User login
CHMP recommends expanding use of drug in CLL

Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
Photo courtesy of GSK
The European Medicines Agency’s Committee for Medicinal Products for Human Use (CHMP) has recommended expanding the approved indication for ofatumumab (Arzerra®).
The CHMP is recommending the drug be approved for use in combination with fludarabine and cyclophosphamide to treat adults with relapsed chronic lymphocytic leukemia (CLL).
The CHMP’s recommendation will be reviewed by the European Commission.
A final decision is expected in the coming months.
Ofatumumab is a monoclonal antibody designed to target CD20. The drug is marketed under a collaboration agreement between Genmab and Novartis.
The European Commission has already approved ofatumumab for the following indications:
- As a single-agent to treat CLL patients who are refractory to fludarabine and alemtuzumab
- For use in combination with chlorambucil or bendamustine in CLL patients who
have not received prior therapy and are not eligible for
fludarabine-based therapy.
COMPLEMENT 2 trial
The CHMP’s recommendation to approve ofatumumab in combination with fludarabine and cyclophosphamide was based on results from the phase 3 COMPLEMENT 2 study. Novartis reported top-line results from this study last April.
The trial enrolled 365 patients with relapsed CLL. The patients were randomized 1:1 to receive up to 6 cycles of ofatumumab in combination with fludarabine and cyclophosphamide or up to 6 cycles of fludarabine and cyclophosphamide alone.
The primary endpoint was progression-free survival, as assessed by an independent review committee.
The median progression-free survival was 28.9 months for patients receiving ofatumumab plus fludarabine and cyclophosphamide, compared to 18.8 months for patients receiving fludarabine and cyclophosphamide alone (hazard ratio=0.67, P=0.0032).
Novartis said the safety profile observed in this study was consistent with other trials of ofatumumab, and no new safety signals were observed. ![]()
NCCN issues challenge to ‘bag’ vincristine
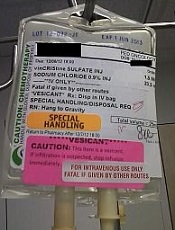
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Photo courtesy of ISMP
PHILADELPHIA—To ensure proper administration of vincristine, the National Comprehensive Cancer Network (NCCN) has issued a challenge to hospitals, medical centers, and oncology practices as part of its “Just Bag It!” campaign.
Vincristine—the “O” for Oncovin in the CHOP regimen—is widely used to treat patients with leukemia or lymphoma.
It is considered an important chemotherapeutic agent. However, if administered incorrectly, vincristine is uniformly fatal, usually within a week, according to the NCCN.
Vincristine is highly neurotoxic and should always be administered intravenously. If it is mistakenly given intrathecally along with other chemotherapy drugs, it causes ascending paralysis, neurological defects, and death.
Therefore, the NCCN recommends always diluting and administering vincristine in a mini IV-drip bag, never through a syringe.
This precaution decreases the chances of improper dosage and makes it impossible to accidentally administer vincristine into the spinal fluid.
The NCCN initiated the safe-handling campaign in response to the death 11 years ago of a 21-year-old patient who received vincristine incorrectly administered into his spinal fluid. He was referred to Robert W. Carlson, MD, NCCN’s chief executive officer, who, at the time, was at Stanford Hospital, not the hospital where the error occurred.
The patient, Christopher Wibeto, had a “likely curable” non-Hodgkin lymphoma and died 4 days later.
“When I first met Christopher, he was doing well,” Dr Carlson said. “He was a delightful young gentleman, very articulate. He was funny. Even in the ICU, he had me chuckling and laughing at what he was saying.”
“But we knew that the medical error would almost certainly lead to his death. Shortly thereafter, I met his parents, Debra and Robin, . . . and had to tell them what the consequences of that medical error were likely to be. And they joined me in Christopher’s room while we talked with him about what the consequences of that medical error were likely to be.”
Making the situation even more painful, Dr Carlson, at that time, was the father of a young son who is now almost the age Christopher was then.
Dr Carlson said he realized that “we needed to come up with systems to assure that this did not happen, not today or tomorrow or ever again.”
Motivated by the tragedy, Dr Carlson spearheaded a national effort to address this mistake when he joined NCCN as CEO, enlisting the help of NCCN’s Best Practices Committee.
The NCCN developed and issued guidelines, and all 27 member institutions have adopted policies in line with the guidelines.
The Institute for Safe Medication Practices (ISMP) has undertaken efforts over more than a decade to implement procedures for safe vincristine administration.
ISMP conducted surveys and follow-up self-assessments regarding use of IV bags for vincristine at oncology practice sites. They found that only about half the institutions surveyed dilute IV vincristine for administration in a small-volume bag.
Some practitioners associate the use of an IV bag with an increased risk of extravasation (when the chemotherapy agent leaks into the tissue surrounding the administration site). Research shows, however, that the risk of extravasation is extremely low (less than 0.5%), regardless of how vincristine is administered.
And cost is not an issue when implementing the mini-bag policy, according to the president of ISMP, Michael R. Cohen, RPh.
“It cost a few pennies more,” he said. “And I mean pennies. I think probably what is an issue is just the age-old habit of putting vincristine in a syringe and being able to change that habit.”
Since the introduction of vincristine use in the 1960s, 125 documented cases of accidental death in the US and abroad have been reported. While the error is relatively rare, it is preventable and unique in its level of mortality.
“It’s hard to understand why this idea of ‘Just Bag It’ hasn’t permeated healthcare at this point,” Cohen said. “Because it is a sure-fire way to prevent this type of error.”
The ISMP, the Joint Commission, the World Health Organization, and the Oncology Nursing Society also recommend the bag-it policy. ![]()
Health Canada approves drug for patients with VTE, NVAF

Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Image by Andre E.X. Brown
Health Canada has approved edoxaban (Lixiana®), an oral factor Xa inhibitor, for use in patients with venous thromboembolism (VTE) or nonvalvular atrial fibrillation (NVAF).
The drug can now be used to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE).
It can also be used to prevent stroke and systemic embolism in adults with NVAF in whom anticoagulation is appropriate.
