User login
FDA approves elotuzumab with pom/dex in refractory myeloma
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.

Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is already approved in combination with lenalidomide and dexamethasone to treat adult myeloma patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from ELOQUENT-3. This phase 2 trial enrolled multiple myeloma patients who had refractory or relapsed disease and had received both lenalidomide and a proteasome inhibitor.
In the trial, patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n = 60) or pomalidomide and dexamethasone (Pd, n = 57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P = .0029); the rate of complete response or stringent complete response was 8.3% and 1.8%, respectively.
Median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (P = .0078).
Serious adverse events occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious adverse events were pneumonia and respiratory tract infection.
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available on the Empliciti website.
Bristol-Myers Squibb and AbbVie are codeveloping elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
BNP levels and mortality in patients with and without heart failure
Clinical question: Does B-type natriuretic peptide (BNP) have prognostic value outside of heart failure (HF) patients?
Background: BNP levels are influenced by both cardiac and extracardiac stimuli and thus might have prognostic value outside of the traditional use to guide therapy for HF patients.
Study design: Retrospective cohort study of the Vanderbilt electronic health record.
Setting: Vanderbilt University Medical Center, Nashville, Tenn.

Synopsis: The study evaluated 30,487 patients with at least two BNP values for 2002-2015. Within this cohort, 62% of patients did not have a HF diagnosis. Risk of death was elevated in all patients regardless of HF status as BNP values rose. An increase from the 25th to 75th percentile in BNP value was associated with an increased risk of death in non-HF patients (hazard ratio, 2.08; 95% confidence interval, 1.99-2.2). Additionally, in a multivariate analysis BNP was the strongest predictor of death, compared with traditional risk factors in both HF and non-HF patients. The main limitation to this study was the use of ICD codes for diagnosis of HF.
Bottom line: BNP has predictive value for risk of death in non-HF patients; as BNP levels rise, regardless of HF status, so does risk of death.
Citation: York MK et al. B-type natriuretic peptide levels and mortality in patients with and without heart failure. J Am Coll Cardiol. 2018 May 15;71(19):2079-88.
Dr. Abramowicz is an assistant professor in the division of hospital medicine, University of Colorado, Denver.
Clinical question: Does B-type natriuretic peptide (BNP) have prognostic value outside of heart failure (HF) patients?
Background: BNP levels are influenced by both cardiac and extracardiac stimuli and thus might have prognostic value outside of the traditional use to guide therapy for HF patients.
Study design: Retrospective cohort study of the Vanderbilt electronic health record.
Setting: Vanderbilt University Medical Center, Nashville, Tenn.
Synopsis: The study evaluated 30,487 patients with at least two BNP values for 2002-2015. Within this cohort, 62% of patients did not have a HF diagnosis. Risk of death was elevated in all patients regardless of HF status as BNP values rose. An increase from the 25th to 75th percentile in BNP value was associated with an increased risk of death in non-HF patients (hazard ratio, 2.08; 95% confidence interval, 1.99-2.2). Additionally, in a multivariate analysis BNP was the strongest predictor of death, compared with traditional risk factors in both HF and non-HF patients. The main limitation to this study was the use of ICD codes for diagnosis of HF.
Bottom line: BNP has predictive value for risk of death in non-HF patients; as BNP levels rise, regardless of HF status, so does risk of death.
Citation: York MK et al. B-type natriuretic peptide levels and mortality in patients with and without heart failure. J Am Coll Cardiol. 2018 May 15;71(19):2079-88.
Dr. Abramowicz is an assistant professor in the division of hospital medicine, University of Colorado, Denver.
Clinical question: Does B-type natriuretic peptide (BNP) have prognostic value outside of heart failure (HF) patients?
Background: BNP levels are influenced by both cardiac and extracardiac stimuli and thus might have prognostic value outside of the traditional use to guide therapy for HF patients.
Study design: Retrospective cohort study of the Vanderbilt electronic health record.
Setting: Vanderbilt University Medical Center, Nashville, Tenn.
Synopsis: The study evaluated 30,487 patients with at least two BNP values for 2002-2015. Within this cohort, 62% of patients did not have a HF diagnosis. Risk of death was elevated in all patients regardless of HF status as BNP values rose. An increase from the 25th to 75th percentile in BNP value was associated with an increased risk of death in non-HF patients (hazard ratio, 2.08; 95% confidence interval, 1.99-2.2). Additionally, in a multivariate analysis BNP was the strongest predictor of death, compared with traditional risk factors in both HF and non-HF patients. The main limitation to this study was the use of ICD codes for diagnosis of HF.
Bottom line: BNP has predictive value for risk of death in non-HF patients; as BNP levels rise, regardless of HF status, so does risk of death.
Citation: York MK et al. B-type natriuretic peptide levels and mortality in patients with and without heart failure. J Am Coll Cardiol. 2018 May 15;71(19):2079-88.
Dr. Abramowicz is an assistant professor in the division of hospital medicine, University of Colorado, Denver.

A Method to His Madness
ANSWER
The correct interpretation includes sinus tachycardia with biatrial enlargement and ST- and T-wave abnormalities consistent with inferolateral ischemia.
Sinus tachycardia is evident from an atrial rate > 100 beats/min, with a consistent PR interval for each P and R wave.
Criteria for right atrial enlargement include tall P waves in leads II, III, and aVF, and for left atrial enlargement, P waves in lead I ≥ 110 ms with terminal negativity of the P wave in lead V1 ≥ 1 mm2. When both criteria are met, the diagnosis is biatrial enlargement.
The presence of ST elevation in lead V4 and T-wave inversions in leads V5 and V6 and limb leads II, III, and aVF suggest inferolateral ischemia.
ANSWER
The correct interpretation includes sinus tachycardia with biatrial enlargement and ST- and T-wave abnormalities consistent with inferolateral ischemia.
Sinus tachycardia is evident from an atrial rate > 100 beats/min, with a consistent PR interval for each P and R wave.
Criteria for right atrial enlargement include tall P waves in leads II, III, and aVF, and for left atrial enlargement, P waves in lead I ≥ 110 ms with terminal negativity of the P wave in lead V1 ≥ 1 mm2. When both criteria are met, the diagnosis is biatrial enlargement.
The presence of ST elevation in lead V4 and T-wave inversions in leads V5 and V6 and limb leads II, III, and aVF suggest inferolateral ischemia.
ANSWER
The correct interpretation includes sinus tachycardia with biatrial enlargement and ST- and T-wave abnormalities consistent with inferolateral ischemia.
Sinus tachycardia is evident from an atrial rate > 100 beats/min, with a consistent PR interval for each P and R wave.
Criteria for right atrial enlargement include tall P waves in leads II, III, and aVF, and for left atrial enlargement, P waves in lead I ≥ 110 ms with terminal negativity of the P wave in lead V1 ≥ 1 mm2. When both criteria are met, the diagnosis is biatrial enlargement.
The presence of ST elevation in lead V4 and T-wave inversions in leads V5 and V6 and limb leads II, III, and aVF suggest inferolateral ischemia.

A 34-year-old man with a history of malignant hypertension is brought to the emergency department via ACLS ambulance with substernal chest pain and a field blood pressure of 228/136 mm Hg. According to the paramedics, the patient’s symptoms include dyspnea, emotional lability, agitation, and violent behavior requiring physical restraints.
The patient is known at your institution (and many others in the area) for his frequent admissions and poor compliance with medications. Today, the patient’s agitation and paranoia prevent you from obtaining a clear history of the present illness. You do learn that he recently started working in a meth lab, where he has had free access to illicit substances.
His toxicology screen is positive for methamphetamines, cocaine, and phencyclidine. Additional lab tests indicate elevated serial troponin levels, diagnostic of non-ST-segment elevation myocardial infarction (NSTEMI), and an elevated B-type natriuretic peptide level, compatible with congestive
A review of his electronic medical record reveals allergies to sulfa and erythromycin. His medication list includes carvedilol (6.25 mg bid), furosemide (40 mg/d), and lisinopril (20 mg bid), with multiple references to noncompliance. You also note that he has had several skin and soft-tissue infections requiring both oral and IV antibiotic therapy.
The patient’s vital signs on arrival include a blood pressure of 210/140 mm Hg; pulse, 110 beats/min; respiratory rate, 20 breaths/min-1; and temperature, 38.2°C. His weight is 147 lb and his height, 69 in.
Physical exam reveals a man who appears agitated, restless, and psychotic. His skin is remarkable for multiple excoriated lesions on both upper and lower extremities. Recent needle tracks are present in both antecubital fossae.
The HEENT exam is remarkable for enlarged pupils and exophthalmos. Punctate lesions are seen on the nasal septum, and the teeth are in poor repair with multiple caries. The neck is supple, with a palpable thyroid and no evidence of jugular venous distention.
Pulmonary sounds reveal coarse rhonchi in both bases, with no evidence of wheezing. The cardiac exam reveals tachycardia with a regular rate and no extra heart sounds or murmurs. Peripheral pulses are strong and equal bilaterally. The abdominal exam is remarkable for diffuse tenderness to palpation without focal pain, as well as a firm liver edge 2 cm below the right costal margin. The neurologic exam is remarkable for agitation, paranoia, and psychosis. There are no focal cranial nerve defects.
Workup—including chest and abdominal CT, echocardiogram, and ECG—is ordered. The ECG reveals a ventricular rate of 105 beats/min; PR interval, 170 ms; QRS duration, 96 ms; QT/QTc interval, 362/478 ms; P axis, 54°; R axis, 81°; and T axis, –59°. What is your interpretation?

Needle aspiration comes first for most breast abscesses
BOSTON – When surgeon Wendy R. Greene, MD, FACS, director of acute and critical care surgery at Emory University, Atlanta, asked a room of about 300 general surgeons at the annual clinical congress of the American College of Surgeons how many use needle aspiration first for breast abscesses, and how many use a scalpel, it was about a 50-50 split.

This divided response is why Dr. Greene addressed in her presentation the right approach to the problem of breast abscesses. In short, “for run-of-the-mill abscesses less than 5 cm, don’t get out the scalpel; get out the needle first,” she said.
New mothers over age 30 years are most at risk, especially if they are past 40 weeks’ gestation.
There certainly are indications for the scalpel first. If the skin overlaying the abscess is dead, shiny, sloughing off, or leaking pus, or if the abscess is larger than 5 cm on ultrasound, a small stab incision is in order, and it should be made at the maximum point of fluctuation, after numbing the surrounding tissue. Put a wipe in place to catch the pus, debride as necessary, and “irrigate, irrigate, irrigate,” Dr. Greene said.
She uses suction to make sure all the pus is out, then injects a lidocaine into the cavity for pain control and lets it rest a few minutes before another round of suction.
Septic, deteriorating patients, and the immunocompromised, need a larger incision and drainage, with IV antibiotics in the hospital, but even in those cases, “avoid placing percutaneous drains; there’s rarely a role for them in modern management of breast abscesses.” Women will have poorer results and poorer cosmesis, Dr. Greene said.
Aggressive drainage isn’t necessary most of the time, and it can destroy healthy tissue and leave new mothers with breastfeeding problems and milk fistulas. There’s also a risk for scarring, deformity, and loss of the ability to lactate.
An 18-21 gauge needle with local anesthetic is usually enough. The lesion should be obvious on ultrasound, and it’s useful to guide the needle and ensure the cavity collapses on aspiration.
Dr. Greene said it also is important to culture milk in new mothers, and culture her infant’s nose and mouth, because cracked skin on the breast can let germs from nursing into the milk ducts.
Women are sent home after aspiration with antibiotics for 7-10 days, ones that are safe for nursing infants. They might be back with another abscess in a few weeks, so it’s important to be patient and ready for ongoing treatment.
The ultimate worry with recurrent cases is that a breast mass is blocking a milk duct, so mammography is often in order for repeat patients, especially with a family history of breast cancer. Wait until the acute infection has died down; a mammograph can be too painful otherwise, Dr. Greene said.
In the meantime, let infants nurse. They “are a great way to help drain the breast,” she said.
Dr. Greene had no relevant disclosures.
BOSTON – When surgeon Wendy R. Greene, MD, FACS, director of acute and critical care surgery at Emory University, Atlanta, asked a room of about 300 general surgeons at the annual clinical congress of the American College of Surgeons how many use needle aspiration first for breast abscesses, and how many use a scalpel, it was about a 50-50 split.
This divided response is why Dr. Greene addressed in her presentation the right approach to the problem of breast abscesses. In short, “for run-of-the-mill abscesses less than 5 cm, don’t get out the scalpel; get out the needle first,” she said.
New mothers over age 30 years are most at risk, especially if they are past 40 weeks’ gestation.
There certainly are indications for the scalpel first. If the skin overlaying the abscess is dead, shiny, sloughing off, or leaking pus, or if the abscess is larger than 5 cm on ultrasound, a small stab incision is in order, and it should be made at the maximum point of fluctuation, after numbing the surrounding tissue. Put a wipe in place to catch the pus, debride as necessary, and “irrigate, irrigate, irrigate,” Dr. Greene said.
She uses suction to make sure all the pus is out, then injects a lidocaine into the cavity for pain control and lets it rest a few minutes before another round of suction.
Septic, deteriorating patients, and the immunocompromised, need a larger incision and drainage, with IV antibiotics in the hospital, but even in those cases, “avoid placing percutaneous drains; there’s rarely a role for them in modern management of breast abscesses.” Women will have poorer results and poorer cosmesis, Dr. Greene said.
Aggressive drainage isn’t necessary most of the time, and it can destroy healthy tissue and leave new mothers with breastfeeding problems and milk fistulas. There’s also a risk for scarring, deformity, and loss of the ability to lactate.
An 18-21 gauge needle with local anesthetic is usually enough. The lesion should be obvious on ultrasound, and it’s useful to guide the needle and ensure the cavity collapses on aspiration.
Dr. Greene said it also is important to culture milk in new mothers, and culture her infant’s nose and mouth, because cracked skin on the breast can let germs from nursing into the milk ducts.
Women are sent home after aspiration with antibiotics for 7-10 days, ones that are safe for nursing infants. They might be back with another abscess in a few weeks, so it’s important to be patient and ready for ongoing treatment.
The ultimate worry with recurrent cases is that a breast mass is blocking a milk duct, so mammography is often in order for repeat patients, especially with a family history of breast cancer. Wait until the acute infection has died down; a mammograph can be too painful otherwise, Dr. Greene said.
In the meantime, let infants nurse. They “are a great way to help drain the breast,” she said.
Dr. Greene had no relevant disclosures.
BOSTON – When surgeon Wendy R. Greene, MD, FACS, director of acute and critical care surgery at Emory University, Atlanta, asked a room of about 300 general surgeons at the annual clinical congress of the American College of Surgeons how many use needle aspiration first for breast abscesses, and how many use a scalpel, it was about a 50-50 split.
This divided response is why Dr. Greene addressed in her presentation the right approach to the problem of breast abscesses. In short, “for run-of-the-mill abscesses less than 5 cm, don’t get out the scalpel; get out the needle first,” she said.
