User login
Criteria-based fibromyalgia diagnosis and rheumatologists’ clinical diagnosis often disagree
particularly the most recent revision of criteria from 2011 that rely on patients’ self-report of symptoms.

Frederick Wolfe, MD, of the National Data Bank for Rheumatic Diseases and the University of Kansas, Wichita, and his colleagues reported in Arthritis Care & Research that clinicians failed to identify almost half of cases that met the 2011 modification of the American College of Rheumatology’s self-report criteria for fibromyalgia during a 3-month period at Rush Medical College, Chicago. This led them to conclude that the “overall agreement between clinicians’ diagnosis of fibromyalgia and diagnosis by fibromyalgia criteria is only fair.”
The findings call into question studies of fibromyalgia based on ICD-10 diagnosis, according to the authors.
Several widely accepted criteria sets have provided definitions and methods of diagnosis, but a number of studies have suggested that fibromyalgia is overdiagnosed, the authors wrote.
“In the current study, we consider underdiagnosis as well as the form of overdiagnosis that lead to diagnosis when fibromyalgia criteria are not satisfied and symptoms are insufficient for diagnosis,” they added.
The research included 497 consecutive patients attending the Rush Medical College rheumatology clinic who completed a questionnaire assessing fibromyalgia diagnostic variables used in the ACR 2010 preliminary diagnostic criteria for fibromyalgia and its 2011 modification for self-report immediately prior to being seen at the clinic. Clinicians were not given instructions on how any diseases were to be diagnosed, including no instructions on using 2011 fibromyalgia criteria.
The results showed that 121 (24.3%) of the patients satisfied the 2011 fibromyalgia criteria. The agreement between clinicians and criteria was 79.2%, but agreement beyond chance was considered “fair” based on a kappa score of 0.41 and a probabilistic benchmark interval of 0.21-0.40. The researchers wrote that this benchmark represents “the probability for each coefficient of falling into the selected benchmark interval along with the cumulative probability of exceeding the predetermined threshold associated with the interval.”
A total of 104 (20.9%) received a clinician ICD-10 diagnosis of fibromyalgia, but only 61 (58.7%) actually satisfied criteria. Physicians failed to identify 60 (49.6%) criteria-positive patients and incorrectly identified 43 (11.4%) criteria-negative patients.
In a subset of 88 patients with RA, agreement was 84.1%, and the kappa score was 0.32, indicating “slight to fair” agreement (probabilistic benchmark interval, 0.00-0.20). Among 13 RA patients with criteria-positive fibromyalgia, 5 were identified by clinicians; among those who were criteria negative, 6 were deemed positive by clinicians.
The authors noted than “even worse” results were obtained in patients with systemic lupus erythematosus, where the agreement between criteria and clinician ICD diagnosis yielded a kappa value of 0.08. In patients with osteoarthritis, the kappa score was 0.51 (probabilistic benchmark interval, 0.21-0.40).
Overall, the results showed that women and patients with more symptoms but fewer pain areas were more likely to receive a clinician’s diagnosis than to satisfy fibromyalgia criteria. “Clinicians gave greater weight in making a diagnosis to being a woman and having increased symptoms and were willing to diagnose patients with lower WPI [Widespread Pain Index] and PSD [polysymptomatic distress] scores,” the researchers noted.
Dr. Wolfe and his associates wrote that it is unclear why the physicians in the study who misclassified fibromyalgia and missed patients with the diagnosis did not do better. “They may have simply misdiagnosed the condition or not accepted the use of the formal diagnostic system as necessary or clinically useful for identifying fibromyalgia in routine care.”
The researchers noted that it is likely that diagnosis in the community by general physicians, for whom the criteria are not as well known, is even more inaccurate. “It is likely that misdiagnosis is a public health problem and one that can lead to overdiagnosis and overtreatment as well as to inappropriate treatment of individuals not recognized to have fibromyalgia symptoms.”
No disclosures or funding were reported.
SOURCE: Wolfe F et al. Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23731.
Despite voicing concern regarding the “self-report nature of fibromyalgia,” Wolfe et al. selected the 2011 self-report fibromyalgia criteria as their “gold standard” for fibromyalgia diagnosis. Their findings therefore should not be surprising.
The authors’ conclusion that expert physicians often misdiagnose fibromyalgia implies that published criteria are superior to expert clinical judgment for individual patient diagnosis.
However, this view fails to take into account the myriad variables seen by the clinician in the clinic. Until biomarkers or genetic testing allows for more objective disease markers, common conditions like fibromyalgia will continue to be symptom-based diagnoses. Therefore, the gold standard diagnostic criteria for fibromyalgia and other common illnesses is expert opinion.
Rheumatologists are the go-to experts for the diagnosis of fibromyalgia, whether or not we readily accept that role.
These comments are adapted from an accompanying editorial by Don Goldenberg, MD, of Oregon Health & Science University, Portland, and Tufts University, Boston (Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23727).
Despite voicing concern regarding the “self-report nature of fibromyalgia,” Wolfe et al. selected the 2011 self-report fibromyalgia criteria as their “gold standard” for fibromyalgia diagnosis. Their findings therefore should not be surprising.
The authors’ conclusion that expert physicians often misdiagnose fibromyalgia implies that published criteria are superior to expert clinical judgment for individual patient diagnosis.
However, this view fails to take into account the myriad variables seen by the clinician in the clinic. Until biomarkers or genetic testing allows for more objective disease markers, common conditions like fibromyalgia will continue to be symptom-based diagnoses. Therefore, the gold standard diagnostic criteria for fibromyalgia and other common illnesses is expert opinion.
Rheumatologists are the go-to experts for the diagnosis of fibromyalgia, whether or not we readily accept that role.
These comments are adapted from an accompanying editorial by Don Goldenberg, MD, of Oregon Health & Science University, Portland, and Tufts University, Boston (Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23727).
Despite voicing concern regarding the “self-report nature of fibromyalgia,” Wolfe et al. selected the 2011 self-report fibromyalgia criteria as their “gold standard” for fibromyalgia diagnosis. Their findings therefore should not be surprising.
The authors’ conclusion that expert physicians often misdiagnose fibromyalgia implies that published criteria are superior to expert clinical judgment for individual patient diagnosis.
However, this view fails to take into account the myriad variables seen by the clinician in the clinic. Until biomarkers or genetic testing allows for more objective disease markers, common conditions like fibromyalgia will continue to be symptom-based diagnoses. Therefore, the gold standard diagnostic criteria for fibromyalgia and other common illnesses is expert opinion.
Rheumatologists are the go-to experts for the diagnosis of fibromyalgia, whether or not we readily accept that role.
These comments are adapted from an accompanying editorial by Don Goldenberg, MD, of Oregon Health & Science University, Portland, and Tufts University, Boston (Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23727).
particularly the most recent revision of criteria from 2011 that rely on patients’ self-report of symptoms.
Frederick Wolfe, MD, of the National Data Bank for Rheumatic Diseases and the University of Kansas, Wichita, and his colleagues reported in Arthritis Care & Research that clinicians failed to identify almost half of cases that met the 2011 modification of the American College of Rheumatology’s self-report criteria for fibromyalgia during a 3-month period at Rush Medical College, Chicago. This led them to conclude that the “overall agreement between clinicians’ diagnosis of fibromyalgia and diagnosis by fibromyalgia criteria is only fair.”
The findings call into question studies of fibromyalgia based on ICD-10 diagnosis, according to the authors.
Several widely accepted criteria sets have provided definitions and methods of diagnosis, but a number of studies have suggested that fibromyalgia is overdiagnosed, the authors wrote.
“In the current study, we consider underdiagnosis as well as the form of overdiagnosis that lead to diagnosis when fibromyalgia criteria are not satisfied and symptoms are insufficient for diagnosis,” they added.
The research included 497 consecutive patients attending the Rush Medical College rheumatology clinic who completed a questionnaire assessing fibromyalgia diagnostic variables used in the ACR 2010 preliminary diagnostic criteria for fibromyalgia and its 2011 modification for self-report immediately prior to being seen at the clinic. Clinicians were not given instructions on how any diseases were to be diagnosed, including no instructions on using 2011 fibromyalgia criteria.
The results showed that 121 (24.3%) of the patients satisfied the 2011 fibromyalgia criteria. The agreement between clinicians and criteria was 79.2%, but agreement beyond chance was considered “fair” based on a kappa score of 0.41 and a probabilistic benchmark interval of 0.21-0.40. The researchers wrote that this benchmark represents “the probability for each coefficient of falling into the selected benchmark interval along with the cumulative probability of exceeding the predetermined threshold associated with the interval.”
A total of 104 (20.9%) received a clinician ICD-10 diagnosis of fibromyalgia, but only 61 (58.7%) actually satisfied criteria. Physicians failed to identify 60 (49.6%) criteria-positive patients and incorrectly identified 43 (11.4%) criteria-negative patients.
In a subset of 88 patients with RA, agreement was 84.1%, and the kappa score was 0.32, indicating “slight to fair” agreement (probabilistic benchmark interval, 0.00-0.20). Among 13 RA patients with criteria-positive fibromyalgia, 5 were identified by clinicians; among those who were criteria negative, 6 were deemed positive by clinicians.
The authors noted than “even worse” results were obtained in patients with systemic lupus erythematosus, where the agreement between criteria and clinician ICD diagnosis yielded a kappa value of 0.08. In patients with osteoarthritis, the kappa score was 0.51 (probabilistic benchmark interval, 0.21-0.40).
Overall, the results showed that women and patients with more symptoms but fewer pain areas were more likely to receive a clinician’s diagnosis than to satisfy fibromyalgia criteria. “Clinicians gave greater weight in making a diagnosis to being a woman and having increased symptoms and were willing to diagnose patients with lower WPI [Widespread Pain Index] and PSD [polysymptomatic distress] scores,” the researchers noted.
Dr. Wolfe and his associates wrote that it is unclear why the physicians in the study who misclassified fibromyalgia and missed patients with the diagnosis did not do better. “They may have simply misdiagnosed the condition or not accepted the use of the formal diagnostic system as necessary or clinically useful for identifying fibromyalgia in routine care.”
The researchers noted that it is likely that diagnosis in the community by general physicians, for whom the criteria are not as well known, is even more inaccurate. “It is likely that misdiagnosis is a public health problem and one that can lead to overdiagnosis and overtreatment as well as to inappropriate treatment of individuals not recognized to have fibromyalgia symptoms.”
No disclosures or funding were reported.
SOURCE: Wolfe F et al. Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23731.
particularly the most recent revision of criteria from 2011 that rely on patients’ self-report of symptoms.
Frederick Wolfe, MD, of the National Data Bank for Rheumatic Diseases and the University of Kansas, Wichita, and his colleagues reported in Arthritis Care & Research that clinicians failed to identify almost half of cases that met the 2011 modification of the American College of Rheumatology’s self-report criteria for fibromyalgia during a 3-month period at Rush Medical College, Chicago. This led them to conclude that the “overall agreement between clinicians’ diagnosis of fibromyalgia and diagnosis by fibromyalgia criteria is only fair.”
The findings call into question studies of fibromyalgia based on ICD-10 diagnosis, according to the authors.
Several widely accepted criteria sets have provided definitions and methods of diagnosis, but a number of studies have suggested that fibromyalgia is overdiagnosed, the authors wrote.
“In the current study, we consider underdiagnosis as well as the form of overdiagnosis that lead to diagnosis when fibromyalgia criteria are not satisfied and symptoms are insufficient for diagnosis,” they added.
The research included 497 consecutive patients attending the Rush Medical College rheumatology clinic who completed a questionnaire assessing fibromyalgia diagnostic variables used in the ACR 2010 preliminary diagnostic criteria for fibromyalgia and its 2011 modification for self-report immediately prior to being seen at the clinic. Clinicians were not given instructions on how any diseases were to be diagnosed, including no instructions on using 2011 fibromyalgia criteria.
The results showed that 121 (24.3%) of the patients satisfied the 2011 fibromyalgia criteria. The agreement between clinicians and criteria was 79.2%, but agreement beyond chance was considered “fair” based on a kappa score of 0.41 and a probabilistic benchmark interval of 0.21-0.40. The researchers wrote that this benchmark represents “the probability for each coefficient of falling into the selected benchmark interval along with the cumulative probability of exceeding the predetermined threshold associated with the interval.”
A total of 104 (20.9%) received a clinician ICD-10 diagnosis of fibromyalgia, but only 61 (58.7%) actually satisfied criteria. Physicians failed to identify 60 (49.6%) criteria-positive patients and incorrectly identified 43 (11.4%) criteria-negative patients.
In a subset of 88 patients with RA, agreement was 84.1%, and the kappa score was 0.32, indicating “slight to fair” agreement (probabilistic benchmark interval, 0.00-0.20). Among 13 RA patients with criteria-positive fibromyalgia, 5 were identified by clinicians; among those who were criteria negative, 6 were deemed positive by clinicians.
The authors noted than “even worse” results were obtained in patients with systemic lupus erythematosus, where the agreement between criteria and clinician ICD diagnosis yielded a kappa value of 0.08. In patients with osteoarthritis, the kappa score was 0.51 (probabilistic benchmark interval, 0.21-0.40).
Overall, the results showed that women and patients with more symptoms but fewer pain areas were more likely to receive a clinician’s diagnosis than to satisfy fibromyalgia criteria. “Clinicians gave greater weight in making a diagnosis to being a woman and having increased symptoms and were willing to diagnose patients with lower WPI [Widespread Pain Index] and PSD [polysymptomatic distress] scores,” the researchers noted.
Dr. Wolfe and his associates wrote that it is unclear why the physicians in the study who misclassified fibromyalgia and missed patients with the diagnosis did not do better. “They may have simply misdiagnosed the condition or not accepted the use of the formal diagnostic system as necessary or clinically useful for identifying fibromyalgia in routine care.”
The researchers noted that it is likely that diagnosis in the community by general physicians, for whom the criteria are not as well known, is even more inaccurate. “It is likely that misdiagnosis is a public health problem and one that can lead to overdiagnosis and overtreatment as well as to inappropriate treatment of individuals not recognized to have fibromyalgia symptoms.”
No disclosures or funding were reported.
SOURCE: Wolfe F et al. Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23731.
FROM ARTHRITIS CARE & RESEARCH
Key clinical point: Clinicians failed to identify nearly 50% of criteria-positive cases
Major finding: Physicians failed to identify 60 (49.6%) criteria-positive patients and incorrectly identified 43 (11.4%) criteria-negative patients.
Study details: A group of 497 consecutive unselected rheumatology clinic attendees who completed a questionnaire assessing fibromyalgia diagnostic variables used in the American College of Rheumatology 2010 preliminary diagnostic criteria for fibromyalgia and its 2011 modification for self-report.
Disclosures: No disclosures or funding were reported.
Source: Wolfe F et al. Arthritis Care Res. 2019 Feb 6. doi: 10.1002/acr.23731.
PTSD, cardiovascular disease link likely caused by higher comorbidity burden
Although PTSD is strongly linked to cardiovascular disease, it is not an independent risk factor, results of a recent analysis suggest.

Instead, the association between PTSD and cardiovascular disease (CVD) is likely explained by factors such as smoking and physical and psychiatric disorders, according to authors of the analysis based on EHR data for more than 4,000 U.S. veterans.
Individuals with PTSD were 41% more likely than those without it to develop cardiovascular disease, according to the researchers, led by Jeffrey F. Scherrer, PhD, of Saint Louis University and the Harry S. Truman Veterans Administration Medical Center in Columbia, Mo.
However, PTSD was not associated with CVD in the study after adjustment for physical, psychiatric, and behavioral conditions, Dr. Scherrer and his colleagues reported in the Journal of the American Heart Association.
“Recognizing that PTSD does not preordain CVD may empower patients to seek care to prevent and/or manage CVD risk factors,” they wrote.
Health behavior change and management of chronic disease can mitigate risk of CVD in patients with or without PTSD, they added.
This result contrasts with earlier work associating PTSD with CVD, the authors wrote. In particular, a few well-designed studies did indicate that the link between PTSD and CVD was weakened, but still significant, when controlling for traditional CVD risk factors such as smoking, diabetes, and hypertension.
In the current study, investigators controlled for a variety of physical and psychiatric conditions, as well as smoking, in data for Veterans Affairs patients, of whom 2,519 had a PTSD diagnosis and 1,659 did not. These patients were 87% male, 60% white, and had an average age of 50 years.
The investigators found that PTSD was significantly associated with incident CVD after adjusting for age, with a hazard ratio of 1.41 (95% confidence interval, 1.21-1.63; P less than .0001).
That association remained significant after adjusting for diabetes, obesity, hypertension, and hyperlipidemia, but the magnitude of the association dropped considerably (HR, 1.23; 95% CI, 1.06-1.44; P less than .007) and dropped out altogether after controlling for smoking, substance abuse, sleep disorders, anxiety, and depression (HR, 0.96; 95% CI, 0.81-1.15; P = .691).
Taken together, these findings suggest the association with incident CVD may be explained by combinations of comorbidities that are more prevalent in patients who have PTSD than in those who do not, Dr. Scherrer and his coauthors wrote.
“Because these conditions are more common in patients with PTSD, closer monitoring for comorbidities may be warranted,” they concluded. “Early detection and effective management may reduce the burden of CVD associated with PTSD.”
One study coinvestigator reported consulting for Noblis Therapeutics and grant-related disclosures with the Department of Veterans Affairs, Department of Defense, and National Institute of Mental Health.
SOURCE: Scherrer JF et al. J Am Heart Assoc. 2019 Feb 13. doi: 10.1161/JAHA.118.011133.
Although PTSD is strongly linked to cardiovascular disease, it is not an independent risk factor, results of a recent analysis suggest.
Instead, the association between PTSD and cardiovascular disease (CVD) is likely explained by factors such as smoking and physical and psychiatric disorders, according to authors of the analysis based on EHR data for more than 4,000 U.S. veterans.
Individuals with PTSD were 41% more likely than those without it to develop cardiovascular disease, according to the researchers, led by Jeffrey F. Scherrer, PhD, of Saint Louis University and the Harry S. Truman Veterans Administration Medical Center in Columbia, Mo.
However, PTSD was not associated with CVD in the study after adjustment for physical, psychiatric, and behavioral conditions, Dr. Scherrer and his colleagues reported in the Journal of the American Heart Association.
“Recognizing that PTSD does not preordain CVD may empower patients to seek care to prevent and/or manage CVD risk factors,” they wrote.
Health behavior change and management of chronic disease can mitigate risk of CVD in patients with or without PTSD, they added.
This result contrasts with earlier work associating PTSD with CVD, the authors wrote. In particular, a few well-designed studies did indicate that the link between PTSD and CVD was weakened, but still significant, when controlling for traditional CVD risk factors such as smoking, diabetes, and hypertension.
In the current study, investigators controlled for a variety of physical and psychiatric conditions, as well as smoking, in data for Veterans Affairs patients, of whom 2,519 had a PTSD diagnosis and 1,659 did not. These patients were 87% male, 60% white, and had an average age of 50 years.
The investigators found that PTSD was significantly associated with incident CVD after adjusting for age, with a hazard ratio of 1.41 (95% confidence interval, 1.21-1.63; P less than .0001).
That association remained significant after adjusting for diabetes, obesity, hypertension, and hyperlipidemia, but the magnitude of the association dropped considerably (HR, 1.23; 95% CI, 1.06-1.44; P less than .007) and dropped out altogether after controlling for smoking, substance abuse, sleep disorders, anxiety, and depression (HR, 0.96; 95% CI, 0.81-1.15; P = .691).
Taken together, these findings suggest the association with incident CVD may be explained by combinations of comorbidities that are more prevalent in patients who have PTSD than in those who do not, Dr. Scherrer and his coauthors wrote.
“Because these conditions are more common in patients with PTSD, closer monitoring for comorbidities may be warranted,” they concluded. “Early detection and effective management may reduce the burden of CVD associated with PTSD.”
One study coinvestigator reported consulting for Noblis Therapeutics and grant-related disclosures with the Department of Veterans Affairs, Department of Defense, and National Institute of Mental Health.
SOURCE: Scherrer JF et al. J Am Heart Assoc. 2019 Feb 13. doi: 10.1161/JAHA.118.011133.