Edoxaban was discovered and developed by Daiichi Sankyo Co., Ltd., but Servier Canada will market the drug in Canada.
Edoxaban has been approved in the US, European Union, Switzerland, Japan, South Korea, Taiwan, and Hong Kong. The drug is marketed as Savaysa® in the US and as Lixiana® elsewhere.
Health Canada’s approval of edoxaban is based on data from a pair of phase 3 trials, ENGAGE AF-TIMI 48 and Hokusai-VTE.
Hokusai-VTE
In the Hokusai-VTE trial, researchers evaluated edoxaban in 4921 patients with DVT and 3319 with PE. Patients received initial treatment with low-molecular-weight heparin and were then randomized to receive edoxaban or warfarin daily for 3 to 12 months.
Overall, edoxaban proved as effective as warfarin. Recurrent, symptomatic VTE occurred in 3.2% and 3.5% of patients, respectively (P<0.001 for non-inferiority).
In addition, the incidence of clinically relevant bleeding was significantly lower in the edoxaban arm than the warfarin arm—8.5% and 10.3%, respectively (P=0.004 for superiority).
ENGAGE-AF TIMI 48
In the ENGAGE AF-TIMI 48 trial, researchers compared edoxaban and warfarin as prophylaxis for stroke or systemic embolism in patients with NVAF.
The trial included 21,105 patients who were randomized to receive warfarin (n=7036), edoxaban at 60 mg (n=7035), or edoxaban at 30 mg (n=7034).
Edoxaban was at least non-inferior to warfarin with regard to efficacy. The annual incidence of stroke or systemic embolism was 1.50% with warfarin, 1.18% with edoxaban at 60 mg (P<0.001 for non-inferiority), and 1.61% with edoxaban at 30 mg (P=0.005 for non-inferiority).
In addition, edoxaban was associated with a significantly lower rate of major and fatal bleeding. The annual incidence of major bleeding was 3.43% with warfarin, 2.75% with edoxaban at 60 mg (P<0.001), and 1.61% with edoxaban at 30 mg (P<0.001).
Fatal bleeds occurred at an annual rate of 0.38% with warfarin, 0.21% with edoxaban at 60 mg (P=0.006), and 0.13% with edoxaban at 30 mg (P<0.001). ![]()
Do Kidney Patients Know an App From a Nap?
Q)It seems that at every conference I attend, a tech/marketing rep stands up to rave about “this” app or “that” online program. My average patient is older than 60 (physiologically 80), has vision issues related to diabetes, hypertension, or cataracts, can’t afford a smartphone, wouldn’t know an app from a nap, and has trouble just managing to eat correctly. What makes these reps think that patients can use technology?
We are often encouraged to incorporate technology into our daily interactions with patients. Meaningful use has us signing up 70-year-old patients for our practice’s patient portal and counting on them to write a message to us so we can receive credit. Our initial response is to groan and ask if the government knows what kind of patients we see.
However, a new article in the Clinical Journal of the American Society of Nephrology suggests that our patients may be more tech savvy than we think.1 The study found that patients with chronic kidney disease (CKD) not only know how to use a smartphone application but also find its implementation useful.
Patients included in the study were, on average, 59 and had stage IV to stage V CKD and an estimated glomerular filtration rate (eGFR) of ≥ 30 mL/min/1.73 m2. The study assessed knowledge of blood pressure, medications, CKD-related symptoms, and CKD-related laboratory tests.
Although 60% of the study cohort had never used a smartphone before, monthly adherence rates were higher than 80%. Outcomes included a statistically significant reduction in blood pressure, which was attributed to patients’ ability to better monitor their health and reduce their anxiety. The smartphone data sets also helped to identify cases of masked hypertension and more than 100 medication errors, 60% of which required intervention. Subsequent visits with providers were found to be more useful as a result, since both patients and providers had better quality information.
An accompanying editorial cautioned, however, that despite these positive findings, we must be mindful that smartphone ownership is less common among lower income patients. Fifty percent of those making less than $30,000 per year own a smartphone, compared with 84% of patients with an annual income of $75,000 or more.2 CKD patients are of varying socioeconomic status, with lower eGFR often corresponding to lower socioeconomic status.
So while medical apps have a future with CKD (and by implication, all) patients, they are not unlike much else in medicine: We must tailor our practice to meet the needs of our patient population. These findings are encouraging for use of smartphone technology, but it is not a “one size fits all” solution. —SM
Sherry Mathes, NP-C
Georgia Nephrology LLC, Lawrenceville, Georgia
1. Ong SW, Jassal SV, Miller JA, et al. Integrating a smartphone-based self-management system into usual care of advanced CKD. Clin J Am Soc Nephrol. 2016;11(6):1054-1062.
2. Desai T, Yee J, Soman S. Smartphone apps: a patient’s new best friend? Clin J Am Soc Nephrol. 2016;11(6):935-937.
Clinician Reviews in partnership with

Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Sherry Mathes, NP-C, who practices at Georgia Nephrology LLC in Lawrenceville, Georgia, and Cynthia Smith, DNP, CNN-NP, FNP-BC, APRN, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Sherry Mathes, NP-C, who practices at Georgia Nephrology LLC in Lawrenceville, Georgia, and Cynthia Smith, DNP, CNN-NP, FNP-BC, APRN, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
Clinician Reviews in partnership with
Renal Consult is edited by Jane S. Davis, CRNP, DNP, a member of the Clinician Reviews editorial board, who is a nurse practitioner in the Division of Nephrology at the University of Alabama at Birmingham and is the communications chairperson for the National Kidney Foundation’s Council of Advanced Practitioners (NKF-CAP); and Kim Zuber, PA-C, MSPS, DFAAPA, a semi-retired PA who works with the American Academy of Nephrology PAs and is a past chair of the NKF-CAP. This month’s responses were authored by Sherry Mathes, NP-C, who practices at Georgia Nephrology LLC in Lawrenceville, Georgia, and Cynthia Smith, DNP, CNN-NP, FNP-BC, APRN, who practices with Renal Consultants PLLC in South Charleston, West Virginia.