New mothers over age 30 years are most at risk, especially if they are past 40 weeks’ gestation.
There certainly are indications for the scalpel first. If the skin overlaying the abscess is dead, shiny, sloughing off, or leaking pus, or if the abscess is larger than 5 cm on ultrasound, a small stab incision is in order, and it should be made at the maximum point of fluctuation, after numbing the surrounding tissue. Put a wipe in place to catch the pus, debride as necessary, and “irrigate, irrigate, irrigate,” Dr. Greene said.
She uses suction to make sure all the pus is out, then injects a lidocaine into the cavity for pain control and lets it rest a few minutes before another round of suction.
Septic, deteriorating patients, and the immunocompromised, need a larger incision and drainage, with IV antibiotics in the hospital, but even in those cases, “avoid placing percutaneous drains; there’s rarely a role for them in modern management of breast abscesses.” Women will have poorer results and poorer cosmesis, Dr. Greene said.
Aggressive drainage isn’t necessary most of the time, and it can destroy healthy tissue and leave new mothers with breastfeeding problems and milk fistulas. There’s also a risk for scarring, deformity, and loss of the ability to lactate.
An 18-21 gauge needle with local anesthetic is usually enough. The lesion should be obvious on ultrasound, and it’s useful to guide the needle and ensure the cavity collapses on aspiration.
Dr. Greene said it also is important to culture milk in new mothers, and culture her infant’s nose and mouth, because cracked skin on the breast can let germs from nursing into the milk ducts.
Women are sent home after aspiration with antibiotics for 7-10 days, ones that are safe for nursing infants. They might be back with another abscess in a few weeks, so it’s important to be patient and ready for ongoing treatment.
The ultimate worry with recurrent cases is that a breast mass is blocking a milk duct, so mammography is often in order for repeat patients, especially with a family history of breast cancer. Wait until the acute infection has died down; a mammograph can be too painful otherwise, Dr. Greene said.
In the meantime, let infants nurse. They “are a great way to help drain the breast,” she said.
Dr. Greene had no relevant disclosures.
EXPERT ANALYSIS FROM THE ACS CLINICAL CONGRESS
Providing Rural Veterans With Access to Exercise Through Gerofit
Clinical video telehealth can be used to deliver functional circuit exercise training to older veterans in remote locations.
Exercise increases endurance, muscle strength, and functional performance with corresponding gains in mobility, survival, and quality of life.1 However, even with these benefits and improvements in clinical outcomes, only 15% of adults aged ≥ 65 years follow current guidelines for exercise.2 Despite their prior military training, the majority of veterans do not meet physical activity recommendations.3 Time, travel, and support are common barriers to exercise participation and adherence—barriers that are further amplified among older adults.
The Veterans Health Administration (VHA) is recognized as a world leader in telehealth service development. Currently, 677,000 veterans have received telehealth services, which represents 12% of the 5.6 million veterans under VHA care.4 Clinical video telehealth (CVT) is widely used within the VHA system to deliver health care that otherwise would not be available to veterans. Veterans who have difficulty traveling to the nearest US Department of Veteran Affairs (VA) medical center (VAMC) can access CVT programs at a participating VHA community-based outpatient clinic (CBOC). The VA has more than 45 CVT programs, including programs for mental health, weight management, cardiology, and dermatology. Outside the VA, cardiac exercise rehabilitation provided by CVT has been shown to be as effective as center-based programs in improving cardiovascular risk factors and functional capacity.5 A VHA exercise program that leveraged CVT resources and was dedicated to older adults with a wide range of comorbid conditions would have a high impact on the health and well-being of older veterans.
Gerofit is a VHA clinical demonstration program of supervised center-based exercise for veterans aged ≥ 65 years. Developed at the Durham VAMC Geriatric Research, Education, and Clinical Center (GRECC) in North Carolina, it has demonstrated improved clinical outcomes, including physical function, mobility, quality of life, and survival.6-10 The program offers veterans individualized exercise in a group setting that focuses on improving endurance, strength, and balance. The exercise prescription is based on the patient’s physical limitations as identified in a physical performance assessment.
With support from VHA Geriatric Extended Care (GEC) and the Office of Rural Health (ORH), Gerofit was implemented in 10 VAMCs across 8 VISNs. However, barriers such as travel time, distance, and transportation limit participation. Previously, we found that rural veterans lack access to exercise programs.11,12 Although some do aerobic exercise (AEX), most do not do resistance training (RT), though they are willing to learn. Access to Gerofit for rural veterans is expanding with recent support from the ORH Enterprise Wide Initiative. Rural program expansion includes several different Gerofit initiatives, many involving CBOCs.
The Salem VAMC Gerofit program sought to adapt the facility-based assessment and exercise procedures into a self-reliant CVT class for its CBOCs. This article describes the development of the Salem VAMC Gerofit CVT program, hereafter referred to as Tele-Gerofit.
Related: Expanding the Scope of Telemedicine in Gastroenterology
Program Design
Gerofit was established in 1986 at the Durham GRECC as an exercise and health promotion program for veterans aged ≥ 65 years.13 Its goal is to prevent or improve functional decline from physical inactivity and age-related conditions. Gerofit targets the geriatric patient population and thus extends beyond cardiac and pulmonary rehabilitation or weight loss programs. The primary exclusion criteria are based on safety issues in the context of a group exercise setting of older adults and include oxygen dependency, unstable cardiac disease, and moderate-to-severe cognitive impairment.
To participate in Gerofit, veterans must be able to perform activities of daily living and self-manage an exercise prescription developed by the exercise instructor based on physical performance testing. These physical performance tests include measures that are independent predictors of disability, loss of independent living, and death, as well as surrogate measures of exercise capacity (eg, strength, endurance, balance).14,15 A novel aspect of Gerofit is that the physical performance assessment is used not only to determine physical limitations, but also to individualize the exercise prescription based on the observed deficits in strength, endurance, or balance. These assessments are performed at initial enrollment; 3 months, 6 months, and 1 year later; and annually after that. Currently, the center-based Gerofit programs administer 5 items of the Senior Fitness Test: 6-minute walk, 10-meter walk (10-MWT), 30-second 1-arm curl, 30-second chair-stand test, and 8-foot up-and-go.15 The side-by-side, semitandem, and tandem standing balance tests from the short physical performance battery also are performed.16 In addition, participants complete a questionnaire that includes items from the physical functioning scale of the 12-Item Short Form Health Survey (SF-12).
After each assessment, the Gerofit exercise instructor reviews the results with the veteran and formulates an individualized exercise prescription along with goals for improvement. Veterans are encouraged to attend supervised center-based exercise sessions 3 times weekly. Classes are offered in a gym or fitness center at the VAMC or in leased space. Each patient uses a cue card that lists an exercise plan personalized for intensity and duration for aerobic exercise (AEX; eg, treadmill walking, stationary bicycling, arm ergometry), RT using dumbbells and weight equipment, and functional exercises for flexibility and balance. Some medical centers also offer yoga, tai chi, or dancing Gerofit classes.
For participants in the Durham Gerofit program, mortality decreased 25% over a decade (hazard ratio, 0.75; 95% CI, 0.61-0.91).9 A substudy that included the Psychological General Well-Being Index found that 81% of participants significantly increased their score after 1 year.7 Observed initial improvement in physical performance has been sustained over 5 years.10,17 One-year results from the recent Gerofit expansion to 6 other VAMCs showed clinically and statistically significantly improved physical performance from baseline to 3-, 6-, and 12-month follow-up.18
Adaptation of Gerofit to CVT Delivery
Initial work. The Greater Los Angeles VAMC Gerofit program conducted a pilot CVT exercise class of 6 veterans at the rural Bakersfield CBOC in California.19 Each week, an exercise instructor broadcast a 60-minute exercise class that included warm-up, RT with bands, progressive balance training, and flexibility. Trained student volunteers from California State University in Bakersfield kinesiology program were on site at the Bakersfield CBOC to perform the assessments and aid in exercises during the CVT sessions. Despite the lack of AEX per se, veterans showed significant improvement in endurance as measured by an increase in the number of steps completed in 2 minutes at the 3-month assessment (P = .049). Although exercises were not delivered in a circuit format, the improved endurance supported the potential for cardiovascular benefit from RT in older adults.
This pilot project also demonstrated that key components of the Gerofit program could be delivered safely by telehealth with onsite supervision. The Miami VA Healthcare System also offers CVT Gerofit exercise classes broadcast to the rural Florida CBOCs of Key Largo and Homestead.11 The exercise activities offered for the Miami CVT participants incorporate components of AEX (calisthenics) and RT (resistance bands). Veterans enjoyed the classes, and adherence was good. However, availability of staff and space are an ongoing challenge.
In Key Largo, 5 veterans participated before the CVT classes were placed on hold owing to the demands of other CVT programs and limited availability of the telehealth clinical technician (TCT). The Homestead CBOC continues to offer CVT Gerofit exercise classes and has 6 regular participants. Notably, the physical space at the Homestead CBOC is smaller than that at the Key Largo CBOC; the Homestead CBOC has adjusted by shifting to exercises performed while standing or sitting, ensuring participants’ safety and satisfaction.
The Baltimore, Maryland VAMC Gerofit program offers other innovative CVT exercise classes, including a tai chi class, and a class with exercise performed while sitting in a chair. Although the Baltimore VAMC CVT exercise classes do not have the scope of the center-based exercise prescriptions, they are unique in that they are broadcast not only to their affiliated CBOCs, but also other Gerofit programs in different VISNs.
Related: Telehealth for Rural Veterans With Neurologic Disorders
Salem VAMC Gerofit Program. The center-based Salem VAMC Gerofit program was established in July 2015. In fiscal year 2017, its dedicated exercise facility had more than 5,000 patient visits. Despite the program’s success, we prioritized establishing Tele-Gerofit because of the medical center’s rural location in southwest Virginia and the large number of veterans who receive care at CBOCs. Therefore, much as with the pilot CVT Gerofit classes in Los Angeles and Miami, the target setting was rural CBOCs. The goal for Salem VAMC Tele-Gerofit was to modify Gerofit delivery to the CVT format and a CBOC setting with minimal modification of the content and personnel requirements of both physical performance testing and exercise training procedures. 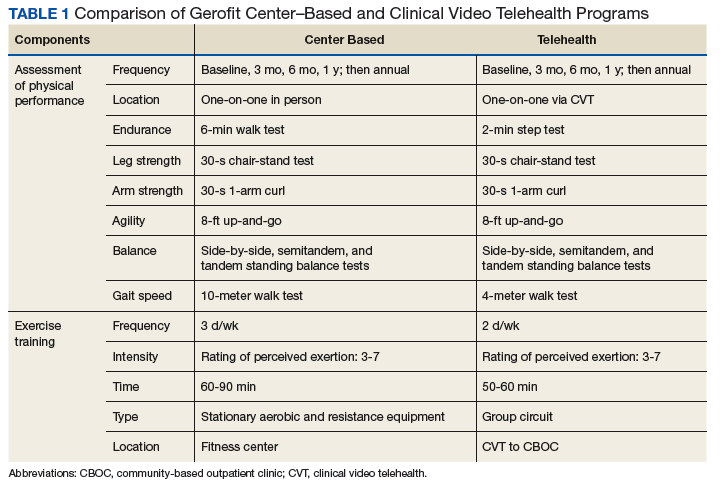
Adjustments for CBOC Setting. The enrollment process for Tele-Gerofit is the same as that for the center-based program. To start, a veteran’s primary care provider reviews the list of eligibility criteria and, if the veteran qualifies, places a consult. A Gerofit team member then contacts the veteran by phone to describe the program and schedule an assessment. At the baseline physical performance assessment, American College of Sports Medicine guidelines on exercise participation, health screening, and exercise intensity are used to evaluate veterans and rank them by their cardiovascular risk.20 All new program participants start with low-intensity exercise and gradually progress to recommended levels of exercise. Before starting an exercise class, participants are instructed on use of the 10-point rating of perceived exertion (RPE).
Each CBOC site is supplied with an RPE poster that is displayed for participants’ use. During a Tele-Gerofit class, the exercise instructor asks participants to periodically report their RPE. This class differs slightly from the center-based exercise sessions in which RPE is primarily assessed when a different exercise is introduced or the duration or intensity of an exercise is increased. The Gerofit instructor monitors exercise and treatment fidelity, but the onsite TCT observes for safety during class. The TCT also takes initial vital signs and sets up the room for the class. Emergency contacts and procedures are posted in each CBOC CVT room and are available to the center-based exercise instructor. Because the CBOCs are not inside medical facilities, some CBOC directors have asked that heart rate monitors be used as an extra safety precaution to ensure that high-risk participants do not exceed a heart rate limit that may be set by their cardiologists.
Modifications to Physical Performance Assessment. Physical performance testing had to be adapted to the small rooms available at the CBOCs. For measuring normal gait speed, the 10-MWT was replaced with the 4-meter walk test (4-MWT). The 4-MWT has excellent test–retest reliability with an intraclass correlation coefficient (ICC) of 0.93, but the discrepancy in gait speed between the 4-MWT and the 10-MWT is such that the tests cannot be used interchangeably.21 For measuring endurance, the 6-minute walk test was replaced with the 2-minute step test (2-MST). In older adults, the 2-MST has a moderate correlation with 6-minute walk distance (r = 0.36; P = .04) and high reliability (ICC, 0.90).15,22 The 30-second 1-arm curl, the 30-second chair-stand test, and the 8-foot up-and-go test are performed without modification and require only dumbbells, a chair without wheels, and a stopwatch.
The exercise instructor at the Salem VAMC conducts physical performance testing by 2-way videoconferencing with the veteran in a room at the CBOC. The TCT at the CBOC assists by measuring and demarcating 4 meters on the floor and a designated height on the wall for knee elevation for 4-MWT and 2-MST, respectively. The TCT remains in the room during the assessment visit. Except for taking vital signs before and after the physical performance assessment, the TCT does not participate in the testing. To date, more than 20 physical performance assessments have been conducted without difficulty at Salem-affiliated CBOCs. The primary challenge has been scheduling the room with CVT equipment (ie, camera and screen) for the 30-minute individual assessment session, which occurs on a rolling basis as individuals are enrolled and followed.
After the assessment is completed, the exercise instructor reviews the results with the participant and provides feedback on areas in need of improvement. However, these education sessions can be lengthy and are best supported by giving the patient a personalized handout.
