Safety Brief
Rollout
The DSB began 3 days per week on Monday, Wednesday and Friday at 0830. The time was moved to 0800 since participants found the later time difficult as it fell in the middle of an hour, potentially conflicting with other meetings and preparation for the daily bed huddle. We recognized that many meetings began right at the start of the DSB. The CEO requested that all 0800 meetings begin with a call in to listen to the DSB. After 2 months, the frequency was increased to 5 days per week, Monday through Friday. The hospital trialed a weekend DSB, however, feedback from participants found this extremely difficult to attend due to leaner weekend staffing models and found that information shared was not impactful. In particular, items were identified on the weekend daily safety briefs but the staff needed to resolve those items were generally not available until Monday.
Refinements
Coaching occurred to help people be more succinct in sharing information that would impact other areas. Information that was relevant only internally to their department was streamlined. The participants were counseled to identify items that had potential impact on other departments or where other departments had resources that might improve operations.
After a year, participating departments requested the addition of the logistics and construction departments to the DSB. The addition of the logistics department offered the opportunity for clinical departments to communicate what equipment was needed to start the day and created the opportunity for logistics to close the feedback loop by giving an estimate on expected time of arrival of equipment. The addition of the construction department helped communicate issues that may impact the organization, and helps to coordinate care to minimally impact patients and operations.
Examples of Safety Improvements
The DSB keeps the departmental leadership aware of problems developing in all areas of the hospital. Upcoming safety risks are identified early so that plans can be put in place to ameliorate them. The expectation of the DSB leader is that a problem that isn’t readily solved during the DSB must be taken to senior administration for resolution. As an example, an issue involving delays in the purchase of a required neonatal ventilator was taken directly to the CEO by the DSB leader, resulting in completion of the purchase within days. Importantly, the requirement to report at the DSB leads to a preoccupation with risk and reporting and leads to transparency among interdependent departments.
Another issue effectively addressed by the DSB was when we received notification of a required mandatory power shutdown for an extended period of time. The local power company informed our facilities management department director that they discovered issues requiring urgent replacement of the transformer within 2 weeks. Facilities management reported this in the morning DSB. The DSB leader requested all stakeholders to stay on the call following completion of the DSB, and plans were set in motion to plan for the shutdown of power. The team agreed to conference call again at noon the same day to continue planning, and the affected building was prepared for the shutdown by the following day.
Another benefit of the DSB is illustrated by our inpatient psychiatry unit, which reports an acuity measure each day on a scale of 1 to 10. The MetroHealth Police Department utilizes the report to adjust their rounding schedule, with increased presence on days with high acuity, which has led to an improvement in morale among psychiatry unit staff.
Challenges and Solutions
Since these reports are available to a wide audience in the organization, it is important to assure the reporters that no repercussions will ensue from any information that they provide. Senior leadership was enlisted to communicate with their departments that no repercussions would occur from reporting. As an example, some managers reported to the DSB development team privately that their supervisors were concerned about reporting of staff shortages on the DSB. As the shortages had patient care implications and affected other clinical departments, the DSB development team met with the involved supervisors to address the need for open reporting. In fact, repeated reporting of shortages in one support department on the DSB resulted in that issue being taken to high levels of administration leading to an increase in their staffing levels.
Scheduling can be a challenge for DSB participants. Holding the DSB at 0800 has led some departments to delegate the reporting or information gathering. For the individual reporting departments, creating a reporting workflow was a challenge. The departments needed to ensure that their DSB report was ready to go by 0800. This timeline forced departments to improve their own interdepartmental communication structure. An unexpected benefit of this requirement is that some departments have created a morning huddle to share information, which has reportedly improved communication and morale. The ambulatory network created a separate shared database for clinics to post concerns meeting DSB reporting criteria. One designated staff member would access this collective information when preparing for the DSB report. While most departments have a senior manager providing their report, this is not a requirement. In many departments, that reporter varies from day to day, although consistently it is someone with some administrative or leadership role in the department.
Conference call technology presented the solution to the problem of acquiring a meeting space for a large group. The DSB is broadcast from one physical location, where the facilitators and leader convene. While this conference room is open to anyone who wants to attend in person, most departments choose to participate through the conference line. The DSB conference call is open to anyone in the organization to access. Typically 35 to 40 phones are accessing the line each DSB. Challenges included callers not muting their phones, creating distracting background noise, and callers placing their phones on hold, which prompted the hospital hold message to play continuously. Multiple repeated reminders via email and at the start of the DSB has rectified this issue for the most part, with occasional reminders made when the issue recurs.
Data Management
Initially, an Excel file was created with columns for each reporting department as well as each item they were asked to report on. This “running” file became cumbersome. Retrieving information on past issues was not automated. Therefore, we enlisted the help of a data analyst to create an Access database. When it was complete, this new database allowed us to save information by individual dates, query number of days to issue resolution, and create reports noting unresolved issues for the leader to reference. Many data points can be queried in the access database. Real-time reports are available at all times and updated with every data entry. The database is able to identify departments not on the daily call and trend information, ie, how many listeners were on the DSB, number of falls, forensic patients in house, number of patients awaiting admission from the ED, number of ambulatory visits scheduled each day, equipment needed, number of cardiac arrest calls, and number of neonatal resuscitations.
At the conclusion of the call, the DSB report is completed and posted to a shared website on the hospital intranet for the entire hospital to access and read. Feedback from participant indicated that they found it cumbersome to access this. The communications department was enlisted to enable easy access and staff can now access the DSB report from the front page of the hospital intranet.
Outcomes
Since initiation of our DSB, we have tracked the average number of minutes spent on each call. When calls began, the average time on the call was 12.4 minutes. With the evolution of the DSB and coaching managers in various departments, the average time on the call is now 9.5 minutes in 2015, despite additional reporting departments joining the DSB.
Summary
The DSB has become an important tool in creating and moving towards a culture of safety and high reliability within the MetroHealth System. Over time, processes have become organized and engrained in all departments. This format has allowed issues to be brought forward timely where immediate attention can be given to achieve resolution in a nonthreatening manner, improving transparency. The fluidity of the DSB allows it to be enhanced and modified as improvements and opportunities are identified in the organization. The DSB has provided opportunities to create situational awareness which allows a look forward to prevention and creates a proactive environment. The results of these efforts has made MetroHealth a safer place for patients, visitors, and employees.
Corresponding author: Anne M. Aulisio, MSN, [email protected].
Financial disclosures: None.
From the MetroHealth Medical Center, Cleveland, OH.
Abstract
- Objective: To describe the process for the creation and development of the Daily Safety Brief (DSB) in our safety net hospital.
- Methods: We developed the DSB, a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis. Reporting issues while they are small oftentimes makes them easier to manage. The average call length with 25 departments reporting is just 9.5 minutes.
- Results: Survey results reveal an overall average improvement in awareness among DSB participants about hospital safety issues. Average days to issue resolution is currently 2.3 days, with open issues tracked and reported on daily.
- Conclusion: The DSB has improved real-time communication and awareness about safety issues in our organization.
As health care organizations strive to ensure a culture of safety for patients and staff, they must also be able to demonstrate reliability in that culture. The concept of highly reliable organizations originated in aviation and military fields due to the high-stakes environment and need for rapid and effective communication across departments. High reliability in health care organizations is described by the Joint Commission as consistent excellence in quality and safety for every patient, every time [1].
Highly reliable organizations put systems in place that makes them resilient with methods that lead to consistent accomplishment of goals and strategies to avoid potentially catastrophic errors [2]. An integral component to success in all high reliability organizations is a method of “Plan-of-the-Day” meetings to keep staff apprised of critical updates throughout the health system impacting care delivery [3]. Leaders at MetroHealth Medical Center believed that a daily safety briefing would help support the hospital’s journey to high reliability. We developed the Daily Safety Brief (DSB), a daily interdepartmental briefing intended to increase the safety of patients, employees, and visitors by improving communication and situational awareness. Situational awareness involves gathering the right information, analyzing it, and making predictions and projections based on the analysis [4]. Reporting issues while they are small oftentimes makes them easier to manage. This article will describe the development and implementation of the DSB in our hospital.
Setting
MetroHealth Medical Center is an academic medical center in Cleveland, OH, affiliated with Case Western Reserve University. Metrohealth is a public safety net hospital with 731 licensed beds and a total of 1,160,773 patient visits in 2014, with 27,933 inpatient stays and 106,000 emergency department (ED) visits. The staff includes 507 physicians, 374 resident physicians, and 1222 nurses.
Program Development
As Metrohealth was contemplating the DSB, a group of senior leaders, including the chief medical officer, visited the Cincinnati Children’s Hospital, which had a DSB process in place. Following that visit, a larger group of physicians and administrators from intake points, procedural areas, and ancillary departments were invited to listen in live to Cincinnati’s DSB. This turned out to be a pivotal step in gaining buy-in. The initial concerns from participants were that this would be another scheduled meeting in an already busy day. What we learned from listening in was that the DSB was conducted in a manner that was succinct and professional. Issues were identified without accusations or unrelated agendas. Following the call, participants discussed how impressed they were and clearly saw the value of the information that was shared. They began to brainstorm about what they could report that would be relevant to the audience.
It was determined that a leader and 2 facilitators would be assigned to each call. The role of the DSB leader is to trigger individual department report outs and to ensure follow-up on unresolved safety issues from the previous DSB. Leaders are recruited by senior leadership and need to be familiar with the effects that issues can have across the health care system. Leaders need to be able to ask pertinent questions, have the credibility to raise concerns, and have access to senior administration when they need to bypass usual administrative channels.
The role of the facilitators, who are all members of the Center for Quality, is to connect to the conference bridge line, to keep the DSB leader on task, and to record all departmental data and pertinent details of the DSB. The facilitators maintain the daily DSB document, which outlines the order in which departments are called to report and identifies for the leader any open items identified in the previous day’s DSB.
The Daily Safety Brief
Rollout
The DSB began 3 days per week on Monday, Wednesday and Friday at 0830. The time was moved to 0800 since participants found the later time difficult as it fell in the middle of an hour, potentially conflicting with other meetings and preparation for the daily bed huddle. We recognized that many meetings began right at the start of the DSB. The CEO requested that all 0800 meetings begin with a call in to listen to the DSB. After 2 months, the frequency was increased to 5 days per week, Monday through Friday. The hospital trialed a weekend DSB, however, feedback from participants found this extremely difficult to attend due to leaner weekend staffing models and found that information shared was not impactful. In particular, items were identified on the weekend daily safety briefs but the staff needed to resolve those items were generally not available until Monday.
Refinements
Coaching occurred to help people be more succinct in sharing information that would impact other areas. Information that was relevant only internally to their department was streamlined. The participants were counseled to identify items that had potential impact on other departments or where other departments had resources that might improve operations.
After a year, participating departments requested the addition of the logistics and construction departments to the DSB. The addition of the logistics department offered the opportunity for clinical departments to communicate what equipment was needed to start the day and created the opportunity for logistics to close the feedback loop by giving an estimate on expected time of arrival of equipment. The addition of the construction department helped communicate issues that may impact the organization, and helps to coordinate care to minimally impact patients and operations.
Examples of Safety Improvements
The DSB keeps the departmental leadership aware of problems developing in all areas of the hospital. Upcoming safety risks are identified early so that plans can be put in place to ameliorate them. The expectation of the DSB leader is that a problem that isn’t readily solved during the DSB must be taken to senior administration for resolution. As an example, an issue involving delays in the purchase of a required neonatal ventilator was taken directly to the CEO by the DSB leader, resulting in completion of the purchase within days. Importantly, the requirement to report at the DSB leads to a preoccupation with risk and reporting and leads to transparency among interdependent departments.
Another issue effectively addressed by the DSB was when we received notification of a required mandatory power shutdown for an extended period of time. The local power company informed our facilities management department director that they discovered issues requiring urgent replacement of the transformer within 2 weeks. Facilities management reported this in the morning DSB. The DSB leader requested all stakeholders to stay on the call following completion of the DSB, and plans were set in motion to plan for the shutdown of power. The team agreed to conference call again at noon the same day to continue planning, and the affected building was prepared for the shutdown by the following day.
Another benefit of the DSB is illustrated by our inpatient psychiatry unit, which reports an acuity measure each day on a scale of 1 to 10. The MetroHealth Police Department utilizes the report to adjust their rounding schedule, with increased presence on days with high acuity, which has led to an improvement in morale among psychiatry unit staff.
Challenges and Solutions
Since these reports are available to a wide audience in the organization, it is important to assure the reporters that no repercussions will ensue from any information that they provide. Senior leadership was enlisted to communicate with their departments that no repercussions would occur from reporting. As an example, some managers reported to the DSB development team privately that their supervisors were concerned about reporting of staff shortages on the DSB. As the shortages had patient care implications and affected other clinical departments, the DSB development team met with the involved supervisors to address the need for open reporting. In fact, repeated reporting of shortages in one support department on the DSB resulted in that issue being taken to high levels of administration leading to an increase in their staffing levels.
Scheduling can be a challenge for DSB participants. Holding the DSB at 0800 has led some departments to delegate the reporting or information gathering. For the individual reporting departments, creating a reporting workflow was a challenge. The departments needed to ensure that their DSB report was ready to go by 0800. This timeline forced departments to improve their own interdepartmental communication structure. An unexpected benefit of this requirement is that some departments have created a morning huddle to share information, which has reportedly improved communication and morale. The ambulatory network created a separate shared database for clinics to post concerns meeting DSB reporting criteria. One designated staff member would access this collective information when preparing for the DSB report. While most departments have a senior manager providing their report, this is not a requirement. In many departments, that reporter varies from day to day, although consistently it is someone with some administrative or leadership role in the department.
Conference call technology presented the solution to the problem of acquiring a meeting space for a large group. The DSB is broadcast from one physical location, where the facilitators and leader convene. While this conference room is open to anyone who wants to attend in person, most departments choose to participate through the conference line. The DSB conference call is open to anyone in the organization to access. Typically 35 to 40 phones are accessing the line each DSB. Challenges included callers not muting their phones, creating distracting background noise, and callers placing their phones on hold, which prompted the hospital hold message to play continuously. Multiple repeated reminders via email and at the start of the DSB has rectified this issue for the most part, with occasional reminders made when the issue recurs.
Data Management
Initially, an Excel file was created with columns for each reporting department as well as each item they were asked to report on. This “running” file became cumbersome. Retrieving information on past issues was not automated. Therefore, we enlisted the help of a data analyst to create an Access database. When it was complete, this new database allowed us to save information by individual dates, query number of days to issue resolution, and create reports noting unresolved issues for the leader to reference. Many data points can be queried in the access database. Real-time reports are available at all times and updated with every data entry. The database is able to identify departments not on the daily call and trend information, ie, how many listeners were on the DSB, number of falls, forensic patients in house, number of patients awaiting admission from the ED, number of ambulatory visits scheduled each day, equipment needed, number of cardiac arrest calls, and number of neonatal resuscitations.
At the conclusion of the call, the DSB report is completed and posted to a shared website on the hospital intranet for the entire hospital to access and read. Feedback from participant indicated that they found it cumbersome to access this. The communications department was enlisted to enable easy access and staff can now access the DSB report from the front page of the hospital intranet.
Outcomes
Since initiation of our DSB, we have tracked the average number of minutes spent on each call. When calls began, the average time on the call was 12.4 minutes. With the evolution of the DSB and coaching managers in various departments, the average time on the call is now 9.5 minutes in 2015, despite additional reporting departments joining the DSB.
Summary
The DSB has become an important tool in creating and moving towards a culture of safety and high reliability within the MetroHealth System. Over time, processes have become organized and engrained in all departments. This format has allowed issues to be brought forward timely where immediate attention can be given to achieve resolution in a nonthreatening manner, improving transparency. The fluidity of the DSB allows it to be enhanced and modified as improvements and opportunities are identified in the organization. The DSB has provided opportunities to create situational awareness which allows a look forward to prevention and creates a proactive environment. The results of these efforts has made MetroHealth a safer place for patients, visitors, and employees.
