User login
Stroke patients benefit from neurologic music therapy
Neurologic music therapy (NMT), a specially designed intervention targeting movement, balance, and cognitive functioning, improves depressive symptoms and increases brain-derived neurotrophic factor (BDNF), early results of a small study suggest.
“We’re really happy with the results,” said lead study author psychotherapist Honey Bryant, a PhD candidate and research assistant at the Centre for Neuroscience Studies, Queen’s University, Kingston, Ont.
“We showed ”
The findings were presented at the virtual XXVI World Congress of Neurology.
Moving with music
With improved stroke survival rates and longer life expectancy, there’s an increasing need for effective post-stroke interventions for neurocognitive impairments and mood disorders, the authors noted.
NMT is an evidence-based treatment system that uses elements of music such as rhythm, melody, and tempo to treat various brain conditions. A trained NMT therapist uses standardized techniques to address goals in the areas of speech, movement, and cognition.
The intervention is not new – it’s been around for a few decades – but there are “minimal papers on NMT and nothing on stroke rehabilitation used in the way we did it,” said Ms. Bryant.
The study included 57 patients, mean age 75 years, receiving rehabilitation following a stroke who were randomly assigned to NMT or passive music listening.
In the NMT group, a music therapist asked participants to choose music beforehand and integrated this into each session.
“Each day was different,” said Ms. Bryant. “For example, if it involved motor movement, the music therapist would say, ‘When I sing this word, raise your arm up.’ For Johnny Cash’s ‘Ring of Fire,’ we made our arms into a circle.”
She explained that the rhythm and timing of the music can affect the motor system and other areas of the brain.
Those in the passive music group listened to a curated list of calming classical and relaxing spa music.
Both groups were offered five 45-minute sessions per week for 2 weeks.
Among other things, researchers used the Hospital Anxiety and Depression Scale (HADS), administered a semistructured interview, and collected blood samples to determine levels of cortisol and BDNF.
After the 2-week intervention, the researchers found participants in the NMT group had a significant mean decrease in depression.
They also had increased cortisol levels, which is not unexpected after a stroke, especially with increased anxiety linked to financial and other stressors, said Ms. Bryant, adding these levels should decrease with treatment.
Recipients of the NMT had significant increases in BDNF, a neurotrophin that plays an important role in neuronal survival and growth, but only in those who attended several consecutive sessions.
Increased plasticity
“We see greater increases in plasticity when the therapy is used intensively, meaning at least four treatments consecutively,” said Ms. Bryant. Participants in the NMT group also reported they “overall felt well,” she added.
She noted NMT can be tailored to individual deficit, “so you can make it solely for motor movement or you can make it solely for language.”
Next steps could include more closely targeting the music to individual preferences and investigating whether the benefits of the intervention extend to other types of brain injury, for example traumatic brain injury, which typically affects younger people, said Ms. Bryant.
“In this study, participants were older and there was an unknown; a lot of them were going back into the community but didn’t know if it was into a retirement home or long-term care.”
It’s unclear if the benefits are sustained after the intervention stops, she said.
There are also the issues of cost and accessibility; in Kingston, there are few music therapists certified in the area of NMT.
Ms. Bryant hopes NMT is eventually included in stroke rehabilitation. “Stroke therapy is typically very intensive on its own; you’re doing it every single day for about a month or 6 weeks,” she said. “It would be interesting to see whether we would see a shorter hospital stay if this is included in stroke rehab.”
Asked to comment, Michael H. Thaut, PhD, professor, faculty of music and faculty of medicine, and Canada research chair in music, neuroscience and health at the University of Toronto, said while these data are preliminary, “they do extend the benefits of NMT in stroke rehabilitation, especially measuring BDNF in addition to having behavioral data.”
However, it’s “unfortunate” the poster didn’t specify which cognitive intervention techniques were used in the study, said Dr. Thaut. “There are nine coded techniques in NMT, including for attention, memory, psychosocial function, and executive function.”
His own study, published in NeuroRehabilitation, focused on training for motor goals in stroke patients. It showed that NMT benefited cognitive functioning and affective responses.
The study was funded by a Queen’s University Research Initiation Grant. Ms. Bryant and Dr. Thaut have not disclosed any relevant financial relationships.
A version of this article first appeared on Medscape.com.
Neurologic music therapy (NMT), a specially designed intervention targeting movement, balance, and cognitive functioning, improves depressive symptoms and increases brain-derived neurotrophic factor (BDNF), early results of a small study suggest.
“We’re really happy with the results,” said lead study author psychotherapist Honey Bryant, a PhD candidate and research assistant at the Centre for Neuroscience Studies, Queen’s University, Kingston, Ont.
“We showed ”
The findings were presented at the virtual XXVI World Congress of Neurology.
Moving with music
With improved stroke survival rates and longer life expectancy, there’s an increasing need for effective post-stroke interventions for neurocognitive impairments and mood disorders, the authors noted.
NMT is an evidence-based treatment system that uses elements of music such as rhythm, melody, and tempo to treat various brain conditions. A trained NMT therapist uses standardized techniques to address goals in the areas of speech, movement, and cognition.
The intervention is not new – it’s been around for a few decades – but there are “minimal papers on NMT and nothing on stroke rehabilitation used in the way we did it,” said Ms. Bryant.
The study included 57 patients, mean age 75 years, receiving rehabilitation following a stroke who were randomly assigned to NMT or passive music listening.
In the NMT group, a music therapist asked participants to choose music beforehand and integrated this into each session.
“Each day was different,” said Ms. Bryant. “For example, if it involved motor movement, the music therapist would say, ‘When I sing this word, raise your arm up.’ For Johnny Cash’s ‘Ring of Fire,’ we made our arms into a circle.”
She explained that the rhythm and timing of the music can affect the motor system and other areas of the brain.
Those in the passive music group listened to a curated list of calming classical and relaxing spa music.
Both groups were offered five 45-minute sessions per week for 2 weeks.
Among other things, researchers used the Hospital Anxiety and Depression Scale (HADS), administered a semistructured interview, and collected blood samples to determine levels of cortisol and BDNF.
After the 2-week intervention, the researchers found participants in the NMT group had a significant mean decrease in depression.
They also had increased cortisol levels, which is not unexpected after a stroke, especially with increased anxiety linked to financial and other stressors, said Ms. Bryant, adding these levels should decrease with treatment.
Recipients of the NMT had significant increases in BDNF, a neurotrophin that plays an important role in neuronal survival and growth, but only in those who attended several consecutive sessions.
Increased plasticity
“We see greater increases in plasticity when the therapy is used intensively, meaning at least four treatments consecutively,” said Ms. Bryant. Participants in the NMT group also reported they “overall felt well,” she added.
She noted NMT can be tailored to individual deficit, “so you can make it solely for motor movement or you can make it solely for language.”
Next steps could include more closely targeting the music to individual preferences and investigating whether the benefits of the intervention extend to other types of brain injury, for example traumatic brain injury, which typically affects younger people, said Ms. Bryant.
“In this study, participants were older and there was an unknown; a lot of them were going back into the community but didn’t know if it was into a retirement home or long-term care.”
It’s unclear if the benefits are sustained after the intervention stops, she said.
There are also the issues of cost and accessibility; in Kingston, there are few music therapists certified in the area of NMT.
Ms. Bryant hopes NMT is eventually included in stroke rehabilitation. “Stroke therapy is typically very intensive on its own; you’re doing it every single day for about a month or 6 weeks,” she said. “It would be interesting to see whether we would see a shorter hospital stay if this is included in stroke rehab.”
Asked to comment, Michael H. Thaut, PhD, professor, faculty of music and faculty of medicine, and Canada research chair in music, neuroscience and health at the University of Toronto, said while these data are preliminary, “they do extend the benefits of NMT in stroke rehabilitation, especially measuring BDNF in addition to having behavioral data.”
However, it’s “unfortunate” the poster didn’t specify which cognitive intervention techniques were used in the study, said Dr. Thaut. “There are nine coded techniques in NMT, including for attention, memory, psychosocial function, and executive function.”
His own study, published in NeuroRehabilitation, focused on training for motor goals in stroke patients. It showed that NMT benefited cognitive functioning and affective responses.
The study was funded by a Queen’s University Research Initiation Grant. Ms. Bryant and Dr. Thaut have not disclosed any relevant financial relationships.
A version of this article first appeared on Medscape.com.
Neurologic music therapy (NMT), a specially designed intervention targeting movement, balance, and cognitive functioning, improves depressive symptoms and increases brain-derived neurotrophic factor (BDNF), early results of a small study suggest.
“We’re really happy with the results,” said lead study author psychotherapist Honey Bryant, a PhD candidate and research assistant at the Centre for Neuroscience Studies, Queen’s University, Kingston, Ont.
“We showed ”
The findings were presented at the virtual XXVI World Congress of Neurology.
Moving with music
With improved stroke survival rates and longer life expectancy, there’s an increasing need for effective post-stroke interventions for neurocognitive impairments and mood disorders, the authors noted.
NMT is an evidence-based treatment system that uses elements of music such as rhythm, melody, and tempo to treat various brain conditions. A trained NMT therapist uses standardized techniques to address goals in the areas of speech, movement, and cognition.
The intervention is not new – it’s been around for a few decades – but there are “minimal papers on NMT and nothing on stroke rehabilitation used in the way we did it,” said Ms. Bryant.
The study included 57 patients, mean age 75 years, receiving rehabilitation following a stroke who were randomly assigned to NMT or passive music listening.
In the NMT group, a music therapist asked participants to choose music beforehand and integrated this into each session.
“Each day was different,” said Ms. Bryant. “For example, if it involved motor movement, the music therapist would say, ‘When I sing this word, raise your arm up.’ For Johnny Cash’s ‘Ring of Fire,’ we made our arms into a circle.”
She explained that the rhythm and timing of the music can affect the motor system and other areas of the brain.
Those in the passive music group listened to a curated list of calming classical and relaxing spa music.
Both groups were offered five 45-minute sessions per week for 2 weeks.
Among other things, researchers used the Hospital Anxiety and Depression Scale (HADS), administered a semistructured interview, and collected blood samples to determine levels of cortisol and BDNF.
After the 2-week intervention, the researchers found participants in the NMT group had a significant mean decrease in depression.
They also had increased cortisol levels, which is not unexpected after a stroke, especially with increased anxiety linked to financial and other stressors, said Ms. Bryant, adding these levels should decrease with treatment.
Recipients of the NMT had significant increases in BDNF, a neurotrophin that plays an important role in neuronal survival and growth, but only in those who attended several consecutive sessions.
Increased plasticity
“We see greater increases in plasticity when the therapy is used intensively, meaning at least four treatments consecutively,” said Ms. Bryant. Participants in the NMT group also reported they “overall felt well,” she added.
She noted NMT can be tailored to individual deficit, “so you can make it solely for motor movement or you can make it solely for language.”
Next steps could include more closely targeting the music to individual preferences and investigating whether the benefits of the intervention extend to other types of brain injury, for example traumatic brain injury, which typically affects younger people, said Ms. Bryant.
“In this study, participants were older and there was an unknown; a lot of them were going back into the community but didn’t know if it was into a retirement home or long-term care.”
It’s unclear if the benefits are sustained after the intervention stops, she said.
There are also the issues of cost and accessibility; in Kingston, there are few music therapists certified in the area of NMT.
Ms. Bryant hopes NMT is eventually included in stroke rehabilitation. “Stroke therapy is typically very intensive on its own; you’re doing it every single day for about a month or 6 weeks,” she said. “It would be interesting to see whether we would see a shorter hospital stay if this is included in stroke rehab.”
Asked to comment, Michael H. Thaut, PhD, professor, faculty of music and faculty of medicine, and Canada research chair in music, neuroscience and health at the University of Toronto, said while these data are preliminary, “they do extend the benefits of NMT in stroke rehabilitation, especially measuring BDNF in addition to having behavioral data.”
However, it’s “unfortunate” the poster didn’t specify which cognitive intervention techniques were used in the study, said Dr. Thaut. “There are nine coded techniques in NMT, including for attention, memory, psychosocial function, and executive function.”
His own study, published in NeuroRehabilitation, focused on training for motor goals in stroke patients. It showed that NMT benefited cognitive functioning and affective responses.