Functional Circuit Exercise. In Tele-Gerofit, exercise training is delivered by CVT broadcast from the Salem VAMC to veterans in a room (equipped with steps, dumbbells, chairs, and bands) at the CBOC. This type of exercise training, which uses only mobile equipment and plyometric (weight-bearing) exercises, is referred to as functional exercise. The AEX includes marching in place, moving on and off a raised step, and body-weight exercises, while RT uses dumbbells, resistance bands, and plyometric exercises (Table 2). 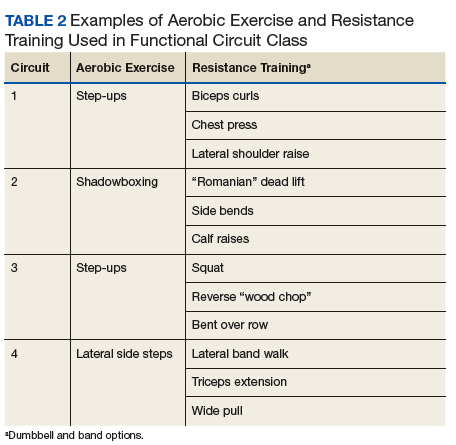
Progression of intensity is achieved by increasing the rate of stepping and the size of the steps (AEX) or the number of repetitions and the weight of the dumbbells or bands (RT). Each veteran exercises at an intensity level that is appropriate for his or her baseline limitations and medical conditions. The exercise instructor uses different forms of the same equipment (eg, heavier dumbbells, higher steps) to vary intensity among individuals while having them perform the same exercises as a group. The challenge is to adjust the pace of the AEX or the timing of the RT repetitions for individuals new to the class.
Delivery of exercise training in the form of circuits allows for a diverse exercise program in a setting with limited space. Circuit training is an exercise modality that consists of a series of different exercises, each usually completed in 30 to 60 seconds, with minimal rest between each type of exercise. Each Tele-Gerofit circuit has a mix of AEX and RT exercises performed for 3 minutes consecutively (Figure). 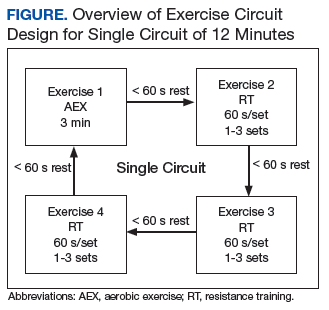
The design of the circuit training can be adjusted based on the number of individuals in the class. Larger classes can be split into 2 groups that alternate between exercise sets, while smaller classes have 1 group performing the same exercise set and then rotating to either the AEX or RT set. Total exercise time to complete the circuit depends on the number of different exercises, number of repetitions, and the rest between repetitions and the different exercises. In this way, total exercise time can be made shorter or longer depending on the veteran’s capacity.
Frequency. Tele-Gerofit exercise classes are currently offered twice weekly and last about 1 hour, which includes warm-up (8-10 minutes), functional circuit training (40 minutes), and cooldown/stretching (8-10 minutes). A challenge for the exercise instructor is the need to provide ongoing clear instructions both to the class and to individuals as needed. As the exercise prescription for each patient is based on physical performance testing, the exercise instructor for the training must be familiar with the test results. Derivation of the exercise prescription in Tele-Gerofit follows the same process as center-based Gerofit.
Each patient is given an exercise prescription written to address any impairments noted in the different domains of the physical performance assessment, scored using age and sex percentiles. For instance, individuals scoring poorly on lower body strength are given specific lower body strengthening exercises. Participants are given an exercise program that guides them toward achieving recommended physical activity guidelines using their RPE to modulate each exercise. Duration and intensity of each type of planned exercise are formally discussed after initial and follow-up assessments. In addition, exercise training is informally progressed throughout the program. For Tele-Gerofit, instructors must design each class with the group in mind while being prepared for modifications and specific changes for individuals.
Discussion
Tele-Gerofit adapts the well-established center-based Gerofit program to be executed without an exercise facility while maintaining the content of the evidence-based procedures. Physical performance testing and exercise training were modified, adding elements necessary for CVT assessments and classes to be broadcast from the Salem VAMC to its affiliated CBOCs. Tele-Gerofit exercises are performed in a circuit style that allows a veteran or small structured groups of veterans to move among exercises and requires less space than traditional group exercise does. Safety and monitoring concerns are addressed with a safety procedure that includes emergency plans for each site, prescreening of enrolled participants, and monitoring of exercise intensity in accordance with national guidelines.1 Similar to the center-based Gerofit program, the exercise prescription is tailored to each veteran’s physical limitations based on initial and ongoing assessment of physical performance. Tele-Gerofit physical performance testing fulfills the same need with only a few modifications using validated measures. Tele-Gerofit assessments are administered by CVT without the need for additional staff on site.
Adaptation of center-based Gerofit exercise classes to Tele-Gerofit is a major innovation. Use of a circuit exercise design was supported by findings in older adults that RT alone, when performed quickly with minimal rest between each set and exercise station, increases both aerobic capacity and strength.23,24 Older adult RT trials that compared circuit RT with traditional RT found that strength gains are comparable between circuit and traditional RT.24-26 Working with adults aged > 60 years, Takeshima and colleagues conducted a trial of circuit exercise with added callisthenic exercises performed in place between RT on exercise machines.27 This dual-modality (AEX+RT) circuit approach was well tolerated and effective, increasing aerobic capacity and strength. Unfortunately, the resistance exercise machines used in those circuit exercise studies and in the center-based Gerofit program are not an option for Tele-Gerofit.
The requirement for an exercise facility was removed by designing Tele-Gerofit exercise to include only functional exercises that rely on body weight or small mobile exercise equipment. Although popular among young adults, functional circuit exercise is understudied in older adults. Recently, a 12-week functional circuit exercise intervention in frail elderly adults demonstrated significant improvements in gait speed and the timed chair-stand test.28 A pilot observational study of Gerofit participants at the Canandaigua VAMC offered 27 veterans functional circuit exercise instead of their traditional exercise facility class and found larger increases in the timed chair-stand test and 6-minute walk distance compared with 11 Gerofit participants in the traditional program.29
This Tele-Gerofit exercise training combines functional and circuit exercise strategies into telehealth delivery. However, its effect on physical performance remains to be demonstrated. To address this question, we are conducting a single-arm pilot study of Tele-Gerofit with CVT broadcast to 3 Salem CBOC affiliates (Wytheville, Staunton, and Danville, Virginia). The goal is to determine the effect on physical performance and collect feasibility data, including attendance rate and patient satisfaction with the video broadcast. In addition, we are planning an effectiveness trial to compare the impact of functional circuit exercise delivered in person (center based, not CVT) with the parent Gerofit exercise program on direct measures of endurance and strength, in addition to physical performance.
Related: Setting and Method of Measurement Affect Blood Pressure Readings in Older Veterans
Implementation research is needed to determine how Tele-Gerofit can be disseminated to other VAMCs and community-based centers beyond CBOCs. Although the cost of the equipment used to implement Tele-Gerofit is minimal, the program requires dedicated and experienced exercise instructors, and the sharing of telehealth resources with other clinical programs. The authors expect that a diverse group of stakeholders is needed across service lines of primary care, geriatrics and extended care, physical medicine and rehabilitation, and telehealth. Of note, this multidisciplinary collaboration is a hallmark of the Gerofit program. The recent success of the implementation of center-based Gerofit in VAMCs across the US demonstrates the program’s flexibility and robust results.18
Plans also include refining strategies for physical performance testing and exercise monitoring. For instance, we would like to adapt telehealth technology for heart rate monitors that can be worn by high-risk veterans at the CBOC and viewed in real time by the exercise instructor.
Conclusion
Gerofit, which is designed to help older veterans maintain independent living and prevent disability, has been demonstrated to improve quality of life and survival. Our goal has been to adapt Gerofit to CVT and provide a supervised, individualized exercise program in a group setting—a program that can be widely disseminated. Salem VAMC Tele-Gerofit is an innovative and prescriptive program that delivers CVT functional circuit exercise training to remote locations without the need for stationary exercise equipment. This approach has the potential to become an effective and feasible exercise strategy for preventing and minimizing disability in the increasing population of older veterans. Work is needed to determine whether Tele-Gerofit provides a rapid translation of Gerofit to clinical practice and improved outcomes with substantial cost savings from reduced hospitalization and institutionalization.
Acknowledgments
Gerofit has been funded by the Veterans Health Affairs Office of Geriatrics and Extended Care Non-Institutional Long-Term Care Funding and Mentored Partnership Program, and the Veterans Health Affairs Office of Rural Health Rural Enterprise-Wide Initiative.
The authors thank Kim Birkett, MPH, for assistance in editing, references, and graphics and the staff at the Wytheville, Staunton, and Danville community-based outpatient clinics for their support.
1. American College of Sports Medicine, Chodzko-Zajko WJ, Proctor DN, et al. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med Sci Sports Exerc. 2009;41(7):1510-1530.
2. Centers for Disease Control and Prevention. Adult participation in aerobic and muscle-strengthening physical activities—United States, 2011. MMWR Morb Mortal Wkly Rep. 2013;62(17):326-330.
3. Littman AJ, Forsberg CW, Koepsell TD. Physical activity in a national sample of veterans. Med Sci Sports Exerc. 2009;41(5):1006-1013.
4. US Department of Veterans Affairs, Office of Rural Health. Annual Report: Thrive 2015. https://www.ruralhealth.va.gov/docs/ORH_Annual_Report_2015_FINAL.pdf. Published 2015. Accessed July 16, 2018.
5. Rawstorn JC, Gant N, Direito A, Beckmann C, Maddison R. Telehealth exercise-based cardiac rehabilitation: a systematic review and meta-analysis. Heart. 2016;102(15):1183-1192.
6. Morey MC. Celebrating 20 years of excellence in exercise for the older veteran. Fed Pract. 2007;24(10):49-65.
7. Cowper PA, Morey MC, Bearon LB, et al. The impact of supervised exercise on the psychological well-being and health status of older veterans. J Appl Gerontol. 1991;10(4):469-485.
8. Morey MC, Cowper PA, Feussner JR, et al. Evaluation of a supervised exercise program in a geriatric population. J Am Geriatr Soc. 1989;37(4):348-354.
9. Morey MC, Pieper CF, Crowley GM, Sullivan RJ, Puglisi CM. Exercise adherence and 10-year mortality in chronically ill older adults. J Am Geriatr Soc. 2002;50(12):1929-1933.
10. Morey MC, Pieper CF, Sullivan RJ Jr, Crowley GM, Cowper PA, Robbins MS. Five-year performance trends for older exercisers: a hierarchical model of endurance, strength, and flexibility. J Am Geriatr Soc. 1996;44(10):1226-1231.
11. Valencia WM, Botros D, Pendlebury D, et al. Proactive reach and telehealth monitoring (Gerofit) enhance resistance exercise at rural setting. Innovat Aging. 2017;1(suppl 1):225.12. Pendlebury D, Botros D VW. Proactive Reach: an innovative access approach to identify & deliver GEROFIT exercise telehealth counseling to rural veterans & enhance CBOC services. J Am Geriatr Soc. 2017(suppl 1):S208. Poster presented at: Annual Scientific Meeting of the American Geriatrics Society; May 18, 2017; San Antonio, TX.
13. Morey MC, Crowley GM, Robbins MS, Cowper PA, Sullivan RJ Jr. The Gerofit program: a VA innovation. South Med J. 1994;87(5):S83-S87.
14. Cooper R, Kuh D, Hardy R; Mortality Review Group; FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: systematic review and meta-analysis. BMJ. 2010;341:c4467.
15. Rikli RE, Jones CJ. Development and validation of a functional fitness test for community-residing older adults. J Aging Phys Act. 1999;7(2):129-161.
16. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol. 1994;49(2):M85-M94.
17. Morey MC, Cowper PA, Feussner JR, et al. Two-year trends in physical performance following supervised exercise among community-dwelling older veterans. J Am Geriatr Soc. 1991;39(10):986-992.
18. Morey MC, Lee CC, Castle S, et al. Should structured exercise be promoted as a model of care? Dissemination of the Department of Veterans Affairs Gerofit program. J Am Geriatr Soc. 2018;66(5):1009-1016.
19. Blanchard E, Castle S, Ines E, et al. Delivering a clinical exercise program to rural veterans via video telehealth. Poster C167 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
20. Riebe D, Ehrman JK, Liguori G, Magal M, eds; American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription. 10th ed. Philadelphia, PA: Wolters Kluwer Health; 2018.
21. Peters DM, Fritz SL, Krotish DE. Assessing the reliability and validity of a shorter walk test compared with the 10-meter walk test for measurements of gait speed in healthy, older adults. J Geriatr Phys Ther. 2013;36(1):24-30.
22. Pedrosa R, Holanda G. Correlation between the walk, 2-minute step and TUG tests among hypertensive older women. Rev Bras Fisioter. 2009;13(3):252-256.
23. Romero-Arenas S, Blazevich AJ, Martinez-Pascual M, et al. Effects of high-resistance circuit training in an elderly population. Exp Gerontol. 2013;48(3):334-340.
24. Brentano MA, Cadore EL, Da Silva EM, et al. Physiological adaptations to strength and circuit training in postmenopausal women with bone loss. J Strength Cond Res. 2008;22(6):1816-1825.
25. Romero-Arenas S, Martinez-Pascual M, Alcaraz PE. Impact of resistance circuit training on neuromuscular, cardiorespiratory and body composition adaptations in the elderly. Aging Dis. 2013;4(5):256-263.
26. Paoli A, Pacelli F, Bargossi AM, et al. Effects of three distinct protocols of fitness training on body composition, strength and blood lactate. J Sports Med Phys Fitness. 2010;50(1):43-51.
27. Takeshima N, Rogers ME, Islam MM, Yamauchi T, Watanabe E, Okada A. Effect of concurrent aerobic and resistance circuit exercise training on fitness in older adults. Eur J Appl Physiol. 2004;93(1-2):173-182.
28. Giné-Garriga M, Guerra M, Pagés E, Manini TM, Jiménez R, Unnithan VB. The effect of functional circuit training on physical frailty in frail older adults: a randomized controlled trial. J Aging Phys Act. 2010;18(4):401-424.
29. Biddle ED, Reynolds P, Kopp T, Cammarata H, Conway P. Implementation of functional training tools elicits improvements in aerobic fitness and lower body strength in older veterans. Poster C169 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
Clinical video telehealth can be used to deliver functional circuit exercise training to older veterans in remote locations.
Clinical video telehealth can be used to deliver functional circuit exercise training to older veterans in remote locations.
Exercise increases endurance, muscle strength, and functional performance with corresponding gains in mobility, survival, and quality of life.1 However, even with these benefits and improvements in clinical outcomes, only 15% of adults aged ≥ 65 years follow current guidelines for exercise.2 Despite their prior military training, the majority of veterans do not meet physical activity recommendations.3 Time, travel, and support are common barriers to exercise participation and adherence—barriers that are further amplified among older adults.
The Veterans Health Administration (VHA) is recognized as a world leader in telehealth service development. Currently, 677,000 veterans have received telehealth services, which represents 12% of the 5.6 million veterans under VHA care.4 Clinical video telehealth (CVT) is widely used within the VHA system to deliver health care that otherwise would not be available to veterans. Veterans who have difficulty traveling to the nearest US Department of Veteran Affairs (VA) medical center (VAMC) can access CVT programs at a participating VHA community-based outpatient clinic (CBOC). The VA has more than 45 CVT programs, including programs for mental health, weight management, cardiology, and dermatology. Outside the VA, cardiac exercise rehabilitation provided by CVT has been shown to be as effective as center-based programs in improving cardiovascular risk factors and functional capacity.5 A VHA exercise program that leveraged CVT resources and was dedicated to older adults with a wide range of comorbid conditions would have a high impact on the health and well-being of older veterans.
