User login
Bimekizumab superior to adalimumab in head-to-head psoriasis study
for treatment of moderate to severe plaque psoriasis in the head-to-head, phase 3 BE SURE trial, Jerry Bagel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“Results demonstrated that bimekizumab was superior to adalimumab over 16 weeks of treatment in terms of the speed, depth, and durability of skin clearance,” reported Dr. Bagel, a dermatologist at the Psoriasis Center of Central New Jersey, East Windsor.
The Food and Drug Administration is now reviewing UCB’s application for marketing approval of bimekizumab for treatment of moderate to severe psoriasis in adults.
BE SURE was a 478-patient, double-blind, phase 3 trial in which patients were randomized to one of three regimens: 320 mg of bimekizumab every 4 weeks; the tumor necrosis factor blocker adalimumab (Humira) at 40 mg every 2 weeks for 24 weeks, followed by a switch to bimekizumab at 320 mg every 4 weeks; or 320 mg of bimekizumab every 4 weeks for 16 weeks, then ratcheting back to dosing every 8 weeks. The trial concluded at week 56, Dr. Bagel explained at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The two coprimary endpoints were the 16-week rates of a 90% improvement from baseline in Psoriasis Area and Severity Index score, or PASI 90 response, and an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear. Bimekizumab every 4 weeks bested adalimumab on both endpoints, with a PASI 90 rate of 86.2%, compared with 47.2%, and a IGA 0/1 rate of 85.3% versus 57.2%. The 16-week PASI 100 response rate was 60.8% with bimekizumab and 23.9% with adalimumab.
The response to bimekizumab was notably fast: already by week 4, the PASI 75 rate was 76.4%, compared with 31.4% with adalimumab. And once patients switched from adalimumab to bimekizumab at week 24, their response rates shot up rapidly. Bimekizumab was equally effective whether dosed at 320 mg every 4 weeks or at maintenance dosing every 8 weeks, such that at week 56 patients in all three study arms had PASI 90 rates of 82%-84%.
The most frequent treatment-emergent adverse events associated with bimekizumab were oral candidiasis, nasopharyngitis, and upper respiratory tract infection. The oral candidiasis, which occurred in 13.2% of patients on bimekizumab every 4 weeks, was mainly mild to moderate, localized, and in no instance led to discontinuation of therapy, according to Dr. Bagel.
“Very impressive data,” commented session comoderator Linda Stein Gold, MD. “This study shows some data that’s potentially unprecedented. Bimekizumab was superior to one of the drugs that we know, we’ve used, and know is very, very effective.”
“Note the speed of this drug,” added comoderator Bruce E. Strober, MD, PhD, of Yale University, New Haven, Conn., and Central Connecticut Dermatology, Cromwell, Conn. “It achieved at week 4 the efficacy that it took adalimumab until week 16 to reach. So it is a very fast drug. Bimekizumab will be the fastest drug you’ve ever, ever worked with.”
“You’ll see in the bimekizumab studies about a fivefold increased frequency of oral candidiasis relative to our more legacy IL-17 inhibitors, such as ixekizumab, secukinumab, and brodalumab. I think that means approximately one in five or one in six patients will have some form of candidiasis when you treat them with bimekizumab,” he said. Therefore, he added, “in some patients you’ll have to manage oral candidiasis. Most affected patients don’t leave the studies, so it’s manageable, but you’ll have to become something of an authority on how to treat with, for example, oral antifungal swish-and-swallow, swish-and-spit, or oral fluconazole. And some of these patients will have recurrent infections.”
It’s a prospect that doesn’t concern Dr. Stein Gold. “This is a side effect that we can treat. We can see it, we’re comfortable with it, and it’s certainly something we can get a handle on,” said Dr. Stein Gold, director of dermatology clinical research at the Henry Ford Health System in Detroit.
BE SURE was funded by UCB. Dr. Bagel reported serving as a speaker for, consultant to, and paid investigator for AbbVie, Celgene, Eli Lilly, Leo Pharma, Novartis, and Ortho Pharmaceuticals. Dr. Stein Gold and Dr. Strober reported having financial relationships with numerous pharmaceutical companies.
MedscapeLIVE! and this news organization are owned by the same parent company.
for treatment of moderate to severe plaque psoriasis in the head-to-head, phase 3 BE SURE trial, Jerry Bagel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“Results demonstrated that bimekizumab was superior to adalimumab over 16 weeks of treatment in terms of the speed, depth, and durability of skin clearance,” reported Dr. Bagel, a dermatologist at the Psoriasis Center of Central New Jersey, East Windsor.
The Food and Drug Administration is now reviewing UCB’s application for marketing approval of bimekizumab for treatment of moderate to severe psoriasis in adults.
BE SURE was a 478-patient, double-blind, phase 3 trial in which patients were randomized to one of three regimens: 320 mg of bimekizumab every 4 weeks; the tumor necrosis factor blocker adalimumab (Humira) at 40 mg every 2 weeks for 24 weeks, followed by a switch to bimekizumab at 320 mg every 4 weeks; or 320 mg of bimekizumab every 4 weeks for 16 weeks, then ratcheting back to dosing every 8 weeks. The trial concluded at week 56, Dr. Bagel explained at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The two coprimary endpoints were the 16-week rates of a 90% improvement from baseline in Psoriasis Area and Severity Index score, or PASI 90 response, and an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear. Bimekizumab every 4 weeks bested adalimumab on both endpoints, with a PASI 90 rate of 86.2%, compared with 47.2%, and a IGA 0/1 rate of 85.3% versus 57.2%. The 16-week PASI 100 response rate was 60.8% with bimekizumab and 23.9% with adalimumab.
The response to bimekizumab was notably fast: already by week 4, the PASI 75 rate was 76.4%, compared with 31.4% with adalimumab. And once patients switched from adalimumab to bimekizumab at week 24, their response rates shot up rapidly. Bimekizumab was equally effective whether dosed at 320 mg every 4 weeks or at maintenance dosing every 8 weeks, such that at week 56 patients in all three study arms had PASI 90 rates of 82%-84%.
The most frequent treatment-emergent adverse events associated with bimekizumab were oral candidiasis, nasopharyngitis, and upper respiratory tract infection. The oral candidiasis, which occurred in 13.2% of patients on bimekizumab every 4 weeks, was mainly mild to moderate, localized, and in no instance led to discontinuation of therapy, according to Dr. Bagel.
