User login
Flu season intensifies: High activity now in 19 states
The effects of the flu became much more widespread in the last full week of 2018 as the number of states with a high level of influenza activity more than doubled from the week before, according to the Centers for Disease Control and Prevention.
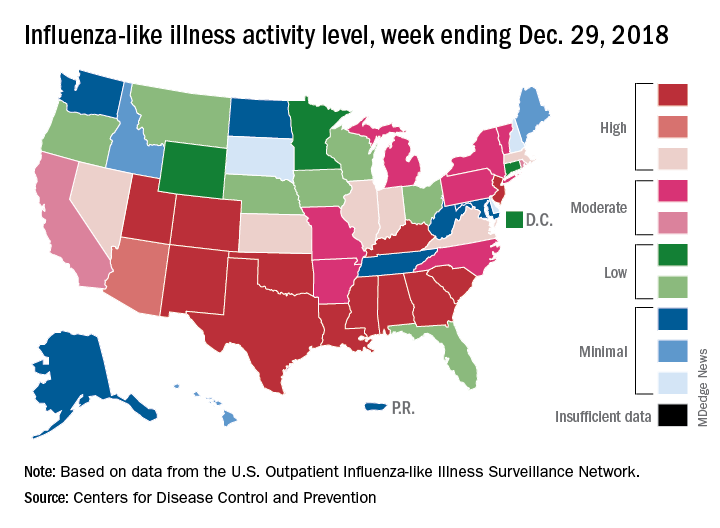
A total of 19 states were in the high range (8-10) on the CDC’s 1-10 scale of influenza-like illness (ILI) activity for the week ending Dec. 29, compared with 9 states the week before, the CDC’s influenza division reported Jan. 4. Of those 19 most-affected states, 12 were at level 10, 1 was at level 9, and 6 were at level 8. Geographic distribution of the virus was reported to be widespread in 24 states, the CDC said.
The proportion of outpatient visits for ILI – defined as fever (temperature of 100° F or greater) and cough and/or sore throat – rose to 4.1% for the week, which was up from 3.3% the previous week and well above the national baseline of 2.2%.
“The increase in the percentage of patient visits for ILI may be influenced in part by a reduction in routine health care visits during the winter holidays,” the report noted. There were 38 influenza deaths reported for the most recent week with available data (the week ending Dec. 22), although reporting for that week was just over 54% complete as of Jan. 4. For the previous weeks, 39 flu-related deaths occurred during the week ending Dec. 15 (reporting 84% complete) and 43 deaths during the week ending Dec. 8 (reporting 94% complete). For the respective weeks of last year’s flu season, total deaths were 359, 165, and 118, CDC data show.
For the week ending Dec. 29, two pediatric deaths were reported, one of which occurred the week before. For the 2018-2019 season so far, 13 flu-related pediatric deaths have been reported, the CDC said.
The effects of the flu became much more widespread in the last full week of 2018 as the number of states with a high level of influenza activity more than doubled from the week before, according to the Centers for Disease Control and Prevention.
A total of 19 states were in the high range (8-10) on the CDC’s 1-10 scale of influenza-like illness (ILI) activity for the week ending Dec. 29, compared with 9 states the week before, the CDC’s influenza division reported Jan. 4. Of those 19 most-affected states, 12 were at level 10, 1 was at level 9, and 6 were at level 8. Geographic distribution of the virus was reported to be widespread in 24 states, the CDC said.
The proportion of outpatient visits for ILI – defined as fever (temperature of 100° F or greater) and cough and/or sore throat – rose to 4.1% for the week, which was up from 3.3% the previous week and well above the national baseline of 2.2%.
“The increase in the percentage of patient visits for ILI may be influenced in part by a reduction in routine health care visits during the winter holidays,” the report noted. There were 38 influenza deaths reported for the most recent week with available data (the week ending Dec. 22), although reporting for that week was just over 54% complete as of Jan. 4. For the previous weeks, 39 flu-related deaths occurred during the week ending Dec. 15 (reporting 84% complete) and 43 deaths during the week ending Dec. 8 (reporting 94% complete). For the respective weeks of last year’s flu season, total deaths were 359, 165, and 118, CDC data show.
For the week ending Dec. 29, two pediatric deaths were reported, one of which occurred the week before. For the 2018-2019 season so far, 13 flu-related pediatric deaths have been reported, the CDC said.
The effects of the flu became much more widespread in the last full week of 2018 as the number of states with a high level of influenza activity more than doubled from the week before, according to the Centers for Disease Control and Prevention.
A total of 19 states were in the high range (8-10) on the CDC’s 1-10 scale of influenza-like illness (ILI) activity for the week ending Dec. 29, compared with 9 states the week before, the CDC’s influenza division reported Jan. 4. Of those 19 most-affected states, 12 were at level 10, 1 was at level 9, and 6 were at level 8. Geographic distribution of the virus was reported to be widespread in 24 states, the CDC said.
The proportion of outpatient visits for ILI – defined as fever (temperature of 100° F or greater) and cough and/or sore throat – rose to 4.1% for the week, which was up from 3.3% the previous week and well above the national baseline of 2.2%.
“The increase in the percentage of patient visits for ILI may be influenced in part by a reduction in routine health care visits during the winter holidays,” the report noted. There were 38 influenza deaths reported for the most recent week with available data (the week ending Dec. 22), although reporting for that week was just over 54% complete as of Jan. 4. For the previous weeks, 39 flu-related deaths occurred during the week ending Dec. 15 (reporting 84% complete) and 43 deaths during the week ending Dec. 8 (reporting 94% complete). For the respective weeks of last year’s flu season, total deaths were 359, 165, and 118, CDC data show.
For the week ending Dec. 29, two pediatric deaths were reported, one of which occurred the week before. For the 2018-2019 season so far, 13 flu-related pediatric deaths have been reported, the CDC said.
Hospital Readmissions Reduction Program may be doing more harm than good
A Medicare program aimed at lowering readmissions to hospitals could be having an adverse effect on mortality.

Results from a retrospective cohort study of hospitalizations for heart failure, acute myocardial infarction, and pneumonia among Medicare beneficiaries aged 65 years and older between April 1, 2005 and March 31, 2015 (covering the period before and after the Medicare Hospital Readmissions Reduction Program was announced in April 2010 and implemented in October 2012) found a significant increase in 30-day post discharge mortality among heart failure and pneumonia patients.
“Most concerning, however, is the possibility that the relationship between the HRRP and postdischarge mortality for heart failure and pneumonia is causal, indicating that the HRRP led to changes in quality of care that adversely affected patients,” Rishi Wadhera, MD, Harvard Medical School, Boston, and his colleagues wrote in a report published Dec. 25, 2018, in JAMA.
They looked at 8.3 million hospitalizations for heart failure, acute MI, and pneumonia, among whom 7.9 million were alive at the time of discharge. There were roughly 270,000 deaths within 30 days of discharge for heart failure; 128,000 for acute MI; and 246,000 for pneumonia.
To examine trends, the timing was divided into four periods: two prior to the announcement of the HRRP (April 2005–September 2007 and October 2007–March 2010); a third covering the time when the HRRP was announced (April 2010–September 2012); and the fourth when HRRP was implemented (October 2012–March 2015).
They found that among patients discharged with heart failure, 30-day mortality was rising even before the announcement of the HRRP, by 0.27% from the first period to the second period. That baseline trend continued when the HRRP was announced, by 0.49%, from second period to third. The difference in change between those periods was 0.22%. After implementation, 30-day mortality increased by 0.52%, with a difference in change from the third period of 0.25%. Both changes were statistically significant.
Among pneumonia patients, postdischarge mortality was stable before HRRP, but significantly increased after HRRP announcement, by 0.26%, with a difference in change from the second period to the third period of 0.22%. After implementation, the 30-day postdischarge mortality was 0.44%, with a significant difference in change of 0.40%.
Acute MI was a different story. Postdischarge mortality decreased significantly after the implementation of the HRRP, by 0.22%. The difference in change was –0.26%.
The authors suggested that “some hospitals may have focused more resources and efforts on reducing or avoiding readmissions than on prioritizing survival.” They add that the increases in heart failure morbidity could be related to patients with more severe heart conditions.
They noted that “although hospitals that reduce readmissions also appear to reduce mortality, this hospital-level concordance does not reflect the change in readmissions and mortality at the level of the patient population, which is arguably of greater importance to individual patients and to public health.”
Further research is needed to understand whether the increase in 30-day postdischarge mortality is a result of the HRRP, the authors concluded.
SOURCE: Wadhera R et al. JAMA. 2018 Dec 25. doi: 10.1001/jama.2018.19232.
Evidence in this study shows that while the Hospital Readmissions Reduction Program my be succeeding in reducing hospital admissions, little evidence is available to show that it is having a positive effect on patient outcomes.