Gerofit is a VHA clinical demonstration program of supervised center-based exercise for veterans aged ≥ 65 years. Developed at the Durham VAMC Geriatric Research, Education, and Clinical Center (GRECC) in North Carolina, it has demonstrated improved clinical outcomes, including physical function, mobility, quality of life, and survival.6-10 The program offers veterans individualized exercise in a group setting that focuses on improving endurance, strength, and balance. The exercise prescription is based on the patient’s physical limitations as identified in a physical performance assessment.
With support from VHA Geriatric Extended Care (GEC) and the Office of Rural Health (ORH), Gerofit was implemented in 10 VAMCs across 8 VISNs. However, barriers such as travel time, distance, and transportation limit participation. Previously, we found that rural veterans lack access to exercise programs.11,12 Although some do aerobic exercise (AEX), most do not do resistance training (RT), though they are willing to learn. Access to Gerofit for rural veterans is expanding with recent support from the ORH Enterprise Wide Initiative. Rural program expansion includes several different Gerofit initiatives, many involving CBOCs.
The Salem VAMC Gerofit program sought to adapt the facility-based assessment and exercise procedures into a self-reliant CVT class for its CBOCs. This article describes the development of the Salem VAMC Gerofit CVT program, hereafter referred to as Tele-Gerofit.
Related: Expanding the Scope of Telemedicine in Gastroenterology
Program Design
Gerofit was established in 1986 at the Durham GRECC as an exercise and health promotion program for veterans aged ≥ 65 years.13 Its goal is to prevent or improve functional decline from physical inactivity and age-related conditions. Gerofit targets the geriatric patient population and thus extends beyond cardiac and pulmonary rehabilitation or weight loss programs. The primary exclusion criteria are based on safety issues in the context of a group exercise setting of older adults and include oxygen dependency, unstable cardiac disease, and moderate-to-severe cognitive impairment.
To participate in Gerofit, veterans must be able to perform activities of daily living and self-manage an exercise prescription developed by the exercise instructor based on physical performance testing. These physical performance tests include measures that are independent predictors of disability, loss of independent living, and death, as well as surrogate measures of exercise capacity (eg, strength, endurance, balance).14,15 A novel aspect of Gerofit is that the physical performance assessment is used not only to determine physical limitations, but also to individualize the exercise prescription based on the observed deficits in strength, endurance, or balance. These assessments are performed at initial enrollment; 3 months, 6 months, and 1 year later; and annually after that. Currently, the center-based Gerofit programs administer 5 items of the Senior Fitness Test: 6-minute walk, 10-meter walk (10-MWT), 30-second 1-arm curl, 30-second chair-stand test, and 8-foot up-and-go.15 The side-by-side, semitandem, and tandem standing balance tests from the short physical performance battery also are performed.16 In addition, participants complete a questionnaire that includes items from the physical functioning scale of the 12-Item Short Form Health Survey (SF-12).
After each assessment, the Gerofit exercise instructor reviews the results with the veteran and formulates an individualized exercise prescription along with goals for improvement. Veterans are encouraged to attend supervised center-based exercise sessions 3 times weekly. Classes are offered in a gym or fitness center at the VAMC or in leased space. Each patient uses a cue card that lists an exercise plan personalized for intensity and duration for aerobic exercise (AEX; eg, treadmill walking, stationary bicycling, arm ergometry), RT using dumbbells and weight equipment, and functional exercises for flexibility and balance. Some medical centers also offer yoga, tai chi, or dancing Gerofit classes.
For participants in the Durham Gerofit program, mortality decreased 25% over a decade (hazard ratio, 0.75; 95% CI, 0.61-0.91).9 A substudy that included the Psychological General Well-Being Index found that 81% of participants significantly increased their score after 1 year.7 Observed initial improvement in physical performance has been sustained over 5 years.10,17 One-year results from the recent Gerofit expansion to 6 other VAMCs showed clinically and statistically significantly improved physical performance from baseline to 3-, 6-, and 12-month follow-up.18
Adaptation of Gerofit to CVT Delivery
Initial work. The Greater Los Angeles VAMC Gerofit program conducted a pilot CVT exercise class of 6 veterans at the rural Bakersfield CBOC in California.19 Each week, an exercise instructor broadcast a 60-minute exercise class that included warm-up, RT with bands, progressive balance training, and flexibility. Trained student volunteers from California State University in Bakersfield kinesiology program were on site at the Bakersfield CBOC to perform the assessments and aid in exercises during the CVT sessions. Despite the lack of AEX per se, veterans showed significant improvement in endurance as measured by an increase in the number of steps completed in 2 minutes at the 3-month assessment (P = .049). Although exercises were not delivered in a circuit format, the improved endurance supported the potential for cardiovascular benefit from RT in older adults.
This pilot project also demonstrated that key components of the Gerofit program could be delivered safely by telehealth with onsite supervision. The Miami VA Healthcare System also offers CVT Gerofit exercise classes broadcast to the rural Florida CBOCs of Key Largo and Homestead.11 The exercise activities offered for the Miami CVT participants incorporate components of AEX (calisthenics) and RT (resistance bands). Veterans enjoyed the classes, and adherence was good. However, availability of staff and space are an ongoing challenge.
In Key Largo, 5 veterans participated before the CVT classes were placed on hold owing to the demands of other CVT programs and limited availability of the telehealth clinical technician (TCT). The Homestead CBOC continues to offer CVT Gerofit exercise classes and has 6 regular participants. Notably, the physical space at the Homestead CBOC is smaller than that at the Key Largo CBOC; the Homestead CBOC has adjusted by shifting to exercises performed while standing or sitting, ensuring participants’ safety and satisfaction.
The Baltimore, Maryland VAMC Gerofit program offers other innovative CVT exercise classes, including a tai chi class, and a class with exercise performed while sitting in a chair. Although the Baltimore VAMC CVT exercise classes do not have the scope of the center-based exercise prescriptions, they are unique in that they are broadcast not only to their affiliated CBOCs, but also other Gerofit programs in different VISNs.
Related: Telehealth for Rural Veterans With Neurologic Disorders
Salem VAMC Gerofit Program. The center-based Salem VAMC Gerofit program was established in July 2015. In fiscal year 2017, its dedicated exercise facility had more than 5,000 patient visits. Despite the program’s success, we prioritized establishing Tele-Gerofit because of the medical center’s rural location in southwest Virginia and the large number of veterans who receive care at CBOCs. Therefore, much as with the pilot CVT Gerofit classes in Los Angeles and Miami, the target setting was rural CBOCs. The goal for Salem VAMC Tele-Gerofit was to modify Gerofit delivery to the CVT format and a CBOC setting with minimal modification of the content and personnel requirements of both physical performance testing and exercise training procedures.
Adjustments for CBOC Setting. The enrollment process for Tele-Gerofit is the same as that for the center-based program. To start, a veteran’s primary care provider reviews the list of eligibility criteria and, if the veteran qualifies, places a consult. A Gerofit team member then contacts the veteran by phone to describe the program and schedule an assessment. At the baseline physical performance assessment, American College of Sports Medicine guidelines on exercise participation, health screening, and exercise intensity are used to evaluate veterans and rank them by their cardiovascular risk.20 All new program participants start with low-intensity exercise and gradually progress to recommended levels of exercise. Before starting an exercise class, participants are instructed on use of the 10-point rating of perceived exertion (RPE).
Each CBOC site is supplied with an RPE poster that is displayed for participants’ use. During a Tele-Gerofit class, the exercise instructor asks participants to periodically report their RPE. This class differs slightly from the center-based exercise sessions in which RPE is primarily assessed when a different exercise is introduced or the duration or intensity of an exercise is increased. The Gerofit instructor monitors exercise and treatment fidelity, but the onsite TCT observes for safety during class. The TCT also takes initial vital signs and sets up the room for the class. Emergency contacts and procedures are posted in each CBOC CVT room and are available to the center-based exercise instructor. Because the CBOCs are not inside medical facilities, some CBOC directors have asked that heart rate monitors be used as an extra safety precaution to ensure that high-risk participants do not exceed a heart rate limit that may be set by their cardiologists.
Modifications to Physical Performance Assessment. Physical performance testing had to be adapted to the small rooms available at the CBOCs. For measuring normal gait speed, the 10-MWT was replaced with the 4-meter walk test (4-MWT). The 4-MWT has excellent test–retest reliability with an intraclass correlation coefficient (ICC) of 0.93, but the discrepancy in gait speed between the 4-MWT and the 10-MWT is such that the tests cannot be used interchangeably.21 For measuring endurance, the 6-minute walk test was replaced with the 2-minute step test (2-MST). In older adults, the 2-MST has a moderate correlation with 6-minute walk distance (r = 0.36; P = .04) and high reliability (ICC, 0.90).15,22 The 30-second 1-arm curl, the 30-second chair-stand test, and the 8-foot up-and-go test are performed without modification and require only dumbbells, a chair without wheels, and a stopwatch.
The exercise instructor at the Salem VAMC conducts physical performance testing by 2-way videoconferencing with the veteran in a room at the CBOC. The TCT at the CBOC assists by measuring and demarcating 4 meters on the floor and a designated height on the wall for knee elevation for 4-MWT and 2-MST, respectively. The TCT remains in the room during the assessment visit. Except for taking vital signs before and after the physical performance assessment, the TCT does not participate in the testing. To date, more than 20 physical performance assessments have been conducted without difficulty at Salem-affiliated CBOCs. The primary challenge has been scheduling the room with CVT equipment (ie, camera and screen) for the 30-minute individual assessment session, which occurs on a rolling basis as individuals are enrolled and followed.
After the assessment is completed, the exercise instructor reviews the results with the participant and provides feedback on areas in need of improvement. However, these education sessions can be lengthy and are best supported by giving the patient a personalized handout.
Functional Circuit Exercise. In Tele-Gerofit, exercise training is delivered by CVT broadcast from the Salem VAMC to veterans in a room (equipped with steps, dumbbells, chairs, and bands) at the CBOC. This type of exercise training, which uses only mobile equipment and plyometric (weight-bearing) exercises, is referred to as functional exercise. The AEX includes marching in place, moving on and off a raised step, and body-weight exercises, while RT uses dumbbells, resistance bands, and plyometric exercises (Table 2).
Progression of intensity is achieved by increasing the rate of stepping and the size of the steps (AEX) or the number of repetitions and the weight of the dumbbells or bands (RT). Each veteran exercises at an intensity level that is appropriate for his or her baseline limitations and medical conditions. The exercise instructor uses different forms of the same equipment (eg, heavier dumbbells, higher steps) to vary intensity among individuals while having them perform the same exercises as a group. The challenge is to adjust the pace of the AEX or the timing of the RT repetitions for individuals new to the class.
Delivery of exercise training in the form of circuits allows for a diverse exercise program in a setting with limited space. Circuit training is an exercise modality that consists of a series of different exercises, each usually completed in 30 to 60 seconds, with minimal rest between each type of exercise. Each Tele-Gerofit circuit has a mix of AEX and RT exercises performed for 3 minutes consecutively (Figure).
The design of the circuit training can be adjusted based on the number of individuals in the class. Larger classes can be split into 2 groups that alternate between exercise sets, while smaller classes have 1 group performing the same exercise set and then rotating to either the AEX or RT set. Total exercise time to complete the circuit depends on the number of different exercises, number of repetitions, and the rest between repetitions and the different exercises. In this way, total exercise time can be made shorter or longer depending on the veteran’s capacity.
Frequency. Tele-Gerofit exercise classes are currently offered twice weekly and last about 1 hour, which includes warm-up (8-10 minutes), functional circuit training (40 minutes), and cooldown/stretching (8-10 minutes). A challenge for the exercise instructor is the need to provide ongoing clear instructions both to the class and to individuals as needed. As the exercise prescription for each patient is based on physical performance testing, the exercise instructor for the training must be familiar with the test results. Derivation of the exercise prescription in Tele-Gerofit follows the same process as center-based Gerofit.
Each patient is given an exercise prescription written to address any impairments noted in the different domains of the physical performance assessment, scored using age and sex percentiles. For instance, individuals scoring poorly on lower body strength are given specific lower body strengthening exercises. Participants are given an exercise program that guides them toward achieving recommended physical activity guidelines using their RPE to modulate each exercise. Duration and intensity of each type of planned exercise are formally discussed after initial and follow-up assessments. In addition, exercise training is informally progressed throughout the program. For Tele-Gerofit, instructors must design each class with the group in mind while being prepared for modifications and specific changes for individuals.
Discussion
Tele-Gerofit adapts the well-established center-based Gerofit program to be executed without an exercise facility while maintaining the content of the evidence-based procedures. Physical performance testing and exercise training were modified, adding elements necessary for CVT assessments and classes to be broadcast from the Salem VAMC to its affiliated CBOCs. Tele-Gerofit exercises are performed in a circuit style that allows a veteran or small structured groups of veterans to move among exercises and requires less space than traditional group exercise does. Safety and monitoring concerns are addressed with a safety procedure that includes emergency plans for each site, prescreening of enrolled participants, and monitoring of exercise intensity in accordance with national guidelines.1 Similar to the center-based Gerofit program, the exercise prescription is tailored to each veteran’s physical limitations based on initial and ongoing assessment of physical performance. Tele-Gerofit physical performance testing fulfills the same need with only a few modifications using validated measures. Tele-Gerofit assessments are administered by CVT without the need for additional staff on site.
Adaptation of center-based Gerofit exercise classes to Tele-Gerofit is a major innovation. Use of a circuit exercise design was supported by findings in older adults that RT alone, when performed quickly with minimal rest between each set and exercise station, increases both aerobic capacity and strength.23,24 Older adult RT trials that compared circuit RT with traditional RT found that strength gains are comparable between circuit and traditional RT.24-26 Working with adults aged > 60 years, Takeshima and colleagues conducted a trial of circuit exercise with added callisthenic exercises performed in place between RT on exercise machines.27 This dual-modality (AEX+RT) circuit approach was well tolerated and effective, increasing aerobic capacity and strength. Unfortunately, the resistance exercise machines used in those circuit exercise studies and in the center-based Gerofit program are not an option for Tele-Gerofit.