The study was funded by a Queen’s University Research Initiation Grant. Ms. Bryant and Dr. Thaut have not disclosed any relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM WCN 2023
AHA joins new cardiovascular certification group ABCVM
T to be known as the American Board of Cardiovascular Medicine (ABCVM).
The ABCVM would be independent of the American Board of Internal Medicine (ABIM), the current organization providing maintenance of certification for cardiologists along with 20 other internal medicine subspecialties. The ABIM’s maintenance of certification process has been widely criticized for many years and has been described as “needlessly burdensome and expensive.”
The AHA will be joining the American College of Cardiology (ACC), Heart Failure Society of America (HFSA), Heart Rhythm Society (HRS), and Society for Cardiovascular Angiography & Interventions (SCAI) in forming the ABCVM.
These four other societies issued a joint statement in September saying that they will apply to the American Board of Medical Specialties (ABMS) to request an independent cardiology board that follows a “new competency-based approach to continuous certification — one that harnesses the knowledge, skills, and attitudes required to sustain professional excellence and care for cardiovascular patients effectively.”
The new board requirements will “de-emphasize timed, high stakes performance exams in the continuous certification process and instead will focus on learning assessments to identify gaps in current knowledge or skills,” the statement noted.
At the time the September statement was issued, the AHA was said to be supportive of the move but was waiting for formal endorsement to join the effort by its board of directors.
That has now happened, with the AHA’s national board of directors voting to provide “full support” for the creation of the proposed ABCVM.
“We enthusiastically join with our colleagues in proposing a new professional certification body to accredit cardiovascular professionals called the American Board of Cardiovascular Medicine,” said the association’s volunteer president Joseph C. Wu, MD. “The new ABCVM will be independent of the ABIM and focus on the specific competency-based trainings and appropriate ongoing certifications that align with and strengthen skills for cardiovascular physicians and enhance quality of care for people with cardiovascular disease,” Wu said.
“The AHA joins the consortium to submit the application to the American Board of Medical Specialties (ABMS) requesting an independent medical board for cardiovascular medicine. The consortium’s robust proposal harnesses the knowledge, skills, and benchmarks appropriate for professional excellence and delivery of effective, high-quality cardiovascular care,” Wu added.
The leaders of the ABCVM will include professional representatives from the consortium of member organizations, with a specific focus on relevant education, trainings, and supports that recognize the increasing specialization in cardiology and the latest advances in the various subspecialties of cardiovascular medicine, the AHA notes in a statement.
Professional certification by ABIM is a condition of employment for physicians practicing in large hospitals or health systems. A dedicated certification board separate from ABIM will help to ensure that cardiovascular professionals are maintaining the expertise appropriate to high-quality care and improved outcomes for their patients, the AHA said.
A version of this article first appeared on Medscape.com.
T to be known as the American Board of Cardiovascular Medicine (ABCVM).
The ABCVM would be independent of the American Board of Internal Medicine (ABIM), the current organization providing maintenance of certification for cardiologists along with 20 other internal medicine subspecialties. The ABIM’s maintenance of certification process has been widely criticized for many years and has been described as “needlessly burdensome and expensive.”
The AHA will be joining the American College of Cardiology (ACC), Heart Failure Society of America (HFSA), Heart Rhythm Society (HRS), and Society for Cardiovascular Angiography & Interventions (SCAI) in forming the ABCVM.
These four other societies issued a joint statement in September saying that they will apply to the American Board of Medical Specialties (ABMS) to request an independent cardiology board that follows a “new competency-based approach to continuous certification — one that harnesses the knowledge, skills, and attitudes required to sustain professional excellence and care for cardiovascular patients effectively.”
The new board requirements will “de-emphasize timed, high stakes performance exams in the continuous certification process and instead will focus on learning assessments to identify gaps in current knowledge or skills,” the statement noted.
At the time the September statement was issued, the AHA was said to be supportive of the move but was waiting for formal endorsement to join the effort by its board of directors.
That has now happened, with the AHA’s national board of directors voting to provide “full support” for the creation of the proposed ABCVM.
“We enthusiastically join with our colleagues in proposing a new professional certification body to accredit cardiovascular professionals called the American Board of Cardiovascular Medicine,” said the association’s volunteer president Joseph C. Wu, MD. “The new ABCVM will be independent of the ABIM and focus on the specific competency-based trainings and appropriate ongoing certifications that align with and strengthen skills for cardiovascular physicians and enhance quality of care for people with cardiovascular disease,” Wu said.
“The AHA joins the consortium to submit the application to the American Board of Medical Specialties (ABMS) requesting an independent medical board for cardiovascular medicine. The consortium’s robust proposal harnesses the knowledge, skills, and benchmarks appropriate for professional excellence and delivery of effective, high-quality cardiovascular care,” Wu added.
The leaders of the ABCVM will include professional representatives from the consortium of member organizations, with a specific focus on relevant education, trainings, and supports that recognize the increasing specialization in cardiology and the latest advances in the various subspecialties of cardiovascular medicine, the AHA notes in a statement.
Professional certification by ABIM is a condition of employment for physicians practicing in large hospitals or health systems. A dedicated certification board separate from ABIM will help to ensure that cardiovascular professionals are maintaining the expertise appropriate to high-quality care and improved outcomes for their patients, the AHA said.
A version of this article first appeared on Medscape.com.
T to be known as the American Board of Cardiovascular Medicine (ABCVM).
The ABCVM would be independent of the American Board of Internal Medicine (ABIM), the current organization providing maintenance of certification for cardiologists along with 20 other internal medicine subspecialties. The ABIM’s maintenance of certification process has been widely criticized for many years and has been described as “needlessly burdensome and expensive.”
The AHA will be joining the American College of Cardiology (ACC), Heart Failure Society of America (HFSA), Heart Rhythm Society (HRS), and Society for Cardiovascular Angiography & Interventions (SCAI) in forming the ABCVM.
These four other societies issued a joint statement in September saying that they will apply to the American Board of Medical Specialties (ABMS) to request an independent cardiology board that follows a “new competency-based approach to continuous certification — one that harnesses the knowledge, skills, and attitudes required to sustain professional excellence and care for cardiovascular patients effectively.”
The new board requirements will “de-emphasize timed, high stakes performance exams in the continuous certification process and instead will focus on learning assessments to identify gaps in current knowledge or skills,” the statement noted.
At the time the September statement was issued, the AHA was said to be supportive of the move but was waiting for formal endorsement to join the effort by its board of directors.
That has now happened, with the AHA’s national board of directors voting to provide “full support” for the creation of the proposed ABCVM.
“We enthusiastically join with our colleagues in proposing a new professional certification body to accredit cardiovascular professionals called the American Board of Cardiovascular Medicine,” said the association’s volunteer president Joseph C. Wu, MD. “The new ABCVM will be independent of the ABIM and focus on the specific competency-based trainings and appropriate ongoing certifications that align with and strengthen skills for cardiovascular physicians and enhance quality of care for people with cardiovascular disease,” Wu said.
“The AHA joins the consortium to submit the application to the American Board of Medical Specialties (ABMS) requesting an independent medical board for cardiovascular medicine. The consortium’s robust proposal harnesses the knowledge, skills, and benchmarks appropriate for professional excellence and delivery of effective, high-quality cardiovascular care,” Wu added.
The leaders of the ABCVM will include professional representatives from the consortium of member organizations, with a specific focus on relevant education, trainings, and supports that recognize the increasing specialization in cardiology and the latest advances in the various subspecialties of cardiovascular medicine, the AHA notes in a statement.
Professional certification by ABIM is a condition of employment for physicians practicing in large hospitals or health systems. A dedicated certification board separate from ABIM will help to ensure that cardiovascular professionals are maintaining the expertise appropriate to high-quality care and improved outcomes for their patients, the AHA said.
A version of this article first appeared on Medscape.com.
Reimagining rehabilitation: In-home physical therapy gets a boost
As the aging population grows and telehealth expands in the wake of the COVID-19 pandemic, an emerging trend of in-home care is reshaping how patients access and receive physical therapy services.
Partnerships between hospitals and home health companies are increasing access to rehabilitation services not only for older adults but also for people in rural areas, those without reliable transportation, and patients with injuries that hinder their driving abilities.
“We find more and more that physical therapy at their home, instead of coming to an outpatient facility, is something more and more folks are requesting,” said Bill Benoit, MBA, chief operating officer of University Hospitals, Cleveland. “In this post-COVID environment, people are getting all different types of services in their home when they’re available, and this is one of them. The pandemic sped up the process of us moving away from the traditional brick and mortar hospital.”
UH recently announced a partnership with Luna Physical Therapy, a company founded in 2018 that provides home services. Luna has teamed up with more than two dozen other hospitals in the United States to offer home-based rehabilitation, according to the company.
The process for arranging in-home therapies through hospital-clinic partnerships is like any other inpatient or outpatient rehabilitation, Mr. Benoit said: A patient meets with a specialist or primary care practitioner, they discuss options, and eventually the clinician recommends physical therapy. The only difference here, he said, is rather than going to a separate facility or a hospital, the patient logs onto a mobile app that matches them with a physical therapist on the basis of their location, needs, and the times they are available.
The prescribing physician oversees the patient’s progress through notes provided by the therapist.
“For the primary care physician or surgeon, they’re not going to see much of a difference,” Mr. Benoit said. “This just adds to that list of options for patients.”
Safer, more productive PT
A study, published in the journal Family Practice, found that 76% of patients who are prescribed physical therapy do not initiate the services after it has been recommended.
Aside from the convenience and expanded accessibility for patients, the home therapy option can be more productive, said Denise Wagner, PT, DPT, a physical therapist with Johns Hopkins, Baltimore.
“Home is safer for many patients, but home is also more engaging and motivating,” she said. “Home health clinicians are experts in using whatever they find in the home environment as equipment; many people have stairs in their home, so we can use the rail as something to hold. If patient likes to walk their dog, we can use putting a leash on dog as balance activity.”
Therapy in the home setting helps physical therapists customize programs to fit each patient’s lifestyle, said Gira Shah, PT, a physical therapist with Providence Home Services in Seattle.
For example, patients generally want to know how to function within their own space – navigate their kitchens to make food or get in and out of their bathtubs. Staying in that space allows therapists to focus on those specific goals, Ms. Shah said. “It’s more of a functional therapy. The beauty of this [is that] as therapists we’re trying to assess, ‘what does the patient need to be independent?’ ”
The consulting firm McKinsey predicts that as much as $265 billion in health care services for Medicare recipients will be provided within the home by 2025.
The obvious question is: Why would hospitals partner with clinics rather than offer in-home services on their own?
The answer, like most things in health care, boils down to money.
The billing and documentation system that they use is more efficient than anything hospitals have, said John Brickley, PT, MA, vice president and physical therapist at MedStar Health, a health care system in Maryland and the Washington, D.C., area. MedStar and Luna announced a partnership last June.
“We would financially fall on our face if we tried to use our own billing systems; it would take too much time,” Mr. Brickley said. “Do we need them from a quality-of-care standpoint? No. They have the type of technology that’s not at our disposal.”
Patients should be aware of the difference between home-based PT and other health services for homebound patients, Mr. Brickley said. Medicare considers a patient homebound if they need the help of another person or medical equipment to leave their home or if their doctor believes their condition would worsen with greater mobility.
From the perspective of an insurance company, a home therapy session arranged by a hospital-clinic partnership is an ambulatory appointment and uses the same charging mechanism as most other visits. For a home health care visit, patients must qualify as homebound.
Home-based PT can be used for conditions including neurologic issues, bone and joint problems, balance, and fall deconditioning and prevention. But if a patient needs heavy equipment that cannot be transported, outpatient services are more practical.
That should be determined by the primary care practitioner or specialist evaluating each patient, said Palak Shah, PT, cofounder and head of clinical services at Luna.
“Primary care physicians play a huge role – that’s where patients express their initial concerns,” she said. “It’s up to them to make patients aware about all the options.”
A version of this article first appeared on Medscape.com.
As the aging population grows and telehealth expands in the wake of the COVID-19 pandemic, an emerging trend of in-home care is reshaping how patients access and receive physical therapy services.