The Centers for Medicare & Medicaid Services needs to reexamine the program and find alternative methods that are both effective at reducing hospital readmissions while at the same time protect patients from unintentional harm, including death.
Gregg C. Fonarow, MD , University of California Medical Center, Los Angeles, in an editorial published in JAMA, Dec. 25, 2018. doi:10.1001/jama.2018.19325 .
Evidence in this study shows that while the Hospital Readmissions Reduction Program my be succeeding in reducing hospital admissions, little evidence is available to show that it is having a positive effect on patient outcomes.
The Centers for Medicare & Medicaid Services needs to reexamine the program and find alternative methods that are both effective at reducing hospital readmissions while at the same time protect patients from unintentional harm, including death.
Gregg C. Fonarow, MD , University of California Medical Center, Los Angeles, in an editorial published in JAMA, Dec. 25, 2018. doi:10.1001/jama.2018.19325 .
Evidence in this study shows that while the Hospital Readmissions Reduction Program my be succeeding in reducing hospital admissions, little evidence is available to show that it is having a positive effect on patient outcomes.
The Centers for Medicare & Medicaid Services needs to reexamine the program and find alternative methods that are both effective at reducing hospital readmissions while at the same time protect patients from unintentional harm, including death.
Gregg C. Fonarow, MD , University of California Medical Center, Los Angeles, in an editorial published in JAMA, Dec. 25, 2018. doi:10.1001/jama.2018.19325 .
A Medicare program aimed at lowering readmissions to hospitals could be having an adverse effect on mortality.
Results from a retrospective cohort study of hospitalizations for heart failure, acute myocardial infarction, and pneumonia among Medicare beneficiaries aged 65 years and older between April 1, 2005 and March 31, 2015 (covering the period before and after the Medicare Hospital Readmissions Reduction Program was announced in April 2010 and implemented in October 2012) found a significant increase in 30-day post discharge mortality among heart failure and pneumonia patients.
“Most concerning, however, is the possibility that the relationship between the HRRP and postdischarge mortality for heart failure and pneumonia is causal, indicating that the HRRP led to changes in quality of care that adversely affected patients,” Rishi Wadhera, MD, Harvard Medical School, Boston, and his colleagues wrote in a report published Dec. 25, 2018, in JAMA.
They looked at 8.3 million hospitalizations for heart failure, acute MI, and pneumonia, among whom 7.9 million were alive at the time of discharge. There were roughly 270,000 deaths within 30 days of discharge for heart failure; 128,000 for acute MI; and 246,000 for pneumonia.
To examine trends, the timing was divided into four periods: two prior to the announcement of the HRRP (April 2005–September 2007 and October 2007–March 2010); a third covering the time when the HRRP was announced (April 2010–September 2012); and the fourth when HRRP was implemented (October 2012–March 2015).
They found that among patients discharged with heart failure, 30-day mortality was rising even before the announcement of the HRRP, by 0.27% from the first period to the second period. That baseline trend continued when the HRRP was announced, by 0.49%, from second period to third. The difference in change between those periods was 0.22%. After implementation, 30-day mortality increased by 0.52%, with a difference in change from the third period of 0.25%. Both changes were statistically significant.
Among pneumonia patients, postdischarge mortality was stable before HRRP, but significantly increased after HRRP announcement, by 0.26%, with a difference in change from the second period to the third period of 0.22%. After implementation, the 30-day postdischarge mortality was 0.44%, with a significant difference in change of 0.40%.
Acute MI was a different story. Postdischarge mortality decreased significantly after the implementation of the HRRP, by 0.22%. The difference in change was –0.26%.
The authors suggested that “some hospitals may have focused more resources and efforts on reducing or avoiding readmissions than on prioritizing survival.” They add that the increases in heart failure morbidity could be related to patients with more severe heart conditions.
They noted that “although hospitals that reduce readmissions also appear to reduce mortality, this hospital-level concordance does not reflect the change in readmissions and mortality at the level of the patient population, which is arguably of greater importance to individual patients and to public health.”
Further research is needed to understand whether the increase in 30-day postdischarge mortality is a result of the HRRP, the authors concluded.
SOURCE: Wadhera R et al. JAMA. 2018 Dec 25. doi: 10.1001/jama.2018.19232.
A Medicare program aimed at lowering readmissions to hospitals could be having an adverse effect on mortality.
Results from a retrospective cohort study of hospitalizations for heart failure, acute myocardial infarction, and pneumonia among Medicare beneficiaries aged 65 years and older between April 1, 2005 and March 31, 2015 (covering the period before and after the Medicare Hospital Readmissions Reduction Program was announced in April 2010 and implemented in October 2012) found a significant increase in 30-day post discharge mortality among heart failure and pneumonia patients.
“Most concerning, however, is the possibility that the relationship between the HRRP and postdischarge mortality for heart failure and pneumonia is causal, indicating that the HRRP led to changes in quality of care that adversely affected patients,” Rishi Wadhera, MD, Harvard Medical School, Boston, and his colleagues wrote in a report published Dec. 25, 2018, in JAMA.
They looked at 8.3 million hospitalizations for heart failure, acute MI, and pneumonia, among whom 7.9 million were alive at the time of discharge. There were roughly 270,000 deaths within 30 days of discharge for heart failure; 128,000 for acute MI; and 246,000 for pneumonia.
To examine trends, the timing was divided into four periods: two prior to the announcement of the HRRP (April 2005–September 2007 and October 2007–March 2010); a third covering the time when the HRRP was announced (April 2010–September 2012); and the fourth when HRRP was implemented (October 2012–March 2015).
They found that among patients discharged with heart failure, 30-day mortality was rising even before the announcement of the HRRP, by 0.27% from the first period to the second period. That baseline trend continued when the HRRP was announced, by 0.49%, from second period to third. The difference in change between those periods was 0.22%. After implementation, 30-day mortality increased by 0.52%, with a difference in change from the third period of 0.25%. Both changes were statistically significant.
Among pneumonia patients, postdischarge mortality was stable before HRRP, but significantly increased after HRRP announcement, by 0.26%, with a difference in change from the second period to the third period of 0.22%. After implementation, the 30-day postdischarge mortality was 0.44%, with a significant difference in change of 0.40%.
Acute MI was a different story. Postdischarge mortality decreased significantly after the implementation of the HRRP, by 0.22%. The difference in change was –0.26%.
The authors suggested that “some hospitals may have focused more resources and efforts on reducing or avoiding readmissions than on prioritizing survival.” They add that the increases in heart failure morbidity could be related to patients with more severe heart conditions.
They noted that “although hospitals that reduce readmissions also appear to reduce mortality, this hospital-level concordance does not reflect the change in readmissions and mortality at the level of the patient population, which is arguably of greater importance to individual patients and to public health.”
Further research is needed to understand whether the increase in 30-day postdischarge mortality is a result of the HRRP, the authors concluded.
SOURCE: Wadhera R et al. JAMA. 2018 Dec 25. doi: 10.1001/jama.2018.19232.
FROM JAMA
Key clinical point:
Major finding: Heart failure patients saw mortality increase 0.52% after HRRP launched.
Study details: A retrospective cohort study across 10 years, including time before and after the implementation of the HRRP.
Disclosures: The Richard A. and Susan F. Smith Center for Outcomes Research in Cardiology funded the study. No relevant conflicts of interest were disclosed.
Source: Wadhera R et al. JAMA 2018 Dec 25. doi: 10.1001/jama.2018.19232.
Tests can identify leukemia risk in newborns with Down syndrome
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.

“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.
“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.
“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
EXPERT ANALYSIS FROM ASH 2018
VHA Suicide Prevention Media Outreach Is Falling Short—But Not for Lack of Money
The VHA’s suicide prevention media outreach activities—including social media postings, public service announcements, paid media, and Suicide Prevention Month activities—shrank markedly in fiscal years 2017 and 2018, according to the Government Accountability Office (GAO).
Since 2010, the primary focus of the outreach campaign has been to raise awareness of the Veterans Crisis Line (VCL), with output falling into 2 main categories: unpaid (eg, social media, public service announcements [PSAs], website) and paid (digital media, such as online keyword searches, and “out-of-home” media, such as billboards).
But between 2016 and the first 10 months of 2018, social media content dropped from 339 pieces to only 47. VHA also had not aired a suicide prevention PSA in more than 1 year, the first time there has been a gap of more than 1 month since June 2012.
In 2015, with a budget of > $4 million, VHA ran 58 advertisements on Google, Bing, and Facebook; 30 billboards; 180 bus advertisements; > 19,000 radio advertisements; 252 print advertisements; and 39 movie theater placements across the US. Fiscal years 2013, 2014, and 2016 were similarly productive.