Corresponding author: Anne M. Aulisio, MSN, [email protected].
Financial disclosures: None.
1. Joint Commission Center for Transforming Healthcare. Available at www.centerfortransforminghealthcare.org.
2. Gamble M. 5 traits of high reliability organizations: how to hardwire each in your organization. Becker’s Hospital Review 29 Apr 2013. Accessed at www.beckershospitalreview.com/hospital-management-administration/5-traits-of-high-reliability-organizations-how-to-hardwire-each-in-your-organization.html.
3. Stockmeier C, Clapper C. Daily check-in for safety: from best practice to common practice. Patient Safety Qual Healthcare 2011:23. Accessed at psqh.com/daily-check-in-for-safety-from-best-practice-to-common-practice.
4. Creating situational awareness: a systems approach. In: Institute of Medicine (US) Forum on Medical and Public Health Preparedness for Catastrophic Events. Medical surge capacity: workshop summary. Washington, DC: National Academies Press; 2010. Accessed at www.ncbi.nlm.nih.gov/books/NBK32859/.
5. TeamSTEPPS. Available at www.ahrq.gov/professionals/education/curriculum-tools/teamstepps/index.html.
1. Joint Commission Center for Transforming Healthcare. Available at www.centerfortransforminghealthcare.org.
2. Gamble M. 5 traits of high reliability organizations: how to hardwire each in your organization. Becker’s Hospital Review 29 Apr 2013. Accessed at www.beckershospitalreview.com/hospital-management-administration/5-traits-of-high-reliability-organizations-how-to-hardwire-each-in-your-organization.html.
3. Stockmeier C, Clapper C. Daily check-in for safety: from best practice to common practice. Patient Safety Qual Healthcare 2011:23. Accessed at psqh.com/daily-check-in-for-safety-from-best-practice-to-common-practice.
4. Creating situational awareness: a systems approach. In: Institute of Medicine (US) Forum on Medical and Public Health Preparedness for Catastrophic Events. Medical surge capacity: workshop summary. Washington, DC: National Academies Press; 2010. Accessed at www.ncbi.nlm.nih.gov/books/NBK32859/.
5. TeamSTEPPS. Available at www.ahrq.gov/professionals/education/curriculum-tools/teamstepps/index.html.
High-Dose Vitamin D Supplementation May Lead to Increased Risk of Falls
Study Overview
Objective. To determine the effectiveness of high-dose vitamin D versus low-dose vitamin D in reducing the risk of functional decline in older adults.
Design. Double-blind randomized controlled trial.
Setting and participants. This single-center study was conducted at the University of Zurich. Home-dwelling adults aged 70 and over were recruited through newspaper advertisement in Zurich from December 2009 to May 2010. Inclusion criteria included maintenance of mobility with or without a walking aid, having the ability to use public transportation to attend clinic visits, and scoring at least 27 on the Mini-Mental State Examination. Exclusion criteria include supplemental vitamin D use exceeding 800 IU per day and unwillingness to discontinue additional calcium and vitamin D supplementation, current cancer, malabsorption syndrome, heavy alcohol consumption, uncontrolled hypocalcemia, severe visual or hearing impairment, use of medications affecting calcium metabolism, diseases causing hypercalcemia, planned travel to sunny locations for longer than 2 months per year, maximum calcium supplement dose of 250 mg/day, use of medications affecting serum 25-hydroxyvitamin D (25[OH]D) level, body mass index ≥ 40, diseases predisposing to falls, hypercalcemia, kidney disease with creatinine clearance < 15, or kidney stone within 10 years prior to enrollment.
Intervention. Participants were randomized to receive either monthly supplementation of 24,000 IU of vitamin D3 per month (low-dose group), 60,000 IU of vitamin D3 once per month (high-dose group), or 24,000 IU of vitamin D3 plus 300 µg of calcifediol once per month. It was hypothesized that higher monthly doses of vitamin D or in combination with calcifediol, which is a liver metabolite approximately 2 to 3 times more potent than vitamin D3, will increase levels of 25(OH)D and reduce the risk of functional decline.
Main outcome measures. Lower extremity function using the Short Physical Performance Battery and 25(OH)D levels at 6 and 12 months. Study nurses called participants monthly to assess falls, adverse events, and adherence to study medications.
Main results. A total of 200 participants were enrolled. Average age was 78 years (SD = 5) and 67% were female; all had a history of falls in the previous year and average baseline 25(OH)D levels ranged from 18.4 to 20.9 ng/mL in the three groups. Adherence to the study medication exceeded 94% throughout the study trial in all treatment groups.
At 6 and 12 months, 25(OH)D levels increased by an average of 12.7 and 11.7 ng/mL in the low-dose group, an average of 18.3 and 19.2 ng/mL in the high-dose group, and an average of 27.6 and 25.8 ng/mL in the calcifediol-added group. The mean changes in physical performance score indicating lower extremity function did not differ significantly among treatment groups (P = 0.26), but for one measure—the 5 successive chair stands—the 2 high-dose groups had less improvement when compared with the low-dose group. At 12 months, 66.9% of the high-dose group and 66.1% in the group with calcifediol fell during the study period, which was more than the low-dose group (47.9%, P = 0.048). The mean number of falls was also higher among the high-dose and calcifediol groups when compared with the low-dose group.
Conclusion. Higher doses of vitamin D were not better than lower doses of vitamin D in improving lower extremity function and were associated with higher risk of falls.
Commentary
Vitamin D deficiency is common among older adults and is associated with sarcopenia, functional decline, falls, and fractures [1,2]. Prior meta-analysis has supported that supplementation with vitamin D may lead to improved outcomes in fracture prevention [3]. However, the US Preventive Services Task Force, using more recent evidence reviews and an updated meta-analysis [4], found evidence lacking regarding the benefit of supplementation with vitamin D in community-dwelling postmenopausal women at doses > 400 IU, found no benefit in this group for doses ≤ 400, and found evidence lacking for supplementation in men or premenopausal women at any dose [5]. At the same time, the USPSTF also recommends exercise or physical therapy and vitamin D supplementation (800 IU daily) to prevent falls in community-dwelling adults ≥ 65 years at increased risk for falls [6]. This is consistent with the Institute of Medicine’s recommendation of 800 IU per day for older adults [7].
The current study attempted to elucidate the potential impact of high-dose vitamin D supplementation, hypothesizing that higher doses will achieve improvement in vitamin D levels and better outcomes in terms of lower extremity function and falls. However, the investigators found that rather than lowering risk of falls, higher-dose vitamin D was associated with elevated risk of falls without the benefit of improving lower extremity function. This is not the first study that has demonstrated that higher doses of vitamin D supplementation may be associated with harm. A prior randomized controlled trial utilizing a different dosing strategy of annual high- dose vitamin D supplementation also found that higher doses were associated with increased risks of falls [8]. Nonetheless, it helps support the notion that in vitamin D supplementation, more is not necessarily better.
The study is not without its drawbacks. The sample size was relatively small and the trial may have been underpowered to detect whether there may be certain patients for whom high-dose vitamin D supplementation may have a role. Also, the study was based in Zurich, which has a relatively uniform population, and study results may not be generalizable to populations in other countries.
Applications for Clinical Practice
The study lends support to the current recommendation of the Institute of Medicine—800 IU a day—for fall prevention, which is equivalent in dose to the 24,000 IU per month utilized in the trial. One of the questions not answered by the study is whether high-dose supplementation for adults who have severe deficiency in vitamin D is beneficial or harmful when compared with lower-dose supplementation. In clinical practice, clinicians often check an initial level of vitamin D and aim for a target level with supplementation. Among those patients with extremely low baseline levels, a lower-dose regimen of 800 IU a day may not yield a normalized level of vitamin D. Further studies are needed to elucidate whether there may be a role for higher-dose supplementation in these individuals. Nonetheless, it is clear that the current evidence does not support the routine use of high-dose vitamin D supplementation; it does not lead to better lower extremity function and may cause harm.
—William Hung, MD, MPH
1. Visser M, Deeg DJ, Lips P; Longitudinal Aging Study Amsterdam. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab 2003;88:5766–72.
2. Cauley JA, Lacroix AZ, Wu L, et al. Serum 25-hydroxy-vitamin D concentrations and risk for hip fractures. Ann Intern Med 2008;149:242–50.
3. Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Fracture prevention with vitamin D supplementation: a meta-analysis of randomized controlled trials. JAMA 2005;293:2257–64.
4. Chung M, Lee J, Terasawa T, et al. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:827–38.
5. Moyer VA, on behalf of the U.S. Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013;158:691–6.
6. Moyer VA et al. Prevention of falls in community-dwelling older adults: U.S. Prevention Services Task Force Recommendation statement. Ann Intern Med 2012; 157:197–204.
7. Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press; 2010.
8. Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010;303:1815.
Study Overview
Objective. To determine the effectiveness of high-dose vitamin D versus low-dose vitamin D in reducing the risk of functional decline in older adults.
Design. Double-blind randomized controlled trial.
Setting and participants. This single-center study was conducted at the University of Zurich. Home-dwelling adults aged 70 and over were recruited through newspaper advertisement in Zurich from December 2009 to May 2010. Inclusion criteria included maintenance of mobility with or without a walking aid, having the ability to use public transportation to attend clinic visits, and scoring at least 27 on the Mini-Mental State Examination. Exclusion criteria include supplemental vitamin D use exceeding 800 IU per day and unwillingness to discontinue additional calcium and vitamin D supplementation, current cancer, malabsorption syndrome, heavy alcohol consumption, uncontrolled hypocalcemia, severe visual or hearing impairment, use of medications affecting calcium metabolism, diseases causing hypercalcemia, planned travel to sunny locations for longer than 2 months per year, maximum calcium supplement dose of 250 mg/day, use of medications affecting serum 25-hydroxyvitamin D (25[OH]D) level, body mass index ≥ 40, diseases predisposing to falls, hypercalcemia, kidney disease with creatinine clearance < 15, or kidney stone within 10 years prior to enrollment.
Intervention. Participants were randomized to receive either monthly supplementation of 24,000 IU of vitamin D3 per month (low-dose group), 60,000 IU of vitamin D3 once per month (high-dose group), or 24,000 IU of vitamin D3 plus 300 µg of calcifediol once per month. It was hypothesized that higher monthly doses of vitamin D or in combination with calcifediol, which is a liver metabolite approximately 2 to 3 times more potent than vitamin D3, will increase levels of 25(OH)D and reduce the risk of functional decline.
Main outcome measures. Lower extremity function using the Short Physical Performance Battery and 25(OH)D levels at 6 and 12 months. Study nurses called participants monthly to assess falls, adverse events, and adherence to study medications.
Main results. A total of 200 participants were enrolled. Average age was 78 years (SD = 5) and 67% were female; all had a history of falls in the previous year and average baseline 25(OH)D levels ranged from 18.4 to 20.9 ng/mL in the three groups. Adherence to the study medication exceeded 94% throughout the study trial in all treatment groups.
At 6 and 12 months, 25(OH)D levels increased by an average of 12.7 and 11.7 ng/mL in the low-dose group, an average of 18.3 and 19.2 ng/mL in the high-dose group, and an average of 27.6 and 25.8 ng/mL in the calcifediol-added group. The mean changes in physical performance score indicating lower extremity function did not differ significantly among treatment groups (P = 0.26), but for one measure—the 5 successive chair stands—the 2 high-dose groups had less improvement when compared with the low-dose group. At 12 months, 66.9% of the high-dose group and 66.1% in the group with calcifediol fell during the study period, which was more than the low-dose group (47.9%, P = 0.048). The mean number of falls was also higher among the high-dose and calcifediol groups when compared with the low-dose group.
Conclusion. Higher doses of vitamin D were not better than lower doses of vitamin D in improving lower extremity function and were associated with higher risk of falls.
Commentary
Vitamin D deficiency is common among older adults and is associated with sarcopenia, functional decline, falls, and fractures [1,2]. Prior meta-analysis has supported that supplementation with vitamin D may lead to improved outcomes in fracture prevention [3]. However, the US Preventive Services Task Force, using more recent evidence reviews and an updated meta-analysis [4], found evidence lacking regarding the benefit of supplementation with vitamin D in community-dwelling postmenopausal women at doses > 400 IU, found no benefit in this group for doses ≤ 400, and found evidence lacking for supplementation in men or premenopausal women at any dose [5]. At the same time, the USPSTF also recommends exercise or physical therapy and vitamin D supplementation (800 IU daily) to prevent falls in community-dwelling adults ≥ 65 years at increased risk for falls [6]. This is consistent with the Institute of Medicine’s recommendation of 800 IU per day for older adults [7].
The current study attempted to elucidate the potential impact of high-dose vitamin D supplementation, hypothesizing that higher doses will achieve improvement in vitamin D levels and better outcomes in terms of lower extremity function and falls. However, the investigators found that rather than lowering risk of falls, higher-dose vitamin D was associated with elevated risk of falls without the benefit of improving lower extremity function. This is not the first study that has demonstrated that higher doses of vitamin D supplementation may be associated with harm. A prior randomized controlled trial utilizing a different dosing strategy of annual high- dose vitamin D supplementation also found that higher doses were associated with increased risks of falls [8]. Nonetheless, it helps support the notion that in vitamin D supplementation, more is not necessarily better.
The study is not without its drawbacks. The sample size was relatively small and the trial may have been underpowered to detect whether there may be certain patients for whom high-dose vitamin D supplementation may have a role. Also, the study was based in Zurich, which has a relatively uniform population, and study results may not be generalizable to populations in other countries.
Applications for Clinical Practice
The study lends support to the current recommendation of the Institute of Medicine—800 IU a day—for fall prevention, which is equivalent in dose to the 24,000 IU per month utilized in the trial. One of the questions not answered by the study is whether high-dose supplementation for adults who have severe deficiency in vitamin D is beneficial or harmful when compared with lower-dose supplementation. In clinical practice, clinicians often check an initial level of vitamin D and aim for a target level with supplementation. Among those patients with extremely low baseline levels, a lower-dose regimen of 800 IU a day may not yield a normalized level of vitamin D. Further studies are needed to elucidate whether there may be a role for higher-dose supplementation in these individuals. Nonetheless, it is clear that the current evidence does not support the routine use of high-dose vitamin D supplementation; it does not lead to better lower extremity function and may cause harm.
—William Hung, MD, MPH
Study Overview
Objective. To determine the effectiveness of high-dose vitamin D versus low-dose vitamin D in reducing the risk of functional decline in older adults.
Design. Double-blind randomized controlled trial.
Setting and participants. This single-center study was conducted at the University of Zurich. Home-dwelling adults aged 70 and over were recruited through newspaper advertisement in Zurich from December 2009 to May 2010. Inclusion criteria included maintenance of mobility with or without a walking aid, having the ability to use public transportation to attend clinic visits, and scoring at least 27 on the Mini-Mental State Examination. Exclusion criteria include supplemental vitamin D use exceeding 800 IU per day and unwillingness to discontinue additional calcium and vitamin D supplementation, current cancer, malabsorption syndrome, heavy alcohol consumption, uncontrolled hypocalcemia, severe visual or hearing impairment, use of medications affecting calcium metabolism, diseases causing hypercalcemia, planned travel to sunny locations for longer than 2 months per year, maximum calcium supplement dose of 250 mg/day, use of medications affecting serum 25-hydroxyvitamin D (25[OH]D) level, body mass index ≥ 40, diseases predisposing to falls, hypercalcemia, kidney disease with creatinine clearance < 15, or kidney stone within 10 years prior to enrollment.