The requirement for an exercise facility was removed by designing Tele-Gerofit exercise to include only functional exercises that rely on body weight or small mobile exercise equipment. Although popular among young adults, functional circuit exercise is understudied in older adults. Recently, a 12-week functional circuit exercise intervention in frail elderly adults demonstrated significant improvements in gait speed and the timed chair-stand test.28 A pilot observational study of Gerofit participants at the Canandaigua VAMC offered 27 veterans functional circuit exercise instead of their traditional exercise facility class and found larger increases in the timed chair-stand test and 6-minute walk distance compared with 11 Gerofit participants in the traditional program.29
This Tele-Gerofit exercise training combines functional and circuit exercise strategies into telehealth delivery. However, its effect on physical performance remains to be demonstrated. To address this question, we are conducting a single-arm pilot study of Tele-Gerofit with CVT broadcast to 3 Salem CBOC affiliates (Wytheville, Staunton, and Danville, Virginia). The goal is to determine the effect on physical performance and collect feasibility data, including attendance rate and patient satisfaction with the video broadcast. In addition, we are planning an effectiveness trial to compare the impact of functional circuit exercise delivered in person (center based, not CVT) with the parent Gerofit exercise program on direct measures of endurance and strength, in addition to physical performance.
Related: Setting and Method of Measurement Affect Blood Pressure Readings in Older Veterans
Implementation research is needed to determine how Tele-Gerofit can be disseminated to other VAMCs and community-based centers beyond CBOCs. Although the cost of the equipment used to implement Tele-Gerofit is minimal, the program requires dedicated and experienced exercise instructors, and the sharing of telehealth resources with other clinical programs. The authors expect that a diverse group of stakeholders is needed across service lines of primary care, geriatrics and extended care, physical medicine and rehabilitation, and telehealth. Of note, this multidisciplinary collaboration is a hallmark of the Gerofit program. The recent success of the implementation of center-based Gerofit in VAMCs across the US demonstrates the program’s flexibility and robust results.18
Plans also include refining strategies for physical performance testing and exercise monitoring. For instance, we would like to adapt telehealth technology for heart rate monitors that can be worn by high-risk veterans at the CBOC and viewed in real time by the exercise instructor.
Conclusion
Gerofit, which is designed to help older veterans maintain independent living and prevent disability, has been demonstrated to improve quality of life and survival. Our goal has been to adapt Gerofit to CVT and provide a supervised, individualized exercise program in a group setting—a program that can be widely disseminated. Salem VAMC Tele-Gerofit is an innovative and prescriptive program that delivers CVT functional circuit exercise training to remote locations without the need for stationary exercise equipment. This approach has the potential to become an effective and feasible exercise strategy for preventing and minimizing disability in the increasing population of older veterans. Work is needed to determine whether Tele-Gerofit provides a rapid translation of Gerofit to clinical practice and improved outcomes with substantial cost savings from reduced hospitalization and institutionalization.
Acknowledgments
Gerofit has been funded by the Veterans Health Affairs Office of Geriatrics and Extended Care Non-Institutional Long-Term Care Funding and Mentored Partnership Program, and the Veterans Health Affairs Office of Rural Health Rural Enterprise-Wide Initiative.
The authors thank Kim Birkett, MPH, for assistance in editing, references, and graphics and the staff at the Wytheville, Staunton, and Danville community-based outpatient clinics for their support.
Exercise increases endurance, muscle strength, and functional performance with corresponding gains in mobility, survival, and quality of life.1 However, even with these benefits and improvements in clinical outcomes, only 15% of adults aged ≥ 65 years follow current guidelines for exercise.2 Despite their prior military training, the majority of veterans do not meet physical activity recommendations.3 Time, travel, and support are common barriers to exercise participation and adherence—barriers that are further amplified among older adults.
The Veterans Health Administration (VHA) is recognized as a world leader in telehealth service development. Currently, 677,000 veterans have received telehealth services, which represents 12% of the 5.6 million veterans under VHA care.4 Clinical video telehealth (CVT) is widely used within the VHA system to deliver health care that otherwise would not be available to veterans. Veterans who have difficulty traveling to the nearest US Department of Veteran Affairs (VA) medical center (VAMC) can access CVT programs at a participating VHA community-based outpatient clinic (CBOC). The VA has more than 45 CVT programs, including programs for mental health, weight management, cardiology, and dermatology. Outside the VA, cardiac exercise rehabilitation provided by CVT has been shown to be as effective as center-based programs in improving cardiovascular risk factors and functional capacity.5 A VHA exercise program that leveraged CVT resources and was dedicated to older adults with a wide range of comorbid conditions would have a high impact on the health and well-being of older veterans.
Gerofit is a VHA clinical demonstration program of supervised center-based exercise for veterans aged ≥ 65 years. Developed at the Durham VAMC Geriatric Research, Education, and Clinical Center (GRECC) in North Carolina, it has demonstrated improved clinical outcomes, including physical function, mobility, quality of life, and survival.6-10 The program offers veterans individualized exercise in a group setting that focuses on improving endurance, strength, and balance. The exercise prescription is based on the patient’s physical limitations as identified in a physical performance assessment.
With support from VHA Geriatric Extended Care (GEC) and the Office of Rural Health (ORH), Gerofit was implemented in 10 VAMCs across 8 VISNs. However, barriers such as travel time, distance, and transportation limit participation. Previously, we found that rural veterans lack access to exercise programs.11,12 Although some do aerobic exercise (AEX), most do not do resistance training (RT), though they are willing to learn. Access to Gerofit for rural veterans is expanding with recent support from the ORH Enterprise Wide Initiative. Rural program expansion includes several different Gerofit initiatives, many involving CBOCs.
The Salem VAMC Gerofit program sought to adapt the facility-based assessment and exercise procedures into a self-reliant CVT class for its CBOCs. This article describes the development of the Salem VAMC Gerofit CVT program, hereafter referred to as Tele-Gerofit.
Related: Expanding the Scope of Telemedicine in Gastroenterology
Program Design
Gerofit was established in 1986 at the Durham GRECC as an exercise and health promotion program for veterans aged ≥ 65 years.13 Its goal is to prevent or improve functional decline from physical inactivity and age-related conditions. Gerofit targets the geriatric patient population and thus extends beyond cardiac and pulmonary rehabilitation or weight loss programs. The primary exclusion criteria are based on safety issues in the context of a group exercise setting of older adults and include oxygen dependency, unstable cardiac disease, and moderate-to-severe cognitive impairment.
To participate in Gerofit, veterans must be able to perform activities of daily living and self-manage an exercise prescription developed by the exercise instructor based on physical performance testing. These physical performance tests include measures that are independent predictors of disability, loss of independent living, and death, as well as surrogate measures of exercise capacity (eg, strength, endurance, balance).14,15 A novel aspect of Gerofit is that the physical performance assessment is used not only to determine physical limitations, but also to individualize the exercise prescription based on the observed deficits in strength, endurance, or balance. These assessments are performed at initial enrollment; 3 months, 6 months, and 1 year later; and annually after that. Currently, the center-based Gerofit programs administer 5 items of the Senior Fitness Test: 6-minute walk, 10-meter walk (10-MWT), 30-second 1-arm curl, 30-second chair-stand test, and 8-foot up-and-go.15 The side-by-side, semitandem, and tandem standing balance tests from the short physical performance battery also are performed.16 In addition, participants complete a questionnaire that includes items from the physical functioning scale of the 12-Item Short Form Health Survey (SF-12).
After each assessment, the Gerofit exercise instructor reviews the results with the veteran and formulates an individualized exercise prescription along with goals for improvement. Veterans are encouraged to attend supervised center-based exercise sessions 3 times weekly. Classes are offered in a gym or fitness center at the VAMC or in leased space. Each patient uses a cue card that lists an exercise plan personalized for intensity and duration for aerobic exercise (AEX; eg, treadmill walking, stationary bicycling, arm ergometry), RT using dumbbells and weight equipment, and functional exercises for flexibility and balance. Some medical centers also offer yoga, tai chi, or dancing Gerofit classes.
For participants in the Durham Gerofit program, mortality decreased 25% over a decade (hazard ratio, 0.75; 95% CI, 0.61-0.91).9 A substudy that included the Psychological General Well-Being Index found that 81% of participants significantly increased their score after 1 year.7 Observed initial improvement in physical performance has been sustained over 5 years.10,17 One-year results from the recent Gerofit expansion to 6 other VAMCs showed clinically and statistically significantly improved physical performance from baseline to 3-, 6-, and 12-month follow-up.18
Adaptation of Gerofit to CVT Delivery
Initial work. The Greater Los Angeles VAMC Gerofit program conducted a pilot CVT exercise class of 6 veterans at the rural Bakersfield CBOC in California.19 Each week, an exercise instructor broadcast a 60-minute exercise class that included warm-up, RT with bands, progressive balance training, and flexibility. Trained student volunteers from California State University in Bakersfield kinesiology program were on site at the Bakersfield CBOC to perform the assessments and aid in exercises during the CVT sessions. Despite the lack of AEX per se, veterans showed significant improvement in endurance as measured by an increase in the number of steps completed in 2 minutes at the 3-month assessment (P = .049). Although exercises were not delivered in a circuit format, the improved endurance supported the potential for cardiovascular benefit from RT in older adults.
This pilot project also demonstrated that key components of the Gerofit program could be delivered safely by telehealth with onsite supervision. The Miami VA Healthcare System also offers CVT Gerofit exercise classes broadcast to the rural Florida CBOCs of Key Largo and Homestead.11 The exercise activities offered for the Miami CVT participants incorporate components of AEX (calisthenics) and RT (resistance bands). Veterans enjoyed the classes, and adherence was good. However, availability of staff and space are an ongoing challenge.
In Key Largo, 5 veterans participated before the CVT classes were placed on hold owing to the demands of other CVT programs and limited availability of the telehealth clinical technician (TCT). The Homestead CBOC continues to offer CVT Gerofit exercise classes and has 6 regular participants. Notably, the physical space at the Homestead CBOC is smaller than that at the Key Largo CBOC; the Homestead CBOC has adjusted by shifting to exercises performed while standing or sitting, ensuring participants’ safety and satisfaction.
The Baltimore, Maryland VAMC Gerofit program offers other innovative CVT exercise classes, including a tai chi class, and a class with exercise performed while sitting in a chair. Although the Baltimore VAMC CVT exercise classes do not have the scope of the center-based exercise prescriptions, they are unique in that they are broadcast not only to their affiliated CBOCs, but also other Gerofit programs in different VISNs.
Related: Telehealth for Rural Veterans With Neurologic Disorders
Salem VAMC Gerofit Program. The center-based Salem VAMC Gerofit program was established in July 2015. In fiscal year 2017, its dedicated exercise facility had more than 5,000 patient visits. Despite the program’s success, we prioritized establishing Tele-Gerofit because of the medical center’s rural location in southwest Virginia and the large number of veterans who receive care at CBOCs. Therefore, much as with the pilot CVT Gerofit classes in Los Angeles and Miami, the target setting was rural CBOCs. The goal for Salem VAMC Tele-Gerofit was to modify Gerofit delivery to the CVT format and a CBOC setting with minimal modification of the content and personnel requirements of both physical performance testing and exercise training procedures.
Adjustments for CBOC Setting. The enrollment process for Tele-Gerofit is the same as that for the center-based program. To start, a veteran’s primary care provider reviews the list of eligibility criteria and, if the veteran qualifies, places a consult. A Gerofit team member then contacts the veteran by phone to describe the program and schedule an assessment. At the baseline physical performance assessment, American College of Sports Medicine guidelines on exercise participation, health screening, and exercise intensity are used to evaluate veterans and rank them by their cardiovascular risk.20 All new program participants start with low-intensity exercise and gradually progress to recommended levels of exercise. Before starting an exercise class, participants are instructed on use of the 10-point rating of perceived exertion (RPE).
Each CBOC site is supplied with an RPE poster that is displayed for participants’ use. During a Tele-Gerofit class, the exercise instructor asks participants to periodically report their RPE. This class differs slightly from the center-based exercise sessions in which RPE is primarily assessed when a different exercise is introduced or the duration or intensity of an exercise is increased. The Gerofit instructor monitors exercise and treatment fidelity, but the onsite TCT observes for safety during class. The TCT also takes initial vital signs and sets up the room for the class. Emergency contacts and procedures are posted in each CBOC CVT room and are available to the center-based exercise instructor. Because the CBOCs are not inside medical facilities, some CBOC directors have asked that heart rate monitors be used as an extra safety precaution to ensure that high-risk participants do not exceed a heart rate limit that may be set by their cardiologists.
Modifications to Physical Performance Assessment. Physical performance testing had to be adapted to the small rooms available at the CBOCs. For measuring normal gait speed, the 10-MWT was replaced with the 4-meter walk test (4-MWT). The 4-MWT has excellent test–retest reliability with an intraclass correlation coefficient (ICC) of 0.93, but the discrepancy in gait speed between the 4-MWT and the 10-MWT is such that the tests cannot be used interchangeably.21 For measuring endurance, the 6-minute walk test was replaced with the 2-minute step test (2-MST). In older adults, the 2-MST has a moderate correlation with 6-minute walk distance (r = 0.36; P = .04) and high reliability (ICC, 0.90).15,22 The 30-second 1-arm curl, the 30-second chair-stand test, and the 8-foot up-and-go test are performed without modification and require only dumbbells, a chair without wheels, and a stopwatch.
The exercise instructor at the Salem VAMC conducts physical performance testing by 2-way videoconferencing with the veteran in a room at the CBOC. The TCT at the CBOC assists by measuring and demarcating 4 meters on the floor and a designated height on the wall for knee elevation for 4-MWT and 2-MST, respectively. The TCT remains in the room during the assessment visit. Except for taking vital signs before and after the physical performance assessment, the TCT does not participate in the testing. To date, more than 20 physical performance assessments have been conducted without difficulty at Salem-affiliated CBOCs. The primary challenge has been scheduling the room with CVT equipment (ie, camera and screen) for the 30-minute individual assessment session, which occurs on a rolling basis as individuals are enrolled and followed.
After the assessment is completed, the exercise instructor reviews the results with the participant and provides feedback on areas in need of improvement. However, these education sessions can be lengthy and are best supported by giving the patient a personalized handout.
Functional Circuit Exercise. In Tele-Gerofit, exercise training is delivered by CVT broadcast from the Salem VAMC to veterans in a room (equipped with steps, dumbbells, chairs, and bands) at the CBOC. This type of exercise training, which uses only mobile equipment and plyometric (weight-bearing) exercises, is referred to as functional exercise. The AEX includes marching in place, moving on and off a raised step, and body-weight exercises, while RT uses dumbbells, resistance bands, and plyometric exercises (Table 2).
Progression of intensity is achieved by increasing the rate of stepping and the size of the steps (AEX) or the number of repetitions and the weight of the dumbbells or bands (RT). Each veteran exercises at an intensity level that is appropriate for his or her baseline limitations and medical conditions. The exercise instructor uses different forms of the same equipment (eg, heavier dumbbells, higher steps) to vary intensity among individuals while having them perform the same exercises as a group. The challenge is to adjust the pace of the AEX or the timing of the RT repetitions for individuals new to the class.
Delivery of exercise training in the form of circuits allows for a diverse exercise program in a setting with limited space. Circuit training is an exercise modality that consists of a series of different exercises, each usually completed in 30 to 60 seconds, with minimal rest between each type of exercise. Each Tele-Gerofit circuit has a mix of AEX and RT exercises performed for 3 minutes consecutively (Figure).