“Very impressive data,” commented session comoderator Linda Stein Gold, MD. “This study shows some data that’s potentially unprecedented. Bimekizumab was superior to one of the drugs that we know, we’ve used, and know is very, very effective.”
“Note the speed of this drug,” added comoderator Bruce E. Strober, MD, PhD, of Yale University, New Haven, Conn., and Central Connecticut Dermatology, Cromwell, Conn. “It achieved at week 4 the efficacy that it took adalimumab until week 16 to reach. So it is a very fast drug. Bimekizumab will be the fastest drug you’ve ever, ever worked with.”
“You’ll see in the bimekizumab studies about a fivefold increased frequency of oral candidiasis relative to our more legacy IL-17 inhibitors, such as ixekizumab, secukinumab, and brodalumab. I think that means approximately one in five or one in six patients will have some form of candidiasis when you treat them with bimekizumab,” he said. Therefore, he added, “in some patients you’ll have to manage oral candidiasis. Most affected patients don’t leave the studies, so it’s manageable, but you’ll have to become something of an authority on how to treat with, for example, oral antifungal swish-and-swallow, swish-and-spit, or oral fluconazole. And some of these patients will have recurrent infections.”
It’s a prospect that doesn’t concern Dr. Stein Gold. “This is a side effect that we can treat. We can see it, we’re comfortable with it, and it’s certainly something we can get a handle on,” said Dr. Stein Gold, director of dermatology clinical research at the Henry Ford Health System in Detroit.
BE SURE was funded by UCB. Dr. Bagel reported serving as a speaker for, consultant to, and paid investigator for AbbVie, Celgene, Eli Lilly, Leo Pharma, Novartis, and Ortho Pharmaceuticals. Dr. Stein Gold and Dr. Strober reported having financial relationships with numerous pharmaceutical companies.
MedscapeLIVE! and this news organization are owned by the same parent company.
for treatment of moderate to severe plaque psoriasis in the head-to-head, phase 3 BE SURE trial, Jerry Bagel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“Results demonstrated that bimekizumab was superior to adalimumab over 16 weeks of treatment in terms of the speed, depth, and durability of skin clearance,” reported Dr. Bagel, a dermatologist at the Psoriasis Center of Central New Jersey, East Windsor.
The Food and Drug Administration is now reviewing UCB’s application for marketing approval of bimekizumab for treatment of moderate to severe psoriasis in adults.
BE SURE was a 478-patient, double-blind, phase 3 trial in which patients were randomized to one of three regimens: 320 mg of bimekizumab every 4 weeks; the tumor necrosis factor blocker adalimumab (Humira) at 40 mg every 2 weeks for 24 weeks, followed by a switch to bimekizumab at 320 mg every 4 weeks; or 320 mg of bimekizumab every 4 weeks for 16 weeks, then ratcheting back to dosing every 8 weeks. The trial concluded at week 56, Dr. Bagel explained at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The two coprimary endpoints were the 16-week rates of a 90% improvement from baseline in Psoriasis Area and Severity Index score, or PASI 90 response, and an Investigator’s Global Assessment (IGA) score of 0 or 1, meaning clear or almost clear. Bimekizumab every 4 weeks bested adalimumab on both endpoints, with a PASI 90 rate of 86.2%, compared with 47.2%, and a IGA 0/1 rate of 85.3% versus 57.2%. The 16-week PASI 100 response rate was 60.8% with bimekizumab and 23.9% with adalimumab.
The response to bimekizumab was notably fast: already by week 4, the PASI 75 rate was 76.4%, compared with 31.4% with adalimumab. And once patients switched from adalimumab to bimekizumab at week 24, their response rates shot up rapidly. Bimekizumab was equally effective whether dosed at 320 mg every 4 weeks or at maintenance dosing every 8 weeks, such that at week 56 patients in all three study arms had PASI 90 rates of 82%-84%.
The most frequent treatment-emergent adverse events associated with bimekizumab were oral candidiasis, nasopharyngitis, and upper respiratory tract infection. The oral candidiasis, which occurred in 13.2% of patients on bimekizumab every 4 weeks, was mainly mild to moderate, localized, and in no instance led to discontinuation of therapy, according to Dr. Bagel.
“Very impressive data,” commented session comoderator Linda Stein Gold, MD. “This study shows some data that’s potentially unprecedented. Bimekizumab was superior to one of the drugs that we know, we’ve used, and know is very, very effective.”
“Note the speed of this drug,” added comoderator Bruce E. Strober, MD, PhD, of Yale University, New Haven, Conn., and Central Connecticut Dermatology, Cromwell, Conn. “It achieved at week 4 the efficacy that it took adalimumab until week 16 to reach. So it is a very fast drug. Bimekizumab will be the fastest drug you’ve ever, ever worked with.”
“You’ll see in the bimekizumab studies about a fivefold increased frequency of oral candidiasis relative to our more legacy IL-17 inhibitors, such as ixekizumab, secukinumab, and brodalumab. I think that means approximately one in five or one in six patients will have some form of candidiasis when you treat them with bimekizumab,” he said. Therefore, he added, “in some patients you’ll have to manage oral candidiasis. Most affected patients don’t leave the studies, so it’s manageable, but you’ll have to become something of an authority on how to treat with, for example, oral antifungal swish-and-swallow, swish-and-spit, or oral fluconazole. And some of these patients will have recurrent infections.”
It’s a prospect that doesn’t concern Dr. Stein Gold. “This is a side effect that we can treat. We can see it, we’re comfortable with it, and it’s certainly something we can get a handle on,” said Dr. Stein Gold, director of dermatology clinical research at the Henry Ford Health System in Detroit.
BE SURE was funded by UCB. Dr. Bagel reported serving as a speaker for, consultant to, and paid investigator for AbbVie, Celgene, Eli Lilly, Leo Pharma, Novartis, and Ortho Pharmaceuticals. Dr. Stein Gold and Dr. Strober reported having financial relationships with numerous pharmaceutical companies.
MedscapeLIVE! and this news organization are owned by the same parent company.
