User login
Pedunculated Tumor on the Posterior Neck
The Diagnosis: Nodular Hidradenoma
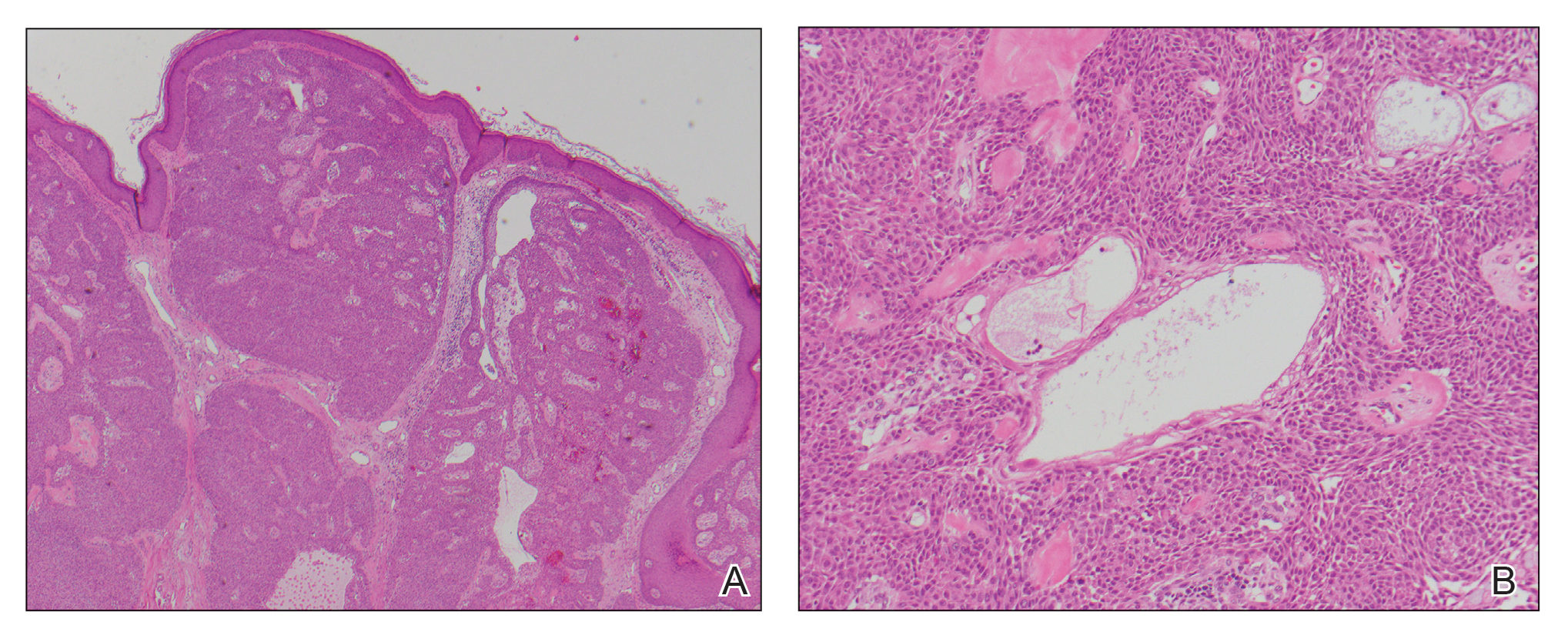
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
The Diagnosis: Nodular Hidradenoma
A biopsy of the nodule showed a large, fungating, well-circumscribed, multilobulated neoplasm composed of primarily monotonous eosinophilic cells in a background of keloidal stroma (Figure). There was a minority population of small, monotonous, clear cells within the lobules, and no glandular structures were noted. Neither cytological nor architectural atypia were evident. MART-1/Melan-A and S-100 stains were negative, consistent with a diagnosis of benign nodular hidradenoma.
Nodular hidradenoma (also known as acrospiroma, solid-cystic hidradenoma, clear cell hidradenoma, and eccrine sweat gland adenoma) is a benign adnexal tumor of the apocrine or eccrine glands.1,2 Nodular hidradenoma can arise at any cutaneous site but most commonly arises on the head and anterior portion of the trunk and rarely on the extremities.2 It presents as a solitary nodular, cystic, or pedunculated mass that can reach up to several centimeters in diameter.2,3 Nodular hidradenoma more commonly affects women compared to men with a ratio of 1.7 to 1 and commonly presents between the third and fifth decades of life, with an average age at presentation of 37.2 years.2,4 There can be associated skin changes, including smoothening, thickening, ulceration, and bluish discoloration. Dermoscopy commonly shows a pinkish homogenous area that extends throughout the entire lesion. This homogenous area less commonly can be bluish, brownish, or pink-blue. Most nodular hidradenomas also can exhibit vascularization, with arborizing telangiectases, polymorphous atypical vessels, and linear irregular vessels being most common; however, this is not specific to nodular hidradenoma.3 Occasionally, tumors can drain serous or hemorrhagic fluid. Nodular hidradenoma commonly is a slow-growing tumor.5 Rapid increase in tumor size can be indicative of malignant transformation, hemorrhage into the tumor, or trauma to the area.2
Histologically, nodular hidradenoma consists of a circumscribed, nonencapsulated, multilobular tumor commonly found in the dermis and sometimes extending into the subcutaneous tissue. There usually is no epidermal attachment, and the overlying epidermis largely is normal. The tumor consists of large multilobulated areas of epithelial cells, tubular lamina, and large cystic areas filled with homogenous eosinophilic material.1 It notably is composed of 2 epithelial cell types: (1) fusiform cells with elongated vesicular nuclei and basophilic cytoplasm, and (2) large polygonal cells with round eccentric nuclei and eosinophilic, periodic acid–Schiff–positive cytoplasm that washes away during fixation, giving the appearance of clear cells.5 Both types of cells are small, monotonous, and void of mitosis or dyskeratosis. Although there can be ducts with apocrine secretion present within the lobulated tumor, they are not consistently found. Due to the varying features that are neither mandatory nor consistent to arrive at this diagnosis, some dermatopathologists view the term hidradenoma as a catch-all term that includes several different types of benign sweat gland tumors. Some authors divide the terminology into apocrine hidradenoma and eccrine hidradenoma based on whether the tumor is composed of solely clear mucinous cells, or poroid and cuticular cells, respectively.
Although nodular hidradenoma classically is a benign tumor, total surgical excision is recommended due to the rare risk for malignant transformation. Rarely, longstanding hidradenomas can metastasize to lymph nodes, bone, or viscera; in these instances, metastatic hidradenoma has a 5-year survival rate of 30%. Recurrence may occur in tumors that are inadequately excised, and the rate of recurrence is estimated to be approximately 10% of surgically excised tumors.5 However, utilization of Mohs micrographic surgery for excision of nodular hidradenoma is associated with a reduced recurrence rate.6
Keloids present as painful, sometimes pruritic, raised scars that extend beyond the boundary of the initial injury, commonly arising on the shoulder, upper arm, and chest. Histopathology reveals nodules of thick hyalinized collagen bundles, keloidal collagen with mucinous ground substance, and few fibroblasts.7
Metastatic renal cell carcinoma to the skin most commonly presents on the face and scalp as a nodular, rapidly growing, round to oval lesion that is flesh colored to reddish purple in a patient with history of renal cell carcinoma.8 Histopathology shows clusters of atypical, nucleated clear cells surrounded by chicken wire vasculature.8,9
Verruca vulgaris is caused by human papillomavirus and most commonly occurs on the hands and feet. It presents as a pink to white, sessile lesion with a verrucous surface and exophytic growths. Histopathology shows acanthosis; hypergranulosis; exophytic projections with a fibrovascular core; inward cupping of the rete ridges; and koilocytes, which are cells with an eccentric, raisinlike nucleus and vacuolated cytoplasm in the granular layer of the epidermis.10
Similar to nodular hidradenoma, nodular melanoma most commonly presents on the head and neck as a symmetric, elevated, amelanotic nodule, but in contrast to nodular hidradenoma, it typically is confined to a smaller diameter.11 Histologically, it is characterized by sheets of atypical, commonly epithelioid melanocytes with a lack of maturation and brisk mitotic activity extending through the epidermis and dermis with lateral extension limited to less than 3 rete ridges.12
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508
- Patterson JW, Weedon D. Tumors of cutaneous appendages. In: Patterson JW, Weedon D. Weedon’s Skin Pathology. 5th ed. Elsevier; 2020:951-1016.