Although PTSD is strongly linked to cardiovascular disease, it is not an independent risk factor, results of a recent analysis suggest.
Instead, the association between PTSD and cardiovascular disease (CVD) is likely explained by factors such as smoking and physical and psychiatric disorders, according to authors of the analysis based on EHR data for more than 4,000 U.S. veterans.
Individuals with PTSD were 41% more likely than those without it to develop cardiovascular disease, according to the researchers, led by Jeffrey F. Scherrer, PhD, of Saint Louis University and the Harry S. Truman Veterans Administration Medical Center in Columbia, Mo.
However, PTSD was not associated with CVD in the study after adjustment for physical, psychiatric, and behavioral conditions, Dr. Scherrer and his colleagues reported in the Journal of the American Heart Association.
“Recognizing that PTSD does not preordain CVD may empower patients to seek care to prevent and/or manage CVD risk factors,” they wrote.
Health behavior change and management of chronic disease can mitigate risk of CVD in patients with or without PTSD, they added.
This result contrasts with earlier work associating PTSD with CVD, the authors wrote. In particular, a few well-designed studies did indicate that the link between PTSD and CVD was weakened, but still significant, when controlling for traditional CVD risk factors such as smoking, diabetes, and hypertension.
In the current study, investigators controlled for a variety of physical and psychiatric conditions, as well as smoking, in data for Veterans Affairs patients, of whom 2,519 had a PTSD diagnosis and 1,659 did not. These patients were 87% male, 60% white, and had an average age of 50 years.
The investigators found that PTSD was significantly associated with incident CVD after adjusting for age, with a hazard ratio of 1.41 (95% confidence interval, 1.21-1.63; P less than .0001).
That association remained significant after adjusting for diabetes, obesity, hypertension, and hyperlipidemia, but the magnitude of the association dropped considerably (HR, 1.23; 95% CI, 1.06-1.44; P less than .007) and dropped out altogether after controlling for smoking, substance abuse, sleep disorders, anxiety, and depression (HR, 0.96; 95% CI, 0.81-1.15; P = .691).
Taken together, these findings suggest the association with incident CVD may be explained by combinations of comorbidities that are more prevalent in patients who have PTSD than in those who do not, Dr. Scherrer and his coauthors wrote.
“Because these conditions are more common in patients with PTSD, closer monitoring for comorbidities may be warranted,” they concluded. “Early detection and effective management may reduce the burden of CVD associated with PTSD.”
One study coinvestigator reported consulting for Noblis Therapeutics and grant-related disclosures with the Department of Veterans Affairs, Department of Defense, and National Institute of Mental Health.
SOURCE: Scherrer JF et al. J Am Heart Assoc. 2019 Feb 13. doi: 10.1161/JAHA.118.011133.
FROM THE JOURNAL OF THE AMERICAN HEART ASSOCIATION
Key clinical point: The link between PTSD and cardiovascular disease may be explained by higher prevalence of comorbidities in individuals with the disorder, rather than the disorder itself.
Major finding: PTSD was significantly associated with incident cardiovascular disease after adjusting for age, but was no longer associated with cardiovascular disease after further adjustment for physical and psychological comorbidities and smoking (hazard ratio, 0.96; 95% confidence interval, 0.81-1.15; P = 0.691).
Study details: A retrospective study of EHR data for Veterans Affairs patients, of whom 2,519 had a PTSD diagnosis and 1,659 did not.
Disclosures: One study author reported financial disclosures related to Noblis Therapeutics, the Department of Veterans Affairs, Department of Defense, the National Institute of Mental Health.
Source: Scherrer JF et al. J Am Heart Assoc. 2019 Feb 13. doi: 10.1161/JAHA.118.011133.
Conservatism spreads in prostate cancer
, the United States now has more than 100 measles cases for the year, e-cigarette use reverses progress in reducing teens’ tobacco use, and consider adopting the MESA 10-year coronary heart disease risk calculator.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
, the United States now has more than 100 measles cases for the year, e-cigarette use reverses progress in reducing teens’ tobacco use, and consider adopting the MESA 10-year coronary heart disease risk calculator.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify
, the United States now has more than 100 measles cases for the year, e-cigarette use reverses progress in reducing teens’ tobacco use, and consider adopting the MESA 10-year coronary heart disease risk calculator.
Amazon Alexa
Apple Podcasts
Google Podcasts
Spotify

Trial to Test Effectiveness of CBT Phone Sessions for Chronic Pain After TBI
As many as 81.5% of veterans may experience chronic pain, pain that lasts beyond the point of healing and for at least 3 months. It is also particularly prevalent among veterans with traumatic brain injury (TBI) , often accompanied by comorbid conditions. Nearly 90% of veterans with a history of TBI have a psychiatric diagnosis, about 75% have insomnia, and 70% have a pain diagnosis, say researchers from University of Washington and Veterans Administration Puget Sound Health Care System (VAPSHCS).
Cognitive behavioral therapy (CBT) has been shown to help reduce pain, as well as pain-related disability and distress, but no randomized controlled trials (RCT) have examined CBT’s efficacy for pain after TBI in veterans, the researchers say.
In response, the VAPSHCS researchers have designed an RCT to compare telephone-based CBT with telephone-delivered pain education for veterans with TBI and chronic pain. The single-center 2-group trial will enroll up to 160 veterans with TBI to examine the relative efficacy of the interventions on average pain intensity, pain interference, sleep, depression, and life satisfaction.
The participants will be drawn from VAPSHCS, and can be enrolled via clinician referral, electronic health record review, and self-referral. Outcome variables will be collected pre-, mid-, and posttreatment, and 6 months following randomization.
Both interventions will consist of 8 hour-long phone sessions over approximately 8 to 12 weeks, scheduled at times convenient for the participants. Both interventions will also use a participant treatment workbook, with session-specific content to be discussed during the telephone sessions, and audio-recordings to augment material covered. Clinicians will make brief “booster” calls 2, 6, and 10 weeks after the final treatment session.
The trial is innovative, the researchers say, in that it is tailored to veterans, through relatable examples, and to those with TBI, by reducing content and providing multiple methods of engaging with information, as well as using known strategies to help with recall. If effective, the intervention could be disseminated throughout the VHA system, potentially to other personnel who have difficulty accessing specialty pain care.
The trial is registered at ClinicalTrials.gov, protocol NCT01768650.
As many as 81.5% of veterans may experience chronic pain, pain that lasts beyond the point of healing and for at least 3 months. It is also particularly prevalent among veterans with traumatic brain injury (TBI) , often accompanied by comorbid conditions. Nearly 90% of veterans with a history of TBI have a psychiatric diagnosis, about 75% have insomnia, and 70% have a pain diagnosis, say researchers from University of Washington and Veterans Administration Puget Sound Health Care System (VAPSHCS).
Cognitive behavioral therapy (CBT) has been shown to help reduce pain, as well as pain-related disability and distress, but no randomized controlled trials (RCT) have examined CBT’s efficacy for pain after TBI in veterans, the researchers say.
In response, the VAPSHCS researchers have designed an RCT to compare telephone-based CBT with telephone-delivered pain education for veterans with TBI and chronic pain. The single-center 2-group trial will enroll up to 160 veterans with TBI to examine the relative efficacy of the interventions on average pain intensity, pain interference, sleep, depression, and life satisfaction.
The participants will be drawn from VAPSHCS, and can be enrolled via clinician referral, electronic health record review, and self-referral. Outcome variables will be collected pre-, mid-, and posttreatment, and 6 months following randomization.
Both interventions will consist of 8 hour-long phone sessions over approximately 8 to 12 weeks, scheduled at times convenient for the participants. Both interventions will also use a participant treatment workbook, with session-specific content to be discussed during the telephone sessions, and audio-recordings to augment material covered. Clinicians will make brief “booster” calls 2, 6, and 10 weeks after the final treatment session.
The trial is innovative, the researchers say, in that it is tailored to veterans, through relatable examples, and to those with TBI, by reducing content and providing multiple methods of engaging with information, as well as using known strategies to help with recall. If effective, the intervention could be disseminated throughout the VHA system, potentially to other personnel who have difficulty accessing specialty pain care.
The trial is registered at ClinicalTrials.gov, protocol NCT01768650.
As many as 81.5% of veterans may experience chronic pain, pain that lasts beyond the point of healing and for at least 3 months. It is also particularly prevalent among veterans with traumatic brain injury (TBI) , often accompanied by comorbid conditions. Nearly 90% of veterans with a history of TBI have a psychiatric diagnosis, about 75% have insomnia, and 70% have a pain diagnosis, say researchers from University of Washington and Veterans Administration Puget Sound Health Care System (VAPSHCS).
Cognitive behavioral therapy (CBT) has been shown to help reduce pain, as well as pain-related disability and distress, but no randomized controlled trials (RCT) have examined CBT’s efficacy for pain after TBI in veterans, the researchers say.
In response, the VAPSHCS researchers have designed an RCT to compare telephone-based CBT with telephone-delivered pain education for veterans with TBI and chronic pain. The single-center 2-group trial will enroll up to 160 veterans with TBI to examine the relative efficacy of the interventions on average pain intensity, pain interference, sleep, depression, and life satisfaction.
The participants will be drawn from VAPSHCS, and can be enrolled via clinician referral, electronic health record review, and self-referral. Outcome variables will be collected pre-, mid-, and posttreatment, and 6 months following randomization.
Both interventions will consist of 8 hour-long phone sessions over approximately 8 to 12 weeks, scheduled at times convenient for the participants. Both interventions will also use a participant treatment workbook, with session-specific content to be discussed during the telephone sessions, and audio-recordings to augment material covered. Clinicians will make brief “booster” calls 2, 6, and 10 weeks after the final treatment session.
The trial is innovative, the researchers say, in that it is tailored to veterans, through relatable examples, and to those with TBI, by reducing content and providing multiple methods of engaging with information, as well as using known strategies to help with recall. If effective, the intervention could be disseminated throughout the VHA system, potentially to other personnel who have difficulty accessing specialty pain care.
The trial is registered at ClinicalTrials.gov, protocol NCT01768650.
FDA panels back intranasal esketamine for refractory depression
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.

Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
“The REMS program that was proposed by the company and seemingly endorsed by the FDA provides adequate protection,” Sanjay J. Mathew, MD, said in an interview. “I think that was one of the reasons it sailed through the panels.”
An important aspect of intranasal ketamine is that, as an N-methyl-D-aspartate receptor antagonist, it is “an entirely new class” for treating depression, said Dr. Mathew. “This is the first approval that does not work on serotonin or norepinephrine or dopamine. This is a big, big development. We can’t overstate that.”
Also, the nasal spray had to beat a placebo and a newly administered antidepressant. “There was a relatively high bar for showing convincing efficacy,” he said. “So if approved, this drug would be prescribed with an oral antidepressant. Intranasal esketamine represents 20 years’ worth of effort. Today was an important day for psychiatry,” he said. “It was an important day for patients with depression.”
Dr. Mathew is the Marjorie Bintliff Johnson and Raleigh White Johnson Jr. Vice Chair for Research and professor in the Menninger department of psychiatry & behavioral sciences at the Baylor College of Medicine in Houston. He has served as a consultant for and has had research funded by Janssen.
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.
Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
ROCKVILLE, MD – If approved for treatment-resistant depression, intranasal esketamine will be strictly regulated in the clinic, with federal monitoring requirements designed to prevent misuse, abuse, or diversion of the drug.
Managed under a Food and Drug Administration Risk Evaluation and Mitigation Strategy (REMS), such a program would establish a stringent post-administration protocol of observation and blood pressure monitoring and require every provider – whether a large health care center or a single clinician – to obtain federal certification to dispense the medication.
At a joint meeting of FDA’s Psychopharmacologic Drugs Advisory and Drug Safety and Risk Management Advisory committees, some members offered a more tempered view while still supporting the approval pathway of the N-methyl-D-aspartate receptor antagonist. By a vote of 14-2, with one abstention, they agreed Feb. 12 that the benefits outweigh the risks of esketamine for treatment-resistant depression.
“I think it has the potential to be a game changer in treatment-resistant depression,” said Walter Dunn, MD, PhD, of the University of California, Los Angeles. “We may someday talk about 2019 in the same way we now talk about the late ’80s, when the first [selective serotonin reuptake inhibitors] were approved.”
Janssen Pharmaceuticals, which is developing the drug, incorporated concerns about misuse from the beginning. Even the delivery device is designed to prevent such issues, a company spokesman said.
Each disposable intranasal delivery device contains 28 mg esketamine; it will come in prepackaged units of one, two, or three devices to deliver the prescribed doses of 28 mg, 56 mg, or 84 mg, respectively. The device does not require priming and, after use, contains only about 30 microliters of residual medication. Its interlocking design, with a glass vial inside the plastic outer assembly, would make it very difficult to pull apart, should anyone want to obtain the residue.
The proposed REMS – the key requirement for approval at this point – would include the following measures:
- Prescriber training on the risks of esketamine and importance of monitoring patients after their dose is administered and the need to register patients
- Administration of esketamine only in certain health care settings that ensure patient monitoring by a health care clinician for 2 hours after administration
- Pharmacies, clinicians, or health care settings that dispense the drug are specially certified to ensure that esketamine is not dispensed directly to patients and that patients are monitored
- Enrollment of patients who are treated with esketamine in a registry to better characterize the risks associated with esketamine administration and inform risk mitigation strategies
After administration, patients would be monitored for at least 2 hours for the common side effects, sedation and dissociation that typically clear within that time. Transient blood pressure fluctuations also can occur shortly after administration and would be monitored until stable. Patients should also be counseled not to drive the day of treatment, and to bring a companion along to drive them home.
Dr. Dunn, however, suggested that some facets of the proposed REMS might create unnecessary barriers for some patients and that stringent monitoring after every single dose – potentially for years – might not be necessary for everyone.
“The REM is certainly important to address the potential for diversion and misuse and adverse effects, but there needs to be a pathway to reduce monitoring requirements” on an individual basis. “If a patient is doing well for a year or so, in remission with no side effects, we should have a way to reduce the need for monitoring. If we make it too much of a burden to go in, get the medication, stay for a couple of hours for monitoring, it’s easy to skip a dose. And we know the number one predictor of relapse is medication nonadherence.”
The facility certification requirement also could curtail access to esketamine, said Steven B. Meisel, PharmD, of Minneapolis.
“How do we define a medically supervised center? Is it somewhere with a nurse onsite? A physician onsite? Does it have to have access to emergency services? This issue of access vs. control and safety is a very important one.”
He posed a clinical conundrum: A patient doing well on regular esketamine who wants to go on an extended trip. Under the proposed REMS, that patient would not be able to access his regular dose, which could only be handled, sorted, and administered by a certified health care clinician. “How are we going to deal with this? There will be great pressure to loosen this up in some manner. But if we allow a patient who’s been doing well on regular treatment with no relapse to have this at home, do we open the way for a teenager to take a bottle or two to a party? Those are real-world issues and must be considered when we establish a REM in a real world that demands access to needed therapy.”
Erring on the side of caution is the responsibility of policymakers, argued Kim Witczak, executive director of Woodymatters, a consumer-driven, nonprofit drug safety organization dedicated to FDA reform. Ms. Witczak was one of two dissenting voices on the vote.
“This has so much potential for so many people who just want a quick fix [for their mood disorders], and the marketing side will see this,” she predicted. “I would want to be very cautious. Once it gets out there into the real world, there will be a lot of people trying to get it. We don’t want to have ‘Esketamines “R” Us’ clinics popping up everywhere.”
The FDA usually follows its panels’ recommendations, which are not binding.
Clinical trial: Randomized study of lap vs. robotic hernia surgery underway
A multicenter, randomized study comparing

In the trial precis, the researchers wrote, “The robotic platform in surgery is growing exponentially. Despite this, the evidence supporting robotics remains limited. Studies demonstrating benefit, such as improved outcomes or decreased hospital length of stay, are largely cohort studies subject to substantial bias. Among randomized controlled trials, none have demonstrated benefit with robotic surgery.”
Study participants will be randomized to two arms, one for laparoscopic hernia repair and the other for robotic repair. Patients in both arms will be treated with a mid-density polypropylene mesh with a one-sided adhesion barrier.
The primary outcomes studied are length of stay in the hospital and readmissions out to 90 days. Secondary outcomes include the occurrence of surgical-site infection, hematoma, seroma, dehiscence, necrosis, nonhealing wound, hernia recurrence, and several cost and quality-of-life measures.
Patients included must be over age 18 and undergoing elective ventral hernia repair deemed appropriate for minimally invasive repair. Exclusions include those unlikely to survive beyond 2 years based on surgeon judgment or are unlikely to follow up. In addition, patients are excluded if they have advanced COPD or heart failure, a history of open abdomen or extensive lysis of adhesions for bowel obstruction, ascites caused by cirrhosis or malignancy, active infection, or a large hernia larger than 12 cm. Estimated enrollment is 120 patients, and the researchers expect the study to end in 2023.
For more details on the study (NT03490266), go to clinicaltrials.gov.
A multicenter, randomized study comparing
In the trial precis, the researchers wrote, “The robotic platform in surgery is growing exponentially. Despite this, the evidence supporting robotics remains limited. Studies demonstrating benefit, such as improved outcomes or decreased hospital length of stay, are largely cohort studies subject to substantial bias. Among randomized controlled trials, none have demonstrated benefit with robotic surgery.”
Study participants will be randomized to two arms, one for laparoscopic hernia repair and the other for robotic repair. Patients in both arms will be treated with a mid-density polypropylene mesh with a one-sided adhesion barrier.
The primary outcomes studied are length of stay in the hospital and readmissions out to 90 days. Secondary outcomes include the occurrence of surgical-site infection, hematoma, seroma, dehiscence, necrosis, nonhealing wound, hernia recurrence, and several cost and quality-of-life measures.
Patients included must be over age 18 and undergoing elective ventral hernia repair deemed appropriate for minimally invasive repair. Exclusions include those unlikely to survive beyond 2 years based on surgeon judgment or are unlikely to follow up. In addition, patients are excluded if they have advanced COPD or heart failure, a history of open abdomen or extensive lysis of adhesions for bowel obstruction, ascites caused by cirrhosis or malignancy, active infection, or a large hernia larger than 12 cm. Estimated enrollment is 120 patients, and the researchers expect the study to end in 2023.
For more details on the study (NT03490266), go to clinicaltrials.gov.
A multicenter, randomized study comparing
In the trial precis, the researchers wrote, “The robotic platform in surgery is growing exponentially. Despite this, the evidence supporting robotics remains limited. Studies demonstrating benefit, such as improved outcomes or decreased hospital length of stay, are largely cohort studies subject to substantial bias. Among randomized controlled trials, none have demonstrated benefit with robotic surgery.”
Study participants will be randomized to two arms, one for laparoscopic hernia repair and the other for robotic repair. Patients in both arms will be treated with a mid-density polypropylene mesh with a one-sided adhesion barrier.
The primary outcomes studied are length of stay in the hospital and readmissions out to 90 days. Secondary outcomes include the occurrence of surgical-site infection, hematoma, seroma, dehiscence, necrosis, nonhealing wound, hernia recurrence, and several cost and quality-of-life measures.
Patients included must be over age 18 and undergoing elective ventral hernia repair deemed appropriate for minimally invasive repair. Exclusions include those unlikely to survive beyond 2 years based on surgeon judgment or are unlikely to follow up. In addition, patients are excluded if they have advanced COPD or heart failure, a history of open abdomen or extensive lysis of adhesions for bowel obstruction, ascites caused by cirrhosis or malignancy, active infection, or a large hernia larger than 12 cm. Estimated enrollment is 120 patients, and the researchers expect the study to end in 2023.
For more details on the study (NT03490266), go to clinicaltrials.gov.
Vaccination and antiviral treatment do not affect stroke risk following shingles
HONOLULU – according to findings from a retrospective study of Medicare beneficiaries with shingles and ischemic stroke.

The findings suggest that primary prevention of shingles through vaccination might be the most effective approach to prevent shingles-associated acute ischemic stroke, said the researchers, who presented the study at the International Stroke Conference sponsored by the American Heart Association.