Meanwhile, in FY 2017, the VHA spent < 10% of its approximately $1.7 million on paid ads on Google and Bing. And as of September 2018, VHA said it had spent only $57,000 of its $6.2 million paid media budget.
The waning outreach is “inconsistent with VA’s strategic goals,” the GAO says, which identify suicide prevention as the agency’s top clinical priority for FY 2018 through 2024.
VHA officials said they had not spent all the available funds due to changes in leadership and organizational realignment of the suicide prevention program. The position of National Director for Suicide Prevention position, for example, was vacant from July 2017 to April 2018. It was filled temporarily for 6 months; the interim director was then hired permanently in April 2018.
Since 2016, the VHA says, the plan has been to have a national strategy for preventing veteran suicides—an average of 20 per day, according to the VA—using a public health approach, focusing less on raising awareness of the VCL and more on reaching veterans before the point of crisis. However, in May 2018, VHA officials told the GAO that they “were just beginning to conceptualize what the suicide prevention outreach campaign should look like moving forward.”
One problem, the GAO says, is that the VHA has not established targets for the majority of the metrics it uses to help gauge the effectiveness of the outreach campaign. As a result, the VHA does not have the information it needs for a full evaluation. The only target the VHA has set is for each PSA to rank in the top 10% of the Nielsen ratings because, it says, that is the only meaningful target available that is accepted industry-wide. (The GAO notes, however: “VHA could use information about how its metrics performed in the past to develop reasonable and meaningful targets for future performance.”)
The GAO has made 2 recommendations to the VA based on its audit:
- The Under Secretary for Health should establish an approach for overseeing its suicide prevention media outreach efforts that includes “clear delineation of roles and responsibilities for those in leadership and contract oversight roles, including during periods of staff turnover or program changes.”
- Officials within the Office of Mental Health and Suicide Prevention (OMHSP) must establish targets for the metrics used to evaluate the campaign’s effectiveness.
In written comments, the VA concurred with the GAO recommendations and provided a time line for addressing them. VA said OMHSP has made organizational improvements, including creating a new organizational structure. It also has plans to work with communications experts to develop metrics, targets, and an evaluation strategy, expected to be completed by April 2019.
The VHA’s suicide prevention media outreach activities—including social media postings, public service announcements, paid media, and Suicide Prevention Month activities—shrank markedly in fiscal years 2017 and 2018, according to the Government Accountability Office (GAO).
Since 2010, the primary focus of the outreach campaign has been to raise awareness of the Veterans Crisis Line (VCL), with output falling into 2 main categories: unpaid (eg, social media, public service announcements [PSAs], website) and paid (digital media, such as online keyword searches, and “out-of-home” media, such as billboards).
But between 2016 and the first 10 months of 2018, social media content dropped from 339 pieces to only 47. VHA also had not aired a suicide prevention PSA in more than 1 year, the first time there has been a gap of more than 1 month since June 2012.
In 2015, with a budget of > $4 million, VHA ran 58 advertisements on Google, Bing, and Facebook; 30 billboards; 180 bus advertisements; > 19,000 radio advertisements; 252 print advertisements; and 39 movie theater placements across the US. Fiscal years 2013, 2014, and 2016 were similarly productive.
Meanwhile, in FY 2017, the VHA spent < 10% of its approximately $1.7 million on paid ads on Google and Bing. And as of September 2018, VHA said it had spent only $57,000 of its $6.2 million paid media budget.
The waning outreach is “inconsistent with VA’s strategic goals,” the GAO says, which identify suicide prevention as the agency’s top clinical priority for FY 2018 through 2024.
VHA officials said they had not spent all the available funds due to changes in leadership and organizational realignment of the suicide prevention program. The position of National Director for Suicide Prevention position, for example, was vacant from July 2017 to April 2018. It was filled temporarily for 6 months; the interim director was then hired permanently in April 2018.
Since 2016, the VHA says, the plan has been to have a national strategy for preventing veteran suicides—an average of 20 per day, according to the VA—using a public health approach, focusing less on raising awareness of the VCL and more on reaching veterans before the point of crisis. However, in May 2018, VHA officials told the GAO that they “were just beginning to conceptualize what the suicide prevention outreach campaign should look like moving forward.”
One problem, the GAO says, is that the VHA has not established targets for the majority of the metrics it uses to help gauge the effectiveness of the outreach campaign. As a result, the VHA does not have the information it needs for a full evaluation. The only target the VHA has set is for each PSA to rank in the top 10% of the Nielsen ratings because, it says, that is the only meaningful target available that is accepted industry-wide. (The GAO notes, however: “VHA could use information about how its metrics performed in the past to develop reasonable and meaningful targets for future performance.”)
The GAO has made 2 recommendations to the VA based on its audit:
- The Under Secretary for Health should establish an approach for overseeing its suicide prevention media outreach efforts that includes “clear delineation of roles and responsibilities for those in leadership and contract oversight roles, including during periods of staff turnover or program changes.”
- Officials within the Office of Mental Health and Suicide Prevention (OMHSP) must establish targets for the metrics used to evaluate the campaign’s effectiveness.
In written comments, the VA concurred with the GAO recommendations and provided a time line for addressing them. VA said OMHSP has made organizational improvements, including creating a new organizational structure. It also has plans to work with communications experts to develop metrics, targets, and an evaluation strategy, expected to be completed by April 2019.
The VHA’s suicide prevention media outreach activities—including social media postings, public service announcements, paid media, and Suicide Prevention Month activities—shrank markedly in fiscal years 2017 and 2018, according to the Government Accountability Office (GAO).
Since 2010, the primary focus of the outreach campaign has been to raise awareness of the Veterans Crisis Line (VCL), with output falling into 2 main categories: unpaid (eg, social media, public service announcements [PSAs], website) and paid (digital media, such as online keyword searches, and “out-of-home” media, such as billboards).
But between 2016 and the first 10 months of 2018, social media content dropped from 339 pieces to only 47. VHA also had not aired a suicide prevention PSA in more than 1 year, the first time there has been a gap of more than 1 month since June 2012.
In 2015, with a budget of > $4 million, VHA ran 58 advertisements on Google, Bing, and Facebook; 30 billboards; 180 bus advertisements; > 19,000 radio advertisements; 252 print advertisements; and 39 movie theater placements across the US. Fiscal years 2013, 2014, and 2016 were similarly productive.
Meanwhile, in FY 2017, the VHA spent < 10% of its approximately $1.7 million on paid ads on Google and Bing. And as of September 2018, VHA said it had spent only $57,000 of its $6.2 million paid media budget.
The waning outreach is “inconsistent with VA’s strategic goals,” the GAO says, which identify suicide prevention as the agency’s top clinical priority for FY 2018 through 2024.
VHA officials said they had not spent all the available funds due to changes in leadership and organizational realignment of the suicide prevention program. The position of National Director for Suicide Prevention position, for example, was vacant from July 2017 to April 2018. It was filled temporarily for 6 months; the interim director was then hired permanently in April 2018.
Since 2016, the VHA says, the plan has been to have a national strategy for preventing veteran suicides—an average of 20 per day, according to the VA—using a public health approach, focusing less on raising awareness of the VCL and more on reaching veterans before the point of crisis. However, in May 2018, VHA officials told the GAO that they “were just beginning to conceptualize what the suicide prevention outreach campaign should look like moving forward.”
One problem, the GAO says, is that the VHA has not established targets for the majority of the metrics it uses to help gauge the effectiveness of the outreach campaign. As a result, the VHA does not have the information it needs for a full evaluation. The only target the VHA has set is for each PSA to rank in the top 10% of the Nielsen ratings because, it says, that is the only meaningful target available that is accepted industry-wide. (The GAO notes, however: “VHA could use information about how its metrics performed in the past to develop reasonable and meaningful targets for future performance.”)
The GAO has made 2 recommendations to the VA based on its audit:
- The Under Secretary for Health should establish an approach for overseeing its suicide prevention media outreach efforts that includes “clear delineation of roles and responsibilities for those in leadership and contract oversight roles, including during periods of staff turnover or program changes.”
- Officials within the Office of Mental Health and Suicide Prevention (OMHSP) must establish targets for the metrics used to evaluate the campaign’s effectiveness.
In written comments, the VA concurred with the GAO recommendations and provided a time line for addressing them. VA said OMHSP has made organizational improvements, including creating a new organizational structure. It also has plans to work with communications experts to develop metrics, targets, and an evaluation strategy, expected to be completed by April 2019.
Armored CAR protects T cells, induces remissions
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.

The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.
The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
SAN DIEGO – A second-generation CD19-specific “armored” chimeric antigen receptor (CAR) T-cell construct was associated with high complete remission rates in diffuse large B-cell lymphoma (DLBCL) and indolent non-Hodgkin lymphoma (NHL) in a phase 1 trial.