FROM INNOVATIONS IN DERMATOLOGY
Clinical Edge Commentary: CML April 2021

ENESTfreedom is an important study that set the current guidelines for TFR. The trial included 190 patients treated with front line nilotinib at 300mg twice daily for two years that had achieved an MR4.5, followed by one year of consolidation maintaining deep molecular response of at least MR4 were allowed to discontinued therapy. A recent 5 year update of the trial showed that 81/190 patients (42.6%) still remained on TFR (MR4) with 76 (40.0%) in MR4.5. From the patients who lost major molecular response (MMR) and were re-treated, 90/91 patients entering this phase (98.9%) regained MMR and 84/91 patients (92.3%) regained MR4.5. More important, no disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously, including a declined incidence of AEs at 96 weeks of the TFR follow up (with low incidence CV AEs), while an expected increase on those in the treatment re-initiation phase. As previously has been described in other trials, Low Sokal risk score, BCR-ABL1 IS levels at 48 weeks of TFR and stable MR4.5 response for the first year of TFR were associated with higher TFR rates.
When looking at patients who can be candidates for TFR, it is important to take in consideration the likelihood of successful discontinuation and educate patients on certain factors that may contribute to good outcomes. The Australian CML group had previously reported the importance of the initial decline rate of BCR-ABL1 measured as halving time as an important factor associated to deep molecular response. The same group, in a very interesting publication, reported the impact of halving time as a strong predictor of sustained TFR post-tyrosine kinase inhibitor (TKI) cessation in CML patients treated with front line TKI therapies.
Out of 115 patients who attempted TFR and had ≥12 months of follow-up, 55% sustained TFR, defined as remaining in major molecular response off TKI therapy for 12 months, similar percentage seen in other studies. However, when the time taken for the BCR-ABL1 value to halve was applied, it became the strongest independent predictor of sustained TFR with 80% in patients with a halving time of <9.35 days (first quartile) compared with only 4% if the halving time was >21.85 days (last quartile) (P < .001). The e14a2 BCR-ABL1 transcript type and duration of TKI exposure before attempting TFR were also independent predictors of sustained TFR. However, the BCR-ABL1 value measured at 3 months of TKI was not an independent predictor of sustained TFR.
ENESTfreedom is an important study that set the current guidelines for TFR. The trial included 190 patients treated with front line nilotinib at 300mg twice daily for two years that had achieved an MR4.5, followed by one year of consolidation maintaining deep molecular response of at least MR4 were allowed to discontinued therapy. A recent 5 year update of the trial showed that 81/190 patients (42.6%) still remained on TFR (MR4) with 76 (40.0%) in MR4.5. From the patients who lost major molecular response (MMR) and were re-treated, 90/91 patients entering this phase (98.9%) regained MMR and 84/91 patients (92.3%) regained MR4.5. More important, no disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously, including a declined incidence of AEs at 96 weeks of the TFR follow up (with low incidence CV AEs), while an expected increase on those in the treatment re-initiation phase. As previously has been described in other trials, Low Sokal risk score, BCR-ABL1 IS levels at 48 weeks of TFR and stable MR4.5 response for the first year of TFR were associated with higher TFR rates.
When looking at patients who can be candidates for TFR, it is important to take in consideration the likelihood of successful discontinuation and educate patients on certain factors that may contribute to good outcomes. The Australian CML group had previously reported the importance of the initial decline rate of BCR-ABL1 measured as halving time as an important factor associated to deep molecular response. The same group, in a very interesting publication, reported the impact of halving time as a strong predictor of sustained TFR post-tyrosine kinase inhibitor (TKI) cessation in CML patients treated with front line TKI therapies.
Out of 115 patients who attempted TFR and had ≥12 months of follow-up, 55% sustained TFR, defined as remaining in major molecular response off TKI therapy for 12 months, similar percentage seen in other studies. However, when the time taken for the BCR-ABL1 value to halve was applied, it became the strongest independent predictor of sustained TFR with 80% in patients with a halving time of <9.35 days (first quartile) compared with only 4% if the halving time was >21.85 days (last quartile) (P < .001). The e14a2 BCR-ABL1 transcript type and duration of TKI exposure before attempting TFR were also independent predictors of sustained TFR. However, the BCR-ABL1 value measured at 3 months of TKI was not an independent predictor of sustained TFR.
ENESTfreedom is an important study that set the current guidelines for TFR. The trial included 190 patients treated with front line nilotinib at 300mg twice daily for two years that had achieved an MR4.5, followed by one year of consolidation maintaining deep molecular response of at least MR4 were allowed to discontinued therapy. A recent 5 year update of the trial showed that 81/190 patients (42.6%) still remained on TFR (MR4) with 76 (40.0%) in MR4.5. From the patients who lost major molecular response (MMR) and were re-treated, 90/91 patients entering this phase (98.9%) regained MMR and 84/91 patients (92.3%) regained MR4.5. More important, no disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously, including a declined incidence of AEs at 96 weeks of the TFR follow up (with low incidence CV AEs), while an expected increase on those in the treatment re-initiation phase. As previously has been described in other trials, Low Sokal risk score, BCR-ABL1 IS levels at 48 weeks of TFR and stable MR4.5 response for the first year of TFR were associated with higher TFR rates.
When looking at patients who can be candidates for TFR, it is important to take in consideration the likelihood of successful discontinuation and educate patients on certain factors that may contribute to good outcomes. The Australian CML group had previously reported the importance of the initial decline rate of BCR-ABL1 measured as halving time as an important factor associated to deep molecular response. The same group, in a very interesting publication, reported the impact of halving time as a strong predictor of sustained TFR post-tyrosine kinase inhibitor (TKI) cessation in CML patients treated with front line TKI therapies.
Out of 115 patients who attempted TFR and had ≥12 months of follow-up, 55% sustained TFR, defined as remaining in major molecular response off TKI therapy for 12 months, similar percentage seen in other studies. However, when the time taken for the BCR-ABL1 value to halve was applied, it became the strongest independent predictor of sustained TFR with 80% in patients with a halving time of <9.35 days (first quartile) compared with only 4% if the halving time was >21.85 days (last quartile) (P < .001). The e14a2 BCR-ABL1 transcript type and duration of TKI exposure before attempting TFR were also independent predictors of sustained TFR. However, the BCR-ABL1 value measured at 3 months of TKI was not an independent predictor of sustained TFR.
Bosutinib is effective, relative safe in elderly CML patients resistant/intolerant to prior TKIs
Key clinical point: Bosutinib was effective and safe as a second or subsequent line of treatment in a real-life cohort of elderly patients with chronic-phase chronic myeloid leukemia (CML-CP) and multiple baseline comorbidities who were resistant or intolerant to previous tyrosine kinase inhibitors (TKIs).