Almost one in three people in the United States will develop shingles, also known as herpes zoster, in their lifetime, according to the Centers for Disease Control and Prevention. Previous research has not simultaneously examined the effect of shingles vaccination and antiviral treatment following shingles onset on the risk of acute ischemic stroke.
Quanhe Yang, PhD, a senior scientist at the CDC, and his colleagues examined data for 35,186 Medicare fee-for-service beneficiaries who were 66 years or older, diagnosed with shingles during 2008-2014, and diagnosed with acute ischemic stroke within a year of shingles diagnosis. Using a self-controlled case series design, the investigators analyzed the association between shingles and stroke. Dr. Yang and his colleagues estimated the incident rate ratio (IRR) by comparing the incidence of stroke during risk periods (i.e., periods following shingles), compared with control periods. To minimize confounding by age, they restricted their analyses to approximately 365 days from the shingles index date.
To investigate how vaccination against shingles with Zostavax and antiviral treatment following shingles affected stroke risk, the researchers classified beneficiaries into the following four groups: Group 1 had no vaccination and no antiviral treatment (49% of beneficiaries), Group 2 had vaccination only (9%), Group 3 had antiviral treatment only (34%), and Group 4 had vaccination and antiviral treatment (8%). The researchers tested for interaction to examine the changes in IRRs across the four groups.
IRRs for stroke progressively declined as time passed from the index shingles date, from 1.61 at 0-14 days following shingles to 1.35 at 15-30 days, 1.16 at 31-90 days, and 1.05 at 91-180 days. The researchers found no evidence that shingles vaccination and antiviral treatment modified the risk of acute ischemic stroke. The association between shingles and risk for acute ischemic stroke was consistent across age groups (i.e., 66-74 years, 75-84 years, and 85 years or older), sex, and race (i.e., non-Hispanic white, non-Hispanic black, and Hispanic, other).
One of the study’s strengths was that its sample was a large national cohort of Medicare fee-for-service beneficiaries, Dr. Yang said. In addition, the study design eliminated all fixed confounding effects. Potential weaknesses, however, included the fact that herpes zoster diagnosis was based on administrative data and that the vaccine’s efficacy declines over time.
The findings suggest that the importance of following the recommended shingles vaccination protocol in the prevention of shingles, Dr. Yang said. Shingrix, a vaccine that the Food and Drug Administration approved in 2017, prevents shingles with an efficacy greater than 90%, he added.
The investigators reported no funding source or disclosures for this study.
SOURCE: Yang Q et al. Circulation. 2019;50(Suppl_1): Abstract 39
HONOLULU – according to findings from a retrospective study of Medicare beneficiaries with shingles and ischemic stroke.
The findings suggest that primary prevention of shingles through vaccination might be the most effective approach to prevent shingles-associated acute ischemic stroke, said the researchers, who presented the study at the International Stroke Conference sponsored by the American Heart Association.
Almost one in three people in the United States will develop shingles, also known as herpes zoster, in their lifetime, according to the Centers for Disease Control and Prevention. Previous research has not simultaneously examined the effect of shingles vaccination and antiviral treatment following shingles onset on the risk of acute ischemic stroke.
Quanhe Yang, PhD, a senior scientist at the CDC, and his colleagues examined data for 35,186 Medicare fee-for-service beneficiaries who were 66 years or older, diagnosed with shingles during 2008-2014, and diagnosed with acute ischemic stroke within a year of shingles diagnosis. Using a self-controlled case series design, the investigators analyzed the association between shingles and stroke. Dr. Yang and his colleagues estimated the incident rate ratio (IRR) by comparing the incidence of stroke during risk periods (i.e., periods following shingles), compared with control periods. To minimize confounding by age, they restricted their analyses to approximately 365 days from the shingles index date.
To investigate how vaccination against shingles with Zostavax and antiviral treatment following shingles affected stroke risk, the researchers classified beneficiaries into the following four groups: Group 1 had no vaccination and no antiviral treatment (49% of beneficiaries), Group 2 had vaccination only (9%), Group 3 had antiviral treatment only (34%), and Group 4 had vaccination and antiviral treatment (8%). The researchers tested for interaction to examine the changes in IRRs across the four groups.
IRRs for stroke progressively declined as time passed from the index shingles date, from 1.61 at 0-14 days following shingles to 1.35 at 15-30 days, 1.16 at 31-90 days, and 1.05 at 91-180 days. The researchers found no evidence that shingles vaccination and antiviral treatment modified the risk of acute ischemic stroke. The association between shingles and risk for acute ischemic stroke was consistent across age groups (i.e., 66-74 years, 75-84 years, and 85 years or older), sex, and race (i.e., non-Hispanic white, non-Hispanic black, and Hispanic, other).
One of the study’s strengths was that its sample was a large national cohort of Medicare fee-for-service beneficiaries, Dr. Yang said. In addition, the study design eliminated all fixed confounding effects. Potential weaknesses, however, included the fact that herpes zoster diagnosis was based on administrative data and that the vaccine’s efficacy declines over time.
The findings suggest that the importance of following the recommended shingles vaccination protocol in the prevention of shingles, Dr. Yang said. Shingrix, a vaccine that the Food and Drug Administration approved in 2017, prevents shingles with an efficacy greater than 90%, he added.
The investigators reported no funding source or disclosures for this study.
SOURCE: Yang Q et al. Circulation. 2019;50(Suppl_1): Abstract 39
HONOLULU – according to findings from a retrospective study of Medicare beneficiaries with shingles and ischemic stroke.
The findings suggest that primary prevention of shingles through vaccination might be the most effective approach to prevent shingles-associated acute ischemic stroke, said the researchers, who presented the study at the International Stroke Conference sponsored by the American Heart Association.
Almost one in three people in the United States will develop shingles, also known as herpes zoster, in their lifetime, according to the Centers for Disease Control and Prevention. Previous research has not simultaneously examined the effect of shingles vaccination and antiviral treatment following shingles onset on the risk of acute ischemic stroke.
Quanhe Yang, PhD, a senior scientist at the CDC, and his colleagues examined data for 35,186 Medicare fee-for-service beneficiaries who were 66 years or older, diagnosed with shingles during 2008-2014, and diagnosed with acute ischemic stroke within a year of shingles diagnosis. Using a self-controlled case series design, the investigators analyzed the association between shingles and stroke. Dr. Yang and his colleagues estimated the incident rate ratio (IRR) by comparing the incidence of stroke during risk periods (i.e., periods following shingles), compared with control periods. To minimize confounding by age, they restricted their analyses to approximately 365 days from the shingles index date.
To investigate how vaccination against shingles with Zostavax and antiviral treatment following shingles affected stroke risk, the researchers classified beneficiaries into the following four groups: Group 1 had no vaccination and no antiviral treatment (49% of beneficiaries), Group 2 had vaccination only (9%), Group 3 had antiviral treatment only (34%), and Group 4 had vaccination and antiviral treatment (8%). The researchers tested for interaction to examine the changes in IRRs across the four groups.
IRRs for stroke progressively declined as time passed from the index shingles date, from 1.61 at 0-14 days following shingles to 1.35 at 15-30 days, 1.16 at 31-90 days, and 1.05 at 91-180 days. The researchers found no evidence that shingles vaccination and antiviral treatment modified the risk of acute ischemic stroke. The association between shingles and risk for acute ischemic stroke was consistent across age groups (i.e., 66-74 years, 75-84 years, and 85 years or older), sex, and race (i.e., non-Hispanic white, non-Hispanic black, and Hispanic, other).
One of the study’s strengths was that its sample was a large national cohort of Medicare fee-for-service beneficiaries, Dr. Yang said. In addition, the study design eliminated all fixed confounding effects. Potential weaknesses, however, included the fact that herpes zoster diagnosis was based on administrative data and that the vaccine’s efficacy declines over time.
The findings suggest that the importance of following the recommended shingles vaccination protocol in the prevention of shingles, Dr. Yang said. Shingrix, a vaccine that the Food and Drug Administration approved in 2017, prevents shingles with an efficacy greater than 90%, he added.
The investigators reported no funding source or disclosures for this study.
SOURCE: Yang Q et al. Circulation. 2019;50(Suppl_1): Abstract 39
REPORTING FROM ISC 2019
Key clinical point: After a patient develops shingles, prior vaccination or treatment with antiviral medication does not change the risk of acute ischemic stroke.
Major finding: Stroke incidence increased by 61% within 14 days after shingles onset.
Study details: A self-controlled case series of 35,186 Medicare beneficiaries with shingles and acute ischemic stroke.
Disclosures: The authors reported no funding source or disclosures for this study.
Source: Yang Q et al. Circulation. 2019;50(Suppl_1), Abstract 39
Aspiration Pneumonia in Older Adults
Aspiration pneumonia refers to an infection of the lung parenchyma in an individual who has inhaled a bolus of endogenous flora that overwhelms the natural defenses of the respiratory system. It primarily affects older adults with almost 80% of cases occurring in those 65 years and older.1 Compared with nonaspiration pneumonia, aspiration pneumonia (whether community acquired or healthcare associated) results in more ICU stays, mechanical ventilation, increased length of hospital stay, and higher mortality.2
The etiology of aspiration pneumonia comes from aspirated bacteria from the oropharynx or stomach.3 However, aspiration alone is a common occurrence and does not always lead to clinical pneumonia. Indeed, one study demonstrated that 45% of “normal subjects” aspirate in their sleep,4 illustrating that our bodies have evolved defense mechanisms to protect us from aspirated bacteria. Thus, it is only when these systems are overwhelmed, after compromise of both glottic closure and the cough reflex in addition to dysphagia,3 that an infection manifests.
ASPIRATION PNEUMONITIS
Aspiration pneumonitis refers to a significant inflammation of the lung parenchyma that results from inhalation of regurgitated gastric contents.5 It can produce fever, cough, wheezing, shortness of breath, hypoxemia, leukocytosis, and a pulmonary infiltrate as well as lead to severe acute respiratory distress syndrome and even death. In the past, the use of antibiotics shortly after aspiration in patients who develop a fever, leukocytosis, or a pulmonary infiltrate was discouraged.5 Empiric antibiotics were recommended only for patients who aspirate gastric contents and who have conditions associated with colonization of gastric contents, such as small-bowel obstruction.5 Yet, it is difficult to distinguish aspiration pneumonitis from pneumonia6 and there are no randomized trials in older adults to help guide their management.
PRESENTATION OF ASPIRATION PNEUMONIA
Pneumonia in older adults can present in an atypical fashion. In one study of community-acquired pneumonia (CAP), the combination of cough, fever, and dyspnea is present in only 31% of patients, although separately, they are present in 67%, 64%, and 71% of patients, respectively. The same study also showed that delirium was present in 45% of patients with CAP.7 Nonrespiratory symptoms were present during the initial presentation of CAP in 55% of patients, with confusion in 42%, and falls in 16% of cases.8 The same is true of aspiration pneumonia where altered mental status is seen in approximately 30% of community-acquired aspiration pneumonia (CAAP) patients and in 19% of continuing care facility patients with aspiration pneumonia.2 Another study that compared CAP, CAAP, and healthcare-associated aspiration pneumonia (HCAAP) showed that confusion is present in 5.1%, 12.7%, and 18.6%, respectively.9 The absence of fever in older adults is shown in studies where fever, defined as greater than or equal to 37.5°C, is absent in 32% of the very old10and in 40% of patients 65 years or older when it was defined as greater than 37°C.8 The inconsistencies regarding typical symptoms of pneumonia in the older adult population are also confirmed in nursing home residents.11 Ultimately, it is important to remember that any infection in older adults, especially in those residing in long-term care facilities, may present with subtle findings such as an acute change in cognitive and functional status.12
Risk Factors for Aspiration Pneumonia
Risk factors for aspiration pneumonia, while not universally agreed upon, are important to recognize as they increase the probability of the diagnosis when present. A 2011 systematic review identified age, male gender, lung disease, dysphagia, and diabetes mellitus (level 2a), as well as severe dementia, angiotensin I-converting enzyme deletion/deletion genotype, and poor oral health (level 2b) as risk factors.13 In 2016, a panel of experts reached a consensus (modified Delphi Method) on the following risk factors for the diagnosis of aspiration pneumonia in nursing home residents: history of dysphagia, choking incident, tube feeding, neurologic disease, and cognitive impairment. The presence of one or more of these risk factors in the appropriate clinical setting may suggest a diagnosis of aspiration pneumonia.14
Radiographic/Ultrasonographic Imaging
In the appropriate scenario, the diagnosis of aspiration pneumonia is supported with an image representative of pneumonia. The pulmonary segment involved in aspiration pneumonia depends on the position of the patient during the aspiration event. If the aspiration event occurs while the patient is in the recumbent position, development of pneumonia is more common in the posterior segments of the upper lobes and the apical segments of the lower lobes; whereas if it occurs while the patient is in an upright position, the location changes to the basal segments of the lower lobes.3
Overall, the sensitivity of a chest X-ray to diagnose pneumonia ranges between 32%-77.7%,15-17 suggesting that a significant proportion of patients suspected of having pneumonia in past research studies, may have been misdiagnosed. Studies using lung ultrasound to identify pneumonia demonstrate a higher sensitivity, but additional research is needed to validate these findings.17-19 Noncontrast CT scans of the chest remain the reference standard for diagnosing pneumonia and currently tend to have the largest impact on diagnosis and subsequent treatment decisions.15,16,20,21 As a result, if radiation exposure risks are not a concern for the patient, we recommend utilizing noncontrast CT imaging whenever the diagnosis is in doubt until future research elucidates the most appropriate approach to imaging.
Diagnosis
Diagnosing aspiration pneumonia is difficult, in part because there is no universal definition or set of diagnostic criteria. The diagnosis of aspiration pneumonia is supported by the fulfillment of three criteria. First, appropriate risk factors for aspiration, as documented above, should be present. Second, there should be evidence of clinical signs and symptoms of pneumonia (typical or atypical). Third, radiographic representation of pneumonia in a dependent pulmonary segment confirms the diagnosis. Once these criteria are met, it is important to distinguish between CAAP and HCAAP with particular attention to risk factors for multidrug-resistant (MDR) organisms and Pseudomonas aeruginosa (PA).
MICROBIOLOGY
Many studies have tried to determine the exact bacterial etiology of aspiration pneumonia as documented in the Table.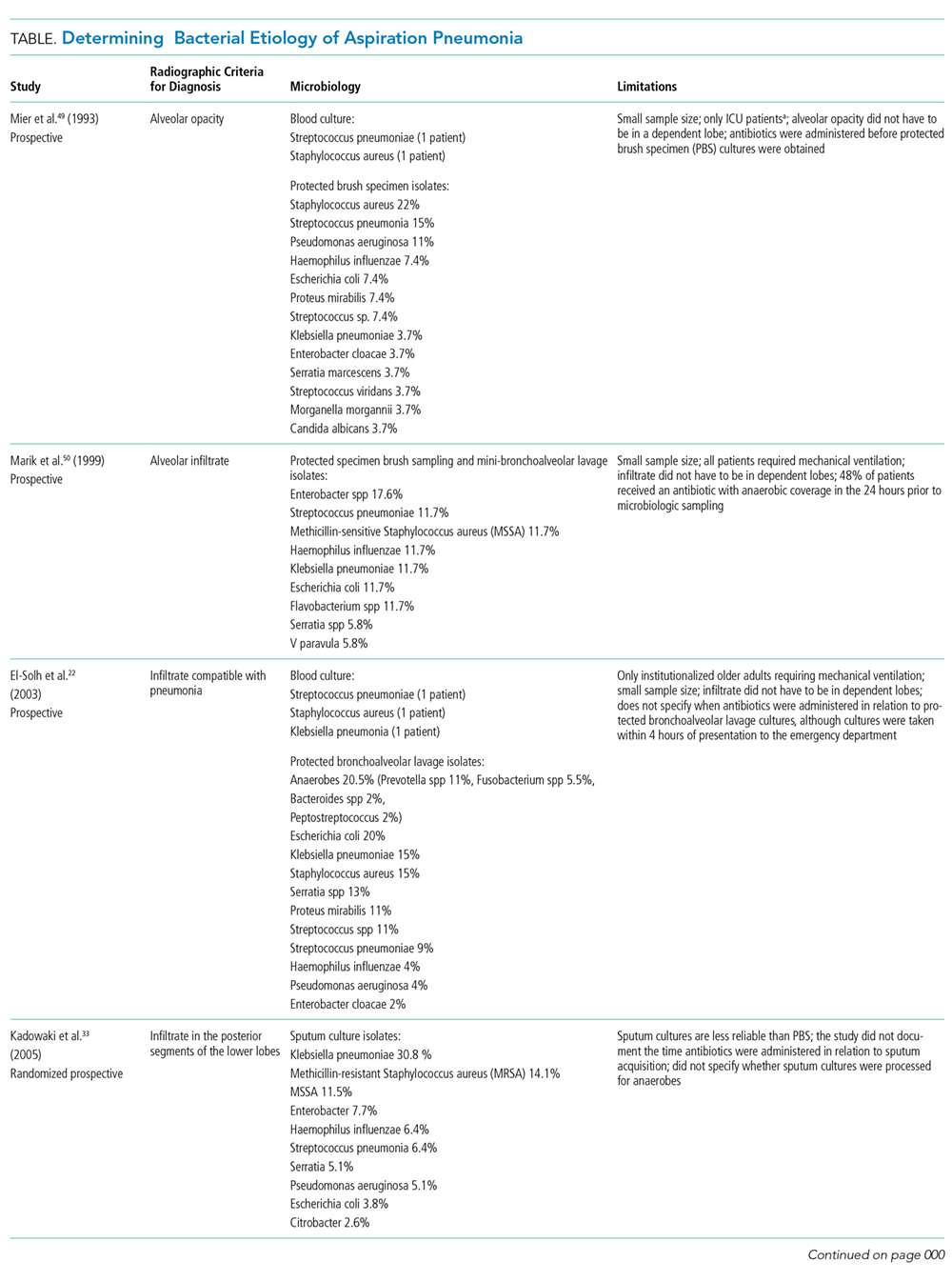
Even when an ideal method is used to obtain a good sample, however, the results are limited by other variables in the study. For example, in studies that use protected brush specimens and protected bronchoalveolar lavage to acquire samples for culture, many patients received antibiotics prior to sampling, and the studies are small (Table). Although anaerobes have traditionally been implicated in aspiration pneumonia, only El-Solh et al.22 were able to culture a significant proportion of anaerobes. The study, however, was limited to institutionalized older adults requiring mechanical ventilation and it did not require the typical radiographic location for aspiration pneumonia. Even under the best circumstances, it is difficult to determine causality because the antibiotics used to treat these cases of aspiration pneumonia cover a broad range of organisms. Based on the studies in the Table, causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods in addition to traditional organisms classically thought to cause aspiration pneumonia-anaerobes. Microbiologic etiology, however, may also be insinuated from the studies discussed in the therapeutic strategies section below as some include antibiotics with limited antimicrobial activity.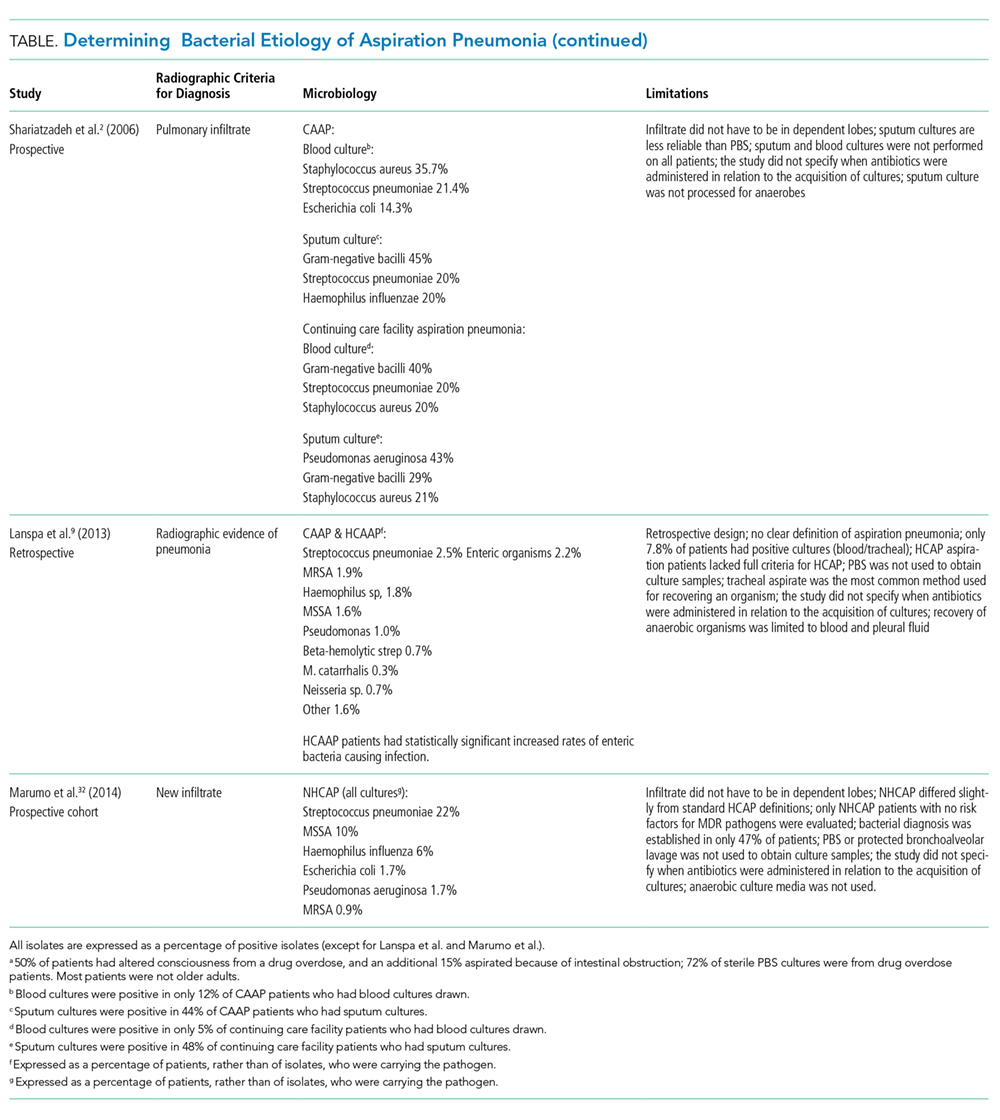
Therapeutic Strategies
The management of aspiration pneumonia has evolved significantly since it was first studied in the 1970s because of the development of antibiotic resistance patterns, newer antibiotics, and increasing information on the diversity of pathogens involved in each subset of aspiration syndromes. The antimicrobial treatment of aspiration pneumonia was classically directed against anaerobic pathogens; treatment of these infections, however, was extrapolated from studies of pulmonary abscesses and other anaerobic pulmonary infections.
A randomized controlled trial in the mid-1980s comparing penicillin and clindamycin demonstrated a significantly improved cure rate in the clindamycin group.23 A follow-up study in 1990 implicated a significant number of penicillin-resistant Bacteroides infections—the majority of these infections were subsequently reclassified as Prevotella melaninogenica—as the cause for high rates of penicillin resistance in lung abscesses and necrotizing pneumonias, further supporting clindamycin as the treatment of choice for these infections.24 Amoxicillin-clavulanic acid (IV and PO regimens), studied in the treatment of community-acquired necrotizing pneumonia/lung abscess, shows good efficacy as well.25 This study also attempted to elucidate the underlying causative organisms in these patients. Organisms associated with CAP as well as anaerobic organisms were isolated, giving more credence to the idea of broader coverage for aspiration pneumonia.
Community-Acquired Aspiration Pneumonia/Healthcare-Associated Aspiration Pneumonia
The importance of making a diagnostic distinction between CAAP versus HCAAP is critical for management strategies. A prospective population-based study demonstrated that ICU length of stay and 30-day mortality is highest for HCAAP, followed by CAAP, and lastly for those with CAP.9 Although some studies use different nomenclature for identifying aspiration pneumonia patients at risk for a wider array of microorganisms, we attempt to standardize the language by using HCAAP. The literature on nonaspiration pneumonia is changing from terms such as CAP and healthcare-associated pneumonia (HCAP) to pneumonia with the risk of MDR organisms. One study proposed a new treatment algorithm for CAP based on the presence or absence of the following six risk factors: prior hospitalization of greater than or equal to two days in the preceding 90 days, immunosuppression, previous antibiotic use within the preceding 90 days, use of gastric acid-suppressive agents, tube feeding, and nonambulatory status.26 A similar approach proposed years earlier for HCAP patients found the following to be risk factors for MDR organisms: hospitalization in the past 90 days, antibiotic therapy in the past six months, poor functional status as defined by activities of daily living score, and immune suppression.27 Other factors, such as structural lung disease, that increase the risk of organisms resistant to standard antibiotic treatment regimens28-31 should be considered in aspiration pneumonia as well. Aspiration pneumonia is following a similar trajectory where the risk of MDR organisms is taking precedence over the environment of acquisition. The final nomenclature will allow the healthcare provider to understand the organisms that need to be targeted when choosing an appropriate antibiotic treatment regimen.
There is evidence supporting the premise that CAAP and nursing home patients with no risk factors for MDR organisms can be treated with standard regimens used for patients with CAP. A prospective cohort study in 2014 did not show any statistically significant differences in clinical outcomes in nursing and healthcare-associated aspiration pneumonia patients (with no risks of MDR organisms) treated with azithromycin versus ampicillin/sulbactam. However, only 36 patients were included in the azithromycin arm, and the therapeutic choices were made by the treating physician.32
A prospective study of 95 long-term care residents reported that of those patients admitted to the ICU with severe aspiration pneumonia, the causative organisms are gram-negative enteric bacilli in 49% of isolates, anaerobes in 16%, and Staphylococcus aureus in 12%.22 This study mentioned that six of seven anaerobic pneumonia cases had inadequate anaerobic coverage yet were effectively treated; based on the organisms represented, however, the antibiotics administered did provide some coverage.22 Prevotella was one of the common anaerobic organisms that could be treated by levofloxacin or ceftriaxone/azithromycin, possibly explaining the success of azithromycin in the study quoted previously.22,32 Therefore, although anaerobic organisms still need to be considered, some may be treated by traditional CAP coverage.22
In a 2005 randomized prospective study of 100 patients aged 71 to 94 years, clindamycin was found to have clinical efficacy equivalent to ampicillin-sulbactam and panipenem in the treatment of mild-to-moderate aspiration pneumonia.33 Most patients in this study are nursing home residents, and 53% of sputum cultures in the clindamycin arm grew gram-negative rods. In contrast to the previous study, the significance of gram-negative rod infections in this population of patients, with less severe infections, is called into question, as clindamycin has no coverage against these organisms. This premise is supported by a more recent study using azithromycin in nursing and healthcare-associated aspiration pneumonia patients, mentioned previously.32 Taken together, these three studies suggest that the severity of aspiration pneumonia may be a risk factor that needs to be taken into account when considering broad-spectrum antimicrobial coverage.
While further research is needed to validate treatment approaches, based on the current literature we propose the following:
CAAP requiring hospitalization but without any of the following-risk for PA or MDR organisms, septic shock, the need for ICU admission, or mechanical ventilation-can be treated with standard CAP therapy that covers anaerobes.26,32-34 Patients with CAAP and either of the following—risk factors for MDR organisms, septic shock, need for ICU admission, or mechanical ventilation—should be considered for broader coverage with vancomycin or linezolid, antipseudomonal antibiotics, and anaerobic coverage. CAAP with specific risk for a PA infection should be considered for two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic, and one has anaerobic coverage).
Severe HCAAP without risk for MDR organisms or PA but with any of the following-septic shock, ICU admission, or mechanical ventilation-can be treated based on the 2016 Infectious Diseases Society of America guideline recommendation for hospital-acquired pneumonia, with a regimen that also provides adequate anaerobic coverage.35 If patients have HCAAP with one or more risk factors for MDR organisms, no septic shock, and no need for ICU admission or mechanical ventilation, provide coverage with a similar regimen. In contrast, HCAAP with risk factors for PA or severe HCAAP causing septic shock, requiring ICU admission, or needing mechanical ventilation, which occurs in the setting of one or more risk factors for MDR organisms, or structural lung disease, should receive two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic and one has anaerobic coverage) in addition to vancomycin or linezolid.
A recent systematic review demonstrates the paucity of studies of ideal methodologic design which complicates the ability to recommend, with confidence, one guideline-based antimicrobial regimen over another.36 Future studies may determine that despite the severity of the infection, if patients do not carry any risk for MDR pathogens or PA, they may only require CAAP coverage. When a patient presents with an acute infection, it is prudent to review previous cultures, and although it may be necessary to treat with broad-spectrum antibiotics initially, it is always important to narrow the spectrum based on reliable culture results. If future studies support the results of many studies cited in this article, we may be using fewer antibiotics with narrower spectrums in the near future.
Prevention
Although the healthcare system has practices in place to prevent aspiration pneumonia, the evidence supporting them are either inconclusive or not of ideal methodological design. Two systematic reviews failed to show statistically significant decreases in rates of aspiration pneumonia or mortality using the standard of care positioning strategies or thickened fluids in patients with chronic dysphagia.37,38 One study showed a decreased incidence of all pneumonia in dysphasic patients with dementia or Parkinson disease when a chin-down posture (with thin liquids) or thickened fluids in a head-neutral position was used. The study, however, has significant limitations, including a lack of a “no treatment” group for comparison, which did not allow investigators to conclude that the decreased incidence was from their interventions.39
There are preventive strategies that show a decreased risk of aspiration pneumonia. Poor oral hygiene seems to be a modifiable risk factor to establish better control of oral flora and decrease aspiration pneumonia. A systematic review of five studies, evaluating the effects of oral healthcare on the incidence of aspiration pneumonia in frail older people, found that tooth brushing after each meal along with cleaning dentures once a day and professional oral healthcare once a week decreases febrile days, pneumonia, and dying from pneumonia.40A two-year historical cohort study using aromatherapy with black pepper oil, followed by application of capsaicin troches, and finally menthol gel, as the first meal, leads to a decreased incidence of pneumonia and febrile days in older adults with dysphagia.41 Well-designed validation studies may establish these practices as the new standard of care for preventing pneumonia in patients with dysphagia.
Feeding Tubes
Multiple studies show that in older adults with advanced dementia there is no survival benefit from percutaneous endoscopic gastrostomy (PEG) tube placement42-44 and more recent systematic reviews also conclude that there is currently no evidence to support the use of PEG tubes in this specific population.45,46 In February 2013, as part of the American Board of Internal Medicine Foundation Choosing Wisely® campaign, the American Geriatrics Society advised providers not to recommend percutaneous feeding tubes in patients with advanced dementia, rather, “offer assisted oral feeding.”47 It is worth noting, however, that none of the studies reviewed were of ideal methodological design, so opinions may change with future studies.
A more recent study compared liquid feeds versus semisolid feeds in patients with PEG tubes. The study shows a 22.2% incidence of aspiration pneumonia in the liquid feed group, which is comparable to prior studies, but the incidence of aspiration pneumonia is only 2.2% in the semisolid feed group (P < .005).48 A benefit of this size warrants future studies for validation.
CONCLUSION
Aspiration pneumonia leads to increased mortality when compared with CAP and HCAP.2 Until future studies validate or refute the current understanding surrounding its management, the following should provide some guidance: aspiration pneumonia should be suspected in any individual with risk factors of aspiration who presents with typical or atypical symptoms of pneumonia. Confirmation of the diagnosis requires an image representative of pneumonia in the typical dependent lung segment on chest X-ray, lung ultrasound, or noncontrast CT scan of the chest. Treatment of aspiration pneumonia should take into account the site of acquisition, severity of illness, and risk for MDR organisms as the causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods, in addition to the traditional organisms classically thought to cause aspiration pneumonia-anaerobes.
Disclosures
The authors have nothing to disclose.
1. Wu CP, Chen YW, Wang MJ, Pinelis E. National trends in admission for aspiration pneumonia in the United States, 2002-2012. Ann Am Thorac Soc. 2017;14(6):874-879. doi: 10.1513/AnnalsATS.201611-867OC. PubMed
2. Reza Shariatzadeh M, Huang JQ, Marrie TJ. Differences in the features of aspiration pneumonia according to site of acquisition: community or continuing care facility. J Am Geriatr Soc. 2006;54(2):296-302. doi: 10.1111/j.1532-5415.2005.00608.x. PubMed
3. Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest. 1975;68(4):560-566. doi: 10.1378/chest.68.4.560. PubMed
4. Huxley EJ, Viroslav J, Gray WR, Pierce AK. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med. 1978;64(4):564-568. doi: 10.1016/0002-9343(78)90574-0. PubMed
5. Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671. doi: 10.1056/NEJM200103013440908. PubMed
6. Raghavendran K, Nemzek J, Napolitano LM, Knight PR. Aspiration-induced lung injury. Crit Care Med. 2011;39(4):818-826. doi: 10.1097/CCM.0b013e31820a856b. PubMed
7. Riquelme R, Torres A, el-Ebiary M, et al. Community-acquired pneumonia in the elderly. Clinical and nutritional aspects. Am J Respir Crit Care Med. 1997;156(6):1908-1914. doi: 10.1164/ajrccm.156.6.9702005. PubMed
8. Venkatesan P, Gladman J, Macfarlane JT, et al. A hospital study of community acquired pneumonia in the elderly. Thorax. 1990;45(4):254-258. doi: 10.1136/thx.45.4.254. PubMed
9. Lanspa MJ, Jones BE, Brown SM, Dean NC. Mortality, morbidity, and disease severity of patients with aspiration pneumonia. J Hosp Med. 2013;8(2):83-90. doi: 10.1002/jhm.1996. PubMed
10. Fernández-Sabé N, Carratalà J, Rosón B, et al. Community-acquired pneumonia in very elderly patients: causative organisms, clinical characteristics, and outcomes. Medicine (Baltimore). 2003;82(3):159-169. doi: 10.1097/01.md.0000076005.64510.87. PubMed
11. Mehr DR, Binder EF, Kruse RL, et al. Clinical findings associated with radiographic pneumonia in nursing home residents. J Fam Pract. 2001;50(11):931-937. PubMed
12. Bentley DW, Bradley S, High K, et al. Practice guideline for evaluation of fever and infection in long-term care facilities. Clin Infect Dis. 2000;31(3):640-653. doi: 10.1086/314013. PubMed
13. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Risk factors for aspiration pneumonia in frail older people: a systematic literature review. J Am Med Dir Assoc. 2011;12(5):344-354. doi: 10.1016/j.jamda.2010.12.099. PubMed
14. Hollaar V, van der Maarel-Wierink C, van der Putten GJ, et al. Defining characteristics and risk indicators for diagnosing nursing home-acquired pneumonia and aspiration pneumonia in nursing home residents, using the electronically-modified Delphi Method. BMC Geriatr. 2016;16:60. doi: 10.1186/s12877-016-0231-4. PubMed
15. Esayag Y, Nikitin I, Bar-Ziv J, et al. Diagnostic value of chest radiographs in bedridden patients suspected of having pneumonia. Am J Med. 2010;123(1):88.e1-88.e5. doi: 10.1016/j.amjmed.2009.09.012. PubMed
16. Claessens YE, Debray MP, Tubach F, et al. Early chest computed tomography scan to assist diagnosis and guide treatment decision for suspected community-acquired pneumonia. Am J Respir Crit Care Med. 2015;192(8):974-982. doi: 10.1164/rccm.201501-0017OC. PubMed
17. Liu XL, Lian R, Tao YK, Gu CD, Zhang GQ. Lung ultrasonography: an effective way to diagnose community-acquired pneumonia. Emerg Med J. 2015;32(6):433-438. doi: 10.1136/emermed-2013-203039. PubMed
18. Bourcier JE, Paquet J, Seinger M, et al. Performance comparison of lung ultrasound and chest x-ray for the diagnosis of pneumonia in the ED. Am J Emerg Med. 2014;32(2):115-118. doi: 10.1016/j.ajem.2013.10.003. PubMed
19. Chavez MA, Shams N, Ellington LE, et al. Lung ultrasound for the diagnosis of pneumonia in adults: a systematic review and meta-analysis. Respir Res. 2014;15:50. doi: 10.1186/1465-9921-15-50. PubMed
20. Syrjälä H, Broas M, Suramo I, Ojala A, Lähde S. High-resolution computed tomography for the diagnosis of community-acquired pneumonia. Clin Infect Dis. 1998;27(2):358-363. doi: 10.1086/514675. PubMed
21. Hayden GE, Wrenn KW. Chest radiograph vs. computed tomography scan in the evaluation for pneumonia. J Emerg Med. 2009;36(3):266-270. doi: 10.1016/j.jemermed.2007.11.042. PubMed
22. El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med. 2003;167(12):1650-1654. doi: 10.1164/rccm.200212-1543OC. PubMed
23. Levison ME, Mangura CT, Lorber B, et al. Clindamycin compared with penicillin for the treatment of anaerobic lung abscess. Ann Intern Med. 1983;98(4):466-471. doi: 10.7326/0003-4819-98-4-466. PubMed
24. Gudiol F, Manresa F, Pallares R, et al. Clindamycin vs penicillin for anaerobic lung infections. High rate of penicillin failures associated with penicillin-resistant Bacteroides melaninogenicus. Arch Intern Med. 1990;150(12):2525-2529. doi: 10.1001/archinte.150.12.2525. PubMed
25. Germaud P, Poirier J, Jacqueme P, et al. Monotherapy using amoxicillin/clavulanic acid as treatment of first choice in community-acquired lung abscess. Apropos of 57 cases. Rev Pneumol Clin. 1993;49(3):137-141. PubMed
26. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995. doi: 10.1164/rccm.201301-0079OC. PubMed
27. Brito V, Niederman MS. Healthcare-associated pneumonia is a heterogeneous disease, and all patients do not need the same broad-spectrum antibiotic therapy as complex nosocomial pneumonia. Curr Opin Infect Dis. 2009;22(3):316-325. doi: 10.1097/QCO.0b013e328329fa4e. PubMed
28. Restrepo MI, Babu BL, Reyes LF, et al. Burden and risk factors for Pseudomonas aeruginosa community-acquired pneumonia: a multinational point prevalence study of hospitalised patients. Eur Respir J. 2018;52(2). doi: 10.1183/13993003.01190-2017. PubMed
29. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44 Supplement 2:S27-S72. doi: 10.1086/511159. PubMed
30. Cillóniz C, Gabarrús A, Ferrer M, et al. Community-acquired pneumonia due to multidrug- and non-multidrug-resistant Pseudomonas aeruginosa. Chest. 2016;150(2):415-425. doi: 10.1016/j.chest.2016.03.042. PubMed
31. Prina E, Ranzani OT, Polverino E, et al. Risk factors associated with potentially antibiotic-resistant pathogens in community-acquired pneumonia. Ann Am Thorac Soc. 2015;12(2):153-160. doi: 10.1513/AnnalsATS.201407-305OC. PubMed
32. Marumo S, Teranishi T, Higami Y, et al. Effectiveness of azithromycin in aspiration pneumonia: a prospective observational study. BMC Infect Dis. 2014;14:685. doi: 10.1186/s12879-014-0685-y. PubMed
33. Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest. 2005;127(4):1276-1282. doi: 10.1378/chest.127.4.1276. PubMed
34. Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383. doi: 10.1093/cid/cit571. PubMed
35. Kalil AC, Metersky ML, Klompas M, et al. Executive Summary: management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):575-582. doi: 10.1093/cid/ciw504. PubMed
36. Bowerman TJ, Zhang J, Waite LM. Antibacterial treatment of aspiration pneumonia in older people: a systematic review. Clin Interv Aging. 2018;13:2201-2213. doi: 10.2147/CIA.S183344. PubMed
37. Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022. doi: 10.1046/j.1365-2389.2003.51318.x. PubMed
38. Andersen UT, Beck AM, Kjaersgaard A, Hansen T, Poulsen I. Systematic review and evidence based recommendations on texture modified foods and thickened fluids for adults (≥18 years) with oropharyngeal dysphagia. Clin Nutr ESPEN. 2013;8(4):e127-e134.
39. Robbins J, Gensler G, Hind J, et al. Comparison of 2 interventions for liquid aspiration on pneumonia incidence: a randomized trial. Ann Intern Med. 2008;148(7):509-518. doi: 10.7326/0003-4819-148-7-200804010-00007. PubMed
40. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Oral health care and aspiration pneumonia in frail older people: a systematic literature review. Gerodontology. 2013;30(1):3-9. doi: 10.1111/j.1741-2358.2012.00637.x. PubMed
41. Ebihara T, Ebihara S, Yamazaki M, et al. Intensive stepwise method for oral intake using a combination of transient receptor potential stimulation and olfactory stimulation inhibits the incidence of pneumonia in dysphagic older adults. J Am Geriatr Soc. 2010;58(1):196-198. doi: 10.1111/j.1532-5415.2009.02648.x. PubMed
42. Sanders DS, Carter MJ, D’Silva J, et al. Survival analysis in percutaneous endoscopic gastrostomy feeding: a worse outcome in patients with dementia. Am J Gastroenterol. 2000;95(6):1472-1475. doi: 10.1111/j.1572-0241.2000.02079.x. PubMed
43. Murphy LM, Lipman TO. Percutaneous endoscopic gastrostomy does not prolong survival in patients with dementia. Arch Intern Med. 2003;163(11):1351-1353. doi: 10.1001/archinte.163.11.1351. PubMed
44. Rimon E, Kagansky N, Levy S. Percutaneous endoscopic gastrostomy; evidence of different prognosis in various patient subgroups. Age Ageing. 2005;34(4):353-357. doi: 10.1093/ageing/afi085. PubMed
45. Candy B, Sampson EL, Jones L. Enteral tube feeding in older people with advanced dementia: findings from a Cochrane systematic review. Int J Palliat Nurs. 2009;15(8):396-404. doi: 10.12968/ijpn.2009.15.8.43799. PubMed
46. Goldberg LS, Altman KW. The role of gastrostomy tube placement in advanced dementia with dysphagia: a critical review. Clin Interv Aging. 2014;9:1733-1739. doi: 10.2147/CIA.S53153. PubMed
47. Workgroup AGSCW. American Geriatrics Society identifies five things that healthcare providers and patients should question. J Am Geriatr Soc. 2013;61(4):622-631. doi: 10.1111/jgs.12226. PubMed
48. Toh Yoon EW, Yoneda K, Nishihara K. Semi-solid feeds may reduce the risk of aspiration pneumonia and shorten postoperative length of stay after percutaneous endoscopic gastrostomy (PEG). Endosc Int Open. 2016;4(12):E1247-E1251. doi: 10.1055/s-0042-117218. PubMed
49. Mier L, Dreyfuss D, Darchy B, et al. Is penicillin-G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intens Care Med. 1993;19(5):279-284. doi: 10.1007/BF01690548. PubMed
50. Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest. 1999;115(1):178-183. doi: 10.1378/chest.115.1.178. PubMed
Aspiration pneumonia refers to an infection of the lung parenchyma in an individual who has inhaled a bolus of endogenous flora that overwhelms the natural defenses of the respiratory system. It primarily affects older adults with almost 80% of cases occurring in those 65 years and older.1 Compared with nonaspiration pneumonia, aspiration pneumonia (whether community acquired or healthcare associated) results in more ICU stays, mechanical ventilation, increased length of hospital stay, and higher mortality.2
The etiology of aspiration pneumonia comes from aspirated bacteria from the oropharynx or stomach.3 However, aspiration alone is a common occurrence and does not always lead to clinical pneumonia. Indeed, one study demonstrated that 45% of “normal subjects” aspirate in their sleep,4 illustrating that our bodies have evolved defense mechanisms to protect us from aspirated bacteria. Thus, it is only when these systems are overwhelmed, after compromise of both glottic closure and the cough reflex in addition to dysphagia,3 that an infection manifests.
ASPIRATION PNEUMONITIS
Aspiration pneumonitis refers to a significant inflammation of the lung parenchyma that results from inhalation of regurgitated gastric contents.5 It can produce fever, cough, wheezing, shortness of breath, hypoxemia, leukocytosis, and a pulmonary infiltrate as well as lead to severe acute respiratory distress syndrome and even death. In the past, the use of antibiotics shortly after aspiration in patients who develop a fever, leukocytosis, or a pulmonary infiltrate was discouraged.5 Empiric antibiotics were recommended only for patients who aspirate gastric contents and who have conditions associated with colonization of gastric contents, such as small-bowel obstruction.5 Yet, it is difficult to distinguish aspiration pneumonitis from pneumonia6 and there are no randomized trials in older adults to help guide their management.
PRESENTATION OF ASPIRATION PNEUMONIA
Pneumonia in older adults can present in an atypical fashion. In one study of community-acquired pneumonia (CAP), the combination of cough, fever, and dyspnea is present in only 31% of patients, although separately, they are present in 67%, 64%, and 71% of patients, respectively. The same study also showed that delirium was present in 45% of patients with CAP.7 Nonrespiratory symptoms were present during the initial presentation of CAP in 55% of patients, with confusion in 42%, and falls in 16% of cases.8 The same is true of aspiration pneumonia where altered mental status is seen in approximately 30% of community-acquired aspiration pneumonia (CAAP) patients and in 19% of continuing care facility patients with aspiration pneumonia.2 Another study that compared CAP, CAAP, and healthcare-associated aspiration pneumonia (HCAAP) showed that confusion is present in 5.1%, 12.7%, and 18.6%, respectively.9 The absence of fever in older adults is shown in studies where fever, defined as greater than or equal to 37.5°C, is absent in 32% of the very old10and in 40% of patients 65 years or older when it was defined as greater than 37°C.8 The inconsistencies regarding typical symptoms of pneumonia in the older adult population are also confirmed in nursing home residents.11 Ultimately, it is important to remember that any infection in older adults, especially in those residing in long-term care facilities, may present with subtle findings such as an acute change in cognitive and functional status.12
Risk Factors for Aspiration Pneumonia
Risk factors for aspiration pneumonia, while not universally agreed upon, are important to recognize as they increase the probability of the diagnosis when present. A 2011 systematic review identified age, male gender, lung disease, dysphagia, and diabetes mellitus (level 2a), as well as severe dementia, angiotensin I-converting enzyme deletion/deletion genotype, and poor oral health (level 2b) as risk factors.13 In 2016, a panel of experts reached a consensus (modified Delphi Method) on the following risk factors for the diagnosis of aspiration pneumonia in nursing home residents: history of dysphagia, choking incident, tube feeding, neurologic disease, and cognitive impairment. The presence of one or more of these risk factors in the appropriate clinical setting may suggest a diagnosis of aspiration pneumonia.14
Radiographic/Ultrasonographic Imaging
In the appropriate scenario, the diagnosis of aspiration pneumonia is supported with an image representative of pneumonia. The pulmonary segment involved in aspiration pneumonia depends on the position of the patient during the aspiration event. If the aspiration event occurs while the patient is in the recumbent position, development of pneumonia is more common in the posterior segments of the upper lobes and the apical segments of the lower lobes; whereas if it occurs while the patient is in an upright position, the location changes to the basal segments of the lower lobes.3
Overall, the sensitivity of a chest X-ray to diagnose pneumonia ranges between 32%-77.7%,15-17 suggesting that a significant proportion of patients suspected of having pneumonia in past research studies, may have been misdiagnosed. Studies using lung ultrasound to identify pneumonia demonstrate a higher sensitivity, but additional research is needed to validate these findings.17-19 Noncontrast CT scans of the chest remain the reference standard for diagnosing pneumonia and currently tend to have the largest impact on diagnosis and subsequent treatment decisions.15,16,20,21 As a result, if radiation exposure risks are not a concern for the patient, we recommend utilizing noncontrast CT imaging whenever the diagnosis is in doubt until future research elucidates the most appropriate approach to imaging.
Diagnosis
Diagnosing aspiration pneumonia is difficult, in part because there is no universal definition or set of diagnostic criteria. The diagnosis of aspiration pneumonia is supported by the fulfillment of three criteria. First, appropriate risk factors for aspiration, as documented above, should be present. Second, there should be evidence of clinical signs and symptoms of pneumonia (typical or atypical). Third, radiographic representation of pneumonia in a dependent pulmonary segment confirms the diagnosis. Once these criteria are met, it is important to distinguish between CAAP and HCAAP with particular attention to risk factors for multidrug-resistant (MDR) organisms and Pseudomonas aeruginosa (PA).
MICROBIOLOGY
Many studies have tried to determine the exact bacterial etiology of aspiration pneumonia as documented in the Table.
Even when an ideal method is used to obtain a good sample, however, the results are limited by other variables in the study. For example, in studies that use protected brush specimens and protected bronchoalveolar lavage to acquire samples for culture, many patients received antibiotics prior to sampling, and the studies are small (Table). Although anaerobes have traditionally been implicated in aspiration pneumonia, only El-Solh et al.22 were able to culture a significant proportion of anaerobes. The study, however, was limited to institutionalized older adults requiring mechanical ventilation and it did not require the typical radiographic location for aspiration pneumonia. Even under the best circumstances, it is difficult to determine causality because the antibiotics used to treat these cases of aspiration pneumonia cover a broad range of organisms. Based on the studies in the Table, causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods in addition to traditional organisms classically thought to cause aspiration pneumonia-anaerobes. Microbiologic etiology, however, may also be insinuated from the studies discussed in the therapeutic strategies section below as some include antibiotics with limited antimicrobial activity.
Therapeutic Strategies
The management of aspiration pneumonia has evolved significantly since it was first studied in the 1970s because of the development of antibiotic resistance patterns, newer antibiotics, and increasing information on the diversity of pathogens involved in each subset of aspiration syndromes. The antimicrobial treatment of aspiration pneumonia was classically directed against anaerobic pathogens; treatment of these infections, however, was extrapolated from studies of pulmonary abscesses and other anaerobic pulmonary infections.
A randomized controlled trial in the mid-1980s comparing penicillin and clindamycin demonstrated a significantly improved cure rate in the clindamycin group.23 A follow-up study in 1990 implicated a significant number of penicillin-resistant Bacteroides infections—the majority of these infections were subsequently reclassified as Prevotella melaninogenica—as the cause for high rates of penicillin resistance in lung abscesses and necrotizing pneumonias, further supporting clindamycin as the treatment of choice for these infections.24 Amoxicillin-clavulanic acid (IV and PO regimens), studied in the treatment of community-acquired necrotizing pneumonia/lung abscess, shows good efficacy as well.25 This study also attempted to elucidate the underlying causative organisms in these patients. Organisms associated with CAP as well as anaerobic organisms were isolated, giving more credence to the idea of broader coverage for aspiration pneumonia.
Community-Acquired Aspiration Pneumonia/Healthcare-Associated Aspiration Pneumonia
The importance of making a diagnostic distinction between CAAP versus HCAAP is critical for management strategies. A prospective population-based study demonstrated that ICU length of stay and 30-day mortality is highest for HCAAP, followed by CAAP, and lastly for those with CAP.9 Although some studies use different nomenclature for identifying aspiration pneumonia patients at risk for a wider array of microorganisms, we attempt to standardize the language by using HCAAP. The literature on nonaspiration pneumonia is changing from terms such as CAP and healthcare-associated pneumonia (HCAP) to pneumonia with the risk of MDR organisms. One study proposed a new treatment algorithm for CAP based on the presence or absence of the following six risk factors: prior hospitalization of greater than or equal to two days in the preceding 90 days, immunosuppression, previous antibiotic use within the preceding 90 days, use of gastric acid-suppressive agents, tube feeding, and nonambulatory status.26 A similar approach proposed years earlier for HCAP patients found the following to be risk factors for MDR organisms: hospitalization in the past 90 days, antibiotic therapy in the past six months, poor functional status as defined by activities of daily living score, and immune suppression.27 Other factors, such as structural lung disease, that increase the risk of organisms resistant to standard antibiotic treatment regimens28-31 should be considered in aspiration pneumonia as well. Aspiration pneumonia is following a similar trajectory where the risk of MDR organisms is taking precedence over the environment of acquisition. The final nomenclature will allow the healthcare provider to understand the organisms that need to be targeted when choosing an appropriate antibiotic treatment regimen.
There is evidence supporting the premise that CAAP and nursing home patients with no risk factors for MDR organisms can be treated with standard regimens used for patients with CAP. A prospective cohort study in 2014 did not show any statistically significant differences in clinical outcomes in nursing and healthcare-associated aspiration pneumonia patients (with no risks of MDR organisms) treated with azithromycin versus ampicillin/sulbactam. However, only 36 patients were included in the azithromycin arm, and the therapeutic choices were made by the treating physician.32
A prospective study of 95 long-term care residents reported that of those patients admitted to the ICU with severe aspiration pneumonia, the causative organisms are gram-negative enteric bacilli in 49% of isolates, anaerobes in 16%, and Staphylococcus aureus in 12%.22 This study mentioned that six of seven anaerobic pneumonia cases had inadequate anaerobic coverage yet were effectively treated; based on the organisms represented, however, the antibiotics administered did provide some coverage.22 Prevotella was one of the common anaerobic organisms that could be treated by levofloxacin or ceftriaxone/azithromycin, possibly explaining the success of azithromycin in the study quoted previously.22,32 Therefore, although anaerobic organisms still need to be considered, some may be treated by traditional CAP coverage.22
In a 2005 randomized prospective study of 100 patients aged 71 to 94 years, clindamycin was found to have clinical efficacy equivalent to ampicillin-sulbactam and panipenem in the treatment of mild-to-moderate aspiration pneumonia.33 Most patients in this study are nursing home residents, and 53% of sputum cultures in the clindamycin arm grew gram-negative rods. In contrast to the previous study, the significance of gram-negative rod infections in this population of patients, with less severe infections, is called into question, as clindamycin has no coverage against these organisms. This premise is supported by a more recent study using azithromycin in nursing and healthcare-associated aspiration pneumonia patients, mentioned previously.32 Taken together, these three studies suggest that the severity of aspiration pneumonia may be a risk factor that needs to be taken into account when considering broad-spectrum antimicrobial coverage.
While further research is needed to validate treatment approaches, based on the current literature we propose the following:
CAAP requiring hospitalization but without any of the following-risk for PA or MDR organisms, septic shock, the need for ICU admission, or mechanical ventilation-can be treated with standard CAP therapy that covers anaerobes.26,32-34 Patients with CAAP and either of the following—risk factors for MDR organisms, septic shock, need for ICU admission, or mechanical ventilation—should be considered for broader coverage with vancomycin or linezolid, antipseudomonal antibiotics, and anaerobic coverage. CAAP with specific risk for a PA infection should be considered for two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic, and one has anaerobic coverage).
Severe HCAAP without risk for MDR organisms or PA but with any of the following-septic shock, ICU admission, or mechanical ventilation-can be treated based on the 2016 Infectious Diseases Society of America guideline recommendation for hospital-acquired pneumonia, with a regimen that also provides adequate anaerobic coverage.35 If patients have HCAAP with one or more risk factors for MDR organisms, no septic shock, and no need for ICU admission or mechanical ventilation, provide coverage with a similar regimen. In contrast, HCAAP with risk factors for PA or severe HCAAP causing septic shock, requiring ICU admission, or needing mechanical ventilation, which occurs in the setting of one or more risk factors for MDR organisms, or structural lung disease, should receive two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic and one has anaerobic coverage) in addition to vancomycin or linezolid.
A recent systematic review demonstrates the paucity of studies of ideal methodologic design which complicates the ability to recommend, with confidence, one guideline-based antimicrobial regimen over another.36 Future studies may determine that despite the severity of the infection, if patients do not carry any risk for MDR pathogens or PA, they may only require CAAP coverage. When a patient presents with an acute infection, it is prudent to review previous cultures, and although it may be necessary to treat with broad-spectrum antibiotics initially, it is always important to narrow the spectrum based on reliable culture results. If future studies support the results of many studies cited in this article, we may be using fewer antibiotics with narrower spectrums in the near future.
Prevention
Although the healthcare system has practices in place to prevent aspiration pneumonia, the evidence supporting them are either inconclusive or not of ideal methodological design. Two systematic reviews failed to show statistically significant decreases in rates of aspiration pneumonia or mortality using the standard of care positioning strategies or thickened fluids in patients with chronic dysphagia.37,38 One study showed a decreased incidence of all pneumonia in dysphasic patients with dementia or Parkinson disease when a chin-down posture (with thin liquids) or thickened fluids in a head-neutral position was used. The study, however, has significant limitations, including a lack of a “no treatment” group for comparison, which did not allow investigators to conclude that the decreased incidence was from their interventions.39
There are preventive strategies that show a decreased risk of aspiration pneumonia. Poor oral hygiene seems to be a modifiable risk factor to establish better control of oral flora and decrease aspiration pneumonia. A systematic review of five studies, evaluating the effects of oral healthcare on the incidence of aspiration pneumonia in frail older people, found that tooth brushing after each meal along with cleaning dentures once a day and professional oral healthcare once a week decreases febrile days, pneumonia, and dying from pneumonia.40A two-year historical cohort study using aromatherapy with black pepper oil, followed by application of capsaicin troches, and finally menthol gel, as the first meal, leads to a decreased incidence of pneumonia and febrile days in older adults with dysphagia.41 Well-designed validation studies may establish these practices as the new standard of care for preventing pneumonia in patients with dysphagia.
Feeding Tubes
Multiple studies show that in older adults with advanced dementia there is no survival benefit from percutaneous endoscopic gastrostomy (PEG) tube placement42-44 and more recent systematic reviews also conclude that there is currently no evidence to support the use of PEG tubes in this specific population.45,46 In February 2013, as part of the American Board of Internal Medicine Foundation Choosing Wisely® campaign, the American Geriatrics Society advised providers not to recommend percutaneous feeding tubes in patients with advanced dementia, rather, “offer assisted oral feeding.”47 It is worth noting, however, that none of the studies reviewed were of ideal methodological design, so opinions may change with future studies.
A more recent study compared liquid feeds versus semisolid feeds in patients with PEG tubes. The study shows a 22.2% incidence of aspiration pneumonia in the liquid feed group, which is comparable to prior studies, but the incidence of aspiration pneumonia is only 2.2% in the semisolid feed group (P < .005).48 A benefit of this size warrants future studies for validation.
CONCLUSION
Aspiration pneumonia leads to increased mortality when compared with CAP and HCAP.2 Until future studies validate or refute the current understanding surrounding its management, the following should provide some guidance: aspiration pneumonia should be suspected in any individual with risk factors of aspiration who presents with typical or atypical symptoms of pneumonia. Confirmation of the diagnosis requires an image representative of pneumonia in the typical dependent lung segment on chest X-ray, lung ultrasound, or noncontrast CT scan of the chest. Treatment of aspiration pneumonia should take into account the site of acquisition, severity of illness, and risk for MDR organisms as the causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods, in addition to the traditional organisms classically thought to cause aspiration pneumonia-anaerobes.
Disclosures
The authors have nothing to disclose.
Aspiration pneumonia refers to an infection of the lung parenchyma in an individual who has inhaled a bolus of endogenous flora that overwhelms the natural defenses of the respiratory system. It primarily affects older adults with almost 80% of cases occurring in those 65 years and older.1 Compared with nonaspiration pneumonia, aspiration pneumonia (whether community acquired or healthcare associated) results in more ICU stays, mechanical ventilation, increased length of hospital stay, and higher mortality.2
The etiology of aspiration pneumonia comes from aspirated bacteria from the oropharynx or stomach.3 However, aspiration alone is a common occurrence and does not always lead to clinical pneumonia. Indeed, one study demonstrated that 45% of “normal subjects” aspirate in their sleep,4 illustrating that our bodies have evolved defense mechanisms to protect us from aspirated bacteria. Thus, it is only when these systems are overwhelmed, after compromise of both glottic closure and the cough reflex in addition to dysphagia,3 that an infection manifests.