The CAR T construct – labeled 1928z-41BBL – also induced “encouraging” complete remission rates in patients with chronic lymphocytic leukemia (CLL) with Richter’s transformation, reported Jae H. Park, MD, of Memorial Sloan Kettering Cancer Center (MSKCC), New York, and his colleagues.
“Interestingly and encouragingly, severe [cytokine release syndrome] was not seen and grade 3 neurotoxicity was observed in less than 10%, with no grade 4 neurotoxicity, so there appears to be a favorable side effect profile,” Dr. Park said at the annual meeting of the American Society of Hematology.
Just as armored cars are designed to protect their valuable contents from people with bad intent, armored CAR T cells are engineered to protect the modified T-cells from a hostile tumor microenvironment and simultaneously recruit non-modified T cells to the target to produce a more robust immune response against malignant cells.
MSKCC investigators had previously shown that in contrast to other CAR T-cell constructs, the 1928z-41BBL configuration, which consists of two signaling domains (CD28 and CD3zeta) and the 4-1BB ligand, hit the sweet spot between tumor-killing function and T-cell persistence (Cancer Cell. 2015 Oct 12;28[4]:415-28).
In the current study, they enrolled 35 adults with relapsed or refractory CD19-positive hematologic malignancies, 29 of whom eventually underwent CAR T-cell infusions. The treated population comprised 14 patients with CLL (4 of whom had Richter’s transformation), 9 with DLBCL, 5 with indolent NHL, and 1 with acute lymphoblastic leukemia.
The patients with CLL had received a median of 5.5 prior lines of therapy, including ibrutinib (Imbruvica) and venetoclax (Venclexta).
There were 15 complete remissions (CR), with CR rates of 78% in DLBCL, 20% in CLL, 67% in CLL with Richter’s transformation, 60% in patients with indolent NHL, as well as CR in the single patient with ALL.
There were eight partial remissions. One patient with CLL had stable disease, and four patients had disease progression (one patient each with DLBCL, CLL, CLL with Richter’s, and indolent NHL).
Dr. Park noted that T cells are being detected in peripheral blood more than 6 months after T-cell infusion.
There were no cases of severe cytokine release syndrome, defined as requiring vasopressors and/or mechanical ventilation for hypoxia, and just three cases of grade 3 neurotoxicity. There were no cases of grade 4 neurotoxicity, no deaths related to neurotoxicity, and no cases of cerebral edema – a serious complication that has been seen in earlier CAR T-cell studies.
Split or multiple infusions of CAR T cells or incorporation of the technique into earlier lines of therapy might generate higher response rates, Dr. Park said.
The study was supported by Juno Therapeutics. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
SOURCE: Park JH et al. ASH 2018, Abstract 224.
REPORTING FROM ASH 2018
Key clinical point: The 1928z-41BBL CAR T-cell construct induced high rates of complete remissions.
Major finding: The CAR T product was associated with a 78% complete remission rate in patients with heavily pretreated diffuse large B-cell lymphoma.
Study details: A phase 1 trial in 29 patients with CD19-positive hematologic malignancies.
Disclosures: Juno Therapeutics supported the study. Dr. Park reported consulting for and research funding from Juno, and financial relationships with other companies.
Source: Park JH et al. ASH 2018, Abstract 224.
Long-term side effects of CAR T cells mostly mild
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.

Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
SAN DIEGO – Longer-term follow-up of patients treated with CD19-targeted chimeric antigen receptor (CAR) T cells for hematologic malignancies indicates that the altered cells are generally safe, with most late events being mild in nature and possibly related to therapies delivered before or after CAR T cells, investigators reported.
Among patients treated with CD19-targeted CAR T cells for relapsed or refractory chronic lymphocytic leukemia (CLL) or non-Hodgkin lymphoma (NHL), the most frequent late adverse event was hypogammaglobulinemia, which occurred in 29 of 48 patients evaluated, reported Ana Cordeiro, MD, from the Fred Hutchinson Cancer Research Center in Seattle.
“Our results suggest that CD19 CAR T cells are safe,” Dr. Cordeiro said at the annual meeting of the American Society of Hematology. “However, continuing with prospective systematic and long-term follow-up of these patients is required for better understanding of these late effects.”
Dr. Cordeiro and colleagues studied a total of 60 patients who were enrolled in a phase 1/2 trial at their center of a CD19-targeted CAR T-cell construct and survived for at least 1 year.
The goal of the study was to describe complications that occurred or persisted beyond 90 days after CAR T-cell infusion.
The cohort included 43 patients treated for NHL and 17 treated for CLL. Patients with CLL were followed for a median of 27.5 months, and patients with NHL were followed for a median of 23.8 months.
As of September 2018, 47 patients were still alive, including 15 patients with CLL (88%) and 32 patients with NHL (74%). Of the 17 patients who died, 10 died from progressive disease (2 from CLL and 8 from NHL), and 3 patients died from nonrelapse causes associated with complications from subsequent allogeneic stem cell transplantation (allo-HCT), including 1 patient from graft-versus-host disease (GVHD) and infection, 1 from infection, and 1 from cerberovascular accident/thrombotic microangiopathy.
Of 38 patients who received additional therapies, 17 had subsequent CAR T-cell infusions under the same protocol, and 16 went on to allo-HCT. Treatments for the remaining five patients were not specified.
Of the 22 patients who did not receive additional treatment for their primary malignancies, 21 were in ongoing complete remission following a single CAR T-cell infusion after a median follow-up of 28 months. However, two patients in this group did require treatment for therapy-related myelodysplastic syndrome (t-MDS). The remaining patient had a small CLL clone at last follow-up.
Late adverse events included the following:
- Late significant cytopenias in three of 19 patients evaluated.
- Late hypogammaglobulinemia in 29 of 48 evaluated patients.
- A total of 138 late infections in 31 of the 60 patients.
- Subsequent malignancies in 10 of the 60 patients, including t-MDS, nonmelanoma skin cancer, and noninvasive bladder cancer.
- Late immune-related events in seven patients.
- Late neurogenic/psychiatric events, including one case each of transient ischemic attack at 3.8 months, encephalopathy and myoclonic seizure in the setting of chemotherapy, and a fatal cerebrovascular accident in the setting of allo-HCT and thrombotic microangiopathy. These patients did not have acute neurotoxicity after CAR T-cell therapy, Dr. Cordeiro noted. In addition, three patients experienced exacerbation of depression or anxiety following infusion.
- GVHD in nine patients at a median time from allo-HCT to first CAR T-cell infusion of 46.3 months (range, 6.7 months to 11 years).
Focusing on those patients who achieve complete remissions after CAR T-cell therapy could help investigators isolate late events that are most likely related to CAR T cells, Dr. Cordeiro said.
Dr. Cordeiro reported having no relevant conflicts of interest.
SOURCE: Cordeiro A et al. ASH 2018, Abstract 223.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: The most frequent adverse event was hypogammaglobulinemia in 60% of evaluable patients.
Study details: Prospective observational study of 60 patients with relapsed/refractory CLL or NHL.
Disclosures: Dr. Cordeiro reported having no relevant conflicts of interest.
Source: Cordeiro A et al. ASH 2018, Abstract 223.
Integrated analysis suggests cladribine’s safety in MS
according to an integrated analysis of several clinical trials published in Multiple Sclerosis and Related Disorders.

The drug causes transient lymphopenia, as is to be expected from its mechanism of action, but most patients who receive cladribine do not have grade 3 or 4 lymphopenia, the authors wrote. In general, cladribine is not associated with an increased risk of infections or malignancy.
Data indicate that cladribine’s mechanism of action contributes to its durable clinical effect, despite the brief treatment periods. Clinical trials have provided information about the treatment’s safety and tolerability. To examine the treatment’s long-term safety, Stuart Cook, MD, of New Jersey Medical School, Newark, and his colleagues pooled data for patients with multiple sclerosis (MS) treated with cladribine tablets (3.5 mg/kg) as monotherapy or placebo in three phase 3 clinical trials (CLARITY, CLARITY Extension, and ORACLE-MS) and followed up in the PREMIERE registry. The investigators called these patients the monotherapy oral cohort.
To investigate the potential associations between cladribine and rarer adverse events such as malignancies, Dr. Cook and his colleagues examined data from patients with MS who received cladribine, regardless of the formulation or route of administration, or placebo (the all-exposed cohort). This cohort included data from the ONWARD trial, in which patients received cladribine tablets or placebo in combination with interferon-beta, as well as data from five trials in which patients received parenteral cladribine or placebo.
The monotherapy oral cohort included 923 patients who received cladribine and 641 who received placebo. The median age at enrollment in this cohort was approximately 36 years. The majority of patients were women and disease characteristics were balanced between arms. The all-exposed cohort included 1,926 patients who received cladribine and 802 who received placebo. The demographic characteristics of this cohort were similar to those of the monotherapy oral cohort.