The design of the circuit training can be adjusted based on the number of individuals in the class. Larger classes can be split into 2 groups that alternate between exercise sets, while smaller classes have 1 group performing the same exercise set and then rotating to either the AEX or RT set. Total exercise time to complete the circuit depends on the number of different exercises, number of repetitions, and the rest between repetitions and the different exercises. In this way, total exercise time can be made shorter or longer depending on the veteran’s capacity.
Frequency. Tele-Gerofit exercise classes are currently offered twice weekly and last about 1 hour, which includes warm-up (8-10 minutes), functional circuit training (40 minutes), and cooldown/stretching (8-10 minutes). A challenge for the exercise instructor is the need to provide ongoing clear instructions both to the class and to individuals as needed. As the exercise prescription for each patient is based on physical performance testing, the exercise instructor for the training must be familiar with the test results. Derivation of the exercise prescription in Tele-Gerofit follows the same process as center-based Gerofit.
Each patient is given an exercise prescription written to address any impairments noted in the different domains of the physical performance assessment, scored using age and sex percentiles. For instance, individuals scoring poorly on lower body strength are given specific lower body strengthening exercises. Participants are given an exercise program that guides them toward achieving recommended physical activity guidelines using their RPE to modulate each exercise. Duration and intensity of each type of planned exercise are formally discussed after initial and follow-up assessments. In addition, exercise training is informally progressed throughout the program. For Tele-Gerofit, instructors must design each class with the group in mind while being prepared for modifications and specific changes for individuals.
Discussion
Tele-Gerofit adapts the well-established center-based Gerofit program to be executed without an exercise facility while maintaining the content of the evidence-based procedures. Physical performance testing and exercise training were modified, adding elements necessary for CVT assessments and classes to be broadcast from the Salem VAMC to its affiliated CBOCs. Tele-Gerofit exercises are performed in a circuit style that allows a veteran or small structured groups of veterans to move among exercises and requires less space than traditional group exercise does. Safety and monitoring concerns are addressed with a safety procedure that includes emergency plans for each site, prescreening of enrolled participants, and monitoring of exercise intensity in accordance with national guidelines.1 Similar to the center-based Gerofit program, the exercise prescription is tailored to each veteran’s physical limitations based on initial and ongoing assessment of physical performance. Tele-Gerofit physical performance testing fulfills the same need with only a few modifications using validated measures. Tele-Gerofit assessments are administered by CVT without the need for additional staff on site.
Adaptation of center-based Gerofit exercise classes to Tele-Gerofit is a major innovation. Use of a circuit exercise design was supported by findings in older adults that RT alone, when performed quickly with minimal rest between each set and exercise station, increases both aerobic capacity and strength.23,24 Older adult RT trials that compared circuit RT with traditional RT found that strength gains are comparable between circuit and traditional RT.24-26 Working with adults aged > 60 years, Takeshima and colleagues conducted a trial of circuit exercise with added callisthenic exercises performed in place between RT on exercise machines.27 This dual-modality (AEX+RT) circuit approach was well tolerated and effective, increasing aerobic capacity and strength. Unfortunately, the resistance exercise machines used in those circuit exercise studies and in the center-based Gerofit program are not an option for Tele-Gerofit.
The requirement for an exercise facility was removed by designing Tele-Gerofit exercise to include only functional exercises that rely on body weight or small mobile exercise equipment. Although popular among young adults, functional circuit exercise is understudied in older adults. Recently, a 12-week functional circuit exercise intervention in frail elderly adults demonstrated significant improvements in gait speed and the timed chair-stand test.28 A pilot observational study of Gerofit participants at the Canandaigua VAMC offered 27 veterans functional circuit exercise instead of their traditional exercise facility class and found larger increases in the timed chair-stand test and 6-minute walk distance compared with 11 Gerofit participants in the traditional program.29
This Tele-Gerofit exercise training combines functional and circuit exercise strategies into telehealth delivery. However, its effect on physical performance remains to be demonstrated. To address this question, we are conducting a single-arm pilot study of Tele-Gerofit with CVT broadcast to 3 Salem CBOC affiliates (Wytheville, Staunton, and Danville, Virginia). The goal is to determine the effect on physical performance and collect feasibility data, including attendance rate and patient satisfaction with the video broadcast. In addition, we are planning an effectiveness trial to compare the impact of functional circuit exercise delivered in person (center based, not CVT) with the parent Gerofit exercise program on direct measures of endurance and strength, in addition to physical performance.
Related: Setting and Method of Measurement Affect Blood Pressure Readings in Older Veterans
Implementation research is needed to determine how Tele-Gerofit can be disseminated to other VAMCs and community-based centers beyond CBOCs. Although the cost of the equipment used to implement Tele-Gerofit is minimal, the program requires dedicated and experienced exercise instructors, and the sharing of telehealth resources with other clinical programs. The authors expect that a diverse group of stakeholders is needed across service lines of primary care, geriatrics and extended care, physical medicine and rehabilitation, and telehealth. Of note, this multidisciplinary collaboration is a hallmark of the Gerofit program. The recent success of the implementation of center-based Gerofit in VAMCs across the US demonstrates the program’s flexibility and robust results.18
Plans also include refining strategies for physical performance testing and exercise monitoring. For instance, we would like to adapt telehealth technology for heart rate monitors that can be worn by high-risk veterans at the CBOC and viewed in real time by the exercise instructor.
Conclusion
Gerofit, which is designed to help older veterans maintain independent living and prevent disability, has been demonstrated to improve quality of life and survival. Our goal has been to adapt Gerofit to CVT and provide a supervised, individualized exercise program in a group setting—a program that can be widely disseminated. Salem VAMC Tele-Gerofit is an innovative and prescriptive program that delivers CVT functional circuit exercise training to remote locations without the need for stationary exercise equipment. This approach has the potential to become an effective and feasible exercise strategy for preventing and minimizing disability in the increasing population of older veterans. Work is needed to determine whether Tele-Gerofit provides a rapid translation of Gerofit to clinical practice and improved outcomes with substantial cost savings from reduced hospitalization and institutionalization.
Acknowledgments
Gerofit has been funded by the Veterans Health Affairs Office of Geriatrics and Extended Care Non-Institutional Long-Term Care Funding and Mentored Partnership Program, and the Veterans Health Affairs Office of Rural Health Rural Enterprise-Wide Initiative.
The authors thank Kim Birkett, MPH, for assistance in editing, references, and graphics and the staff at the Wytheville, Staunton, and Danville community-based outpatient clinics for their support.
1. American College of Sports Medicine, Chodzko-Zajko WJ, Proctor DN, et al. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med Sci Sports Exerc. 2009;41(7):1510-1530.
2. Centers for Disease Control and Prevention. Adult participation in aerobic and muscle-strengthening physical activities—United States, 2011. MMWR Morb Mortal Wkly Rep. 2013;62(17):326-330.
3. Littman AJ, Forsberg CW, Koepsell TD. Physical activity in a national sample of veterans. Med Sci Sports Exerc. 2009;41(5):1006-1013.
4. US Department of Veterans Affairs, Office of Rural Health. Annual Report: Thrive 2015. https://www.ruralhealth.va.gov/docs/ORH_Annual_Report_2015_FINAL.pdf. Published 2015. Accessed July 16, 2018.
5. Rawstorn JC, Gant N, Direito A, Beckmann C, Maddison R. Telehealth exercise-based cardiac rehabilitation: a systematic review and meta-analysis. Heart. 2016;102(15):1183-1192.
6. Morey MC. Celebrating 20 years of excellence in exercise for the older veteran. Fed Pract. 2007;24(10):49-65.
7. Cowper PA, Morey MC, Bearon LB, et al. The impact of supervised exercise on the psychological well-being and health status of older veterans. J Appl Gerontol. 1991;10(4):469-485.
8. Morey MC, Cowper PA, Feussner JR, et al. Evaluation of a supervised exercise program in a geriatric population. J Am Geriatr Soc. 1989;37(4):348-354.
9. Morey MC, Pieper CF, Crowley GM, Sullivan RJ, Puglisi CM. Exercise adherence and 10-year mortality in chronically ill older adults. J Am Geriatr Soc. 2002;50(12):1929-1933.
10. Morey MC, Pieper CF, Sullivan RJ Jr, Crowley GM, Cowper PA, Robbins MS. Five-year performance trends for older exercisers: a hierarchical model of endurance, strength, and flexibility. J Am Geriatr Soc. 1996;44(10):1226-1231.
11. Valencia WM, Botros D, Pendlebury D, et al. Proactive reach and telehealth monitoring (Gerofit) enhance resistance exercise at rural setting. Innovat Aging. 2017;1(suppl 1):225.12. Pendlebury D, Botros D VW. Proactive Reach: an innovative access approach to identify & deliver GEROFIT exercise telehealth counseling to rural veterans & enhance CBOC services. J Am Geriatr Soc. 2017(suppl 1):S208. Poster presented at: Annual Scientific Meeting of the American Geriatrics Society; May 18, 2017; San Antonio, TX.
13. Morey MC, Crowley GM, Robbins MS, Cowper PA, Sullivan RJ Jr. The Gerofit program: a VA innovation. South Med J. 1994;87(5):S83-S87.
14. Cooper R, Kuh D, Hardy R; Mortality Review Group; FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: systematic review and meta-analysis. BMJ. 2010;341:c4467.
15. Rikli RE, Jones CJ. Development and validation of a functional fitness test for community-residing older adults. J Aging Phys Act. 1999;7(2):129-161.
16. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol. 1994;49(2):M85-M94.
17. Morey MC, Cowper PA, Feussner JR, et al. Two-year trends in physical performance following supervised exercise among community-dwelling older veterans. J Am Geriatr Soc. 1991;39(10):986-992.
18. Morey MC, Lee CC, Castle S, et al. Should structured exercise be promoted as a model of care? Dissemination of the Department of Veterans Affairs Gerofit program. J Am Geriatr Soc. 2018;66(5):1009-1016.
19. Blanchard E, Castle S, Ines E, et al. Delivering a clinical exercise program to rural veterans via video telehealth. Poster C167 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
20. Riebe D, Ehrman JK, Liguori G, Magal M, eds; American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription. 10th ed. Philadelphia, PA: Wolters Kluwer Health; 2018.
21. Peters DM, Fritz SL, Krotish DE. Assessing the reliability and validity of a shorter walk test compared with the 10-meter walk test for measurements of gait speed in healthy, older adults. J Geriatr Phys Ther. 2013;36(1):24-30.
22. Pedrosa R, Holanda G. Correlation between the walk, 2-minute step and TUG tests among hypertensive older women. Rev Bras Fisioter. 2009;13(3):252-256.
23. Romero-Arenas S, Blazevich AJ, Martinez-Pascual M, et al. Effects of high-resistance circuit training in an elderly population. Exp Gerontol. 2013;48(3):334-340.
24. Brentano MA, Cadore EL, Da Silva EM, et al. Physiological adaptations to strength and circuit training in postmenopausal women with bone loss. J Strength Cond Res. 2008;22(6):1816-1825.
25. Romero-Arenas S, Martinez-Pascual M, Alcaraz PE. Impact of resistance circuit training on neuromuscular, cardiorespiratory and body composition adaptations in the elderly. Aging Dis. 2013;4(5):256-263.
26. Paoli A, Pacelli F, Bargossi AM, et al. Effects of three distinct protocols of fitness training on body composition, strength and blood lactate. J Sports Med Phys Fitness. 2010;50(1):43-51.
27. Takeshima N, Rogers ME, Islam MM, Yamauchi T, Watanabe E, Okada A. Effect of concurrent aerobic and resistance circuit exercise training on fitness in older adults. Eur J Appl Physiol. 2004;93(1-2):173-182.
28. Giné-Garriga M, Guerra M, Pagés E, Manini TM, Jiménez R, Unnithan VB. The effect of functional circuit training on physical frailty in frail older adults: a randomized controlled trial. J Aging Phys Act. 2010;18(4):401-424.
29. Biddle ED, Reynolds P, Kopp T, Cammarata H, Conway P. Implementation of functional training tools elicits improvements in aerobic fitness and lower body strength in older veterans. Poster C169 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
1. American College of Sports Medicine, Chodzko-Zajko WJ, Proctor DN, et al. American College of Sports Medicine position stand. Exercise and physical activity for older adults. Med Sci Sports Exerc. 2009;41(7):1510-1530.
2. Centers for Disease Control and Prevention. Adult participation in aerobic and muscle-strengthening physical activities—United States, 2011. MMWR Morb Mortal Wkly Rep. 2013;62(17):326-330.
3. Littman AJ, Forsberg CW, Koepsell TD. Physical activity in a national sample of veterans. Med Sci Sports Exerc. 2009;41(5):1006-1013.
4. US Department of Veterans Affairs, Office of Rural Health. Annual Report: Thrive 2015. https://www.ruralhealth.va.gov/docs/ORH_Annual_Report_2015_FINAL.pdf. Published 2015. Accessed July 16, 2018.
5. Rawstorn JC, Gant N, Direito A, Beckmann C, Maddison R. Telehealth exercise-based cardiac rehabilitation: a systematic review and meta-analysis. Heart. 2016;102(15):1183-1192.
6. Morey MC. Celebrating 20 years of excellence in exercise for the older veteran. Fed Pract. 2007;24(10):49-65.
7. Cowper PA, Morey MC, Bearon LB, et al. The impact of supervised exercise on the psychological well-being and health status of older veterans. J Appl Gerontol. 1991;10(4):469-485.
8. Morey MC, Cowper PA, Feussner JR, et al. Evaluation of a supervised exercise program in a geriatric population. J Am Geriatr Soc. 1989;37(4):348-354.
9. Morey MC, Pieper CF, Crowley GM, Sullivan RJ, Puglisi CM. Exercise adherence and 10-year mortality in chronically ill older adults. J Am Geriatr Soc. 2002;50(12):1929-1933.
10. Morey MC, Pieper CF, Sullivan RJ Jr, Crowley GM, Cowper PA, Robbins MS. Five-year performance trends for older exercisers: a hierarchical model of endurance, strength, and flexibility. J Am Geriatr Soc. 1996;44(10):1226-1231.
11. Valencia WM, Botros D, Pendlebury D, et al. Proactive reach and telehealth monitoring (Gerofit) enhance resistance exercise at rural setting. Innovat Aging. 2017;1(suppl 1):225.12. Pendlebury D, Botros D VW. Proactive Reach: an innovative access approach to identify & deliver GEROFIT exercise telehealth counseling to rural veterans & enhance CBOC services. J Am Geriatr Soc. 2017(suppl 1):S208. Poster presented at: Annual Scientific Meeting of the American Geriatrics Society; May 18, 2017; San Antonio, TX.
13. Morey MC, Crowley GM, Robbins MS, Cowper PA, Sullivan RJ Jr. The Gerofit program: a VA innovation. South Med J. 1994;87(5):S83-S87.