Q)It seems that at every conference I attend, a tech/marketing rep stands up to rave about “this” app or “that” online program. My average patient is older than 60 (physiologically 80), has vision issues related to diabetes, hypertension, or cataracts, can’t afford a smartphone, wouldn’t know an app from a nap, and has trouble just managing to eat correctly. What makes these reps think that patients can use technology?
We are often encouraged to incorporate technology into our daily interactions with patients. Meaningful use has us signing up 70-year-old patients for our practice’s patient portal and counting on them to write a message to us so we can receive credit. Our initial response is to groan and ask if the government knows what kind of patients we see.
However, a new article in the Clinical Journal of the American Society of Nephrology suggests that our patients may be more tech savvy than we think.1 The study found that patients with chronic kidney disease (CKD) not only know how to use a smartphone application but also find its implementation useful.
Patients included in the study were, on average, 59 and had stage IV to stage V CKD and an estimated glomerular filtration rate (eGFR) of ≥ 30 mL/min/1.73 m2. The study assessed knowledge of blood pressure, medications, CKD-related symptoms, and CKD-related laboratory tests.
Although 60% of the study cohort had never used a smartphone before, monthly adherence rates were higher than 80%. Outcomes included a statistically significant reduction in blood pressure, which was attributed to patients’ ability to better monitor their health and reduce their anxiety. The smartphone data sets also helped to identify cases of masked hypertension and more than 100 medication errors, 60% of which required intervention. Subsequent visits with providers were found to be more useful as a result, since both patients and providers had better quality information.
An accompanying editorial cautioned, however, that despite these positive findings, we must be mindful that smartphone ownership is less common among lower income patients. Fifty percent of those making less than $30,000 per year own a smartphone, compared with 84% of patients with an annual income of $75,000 or more.2 CKD patients are of varying socioeconomic status, with lower eGFR often corresponding to lower socioeconomic status.
So while medical apps have a future with CKD (and by implication, all) patients, they are not unlike much else in medicine: We must tailor our practice to meet the needs of our patient population. These findings are encouraging for use of smartphone technology, but it is not a “one size fits all” solution. —SM
Sherry Mathes, NP-C
Georgia Nephrology LLC, Lawrenceville, Georgia
Q)It seems that at every conference I attend, a tech/marketing rep stands up to rave about “this” app or “that” online program. My average patient is older than 60 (physiologically 80), has vision issues related to diabetes, hypertension, or cataracts, can’t afford a smartphone, wouldn’t know an app from a nap, and has trouble just managing to eat correctly. What makes these reps think that patients can use technology?
We are often encouraged to incorporate technology into our daily interactions with patients. Meaningful use has us signing up 70-year-old patients for our practice’s patient portal and counting on them to write a message to us so we can receive credit. Our initial response is to groan and ask if the government knows what kind of patients we see.
However, a new article in the Clinical Journal of the American Society of Nephrology suggests that our patients may be more tech savvy than we think.1 The study found that patients with chronic kidney disease (CKD) not only know how to use a smartphone application but also find its implementation useful.
Patients included in the study were, on average, 59 and had stage IV to stage V CKD and an estimated glomerular filtration rate (eGFR) of ≥ 30 mL/min/1.73 m2. The study assessed knowledge of blood pressure, medications, CKD-related symptoms, and CKD-related laboratory tests.
Although 60% of the study cohort had never used a smartphone before, monthly adherence rates were higher than 80%. Outcomes included a statistically significant reduction in blood pressure, which was attributed to patients’ ability to better monitor their health and reduce their anxiety. The smartphone data sets also helped to identify cases of masked hypertension and more than 100 medication errors, 60% of which required intervention. Subsequent visits with providers were found to be more useful as a result, since both patients and providers had better quality information.
An accompanying editorial cautioned, however, that despite these positive findings, we must be mindful that smartphone ownership is less common among lower income patients. Fifty percent of those making less than $30,000 per year own a smartphone, compared with 84% of patients with an annual income of $75,000 or more.2 CKD patients are of varying socioeconomic status, with lower eGFR often corresponding to lower socioeconomic status.
So while medical apps have a future with CKD (and by implication, all) patients, they are not unlike much else in medicine: We must tailor our practice to meet the needs of our patient population. These findings are encouraging for use of smartphone technology, but it is not a “one size fits all” solution. —SM
Sherry Mathes, NP-C
Georgia Nephrology LLC, Lawrenceville, Georgia
1. Ong SW, Jassal SV, Miller JA, et al. Integrating a smartphone-based self-management system into usual care of advanced CKD. Clin J Am Soc Nephrol. 2016;11(6):1054-1062.
2. Desai T, Yee J, Soman S. Smartphone apps: a patient’s new best friend? Clin J Am Soc Nephrol. 2016;11(6):935-937.
1. Ong SW, Jassal SV, Miller JA, et al. Integrating a smartphone-based self-management system into usual care of advanced CKD. Clin J Am Soc Nephrol. 2016;11(6):1054-1062.
2. Desai T, Yee J, Soman S. Smartphone apps: a patient’s new best friend? Clin J Am Soc Nephrol. 2016;11(6):935-937.
High protein intake moderately associated with improved breast cancer survival
Higher protein intake, particularly protein from animal sources, is associated with a modest but lower risk of breast cancer recurrence and death, regardless of insulin receptor status.
Using information gathered through biennial questionnaires from 6,348 women who were diagnosed with stage I to III breast cancer between 1976 and 2004, investigators found a significant inverse association between total protein intake and distant breast cancer recurrence (P = .02). This association was driven specifically by protein from animal sources (P = .003) rather than vegetable sources.
Pathology records were reviewed and histology samples were analyzed for insulin receptor and estrogen receptor expression. Associations between breast cancer recurrence and protein intake, amino acids, or protein-containing food groups did not differ by tumor receptor status or body mass index at time of cancer diagnosis. Given that the association between protein intake and recurrence was not confined to tumors expressing insulin receptors, “It is difficult to invoke the insulin pathway as a mechanism to explain these findings,” the investigators wrote.
Given the only “modest survival advantage” of higher protein intake among women with breast cancer, and given the “challenges involved in randomized trials of diet, this association is unlikely to ever be definitively tested in a randomized trial,” Dr. Holmes and her associates wrote.
“However, the modest survival advantage with higher protein intake has been found in several studies, and we feel it is important that patients with breast cancer and their clinicians know this. At the least, it may provide reassurance that consuming protein-containing foods is not likely to increase the risk of breast cancer recurrence,” the researchers concluded.
This study was sponsored by grants from the National Institutes of Health. Dr. Holmes and one other investigator reported receiving financial compensation from Bayer HealthCare Pharmaceuticals.
[email protected]
On Twitter @jessnicolecraig
Higher protein intake, particularly protein from animal sources, is associated with a modest but lower risk of breast cancer recurrence and death, regardless of insulin receptor status.
Using information gathered through biennial questionnaires from 6,348 women who were diagnosed with stage I to III breast cancer between 1976 and 2004, investigators found a significant inverse association between total protein intake and distant breast cancer recurrence (P = .02). This association was driven specifically by protein from animal sources (P = .003) rather than vegetable sources.
Pathology records were reviewed and histology samples were analyzed for insulin receptor and estrogen receptor expression. Associations between breast cancer recurrence and protein intake, amino acids, or protein-containing food groups did not differ by tumor receptor status or body mass index at time of cancer diagnosis. Given that the association between protein intake and recurrence was not confined to tumors expressing insulin receptors, “It is difficult to invoke the insulin pathway as a mechanism to explain these findings,” the investigators wrote.
Given the only “modest survival advantage” of higher protein intake among women with breast cancer, and given the “challenges involved in randomized trials of diet, this association is unlikely to ever be definitively tested in a randomized trial,” Dr. Holmes and her associates wrote.
“However, the modest survival advantage with higher protein intake has been found in several studies, and we feel it is important that patients with breast cancer and their clinicians know this. At the least, it may provide reassurance that consuming protein-containing foods is not likely to increase the risk of breast cancer recurrence,” the researchers concluded.
This study was sponsored by grants from the National Institutes of Health. Dr. Holmes and one other investigator reported receiving financial compensation from Bayer HealthCare Pharmaceuticals.
[email protected]
On Twitter @jessnicolecraig
Higher protein intake, particularly protein from animal sources, is associated with a modest but lower risk of breast cancer recurrence and death, regardless of insulin receptor status.
Using information gathered through biennial questionnaires from 6,348 women who were diagnosed with stage I to III breast cancer between 1976 and 2004, investigators found a significant inverse association between total protein intake and distant breast cancer recurrence (P = .02). This association was driven specifically by protein from animal sources (P = .003) rather than vegetable sources.
Pathology records were reviewed and histology samples were analyzed for insulin receptor and estrogen receptor expression. Associations between breast cancer recurrence and protein intake, amino acids, or protein-containing food groups did not differ by tumor receptor status or body mass index at time of cancer diagnosis. Given that the association between protein intake and recurrence was not confined to tumors expressing insulin receptors, “It is difficult to invoke the insulin pathway as a mechanism to explain these findings,” the investigators wrote.
Given the only “modest survival advantage” of higher protein intake among women with breast cancer, and given the “challenges involved in randomized trials of diet, this association is unlikely to ever be definitively tested in a randomized trial,” Dr. Holmes and her associates wrote.
“However, the modest survival advantage with higher protein intake has been found in several studies, and we feel it is important that patients with breast cancer and their clinicians know this. At the least, it may provide reassurance that consuming protein-containing foods is not likely to increase the risk of breast cancer recurrence,” the researchers concluded.
This study was sponsored by grants from the National Institutes of Health. Dr. Holmes and one other investigator reported receiving financial compensation from Bayer HealthCare Pharmaceuticals.
[email protected]
On Twitter @jessnicolecraig
FROM THE JOURNAL OF CLINICAL ONCOLOGY
Key clinical point:
Major finding: The 5-year recurrence-free survival for women in the highest quintile of protein consumption was 94.0%, while those in the lowest quintile of protein consumption had 5-year recurrence-free survival of 92.1%.
Data source: Biennial questionnaires for 6,348 women diagnosed with any stage breast cancer between 1976 and 2004.
Disclosures: This study was sponsored by grants from the National Institutes of Health. Dr. Holmes and one coinvestigator reported receiving financial compensation from Bayer HealthCare Pharmaceuticals.
Study offers reassuring data on certolizumab use in pregnancy
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.

Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
VIENNA – Data from a large prospective registry of pregnancy outcomes in women on certolizumab are reassuring to date, Alexa B. Kimball, MD, reported at the annual congress of the European Academy of Dermatology and Venereology.
“This unique dataset of pregnancies exposed to a single agent suggests that exposure is really not a problem, although we’ll continue to collect prospective data and anticipate doing so in women with psoriasis going forward,” said Dr. Kimball, professor of dermatology at Harvard Medical School, Boston.
Certolizumab is a tumor necrosis factor–alpha inhibitor currently approved in the United States for the treatment of Crohn’s disease, rheumatoid arthritis, ankylosing spondylitis, and psoriatic arthritis. It is now in clinical trials for psoriasis, and Dr. Kimball said she expects that it will eventually receive an indication for that disease as well. In the interim, she switches her psoriasis patients who are pregnant or plan to become so to either off-label certolizumab or etanercept (Enbrel). She does the same for her psoriatic arthritis patients, although in that situation certolizumab is on-label therapy.
The reason she turns to etanercept or certolizumab in pregnancy or anticipated pregnancy is that these two biologics, unlike others, don’t cross the placenta in the third trimester.
“I’m concerned about the selective uptake of other monoclonal antibodies in the third trimester. We do see in babies born to moms exposed to these other drugs – like ustekinumab, infliximab, and adalimumab – that the baby’s drug blood levels at birth are higher than the mom’s, so you have potentially put them at some risk for infections unnecessarily and maybe have affected how their immune system develops. So if a woman comes to me who is pregnant and on a biologic agent I would either stop treatment in the second trimester or switch to etanercept or certolizumab,” the dermatologist said.
Of the 256 pregnancies prospectively followed in the UCB certolizumab registry, 80.9% resulted in live births, and 10.2% ended in spontaneous abortion or miscarriage. In addition, the induced abortion rate was 8.6%, and there was a single stillbirth.
Of note, the mean age at pregnancy was 31 years, and 29% of the women became pregnant at age 35 or older. In contrast, the mean age at first pregnancy in the general population is 26, and only about 10% are 35 or older. These data are consistent with Dr. Kimball’s own clinical experience, which is that women with moderate to severe psoriasis or psoriatic arthritis often have trouble conceiving, and if they eventually succeed it’s often at a more advanced age.
The rate of maternal complications in this series was unremarkable: preeclampsia in 3.5%, infection in 3.9%, disease flare in 5.1%, and gestational diabetes in 3.1%. The median gestational age at birth was 39 weeks. The rate of early preterm birth before 32 weeks was 3.5%, with 12.6% of babies arriving at 32-36 weeks. Considering these are older moms being treated for serious underlying systemic inflammatory diseases, these numbers look good, according to Dr. Kimball.
Most women were exposed to certolizumab during the first trimester at least, and many were on the drug throughout pregnancy.
She said about one-third of psoriasis patients experience improvement in their skin diseases during pregnancy.
“If they’re doing really well, I see no reason to keep them on systemic therapy during pregnancy. But I will say that I see a lot of very bad postpartum flares, and I’m quite cautious about that. For the women with psoriatic arthritis there may not be a choice; you may need to continue to treat them all the way through their pregnancy to keep from getting permanent joint destruction,” the dermatologist said.
Dr. Kimball reported receiving research grants from and serving as a consultant to UCB and numerous other pharmaceutical companies.
AT THE EADV CONGRESS
Key clinical point:
Major finding: The rate of major congenital malformations in a large prospective series of pregnancies in women on certolizumab was reassuringly low at 4.2%, with no pattern of malformations being seen.
Data source: This was a report on maternal and fetal outcomes of 256 prospectively followed pregnancies in women on certolizumab.
Disclosures: The presenter reported receiving research funds from and serving as a consultant to UCB, which markets certolizumab and maintains the pregnancy registry.
VIDEO: Topical antifungals win with patients
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
LAS VEGAS – New topical treatment options for onychomycosis represent significant improvements over older agents, and may approach the success seen with oral drugs, according to Dr. Theodore Rosen, professor of dermatology, Baylor College of Medicine, Houston.
Efinaconazole and tavaborole both permeate the nail and allow for spreading to the lateral nail folds and hyponychium, Dr. Rosen said in a video interview at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. Moreover, the topical treatments are popular with patients. Even if patients are not 100% clear, they are usually happy if their condition improves enough to wear sandals with confidence, Dr. Rosen added in the interview.
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
EXPERT ANALYSIS FROM SDEF LAS VEGAS SEMINAR

Tobacco-related cancer incidence, mortality drop
Tobacco-related cancer incidence and mortality rates dropped from 2004 to 2013, the Centers for Disease Control and Prevention reported in the Morbidity and Mortality Weekly Report published on Nov. 11, 2016.
Overall, tobacco-related invasive cancer incidence decreased from 206 cases per 100,000 during 2004-2008 to 193 cases per 100,000 during 2009-2013.![]()
The announcement of this data marks a continuation of the downward trend that has been observed since the 1990s, lead author S. Jane Henley, MSPH, and her associates at the CDC wrote in the report (MMWR. 2016 Nov 11;65[44]:1212-8).
Despite this continued decline in tobacco-related cancer incidence and mortality rates, the tobacco-related cancer burden remains high, and disparities in the rates and decline of tobacco-related cancer persists.
“Tobacco use remains the leading preventable cause of disease and death in the United States,” reported Henley and her associates. For each year between 2009 and 2013, an estimated 660,000 Americans were diagnosed with tobacco-related cancer, and an estimated 343,000 people died from those cancers, according to the investigators’ analysis of data collected by the United States Cancer Statistics working group, which compiles data from multiple nationwide sources including the National Program of Cancer Registries and the National Cancer Institute’s Surveillance, Epidemiology, and End Results program.
Tobacco-related cancer incidence and deaths were higher among men than women and higher among blacks than any other ethnic group. However, the cancer incidence and mortality rates also declined the fastest among men and blacks, compared with women and other ethnic groups, respectively.
Cancer incidence and death were also highest and decreased the most slowly in counties with lower educational attainment or highest poverty. Conversely, cancer incidence and mortality was the lowest and decreased the most quickly in metropolitan areas with populations greater than 1 million people.
Given that an estimated 40% of cancers diagnosed in the country and 3 in 10 cancer deaths are attributable to cigarette smoking and the use of smokeless tobacco, it is imperative that the CDC implement programs to help the almost 6 million smokers quit, CDC director Tom Frieden, MD, said in an associated telebriefing. Most people who smoke want to quit, and the health care system should do all it can to help them, Dr. Frieden said. At the same time, he echoed a claim from Henley’s paper, which said many tobacco-related cancers could be prevented by reducing tobacco use through implementation of evidence-based tobacco prevention and control interventions, such as increasing tobacco product prices, enforcing smoke-free laws, and promoting anti-tobacco mass media campaigns. These programs should be tailored to local geographic areas and demographics given the continued inconsistent progress and persistent disparities in tobacco-related cancer incidence and mortality, Dr. Frieden added.
[email protected]
On Twitter @jessnicolecraig
Tobacco-related cancer incidence and mortality rates dropped from 2004 to 2013, the Centers for Disease Control and Prevention reported in the Morbidity and Mortality Weekly Report published on Nov. 11, 2016.
Overall, tobacco-related invasive cancer incidence decreased from 206 cases per 100,000 during 2004-2008 to 193 cases per 100,000 during 2009-2013.![]()
The announcement of this data marks a continuation of the downward trend that has been observed since the 1990s, lead author S. Jane Henley, MSPH, and her associates at the CDC wrote in the report (MMWR. 2016 Nov 11;65[44]:1212-8).
Despite this continued decline in tobacco-related cancer incidence and mortality rates, the tobacco-related cancer burden remains high, and disparities in the rates and decline of tobacco-related cancer persists.
“Tobacco use remains the leading preventable cause of disease and death in the United States,” reported Henley and her associates. For each year between 2009 and 2013, an estimated 660,000 Americans were diagnosed with tobacco-related cancer, and an estimated 343,000 people died from those cancers, according to the investigators’ analysis of data collected by the United States Cancer Statistics working group, which compiles data from multiple nationwide sources including the National Program of Cancer Registries and the National Cancer Institute’s Surveillance, Epidemiology, and End Results program.
Tobacco-related cancer incidence and deaths were higher among men than women and higher among blacks than any other ethnic group. However, the cancer incidence and mortality rates also declined the fastest among men and blacks, compared with women and other ethnic groups, respectively.
Cancer incidence and death were also highest and decreased the most slowly in counties with lower educational attainment or highest poverty. Conversely, cancer incidence and mortality was the lowest and decreased the most quickly in metropolitan areas with populations greater than 1 million people.
Given that an estimated 40% of cancers diagnosed in the country and 3 in 10 cancer deaths are attributable to cigarette smoking and the use of smokeless tobacco, it is imperative that the CDC implement programs to help the almost 6 million smokers quit, CDC director Tom Frieden, MD, said in an associated telebriefing. Most people who smoke want to quit, and the health care system should do all it can to help them, Dr. Frieden said. At the same time, he echoed a claim from Henley’s paper, which said many tobacco-related cancers could be prevented by reducing tobacco use through implementation of evidence-based tobacco prevention and control interventions, such as increasing tobacco product prices, enforcing smoke-free laws, and promoting anti-tobacco mass media campaigns. These programs should be tailored to local geographic areas and demographics given the continued inconsistent progress and persistent disparities in tobacco-related cancer incidence and mortality, Dr. Frieden added.
[email protected]
On Twitter @jessnicolecraig
Tobacco-related cancer incidence and mortality rates dropped from 2004 to 2013, the Centers for Disease Control and Prevention reported in the Morbidity and Mortality Weekly Report published on Nov. 11, 2016.
Overall, tobacco-related invasive cancer incidence decreased from 206 cases per 100,000 during 2004-2008 to 193 cases per 100,000 during 2009-2013.![]()
The announcement of this data marks a continuation of the downward trend that has been observed since the 1990s, lead author S. Jane Henley, MSPH, and her associates at the CDC wrote in the report (MMWR. 2016 Nov 11;65[44]:1212-8).
Despite this continued decline in tobacco-related cancer incidence and mortality rates, the tobacco-related cancer burden remains high, and disparities in the rates and decline of tobacco-related cancer persists.
“Tobacco use remains the leading preventable cause of disease and death in the United States,” reported Henley and her associates. For each year between 2009 and 2013, an estimated 660,000 Americans were diagnosed with tobacco-related cancer, and an estimated 343,000 people died from those cancers, according to the investigators’ analysis of data collected by the United States Cancer Statistics working group, which compiles data from multiple nationwide sources including the National Program of Cancer Registries and the National Cancer Institute’s Surveillance, Epidemiology, and End Results program.
Tobacco-related cancer incidence and deaths were higher among men than women and higher among blacks than any other ethnic group. However, the cancer incidence and mortality rates also declined the fastest among men and blacks, compared with women and other ethnic groups, respectively.
Cancer incidence and death were also highest and decreased the most slowly in counties with lower educational attainment or highest poverty. Conversely, cancer incidence and mortality was the lowest and decreased the most quickly in metropolitan areas with populations greater than 1 million people.
Given that an estimated 40% of cancers diagnosed in the country and 3 in 10 cancer deaths are attributable to cigarette smoking and the use of smokeless tobacco, it is imperative that the CDC implement programs to help the almost 6 million smokers quit, CDC director Tom Frieden, MD, said in an associated telebriefing. Most people who smoke want to quit, and the health care system should do all it can to help them, Dr. Frieden said. At the same time, he echoed a claim from Henley’s paper, which said many tobacco-related cancers could be prevented by reducing tobacco use through implementation of evidence-based tobacco prevention and control interventions, such as increasing tobacco product prices, enforcing smoke-free laws, and promoting anti-tobacco mass media campaigns. These programs should be tailored to local geographic areas and demographics given the continued inconsistent progress and persistent disparities in tobacco-related cancer incidence and mortality, Dr. Frieden added.
[email protected]
On Twitter @jessnicolecraig
FROM MMWR
Key clinical point:
Major finding: Tobacco-related cancer mortality dropped from 108 deaths per 100,000 during 2004-2008 to 100 per 100,000 during 2009-2013.
Data source: Retrospective analysis of United States Cancer Statistics data for 2004 to 2013.
Disclosures: This study was sponsored by the Centers for Disease Control and Prevention. The authors’ disclosures were not reported.
PCR will improve diagnostic testing for dermatophytes, expert says
LAS VEGAS – Until polymerase chain reaction (PCR) testing for diagnosing dermatophyte infections becomes available in the United States, the options remain the KOH test, periodic acid–Schiff (PAS) stain, and culture, Dr. Theodore Rosen said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
PCR testing is both the most sensitive and most specific method to diagnose dermatophyte infections and eventually will become the gold standard for diagnosing such infections, noted Dr. Rosen, professor of dermatology, Baylor College of Medicine, Houston. A commercial PCR kit to diagnose dermatophytes, manufactured by a Danish company, is available outside of the United States but currently is not approved in this country.

He listed several studies that compared these methods, including a retrospective trial that analyzed the reliability of different tests in verifying a clinical diagnosis of onychomycosis in 108 patients (J Am Podiatr Med Assoc. 2015 Nov;105[6]:503-8). When toenail clippings from the study participants were tested, PAS produced the most consistent positive results (60%, compared with 43.5% for KOH and 39.8% for culture). Compared with the KOH test, PAS also had a higher sensitivity (0.79 vs. 0.64) for confirming fungal infection.
Dr. Rosen said that PCR testing should become available in the United States in the future. “PCR is not only good because we know that the fungus is there, but it can be very specific,” he said. A small percentage of onychomycosis cases are due to Candida, most often affecting the fingernails of people who have their hands in water frequently, such as bartenders and housekeepers.
“And then there’s another small percentage that are due to nondermatophyte molds, the kind of things that are everywhere in the environment,” said Dr. Rosen. Describing the appearance of these types of cases, he said, “sometimes they look different … they’re darker. The nails are a little more heavily affected.”
Dr. Rosen listed several PCR studies, including one that compared PCR with conventional diagnostic methods in 107 nail specimens of patients with clinically suspected onychomycosis. The study found that PCR use increased the diagnosis of specimens positive for dermatophytes by almost 40%. PCR was positive in 72% of the specimens, compared with 57% of the fungal cultures. In addition, PCR detected dermatophytes in 39 specimens that cultures missed (Australas J Dermatol. 2013 May;54[2]:105-8).
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
LAS VEGAS – Until polymerase chain reaction (PCR) testing for diagnosing dermatophyte infections becomes available in the United States, the options remain the KOH test, periodic acid–Schiff (PAS) stain, and culture, Dr. Theodore Rosen said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
PCR testing is both the most sensitive and most specific method to diagnose dermatophyte infections and eventually will become the gold standard for diagnosing such infections, noted Dr. Rosen, professor of dermatology, Baylor College of Medicine, Houston. A commercial PCR kit to diagnose dermatophytes, manufactured by a Danish company, is available outside of the United States but currently is not approved in this country.
He listed several studies that compared these methods, including a retrospective trial that analyzed the reliability of different tests in verifying a clinical diagnosis of onychomycosis in 108 patients (J Am Podiatr Med Assoc. 2015 Nov;105[6]:503-8). When toenail clippings from the study participants were tested, PAS produced the most consistent positive results (60%, compared with 43.5% for KOH and 39.8% for culture). Compared with the KOH test, PAS also had a higher sensitivity (0.79 vs. 0.64) for confirming fungal infection.
Dr. Rosen said that PCR testing should become available in the United States in the future. “PCR is not only good because we know that the fungus is there, but it can be very specific,” he said. A small percentage of onychomycosis cases are due to Candida, most often affecting the fingernails of people who have their hands in water frequently, such as bartenders and housekeepers.
“And then there’s another small percentage that are due to nondermatophyte molds, the kind of things that are everywhere in the environment,” said Dr. Rosen. Describing the appearance of these types of cases, he said, “sometimes they look different … they’re darker. The nails are a little more heavily affected.”
Dr. Rosen listed several PCR studies, including one that compared PCR with conventional diagnostic methods in 107 nail specimens of patients with clinically suspected onychomycosis. The study found that PCR use increased the diagnosis of specimens positive for dermatophytes by almost 40%. PCR was positive in 72% of the specimens, compared with 57% of the fungal cultures. In addition, PCR detected dermatophytes in 39 specimens that cultures missed (Australas J Dermatol. 2013 May;54[2]:105-8).
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
LAS VEGAS – Until polymerase chain reaction (PCR) testing for diagnosing dermatophyte infections becomes available in the United States, the options remain the KOH test, periodic acid–Schiff (PAS) stain, and culture, Dr. Theodore Rosen said at the Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar.
PCR testing is both the most sensitive and most specific method to diagnose dermatophyte infections and eventually will become the gold standard for diagnosing such infections, noted Dr. Rosen, professor of dermatology, Baylor College of Medicine, Houston. A commercial PCR kit to diagnose dermatophytes, manufactured by a Danish company, is available outside of the United States but currently is not approved in this country.
He listed several studies that compared these methods, including a retrospective trial that analyzed the reliability of different tests in verifying a clinical diagnosis of onychomycosis in 108 patients (J Am Podiatr Med Assoc. 2015 Nov;105[6]:503-8). When toenail clippings from the study participants were tested, PAS produced the most consistent positive results (60%, compared with 43.5% for KOH and 39.8% for culture). Compared with the KOH test, PAS also had a higher sensitivity (0.79 vs. 0.64) for confirming fungal infection.
Dr. Rosen said that PCR testing should become available in the United States in the future. “PCR is not only good because we know that the fungus is there, but it can be very specific,” he said. A small percentage of onychomycosis cases are due to Candida, most often affecting the fingernails of people who have their hands in water frequently, such as bartenders and housekeepers.
“And then there’s another small percentage that are due to nondermatophyte molds, the kind of things that are everywhere in the environment,” said Dr. Rosen. Describing the appearance of these types of cases, he said, “sometimes they look different … they’re darker. The nails are a little more heavily affected.”
Dr. Rosen listed several PCR studies, including one that compared PCR with conventional diagnostic methods in 107 nail specimens of patients with clinically suspected onychomycosis. The study found that PCR use increased the diagnosis of specimens positive for dermatophytes by almost 40%. PCR was positive in 72% of the specimens, compared with 57% of the fungal cultures. In addition, PCR detected dermatophytes in 39 specimens that cultures missed (Australas J Dermatol. 2013 May;54[2]:105-8).
SDEF and this news organization are owned by the same parent company.
Dr. Rosen disclosed being a paid participant on the scientific advisory boards for Anacor and Valeant.
EXPERT ANALYSIS FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
Misvalued codes lead to small cut in Medicare fee schedule
Instead of getting a 0.5% pay increase, doctors will lose 0.18% of their Medicare payments because the Centers for Medicare & Medicaid Services could not find enough misvalued services in the 2017 Medicare physician fee schedule.
A health reform law called the Protecting Access to Medicare Act of 2014 (H.R. 4302) was designed to hold down Medicare spending by requiring CMS to identify misvalued codes based on up to 16 criteria. Once identified, the value of those codes was to be lowered by CMS by an amount equal to 1% of the overall fee schedule so that physician pay could be increased by 1%. Codes up for consideration included codes that were the fastest-growing, codes for maturing technologies, codes for services that have undergone a change in service site, and codes with a significant difference in value based on service site.
A total of 19 potentially misvalued codes were found, including biopsy of finger or toe nail; injection of carpal tunnel; change of stomach feeding, accessed through skin; irrigation of vagina and/or application of drug to treat infection; wound closure utilizing tissue adhesive(s) only; and injection of anesthetic agent and/or steroid drug into nerve of foot. Lowering the value of those codes reduced the physician fee schedule by 0.32%, causing the 0.18% pay cut.
It is disappointing that a 0.5% fee schedule increase could not be accomplished, according to officials at the American Academy of Family Physicians.
“What I have to do in my office visit has greatly increased, whereas what it takes [specialists] to do their procedure has decreased. There needs to be an adjustment in the fee schedule,” AAFP President John Meigs, MD, said in an interview. This legislation “was supposed to spur Medicare and CMS to start making some adjustments and with these adjustments, give that tiny little pay increase to the fee schedule. That leads to the frustration of primary care physicians and family medicine physicians. We were promised a tiny pay increase and we are not going to get it.”

“CMS is paying lip service to increasing value of primary care, increasing the value of cognitive-based services, and they did come up with some new codes,” Dr. Meigs said. “They did some things with paying for diabetic patients that are things that they should have been paying for a long time ago. But the administrative hassle and regulatory burden of trying to do the documentation they require to actually get paid for some of these codes have a lot of physicians throwing their hands up saying, ‘it’s not worth my time to spend $5 to make a nickel.’ ”
Instead of getting a 0.5% pay increase, doctors will lose 0.18% of their Medicare payments because the Centers for Medicare & Medicaid Services could not find enough misvalued services in the 2017 Medicare physician fee schedule.
A health reform law called the Protecting Access to Medicare Act of 2014 (H.R. 4302) was designed to hold down Medicare spending by requiring CMS to identify misvalued codes based on up to 16 criteria. Once identified, the value of those codes was to be lowered by CMS by an amount equal to 1% of the overall fee schedule so that physician pay could be increased by 1%. Codes up for consideration included codes that were the fastest-growing, codes for maturing technologies, codes for services that have undergone a change in service site, and codes with a significant difference in value based on service site.
A total of 19 potentially misvalued codes were found, including biopsy of finger or toe nail; injection of carpal tunnel; change of stomach feeding, accessed through skin; irrigation of vagina and/or application of drug to treat infection; wound closure utilizing tissue adhesive(s) only; and injection of anesthetic agent and/or steroid drug into nerve of foot. Lowering the value of those codes reduced the physician fee schedule by 0.32%, causing the 0.18% pay cut.
It is disappointing that a 0.5% fee schedule increase could not be accomplished, according to officials at the American Academy of Family Physicians.
“What I have to do in my office visit has greatly increased, whereas what it takes [specialists] to do their procedure has decreased. There needs to be an adjustment in the fee schedule,” AAFP President John Meigs, MD, said in an interview. This legislation “was supposed to spur Medicare and CMS to start making some adjustments and with these adjustments, give that tiny little pay increase to the fee schedule. That leads to the frustration of primary care physicians and family medicine physicians. We were promised a tiny pay increase and we are not going to get it.”
“CMS is paying lip service to increasing value of primary care, increasing the value of cognitive-based services, and they did come up with some new codes,” Dr. Meigs said. “They did some things with paying for diabetic patients that are things that they should have been paying for a long time ago. But the administrative hassle and regulatory burden of trying to do the documentation they require to actually get paid for some of these codes have a lot of physicians throwing their hands up saying, ‘it’s not worth my time to spend $5 to make a nickel.’ ”
Instead of getting a 0.5% pay increase, doctors will lose 0.18% of their Medicare payments because the Centers for Medicare & Medicaid Services could not find enough misvalued services in the 2017 Medicare physician fee schedule.
A health reform law called the Protecting Access to Medicare Act of 2014 (H.R. 4302) was designed to hold down Medicare spending by requiring CMS to identify misvalued codes based on up to 16 criteria. Once identified, the value of those codes was to be lowered by CMS by an amount equal to 1% of the overall fee schedule so that physician pay could be increased by 1%. Codes up for consideration included codes that were the fastest-growing, codes for maturing technologies, codes for services that have undergone a change in service site, and codes with a significant difference in value based on service site.
A total of 19 potentially misvalued codes were found, including biopsy of finger or toe nail; injection of carpal tunnel; change of stomach feeding, accessed through skin; irrigation of vagina and/or application of drug to treat infection; wound closure utilizing tissue adhesive(s) only; and injection of anesthetic agent and/or steroid drug into nerve of foot. Lowering the value of those codes reduced the physician fee schedule by 0.32%, causing the 0.18% pay cut.
It is disappointing that a 0.5% fee schedule increase could not be accomplished, according to officials at the American Academy of Family Physicians.
“What I have to do in my office visit has greatly increased, whereas what it takes [specialists] to do their procedure has decreased. There needs to be an adjustment in the fee schedule,” AAFP President John Meigs, MD, said in an interview. This legislation “was supposed to spur Medicare and CMS to start making some adjustments and with these adjustments, give that tiny little pay increase to the fee schedule. That leads to the frustration of primary care physicians and family medicine physicians. We were promised a tiny pay increase and we are not going to get it.”
“CMS is paying lip service to increasing value of primary care, increasing the value of cognitive-based services, and they did come up with some new codes,” Dr. Meigs said. “They did some things with paying for diabetic patients that are things that they should have been paying for a long time ago. But the administrative hassle and regulatory burden of trying to do the documentation they require to actually get paid for some of these codes have a lot of physicians throwing their hands up saying, ‘it’s not worth my time to spend $5 to make a nickel.’ ”