In the monotherapy oral cohort, the incidence rate of treatment-emergent adverse events, in adjusted adverse events per 100 patient-years, was 103.29 for the active group versus 94.26 for controls. Lymphopenia (7.94 vs. 1.06) and decreased lymphocyte count (0.78 vs. 0.10) occurred more frequently in the active group than in the placebo group.
Herpes zoster also occurred more frequently in the cladribine group (0.83 vs. 0.20). However, cladribine was not associated with systemic serious disseminated herpes zoster. Furthermore, the investigators found no overall increased risk of infections, including opportunistic infections, with cladribine tablets versus placebo, except for herpes zoster.
In addition, Dr. Cook and his colleagues found no increase in malignancy rates among patients who received cladribine, compared with those who received placebo. They also found no increase in the incidence of malignancies over time in the cladribine group.
The adverse event profile for cladribine tablets (3.5 mg/kg) as monotherapy has been well characterized, the investigators wrote. The results of this analysis are broadly similar to the short-term adverse event results reported in the individual trials.
EMD Serono and Merck Serono, two affiliates of Merck in Darmstadt, Germany, sponsored the study. Merck manufactures cladribine.
SOURCE: Cook S et al. Mult Scler Relat Disord. 2018 Nov 18. doi: 10.1016/j.msard.2018.11.021.
according to an integrated analysis of several clinical trials published in Multiple Sclerosis and Related Disorders.
The drug causes transient lymphopenia, as is to be expected from its mechanism of action, but most patients who receive cladribine do not have grade 3 or 4 lymphopenia, the authors wrote. In general, cladribine is not associated with an increased risk of infections or malignancy.
Data indicate that cladribine’s mechanism of action contributes to its durable clinical effect, despite the brief treatment periods. Clinical trials have provided information about the treatment’s safety and tolerability. To examine the treatment’s long-term safety, Stuart Cook, MD, of New Jersey Medical School, Newark, and his colleagues pooled data for patients with multiple sclerosis (MS) treated with cladribine tablets (3.5 mg/kg) as monotherapy or placebo in three phase 3 clinical trials (CLARITY, CLARITY Extension, and ORACLE-MS) and followed up in the PREMIERE registry. The investigators called these patients the monotherapy oral cohort.
To investigate the potential associations between cladribine and rarer adverse events such as malignancies, Dr. Cook and his colleagues examined data from patients with MS who received cladribine, regardless of the formulation or route of administration, or placebo (the all-exposed cohort). This cohort included data from the ONWARD trial, in which patients received cladribine tablets or placebo in combination with interferon-beta, as well as data from five trials in which patients received parenteral cladribine or placebo.
The monotherapy oral cohort included 923 patients who received cladribine and 641 who received placebo. The median age at enrollment in this cohort was approximately 36 years. The majority of patients were women and disease characteristics were balanced between arms. The all-exposed cohort included 1,926 patients who received cladribine and 802 who received placebo. The demographic characteristics of this cohort were similar to those of the monotherapy oral cohort.
In the monotherapy oral cohort, the incidence rate of treatment-emergent adverse events, in adjusted adverse events per 100 patient-years, was 103.29 for the active group versus 94.26 for controls. Lymphopenia (7.94 vs. 1.06) and decreased lymphocyte count (0.78 vs. 0.10) occurred more frequently in the active group than in the placebo group.
Herpes zoster also occurred more frequently in the cladribine group (0.83 vs. 0.20). However, cladribine was not associated with systemic serious disseminated herpes zoster. Furthermore, the investigators found no overall increased risk of infections, including opportunistic infections, with cladribine tablets versus placebo, except for herpes zoster.
In addition, Dr. Cook and his colleagues found no increase in malignancy rates among patients who received cladribine, compared with those who received placebo. They also found no increase in the incidence of malignancies over time in the cladribine group.
The adverse event profile for cladribine tablets (3.5 mg/kg) as monotherapy has been well characterized, the investigators wrote. The results of this analysis are broadly similar to the short-term adverse event results reported in the individual trials.
EMD Serono and Merck Serono, two affiliates of Merck in Darmstadt, Germany, sponsored the study. Merck manufactures cladribine.
SOURCE: Cook S et al. Mult Scler Relat Disord. 2018 Nov 18. doi: 10.1016/j.msard.2018.11.021.
according to an integrated analysis of several clinical trials published in Multiple Sclerosis and Related Disorders.
The drug causes transient lymphopenia, as is to be expected from its mechanism of action, but most patients who receive cladribine do not have grade 3 or 4 lymphopenia, the authors wrote. In general, cladribine is not associated with an increased risk of infections or malignancy.
Data indicate that cladribine’s mechanism of action contributes to its durable clinical effect, despite the brief treatment periods. Clinical trials have provided information about the treatment’s safety and tolerability. To examine the treatment’s long-term safety, Stuart Cook, MD, of New Jersey Medical School, Newark, and his colleagues pooled data for patients with multiple sclerosis (MS) treated with cladribine tablets (3.5 mg/kg) as monotherapy or placebo in three phase 3 clinical trials (CLARITY, CLARITY Extension, and ORACLE-MS) and followed up in the PREMIERE registry. The investigators called these patients the monotherapy oral cohort.
To investigate the potential associations between cladribine and rarer adverse events such as malignancies, Dr. Cook and his colleagues examined data from patients with MS who received cladribine, regardless of the formulation or route of administration, or placebo (the all-exposed cohort). This cohort included data from the ONWARD trial, in which patients received cladribine tablets or placebo in combination with interferon-beta, as well as data from five trials in which patients received parenteral cladribine or placebo.
The monotherapy oral cohort included 923 patients who received cladribine and 641 who received placebo. The median age at enrollment in this cohort was approximately 36 years. The majority of patients were women and disease characteristics were balanced between arms. The all-exposed cohort included 1,926 patients who received cladribine and 802 who received placebo. The demographic characteristics of this cohort were similar to those of the monotherapy oral cohort.
In the monotherapy oral cohort, the incidence rate of treatment-emergent adverse events, in adjusted adverse events per 100 patient-years, was 103.29 for the active group versus 94.26 for controls. Lymphopenia (7.94 vs. 1.06) and decreased lymphocyte count (0.78 vs. 0.10) occurred more frequently in the active group than in the placebo group.
Herpes zoster also occurred more frequently in the cladribine group (0.83 vs. 0.20). However, cladribine was not associated with systemic serious disseminated herpes zoster. Furthermore, the investigators found no overall increased risk of infections, including opportunistic infections, with cladribine tablets versus placebo, except for herpes zoster.
In addition, Dr. Cook and his colleagues found no increase in malignancy rates among patients who received cladribine, compared with those who received placebo. They also found no increase in the incidence of malignancies over time in the cladribine group.
The adverse event profile for cladribine tablets (3.5 mg/kg) as monotherapy has been well characterized, the investigators wrote. The results of this analysis are broadly similar to the short-term adverse event results reported in the individual trials.
EMD Serono and Merck Serono, two affiliates of Merck in Darmstadt, Germany, sponsored the study. Merck manufactures cladribine.
SOURCE: Cook S et al. Mult Scler Relat Disord. 2018 Nov 18. doi: 10.1016/j.msard.2018.11.021.
FROM MULTIPLE SCLEROSIS AND RELATED DISORDERS
Key clinical point: The safety results of an integrated analysis of cladribine therapy are broadly similar to those of previous trials.
Major finding: The incidence rate of treatment-emergent adverse events was similar between cladribine and placebo.
Study details: An integrated analysis including 4,292 patients with relapsing remitting multiple sclerosis.
Disclosures: EMD Serono and Merck Serono, two affiliates of Merck in Darmstadt, Germany, sponsored the study. Merck manufactures cladribine.
Source: Cook S et al. Mult Scler Relat Disord. 2018 Nov 18. doi: 10.1016/j.msard.2018.11.021.
White matter volume reduced in first-episode depression
Decrements ‘seem to vanish’ after 2-year follow-up
White matter volume was significantly reduced initially in patients with first-episode depression, but not after a 2-year follow-up, according to Mar Carceller-Sindreu, MD, of the department of psychiatry at Hospital de la Santa Creu i Sant Pau in Barcelona, and her associates.
A total of 33 patients with first-episode depression and 33 healthy controls were included in the study. Among them, 27 first-episode depression patients and 17 controls completed the 2-year follow-up. They underwent structural MRIs at baseline and at follow-up, the Hamilton Depressive Rating Scale was administered throughout the study period, and whole-brain, voxel-based morphometry was used to measure white matter and gray matter, Dr. Carceller-Sindreu and her associates wrote. The report is in the Journal of Affective Disorders.
At baseline, the white matter volume in the prefrontal cortex of patients with first-episode depression was significantly lower than in healthy controls; no difference was seen in gray matter volume. At the 2-year follow-up, no difference was seen in either white matter or gray matter volume. In other words, the baseline differences “seem to vanish, as if they were normalized,” Dr. Carceller-Sindreu said. The normalization of white matter might have been caused by treatment normalization or attributable to lack of study power, she and her associates noted.
In addition, patients who had recurring depression over the study period had higher white matter volume in the left posterior corona radiata and right posterior thalamic radiation at follow-up, compared with patients whose depression did not recur. This finding “could represent compensatory effects to cope with the disease,” the investigators wrote.
“, so as to unveil many of the open questions,” they concluded.
The study was supported by the Spanish FIS grant, the European Regional Development Fund, and the CERCA Programme. Two study authors reported conflicts of interest with numerous pharmaceutical companies.
SOURCE: Carceller-Sindreu M et al. J Affect Disord. 2018 Nov 13. doi: 10.1016/j.jad.2018.11.085.
Decrements ‘seem to vanish’ after 2-year follow-up
Decrements ‘seem to vanish’ after 2-year follow-up
White matter volume was significantly reduced initially in patients with first-episode depression, but not after a 2-year follow-up, according to Mar Carceller-Sindreu, MD, of the department of psychiatry at Hospital de la Santa Creu i Sant Pau in Barcelona, and her associates.
A total of 33 patients with first-episode depression and 33 healthy controls were included in the study. Among them, 27 first-episode depression patients and 17 controls completed the 2-year follow-up. They underwent structural MRIs at baseline and at follow-up, the Hamilton Depressive Rating Scale was administered throughout the study period, and whole-brain, voxel-based morphometry was used to measure white matter and gray matter, Dr. Carceller-Sindreu and her associates wrote. The report is in the Journal of Affective Disorders.
At baseline, the white matter volume in the prefrontal cortex of patients with first-episode depression was significantly lower than in healthy controls; no difference was seen in gray matter volume. At the 2-year follow-up, no difference was seen in either white matter or gray matter volume. In other words, the baseline differences “seem to vanish, as if they were normalized,” Dr. Carceller-Sindreu said. The normalization of white matter might have been caused by treatment normalization or attributable to lack of study power, she and her associates noted.
In addition, patients who had recurring depression over the study period had higher white matter volume in the left posterior corona radiata and right posterior thalamic radiation at follow-up, compared with patients whose depression did not recur. This finding “could represent compensatory effects to cope with the disease,” the investigators wrote.
“, so as to unveil many of the open questions,” they concluded.
The study was supported by the Spanish FIS grant, the European Regional Development Fund, and the CERCA Programme. Two study authors reported conflicts of interest with numerous pharmaceutical companies.
SOURCE: Carceller-Sindreu M et al. J Affect Disord. 2018 Nov 13. doi: 10.1016/j.jad.2018.11.085.
White matter volume was significantly reduced initially in patients with first-episode depression, but not after a 2-year follow-up, according to Mar Carceller-Sindreu, MD, of the department of psychiatry at Hospital de la Santa Creu i Sant Pau in Barcelona, and her associates.
A total of 33 patients with first-episode depression and 33 healthy controls were included in the study. Among them, 27 first-episode depression patients and 17 controls completed the 2-year follow-up. They underwent structural MRIs at baseline and at follow-up, the Hamilton Depressive Rating Scale was administered throughout the study period, and whole-brain, voxel-based morphometry was used to measure white matter and gray matter, Dr. Carceller-Sindreu and her associates wrote. The report is in the Journal of Affective Disorders.
At baseline, the white matter volume in the prefrontal cortex of patients with first-episode depression was significantly lower than in healthy controls; no difference was seen in gray matter volume. At the 2-year follow-up, no difference was seen in either white matter or gray matter volume. In other words, the baseline differences “seem to vanish, as if they were normalized,” Dr. Carceller-Sindreu said. The normalization of white matter might have been caused by treatment normalization or attributable to lack of study power, she and her associates noted.
In addition, patients who had recurring depression over the study period had higher white matter volume in the left posterior corona radiata and right posterior thalamic radiation at follow-up, compared with patients whose depression did not recur. This finding “could represent compensatory effects to cope with the disease,” the investigators wrote.
“, so as to unveil many of the open questions,” they concluded.
The study was supported by the Spanish FIS grant, the European Regional Development Fund, and the CERCA Programme. Two study authors reported conflicts of interest with numerous pharmaceutical companies.
SOURCE: Carceller-Sindreu M et al. J Affect Disord. 2018 Nov 13. doi: 10.1016/j.jad.2018.11.085.
FROM THE JOURNAL OF AFFECTIVE DISORDERS
Expert calls for better ways to preserve beta cell function in youth
LOS ANGELES –

At the same time, the SEARCH for Diabetes in Youth trial showed that the incidence of T2DM in U.S. youth continues to rise, especially among Native Americans and non-Hispanic blacks (P less than .001 for both associations; N Engl J Med. 2017;376:1419-29). In addition, the earlier Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study showed that rapid treatment failure in youth-onset T2DM was associated with loss of beta cell function (N Engl J Med. 2012;366:2247-56).
“Early treatment of youth with impaired glucose tolerance or type 2 diabetes may require other medications alone or in combination or for longer periods of time to combat the severe insulin resistance of puberty and arrest progressive loss of beta cell function,” Sonia Caprio, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease.
She based her remarks on a review of the recently completed multicenter Restoring Insulin Secretion (RISE) Pediatric Medication Study, (Diabetes Care 2018;41[8]:1717-25). It set out to answer the following question: In adolescents with impaired glucose tolerance or recently diagnosed T2DM, can beta cell function be preserved or improved during 12 months of active treatment and maintained for 3 months following the withdrawal of therapy?
To find out, Dr. Caprio, a pediatric endocrinologist at Yale University, New Haven, Conn., and her colleagues enrolled 91 youth who were randomized to one of two treatment arms: metformin alone titrated over 4 weeks from 500 mg/day to a 1,000 mg twice daily dose (modified if necessary due to GI symptoms), or to glargine followed by metformin. This group received once-daily insulin glargine, titrated twice weekly over 1 month based on daily self-monitoring of blood glucose to a goal of 80-90 mg/dL. Glargine was discontinued after 3 months and metformin was titrated. Beta-cell function (insulin sensitivity paired with beta-cell responses) was assessed by the two-step hyperglycemic clamp at baseline, 12 months (on treatment), and 15 months (3 months off treatment). All clinical data were collected 3 months after discontinuation of active treatment.
Dr. Caprio described the two-step hyperglycemic clamp as “a robust approach to quantification of insulin sensitivity and beta-cell responses to both glucose and the nonglucose secretagogue arginine. It provides mechanistic insights into how the tested interventions affected two key metabolic defects of type 2 diabetes: insulin sensitivity and beta cell responses.”
The mean age of patients was 14 years, 71% were female, their mean body mass index was 37 kg/m2. The researchers observed no significant differences between treatment groups at baseline, 12 months, or 15 months in beta cell function, BMI percentile, hemoglobin A1c, fasting glucose, or oral glucose tolerance test 2-hour glucose results. In both treatment groups, clamp-measured beta cell function was significantly lower at 12 and 15 months, compared with baseline. HbA1c fell transiently at 6 months within both groups. BMI was higher in the glargine followed by metformin versus metformin alone group between 3 and 9 months. Only 5% of participants discontinued the interventions, and both treatments were well tolerated.
“These findings are discouraging,” Dr. Caprio said. “They contrast with previous studies in adults showing an improvement in beta cell function with metformin or insulin for type 2 diabetes prevention and treatment.” Results of the RISE Pediatric Medication Study “call for further studies to better understand the physiology underlying beta cell dysfunction in youth to identify effective treatment options. Better approaches to prevent and treat obesity in youth are critically needed.”
Dr. Caprio reported having no disclosures.
LOS ANGELES –
At the same time, the SEARCH for Diabetes in Youth trial showed that the incidence of T2DM in U.S. youth continues to rise, especially among Native Americans and non-Hispanic blacks (P less than .001 for both associations; N Engl J Med. 2017;376:1419-29). In addition, the earlier Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study showed that rapid treatment failure in youth-onset T2DM was associated with loss of beta cell function (N Engl J Med. 2012;366:2247-56).
“Early treatment of youth with impaired glucose tolerance or type 2 diabetes may require other medications alone or in combination or for longer periods of time to combat the severe insulin resistance of puberty and arrest progressive loss of beta cell function,” Sonia Caprio, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease.
She based her remarks on a review of the recently completed multicenter Restoring Insulin Secretion (RISE) Pediatric Medication Study, (Diabetes Care 2018;41[8]:1717-25). It set out to answer the following question: In adolescents with impaired glucose tolerance or recently diagnosed T2DM, can beta cell function be preserved or improved during 12 months of active treatment and maintained for 3 months following the withdrawal of therapy?