14. Cooper R, Kuh D, Hardy R; Mortality Review Group; FALCon and HALCyon Study Teams. Objectively measured physical capability levels and mortality: systematic review and meta-analysis. BMJ. 2010;341:c4467.
15. Rikli RE, Jones CJ. Development and validation of a functional fitness test for community-residing older adults. J Aging Phys Act. 1999;7(2):129-161.
16. Guralnik JM, Simonsick EM, Ferrucci L, et al. A short physical performance battery assessing lower extremity function: association with self-reported disability and prediction of mortality and nursing home admission. J Gerontol. 1994;49(2):M85-M94.
17. Morey MC, Cowper PA, Feussner JR, et al. Two-year trends in physical performance following supervised exercise among community-dwelling older veterans. J Am Geriatr Soc. 1991;39(10):986-992.
18. Morey MC, Lee CC, Castle S, et al. Should structured exercise be promoted as a model of care? Dissemination of the Department of Veterans Affairs Gerofit program. J Am Geriatr Soc. 2018;66(5):1009-1016.
19. Blanchard E, Castle S, Ines E, et al. Delivering a clinical exercise program to rural veterans via video telehealth. Poster C167 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
20. Riebe D, Ehrman JK, Liguori G, Magal M, eds; American College of Sports Medicine. ACSM’s Guidelines for Exercise Testing and Prescription. 10th ed. Philadelphia, PA: Wolters Kluwer Health; 2018.
21. Peters DM, Fritz SL, Krotish DE. Assessing the reliability and validity of a shorter walk test compared with the 10-meter walk test for measurements of gait speed in healthy, older adults. J Geriatr Phys Ther. 2013;36(1):24-30.
22. Pedrosa R, Holanda G. Correlation between the walk, 2-minute step and TUG tests among hypertensive older women. Rev Bras Fisioter. 2009;13(3):252-256.
23. Romero-Arenas S, Blazevich AJ, Martinez-Pascual M, et al. Effects of high-resistance circuit training in an elderly population. Exp Gerontol. 2013;48(3):334-340.
24. Brentano MA, Cadore EL, Da Silva EM, et al. Physiological adaptations to strength and circuit training in postmenopausal women with bone loss. J Strength Cond Res. 2008;22(6):1816-1825.
25. Romero-Arenas S, Martinez-Pascual M, Alcaraz PE. Impact of resistance circuit training on neuromuscular, cardiorespiratory and body composition adaptations in the elderly. Aging Dis. 2013;4(5):256-263.
26. Paoli A, Pacelli F, Bargossi AM, et al. Effects of three distinct protocols of fitness training on body composition, strength and blood lactate. J Sports Med Phys Fitness. 2010;50(1):43-51.
27. Takeshima N, Rogers ME, Islam MM, Yamauchi T, Watanabe E, Okada A. Effect of concurrent aerobic and resistance circuit exercise training on fitness in older adults. Eur J Appl Physiol. 2004;93(1-2):173-182.
28. Giné-Garriga M, Guerra M, Pagés E, Manini TM, Jiménez R, Unnithan VB. The effect of functional circuit training on physical frailty in frail older adults: a randomized controlled trial. J Aging Phys Act. 2010;18(4):401-424.
29. Biddle ED, Reynolds P, Kopp T, Cammarata H, Conway P. Implementation of functional training tools elicits improvements in aerobic fitness and lower body strength in older veterans. Poster C169 presented at: Annual Scientific Meeting of the American Geriatrics Society; May 19-21, 2016; Long Beach, CA.
Low Spinal Cord Volume Associated With Increased MS Disability
Total and regional brain volumes may differ between patients whose level of disability does not correspond with their lesion load.
BERLIN—Spinal cord volume deficits in patients with multiple sclerosis (MS) may explain clinical disability that appears out of proportion to lesion load on brain imaging, according to research presented at ECTRIMS 2018.
In a pool of 362 patients with mild to moderate MS-related disability and identical white matter lesion load identified by MRI, those with higher disability had significantly lower spinal cord volumes, compared with those with mild disability.
An Analysis of Patient Records
Some patients with MS have a relatively high level of disability, but a low burden of white matter intracerebral lesions on MRI. Little is known about spinal cord volume in patients with MS and a pronounced dissociation between intracerebral lesion load and disability, said Michaela Andelova, MD, of Charles University in Prague.

Dr. Andelova and her colleagues hypothesized that spinal cord volume would differ between patients with varying levels of disability despite identical white matter lesion load. To test this hypothesis, they looked at records for 1,245 patients with relapsing-remitting MS. They divided them into three groups by severity of clinical disability and extent of cerebral T2 hyperintense lesion load. The investigators identified 53 patients whose total volume of T2-weighted hyperintense lesions was less than 3 mL, but whose Expanded Disability Status Scale (EDSS) scores were at least 3.5. They called them the low lesion load–high disability (LLHD) group.
The researchers then identified another 71 patients with a volume of T2-weighted hyperintensities greater than 9 mL, but whose EDSS score was less than 1.5. They called them the high lesion load–low disability (HLLD) group.
The remaining 1,121 patients, who did not have these paradoxical associations, were analyzed separately.The investigators measured mean upper cervical cord area (MUCCA) for all patients. On the basis of images acquired by a 3-T MRI scanner, they used an in-house, semiautomated method to calculate MUCCA as the mean sum of spinal cord area in 21 slices centered at the C3–C4 intervertebral disk.
Mean Upper Cervical Cord Area Correlated With Disability
“Despite higher disability, LLHD patients demonstrated significantly higher normalized total brain volume, higher normalized volumes of the thalamus and callosum, and smaller lateral ventricles than the HLLD group,” said Dr. Andelova and her collaborators.
However, the LLHD patients had MUCCA values that were significantly lower than those of the other groups. The nonparadoxical group’s mean MUCCA was 84.02 mm2, while the HLLD group had a mean MUCCA of 85.75 mm2. The difference between these groups was not statistically significant. By contrast, the LLHD group’s mean MUCCA was significantly smaller than that of the other groups, at 80.40 mm2.
Next, Dr. Andelova and her colleagues compared 362 patients with moderate disability (ie, EDSS scores between 3.5 and 6.5) with matched patients who had mild MS-related disability (ie, EDSS score less than 3) and identical cerebral lesion loads. They found that MUCCA was significantly smaller in the group with moderate disability (78.86 mm2 vs 84.44 mm2).
In addition to having identical lesion loads, the mild and moderate disability groups had similar normalized total brain volume and regional brain volumes. The group with moderate disability had slightly less white matter volume, said Dr. Andelova. All differences between groups retained statistical significance after adjustment for potential confounders such as age, sex, and duration of disease.
“Reduced spinal cord volume may explain part of the clinical–radiologic paradox in patients who have high disability despite low intracranial lesion load,” said the researchers. “In line with this finding, relatively preserved spinal cord volume may be associated with functional reserve and less physical disability in patients with low disability despite high cerebral lesion load.”
Dr. Andelova and her collaborators plan to examine cerebral lesion distribution and perform quantitative MRI investigations of lesion distribution. They intend to seek potential associations between various distribution patterns and accelerated spinal atrophy.
—Kari Oakes
Total and regional brain volumes may differ between patients whose level of disability does not correspond with their lesion load.
Total and regional brain volumes may differ between patients whose level of disability does not correspond with their lesion load.
BERLIN—Spinal cord volume deficits in patients with multiple sclerosis (MS) may explain clinical disability that appears out of proportion to lesion load on brain imaging, according to research presented at ECTRIMS 2018.
In a pool of 362 patients with mild to moderate MS-related disability and identical white matter lesion load identified by MRI, those with higher disability had significantly lower spinal cord volumes, compared with those with mild disability.
An Analysis of Patient Records
Some patients with MS have a relatively high level of disability, but a low burden of white matter intracerebral lesions on MRI. Little is known about spinal cord volume in patients with MS and a pronounced dissociation between intracerebral lesion load and disability, said Michaela Andelova, MD, of Charles University in Prague.
Dr. Andelova and her colleagues hypothesized that spinal cord volume would differ between patients with varying levels of disability despite identical white matter lesion load. To test this hypothesis, they looked at records for 1,245 patients with relapsing-remitting MS. They divided them into three groups by severity of clinical disability and extent of cerebral T2 hyperintense lesion load. The investigators identified 53 patients whose total volume of T2-weighted hyperintense lesions was less than 3 mL, but whose Expanded Disability Status Scale (EDSS) scores were at least 3.5. They called them the low lesion load–high disability (LLHD) group.
The researchers then identified another 71 patients with a volume of T2-weighted hyperintensities greater than 9 mL, but whose EDSS score was less than 1.5. They called them the high lesion load–low disability (HLLD) group.
The remaining 1,121 patients, who did not have these paradoxical associations, were analyzed separately.The investigators measured mean upper cervical cord area (MUCCA) for all patients. On the basis of images acquired by a 3-T MRI scanner, they used an in-house, semiautomated method to calculate MUCCA as the mean sum of spinal cord area in 21 slices centered at the C3–C4 intervertebral disk.
Mean Upper Cervical Cord Area Correlated With Disability
“Despite higher disability, LLHD patients demonstrated significantly higher normalized total brain volume, higher normalized volumes of the thalamus and callosum, and smaller lateral ventricles than the HLLD group,” said Dr. Andelova and her collaborators.
However, the LLHD patients had MUCCA values that were significantly lower than those of the other groups. The nonparadoxical group’s mean MUCCA was 84.02 mm2, while the HLLD group had a mean MUCCA of 85.75 mm2. The difference between these groups was not statistically significant. By contrast, the LLHD group’s mean MUCCA was significantly smaller than that of the other groups, at 80.40 mm2.
Next, Dr. Andelova and her colleagues compared 362 patients with moderate disability (ie, EDSS scores between 3.5 and 6.5) with matched patients who had mild MS-related disability (ie, EDSS score less than 3) and identical cerebral lesion loads. They found that MUCCA was significantly smaller in the group with moderate disability (78.86 mm2 vs 84.44 mm2).
In addition to having identical lesion loads, the mild and moderate disability groups had similar normalized total brain volume and regional brain volumes. The group with moderate disability had slightly less white matter volume, said Dr. Andelova. All differences between groups retained statistical significance after adjustment for potential confounders such as age, sex, and duration of disease.
“Reduced spinal cord volume may explain part of the clinical–radiologic paradox in patients who have high disability despite low intracranial lesion load,” said the researchers. “In line with this finding, relatively preserved spinal cord volume may be associated with functional reserve and less physical disability in patients with low disability despite high cerebral lesion load.”
Dr. Andelova and her collaborators plan to examine cerebral lesion distribution and perform quantitative MRI investigations of lesion distribution. They intend to seek potential associations between various distribution patterns and accelerated spinal atrophy.
—Kari Oakes
BERLIN—Spinal cord volume deficits in patients with multiple sclerosis (MS) may explain clinical disability that appears out of proportion to lesion load on brain imaging, according to research presented at ECTRIMS 2018.
In a pool of 362 patients with mild to moderate MS-related disability and identical white matter lesion load identified by MRI, those with higher disability had significantly lower spinal cord volumes, compared with those with mild disability.
An Analysis of Patient Records
Some patients with MS have a relatively high level of disability, but a low burden of white matter intracerebral lesions on MRI. Little is known about spinal cord volume in patients with MS and a pronounced dissociation between intracerebral lesion load and disability, said Michaela Andelova, MD, of Charles University in Prague.
Dr. Andelova and her colleagues hypothesized that spinal cord volume would differ between patients with varying levels of disability despite identical white matter lesion load. To test this hypothesis, they looked at records for 1,245 patients with relapsing-remitting MS. They divided them into three groups by severity of clinical disability and extent of cerebral T2 hyperintense lesion load. The investigators identified 53 patients whose total volume of T2-weighted hyperintense lesions was less than 3 mL, but whose Expanded Disability Status Scale (EDSS) scores were at least 3.5. They called them the low lesion load–high disability (LLHD) group.
The researchers then identified another 71 patients with a volume of T2-weighted hyperintensities greater than 9 mL, but whose EDSS score was less than 1.5. They called them the high lesion load–low disability (HLLD) group.
The remaining 1,121 patients, who did not have these paradoxical associations, were analyzed separately.The investigators measured mean upper cervical cord area (MUCCA) for all patients. On the basis of images acquired by a 3-T MRI scanner, they used an in-house, semiautomated method to calculate MUCCA as the mean sum of spinal cord area in 21 slices centered at the C3–C4 intervertebral disk.
Mean Upper Cervical Cord Area Correlated With Disability
“Despite higher disability, LLHD patients demonstrated significantly higher normalized total brain volume, higher normalized volumes of the thalamus and callosum, and smaller lateral ventricles than the HLLD group,” said Dr. Andelova and her collaborators.
However, the LLHD patients had MUCCA values that were significantly lower than those of the other groups. The nonparadoxical group’s mean MUCCA was 84.02 mm2, while the HLLD group had a mean MUCCA of 85.75 mm2. The difference between these groups was not statistically significant. By contrast, the LLHD group’s mean MUCCA was significantly smaller than that of the other groups, at 80.40 mm2.
Next, Dr. Andelova and her colleagues compared 362 patients with moderate disability (ie, EDSS scores between 3.5 and 6.5) with matched patients who had mild MS-related disability (ie, EDSS score less than 3) and identical cerebral lesion loads. They found that MUCCA was significantly smaller in the group with moderate disability (78.86 mm2 vs 84.44 mm2).
In addition to having identical lesion loads, the mild and moderate disability groups had similar normalized total brain volume and regional brain volumes. The group with moderate disability had slightly less white matter volume, said Dr. Andelova. All differences between groups retained statistical significance after adjustment for potential confounders such as age, sex, and duration of disease.
“Reduced spinal cord volume may explain part of the clinical–radiologic paradox in patients who have high disability despite low intracranial lesion load,” said the researchers. “In line with this finding, relatively preserved spinal cord volume may be associated with functional reserve and less physical disability in patients with low disability despite high cerebral lesion load.”
Dr. Andelova and her collaborators plan to examine cerebral lesion distribution and perform quantitative MRI investigations of lesion distribution. They intend to seek potential associations between various distribution patterns and accelerated spinal atrophy.
—Kari Oakes
Most nonemergent diagnoses can’t be predicted
Also today, opioids have a negative effect on breathing during sleep, the American Academy of Pediatrics renews its public health approach regarding gun injury prevention, and fever and intestinal symptoms can delay diagnosis of Kawasaki disease in children.
Also today, opioids have a negative effect on breathing during sleep, the American Academy of Pediatrics renews its public health approach regarding gun injury prevention, and fever and intestinal symptoms can delay diagnosis of Kawasaki disease in children.
Also today, opioids have a negative effect on breathing during sleep, the American Academy of Pediatrics renews its public health approach regarding gun injury prevention, and fever and intestinal symptoms can delay diagnosis of Kawasaki disease in children.