Major finding: Overall, rates of cytogenic response, molecular response, 3-year event-free survival, and 3-year overall survival were 81.2%, 66.6%, 60.9%, and 86.4%, respectively. Grade 3/4 hematological and extra-hematological toxicities were reported in 6.9% and 18.8% of patients, respectively, with 11.9% of patients permanently discontinuing bosutinib because of toxicity.
Study details: Findings are from a retrospective analysis of 101 elderly (age, older than 65 years) patients with CML-CP treated with bosutinib in second or subsequent line. Patients switched to bosutinib because of intolerance (n=46) or resistance (n=55) to previous TKI therapies.
Disclosures: This study did not receive any type of funding. Some investigators including the lead author reported consulting for or receiving honoraria from various pharmaceutical companies.
Source: Latagliata R et al. Hematol Oncol. 2021 Feb 22. doi: 10.1002/hon.2851.
Key clinical point: Bosutinib was effective and safe as a second or subsequent line of treatment in a real-life cohort of elderly patients with chronic-phase chronic myeloid leukemia (CML-CP) and multiple baseline comorbidities who were resistant or intolerant to previous tyrosine kinase inhibitors (TKIs).
Major finding: Overall, rates of cytogenic response, molecular response, 3-year event-free survival, and 3-year overall survival were 81.2%, 66.6%, 60.9%, and 86.4%, respectively. Grade 3/4 hematological and extra-hematological toxicities were reported in 6.9% and 18.8% of patients, respectively, with 11.9% of patients permanently discontinuing bosutinib because of toxicity.
Study details: Findings are from a retrospective analysis of 101 elderly (age, older than 65 years) patients with CML-CP treated with bosutinib in second or subsequent line. Patients switched to bosutinib because of intolerance (n=46) or resistance (n=55) to previous TKI therapies.
Disclosures: This study did not receive any type of funding. Some investigators including the lead author reported consulting for or receiving honoraria from various pharmaceutical companies.
Source: Latagliata R et al. Hematol Oncol. 2021 Feb 22. doi: 10.1002/hon.2851.
Key clinical point: Bosutinib was effective and safe as a second or subsequent line of treatment in a real-life cohort of elderly patients with chronic-phase chronic myeloid leukemia (CML-CP) and multiple baseline comorbidities who were resistant or intolerant to previous tyrosine kinase inhibitors (TKIs).
Major finding: Overall, rates of cytogenic response, molecular response, 3-year event-free survival, and 3-year overall survival were 81.2%, 66.6%, 60.9%, and 86.4%, respectively. Grade 3/4 hematological and extra-hematological toxicities were reported in 6.9% and 18.8% of patients, respectively, with 11.9% of patients permanently discontinuing bosutinib because of toxicity.
Study details: Findings are from a retrospective analysis of 101 elderly (age, older than 65 years) patients with CML-CP treated with bosutinib in second or subsequent line. Patients switched to bosutinib because of intolerance (n=46) or resistance (n=55) to previous TKI therapies.
Disclosures: This study did not receive any type of funding. Some investigators including the lead author reported consulting for or receiving honoraria from various pharmaceutical companies.
Source: Latagliata R et al. Hematol Oncol. 2021 Feb 22. doi: 10.1002/hon.2851.
Hedgehog inhibitor alternative dosing advantageous for BCC
in a successful effort to maintain efficacy while reducing treatment discontinuation caused by unacceptable side effects, Vishal Patel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“It’s the tolerability issues that make these drugs very difficult to prescribe and use regularly. What we’ve seen in the last few years is that a lot of alternative dosing regimens have been published that have been both effective at treating the tumor and keeping the tumor clear and at bay while lowering the side-effect profile,” explained Dr. Patel, a Mohs surgeon and director of the cutaneous oncology program at the George Washington University Cancer Center in Washington, D.C.
Product labeling for the two available hedgehog pathway inhibitors, vismodegib (Erivedge) and sonidegib (Odomzo), calls for once-daily therapy until disease progression or unacceptable toxicity. Studies show that, when used in this way, these agents achieve objective response rates in the 40% range for patients with locally advanced BCC and 15%-33% for those with metastatic BCC.
“The critical thing in these patients is not that the drugs work – although they can work in quite remarkable ways – but rather it’s that nearly all patients experience at least one side effect. And grade 3 or 4 adverse effects that can lead to cessation of drug occur in about 25% of patients,” he said at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The classic side effects of the hedgehog pathway inhibitors are muscle spasms, hair loss, fatigue, loss of taste, diarrhea, and weight loss.
Among the alternative dosing regimens that have been published with good results, mostly in single-center retrospective case series, are a weekdays-on/weekends-off strategy at the Cleveland Clinic and an Italian approach entailing an initial 3-4 months of daily therapy followed by a switch to alternate-day therapy.
But Dr. Patel favors a different off-label regimen in lieu of Food and Drug Administration–recommended daily dosing indefinitely. It takes advantage of the fact that most patients don’t begin to get the classic side effects until about the 3-month mark.
“What we’ve begun to recommend as a much better option for patients who need to be on the drug potentially forever is that the drug is dosed daily for 3 months to shrink the tumor and get the optimal effect, and then at that point we taper the dose down to every other day, then every third day, or even up to a week as long as the tumor continues to stay at bay. If there’s any sign of recurrence or a scouting biopsy shows tumor, we reinstitute the daily medicine,” the dermatologist said.
This strategy requires careful monitoring for emergence of the typical side effects. Also, an important caveat regarding sonidegib is that it shouldn’t be given concomitantly with medications that are moderate or strong inhibitors of CYP3A, so it’s essential to get a complete medical history when giving this drug, Dr. Patel noted.
He reported having no financial conflicts regarding his presentation.
MedscapeLIVE! and this news organization are owned by the same parent company.
in a successful effort to maintain efficacy while reducing treatment discontinuation caused by unacceptable side effects, Vishal Patel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“It’s the tolerability issues that make these drugs very difficult to prescribe and use regularly. What we’ve seen in the last few years is that a lot of alternative dosing regimens have been published that have been both effective at treating the tumor and keeping the tumor clear and at bay while lowering the side-effect profile,” explained Dr. Patel, a Mohs surgeon and director of the cutaneous oncology program at the George Washington University Cancer Center in Washington, D.C.