- Ngo N, Susa M, Nakagawa T, et al. Malignant transformation of nodular hidradenoma in the lower leg. Case Rep Oncol. 2018;11:298-304. doi:10.1159/000489255
- Zaballos P, Gómez-Martín I, Martin JM, et al. Dermoscopy of adnexal tumors. Dermatol Clin. 2018;36:397-412. doi:10.1016/j .det.2018.05.007
- Hernández-Pérez E, Cestoni-Parducci R. Nodular hidradenoma and hidradenocarcinoma: a 10-year review. J Am Acad Dermatol. 1985; 12:15-20. doi:10.1016/s0190-9622(85)70002-3
- Stratigos AJ, Olbricht S, Kwan TH, et al. Nodular hidradenoma. Dermatol Surg. 1998;24:387-391. doi:10.1111/j.1524-4725.1998.tb04173.x
- Yavel R, Hinshaw M, Rao V, et al. Hidradenomas and a hidradenocarcinoma of the scalp managed using Mohs micrographic surgery and a multidisciplinary approach. Dermatol Surg. 2009;35:273-281. doi:10.1111/j.1524-4725.2008.34424.x
- Lee JY-Y, Yang C-C, Chao S-C, et al. Histopathological differential diagnosis of keloid and hypertrophic scar. Am J Dermatopathol. 2004;26:379-384. doi:10.1097/00000372-200410000-00006
- Ferhatoglu MF, Senol K, Filiz AI. Skin metastasis of renal cell carcinoma: a case report. Cureus. 2018;10:E3614. doi:10.7759/cureus.3614
- Jaitly V, Jahan-Tigh R, Belousova T, et al. Case report and literature review of nodular hidradenoma, a rare adnexal tumor that mimics breast carcinoma, in a 20-year-old woman. Lab Med. 2019;50:320-325. doi:10.1093/labmed/lmy084
- Betz SJ. HPV-related papillary lesions of the oral mucosa: a review. Head Neck Pathol. 2019;13:80-90. doi:10.1007/s12105-019-01003-7
- Kalkhoran S, Milne O, Zalaudek I, et al. Historical, clinical, and dermoscopic characteristics of thin nodular melanoma. Arch Dermatol. 2010;146:311-318. doi:10.1001/archdermatol.2009.369
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19(suppl 2):S34-S40. doi:10.1038 /modpathol.3800508

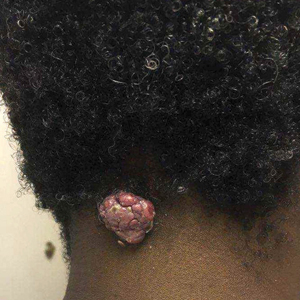
A 56-year-old man presented with a progressively enlarging lesion on the posterior neck of 8 months’ duration. He reported localized pruritus of the lesion that improved with triamcinolone cream 0.05% and oral hydroxyzine as well as occasional irritation of the mass with oozing of clear fluid and blood. He denied associated pain and constitutional symptoms. Physical examination revealed a 2.5-cm, nodular, pedunculated, rubbery mass with foci of crusting on the central posterior neck. The mass was flesh colored to pink, and no lymphadenopathy was noted on physical examination.
Clinical Edge Journal Scan Commentary: RA October 2021

Cigarette smoking is a well-known modifiable risk factor for the development of rheumatoid arthritis (RA). Studies have suggested not only an elevated risk but possible pathogenetic role in the development of autoantibodies, as well as effects on disease outcomes. Passive cigarette smoking has also been proposed as a potential risk factor for RA, though studies are harder to evaluate. This review of prospective data from the Nurses Health Study (NHS) by Yoshida et al looks at incident RA among women enrolled in the study and the influence of in utero, childhood, and adulthood exposure to cigarettes. Childhood exposure to parental smoking was associated with seropositive RA (hazard ratio 1.75) even after controlling for adult personal smoking, and maternal smoking during pregnancy was associated with RA, though the latter effect was not seen after controlling for subsequent smoking exposure. As the authors point out, verifiable prospective data is difficult to obtain regarding exposure to smoking in utero or in childhood and recall bias is possible in obtaining historical information in this prospective study given the use of questionnaires, though it remains plausible given prior studies on the association of personal smoking with RA.
The involvement of gut microbiota in development of autoimmunity has also been postulated but not well-explained. Several recent studies have examined the impact of antibiotic use on the development of RA, including a recent large UK-based case-control study suggesting an increase in RA incidence in people with antibiotic exposure. While a systematic review is ongoing, this prospective cohort study by Liu et al also examines data from NHSI and NHSII and RA risk in patients exposed to antibiotics, stratified by duration of use (none, ≤14 days, ≥15 days). It is reassuring that in this study neither short term (≤14 days) nor long term (≥15 days) antibiotic use was associated with RA risk. Comparison with prior studies with prescription data, however, is limited given the use of questionnaires to establish duration of recent antibiotic exposure.
Fatigue is a common symptom of RA and has a high impact on quality of life in terms of function. The study by Holten et al examines data from the ARCTIC trial in terms of associations between disease activity and fatigue in early RA, as well as change in fatigue with therapy for RA. Fatigue was measured via a visual analog scale (VAS) and did decrease with therapy from baseline; 80% of patients in the study had moderate or high disease activity based on disease activity score (DAS) at baseline and 69% of patients reported fatigue, while 9% of patients had moderate or high disease activity based on DAS at 24 months and 38% reported fatigue. Interestingly, patients who were in remission (per DAS) at 6 months had a reduced risk of fatigue at 24 months. It is hard to interpret this information in a granular way as fatigue is not measured in a standardized way across clinical studies and the only instrument of measure in the ARCTIC trial was the VAS. An alternate view, for example examining the impact of baseline fatigue on response to therapy, may also be reasonable, or fatigue may be a residual symptom similar to chronic myofascial or “non-inflammatory” pain not responsive to treatment in RA.
Finally, another associated extra-articular manifestation of RA is bronchiectasis. Martin et al performed a systematic review and meta-analysis of the literature and found that the prevalence of bronchiectasis was about 18% in RA patients, suggesting that it is more common than previously thought. However, inclusion of CT imaging may detect subclinical bronchiectasis and other secondary causes were not determined. Still, given the effects on quality of life and mortality, further research into causes and risk factors for bronchiectasis in RA is warranted.
Cigarette smoking is a well-known modifiable risk factor for the development of rheumatoid arthritis (RA). Studies have suggested not only an elevated risk but possible pathogenetic role in the development of autoantibodies, as well as effects on disease outcomes. Passive cigarette smoking has also been proposed as a potential risk factor for RA, though studies are harder to evaluate. This review of prospective data from the Nurses Health Study (NHS) by Yoshida et al looks at incident RA among women enrolled in the study and the influence of in utero, childhood, and adulthood exposure to cigarettes. Childhood exposure to parental smoking was associated with seropositive RA (hazard ratio 1.75) even after controlling for adult personal smoking, and maternal smoking during pregnancy was associated with RA, though the latter effect was not seen after controlling for subsequent smoking exposure. As the authors point out, verifiable prospective data is difficult to obtain regarding exposure to smoking in utero or in childhood and recall bias is possible in obtaining historical information in this prospective study given the use of questionnaires, though it remains plausible given prior studies on the association of personal smoking with RA.
The involvement of gut microbiota in development of autoimmunity has also been postulated but not well-explained. Several recent studies have examined the impact of antibiotic use on the development of RA, including a recent large UK-based case-control study suggesting an increase in RA incidence in people with antibiotic exposure. While a systematic review is ongoing, this prospective cohort study by Liu et al also examines data from NHSI and NHSII and RA risk in patients exposed to antibiotics, stratified by duration of use (none, ≤14 days, ≥15 days). It is reassuring that in this study neither short term (≤14 days) nor long term (≥15 days) antibiotic use was associated with RA risk. Comparison with prior studies with prescription data, however, is limited given the use of questionnaires to establish duration of recent antibiotic exposure.
Fatigue is a common symptom of RA and has a high impact on quality of life in terms of function. The study by Holten et al examines data from the ARCTIC trial in terms of associations between disease activity and fatigue in early RA, as well as change in fatigue with therapy for RA. Fatigue was measured via a visual analog scale (VAS) and did decrease with therapy from baseline; 80% of patients in the study had moderate or high disease activity based on disease activity score (DAS) at baseline and 69% of patients reported fatigue, while 9% of patients had moderate or high disease activity based on DAS at 24 months and 38% reported fatigue. Interestingly, patients who were in remission (per DAS) at 6 months had a reduced risk of fatigue at 24 months. It is hard to interpret this information in a granular way as fatigue is not measured in a standardized way across clinical studies and the only instrument of measure in the ARCTIC trial was the VAS. An alternate view, for example examining the impact of baseline fatigue on response to therapy, may also be reasonable, or fatigue may be a residual symptom similar to chronic myofascial or “non-inflammatory” pain not responsive to treatment in RA.
Finally, another associated extra-articular manifestation of RA is bronchiectasis. Martin et al performed a systematic review and meta-analysis of the literature and found that the prevalence of bronchiectasis was about 18% in RA patients, suggesting that it is more common than previously thought. However, inclusion of CT imaging may detect subclinical bronchiectasis and other secondary causes were not determined. Still, given the effects on quality of life and mortality, further research into causes and risk factors for bronchiectasis in RA is warranted.