To find out, Dr. Caprio, a pediatric endocrinologist at Yale University, New Haven, Conn., and her colleagues enrolled 91 youth who were randomized to one of two treatment arms: metformin alone titrated over 4 weeks from 500 mg/day to a 1,000 mg twice daily dose (modified if necessary due to GI symptoms), or to glargine followed by metformin. This group received once-daily insulin glargine, titrated twice weekly over 1 month based on daily self-monitoring of blood glucose to a goal of 80-90 mg/dL. Glargine was discontinued after 3 months and metformin was titrated. Beta-cell function (insulin sensitivity paired with beta-cell responses) was assessed by the two-step hyperglycemic clamp at baseline, 12 months (on treatment), and 15 months (3 months off treatment). All clinical data were collected 3 months after discontinuation of active treatment.
Dr. Caprio described the two-step hyperglycemic clamp as “a robust approach to quantification of insulin sensitivity and beta-cell responses to both glucose and the nonglucose secretagogue arginine. It provides mechanistic insights into how the tested interventions affected two key metabolic defects of type 2 diabetes: insulin sensitivity and beta cell responses.”
The mean age of patients was 14 years, 71% were female, their mean body mass index was 37 kg/m2. The researchers observed no significant differences between treatment groups at baseline, 12 months, or 15 months in beta cell function, BMI percentile, hemoglobin A1c, fasting glucose, or oral glucose tolerance test 2-hour glucose results. In both treatment groups, clamp-measured beta cell function was significantly lower at 12 and 15 months, compared with baseline. HbA1c fell transiently at 6 months within both groups. BMI was higher in the glargine followed by metformin versus metformin alone group between 3 and 9 months. Only 5% of participants discontinued the interventions, and both treatments were well tolerated.
“These findings are discouraging,” Dr. Caprio said. “They contrast with previous studies in adults showing an improvement in beta cell function with metformin or insulin for type 2 diabetes prevention and treatment.” Results of the RISE Pediatric Medication Study “call for further studies to better understand the physiology underlying beta cell dysfunction in youth to identify effective treatment options. Better approaches to prevent and treat obesity in youth are critically needed.”
Dr. Caprio reported having no disclosures.
LOS ANGELES –
At the same time, the SEARCH for Diabetes in Youth trial showed that the incidence of T2DM in U.S. youth continues to rise, especially among Native Americans and non-Hispanic blacks (P less than .001 for both associations; N Engl J Med. 2017;376:1419-29). In addition, the earlier Treatment Options for Type 2 Diabetes in Adolescents and Youth (TODAY) study showed that rapid treatment failure in youth-onset T2DM was associated with loss of beta cell function (N Engl J Med. 2012;366:2247-56).
“Early treatment of youth with impaired glucose tolerance or type 2 diabetes may require other medications alone or in combination or for longer periods of time to combat the severe insulin resistance of puberty and arrest progressive loss of beta cell function,” Sonia Caprio, MD, said at the World Congress on Insulin Resistance, Diabetes & Cardiovascular Disease.
She based her remarks on a review of the recently completed multicenter Restoring Insulin Secretion (RISE) Pediatric Medication Study, (Diabetes Care 2018;41[8]:1717-25). It set out to answer the following question: In adolescents with impaired glucose tolerance or recently diagnosed T2DM, can beta cell function be preserved or improved during 12 months of active treatment and maintained for 3 months following the withdrawal of therapy?
To find out, Dr. Caprio, a pediatric endocrinologist at Yale University, New Haven, Conn., and her colleagues enrolled 91 youth who were randomized to one of two treatment arms: metformin alone titrated over 4 weeks from 500 mg/day to a 1,000 mg twice daily dose (modified if necessary due to GI symptoms), or to glargine followed by metformin. This group received once-daily insulin glargine, titrated twice weekly over 1 month based on daily self-monitoring of blood glucose to a goal of 80-90 mg/dL. Glargine was discontinued after 3 months and metformin was titrated. Beta-cell function (insulin sensitivity paired with beta-cell responses) was assessed by the two-step hyperglycemic clamp at baseline, 12 months (on treatment), and 15 months (3 months off treatment). All clinical data were collected 3 months after discontinuation of active treatment.
Dr. Caprio described the two-step hyperglycemic clamp as “a robust approach to quantification of insulin sensitivity and beta-cell responses to both glucose and the nonglucose secretagogue arginine. It provides mechanistic insights into how the tested interventions affected two key metabolic defects of type 2 diabetes: insulin sensitivity and beta cell responses.”
The mean age of patients was 14 years, 71% were female, their mean body mass index was 37 kg/m2. The researchers observed no significant differences between treatment groups at baseline, 12 months, or 15 months in beta cell function, BMI percentile, hemoglobin A1c, fasting glucose, or oral glucose tolerance test 2-hour glucose results. In both treatment groups, clamp-measured beta cell function was significantly lower at 12 and 15 months, compared with baseline. HbA1c fell transiently at 6 months within both groups. BMI was higher in the glargine followed by metformin versus metformin alone group between 3 and 9 months. Only 5% of participants discontinued the interventions, and both treatments were well tolerated.
“These findings are discouraging,” Dr. Caprio said. “They contrast with previous studies in adults showing an improvement in beta cell function with metformin or insulin for type 2 diabetes prevention and treatment.” Results of the RISE Pediatric Medication Study “call for further studies to better understand the physiology underlying beta cell dysfunction in youth to identify effective treatment options. Better approaches to prevent and treat obesity in youth are critically needed.”
Dr. Caprio reported having no disclosures.
EXPERT ANALYSIS FROM WCIRDC 2018
Synthetic opioids drive spike in U.S. fatal drug overdoses
New federal statistics suggest that the opioid epidemic in the United States is evolving as physicians crack down on the use of prescription painkillers: Fatal drug overdose deaths rose by 12% from 2016 to 2017, boosted by a wave of fatalities linked to illicit synthetic opioids like fentanyl that are now linked to an estimated 60% of opioid-related deaths.

“Overall, the overdose epidemic continues to worsen, and it has grown increasingly complex by coinvolvement of prescription and illicit drugs,” Lawrence Scholl, PhD, MPH, and his associates at the Centers for Disease Control & Prevention wrote in the Morbidity and Mortality Weekly Report.
The new statistics provide more evidence that 2017 marked “a sharp increase in what has characterized as the third wave of the opioid epidemic,” said drug and health policy researcher Stephen Crystal, PhD, of Rutgers University, New Brunswick, N.J., in an interview. He was referring to a wave that experts believe started in 2013 amid a spike in U.S. overdose deaths from fentanyl and other synthetic opioids.
The new report analyzes fatal drug overdose data from 2013 to 2017. According to the findings, the total number of those overdoses rose to 70,237 in 2017, up from 63,632 in 2016. The highest drug overdose death rates in 2017 were in West Virginia, followed by Ohio, Pennsylvania, and the District of Columbia.
Some statistics did not change much from 2016 to 2017: About two-thirds of the drug overdose deaths were linked to opioids in both years, and the death rate of cases linked to prescription drugs and heroin remained steady. (Death rates in the report were age adjusted.)
However, the percentage of fatal overdose cases linked to synthetic opioids grew 45% from 2016 to 2017. Overall, 60% of opioid-related fatal overdoses in 2017 involved synthetic opioids.
The report identifies increases in several areas from 2016 to 2017. Opioid-related drug overdose deaths among black people rose by 25%, and an analysis of data from 34 states and the District of Columbia found the highest increases in death rates in North Carolina (29%), Ohio (19%), and Maine (19%).
In regard to deaths linked to synthetic opioids specifically, the highest death rates in 2017 were in West Virginia (37 per 100,000), Ohio (32 per 100,000), and New Hampshire (30 per 100,000).
“Part of what we’re seeing in these increased numbers are individuals who have pain, can’t get prescribed opioids, and turn to street drugs,” Dr. Crystal said, adding that “abruptly cutting patients off is not good, and leaving patients with a lot of untreated pain is not good. If people are going to be discontinued [from opioids] or have their doses reduced, the taper needs to be done very slowly and carefully.”
Synthetic opioids were not the only drugs that are driving up fatal overdoses, as the death rates of cases linked to cocaine and psychostimulants (such as methamphetamine) jumped by more than a third in 2017.
“The most important thing these numbers are telling me is that it’s becoming more and more attractive to drug dealers to put fentanyl in the heroin, cocaine, and other drugs they sell,” Dr. Crystal said. “When that happens, dependence on street drugs becomes much more deadly. It’s almost impossible to get the dose right. Every time you shoot up, you’re taking a chance that you’ll overdose.”