Product labeling for the two available hedgehog pathway inhibitors, vismodegib (Erivedge) and sonidegib (Odomzo), calls for once-daily therapy until disease progression or unacceptable toxicity. Studies show that, when used in this way, these agents achieve objective response rates in the 40% range for patients with locally advanced BCC and 15%-33% for those with metastatic BCC.
“The critical thing in these patients is not that the drugs work – although they can work in quite remarkable ways – but rather it’s that nearly all patients experience at least one side effect. And grade 3 or 4 adverse effects that can lead to cessation of drug occur in about 25% of patients,” he said at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The classic side effects of the hedgehog pathway inhibitors are muscle spasms, hair loss, fatigue, loss of taste, diarrhea, and weight loss.
Among the alternative dosing regimens that have been published with good results, mostly in single-center retrospective case series, are a weekdays-on/weekends-off strategy at the Cleveland Clinic and an Italian approach entailing an initial 3-4 months of daily therapy followed by a switch to alternate-day therapy.
But Dr. Patel favors a different off-label regimen in lieu of Food and Drug Administration–recommended daily dosing indefinitely. It takes advantage of the fact that most patients don’t begin to get the classic side effects until about the 3-month mark.
“What we’ve begun to recommend as a much better option for patients who need to be on the drug potentially forever is that the drug is dosed daily for 3 months to shrink the tumor and get the optimal effect, and then at that point we taper the dose down to every other day, then every third day, or even up to a week as long as the tumor continues to stay at bay. If there’s any sign of recurrence or a scouting biopsy shows tumor, we reinstitute the daily medicine,” the dermatologist said.
This strategy requires careful monitoring for emergence of the typical side effects. Also, an important caveat regarding sonidegib is that it shouldn’t be given concomitantly with medications that are moderate or strong inhibitors of CYP3A, so it’s essential to get a complete medical history when giving this drug, Dr. Patel noted.
He reported having no financial conflicts regarding his presentation.
MedscapeLIVE! and this news organization are owned by the same parent company.
in a successful effort to maintain efficacy while reducing treatment discontinuation caused by unacceptable side effects, Vishal Patel, MD, said at Innovations in Dermatology: Virtual Spring Conference 2021.
“It’s the tolerability issues that make these drugs very difficult to prescribe and use regularly. What we’ve seen in the last few years is that a lot of alternative dosing regimens have been published that have been both effective at treating the tumor and keeping the tumor clear and at bay while lowering the side-effect profile,” explained Dr. Patel, a Mohs surgeon and director of the cutaneous oncology program at the George Washington University Cancer Center in Washington, D.C.
Product labeling for the two available hedgehog pathway inhibitors, vismodegib (Erivedge) and sonidegib (Odomzo), calls for once-daily therapy until disease progression or unacceptable toxicity. Studies show that, when used in this way, these agents achieve objective response rates in the 40% range for patients with locally advanced BCC and 15%-33% for those with metastatic BCC.
“The critical thing in these patients is not that the drugs work – although they can work in quite remarkable ways – but rather it’s that nearly all patients experience at least one side effect. And grade 3 or 4 adverse effects that can lead to cessation of drug occur in about 25% of patients,” he said at the conference sponsored by MedscapeLIVE! and the producers of the Hawaii Dermatology Seminar and Caribbean Dermatology Symposium.
The classic side effects of the hedgehog pathway inhibitors are muscle spasms, hair loss, fatigue, loss of taste, diarrhea, and weight loss.
Among the alternative dosing regimens that have been published with good results, mostly in single-center retrospective case series, are a weekdays-on/weekends-off strategy at the Cleveland Clinic and an Italian approach entailing an initial 3-4 months of daily therapy followed by a switch to alternate-day therapy.
But Dr. Patel favors a different off-label regimen in lieu of Food and Drug Administration–recommended daily dosing indefinitely. It takes advantage of the fact that most patients don’t begin to get the classic side effects until about the 3-month mark.
“What we’ve begun to recommend as a much better option for patients who need to be on the drug potentially forever is that the drug is dosed daily for 3 months to shrink the tumor and get the optimal effect, and then at that point we taper the dose down to every other day, then every third day, or even up to a week as long as the tumor continues to stay at bay. If there’s any sign of recurrence or a scouting biopsy shows tumor, we reinstitute the daily medicine,” the dermatologist said.
This strategy requires careful monitoring for emergence of the typical side effects. Also, an important caveat regarding sonidegib is that it shouldn’t be given concomitantly with medications that are moderate or strong inhibitors of CYP3A, so it’s essential to get a complete medical history when giving this drug, Dr. Patel noted.
He reported having no financial conflicts regarding his presentation.
MedscapeLIVE! and this news organization are owned by the same parent company.
FROM INNOVATIONS IN DERMATOLOGY
Quality of life and health state utility in patients with CML in real-life setting
Key clinical point: Even after effective treatment with tyrosine kinase inhibitors, patients with chronic myeloid leukemia (CML) had notably altered global quality of life (QoL) and health utility scores.
Major finding: Compared with the general population, patients with CML had notably affected QoL in terms of social, role, and cognitive functioning with a mean difference of −16.0, −13.1, and −11.7, respectively. The utility score had a deviation from the reference norm of −0.15 on an average (standard deviation, 0.25) compared with the general population.
Study details: Findings are from a prospective web-based survey that assessed QoL and health utility scores in 383 patients with CML (92% in chronic-phase) in France.
Disclosures: This study was funded by the French National Agency for Research and the French Institute for Public Health Research. The authors declared no conflicts of interest.
Source: Foulon S et al. Qual Life Res. 2021 Mar 2. doi: 10.1007/s11136-021-02794-5.
Key clinical point: Even after effective treatment with tyrosine kinase inhibitors, patients with chronic myeloid leukemia (CML) had notably altered global quality of life (QoL) and health utility scores.
Major finding: Compared with the general population, patients with CML had notably affected QoL in terms of social, role, and cognitive functioning with a mean difference of −16.0, −13.1, and −11.7, respectively. The utility score had a deviation from the reference norm of −0.15 on an average (standard deviation, 0.25) compared with the general population.
Study details: Findings are from a prospective web-based survey that assessed QoL and health utility scores in 383 patients with CML (92% in chronic-phase) in France.
Disclosures: This study was funded by the French National Agency for Research and the French Institute for Public Health Research. The authors declared no conflicts of interest.
Source: Foulon S et al. Qual Life Res. 2021 Mar 2. doi: 10.1007/s11136-021-02794-5.