Cigarette smoking is a well-known modifiable risk factor for the development of rheumatoid arthritis (RA). Studies have suggested not only an elevated risk but possible pathogenetic role in the development of autoantibodies, as well as effects on disease outcomes. Passive cigarette smoking has also been proposed as a potential risk factor for RA, though studies are harder to evaluate. This review of prospective data from the Nurses Health Study (NHS) by Yoshida et al looks at incident RA among women enrolled in the study and the influence of in utero, childhood, and adulthood exposure to cigarettes. Childhood exposure to parental smoking was associated with seropositive RA (hazard ratio 1.75) even after controlling for adult personal smoking, and maternal smoking during pregnancy was associated with RA, though the latter effect was not seen after controlling for subsequent smoking exposure. As the authors point out, verifiable prospective data is difficult to obtain regarding exposure to smoking in utero or in childhood and recall bias is possible in obtaining historical information in this prospective study given the use of questionnaires, though it remains plausible given prior studies on the association of personal smoking with RA.
The involvement of gut microbiota in development of autoimmunity has also been postulated but not well-explained. Several recent studies have examined the impact of antibiotic use on the development of RA, including a recent large UK-based case-control study suggesting an increase in RA incidence in people with antibiotic exposure. While a systematic review is ongoing, this prospective cohort study by Liu et al also examines data from NHSI and NHSII and RA risk in patients exposed to antibiotics, stratified by duration of use (none, ≤14 days, ≥15 days). It is reassuring that in this study neither short term (≤14 days) nor long term (≥15 days) antibiotic use was associated with RA risk. Comparison with prior studies with prescription data, however, is limited given the use of questionnaires to establish duration of recent antibiotic exposure.
Fatigue is a common symptom of RA and has a high impact on quality of life in terms of function. The study by Holten et al examines data from the ARCTIC trial in terms of associations between disease activity and fatigue in early RA, as well as change in fatigue with therapy for RA. Fatigue was measured via a visual analog scale (VAS) and did decrease with therapy from baseline; 80% of patients in the study had moderate or high disease activity based on disease activity score (DAS) at baseline and 69% of patients reported fatigue, while 9% of patients had moderate or high disease activity based on DAS at 24 months and 38% reported fatigue. Interestingly, patients who were in remission (per DAS) at 6 months had a reduced risk of fatigue at 24 months. It is hard to interpret this information in a granular way as fatigue is not measured in a standardized way across clinical studies and the only instrument of measure in the ARCTIC trial was the VAS. An alternate view, for example examining the impact of baseline fatigue on response to therapy, may also be reasonable, or fatigue may be a residual symptom similar to chronic myofascial or “non-inflammatory” pain not responsive to treatment in RA.
Finally, another associated extra-articular manifestation of RA is bronchiectasis. Martin et al performed a systematic review and meta-analysis of the literature and found that the prevalence of bronchiectasis was about 18% in RA patients, suggesting that it is more common than previously thought. However, inclusion of CT imaging may detect subclinical bronchiectasis and other secondary causes were not determined. Still, given the effects on quality of life and mortality, further research into causes and risk factors for bronchiectasis in RA is warranted.
Nutritious meals, more fruits and vegetables boost children’s mental and emotional health
Good nutrition has long been linked to better behavior and academic performance in schoolchildren, as longstanding breakfast and lunch programs in U.S. schools attest. Now British researchers report that nutrition, a modifiable risk factor that can adversely impact mental health, should be part of public health strategies to boost children’s psychological wellness.
In a cross-sectional study published online Sept. 27 in BMJ Nutrition, Prevention & Health, a team from the University of East Anglia in Norwich, England, found a nutritious breakfast and lunch were linked to emotional well-being in schoolchildren of both primary and secondary school age. They also found that some school kids ate neither breakfast nor lunch.
In particular, eating more fruits and vegetables was significantly associated with better mental health in secondary schoolchildren, while a nutritious breakfast and lunch were linked to emotional well-being in students across the age spectrum, according to senior lecturer Richard P. Hayhoe, PhD, of East Anglia University and Anglia Ruskin University in Norwich and colleagues.
They found that primary school pupils who ate only a snack for breakfast had mental well-being scores 5.50 units lower than those eating a substantial breakfast, while having no lunch was tied to scores more than 6 units lower.
“The importance of good-quality nutrition for childhood growth and development is well established,” the authors wrote. “As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being.”
Their current analysis examined data on 7,570 secondary and 1,253 primary school children from 50 schools participating in the Norfolk Children and Young People Health and Well-being Survey 2017.
Multivariable linear regression measured the association between nutritional factors and mental well-being assessed by the Warwick-Edinburgh Mental Well-being Scale for secondary school pupils or by the Stirling Children’s Well-being Scale for primary school pupils. All analyses were adjusted for covariates including demographic, health variables, living/home situations, and adverse experiences.
“The 2017 survey provided a means for Norfolk children and young people to share their feelings on topics such as healthy lifestyles and nutrition, relationships, school experiences, bullying, and their mental well-being,” Dr. Hayhoe said in an interview. “Initial analysis of the data suggested an association between nutrition and well-being and so we decided to investigate this further.”
Dr. Hayhoe added that, as in the United States, youngsters in England get a high proportion of their daily calories from ultraprocessed convenience foods of lesser nutritional value.
“But what we didn’t know was whether the dietary habits of children in our survey had any association with their mental well-being,” he said. “Our current findings suggest that increasing fruit and vegetable consumption and ensuring all schoolchildren eat a nutritional breakfast and lunch may be of benefit to their mental well-being.”
His group cautions, however, that this is an observational study that cannot establish direct causation.
“This study provides the first insights into how fruit and vegetable intake affects children’s mental health, and contributes to the emerging evidence around ‘food and mood,’ ” said Sumantra Ray, MD, executive director of the NNEdPro Global Centre for Nutrition and Health in Cambridge, England.
“The findings are timely, not only because of the impact the pandemic has had on mental well-being, food security, and diet quality, especially in school children, but also in light of the recently published National Food Strategy for England, which highlighted gaps in school meal provision,” added Dr. Ray, who was not involved in the study.
Study results
In total, 10,853 schoolchildren completed the survey: 9% of Norfolk primary school children aged 9-11 and 22% of secondary school students, with approximately 6% of these in the 17- and 18-year-old age bracket. Comprehensive dietary questions explored fruit and vegetable intake, as well as type of breakfast and lunch eaten, alcohol intake, eligibility for free school meals, and satisfaction with weight.
The survey also gathered information on parameters ranging from having one’s own bedroom and bed and exposure to violence or discord in the home.
“Some of these were found to be associated with lower mental well-being scores, but we did not specifically investigate the interaction between these factors and the nutritional factors,” Dr. Hayhoe said. However, the difference in mental well-being between children who ate the most fruit and vegetables and those who ate the least was on a similar scale to those reporting daily, or almost daily, arguing or violence at home, he said.
Average mental health was assessed using validated age-appropriate measures. The mean mental health score of participants was 46.6 out of 70 for secondary school students and 46 out of 60 for primary school pupils.
Among the survey findings were:
- Just 25% of secondary school participants and 28.5% of primary school pupils reported eating the recommended five portions of fruits and vegetables a day, with 10% and 9%, respectively, eating none.
- 21% of secondary and 12% of primary school pupils consumed only a non–energy drink or nothing for breakfast, while 11.5% of secondary schoolchildren ate no lunch. In one high school class of 30, for example, four had nothing to eat or drink before starting classes in the morning, and three had nothing to eat or drink before starting classes in the afternoon.
- Higher combined fruit and vegetable intake was significantly associated in dose-related fashion with higher mental health scores: 3.73 (95% confidence interval, 2.94- 4.53) units higher in those consuming five or more fruits and vegetables (P < .001), compared with none.
- Breakfast or lunch type also correlated with significant differences in well-being scores. Compared with children consuming a conventional breakfast (porridge, toast, cereal, yogurt, fruit, or a cooked meal), those eating no breakfast had mean well-being scores that were 2.73 (95% CI, 2.11-3.35) units lower (P < .001). Those consuming only an energy drink scored even worse: 3.14 (95% CI, 1.20- 5.09) units lower (P = .002).
- Skipping lunch resulted in a 2.95-unit drop in well-being score (95% CI, 2.22-3.68, P < .001), compared with consuming a packed lunch.
In terms of the amounts of fruits and vegetables consumed, one or two daily portions were associated with a score 1.42 units higher, while three or four portions correlated with a score 2.34 units higher. Those eating five or more portions scored 3.73 units higher.
- For primary school pupils, eating only a snack for breakfast was associated with a score 5.50 units lower, and consuming only a non–energy drink was tied to a score 2.67 units lower than eating a conventional breakfast. Not eating any breakfast was associated with a score 3.62 units lower.
- Eating school food versus a packed lunch was associated with a score 1.27 units lower, although this wasn’t statistically significant. Having no lunch was associated with a score 6.08 units lower, although only a few children fell into this group.
“As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being,” the authors wrote, calling for further investigation of the association between nutrition and mental well-being.
This study was commissioned by Norfolk County Council Public Health and the Norfolk Safeguarding Children Board. The University of East Anglia and Social Care Partners provided funding to support Dr. Hayhoe’s work on this project.
Some coauthors are employed by the Norfolk County Council that commissioned the survey.