Intervention. Participants were randomized to receive either monthly supplementation of 24,000 IU of vitamin D3 per month (low-dose group), 60,000 IU of vitamin D3 once per month (high-dose group), or 24,000 IU of vitamin D3 plus 300 µg of calcifediol once per month. It was hypothesized that higher monthly doses of vitamin D or in combination with calcifediol, which is a liver metabolite approximately 2 to 3 times more potent than vitamin D3, will increase levels of 25(OH)D and reduce the risk of functional decline.
Main outcome measures. Lower extremity function using the Short Physical Performance Battery and 25(OH)D levels at 6 and 12 months. Study nurses called participants monthly to assess falls, adverse events, and adherence to study medications.
Main results. A total of 200 participants were enrolled. Average age was 78 years (SD = 5) and 67% were female; all had a history of falls in the previous year and average baseline 25(OH)D levels ranged from 18.4 to 20.9 ng/mL in the three groups. Adherence to the study medication exceeded 94% throughout the study trial in all treatment groups.
At 6 and 12 months, 25(OH)D levels increased by an average of 12.7 and 11.7 ng/mL in the low-dose group, an average of 18.3 and 19.2 ng/mL in the high-dose group, and an average of 27.6 and 25.8 ng/mL in the calcifediol-added group. The mean changes in physical performance score indicating lower extremity function did not differ significantly among treatment groups (P = 0.26), but for one measure—the 5 successive chair stands—the 2 high-dose groups had less improvement when compared with the low-dose group. At 12 months, 66.9% of the high-dose group and 66.1% in the group with calcifediol fell during the study period, which was more than the low-dose group (47.9%, P = 0.048). The mean number of falls was also higher among the high-dose and calcifediol groups when compared with the low-dose group.
Conclusion. Higher doses of vitamin D were not better than lower doses of vitamin D in improving lower extremity function and were associated with higher risk of falls.
Commentary
Vitamin D deficiency is common among older adults and is associated with sarcopenia, functional decline, falls, and fractures [1,2]. Prior meta-analysis has supported that supplementation with vitamin D may lead to improved outcomes in fracture prevention [3]. However, the US Preventive Services Task Force, using more recent evidence reviews and an updated meta-analysis [4], found evidence lacking regarding the benefit of supplementation with vitamin D in community-dwelling postmenopausal women at doses > 400 IU, found no benefit in this group for doses ≤ 400, and found evidence lacking for supplementation in men or premenopausal women at any dose [5]. At the same time, the USPSTF also recommends exercise or physical therapy and vitamin D supplementation (800 IU daily) to prevent falls in community-dwelling adults ≥ 65 years at increased risk for falls [6]. This is consistent with the Institute of Medicine’s recommendation of 800 IU per day for older adults [7].
The current study attempted to elucidate the potential impact of high-dose vitamin D supplementation, hypothesizing that higher doses will achieve improvement in vitamin D levels and better outcomes in terms of lower extremity function and falls. However, the investigators found that rather than lowering risk of falls, higher-dose vitamin D was associated with elevated risk of falls without the benefit of improving lower extremity function. This is not the first study that has demonstrated that higher doses of vitamin D supplementation may be associated with harm. A prior randomized controlled trial utilizing a different dosing strategy of annual high- dose vitamin D supplementation also found that higher doses were associated with increased risks of falls [8]. Nonetheless, it helps support the notion that in vitamin D supplementation, more is not necessarily better.
The study is not without its drawbacks. The sample size was relatively small and the trial may have been underpowered to detect whether there may be certain patients for whom high-dose vitamin D supplementation may have a role. Also, the study was based in Zurich, which has a relatively uniform population, and study results may not be generalizable to populations in other countries.
Applications for Clinical Practice
The study lends support to the current recommendation of the Institute of Medicine—800 IU a day—for fall prevention, which is equivalent in dose to the 24,000 IU per month utilized in the trial. One of the questions not answered by the study is whether high-dose supplementation for adults who have severe deficiency in vitamin D is beneficial or harmful when compared with lower-dose supplementation. In clinical practice, clinicians often check an initial level of vitamin D and aim for a target level with supplementation. Among those patients with extremely low baseline levels, a lower-dose regimen of 800 IU a day may not yield a normalized level of vitamin D. Further studies are needed to elucidate whether there may be a role for higher-dose supplementation in these individuals. Nonetheless, it is clear that the current evidence does not support the routine use of high-dose vitamin D supplementation; it does not lead to better lower extremity function and may cause harm.
—William Hung, MD, MPH
1. Visser M, Deeg DJ, Lips P; Longitudinal Aging Study Amsterdam. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab 2003;88:5766–72.
2. Cauley JA, Lacroix AZ, Wu L, et al. Serum 25-hydroxy-vitamin D concentrations and risk for hip fractures. Ann Intern Med 2008;149:242–50.
3. Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Fracture prevention with vitamin D supplementation: a meta-analysis of randomized controlled trials. JAMA 2005;293:2257–64.
4. Chung M, Lee J, Terasawa T, et al. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:827–38.
5. Moyer VA, on behalf of the U.S. Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013;158:691–6.
6. Moyer VA et al. Prevention of falls in community-dwelling older adults: U.S. Prevention Services Task Force Recommendation statement. Ann Intern Med 2012; 157:197–204.
7. Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press; 2010.
8. Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010;303:1815.
1. Visser M, Deeg DJ, Lips P; Longitudinal Aging Study Amsterdam. Low vitamin D and high parathyroid hormone levels as determinants of loss of muscle strength and muscle mass (sarcopenia): the Longitudinal Aging Study Amsterdam. J Clin Endocrinol Metab 2003;88:5766–72.
2. Cauley JA, Lacroix AZ, Wu L, et al. Serum 25-hydroxy-vitamin D concentrations and risk for hip fractures. Ann Intern Med 2008;149:242–50.
3. Bischoff-Ferrari HA, Willett WC, Wong JB, et al. Fracture prevention with vitamin D supplementation: a meta-analysis of randomized controlled trials. JAMA 2005;293:2257–64.
4. Chung M, Lee J, Terasawa T, et al. Vitamin D with or without calcium supplementation for prevention of cancer and fractures: an updated meta-analysis for the U.S. Preventive Services Task Force. Ann Intern Med 2011;155:827–38.
5. Moyer VA, on behalf of the U.S. Preventive Services Task Force. Vitamin D and calcium supplementation to prevent fractures in adults: U.S. Preventive Services Task Force Recommendation Statement. Ann Intern Med 2013;158:691–6.
6. Moyer VA et al. Prevention of falls in community-dwelling older adults: U.S. Prevention Services Task Force Recommendation statement. Ann Intern Med 2012; 157:197–204.
7. Institute of Medicine. Dietary reference intakes for calcium and vitamin D. Washington, DC: National Academies Press; 2010.
8. Sanders KM, Stuart AL, Williamson EJ, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA 2010;303:1815.
The Role of Health Literacy and Patient Activation in Predicting Patient Health Information Seeking and Sharing
Study Overview
Objective. To assess how patients look for patient-obtained medication information (POMI) to prepare for a clinical appointment, whether they share those findings with their provider, and how health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI.
Design. Cross-sectional survey-based study.
Setting and participants. The study took place over 1 week at 2 academic medical centers located in Las Vegas, Nevada, and Washington, DC. At a central waiting area at each facility, patients aged 18 and older waiting for their clinical appointment were invited to complete a survey, either on a computer tablet or with paper and pencil, before and after their appointment.
Measures and analysis. The pre-survey included demographic measures (age, gender, education, and ethnicity), the reason for the visit (routine care, sick visit, follow-up after survey, and follow-up after emergency room visit), and an item to assess self-report of perceived general health (from poor to excellent). Health literacy was assessed by a self-report measure that included subscales for the 3 dimensions of health literacy: functional, communicative, and critical health literacy [1]; together, these capture the ability of patients to retain health knowledge, gather and communicate health concepts, and apply health information. Patient activation was scored using the Patient Activation Measure (13 Likert-style items, total scale range 0–100); patient activation combines a patient’s self-reported knowledge, skill, and confidence for self-management of general health or a chronic condition [2]. Information seeking was measured by time spent (did not look for information, 1 hour, 2 hours, 3 hours, or more than 3 hours), and information channels used to look for POMI (eg, magazines/newspapers, internet website or search engine) were presented dichotomously (yes/no).
The post-survey first asked whether the participant shared information with their provider (yes/no). If the participant said yes, 4 items assessed their perception of the provider’s response, including amount of time spent discussing POMI, how seriously the provider considered the information, and overall reaction (scored as a mean, each item measured from 1–5, with 5 indicating the most positive reactions). For hypothesis testing, logistic regression models were used to test the effects of the independent variables. To explore the relationship between health literacy/patient activation and physician response, correlations were calculated.
Main results. Over 400 patients were asked to participate, and of these a total of 243 (60.75%) patients were eligible, consented, and completed surveys. Participants were predominantly white (57.6%), female (63%), had some college education or higher (80.2%), and had a clinical appointment for routine care (69.3%). The mean age was 47.04 years (SD, 15.78), the mean health status was 3.20 (SD, 0.94), and the mean Patient Activation Measure was 72.43 (SD, 16.00).
More than half of participants (58.26%) who responded to the item about information seeking indicated seeking POMI prior to their clinical appointment. Of these, the majority (88.7%) reported using the internet, particularly WebMD, as an information channel. Significant predictors of information seeking included age (P = 0.01, OR = 0.973), communicative health literacy (P = 0.01, or = 1.975), and critical health literacy (P = 0.05, OR = 1.518). Lower age, higher communicative health literacy, and higher critical health literacy increased the likelihood of the patient seeking POMI prior to the clinical appointment. Other assessed predictors were not significant, including gender, functional health literacy, patient activation, reason for visit, and reported health status.
58.2% of the 141 information-seeking patients talked to their health care provider about the information they found. However, no predictor variables included in a logistic regression analysis were significant, including age, gender, reason for visit, reported health status, functional health literacy, communicative health literacy, critical health literacy, and patient activation. For the research question (how do health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI), the mean score on the 4-item measure was 4.08 (SD, 0.90), indicating a generally positive response; most reported the physician response was good or higher. Patient activation correlated positively with perceived physician response (r = 0.245, P = 0.03).
Conclusion. The lack of data to predict who will introduce POMI at the medical visit is disconcerting. Providers might consider directly asking or passively surveying what outside information sources the patient has engaged with, regardless of whether patient introduces the information or does not introduce it.
Commentary
Patient engagement plays an important role in health care [3]. Activated patients often have skills and confidence to engage in their health care and with their provider, which often contributes to better health outcomes and care experiences [2,4] as well as lower health care costs [5]. Health information is needed to make informed decisions, manage health, and practice healthy behaviors [6], and patients are increasingly taking an active role in seeking out medical or health information outside of the clinical encounter in order to make shared health decisions with their provider [7]. Indeed, one of the Healthy People 2020 goals is to “Use health communication strategies and health information technology to improve population health outcomes and health care quality, and to achieve health equity” [8].
However, seeking POMI requires health literacy skills and supportive relationships, particularly when navigating the many channels and complexities of publicly available health information [8]. This is especially true on the internet, where there is often varying accuracy and clarity of information presented. According to 2011 data from the Pew Research Center [9], 74% of adults in the United States use the internet, and of those adults 80% have looked online for health information; 34% have read another person’s commentary or experience about health or medical issues on an online news group, website, or blog; 25% have watched an online video about health or medical issues; and 24% have consulted online reviews of particular drugs or medical treatments.
A general strength of this study was the cross-sectional design, which allowed for surveying patients around attitudes, motivations, and behaviors immediately before and after their clinical encounter. According to the authors, this study design was aimed to extend knowledge around information seeking and provider discussions that have occurred distally and relied on patient long-term recall. Additionally, this study surveyed a variety of patients (not limited to either primary or specialist appointments) at 2 different academic medical centers, and gave patients a choice to either take the survey on a computer tablet or traditional paper and pencil. Further, the authors assessed the reliability of scales used and included a number of predictor variables in the logistic regression models for hypothesis testing.
The authors acknowledged several limitations, including the use of convenience sampling and self-reported data with volunteer participants, which can result in self-selection bias and social desirability bias. As study participants were self-selecting, low health literacy patients may have been more likely to not volunteer to take the survey, which might explain the relatively high mean scores on the health literacy measures. Further, participants were mostly white, female, college-educated, health literate, and scheduled for a routine visit, which limits the generalizability of the study findings and the ability to identify significant predictors.
Regarding the study design, pre-/post-tests are usually used to measure the change in a situation, phenomenon, problem, or attitude. However, as the authors did not aim to measure any change during the clinical encounter itself, the use of only a post-test may have been more appropriate. The use of a pre-/post-test design may have increased the likelihood of patients both recalling POMI before the encounter and then sharing POMI with their provider. Also, in the post-survey, the authors only asked follow-up questions of patients that shared POMI with their provider. An open-response question could have been included to explore further why some patients chose not to introduce POMI during the clinical encounter. Lastly, the authors may have been able to reach more patients with lower health literacy if surveys were administered at public hospitals as opposed to academic medical centers. While some providers may perceive that patients in academic medical centers are more complex or may have limited access to care [10], patients at public hospitals and safety net hospitals tend to be of lower income and have limited or no insurance [11,12].
Applications For Clinical Practice
There are documented communication-enhancing techniques and strategies that providers and other health professionals use, particularly among patients with low health literacy [13]. Based on this study, the authors conclude that providers may try another strategy of directly asking or passively surveying any POMI, regardless of whether the patient initiates this conversation. Other research has acknowledged that recognition of health literacy status allows for the use of appropriate communication tools [14]. However, providers need to recognize barriers to health information seeking, particularly among minorities and underserved populations [15], as well as the potential for embarrassment that patients might experience as a result of revealing misunderstandings of health information or general reading difficulties [16]. This study highlights the need for further research to identify predictors of health information seeking and especially health information sharing by patients during the clinical encounter.
—Katrina F. Mateo, MPH
1. Nutbeam D. Health literacy as a public health goal: a challenge for contemporary health education and communication strategies into the 21st century. Health Promot Int 2000;15:259–67.
2. Greene J, Hibbard JH. Why does patient activation matter? an examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med 2011;27:520–6.
3. Coulter A. Patient engagement--what works? J Ambul Care Manage 2012;35:80–9.
4. Hibbard JH, Greene J. What the evidence shows about patient activation: better health outcomes and care experiences; fewer data on costs. Health Aff (Millwood) 2013;32:207–14.
5. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients’ “scores”. Health Aff (Millwood) 2013;32:216–22.
6. Nelson DE, Kreps GL, Hesse BW, et al. The Health Information National Trends Survey (HINTS): development, design, and dissemination. J Health Commun 2004;9:443–60.
7. Truog RD. Patients and doctors--evolution of a relationship. N Engl J Med 2012;366:581–5.
8. Office of Disease Prevention and Health Promotion. Health Communication and Health Information Technology. Available at www.healthypeople.gov/2020/topics-objectives/topic/health-communication-and-health-information-technology.
9. Fox S. Social media in context. Pew Research Center. 2011. Available at www.pewinternet.org/2011/05/12/social-media-in-context/.
10. Christmas C, Durso SC, Kravet SJ, Wright SM. Advantages and challenges of working as a clinician in an academic department of medicine: academic clinicians’ perspectives. J Grad Med Educ 2010;2:478–84.
11. Kane NM, Singer SJ, Clark JR, et al. Strained local and state government finances among current realities that threaten public hospitals’ profitability. Health Aff (Millwood) 2012;31:1680–9.
12. Felland LE, Stark L. Local public hospitals: changing with the times. Res Brief 2012;(25):1–13.
13. Schwartzberg JG, Cowett A, VanGeest J, Wolf MS. Communication techniques for patients with low health literacy: a survey of physicians, nurses, and pharmacists. Am J Health Behav 2007;31 Suppl 1:S96–104.
14. Stocks NP, Hill CL, Gravier S, et al. Health literacy--a new concept for general practice? Aust Fam Physician 2009;38:144–7.
15. Warren J, Kvasny L, Hecht M, et al. Barriers, control and identity in health information seeking among African American women. J Health Dispar Res Pract 2012;3(3).
16. Wolf MS, Williams MV, Parker RM, et al. Patients’ shame and attitudes toward discussing the results of literacy screening. J Health Commun 2007;12:721–32.
Study Overview
Objective. To assess how patients look for patient-obtained medication information (POMI) to prepare for a clinical appointment, whether they share those findings with their provider, and how health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI.
Design. Cross-sectional survey-based study.
Setting and participants. The study took place over 1 week at 2 academic medical centers located in Las Vegas, Nevada, and Washington, DC. At a central waiting area at each facility, patients aged 18 and older waiting for their clinical appointment were invited to complete a survey, either on a computer tablet or with paper and pencil, before and after their appointment.
Measures and analysis. The pre-survey included demographic measures (age, gender, education, and ethnicity), the reason for the visit (routine care, sick visit, follow-up after survey, and follow-up after emergency room visit), and an item to assess self-report of perceived general health (from poor to excellent). Health literacy was assessed by a self-report measure that included subscales for the 3 dimensions of health literacy: functional, communicative, and critical health literacy [1]; together, these capture the ability of patients to retain health knowledge, gather and communicate health concepts, and apply health information. Patient activation was scored using the Patient Activation Measure (13 Likert-style items, total scale range 0–100); patient activation combines a patient’s self-reported knowledge, skill, and confidence for self-management of general health or a chronic condition [2]. Information seeking was measured by time spent (did not look for information, 1 hour, 2 hours, 3 hours, or more than 3 hours), and information channels used to look for POMI (eg, magazines/newspapers, internet website or search engine) were presented dichotomously (yes/no).
The post-survey first asked whether the participant shared information with their provider (yes/no). If the participant said yes, 4 items assessed their perception of the provider’s response, including amount of time spent discussing POMI, how seriously the provider considered the information, and overall reaction (scored as a mean, each item measured from 1–5, with 5 indicating the most positive reactions). For hypothesis testing, logistic regression models were used to test the effects of the independent variables. To explore the relationship between health literacy/patient activation and physician response, correlations were calculated.
Main results. Over 400 patients were asked to participate, and of these a total of 243 (60.75%) patients were eligible, consented, and completed surveys. Participants were predominantly white (57.6%), female (63%), had some college education or higher (80.2%), and had a clinical appointment for routine care (69.3%). The mean age was 47.04 years (SD, 15.78), the mean health status was 3.20 (SD, 0.94), and the mean Patient Activation Measure was 72.43 (SD, 16.00).
More than half of participants (58.26%) who responded to the item about information seeking indicated seeking POMI prior to their clinical appointment. Of these, the majority (88.7%) reported using the internet, particularly WebMD, as an information channel. Significant predictors of information seeking included age (P = 0.01, OR = 0.973), communicative health literacy (P = 0.01, or = 1.975), and critical health literacy (P = 0.05, OR = 1.518). Lower age, higher communicative health literacy, and higher critical health literacy increased the likelihood of the patient seeking POMI prior to the clinical appointment. Other assessed predictors were not significant, including gender, functional health literacy, patient activation, reason for visit, and reported health status.
58.2% of the 141 information-seeking patients talked to their health care provider about the information they found. However, no predictor variables included in a logistic regression analysis were significant, including age, gender, reason for visit, reported health status, functional health literacy, communicative health literacy, critical health literacy, and patient activation. For the research question (how do health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI), the mean score on the 4-item measure was 4.08 (SD, 0.90), indicating a generally positive response; most reported the physician response was good or higher. Patient activation correlated positively with perceived physician response (r = 0.245, P = 0.03).
Conclusion. The lack of data to predict who will introduce POMI at the medical visit is disconcerting. Providers might consider directly asking or passively surveying what outside information sources the patient has engaged with, regardless of whether patient introduces the information or does not introduce it.
Commentary
Patient engagement plays an important role in health care [3]. Activated patients often have skills and confidence to engage in their health care and with their provider, which often contributes to better health outcomes and care experiences [2,4] as well as lower health care costs [5]. Health information is needed to make informed decisions, manage health, and practice healthy behaviors [6], and patients are increasingly taking an active role in seeking out medical or health information outside of the clinical encounter in order to make shared health decisions with their provider [7]. Indeed, one of the Healthy People 2020 goals is to “Use health communication strategies and health information technology to improve population health outcomes and health care quality, and to achieve health equity” [8].
However, seeking POMI requires health literacy skills and supportive relationships, particularly when navigating the many channels and complexities of publicly available health information [8]. This is especially true on the internet, where there is often varying accuracy and clarity of information presented. According to 2011 data from the Pew Research Center [9], 74% of adults in the United States use the internet, and of those adults 80% have looked online for health information; 34% have read another person’s commentary or experience about health or medical issues on an online news group, website, or blog; 25% have watched an online video about health or medical issues; and 24% have consulted online reviews of particular drugs or medical treatments.
A general strength of this study was the cross-sectional design, which allowed for surveying patients around attitudes, motivations, and behaviors immediately before and after their clinical encounter. According to the authors, this study design was aimed to extend knowledge around information seeking and provider discussions that have occurred distally and relied on patient long-term recall. Additionally, this study surveyed a variety of patients (not limited to either primary or specialist appointments) at 2 different academic medical centers, and gave patients a choice to either take the survey on a computer tablet or traditional paper and pencil. Further, the authors assessed the reliability of scales used and included a number of predictor variables in the logistic regression models for hypothesis testing.
The authors acknowledged several limitations, including the use of convenience sampling and self-reported data with volunteer participants, which can result in self-selection bias and social desirability bias. As study participants were self-selecting, low health literacy patients may have been more likely to not volunteer to take the survey, which might explain the relatively high mean scores on the health literacy measures. Further, participants were mostly white, female, college-educated, health literate, and scheduled for a routine visit, which limits the generalizability of the study findings and the ability to identify significant predictors.
Regarding the study design, pre-/post-tests are usually used to measure the change in a situation, phenomenon, problem, or attitude. However, as the authors did not aim to measure any change during the clinical encounter itself, the use of only a post-test may have been more appropriate. The use of a pre-/post-test design may have increased the likelihood of patients both recalling POMI before the encounter and then sharing POMI with their provider. Also, in the post-survey, the authors only asked follow-up questions of patients that shared POMI with their provider. An open-response question could have been included to explore further why some patients chose not to introduce POMI during the clinical encounter. Lastly, the authors may have been able to reach more patients with lower health literacy if surveys were administered at public hospitals as opposed to academic medical centers. While some providers may perceive that patients in academic medical centers are more complex or may have limited access to care [10], patients at public hospitals and safety net hospitals tend to be of lower income and have limited or no insurance [11,12].
Applications For Clinical Practice
There are documented communication-enhancing techniques and strategies that providers and other health professionals use, particularly among patients with low health literacy [13]. Based on this study, the authors conclude that providers may try another strategy of directly asking or passively surveying any POMI, regardless of whether the patient initiates this conversation. Other research has acknowledged that recognition of health literacy status allows for the use of appropriate communication tools [14]. However, providers need to recognize barriers to health information seeking, particularly among minorities and underserved populations [15], as well as the potential for embarrassment that patients might experience as a result of revealing misunderstandings of health information or general reading difficulties [16]. This study highlights the need for further research to identify predictors of health information seeking and especially health information sharing by patients during the clinical encounter.
—Katrina F. Mateo, MPH
Study Overview
Objective. To assess how patients look for patient-obtained medication information (POMI) to prepare for a clinical appointment, whether they share those findings with their provider, and how health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI.
Design. Cross-sectional survey-based study.
Setting and participants. The study took place over 1 week at 2 academic medical centers located in Las Vegas, Nevada, and Washington, DC. At a central waiting area at each facility, patients aged 18 and older waiting for their clinical appointment were invited to complete a survey, either on a computer tablet or with paper and pencil, before and after their appointment.
Measures and analysis. The pre-survey included demographic measures (age, gender, education, and ethnicity), the reason for the visit (routine care, sick visit, follow-up after survey, and follow-up after emergency room visit), and an item to assess self-report of perceived general health (from poor to excellent). Health literacy was assessed by a self-report measure that included subscales for the 3 dimensions of health literacy: functional, communicative, and critical health literacy [1]; together, these capture the ability of patients to retain health knowledge, gather and communicate health concepts, and apply health information. Patient activation was scored using the Patient Activation Measure (13 Likert-style items, total scale range 0–100); patient activation combines a patient’s self-reported knowledge, skill, and confidence for self-management of general health or a chronic condition [2]. Information seeking was measured by time spent (did not look for information, 1 hour, 2 hours, 3 hours, or more than 3 hours), and information channels used to look for POMI (eg, magazines/newspapers, internet website or search engine) were presented dichotomously (yes/no).
The post-survey first asked whether the participant shared information with their provider (yes/no). If the participant said yes, 4 items assessed their perception of the provider’s response, including amount of time spent discussing POMI, how seriously the provider considered the information, and overall reaction (scored as a mean, each item measured from 1–5, with 5 indicating the most positive reactions). For hypothesis testing, logistic regression models were used to test the effects of the independent variables. To explore the relationship between health literacy/patient activation and physician response, correlations were calculated.
Main results. Over 400 patients were asked to participate, and of these a total of 243 (60.75%) patients were eligible, consented, and completed surveys. Participants were predominantly white (57.6%), female (63%), had some college education or higher (80.2%), and had a clinical appointment for routine care (69.3%). The mean age was 47.04 years (SD, 15.78), the mean health status was 3.20 (SD, 0.94), and the mean Patient Activation Measure was 72.43 (SD, 16.00).
More than half of participants (58.26%) who responded to the item about information seeking indicated seeking POMI prior to their clinical appointment. Of these, the majority (88.7%) reported using the internet, particularly WebMD, as an information channel. Significant predictors of information seeking included age (P = 0.01, OR = 0.973), communicative health literacy (P = 0.01, or = 1.975), and critical health literacy (P = 0.05, OR = 1.518). Lower age, higher communicative health literacy, and higher critical health literacy increased the likelihood of the patient seeking POMI prior to the clinical appointment. Other assessed predictors were not significant, including gender, functional health literacy, patient activation, reason for visit, and reported health status.
58.2% of the 141 information-seeking patients talked to their health care provider about the information they found. However, no predictor variables included in a logistic regression analysis were significant, including age, gender, reason for visit, reported health status, functional health literacy, communicative health literacy, critical health literacy, and patient activation. For the research question (how do health literacy and patient activation relate to a patient’s perception of the physician’s reaction to POMI), the mean score on the 4-item measure was 4.08 (SD, 0.90), indicating a generally positive response; most reported the physician response was good or higher. Patient activation correlated positively with perceived physician response (r = 0.245, P = 0.03).
Conclusion. The lack of data to predict who will introduce POMI at the medical visit is disconcerting. Providers might consider directly asking or passively surveying what outside information sources the patient has engaged with, regardless of whether patient introduces the information or does not introduce it.
Commentary
Patient engagement plays an important role in health care [3]. Activated patients often have skills and confidence to engage in their health care and with their provider, which often contributes to better health outcomes and care experiences [2,4] as well as lower health care costs [5]. Health information is needed to make informed decisions, manage health, and practice healthy behaviors [6], and patients are increasingly taking an active role in seeking out medical or health information outside of the clinical encounter in order to make shared health decisions with their provider [7]. Indeed, one of the Healthy People 2020 goals is to “Use health communication strategies and health information technology to improve population health outcomes and health care quality, and to achieve health equity” [8].
However, seeking POMI requires health literacy skills and supportive relationships, particularly when navigating the many channels and complexities of publicly available health information [8]. This is especially true on the internet, where there is often varying accuracy and clarity of information presented. According to 2011 data from the Pew Research Center [9], 74% of adults in the United States use the internet, and of those adults 80% have looked online for health information; 34% have read another person’s commentary or experience about health or medical issues on an online news group, website, or blog; 25% have watched an online video about health or medical issues; and 24% have consulted online reviews of particular drugs or medical treatments.
A general strength of this study was the cross-sectional design, which allowed for surveying patients around attitudes, motivations, and behaviors immediately before and after their clinical encounter. According to the authors, this study design was aimed to extend knowledge around information seeking and provider discussions that have occurred distally and relied on patient long-term recall. Additionally, this study surveyed a variety of patients (not limited to either primary or specialist appointments) at 2 different academic medical centers, and gave patients a choice to either take the survey on a computer tablet or traditional paper and pencil. Further, the authors assessed the reliability of scales used and included a number of predictor variables in the logistic regression models for hypothesis testing.
The authors acknowledged several limitations, including the use of convenience sampling and self-reported data with volunteer participants, which can result in self-selection bias and social desirability bias. As study participants were self-selecting, low health literacy patients may have been more likely to not volunteer to take the survey, which might explain the relatively high mean scores on the health literacy measures. Further, participants were mostly white, female, college-educated, health literate, and scheduled for a routine visit, which limits the generalizability of the study findings and the ability to identify significant predictors.
Regarding the study design, pre-/post-tests are usually used to measure the change in a situation, phenomenon, problem, or attitude. However, as the authors did not aim to measure any change during the clinical encounter itself, the use of only a post-test may have been more appropriate. The use of a pre-/post-test design may have increased the likelihood of patients both recalling POMI before the encounter and then sharing POMI with their provider. Also, in the post-survey, the authors only asked follow-up questions of patients that shared POMI with their provider. An open-response question could have been included to explore further why some patients chose not to introduce POMI during the clinical encounter. Lastly, the authors may have been able to reach more patients with lower health literacy if surveys were administered at public hospitals as opposed to academic medical centers. While some providers may perceive that patients in academic medical centers are more complex or may have limited access to care [10], patients at public hospitals and safety net hospitals tend to be of lower income and have limited or no insurance [11,12].
Applications For Clinical Practice
There are documented communication-enhancing techniques and strategies that providers and other health professionals use, particularly among patients with low health literacy [13]. Based on this study, the authors conclude that providers may try another strategy of directly asking or passively surveying any POMI, regardless of whether the patient initiates this conversation. Other research has acknowledged that recognition of health literacy status allows for the use of appropriate communication tools [14]. However, providers need to recognize barriers to health information seeking, particularly among minorities and underserved populations [15], as well as the potential for embarrassment that patients might experience as a result of revealing misunderstandings of health information or general reading difficulties [16]. This study highlights the need for further research to identify predictors of health information seeking and especially health information sharing by patients during the clinical encounter.
—Katrina F. Mateo, MPH
1. Nutbeam D. Health literacy as a public health goal: a challenge for contemporary health education and communication strategies into the 21st century. Health Promot Int 2000;15:259–67.
2. Greene J, Hibbard JH. Why does patient activation matter? an examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med 2011;27:520–6.
3. Coulter A. Patient engagement--what works? J Ambul Care Manage 2012;35:80–9.
4. Hibbard JH, Greene J. What the evidence shows about patient activation: better health outcomes and care experiences; fewer data on costs. Health Aff (Millwood) 2013;32:207–14.
5. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients’ “scores”. Health Aff (Millwood) 2013;32:216–22.
6. Nelson DE, Kreps GL, Hesse BW, et al. The Health Information National Trends Survey (HINTS): development, design, and dissemination. J Health Commun 2004;9:443–60.
7. Truog RD. Patients and doctors--evolution of a relationship. N Engl J Med 2012;366:581–5.
8. Office of Disease Prevention and Health Promotion. Health Communication and Health Information Technology. Available at www.healthypeople.gov/2020/topics-objectives/topic/health-communication-and-health-information-technology.
9. Fox S. Social media in context. Pew Research Center. 2011. Available at www.pewinternet.org/2011/05/12/social-media-in-context/.
10. Christmas C, Durso SC, Kravet SJ, Wright SM. Advantages and challenges of working as a clinician in an academic department of medicine: academic clinicians’ perspectives. J Grad Med Educ 2010;2:478–84.
11. Kane NM, Singer SJ, Clark JR, et al. Strained local and state government finances among current realities that threaten public hospitals’ profitability. Health Aff (Millwood) 2012;31:1680–9.
12. Felland LE, Stark L. Local public hospitals: changing with the times. Res Brief 2012;(25):1–13.
13. Schwartzberg JG, Cowett A, VanGeest J, Wolf MS. Communication techniques for patients with low health literacy: a survey of physicians, nurses, and pharmacists. Am J Health Behav 2007;31 Suppl 1:S96–104.
14. Stocks NP, Hill CL, Gravier S, et al. Health literacy--a new concept for general practice? Aust Fam Physician 2009;38:144–7.
15. Warren J, Kvasny L, Hecht M, et al. Barriers, control and identity in health information seeking among African American women. J Health Dispar Res Pract 2012;3(3).
16. Wolf MS, Williams MV, Parker RM, et al. Patients’ shame and attitudes toward discussing the results of literacy screening. J Health Commun 2007;12:721–32.
1. Nutbeam D. Health literacy as a public health goal: a challenge for contemporary health education and communication strategies into the 21st century. Health Promot Int 2000;15:259–67.
2. Greene J, Hibbard JH. Why does patient activation matter? an examination of the relationships between patient activation and health-related outcomes. J Gen Intern Med 2011;27:520–6.
3. Coulter A. Patient engagement--what works? J Ambul Care Manage 2012;35:80–9.
4. Hibbard JH, Greene J. What the evidence shows about patient activation: better health outcomes and care experiences; fewer data on costs. Health Aff (Millwood) 2013;32:207–14.
5. Hibbard JH, Greene J, Overton V. Patients with lower activation associated with higher costs; delivery systems should know their patients’ “scores”. Health Aff (Millwood) 2013;32:216–22.
6. Nelson DE, Kreps GL, Hesse BW, et al. The Health Information National Trends Survey (HINTS): development, design, and dissemination. J Health Commun 2004;9:443–60.
7. Truog RD. Patients and doctors--evolution of a relationship. N Engl J Med 2012;366:581–5.
8. Office of Disease Prevention and Health Promotion. Health Communication and Health Information Technology. Available at www.healthypeople.gov/2020/topics-objectives/topic/health-communication-and-health-information-technology.
9. Fox S. Social media in context. Pew Research Center. 2011. Available at www.pewinternet.org/2011/05/12/social-media-in-context/.
10. Christmas C, Durso SC, Kravet SJ, Wright SM. Advantages and challenges of working as a clinician in an academic department of medicine: academic clinicians’ perspectives. J Grad Med Educ 2010;2:478–84.
11. Kane NM, Singer SJ, Clark JR, et al. Strained local and state government finances among current realities that threaten public hospitals’ profitability. Health Aff (Millwood) 2012;31:1680–9.
12. Felland LE, Stark L. Local public hospitals: changing with the times. Res Brief 2012;(25):1–13.
13. Schwartzberg JG, Cowett A, VanGeest J, Wolf MS. Communication techniques for patients with low health literacy: a survey of physicians, nurses, and pharmacists. Am J Health Behav 2007;31 Suppl 1:S96–104.
14. Stocks NP, Hill CL, Gravier S, et al. Health literacy--a new concept for general practice? Aust Fam Physician 2009;38:144–7.
15. Warren J, Kvasny L, Hecht M, et al. Barriers, control and identity in health information seeking among African American women. J Health Dispar Res Pract 2012;3(3).
16. Wolf MS, Williams MV, Parker RM, et al. Patients’ shame and attitudes toward discussing the results of literacy screening. J Health Commun 2007;12:721–32.
Induction ALL treatment can cause bone loss
of cancellous bone
Patients with acute lymphoblastic leukemia (ALL) may experience significant bone loss much earlier than previously assumed, according to a study published in the journal Bone.
Investigators analyzed a cohort of adolescent and young adult ALL patients before and after their first month of chemotherapy and observed “significant alterations” to cancellous and cortical bone in this short period of time.
Previous studies to determine the changes to bone density during ALL therapy had focused on the cumulative effects of chemotherapy after months or even years of treatment.
“In clinic, we would see patients with fractures and vertebral compression during the very first few weeks of treatment,” said study author Etan Orgel, MD, of Children’s Hospital Los Angeles in California.
“But we were unaware of any study that specifically examined bone before chemotherapy and immediately after the first 30 days of treatment, which would allow us to understand the impact of this early treatment phase.”
So Dr Orgel and his colleagues conducted a prospective study of 38 patients, ages 10 to 21, who were newly diagnosed with ALL.
The team used quantitative computerized tomography (QCT) to assess leukemia-related changes to bone at diagnosis and then the subsequent effects of the induction phase of chemotherapy.
All of the patients received a 28-day induction regimen consisting of vincristine, pegylated L-asparaginase, anthracycline (daunorubicin or doxorubicin), and a glucocorticoid (either prednisone at 60 mg/m2/day for 28 days or dexamethasone at 10 mg/m2/day for 14 days).
The investigators compared the patients to age- and sex-matched controls and found that leukemia did not dramatically alter the properties of bone before chemotherapy.
However, QCT revealed significant changes during the 30-day induction phase in the 35 patients who were well enough to undergo imaging after treatment.
The patients experienced a significant decrease in cancellous volumetric bone mineral density, which was measured in the spine. The median decrease was 27% (P<0.001).
There was no significant change in cortical volumetric bone mineral density, which was measured in the tibia (−0.0%, P=0.860) or femur (−0.7%, P=0.290).
But there was significant cortical thinning in the tibia. The average cortical thickness decreased 1.2% (P<0.001), and the cortical area decreased 0.4% (P=0.014).
The femur was less affected, the investigators said. There was a decrease in average cortical thickness, but this was not significant (-0.3%, P=0.740).
To help clinicians relate to these findings, the investigators also measured bone mineral density using the older but more widely available technique of dual-energy x-ray absorptiometry. They found that it underestimated these changes as compared to QCT measurements.
“Now that we know how soon bone toxicity occurs, we need to re-evaluate our approaches to managing these changes and focus research efforts on new ways to mitigate this common yet significant adverse effect,” said study author Steven Mittelman, MD, PhD, of Children’s Hospital Los Angeles. ![]()
of cancellous bone
Patients with acute lymphoblastic leukemia (ALL) may experience significant bone loss much earlier than previously assumed, according to a study published in the journal Bone.
Investigators analyzed a cohort of adolescent and young adult ALL patients before and after their first month of chemotherapy and observed “significant alterations” to cancellous and cortical bone in this short period of time.
Previous studies to determine the changes to bone density during ALL therapy had focused on the cumulative effects of chemotherapy after months or even years of treatment.
“In clinic, we would see patients with fractures and vertebral compression during the very first few weeks of treatment,” said study author Etan Orgel, MD, of Children’s Hospital Los Angeles in California.
“But we were unaware of any study that specifically examined bone before chemotherapy and immediately after the first 30 days of treatment, which would allow us to understand the impact of this early treatment phase.”
So Dr Orgel and his colleagues conducted a prospective study of 38 patients, ages 10 to 21, who were newly diagnosed with ALL.
The team used quantitative computerized tomography (QCT) to assess leukemia-related changes to bone at diagnosis and then the subsequent effects of the induction phase of chemotherapy.
All of the patients received a 28-day induction regimen consisting of vincristine, pegylated L-asparaginase, anthracycline (daunorubicin or doxorubicin), and a glucocorticoid (either prednisone at 60 mg/m2/day for 28 days or dexamethasone at 10 mg/m2/day for 14 days).
The investigators compared the patients to age- and sex-matched controls and found that leukemia did not dramatically alter the properties of bone before chemotherapy.
However, QCT revealed significant changes during the 30-day induction phase in the 35 patients who were well enough to undergo imaging after treatment.
The patients experienced a significant decrease in cancellous volumetric bone mineral density, which was measured in the spine. The median decrease was 27% (P<0.001).
There was no significant change in cortical volumetric bone mineral density, which was measured in the tibia (−0.0%, P=0.860) or femur (−0.7%, P=0.290).
But there was significant cortical thinning in the tibia. The average cortical thickness decreased 1.2% (P<0.001), and the cortical area decreased 0.4% (P=0.014).
The femur was less affected, the investigators said. There was a decrease in average cortical thickness, but this was not significant (-0.3%, P=0.740).
To help clinicians relate to these findings, the investigators also measured bone mineral density using the older but more widely available technique of dual-energy x-ray absorptiometry. They found that it underestimated these changes as compared to QCT measurements.
“Now that we know how soon bone toxicity occurs, we need to re-evaluate our approaches to managing these changes and focus research efforts on new ways to mitigate this common yet significant adverse effect,” said study author Steven Mittelman, MD, PhD, of Children’s Hospital Los Angeles. ![]()
of cancellous bone
Patients with acute lymphoblastic leukemia (ALL) may experience significant bone loss much earlier than previously assumed, according to a study published in the journal Bone.
Investigators analyzed a cohort of adolescent and young adult ALL patients before and after their first month of chemotherapy and observed “significant alterations” to cancellous and cortical bone in this short period of time.
Previous studies to determine the changes to bone density during ALL therapy had focused on the cumulative effects of chemotherapy after months or even years of treatment.
“In clinic, we would see patients with fractures and vertebral compression during the very first few weeks of treatment,” said study author Etan Orgel, MD, of Children’s Hospital Los Angeles in California.
“But we were unaware of any study that specifically examined bone before chemotherapy and immediately after the first 30 days of treatment, which would allow us to understand the impact of this early treatment phase.”
So Dr Orgel and his colleagues conducted a prospective study of 38 patients, ages 10 to 21, who were newly diagnosed with ALL.
The team used quantitative computerized tomography (QCT) to assess leukemia-related changes to bone at diagnosis and then the subsequent effects of the induction phase of chemotherapy.
All of the patients received a 28-day induction regimen consisting of vincristine, pegylated L-asparaginase, anthracycline (daunorubicin or doxorubicin), and a glucocorticoid (either prednisone at 60 mg/m2/day for 28 days or dexamethasone at 10 mg/m2/day for 14 days).
The investigators compared the patients to age- and sex-matched controls and found that leukemia did not dramatically alter the properties of bone before chemotherapy.
However, QCT revealed significant changes during the 30-day induction phase in the 35 patients who were well enough to undergo imaging after treatment.
The patients experienced a significant decrease in cancellous volumetric bone mineral density, which was measured in the spine. The median decrease was 27% (P<0.001).
There was no significant change in cortical volumetric bone mineral density, which was measured in the tibia (−0.0%, P=0.860) or femur (−0.7%, P=0.290).
But there was significant cortical thinning in the tibia. The average cortical thickness decreased 1.2% (P<0.001), and the cortical area decreased 0.4% (P=0.014).
The femur was less affected, the investigators said. There was a decrease in average cortical thickness, but this was not significant (-0.3%, P=0.740).
To help clinicians relate to these findings, the investigators also measured bone mineral density using the older but more widely available technique of dual-energy x-ray absorptiometry. They found that it underestimated these changes as compared to QCT measurements.
“Now that we know how soon bone toxicity occurs, we need to re-evaluate our approaches to managing these changes and focus research efforts on new ways to mitigate this common yet significant adverse effect,” said study author Steven Mittelman, MD, PhD, of Children’s Hospital Los Angeles. ![]()
Elephant in the room of dermatology
I have become increasingly dismayed by reports of dermatologists who allow their nurse practitioners and physician assistants to practice independently.
That is, the employing dermatologists only see the patients, new or established, if they are asked to, and often are not even on the premises. In fact, they might be thousands of miles away.

A little background is in order. Physician assistants and nurse practitioners are formally trained in primary care, not in dermatology, although there are currently three 1-year programs to help them specialize in dermatology. When Congress authorized their independent payment in 1997, they envisioned primary care nurses traveling the hills, hollers, and inner cities improving health care. Unfortunately, this hasn’t happened, and instead they have moved into suburban America, and increasingly, are practicing specialty medicine.
It can be argued that decreased access to primary care, which was the reason midlevels were created, is more important than is access to dermatology. In particular, extenders have targeted office-based specialties such as dermatology, but also neurology and pain medicine. These specialties are office based, and credentialing by hospitals is not required to bill insurance plans. Also, these specialties have good-paying, seemingly simple, small procedures. They have accomplished this with the avid help of dermatologists, I might add. There will be an estimated 10,000 “dermatology” nurse practitioners and physician assistants next year.
Let me be clear: I am not opposed to a dermatology extender who works closely with a dermatologist and does intake histories and physicals, then staffs with the physician, assists with surgery, or sees routine follow-ups (think acne, psoriasis, atopy, suture removal, and warts) on an established protocol with the full knowledge of the patient.
This is not what we are seeing. We have nurse practitioners buying retiring dermatologists’ practices, physician assistants independently setting up remote clinics then hiring “supervising” dermatologists to visit once a week to sign and review charts, and independent “dermatology” clinics with a doctor thousands of miles away available, if really needed, by telephone or the Internet. (This is not really telemedicine, is it?) These extenders are listed as dermatologists on the Internet, or they hide behind the name of a dermatologist, and when you call their offices, and ask if you will see a “real” dermatologist, the answer is often “Oh, don’t worry, our nurse or PA specializes in dermatology.”
I think this is grossly unfair to patients, who, when they call the dermatology center listed on the Internet, can’t conceive that their dermatology appointment is not being made with a dermatologist, not even being made with a physician, but with a nurse practitioner or physician assistant! I also think it does a huge disservice to the specialty. The “collaborating” dermatologists enabling these extenders are renting out the good name of our specialty.
Patients seen by these extenders may also be subject to unnecessary biopsies. (If you don’t know what it is, you must biopsy it!) It also results in additional charges for pathology and additional medical misadventures.
In addition, I think it is unfair to the extender who is put in this situation. They may have worked in a dermatologist’s office for a few months or even years but are now being asked to pretend to be something they are not and being expected to perform at the level of a medical professional who has had many years of intense, focused training in dermatology. If they are not uncomfortable being thrust into this situation, then they are delusional.
I think it is unfair to the medical system who pays for the less informed opinion and unnecessary procedures. This is not a “good value” except to the rent-seeking dermatologists, who are the front men for this money-making deception.
This is an elephant in the room of dermatology, and I think we have to confront it. This trivializes our specialty and helps allow other specialties and regulatory agencies to ignore or exclude us from the networks and from the conversation. Unfortunately, many of our members and some of our leadership have been corrupted by the “easy money” or “easy time” afforded by this cheapening of the specialty.
Let me give you some examples. The rent seekers piously claim, “there is a shortage of dermatologists, and we are just trying to help save the world.” They also say “my nurses are special and great.”
The honest ones in private practice, however, say, “Man, I make $200,000 a year off my PA while I am off.” The honest academics say, “Listen, I work in a gulag, and I would never be able to travel if my nurses didn’t see my patients.” Time is money, and the academic who gets an extra 10 hours a week out of clinic is benefiting as much as the guy who makes $200,000 a year.
Of course, this situation is not sustainable and will become less viable because of a coming tsunami of malpractice claims, more focused insurer benchmarks revealing excessive test ordering and minor procedures, and patients getting wise. One obvious solution to this would be to reimburse extenders only for evaluation and management codes, which will take a change in law.
Meanwhile, I encourage all of you who feel the same nausea I do to ask candidates for your state and national dermatology organizations if they employ unsupervised extenders. Then check their website for the names of the extenders they employ. Then go to the Medicare data and look up their extenders and see if they bill independently for dermatologic procedures. I think you will be very disturbed, as I was and am.
I hope this stimulates a little self-examination among dermatologists.
Dr. Coldiron is a past-president of the American Academy of Dermatology. He is currently in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].
I have become increasingly dismayed by reports of dermatologists who allow their nurse practitioners and physician assistants to practice independently.
That is, the employing dermatologists only see the patients, new or established, if they are asked to, and often are not even on the premises. In fact, they might be thousands of miles away.
A little background is in order. Physician assistants and nurse practitioners are formally trained in primary care, not in dermatology, although there are currently three 1-year programs to help them specialize in dermatology. When Congress authorized their independent payment in 1997, they envisioned primary care nurses traveling the hills, hollers, and inner cities improving health care. Unfortunately, this hasn’t happened, and instead they have moved into suburban America, and increasingly, are practicing specialty medicine.
It can be argued that decreased access to primary care, which was the reason midlevels were created, is more important than is access to dermatology. In particular, extenders have targeted office-based specialties such as dermatology, but also neurology and pain medicine. These specialties are office based, and credentialing by hospitals is not required to bill insurance plans. Also, these specialties have good-paying, seemingly simple, small procedures. They have accomplished this with the avid help of dermatologists, I might add. There will be an estimated 10,000 “dermatology” nurse practitioners and physician assistants next year.
Let me be clear: I am not opposed to a dermatology extender who works closely with a dermatologist and does intake histories and physicals, then staffs with the physician, assists with surgery, or sees routine follow-ups (think acne, psoriasis, atopy, suture removal, and warts) on an established protocol with the full knowledge of the patient.
This is not what we are seeing. We have nurse practitioners buying retiring dermatologists’ practices, physician assistants independently setting up remote clinics then hiring “supervising” dermatologists to visit once a week to sign and review charts, and independent “dermatology” clinics with a doctor thousands of miles away available, if really needed, by telephone or the Internet. (This is not really telemedicine, is it?) These extenders are listed as dermatologists on the Internet, or they hide behind the name of a dermatologist, and when you call their offices, and ask if you will see a “real” dermatologist, the answer is often “Oh, don’t worry, our nurse or PA specializes in dermatology.”
I think this is grossly unfair to patients, who, when they call the dermatology center listed on the Internet, can’t conceive that their dermatology appointment is not being made with a dermatologist, not even being made with a physician, but with a nurse practitioner or physician assistant! I also think it does a huge disservice to the specialty. The “collaborating” dermatologists enabling these extenders are renting out the good name of our specialty.
Patients seen by these extenders may also be subject to unnecessary biopsies. (If you don’t know what it is, you must biopsy it!) It also results in additional charges for pathology and additional medical misadventures.
In addition, I think it is unfair to the extender who is put in this situation. They may have worked in a dermatologist’s office for a few months or even years but are now being asked to pretend to be something they are not and being expected to perform at the level of a medical professional who has had many years of intense, focused training in dermatology. If they are not uncomfortable being thrust into this situation, then they are delusional.
I think it is unfair to the medical system who pays for the less informed opinion and unnecessary procedures. This is not a “good value” except to the rent-seeking dermatologists, who are the front men for this money-making deception.
This is an elephant in the room of dermatology, and I think we have to confront it. This trivializes our specialty and helps allow other specialties and regulatory agencies to ignore or exclude us from the networks and from the conversation. Unfortunately, many of our members and some of our leadership have been corrupted by the “easy money” or “easy time” afforded by this cheapening of the specialty.
Let me give you some examples. The rent seekers piously claim, “there is a shortage of dermatologists, and we are just trying to help save the world.” They also say “my nurses are special and great.”
The honest ones in private practice, however, say, “Man, I make $200,000 a year off my PA while I am off.” The honest academics say, “Listen, I work in a gulag, and I would never be able to travel if my nurses didn’t see my patients.” Time is money, and the academic who gets an extra 10 hours a week out of clinic is benefiting as much as the guy who makes $200,000 a year.
Of course, this situation is not sustainable and will become less viable because of a coming tsunami of malpractice claims, more focused insurer benchmarks revealing excessive test ordering and minor procedures, and patients getting wise. One obvious solution to this would be to reimburse extenders only for evaluation and management codes, which will take a change in law.
Meanwhile, I encourage all of you who feel the same nausea I do to ask candidates for your state and national dermatology organizations if they employ unsupervised extenders. Then check their website for the names of the extenders they employ. Then go to the Medicare data and look up their extenders and see if they bill independently for dermatologic procedures. I think you will be very disturbed, as I was and am.
I hope this stimulates a little self-examination among dermatologists.
Dr. Coldiron is a past-president of the American Academy of Dermatology. He is currently in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].
I have become increasingly dismayed by reports of dermatologists who allow their nurse practitioners and physician assistants to practice independently.
That is, the employing dermatologists only see the patients, new or established, if they are asked to, and often are not even on the premises. In fact, they might be thousands of miles away.
A little background is in order. Physician assistants and nurse practitioners are formally trained in primary care, not in dermatology, although there are currently three 1-year programs to help them specialize in dermatology. When Congress authorized their independent payment in 1997, they envisioned primary care nurses traveling the hills, hollers, and inner cities improving health care. Unfortunately, this hasn’t happened, and instead they have moved into suburban America, and increasingly, are practicing specialty medicine.
It can be argued that decreased access to primary care, which was the reason midlevels were created, is more important than is access to dermatology. In particular, extenders have targeted office-based specialties such as dermatology, but also neurology and pain medicine. These specialties are office based, and credentialing by hospitals is not required to bill insurance plans. Also, these specialties have good-paying, seemingly simple, small procedures. They have accomplished this with the avid help of dermatologists, I might add. There will be an estimated 10,000 “dermatology” nurse practitioners and physician assistants next year.
Let me be clear: I am not opposed to a dermatology extender who works closely with a dermatologist and does intake histories and physicals, then staffs with the physician, assists with surgery, or sees routine follow-ups (think acne, psoriasis, atopy, suture removal, and warts) on an established protocol with the full knowledge of the patient.
This is not what we are seeing. We have nurse practitioners buying retiring dermatologists’ practices, physician assistants independently setting up remote clinics then hiring “supervising” dermatologists to visit once a week to sign and review charts, and independent “dermatology” clinics with a doctor thousands of miles away available, if really needed, by telephone or the Internet. (This is not really telemedicine, is it?) These extenders are listed as dermatologists on the Internet, or they hide behind the name of a dermatologist, and when you call their offices, and ask if you will see a “real” dermatologist, the answer is often “Oh, don’t worry, our nurse or PA specializes in dermatology.”
I think this is grossly unfair to patients, who, when they call the dermatology center listed on the Internet, can’t conceive that their dermatology appointment is not being made with a dermatologist, not even being made with a physician, but with a nurse practitioner or physician assistant! I also think it does a huge disservice to the specialty. The “collaborating” dermatologists enabling these extenders are renting out the good name of our specialty.
Patients seen by these extenders may also be subject to unnecessary biopsies. (If you don’t know what it is, you must biopsy it!) It also results in additional charges for pathology and additional medical misadventures.
In addition, I think it is unfair to the extender who is put in this situation. They may have worked in a dermatologist’s office for a few months or even years but are now being asked to pretend to be something they are not and being expected to perform at the level of a medical professional who has had many years of intense, focused training in dermatology. If they are not uncomfortable being thrust into this situation, then they are delusional.
I think it is unfair to the medical system who pays for the less informed opinion and unnecessary procedures. This is not a “good value” except to the rent-seeking dermatologists, who are the front men for this money-making deception.
This is an elephant in the room of dermatology, and I think we have to confront it. This trivializes our specialty and helps allow other specialties and regulatory agencies to ignore or exclude us from the networks and from the conversation. Unfortunately, many of our members and some of our leadership have been corrupted by the “easy money” or “easy time” afforded by this cheapening of the specialty.
Let me give you some examples. The rent seekers piously claim, “there is a shortage of dermatologists, and we are just trying to help save the world.” They also say “my nurses are special and great.”
The honest ones in private practice, however, say, “Man, I make $200,000 a year off my PA while I am off.” The honest academics say, “Listen, I work in a gulag, and I would never be able to travel if my nurses didn’t see my patients.” Time is money, and the academic who gets an extra 10 hours a week out of clinic is benefiting as much as the guy who makes $200,000 a year.
Of course, this situation is not sustainable and will become less viable because of a coming tsunami of malpractice claims, more focused insurer benchmarks revealing excessive test ordering and minor procedures, and patients getting wise. One obvious solution to this would be to reimburse extenders only for evaluation and management codes, which will take a change in law.
Meanwhile, I encourage all of you who feel the same nausea I do to ask candidates for your state and national dermatology organizations if they employ unsupervised extenders. Then check their website for the names of the extenders they employ. Then go to the Medicare data and look up their extenders and see if they bill independently for dermatologic procedures. I think you will be very disturbed, as I was and am.
I hope this stimulates a little self-examination among dermatologists.
Dr. Coldiron is a past-president of the American Academy of Dermatology. He is currently in private practice but maintains a clinical assistant professorship at the University of Cincinnati. He cares for patients, teaches medical students and residents, and has several active clinical research projects. Dr. Coldiron is the author of more than 80 scientific letters, papers, and several book chapters, and he speaks frequently on a variety of topics. Reach him at [email protected].

End-stage renal disease risk in lupus nephritis remains unchanged of late
The world health community has lost ground in its fight to reduce end-stage renal disease in patients with lupus nephritis, a systematic review and meta-analysis concluded.
The risk of end-stage renal disease (ESRD) at 5 years of lupus nephritis decreased substantially from the 1970s, when it was 16%, to the mid-1990s, when it plateaued at 11%.
ESRD risks at 10 years and 15 years declined more sharply in the 1970s and 1980s but also plateaued in the mid-1990s at 17% and 22%, respectively.
This plateau was followed by a notable increase in risk in the late 2000s, particularly in the 10-year and 15-year estimates, Dr. Maria Tektonidou of the University of Athens and her coauthors reported (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39594).
“Despite extensive use of immunosuppressive medications through the 2000s, we did not find continued improvement in ESRD risks, but instead a slight increase in risks in the late 2000s,” they wrote.
The increase did not appear to be related to greater representation in recent studies of ethnic minorities, who may be more likely to develop ESRD. In the main analysis involving 148 of the 187 studies, “trends suggest this increase may have been temporary, but further follow-up will be needed to determine if this is sustained,” the investigators added.
Notably, patients with class-IV lupus nephritis had the greatest risk of ESRD during the 2000s, with a 15-year risk of 44%.
The 15-year risk of ESRD also was higher by 10 percentage points in developing countries than in developed countries during the 2000s.
The trends are worrisome because ESRD is a costly complication of lupus nephritis, which affects more than half of all patients with systemic lupus erythematosus (SLE). Patients with lupus nephritis have a 26-fold increased risk of death and estimated annual health care costs between $43,000 and $107,000 per patient, the authors noted.
The systematic review and Bayesian meta-analysis included 187 studies reporting on 18,309 adults with lupus nephritis from 1971 to 2015. The main analysis of ESRD risk included 102 studies from developed countries and 46 studies from developing countries.
Across all studies, 86% of patients were women, 32% had elevated serum creatinine levels at study entry, and proteinuria averaged 3.6 g daily. The average age was 31.2 years and mean duration of lupus nephritis was 2.7 years.
The proportion of patients treated with glucocorticoids alone in the studies declined from 54% in 1966 to 9% in 2010, while use of immunosuppressive therapies increased.
The decrease in ESRD risks early on coincided with increased use of immunosuppressives, particularly cyclophosphamide, and better control of hypertension and proteinuria. As for why those gains have stalled, Dr. Tektonidou and her colleagues said it’s possible that the limits of effectiveness of current treatments have been reached and better outcomes will require new therapies. “It is also possible that the plateau primarily reflects lack of progress in the way currently available and effective treatments are deployed,” they added. “This includes health system factors that result in delays in treatment initiation, and patient and health system factors that result in treatment interruptions and reduced adherence.”

In an accompanying editorial, Dr. Candace Feldman and Dr. Karen Costenbader, both of Brigham and Women’s Hospital in Boston, wrote, “While we have made advances over the past 50 years in our understanding of immunosuppressive medications, there have been few meaningful improvements in other domains that contribute to ESRD and to the persistent and disproportionate burden among vulnerable populations” (Ann Rheum Dis. 2016 Jan 27. doi: 10.1002/art.39593).
Despite the clear importance of medication adherence to SLE care, a recent systematic review of adherence interventions in rheumatic diseases (Ann Rheum Dis. 2015 Feb 9. doi: 10.1136/annrheumdis-2014-206593) found few SLE-specific interventions overall and none that significantly improved adherence outcomes, Dr. Feldman and Dr. Costenbader pointed out.
Dr. Tektonidou and her associates acknowledged that the new systematic review and meta-analysis were limited by the inability to estimate risks beyond 15 years and because the findings were similar only when observational studies were considered. Factors associated with ESRD, such as renal flares and uncontrolled hypertension, were not examined, and few studies were judged to be of high-quality.
Still, the results can be used to counsel patients on risks of ESRD and also will provide benchmarks to judge the effectiveness of future treatments, the authors concluded.
Dr. Feldman and Dr. Costenbader disagreed with this conclusion, citing various study limitations and the many nuanced factors that play into a patient’s risk of developing ESRD.
“This study should rather be used to provide a broad overview of our understanding of changes in SLE ESRD over time, rather than data to counsel an individual patient on his/her risks,” they wrote.
The study was supported by the intramural research program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors reported having no conflicts of interest.
The world health community has lost ground in its fight to reduce end-stage renal disease in patients with lupus nephritis, a systematic review and meta-analysis concluded.
The risk of end-stage renal disease (ESRD) at 5 years of lupus nephritis decreased substantially from the 1970s, when it was 16%, to the mid-1990s, when it plateaued at 11%.
ESRD risks at 10 years and 15 years declined more sharply in the 1970s and 1980s but also plateaued in the mid-1990s at 17% and 22%, respectively.
This plateau was followed by a notable increase in risk in the late 2000s, particularly in the 10-year and 15-year estimates, Dr. Maria Tektonidou of the University of Athens and her coauthors reported (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39594).
“Despite extensive use of immunosuppressive medications through the 2000s, we did not find continued improvement in ESRD risks, but instead a slight increase in risks in the late 2000s,” they wrote.
The increase did not appear to be related to greater representation in recent studies of ethnic minorities, who may be more likely to develop ESRD. In the main analysis involving 148 of the 187 studies, “trends suggest this increase may have been temporary, but further follow-up will be needed to determine if this is sustained,” the investigators added.
Notably, patients with class-IV lupus nephritis had the greatest risk of ESRD during the 2000s, with a 15-year risk of 44%.
The 15-year risk of ESRD also was higher by 10 percentage points in developing countries than in developed countries during the 2000s.
The trends are worrisome because ESRD is a costly complication of lupus nephritis, which affects more than half of all patients with systemic lupus erythematosus (SLE). Patients with lupus nephritis have a 26-fold increased risk of death and estimated annual health care costs between $43,000 and $107,000 per patient, the authors noted.
The systematic review and Bayesian meta-analysis included 187 studies reporting on 18,309 adults with lupus nephritis from 1971 to 2015. The main analysis of ESRD risk included 102 studies from developed countries and 46 studies from developing countries.
Across all studies, 86% of patients were women, 32% had elevated serum creatinine levels at study entry, and proteinuria averaged 3.6 g daily. The average age was 31.2 years and mean duration of lupus nephritis was 2.7 years.
The proportion of patients treated with glucocorticoids alone in the studies declined from 54% in 1966 to 9% in 2010, while use of immunosuppressive therapies increased.
The decrease in ESRD risks early on coincided with increased use of immunosuppressives, particularly cyclophosphamide, and better control of hypertension and proteinuria. As for why those gains have stalled, Dr. Tektonidou and her colleagues said it’s possible that the limits of effectiveness of current treatments have been reached and better outcomes will require new therapies. “It is also possible that the plateau primarily reflects lack of progress in the way currently available and effective treatments are deployed,” they added. “This includes health system factors that result in delays in treatment initiation, and patient and health system factors that result in treatment interruptions and reduced adherence.”
In an accompanying editorial, Dr. Candace Feldman and Dr. Karen Costenbader, both of Brigham and Women’s Hospital in Boston, wrote, “While we have made advances over the past 50 years in our understanding of immunosuppressive medications, there have been few meaningful improvements in other domains that contribute to ESRD and to the persistent and disproportionate burden among vulnerable populations” (Ann Rheum Dis. 2016 Jan 27. doi: 10.1002/art.39593).
Despite the clear importance of medication adherence to SLE care, a recent systematic review of adherence interventions in rheumatic diseases (Ann Rheum Dis. 2015 Feb 9. doi: 10.1136/annrheumdis-2014-206593) found few SLE-specific interventions overall and none that significantly improved adherence outcomes, Dr. Feldman and Dr. Costenbader pointed out.
Dr. Tektonidou and her associates acknowledged that the new systematic review and meta-analysis were limited by the inability to estimate risks beyond 15 years and because the findings were similar only when observational studies were considered. Factors associated with ESRD, such as renal flares and uncontrolled hypertension, were not examined, and few studies were judged to be of high-quality.
Still, the results can be used to counsel patients on risks of ESRD and also will provide benchmarks to judge the effectiveness of future treatments, the authors concluded.
Dr. Feldman and Dr. Costenbader disagreed with this conclusion, citing various study limitations and the many nuanced factors that play into a patient’s risk of developing ESRD.
“This study should rather be used to provide a broad overview of our understanding of changes in SLE ESRD over time, rather than data to counsel an individual patient on his/her risks,” they wrote.
The study was supported by the intramural research program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors reported having no conflicts of interest.
The world health community has lost ground in its fight to reduce end-stage renal disease in patients with lupus nephritis, a systematic review and meta-analysis concluded.
The risk of end-stage renal disease (ESRD) at 5 years of lupus nephritis decreased substantially from the 1970s, when it was 16%, to the mid-1990s, when it plateaued at 11%.
ESRD risks at 10 years and 15 years declined more sharply in the 1970s and 1980s but also plateaued in the mid-1990s at 17% and 22%, respectively.
This plateau was followed by a notable increase in risk in the late 2000s, particularly in the 10-year and 15-year estimates, Dr. Maria Tektonidou of the University of Athens and her coauthors reported (Arthritis Rheumatol. 2016 Jan 27. doi: 10.1002/art.39594).
“Despite extensive use of immunosuppressive medications through the 2000s, we did not find continued improvement in ESRD risks, but instead a slight increase in risks in the late 2000s,” they wrote.
The increase did not appear to be related to greater representation in recent studies of ethnic minorities, who may be more likely to develop ESRD. In the main analysis involving 148 of the 187 studies, “trends suggest this increase may have been temporary, but further follow-up will be needed to determine if this is sustained,” the investigators added.
Notably, patients with class-IV lupus nephritis had the greatest risk of ESRD during the 2000s, with a 15-year risk of 44%.
The 15-year risk of ESRD also was higher by 10 percentage points in developing countries than in developed countries during the 2000s.
The trends are worrisome because ESRD is a costly complication of lupus nephritis, which affects more than half of all patients with systemic lupus erythematosus (SLE). Patients with lupus nephritis have a 26-fold increased risk of death and estimated annual health care costs between $43,000 and $107,000 per patient, the authors noted.
The systematic review and Bayesian meta-analysis included 187 studies reporting on 18,309 adults with lupus nephritis from 1971 to 2015. The main analysis of ESRD risk included 102 studies from developed countries and 46 studies from developing countries.
Across all studies, 86% of patients were women, 32% had elevated serum creatinine levels at study entry, and proteinuria averaged 3.6 g daily. The average age was 31.2 years and mean duration of lupus nephritis was 2.7 years.
The proportion of patients treated with glucocorticoids alone in the studies declined from 54% in 1966 to 9% in 2010, while use of immunosuppressive therapies increased.
The decrease in ESRD risks early on coincided with increased use of immunosuppressives, particularly cyclophosphamide, and better control of hypertension and proteinuria. As for why those gains have stalled, Dr. Tektonidou and her colleagues said it’s possible that the limits of effectiveness of current treatments have been reached and better outcomes will require new therapies. “It is also possible that the plateau primarily reflects lack of progress in the way currently available and effective treatments are deployed,” they added. “This includes health system factors that result in delays in treatment initiation, and patient and health system factors that result in treatment interruptions and reduced adherence.”
In an accompanying editorial, Dr. Candace Feldman and Dr. Karen Costenbader, both of Brigham and Women’s Hospital in Boston, wrote, “While we have made advances over the past 50 years in our understanding of immunosuppressive medications, there have been few meaningful improvements in other domains that contribute to ESRD and to the persistent and disproportionate burden among vulnerable populations” (Ann Rheum Dis. 2016 Jan 27. doi: 10.1002/art.39593).
Despite the clear importance of medication adherence to SLE care, a recent systematic review of adherence interventions in rheumatic diseases (Ann Rheum Dis. 2015 Feb 9. doi: 10.1136/annrheumdis-2014-206593) found few SLE-specific interventions overall and none that significantly improved adherence outcomes, Dr. Feldman and Dr. Costenbader pointed out.
Dr. Tektonidou and her associates acknowledged that the new systematic review and meta-analysis were limited by the inability to estimate risks beyond 15 years and because the findings were similar only when observational studies were considered. Factors associated with ESRD, such as renal flares and uncontrolled hypertension, were not examined, and few studies were judged to be of high-quality.
Still, the results can be used to counsel patients on risks of ESRD and also will provide benchmarks to judge the effectiveness of future treatments, the authors concluded.
Dr. Feldman and Dr. Costenbader disagreed with this conclusion, citing various study limitations and the many nuanced factors that play into a patient’s risk of developing ESRD.
“This study should rather be used to provide a broad overview of our understanding of changes in SLE ESRD over time, rather than data to counsel an individual patient on his/her risks,” they wrote.
The study was supported by the intramural research program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors reported having no conflicts of interest.
FROM ARTHRITIS & RHEUMATOLOGY
Key clinical point: The risk of end-stage renal disease in lupus nephritis decreased from the 1970s to the mid-1990s but has since remained largely unchanged.
Major finding: Patients with class-IV lupus nephritis had the greatest risk of ESRD during the 2000s, with a 15-year risk of 44%.
Data source: Systematic review and Bayesian meta-analysis of 18,309 adults with lupus nephritis.
Disclosures: The study was supported by the intramural research program of the National Institute of Arthritis and Musculoskeletal and Skin Diseases. The authors reported having no conflicts of interest.

Novel test detects low levels of residual CML
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.

Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
A novel DNA-based test may prove useful for identifying which chronic myeloid leukemia patients with undetectable BCR-ABL1 transcripts can safely discontinue tyrosine kinase inhibitor (TKI) therapy, according to Mary Alikian, Ph.D., of Hammersmith Hospital, London, and her colleagues.
The test can quantify very low levels of residual disease in peripheral blood samples from patients with CML in whom BCR-ABL1 transcripts were undetectable using reverse transcription quantitative polymerase chain reaction (RT-qPCR), the researchers reported in a study published online in the Journal of Molecular Diagnostics.
Their personalized DNA-based digital PCR method rapidly identifies t(9;22) fusion junctions using targeted next-generation sequencing and generates high-performance DNA-based hydrolysis probe assays that are specific to the unique molecular footprint of each patient’s CML clone. The researchers further enhanced the sensitivity of the DNA-based approach by optimizing the technique for use on a digital PCR (dPCR) platform, which provides absolute molecular quantification without the need for a standard curve. This approach avoids laborious breakpoint mapping and improves sensitivity.
The researchers successfully mapped genomic breakpoints in all samples from 32 patients with early-stage disease. Using DNA-based dPCR, disease was quantified in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%), the researchers reported (J Mol Diagn. 2016;18:176e189).
Of CML patients who achieve sustained undetectable BCR-ABL1 transcripts on TKI therapy, about 60% experience the return of detectable disease after stopping TKIs and have to restart treatment. An improved method of identifying patients with the lowest likelihood of relapse would allow safe withdrawal of TKI therapy for the 40% of patients who would remain disease free.
The researchers are currently investigating the impact of residual-disease level as assessed by dPCR at the time of treatment withdrawal on outcome within the UK-based DESTINY clinical trial (Deescalation and Stopping Treatment of Imatinib, Nilotinib or Sprycel in Chronic Myeloid Leukaemia). “If validated in clinical trials of stopping TKI, the technique will permit a more personalized approach to recommendations for dose reduction or drug cessation in individual patients, ensuring that therapy is withdrawn only from patients with the highest likelihood of long-term remission,” they wrote.
Identifying genomic breakpoints as soon as CML is diagnosed would allow for the design and optimization of a patient-specific assay. Patients’ response to therapy would then be monitored via standard RT-qPCR until they have reached molecular response. Thereafter, routine monitoring would be augmented with DNA quantification by dPCR and would benefit from the publication of standardized guidelines, as with RT-qPCR.
In the future, it will therefore be important to explore not only whether the risk of relapse after withdrawal is a feature of the number of residual CML cells but also whether it relates to the degree of transcriptional activity in those cells, the researchers wrote. “We observed that 8% (3 of 36) of the samples were positive by RNA-based but negative by DNA-based methods. Conversely, in samples with detectable BCR-ABL1 DNA, there was heterogeneity in the detectability of transcript by RT-dPCR that appeared to be unrelated to the amount of BCR-ABL1 DNA detected. It should be borne in mind that RT and cDNA synthesis steps remain a potential source of variation affecting cDNA concentration, and therefore these results should be interpreted with caution.”
The researchers had no relevant disclosures. The study was supported by Leading Leukemia Research (LEUKA) charity grant 06/Q0406/47, the National Institute for Health Research Biomedical Research Center Funding Scheme, and the Imperial College High Performance Computing Service.
On Twitter @maryjodales
FROM THE JOURNAL OF MOLECULAR DIAGNOSTICS
Key clinical point: A novel test may predict which CML patients in remission can safely stop TKI therapy.
Major finding: Digital PCR for BCR-ABL1 DNA detected persistent disease in 81% of the molecular-remission samples, outperforming both RT-dPCR (25%) and DNA-based quantitative PCR (19%).
Data source: DNA-based dPCR measures in 46 follow-up samples from 6 of the 32 patients, including 36 samples that were in deep molecular remission.
Disclosures: The researchers had no relevant disclosures.

Rivaroxaban trial results hold up, EMA says

The European Medicines Agency (EMA) has concluded that a defect discovered in a system used to measure international normalized ratios (INRs) in the ROCKET AF study does not change the study’s overall conclusions.
The Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips) was recalled in December 2014 after it was found to produce falsely low test results.
A recent investigation by The BMJ suggested this defect may have impacted the results of ROCKET AF, in which researchers compared warfarin to rivaroxaban (Xarelto) in patients with non-valvular atrial fibrillation (NVAF).
However, the EMA said further analyses of the study suggest the issue with the INRatio system did not affect the overall safety or benefit-risk balance of rivaroxaban. So rivaroxaban can continue to be used as before, in line with the current prescribing information.
The ROCKET AF study was the main clinical trial underpinning the use of rivaroxaban in patients with NVAF.
The results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism in these patients. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
The INRatio system was used to measure INRs in study subjects taking warfarin. Because of the defect, there were concerns that the system could have provided lower INR values in some patients in the warfarin group.
The lower values could, in turn, have led investigators to give too high a dose in the warfarin group, increasing their risk of bleeding and therefore giving a false impression of the comparative safety of rivaroxaban.
So the EMA’s Committee for Medicinal Products for Human Use (CHMP) assessed further analyses of the ROCKET AF study data, taking into account the defect of the INRatio system.
The CHMP concluded that any incorrect measurements obtained with the defective system would have had a marginal effect on the study results, and the safety of rivaroxaban remains unchanged.
In addition, the CHMP said data from other large studies confirmed the comparative safety of rivaroxaban and showed similar rates of bleeding in their warfarin groups.
The CHMP therefore concluded that the benefit-risk balance of rivaroxaban in patients with NVAF remains unchanged.
The CHMP’s assessment report, which includes detailed information on the analyses performed, will be published on the EMA’s website soon.
The EMA started investigating this issue as soon as it was informed of the defect of the INRatio system by the marketing authorization holder of rivaroxaban, Bayer Pharma AG, in September 2015.
Bayer said that, although the INRatio system was recalled in December 2014, the company and its development partner, Janssen, did not become aware of the defect until September 2015. ![]()
The European Medicines Agency (EMA) has concluded that a defect discovered in a system used to measure international normalized ratios (INRs) in the ROCKET AF study does not change the study’s overall conclusions.
The Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips) was recalled in December 2014 after it was found to produce falsely low test results.
A recent investigation by The BMJ suggested this defect may have impacted the results of ROCKET AF, in which researchers compared warfarin to rivaroxaban (Xarelto) in patients with non-valvular atrial fibrillation (NVAF).
However, the EMA said further analyses of the study suggest the issue with the INRatio system did not affect the overall safety or benefit-risk balance of rivaroxaban. So rivaroxaban can continue to be used as before, in line with the current prescribing information.
The ROCKET AF study was the main clinical trial underpinning the use of rivaroxaban in patients with NVAF.
The results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism in these patients. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
The INRatio system was used to measure INRs in study subjects taking warfarin. Because of the defect, there were concerns that the system could have provided lower INR values in some patients in the warfarin group.
The lower values could, in turn, have led investigators to give too high a dose in the warfarin group, increasing their risk of bleeding and therefore giving a false impression of the comparative safety of rivaroxaban.
So the EMA’s Committee for Medicinal Products for Human Use (CHMP) assessed further analyses of the ROCKET AF study data, taking into account the defect of the INRatio system.
The CHMP concluded that any incorrect measurements obtained with the defective system would have had a marginal effect on the study results, and the safety of rivaroxaban remains unchanged.
In addition, the CHMP said data from other large studies confirmed the comparative safety of rivaroxaban and showed similar rates of bleeding in their warfarin groups.
The CHMP therefore concluded that the benefit-risk balance of rivaroxaban in patients with NVAF remains unchanged.
The CHMP’s assessment report, which includes detailed information on the analyses performed, will be published on the EMA’s website soon.
The EMA started investigating this issue as soon as it was informed of the defect of the INRatio system by the marketing authorization holder of rivaroxaban, Bayer Pharma AG, in September 2015.
Bayer said that, although the INRatio system was recalled in December 2014, the company and its development partner, Janssen, did not become aware of the defect until September 2015. ![]()
The European Medicines Agency (EMA) has concluded that a defect discovered in a system used to measure international normalized ratios (INRs) in the ROCKET AF study does not change the study’s overall conclusions.
The Alere INRatio Monitor System (INRatio Monitor or INRatio2 Monitor and INRatio Test Strips) was recalled in December 2014 after it was found to produce falsely low test results.
A recent investigation by The BMJ suggested this defect may have impacted the results of ROCKET AF, in which researchers compared warfarin to rivaroxaban (Xarelto) in patients with non-valvular atrial fibrillation (NVAF).