ASPIRATION PNEUMONITIS
Aspiration pneumonitis refers to a significant inflammation of the lung parenchyma that results from inhalation of regurgitated gastric contents.5 It can produce fever, cough, wheezing, shortness of breath, hypoxemia, leukocytosis, and a pulmonary infiltrate as well as lead to severe acute respiratory distress syndrome and even death. In the past, the use of antibiotics shortly after aspiration in patients who develop a fever, leukocytosis, or a pulmonary infiltrate was discouraged.5 Empiric antibiotics were recommended only for patients who aspirate gastric contents and who have conditions associated with colonization of gastric contents, such as small-bowel obstruction.5 Yet, it is difficult to distinguish aspiration pneumonitis from pneumonia6 and there are no randomized trials in older adults to help guide their management.
PRESENTATION OF ASPIRATION PNEUMONIA
Pneumonia in older adults can present in an atypical fashion. In one study of community-acquired pneumonia (CAP), the combination of cough, fever, and dyspnea is present in only 31% of patients, although separately, they are present in 67%, 64%, and 71% of patients, respectively. The same study also showed that delirium was present in 45% of patients with CAP.7 Nonrespiratory symptoms were present during the initial presentation of CAP in 55% of patients, with confusion in 42%, and falls in 16% of cases.8 The same is true of aspiration pneumonia where altered mental status is seen in approximately 30% of community-acquired aspiration pneumonia (CAAP) patients and in 19% of continuing care facility patients with aspiration pneumonia.2 Another study that compared CAP, CAAP, and healthcare-associated aspiration pneumonia (HCAAP) showed that confusion is present in 5.1%, 12.7%, and 18.6%, respectively.9 The absence of fever in older adults is shown in studies where fever, defined as greater than or equal to 37.5°C, is absent in 32% of the very old10and in 40% of patients 65 years or older when it was defined as greater than 37°C.8 The inconsistencies regarding typical symptoms of pneumonia in the older adult population are also confirmed in nursing home residents.11 Ultimately, it is important to remember that any infection in older adults, especially in those residing in long-term care facilities, may present with subtle findings such as an acute change in cognitive and functional status.12
Risk Factors for Aspiration Pneumonia
Risk factors for aspiration pneumonia, while not universally agreed upon, are important to recognize as they increase the probability of the diagnosis when present. A 2011 systematic review identified age, male gender, lung disease, dysphagia, and diabetes mellitus (level 2a), as well as severe dementia, angiotensin I-converting enzyme deletion/deletion genotype, and poor oral health (level 2b) as risk factors.13 In 2016, a panel of experts reached a consensus (modified Delphi Method) on the following risk factors for the diagnosis of aspiration pneumonia in nursing home residents: history of dysphagia, choking incident, tube feeding, neurologic disease, and cognitive impairment. The presence of one or more of these risk factors in the appropriate clinical setting may suggest a diagnosis of aspiration pneumonia.14
Radiographic/Ultrasonographic Imaging
In the appropriate scenario, the diagnosis of aspiration pneumonia is supported with an image representative of pneumonia. The pulmonary segment involved in aspiration pneumonia depends on the position of the patient during the aspiration event. If the aspiration event occurs while the patient is in the recumbent position, development of pneumonia is more common in the posterior segments of the upper lobes and the apical segments of the lower lobes; whereas if it occurs while the patient is in an upright position, the location changes to the basal segments of the lower lobes.3
Overall, the sensitivity of a chest X-ray to diagnose pneumonia ranges between 32%-77.7%,15-17 suggesting that a significant proportion of patients suspected of having pneumonia in past research studies, may have been misdiagnosed. Studies using lung ultrasound to identify pneumonia demonstrate a higher sensitivity, but additional research is needed to validate these findings.17-19 Noncontrast CT scans of the chest remain the reference standard for diagnosing pneumonia and currently tend to have the largest impact on diagnosis and subsequent treatment decisions.15,16,20,21 As a result, if radiation exposure risks are not a concern for the patient, we recommend utilizing noncontrast CT imaging whenever the diagnosis is in doubt until future research elucidates the most appropriate approach to imaging.
Diagnosis
Diagnosing aspiration pneumonia is difficult, in part because there is no universal definition or set of diagnostic criteria. The diagnosis of aspiration pneumonia is supported by the fulfillment of three criteria. First, appropriate risk factors for aspiration, as documented above, should be present. Second, there should be evidence of clinical signs and symptoms of pneumonia (typical or atypical). Third, radiographic representation of pneumonia in a dependent pulmonary segment confirms the diagnosis. Once these criteria are met, it is important to distinguish between CAAP and HCAAP with particular attention to risk factors for multidrug-resistant (MDR) organisms and Pseudomonas aeruginosa (PA).
MICROBIOLOGY
Many studies have tried to determine the exact bacterial etiology of aspiration pneumonia as documented in the Table.
Even when an ideal method is used to obtain a good sample, however, the results are limited by other variables in the study. For example, in studies that use protected brush specimens and protected bronchoalveolar lavage to acquire samples for culture, many patients received antibiotics prior to sampling, and the studies are small (Table). Although anaerobes have traditionally been implicated in aspiration pneumonia, only El-Solh et al.22 were able to culture a significant proportion of anaerobes. The study, however, was limited to institutionalized older adults requiring mechanical ventilation and it did not require the typical radiographic location for aspiration pneumonia. Even under the best circumstances, it is difficult to determine causality because the antibiotics used to treat these cases of aspiration pneumonia cover a broad range of organisms. Based on the studies in the Table, causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods in addition to traditional organisms classically thought to cause aspiration pneumonia-anaerobes. Microbiologic etiology, however, may also be insinuated from the studies discussed in the therapeutic strategies section below as some include antibiotics with limited antimicrobial activity.
Therapeutic Strategies
The management of aspiration pneumonia has evolved significantly since it was first studied in the 1970s because of the development of antibiotic resistance patterns, newer antibiotics, and increasing information on the diversity of pathogens involved in each subset of aspiration syndromes. The antimicrobial treatment of aspiration pneumonia was classically directed against anaerobic pathogens; treatment of these infections, however, was extrapolated from studies of pulmonary abscesses and other anaerobic pulmonary infections.
A randomized controlled trial in the mid-1980s comparing penicillin and clindamycin demonstrated a significantly improved cure rate in the clindamycin group.23 A follow-up study in 1990 implicated a significant number of penicillin-resistant Bacteroides infections—the majority of these infections were subsequently reclassified as Prevotella melaninogenica—as the cause for high rates of penicillin resistance in lung abscesses and necrotizing pneumonias, further supporting clindamycin as the treatment of choice for these infections.24 Amoxicillin-clavulanic acid (IV and PO regimens), studied in the treatment of community-acquired necrotizing pneumonia/lung abscess, shows good efficacy as well.25 This study also attempted to elucidate the underlying causative organisms in these patients. Organisms associated with CAP as well as anaerobic organisms were isolated, giving more credence to the idea of broader coverage for aspiration pneumonia.
Community-Acquired Aspiration Pneumonia/Healthcare-Associated Aspiration Pneumonia
The importance of making a diagnostic distinction between CAAP versus HCAAP is critical for management strategies. A prospective population-based study demonstrated that ICU length of stay and 30-day mortality is highest for HCAAP, followed by CAAP, and lastly for those with CAP.9 Although some studies use different nomenclature for identifying aspiration pneumonia patients at risk for a wider array of microorganisms, we attempt to standardize the language by using HCAAP. The literature on nonaspiration pneumonia is changing from terms such as CAP and healthcare-associated pneumonia (HCAP) to pneumonia with the risk of MDR organisms. One study proposed a new treatment algorithm for CAP based on the presence or absence of the following six risk factors: prior hospitalization of greater than or equal to two days in the preceding 90 days, immunosuppression, previous antibiotic use within the preceding 90 days, use of gastric acid-suppressive agents, tube feeding, and nonambulatory status.26 A similar approach proposed years earlier for HCAP patients found the following to be risk factors for MDR organisms: hospitalization in the past 90 days, antibiotic therapy in the past six months, poor functional status as defined by activities of daily living score, and immune suppression.27 Other factors, such as structural lung disease, that increase the risk of organisms resistant to standard antibiotic treatment regimens28-31 should be considered in aspiration pneumonia as well. Aspiration pneumonia is following a similar trajectory where the risk of MDR organisms is taking precedence over the environment of acquisition. The final nomenclature will allow the healthcare provider to understand the organisms that need to be targeted when choosing an appropriate antibiotic treatment regimen.
There is evidence supporting the premise that CAAP and nursing home patients with no risk factors for MDR organisms can be treated with standard regimens used for patients with CAP. A prospective cohort study in 2014 did not show any statistically significant differences in clinical outcomes in nursing and healthcare-associated aspiration pneumonia patients (with no risks of MDR organisms) treated with azithromycin versus ampicillin/sulbactam. However, only 36 patients were included in the azithromycin arm, and the therapeutic choices were made by the treating physician.32
A prospective study of 95 long-term care residents reported that of those patients admitted to the ICU with severe aspiration pneumonia, the causative organisms are gram-negative enteric bacilli in 49% of isolates, anaerobes in 16%, and Staphylococcus aureus in 12%.22 This study mentioned that six of seven anaerobic pneumonia cases had inadequate anaerobic coverage yet were effectively treated; based on the organisms represented, however, the antibiotics administered did provide some coverage.22 Prevotella was one of the common anaerobic organisms that could be treated by levofloxacin or ceftriaxone/azithromycin, possibly explaining the success of azithromycin in the study quoted previously.22,32 Therefore, although anaerobic organisms still need to be considered, some may be treated by traditional CAP coverage.22
In a 2005 randomized prospective study of 100 patients aged 71 to 94 years, clindamycin was found to have clinical efficacy equivalent to ampicillin-sulbactam and panipenem in the treatment of mild-to-moderate aspiration pneumonia.33 Most patients in this study are nursing home residents, and 53% of sputum cultures in the clindamycin arm grew gram-negative rods. In contrast to the previous study, the significance of gram-negative rod infections in this population of patients, with less severe infections, is called into question, as clindamycin has no coverage against these organisms. This premise is supported by a more recent study using azithromycin in nursing and healthcare-associated aspiration pneumonia patients, mentioned previously.32 Taken together, these three studies suggest that the severity of aspiration pneumonia may be a risk factor that needs to be taken into account when considering broad-spectrum antimicrobial coverage.
While further research is needed to validate treatment approaches, based on the current literature we propose the following:
CAAP requiring hospitalization but without any of the following-risk for PA or MDR organisms, septic shock, the need for ICU admission, or mechanical ventilation-can be treated with standard CAP therapy that covers anaerobes.26,32-34 Patients with CAAP and either of the following—risk factors for MDR organisms, septic shock, need for ICU admission, or mechanical ventilation—should be considered for broader coverage with vancomycin or linezolid, antipseudomonal antibiotics, and anaerobic coverage. CAAP with specific risk for a PA infection should be considered for two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic, and one has anaerobic coverage).
Severe HCAAP without risk for MDR organisms or PA but with any of the following-septic shock, ICU admission, or mechanical ventilation-can be treated based on the 2016 Infectious Diseases Society of America guideline recommendation for hospital-acquired pneumonia, with a regimen that also provides adequate anaerobic coverage.35 If patients have HCAAP with one or more risk factors for MDR organisms, no septic shock, and no need for ICU admission or mechanical ventilation, provide coverage with a similar regimen. In contrast, HCAAP with risk factors for PA or severe HCAAP causing septic shock, requiring ICU admission, or needing mechanical ventilation, which occurs in the setting of one or more risk factors for MDR organisms, or structural lung disease, should receive two antipseudomonal antibiotics (where only one can be a beta-lactam antibiotic and one has anaerobic coverage) in addition to vancomycin or linezolid.
A recent systematic review demonstrates the paucity of studies of ideal methodologic design which complicates the ability to recommend, with confidence, one guideline-based antimicrobial regimen over another.36 Future studies may determine that despite the severity of the infection, if patients do not carry any risk for MDR pathogens or PA, they may only require CAAP coverage. When a patient presents with an acute infection, it is prudent to review previous cultures, and although it may be necessary to treat with broad-spectrum antibiotics initially, it is always important to narrow the spectrum based on reliable culture results. If future studies support the results of many studies cited in this article, we may be using fewer antibiotics with narrower spectrums in the near future.
Prevention
Although the healthcare system has practices in place to prevent aspiration pneumonia, the evidence supporting them are either inconclusive or not of ideal methodological design. Two systematic reviews failed to show statistically significant decreases in rates of aspiration pneumonia or mortality using the standard of care positioning strategies or thickened fluids in patients with chronic dysphagia.37,38 One study showed a decreased incidence of all pneumonia in dysphasic patients with dementia or Parkinson disease when a chin-down posture (with thin liquids) or thickened fluids in a head-neutral position was used. The study, however, has significant limitations, including a lack of a “no treatment” group for comparison, which did not allow investigators to conclude that the decreased incidence was from their interventions.39
There are preventive strategies that show a decreased risk of aspiration pneumonia. Poor oral hygiene seems to be a modifiable risk factor to establish better control of oral flora and decrease aspiration pneumonia. A systematic review of five studies, evaluating the effects of oral healthcare on the incidence of aspiration pneumonia in frail older people, found that tooth brushing after each meal along with cleaning dentures once a day and professional oral healthcare once a week decreases febrile days, pneumonia, and dying from pneumonia.40A two-year historical cohort study using aromatherapy with black pepper oil, followed by application of capsaicin troches, and finally menthol gel, as the first meal, leads to a decreased incidence of pneumonia and febrile days in older adults with dysphagia.41 Well-designed validation studies may establish these practices as the new standard of care for preventing pneumonia in patients with dysphagia.
Feeding Tubes
Multiple studies show that in older adults with advanced dementia there is no survival benefit from percutaneous endoscopic gastrostomy (PEG) tube placement42-44 and more recent systematic reviews also conclude that there is currently no evidence to support the use of PEG tubes in this specific population.45,46 In February 2013, as part of the American Board of Internal Medicine Foundation Choosing Wisely® campaign, the American Geriatrics Society advised providers not to recommend percutaneous feeding tubes in patients with advanced dementia, rather, “offer assisted oral feeding.”47 It is worth noting, however, that none of the studies reviewed were of ideal methodological design, so opinions may change with future studies.
A more recent study compared liquid feeds versus semisolid feeds in patients with PEG tubes. The study shows a 22.2% incidence of aspiration pneumonia in the liquid feed group, which is comparable to prior studies, but the incidence of aspiration pneumonia is only 2.2% in the semisolid feed group (P < .005).48 A benefit of this size warrants future studies for validation.
CONCLUSION
Aspiration pneumonia leads to increased mortality when compared with CAP and HCAP.2 Until future studies validate or refute the current understanding surrounding its management, the following should provide some guidance: aspiration pneumonia should be suspected in any individual with risk factors of aspiration who presents with typical or atypical symptoms of pneumonia. Confirmation of the diagnosis requires an image representative of pneumonia in the typical dependent lung segment on chest X-ray, lung ultrasound, or noncontrast CT scan of the chest. Treatment of aspiration pneumonia should take into account the site of acquisition, severity of illness, and risk for MDR organisms as the causative organisms may include Streptococcus pneumoniae, Haemophilus influenzae, Staphylococcus aureus, and gram-negative rods, in addition to the traditional organisms classically thought to cause aspiration pneumonia-anaerobes.
Disclosures
The authors have nothing to disclose.
1. Wu CP, Chen YW, Wang MJ, Pinelis E. National trends in admission for aspiration pneumonia in the United States, 2002-2012. Ann Am Thorac Soc. 2017;14(6):874-879. doi: 10.1513/AnnalsATS.201611-867OC. PubMed
2. Reza Shariatzadeh M, Huang JQ, Marrie TJ. Differences in the features of aspiration pneumonia according to site of acquisition: community or continuing care facility. J Am Geriatr Soc. 2006;54(2):296-302. doi: 10.1111/j.1532-5415.2005.00608.x. PubMed
3. Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest. 1975;68(4):560-566. doi: 10.1378/chest.68.4.560. PubMed
4. Huxley EJ, Viroslav J, Gray WR, Pierce AK. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med. 1978;64(4):564-568. doi: 10.1016/0002-9343(78)90574-0. PubMed
5. Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671. doi: 10.1056/NEJM200103013440908. PubMed
6. Raghavendran K, Nemzek J, Napolitano LM, Knight PR. Aspiration-induced lung injury. Crit Care Med. 2011;39(4):818-826. doi: 10.1097/CCM.0b013e31820a856b. PubMed
7. Riquelme R, Torres A, el-Ebiary M, et al. Community-acquired pneumonia in the elderly. Clinical and nutritional aspects. Am J Respir Crit Care Med. 1997;156(6):1908-1914. doi: 10.1164/ajrccm.156.6.9702005. PubMed
8. Venkatesan P, Gladman J, Macfarlane JT, et al. A hospital study of community acquired pneumonia in the elderly. Thorax. 1990;45(4):254-258. doi: 10.1136/thx.45.4.254. PubMed
9. Lanspa MJ, Jones BE, Brown SM, Dean NC. Mortality, morbidity, and disease severity of patients with aspiration pneumonia. J Hosp Med. 2013;8(2):83-90. doi: 10.1002/jhm.1996. PubMed
10. Fernández-Sabé N, Carratalà J, Rosón B, et al. Community-acquired pneumonia in very elderly patients: causative organisms, clinical characteristics, and outcomes. Medicine (Baltimore). 2003;82(3):159-169. doi: 10.1097/01.md.0000076005.64510.87. PubMed
11. Mehr DR, Binder EF, Kruse RL, et al. Clinical findings associated with radiographic pneumonia in nursing home residents. J Fam Pract. 2001;50(11):931-937. PubMed
12. Bentley DW, Bradley S, High K, et al. Practice guideline for evaluation of fever and infection in long-term care facilities. Clin Infect Dis. 2000;31(3):640-653. doi: 10.1086/314013. PubMed
13. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Risk factors for aspiration pneumonia in frail older people: a systematic literature review. J Am Med Dir Assoc. 2011;12(5):344-354. doi: 10.1016/j.jamda.2010.12.099. PubMed
14. Hollaar V, van der Maarel-Wierink C, van der Putten GJ, et al. Defining characteristics and risk indicators for diagnosing nursing home-acquired pneumonia and aspiration pneumonia in nursing home residents, using the electronically-modified Delphi Method. BMC Geriatr. 2016;16:60. doi: 10.1186/s12877-016-0231-4. PubMed
15. Esayag Y, Nikitin I, Bar-Ziv J, et al. Diagnostic value of chest radiographs in bedridden patients suspected of having pneumonia. Am J Med. 2010;123(1):88.e1-88.e5. doi: 10.1016/j.amjmed.2009.09.012. PubMed
16. Claessens YE, Debray MP, Tubach F, et al. Early chest computed tomography scan to assist diagnosis and guide treatment decision for suspected community-acquired pneumonia. Am J Respir Crit Care Med. 2015;192(8):974-982. doi: 10.1164/rccm.201501-0017OC. PubMed
17. Liu XL, Lian R, Tao YK, Gu CD, Zhang GQ. Lung ultrasonography: an effective way to diagnose community-acquired pneumonia. Emerg Med J. 2015;32(6):433-438. doi: 10.1136/emermed-2013-203039. PubMed
18. Bourcier JE, Paquet J, Seinger M, et al. Performance comparison of lung ultrasound and chest x-ray for the diagnosis of pneumonia in the ED. Am J Emerg Med. 2014;32(2):115-118. doi: 10.1016/j.ajem.2013.10.003. PubMed
19. Chavez MA, Shams N, Ellington LE, et al. Lung ultrasound for the diagnosis of pneumonia in adults: a systematic review and meta-analysis. Respir Res. 2014;15:50. doi: 10.1186/1465-9921-15-50. PubMed
20. Syrjälä H, Broas M, Suramo I, Ojala A, Lähde S. High-resolution computed tomography for the diagnosis of community-acquired pneumonia. Clin Infect Dis. 1998;27(2):358-363. doi: 10.1086/514675. PubMed
21. Hayden GE, Wrenn KW. Chest radiograph vs. computed tomography scan in the evaluation for pneumonia. J Emerg Med. 2009;36(3):266-270. doi: 10.1016/j.jemermed.2007.11.042. PubMed
22. El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med. 2003;167(12):1650-1654. doi: 10.1164/rccm.200212-1543OC. PubMed
23. Levison ME, Mangura CT, Lorber B, et al. Clindamycin compared with penicillin for the treatment of anaerobic lung abscess. Ann Intern Med. 1983;98(4):466-471. doi: 10.7326/0003-4819-98-4-466. PubMed
24. Gudiol F, Manresa F, Pallares R, et al. Clindamycin vs penicillin for anaerobic lung infections. High rate of penicillin failures associated with penicillin-resistant Bacteroides melaninogenicus. Arch Intern Med. 1990;150(12):2525-2529. doi: 10.1001/archinte.150.12.2525. PubMed
25. Germaud P, Poirier J, Jacqueme P, et al. Monotherapy using amoxicillin/clavulanic acid as treatment of first choice in community-acquired lung abscess. Apropos of 57 cases. Rev Pneumol Clin. 1993;49(3):137-141. PubMed
26. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995. doi: 10.1164/rccm.201301-0079OC. PubMed
27. Brito V, Niederman MS. Healthcare-associated pneumonia is a heterogeneous disease, and all patients do not need the same broad-spectrum antibiotic therapy as complex nosocomial pneumonia. Curr Opin Infect Dis. 2009;22(3):316-325. doi: 10.1097/QCO.0b013e328329fa4e. PubMed
28. Restrepo MI, Babu BL, Reyes LF, et al. Burden and risk factors for Pseudomonas aeruginosa community-acquired pneumonia: a multinational point prevalence study of hospitalised patients. Eur Respir J. 2018;52(2). doi: 10.1183/13993003.01190-2017. PubMed
29. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44 Supplement 2:S27-S72. doi: 10.1086/511159. PubMed
30. Cillóniz C, Gabarrús A, Ferrer M, et al. Community-acquired pneumonia due to multidrug- and non-multidrug-resistant Pseudomonas aeruginosa. Chest. 2016;150(2):415-425. doi: 10.1016/j.chest.2016.03.042. PubMed
31. Prina E, Ranzani OT, Polverino E, et al. Risk factors associated with potentially antibiotic-resistant pathogens in community-acquired pneumonia. Ann Am Thorac Soc. 2015;12(2):153-160. doi: 10.1513/AnnalsATS.201407-305OC. PubMed
32. Marumo S, Teranishi T, Higami Y, et al. Effectiveness of azithromycin in aspiration pneumonia: a prospective observational study. BMC Infect Dis. 2014;14:685. doi: 10.1186/s12879-014-0685-y. PubMed
33. Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest. 2005;127(4):1276-1282. doi: 10.1378/chest.127.4.1276. PubMed
34. Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383. doi: 10.1093/cid/cit571. PubMed
35. Kalil AC, Metersky ML, Klompas M, et al. Executive Summary: management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):575-582. doi: 10.1093/cid/ciw504. PubMed
36. Bowerman TJ, Zhang J, Waite LM. Antibacterial treatment of aspiration pneumonia in older people: a systematic review. Clin Interv Aging. 2018;13:2201-2213. doi: 10.2147/CIA.S183344. PubMed
37. Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022. doi: 10.1046/j.1365-2389.2003.51318.x. PubMed
38. Andersen UT, Beck AM, Kjaersgaard A, Hansen T, Poulsen I. Systematic review and evidence based recommendations on texture modified foods and thickened fluids for adults (≥18 years) with oropharyngeal dysphagia. Clin Nutr ESPEN. 2013;8(4):e127-e134.
39. Robbins J, Gensler G, Hind J, et al. Comparison of 2 interventions for liquid aspiration on pneumonia incidence: a randomized trial. Ann Intern Med. 2008;148(7):509-518. doi: 10.7326/0003-4819-148-7-200804010-00007. PubMed
40. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Oral health care and aspiration pneumonia in frail older people: a systematic literature review. Gerodontology. 2013;30(1):3-9. doi: 10.1111/j.1741-2358.2012.00637.x. PubMed
41. Ebihara T, Ebihara S, Yamazaki M, et al. Intensive stepwise method for oral intake using a combination of transient receptor potential stimulation and olfactory stimulation inhibits the incidence of pneumonia in dysphagic older adults. J Am Geriatr Soc. 2010;58(1):196-198. doi: 10.1111/j.1532-5415.2009.02648.x. PubMed
42. Sanders DS, Carter MJ, D’Silva J, et al. Survival analysis in percutaneous endoscopic gastrostomy feeding: a worse outcome in patients with dementia. Am J Gastroenterol. 2000;95(6):1472-1475. doi: 10.1111/j.1572-0241.2000.02079.x. PubMed
43. Murphy LM, Lipman TO. Percutaneous endoscopic gastrostomy does not prolong survival in patients with dementia. Arch Intern Med. 2003;163(11):1351-1353. doi: 10.1001/archinte.163.11.1351. PubMed
44. Rimon E, Kagansky N, Levy S. Percutaneous endoscopic gastrostomy; evidence of different prognosis in various patient subgroups. Age Ageing. 2005;34(4):353-357. doi: 10.1093/ageing/afi085. PubMed
45. Candy B, Sampson EL, Jones L. Enteral tube feeding in older people with advanced dementia: findings from a Cochrane systematic review. Int J Palliat Nurs. 2009;15(8):396-404. doi: 10.12968/ijpn.2009.15.8.43799. PubMed
46. Goldberg LS, Altman KW. The role of gastrostomy tube placement in advanced dementia with dysphagia: a critical review. Clin Interv Aging. 2014;9:1733-1739. doi: 10.2147/CIA.S53153. PubMed
47. Workgroup AGSCW. American Geriatrics Society identifies five things that healthcare providers and patients should question. J Am Geriatr Soc. 2013;61(4):622-631. doi: 10.1111/jgs.12226. PubMed
48. Toh Yoon EW, Yoneda K, Nishihara K. Semi-solid feeds may reduce the risk of aspiration pneumonia and shorten postoperative length of stay after percutaneous endoscopic gastrostomy (PEG). Endosc Int Open. 2016;4(12):E1247-E1251. doi: 10.1055/s-0042-117218. PubMed
49. Mier L, Dreyfuss D, Darchy B, et al. Is penicillin-G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intens Care Med. 1993;19(5):279-284. doi: 10.1007/BF01690548. PubMed
50. Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest. 1999;115(1):178-183. doi: 10.1378/chest.115.1.178. PubMed
1. Wu CP, Chen YW, Wang MJ, Pinelis E. National trends in admission for aspiration pneumonia in the United States, 2002-2012. Ann Am Thorac Soc. 2017;14(6):874-879. doi: 10.1513/AnnalsATS.201611-867OC. PubMed
2. Reza Shariatzadeh M, Huang JQ, Marrie TJ. Differences in the features of aspiration pneumonia according to site of acquisition: community or continuing care facility. J Am Geriatr Soc. 2006;54(2):296-302. doi: 10.1111/j.1532-5415.2005.00608.x. PubMed
3. Bartlett JG, Gorbach SL. The triple threat of aspiration pneumonia. Chest. 1975;68(4):560-566. doi: 10.1378/chest.68.4.560. PubMed
4. Huxley EJ, Viroslav J, Gray WR, Pierce AK. Pharyngeal aspiration in normal adults and patients with depressed consciousness. Am J Med. 1978;64(4):564-568. doi: 10.1016/0002-9343(78)90574-0. PubMed
5. Marik PE. Aspiration pneumonitis and aspiration pneumonia. N Engl J Med. 2001;344(9):665-671. doi: 10.1056/NEJM200103013440908. PubMed
6. Raghavendran K, Nemzek J, Napolitano LM, Knight PR. Aspiration-induced lung injury. Crit Care Med. 2011;39(4):818-826. doi: 10.1097/CCM.0b013e31820a856b. PubMed
7. Riquelme R, Torres A, el-Ebiary M, et al. Community-acquired pneumonia in the elderly. Clinical and nutritional aspects. Am J Respir Crit Care Med. 1997;156(6):1908-1914. doi: 10.1164/ajrccm.156.6.9702005. PubMed
8. Venkatesan P, Gladman J, Macfarlane JT, et al. A hospital study of community acquired pneumonia in the elderly. Thorax. 1990;45(4):254-258. doi: 10.1136/thx.45.4.254. PubMed
9. Lanspa MJ, Jones BE, Brown SM, Dean NC. Mortality, morbidity, and disease severity of patients with aspiration pneumonia. J Hosp Med. 2013;8(2):83-90. doi: 10.1002/jhm.1996. PubMed
10. Fernández-Sabé N, Carratalà J, Rosón B, et al. Community-acquired pneumonia in very elderly patients: causative organisms, clinical characteristics, and outcomes. Medicine (Baltimore). 2003;82(3):159-169. doi: 10.1097/01.md.0000076005.64510.87. PubMed
11. Mehr DR, Binder EF, Kruse RL, et al. Clinical findings associated with radiographic pneumonia in nursing home residents. J Fam Pract. 2001;50(11):931-937. PubMed
12. Bentley DW, Bradley S, High K, et al. Practice guideline for evaluation of fever and infection in long-term care facilities. Clin Infect Dis. 2000;31(3):640-653. doi: 10.1086/314013. PubMed
13. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Risk factors for aspiration pneumonia in frail older people: a systematic literature review. J Am Med Dir Assoc. 2011;12(5):344-354. doi: 10.1016/j.jamda.2010.12.099. PubMed
14. Hollaar V, van der Maarel-Wierink C, van der Putten GJ, et al. Defining characteristics and risk indicators for diagnosing nursing home-acquired pneumonia and aspiration pneumonia in nursing home residents, using the electronically-modified Delphi Method. BMC Geriatr. 2016;16:60. doi: 10.1186/s12877-016-0231-4. PubMed
15. Esayag Y, Nikitin I, Bar-Ziv J, et al. Diagnostic value of chest radiographs in bedridden patients suspected of having pneumonia. Am J Med. 2010;123(1):88.e1-88.e5. doi: 10.1016/j.amjmed.2009.09.012. PubMed
16. Claessens YE, Debray MP, Tubach F, et al. Early chest computed tomography scan to assist diagnosis and guide treatment decision for suspected community-acquired pneumonia. Am J Respir Crit Care Med. 2015;192(8):974-982. doi: 10.1164/rccm.201501-0017OC. PubMed
17. Liu XL, Lian R, Tao YK, Gu CD, Zhang GQ. Lung ultrasonography: an effective way to diagnose community-acquired pneumonia. Emerg Med J. 2015;32(6):433-438. doi: 10.1136/emermed-2013-203039. PubMed
18. Bourcier JE, Paquet J, Seinger M, et al. Performance comparison of lung ultrasound and chest x-ray for the diagnosis of pneumonia in the ED. Am J Emerg Med. 2014;32(2):115-118. doi: 10.1016/j.ajem.2013.10.003. PubMed
19. Chavez MA, Shams N, Ellington LE, et al. Lung ultrasound for the diagnosis of pneumonia in adults: a systematic review and meta-analysis. Respir Res. 2014;15:50. doi: 10.1186/1465-9921-15-50. PubMed
20. Syrjälä H, Broas M, Suramo I, Ojala A, Lähde S. High-resolution computed tomography for the diagnosis of community-acquired pneumonia. Clin Infect Dis. 1998;27(2):358-363. doi: 10.1086/514675. PubMed
21. Hayden GE, Wrenn KW. Chest radiograph vs. computed tomography scan in the evaluation for pneumonia. J Emerg Med. 2009;36(3):266-270. doi: 10.1016/j.jemermed.2007.11.042. PubMed
22. El-Solh AA, Pietrantoni C, Bhat A, et al. Microbiology of severe aspiration pneumonia in institutionalized elderly. Am J Respir Crit Care Med. 2003;167(12):1650-1654. doi: 10.1164/rccm.200212-1543OC. PubMed
23. Levison ME, Mangura CT, Lorber B, et al. Clindamycin compared with penicillin for the treatment of anaerobic lung abscess. Ann Intern Med. 1983;98(4):466-471. doi: 10.7326/0003-4819-98-4-466. PubMed
24. Gudiol F, Manresa F, Pallares R, et al. Clindamycin vs penicillin for anaerobic lung infections. High rate of penicillin failures associated with penicillin-resistant Bacteroides melaninogenicus. Arch Intern Med. 1990;150(12):2525-2529. doi: 10.1001/archinte.150.12.2525. PubMed
25. Germaud P, Poirier J, Jacqueme P, et al. Monotherapy using amoxicillin/clavulanic acid as treatment of first choice in community-acquired lung abscess. Apropos of 57 cases. Rev Pneumol Clin. 1993;49(3):137-141. PubMed
26. Shindo Y, Ito R, Kobayashi D, et al. Risk factors for drug-resistant pathogens in community-acquired and healthcare-associated pneumonia. Am J Respir Crit Care Med. 2013;188(8):985-995. doi: 10.1164/rccm.201301-0079OC. PubMed
27. Brito V, Niederman MS. Healthcare-associated pneumonia is a heterogeneous disease, and all patients do not need the same broad-spectrum antibiotic therapy as complex nosocomial pneumonia. Curr Opin Infect Dis. 2009;22(3):316-325. doi: 10.1097/QCO.0b013e328329fa4e. PubMed
28. Restrepo MI, Babu BL, Reyes LF, et al. Burden and risk factors for Pseudomonas aeruginosa community-acquired pneumonia: a multinational point prevalence study of hospitalised patients. Eur Respir J. 2018;52(2). doi: 10.1183/13993003.01190-2017. PubMed
29. Mandell LA, Wunderink RG, Anzueto A, et al. Infectious Diseases Society of America/American Thoracic Society consensus guidelines on the management of community-acquired pneumonia in adults. Clin Infect Dis. 2007;44 Supplement 2:S27-S72. doi: 10.1086/511159. PubMed
30. Cillóniz C, Gabarrús A, Ferrer M, et al. Community-acquired pneumonia due to multidrug- and non-multidrug-resistant Pseudomonas aeruginosa. Chest. 2016;150(2):415-425. doi: 10.1016/j.chest.2016.03.042. PubMed
31. Prina E, Ranzani OT, Polverino E, et al. Risk factors associated with potentially antibiotic-resistant pathogens in community-acquired pneumonia. Ann Am Thorac Soc. 2015;12(2):153-160. doi: 10.1513/AnnalsATS.201407-305OC. PubMed
32. Marumo S, Teranishi T, Higami Y, et al. Effectiveness of azithromycin in aspiration pneumonia: a prospective observational study. BMC Infect Dis. 2014;14:685. doi: 10.1186/s12879-014-0685-y. PubMed
33. Kadowaki M, Demura Y, Mizuno S, et al. Reappraisal of clindamycin IV monotherapy for treatment of mild-to-moderate aspiration pneumonia in elderly patients. Chest. 2005;127(4):1276-1282. doi: 10.1378/chest.127.4.1276. PubMed
34. Maruyama T, Fujisawa T, Okuno M, et al. A new strategy for healthcare-associated pneumonia: a 2-year prospective multicenter cohort study using risk factors for multidrug-resistant pathogens to select initial empiric therapy. Clin Infect Dis. 2013;57(10):1373-1383. doi: 10.1093/cid/cit571. PubMed
35. Kalil AC, Metersky ML, Klompas M, et al. Executive Summary: management of adults with hospital-acquired and ventilator-associated pneumonia: 2016 clinical practice guidelines by the Infectious Diseases Society of America and the American Thoracic Society. Clin Infect Dis. 2016;63(5):575-582. doi: 10.1093/cid/ciw504. PubMed
36. Bowerman TJ, Zhang J, Waite LM. Antibacterial treatment of aspiration pneumonia in older people: a systematic review. Clin Interv Aging. 2018;13:2201-2213. doi: 10.2147/CIA.S183344. PubMed
37. Loeb MB, Becker M, Eady A, Walker-Dilks C. Interventions to prevent aspiration pneumonia in older adults: a systematic review. J Am Geriatr Soc. 2003;51(7):1018-1022. doi: 10.1046/j.1365-2389.2003.51318.x. PubMed
38. Andersen UT, Beck AM, Kjaersgaard A, Hansen T, Poulsen I. Systematic review and evidence based recommendations on texture modified foods and thickened fluids for adults (≥18 years) with oropharyngeal dysphagia. Clin Nutr ESPEN. 2013;8(4):e127-e134.
39. Robbins J, Gensler G, Hind J, et al. Comparison of 2 interventions for liquid aspiration on pneumonia incidence: a randomized trial. Ann Intern Med. 2008;148(7):509-518. doi: 10.7326/0003-4819-148-7-200804010-00007. PubMed
40. van der Maarel-Wierink CD, Vanobbergen JN, Bronkhorst EM, Schols JM, de Baat C. Oral health care and aspiration pneumonia in frail older people: a systematic literature review. Gerodontology. 2013;30(1):3-9. doi: 10.1111/j.1741-2358.2012.00637.x. PubMed
41. Ebihara T, Ebihara S, Yamazaki M, et al. Intensive stepwise method for oral intake using a combination of transient receptor potential stimulation and olfactory stimulation inhibits the incidence of pneumonia in dysphagic older adults. J Am Geriatr Soc. 2010;58(1):196-198. doi: 10.1111/j.1532-5415.2009.02648.x. PubMed
42. Sanders DS, Carter MJ, D’Silva J, et al. Survival analysis in percutaneous endoscopic gastrostomy feeding: a worse outcome in patients with dementia. Am J Gastroenterol. 2000;95(6):1472-1475. doi: 10.1111/j.1572-0241.2000.02079.x. PubMed
43. Murphy LM, Lipman TO. Percutaneous endoscopic gastrostomy does not prolong survival in patients with dementia. Arch Intern Med. 2003;163(11):1351-1353. doi: 10.1001/archinte.163.11.1351. PubMed
44. Rimon E, Kagansky N, Levy S. Percutaneous endoscopic gastrostomy; evidence of different prognosis in various patient subgroups. Age Ageing. 2005;34(4):353-357. doi: 10.1093/ageing/afi085. PubMed
45. Candy B, Sampson EL, Jones L. Enteral tube feeding in older people with advanced dementia: findings from a Cochrane systematic review. Int J Palliat Nurs. 2009;15(8):396-404. doi: 10.12968/ijpn.2009.15.8.43799. PubMed
46. Goldberg LS, Altman KW. The role of gastrostomy tube placement in advanced dementia with dysphagia: a critical review. Clin Interv Aging. 2014;9:1733-1739. doi: 10.2147/CIA.S53153. PubMed
47. Workgroup AGSCW. American Geriatrics Society identifies five things that healthcare providers and patients should question. J Am Geriatr Soc. 2013;61(4):622-631. doi: 10.1111/jgs.12226. PubMed
48. Toh Yoon EW, Yoneda K, Nishihara K. Semi-solid feeds may reduce the risk of aspiration pneumonia and shorten postoperative length of stay after percutaneous endoscopic gastrostomy (PEG). Endosc Int Open. 2016;4(12):E1247-E1251. doi: 10.1055/s-0042-117218. PubMed
49. Mier L, Dreyfuss D, Darchy B, et al. Is penicillin-G an adequate initial treatment for aspiration pneumonia? A prospective evaluation using a protected specimen brush and quantitative cultures. Intens Care Med. 1993;19(5):279-284. doi: 10.1007/BF01690548. PubMed
50. Marik PE, Careau P. The role of anaerobes in patients with ventilator-associated pneumonia and aspiration pneumonia: a prospective study. Chest. 1999;115(1):178-183. doi: 10.1378/chest.115.1.178. PubMed
© 2019 Society of Hospital Medicine
Venous thromboembolism risk elevated in ankylosing spondylitis patients
Newly diagnosed ankylosing spondylitis (AS) patients are at increased risk for venous thromboembolism (VTE), especially during the first year after diagnosis, according to a population-based study of 7,190 cases.

In a study published in Annals of the Rheumatic Diseases, the researchers identified 7,190 incident cases of AS among adults using a health care database of residents of British Columbia and matched them for age, sex, and entry time into the cohort with 71,900 healthy individuals from the general population over a mean follow-up time of 6.2 years.