Good nutrition has long been linked to better behavior and academic performance in schoolchildren, as longstanding breakfast and lunch programs in U.S. schools attest. Now British researchers report that nutrition, a modifiable risk factor that can adversely impact mental health, should be part of public health strategies to boost children’s psychological wellness.
In a cross-sectional study published online Sept. 27 in BMJ Nutrition, Prevention & Health, a team from the University of East Anglia in Norwich, England, found a nutritious breakfast and lunch were linked to emotional well-being in schoolchildren of both primary and secondary school age. They also found that some school kids ate neither breakfast nor lunch.
In particular, eating more fruits and vegetables was significantly associated with better mental health in secondary schoolchildren, while a nutritious breakfast and lunch were linked to emotional well-being in students across the age spectrum, according to senior lecturer Richard P. Hayhoe, PhD, of East Anglia University and Anglia Ruskin University in Norwich and colleagues.
They found that primary school pupils who ate only a snack for breakfast had mental well-being scores 5.50 units lower than those eating a substantial breakfast, while having no lunch was tied to scores more than 6 units lower.
“The importance of good-quality nutrition for childhood growth and development is well established,” the authors wrote. “As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being.”
Their current analysis examined data on 7,570 secondary and 1,253 primary school children from 50 schools participating in the Norfolk Children and Young People Health and Well-being Survey 2017.
Multivariable linear regression measured the association between nutritional factors and mental well-being assessed by the Warwick-Edinburgh Mental Well-being Scale for secondary school pupils or by the Stirling Children’s Well-being Scale for primary school pupils. All analyses were adjusted for covariates including demographic, health variables, living/home situations, and adverse experiences.
“The 2017 survey provided a means for Norfolk children and young people to share their feelings on topics such as healthy lifestyles and nutrition, relationships, school experiences, bullying, and their mental well-being,” Dr. Hayhoe said in an interview. “Initial analysis of the data suggested an association between nutrition and well-being and so we decided to investigate this further.”
Dr. Hayhoe added that, as in the United States, youngsters in England get a high proportion of their daily calories from ultraprocessed convenience foods of lesser nutritional value.
“But what we didn’t know was whether the dietary habits of children in our survey had any association with their mental well-being,” he said. “Our current findings suggest that increasing fruit and vegetable consumption and ensuring all schoolchildren eat a nutritional breakfast and lunch may be of benefit to their mental well-being.”
His group cautions, however, that this is an observational study that cannot establish direct causation.
“This study provides the first insights into how fruit and vegetable intake affects children’s mental health, and contributes to the emerging evidence around ‘food and mood,’ ” said Sumantra Ray, MD, executive director of the NNEdPro Global Centre for Nutrition and Health in Cambridge, England.
“The findings are timely, not only because of the impact the pandemic has had on mental well-being, food security, and diet quality, especially in school children, but also in light of the recently published National Food Strategy for England, which highlighted gaps in school meal provision,” added Dr. Ray, who was not involved in the study.
Study results
In total, 10,853 schoolchildren completed the survey: 9% of Norfolk primary school children aged 9-11 and 22% of secondary school students, with approximately 6% of these in the 17- and 18-year-old age bracket. Comprehensive dietary questions explored fruit and vegetable intake, as well as type of breakfast and lunch eaten, alcohol intake, eligibility for free school meals, and satisfaction with weight.
The survey also gathered information on parameters ranging from having one’s own bedroom and bed and exposure to violence or discord in the home.
“Some of these were found to be associated with lower mental well-being scores, but we did not specifically investigate the interaction between these factors and the nutritional factors,” Dr. Hayhoe said. However, the difference in mental well-being between children who ate the most fruit and vegetables and those who ate the least was on a similar scale to those reporting daily, or almost daily, arguing or violence at home, he said.
Average mental health was assessed using validated age-appropriate measures. The mean mental health score of participants was 46.6 out of 70 for secondary school students and 46 out of 60 for primary school pupils.
Among the survey findings were:
- Just 25% of secondary school participants and 28.5% of primary school pupils reported eating the recommended five portions of fruits and vegetables a day, with 10% and 9%, respectively, eating none.
- 21% of secondary and 12% of primary school pupils consumed only a non–energy drink or nothing for breakfast, while 11.5% of secondary schoolchildren ate no lunch. In one high school class of 30, for example, four had nothing to eat or drink before starting classes in the morning, and three had nothing to eat or drink before starting classes in the afternoon.
- Higher combined fruit and vegetable intake was significantly associated in dose-related fashion with higher mental health scores: 3.73 (95% confidence interval, 2.94- 4.53) units higher in those consuming five or more fruits and vegetables (P < .001), compared with none.
- Breakfast or lunch type also correlated with significant differences in well-being scores. Compared with children consuming a conventional breakfast (porridge, toast, cereal, yogurt, fruit, or a cooked meal), those eating no breakfast had mean well-being scores that were 2.73 (95% CI, 2.11-3.35) units lower (P < .001). Those consuming only an energy drink scored even worse: 3.14 (95% CI, 1.20- 5.09) units lower (P = .002).
- Skipping lunch resulted in a 2.95-unit drop in well-being score (95% CI, 2.22-3.68, P < .001), compared with consuming a packed lunch.
In terms of the amounts of fruits and vegetables consumed, one or two daily portions were associated with a score 1.42 units higher, while three or four portions correlated with a score 2.34 units higher. Those eating five or more portions scored 3.73 units higher.
- For primary school pupils, eating only a snack for breakfast was associated with a score 5.50 units lower, and consuming only a non–energy drink was tied to a score 2.67 units lower than eating a conventional breakfast. Not eating any breakfast was associated with a score 3.62 units lower.
- Eating school food versus a packed lunch was associated with a score 1.27 units lower, although this wasn’t statistically significant. Having no lunch was associated with a score 6.08 units lower, although only a few children fell into this group.
“As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being,” the authors wrote, calling for further investigation of the association between nutrition and mental well-being.
This study was commissioned by Norfolk County Council Public Health and the Norfolk Safeguarding Children Board. The University of East Anglia and Social Care Partners provided funding to support Dr. Hayhoe’s work on this project.
Some coauthors are employed by the Norfolk County Council that commissioned the survey.
Good nutrition has long been linked to better behavior and academic performance in schoolchildren, as longstanding breakfast and lunch programs in U.S. schools attest. Now British researchers report that nutrition, a modifiable risk factor that can adversely impact mental health, should be part of public health strategies to boost children’s psychological wellness.
In a cross-sectional study published online Sept. 27 in BMJ Nutrition, Prevention & Health, a team from the University of East Anglia in Norwich, England, found a nutritious breakfast and lunch were linked to emotional well-being in schoolchildren of both primary and secondary school age. They also found that some school kids ate neither breakfast nor lunch.
In particular, eating more fruits and vegetables was significantly associated with better mental health in secondary schoolchildren, while a nutritious breakfast and lunch were linked to emotional well-being in students across the age spectrum, according to senior lecturer Richard P. Hayhoe, PhD, of East Anglia University and Anglia Ruskin University in Norwich and colleagues.
They found that primary school pupils who ate only a snack for breakfast had mental well-being scores 5.50 units lower than those eating a substantial breakfast, while having no lunch was tied to scores more than 6 units lower.
“The importance of good-quality nutrition for childhood growth and development is well established,” the authors wrote. “As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being.”
Their current analysis examined data on 7,570 secondary and 1,253 primary school children from 50 schools participating in the Norfolk Children and Young People Health and Well-being Survey 2017.
Multivariable linear regression measured the association between nutritional factors and mental well-being assessed by the Warwick-Edinburgh Mental Well-being Scale for secondary school pupils or by the Stirling Children’s Well-being Scale for primary school pupils. All analyses were adjusted for covariates including demographic, health variables, living/home situations, and adverse experiences.
“The 2017 survey provided a means for Norfolk children and young people to share their feelings on topics such as healthy lifestyles and nutrition, relationships, school experiences, bullying, and their mental well-being,” Dr. Hayhoe said in an interview. “Initial analysis of the data suggested an association between nutrition and well-being and so we decided to investigate this further.”
Dr. Hayhoe added that, as in the United States, youngsters in England get a high proportion of their daily calories from ultraprocessed convenience foods of lesser nutritional value.
“But what we didn’t know was whether the dietary habits of children in our survey had any association with their mental well-being,” he said. “Our current findings suggest that increasing fruit and vegetable consumption and ensuring all schoolchildren eat a nutritional breakfast and lunch may be of benefit to their mental well-being.”
His group cautions, however, that this is an observational study that cannot establish direct causation.
“This study provides the first insights into how fruit and vegetable intake affects children’s mental health, and contributes to the emerging evidence around ‘food and mood,’ ” said Sumantra Ray, MD, executive director of the NNEdPro Global Centre for Nutrition and Health in Cambridge, England.
“The findings are timely, not only because of the impact the pandemic has had on mental well-being, food security, and diet quality, especially in school children, but also in light of the recently published National Food Strategy for England, which highlighted gaps in school meal provision,” added Dr. Ray, who was not involved in the study.
Study results
In total, 10,853 schoolchildren completed the survey: 9% of Norfolk primary school children aged 9-11 and 22% of secondary school students, with approximately 6% of these in the 17- and 18-year-old age bracket. Comprehensive dietary questions explored fruit and vegetable intake, as well as type of breakfast and lunch eaten, alcohol intake, eligibility for free school meals, and satisfaction with weight.
The survey also gathered information on parameters ranging from having one’s own bedroom and bed and exposure to violence or discord in the home.
“Some of these were found to be associated with lower mental well-being scores, but we did not specifically investigate the interaction between these factors and the nutritional factors,” Dr. Hayhoe said. However, the difference in mental well-being between children who ate the most fruit and vegetables and those who ate the least was on a similar scale to those reporting daily, or almost daily, arguing or violence at home, he said.
Average mental health was assessed using validated age-appropriate measures. The mean mental health score of participants was 46.6 out of 70 for secondary school students and 46 out of 60 for primary school pupils.
Among the survey findings were:
- Just 25% of secondary school participants and 28.5% of primary school pupils reported eating the recommended five portions of fruits and vegetables a day, with 10% and 9%, respectively, eating none.
- 21% of secondary and 12% of primary school pupils consumed only a non–energy drink or nothing for breakfast, while 11.5% of secondary schoolchildren ate no lunch. In one high school class of 30, for example, four had nothing to eat or drink before starting classes in the morning, and three had nothing to eat or drink before starting classes in the afternoon.
- Higher combined fruit and vegetable intake was significantly associated in dose-related fashion with higher mental health scores: 3.73 (95% confidence interval, 2.94- 4.53) units higher in those consuming five or more fruits and vegetables (P < .001), compared with none.
- Breakfast or lunch type also correlated with significant differences in well-being scores. Compared with children consuming a conventional breakfast (porridge, toast, cereal, yogurt, fruit, or a cooked meal), those eating no breakfast had mean well-being scores that were 2.73 (95% CI, 2.11-3.35) units lower (P < .001). Those consuming only an energy drink scored even worse: 3.14 (95% CI, 1.20- 5.09) units lower (P = .002).
- Skipping lunch resulted in a 2.95-unit drop in well-being score (95% CI, 2.22-3.68, P < .001), compared with consuming a packed lunch.
In terms of the amounts of fruits and vegetables consumed, one or two daily portions were associated with a score 1.42 units higher, while three or four portions correlated with a score 2.34 units higher. Those eating five or more portions scored 3.73 units higher.
- For primary school pupils, eating only a snack for breakfast was associated with a score 5.50 units lower, and consuming only a non–energy drink was tied to a score 2.67 units lower than eating a conventional breakfast. Not eating any breakfast was associated with a score 3.62 units lower.
- Eating school food versus a packed lunch was associated with a score 1.27 units lower, although this wasn’t statistically significant. Having no lunch was associated with a score 6.08 units lower, although only a few children fell into this group.
“As a potentially modifiable factor, both at an individual and societal level, nutrition may therefore represent an important public health target for strategies to address childhood mental well-being,” the authors wrote, calling for further investigation of the association between nutrition and mental well-being.
This study was commissioned by Norfolk County Council Public Health and the Norfolk Safeguarding Children Board. The University of East Anglia and Social Care Partners provided funding to support Dr. Hayhoe’s work on this project.
Some coauthors are employed by the Norfolk County Council that commissioned the survey.
BMJ NUTRITION, PREVENTION & HEALTH
‘Alarming’ increase in fake pills laced with fentanyl, methamphetamine, DEA warns
The U.S. Drug Enforcement Administration has issued a public safety alert over an “alarming” increase in fake prescription pills laced with the synthetic opioid fentanyl or the stimulant methamphetamine.
“The United States is facing an unprecedented crisis of overdose deaths fueled by illegally manufactured fentanyl and methamphetamine,” DEA Administrator Anne Milgram said in the alert.
“Counterfeit pills that contain these dangerous and extremely addictive drugs are more lethal and more accessible than ever before. DEA is focusing resources on taking down the violent drug traffickers causing the greatest harm and posing the greatest threat to the safety and health of Americans,” Ms. Milgram said.
Criminal drug networks are mass-producing fake fentanyl- and methamphetamine-laced pills and deceptively marketing them as legitimate prescription pills, the DEA warns.
such as oxycodone (Oxycontin, Percocet), hydrocodone (Vicodin), and alprazolam (Xanax); or stimulants like amphetamines (Adderall).
The agency has seized fake pills in every U.S. state. More than 9.5 million fake pills have been seized so far this year – more than the last 2 years combined.
The number of seized counterfeit pills with fentanyl has jumped nearly 430% since 2019. DEA lab tests reveal that two out of every five pills with fentanyl contain a potentially lethal dose.
These deadly pills are widely accessible and often sold on social media and e-commerce platforms – making them available to anyone with a smartphone, including minors, the DEA warns.
More than 93,000 people died of a drug overdose in the United States last year, according to federal statistics, and fentanyl is the primary driver of this alarming increase in overdose deaths, the DEA says.
The agency has launched a “One Pill Can Kill” public awareness campaign to educate the public of the dangers of counterfeit pills purchased outside of a licensed pharmacy. These pills are “illegal, dangerous, and potentially lethal,” the DEA warns.
This alert does not apply to legitimate pharmaceutical medications prescribed by doctors and dispensed by licensed pharmacists, the DEA says.
“The legitimate prescription supply chain is not impacted. Anyone filling a prescription at a licensed pharmacy can be confident that the medications they receive are safe when taken as directed by a medical professional,” the agency says.
A version of this article first appeared on Medscape.com.
The U.S. Drug Enforcement Administration has issued a public safety alert over an “alarming” increase in fake prescription pills laced with the synthetic opioid fentanyl or the stimulant methamphetamine.
“The United States is facing an unprecedented crisis of overdose deaths fueled by illegally manufactured fentanyl and methamphetamine,” DEA Administrator Anne Milgram said in the alert.
“Counterfeit pills that contain these dangerous and extremely addictive drugs are more lethal and more accessible than ever before. DEA is focusing resources on taking down the violent drug traffickers causing the greatest harm and posing the greatest threat to the safety and health of Americans,” Ms. Milgram said.
Criminal drug networks are mass-producing fake fentanyl- and methamphetamine-laced pills and deceptively marketing them as legitimate prescription pills, the DEA warns.
such as oxycodone (Oxycontin, Percocet), hydrocodone (Vicodin), and alprazolam (Xanax); or stimulants like amphetamines (Adderall).
The agency has seized fake pills in every U.S. state. More than 9.5 million fake pills have been seized so far this year – more than the last 2 years combined.
The number of seized counterfeit pills with fentanyl has jumped nearly 430% since 2019. DEA lab tests reveal that two out of every five pills with fentanyl contain a potentially lethal dose.
These deadly pills are widely accessible and often sold on social media and e-commerce platforms – making them available to anyone with a smartphone, including minors, the DEA warns.
More than 93,000 people died of a drug overdose in the United States last year, according to federal statistics, and fentanyl is the primary driver of this alarming increase in overdose deaths, the DEA says.
The agency has launched a “One Pill Can Kill” public awareness campaign to educate the public of the dangers of counterfeit pills purchased outside of a licensed pharmacy. These pills are “illegal, dangerous, and potentially lethal,” the DEA warns.
This alert does not apply to legitimate pharmaceutical medications prescribed by doctors and dispensed by licensed pharmacists, the DEA says.
“The legitimate prescription supply chain is not impacted. Anyone filling a prescription at a licensed pharmacy can be confident that the medications they receive are safe when taken as directed by a medical professional,” the agency says.
A version of this article first appeared on Medscape.com.
The U.S. Drug Enforcement Administration has issued a public safety alert over an “alarming” increase in fake prescription pills laced with the synthetic opioid fentanyl or the stimulant methamphetamine.
“The United States is facing an unprecedented crisis of overdose deaths fueled by illegally manufactured fentanyl and methamphetamine,” DEA Administrator Anne Milgram said in the alert.
“Counterfeit pills that contain these dangerous and extremely addictive drugs are more lethal and more accessible than ever before. DEA is focusing resources on taking down the violent drug traffickers causing the greatest harm and posing the greatest threat to the safety and health of Americans,” Ms. Milgram said.
Criminal drug networks are mass-producing fake fentanyl- and methamphetamine-laced pills and deceptively marketing them as legitimate prescription pills, the DEA warns.
such as oxycodone (Oxycontin, Percocet), hydrocodone (Vicodin), and alprazolam (Xanax); or stimulants like amphetamines (Adderall).
The agency has seized fake pills in every U.S. state. More than 9.5 million fake pills have been seized so far this year – more than the last 2 years combined.
The number of seized counterfeit pills with fentanyl has jumped nearly 430% since 2019. DEA lab tests reveal that two out of every five pills with fentanyl contain a potentially lethal dose.
These deadly pills are widely accessible and often sold on social media and e-commerce platforms – making them available to anyone with a smartphone, including minors, the DEA warns.
More than 93,000 people died of a drug overdose in the United States last year, according to federal statistics, and fentanyl is the primary driver of this alarming increase in overdose deaths, the DEA says.