Key clinical point: Even after effective treatment with tyrosine kinase inhibitors, patients with chronic myeloid leukemia (CML) had notably altered global quality of life (QoL) and health utility scores.
Major finding: Compared with the general population, patients with CML had notably affected QoL in terms of social, role, and cognitive functioning with a mean difference of −16.0, −13.1, and −11.7, respectively. The utility score had a deviation from the reference norm of −0.15 on an average (standard deviation, 0.25) compared with the general population.
Study details: Findings are from a prospective web-based survey that assessed QoL and health utility scores in 383 patients with CML (92% in chronic-phase) in France.
Disclosures: This study was funded by the French National Agency for Research and the French Institute for Public Health Research. The authors declared no conflicts of interest.
Source: Foulon S et al. Qual Life Res. 2021 Mar 2. doi: 10.1007/s11136-021-02794-5.
Dasatinib/nivolumab combo safe but shows no meaningful activity in previously treated CML
Key clinical point: Combination of dasatinib and nivolumab was safe but did not show any clinical activity in patients with difficult-to-treat chronic myeloid leukemia (CML) in chronic phase (CP) or accelerated phase (AP) who received at least 2 prior tyrosine kinase inhibitors (TKIs).
Major finding: No drug limiting toxicities were observed even with the maximum dose of both drugs. The most common grade 3 or higher adverse event (AE) of any cause was diarrhea (13%), of which only 3% were considered drug related. Serious AEs occurred in 48% of patients; however, only 2 were considered drug related. The overall response rate was low with none being durable.
Study details: Findings are from a phase 1b dose-escalation study involving 31 patients with CML in CP (n=24) or AP (n=7) who received 2 or more prior TKIs and showed progression, resistance, suboptimal response, or intolerance to the most recent therapy.
Disclosures: This trial was funded by Bristol Myers Squibb, Princeton, NJ, USA. The lead author along with other authors declared receiving grants, advisory boards, honoraria, and/or research funding from various pharmaceutical companies including Bristol Myers Squibb. R Swanink and PM Regueira were employees of Bristol Myers Squibb.
Source: Martínez-López J et al. Leuk Lymphoma. 2021 Mar 2. doi: 10.1080/10428194.2021.1889536.
Key clinical point: Combination of dasatinib and nivolumab was safe but did not show any clinical activity in patients with difficult-to-treat chronic myeloid leukemia (CML) in chronic phase (CP) or accelerated phase (AP) who received at least 2 prior tyrosine kinase inhibitors (TKIs).
Major finding: No drug limiting toxicities were observed even with the maximum dose of both drugs. The most common grade 3 or higher adverse event (AE) of any cause was diarrhea (13%), of which only 3% were considered drug related. Serious AEs occurred in 48% of patients; however, only 2 were considered drug related. The overall response rate was low with none being durable.
Study details: Findings are from a phase 1b dose-escalation study involving 31 patients with CML in CP (n=24) or AP (n=7) who received 2 or more prior TKIs and showed progression, resistance, suboptimal response, or intolerance to the most recent therapy.
Disclosures: This trial was funded by Bristol Myers Squibb, Princeton, NJ, USA. The lead author along with other authors declared receiving grants, advisory boards, honoraria, and/or research funding from various pharmaceutical companies including Bristol Myers Squibb. R Swanink and PM Regueira were employees of Bristol Myers Squibb.
Source: Martínez-López J et al. Leuk Lymphoma. 2021 Mar 2. doi: 10.1080/10428194.2021.1889536.
Key clinical point: Combination of dasatinib and nivolumab was safe but did not show any clinical activity in patients with difficult-to-treat chronic myeloid leukemia (CML) in chronic phase (CP) or accelerated phase (AP) who received at least 2 prior tyrosine kinase inhibitors (TKIs).
Major finding: No drug limiting toxicities were observed even with the maximum dose of both drugs. The most common grade 3 or higher adverse event (AE) of any cause was diarrhea (13%), of which only 3% were considered drug related. Serious AEs occurred in 48% of patients; however, only 2 were considered drug related. The overall response rate was low with none being durable.
Study details: Findings are from a phase 1b dose-escalation study involving 31 patients with CML in CP (n=24) or AP (n=7) who received 2 or more prior TKIs and showed progression, resistance, suboptimal response, or intolerance to the most recent therapy.
Disclosures: This trial was funded by Bristol Myers Squibb, Princeton, NJ, USA. The lead author along with other authors declared receiving grants, advisory boards, honoraria, and/or research funding from various pharmaceutical companies including Bristol Myers Squibb. R Swanink and PM Regueira were employees of Bristol Myers Squibb.
Source: Martínez-López J et al. Leuk Lymphoma. 2021 Mar 2. doi: 10.1080/10428194.2021.1889536.
CML-CP: Imatinib shows long-term efficacy after interferon therapy failure
Key clinical point: Imatinib treatment following interferon-α failure showed long-term efficacy in a real-life setting of patients with late chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: After a median of 4.5 months of therapy initiation, 84% of patients achieved complete cytogenic response as their best cytogenetic response. The estimated 18-year overall survival, event-free survival, and progression-free survival were 64.8%, 69%, and 64.4%, respectively. Imatinib discontinuation and CML-related deaths occurred in 36% and 34% of patients, respectively, and 86% of patients were in treatment-free remission at the last follow-up.
Study details: Findings are from a retrospective analysis of 139 patients with late CML-CP treated with imatinib following interferon-α therapy failure followed up for a median of 16.6 years.
Disclosures: No specific funding source was identified. The authors declared no conflicts of interest.
Source: Pepe S et al. Leuk Lymphoma. 2021 Mar 16. doi: 10.1080/10428194.2021.1901094.
Key clinical point: Imatinib treatment following interferon-α failure showed long-term efficacy in a real-life setting of patients with late chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: After a median of 4.5 months of therapy initiation, 84% of patients achieved complete cytogenic response as their best cytogenetic response. The estimated 18-year overall survival, event-free survival, and progression-free survival were 64.8%, 69%, and 64.4%, respectively. Imatinib discontinuation and CML-related deaths occurred in 36% and 34% of patients, respectively, and 86% of patients were in treatment-free remission at the last follow-up.
Study details: Findings are from a retrospective analysis of 139 patients with late CML-CP treated with imatinib following interferon-α therapy failure followed up for a median of 16.6 years.