The incidence rate of VTE overall per 1,000 person-years was 1.56 among AS patients, compared with 0.77 in a control cohort from the general population. The incidence rates for DVT were 1.06 in AS patients and 0.50 in controls; incidence rates for PE were 0.79 in AS patients and 0.40 in controls.
The adjusted hazard ratios for VTE overall and DVT were similar and statistically significant in AS patients at 1.53 and 1.62, respectively, versus controls. But the adjusted hazard ratio of 1.36 for PE did not reach statistical significance. The adjusted risks of VTE overall, PE, and DVT were highest in the first year of diagnosis, reaching twofold greater risk for all, but none of the risks were statistically significant.
More research is needed to better identify subsets of AS patients at increased risk for VTE, and to assess whether treatment of inflammation can mitigate this risk, but in the meantime clinicians should be alert to the possibility of life-threatening complications from DVT and PE in their AS patients, especially soon after diagnosis, the researchers said.
The findings are supported by the study’s large sample size but are also limited by several factors, including the observational nature of the study and an inability to account for use of NSAIDs, the researchers noted.
“These results call for awareness of this complication, increased vigilance, and preventive intervention by controlling the inflammatory process or by anticoagulation in a high-risk AS population,” they concluded.
The study was supported in part by grants from the Canadian Arthritis Network, the Arthritis Society of Canada, the British Columbia Lupus Society, and the Canadian Institutes for Health Research. The researchers had no financial conflicts to disclose.
SOURCE: Aviña-Zubieta JA et al. Ann Rheum Dis. 2019 Feb 8. doi: 10.1136/annrheumdis-2018-214388.
Newly diagnosed ankylosing spondylitis (AS) patients are at increased risk for venous thromboembolism (VTE), especially during the first year after diagnosis, according to a population-based study of 7,190 cases.
In a study published in Annals of the Rheumatic Diseases, the researchers identified 7,190 incident cases of AS among adults using a health care database of residents of British Columbia and matched them for age, sex, and entry time into the cohort with 71,900 healthy individuals from the general population over a mean follow-up time of 6.2 years.
The incidence rate of VTE overall per 1,000 person-years was 1.56 among AS patients, compared with 0.77 in a control cohort from the general population. The incidence rates for DVT were 1.06 in AS patients and 0.50 in controls; incidence rates for PE were 0.79 in AS patients and 0.40 in controls.
The adjusted hazard ratios for VTE overall and DVT were similar and statistically significant in AS patients at 1.53 and 1.62, respectively, versus controls. But the adjusted hazard ratio of 1.36 for PE did not reach statistical significance. The adjusted risks of VTE overall, PE, and DVT were highest in the first year of diagnosis, reaching twofold greater risk for all, but none of the risks were statistically significant.
More research is needed to better identify subsets of AS patients at increased risk for VTE, and to assess whether treatment of inflammation can mitigate this risk, but in the meantime clinicians should be alert to the possibility of life-threatening complications from DVT and PE in their AS patients, especially soon after diagnosis, the researchers said.
The findings are supported by the study’s large sample size but are also limited by several factors, including the observational nature of the study and an inability to account for use of NSAIDs, the researchers noted.
“These results call for awareness of this complication, increased vigilance, and preventive intervention by controlling the inflammatory process or by anticoagulation in a high-risk AS population,” they concluded.
The study was supported in part by grants from the Canadian Arthritis Network, the Arthritis Society of Canada, the British Columbia Lupus Society, and the Canadian Institutes for Health Research. The researchers had no financial conflicts to disclose.
SOURCE: Aviña-Zubieta JA et al. Ann Rheum Dis. 2019 Feb 8. doi: 10.1136/annrheumdis-2018-214388.
Newly diagnosed ankylosing spondylitis (AS) patients are at increased risk for venous thromboembolism (VTE), especially during the first year after diagnosis, according to a population-based study of 7,190 cases.
In a study published in Annals of the Rheumatic Diseases, the researchers identified 7,190 incident cases of AS among adults using a health care database of residents of British Columbia and matched them for age, sex, and entry time into the cohort with 71,900 healthy individuals from the general population over a mean follow-up time of 6.2 years.
The incidence rate of VTE overall per 1,000 person-years was 1.56 among AS patients, compared with 0.77 in a control cohort from the general population. The incidence rates for DVT were 1.06 in AS patients and 0.50 in controls; incidence rates for PE were 0.79 in AS patients and 0.40 in controls.
The adjusted hazard ratios for VTE overall and DVT were similar and statistically significant in AS patients at 1.53 and 1.62, respectively, versus controls. But the adjusted hazard ratio of 1.36 for PE did not reach statistical significance. The adjusted risks of VTE overall, PE, and DVT were highest in the first year of diagnosis, reaching twofold greater risk for all, but none of the risks were statistically significant.
More research is needed to better identify subsets of AS patients at increased risk for VTE, and to assess whether treatment of inflammation can mitigate this risk, but in the meantime clinicians should be alert to the possibility of life-threatening complications from DVT and PE in their AS patients, especially soon after diagnosis, the researchers said.
The findings are supported by the study’s large sample size but are also limited by several factors, including the observational nature of the study and an inability to account for use of NSAIDs, the researchers noted.
“These results call for awareness of this complication, increased vigilance, and preventive intervention by controlling the inflammatory process or by anticoagulation in a high-risk AS population,” they concluded.
The study was supported in part by grants from the Canadian Arthritis Network, the Arthritis Society of Canada, the British Columbia Lupus Society, and the Canadian Institutes for Health Research. The researchers had no financial conflicts to disclose.
SOURCE: Aviña-Zubieta JA et al. Ann Rheum Dis. 2019 Feb 8. doi: 10.1136/annrheumdis-2018-214388.
FROM ANNALS OF THE RHEUMATIC DISEASES
Key clinical point: Newly diagnosed AS patients demonstrated increased risk of venous thromboembolism, including deep vein thrombosis and pulmonary embolism, compared with controls.
Major finding: The relative risk for deep vein thrombosis was 63% higher for AS patients versus controls, but a 39% higher risk of pulmonary embolism did not reach statistical significance.
Study details: A population-based study including 7,190 incident AS cases and 71,900 matched controls from a health care database of residents of British Columbia.
Disclosures: The study was supported in part by grants from the Canadian Arthritis Network, the Arthritis Society of Canada, the British Columbia Lupus Society, and the Canadian Institutes for Health Research. The researchers had no financial conflicts to disclose.
Source: Aviña-Zubieta JA et al. Ann Rheum Dis. 2019 Feb 8. doi: 10.1136/annrheumdis-2018-214388.
Short sleep linked with high homocysteine for some populations
Short sleep’s association with cardiovascular risk may be mediated in part by elevated homocysteine levels, suggests a new analysis of data from the 2005-2006 National Health and Nutrition Examination Survey (NHANES).

The study, published in the Journal of Clinical Sleep Medicine, found that elevated homocysteine levels were only associated with short sleep duration for some populations, including women, non-Hispanic white individuals, and participants with obesity.
A total of 4,480 NHANES participants had serum homocysteine levels on record and were included in the study; of these, those with self-reported sleep duration of 7 hours had the lowest serum homocysteine levels. Those with the shortest sleep duration – 5 hours or less per night – had the highest homocysteine levels.
When participants were broken into subgroups by such factors as sex, ethnicity/race, and body mass index, the association between extremely short sleep and elevated homocysteine levels was retained for three groups: women, non-Hispanic white participants, and those with BMIs of 30 kg/m2 and higher.
“[T]his finding might suggest increased vulnerability to cardiovascular risk or other atherothrombotic events in these groups in the context of short sleep,” wrote Tien-Yu Chen, MD, of Tri-Service General Hospital, Taipei, Taiwan, and coauthors in the abstract accompanying the study.
In the NHANES questionnaire, participants were asked how much sleep they usually got, in whole hours. Answers were grouped into 5 hours or less, 6 hours, 7 hours, or 8 hours, and 9 hours or more. Serum homocysteine was measured once for each study participant.
Using multivariate linear regression, homocysteine was considered the dependent, continuous variable, and the association between sleep duration and homocysteine was assessed using three models that accounted for confounders. The first and simplest model accounted for age, sex, and race/ethnicity. The second model added BMI, several cardiometabolic laboratory values, and vitamin B6, vitamin B12, and folate levels. The third model included all previous factors and added patient characteristics and comorbidities, such as sleep disorders, mental health service use, cardiovascular disease and cancer diagnoses, and alcohol and tobacco use.
In their analysis, Dr. Chen and colleagues dichotomized homocysteine levels to above or below the 75th percentile of the log homocysteine level, which fell at 9.74 nmol/L.
After adjustment, women, but not men, had an association between short sleep and increased odds of elevated homocysteine (odds ratio, 2.691; P = .010). This association “persisted in fully adjusted models,” wrote Dr. Chen and coauthors.
For individuals with obesity (BMI of 30 or greater), the association between elevated homocysteine and extremely short sleep (5 hours or less) persisted in fully adjusted models (beta = .062; P = .039 for model 3).
When looking at ethnicity, the association between extremely short sleep and elevated homocysteine was only seen among non-Hispanic white participants; again, this association was seen after full adjustment for confounders (beta = .068; P = .032). Small sample sizes limited some of the racial/ethnic analyses, noted the investigators.
Homocysteine, explained Dr. Chen and coauthors, is associated with a variety atherogenic changes, and elevated levels are associated with increased risk for cardiovascular disease and mortality. Short sleep is also associated with increased cardiovascular risk, as is long sleep in some studies.
However, though preliminary work had shown that short sleep had an association with homocysteine levels, the relationship is unclear since that study had many potential cardiovascular confounders, said Dr. Chen and coauthors.
The association between extremely short sleep duration and cardiovascular events has been well established, with increased inflammation playing a potential role, although the reasons for the association are still being elucidated. “Because increased homocysteine levels are considered an independent risk factor for cardiovascular diseases, further studies are needed to better understand the relationships among short sleep duration, homocysteine levels, and cardiovascular events,” the investigators wrote.
Whether menstrual variations in serum homocysteine and sleep may have played a part in the significant association seen in women, but not men, was not ascertainable from the NHANES data, which introduces possible confounding, the authors noted.
Similarly, there may be ethnic differences in baseline serum homocysteine levels, said Dr. Chen and his colleagues.
The study’s strengths include the large sample size and ability to control for many demographic and individual characteristics, including comorbidities. However, sleep duration was based on self-report and did not include information about napping or sleep-wake times. Also, sleep quality was not assessed beyond a question about snoring or snorting and a question about a prior diagnosis of a sleep disorder.
“Further longitudinal investigations concerning the effect of sleep deprivation on homocysteine alteration might help provide a better understanding of the pathogenesis of cardiometabolic risk,” concluded Dr. Chen and colleagues.
One of the coauthors reported financial relationships with multiple pharmaceutical companies and UpToDate. The authors reported no external sources of funding.
SOURCE: Chen T-Y et al. J Clin Sleep Med. 2019;15(1):139-48.
Short sleep’s association with cardiovascular risk may be mediated in part by elevated homocysteine levels, suggests a new analysis of data from the 2005-2006 National Health and Nutrition Examination Survey (NHANES).
The study, published in the Journal of Clinical Sleep Medicine, found that elevated homocysteine levels were only associated with short sleep duration for some populations, including women, non-Hispanic white individuals, and participants with obesity.
A total of 4,480 NHANES participants had serum homocysteine levels on record and were included in the study; of these, those with self-reported sleep duration of 7 hours had the lowest serum homocysteine levels. Those with the shortest sleep duration – 5 hours or less per night – had the highest homocysteine levels.
When participants were broken into subgroups by such factors as sex, ethnicity/race, and body mass index, the association between extremely short sleep and elevated homocysteine levels was retained for three groups: women, non-Hispanic white participants, and those with BMIs of 30 kg/m2 and higher.
“[T]his finding might suggest increased vulnerability to cardiovascular risk or other atherothrombotic events in these groups in the context of short sleep,” wrote Tien-Yu Chen, MD, of Tri-Service General Hospital, Taipei, Taiwan, and coauthors in the abstract accompanying the study.
In the NHANES questionnaire, participants were asked how much sleep they usually got, in whole hours. Answers were grouped into 5 hours or less, 6 hours, 7 hours, or 8 hours, and 9 hours or more. Serum homocysteine was measured once for each study participant.
Using multivariate linear regression, homocysteine was considered the dependent, continuous variable, and the association between sleep duration and homocysteine was assessed using three models that accounted for confounders. The first and simplest model accounted for age, sex, and race/ethnicity. The second model added BMI, several cardiometabolic laboratory values, and vitamin B6, vitamin B12, and folate levels. The third model included all previous factors and added patient characteristics and comorbidities, such as sleep disorders, mental health service use, cardiovascular disease and cancer diagnoses, and alcohol and tobacco use.
In their analysis, Dr. Chen and colleagues dichotomized homocysteine levels to above or below the 75th percentile of the log homocysteine level, which fell at 9.74 nmol/L.
After adjustment, women, but not men, had an association between short sleep and increased odds of elevated homocysteine (odds ratio, 2.691; P = .010). This association “persisted in fully adjusted models,” wrote Dr. Chen and coauthors.
For individuals with obesity (BMI of 30 or greater), the association between elevated homocysteine and extremely short sleep (5 hours or less) persisted in fully adjusted models (beta = .062; P = .039 for model 3).
When looking at ethnicity, the association between extremely short sleep and elevated homocysteine was only seen among non-Hispanic white participants; again, this association was seen after full adjustment for confounders (beta = .068; P = .032). Small sample sizes limited some of the racial/ethnic analyses, noted the investigators.
Homocysteine, explained Dr. Chen and coauthors, is associated with a variety atherogenic changes, and elevated levels are associated with increased risk for cardiovascular disease and mortality. Short sleep is also associated with increased cardiovascular risk, as is long sleep in some studies.
However, though preliminary work had shown that short sleep had an association with homocysteine levels, the relationship is unclear since that study had many potential cardiovascular confounders, said Dr. Chen and coauthors.
The association between extremely short sleep duration and cardiovascular events has been well established, with increased inflammation playing a potential role, although the reasons for the association are still being elucidated. “Because increased homocysteine levels are considered an independent risk factor for cardiovascular diseases, further studies are needed to better understand the relationships among short sleep duration, homocysteine levels, and cardiovascular events,” the investigators wrote.
Whether menstrual variations in serum homocysteine and sleep may have played a part in the significant association seen in women, but not men, was not ascertainable from the NHANES data, which introduces possible confounding, the authors noted.
Similarly, there may be ethnic differences in baseline serum homocysteine levels, said Dr. Chen and his colleagues.
The study’s strengths include the large sample size and ability to control for many demographic and individual characteristics, including comorbidities. However, sleep duration was based on self-report and did not include information about napping or sleep-wake times. Also, sleep quality was not assessed beyond a question about snoring or snorting and a question about a prior diagnosis of a sleep disorder.
“Further longitudinal investigations concerning the effect of sleep deprivation on homocysteine alteration might help provide a better understanding of the pathogenesis of cardiometabolic risk,” concluded Dr. Chen and colleagues.
One of the coauthors reported financial relationships with multiple pharmaceutical companies and UpToDate. The authors reported no external sources of funding.
SOURCE: Chen T-Y et al. J Clin Sleep Med. 2019;15(1):139-48.
Short sleep’s association with cardiovascular risk may be mediated in part by elevated homocysteine levels, suggests a new analysis of data from the 2005-2006 National Health and Nutrition Examination Survey (NHANES).
The study, published in the Journal of Clinical Sleep Medicine, found that elevated homocysteine levels were only associated with short sleep duration for some populations, including women, non-Hispanic white individuals, and participants with obesity.
A total of 4,480 NHANES participants had serum homocysteine levels on record and were included in the study; of these, those with self-reported sleep duration of 7 hours had the lowest serum homocysteine levels. Those with the shortest sleep duration – 5 hours or less per night – had the highest homocysteine levels.
When participants were broken into subgroups by such factors as sex, ethnicity/race, and body mass index, the association between extremely short sleep and elevated homocysteine levels was retained for three groups: women, non-Hispanic white participants, and those with BMIs of 30 kg/m2 and higher.
“[T]his finding might suggest increased vulnerability to cardiovascular risk or other atherothrombotic events in these groups in the context of short sleep,” wrote Tien-Yu Chen, MD, of Tri-Service General Hospital, Taipei, Taiwan, and coauthors in the abstract accompanying the study.
In the NHANES questionnaire, participants were asked how much sleep they usually got, in whole hours. Answers were grouped into 5 hours or less, 6 hours, 7 hours, or 8 hours, and 9 hours or more. Serum homocysteine was measured once for each study participant.
Using multivariate linear regression, homocysteine was considered the dependent, continuous variable, and the association between sleep duration and homocysteine was assessed using three models that accounted for confounders. The first and simplest model accounted for age, sex, and race/ethnicity. The second model added BMI, several cardiometabolic laboratory values, and vitamin B6, vitamin B12, and folate levels. The third model included all previous factors and added patient characteristics and comorbidities, such as sleep disorders, mental health service use, cardiovascular disease and cancer diagnoses, and alcohol and tobacco use.
In their analysis, Dr. Chen and colleagues dichotomized homocysteine levels to above or below the 75th percentile of the log homocysteine level, which fell at 9.74 nmol/L.
After adjustment, women, but not men, had an association between short sleep and increased odds of elevated homocysteine (odds ratio, 2.691; P = .010). This association “persisted in fully adjusted models,” wrote Dr. Chen and coauthors.
For individuals with obesity (BMI of 30 or greater), the association between elevated homocysteine and extremely short sleep (5 hours or less) persisted in fully adjusted models (beta = .062; P = .039 for model 3).
When looking at ethnicity, the association between extremely short sleep and elevated homocysteine was only seen among non-Hispanic white participants; again, this association was seen after full adjustment for confounders (beta = .068; P = .032). Small sample sizes limited some of the racial/ethnic analyses, noted the investigators.
Homocysteine, explained Dr. Chen and coauthors, is associated with a variety atherogenic changes, and elevated levels are associated with increased risk for cardiovascular disease and mortality. Short sleep is also associated with increased cardiovascular risk, as is long sleep in some studies.
However, though preliminary work had shown that short sleep had an association with homocysteine levels, the relationship is unclear since that study had many potential cardiovascular confounders, said Dr. Chen and coauthors.
The association between extremely short sleep duration and cardiovascular events has been well established, with increased inflammation playing a potential role, although the reasons for the association are still being elucidated. “Because increased homocysteine levels are considered an independent risk factor for cardiovascular diseases, further studies are needed to better understand the relationships among short sleep duration, homocysteine levels, and cardiovascular events,” the investigators wrote.
Whether menstrual variations in serum homocysteine and sleep may have played a part in the significant association seen in women, but not men, was not ascertainable from the NHANES data, which introduces possible confounding, the authors noted.
Similarly, there may be ethnic differences in baseline serum homocysteine levels, said Dr. Chen and his colleagues.
The study’s strengths include the large sample size and ability to control for many demographic and individual characteristics, including comorbidities. However, sleep duration was based on self-report and did not include information about napping or sleep-wake times. Also, sleep quality was not assessed beyond a question about snoring or snorting and a question about a prior diagnosis of a sleep disorder.
“Further longitudinal investigations concerning the effect of sleep deprivation on homocysteine alteration might help provide a better understanding of the pathogenesis of cardiometabolic risk,” concluded Dr. Chen and colleagues.
One of the coauthors reported financial relationships with multiple pharmaceutical companies and UpToDate. The authors reported no external sources of funding.
SOURCE: Chen T-Y et al. J Clin Sleep Med. 2019;15(1):139-48.
FROM THE JOURNAL OF CLINICAL SLEEP MEDICINE
Key clinical point: Extreme short sleep was associated with high homocysteine levels.
Major finding: In women, extreme short sleep was associated with an odds ratio of 2.691 for elevated homocysteine.
Study details: Analysis of data from 4,480 NHANES participants.
Disclosures: One coauthor reported relationships with multiple pharmaceutical companies and UpToDate. The authors reported no outside sources of funding.
Source: Chen T-Y et al. J Clin Sleep Med. 2019;15(1):139-48.