Partnerships between hospitals and home health companies are increasing access to rehabilitation services not only for older adults but also for people in rural areas, those without reliable transportation, and patients with injuries that hinder their driving abilities.
“We find more and more that physical therapy at their home, instead of coming to an outpatient facility, is something more and more folks are requesting,” said Bill Benoit, MBA, chief operating officer of University Hospitals, Cleveland. “In this post-COVID environment, people are getting all different types of services in their home when they’re available, and this is one of them. The pandemic sped up the process of us moving away from the traditional brick and mortar hospital.”
UH recently announced a partnership with Luna Physical Therapy, a company founded in 2018 that provides home services. Luna has teamed up with more than two dozen other hospitals in the United States to offer home-based rehabilitation, according to the company.
The process for arranging in-home therapies through hospital-clinic partnerships is like any other inpatient or outpatient rehabilitation, Mr. Benoit said: A patient meets with a specialist or primary care practitioner, they discuss options, and eventually the clinician recommends physical therapy. The only difference here, he said, is rather than going to a separate facility or a hospital, the patient logs onto a mobile app that matches them with a physical therapist on the basis of their location, needs, and the times they are available.
The prescribing physician oversees the patient’s progress through notes provided by the therapist.
“For the primary care physician or surgeon, they’re not going to see much of a difference,” Mr. Benoit said. “This just adds to that list of options for patients.”
Safer, more productive PT
A study, published in the journal Family Practice, found that 76% of patients who are prescribed physical therapy do not initiate the services after it has been recommended.
Aside from the convenience and expanded accessibility for patients, the home therapy option can be more productive, said Denise Wagner, PT, DPT, a physical therapist with Johns Hopkins, Baltimore.
“Home is safer for many patients, but home is also more engaging and motivating,” she said. “Home health clinicians are experts in using whatever they find in the home environment as equipment; many people have stairs in their home, so we can use the rail as something to hold. If patient likes to walk their dog, we can use putting a leash on dog as balance activity.”
Therapy in the home setting helps physical therapists customize programs to fit each patient’s lifestyle, said Gira Shah, PT, a physical therapist with Providence Home Services in Seattle.
For example, patients generally want to know how to function within their own space – navigate their kitchens to make food or get in and out of their bathtubs. Staying in that space allows therapists to focus on those specific goals, Ms. Shah said. “It’s more of a functional therapy. The beauty of this [is that] as therapists we’re trying to assess, ‘what does the patient need to be independent?’ ”
The consulting firm McKinsey predicts that as much as $265 billion in health care services for Medicare recipients will be provided within the home by 2025.
The obvious question is: Why would hospitals partner with clinics rather than offer in-home services on their own?
The answer, like most things in health care, boils down to money.
The billing and documentation system that they use is more efficient than anything hospitals have, said John Brickley, PT, MA, vice president and physical therapist at MedStar Health, a health care system in Maryland and the Washington, D.C., area. MedStar and Luna announced a partnership last June.
“We would financially fall on our face if we tried to use our own billing systems; it would take too much time,” Mr. Brickley said. “Do we need them from a quality-of-care standpoint? No. They have the type of technology that’s not at our disposal.”
Patients should be aware of the difference between home-based PT and other health services for homebound patients, Mr. Brickley said. Medicare considers a patient homebound if they need the help of another person or medical equipment to leave their home or if their doctor believes their condition would worsen with greater mobility.
From the perspective of an insurance company, a home therapy session arranged by a hospital-clinic partnership is an ambulatory appointment and uses the same charging mechanism as most other visits. For a home health care visit, patients must qualify as homebound.
Home-based PT can be used for conditions including neurologic issues, bone and joint problems, balance, and fall deconditioning and prevention. But if a patient needs heavy equipment that cannot be transported, outpatient services are more practical.
That should be determined by the primary care practitioner or specialist evaluating each patient, said Palak Shah, PT, cofounder and head of clinical services at Luna.
“Primary care physicians play a huge role – that’s where patients express their initial concerns,” she said. “It’s up to them to make patients aware about all the options.”
A version of this article first appeared on Medscape.com.
As the aging population grows and telehealth expands in the wake of the COVID-19 pandemic, an emerging trend of in-home care is reshaping how patients access and receive physical therapy services.
Partnerships between hospitals and home health companies are increasing access to rehabilitation services not only for older adults but also for people in rural areas, those without reliable transportation, and patients with injuries that hinder their driving abilities.
“We find more and more that physical therapy at their home, instead of coming to an outpatient facility, is something more and more folks are requesting,” said Bill Benoit, MBA, chief operating officer of University Hospitals, Cleveland. “In this post-COVID environment, people are getting all different types of services in their home when they’re available, and this is one of them. The pandemic sped up the process of us moving away from the traditional brick and mortar hospital.”
UH recently announced a partnership with Luna Physical Therapy, a company founded in 2018 that provides home services. Luna has teamed up with more than two dozen other hospitals in the United States to offer home-based rehabilitation, according to the company.
The process for arranging in-home therapies through hospital-clinic partnerships is like any other inpatient or outpatient rehabilitation, Mr. Benoit said: A patient meets with a specialist or primary care practitioner, they discuss options, and eventually the clinician recommends physical therapy. The only difference here, he said, is rather than going to a separate facility or a hospital, the patient logs onto a mobile app that matches them with a physical therapist on the basis of their location, needs, and the times they are available.
The prescribing physician oversees the patient’s progress through notes provided by the therapist.
“For the primary care physician or surgeon, they’re not going to see much of a difference,” Mr. Benoit said. “This just adds to that list of options for patients.”
Safer, more productive PT
A study, published in the journal Family Practice, found that 76% of patients who are prescribed physical therapy do not initiate the services after it has been recommended.
Aside from the convenience and expanded accessibility for patients, the home therapy option can be more productive, said Denise Wagner, PT, DPT, a physical therapist with Johns Hopkins, Baltimore.
“Home is safer for many patients, but home is also more engaging and motivating,” she said. “Home health clinicians are experts in using whatever they find in the home environment as equipment; many people have stairs in their home, so we can use the rail as something to hold. If patient likes to walk their dog, we can use putting a leash on dog as balance activity.”
Therapy in the home setting helps physical therapists customize programs to fit each patient’s lifestyle, said Gira Shah, PT, a physical therapist with Providence Home Services in Seattle.
For example, patients generally want to know how to function within their own space – navigate their kitchens to make food or get in and out of their bathtubs. Staying in that space allows therapists to focus on those specific goals, Ms. Shah said. “It’s more of a functional therapy. The beauty of this [is that] as therapists we’re trying to assess, ‘what does the patient need to be independent?’ ”
The consulting firm McKinsey predicts that as much as $265 billion in health care services for Medicare recipients will be provided within the home by 2025.
The obvious question is: Why would hospitals partner with clinics rather than offer in-home services on their own?
The answer, like most things in health care, boils down to money.
The billing and documentation system that they use is more efficient than anything hospitals have, said John Brickley, PT, MA, vice president and physical therapist at MedStar Health, a health care system in Maryland and the Washington, D.C., area. MedStar and Luna announced a partnership last June.
“We would financially fall on our face if we tried to use our own billing systems; it would take too much time,” Mr. Brickley said. “Do we need them from a quality-of-care standpoint? No. They have the type of technology that’s not at our disposal.”
Patients should be aware of the difference between home-based PT and other health services for homebound patients, Mr. Brickley said. Medicare considers a patient homebound if they need the help of another person or medical equipment to leave their home or if their doctor believes their condition would worsen with greater mobility.
From the perspective of an insurance company, a home therapy session arranged by a hospital-clinic partnership is an ambulatory appointment and uses the same charging mechanism as most other visits. For a home health care visit, patients must qualify as homebound.
Home-based PT can be used for conditions including neurologic issues, bone and joint problems, balance, and fall deconditioning and prevention. But if a patient needs heavy equipment that cannot be transported, outpatient services are more practical.
That should be determined by the primary care practitioner or specialist evaluating each patient, said Palak Shah, PT, cofounder and head of clinical services at Luna.
“Primary care physicians play a huge role – that’s where patients express their initial concerns,” she said. “It’s up to them to make patients aware about all the options.”
A version of this article first appeared on Medscape.com.
Pustular Eruption on the Face
The Diagnosis: Eczema Herpeticum
The patient’s condition with worsening facial edema and notable pain prompted a bedside Tzanck smear using a sample from the base of a deroofed forehead vesicle. In addition, a swab of a deroofed lesion was sent for herpes simplex virus and varicella-zoster virus (VZV) polymerase chain reaction (PCR) testing. The Tzanck smear demonstrated ballooning multinucleated syncytial giant cells and eosinophilic inclusion bodies (Figure), which are characteristic of certain herpesviruses including herpes simplex virus and VZV. He was started on intravenous acyclovir while PCR results were pending; the PCR test later confirmed positivity for herpes simplex virus type 1. Treatment was transitioned to oral valacyclovir once the lesions started crusting over. Notable healing and epithelialization of the lesions occurred during his hospital stay, and he was discharged home 5 days after starting treatment. He was counseled on autoinoculation, advised that he was considered infectious until all lesions had crusted over, and encouraged to employ frequent handwashing. Complete resolution of eczema herpeticum (EH) was noted at 3-week follow-up.
.")
Eczema herpeticum (also known as Kaposi varicelliform eruption) is a potentially life-threatening disseminated cutaneous infection caused by herpes simplex virus types 1 and 2 in patients with pre-existing skin disease.1 It typically presents as a complication of atopic dermatitis (AD) but also has been identified as a rare complication in other conditions that disrupt the normal skin barrier, including mycosis fungoides, pemphigus foliaceus, pemphigus vulgaris, Darier disease, pityriasis rubra pilaris, contact dermatitis, and seborrheic dermatitis.1-4
The pathogenesis of EH is multifactorial. Disruption of the stratum corneum; impaired natural killer cell function; early-onset, untreated, or severe AD; disrupted skin microbiota with skewed colonization by Staphylococcus aureus; immunosuppressive AD therapies such as calcineurin inhibitors; eosinophilia; and helper T cell (TH2) cytokine predominance all have been suggested to play a role in the development of EH.5-8
As seen in our patient, EH presents with a sudden eruption of painful or pruritic, grouped, monomorphic, domeshaped vesicles with background swelling and erythema typically on the head, neck, and trunk. Vesicles then progress to punched-out erosions with overlying hemorrhagic crusting that can coalesce to form large denuded areas susceptible to superinfection with bacteria.9 Other accompanying symptoms include high fever, chills, malaise, and lymphadenopathy. Associated inflammation, classically described as erythema, may be difficult to discern in patients with darker skin and appears as hyperpigmentation; therefore, identification of clusters of monomorphic vesicles in areas of pre-existing dermatitis is particularly important for clinical diagnosis in people with darker skin types.
Various tests are available to confirm diagnosis in ambiguous cases. Bedside Tzanck smears can be performed rapidly and are considered positive if characteristic multinucleated giant cells are noted; however, they do not differentiate between the various herpesviruses. Direct fluorescent antibody testing of scraped lesions and viral cultures of swabbed vesicular fluid are equally effective in distinguishing between herpes simplex virus type 1, herpes simplex virus type 2, and VZV; PCR confirms the diagnosis with high specificity and sensitivity.10
In our patient, the initial differential diagnosis included EH, acute generalized exanthematous pustulosis, allergic contact dermatitis, and Orthopoxvirus infection. The positive Tzanck smear reduced the likelihood of a nonviral etiology. Additionally, worsening of the rash despite discontinuation of medications and utilization of topical steroids argued against acute generalized exanthematous pustulosis and allergic contact dermatitis. The laboratory findings reduced the likelihood of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and PCR findings ultimately ruled out Orthopoxvirus infections. Additional differential diagnoses for EH include dermatitis herpetiformis; primary VZV infection; hand, foot, and mouth disease; disseminated zoster infection; disseminated molluscum contagiosum; and eczema coxsackium.
Complications of EH include scarring; herpetic keratitis due to corneal infection, which if left untreated can progress to blindness; and rarely death due to multiorgan failure or septicemia.11 The traditional smallpox vaccine (ACAM2000) is contraindicated in patients with AD and EH, even when AD is in remission. These patients should avoid contact with recently vaccinated individuals.12 An alternative vaccine—Jynneos (Bavarian Nordic)—is available for these patients and their family members.13 Clinicians should be aware of this guideline, especially given the recent mpox (monkeypox) outbreaks.
Mild cases of EH are more common, may sometimes go unnoticed, and self-resolve in healthy patients. Severe cases may require systemic antiviral therapy. Acyclovir and its prodrug valacyclovir are standard treatments for EH. Alternatively, foscarnet or cidofovir can be used in the treatment of acyclovir-resistant thymidine kinase– deficient herpes simplex virus and other acyclovirresistant cases.14 Any secondary bacterial superinfections, usually due to staphylococcal or streptococcal bacteria, should be treated with antibiotics. A thorough ophthalmologic evaluation should be performed for patients with periocular involvement of EH. Empiric treatment should be started immediately, given a relative low toxicity of systemic antiviral therapy and high morbidity and mortality associated with untreated widespread EH.
It is important to maintain a high index of clinical suspicion for EH, especially in patients with pre-existing conditions such as AD who present with systemic symptoms and facial vesicles, pustules, or erosions to ensure prompt diagnosis and appropriate treatment.
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Tayabali K, Pothiwalla H, Lowitt M. Eczema herpeticum in Darier’s disease: a topical storm. J Community Hosp Intern Med Perspect. 2019;9:347. doi:10.1080/20009666.2019.1650590
- Cavalié M, Giacchero D, Cardot-Leccia N, et al. Kaposi’s varicelliform eruption in a patient with pityriasis rubra pilaris (pityriasis rubra pilaris herpeticum). J Eur Acad Dermatol Venereol. 2013;27:1585-1586. doi:10.1111/JDV.12120
- Lee GH, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey disease. Ann Dermatol. 2009;21:311-314. doi:10.5021/ad.2009.21.3.311
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum— a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients. J Eur Acad Dermatol Venereol. 2020;34:1074-1079. doi:10.1111/JDV.16090
- Kawakami Y, Ando T, Lee J-R, et al. Defective natural killer cell activity in a mouse model of eczema herpeticum. J Allergy Clin Immunol. 2017;139:997-1006.e10. doi:10.1016/j.jaci.2016.06.034
- Beck L, Latchney L, Zaccaro D, et al. Biomarkers of disease severity and Th2 polarity are predictors of risk for eczema herpeticum. J Allergy Clin Immunol. 2008;121:S37-S37. doi:10.1016/j.jaci.2007.12.152
- Kim M, Jung M, Hong SP, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol. 2010; 19:501-510. doi:10.1111/J.1600-0625.2009.00941.X
- Karray M, Kwan E, Souissi A. Kaposi varicelliform eruption. StatPearls [Internet]. StatPearls Publishing; 2023. https://www.ncbi.nlm.nih.gov/books/NBK482432/
- Dominguez SR, Pretty K, Hengartner R, et al. Comparison of herpes simplex virus PCR with culture for virus detection in multisource surface swab specimens from neonates [published online September 25, 2018]. J Clin Microbiol. doi:10.1128/JCM.00632-18
- Feye F, De Halleux C, Gillet JB, et al. Exacerbation of atopic dermatitis in the emergency department. Eur J Emerg Med. 2004;11:49-52. doi:10.1097/00063110-200412000-00014
- Casey C, Vellozzi C, Mootrey GT, et al; Vaccinia Case Definition Development Working Group; Advisory Committee on Immunization Practices-Armed Forces Epidemiological Board Smallpox Vaccine Safety Working Group. Surveillance guidelines for smallpox vaccine (vaccinia) adverse reactions. MMWR Recomm Rep. 2006;55:1-16.
- Rao AK, Petersen BW, Whitehill F, et al. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for preexposure vaccination of persons at risk for occupational exposure to orthopoxviruses: recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585 /MMWR.MM7122E1
- Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459. doi:10.1128/AAC.00615-10
The Diagnosis: Eczema Herpeticum
The patient’s condition with worsening facial edema and notable pain prompted a bedside Tzanck smear using a sample from the base of a deroofed forehead vesicle. In addition, a swab of a deroofed lesion was sent for herpes simplex virus and varicella-zoster virus (VZV) polymerase chain reaction (PCR) testing. The Tzanck smear demonstrated ballooning multinucleated syncytial giant cells and eosinophilic inclusion bodies (Figure), which are characteristic of certain herpesviruses including herpes simplex virus and VZV. He was started on intravenous acyclovir while PCR results were pending; the PCR test later confirmed positivity for herpes simplex virus type 1. Treatment was transitioned to oral valacyclovir once the lesions started crusting over. Notable healing and epithelialization of the lesions occurred during his hospital stay, and he was discharged home 5 days after starting treatment. He was counseled on autoinoculation, advised that he was considered infectious until all lesions had crusted over, and encouraged to employ frequent handwashing. Complete resolution of eczema herpeticum (EH) was noted at 3-week follow-up.
Eczema herpeticum (also known as Kaposi varicelliform eruption) is a potentially life-threatening disseminated cutaneous infection caused by herpes simplex virus types 1 and 2 in patients with pre-existing skin disease.1 It typically presents as a complication of atopic dermatitis (AD) but also has been identified as a rare complication in other conditions that disrupt the normal skin barrier, including mycosis fungoides, pemphigus foliaceus, pemphigus vulgaris, Darier disease, pityriasis rubra pilaris, contact dermatitis, and seborrheic dermatitis.1-4
The pathogenesis of EH is multifactorial. Disruption of the stratum corneum; impaired natural killer cell function; early-onset, untreated, or severe AD; disrupted skin microbiota with skewed colonization by Staphylococcus aureus; immunosuppressive AD therapies such as calcineurin inhibitors; eosinophilia; and helper T cell (TH2) cytokine predominance all have been suggested to play a role in the development of EH.5-8
As seen in our patient, EH presents with a sudden eruption of painful or pruritic, grouped, monomorphic, domeshaped vesicles with background swelling and erythema typically on the head, neck, and trunk. Vesicles then progress to punched-out erosions with overlying hemorrhagic crusting that can coalesce to form large denuded areas susceptible to superinfection with bacteria.9 Other accompanying symptoms include high fever, chills, malaise, and lymphadenopathy. Associated inflammation, classically described as erythema, may be difficult to discern in patients with darker skin and appears as hyperpigmentation; therefore, identification of clusters of monomorphic vesicles in areas of pre-existing dermatitis is particularly important for clinical diagnosis in people with darker skin types.
Various tests are available to confirm diagnosis in ambiguous cases. Bedside Tzanck smears can be performed rapidly and are considered positive if characteristic multinucleated giant cells are noted; however, they do not differentiate between the various herpesviruses. Direct fluorescent antibody testing of scraped lesions and viral cultures of swabbed vesicular fluid are equally effective in distinguishing between herpes simplex virus type 1, herpes simplex virus type 2, and VZV; PCR confirms the diagnosis with high specificity and sensitivity.10
In our patient, the initial differential diagnosis included EH, acute generalized exanthematous pustulosis, allergic contact dermatitis, and Orthopoxvirus infection. The positive Tzanck smear reduced the likelihood of a nonviral etiology. Additionally, worsening of the rash despite discontinuation of medications and utilization of topical steroids argued against acute generalized exanthematous pustulosis and allergic contact dermatitis. The laboratory findings reduced the likelihood of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and PCR findings ultimately ruled out Orthopoxvirus infections. Additional differential diagnoses for EH include dermatitis herpetiformis; primary VZV infection; hand, foot, and mouth disease; disseminated zoster infection; disseminated molluscum contagiosum; and eczema coxsackium.
Complications of EH include scarring; herpetic keratitis due to corneal infection, which if left untreated can progress to blindness; and rarely death due to multiorgan failure or septicemia.11 The traditional smallpox vaccine (ACAM2000) is contraindicated in patients with AD and EH, even when AD is in remission. These patients should avoid contact with recently vaccinated individuals.12 An alternative vaccine—Jynneos (Bavarian Nordic)—is available for these patients and their family members.13 Clinicians should be aware of this guideline, especially given the recent mpox (monkeypox) outbreaks.
Mild cases of EH are more common, may sometimes go unnoticed, and self-resolve in healthy patients. Severe cases may require systemic antiviral therapy. Acyclovir and its prodrug valacyclovir are standard treatments for EH. Alternatively, foscarnet or cidofovir can be used in the treatment of acyclovir-resistant thymidine kinase– deficient herpes simplex virus and other acyclovirresistant cases.14 Any secondary bacterial superinfections, usually due to staphylococcal or streptococcal bacteria, should be treated with antibiotics. A thorough ophthalmologic evaluation should be performed for patients with periocular involvement of EH. Empiric treatment should be started immediately, given a relative low toxicity of systemic antiviral therapy and high morbidity and mortality associated with untreated widespread EH.
It is important to maintain a high index of clinical suspicion for EH, especially in patients with pre-existing conditions such as AD who present with systemic symptoms and facial vesicles, pustules, or erosions to ensure prompt diagnosis and appropriate treatment.
The Diagnosis: Eczema Herpeticum
The patient’s condition with worsening facial edema and notable pain prompted a bedside Tzanck smear using a sample from the base of a deroofed forehead vesicle. In addition, a swab of a deroofed lesion was sent for herpes simplex virus and varicella-zoster virus (VZV) polymerase chain reaction (PCR) testing. The Tzanck smear demonstrated ballooning multinucleated syncytial giant cells and eosinophilic inclusion bodies (Figure), which are characteristic of certain herpesviruses including herpes simplex virus and VZV. He was started on intravenous acyclovir while PCR results were pending; the PCR test later confirmed positivity for herpes simplex virus type 1. Treatment was transitioned to oral valacyclovir once the lesions started crusting over. Notable healing and epithelialization of the lesions occurred during his hospital stay, and he was discharged home 5 days after starting treatment. He was counseled on autoinoculation, advised that he was considered infectious until all lesions had crusted over, and encouraged to employ frequent handwashing. Complete resolution of eczema herpeticum (EH) was noted at 3-week follow-up.
Eczema herpeticum (also known as Kaposi varicelliform eruption) is a potentially life-threatening disseminated cutaneous infection caused by herpes simplex virus types 1 and 2 in patients with pre-existing skin disease.1 It typically presents as a complication of atopic dermatitis (AD) but also has been identified as a rare complication in other conditions that disrupt the normal skin barrier, including mycosis fungoides, pemphigus foliaceus, pemphigus vulgaris, Darier disease, pityriasis rubra pilaris, contact dermatitis, and seborrheic dermatitis.1-4
The pathogenesis of EH is multifactorial. Disruption of the stratum corneum; impaired natural killer cell function; early-onset, untreated, or severe AD; disrupted skin microbiota with skewed colonization by Staphylococcus aureus; immunosuppressive AD therapies such as calcineurin inhibitors; eosinophilia; and helper T cell (TH2) cytokine predominance all have been suggested to play a role in the development of EH.5-8
As seen in our patient, EH presents with a sudden eruption of painful or pruritic, grouped, monomorphic, domeshaped vesicles with background swelling and erythema typically on the head, neck, and trunk. Vesicles then progress to punched-out erosions with overlying hemorrhagic crusting that can coalesce to form large denuded areas susceptible to superinfection with bacteria.9 Other accompanying symptoms include high fever, chills, malaise, and lymphadenopathy. Associated inflammation, classically described as erythema, may be difficult to discern in patients with darker skin and appears as hyperpigmentation; therefore, identification of clusters of monomorphic vesicles in areas of pre-existing dermatitis is particularly important for clinical diagnosis in people with darker skin types.
Various tests are available to confirm diagnosis in ambiguous cases. Bedside Tzanck smears can be performed rapidly and are considered positive if characteristic multinucleated giant cells are noted; however, they do not differentiate between the various herpesviruses. Direct fluorescent antibody testing of scraped lesions and viral cultures of swabbed vesicular fluid are equally effective in distinguishing between herpes simplex virus type 1, herpes simplex virus type 2, and VZV; PCR confirms the diagnosis with high specificity and sensitivity.10
In our patient, the initial differential diagnosis included EH, acute generalized exanthematous pustulosis, allergic contact dermatitis, and Orthopoxvirus infection. The positive Tzanck smear reduced the likelihood of a nonviral etiology. Additionally, worsening of the rash despite discontinuation of medications and utilization of topical steroids argued against acute generalized exanthematous pustulosis and allergic contact dermatitis. The laboratory findings reduced the likelihood of drug reaction with eosinophilia and systemic symptoms (DRESS) syndrome, and PCR findings ultimately ruled out Orthopoxvirus infections. Additional differential diagnoses for EH include dermatitis herpetiformis; primary VZV infection; hand, foot, and mouth disease; disseminated zoster infection; disseminated molluscum contagiosum; and eczema coxsackium.
Complications of EH include scarring; herpetic keratitis due to corneal infection, which if left untreated can progress to blindness; and rarely death due to multiorgan failure or septicemia.11 The traditional smallpox vaccine (ACAM2000) is contraindicated in patients with AD and EH, even when AD is in remission. These patients should avoid contact with recently vaccinated individuals.12 An alternative vaccine—Jynneos (Bavarian Nordic)—is available for these patients and their family members.13 Clinicians should be aware of this guideline, especially given the recent mpox (monkeypox) outbreaks.
Mild cases of EH are more common, may sometimes go unnoticed, and self-resolve in healthy patients. Severe cases may require systemic antiviral therapy. Acyclovir and its prodrug valacyclovir are standard treatments for EH. Alternatively, foscarnet or cidofovir can be used in the treatment of acyclovir-resistant thymidine kinase– deficient herpes simplex virus and other acyclovirresistant cases.14 Any secondary bacterial superinfections, usually due to staphylococcal or streptococcal bacteria, should be treated with antibiotics. A thorough ophthalmologic evaluation should be performed for patients with periocular involvement of EH. Empiric treatment should be started immediately, given a relative low toxicity of systemic antiviral therapy and high morbidity and mortality associated with untreated widespread EH.
It is important to maintain a high index of clinical suspicion for EH, especially in patients with pre-existing conditions such as AD who present with systemic symptoms and facial vesicles, pustules, or erosions to ensure prompt diagnosis and appropriate treatment.
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Tayabali K, Pothiwalla H, Lowitt M. Eczema herpeticum in Darier’s disease: a topical storm. J Community Hosp Intern Med Perspect. 2019;9:347. doi:10.1080/20009666.2019.1650590
- Cavalié M, Giacchero D, Cardot-Leccia N, et al. Kaposi’s varicelliform eruption in a patient with pityriasis rubra pilaris (pityriasis rubra pilaris herpeticum). J Eur Acad Dermatol Venereol. 2013;27:1585-1586. doi:10.1111/JDV.12120
- Lee GH, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey disease. Ann Dermatol. 2009;21:311-314. doi:10.5021/ad.2009.21.3.311
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum— a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients. J Eur Acad Dermatol Venereol. 2020;34:1074-1079. doi:10.1111/JDV.16090
- Kawakami Y, Ando T, Lee J-R, et al. Defective natural killer cell activity in a mouse model of eczema herpeticum. J Allergy Clin Immunol. 2017;139:997-1006.e10. doi:10.1016/j.jaci.2016.06.034
- Beck L, Latchney L, Zaccaro D, et al. Biomarkers of disease severity and Th2 polarity are predictors of risk for eczema herpeticum. J Allergy Clin Immunol. 2008;121:S37-S37. doi:10.1016/j.jaci.2007.12.152
- Kim M, Jung M, Hong SP, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol. 2010; 19:501-510. doi:10.1111/J.1600-0625.2009.00941.X
- Karray M, Kwan E, Souissi A. Kaposi varicelliform eruption. StatPearls [Internet]. StatPearls Publishing; 2023. https://www.ncbi.nlm.nih.gov/books/NBK482432/
- Dominguez SR, Pretty K, Hengartner R, et al. Comparison of herpes simplex virus PCR with culture for virus detection in multisource surface swab specimens from neonates [published online September 25, 2018]. J Clin Microbiol. doi:10.1128/JCM.00632-18
- Feye F, De Halleux C, Gillet JB, et al. Exacerbation of atopic dermatitis in the emergency department. Eur J Emerg Med. 2004;11:49-52. doi:10.1097/00063110-200412000-00014
- Casey C, Vellozzi C, Mootrey GT, et al; Vaccinia Case Definition Development Working Group; Advisory Committee on Immunization Practices-Armed Forces Epidemiological Board Smallpox Vaccine Safety Working Group. Surveillance guidelines for smallpox vaccine (vaccinia) adverse reactions. MMWR Recomm Rep. 2006;55:1-16.
- Rao AK, Petersen BW, Whitehill F, et al. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for preexposure vaccination of persons at risk for occupational exposure to orthopoxviruses: recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585 /MMWR.MM7122E1
- Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459. doi:10.1128/AAC.00615-10
- Baaniya B, Agrawal S. Kaposi varicelliform eruption in a patient with pemphigus vulgaris: a case report and review of the literature. Case Rep Dermatol Med. 2020;2020:6695342. doi:10.1155/2020/6695342
- Tayabali K, Pothiwalla H, Lowitt M. Eczema herpeticum in Darier’s disease: a topical storm. J Community Hosp Intern Med Perspect. 2019;9:347. doi:10.1080/20009666.2019.1650590
- Cavalié M, Giacchero D, Cardot-Leccia N, et al. Kaposi’s varicelliform eruption in a patient with pityriasis rubra pilaris (pityriasis rubra pilaris herpeticum). J Eur Acad Dermatol Venereol. 2013;27:1585-1586. doi:10.1111/JDV.12120
- Lee GH, Kim YM, Lee SY, et al. A case of eczema herpeticum with Hailey-Hailey disease. Ann Dermatol. 2009;21:311-314. doi:10.5021/ad.2009.21.3.311
- Seegräber M, Worm M, Werfel T, et al. Recurrent eczema herpeticum— a retrospective European multicenter study evaluating the clinical characteristics of eczema herpeticum cases in atopic dermatitis patients. J Eur Acad Dermatol Venereol. 2020;34:1074-1079. doi:10.1111/JDV.16090
- Kawakami Y, Ando T, Lee J-R, et al. Defective natural killer cell activity in a mouse model of eczema herpeticum. J Allergy Clin Immunol. 2017;139:997-1006.e10. doi:10.1016/j.jaci.2016.06.034
- Beck L, Latchney L, Zaccaro D, et al. Biomarkers of disease severity and Th2 polarity are predictors of risk for eczema herpeticum. J Allergy Clin Immunol. 2008;121:S37-S37. doi:10.1016/j.jaci.2007.12.152
- Kim M, Jung M, Hong SP, et al. Topical calcineurin inhibitors compromise stratum corneum integrity, epidermal permeability and antimicrobial barrier function. Exp Dermatol. 2010; 19:501-510. doi:10.1111/J.1600-0625.2009.00941.X
- Karray M, Kwan E, Souissi A. Kaposi varicelliform eruption. StatPearls [Internet]. StatPearls Publishing; 2023. https://www.ncbi.nlm.nih.gov/books/NBK482432/
- Dominguez SR, Pretty K, Hengartner R, et al. Comparison of herpes simplex virus PCR with culture for virus detection in multisource surface swab specimens from neonates [published online September 25, 2018]. J Clin Microbiol. doi:10.1128/JCM.00632-18
- Feye F, De Halleux C, Gillet JB, et al. Exacerbation of atopic dermatitis in the emergency department. Eur J Emerg Med. 2004;11:49-52. doi:10.1097/00063110-200412000-00014
- Casey C, Vellozzi C, Mootrey GT, et al; Vaccinia Case Definition Development Working Group; Advisory Committee on Immunization Practices-Armed Forces Epidemiological Board Smallpox Vaccine Safety Working Group. Surveillance guidelines for smallpox vaccine (vaccinia) adverse reactions. MMWR Recomm Rep. 2006;55:1-16.
- Rao AK, Petersen BW, Whitehill F, et al. Use of JYNNEOS (Smallpox and Monkeypox Vaccine, Live, Nonreplicating) for preexposure vaccination of persons at risk for occupational exposure to orthopoxviruses: recommendations of the Advisory Committee on Immunization Practices—United States, 2022. MMWR Morb Mortal Wkly Rep. 2022;71:734-742. doi:10.15585 /MMWR.MM7122E1
- Piret J, Boivin G. Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob Agents Chemother. 2011;55:459. doi:10.1128/AAC.00615-10
A 52-year-old man developed a sudden eruption of small pustules on background erythema and edema covering the forehead, nasal bridge, periorbital region, cheeks, and perioral region on day 3 of hospitalization in the intensive care unit for management of septic shock secondary to a complicated urinary tract infection. He had a medical history of benign prostatic hyperplasia, sarcoidosis, and atopic dermatitis. He initially presented to the emergency department with fever, chills, and dysuria of 2 days’ duration. Because he received ceftriaxone, vancomycin, ciprofloxacin, and tamsulosin while hospitalized for the infection, the primary medical team suspected a drug reaction and empirically started applying hydrocortisone cream 2.5%. The rash continued to spread over the ensuing day, prompting a dermatology consultation to rule out a drug eruption and to help guide further management. The patient was in substantial distress and pain. Physical examination revealed numerous discrete and confluent monomorphic pustules on background erythema with faint collarettes of scale covering most of the face. Substantial periorbital and facial edema forced the eyes closed. There was no mucous membrane involvement. A review of systems was negative for dyspnea and dysphagia, and the rash was not present elsewhere on the body. Ophthalmologic evaluation revealed no ocular involvement or vision changes. Laboratory studies demonstrated neutrophilia (17.27×109 cells/L [reference range, 2.0–6.9×109 cells/L]). The eosinophil count, blood urea nitrogen/creatinine, and liver function tests were within reference range.


U.S. study finds unexpectedly high prevalence of myasthenia gravis
PHOENIX – The prevalence is higher than what has been seen in other studies, which could represent a true difference in prevalence, or reflect limitations of the database.
Worldwide estimates suggest that myasthenia gravis affects 700,000 people globally, with incidence rates ranging between 6.3 and 29 per 1,000,000 person-years in Europe and a prevalence between 111.7 and 361 per 1,000,000. Data from Australia, Taiwan, and South Korea also show evidence of increased prevalence in recent years.
However, there is little data about the prevalence of myasthenia gravis in the United States, or about differences between racial groups, according to Bhaskar Roy, MBBS, who presented the study at the 2023 annual meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). He noted that most studies are outdated, and the most recent study focused on ocular myasthenia gravis.
True incidence or artifact?
The finding is surprising and may be an artifact of the immature nature of the All of Us database, according to Srikanth Muppidi, MD, who asked about the limitation during the Q&A session following the talk. “The incidence of 0.13 is definitely higher than what we would think would be the true incidence of myasthenia gravis from [clinical experience]. It’s possible that our understanding of true incidence is wrong and this is the actual incidence. What I would like them to do, and I think they’re trying to do, is to look at this finding [and compare it with] other more mature databases and other regional databases. One of the current challenges of All of Us is that our patients are basically being recruited from some parts of the country, and the middle of the country has hardly any presence in the database, so it becomes really challenging to understand it,” Dr. Muppidi said in an interview.
However, Dr. Muppidi, who is a clinical professor of neurology at Stanford (Calif.) Medicine, noted that the All of Us database is still growing. When it has recruited more patients with a diverse population, “it [will be a] valuable source for rare diseases to try to understand true incidence of those diseases,” he said.
Understanding the true prevalence
Dr. Roy recognized the geographic limitations of the database. “Some states, particularly Massachusetts, New York, and California, had a lot of patients in the database, where there were no patients from many states,” said Dr. Roy, associate professor of neurology at Yale University, New Haven, Conn.
He said that the group is working with other databases, including UK Biobank. “The goal is to incorporate all of these databases together [to determine the true incidence],” said Dr. Roy.
It’s critical to understand the true prevalence of myasthenia gravis since new therapies are in development and coming to market. “I worry that myasthenia gravis might be considered less common than it truly is, and that will limit growth of the field if the feeling is that there are not that many [myasthenia gravis patients] in the country,” said Dr. Muppidi.
The study included data from 369,297 adult patients, using Systematized Nomenclature of Medicine (SNOMED) and International Classification of Diseases (ICD) codes to identify patients with myasthenia gravis. There were 479 cases of myasthenia gravis, for a prevalence of 0.13 (95% confidence interval [CI], 0.12-0.14). Of myasthenia gravis patients, 65% were female and the mean age was 64 years. The prevalence of myasthenia gravis in White individuals was 0.16 (95% CI, 0.15-0.18), of which 63% were female, and the mean age was 66 years. The prevalence among Black individuals was 0.078 (95% CI, 0.060-0.10), with 77% of the population female and a mean age of 58 years. The prevalence in Hispanics was 0.091 (95% CI, 0.070-0.12), with 80% female and a mean age of 58 years. Among Asians, the prevalence was 0.056 (95% CI, 0.025-0.12) and 57% were female, with a mean age of 58 years.
The researchers also looked at the EXPLORE-MG database drawn from Yale (n = 3,269,000), which showed a much lower overall myasthenia gravis prevalence of 0.019 (95% CI, 0.017-0.020), a female proportion of 46.8%, and a mean age of 56.6 years. Notably, EXPLORE-MG had a lower proportion of women and a younger population than All of Us.
The researchers compared data from All of Us with other databases for other conditions. The prevalence of ALS was the same as in other conditions, while diabetic neuropathy was significantly lower (2.7 versus 28.5-50 among diabetic patients) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) was higher (0.084 versus 0.028).
Dr. Muppidi has been on advisory boards for Alexion, Argenx, UBC, and Immunovant. Dr. Roy has consulted for Alexion, Takeda Pharmaceuticals, and Argenx and owns stock in Cabaletta Bio. He has received research support from Takeda, Abcuro, and Argenx.
PHOENIX – The prevalence is higher than what has been seen in other studies, which could represent a true difference in prevalence, or reflect limitations of the database.
Worldwide estimates suggest that myasthenia gravis affects 700,000 people globally, with incidence rates ranging between 6.3 and 29 per 1,000,000 person-years in Europe and a prevalence between 111.7 and 361 per 1,000,000. Data from Australia, Taiwan, and South Korea also show evidence of increased prevalence in recent years.
However, there is little data about the prevalence of myasthenia gravis in the United States, or about differences between racial groups, according to Bhaskar Roy, MBBS, who presented the study at the 2023 annual meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). He noted that most studies are outdated, and the most recent study focused on ocular myasthenia gravis.
True incidence or artifact?
The finding is surprising and may be an artifact of the immature nature of the All of Us database, according to Srikanth Muppidi, MD, who asked about the limitation during the Q&A session following the talk. “The incidence of 0.13 is definitely higher than what we would think would be the true incidence of myasthenia gravis from [clinical experience]. It’s possible that our understanding of true incidence is wrong and this is the actual incidence. What I would like them to do, and I think they’re trying to do, is to look at this finding [and compare it with] other more mature databases and other regional databases. One of the current challenges of All of Us is that our patients are basically being recruited from some parts of the country, and the middle of the country has hardly any presence in the database, so it becomes really challenging to understand it,” Dr. Muppidi said in an interview.
However, Dr. Muppidi, who is a clinical professor of neurology at Stanford (Calif.) Medicine, noted that the All of Us database is still growing. When it has recruited more patients with a diverse population, “it [will be a] valuable source for rare diseases to try to understand true incidence of those diseases,” he said.
Understanding the true prevalence
Dr. Roy recognized the geographic limitations of the database. “Some states, particularly Massachusetts, New York, and California, had a lot of patients in the database, where there were no patients from many states,” said Dr. Roy, associate professor of neurology at Yale University, New Haven, Conn.
He said that the group is working with other databases, including UK Biobank. “The goal is to incorporate all of these databases together [to determine the true incidence],” said Dr. Roy.
It’s critical to understand the true prevalence of myasthenia gravis since new therapies are in development and coming to market. “I worry that myasthenia gravis might be considered less common than it truly is, and that will limit growth of the field if the feeling is that there are not that many [myasthenia gravis patients] in the country,” said Dr. Muppidi.
The study included data from 369,297 adult patients, using Systematized Nomenclature of Medicine (SNOMED) and International Classification of Diseases (ICD) codes to identify patients with myasthenia gravis. There were 479 cases of myasthenia gravis, for a prevalence of 0.13 (95% confidence interval [CI], 0.12-0.14). Of myasthenia gravis patients, 65% were female and the mean age was 64 years. The prevalence of myasthenia gravis in White individuals was 0.16 (95% CI, 0.15-0.18), of which 63% were female, and the mean age was 66 years. The prevalence among Black individuals was 0.078 (95% CI, 0.060-0.10), with 77% of the population female and a mean age of 58 years. The prevalence in Hispanics was 0.091 (95% CI, 0.070-0.12), with 80% female and a mean age of 58 years. Among Asians, the prevalence was 0.056 (95% CI, 0.025-0.12) and 57% were female, with a mean age of 58 years.
The researchers also looked at the EXPLORE-MG database drawn from Yale (n = 3,269,000), which showed a much lower overall myasthenia gravis prevalence of 0.019 (95% CI, 0.017-0.020), a female proportion of 46.8%, and a mean age of 56.6 years. Notably, EXPLORE-MG had a lower proportion of women and a younger population than All of Us.
The researchers compared data from All of Us with other databases for other conditions. The prevalence of ALS was the same as in other conditions, while diabetic neuropathy was significantly lower (2.7 versus 28.5-50 among diabetic patients) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) was higher (0.084 versus 0.028).
Dr. Muppidi has been on advisory boards for Alexion, Argenx, UBC, and Immunovant. Dr. Roy has consulted for Alexion, Takeda Pharmaceuticals, and Argenx and owns stock in Cabaletta Bio. He has received research support from Takeda, Abcuro, and Argenx.
PHOENIX – The prevalence is higher than what has been seen in other studies, which could represent a true difference in prevalence, or reflect limitations of the database.
Worldwide estimates suggest that myasthenia gravis affects 700,000 people globally, with incidence rates ranging between 6.3 and 29 per 1,000,000 person-years in Europe and a prevalence between 111.7 and 361 per 1,000,000. Data from Australia, Taiwan, and South Korea also show evidence of increased prevalence in recent years.
However, there is little data about the prevalence of myasthenia gravis in the United States, or about differences between racial groups, according to Bhaskar Roy, MBBS, who presented the study at the 2023 annual meeting of the American Association of Neuromuscular & Electrodiagnostic Medicine (AANEM). He noted that most studies are outdated, and the most recent study focused on ocular myasthenia gravis.
True incidence or artifact?
The finding is surprising and may be an artifact of the immature nature of the All of Us database, according to Srikanth Muppidi, MD, who asked about the limitation during the Q&A session following the talk. “The incidence of 0.13 is definitely higher than what we would think would be the true incidence of myasthenia gravis from [clinical experience]. It’s possible that our understanding of true incidence is wrong and this is the actual incidence. What I would like them to do, and I think they’re trying to do, is to look at this finding [and compare it with] other more mature databases and other regional databases. One of the current challenges of All of Us is that our patients are basically being recruited from some parts of the country, and the middle of the country has hardly any presence in the database, so it becomes really challenging to understand it,” Dr. Muppidi said in an interview.
However, Dr. Muppidi, who is a clinical professor of neurology at Stanford (Calif.) Medicine, noted that the All of Us database is still growing. When it has recruited more patients with a diverse population, “it [will be a] valuable source for rare diseases to try to understand true incidence of those diseases,” he said.
Understanding the true prevalence
Dr. Roy recognized the geographic limitations of the database. “Some states, particularly Massachusetts, New York, and California, had a lot of patients in the database, where there were no patients from many states,” said Dr. Roy, associate professor of neurology at Yale University, New Haven, Conn.
He said that the group is working with other databases, including UK Biobank. “The goal is to incorporate all of these databases together [to determine the true incidence],” said Dr. Roy.
It’s critical to understand the true prevalence of myasthenia gravis since new therapies are in development and coming to market. “I worry that myasthenia gravis might be considered less common than it truly is, and that will limit growth of the field if the feeling is that there are not that many [myasthenia gravis patients] in the country,” said Dr. Muppidi.
The study included data from 369,297 adult patients, using Systematized Nomenclature of Medicine (SNOMED) and International Classification of Diseases (ICD) codes to identify patients with myasthenia gravis. There were 479 cases of myasthenia gravis, for a prevalence of 0.13 (95% confidence interval [CI], 0.12-0.14). Of myasthenia gravis patients, 65% were female and the mean age was 64 years. The prevalence of myasthenia gravis in White individuals was 0.16 (95% CI, 0.15-0.18), of which 63% were female, and the mean age was 66 years. The prevalence among Black individuals was 0.078 (95% CI, 0.060-0.10), with 77% of the population female and a mean age of 58 years. The prevalence in Hispanics was 0.091 (95% CI, 0.070-0.12), with 80% female and a mean age of 58 years. Among Asians, the prevalence was 0.056 (95% CI, 0.025-0.12) and 57% were female, with a mean age of 58 years.
The researchers also looked at the EXPLORE-MG database drawn from Yale (n = 3,269,000), which showed a much lower overall myasthenia gravis prevalence of 0.019 (95% CI, 0.017-0.020), a female proportion of 46.8%, and a mean age of 56.6 years. Notably, EXPLORE-MG had a lower proportion of women and a younger population than All of Us.
The researchers compared data from All of Us with other databases for other conditions. The prevalence of ALS was the same as in other conditions, while diabetic neuropathy was significantly lower (2.7 versus 28.5-50 among diabetic patients) and chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) was higher (0.084 versus 0.028).
Dr. Muppidi has been on advisory boards for Alexion, Argenx, UBC, and Immunovant. Dr. Roy has consulted for Alexion, Takeda Pharmaceuticals, and Argenx and owns stock in Cabaletta Bio. He has received research support from Takeda, Abcuro, and Argenx.
AT AANEM 2023
Topical ivermectin study sheds light on dysbiosis in rosacea
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
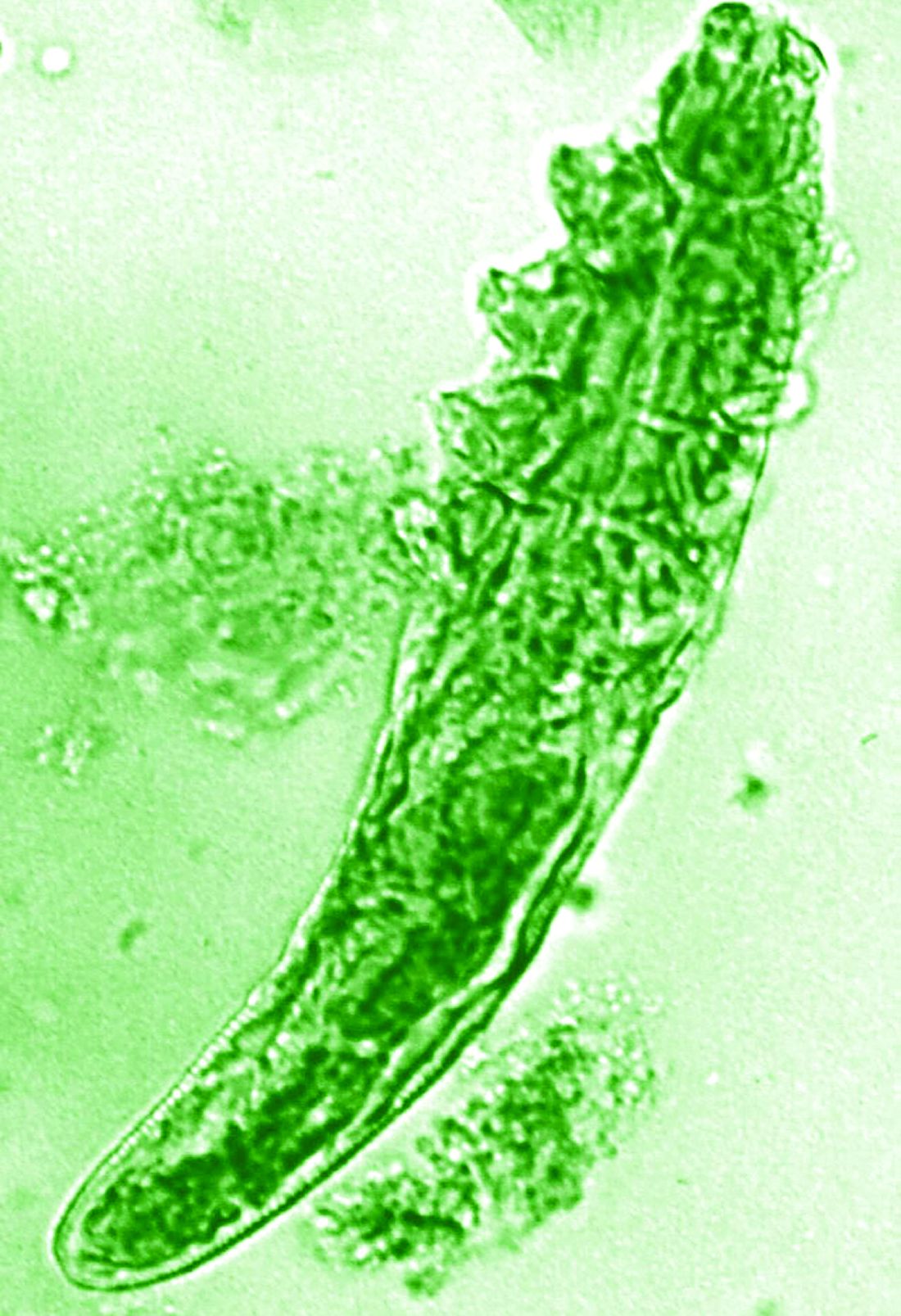
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
, according to a report presented at the recent European Academy of Dermatology and Venereology (EADV) 2023 Congress.
“This is the first hint that the host’s cutaneous microbiome plays a secondary role in the immunopathogenesis of rosacea,” said Bernard Homey, MD, director of the department of dermatology at University Hospital Düsseldorf in Germany.
“In rosacea, we are well aware of trigger factors such as stress, UV light, heat, cold, food, and alcohol,” he said. “We are also well aware that there is an increase in Demodex mites in the pilosebaceous unit.”
Research over the past decade has also started to look at the potential role of the skin microbiome in the disease process, but answers have remained “largely elusive,” Dr. Homey said.
Ivermectin helps, but how?
Ivermectin 1% cream (Soolantra) has been approved by the U.S. Food and Drug Administration since 2014 for the treatment of the inflammatory lesions that are characteristic of rosacea, but its mechanism of action is not clear.
Dr. Homey presented the results of a study of 61 patients designed to look at how ivermectin might be working in the treatment of people with rosacea and investigate if there was any relation to the skin microbiome and transcriptome of patients.
The trial included 41 individuals with papulopustular rosacea and 20 individuals who did not have rosacea. For all patients, surface skin biopsies were performed twice 30 days apart using cyanoacrylate glue; patients with rosacea were treated with topical ivermectin 1% between biopsies. Skin samples obtained at day 0 and day 30 were examined under the microscope, and Demodex counts (mites/cm2) of skin and RNA sequencing of the cutaneous microbiome were undertaken.
The mean age of the patients with rosacea was 54.9 years, and the mean Demodex counts before and after treatment were a respective 7.2 cm2 and 0.9 cm2.
Using the Investigator’s General Assessment to assess the severity of rosacea, Homey reported that 43.9% of patients with rosacea had a decrease in scores at day 30, indicating improvement.
In addition, topical ivermectin resulted in a marked or total decrease in Demodex mite density for 87.5% of patients (n = 24) who were identified as having the mites.
Skin microbiome changes seen
As a form of quality control, skin microbiome changes among the patients were compared with control patients using 16S rRNA sequencing.
“The taxa we find within the cutaneous niche of inflammatory lesions of rosacea patients are significantly different from healthy volunteers,” Dr. Homey said.
Cutibacterium species are predominant in healthy control persons but are not present when there is inflammation in patients with rosacea. Instead, staphylococcus species “take over the niche, similar to atopic dermatitis,” he noted.
Looking at how treatment with ivermectin influences the organisms, the decrease in C. acnes seen in patients with rosacea persisted despite treatment, and the abundance of Staphylococcus epidermidis, S. hominis, and S. capitis increased further. This suggests a possible protective or homeostatic role of C. acnes but a pathogenic role for staphylococci, explained Dr. Homey.
“Surprisingly, although inflammatory lesions decrease, patients get better, the cutaneous microbiome does not revert to homeostatic conditions during topical ivermectin treatment,” he observed.
There is, of course, variability among individuals.
Dr. Homey also reported that Snodgrassella alvi – a microorganism believed to reside in the gut of Demodex folliculorum mites – was found in the skin microbiome of patients with rosacea before but not after ivermectin treatment. This may mean that this microorganism could be partially triggering inflammation in rosacea patients.
Looking at the transcriptome of patients, Dr. Homey said that there was downregulation of distinct genes that might make for more favorable conditions for Demodex mites.
Moreover, insufficient upregulation of interleukin-17 pathways might be working together with barrier defects in the skin and metabolic changes to “pave the way” for colonization by S. epidermidis.
Pulling it together
Dr. Homey and associates conclude in their abstract that the findings “support that rosacea lesions are associated with dysbiosis.”
Although treatment with ivermectin did not normalize the skin’s microbiome, it was associated with a decrease in Demodex mite density and the reduction of microbes associated with Demodex.
Margarida Gonçalo, MD, PhD, professor of dermatology at the University of Coimbra in Portugal, who cochaired the late-breaking news session where the data were presented, asked whether healthy and affected skin in patients with rosacea had been compared, rather than comparing the skin of rosacea lesions with healthy control samples.
“No, we did not this, as this is methodologically a little bit more difficult,” Dr. Homey responded.
Also cochairing the session was Michel Gilliet, MD, chair of the department of dermatology at the University Hospital CHUV in Lausanne, Switzerland. He commented that these “data suggest that there’s an intimate link between Demodex and the skin microbiota and dysbiosis in in rosacea.”
Dr. Gilliet added: “You have a whole dysbiosis going on in rosacea, which is probably only dependent on these bacteria.”
It would be “very interesting,” as a “proof-of-concept” study, to look at whether depleting Demodex would also delete S. alvi, he suggested.
The study was funded by Galderma. Dr. Homey has acted as a consultant, speaker or investigator for many pharmaceutical companies including Galderma.
A version of this article first appeared on Medscape.com.
FROM EADV 2023
AGA provides leadership development for women in GI
As a part of AGA’s ongoing goal to support women in GI and advance gender equity in gastroenterology, we hosted nearly 60 women executives in GI for the inaugural Women’s Executive Leadership Conference held recently in Denver.
– academia, hospital systems, and private practice – for seminars on strengthening leadership skills and career progression, along with opportunities for networking and socializing with other women in the AGA Gastro Squad.

Women on the AGA governing board, including Kim E. Barrett, PhD, AGAF and Sheryl Pfeil, MD, AGAF, led sessions on how to best communicate as a leader and pathways to society leadership. In addition, other leaders such as Aja McCutchen, MD and Gyongyi Szabo, MD, PhD, shared their best practices for leadership and managing others.
Thank you to Fasiha Kanwal, MD, MSHS, and Aimee Lucas, MD, MS, cochairs of the AGA Women’s Executive Leadership Conference, for leading the weekend, and to everyone who contributed to a productive weekend. Stay tuned for more opportunities to engage with the AGA Gastro Squad.
As a part of AGA’s ongoing goal to support women in GI and advance gender equity in gastroenterology, we hosted nearly 60 women executives in GI for the inaugural Women’s Executive Leadership Conference held recently in Denver.
– academia, hospital systems, and private practice – for seminars on strengthening leadership skills and career progression, along with opportunities for networking and socializing with other women in the AGA Gastro Squad.
Women on the AGA governing board, including Kim E. Barrett, PhD, AGAF and Sheryl Pfeil, MD, AGAF, led sessions on how to best communicate as a leader and pathways to society leadership. In addition, other leaders such as Aja McCutchen, MD and Gyongyi Szabo, MD, PhD, shared their best practices for leadership and managing others.
Thank you to Fasiha Kanwal, MD, MSHS, and Aimee Lucas, MD, MS, cochairs of the AGA Women’s Executive Leadership Conference, for leading the weekend, and to everyone who contributed to a productive weekend. Stay tuned for more opportunities to engage with the AGA Gastro Squad.
As a part of AGA’s ongoing goal to support women in GI and advance gender equity in gastroenterology, we hosted nearly 60 women executives in GI for the inaugural Women’s Executive Leadership Conference held recently in Denver.
– academia, hospital systems, and private practice – for seminars on strengthening leadership skills and career progression, along with opportunities for networking and socializing with other women in the AGA Gastro Squad.
Women on the AGA governing board, including Kim E. Barrett, PhD, AGAF and Sheryl Pfeil, MD, AGAF, led sessions on how to best communicate as a leader and pathways to society leadership. In addition, other leaders such as Aja McCutchen, MD and Gyongyi Szabo, MD, PhD, shared their best practices for leadership and managing others.
Thank you to Fasiha Kanwal, MD, MSHS, and Aimee Lucas, MD, MS, cochairs of the AGA Women’s Executive Leadership Conference, for leading the weekend, and to everyone who contributed to a productive weekend. Stay tuned for more opportunities to engage with the AGA Gastro Squad.
A letter from Michael Camilleri, MD, DSc, AGAF, AGA Research Foundation chair and past AGA Institute president
And you understand the tremendous value of research to advance patient care.
We are in a time of major scientific breakthroughs; however, there is a growing gap in federal funding for research. Without gastroenterology and hepatology research, there would be no discoveries to develop new diagnostic and therapeutic approaches and to improve our understanding of the pathogenesis of digestive diseases.
The AGA Research Foundation funds promising GI investigators who don’t receive funding at crucial times in their early careers. The research of these talented individuals, while important to the field, could end prematurely if they are left unfunded. That’s something the fields of gastroenterology and hepatology can’t afford, and that’s why, as an AGA member, I’m making a year-end donation to the AGA Research Foundation. You can help fill the funding gap and protect the next generation of investigators by joining me in supporting the AGA Research Foundation through a personal year-end gift.
Gifts to the AGA Research Foundation this past year directly supported 71 investigators. Despite this success, close to 245 other promising research proposals were not funded.
We must continue to foster the careers of talented scientists and clinicians, and protect the GI research pipeline. A financial contribution to the AGA Research Foundation is the opportunity for you to help foster the careers of talented scientists and protect the GI research pipeline.
Help make a difference. You can make your tax-deductible donation online at www.gastro.org/donateonline; by phone at 301-222-4002; or, by mail:
AGA Research Foundation
4930 Del Ray Avenue
Bethesda, MD 20814
All gifts are tax-deductible to the fullest extent of U.S. law.
Thank you for your support and best wishes for a happy, healthy holiday season and prosperous New Year.
And you understand the tremendous value of research to advance patient care.
We are in a time of major scientific breakthroughs; however, there is a growing gap in federal funding for research. Without gastroenterology and hepatology research, there would be no discoveries to develop new diagnostic and therapeutic approaches and to improve our understanding of the pathogenesis of digestive diseases.
The AGA Research Foundation funds promising GI investigators who don’t receive funding at crucial times in their early careers. The research of these talented individuals, while important to the field, could end prematurely if they are left unfunded. That’s something the fields of gastroenterology and hepatology can’t afford, and that’s why, as an AGA member, I’m making a year-end donation to the AGA Research Foundation. You can help fill the funding gap and protect the next generation of investigators by joining me in supporting the AGA Research Foundation through a personal year-end gift.
Gifts to the AGA Research Foundation this past year directly supported 71 investigators. Despite this success, close to 245 other promising research proposals were not funded.
We must continue to foster the careers of talented scientists and clinicians, and protect the GI research pipeline. A financial contribution to the AGA Research Foundation is the opportunity for you to help foster the careers of talented scientists and protect the GI research pipeline.
Help make a difference. You can make your tax-deductible donation online at www.gastro.org/donateonline; by phone at 301-222-4002; or, by mail:
AGA Research Foundation
4930 Del Ray Avenue
Bethesda, MD 20814
All gifts are tax-deductible to the fullest extent of U.S. law.
Thank you for your support and best wishes for a happy, healthy holiday season and prosperous New Year.
And you understand the tremendous value of research to advance patient care.
We are in a time of major scientific breakthroughs; however, there is a growing gap in federal funding for research. Without gastroenterology and hepatology research, there would be no discoveries to develop new diagnostic and therapeutic approaches and to improve our understanding of the pathogenesis of digestive diseases.
The AGA Research Foundation funds promising GI investigators who don’t receive funding at crucial times in their early careers. The research of these talented individuals, while important to the field, could end prematurely if they are left unfunded. That’s something the fields of gastroenterology and hepatology can’t afford, and that’s why, as an AGA member, I’m making a year-end donation to the AGA Research Foundation. You can help fill the funding gap and protect the next generation of investigators by joining me in supporting the AGA Research Foundation through a personal year-end gift.
Gifts to the AGA Research Foundation this past year directly supported 71 investigators. Despite this success, close to 245 other promising research proposals were not funded.
We must continue to foster the careers of talented scientists and clinicians, and protect the GI research pipeline. A financial contribution to the AGA Research Foundation is the opportunity for you to help foster the careers of talented scientists and protect the GI research pipeline.
Help make a difference. You can make your tax-deductible donation online at www.gastro.org/donateonline; by phone at 301-222-4002; or, by mail:
AGA Research Foundation
4930 Del Ray Avenue
Bethesda, MD 20814
All gifts are tax-deductible to the fullest extent of U.S. law.
Thank you for your support and best wishes for a happy, healthy holiday season and prosperous New Year.
Papules on lip

Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120
Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
Pathology showed noncaseating granulomas consistent with cutaneous sarcoidosis. Based on these biopsy findings, a chest x-ray was ordered, and it confirmed a pulmonary sarcoid. A multidisciplinary work-up (including cardiac evaluation, continued rheumatologic care, and evaluation by Hematology) addressed this new finding.
Sarcoidosis is a multisystem inflammatory disorder characterized by the development of granulomas that can arise in any organ, but frequently involve the skin and lungs. Patients with cutaneous disease develop smooth skin lesions, including flesh-colored to pink or brown papules on the face. Genetic and environmental factors are both thought to contribute to the disease.
Race is a significant factor in the development of disease. Hispanic and Asian patients are significantly less likely to develop the disease compared to White or Black patients. In the Black Women’s Health Study, incidence in Black women was 71 per 100,000.1 Women are more likely to be affected than men.1
Many patients with sarcoidosis have a mild course, but for others the disease may progress on the skin or include pulmonary, renal, neurologic, or cardiac disease. Sometimes sarcoidosis is fatal. Recurrence can occur at any point later in life. Race influences disease severity as well as incidence, with hospitalization being 9 times as likely in Black patients compared with White patients.2 One recent study puts sarcoidosis mortality rates for Black women at 10 per million compared with 3 per million in Black men, and 1 per million in White women or men.3
Patients with disease limited to the skin may be treated with topical steroids such as clobetasol 0.05% cream or ointment or intralesional triamcinolone 10 mg/mL injected into affected lesions every 2 to 4 weeks. With pulmonary or other systemic disease, treatment may include various disease-modifying agents including prednisone, methotrexate, hydroxychloroquine, and TNF-alpha inhibitors. Because of the long-term adverse effects of systemic steroids, these agents are reserved for instances when pulmonary function is significantly impacted.
This patient had a reassuring cardiac and hematology work-up. Her pulmonary function was impacted sufficiently enough that her pulmonologist added a course of prednisone 10 mg daily tapered over 6 weeks. She had been on hydroxychloroquine 200 mg twice daily prior to the diagnosis of sarcoidosis for presumed mixed connective tissue disease and was continued on it for sarcoidosis after completing the prednisone taper. With these treatments, her facial lesions cleared and her breathing symptoms and fatigue improved. She remains under surveillance with a multidisciplinary team.
Photos and text for Photo Rounds Friday courtesy of Jonathan Karnes, MD (copyright retained). Dr. Karnes is the medical director of MDFMR Dermatology Services, Augusta, ME.
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120
1. Cozier Y, Berman J, Palmer J, et al. Sarcoidosis in black women in the United States: data from the Black Women's Health Study. Chest. 2011;139:144-150. doi: 10.1378/chest.10-0413
2. Foreman MG, Mannino DM, Kamugisha L, et al. Hospitalization for patients with sarcoidosis: 1979-2000. Sarcoidosis Vasc Diffuse Lung Dis. 2006;23:124-129.
3. Mirsaeidi M, Machado R, Schraufnagel D, et al. Racial difference in sarcoidosis mortality in the United States. Chest. 2015; 147: 438-449. doi: 10.1378/chest.14-1120

Low vitamin D linked to paclitaxel-induced peripheral neuropathy
TOPLINE:
, suggesting that correcting levels before treatment might help prevent the condition.
METHODOLOGY:
- Past studies have suggested an association between vitamin D insufficiency and paclitaxel-induced peripheral neuropathy, a largely untreatable and sometimes permanent side effect of chemotherapy.
- To confirm the association, investigators reviewed data and samples from 1,191 women in the phase 3 SWOG S0221 trial, which compared weekly and biweekly paclitaxel regimens for early-stage breast cancer.
- Using serum samples collected at baseline, the team evaluated the relationship between insufficient vitamin D levels (20 ng/mL or less) before treatment and grade 3 or higher sensory chemotherapy-induced peripheral neuropathy.
TAKEAWAY:
- Overall, 33.3% of the women had insufficient vitamin D levels at baseline, and 16.4% developed grade 3 or worse sensory chemotherapy-induced peripheral neuropathy.
- The incidence of peripheral neuropathy of grade 3 or greater was higher among patients with pretreatment vitamin D insufficiency (20.7% vs. 14.2%; odds ratio, 1.57; P = .005).
- The association grew stronger after adjusting for age and paclitaxel schedule (adjusted OR, 1.65; P = .003), but not after adjusting for race (adjusted OR, 1.39; P = .066).
IN PRACTICE:
The study “confirms that patients with pretreatment vitamin D insufficiency have a higher incidence of [chemotherapy-induced peripheral neuropathy],” the authors concluded. These results also “suggest that vitamin D supplementation in patients with lower levels of vitamin D may reduce peripheral neuropathy, and particularly high-grade peripheral neuropathy, which would improve these patients’ long-term quality of life,” senior researcher Daniel L. Hertz, PharmD, PhD, University of Michigan College of Pharmacy, Ann Arbor, said in a press release.
SOURCE:
The study, led by Ciao-Sin Chen, PharmD, of the University of Michigan, Ann Arbor, was published in the Journal of the National Comprehensive Cancer Network.
LIMITATIONS:
The trial did not collect data on other peripheral neuropathy risk factors, including preexisting peripheral neuropathy and diabetes. The study included a limited number of non-White participants (16%); larger numbers are needed to elucidate a potential interplay between race, vitamin D, and chemotherapy-induced peripheral neuropathy. The researchers also did not collect data on grade 1 and 2 chemotherapy-induced peripheral neuropathy.
DISCLOSURES:
The study was funded by Amgen, the American Cancer Society, and others. The investigators disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
, suggesting that correcting levels before treatment might help prevent the condition.
METHODOLOGY:
- Past studies have suggested an association between vitamin D insufficiency and paclitaxel-induced peripheral neuropathy, a largely untreatable and sometimes permanent side effect of chemotherapy.
- To confirm the association, investigators reviewed data and samples from 1,191 women in the phase 3 SWOG S0221 trial, which compared weekly and biweekly paclitaxel regimens for early-stage breast cancer.
- Using serum samples collected at baseline, the team evaluated the relationship between insufficient vitamin D levels (20 ng/mL or less) before treatment and grade 3 or higher sensory chemotherapy-induced peripheral neuropathy.
TAKEAWAY:
- Overall, 33.3% of the women had insufficient vitamin D levels at baseline, and 16.4% developed grade 3 or worse sensory chemotherapy-induced peripheral neuropathy.
- The incidence of peripheral neuropathy of grade 3 or greater was higher among patients with pretreatment vitamin D insufficiency (20.7% vs. 14.2%; odds ratio, 1.57; P = .005).
- The association grew stronger after adjusting for age and paclitaxel schedule (adjusted OR, 1.65; P = .003), but not after adjusting for race (adjusted OR, 1.39; P = .066).
IN PRACTICE:
The study “confirms that patients with pretreatment vitamin D insufficiency have a higher incidence of [chemotherapy-induced peripheral neuropathy],” the authors concluded. These results also “suggest that vitamin D supplementation in patients with lower levels of vitamin D may reduce peripheral neuropathy, and particularly high-grade peripheral neuropathy, which would improve these patients’ long-term quality of life,” senior researcher Daniel L. Hertz, PharmD, PhD, University of Michigan College of Pharmacy, Ann Arbor, said in a press release.
SOURCE:
The study, led by Ciao-Sin Chen, PharmD, of the University of Michigan, Ann Arbor, was published in the Journal of the National Comprehensive Cancer Network.
LIMITATIONS:
The trial did not collect data on other peripheral neuropathy risk factors, including preexisting peripheral neuropathy and diabetes. The study included a limited number of non-White participants (16%); larger numbers are needed to elucidate a potential interplay between race, vitamin D, and chemotherapy-induced peripheral neuropathy. The researchers also did not collect data on grade 1 and 2 chemotherapy-induced peripheral neuropathy.
DISCLOSURES:
The study was funded by Amgen, the American Cancer Society, and others. The investigators disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
TOPLINE:
, suggesting that correcting levels before treatment might help prevent the condition.
METHODOLOGY:
- Past studies have suggested an association between vitamin D insufficiency and paclitaxel-induced peripheral neuropathy, a largely untreatable and sometimes permanent side effect of chemotherapy.
- To confirm the association, investigators reviewed data and samples from 1,191 women in the phase 3 SWOG S0221 trial, which compared weekly and biweekly paclitaxel regimens for early-stage breast cancer.
- Using serum samples collected at baseline, the team evaluated the relationship between insufficient vitamin D levels (20 ng/mL or less) before treatment and grade 3 or higher sensory chemotherapy-induced peripheral neuropathy.
TAKEAWAY:
- Overall, 33.3% of the women had insufficient vitamin D levels at baseline, and 16.4% developed grade 3 or worse sensory chemotherapy-induced peripheral neuropathy.
- The incidence of peripheral neuropathy of grade 3 or greater was higher among patients with pretreatment vitamin D insufficiency (20.7% vs. 14.2%; odds ratio, 1.57; P = .005).
- The association grew stronger after adjusting for age and paclitaxel schedule (adjusted OR, 1.65; P = .003), but not after adjusting for race (adjusted OR, 1.39; P = .066).
IN PRACTICE:
The study “confirms that patients with pretreatment vitamin D insufficiency have a higher incidence of [chemotherapy-induced peripheral neuropathy],” the authors concluded. These results also “suggest that vitamin D supplementation in patients with lower levels of vitamin D may reduce peripheral neuropathy, and particularly high-grade peripheral neuropathy, which would improve these patients’ long-term quality of life,” senior researcher Daniel L. Hertz, PharmD, PhD, University of Michigan College of Pharmacy, Ann Arbor, said in a press release.
SOURCE:
The study, led by Ciao-Sin Chen, PharmD, of the University of Michigan, Ann Arbor, was published in the Journal of the National Comprehensive Cancer Network.
LIMITATIONS:
The trial did not collect data on other peripheral neuropathy risk factors, including preexisting peripheral neuropathy and diabetes. The study included a limited number of non-White participants (16%); larger numbers are needed to elucidate a potential interplay between race, vitamin D, and chemotherapy-induced peripheral neuropathy. The researchers also did not collect data on grade 1 and 2 chemotherapy-induced peripheral neuropathy.
DISCLOSURES:
The study was funded by Amgen, the American Cancer Society, and others. The investigators disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.