However, the EMA said further analyses of the study suggest the issue with the INRatio system did not affect the overall safety or benefit-risk balance of rivaroxaban. So rivaroxaban can continue to be used as before, in line with the current prescribing information.
The ROCKET AF study was the main clinical trial underpinning the use of rivaroxaban in patients with NVAF.
The results suggested rivaroxaban was noninferior to warfarin for preventing stroke or systemic embolism in these patients. And there was no significant difference between the treatment arms with regard to major or nonmajor clinically relevant bleeding.
The INRatio system was used to measure INRs in study subjects taking warfarin. Because of the defect, there were concerns that the system could have provided lower INR values in some patients in the warfarin group.
The lower values could, in turn, have led investigators to give too high a dose in the warfarin group, increasing their risk of bleeding and therefore giving a false impression of the comparative safety of rivaroxaban.
So the EMA’s Committee for Medicinal Products for Human Use (CHMP) assessed further analyses of the ROCKET AF study data, taking into account the defect of the INRatio system.
The CHMP concluded that any incorrect measurements obtained with the defective system would have had a marginal effect on the study results, and the safety of rivaroxaban remains unchanged.
In addition, the CHMP said data from other large studies confirmed the comparative safety of rivaroxaban and showed similar rates of bleeding in their warfarin groups.
The CHMP therefore concluded that the benefit-risk balance of rivaroxaban in patients with NVAF remains unchanged.
The CHMP’s assessment report, which includes detailed information on the analyses performed, will be published on the EMA’s website soon.
The EMA started investigating this issue as soon as it was informed of the defect of the INRatio system by the marketing authorization holder of rivaroxaban, Bayer Pharma AG, in September 2015.
Bayer said that, although the INRatio system was recalled in December 2014, the company and its development partner, Janssen, did not become aware of the defect until September 2015. ![]()
A bone to pick
I have had a long and circuitous relationship with the radius. When I was 9 years old, I slipped on the wet grass of our front yard in an attempt to make a highlight-reel baseball catch and landed awkwardly on my left arm. After I continued to complain for a day and a half, my mother took me to see our pediatrician, Dr. Blum. After feeling up and down my forearm and asking me to squeeze his fingers, he pronounced me well.
A week and a half later, when I was still favoring what is my dominant arm, we returned to Dr. Blum. Apparently still unimpressed with my physical exam, he begrudgingly ordered an x-ray. Hurrah! I had a fracture! But then he announced that we would treat it with a splint and an ace wrap. Come on! Everyone knows that if you break a bone you get a cast. From then on he was Dr. Bum to me.

For the next 2 weeks, I was forbidden to play sports, meanwhile losing serious credibility points with my peers who suspected that I was a wimp and had fabricated the whole story. And of course, does anyone go around asking their friends to sign his ace wrap ... really? It took me years of overcompensation to regain even a hint of preteen machismo.
During my last year of medical school, I leapt at the opportunity to take an elective in pediatric orthopedics. It was great! With coaching from the residents, I learned when to suspect a buckle fracture of the radius, identify it on x-ray, and best of all, how to apply a cast.
When I finally entered practice here in Brunswick, we were seriously short of specialists, including orthopedists. When it was discovered that I knew how to apply a forearm cast, the orthopedists encouraged me to treat my own patients with simple forearm fractures. They were more than busy enough with really exciting stuff.
As an artist at heart, the chance to mold in plaster and plastic was a special treat. I took great pride in my creations, and making a beautiful crafted cast was sometimes the high point of my day. No splints or wimpy ace wraps for my patients!
Parents loved the one-stop shopping. History, exam, x-ray, casting, and out the door in less than an hour. Because I was the only primary care physician in town who was casting fractures, I occasionally had to remind the emergency room physicians to send me my patients with buckle fractures instead of knee-jerking a referral to an orthopedist.
But then about 10 years ago, some party-pooping orthopedists from who-knows-where looked at a very large series of pediatric patients with buckle fractures of the radius and discovered that those patients treated with splints had at least as good results as those who had been casted. And ... the patients and parents preferred the splints. I had to admit that maybe Dr. Blum wasn’t such a bum after all. Sadly, I had to respond to the evidence by giving up my hobby except when a child’s temperament or past history of injury suggested that he or she might benefit from the extra protection a cast could afford.
A recent study from Toronto published in Pediatrics, “Primary Care Physician Follow-up of Distal Radius Buckle Fractures,” by Koelink et al., makes me wonder whether a splint and ace wrap may even be overtreatment (Pediatrics. 2016 Jan;137[1]:11-9. doi: 10.1542/peds.2015-2262). In this review of 200 pediatric patients with distal radius buckle fractures, the investigators found that regardless of whether the primary care physician discussed how long to use the splint or when to return to activity, more than two-thirds of the patients wore their splints less than 3 weeks. Despite what the authors considered suboptimal primary care physician guidance, 99% of the patients returned to usual activities within 4 weeks.
My mother and Dr. Blum were on the right track from the beginning. They just needed to ignore my complaints a few days longer.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
I have had a long and circuitous relationship with the radius. When I was 9 years old, I slipped on the wet grass of our front yard in an attempt to make a highlight-reel baseball catch and landed awkwardly on my left arm. After I continued to complain for a day and a half, my mother took me to see our pediatrician, Dr. Blum. After feeling up and down my forearm and asking me to squeeze his fingers, he pronounced me well.
A week and a half later, when I was still favoring what is my dominant arm, we returned to Dr. Blum. Apparently still unimpressed with my physical exam, he begrudgingly ordered an x-ray. Hurrah! I had a fracture! But then he announced that we would treat it with a splint and an ace wrap. Come on! Everyone knows that if you break a bone you get a cast. From then on he was Dr. Bum to me.
For the next 2 weeks, I was forbidden to play sports, meanwhile losing serious credibility points with my peers who suspected that I was a wimp and had fabricated the whole story. And of course, does anyone go around asking their friends to sign his ace wrap ... really? It took me years of overcompensation to regain even a hint of preteen machismo.
During my last year of medical school, I leapt at the opportunity to take an elective in pediatric orthopedics. It was great! With coaching from the residents, I learned when to suspect a buckle fracture of the radius, identify it on x-ray, and best of all, how to apply a cast.
When I finally entered practice here in Brunswick, we were seriously short of specialists, including orthopedists. When it was discovered that I knew how to apply a forearm cast, the orthopedists encouraged me to treat my own patients with simple forearm fractures. They were more than busy enough with really exciting stuff.
As an artist at heart, the chance to mold in plaster and plastic was a special treat. I took great pride in my creations, and making a beautiful crafted cast was sometimes the high point of my day. No splints or wimpy ace wraps for my patients!
Parents loved the one-stop shopping. History, exam, x-ray, casting, and out the door in less than an hour. Because I was the only primary care physician in town who was casting fractures, I occasionally had to remind the emergency room physicians to send me my patients with buckle fractures instead of knee-jerking a referral to an orthopedist.
But then about 10 years ago, some party-pooping orthopedists from who-knows-where looked at a very large series of pediatric patients with buckle fractures of the radius and discovered that those patients treated with splints had at least as good results as those who had been casted. And ... the patients and parents preferred the splints. I had to admit that maybe Dr. Blum wasn’t such a bum after all. Sadly, I had to respond to the evidence by giving up my hobby except when a child’s temperament or past history of injury suggested that he or she might benefit from the extra protection a cast could afford.
A recent study from Toronto published in Pediatrics, “Primary Care Physician Follow-up of Distal Radius Buckle Fractures,” by Koelink et al., makes me wonder whether a splint and ace wrap may even be overtreatment (Pediatrics. 2016 Jan;137[1]:11-9. doi: 10.1542/peds.2015-2262). In this review of 200 pediatric patients with distal radius buckle fractures, the investigators found that regardless of whether the primary care physician discussed how long to use the splint or when to return to activity, more than two-thirds of the patients wore their splints less than 3 weeks. Despite what the authors considered suboptimal primary care physician guidance, 99% of the patients returned to usual activities within 4 weeks.
My mother and Dr. Blum were on the right track from the beginning. They just needed to ignore my complaints a few days longer.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”
I have had a long and circuitous relationship with the radius. When I was 9 years old, I slipped on the wet grass of our front yard in an attempt to make a highlight-reel baseball catch and landed awkwardly on my left arm. After I continued to complain for a day and a half, my mother took me to see our pediatrician, Dr. Blum. After feeling up and down my forearm and asking me to squeeze his fingers, he pronounced me well.
A week and a half later, when I was still favoring what is my dominant arm, we returned to Dr. Blum. Apparently still unimpressed with my physical exam, he begrudgingly ordered an x-ray. Hurrah! I had a fracture! But then he announced that we would treat it with a splint and an ace wrap. Come on! Everyone knows that if you break a bone you get a cast. From then on he was Dr. Bum to me.
For the next 2 weeks, I was forbidden to play sports, meanwhile losing serious credibility points with my peers who suspected that I was a wimp and had fabricated the whole story. And of course, does anyone go around asking their friends to sign his ace wrap ... really? It took me years of overcompensation to regain even a hint of preteen machismo.
During my last year of medical school, I leapt at the opportunity to take an elective in pediatric orthopedics. It was great! With coaching from the residents, I learned when to suspect a buckle fracture of the radius, identify it on x-ray, and best of all, how to apply a cast.
When I finally entered practice here in Brunswick, we were seriously short of specialists, including orthopedists. When it was discovered that I knew how to apply a forearm cast, the orthopedists encouraged me to treat my own patients with simple forearm fractures. They were more than busy enough with really exciting stuff.
As an artist at heart, the chance to mold in plaster and plastic was a special treat. I took great pride in my creations, and making a beautiful crafted cast was sometimes the high point of my day. No splints or wimpy ace wraps for my patients!
Parents loved the one-stop shopping. History, exam, x-ray, casting, and out the door in less than an hour. Because I was the only primary care physician in town who was casting fractures, I occasionally had to remind the emergency room physicians to send me my patients with buckle fractures instead of knee-jerking a referral to an orthopedist.
But then about 10 years ago, some party-pooping orthopedists from who-knows-where looked at a very large series of pediatric patients with buckle fractures of the radius and discovered that those patients treated with splints had at least as good results as those who had been casted. And ... the patients and parents preferred the splints. I had to admit that maybe Dr. Blum wasn’t such a bum after all. Sadly, I had to respond to the evidence by giving up my hobby except when a child’s temperament or past history of injury suggested that he or she might benefit from the extra protection a cast could afford.
A recent study from Toronto published in Pediatrics, “Primary Care Physician Follow-up of Distal Radius Buckle Fractures,” by Koelink et al., makes me wonder whether a splint and ace wrap may even be overtreatment (Pediatrics. 2016 Jan;137[1]:11-9. doi: 10.1542/peds.2015-2262). In this review of 200 pediatric patients with distal radius buckle fractures, the investigators found that regardless of whether the primary care physician discussed how long to use the splint or when to return to activity, more than two-thirds of the patients wore their splints less than 3 weeks. Despite what the authors considered suboptimal primary care physician guidance, 99% of the patients returned to usual activities within 4 weeks.
My mother and Dr. Blum were on the right track from the beginning. They just needed to ignore my complaints a few days longer.
Dr. Wilkoff practiced primary care pediatrics in Brunswick, Maine, for nearly 40 years. He has authored several books on behavioral pediatrics including “How to Say No to Your Toddler.”

U.S. flu activity steady in late January
Overall influenza-like illness (ILI) activity underwent a small increase in the United States during week 16 of the 2015-2016 flu season, but Puerto Rico took a significant turn for the worse as activity there rose to the highest possible level, the Centers for Disease Control and Prevention reported Feb. 5.
Puerto Rico, which had been at level 8 on the CDC’s 1-10 scale of flu activity for the previous 3 weeks, jumped up to level 10 for the week ending Jan. 30, 2016. Meanwhile, the main measure for national flu activity, the proportion of outpatient visits for ILI, was 2.2% for the second week in a row, just above the national baseline of 2.1%, the CDC said.
The state with the highest level of flu activity (level 7) for the week was Connecticut, with Arkansas the only other state in the “moderate” range at level 6. States in the “low” range were Florida, Hawaii, Illinois, Maryland, Nevada, New Jersey, and Pennsylvania at level 4 and Georgia, Idaho, Louisiana, Massachusetts, Mississippi, New Mexico, and Virginia at level 3. All told, there were 22 states with activity above level 1, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
There were two influenza-associated pediatric deaths reported during week 16, although one death actually occurred during the week ending Jan. 16. There have been nine flu-associated pediatric deaths reported so far during the 2015-2016 season.
The hospitalization rate for the season so far is 2.6/100,000 population, up from 2.1/100,000 last week. Adults aged 65 years and older had the highest rate at 8.5/100,000, with children aged 0-4 years next at 3.8/100,000.
Overall influenza-like illness (ILI) activity underwent a small increase in the United States during week 16 of the 2015-2016 flu season, but Puerto Rico took a significant turn for the worse as activity there rose to the highest possible level, the Centers for Disease Control and Prevention reported Feb. 5.
Puerto Rico, which had been at level 8 on the CDC’s 1-10 scale of flu activity for the previous 3 weeks, jumped up to level 10 for the week ending Jan. 30, 2016. Meanwhile, the main measure for national flu activity, the proportion of outpatient visits for ILI, was 2.2% for the second week in a row, just above the national baseline of 2.1%, the CDC said.
The state with the highest level of flu activity (level 7) for the week was Connecticut, with Arkansas the only other state in the “moderate” range at level 6. States in the “low” range were Florida, Hawaii, Illinois, Maryland, Nevada, New Jersey, and Pennsylvania at level 4 and Georgia, Idaho, Louisiana, Massachusetts, Mississippi, New Mexico, and Virginia at level 3. All told, there were 22 states with activity above level 1, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
There were two influenza-associated pediatric deaths reported during week 16, although one death actually occurred during the week ending Jan. 16. There have been nine flu-associated pediatric deaths reported so far during the 2015-2016 season.
The hospitalization rate for the season so far is 2.6/100,000 population, up from 2.1/100,000 last week. Adults aged 65 years and older had the highest rate at 8.5/100,000, with children aged 0-4 years next at 3.8/100,000.
Overall influenza-like illness (ILI) activity underwent a small increase in the United States during week 16 of the 2015-2016 flu season, but Puerto Rico took a significant turn for the worse as activity there rose to the highest possible level, the Centers for Disease Control and Prevention reported Feb. 5.
Puerto Rico, which had been at level 8 on the CDC’s 1-10 scale of flu activity for the previous 3 weeks, jumped up to level 10 for the week ending Jan. 30, 2016. Meanwhile, the main measure for national flu activity, the proportion of outpatient visits for ILI, was 2.2% for the second week in a row, just above the national baseline of 2.1%, the CDC said.
The state with the highest level of flu activity (level 7) for the week was Connecticut, with Arkansas the only other state in the “moderate” range at level 6. States in the “low” range were Florida, Hawaii, Illinois, Maryland, Nevada, New Jersey, and Pennsylvania at level 4 and Georgia, Idaho, Louisiana, Massachusetts, Mississippi, New Mexico, and Virginia at level 3. All told, there were 22 states with activity above level 1, according to data from the CDC’s Outpatient Influenza-like Illness Surveillance Network.
There were two influenza-associated pediatric deaths reported during week 16, although one death actually occurred during the week ending Jan. 16. There have been nine flu-associated pediatric deaths reported so far during the 2015-2016 season.
The hospitalization rate for the season so far is 2.6/100,000 population, up from 2.1/100,000 last week. Adults aged 65 years and older had the highest rate at 8.5/100,000, with children aged 0-4 years next at 3.8/100,000.