Disclosures: No specific funding source was identified. The authors declared no conflicts of interest.
Source: Pepe S et al. Leuk Lymphoma. 2021 Mar 16. doi: 10.1080/10428194.2021.1901094.
Key clinical point: Imatinib treatment following interferon-α failure showed long-term efficacy in a real-life setting of patients with late chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: After a median of 4.5 months of therapy initiation, 84% of patients achieved complete cytogenic response as their best cytogenetic response. The estimated 18-year overall survival, event-free survival, and progression-free survival were 64.8%, 69%, and 64.4%, respectively. Imatinib discontinuation and CML-related deaths occurred in 36% and 34% of patients, respectively, and 86% of patients were in treatment-free remission at the last follow-up.
Study details: Findings are from a retrospective analysis of 139 patients with late CML-CP treated with imatinib following interferon-α therapy failure followed up for a median of 16.6 years.
Disclosures: No specific funding source was identified. The authors declared no conflicts of interest.
Source: Pepe S et al. Leuk Lymphoma. 2021 Mar 16. doi: 10.1080/10428194.2021.1901094.
Dasatinib vs nilotinib as second-line therapy for CML-CP in real-life setting
Key clinical point: Dasatinib and nilotinib as second-line treatments were equally effective, with a high molecular response (MR) and a tolerable safety profile in real-life patients with chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: At 12 months, 47% and 38% of patients had a major MR and 18.2% and 16.6% had a deep MR in the dasatinib and nilotinib groups, respectively (P = .481). Grade 3-4 adverse events were more frequent in dasatinib vs. nilotinib groups (P = .003) with no effect on MR.
Study details: Findings are from a retrospective analysis of 131 patients with CML-CP who switched to second-line treatment with either dasatinib (n=72) or nilotinib (n=59) after frontline imatinib intolerance/resistance.
Disclosures: The authors did not declare any source of funding. M Breccia, M Martelli, and F Efficace reported receiving honoraria, personal fees, and grants from and being on the advisory board for various pharmaceutical companies. Other authors had no disclosures.
Source: Scalzulli E et al. Ann Hematol. 2021 Mar 7. doi: 10.1007/s00277-021-04477-0.
Key clinical point: Dasatinib and nilotinib as second-line treatments were equally effective, with a high molecular response (MR) and a tolerable safety profile in real-life patients with chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: At 12 months, 47% and 38% of patients had a major MR and 18.2% and 16.6% had a deep MR in the dasatinib and nilotinib groups, respectively (P = .481). Grade 3-4 adverse events were more frequent in dasatinib vs. nilotinib groups (P = .003) with no effect on MR.
Study details: Findings are from a retrospective analysis of 131 patients with CML-CP who switched to second-line treatment with either dasatinib (n=72) or nilotinib (n=59) after frontline imatinib intolerance/resistance.
Disclosures: The authors did not declare any source of funding. M Breccia, M Martelli, and F Efficace reported receiving honoraria, personal fees, and grants from and being on the advisory board for various pharmaceutical companies. Other authors had no disclosures.
Source: Scalzulli E et al. Ann Hematol. 2021 Mar 7. doi: 10.1007/s00277-021-04477-0.
Key clinical point: Dasatinib and nilotinib as second-line treatments were equally effective, with a high molecular response (MR) and a tolerable safety profile in real-life patients with chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: At 12 months, 47% and 38% of patients had a major MR and 18.2% and 16.6% had a deep MR in the dasatinib and nilotinib groups, respectively (P = .481). Grade 3-4 adverse events were more frequent in dasatinib vs. nilotinib groups (P = .003) with no effect on MR.
Study details: Findings are from a retrospective analysis of 131 patients with CML-CP who switched to second-line treatment with either dasatinib (n=72) or nilotinib (n=59) after frontline imatinib intolerance/resistance.
Disclosures: The authors did not declare any source of funding. M Breccia, M Martelli, and F Efficace reported receiving honoraria, personal fees, and grants from and being on the advisory board for various pharmaceutical companies. Other authors had no disclosures.
Source: Scalzulli E et al. Ann Hematol. 2021 Mar 7. doi: 10.1007/s00277-021-04477-0.
CML-CP: Sustained long-term high treatment-free remission rates following frontline nilotinib
Key clinical point: More than 3 years of frontline nilotinib treatment was effective with sustained long-term high treatment-free remission (TFR) rates and manageable safety in patients with Philadelphia chromosome-positive (Ph+) chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: Among patients who entered TFR, 41.6% remained in major molecular response (MMR), with 40.0% in MR4.5, whereas 98.9% of patients who lost MMR regained after treatment reinitiation. No disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously.
Study details: Findings are from a 5-year follow-up of phase 2 ENESTfreedom trial including 190 adult patients with Ph+ CML-CP who received at least 2 years of frontline nilotinib treatment, achieved MR4.5, and attempted TFR after undergoing a 1-year nilotinib treatment consolidation phase.
Disclosures: This study was funded by Novartis Pharmaceuticals. The lead author along with other authors reported ties with various pharmaceutical companies including Novartis. P Aimone, S Li, and K Titorenko reported being employees of Novartis.
Source: Radich JP et al. Leukemia. 2021 Mar 11. doi: 10.1038/s41375-021-01205-5.
Key clinical point: More than 3 years of frontline nilotinib treatment was effective with sustained long-term high treatment-free remission (TFR) rates and manageable safety in patients with Philadelphia chromosome-positive (Ph+) chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: Among patients who entered TFR, 41.6% remained in major molecular response (MMR), with 40.0% in MR4.5, whereas 98.9% of patients who lost MMR regained after treatment reinitiation. No disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously.
Study details: Findings are from a 5-year follow-up of phase 2 ENESTfreedom trial including 190 adult patients with Ph+ CML-CP who received at least 2 years of frontline nilotinib treatment, achieved MR4.5, and attempted TFR after undergoing a 1-year nilotinib treatment consolidation phase.
Disclosures: This study was funded by Novartis Pharmaceuticals. The lead author along with other authors reported ties with various pharmaceutical companies including Novartis. P Aimone, S Li, and K Titorenko reported being employees of Novartis.
Source: Radich JP et al. Leukemia. 2021 Mar 11. doi: 10.1038/s41375-021-01205-5.
Key clinical point: More than 3 years of frontline nilotinib treatment was effective with sustained long-term high treatment-free remission (TFR) rates and manageable safety in patients with Philadelphia chromosome-positive (Ph+) chronic-phase chronic myeloid leukemia (CML-CP).