FDA approves elotuzumab combo for rel/ref MM
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
The U.S. Food and Drug Administration (FDA) has approved elotuzumab (Empliciti®) in combination with pomalidomide and dexamethasone.
The combination is now approved for use in adults with multiple myeloma (MM) who have received at least two prior therapies, including lenalidomide and a proteasome inhibitor.
Elotuzumab is also FDA-approved in combination with lenalidomide and dexamethasone to treat adult MM patients who have received one to three prior therapies.
The FDA’s latest approval of elotuzumab is based on results from the phase 2 ELOQUENT-3 trial, which were presented at the 23rd Congress of the European Hematology Association in June.
ELOQUENT-3 enrolled MM patients who had refractory or relapsed and refractory MM and had received both lenalidomide and a proteasome inhibitor.
The patients were randomized to receive elotuzumab plus pomalidomide and dexamethasone (EPd, n=60) or pomalidomide and dexamethasone (Pd, n=57) in 28-day cycles until disease progression or unacceptable toxicity.
The overall response rate was 53.3% in the EPd arm and 26.3% in the Pd arm (P=0.0029). The rate of complete response or stringent complete response was 8.3% in the EPd arm and 1.8% in the Pd arm.
The median progression-free survival was 10.25 months with EPd and 4.67 months with Pd (hazard ratio=0.54, P=0.0078).
Serious adverse events (AEs) occurred in 22% of patients in the EPd arm and 15% in the Pd arm. The most frequent serious AEs (in the EPd and Pd arms, respectively) were pneumonia (13% and 11%) and respiratory tract infection (7% and 3.6%).
AEs occurring in at least 10% of patients in the EPd arm and at least 5% of those in the Pd arm (respectively) included:
- Constipation (22% and 11%)
- Hyperglycemia (20% and 15%)
- Pneumonia (18% and 13%)
- Diarrhea (18% and 9%)
- Respiratory tract infection (17% and 9%)
- Bone pain (15% and 9%)
- Dyspnea (15% and 7%)
- Muscle spasms (13% and 5%)
- Peripheral edema (13% and 7%)
- Lymphopenia (10% and 1.8%).
Additional results from ELOQUENT-3 can be found in the full prescribing information for elotuzumab, which is available at www.empliciti.com.
Bristol-Myers Squibb and AbbVie are co-developing elotuzumab, with Bristol-Myers Squibb solely responsible for commercial activities.
Underused PV treatments save lives, doc says

Researchers have found evidence to suggest that phlebotomy and hydroxyurea (HU) provide real-world benefits for older patients with polycythemia vera (PV), but both treatments are underused.
In a study of more than 800 PV patients, phlebotomy and HU treatment were both associated with lower risks of death and thrombosis.
However, 39% of patients didn’t receive HU, and 36% didn’t undergo phlebotomy.
“Our study highlights the value of adhering to PV treatment guidelines,” said study author Nikolai A. Podoltsev, MD, PhD, of Yale Cancer Center in New Haven, Connecticut.
“Use of the two recommended treatments saves lives.”
Dr. Podoltsev and his colleagues described this survival benefit in Blood Advances.
The researchers studied data from the linked Surveillance, Epidemiology, and End Results–Medicare database. They collected information on 820 older adults diagnosed with PV from 2007 to 2013.
The patients’ median age was 77 (range, 71-83), 57% were female, 91.2% were white, 12.7% had a disability, and 13.2% had a prior thrombotic event.
Patients received the following PV treatments:
- Both phlebotomy and HU concurrently or sequentially (41.1%)
- Phlebotomy only (23.0%)
- HU only (19.6%)
- Neither phlebotomy nor HU (16.3%).
Survival
The median follow-up was 2.83 years. During that time, 37.2% of patients (n=305) died.
The median survival was 6.29 years for phlebotomy recipients and 4.50 years for non-recipients (P<0.01). The median survival was 6.02 years for HU recipients and 5.25 years for non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with decreased mortality. The hazard ratio (HR) for death was 0.65 (P<0.01) for phlebotomy recipients.
Increasing phlebotomy intensity (the number of phlebotomies per year) was also associated with decreased mortality, with an HR of 0.71 (P<0.01).
A higher proportion of days covered (PDC) by HU treatment was associated with decreased mortality as well.
The researchers said every 10% increase of HU PDC was associated with an 8% to 9% lower risk of death. The HR was 0.92 in a model where phlebotomy was a binary variable and 0.91 in a model that included the frequency of phlebotomy (P<0.01 for both).
Thrombosis
In all, 36.1% of patients (n=296) had a thrombotic event, which includes venous and arterial thrombosis.
The incidence of thrombosis was 29.3% (n=142) in phlebotomy recipients and 46.0% (n=154) in non-recipients (P<0.01). The incidence was 27.6% (n=118) in HU recipients and 45.4% (n=178) in non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with a decreased risk of thrombosis, with an HR of 0.52 (P<0.01).
Increasing phlebotomy intensity was associated with a decreased risk of thrombosis as well, with an HR of 0.46 (P<0.01).
And every 10% increase of HU PDC was associated with an 8% lower risk of thrombosis. The HR was 0.92 in both models (P<0.01 for both).
“All of the patients we studied were high-risk for clot development, and we now know from our findings that guideline-recommended treatments reduce the risk of both thrombosis and death,” Dr. Podoltsev said.
“We hope that our research will raise clinicians’ awareness of and adherence to the guidelines and improve the outcomes of PV patients in the future.”
This research was supported by the Frederick A. DeLuca Foundation. Study authors reported relationships with 24 pharmaceutical companies.
Researchers have found evidence to suggest that phlebotomy and hydroxyurea (HU) provide real-world benefits for older patients with polycythemia vera (PV), but both treatments are underused.
In a study of more than 800 PV patients, phlebotomy and HU treatment were both associated with lower risks of death and thrombosis.
However, 39% of patients didn’t receive HU, and 36% didn’t undergo phlebotomy.
“Our study highlights the value of adhering to PV treatment guidelines,” said study author Nikolai A. Podoltsev, MD, PhD, of Yale Cancer Center in New Haven, Connecticut.
“Use of the two recommended treatments saves lives.”
Dr. Podoltsev and his colleagues described this survival benefit in Blood Advances.
The researchers studied data from the linked Surveillance, Epidemiology, and End Results–Medicare database. They collected information on 820 older adults diagnosed with PV from 2007 to 2013.
The patients’ median age was 77 (range, 71-83), 57% were female, 91.2% were white, 12.7% had a disability, and 13.2% had a prior thrombotic event.
Patients received the following PV treatments:
- Both phlebotomy and HU concurrently or sequentially (41.1%)
- Phlebotomy only (23.0%)
- HU only (19.6%)
- Neither phlebotomy nor HU (16.3%).
Survival
The median follow-up was 2.83 years. During that time, 37.2% of patients (n=305) died.
The median survival was 6.29 years for phlebotomy recipients and 4.50 years for non-recipients (P<0.01). The median survival was 6.02 years for HU recipients and 5.25 years for non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with decreased mortality. The hazard ratio (HR) for death was 0.65 (P<0.01) for phlebotomy recipients.
Increasing phlebotomy intensity (the number of phlebotomies per year) was also associated with decreased mortality, with an HR of 0.71 (P<0.01).
A higher proportion of days covered (PDC) by HU treatment was associated with decreased mortality as well.
The researchers said every 10% increase of HU PDC was associated with an 8% to 9% lower risk of death. The HR was 0.92 in a model where phlebotomy was a binary variable and 0.91 in a model that included the frequency of phlebotomy (P<0.01 for both).
Thrombosis
In all, 36.1% of patients (n=296) had a thrombotic event, which includes venous and arterial thrombosis.
The incidence of thrombosis was 29.3% (n=142) in phlebotomy recipients and 46.0% (n=154) in non-recipients (P<0.01). The incidence was 27.6% (n=118) in HU recipients and 45.4% (n=178) in non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with a decreased risk of thrombosis, with an HR of 0.52 (P<0.01).
Increasing phlebotomy intensity was associated with a decreased risk of thrombosis as well, with an HR of 0.46 (P<0.01).
And every 10% increase of HU PDC was associated with an 8% lower risk of thrombosis. The HR was 0.92 in both models (P<0.01 for both).
“All of the patients we studied were high-risk for clot development, and we now know from our findings that guideline-recommended treatments reduce the risk of both thrombosis and death,” Dr. Podoltsev said.
“We hope that our research will raise clinicians’ awareness of and adherence to the guidelines and improve the outcomes of PV patients in the future.”
This research was supported by the Frederick A. DeLuca Foundation. Study authors reported relationships with 24 pharmaceutical companies.
Researchers have found evidence to suggest that phlebotomy and hydroxyurea (HU) provide real-world benefits for older patients with polycythemia vera (PV), but both treatments are underused.
In a study of more than 800 PV patients, phlebotomy and HU treatment were both associated with lower risks of death and thrombosis.
However, 39% of patients didn’t receive HU, and 36% didn’t undergo phlebotomy.
“Our study highlights the value of adhering to PV treatment guidelines,” said study author Nikolai A. Podoltsev, MD, PhD, of Yale Cancer Center in New Haven, Connecticut.
“Use of the two recommended treatments saves lives.”
Dr. Podoltsev and his colleagues described this survival benefit in Blood Advances.
The researchers studied data from the linked Surveillance, Epidemiology, and End Results–Medicare database. They collected information on 820 older adults diagnosed with PV from 2007 to 2013.
The patients’ median age was 77 (range, 71-83), 57% were female, 91.2% were white, 12.7% had a disability, and 13.2% had a prior thrombotic event.
Patients received the following PV treatments:
- Both phlebotomy and HU concurrently or sequentially (41.1%)
- Phlebotomy only (23.0%)
- HU only (19.6%)
- Neither phlebotomy nor HU (16.3%).
Survival
The median follow-up was 2.83 years. During that time, 37.2% of patients (n=305) died.
The median survival was 6.29 years for phlebotomy recipients and 4.50 years for non-recipients (P<0.01). The median survival was 6.02 years for HU recipients and 5.25 years for non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with decreased mortality. The hazard ratio (HR) for death was 0.65 (P<0.01) for phlebotomy recipients.
Increasing phlebotomy intensity (the number of phlebotomies per year) was also associated with decreased mortality, with an HR of 0.71 (P<0.01).
A higher proportion of days covered (PDC) by HU treatment was associated with decreased mortality as well.
The researchers said every 10% increase of HU PDC was associated with an 8% to 9% lower risk of death. The HR was 0.92 in a model where phlebotomy was a binary variable and 0.91 in a model that included the frequency of phlebotomy (P<0.01 for both).
Thrombosis
In all, 36.1% of patients (n=296) had a thrombotic event, which includes venous and arterial thrombosis.
The incidence of thrombosis was 29.3% (n=142) in phlebotomy recipients and 46.0% (n=154) in non-recipients (P<0.01). The incidence was 27.6% (n=118) in HU recipients and 45.4% (n=178) in non-recipients (P<0.01).
In a multivariable analysis, receipt of phlebotomy was associated with a decreased risk of thrombosis, with an HR of 0.52 (P<0.01).
Increasing phlebotomy intensity was associated with a decreased risk of thrombosis as well, with an HR of 0.46 (P<0.01).
And every 10% increase of HU PDC was associated with an 8% lower risk of thrombosis. The HR was 0.92 in both models (P<0.01 for both).
“All of the patients we studied were high-risk for clot development, and we now know from our findings that guideline-recommended treatments reduce the risk of both thrombosis and death,” Dr. Podoltsev said.
“We hope that our research will raise clinicians’ awareness of and adherence to the guidelines and improve the outcomes of PV patients in the future.”
This research was supported by the Frederick A. DeLuca Foundation. Study authors reported relationships with 24 pharmaceutical companies.
Quizartinib receives accelerated assessment for AML

The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).
The European Medicines Agency has granted accelerated assessment to the marketing authorization application (MAA) for the FLT3 inhibitor quizartinib.
With this MAA, Daiichi Sankyo Company, Ltd., is seeking authorization for quizartinib to treat adults with FLT3-ITD-positive, relapsed or refractory acute myeloid leukemia (AML).
Accelerated assessment is given to products expected to be of major interest for public health and therapeutic innovation, and it can reduce the review timeline from 210 days to 150 days.
Quizartinib also has orphan drug designation from the European Commission.
The MAA for quizartinib is based on the phase 3 QuANTUM-R study. Results from this trial were presented at the 23rd Congress of the European Hematology Association in June.
QuANTUM-R enrolled adults with FLT3-ITD AML (at least 3% FLT3-ITD allelic ratio) who had refractory disease or had relapsed within 6 months of their first complete response (CR).
Patients were randomized to receive once-daily treatment with quizartinib (n=245) or a salvage chemotherapy regimen (n=122)—low-dose cytarabine (LoDAC, n=29); combination mitoxantrone, etoposide, and cytarabine (MEC, n=40); or combination fludarabine, cytarabine, and idarubicin (FLAG-IDA, n=53).
Patients who responded to treatment could proceed to hematopoietic stem cell transplant (HSCT), and those in the quizartinib arm could resume quizartinib after HSCT.
In all, 241 patients received quizartinib, and 94 received salvage chemotherapy—LoDAC (n=22), MEC (n=25), and FLAG-IDA (n=47). Of the 28 patients in the chemotherapy group who were not treated, most withdrew consent.
Thirty-two percent of quizartinib-treated patients and 12% of the chemotherapy group went on to HSCT.
Efficacy
The median follow-up was 23.5 months. The efficacy results include all randomized patients.
The overall response rate was 69% in the quizartinib arm and 30% in the chemotherapy arm.
The composite CR rate was 48% in the quizartinib arm and 27% in the chemotherapy arm. This includes:
- The CR rate (4% and 1%, respectively)
- The rate of CR with incomplete platelet recovery (4% and 0%, respectively)
- The rate of CR with incomplete hematologic recovery (40% and 26%, respectively).
The median event-free survival was 6.0 weeks in the quizartinib arm and 3.7 weeks in the chemotherapy arm (hazard ratio=0.90, P=0.1071).
The median overall survival was 6.2 months in the quizartinib arm and 4.7 months in the chemotherapy arm (hazard ratio=0.76, P=0.0177). The 1-year overall survival rate was 27% and 20%, respectively.
Safety
The safety results include only patients who received their assigned treatment.
Grade 3 or higher hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Thrombocytopenia (35% and 34%)
- Anemia (30% and 29%)
- Neutropenia (32% and 25%)
- Febrile neutropenia (31% and 21%)
- Leukopenia (17% and 16%).
Grade 3 or higher non-hematologic treatment-emergent adverse events occurring in at least 5% of patients (in the quizartinib and chemotherapy groups, respectively) included:
- Sepsis/septic shock (16% and 18%)
- Hypokalemia (12% and 9%)
- Pneumonia (12% and 9%)
- Fatigue (8% and 1%)
- Dyspnea (5% for both)
- Hypophosphatemia (5% for both).