The report had limitations, including the fact that details about drug use were missing from 12% (2016) and 15% (2017) of death certificates in fatal overdose cases. By state, the percentages of those death certificates that included drug information ranged from as little as 55% to 99%.
There’s some possible positive news: The report points to preliminary data from 2018 suggesting that the number of annual drug overdose deaths may be leveling off – although it says more analysis is needed to confirm the trend.
Dr. Crystal, however, is not celebrating. “I don’t see this as a good news story, really,” he said, adding that there’s “a little too much of people patting themselves on the back” because they’re proud of cutbacks in opioid prescriptions.
“This doesn’t have to do with the huge number of people who got started with opioids years ago” and are now at risk of using street drugs, he said. “We haven’t engaged that population at the rate we need to. And flattening out at 70,000 drug overdoses a year is not a good news story.”
Dr. Crystal reported no relevant disclosures.
SOURCE: Scholl L et al. MMWR. 2019 Jan 4;67(5152):1419-27.
New federal statistics suggest that the opioid epidemic in the United States is evolving as physicians crack down on the use of prescription painkillers: Fatal drug overdose deaths rose by 12% from 2016 to 2017, boosted by a wave of fatalities linked to illicit synthetic opioids like fentanyl that are now linked to an estimated 60% of opioid-related deaths.
“Overall, the overdose epidemic continues to worsen, and it has grown increasingly complex by coinvolvement of prescription and illicit drugs,” Lawrence Scholl, PhD, MPH, and his associates at the Centers for Disease Control & Prevention wrote in the Morbidity and Mortality Weekly Report.
The new statistics provide more evidence that 2017 marked “a sharp increase in what has characterized as the third wave of the opioid epidemic,” said drug and health policy researcher Stephen Crystal, PhD, of Rutgers University, New Brunswick, N.J., in an interview. He was referring to a wave that experts believe started in 2013 amid a spike in U.S. overdose deaths from fentanyl and other synthetic opioids.
The new report analyzes fatal drug overdose data from 2013 to 2017. According to the findings, the total number of those overdoses rose to 70,237 in 2017, up from 63,632 in 2016. The highest drug overdose death rates in 2017 were in West Virginia, followed by Ohio, Pennsylvania, and the District of Columbia.
Some statistics did not change much from 2016 to 2017: About two-thirds of the drug overdose deaths were linked to opioids in both years, and the death rate of cases linked to prescription drugs and heroin remained steady. (Death rates in the report were age adjusted.)
However, the percentage of fatal overdose cases linked to synthetic opioids grew 45% from 2016 to 2017. Overall, 60% of opioid-related fatal overdoses in 2017 involved synthetic opioids.
The report identifies increases in several areas from 2016 to 2017. Opioid-related drug overdose deaths among black people rose by 25%, and an analysis of data from 34 states and the District of Columbia found the highest increases in death rates in North Carolina (29%), Ohio (19%), and Maine (19%).
In regard to deaths linked to synthetic opioids specifically, the highest death rates in 2017 were in West Virginia (37 per 100,000), Ohio (32 per 100,000), and New Hampshire (30 per 100,000).
“Part of what we’re seeing in these increased numbers are individuals who have pain, can’t get prescribed opioids, and turn to street drugs,” Dr. Crystal said, adding that “abruptly cutting patients off is not good, and leaving patients with a lot of untreated pain is not good. If people are going to be discontinued [from opioids] or have their doses reduced, the taper needs to be done very slowly and carefully.”
Synthetic opioids were not the only drugs that are driving up fatal overdoses, as the death rates of cases linked to cocaine and psychostimulants (such as methamphetamine) jumped by more than a third in 2017.
“The most important thing these numbers are telling me is that it’s becoming more and more attractive to drug dealers to put fentanyl in the heroin, cocaine, and other drugs they sell,” Dr. Crystal said. “When that happens, dependence on street drugs becomes much more deadly. It’s almost impossible to get the dose right. Every time you shoot up, you’re taking a chance that you’ll overdose.”
The report had limitations, including the fact that details about drug use were missing from 12% (2016) and 15% (2017) of death certificates in fatal overdose cases. By state, the percentages of those death certificates that included drug information ranged from as little as 55% to 99%.
There’s some possible positive news: The report points to preliminary data from 2018 suggesting that the number of annual drug overdose deaths may be leveling off – although it says more analysis is needed to confirm the trend.
Dr. Crystal, however, is not celebrating. “I don’t see this as a good news story, really,” he said, adding that there’s “a little too much of people patting themselves on the back” because they’re proud of cutbacks in opioid prescriptions.
“This doesn’t have to do with the huge number of people who got started with opioids years ago” and are now at risk of using street drugs, he said. “We haven’t engaged that population at the rate we need to. And flattening out at 70,000 drug overdoses a year is not a good news story.”
Dr. Crystal reported no relevant disclosures.
SOURCE: Scholl L et al. MMWR. 2019 Jan 4;67(5152):1419-27.
New federal statistics suggest that the opioid epidemic in the United States is evolving as physicians crack down on the use of prescription painkillers: Fatal drug overdose deaths rose by 12% from 2016 to 2017, boosted by a wave of fatalities linked to illicit synthetic opioids like fentanyl that are now linked to an estimated 60% of opioid-related deaths.
“Overall, the overdose epidemic continues to worsen, and it has grown increasingly complex by coinvolvement of prescription and illicit drugs,” Lawrence Scholl, PhD, MPH, and his associates at the Centers for Disease Control & Prevention wrote in the Morbidity and Mortality Weekly Report.
The new statistics provide more evidence that 2017 marked “a sharp increase in what has characterized as the third wave of the opioid epidemic,” said drug and health policy researcher Stephen Crystal, PhD, of Rutgers University, New Brunswick, N.J., in an interview. He was referring to a wave that experts believe started in 2013 amid a spike in U.S. overdose deaths from fentanyl and other synthetic opioids.
The new report analyzes fatal drug overdose data from 2013 to 2017. According to the findings, the total number of those overdoses rose to 70,237 in 2017, up from 63,632 in 2016. The highest drug overdose death rates in 2017 were in West Virginia, followed by Ohio, Pennsylvania, and the District of Columbia.
Some statistics did not change much from 2016 to 2017: About two-thirds of the drug overdose deaths were linked to opioids in both years, and the death rate of cases linked to prescription drugs and heroin remained steady. (Death rates in the report were age adjusted.)
However, the percentage of fatal overdose cases linked to synthetic opioids grew 45% from 2016 to 2017. Overall, 60% of opioid-related fatal overdoses in 2017 involved synthetic opioids.
The report identifies increases in several areas from 2016 to 2017. Opioid-related drug overdose deaths among black people rose by 25%, and an analysis of data from 34 states and the District of Columbia found the highest increases in death rates in North Carolina (29%), Ohio (19%), and Maine (19%).
In regard to deaths linked to synthetic opioids specifically, the highest death rates in 2017 were in West Virginia (37 per 100,000), Ohio (32 per 100,000), and New Hampshire (30 per 100,000).
“Part of what we’re seeing in these increased numbers are individuals who have pain, can’t get prescribed opioids, and turn to street drugs,” Dr. Crystal said, adding that “abruptly cutting patients off is not good, and leaving patients with a lot of untreated pain is not good. If people are going to be discontinued [from opioids] or have their doses reduced, the taper needs to be done very slowly and carefully.”
Synthetic opioids were not the only drugs that are driving up fatal overdoses, as the death rates of cases linked to cocaine and psychostimulants (such as methamphetamine) jumped by more than a third in 2017.
“The most important thing these numbers are telling me is that it’s becoming more and more attractive to drug dealers to put fentanyl in the heroin, cocaine, and other drugs they sell,” Dr. Crystal said. “When that happens, dependence on street drugs becomes much more deadly. It’s almost impossible to get the dose right. Every time you shoot up, you’re taking a chance that you’ll overdose.”
The report had limitations, including the fact that details about drug use were missing from 12% (2016) and 15% (2017) of death certificates in fatal overdose cases. By state, the percentages of those death certificates that included drug information ranged from as little as 55% to 99%.
There’s some possible positive news: The report points to preliminary data from 2018 suggesting that the number of annual drug overdose deaths may be leveling off – although it says more analysis is needed to confirm the trend.
Dr. Crystal, however, is not celebrating. “I don’t see this as a good news story, really,” he said, adding that there’s “a little too much of people patting themselves on the back” because they’re proud of cutbacks in opioid prescriptions.
“This doesn’t have to do with the huge number of people who got started with opioids years ago” and are now at risk of using street drugs, he said. “We haven’t engaged that population at the rate we need to. And flattening out at 70,000 drug overdoses a year is not a good news story.”
Dr. Crystal reported no relevant disclosures.
SOURCE: Scholl L et al. MMWR. 2019 Jan 4;67(5152):1419-27.
FROM MMWR