Major finding: Among patients who entered TFR, 41.6% remained in major molecular response (MMR), with 40.0% in MR4.5, whereas 98.9% of patients who lost MMR regained after treatment reinitiation. No disease progression or CML-related deaths were observed, and the adverse event profile was consistent with that reported previously.
Study details: Findings are from a 5-year follow-up of phase 2 ENESTfreedom trial including 190 adult patients with Ph+ CML-CP who received at least 2 years of frontline nilotinib treatment, achieved MR4.5, and attempted TFR after undergoing a 1-year nilotinib treatment consolidation phase.
Disclosures: This study was funded by Novartis Pharmaceuticals. The lead author along with other authors reported ties with various pharmaceutical companies including Novartis. P Aimone, S Li, and K Titorenko reported being employees of Novartis.
Source: Radich JP et al. Leukemia. 2021 Mar 11. doi: 10.1038/s41375-021-01205-5.
Cardiovascular disease remains leading cause of type 2 diabetes mortality
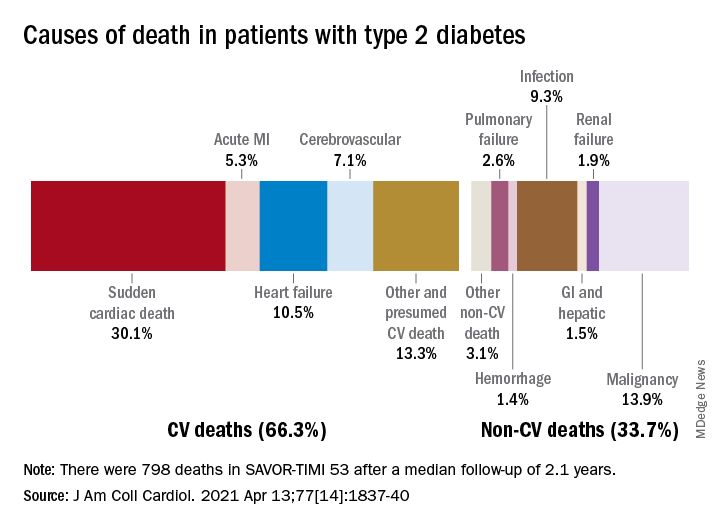
Two-thirds (66.3%) of all 798 deaths after a median 2.1 years of follow-up were caused by one of five cardiovascular (CV) conditions, with sudden cardiac death accounting for the largest share (30.1%) of the total, Ilaria Cavallari, MD, PhD, and associates said in the Journal of the American College of Cardiology.
Most common among the non-CV causes was malignancy at 13.9% of all deaths in a T2DM population at high/very high risk for CV disease (n = 16,492), followed by infection (9.3%), the members of the TIMI Study Group noted.
After variables independently associated with overall mortality were identified, a subdistribution of competing risks was constructed using a competing-risk analysis based on the proportional hazards model, they explained.
Prior heart failure was the clinical variable most associated with CV death and could, along with older age, worse glycemic control, prior CV events, peripheral artery disease, and kidney complications, “identify a subgroup of T2DM patients at high risk of mortality who are likely to achieve the greatest benefit from aggressive management of modifiable risk factors and newer glucose-lowering agents,” the investigators wrote.
It was a pair of laboratory measurements, however, that had the largest subdistribution hazard ratios. “Interestingly, the magnitude of associations of abnormal N-terminal pro–B-type natriuretic peptide [sHR, 2.82] and high-sensitivity troponin T [sHR, 2.46] measured in a stable population were greater than clinical variables in the prediction of all causes of death,” Dr. Cavallari and associates said.
Two-thirds (66.3%) of all 798 deaths after a median 2.1 years of follow-up were caused by one of five cardiovascular (CV) conditions, with sudden cardiac death accounting for the largest share (30.1%) of the total, Ilaria Cavallari, MD, PhD, and associates said in the Journal of the American College of Cardiology.
Most common among the non-CV causes was malignancy at 13.9% of all deaths in a T2DM population at high/very high risk for CV disease (n = 16,492), followed by infection (9.3%), the members of the TIMI Study Group noted.
After variables independently associated with overall mortality were identified, a subdistribution of competing risks was constructed using a competing-risk analysis based on the proportional hazards model, they explained.
Prior heart failure was the clinical variable most associated with CV death and could, along with older age, worse glycemic control, prior CV events, peripheral artery disease, and kidney complications, “identify a subgroup of T2DM patients at high risk of mortality who are likely to achieve the greatest benefit from aggressive management of modifiable risk factors and newer glucose-lowering agents,” the investigators wrote.
It was a pair of laboratory measurements, however, that had the largest subdistribution hazard ratios. “Interestingly, the magnitude of associations of abnormal N-terminal pro–B-type natriuretic peptide [sHR, 2.82] and high-sensitivity troponin T [sHR, 2.46] measured in a stable population were greater than clinical variables in the prediction of all causes of death,” Dr. Cavallari and associates said.
Two-thirds (66.3%) of all 798 deaths after a median 2.1 years of follow-up were caused by one of five cardiovascular (CV) conditions, with sudden cardiac death accounting for the largest share (30.1%) of the total, Ilaria Cavallari, MD, PhD, and associates said in the Journal of the American College of Cardiology.
Most common among the non-CV causes was malignancy at 13.9% of all deaths in a T2DM population at high/very high risk for CV disease (n = 16,492), followed by infection (9.3%), the members of the TIMI Study Group noted.
After variables independently associated with overall mortality were identified, a subdistribution of competing risks was constructed using a competing-risk analysis based on the proportional hazards model, they explained.
Prior heart failure was the clinical variable most associated with CV death and could, along with older age, worse glycemic control, prior CV events, peripheral artery disease, and kidney complications, “identify a subgroup of T2DM patients at high risk of mortality who are likely to achieve the greatest benefit from aggressive management of modifiable risk factors and newer glucose-lowering agents,” the investigators wrote.
It was a pair of laboratory measurements, however, that had the largest subdistribution hazard ratios. “Interestingly, the magnitude of associations of abnormal N-terminal pro–B-type natriuretic peptide [sHR, 2.82] and high-sensitivity troponin T [sHR, 2.46] measured in a stable population were greater than clinical variables in the prediction of all causes of death,” Dr. Cavallari and associates said.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY