User login
Method may produce better CAR-NKTs

Photo by Aaron Logan
Researchers say they have discovered a method for expanding natural killer T cells (NKTs) that ensures their persistence, thereby making NKTs more attractive as chimeric antigen receptor (CAR) carriers for cancer immunotherapy.
When transduced with a CD19-specific CAR, the researchers’ persistent NKTs produced sustained tumor regression in a mouse model of B-cell lymphoma.
The team described this work in the Journal of Clinical Investigation.
“NKT technology is quite powerful and offers a significant potential for treatment of cancer,” said study author Leonid Metelitsa, MD, PhD, of Baylor College of Medicine in Houston, Texas.
“But for it to be most effective, we have to find the best way to expand the cells ex vivo while preserving their ability to persist once delivered back to patients. If they can persist in the body for a long time, they have much longer therapeutic activity, and this is essential for fighting cancer.”
Molecule affects persistence
The researchers noted that central memory T cells are known for their ability to proliferate and persist, and these cells are characterized by expression of the surface molecule CD62L.
In this study, the team found that NKT cells freshly derived from blood did not express CD62L, or it was expressed at a very low level. However, after the researchers expanded NKT cells, they found that CD62L was expressed at higher levels.
“We consistently identified a subset of cells present at very high numbers after the first 12 days of expansion, and critical to this subset of cells was the presence of CD62L,” Dr Metelitsa said. “In fact, they became the majority of the new cells.”
In addition, Dr Metelitsa said that CD62L-positive NKT cells were responsible for further propagation of NKTs in culture, which is important for achieving large numbers of cells. However, extensive culture led to the eventual decline of CD62L expression in NKTs.
To test the role of CD62L in NKT-cell persistence, the researchers delivered CD62L-positive and CD62L-negative NKTs to immune-deficient NSG mice. They found that CD62L-positive NKTs persisted 5 times longer than CD62L-negative NKTs.
CAR-NKTs fight lymphoma
Next, the researchers transduced CD62L-positive and CD62L-negative NKTs with a CD19-specific CAR and delivered these cells to mice with B-cell lymphoma.
The team found that both CD62L-positve and CD62L-negative CAR-NKTs prolonged the survival of mice, when compared to controls (P<0.001).
However, only the CD62L-positive CAR-NKTs induced sustained tumor regression. Seven of 9 mice that received CD62L-positive CAR-NKTs lived, and 5 were tumor-free for at least 3 months. But all 10 mice that received CD62L-negative CAR-NKTs ultimately succumbed to tumor progression (P<0.001).
Costimulation improves NKTs/CAR-NKTs
The researchers then turned their focus to costimulation of NKTs in order to maintain the subset with a high percentage of CD62L-positive cells in prolonged culture. Costimulation involves the interaction of receptors on NKTs with activating molecules on an antigen-presenting cell to increase the NKTs’ immune functions.
“We have known that costimulation is an important part of immune response and immunotherapy, but, in this case, we did not know which costimulatory molecules would be important for the expansion and persistence of CD62L-positive NKT cells,” said Gengwen Tian, MD, of the Baylor College of Medicine.
After testing more than 100 combinations, the researchers discovered that combining an antigen-presenting molecule—CD1d—with 3 costimulatory molecules—CD86, 4-1BBL, and OX40L—induced prolonged persistence and better therapeutic activity of NKTs and CAR-NKTs in mouse models.
“When we developed an antigen-presenting cell clone that expressed CD1d with all of these costimulatory molecules at certain levels, NKT cells maintained a high percentage of CD62L even in a prolonged culture,” Dr Metelitsa said.
The researchers conducted in vivo testing of CAR-NKT cells that were expanded with the original method or with the costimulation method. And they found that costimulated cells had significantly higher therapeutic activity in mouse models of neuroblastoma and lymphoma.
“Our goal now is to optimize our NKT cell expansion protocol so that we can obtain FDA approval to initiate clinical trials,” Dr Metelitsa said. ![]()
Photo by Aaron Logan
Researchers say they have discovered a method for expanding natural killer T cells (NKTs) that ensures their persistence, thereby making NKTs more attractive as chimeric antigen receptor (CAR) carriers for cancer immunotherapy.
When transduced with a CD19-specific CAR, the researchers’ persistent NKTs produced sustained tumor regression in a mouse model of B-cell lymphoma.
The team described this work in the Journal of Clinical Investigation.
“NKT technology is quite powerful and offers a significant potential for treatment of cancer,” said study author Leonid Metelitsa, MD, PhD, of Baylor College of Medicine in Houston, Texas.
“But for it to be most effective, we have to find the best way to expand the cells ex vivo while preserving their ability to persist once delivered back to patients. If they can persist in the body for a long time, they have much longer therapeutic activity, and this is essential for fighting cancer.”
Molecule affects persistence
The researchers noted that central memory T cells are known for their ability to proliferate and persist, and these cells are characterized by expression of the surface molecule CD62L.
In this study, the team found that NKT cells freshly derived from blood did not express CD62L, or it was expressed at a very low level. However, after the researchers expanded NKT cells, they found that CD62L was expressed at higher levels.
“We consistently identified a subset of cells present at very high numbers after the first 12 days of expansion, and critical to this subset of cells was the presence of CD62L,” Dr Metelitsa said. “In fact, they became the majority of the new cells.”
In addition, Dr Metelitsa said that CD62L-positive NKT cells were responsible for further propagation of NKTs in culture, which is important for achieving large numbers of cells. However, extensive culture led to the eventual decline of CD62L expression in NKTs.
To test the role of CD62L in NKT-cell persistence, the researchers delivered CD62L-positive and CD62L-negative NKTs to immune-deficient NSG mice. They found that CD62L-positive NKTs persisted 5 times longer than CD62L-negative NKTs.
CAR-NKTs fight lymphoma
Next, the researchers transduced CD62L-positive and CD62L-negative NKTs with a CD19-specific CAR and delivered these cells to mice with B-cell lymphoma.
The team found that both CD62L-positve and CD62L-negative CAR-NKTs prolonged the survival of mice, when compared to controls (P<0.001).
However, only the CD62L-positive CAR-NKTs induced sustained tumor regression. Seven of 9 mice that received CD62L-positive CAR-NKTs lived, and 5 were tumor-free for at least 3 months. But all 10 mice that received CD62L-negative CAR-NKTs ultimately succumbed to tumor progression (P<0.001).
Costimulation improves NKTs/CAR-NKTs
The researchers then turned their focus to costimulation of NKTs in order to maintain the subset with a high percentage of CD62L-positive cells in prolonged culture. Costimulation involves the interaction of receptors on NKTs with activating molecules on an antigen-presenting cell to increase the NKTs’ immune functions.
“We have known that costimulation is an important part of immune response and immunotherapy, but, in this case, we did not know which costimulatory molecules would be important for the expansion and persistence of CD62L-positive NKT cells,” said Gengwen Tian, MD, of the Baylor College of Medicine.
After testing more than 100 combinations, the researchers discovered that combining an antigen-presenting molecule—CD1d—with 3 costimulatory molecules—CD86, 4-1BBL, and OX40L—induced prolonged persistence and better therapeutic activity of NKTs and CAR-NKTs in mouse models.
“When we developed an antigen-presenting cell clone that expressed CD1d with all of these costimulatory molecules at certain levels, NKT cells maintained a high percentage of CD62L even in a prolonged culture,” Dr Metelitsa said.
The researchers conducted in vivo testing of CAR-NKT cells that were expanded with the original method or with the costimulation method. And they found that costimulated cells had significantly higher therapeutic activity in mouse models of neuroblastoma and lymphoma.
“Our goal now is to optimize our NKT cell expansion protocol so that we can obtain FDA approval to initiate clinical trials,” Dr Metelitsa said. ![]()
Photo by Aaron Logan
Researchers say they have discovered a method for expanding natural killer T cells (NKTs) that ensures their persistence, thereby making NKTs more attractive as chimeric antigen receptor (CAR) carriers for cancer immunotherapy.
When transduced with a CD19-specific CAR, the researchers’ persistent NKTs produced sustained tumor regression in a mouse model of B-cell lymphoma.
The team described this work in the Journal of Clinical Investigation.
“NKT technology is quite powerful and offers a significant potential for treatment of cancer,” said study author Leonid Metelitsa, MD, PhD, of Baylor College of Medicine in Houston, Texas.
“But for it to be most effective, we have to find the best way to expand the cells ex vivo while preserving their ability to persist once delivered back to patients. If they can persist in the body for a long time, they have much longer therapeutic activity, and this is essential for fighting cancer.”
Molecule affects persistence
The researchers noted that central memory T cells are known for their ability to proliferate and persist, and these cells are characterized by expression of the surface molecule CD62L.
In this study, the team found that NKT cells freshly derived from blood did not express CD62L, or it was expressed at a very low level. However, after the researchers expanded NKT cells, they found that CD62L was expressed at higher levels.
“We consistently identified a subset of cells present at very high numbers after the first 12 days of expansion, and critical to this subset of cells was the presence of CD62L,” Dr Metelitsa said. “In fact, they became the majority of the new cells.”
In addition, Dr Metelitsa said that CD62L-positive NKT cells were responsible for further propagation of NKTs in culture, which is important for achieving large numbers of cells. However, extensive culture led to the eventual decline of CD62L expression in NKTs.
To test the role of CD62L in NKT-cell persistence, the researchers delivered CD62L-positive and CD62L-negative NKTs to immune-deficient NSG mice. They found that CD62L-positive NKTs persisted 5 times longer than CD62L-negative NKTs.
CAR-NKTs fight lymphoma
Next, the researchers transduced CD62L-positive and CD62L-negative NKTs with a CD19-specific CAR and delivered these cells to mice with B-cell lymphoma.
The team found that both CD62L-positve and CD62L-negative CAR-NKTs prolonged the survival of mice, when compared to controls (P<0.001).
However, only the CD62L-positive CAR-NKTs induced sustained tumor regression. Seven of 9 mice that received CD62L-positive CAR-NKTs lived, and 5 were tumor-free for at least 3 months. But all 10 mice that received CD62L-negative CAR-NKTs ultimately succumbed to tumor progression (P<0.001).
Costimulation improves NKTs/CAR-NKTs
The researchers then turned their focus to costimulation of NKTs in order to maintain the subset with a high percentage of CD62L-positive cells in prolonged culture. Costimulation involves the interaction of receptors on NKTs with activating molecules on an antigen-presenting cell to increase the NKTs’ immune functions.
“We have known that costimulation is an important part of immune response and immunotherapy, but, in this case, we did not know which costimulatory molecules would be important for the expansion and persistence of CD62L-positive NKT cells,” said Gengwen Tian, MD, of the Baylor College of Medicine.
After testing more than 100 combinations, the researchers discovered that combining an antigen-presenting molecule—CD1d—with 3 costimulatory molecules—CD86, 4-1BBL, and OX40L—induced prolonged persistence and better therapeutic activity of NKTs and CAR-NKTs in mouse models.
“When we developed an antigen-presenting cell clone that expressed CD1d with all of these costimulatory molecules at certain levels, NKT cells maintained a high percentage of CD62L even in a prolonged culture,” Dr Metelitsa said.
The researchers conducted in vivo testing of CAR-NKT cells that were expanded with the original method or with the costimulation method. And they found that costimulated cells had significantly higher therapeutic activity in mouse models of neuroblastoma and lymphoma.
“Our goal now is to optimize our NKT cell expansion protocol so that we can obtain FDA approval to initiate clinical trials,” Dr Metelitsa said. ![]()
HIV patients undertreated for lymphoma, other cancers
cultured lymphocyte
Image courtesy of the CDC
A new study suggests that cancer patients with HIV are less likely to receive cancer treatment, regardless of insurance status and comorbidities.
Patients with HIV were less likely than their HIV-free peers to receive treatment for Hodgkin lymphoma, diffuse large B-cell lymphoma, and 7 solid tumor malignancies.
Gita Suneja, MD, of the University of Utah in Salt Lake City, and her colleagues reported these findings in Cancer.
The team used the National Cancer Data Base to study non-elderly US adults diagnosed with 10 common cancers from 2003 to 2011. There were a total of 10,265 HIV-infected patients and 2,219,232 HIV-free patients.
The researchers examined associations between HIV status and lack of cancer treatment, taking into account insurance status and comorbidities.
The results showed a lack of treatment among HIV patients for all of the cancers studied except anal cancer (relative risk [RR]=1.20, P=0.333).
So HIV-infected patients were more likely to lack cancer treatment for:
- Hodgkin lymphoma (RR=1.92, P<0.001)
- Diffuse large B-cell lymphoma (RR=1.82, P<0.001)
- Head and neck cancer (RR=1.48, P=0.013)
- Upper gastrointestinal tract cancer (RR=2.62, P<0.001)
- Colorectal cancer (RR=1.70, P=0.006)
- Lung cancer (RR=2.46, P<0.001)
- Breast cancer (RR=2.14, P=0.015)
- Cervical cancer (RR=2.81, P<0.001)
- Prostate cancer (RR=2.16, P<0.001).
The researchers said factors that predicted a lack of cancer treatment among HIV-infected individuals varied between those with solid tumors and those with lymphomas.
Advanced stage at the time of cancer diagnosis (stage IV vs stage I) meant HIV patients with solid tumors were less likely to receive cancer treatment, but lymphoma patients were more likely to receive cancer treatment.
Having a higher modified Charlson-Deyo comorbidity score (1 or 2+ vs 0) predicted a lack of cancer treatment for HIV-infected patients with lymphoma but not those with solid tumors.
And older age (45-64 vs 18-44) was associated with a lack of treatment for HIV-infected patients regardless of cancer type, but this was only significant for lymphoma patients.
For the entire cohort, black race (vs white) and a lack of private insurance (Medicaid, Medicare, uninsured, or unknown insurance status) were significant predictors for a lack of cancer treatment among HIV patients.
Still, the researchers noted that, even among privately insured patients, HIV-infected individuals were less likely to receive cancer treatment.
Dr Suneja and her colleagues said several factors may contribute to these findings. For one, HIV-infected patients have historically been excluded from cancer clinical trials, thereby limiting the applicability of trial results for this population.
In addition, cancer treatment guidelines specific to HIV-infected patients are not available for most cancer types. And clinicians may lack experience treating HIV-infected patients with cancer.
Furthermore, the psychosocial and economic challenges associated with the dual management of cancer and HIV treatment may make adherence to treatment a challenge. ![]()
cultured lymphocyte
Image courtesy of the CDC
A new study suggests that cancer patients with HIV are less likely to receive cancer treatment, regardless of insurance status and comorbidities.
Patients with HIV were less likely than their HIV-free peers to receive treatment for Hodgkin lymphoma, diffuse large B-cell lymphoma, and 7 solid tumor malignancies.
Gita Suneja, MD, of the University of Utah in Salt Lake City, and her colleagues reported these findings in Cancer.
The team used the National Cancer Data Base to study non-elderly US adults diagnosed with 10 common cancers from 2003 to 2011. There were a total of 10,265 HIV-infected patients and 2,219,232 HIV-free patients.
The researchers examined associations between HIV status and lack of cancer treatment, taking into account insurance status and comorbidities.
The results showed a lack of treatment among HIV patients for all of the cancers studied except anal cancer (relative risk [RR]=1.20, P=0.333).
So HIV-infected patients were more likely to lack cancer treatment for:
- Hodgkin lymphoma (RR=1.92, P<0.001)
- Diffuse large B-cell lymphoma (RR=1.82, P<0.001)
- Head and neck cancer (RR=1.48, P=0.013)
- Upper gastrointestinal tract cancer (RR=2.62, P<0.001)
- Colorectal cancer (RR=1.70, P=0.006)
- Lung cancer (RR=2.46, P<0.001)
- Breast cancer (RR=2.14, P=0.015)
- Cervical cancer (RR=2.81, P<0.001)
- Prostate cancer (RR=2.16, P<0.001).
The researchers said factors that predicted a lack of cancer treatment among HIV-infected individuals varied between those with solid tumors and those with lymphomas.
Advanced stage at the time of cancer diagnosis (stage IV vs stage I) meant HIV patients with solid tumors were less likely to receive cancer treatment, but lymphoma patients were more likely to receive cancer treatment.
Having a higher modified Charlson-Deyo comorbidity score (1 or 2+ vs 0) predicted a lack of cancer treatment for HIV-infected patients with lymphoma but not those with solid tumors.
And older age (45-64 vs 18-44) was associated with a lack of treatment for HIV-infected patients regardless of cancer type, but this was only significant for lymphoma patients.
For the entire cohort, black race (vs white) and a lack of private insurance (Medicaid, Medicare, uninsured, or unknown insurance status) were significant predictors for a lack of cancer treatment among HIV patients.
Still, the researchers noted that, even among privately insured patients, HIV-infected individuals were less likely to receive cancer treatment.
Dr Suneja and her colleagues said several factors may contribute to these findings. For one, HIV-infected patients have historically been excluded from cancer clinical trials, thereby limiting the applicability of trial results for this population.
In addition, cancer treatment guidelines specific to HIV-infected patients are not available for most cancer types. And clinicians may lack experience treating HIV-infected patients with cancer.
Furthermore, the psychosocial and economic challenges associated with the dual management of cancer and HIV treatment may make adherence to treatment a challenge. ![]()
cultured lymphocyte
Image courtesy of the CDC
A new study suggests that cancer patients with HIV are less likely to receive cancer treatment, regardless of insurance status and comorbidities.
Patients with HIV were less likely than their HIV-free peers to receive treatment for Hodgkin lymphoma, diffuse large B-cell lymphoma, and 7 solid tumor malignancies.
Gita Suneja, MD, of the University of Utah in Salt Lake City, and her colleagues reported these findings in Cancer.
The team used the National Cancer Data Base to study non-elderly US adults diagnosed with 10 common cancers from 2003 to 2011. There were a total of 10,265 HIV-infected patients and 2,219,232 HIV-free patients.
The researchers examined associations between HIV status and lack of cancer treatment, taking into account insurance status and comorbidities.
The results showed a lack of treatment among HIV patients for all of the cancers studied except anal cancer (relative risk [RR]=1.20, P=0.333).
So HIV-infected patients were more likely to lack cancer treatment for:
- Hodgkin lymphoma (RR=1.92, P<0.001)
- Diffuse large B-cell lymphoma (RR=1.82, P<0.001)
- Head and neck cancer (RR=1.48, P=0.013)
- Upper gastrointestinal tract cancer (RR=2.62, P<0.001)
- Colorectal cancer (RR=1.70, P=0.006)
- Lung cancer (RR=2.46, P<0.001)
- Breast cancer (RR=2.14, P=0.015)
- Cervical cancer (RR=2.81, P<0.001)
- Prostate cancer (RR=2.16, P<0.001).
The researchers said factors that predicted a lack of cancer treatment among HIV-infected individuals varied between those with solid tumors and those with lymphomas.
Advanced stage at the time of cancer diagnosis (stage IV vs stage I) meant HIV patients with solid tumors were less likely to receive cancer treatment, but lymphoma patients were more likely to receive cancer treatment.
Having a higher modified Charlson-Deyo comorbidity score (1 or 2+ vs 0) predicted a lack of cancer treatment for HIV-infected patients with lymphoma but not those with solid tumors.
And older age (45-64 vs 18-44) was associated with a lack of treatment for HIV-infected patients regardless of cancer type, but this was only significant for lymphoma patients.
For the entire cohort, black race (vs white) and a lack of private insurance (Medicaid, Medicare, uninsured, or unknown insurance status) were significant predictors for a lack of cancer treatment among HIV patients.
Still, the researchers noted that, even among privately insured patients, HIV-infected individuals were less likely to receive cancer treatment.
Dr Suneja and her colleagues said several factors may contribute to these findings. For one, HIV-infected patients have historically been excluded from cancer clinical trials, thereby limiting the applicability of trial results for this population.
In addition, cancer treatment guidelines specific to HIV-infected patients are not available for most cancer types. And clinicians may lack experience treating HIV-infected patients with cancer.
Furthermore, the psychosocial and economic challenges associated with the dual management of cancer and HIV treatment may make adherence to treatment a challenge. ![]()
PD-1 inhibitor granted accelerated approval for cHL

Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Photo courtesy of Business Wire
The US Food and Drug Administration (FDA) has granted accelerated approval for the PD-1 inhibitor nivolumab (Opdivo) to treat classical Hodgkin lymphoma (cHL).
The drug is approved to treat patients with relapsed or refractory cHL who have received an autologous hematopoietic stem cell transplant (HSCT) and post-transplant brentuximab vedotin.
Nivolumab received accelerated approval because it has not yet shown a clinical benefit in these patients. The FDA’s accelerated approval program allows conditional approval of a drug that fills an unmet medical need for a serious condition.
Accelerated approval is based on a surrogate or intermediate endpoint—in this case, overall response rate—that is reasonably likely to predict clinical benefit. Continued approval of nivolumab for the aforementioned indication may be contingent upon verification of clinical benefit in confirmatory trials.
The FDA previously granted nivolumab breakthrough therapy designation, priority review status, and orphan drug designation.
Dosing and precautions
The recommended dose and schedule of nivolumab for cHL patients is 3 mg/kg intravenously every 2 weeks until disease progression or unacceptable toxicity.
The FDA added a new “Warning and Precaution” to the label for nivolumab, regarding complications of allogeneic HSCT after nivolumab.
Transplant-related deaths have occurred. So the FDA said healthcare professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions.
The FDA has required the manufacturer of nivolumab, Bristol-Myers Squibb, to further study the safety of allogeneic HSCT after nivolumab.
Full prescribing information for the drug is available here.
Trials of nivolumab
The FDA granted nivolumab accelerated approval in cHL patients based on the results of 2 single-arm, multicenter trials—the phase 1 Checkmate 039 trial (presented at ICML last year) and the phase 2 CheckMate 205 trial (to be presented at ASCO 2016).
Efficacy
Thus far, researchers have evaluated the efficacy of nivolumab in 95 cHL patients from both trials. All of these patients previously received an autologous HSCT and post-transplant brentuximab vedotin. They received a median of 5 prior systemic regimens (range, 3 to 15).
The patients received a median of 17 doses of nivolumab (range, 3 to 48). The overall response rate was 65%, and the complete response rate was 7%.
The median time to response was 2.1 months (range, 0.7 to 5.7), and the estimated median duration of response was 8.7 months (range, 0+ to 23.1+).
Safety
Researchers evaluated the safety of nivolumab in 263 patients with relapsed or refractory cHL. Ninety-eight percent of these patients had received an autologous HSCT. The patients received a median of 10 doses of nivolumab (range, 1 to 48) at the approved dose and schedule.
The most common (≥20%) adverse events (AEs) of any grade were fatigue, upper respiratory tract infection, cough, pyrexia, and diarrhea.
Additional common (≥10%) AEs included rash, pruritus, musculoskeletal pain, nausea, vomiting, abdominal pain, headache, peripheral neuropathy, arthralgia, dyspnea, infusion-related reactions, and hypothyroidism or thyroiditis.
Serious AEs were reported in 21% of patients. The most common, reported in 1% to 3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash. ![]()
Readmission Rates and Mortality Measures
The Centers for Medicare & Medicaid Services (CMS) have sought to reduce readmissions in the 30 days following hospital discharge through penalties applied to hospitals with readmission rates that are higher than expected. Expected readmission rates for Medicare fee‐for‐service beneficiaries are calculated from models that use patient‐level administrative data to account for patient morbidities. Readmitted patients are defined as those who are discharged from the hospital alive and then rehospitalized at any acute care facility within 30 days of discharge. These models explicitly exclude sociodemographic variables that may impact quality of and access to outpatient care. Specific exclusions are also applied based on diagnosis codes so as to avoid penalizing hospitals for rehospitalizations that are likely to have been planned.
More recently, a hospital‐wide readmission measure has been developed, which seeks to provide a comprehensive view of each hospital's readmission rate by including the vast majority of Medicare patients. Like the condition‐specific readmission measures, the hospital‐wide readmission measure also excludes sociodemographic variables and incorporates specific condition‐based exclusions so as to avoid counting planned rehospitalizations (e.g., an admission for cholecystectomy following an admission for biliary sepsis). Although not currently used for pay‐for‐performance, this measure has been included in the CMS Star Report along with other readmission measures.[1] CMS does not currently disseminate a hospital‐wide mortality measure, but does disseminate hospital‐level adjusted 30‐day mortality rates for Medicare beneficiaries with discharge diagnoses of stroke, heart failure, myocardial infarction (MI), chronic obstructive pulmonary disease (COPD) and pneumonia, and principal procedure of coronary artery bypass grafting (CABG).
It is conceivable that aggressive efforts to reduce readmissions might delay life‐saving acute care in some scenarios,[2] and there is prior evidence that heart failure readmissions are inversely (but weakly) related to heart failure mortality.[3] It is also plausible that keeping tenuous patients alive until discharge might result in higher readmission rates. We sought to examine the relationship between hospital‐wide adjusted 30‐day readmissions and death rates across the acute care hospitals in the United States. Lacking a measure of hospital‐wide death rates, we examined the relation between hospital‐wide readmissions and each of the 6 condition‐specific mortality measures. For comparison, we also examined the relationships between condition‐specific readmission rates and mortality rates.
METHODS
We used publically available data published by CMS from July 1, 2011 through June 30, 2014.[4] These data are provided at the hospital level, without any patient‐level data. We included 4452 acute care facilities based on having hospital‐wide readmission rates, but not all facilities contributed data for each mortality measure. We excluded from analysis on a measure‐by‐measure basis those facilities for which outcomes were absent, without imputing missing outcome measures, because low volume of a given condition was the main reason for not reporting a measure. For each mortality measure, we constructed a logistic regression model to quantify the odds of performing in the lowest (best) mortality tertile as a function of hospital‐wide readmission tertile. To account for patient volumes, we included in each model the number of eligible patients at each hospital with the specified condition. We repeated these analyses using condition‐specific readmission rates (rather than the hospital‐wide readmission rates) as the independent variable. Specifications for CMS models for mortality and readmissions are publically available.[5]
RESULTS
After adjustment for patient volumes, hospitals in the highest hospital‐wide readmission tertile were more likely to perform in the lowest (best) mortality tertile for 3 of the 6 mortality measures: heart failure, COPD, and stroke (P 0.001 for all). For MI, CABG and pneumonia, there was no significant association between high hospital‐wide readmission rates and low mortality (Table 1). Using condition‐specific readmission rates, there remained an inverse association between readmissions and mortality for heart failure and stroke, but not for COPD. In contrast, hospitals with the highest CABG‐specific readmission rates were significantly less likely to have low CABG‐specific mortality (P 0.001).
| Hospital‐Wide Readmission Rate Tertile [Range of Adjusted Readmission Rates, %]* | |||
|---|---|---|---|
|
1st Tertile, n = 1359 [11.3%‐14.8%], Adjusted Odds Ratio (95% CI) |
2nd Tertile, n = 1785 [14.9%‐15.5%], Adjusted Odds Ratio (95% CI) |
3rd Tertile, n = 1308 [15.6%‐19.8%], Adjusted Odds Ratio (95% CI) |
|
| |||
| Mortality measure (no. of hospitals reporting) | |||
| Acute myocardial infarction (n = 2415) | 1.00 (referent) | 0.88 (0.711.09) | 1.02 (0.831.25) |
| Pneumonia (n = 4067) | 1.00 (referent) | 0.83 (0.710.98) | 1.11 (0.941.31) |
| Heart failure (n = 3668) | 1.00 (referent) | 1.21 (1.021.45) | 1.94 (1.632.30) |
| Stroke (n = 2754) | 1.00 (referent) | 1.13 (0.931.38) | 1.48 (1.221.79) |
| Chronic obstructive pulmonary disease (n = 3633) | 1.00 (referent) | 1.12 (0.951.33) | 1.73 (1.462.05) |
| Coronary artery bypass (n = 1058) | 1.00 (referent) | 0.87 (0.631.19) | 0.99 (0.741.34) |
| Condition‐specific readmission rate tertile | |||
| Mortality measure | |||
| Acute myocardial infarction | 1.00 (referent) | 0.88 (0.711.08) | 0.79 (0.640.99) |
| Pneumonia | 1.00 (referent) | 0.91 (0.781.07) | 0.89 (0.761.04) |
| Heart failure | 1.00 (referent) | 1.15 (0.961.36) | 1.56 (1.311.86) |
| Stroke | 1.00 (referent) | 1.65 (1.342.03) | 1.70 (1.232.35) |
| Chronic obstructive pulmonary disease | 1.00 (referent) | 0.83 (0.700.98) | 0.84 (0.710.99) |
| Coronary artery bypass | 1.00 (referent) | 0.59 (0.440.80) | 0.47 (0.340.64) |
DISCUSSION
We found that higher hospital‐wide readmission rates were associated with lower mortality at the hospital level for 3 of the 6 mortality measures we examined. The findings for heart failure parallel the findings of Krumholz and colleagues who examined 3 of these 6 measures (MI, pneumonia, and heart failure) in relation to readmissions for these specific populations.[3] This prior analysis, however, did not include the 3 more recently reported mortality measures (COPD, stroke, and CABG) and did not use hospital‐wide readmissions.
Causal mechanisms underlying the associations between mortality and readmission at the hospital level deserve further exploration. It is certainly possible that global efforts to keep patients out of the hospital might, in some instances, place patients at risk by delaying necessary acute care.[2] It is also possible that unmeasured variables, particularly access to hospice and palliative care services that might facilitate good deaths, could be associated with both reduced readmissions and higher death rates. Additionally, because deceased patients cannot be readmitted, one might expect that readmissions and mortality might be inversely associated, particularly for conditions with a high postdischarge mortality rate. Similarly, a hospital that does a particularly good job keeping chronically ill patients alive until discharge might exhibit a higher readmission rate than a hospital that is less adept at keeping tenuous patients alive until discharge.
Regardless of the mechanisms of these findings, we present these data to raise the concern that using readmission rates, particularly hospital‐wide readmission rates, as a measure of hospital quality is inherently problematic. It is particularly problematic that CMS has applied equal weight to readmissions and mortality in the Star Report.[1] High readmission rates may result from complications and poor handoffs, but may also stem from the legitimate need to care for chronically ill patients in a high‐intensity setting, particularly fragile patients who have been kept alive against the odds. In conclusion, caution is warranted in viewing readmissions as a quality metric until the associations we describe are better explained using patient‐level data and more robust adjustment than is possible with these publically available data.
Disclosures: Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. There was no financial support for this work. Contributions of the authors are as follows: drafting manuscript (Brotman), revision of manuscript for important intellectual content (brotman, Hoyer, Lepley, Deutschendorf, Leung), acquisition of data (Deutschendorf, Leung, Lepley), interpretation of data (Brotman, Hoyer, Lepley, Deutschendorf, Leung), data analysis (Brotman, Hoyer).
- Centers for Medicare and Medicaid Services. Available at: https://www.cms.gov/Outreach-and-Education/Outreach/NPC/Downloads/2015-08-13-Star-Ratings-Presentation.pdf. Accessed September 2015.
- , , , et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med. 2012;156(10):673–683.
- , , , et al. Relationship between hospital readmission and mortality rates for patients hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2013;309(6):587–593.
- Centers for Medicare and Medicaid Services. Hospital compare datasets. Available at: https://data.medicare.gov/data/hospital‐compare. Accessed September 2015.
- Centers for Medicare and Medicaid Services. Hospital quality initiative. Available at: https://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/HospitalQualityInits. Accessed September 2015.
The Centers for Medicare & Medicaid Services (CMS) have sought to reduce readmissions in the 30 days following hospital discharge through penalties applied to hospitals with readmission rates that are higher than expected. Expected readmission rates for Medicare fee‐for‐service beneficiaries are calculated from models that use patient‐level administrative data to account for patient morbidities. Readmitted patients are defined as those who are discharged from the hospital alive and then rehospitalized at any acute care facility within 30 days of discharge. These models explicitly exclude sociodemographic variables that may impact quality of and access to outpatient care. Specific exclusions are also applied based on diagnosis codes so as to avoid penalizing hospitals for rehospitalizations that are likely to have been planned.
More recently, a hospital‐wide readmission measure has been developed, which seeks to provide a comprehensive view of each hospital's readmission rate by including the vast majority of Medicare patients. Like the condition‐specific readmission measures, the hospital‐wide readmission measure also excludes sociodemographic variables and incorporates specific condition‐based exclusions so as to avoid counting planned rehospitalizations (e.g., an admission for cholecystectomy following an admission for biliary sepsis). Although not currently used for pay‐for‐performance, this measure has been included in the CMS Star Report along with other readmission measures.[1] CMS does not currently disseminate a hospital‐wide mortality measure, but does disseminate hospital‐level adjusted 30‐day mortality rates for Medicare beneficiaries with discharge diagnoses of stroke, heart failure, myocardial infarction (MI), chronic obstructive pulmonary disease (COPD) and pneumonia, and principal procedure of coronary artery bypass grafting (CABG).
It is conceivable that aggressive efforts to reduce readmissions might delay life‐saving acute care in some scenarios,[2] and there is prior evidence that heart failure readmissions are inversely (but weakly) related to heart failure mortality.[3] It is also plausible that keeping tenuous patients alive until discharge might result in higher readmission rates. We sought to examine the relationship between hospital‐wide adjusted 30‐day readmissions and death rates across the acute care hospitals in the United States. Lacking a measure of hospital‐wide death rates, we examined the relation between hospital‐wide readmissions and each of the 6 condition‐specific mortality measures. For comparison, we also examined the relationships between condition‐specific readmission rates and mortality rates.
METHODS
We used publically available data published by CMS from July 1, 2011 through June 30, 2014.[4] These data are provided at the hospital level, without any patient‐level data. We included 4452 acute care facilities based on having hospital‐wide readmission rates, but not all facilities contributed data for each mortality measure. We excluded from analysis on a measure‐by‐measure basis those facilities for which outcomes were absent, without imputing missing outcome measures, because low volume of a given condition was the main reason for not reporting a measure. For each mortality measure, we constructed a logistic regression model to quantify the odds of performing in the lowest (best) mortality tertile as a function of hospital‐wide readmission tertile. To account for patient volumes, we included in each model the number of eligible patients at each hospital with the specified condition. We repeated these analyses using condition‐specific readmission rates (rather than the hospital‐wide readmission rates) as the independent variable. Specifications for CMS models for mortality and readmissions are publically available.[5]
RESULTS
After adjustment for patient volumes, hospitals in the highest hospital‐wide readmission tertile were more likely to perform in the lowest (best) mortality tertile for 3 of the 6 mortality measures: heart failure, COPD, and stroke (P 0.001 for all). For MI, CABG and pneumonia, there was no significant association between high hospital‐wide readmission rates and low mortality (Table 1). Using condition‐specific readmission rates, there remained an inverse association between readmissions and mortality for heart failure and stroke, but not for COPD. In contrast, hospitals with the highest CABG‐specific readmission rates were significantly less likely to have low CABG‐specific mortality (P 0.001).
| Hospital‐Wide Readmission Rate Tertile [Range of Adjusted Readmission Rates, %]* | |||
|---|---|---|---|
|
1st Tertile, n = 1359 [11.3%‐14.8%], Adjusted Odds Ratio (95% CI) |
2nd Tertile, n = 1785 [14.9%‐15.5%], Adjusted Odds Ratio (95% CI) |
3rd Tertile, n = 1308 [15.6%‐19.8%], Adjusted Odds Ratio (95% CI) |
|
| |||
| Mortality measure (no. of hospitals reporting) | |||
| Acute myocardial infarction (n = 2415) | 1.00 (referent) | 0.88 (0.711.09) | 1.02 (0.831.25) |
| Pneumonia (n = 4067) | 1.00 (referent) | 0.83 (0.710.98) | 1.11 (0.941.31) |
| Heart failure (n = 3668) | 1.00 (referent) | 1.21 (1.021.45) | 1.94 (1.632.30) |
| Stroke (n = 2754) | 1.00 (referent) | 1.13 (0.931.38) | 1.48 (1.221.79) |
| Chronic obstructive pulmonary disease (n = 3633) | 1.00 (referent) | 1.12 (0.951.33) | 1.73 (1.462.05) |
| Coronary artery bypass (n = 1058) | 1.00 (referent) | 0.87 (0.631.19) | 0.99 (0.741.34) |
| Condition‐specific readmission rate tertile | |||
| Mortality measure | |||
| Acute myocardial infarction | 1.00 (referent) | 0.88 (0.711.08) | 0.79 (0.640.99) |
| Pneumonia | 1.00 (referent) | 0.91 (0.781.07) | 0.89 (0.761.04) |
| Heart failure | 1.00 (referent) | 1.15 (0.961.36) | 1.56 (1.311.86) |
| Stroke | 1.00 (referent) | 1.65 (1.342.03) | 1.70 (1.232.35) |
| Chronic obstructive pulmonary disease | 1.00 (referent) | 0.83 (0.700.98) | 0.84 (0.710.99) |
| Coronary artery bypass | 1.00 (referent) | 0.59 (0.440.80) | 0.47 (0.340.64) |
DISCUSSION
We found that higher hospital‐wide readmission rates were associated with lower mortality at the hospital level for 3 of the 6 mortality measures we examined. The findings for heart failure parallel the findings of Krumholz and colleagues who examined 3 of these 6 measures (MI, pneumonia, and heart failure) in relation to readmissions for these specific populations.[3] This prior analysis, however, did not include the 3 more recently reported mortality measures (COPD, stroke, and CABG) and did not use hospital‐wide readmissions.
Causal mechanisms underlying the associations between mortality and readmission at the hospital level deserve further exploration. It is certainly possible that global efforts to keep patients out of the hospital might, in some instances, place patients at risk by delaying necessary acute care.[2] It is also possible that unmeasured variables, particularly access to hospice and palliative care services that might facilitate good deaths, could be associated with both reduced readmissions and higher death rates. Additionally, because deceased patients cannot be readmitted, one might expect that readmissions and mortality might be inversely associated, particularly for conditions with a high postdischarge mortality rate. Similarly, a hospital that does a particularly good job keeping chronically ill patients alive until discharge might exhibit a higher readmission rate than a hospital that is less adept at keeping tenuous patients alive until discharge.
Regardless of the mechanisms of these findings, we present these data to raise the concern that using readmission rates, particularly hospital‐wide readmission rates, as a measure of hospital quality is inherently problematic. It is particularly problematic that CMS has applied equal weight to readmissions and mortality in the Star Report.[1] High readmission rates may result from complications and poor handoffs, but may also stem from the legitimate need to care for chronically ill patients in a high‐intensity setting, particularly fragile patients who have been kept alive against the odds. In conclusion, caution is warranted in viewing readmissions as a quality metric until the associations we describe are better explained using patient‐level data and more robust adjustment than is possible with these publically available data.
Disclosures: Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. There was no financial support for this work. Contributions of the authors are as follows: drafting manuscript (Brotman), revision of manuscript for important intellectual content (brotman, Hoyer, Lepley, Deutschendorf, Leung), acquisition of data (Deutschendorf, Leung, Lepley), interpretation of data (Brotman, Hoyer, Lepley, Deutschendorf, Leung), data analysis (Brotman, Hoyer).
The Centers for Medicare & Medicaid Services (CMS) have sought to reduce readmissions in the 30 days following hospital discharge through penalties applied to hospitals with readmission rates that are higher than expected. Expected readmission rates for Medicare fee‐for‐service beneficiaries are calculated from models that use patient‐level administrative data to account for patient morbidities. Readmitted patients are defined as those who are discharged from the hospital alive and then rehospitalized at any acute care facility within 30 days of discharge. These models explicitly exclude sociodemographic variables that may impact quality of and access to outpatient care. Specific exclusions are also applied based on diagnosis codes so as to avoid penalizing hospitals for rehospitalizations that are likely to have been planned.
More recently, a hospital‐wide readmission measure has been developed, which seeks to provide a comprehensive view of each hospital's readmission rate by including the vast majority of Medicare patients. Like the condition‐specific readmission measures, the hospital‐wide readmission measure also excludes sociodemographic variables and incorporates specific condition‐based exclusions so as to avoid counting planned rehospitalizations (e.g., an admission for cholecystectomy following an admission for biliary sepsis). Although not currently used for pay‐for‐performance, this measure has been included in the CMS Star Report along with other readmission measures.[1] CMS does not currently disseminate a hospital‐wide mortality measure, but does disseminate hospital‐level adjusted 30‐day mortality rates for Medicare beneficiaries with discharge diagnoses of stroke, heart failure, myocardial infarction (MI), chronic obstructive pulmonary disease (COPD) and pneumonia, and principal procedure of coronary artery bypass grafting (CABG).
It is conceivable that aggressive efforts to reduce readmissions might delay life‐saving acute care in some scenarios,[2] and there is prior evidence that heart failure readmissions are inversely (but weakly) related to heart failure mortality.[3] It is also plausible that keeping tenuous patients alive until discharge might result in higher readmission rates. We sought to examine the relationship between hospital‐wide adjusted 30‐day readmissions and death rates across the acute care hospitals in the United States. Lacking a measure of hospital‐wide death rates, we examined the relation between hospital‐wide readmissions and each of the 6 condition‐specific mortality measures. For comparison, we also examined the relationships between condition‐specific readmission rates and mortality rates.
METHODS
We used publically available data published by CMS from July 1, 2011 through June 30, 2014.[4] These data are provided at the hospital level, without any patient‐level data. We included 4452 acute care facilities based on having hospital‐wide readmission rates, but not all facilities contributed data for each mortality measure. We excluded from analysis on a measure‐by‐measure basis those facilities for which outcomes were absent, without imputing missing outcome measures, because low volume of a given condition was the main reason for not reporting a measure. For each mortality measure, we constructed a logistic regression model to quantify the odds of performing in the lowest (best) mortality tertile as a function of hospital‐wide readmission tertile. To account for patient volumes, we included in each model the number of eligible patients at each hospital with the specified condition. We repeated these analyses using condition‐specific readmission rates (rather than the hospital‐wide readmission rates) as the independent variable. Specifications for CMS models for mortality and readmissions are publically available.[5]
RESULTS
After adjustment for patient volumes, hospitals in the highest hospital‐wide readmission tertile were more likely to perform in the lowest (best) mortality tertile for 3 of the 6 mortality measures: heart failure, COPD, and stroke (P 0.001 for all). For MI, CABG and pneumonia, there was no significant association between high hospital‐wide readmission rates and low mortality (Table 1). Using condition‐specific readmission rates, there remained an inverse association between readmissions and mortality for heart failure and stroke, but not for COPD. In contrast, hospitals with the highest CABG‐specific readmission rates were significantly less likely to have low CABG‐specific mortality (P 0.001).
| Hospital‐Wide Readmission Rate Tertile [Range of Adjusted Readmission Rates, %]* | |||
|---|---|---|---|
|
1st Tertile, n = 1359 [11.3%‐14.8%], Adjusted Odds Ratio (95% CI) |
2nd Tertile, n = 1785 [14.9%‐15.5%], Adjusted Odds Ratio (95% CI) |
3rd Tertile, n = 1308 [15.6%‐19.8%], Adjusted Odds Ratio (95% CI) |
|
| |||
| Mortality measure (no. of hospitals reporting) | |||
| Acute myocardial infarction (n = 2415) | 1.00 (referent) | 0.88 (0.711.09) | 1.02 (0.831.25) |
| Pneumonia (n = 4067) | 1.00 (referent) | 0.83 (0.710.98) | 1.11 (0.941.31) |
| Heart failure (n = 3668) | 1.00 (referent) | 1.21 (1.021.45) | 1.94 (1.632.30) |
| Stroke (n = 2754) | 1.00 (referent) | 1.13 (0.931.38) | 1.48 (1.221.79) |
| Chronic obstructive pulmonary disease (n = 3633) | 1.00 (referent) | 1.12 (0.951.33) | 1.73 (1.462.05) |
| Coronary artery bypass (n = 1058) | 1.00 (referent) | 0.87 (0.631.19) | 0.99 (0.741.34) |
| Condition‐specific readmission rate tertile | |||
| Mortality measure | |||
| Acute myocardial infarction | 1.00 (referent) | 0.88 (0.711.08) | 0.79 (0.640.99) |
| Pneumonia | 1.00 (referent) | 0.91 (0.781.07) | 0.89 (0.761.04) |
| Heart failure | 1.00 (referent) | 1.15 (0.961.36) | 1.56 (1.311.86) |
| Stroke | 1.00 (referent) | 1.65 (1.342.03) | 1.70 (1.232.35) |
| Chronic obstructive pulmonary disease | 1.00 (referent) | 0.83 (0.700.98) | 0.84 (0.710.99) |
| Coronary artery bypass | 1.00 (referent) | 0.59 (0.440.80) | 0.47 (0.340.64) |
DISCUSSION
We found that higher hospital‐wide readmission rates were associated with lower mortality at the hospital level for 3 of the 6 mortality measures we examined. The findings for heart failure parallel the findings of Krumholz and colleagues who examined 3 of these 6 measures (MI, pneumonia, and heart failure) in relation to readmissions for these specific populations.[3] This prior analysis, however, did not include the 3 more recently reported mortality measures (COPD, stroke, and CABG) and did not use hospital‐wide readmissions.
Causal mechanisms underlying the associations between mortality and readmission at the hospital level deserve further exploration. It is certainly possible that global efforts to keep patients out of the hospital might, in some instances, place patients at risk by delaying necessary acute care.[2] It is also possible that unmeasured variables, particularly access to hospice and palliative care services that might facilitate good deaths, could be associated with both reduced readmissions and higher death rates. Additionally, because deceased patients cannot be readmitted, one might expect that readmissions and mortality might be inversely associated, particularly for conditions with a high postdischarge mortality rate. Similarly, a hospital that does a particularly good job keeping chronically ill patients alive until discharge might exhibit a higher readmission rate than a hospital that is less adept at keeping tenuous patients alive until discharge.
Regardless of the mechanisms of these findings, we present these data to raise the concern that using readmission rates, particularly hospital‐wide readmission rates, as a measure of hospital quality is inherently problematic. It is particularly problematic that CMS has applied equal weight to readmissions and mortality in the Star Report.[1] High readmission rates may result from complications and poor handoffs, but may also stem from the legitimate need to care for chronically ill patients in a high‐intensity setting, particularly fragile patients who have been kept alive against the odds. In conclusion, caution is warranted in viewing readmissions as a quality metric until the associations we describe are better explained using patient‐level data and more robust adjustment than is possible with these publically available data.
Disclosures: Dr. Daniel J. Brotman had full access to the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. There was no financial support for this work. Contributions of the authors are as follows: drafting manuscript (Brotman), revision of manuscript for important intellectual content (brotman, Hoyer, Lepley, Deutschendorf, Leung), acquisition of data (Deutschendorf, Leung, Lepley), interpretation of data (Brotman, Hoyer, Lepley, Deutschendorf, Leung), data analysis (Brotman, Hoyer).
- Centers for Medicare and Medicaid Services. Available at: https://www.cms.gov/Outreach-and-Education/Outreach/NPC/Downloads/2015-08-13-Star-Ratings-Presentation.pdf. Accessed September 2015.
- , , , et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med. 2012;156(10):673–683.
- , , , et al. Relationship between hospital readmission and mortality rates for patients hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2013;309(6):587–593.
- Centers for Medicare and Medicaid Services. Hospital compare datasets. Available at: https://data.medicare.gov/data/hospital‐compare. Accessed September 2015.
- Centers for Medicare and Medicaid Services. Hospital quality initiative. Available at: https://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/HospitalQualityInits. Accessed September 2015.
- Centers for Medicare and Medicaid Services. Available at: https://www.cms.gov/Outreach-and-Education/Outreach/NPC/Downloads/2015-08-13-Star-Ratings-Presentation.pdf. Accessed September 2015.
- , , , et al. A comprehensive care management program to prevent chronic obstructive pulmonary disease hospitalizations: a randomized, controlled trial. Ann Intern Med. 2012;156(10):673–683.
- , , , et al. Relationship between hospital readmission and mortality rates for patients hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2013;309(6):587–593.
- Centers for Medicare and Medicaid Services. Hospital compare datasets. Available at: https://data.medicare.gov/data/hospital‐compare. Accessed September 2015.
- Centers for Medicare and Medicaid Services. Hospital quality initiative. Available at: https://www.cms.gov/Medicare/Quality‐Initiatives‐Patient‐Assessment‐Instruments/HospitalQualityInits. Accessed September 2015.
SCHOLAR Project
The structure and function of academic hospital medicine programs (AHPs) has evolved significantly with the growth of hospital medicine.[1, 2, 3, 4] Many AHPs formed in response to regulatory and financial changes, which drove demand for increased trainee oversight, improved clinical efficiency, and growth in nonteaching services staffed by hospitalists. Differences in local organizational contexts and needs have contributed to great variability in AHP program design and operations. As AHPs have become more established, the need to engage academic hospitalists in scholarship and activities that support professional development and promotion has been recognized. Defining sustainable and successful positions for academic hospitalists is a priority called for by leaders in the field.[5, 6]
In this rapidly evolving context, AHPs have employed a variety of approaches to organizing clinical and academic faculty roles, without guiding evidence or consensus‐based performance benchmarks. A number of AHPs have achieved success along traditional academic metrics of research, scholarship, and education. Currently, it is not known whether specific approaches to AHP organization, structure, or definition of faculty roles are associated with achievement of more traditional markers of academic success.
The Academic Committee of the Society of Hospital Medicine (SHM), and the Academic Hospitalist Task Force of the Society of General Internal Medicine (SGIM) had separately initiated projects to explore characteristics associated with success in AHPs. In 2012, these organizations combined efforts to jointly develop and implement the SCHOLAR (SuCcessful HOspitaLists in Academics and Research) project. The goals were to identify successful AHPs using objective criteria, and to then study those groups in greater detail to generate insights that would be broadly relevant to the field. Efforts to clarify the factors within AHPs linked to success by traditional academic metrics will benefit hospitalists, their leaders, and key stakeholders striving to achieve optimal balance between clinical and academic roles. We describe the initial work of the SCHOLAR project, our definitions of academic success in AHPs, and the characteristics of a cohort of exemplary AHPs who achieved the highest levels on these metrics.
METHODS
Defining Success
The 11 members of the SCHOLAR project held a variety of clinical and academic roles within a geographically diverse group of AHPs. We sought to create a functional definition of success applicable to AHPs. As no gold standard currently exists, we used a consensus process among task force members to arrive at a definition that was quantifiable, feasible, and meaningful. The first step was brainstorming on conference calls held 1 to 2 times monthly over 4 months. Potential defining characteristics that emerged from these discussions related to research, teaching, and administrative activities. When potential characteristics were proposed, we considered how to operationalize each one. Each characteristic was discussed until there was consensus from the entire group. Those around education and administration were the most complex, as many roles are locally driven and defined, and challenging to quantify. For this reason, we focused on promotion as a more global approach to assessing academic hospitalist success in these areas. Although criteria for academic advancement also vary across institutions, we felt that promotion generally reflected having met some threshold of academic success. We also wanted to recognize that scholarship occurs outside the context of funded research. Ultimately, 3 key domains emerged: research grant funding, faculty promotion, and scholarship.
After these 3 domains were identified, the group sought to define quantitative metrics to assess performance. These discussions occurred on subsequent calls over a 4‐month period. Between calls, group members gathered additional information to facilitate assessment of the feasibility of proposed metrics, reporting on progress via email. Again, group consensus was sought for each metric considered. Data on grant funding and successful promotions were available from a previous survey conducted through the SHM in 2011. Leaders from 170 AHPs were contacted, with 50 providing complete responses to the 21‐item questionnaire (see Supporting Information, Appendix 1, in the online version of this article). Results of the survey, heretofore referred to as the Leaders of Academic Hospitalist Programs survey (LAHP‐50), have been described elsewhere.[7] For the purposes of this study, we used the self‐reported data about grant funding and promotions contained in the survey to reflect the current state of the field. Although the survey response rate was approximately 30%, the survey was not anonymous, and many reputationally prominent academic hospitalist programs were represented. For these reasons, the group members felt that the survey results were relevant for the purposes of assessing academic success.
In the LAHP‐50, funding was defined as principal investigator or coinvestigator roles on federally and nonfederally funded research, clinical trials, internal grants, and any other extramurally funded projects. Mean and median funding for the overall sample was calculated. Through a separate question, each program's total faculty full‐time equivalent (FTE) count was reported, allowing us to adjust for group size by assessing both total funding per group and funding/FTE for each responding AHP.
Promotions were defined by the self‐reported number of faculty at each of the following ranks: instructor, assistant professor, associate professor, full professor, and professor above scale/emeritus. In addition, a category of nonacademic track (eg, adjunct faculty, clinical associate) was included to capture hospitalists that did not fit into the traditional promotions categories. We did not distinguish between tenure‐track and nontenure‐track academic ranks. LAHP‐50 survey respondents reported the number of faculty in their group at each academic rank. Given that the majority of academic hospitalists hold a rank of assistant professor or lower,[6, 8, 9] and that the number of full professors was only 3% in the LAHP‐50 cohort, we combined the faculty at the associate and full professor ranks, defining successfully promoted faculty as the percent of hospitalists above the rank of assistant professor.
We created a new metric to assess scholarly output. We had considerable discussion of ways to assess the numbers of peer‐reviewed manuscripts generated by AHPs. However, the group had concerns about the feasibility of identification and attribution of authors to specific AHPs through literature searches. We considered examining only publications in the Journal of Hospital Medicine and the Journal of General Internal Medicine, but felt that this would exclude significant work published by hospitalists in fields of medical education or health services research that would more likely appear in alternate journals. Instead, we quantified scholarship based on the number of abstracts presented at national meetings. We focused on meetings of the SHM and SGIM as the primary professional societies representing hospital medicine. The group felt that even work published outside of the journals of our professional societies would likely be presented at those meetings. We used the following strategy: We reviewed research abstracts accepted for presentation as posters or oral abstracts at the 2010 and 2011 SHM national meetings, and research abstracts with a primary or secondary category of hospital medicine at the 2010 and 2011 SGIM national meetings. By including submissions at both SGIM and SHM meetings, we accounted for the fact that some programs may gravitate more to one society meeting or another. We did not include abstracts in the clinical vignettes or innovations categories. We tallied the number of abstracts by group affiliation of the authors for each of the 4 meetings above and created a cumulative total per group for the 2‐year period. Abstracts with authors from different AHPs were counted once for each individual group. Members of the study group reviewed abstracts from each of the meetings in pairs. Reviewers worked separately and compared tallies of results to ensure consistent tabulations. Internet searches were conducted to identify or confirm author affiliations if it was not apparent in the abstract author list. Abstract tallies were compiled without regard to whether programs had completed the LAHP‐50 survey; thus, we collected data on programs that did not respond to the LAHP‐50 survey.
Identification of the SCHOLAR Cohort
To identify our cohort of top‐performing AHPs, we combined the funding and promotions data from the LAHP‐50 sample with the abstract data. We limited our sample to adult hospital medicine groups to reduce heterogeneity. We created rank lists of programs in each category (grant funding, successful promotions, and scholarship), using data from the LAHP‐50 survey to rank programs on funding and promotions, and data from our abstract counts to rank on scholarship. We limited the top‐performing list in each category to 10 institutions as a cutoff. Because we set a threshold of at least $1 million in total funding, we identified only 9 top performing AHPs with regard to grant funding. We also calculated mean funding/FTE. We chose to rank programs only by funding/FTE rather than total funding per program to better account for group size. For successful promotions, we ranked programs by the percentage of senior faculty. For abstract counts, we included programs whose faculty presented abstracts at a minimum of 2 separate meetings, and ranked programs based on the total number of abstracts per group.
This process resulted in separate lists of top performing programs in each of the 3 domains we associated with academic success, arranged in descending order by grant dollars/FTE, percent of senior faculty, and abstract counts (Table 1). Seventeen different programs were represented across these 3 top 10 lists. One program appeared on all 3 lists, 8 programs appeared on 2 lists, and the remainder appeared on a single list (Table 2). Seven of these programs were identified solely based on abstract presentations, diversifying our top groups beyond only those who completed the LAHP‐50 survey. We considered all of these programs to represent high performance in academic hospital medicine. The group selected this inclusive approach because we recognized that any 1 metric was potentially limited, and we sought to identify diverse pathways to success.
| Funding | Promotions | Scholarship | |
|---|---|---|---|
| Grant $/FTE | Total Grant $ | Senior Faculty, No. (%) | Total Abstract Count |
| |||
| $1,409,090 | $15,500,000 | 3 (60%) | 23 |
| $1,000,000 | $9,000,000 | 3 (60%) | 21 |
| $750,000 | $8,000,000 | 4 (57%) | 20 |
| $478,609 | $6,700,535 | 9 (53%) | 15 |
| $347,826 | $3,000,000 | 8 (44%) | 11 |
| $86,956 | $3,000,000 | 14 (41%) | 11 |
| $66,666 | $2,000,000 | 17 (36%) | 10 |
| $46,153 | $1,500,000 | 9 (33%) | 10 |
| $38,461 | $1,000,000 | 2 (33%) | 9 |
| 4 (31%) | 9 | ||
| Selection Criteria for SCHOLAR Cohort | No. of Programs |
|---|---|
| |
| Abstracts, funding, and promotions | 1 |
| Abstracts plus promotions | 4 |
| Abstracts plus funding | 3 |
| Funding plus promotion | 1 |
| Funding only | 1 |
| Abstract only | 7 |
| Total | 17 |
| Top 10 abstract count | |
| 4 meetings | 2 |
| 3 meetings | 2 |
| 2 meetings | 6 |
The 17 unique adult AHPs appearing on at least 1 of the top 10 lists comprised the SCHOLAR cohort of programs that we studied in greater detail. Data reflecting program demographics were solicited directly from leaders of the AHPs identified in the SCHOLAR cohort, including size and age of program, reporting structure, number of faculty at various academic ranks (for programs that did not complete the LAHP‐50 survey), and number of faculty with fellowship training (defined as any postresidency fellowship program).
Subsequently, we performed comparative analyses between the programs in the SCHOLAR cohort to the general population of AHPs reflected by the LAHP‐50 sample. Because abstract presentations were not recorded in the original LAHP‐50 survey instrument, it was not possible to perform a benchmarking comparison for the scholarship domain.
Data Analysis
To measure the success of the SCHOLAR cohort we compared the grant funding and proportion of successfully promoted faculty at the SCHOLAR programs to those in the overall LAHP‐50 sample. Differences in mean and median grant funding were compared using t tests and Mann‐Whitney rank sum tests. Proportion of promoted faculty were compared using 2 tests. A 2‐tailed of 0.05 was used to test significance of differences.
RESULTS
Demographics
Among the AHPs in the SCHOLAR cohort, the mean program age was 13.2 years (range, 618 years), and the mean program size was 36 faculty (range, 1895; median, 28). On average, 15% of faculty members at SCHOLAR programs were fellowship trained (range, 0%37%). Reporting structure among the SCHOLAR programs was as follows: 53% were an independent division or section of the department of medicine; 29% were a section within general internal medicine, and 18% were an independent clinical group.
Grant Funding
Table 3 compares grant funding in the SCHOLAR programs to programs in the overall LAHP‐50 sample. Mean funding per group and mean funding per FTE were significantly higher in the SCHOLAR group than in the overall sample.
| Funding (Millions) | ||
|---|---|---|
| LAHP‐50 Overall Sample | SCHOLAR | |
| ||
| Median grant funding/AHP | 0.060 | 1.500* |
| Mean grant funding/AHP | 1.147 (015) | 3.984* (015) |
| Median grant funding/FTE | 0.004 | 0.038* |
| Mean grant funding/FTE | 0.095 (01.4) | 0.364* (01.4) |
Thirteen of the SCHOLAR programs were represented in the initial LAHP‐50, but 2 did not report a dollar amount for grants and contracts. Therefore, data for total grant funding were available for only 65% (11 of 17) of the programs in the SCHOLAR cohort. Of note, 28% of AHPs in the overall LAHP‐50 sample reported no external funding sources.
Faculty Promotion
Figure 1 demonstrates the proportion of faculty at various academic ranks. The percent of faculty above the rank of assistant professor in the SCHOLAR programs exceeded those in the overall LAHP‐50 by 5% (17.9% vs 12.8%, P = 0.01). Of note, 6% of the hospitalists at AHPs in the SCHOLAR programs were on nonfaculty tracks.
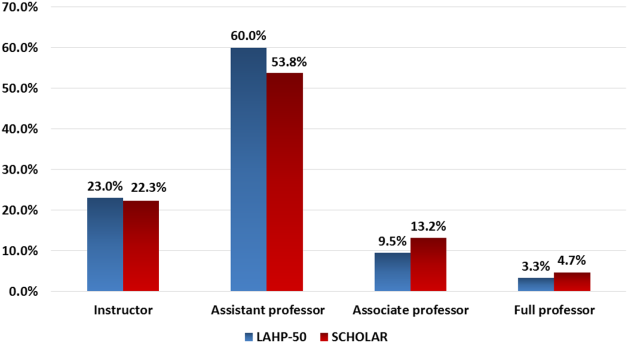
Scholarship
Mean abstract output over the 2‐year period measured was 10.8 (range, 323) in the SCHOLAR cohort. Because we did not collect these data for the LAHP‐50 group, comparative analyses were not possible.
DISCUSSION
Using a definition of academic success that incorporated metrics of grant funding, faculty promotion, and scholarly output, we identified a unique subset of successful AHPsthe SCHOLAR cohort. The programs represented in the SCHOLAR cohort were generally large and relatively mature. Despite this, the cohort consisted of mostly junior faculty, had a paucity of fellowship‐trained hospitalists, and not all reported grant funding.
Prior published work reported complementary findings.[6, 8, 9] A survey of 20 large, well‐established academic hospitalist programs in 2008 found that the majority of hospitalists were junior faculty with a limited publication portfolio. Of the 266 respondents in that study, 86% reported an academic rank at or below assistant professor; funding was not explored.[9] Our similar findings 4 years later add to this work by demonstrating trends over time, and suggest that progress toward creating successful pathways for academic advancement has been slow. In a 2012 survey of the SHM membership, 28% of hospitalists with academic appointments reported no current or future plans to engage in research.[8] These findings suggest that faculty in AHPs may define scholarship through nontraditional pathways, or in some cases choose not to pursue or prioritize scholarship altogether.
Our findings also add to the literature with regard to our assessment of funding, which was variable across the SCHOLAR group. The broad range of funding in the SCHOLAR programs for which we have data (grant dollars $0$15 million per program) suggests that opportunities to improve supported scholarship remain, even among a selected cohort of successful AHPs. The predominance of junior faculty in the SCHOLAR programs may be a reason for this variation. Junior faculty may be engaged in research with funding directed to senior mentors outside their AHP. Alternatively, they may pursue meaningful local hospital quality improvement or educational innovations not supported by external grants, or hold leadership roles in education, quality, or information technology that allow for advancement and promotion without external grant funding. As the scope and impact of these roles increases, senior leaders with alternate sources of support may rely less on research funds; this too may explain some of the differences. Our findings are congruent with results of a study that reviewed original research published by hospitalists, and concluded that the majority of hospitalist research was not externally funded.[8] Our approach for assessing grant funding by adjusting for FTE had the potential to inadvertently favor smaller well‐funded groups over larger ones; however, programs in our sample were similarly represented when ranked by funding/FTE or total grant dollars. As many successful AHPs do concentrate their research funding among a core of focused hospitalist researchers, our definition may not be the ideal metric for some programs.
We chose to define scholarship based on abstract output, rather than peer‐reviewed publications. Although this choice was necessary from a feasibility perspective, it may have excluded programs that prioritize peer‐reviewed publications over abstracts. Although we were unable to incorporate a search strategy to accurately and comprehensively track the publication output attributed specifically to hospitalist researchers and quantify it by program, others have since defined such an approach.[8] However, tracking abstracts theoretically allowed insights into a larger volume of innovative and creative work generated by top AHPs by potentially including work in the earlier stages of development.
We used a consensus‐based definition of success to define our SCHOLAR cohort. There are other ways to measure academic success, which if applied, may have yielded a different sample of programs. For example, over half of the original research articles published in the Journal of Hospital Medicine over a 7‐year span were generated from 5 academic centers.[8] This definition of success may be equally credible, though we note that 4 of these 5 programs were also included in the SCHOLAR cohort. We feel our broader approach was more reflective of the variety of pathways to success available to academic hospitalists. Before our metrics are applied as a benchmarking tool, however, they should ideally be combined with factors not measured in our study to ensure a more comprehensive or balanced reflection of academic success. Factors such as mentorship, level of hospitalist engagement,[10] prevalence of leadership opportunities, operational and fiscal infrastructure, and the impact of local quality, safety, and value efforts should be considered.
Comparison of successfully promoted faculty at AHPs across the country is inherently limited by the wide variation in promotion standards across different institutions; controlling for such differences was not possible with our methodology. For example, it appears that several programs with relatively few senior faculty may have met metrics leading to their inclusion in the SCHOLAR group because of their small program size. Future benchmarking efforts for promotion at AHPs should take scaling into account and consider both total number as well as percentage of senior faculty when evaluating success.
Our methodology has several limitations. Survey data were self‐reported and not independently validated, and as such are subject to recall and reporting biases. Response bias inherently excluded some AHPs that may have met our grant funding or promotions criteria had they participated in the initial LAHP‐50 survey, though we identified and included additional programs through our scholarship metric, increasing the representativeness of the SCHOLAR cohort. Given the dynamic nature of the field, the age of the data we relied upon for analysis limits the generalizability of our specific benchmarks to current practice. However, the development of academic success occurs over the long‐term, and published data on academic hospitalist productivity are consistent with this slower time course.[8] Despite these limitations, our data inform the general topic of gauging performance of AHPs, underscoring the challenges of developing and applying metrics of success, and highlight the variability of performance on selected metrics even among a relatively small group of 17 programs.
In conclusion, we have created a method to quantify academic success that may be useful to academic hospitalists and their group leaders as they set targets for improvement in the field. Even among our SCHOLAR cohort, room for ongoing improvement in development of funded scholarship and a core of senior faculty exists. Further investigation into the unique features of successful groups will offer insight to leaders in academic hospital medicine regarding infrastructure and processes that should be embraced to raise the bar for all AHPs. In addition, efforts to further define and validate nontraditional approaches to scholarship that allow for successful promotion at AHPs would be informative. We view our work less as a singular approach to benchmarking standards for AHPs, and more a call to action to continue efforts to balance scholarly activity and broad professional development of academic hospitalists with increasing clinical demands.
Acknowledgements
The authors thank all of the AHP leaders who participated in the SCHOLAR project. They also thank the Society of Hospital Medicine and Society of General Internal Medicine and the SHM Academic Committee and SGIM Academic Hospitalist Task Force for their support of this work.
Disclosures
The work reported here was supported by the Department of Veterans Affairs, Veterans Health Administration, South Texas Veterans Health Care System. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs. The authors report no conflicts of interest.
- , , , , . Characteristics of primary care providers who adopted the hospitalist model from 2001 to 2009. J Hosp Med. 2015;10(2):75–82.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , , , . Updating threshold‐based identification of hospitalists in 2012 Medicare pay data. J Hosp Med. 2016;11(1):45–47.
- , , , . Use of hospitalists by Medicare beneficiaries: a national picture. Medicare Medicaid Res Rev. 2014;4(2).
- , , , , , . Challenges and opportunities in Academic Hospital Medicine: report from the Academic Hospital Medicine Summit. J Hosp Med. 2009;4(4):240–246.
- , , , . Survey of US academic hospitalist leaders about mentorship and academic activities in hospitalist groups. J Hosp Med. 2011;6(1):5–9.
- , , , , , . The structure of hospital medicine programs at academic medical centers [abstract]. J Hosp Med. 2012;7(suppl 2):s92.
- , , , , . Research and publication trends in hospital medicine. J Hosp Med. 2014;9(3):148–154.
- , , , , , . Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23–27.
- , , , et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123–128.
The structure and function of academic hospital medicine programs (AHPs) has evolved significantly with the growth of hospital medicine.[1, 2, 3, 4] Many AHPs formed in response to regulatory and financial changes, which drove demand for increased trainee oversight, improved clinical efficiency, and growth in nonteaching services staffed by hospitalists. Differences in local organizational contexts and needs have contributed to great variability in AHP program design and operations. As AHPs have become more established, the need to engage academic hospitalists in scholarship and activities that support professional development and promotion has been recognized. Defining sustainable and successful positions for academic hospitalists is a priority called for by leaders in the field.[5, 6]
In this rapidly evolving context, AHPs have employed a variety of approaches to organizing clinical and academic faculty roles, without guiding evidence or consensus‐based performance benchmarks. A number of AHPs have achieved success along traditional academic metrics of research, scholarship, and education. Currently, it is not known whether specific approaches to AHP organization, structure, or definition of faculty roles are associated with achievement of more traditional markers of academic success.
The Academic Committee of the Society of Hospital Medicine (SHM), and the Academic Hospitalist Task Force of the Society of General Internal Medicine (SGIM) had separately initiated projects to explore characteristics associated with success in AHPs. In 2012, these organizations combined efforts to jointly develop and implement the SCHOLAR (SuCcessful HOspitaLists in Academics and Research) project. The goals were to identify successful AHPs using objective criteria, and to then study those groups in greater detail to generate insights that would be broadly relevant to the field. Efforts to clarify the factors within AHPs linked to success by traditional academic metrics will benefit hospitalists, their leaders, and key stakeholders striving to achieve optimal balance between clinical and academic roles. We describe the initial work of the SCHOLAR project, our definitions of academic success in AHPs, and the characteristics of a cohort of exemplary AHPs who achieved the highest levels on these metrics.
METHODS
Defining Success
The 11 members of the SCHOLAR project held a variety of clinical and academic roles within a geographically diverse group of AHPs. We sought to create a functional definition of success applicable to AHPs. As no gold standard currently exists, we used a consensus process among task force members to arrive at a definition that was quantifiable, feasible, and meaningful. The first step was brainstorming on conference calls held 1 to 2 times monthly over 4 months. Potential defining characteristics that emerged from these discussions related to research, teaching, and administrative activities. When potential characteristics were proposed, we considered how to operationalize each one. Each characteristic was discussed until there was consensus from the entire group. Those around education and administration were the most complex, as many roles are locally driven and defined, and challenging to quantify. For this reason, we focused on promotion as a more global approach to assessing academic hospitalist success in these areas. Although criteria for academic advancement also vary across institutions, we felt that promotion generally reflected having met some threshold of academic success. We also wanted to recognize that scholarship occurs outside the context of funded research. Ultimately, 3 key domains emerged: research grant funding, faculty promotion, and scholarship.
After these 3 domains were identified, the group sought to define quantitative metrics to assess performance. These discussions occurred on subsequent calls over a 4‐month period. Between calls, group members gathered additional information to facilitate assessment of the feasibility of proposed metrics, reporting on progress via email. Again, group consensus was sought for each metric considered. Data on grant funding and successful promotions were available from a previous survey conducted through the SHM in 2011. Leaders from 170 AHPs were contacted, with 50 providing complete responses to the 21‐item questionnaire (see Supporting Information, Appendix 1, in the online version of this article). Results of the survey, heretofore referred to as the Leaders of Academic Hospitalist Programs survey (LAHP‐50), have been described elsewhere.[7] For the purposes of this study, we used the self‐reported data about grant funding and promotions contained in the survey to reflect the current state of the field. Although the survey response rate was approximately 30%, the survey was not anonymous, and many reputationally prominent academic hospitalist programs were represented. For these reasons, the group members felt that the survey results were relevant for the purposes of assessing academic success.
In the LAHP‐50, funding was defined as principal investigator or coinvestigator roles on federally and nonfederally funded research, clinical trials, internal grants, and any other extramurally funded projects. Mean and median funding for the overall sample was calculated. Through a separate question, each program's total faculty full‐time equivalent (FTE) count was reported, allowing us to adjust for group size by assessing both total funding per group and funding/FTE for each responding AHP.
Promotions were defined by the self‐reported number of faculty at each of the following ranks: instructor, assistant professor, associate professor, full professor, and professor above scale/emeritus. In addition, a category of nonacademic track (eg, adjunct faculty, clinical associate) was included to capture hospitalists that did not fit into the traditional promotions categories. We did not distinguish between tenure‐track and nontenure‐track academic ranks. LAHP‐50 survey respondents reported the number of faculty in their group at each academic rank. Given that the majority of academic hospitalists hold a rank of assistant professor or lower,[6, 8, 9] and that the number of full professors was only 3% in the LAHP‐50 cohort, we combined the faculty at the associate and full professor ranks, defining successfully promoted faculty as the percent of hospitalists above the rank of assistant professor.
We created a new metric to assess scholarly output. We had considerable discussion of ways to assess the numbers of peer‐reviewed manuscripts generated by AHPs. However, the group had concerns about the feasibility of identification and attribution of authors to specific AHPs through literature searches. We considered examining only publications in the Journal of Hospital Medicine and the Journal of General Internal Medicine, but felt that this would exclude significant work published by hospitalists in fields of medical education or health services research that would more likely appear in alternate journals. Instead, we quantified scholarship based on the number of abstracts presented at national meetings. We focused on meetings of the SHM and SGIM as the primary professional societies representing hospital medicine. The group felt that even work published outside of the journals of our professional societies would likely be presented at those meetings. We used the following strategy: We reviewed research abstracts accepted for presentation as posters or oral abstracts at the 2010 and 2011 SHM national meetings, and research abstracts with a primary or secondary category of hospital medicine at the 2010 and 2011 SGIM national meetings. By including submissions at both SGIM and SHM meetings, we accounted for the fact that some programs may gravitate more to one society meeting or another. We did not include abstracts in the clinical vignettes or innovations categories. We tallied the number of abstracts by group affiliation of the authors for each of the 4 meetings above and created a cumulative total per group for the 2‐year period. Abstracts with authors from different AHPs were counted once for each individual group. Members of the study group reviewed abstracts from each of the meetings in pairs. Reviewers worked separately and compared tallies of results to ensure consistent tabulations. Internet searches were conducted to identify or confirm author affiliations if it was not apparent in the abstract author list. Abstract tallies were compiled without regard to whether programs had completed the LAHP‐50 survey; thus, we collected data on programs that did not respond to the LAHP‐50 survey.
Identification of the SCHOLAR Cohort
To identify our cohort of top‐performing AHPs, we combined the funding and promotions data from the LAHP‐50 sample with the abstract data. We limited our sample to adult hospital medicine groups to reduce heterogeneity. We created rank lists of programs in each category (grant funding, successful promotions, and scholarship), using data from the LAHP‐50 survey to rank programs on funding and promotions, and data from our abstract counts to rank on scholarship. We limited the top‐performing list in each category to 10 institutions as a cutoff. Because we set a threshold of at least $1 million in total funding, we identified only 9 top performing AHPs with regard to grant funding. We also calculated mean funding/FTE. We chose to rank programs only by funding/FTE rather than total funding per program to better account for group size. For successful promotions, we ranked programs by the percentage of senior faculty. For abstract counts, we included programs whose faculty presented abstracts at a minimum of 2 separate meetings, and ranked programs based on the total number of abstracts per group.
This process resulted in separate lists of top performing programs in each of the 3 domains we associated with academic success, arranged in descending order by grant dollars/FTE, percent of senior faculty, and abstract counts (Table 1). Seventeen different programs were represented across these 3 top 10 lists. One program appeared on all 3 lists, 8 programs appeared on 2 lists, and the remainder appeared on a single list (Table 2). Seven of these programs were identified solely based on abstract presentations, diversifying our top groups beyond only those who completed the LAHP‐50 survey. We considered all of these programs to represent high performance in academic hospital medicine. The group selected this inclusive approach because we recognized that any 1 metric was potentially limited, and we sought to identify diverse pathways to success.
| Funding | Promotions | Scholarship | |
|---|---|---|---|
| Grant $/FTE | Total Grant $ | Senior Faculty, No. (%) | Total Abstract Count |
| |||
| $1,409,090 | $15,500,000 | 3 (60%) | 23 |
| $1,000,000 | $9,000,000 | 3 (60%) | 21 |
| $750,000 | $8,000,000 | 4 (57%) | 20 |
| $478,609 | $6,700,535 | 9 (53%) | 15 |
| $347,826 | $3,000,000 | 8 (44%) | 11 |
| $86,956 | $3,000,000 | 14 (41%) | 11 |
| $66,666 | $2,000,000 | 17 (36%) | 10 |
| $46,153 | $1,500,000 | 9 (33%) | 10 |
| $38,461 | $1,000,000 | 2 (33%) | 9 |
| 4 (31%) | 9 | ||
| Selection Criteria for SCHOLAR Cohort | No. of Programs |
|---|---|
| |
| Abstracts, funding, and promotions | 1 |
| Abstracts plus promotions | 4 |
| Abstracts plus funding | 3 |
| Funding plus promotion | 1 |
| Funding only | 1 |
| Abstract only | 7 |
| Total | 17 |
| Top 10 abstract count | |
| 4 meetings | 2 |
| 3 meetings | 2 |
| 2 meetings | 6 |
The 17 unique adult AHPs appearing on at least 1 of the top 10 lists comprised the SCHOLAR cohort of programs that we studied in greater detail. Data reflecting program demographics were solicited directly from leaders of the AHPs identified in the SCHOLAR cohort, including size and age of program, reporting structure, number of faculty at various academic ranks (for programs that did not complete the LAHP‐50 survey), and number of faculty with fellowship training (defined as any postresidency fellowship program).
Subsequently, we performed comparative analyses between the programs in the SCHOLAR cohort to the general population of AHPs reflected by the LAHP‐50 sample. Because abstract presentations were not recorded in the original LAHP‐50 survey instrument, it was not possible to perform a benchmarking comparison for the scholarship domain.
Data Analysis
To measure the success of the SCHOLAR cohort we compared the grant funding and proportion of successfully promoted faculty at the SCHOLAR programs to those in the overall LAHP‐50 sample. Differences in mean and median grant funding were compared using t tests and Mann‐Whitney rank sum tests. Proportion of promoted faculty were compared using 2 tests. A 2‐tailed of 0.05 was used to test significance of differences.
RESULTS
Demographics
Among the AHPs in the SCHOLAR cohort, the mean program age was 13.2 years (range, 618 years), and the mean program size was 36 faculty (range, 1895; median, 28). On average, 15% of faculty members at SCHOLAR programs were fellowship trained (range, 0%37%). Reporting structure among the SCHOLAR programs was as follows: 53% were an independent division or section of the department of medicine; 29% were a section within general internal medicine, and 18% were an independent clinical group.
Grant Funding
Table 3 compares grant funding in the SCHOLAR programs to programs in the overall LAHP‐50 sample. Mean funding per group and mean funding per FTE were significantly higher in the SCHOLAR group than in the overall sample.
| Funding (Millions) | ||
|---|---|---|
| LAHP‐50 Overall Sample | SCHOLAR | |
| ||
| Median grant funding/AHP | 0.060 | 1.500* |
| Mean grant funding/AHP | 1.147 (015) | 3.984* (015) |
| Median grant funding/FTE | 0.004 | 0.038* |
| Mean grant funding/FTE | 0.095 (01.4) | 0.364* (01.4) |
Thirteen of the SCHOLAR programs were represented in the initial LAHP‐50, but 2 did not report a dollar amount for grants and contracts. Therefore, data for total grant funding were available for only 65% (11 of 17) of the programs in the SCHOLAR cohort. Of note, 28% of AHPs in the overall LAHP‐50 sample reported no external funding sources.
Faculty Promotion
Figure 1 demonstrates the proportion of faculty at various academic ranks. The percent of faculty above the rank of assistant professor in the SCHOLAR programs exceeded those in the overall LAHP‐50 by 5% (17.9% vs 12.8%, P = 0.01). Of note, 6% of the hospitalists at AHPs in the SCHOLAR programs were on nonfaculty tracks.
Scholarship
Mean abstract output over the 2‐year period measured was 10.8 (range, 323) in the SCHOLAR cohort. Because we did not collect these data for the LAHP‐50 group, comparative analyses were not possible.
DISCUSSION
Using a definition of academic success that incorporated metrics of grant funding, faculty promotion, and scholarly output, we identified a unique subset of successful AHPsthe SCHOLAR cohort. The programs represented in the SCHOLAR cohort were generally large and relatively mature. Despite this, the cohort consisted of mostly junior faculty, had a paucity of fellowship‐trained hospitalists, and not all reported grant funding.
Prior published work reported complementary findings.[6, 8, 9] A survey of 20 large, well‐established academic hospitalist programs in 2008 found that the majority of hospitalists were junior faculty with a limited publication portfolio. Of the 266 respondents in that study, 86% reported an academic rank at or below assistant professor; funding was not explored.[9] Our similar findings 4 years later add to this work by demonstrating trends over time, and suggest that progress toward creating successful pathways for academic advancement has been slow. In a 2012 survey of the SHM membership, 28% of hospitalists with academic appointments reported no current or future plans to engage in research.[8] These findings suggest that faculty in AHPs may define scholarship through nontraditional pathways, or in some cases choose not to pursue or prioritize scholarship altogether.
Our findings also add to the literature with regard to our assessment of funding, which was variable across the SCHOLAR group. The broad range of funding in the SCHOLAR programs for which we have data (grant dollars $0$15 million per program) suggests that opportunities to improve supported scholarship remain, even among a selected cohort of successful AHPs. The predominance of junior faculty in the SCHOLAR programs may be a reason for this variation. Junior faculty may be engaged in research with funding directed to senior mentors outside their AHP. Alternatively, they may pursue meaningful local hospital quality improvement or educational innovations not supported by external grants, or hold leadership roles in education, quality, or information technology that allow for advancement and promotion without external grant funding. As the scope and impact of these roles increases, senior leaders with alternate sources of support may rely less on research funds; this too may explain some of the differences. Our findings are congruent with results of a study that reviewed original research published by hospitalists, and concluded that the majority of hospitalist research was not externally funded.[8] Our approach for assessing grant funding by adjusting for FTE had the potential to inadvertently favor smaller well‐funded groups over larger ones; however, programs in our sample were similarly represented when ranked by funding/FTE or total grant dollars. As many successful AHPs do concentrate their research funding among a core of focused hospitalist researchers, our definition may not be the ideal metric for some programs.
We chose to define scholarship based on abstract output, rather than peer‐reviewed publications. Although this choice was necessary from a feasibility perspective, it may have excluded programs that prioritize peer‐reviewed publications over abstracts. Although we were unable to incorporate a search strategy to accurately and comprehensively track the publication output attributed specifically to hospitalist researchers and quantify it by program, others have since defined such an approach.[8] However, tracking abstracts theoretically allowed insights into a larger volume of innovative and creative work generated by top AHPs by potentially including work in the earlier stages of development.
We used a consensus‐based definition of success to define our SCHOLAR cohort. There are other ways to measure academic success, which if applied, may have yielded a different sample of programs. For example, over half of the original research articles published in the Journal of Hospital Medicine over a 7‐year span were generated from 5 academic centers.[8] This definition of success may be equally credible, though we note that 4 of these 5 programs were also included in the SCHOLAR cohort. We feel our broader approach was more reflective of the variety of pathways to success available to academic hospitalists. Before our metrics are applied as a benchmarking tool, however, they should ideally be combined with factors not measured in our study to ensure a more comprehensive or balanced reflection of academic success. Factors such as mentorship, level of hospitalist engagement,[10] prevalence of leadership opportunities, operational and fiscal infrastructure, and the impact of local quality, safety, and value efforts should be considered.
Comparison of successfully promoted faculty at AHPs across the country is inherently limited by the wide variation in promotion standards across different institutions; controlling for such differences was not possible with our methodology. For example, it appears that several programs with relatively few senior faculty may have met metrics leading to their inclusion in the SCHOLAR group because of their small program size. Future benchmarking efforts for promotion at AHPs should take scaling into account and consider both total number as well as percentage of senior faculty when evaluating success.
Our methodology has several limitations. Survey data were self‐reported and not independently validated, and as such are subject to recall and reporting biases. Response bias inherently excluded some AHPs that may have met our grant funding or promotions criteria had they participated in the initial LAHP‐50 survey, though we identified and included additional programs through our scholarship metric, increasing the representativeness of the SCHOLAR cohort. Given the dynamic nature of the field, the age of the data we relied upon for analysis limits the generalizability of our specific benchmarks to current practice. However, the development of academic success occurs over the long‐term, and published data on academic hospitalist productivity are consistent with this slower time course.[8] Despite these limitations, our data inform the general topic of gauging performance of AHPs, underscoring the challenges of developing and applying metrics of success, and highlight the variability of performance on selected metrics even among a relatively small group of 17 programs.
In conclusion, we have created a method to quantify academic success that may be useful to academic hospitalists and their group leaders as they set targets for improvement in the field. Even among our SCHOLAR cohort, room for ongoing improvement in development of funded scholarship and a core of senior faculty exists. Further investigation into the unique features of successful groups will offer insight to leaders in academic hospital medicine regarding infrastructure and processes that should be embraced to raise the bar for all AHPs. In addition, efforts to further define and validate nontraditional approaches to scholarship that allow for successful promotion at AHPs would be informative. We view our work less as a singular approach to benchmarking standards for AHPs, and more a call to action to continue efforts to balance scholarly activity and broad professional development of academic hospitalists with increasing clinical demands.
Acknowledgements
The authors thank all of the AHP leaders who participated in the SCHOLAR project. They also thank the Society of Hospital Medicine and Society of General Internal Medicine and the SHM Academic Committee and SGIM Academic Hospitalist Task Force for their support of this work.
Disclosures
The work reported here was supported by the Department of Veterans Affairs, Veterans Health Administration, South Texas Veterans Health Care System. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs. The authors report no conflicts of interest.
The structure and function of academic hospital medicine programs (AHPs) has evolved significantly with the growth of hospital medicine.[1, 2, 3, 4] Many AHPs formed in response to regulatory and financial changes, which drove demand for increased trainee oversight, improved clinical efficiency, and growth in nonteaching services staffed by hospitalists. Differences in local organizational contexts and needs have contributed to great variability in AHP program design and operations. As AHPs have become more established, the need to engage academic hospitalists in scholarship and activities that support professional development and promotion has been recognized. Defining sustainable and successful positions for academic hospitalists is a priority called for by leaders in the field.[5, 6]
In this rapidly evolving context, AHPs have employed a variety of approaches to organizing clinical and academic faculty roles, without guiding evidence or consensus‐based performance benchmarks. A number of AHPs have achieved success along traditional academic metrics of research, scholarship, and education. Currently, it is not known whether specific approaches to AHP organization, structure, or definition of faculty roles are associated with achievement of more traditional markers of academic success.
The Academic Committee of the Society of Hospital Medicine (SHM), and the Academic Hospitalist Task Force of the Society of General Internal Medicine (SGIM) had separately initiated projects to explore characteristics associated with success in AHPs. In 2012, these organizations combined efforts to jointly develop and implement the SCHOLAR (SuCcessful HOspitaLists in Academics and Research) project. The goals were to identify successful AHPs using objective criteria, and to then study those groups in greater detail to generate insights that would be broadly relevant to the field. Efforts to clarify the factors within AHPs linked to success by traditional academic metrics will benefit hospitalists, their leaders, and key stakeholders striving to achieve optimal balance between clinical and academic roles. We describe the initial work of the SCHOLAR project, our definitions of academic success in AHPs, and the characteristics of a cohort of exemplary AHPs who achieved the highest levels on these metrics.
METHODS
Defining Success
The 11 members of the SCHOLAR project held a variety of clinical and academic roles within a geographically diverse group of AHPs. We sought to create a functional definition of success applicable to AHPs. As no gold standard currently exists, we used a consensus process among task force members to arrive at a definition that was quantifiable, feasible, and meaningful. The first step was brainstorming on conference calls held 1 to 2 times monthly over 4 months. Potential defining characteristics that emerged from these discussions related to research, teaching, and administrative activities. When potential characteristics were proposed, we considered how to operationalize each one. Each characteristic was discussed until there was consensus from the entire group. Those around education and administration were the most complex, as many roles are locally driven and defined, and challenging to quantify. For this reason, we focused on promotion as a more global approach to assessing academic hospitalist success in these areas. Although criteria for academic advancement also vary across institutions, we felt that promotion generally reflected having met some threshold of academic success. We also wanted to recognize that scholarship occurs outside the context of funded research. Ultimately, 3 key domains emerged: research grant funding, faculty promotion, and scholarship.
After these 3 domains were identified, the group sought to define quantitative metrics to assess performance. These discussions occurred on subsequent calls over a 4‐month period. Between calls, group members gathered additional information to facilitate assessment of the feasibility of proposed metrics, reporting on progress via email. Again, group consensus was sought for each metric considered. Data on grant funding and successful promotions were available from a previous survey conducted through the SHM in 2011. Leaders from 170 AHPs were contacted, with 50 providing complete responses to the 21‐item questionnaire (see Supporting Information, Appendix 1, in the online version of this article). Results of the survey, heretofore referred to as the Leaders of Academic Hospitalist Programs survey (LAHP‐50), have been described elsewhere.[7] For the purposes of this study, we used the self‐reported data about grant funding and promotions contained in the survey to reflect the current state of the field. Although the survey response rate was approximately 30%, the survey was not anonymous, and many reputationally prominent academic hospitalist programs were represented. For these reasons, the group members felt that the survey results were relevant for the purposes of assessing academic success.
In the LAHP‐50, funding was defined as principal investigator or coinvestigator roles on federally and nonfederally funded research, clinical trials, internal grants, and any other extramurally funded projects. Mean and median funding for the overall sample was calculated. Through a separate question, each program's total faculty full‐time equivalent (FTE) count was reported, allowing us to adjust for group size by assessing both total funding per group and funding/FTE for each responding AHP.
Promotions were defined by the self‐reported number of faculty at each of the following ranks: instructor, assistant professor, associate professor, full professor, and professor above scale/emeritus. In addition, a category of nonacademic track (eg, adjunct faculty, clinical associate) was included to capture hospitalists that did not fit into the traditional promotions categories. We did not distinguish between tenure‐track and nontenure‐track academic ranks. LAHP‐50 survey respondents reported the number of faculty in their group at each academic rank. Given that the majority of academic hospitalists hold a rank of assistant professor or lower,[6, 8, 9] and that the number of full professors was only 3% in the LAHP‐50 cohort, we combined the faculty at the associate and full professor ranks, defining successfully promoted faculty as the percent of hospitalists above the rank of assistant professor.
We created a new metric to assess scholarly output. We had considerable discussion of ways to assess the numbers of peer‐reviewed manuscripts generated by AHPs. However, the group had concerns about the feasibility of identification and attribution of authors to specific AHPs through literature searches. We considered examining only publications in the Journal of Hospital Medicine and the Journal of General Internal Medicine, but felt that this would exclude significant work published by hospitalists in fields of medical education or health services research that would more likely appear in alternate journals. Instead, we quantified scholarship based on the number of abstracts presented at national meetings. We focused on meetings of the SHM and SGIM as the primary professional societies representing hospital medicine. The group felt that even work published outside of the journals of our professional societies would likely be presented at those meetings. We used the following strategy: We reviewed research abstracts accepted for presentation as posters or oral abstracts at the 2010 and 2011 SHM national meetings, and research abstracts with a primary or secondary category of hospital medicine at the 2010 and 2011 SGIM national meetings. By including submissions at both SGIM and SHM meetings, we accounted for the fact that some programs may gravitate more to one society meeting or another. We did not include abstracts in the clinical vignettes or innovations categories. We tallied the number of abstracts by group affiliation of the authors for each of the 4 meetings above and created a cumulative total per group for the 2‐year period. Abstracts with authors from different AHPs were counted once for each individual group. Members of the study group reviewed abstracts from each of the meetings in pairs. Reviewers worked separately and compared tallies of results to ensure consistent tabulations. Internet searches were conducted to identify or confirm author affiliations if it was not apparent in the abstract author list. Abstract tallies were compiled without regard to whether programs had completed the LAHP‐50 survey; thus, we collected data on programs that did not respond to the LAHP‐50 survey.
Identification of the SCHOLAR Cohort
To identify our cohort of top‐performing AHPs, we combined the funding and promotions data from the LAHP‐50 sample with the abstract data. We limited our sample to adult hospital medicine groups to reduce heterogeneity. We created rank lists of programs in each category (grant funding, successful promotions, and scholarship), using data from the LAHP‐50 survey to rank programs on funding and promotions, and data from our abstract counts to rank on scholarship. We limited the top‐performing list in each category to 10 institutions as a cutoff. Because we set a threshold of at least $1 million in total funding, we identified only 9 top performing AHPs with regard to grant funding. We also calculated mean funding/FTE. We chose to rank programs only by funding/FTE rather than total funding per program to better account for group size. For successful promotions, we ranked programs by the percentage of senior faculty. For abstract counts, we included programs whose faculty presented abstracts at a minimum of 2 separate meetings, and ranked programs based on the total number of abstracts per group.
This process resulted in separate lists of top performing programs in each of the 3 domains we associated with academic success, arranged in descending order by grant dollars/FTE, percent of senior faculty, and abstract counts (Table 1). Seventeen different programs were represented across these 3 top 10 lists. One program appeared on all 3 lists, 8 programs appeared on 2 lists, and the remainder appeared on a single list (Table 2). Seven of these programs were identified solely based on abstract presentations, diversifying our top groups beyond only those who completed the LAHP‐50 survey. We considered all of these programs to represent high performance in academic hospital medicine. The group selected this inclusive approach because we recognized that any 1 metric was potentially limited, and we sought to identify diverse pathways to success.
| Funding | Promotions | Scholarship | |
|---|---|---|---|
| Grant $/FTE | Total Grant $ | Senior Faculty, No. (%) | Total Abstract Count |
| |||
| $1,409,090 | $15,500,000 | 3 (60%) | 23 |
| $1,000,000 | $9,000,000 | 3 (60%) | 21 |
| $750,000 | $8,000,000 | 4 (57%) | 20 |
| $478,609 | $6,700,535 | 9 (53%) | 15 |
| $347,826 | $3,000,000 | 8 (44%) | 11 |
| $86,956 | $3,000,000 | 14 (41%) | 11 |
| $66,666 | $2,000,000 | 17 (36%) | 10 |
| $46,153 | $1,500,000 | 9 (33%) | 10 |
| $38,461 | $1,000,000 | 2 (33%) | 9 |
| 4 (31%) | 9 | ||
| Selection Criteria for SCHOLAR Cohort | No. of Programs |
|---|---|
| |
| Abstracts, funding, and promotions | 1 |
| Abstracts plus promotions | 4 |
| Abstracts plus funding | 3 |
| Funding plus promotion | 1 |
| Funding only | 1 |
| Abstract only | 7 |
| Total | 17 |
| Top 10 abstract count | |
| 4 meetings | 2 |
| 3 meetings | 2 |
| 2 meetings | 6 |
The 17 unique adult AHPs appearing on at least 1 of the top 10 lists comprised the SCHOLAR cohort of programs that we studied in greater detail. Data reflecting program demographics were solicited directly from leaders of the AHPs identified in the SCHOLAR cohort, including size and age of program, reporting structure, number of faculty at various academic ranks (for programs that did not complete the LAHP‐50 survey), and number of faculty with fellowship training (defined as any postresidency fellowship program).
Subsequently, we performed comparative analyses between the programs in the SCHOLAR cohort to the general population of AHPs reflected by the LAHP‐50 sample. Because abstract presentations were not recorded in the original LAHP‐50 survey instrument, it was not possible to perform a benchmarking comparison for the scholarship domain.
Data Analysis
To measure the success of the SCHOLAR cohort we compared the grant funding and proportion of successfully promoted faculty at the SCHOLAR programs to those in the overall LAHP‐50 sample. Differences in mean and median grant funding were compared using t tests and Mann‐Whitney rank sum tests. Proportion of promoted faculty were compared using 2 tests. A 2‐tailed of 0.05 was used to test significance of differences.
RESULTS
Demographics
Among the AHPs in the SCHOLAR cohort, the mean program age was 13.2 years (range, 618 years), and the mean program size was 36 faculty (range, 1895; median, 28). On average, 15% of faculty members at SCHOLAR programs were fellowship trained (range, 0%37%). Reporting structure among the SCHOLAR programs was as follows: 53% were an independent division or section of the department of medicine; 29% were a section within general internal medicine, and 18% were an independent clinical group.
Grant Funding
Table 3 compares grant funding in the SCHOLAR programs to programs in the overall LAHP‐50 sample. Mean funding per group and mean funding per FTE were significantly higher in the SCHOLAR group than in the overall sample.
| Funding (Millions) | ||
|---|---|---|
| LAHP‐50 Overall Sample | SCHOLAR | |
| ||
| Median grant funding/AHP | 0.060 | 1.500* |
| Mean grant funding/AHP | 1.147 (015) | 3.984* (015) |
| Median grant funding/FTE | 0.004 | 0.038* |
| Mean grant funding/FTE | 0.095 (01.4) | 0.364* (01.4) |
Thirteen of the SCHOLAR programs were represented in the initial LAHP‐50, but 2 did not report a dollar amount for grants and contracts. Therefore, data for total grant funding were available for only 65% (11 of 17) of the programs in the SCHOLAR cohort. Of note, 28% of AHPs in the overall LAHP‐50 sample reported no external funding sources.
Faculty Promotion
Figure 1 demonstrates the proportion of faculty at various academic ranks. The percent of faculty above the rank of assistant professor in the SCHOLAR programs exceeded those in the overall LAHP‐50 by 5% (17.9% vs 12.8%, P = 0.01). Of note, 6% of the hospitalists at AHPs in the SCHOLAR programs were on nonfaculty tracks.
Scholarship
Mean abstract output over the 2‐year period measured was 10.8 (range, 323) in the SCHOLAR cohort. Because we did not collect these data for the LAHP‐50 group, comparative analyses were not possible.
DISCUSSION
Using a definition of academic success that incorporated metrics of grant funding, faculty promotion, and scholarly output, we identified a unique subset of successful AHPsthe SCHOLAR cohort. The programs represented in the SCHOLAR cohort were generally large and relatively mature. Despite this, the cohort consisted of mostly junior faculty, had a paucity of fellowship‐trained hospitalists, and not all reported grant funding.
Prior published work reported complementary findings.[6, 8, 9] A survey of 20 large, well‐established academic hospitalist programs in 2008 found that the majority of hospitalists were junior faculty with a limited publication portfolio. Of the 266 respondents in that study, 86% reported an academic rank at or below assistant professor; funding was not explored.[9] Our similar findings 4 years later add to this work by demonstrating trends over time, and suggest that progress toward creating successful pathways for academic advancement has been slow. In a 2012 survey of the SHM membership, 28% of hospitalists with academic appointments reported no current or future plans to engage in research.[8] These findings suggest that faculty in AHPs may define scholarship through nontraditional pathways, or in some cases choose not to pursue or prioritize scholarship altogether.
Our findings also add to the literature with regard to our assessment of funding, which was variable across the SCHOLAR group. The broad range of funding in the SCHOLAR programs for which we have data (grant dollars $0$15 million per program) suggests that opportunities to improve supported scholarship remain, even among a selected cohort of successful AHPs. The predominance of junior faculty in the SCHOLAR programs may be a reason for this variation. Junior faculty may be engaged in research with funding directed to senior mentors outside their AHP. Alternatively, they may pursue meaningful local hospital quality improvement or educational innovations not supported by external grants, or hold leadership roles in education, quality, or information technology that allow for advancement and promotion without external grant funding. As the scope and impact of these roles increases, senior leaders with alternate sources of support may rely less on research funds; this too may explain some of the differences. Our findings are congruent with results of a study that reviewed original research published by hospitalists, and concluded that the majority of hospitalist research was not externally funded.[8] Our approach for assessing grant funding by adjusting for FTE had the potential to inadvertently favor smaller well‐funded groups over larger ones; however, programs in our sample were similarly represented when ranked by funding/FTE or total grant dollars. As many successful AHPs do concentrate their research funding among a core of focused hospitalist researchers, our definition may not be the ideal metric for some programs.
We chose to define scholarship based on abstract output, rather than peer‐reviewed publications. Although this choice was necessary from a feasibility perspective, it may have excluded programs that prioritize peer‐reviewed publications over abstracts. Although we were unable to incorporate a search strategy to accurately and comprehensively track the publication output attributed specifically to hospitalist researchers and quantify it by program, others have since defined such an approach.[8] However, tracking abstracts theoretically allowed insights into a larger volume of innovative and creative work generated by top AHPs by potentially including work in the earlier stages of development.
We used a consensus‐based definition of success to define our SCHOLAR cohort. There are other ways to measure academic success, which if applied, may have yielded a different sample of programs. For example, over half of the original research articles published in the Journal of Hospital Medicine over a 7‐year span were generated from 5 academic centers.[8] This definition of success may be equally credible, though we note that 4 of these 5 programs were also included in the SCHOLAR cohort. We feel our broader approach was more reflective of the variety of pathways to success available to academic hospitalists. Before our metrics are applied as a benchmarking tool, however, they should ideally be combined with factors not measured in our study to ensure a more comprehensive or balanced reflection of academic success. Factors such as mentorship, level of hospitalist engagement,[10] prevalence of leadership opportunities, operational and fiscal infrastructure, and the impact of local quality, safety, and value efforts should be considered.
Comparison of successfully promoted faculty at AHPs across the country is inherently limited by the wide variation in promotion standards across different institutions; controlling for such differences was not possible with our methodology. For example, it appears that several programs with relatively few senior faculty may have met metrics leading to their inclusion in the SCHOLAR group because of their small program size. Future benchmarking efforts for promotion at AHPs should take scaling into account and consider both total number as well as percentage of senior faculty when evaluating success.
Our methodology has several limitations. Survey data were self‐reported and not independently validated, and as such are subject to recall and reporting biases. Response bias inherently excluded some AHPs that may have met our grant funding or promotions criteria had they participated in the initial LAHP‐50 survey, though we identified and included additional programs through our scholarship metric, increasing the representativeness of the SCHOLAR cohort. Given the dynamic nature of the field, the age of the data we relied upon for analysis limits the generalizability of our specific benchmarks to current practice. However, the development of academic success occurs over the long‐term, and published data on academic hospitalist productivity are consistent with this slower time course.[8] Despite these limitations, our data inform the general topic of gauging performance of AHPs, underscoring the challenges of developing and applying metrics of success, and highlight the variability of performance on selected metrics even among a relatively small group of 17 programs.
In conclusion, we have created a method to quantify academic success that may be useful to academic hospitalists and their group leaders as they set targets for improvement in the field. Even among our SCHOLAR cohort, room for ongoing improvement in development of funded scholarship and a core of senior faculty exists. Further investigation into the unique features of successful groups will offer insight to leaders in academic hospital medicine regarding infrastructure and processes that should be embraced to raise the bar for all AHPs. In addition, efforts to further define and validate nontraditional approaches to scholarship that allow for successful promotion at AHPs would be informative. We view our work less as a singular approach to benchmarking standards for AHPs, and more a call to action to continue efforts to balance scholarly activity and broad professional development of academic hospitalists with increasing clinical demands.
Acknowledgements
The authors thank all of the AHP leaders who participated in the SCHOLAR project. They also thank the Society of Hospital Medicine and Society of General Internal Medicine and the SHM Academic Committee and SGIM Academic Hospitalist Task Force for their support of this work.
Disclosures
The work reported here was supported by the Department of Veterans Affairs, Veterans Health Administration, South Texas Veterans Health Care System. The views expressed in this article are those of the authors and do not necessarily reflect the position or policy of the Department of Veterans Affairs. The authors report no conflicts of interest.
- , , , , . Characteristics of primary care providers who adopted the hospitalist model from 2001 to 2009. J Hosp Med. 2015;10(2):75–82.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , , , . Updating threshold‐based identification of hospitalists in 2012 Medicare pay data. J Hosp Med. 2016;11(1):45–47.
- , , , . Use of hospitalists by Medicare beneficiaries: a national picture. Medicare Medicaid Res Rev. 2014;4(2).
- , , , , , . Challenges and opportunities in Academic Hospital Medicine: report from the Academic Hospital Medicine Summit. J Hosp Med. 2009;4(4):240–246.
- , , , . Survey of US academic hospitalist leaders about mentorship and academic activities in hospitalist groups. J Hosp Med. 2011;6(1):5–9.
- , , , , , . The structure of hospital medicine programs at academic medical centers [abstract]. J Hosp Med. 2012;7(suppl 2):s92.
- , , , , . Research and publication trends in hospital medicine. J Hosp Med. 2014;9(3):148–154.
- , , , , , . Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23–27.
- , , , et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123–128.
- , , , , . Characteristics of primary care providers who adopted the hospitalist model from 2001 to 2009. J Hosp Med. 2015;10(2):75–82.
- , , , . Growth in the care of older patients by hospitalists in the United States. N Engl J Med. 2009;360(11):1102–1112.
- , , , , . Updating threshold‐based identification of hospitalists in 2012 Medicare pay data. J Hosp Med. 2016;11(1):45–47.
- , , , . Use of hospitalists by Medicare beneficiaries: a national picture. Medicare Medicaid Res Rev. 2014;4(2).
- , , , , , . Challenges and opportunities in Academic Hospital Medicine: report from the Academic Hospital Medicine Summit. J Hosp Med. 2009;4(4):240–246.
- , , , . Survey of US academic hospitalist leaders about mentorship and academic activities in hospitalist groups. J Hosp Med. 2011;6(1):5–9.
- , , , , , . The structure of hospital medicine programs at academic medical centers [abstract]. J Hosp Med. 2012;7(suppl 2):s92.
- , , , , . Research and publication trends in hospital medicine. J Hosp Med. 2014;9(3):148–154.
- , , , , , . Mentorship, productivity, and promotion among academic hospitalists. J Gen Intern Med. 2012;27(1):23–27.
- , , , et al. The key principles and characteristics of an effective hospital medicine group: an assessment guide for hospitals and hospitalists. J Hosp Med. 2014;9(2):123–128.
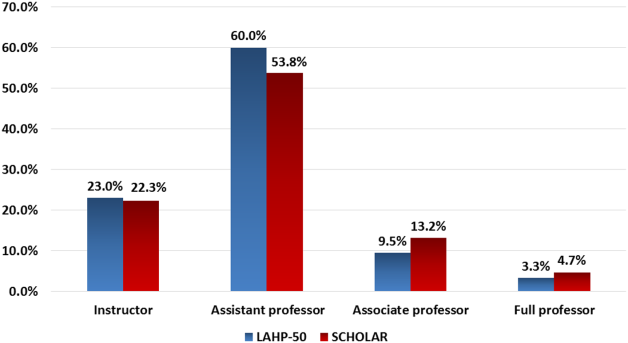
Doctors ask Congress to stop Part B drug payment test
WASHINGTON – Physician organizations are calling on Congress to stop a proposed federal regulation that would test how Medicare pays for drugs administered in a physician’s office.
Subcommittee member Rep. Larry Bucshon (R-Ind.), a cardiothoracic surgeon, called the premise of the proposed rule – that physicians are making prescribing decisions based on the cost of drugs – “almost an insult to the medical profession.” He has introduced legislation, H.R. 5122, that would require the Centers for Medicare & Medicaid Services to rescind the proposed rule.
CMS argues in the proposed rule that the current payment formula – average sales price (ASP) plus 6% – incentivizes the use of expensive drugs over lower-cost alternatives. Therefore, the agency seeks to run a test – half of doctors would continue to receive ASP plus 6%, while others would receive ASP plus 2.5% and a flat fee of $16.80. The proposed rule also would test other value-based tools. At the May 17 hearing, physicians presented their concerns regarding the proposed rule.

“CMS has yet to produce any evidence indicating that physician prescribing patterns show any correlation to that of choosing higher priced drugs as opposed to appropriate therapeutic treatment for patients,” said Dr. Debra A. Patt, vice president of Texas Oncology. Dr. Patt testified on behalf of the American Society of Clinical Oncology, the Community Oncology Alliance, and the U.S. Oncology Network. “Additionally, there is no evidence that the payment changes contemplated by CMS’s model will improve the quality of care, or for that matter, ensure patients have access to the same level of care they are currently receiving.”Dr. Michael Schweitz, national advocacy chair of the Coalition of State Rheumatology Organizations (CSRO), cited CMS’s admission that this rule will likely have no impact on ASP.
“While we appreciate CMS’s attention to the topic of drug costs, we feel that this proposal is misguided,” Dr. Schweitz testified. “As CMS acknowledges in the rule, the proposed approach ‘does not directly address the manufacturer’s ASP, which is a more significant driver of drug expenditures than the add-on payment amount for Part B drugs.’ Given that a slash to the ASP add-on is unlikely to actually lower costs for patients ... and may jeopardize access, we have requested that CMS withdraw the model and we urge the committee to do the same.”
Dr. Patt also questioned ASP, noting that it is simply an average, and the prices actually paid by rural and small group providers could be higher than larger group and hospital practices – and that difference puts the smaller and rural practices at a potentially significant financial disadvantage.
“Average sales price is by its very nature an average,” Dr. Patt testified. “Some people will pay higher amounts for procurement and some people will pay lower amounts. Larger hospital systems and larger practices have the ability to have contracting arrangements where they purchase at a lower price. … Smaller practices disproportionately pay a higher amount.”
Small practices that are slated to receive ASP plus 2.5% and that flat fee “could be losing money on all of the drugs that they buy. It will be impossible for smaller practices in rural areas to be open,” she added, noting that such a situation could cause care to be shifted to hospital outpatient departments, raising costs as well as access issues. “Cutting provider reimbursement without addressing the ASP, the actual cost of the drug in the first place, is just the wrong approach,” Rep. Bucshon said.
According to the proposed rule, whether or not a practice is in the test or control group would be based on ZIP code. Dr. Patt also noted that if she were in a test ZIP code and a neighboring ZIP code was not, she might have to refer patients to that area still receiving ASP plus 6% and “not at my center.”
Witnesses at the hearing also condemned the way CMS devised the proposed tests. Unlike the recent work on the Medicare Access and CHIP Reauthorization Act regulations and more specialized programs like the Oncology Care Model, Dr. Schweitz and Dr. Patt both testified that CMS made no outreach to the provider community with regards to getting their input before issuing the proposed rule.
They also took exception to the scope of the proposed test, which covers 49 of the 50 states (Maryland is excluded) and provides no mechanism for physicians to opt out under the proposed rule.
“I do see this as an experiment, but we conduct clinical research at our cancer and patients have informed consent,” Dr Patt said.
Dr. Schweitz agreed. “When you look at the goals of this plan, initially it appeared that it was to direct a way to save costs. But in meeting with [the Center for Medicare & Medicaid Innovation], we were advised that this is budget neutral. … So the goal of the program is to collect information which makes it a study, a test. So if the goal is to collect information, and the patients are part of that process, they should be signing informed consent.”
Several physician organizations have called on CMS to withdraw the proposal.
“We are deeply concerned that because the new methodology will frequently not properly cover the cost of physician administration of infused drugs, they will be forced to stop offering patients the ability to receive infusion treatments,” the American College of Rheumatology wrote in comments submitted on the proposed rule. Likewise, CSRO “must oppose the Part B drug payment model as it suffers from serious procedural and substantive flaws that we believe render it unworkable – and it does nothing to actually address drug prices,” according its comments.
While the proposal has garnered backlash from several directions, rheumatologists are seeing it as particularly burdensome because of the high price of medications with very limited options to substitute for lower-cost alternatives.
“Although we certainly seek to control costs for patients and Medicare whenever possible, the proposed new methodology does not adequately consider the higher average cost many of our physicians have acquiring, handling, administering, and billing for drugs and biologics,” according to the comments submitted by the ACR.
Indeed, comments from CSRO point out that when factoring in budget sequestration, the actual reimbursement physicians are receiving is ASP plus 4.4%, and doctors are actually losing money on certain drug purchases.
Of additional concern is that the proposed rule does not address ASP itself.
“A far greater concern than the add-on percentage is the underlying ASP, and the steep, fast price increases that these medications show each quarter, according to comments from the CSRO.
From 2007 to 2016, first-quarter ASP for infliximab rose from $53.73 to $79.90; ASP for abatacept rose from $18.70 to $39.44, according to CSRO comments. “These ASP increases are unsustainable for both the Medicare program and its beneficiaries, and we would like to work with CMS to explore actual solutions to stem the increases in those underlying prices.”
In its comments, ACR proposed a number of potential paths forward, starting with certain practices that should be exempted from the proposed demonstration: physician groups with 25 or fewer physicians; physician-owned practices that are located in rural and medically underserved areas; reimbursement changes for drugs and biologics that do not have an alternative with more than a 20% ASP differential; and drugs and biologics where there are three or fewer members of the drug class or biologics.
ACR also proposed altering the add-on formula that takes into account the costs of storing and administering supplies.
“For example, CMS could use a formula for reimbursement of ASP plus 6% or $500 (whichever is lower),” ACR said in its comments. “This formula would allow CMS to effectively target spending on expensive drugs, while leaving in place reimbursement rates for cheaper drugs.”
Additionally, ACR called for CMS to delay the testing of more value-based tools until it understands the impact of the ASP changes that are to be tested under this proposal.
CSRO does not have any specific policy recommendations to replace or modify the proposed rule, but rather calls for CMS to bring together all stakeholders, including patients, providers, payers, and manufacturers to devise a system that would work to the benefit of all while ensuring the best outcomes for patients, Dr. Schweitz said in an interview.
WASHINGTON – Physician organizations are calling on Congress to stop a proposed federal regulation that would test how Medicare pays for drugs administered in a physician’s office.
Subcommittee member Rep. Larry Bucshon (R-Ind.), a cardiothoracic surgeon, called the premise of the proposed rule – that physicians are making prescribing decisions based on the cost of drugs – “almost an insult to the medical profession.” He has introduced legislation, H.R. 5122, that would require the Centers for Medicare & Medicaid Services to rescind the proposed rule.
CMS argues in the proposed rule that the current payment formula – average sales price (ASP) plus 6% – incentivizes the use of expensive drugs over lower-cost alternatives. Therefore, the agency seeks to run a test – half of doctors would continue to receive ASP plus 6%, while others would receive ASP plus 2.5% and a flat fee of $16.80. The proposed rule also would test other value-based tools. At the May 17 hearing, physicians presented their concerns regarding the proposed rule.
“CMS has yet to produce any evidence indicating that physician prescribing patterns show any correlation to that of choosing higher priced drugs as opposed to appropriate therapeutic treatment for patients,” said Dr. Debra A. Patt, vice president of Texas Oncology. Dr. Patt testified on behalf of the American Society of Clinical Oncology, the Community Oncology Alliance, and the U.S. Oncology Network. “Additionally, there is no evidence that the payment changes contemplated by CMS’s model will improve the quality of care, or for that matter, ensure patients have access to the same level of care they are currently receiving.”Dr. Michael Schweitz, national advocacy chair of the Coalition of State Rheumatology Organizations (CSRO), cited CMS’s admission that this rule will likely have no impact on ASP.
“While we appreciate CMS’s attention to the topic of drug costs, we feel that this proposal is misguided,” Dr. Schweitz testified. “As CMS acknowledges in the rule, the proposed approach ‘does not directly address the manufacturer’s ASP, which is a more significant driver of drug expenditures than the add-on payment amount for Part B drugs.’ Given that a slash to the ASP add-on is unlikely to actually lower costs for patients ... and may jeopardize access, we have requested that CMS withdraw the model and we urge the committee to do the same.”
Dr. Patt also questioned ASP, noting that it is simply an average, and the prices actually paid by rural and small group providers could be higher than larger group and hospital practices – and that difference puts the smaller and rural practices at a potentially significant financial disadvantage.
“Average sales price is by its very nature an average,” Dr. Patt testified. “Some people will pay higher amounts for procurement and some people will pay lower amounts. Larger hospital systems and larger practices have the ability to have contracting arrangements where they purchase at a lower price. … Smaller practices disproportionately pay a higher amount.”
Small practices that are slated to receive ASP plus 2.5% and that flat fee “could be losing money on all of the drugs that they buy. It will be impossible for smaller practices in rural areas to be open,” she added, noting that such a situation could cause care to be shifted to hospital outpatient departments, raising costs as well as access issues. “Cutting provider reimbursement without addressing the ASP, the actual cost of the drug in the first place, is just the wrong approach,” Rep. Bucshon said.
According to the proposed rule, whether or not a practice is in the test or control group would be based on ZIP code. Dr. Patt also noted that if she were in a test ZIP code and a neighboring ZIP code was not, she might have to refer patients to that area still receiving ASP plus 6% and “not at my center.”
Witnesses at the hearing also condemned the way CMS devised the proposed tests. Unlike the recent work on the Medicare Access and CHIP Reauthorization Act regulations and more specialized programs like the Oncology Care Model, Dr. Schweitz and Dr. Patt both testified that CMS made no outreach to the provider community with regards to getting their input before issuing the proposed rule.
They also took exception to the scope of the proposed test, which covers 49 of the 50 states (Maryland is excluded) and provides no mechanism for physicians to opt out under the proposed rule.
“I do see this as an experiment, but we conduct clinical research at our cancer and patients have informed consent,” Dr Patt said.
Dr. Schweitz agreed. “When you look at the goals of this plan, initially it appeared that it was to direct a way to save costs. But in meeting with [the Center for Medicare & Medicaid Innovation], we were advised that this is budget neutral. … So the goal of the program is to collect information which makes it a study, a test. So if the goal is to collect information, and the patients are part of that process, they should be signing informed consent.”
Several physician organizations have called on CMS to withdraw the proposal.
“We are deeply concerned that because the new methodology will frequently not properly cover the cost of physician administration of infused drugs, they will be forced to stop offering patients the ability to receive infusion treatments,” the American College of Rheumatology wrote in comments submitted on the proposed rule. Likewise, CSRO “must oppose the Part B drug payment model as it suffers from serious procedural and substantive flaws that we believe render it unworkable – and it does nothing to actually address drug prices,” according its comments.
While the proposal has garnered backlash from several directions, rheumatologists are seeing it as particularly burdensome because of the high price of medications with very limited options to substitute for lower-cost alternatives.
“Although we certainly seek to control costs for patients and Medicare whenever possible, the proposed new methodology does not adequately consider the higher average cost many of our physicians have acquiring, handling, administering, and billing for drugs and biologics,” according to the comments submitted by the ACR.
Indeed, comments from CSRO point out that when factoring in budget sequestration, the actual reimbursement physicians are receiving is ASP plus 4.4%, and doctors are actually losing money on certain drug purchases.
Of additional concern is that the proposed rule does not address ASP itself.
“A far greater concern than the add-on percentage is the underlying ASP, and the steep, fast price increases that these medications show each quarter, according to comments from the CSRO.
From 2007 to 2016, first-quarter ASP for infliximab rose from $53.73 to $79.90; ASP for abatacept rose from $18.70 to $39.44, according to CSRO comments. “These ASP increases are unsustainable for both the Medicare program and its beneficiaries, and we would like to work with CMS to explore actual solutions to stem the increases in those underlying prices.”
In its comments, ACR proposed a number of potential paths forward, starting with certain practices that should be exempted from the proposed demonstration: physician groups with 25 or fewer physicians; physician-owned practices that are located in rural and medically underserved areas; reimbursement changes for drugs and biologics that do not have an alternative with more than a 20% ASP differential; and drugs and biologics where there are three or fewer members of the drug class or biologics.
ACR also proposed altering the add-on formula that takes into account the costs of storing and administering supplies.
“For example, CMS could use a formula for reimbursement of ASP plus 6% or $500 (whichever is lower),” ACR said in its comments. “This formula would allow CMS to effectively target spending on expensive drugs, while leaving in place reimbursement rates for cheaper drugs.”
Additionally, ACR called for CMS to delay the testing of more value-based tools until it understands the impact of the ASP changes that are to be tested under this proposal.
CSRO does not have any specific policy recommendations to replace or modify the proposed rule, but rather calls for CMS to bring together all stakeholders, including patients, providers, payers, and manufacturers to devise a system that would work to the benefit of all while ensuring the best outcomes for patients, Dr. Schweitz said in an interview.
WASHINGTON – Physician organizations are calling on Congress to stop a proposed federal regulation that would test how Medicare pays for drugs administered in a physician’s office.
Subcommittee member Rep. Larry Bucshon (R-Ind.), a cardiothoracic surgeon, called the premise of the proposed rule – that physicians are making prescribing decisions based on the cost of drugs – “almost an insult to the medical profession.” He has introduced legislation, H.R. 5122, that would require the Centers for Medicare & Medicaid Services to rescind the proposed rule.
CMS argues in the proposed rule that the current payment formula – average sales price (ASP) plus 6% – incentivizes the use of expensive drugs over lower-cost alternatives. Therefore, the agency seeks to run a test – half of doctors would continue to receive ASP plus 6%, while others would receive ASP plus 2.5% and a flat fee of $16.80. The proposed rule also would test other value-based tools. At the May 17 hearing, physicians presented their concerns regarding the proposed rule.
“CMS has yet to produce any evidence indicating that physician prescribing patterns show any correlation to that of choosing higher priced drugs as opposed to appropriate therapeutic treatment for patients,” said Dr. Debra A. Patt, vice president of Texas Oncology. Dr. Patt testified on behalf of the American Society of Clinical Oncology, the Community Oncology Alliance, and the U.S. Oncology Network. “Additionally, there is no evidence that the payment changes contemplated by CMS’s model will improve the quality of care, or for that matter, ensure patients have access to the same level of care they are currently receiving.”Dr. Michael Schweitz, national advocacy chair of the Coalition of State Rheumatology Organizations (CSRO), cited CMS’s admission that this rule will likely have no impact on ASP.
“While we appreciate CMS’s attention to the topic of drug costs, we feel that this proposal is misguided,” Dr. Schweitz testified. “As CMS acknowledges in the rule, the proposed approach ‘does not directly address the manufacturer’s ASP, which is a more significant driver of drug expenditures than the add-on payment amount for Part B drugs.’ Given that a slash to the ASP add-on is unlikely to actually lower costs for patients ... and may jeopardize access, we have requested that CMS withdraw the model and we urge the committee to do the same.”
Dr. Patt also questioned ASP, noting that it is simply an average, and the prices actually paid by rural and small group providers could be higher than larger group and hospital practices – and that difference puts the smaller and rural practices at a potentially significant financial disadvantage.
“Average sales price is by its very nature an average,” Dr. Patt testified. “Some people will pay higher amounts for procurement and some people will pay lower amounts. Larger hospital systems and larger practices have the ability to have contracting arrangements where they purchase at a lower price. … Smaller practices disproportionately pay a higher amount.”
Small practices that are slated to receive ASP plus 2.5% and that flat fee “could be losing money on all of the drugs that they buy. It will be impossible for smaller practices in rural areas to be open,” she added, noting that such a situation could cause care to be shifted to hospital outpatient departments, raising costs as well as access issues. “Cutting provider reimbursement without addressing the ASP, the actual cost of the drug in the first place, is just the wrong approach,” Rep. Bucshon said.
According to the proposed rule, whether or not a practice is in the test or control group would be based on ZIP code. Dr. Patt also noted that if she were in a test ZIP code and a neighboring ZIP code was not, she might have to refer patients to that area still receiving ASP plus 6% and “not at my center.”
Witnesses at the hearing also condemned the way CMS devised the proposed tests. Unlike the recent work on the Medicare Access and CHIP Reauthorization Act regulations and more specialized programs like the Oncology Care Model, Dr. Schweitz and Dr. Patt both testified that CMS made no outreach to the provider community with regards to getting their input before issuing the proposed rule.
They also took exception to the scope of the proposed test, which covers 49 of the 50 states (Maryland is excluded) and provides no mechanism for physicians to opt out under the proposed rule.
“I do see this as an experiment, but we conduct clinical research at our cancer and patients have informed consent,” Dr Patt said.
Dr. Schweitz agreed. “When you look at the goals of this plan, initially it appeared that it was to direct a way to save costs. But in meeting with [the Center for Medicare & Medicaid Innovation], we were advised that this is budget neutral. … So the goal of the program is to collect information which makes it a study, a test. So if the goal is to collect information, and the patients are part of that process, they should be signing informed consent.”
Several physician organizations have called on CMS to withdraw the proposal.
“We are deeply concerned that because the new methodology will frequently not properly cover the cost of physician administration of infused drugs, they will be forced to stop offering patients the ability to receive infusion treatments,” the American College of Rheumatology wrote in comments submitted on the proposed rule. Likewise, CSRO “must oppose the Part B drug payment model as it suffers from serious procedural and substantive flaws that we believe render it unworkable – and it does nothing to actually address drug prices,” according its comments.
While the proposal has garnered backlash from several directions, rheumatologists are seeing it as particularly burdensome because of the high price of medications with very limited options to substitute for lower-cost alternatives.
“Although we certainly seek to control costs for patients and Medicare whenever possible, the proposed new methodology does not adequately consider the higher average cost many of our physicians have acquiring, handling, administering, and billing for drugs and biologics,” according to the comments submitted by the ACR.
Indeed, comments from CSRO point out that when factoring in budget sequestration, the actual reimbursement physicians are receiving is ASP plus 4.4%, and doctors are actually losing money on certain drug purchases.
Of additional concern is that the proposed rule does not address ASP itself.
“A far greater concern than the add-on percentage is the underlying ASP, and the steep, fast price increases that these medications show each quarter, according to comments from the CSRO.
From 2007 to 2016, first-quarter ASP for infliximab rose from $53.73 to $79.90; ASP for abatacept rose from $18.70 to $39.44, according to CSRO comments. “These ASP increases are unsustainable for both the Medicare program and its beneficiaries, and we would like to work with CMS to explore actual solutions to stem the increases in those underlying prices.”
In its comments, ACR proposed a number of potential paths forward, starting with certain practices that should be exempted from the proposed demonstration: physician groups with 25 or fewer physicians; physician-owned practices that are located in rural and medically underserved areas; reimbursement changes for drugs and biologics that do not have an alternative with more than a 20% ASP differential; and drugs and biologics where there are three or fewer members of the drug class or biologics.
ACR also proposed altering the add-on formula that takes into account the costs of storing and administering supplies.
“For example, CMS could use a formula for reimbursement of ASP plus 6% or $500 (whichever is lower),” ACR said in its comments. “This formula would allow CMS to effectively target spending on expensive drugs, while leaving in place reimbursement rates for cheaper drugs.”
Additionally, ACR called for CMS to delay the testing of more value-based tools until it understands the impact of the ASP changes that are to be tested under this proposal.
CSRO does not have any specific policy recommendations to replace or modify the proposed rule, but rather calls for CMS to bring together all stakeholders, including patients, providers, payers, and manufacturers to devise a system that would work to the benefit of all while ensuring the best outcomes for patients, Dr. Schweitz said in an interview.
AT HOUSE ENERGY AND COMMERCE HEALTH SUBCOMMITTEE HEARING

Stroke risk rises quickly in recent-onset atrial fib
CHICAGO – The stroke risk in patients with recent-onset atrial fibrillation is similar to that of patients with longer-standing atrial fibrillation, according to a new secondary analysis of the landmark ARISTOTLE trial.
“Our key message is that patients with recent-onset atrial fibrillation had a similar risk of stroke but higher mortality than patients with remotely diagnosed atrial fibrillation, suggesting that patients with recently diagnosed atrial fibrillation are not at low risk and therefore warrant stroke prevention strategies,” Dr. Patricia O. Guimaraes said in presenting the findings at the annual meeting of the American College of Cardiology.

“Sometimes we as physicians hesitate in beginning oral anticoagulation therapy for patients that we just diagnosed. And of course patients are often afraid of anticoagulation therapy. But once they present with atrial fibrillation they are already at risk, and that’s why we need to anticoagulate them promptly,” added Dr. Guimaraes of the Duke Clinical Research Institute in Durham, N.C.
The benefits of apixaban (Eliquis) over warfarin seen in the overall randomized ARISTOTLE trial (N Engl J Med. 2011; 365:981-92) were preserved in the recent-onset subset of the atrial fibrillation (AF) study population, she noted.
The rationale for this new post hoc analysis of ARISTOTLE is that virtually all of the evidence supporting anticoagulation for stroke prevention in AF is based on studies conducted in patients with permanent, persistent, or long-standing paroxysmal AF. Much less is known about stroke risk and the benefits of anticoagulation in patients with recent-onset AF, Dr. Guimaraes explained.
The 1,899 ARISTOTLE participants with AF onset within 30 days prior to enrollment comprised 10.5% of the total study population, all of whom had AF and at least one other stroke risk factor. The recent-onset subgroup was the same age as the 16,241 subjects in this analysis who had longer-standing AF, but the recent-onset group included a higher proportion of women, had a lower prevalence of CAD, and their cardiovascular risk factor profile differed from that of the remote-onset AF group.
The composite endpoint of stroke, systemic embolism, major bleeding, or all-cause mortality occurred at a rate of 8.69%/year in the recent-onset AF group, compared with 6.43%/year in the remote-onset group. However, in a multivariate regression analysis adjusted for potential confounders, the only significant differences in outcome between the two groups were in all-cause mortality – 5.15%/year in the recent-onset group, 3.15% in the remote-onset AF patients – and in the composite of stroke, systemic embolism, or all-cause mortality, which had an incidence of 6.46%/year in the recent-onset group, compared with 4.57%/year in remote-onset patients.
Turning to the impact of apixaban, Dr. Guimaraes noted that, as previously reported in the overall study, the primary endpoint of stroke or systemic embolism occurred in the apixaban group at a rate of 1.27%/year, compared with 1.6%/year with warfarin, for a 21% relative risk reduction in favor of the newer agent. She and her coinvestigators determined that in the remote-onset AF subgroup the relative risk reduction was 20%, while in the recent-onset subgroup the size of the effect was similar at 22%.
The composite safety endpoint of major or clinically relevant bleeding occurred in the remote-onset patients at a rate of 3.97%/year with apixaban versus 5.97%/year with warfarin, for a 33% relative risk reduction favoring the novel agent. In the recent-onset group, the rates were 5.04%/year with apixaban, compared with 6.4%/year with warfarin, for a 22% relative risk reduction.
Dr. Guimaraes observed an important limitation of this post hoc analysis is that the remote-onset AF group may have been selected for improved survival, since they didn’t die in the first 30 days after diagnosis.
Session co-chair Dr. Brian Olshansky commented that this analysis, which highlights the risks of recent-onset AF, argues for a strategy whereby a patient who presents to the ED with new-onset AF should get sent home on apixaban rather than being hospitalized for several days in order to be stabilized on warfarin.
“With recent-onset atrial fibrillation it’s going to take you several days to get anticoagulated with warfarin, whereas you’re immediately anticoagulated with apixaban,” said Dr. Olshansky, emeritus professor of internal medicine at the University of Iowa, Iowa City.
The ARISTOTLE trial was supported by Bristol-Myers Squibb and Pfizer. Dr. Guimaraes reported having no financial conflicts of interest.
CHICAGO – The stroke risk in patients with recent-onset atrial fibrillation is similar to that of patients with longer-standing atrial fibrillation, according to a new secondary analysis of the landmark ARISTOTLE trial.
“Our key message is that patients with recent-onset atrial fibrillation had a similar risk of stroke but higher mortality than patients with remotely diagnosed atrial fibrillation, suggesting that patients with recently diagnosed atrial fibrillation are not at low risk and therefore warrant stroke prevention strategies,” Dr. Patricia O. Guimaraes said in presenting the findings at the annual meeting of the American College of Cardiology.
“Sometimes we as physicians hesitate in beginning oral anticoagulation therapy for patients that we just diagnosed. And of course patients are often afraid of anticoagulation therapy. But once they present with atrial fibrillation they are already at risk, and that’s why we need to anticoagulate them promptly,” added Dr. Guimaraes of the Duke Clinical Research Institute in Durham, N.C.
The benefits of apixaban (Eliquis) over warfarin seen in the overall randomized ARISTOTLE trial (N Engl J Med. 2011; 365:981-92) were preserved in the recent-onset subset of the atrial fibrillation (AF) study population, she noted.
The rationale for this new post hoc analysis of ARISTOTLE is that virtually all of the evidence supporting anticoagulation for stroke prevention in AF is based on studies conducted in patients with permanent, persistent, or long-standing paroxysmal AF. Much less is known about stroke risk and the benefits of anticoagulation in patients with recent-onset AF, Dr. Guimaraes explained.
The 1,899 ARISTOTLE participants with AF onset within 30 days prior to enrollment comprised 10.5% of the total study population, all of whom had AF and at least one other stroke risk factor. The recent-onset subgroup was the same age as the 16,241 subjects in this analysis who had longer-standing AF, but the recent-onset group included a higher proportion of women, had a lower prevalence of CAD, and their cardiovascular risk factor profile differed from that of the remote-onset AF group.
The composite endpoint of stroke, systemic embolism, major bleeding, or all-cause mortality occurred at a rate of 8.69%/year in the recent-onset AF group, compared with 6.43%/year in the remote-onset group. However, in a multivariate regression analysis adjusted for potential confounders, the only significant differences in outcome between the two groups were in all-cause mortality – 5.15%/year in the recent-onset group, 3.15% in the remote-onset AF patients – and in the composite of stroke, systemic embolism, or all-cause mortality, which had an incidence of 6.46%/year in the recent-onset group, compared with 4.57%/year in remote-onset patients.
Turning to the impact of apixaban, Dr. Guimaraes noted that, as previously reported in the overall study, the primary endpoint of stroke or systemic embolism occurred in the apixaban group at a rate of 1.27%/year, compared with 1.6%/year with warfarin, for a 21% relative risk reduction in favor of the newer agent. She and her coinvestigators determined that in the remote-onset AF subgroup the relative risk reduction was 20%, while in the recent-onset subgroup the size of the effect was similar at 22%.
The composite safety endpoint of major or clinically relevant bleeding occurred in the remote-onset patients at a rate of 3.97%/year with apixaban versus 5.97%/year with warfarin, for a 33% relative risk reduction favoring the novel agent. In the recent-onset group, the rates were 5.04%/year with apixaban, compared with 6.4%/year with warfarin, for a 22% relative risk reduction.
Dr. Guimaraes observed an important limitation of this post hoc analysis is that the remote-onset AF group may have been selected for improved survival, since they didn’t die in the first 30 days after diagnosis.
Session co-chair Dr. Brian Olshansky commented that this analysis, which highlights the risks of recent-onset AF, argues for a strategy whereby a patient who presents to the ED with new-onset AF should get sent home on apixaban rather than being hospitalized for several days in order to be stabilized on warfarin.
“With recent-onset atrial fibrillation it’s going to take you several days to get anticoagulated with warfarin, whereas you’re immediately anticoagulated with apixaban,” said Dr. Olshansky, emeritus professor of internal medicine at the University of Iowa, Iowa City.
The ARISTOTLE trial was supported by Bristol-Myers Squibb and Pfizer. Dr. Guimaraes reported having no financial conflicts of interest.
CHICAGO – The stroke risk in patients with recent-onset atrial fibrillation is similar to that of patients with longer-standing atrial fibrillation, according to a new secondary analysis of the landmark ARISTOTLE trial.
“Our key message is that patients with recent-onset atrial fibrillation had a similar risk of stroke but higher mortality than patients with remotely diagnosed atrial fibrillation, suggesting that patients with recently diagnosed atrial fibrillation are not at low risk and therefore warrant stroke prevention strategies,” Dr. Patricia O. Guimaraes said in presenting the findings at the annual meeting of the American College of Cardiology.
“Sometimes we as physicians hesitate in beginning oral anticoagulation therapy for patients that we just diagnosed. And of course patients are often afraid of anticoagulation therapy. But once they present with atrial fibrillation they are already at risk, and that’s why we need to anticoagulate them promptly,” added Dr. Guimaraes of the Duke Clinical Research Institute in Durham, N.C.
The benefits of apixaban (Eliquis) over warfarin seen in the overall randomized ARISTOTLE trial (N Engl J Med. 2011; 365:981-92) were preserved in the recent-onset subset of the atrial fibrillation (AF) study population, she noted.
The rationale for this new post hoc analysis of ARISTOTLE is that virtually all of the evidence supporting anticoagulation for stroke prevention in AF is based on studies conducted in patients with permanent, persistent, or long-standing paroxysmal AF. Much less is known about stroke risk and the benefits of anticoagulation in patients with recent-onset AF, Dr. Guimaraes explained.
The 1,899 ARISTOTLE participants with AF onset within 30 days prior to enrollment comprised 10.5% of the total study population, all of whom had AF and at least one other stroke risk factor. The recent-onset subgroup was the same age as the 16,241 subjects in this analysis who had longer-standing AF, but the recent-onset group included a higher proportion of women, had a lower prevalence of CAD, and their cardiovascular risk factor profile differed from that of the remote-onset AF group.
The composite endpoint of stroke, systemic embolism, major bleeding, or all-cause mortality occurred at a rate of 8.69%/year in the recent-onset AF group, compared with 6.43%/year in the remote-onset group. However, in a multivariate regression analysis adjusted for potential confounders, the only significant differences in outcome between the two groups were in all-cause mortality – 5.15%/year in the recent-onset group, 3.15% in the remote-onset AF patients – and in the composite of stroke, systemic embolism, or all-cause mortality, which had an incidence of 6.46%/year in the recent-onset group, compared with 4.57%/year in remote-onset patients.
Turning to the impact of apixaban, Dr. Guimaraes noted that, as previously reported in the overall study, the primary endpoint of stroke or systemic embolism occurred in the apixaban group at a rate of 1.27%/year, compared with 1.6%/year with warfarin, for a 21% relative risk reduction in favor of the newer agent. She and her coinvestigators determined that in the remote-onset AF subgroup the relative risk reduction was 20%, while in the recent-onset subgroup the size of the effect was similar at 22%.
The composite safety endpoint of major or clinically relevant bleeding occurred in the remote-onset patients at a rate of 3.97%/year with apixaban versus 5.97%/year with warfarin, for a 33% relative risk reduction favoring the novel agent. In the recent-onset group, the rates were 5.04%/year with apixaban, compared with 6.4%/year with warfarin, for a 22% relative risk reduction.
Dr. Guimaraes observed an important limitation of this post hoc analysis is that the remote-onset AF group may have been selected for improved survival, since they didn’t die in the first 30 days after diagnosis.
Session co-chair Dr. Brian Olshansky commented that this analysis, which highlights the risks of recent-onset AF, argues for a strategy whereby a patient who presents to the ED with new-onset AF should get sent home on apixaban rather than being hospitalized for several days in order to be stabilized on warfarin.
“With recent-onset atrial fibrillation it’s going to take you several days to get anticoagulated with warfarin, whereas you’re immediately anticoagulated with apixaban,” said Dr. Olshansky, emeritus professor of internal medicine at the University of Iowa, Iowa City.
The ARISTOTLE trial was supported by Bristol-Myers Squibb and Pfizer. Dr. Guimaraes reported having no financial conflicts of interest.
Key clinical point: Don’t delay starting oral anticoagulation in patients with recent-onset atrial fibrillation.
Major finding: All-cause mortality occurred at a rate of 5.15%/year in patients started on apixaban or warfarin within 30 days following diagnosis of atrial fibrillation, compared with 3.15%/year in those with longer-duration atrial fibrillation.
Data source: This was a secondary post hoc analysis of 18,140 participants in the randomized, double-blind, prospective ARISTOTLE trial of apixaban versus warfarin for stroke prevention.
Disclosures: The study presenter reported having no financial conflicts of interest.

Screen and treat MS patients for emotional distress, depression
ATLANTA – Nearly half of 2,100 multiple sclerosis (MS) patients enrolled at baseline in the Sonya Slifka Longitudinal Multiple Sclerosis Study reported experiencing emotional distress, and 9% of those patients reported difficulties with accessing mental health services.
Younger patients, those with more recently diagnosed illness, and those with more frequent MS relapses were more likely to experience emotional distress, Dr. Laura Safar of Brigham and Women’s Hospital, Boston, said at the annual meeting of the American Psychiatric Association.
The Sonya Slifka study was an 8-year population-based cohort study that, at last report, included more than 4,000 MS patients from across the United States, with varying disease duration. Dr. Safar reported on baseline mental health data from the study.
Patients with MS may experience symptoms involving any part of the central nervous system, including psychiatric symptoms such as depression, anxiety, and cognitive disorders, she said, noting that these are highly prevalent, but often go unrecognized and untreated by primary care doctors and neurologists.
Prior studies have suggested that depression occurs in 25%-80% of patients, depending on the study setting. The rates are higher in those with MS than in the general population or in those with other neurologic and chronic medical conditions, she said.
In the Sonya Slifka study, 77% of patients were women with an average age of 50 years and disease duration ranging from 1 week to 64 years. Most were white. The disease distribution was representative of that seen in the general MS population, with most (57%) having relapsing-remitting disease, 25% having secondary progressive disease, and the remaining patients having primary progressive disease or progressing-relapsing disease (Mult Scler. 2006 Feb;12[1]:24-38).
Reported disability levels varied from none/very mild to severe and requiring a wheelchair or scooter or being bedridden.
Of the 48% of patients reporting emotional distress, most reported having mild to moderate distress, but 40% reported severe distress.
Nearly half (46%) of patients reported accomplishing less than normal because of emotional difficulties, and 31% said they worked less carefully than usual.
Emotional distress was more common in patients who were younger, divorced or never married, unemployed, and in those with lower education and income levels. Emotional distress was associated with shorter duration of illness, with having multiple relapses in the prior year (highest rates were among those with five or more relapses), and with moderate disability level.
Emotional distress was also associated with poorer perceived general health, and those with higher levels of emotional distress tended to experience all or many of the symptoms on the baseline questionnaire.
Further, those with emotional distress tended to lack health insurance and to have problems accessing health care and necessary prescription medications. About one-fourth of the patients had seen a mental health professional in the prior year and nearly 8% wanted to; 2% said they had been referred to a mental health professional, and 93% of these patients had emotional distress, including 6% with severe distress.
Reasons given by these patients for not seeing a mental health professional were cost, difficulty getting an appointment, and too long of a wait.
The findings suggest that in clinical settings it is important to screen MS patients for emotional distress and depression and to treat or refer accordingly, Dr. Safar said.
“As we know from other medical illnesses, this will improve adherence to MS treatment and will improve the prognosis,” she added.
Dr. Safar reported having no disclosures.
ATLANTA – Nearly half of 2,100 multiple sclerosis (MS) patients enrolled at baseline in the Sonya Slifka Longitudinal Multiple Sclerosis Study reported experiencing emotional distress, and 9% of those patients reported difficulties with accessing mental health services.
Younger patients, those with more recently diagnosed illness, and those with more frequent MS relapses were more likely to experience emotional distress, Dr. Laura Safar of Brigham and Women’s Hospital, Boston, said at the annual meeting of the American Psychiatric Association.
The Sonya Slifka study was an 8-year population-based cohort study that, at last report, included more than 4,000 MS patients from across the United States, with varying disease duration. Dr. Safar reported on baseline mental health data from the study.
Patients with MS may experience symptoms involving any part of the central nervous system, including psychiatric symptoms such as depression, anxiety, and cognitive disorders, she said, noting that these are highly prevalent, but often go unrecognized and untreated by primary care doctors and neurologists.
Prior studies have suggested that depression occurs in 25%-80% of patients, depending on the study setting. The rates are higher in those with MS than in the general population or in those with other neurologic and chronic medical conditions, she said.
In the Sonya Slifka study, 77% of patients were women with an average age of 50 years and disease duration ranging from 1 week to 64 years. Most were white. The disease distribution was representative of that seen in the general MS population, with most (57%) having relapsing-remitting disease, 25% having secondary progressive disease, and the remaining patients having primary progressive disease or progressing-relapsing disease (Mult Scler. 2006 Feb;12[1]:24-38).
Reported disability levels varied from none/very mild to severe and requiring a wheelchair or scooter or being bedridden.
Of the 48% of patients reporting emotional distress, most reported having mild to moderate distress, but 40% reported severe distress.
Nearly half (46%) of patients reported accomplishing less than normal because of emotional difficulties, and 31% said they worked less carefully than usual.
Emotional distress was more common in patients who were younger, divorced or never married, unemployed, and in those with lower education and income levels. Emotional distress was associated with shorter duration of illness, with having multiple relapses in the prior year (highest rates were among those with five or more relapses), and with moderate disability level.
Emotional distress was also associated with poorer perceived general health, and those with higher levels of emotional distress tended to experience all or many of the symptoms on the baseline questionnaire.
Further, those with emotional distress tended to lack health insurance and to have problems accessing health care and necessary prescription medications. About one-fourth of the patients had seen a mental health professional in the prior year and nearly 8% wanted to; 2% said they had been referred to a mental health professional, and 93% of these patients had emotional distress, including 6% with severe distress.
Reasons given by these patients for not seeing a mental health professional were cost, difficulty getting an appointment, and too long of a wait.
The findings suggest that in clinical settings it is important to screen MS patients for emotional distress and depression and to treat or refer accordingly, Dr. Safar said.
“As we know from other medical illnesses, this will improve adherence to MS treatment and will improve the prognosis,” she added.
Dr. Safar reported having no disclosures.
ATLANTA – Nearly half of 2,100 multiple sclerosis (MS) patients enrolled at baseline in the Sonya Slifka Longitudinal Multiple Sclerosis Study reported experiencing emotional distress, and 9% of those patients reported difficulties with accessing mental health services.
Younger patients, those with more recently diagnosed illness, and those with more frequent MS relapses were more likely to experience emotional distress, Dr. Laura Safar of Brigham and Women’s Hospital, Boston, said at the annual meeting of the American Psychiatric Association.
The Sonya Slifka study was an 8-year population-based cohort study that, at last report, included more than 4,000 MS patients from across the United States, with varying disease duration. Dr. Safar reported on baseline mental health data from the study.
Patients with MS may experience symptoms involving any part of the central nervous system, including psychiatric symptoms such as depression, anxiety, and cognitive disorders, she said, noting that these are highly prevalent, but often go unrecognized and untreated by primary care doctors and neurologists.
Prior studies have suggested that depression occurs in 25%-80% of patients, depending on the study setting. The rates are higher in those with MS than in the general population or in those with other neurologic and chronic medical conditions, she said.
In the Sonya Slifka study, 77% of patients were women with an average age of 50 years and disease duration ranging from 1 week to 64 years. Most were white. The disease distribution was representative of that seen in the general MS population, with most (57%) having relapsing-remitting disease, 25% having secondary progressive disease, and the remaining patients having primary progressive disease or progressing-relapsing disease (Mult Scler. 2006 Feb;12[1]:24-38).
Reported disability levels varied from none/very mild to severe and requiring a wheelchair or scooter or being bedridden.
Of the 48% of patients reporting emotional distress, most reported having mild to moderate distress, but 40% reported severe distress.
Nearly half (46%) of patients reported accomplishing less than normal because of emotional difficulties, and 31% said they worked less carefully than usual.
Emotional distress was more common in patients who were younger, divorced or never married, unemployed, and in those with lower education and income levels. Emotional distress was associated with shorter duration of illness, with having multiple relapses in the prior year (highest rates were among those with five or more relapses), and with moderate disability level.
Emotional distress was also associated with poorer perceived general health, and those with higher levels of emotional distress tended to experience all or many of the symptoms on the baseline questionnaire.
Further, those with emotional distress tended to lack health insurance and to have problems accessing health care and necessary prescription medications. About one-fourth of the patients had seen a mental health professional in the prior year and nearly 8% wanted to; 2% said they had been referred to a mental health professional, and 93% of these patients had emotional distress, including 6% with severe distress.
Reasons given by these patients for not seeing a mental health professional were cost, difficulty getting an appointment, and too long of a wait.
The findings suggest that in clinical settings it is important to screen MS patients for emotional distress and depression and to treat or refer accordingly, Dr. Safar said.
“As we know from other medical illnesses, this will improve adherence to MS treatment and will improve the prognosis,” she added.
Dr. Safar reported having no disclosures.
AT THE APA ANNUAL MEETING
Key clinical point: Nearly half of 2,100 multiple sclerosis patients enrolled at baseline in the Sonya Slifka Longitudinal Multiple Sclerosis Study reported experiencing emotional distress.
Major finding: 48% of patients reported emotional distress at baseline, and 40% of those reported severe distress.
Data source: The initial 2,100 patients in a longitudinal cohort study.
Disclosures: Dr. Safar reported having no disclosures.
Blood Test Detects Concussion Up to One Week Later
Doctors can detect evidence of a concussion up to one week after a patient is injured by using a simple blood test, according to a report published online ahead of print March 28 in JAMA Neurology. Researchers tested two blood biomarkers—glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase L1 (UCH-L1)—separately and together in patients with mild and moderate traumatic brain injury (TBI) within seven days of the injury. They examined the blood biomarkers with respect to diagnostic precision of TBI, presence of traumatic intracranial lesions detectable by CT, and need for neurosurgical intervention. Linda Papa, MDCM, MSc, and colleagues reported that GFAP performed consistently in detecting mild to moderate TBI, CT lesions, and the need for neurosurgical interventions across seven days. UCH-L1, they said, performed best in the early postinjury period.

Linda Papa, MDCM, MSc
“We have so many diagnostic blood tests for different parts of the body, like the heart, liver and kidneys, but there’s never been a reliable blood test to identify trauma in the brain,” said Dr. Papa, an emergency medicine physician at Orlando Health in Florid and lead author of the study. “We think this particular test could change that.”
Dr. Papa and colleagues designed a prospective cohort study that enrolled adults with trauma seen at a level 1 trauma center from March 1, 2010, to March 5, 2014. All patients underwent screening to determine whether they had experienced mild or moderate TBI, which was defined as blunt head trauma with loss of consciousness, amnesia, or disorientation and a Glasgow Coma Scale score of 9 to 15. Of 3,025 patients assessed, 1,030 met eligibility criteria for enrollment; 446 declined participation. Initial blood samples were obtained in 584 patients enrolled within four hours of injury. Repeated blood sampling was conducted every four hours up to 24 hours postinjury, and then every 12 hours thereafter until 180 hours postinjury.
A total of 1,831 blood samples were drawn from 584 patients (mean age, 40; 62% male) over seven days. Both GFAP and UCH-L1 were detectible within one hour of injury. GFAP peaked at 20 hours postinjury and slowly declined over 72 hours. UCH-L1 rose rapidly and peaked at eight hours postinjury, then declined rapidly over 48 hours.
Over the course of one week, GFAP demonstrated a diagnostic range of areas under the curve for detecting mild to moderate TBI of 0.73 to 0.94, and UCH-L1 demonstrated a diagnostic range of 0.30 to 0.67. For detecting intracranial lesions on CT, the diagnostic ranges of areas under the curve were 0.80 to 0.97 for GFAP and 0.31 to 0.77 for UCH-L1. For distinguishing patients with and without the need for a neurosurgical intervention, the range for GFAP was 0.91 to 100 and the range for UCH-L1 was 0.50 to 0.92.
“In the context of developing a point-of-care test, the early and rapid rise of UCH-L1 could be used to detect TBI immediately at the scene of injury in settings such as in the ambulance, on the playing field, or at the battlefield,” the researchers wrote. “The longer half-life of GFAP makes it a favorable biomarker to use in both the acute and subacute phases of injury because it is able to detect CT lesions for up to seven days after injury. Although its rise is not as rapid as [that of] UCH-L1, it performs well for detecting mild TBI and CT lesions within one hour of injury.”
—Glenn S. Williams
Suggested Reading
Papa L, Brophy GM, Welch RD, et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].
Bogoslovsky T, Diaz-Arrastia R. Dissecting temporal profiles of neuronal and axonal damage after mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].
Doctors can detect evidence of a concussion up to one week after a patient is injured by using a simple blood test, according to a report published online ahead of print March 28 in JAMA Neurology. Researchers tested two blood biomarkers—glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase L1 (UCH-L1)—separately and together in patients with mild and moderate traumatic brain injury (TBI) within seven days of the injury. They examined the blood biomarkers with respect to diagnostic precision of TBI, presence of traumatic intracranial lesions detectable by CT, and need for neurosurgical intervention. Linda Papa, MDCM, MSc, and colleagues reported that GFAP performed consistently in detecting mild to moderate TBI, CT lesions, and the need for neurosurgical interventions across seven days. UCH-L1, they said, performed best in the early postinjury period.
Linda Papa, MDCM, MSc
“We have so many diagnostic blood tests for different parts of the body, like the heart, liver and kidneys, but there’s never been a reliable blood test to identify trauma in the brain,” said Dr. Papa, an emergency medicine physician at Orlando Health in Florid and lead author of the study. “We think this particular test could change that.”
Dr. Papa and colleagues designed a prospective cohort study that enrolled adults with trauma seen at a level 1 trauma center from March 1, 2010, to March 5, 2014. All patients underwent screening to determine whether they had experienced mild or moderate TBI, which was defined as blunt head trauma with loss of consciousness, amnesia, or disorientation and a Glasgow Coma Scale score of 9 to 15. Of 3,025 patients assessed, 1,030 met eligibility criteria for enrollment; 446 declined participation. Initial blood samples were obtained in 584 patients enrolled within four hours of injury. Repeated blood sampling was conducted every four hours up to 24 hours postinjury, and then every 12 hours thereafter until 180 hours postinjury.
A total of 1,831 blood samples were drawn from 584 patients (mean age, 40; 62% male) over seven days. Both GFAP and UCH-L1 were detectible within one hour of injury. GFAP peaked at 20 hours postinjury and slowly declined over 72 hours. UCH-L1 rose rapidly and peaked at eight hours postinjury, then declined rapidly over 48 hours.
Over the course of one week, GFAP demonstrated a diagnostic range of areas under the curve for detecting mild to moderate TBI of 0.73 to 0.94, and UCH-L1 demonstrated a diagnostic range of 0.30 to 0.67. For detecting intracranial lesions on CT, the diagnostic ranges of areas under the curve were 0.80 to 0.97 for GFAP and 0.31 to 0.77 for UCH-L1. For distinguishing patients with and without the need for a neurosurgical intervention, the range for GFAP was 0.91 to 100 and the range for UCH-L1 was 0.50 to 0.92.
“In the context of developing a point-of-care test, the early and rapid rise of UCH-L1 could be used to detect TBI immediately at the scene of injury in settings such as in the ambulance, on the playing field, or at the battlefield,” the researchers wrote. “The longer half-life of GFAP makes it a favorable biomarker to use in both the acute and subacute phases of injury because it is able to detect CT lesions for up to seven days after injury. Although its rise is not as rapid as [that of] UCH-L1, it performs well for detecting mild TBI and CT lesions within one hour of injury.”
—Glenn S. Williams
Doctors can detect evidence of a concussion up to one week after a patient is injured by using a simple blood test, according to a report published online ahead of print March 28 in JAMA Neurology. Researchers tested two blood biomarkers—glial fibrillary acidic protein (GFAP) and ubiquitin C-terminal hydrolase L1 (UCH-L1)—separately and together in patients with mild and moderate traumatic brain injury (TBI) within seven days of the injury. They examined the blood biomarkers with respect to diagnostic precision of TBI, presence of traumatic intracranial lesions detectable by CT, and need for neurosurgical intervention. Linda Papa, MDCM, MSc, and colleagues reported that GFAP performed consistently in detecting mild to moderate TBI, CT lesions, and the need for neurosurgical interventions across seven days. UCH-L1, they said, performed best in the early postinjury period.
Linda Papa, MDCM, MSc
“We have so many diagnostic blood tests for different parts of the body, like the heart, liver and kidneys, but there’s never been a reliable blood test to identify trauma in the brain,” said Dr. Papa, an emergency medicine physician at Orlando Health in Florid and lead author of the study. “We think this particular test could change that.”
Dr. Papa and colleagues designed a prospective cohort study that enrolled adults with trauma seen at a level 1 trauma center from March 1, 2010, to March 5, 2014. All patients underwent screening to determine whether they had experienced mild or moderate TBI, which was defined as blunt head trauma with loss of consciousness, amnesia, or disorientation and a Glasgow Coma Scale score of 9 to 15. Of 3,025 patients assessed, 1,030 met eligibility criteria for enrollment; 446 declined participation. Initial blood samples were obtained in 584 patients enrolled within four hours of injury. Repeated blood sampling was conducted every four hours up to 24 hours postinjury, and then every 12 hours thereafter until 180 hours postinjury.
A total of 1,831 blood samples were drawn from 584 patients (mean age, 40; 62% male) over seven days. Both GFAP and UCH-L1 were detectible within one hour of injury. GFAP peaked at 20 hours postinjury and slowly declined over 72 hours. UCH-L1 rose rapidly and peaked at eight hours postinjury, then declined rapidly over 48 hours.
Over the course of one week, GFAP demonstrated a diagnostic range of areas under the curve for detecting mild to moderate TBI of 0.73 to 0.94, and UCH-L1 demonstrated a diagnostic range of 0.30 to 0.67. For detecting intracranial lesions on CT, the diagnostic ranges of areas under the curve were 0.80 to 0.97 for GFAP and 0.31 to 0.77 for UCH-L1. For distinguishing patients with and without the need for a neurosurgical intervention, the range for GFAP was 0.91 to 100 and the range for UCH-L1 was 0.50 to 0.92.
“In the context of developing a point-of-care test, the early and rapid rise of UCH-L1 could be used to detect TBI immediately at the scene of injury in settings such as in the ambulance, on the playing field, or at the battlefield,” the researchers wrote. “The longer half-life of GFAP makes it a favorable biomarker to use in both the acute and subacute phases of injury because it is able to detect CT lesions for up to seven days after injury. Although its rise is not as rapid as [that of] UCH-L1, it performs well for detecting mild TBI and CT lesions within one hour of injury.”
—Glenn S. Williams
Suggested Reading
Papa L, Brophy GM, Welch RD, et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].
Bogoslovsky T, Diaz-Arrastia R. Dissecting temporal profiles of neuronal and axonal damage after mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].
Suggested Reading
Papa L, Brophy GM, Welch RD, et al. Time course and diagnostic accuracy of glial and neuronal blood biomarkers GFAP and UCH-L1 in a large cohort of trauma patients with and without mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].
Bogoslovsky T, Diaz-Arrastia R. Dissecting temporal profiles of neuronal and axonal damage after mild traumatic brain injury. JAMA Neurol. 2016 March 28 [Epub ahead of print].

FDA grants accelerated approval to nivolumab for Hodgkin lymphoma
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
On Twitter @NikolaidesLaura
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
On Twitter @NikolaidesLaura
The Food and Drug Administration has granted accelerated approval to nivolumab for the treatment of patients with classical Hodgkin lymphoma (cHL) that has relapsed or progressed after autologous hematopoietic stem cell transplantation (HSCT) and posttransplantation brentuximab vedotin.
Approval was based on a 65% objective response rate in 95 patients treated with nivolumab following autologous HSCT and posttransplantation brentuximab vedotin. All patients in the single-arm, multicenter trial had relapsed or refractory cHL and were enrolled regardless of PD-L1 expression status. Patients received a median of 17 doses of nivolumab, the FDA said in a written statement.
![]()
The median time to response was 2.1 months (range, 0.7-5.7 months). The estimated median duration of response was 8.7 months.
The FDA also issued a warning for complications of allogeneic HSCT after nivolumab, reporting that transplant-related deaths have occurred. Health care professionals should follow patients closely for early evidence of transplant-related complications, such as hyperacute graft-versus-host disease (GVHD), severe acute GVHD, steroid-requiring febrile syndrome, hepatic veno-occlusive disease, and other immune-mediated adverse reactions, they said.
The most common adverse reactions in a second single-arm study used to evaluate safety (n = 263) were upper respiratory tract infection, cough, pyrexia, and diarrhea. Other immune-mediated adverse reactions, occurring in 1%-5% of patients, included rash, pneumonitis, hepatitis, hyperthyroidism, and colitis. The most common serious adverse reactions, which were reported in 1%-3% of patients, were pneumonia, pleural effusion, pneumonitis, pyrexia, infusion-related reaction, and rash.
Nivolumab is marketed as Opdivo by Bristol-Myers Squibb and has been previously approved to treat advanced renal cell carcinoma, lung cancer, and melanoma.
On Twitter @NikolaidesLaura