The agency has launched a “One Pill Can Kill” public awareness campaign to educate the public of the dangers of counterfeit pills purchased outside of a licensed pharmacy. These pills are “illegal, dangerous, and potentially lethal,” the DEA warns.
This alert does not apply to legitimate pharmaceutical medications prescribed by doctors and dispensed by licensed pharmacists, the DEA says.
“The legitimate prescription supply chain is not impacted. Anyone filling a prescription at a licensed pharmacy can be confident that the medications they receive are safe when taken as directed by a medical professional,” the agency says.
A version of this article first appeared on Medscape.com.
Heart failure hospitalization risk lower with SGLT2 inhibitors than GLP-1 RAs

Around a 30% reduction in the risk for being hospitalized for heart failure was achieved in people with type 2 diabetes who were treated with a SGLT2 inhibitor over a GLP-1 RA regardless of whether the patients had a preexisting heart condition.
The findings, published in the Annals of Internal Medicine, also showed a 10% lower risk for myocardial infarction or stroke among those treated with a SGLT2 inhibitor who had preexisting cardiovascular disease (CVD), although there was no difference in risk between the two classes of drugs in those without preexisting CVD.
“These findings are important as they suggest that SGLT2 [inhibitors] and GLP-1 RA offer similar benefits in preventing myocardial infarction and stroke in patients with diabetes,” said study investigator Elisabetta Patorno, MD, DrPH, of Brigham and Women’s Hospital and Harvard Medical School in Boston, in an interview.
They also show “that SGLT2 [inhibitors] offer greater efficacy in preventing heart failure, which supports the existing guidelines,” she added.
Paul S. Jellinger, MD, MACE, of the Center for Diabetes and Endocrine Care in Hollywood, Fla., said these data were likely to be “additive to guidelines but not transformative.” The overall analysis results were “not surprising.” It was not unexpected that SGLT2 inhibitors provided a robust chronic heart failure (CHF) benefit, particularly in individuals with history of CVD, he said.
Dr. Jellinger, a clinical endocrinologist and professor of clinical medicine on the voluntary faculty at the University of Miami, observed, however, that “the similar CVD benefit in both drug classes in patients without known CVD adds to our knowledge in this somewhat controversial area and may be useful to the clinician in evaluating therapy in a diabetic individual without evidence of or at high risk for CHF.”
Furthermore, “the study also reminds us that, as demonstrated in published meta-analysis, there is also a modest CHF benefit associated with GLP-1 RA treatment particularly in patients with a history of CVD.”
Addressing the knowledge gap
Thanks to the results of many large-scale, prospective, cardiovascular outcomes studies, both SGLT2 inhibitors and GLP1 RA have been recommended as treatment for people with diabetes who have established CVD. But with no direct head-to-head trials having been conducted, there is a gap in knowledge and there is currently little guidance for physicians on which drug class to choose for an individual patient.
To try to clarify things, Dr. Patorno and associates looked at data from more than 370,000 people with type 2 diabetes who had been treated between April 2013 and December 2017 with either a SGLT2 inhibitor (canagliflozin, dapagliflozin, or empagliflozin) or GLP-1 RA (albiglutide, dulaglutide, exenatide, or liraglutide).
One-to-one propensity score matching was used to create the study groups: participants were first grouped according to their history of CVD, and then by the class of drug they had been prescribed. The primary outcomes were hospitalization for MI, stroke, or heart failure.
Comparing the initiation of a SGLT2 inhibitor with GLP-1 RA therapy, the hazard ratios (HRs) for MI or stroke in patients with and without a history of CVD were a respective 0.90 (95% CI, 0.82 to 0.98) and 1.07 (0.97 to 1.18).
The corresponding hazard ratios for heart failure hospitalizations were 0.71 (0.64 to 0.79) and 0.69 (0.56 to 0.85).
Real-world studies are of ‘increasing value’
“As in other not-randomized studies based on real-world data, residual confounding cannot be completely ruled out,” Dr. Patorno acknowledged. She added, however that “state-of-the-art methodological strategies were implemented to minimize this possibility.”
Limitations notwithstanding, “real world studies are demonstrating increasing value,” observed Dr. Jellinger. Further large-scale cardiovascular outcomes trials that directly compare these two drug classes “are unlikely given the depth of information available now,” Dr. Jellinger suggested.
“This head-to-head retrospective study may be as close as we get and does represent the first effort at a comparison of these two classes.”
Dr. Patorno said of the potential clinical implications: “Because the two classes are equally effective for stroke and myocardial infarction, but the SGLT2 inhibitors are superior for heart failure, when considered in aggregate, SGLT2 inhibitors are likely to prevent more of these adverse cardiovascular events than GLP-1 RA.”
The study received no commercial funding and was supported by the Brigham and Women’s Hospital and Harvard Medical School Division of Pharmacoepidemiology and Pharmacoeconomics.
Dr. Patorno reported no conflicts of interest. Dr. Jellinger is on the speaker’s bureau for Astra Zeneca, Amgen, and Esperion, and has served on advisory boards for Corcept and Regeneron.
Around a 30% reduction in the risk for being hospitalized for heart failure was achieved in people with type 2 diabetes who were treated with a SGLT2 inhibitor over a GLP-1 RA regardless of whether the patients had a preexisting heart condition.
The findings, published in the Annals of Internal Medicine, also showed a 10% lower risk for myocardial infarction or stroke among those treated with a SGLT2 inhibitor who had preexisting cardiovascular disease (CVD), although there was no difference in risk between the two classes of drugs in those without preexisting CVD.
“These findings are important as they suggest that SGLT2 [inhibitors] and GLP-1 RA offer similar benefits in preventing myocardial infarction and stroke in patients with diabetes,” said study investigator Elisabetta Patorno, MD, DrPH, of Brigham and Women’s Hospital and Harvard Medical School in Boston, in an interview.
They also show “that SGLT2 [inhibitors] offer greater efficacy in preventing heart failure, which supports the existing guidelines,” she added.
Paul S. Jellinger, MD, MACE, of the Center for Diabetes and Endocrine Care in Hollywood, Fla., said these data were likely to be “additive to guidelines but not transformative.” The overall analysis results were “not surprising.” It was not unexpected that SGLT2 inhibitors provided a robust chronic heart failure (CHF) benefit, particularly in individuals with history of CVD, he said.
Dr. Jellinger, a clinical endocrinologist and professor of clinical medicine on the voluntary faculty at the University of Miami, observed, however, that “the similar CVD benefit in both drug classes in patients without known CVD adds to our knowledge in this somewhat controversial area and may be useful to the clinician in evaluating therapy in a diabetic individual without evidence of or at high risk for CHF.”
Furthermore, “the study also reminds us that, as demonstrated in published meta-analysis, there is also a modest CHF benefit associated with GLP-1 RA treatment particularly in patients with a history of CVD.”
Addressing the knowledge gap
Thanks to the results of many large-scale, prospective, cardiovascular outcomes studies, both SGLT2 inhibitors and GLP1 RA have been recommended as treatment for people with diabetes who have established CVD. But with no direct head-to-head trials having been conducted, there is a gap in knowledge and there is currently little guidance for physicians on which drug class to choose for an individual patient.
To try to clarify things, Dr. Patorno and associates looked at data from more than 370,000 people with type 2 diabetes who had been treated between April 2013 and December 2017 with either a SGLT2 inhibitor (canagliflozin, dapagliflozin, or empagliflozin) or GLP-1 RA (albiglutide, dulaglutide, exenatide, or liraglutide).
One-to-one propensity score matching was used to create the study groups: participants were first grouped according to their history of CVD, and then by the class of drug they had been prescribed. The primary outcomes were hospitalization for MI, stroke, or heart failure.
Comparing the initiation of a SGLT2 inhibitor with GLP-1 RA therapy, the hazard ratios (HRs) for MI or stroke in patients with and without a history of CVD were a respective 0.90 (95% CI, 0.82 to 0.98) and 1.07 (0.97 to 1.18).
The corresponding hazard ratios for heart failure hospitalizations were 0.71 (0.64 to 0.79) and 0.69 (0.56 to 0.85).
Real-world studies are of ‘increasing value’
“As in other not-randomized studies based on real-world data, residual confounding cannot be completely ruled out,” Dr. Patorno acknowledged. She added, however that “state-of-the-art methodological strategies were implemented to minimize this possibility.”
Limitations notwithstanding, “real world studies are demonstrating increasing value,” observed Dr. Jellinger. Further large-scale cardiovascular outcomes trials that directly compare these two drug classes “are unlikely given the depth of information available now,” Dr. Jellinger suggested.
“This head-to-head retrospective study may be as close as we get and does represent the first effort at a comparison of these two classes.”
Dr. Patorno said of the potential clinical implications: “Because the two classes are equally effective for stroke and myocardial infarction, but the SGLT2 inhibitors are superior for heart failure, when considered in aggregate, SGLT2 inhibitors are likely to prevent more of these adverse cardiovascular events than GLP-1 RA.”
The study received no commercial funding and was supported by the Brigham and Women’s Hospital and Harvard Medical School Division of Pharmacoepidemiology and Pharmacoeconomics.
Dr. Patorno reported no conflicts of interest. Dr. Jellinger is on the speaker’s bureau for Astra Zeneca, Amgen, and Esperion, and has served on advisory boards for Corcept and Regeneron.
Around a 30% reduction in the risk for being hospitalized for heart failure was achieved in people with type 2 diabetes who were treated with a SGLT2 inhibitor over a GLP-1 RA regardless of whether the patients had a preexisting heart condition.
The findings, published in the Annals of Internal Medicine, also showed a 10% lower risk for myocardial infarction or stroke among those treated with a SGLT2 inhibitor who had preexisting cardiovascular disease (CVD), although there was no difference in risk between the two classes of drugs in those without preexisting CVD.
“These findings are important as they suggest that SGLT2 [inhibitors] and GLP-1 RA offer similar benefits in preventing myocardial infarction and stroke in patients with diabetes,” said study investigator Elisabetta Patorno, MD, DrPH, of Brigham and Women’s Hospital and Harvard Medical School in Boston, in an interview.
They also show “that SGLT2 [inhibitors] offer greater efficacy in preventing heart failure, which supports the existing guidelines,” she added.
Paul S. Jellinger, MD, MACE, of the Center for Diabetes and Endocrine Care in Hollywood, Fla., said these data were likely to be “additive to guidelines but not transformative.” The overall analysis results were “not surprising.” It was not unexpected that SGLT2 inhibitors provided a robust chronic heart failure (CHF) benefit, particularly in individuals with history of CVD, he said.
Dr. Jellinger, a clinical endocrinologist and professor of clinical medicine on the voluntary faculty at the University of Miami, observed, however, that “the similar CVD benefit in both drug classes in patients without known CVD adds to our knowledge in this somewhat controversial area and may be useful to the clinician in evaluating therapy in a diabetic individual without evidence of or at high risk for CHF.”
Furthermore, “the study also reminds us that, as demonstrated in published meta-analysis, there is also a modest CHF benefit associated with GLP-1 RA treatment particularly in patients with a history of CVD.”
Addressing the knowledge gap
Thanks to the results of many large-scale, prospective, cardiovascular outcomes studies, both SGLT2 inhibitors and GLP1 RA have been recommended as treatment for people with diabetes who have established CVD. But with no direct head-to-head trials having been conducted, there is a gap in knowledge and there is currently little guidance for physicians on which drug class to choose for an individual patient.
To try to clarify things, Dr. Patorno and associates looked at data from more than 370,000 people with type 2 diabetes who had been treated between April 2013 and December 2017 with either a SGLT2 inhibitor (canagliflozin, dapagliflozin, or empagliflozin) or GLP-1 RA (albiglutide, dulaglutide, exenatide, or liraglutide).
One-to-one propensity score matching was used to create the study groups: participants were first grouped according to their history of CVD, and then by the class of drug they had been prescribed. The primary outcomes were hospitalization for MI, stroke, or heart failure.
Comparing the initiation of a SGLT2 inhibitor with GLP-1 RA therapy, the hazard ratios (HRs) for MI or stroke in patients with and without a history of CVD were a respective 0.90 (95% CI, 0.82 to 0.98) and 1.07 (0.97 to 1.18).
The corresponding hazard ratios for heart failure hospitalizations were 0.71 (0.64 to 0.79) and 0.69 (0.56 to 0.85).
Real-world studies are of ‘increasing value’
“As in other not-randomized studies based on real-world data, residual confounding cannot be completely ruled out,” Dr. Patorno acknowledged. She added, however that “state-of-the-art methodological strategies were implemented to minimize this possibility.”
Limitations notwithstanding, “real world studies are demonstrating increasing value,” observed Dr. Jellinger. Further large-scale cardiovascular outcomes trials that directly compare these two drug classes “are unlikely given the depth of information available now,” Dr. Jellinger suggested.
“This head-to-head retrospective study may be as close as we get and does represent the first effort at a comparison of these two classes.”
Dr. Patorno said of the potential clinical implications: “Because the two classes are equally effective for stroke and myocardial infarction, but the SGLT2 inhibitors are superior for heart failure, when considered in aggregate, SGLT2 inhibitors are likely to prevent more of these adverse cardiovascular events than GLP-1 RA.”
The study received no commercial funding and was supported by the Brigham and Women’s Hospital and Harvard Medical School Division of Pharmacoepidemiology and Pharmacoeconomics.
Dr. Patorno reported no conflicts of interest. Dr. Jellinger is on the speaker’s bureau for Astra Zeneca, Amgen, and Esperion, and has served on advisory boards for Corcept and Regeneron.
FROM ANNALS OF INTERNAL MEDICINE
Physical activity paradoxically tied to higher coronary calcium
new observational data suggest.
In a prospective cohort study of Korean men and women 18 years and older, participants who were the most physically active had the fastest progression of their coronary artery calcium (CAC) scores at 5 years, compared with those who were the least physically active.
“People who exercise may have an increase in their coronary calcium levels, but this is not necessarily bad news. This may mean that atherosclerotic lesions in the coronary arteries are becoming more stable and less dangerous, but we need additional research to understand these changes,” Eliseo Guallar, MD, PhD, professor, Johns Hopkins Bloomberg School of Public Health, Baltimore, the study’s corresponding author, said in an interview.
This paradoxical effect notwithstanding, doctors should continue to advise their patients to follow the physical activity guidelines for Americans that were published in 2018, Dr. Guallar said.
“Physical activity is a key component of a healthy lifestyle. Our analysis can be useful, however, if someone starts exercising and sees that his or her coronary calcium score goes up,” he said.
The study is published online September 20 in Heart.
The degree of build-up of calcium deposits in the coronary arteries is used to determine future cardiovascular disease risk and to guide treatment to prevent myocardial infarction and stroke. A CAC score of at least 100 Agatston units indicates that treatment with statins is warranted, the researchers write.
In the current study, investigators — led by Ki-Chul Sung, MD, Sungkyunkwan University School of Medicine, Seoul, Korea, and Yun Soo Hong, MD, Johns Hopkins Bloomberg School of Public Health, Baltimore — explored the link between different degrees of physical activity and the progression of CAC scores in healthy adults.
“While physical activity improves a wide array of cardiovascular and metabolic biomarkers, endurance athletes were more likely to have a coronary artery calcium (CAC) score >300 Agatston units or coronary plaques compared with sedentary men with a similar risk profile. It is not clear if exercise may itself be associated with calcification of the arteries,” the authors write.
The researchers studied 25,485 participants (22,741 men and 2,744 women) who were part of the Kangbuk Samsung Health Study. All were free of cardiovascular disease at study entry and underwent comprehensive health screening exams at one of two major health centers in Seoul and Suwon, South Korea, between March 1, 2011, and December 31, 2017.
At each exam, participants filled out a questionnaire that included questions on medical and family history, smoking habits, alcohol intake, and education level.
Participants were also quizzed at baseline about their physical activity, using the Korean version of the International Physical Activity Questionnaire Short Form (IPAQ-SF).
On the basis of that, they were categorized into one of three categories: inactive; moderately active, defined as at least 3 days of vigorous-intensity activity for at least 20 min/day or at least 5 days of moderate-intensity activity or walking for at least 30 min/ day or at least 5 days of any combination of walking and moderate- or vigorous-intensity activities, attaining at least 600 MET-min/week; or health-enhancing physically active (HEPA), defined as at least 3 days of vigorous-intensity activity, attaining at least 1,500 MET-min/week or 7 days of any combination of walking or moderate- or vigorous-intensity activities, attaining at least 3000 MET-min/week.
Of the study participants, 47% were classified as inactive, 38% as moderately active, and 15% as HEPA.
Those who were more physically active tended to be older and less likely to smoke than less physically active participants. They also had lower total cholesterol, more hypertension, and existing evidence of calcium deposits in their coronary arteries.
A graded association between physical activity level and the prevalence and progression of coronary artery calcification was seen, irrespective of CAC scores at the start of monitoring.
At baseline, the estimated adjusted average baseline CAC scores in inactive participants was 9.45 (95% CI, 8.76 - 10.14), in moderately active participants was 10.20 (95% CI, 9.40 - 11.00), and in HEPA participants was 12.04 (95% CI, 10.81 - 13.26).
Compared with the least active participants, the estimated adjusted 5-year average increases in CAC was 3.20 (95% CI, 0.72 - 5.69) in moderately active participants and 8.16 (95% CI, 4.80 - 11.53) in HEPA participants.
A higher level of physical activity was associated with faster progression of CAC scores, both in participants with CAC score of 0 at baseline and in those with prevalent CAC.
The authors note there are several limitations to consider when interpreting their findings. These include the absence of an objective assessment of physical activity, the inability to evaluate the association between physical activity and CAC levels with incident cardiovascular events because of a lack of data, and the lack of information on incident myocardial infarction, stroke, CAC density, or volume.
Physical activity might increase coronary atherosclerosis through mechanical stress and vessel wall injury of coronary arteries; physiologic responses during exercise, such as increased blood pressure; increased parathyroid hormone levels; and changes in coronary hemodynamics and inflammation. “In addition, other factors, such as diet, vitamins, and minerals, may change with physical activity,” the authors write.
“The second possibility is that physical activity may increase CAC scores without increasing cardiovascular disease risk,” they write.
“The cardiovascular benefits of physical activity are unquestionable,” the authors emphasize, adding that the national guidelines recommend at least 150 to 300 minutes per week of moderate-intensity or 75 to 150 minutes per week of vigorous-intensity aerobic physical activity.
“Patients and physicians, however, need to consider that engaging in physical activity may accelerate the progression of coronary calcium, possibly due to plaque healing, stabilization and calcification,” they conclude.
Dr. Guallar added: “We would like to link our research to clinical outcomes, so that we can really be sure that the increase in coronary calcium scores does not imply an increase in risk.”
“Do these findings mean that we should stop using coronary artery calcium scores to assess coronary artery disease?” ask Gaurav Gulsin, MD, and Alastair James Moss, MD, University of Leicester, United Kingdom, in an accompanying editorial.
The study highlights the complexity of interpreting CAC scores in patients who have implemented recommendations for physical activity or started statin therapy, they note.
“While proponents would argue that it is an effective tool to screen for subclinical atherosclerosis in asymptomatic individuals, clinicians should be cautious regarding the overuse of this test in otherwise healthy individuals. The coronary artery calcium paradox should not result in paradoxical care for our patients,” Dr. Gulsin and Dr. Moss conclude.
Dr. Sung, Dr. Hong, and the other study authors report no relevant financial relationships. The British Heart Foundation provides funding support for Dr. Gulsin and Dr. Moss.
A version of this article first appeared on Medscape.com.
new observational data suggest.
In a prospective cohort study of Korean men and women 18 years and older, participants who were the most physically active had the fastest progression of their coronary artery calcium (CAC) scores at 5 years, compared with those who were the least physically active.
“People who exercise may have an increase in their coronary calcium levels, but this is not necessarily bad news. This may mean that atherosclerotic lesions in the coronary arteries are becoming more stable and less dangerous, but we need additional research to understand these changes,” Eliseo Guallar, MD, PhD, professor, Johns Hopkins Bloomberg School of Public Health, Baltimore, the study’s corresponding author, said in an interview.
This paradoxical effect notwithstanding, doctors should continue to advise their patients to follow the physical activity guidelines for Americans that were published in 2018, Dr. Guallar said.
“Physical activity is a key component of a healthy lifestyle. Our analysis can be useful, however, if someone starts exercising and sees that his or her coronary calcium score goes up,” he said.
The study is published online September 20 in Heart.
The degree of build-up of calcium deposits in the coronary arteries is used to determine future cardiovascular disease risk and to guide treatment to prevent myocardial infarction and stroke. A CAC score of at least 100 Agatston units indicates that treatment with statins is warranted, the researchers write.
In the current study, investigators — led by Ki-Chul Sung, MD, Sungkyunkwan University School of Medicine, Seoul, Korea, and Yun Soo Hong, MD, Johns Hopkins Bloomberg School of Public Health, Baltimore — explored the link between different degrees of physical activity and the progression of CAC scores in healthy adults.
“While physical activity improves a wide array of cardiovascular and metabolic biomarkers, endurance athletes were more likely to have a coronary artery calcium (CAC) score >300 Agatston units or coronary plaques compared with sedentary men with a similar risk profile. It is not clear if exercise may itself be associated with calcification of the arteries,” the authors write.
The researchers studied 25,485 participants (22,741 men and 2,744 women) who were part of the Kangbuk Samsung Health Study. All were free of cardiovascular disease at study entry and underwent comprehensive health screening exams at one of two major health centers in Seoul and Suwon, South Korea, between March 1, 2011, and December 31, 2017.
At each exam, participants filled out a questionnaire that included questions on medical and family history, smoking habits, alcohol intake, and education level.
Participants were also quizzed at baseline about their physical activity, using the Korean version of the International Physical Activity Questionnaire Short Form (IPAQ-SF).
On the basis of that, they were categorized into one of three categories: inactive; moderately active, defined as at least 3 days of vigorous-intensity activity for at least 20 min/day or at least 5 days of moderate-intensity activity or walking for at least 30 min/ day or at least 5 days of any combination of walking and moderate- or vigorous-intensity activities, attaining at least 600 MET-min/week; or health-enhancing physically active (HEPA), defined as at least 3 days of vigorous-intensity activity, attaining at least 1,500 MET-min/week or 7 days of any combination of walking or moderate- or vigorous-intensity activities, attaining at least 3000 MET-min/week.
Of the study participants, 47% were classified as inactive, 38% as moderately active, and 15% as HEPA.
Those who were more physically active tended to be older and less likely to smoke than less physically active participants. They also had lower total cholesterol, more hypertension, and existing evidence of calcium deposits in their coronary arteries.
A graded association between physical activity level and the prevalence and progression of coronary artery calcification was seen, irrespective of CAC scores at the start of monitoring.
At baseline, the estimated adjusted average baseline CAC scores in inactive participants was 9.45 (95% CI, 8.76 - 10.14), in moderately active participants was 10.20 (95% CI, 9.40 - 11.00), and in HEPA participants was 12.04 (95% CI, 10.81 - 13.26).
Compared with the least active participants, the estimated adjusted 5-year average increases in CAC was 3.20 (95% CI, 0.72 - 5.69) in moderately active participants and 8.16 (95% CI, 4.80 - 11.53) in HEPA participants.
A higher level of physical activity was associated with faster progression of CAC scores, both in participants with CAC score of 0 at baseline and in those with prevalent CAC.
The authors note there are several limitations to consider when interpreting their findings. These include the absence of an objective assessment of physical activity, the inability to evaluate the association between physical activity and CAC levels with incident cardiovascular events because of a lack of data, and the lack of information on incident myocardial infarction, stroke, CAC density, or volume.
Physical activity might increase coronary atherosclerosis through mechanical stress and vessel wall injury of coronary arteries; physiologic responses during exercise, such as increased blood pressure; increased parathyroid hormone levels; and changes in coronary hemodynamics and inflammation. “In addition, other factors, such as diet, vitamins, and minerals, may change with physical activity,” the authors write.
“The second possibility is that physical activity may increase CAC scores without increasing cardiovascular disease risk,” they write.
“The cardiovascular benefits of physical activity are unquestionable,” the authors emphasize, adding that the national guidelines recommend at least 150 to 300 minutes per week of moderate-intensity or 75 to 150 minutes per week of vigorous-intensity aerobic physical activity.
“Patients and physicians, however, need to consider that engaging in physical activity may accelerate the progression of coronary calcium, possibly due to plaque healing, stabilization and calcification,” they conclude.
Dr. Guallar added: “We would like to link our research to clinical outcomes, so that we can really be sure that the increase in coronary calcium scores does not imply an increase in risk.”
“Do these findings mean that we should stop using coronary artery calcium scores to assess coronary artery disease?” ask Gaurav Gulsin, MD, and Alastair James Moss, MD, University of Leicester, United Kingdom, in an accompanying editorial.
The study highlights the complexity of interpreting CAC scores in patients who have implemented recommendations for physical activity or started statin therapy, they note.
“While proponents would argue that it is an effective tool to screen for subclinical atherosclerosis in asymptomatic individuals, clinicians should be cautious regarding the overuse of this test in otherwise healthy individuals. The coronary artery calcium paradox should not result in paradoxical care for our patients,” Dr. Gulsin and Dr. Moss conclude.
Dr. Sung, Dr. Hong, and the other study authors report no relevant financial relationships. The British Heart Foundation provides funding support for Dr. Gulsin and Dr. Moss.
A version of this article first appeared on Medscape.com.
new observational data suggest.
In a prospective cohort study of Korean men and women 18 years and older, participants who were the most physically active had the fastest progression of their coronary artery calcium (CAC) scores at 5 years, compared with those who were the least physically active.
“People who exercise may have an increase in their coronary calcium levels, but this is not necessarily bad news. This may mean that atherosclerotic lesions in the coronary arteries are becoming more stable and less dangerous, but we need additional research to understand these changes,” Eliseo Guallar, MD, PhD, professor, Johns Hopkins Bloomberg School of Public Health, Baltimore, the study’s corresponding author, said in an interview.
This paradoxical effect notwithstanding, doctors should continue to advise their patients to follow the physical activity guidelines for Americans that were published in 2018, Dr. Guallar said.
“Physical activity is a key component of a healthy lifestyle. Our analysis can be useful, however, if someone starts exercising and sees that his or her coronary calcium score goes up,” he said.
The study is published online September 20 in Heart.
The degree of build-up of calcium deposits in the coronary arteries is used to determine future cardiovascular disease risk and to guide treatment to prevent myocardial infarction and stroke. A CAC score of at least 100 Agatston units indicates that treatment with statins is warranted, the researchers write.
In the current study, investigators — led by Ki-Chul Sung, MD, Sungkyunkwan University School of Medicine, Seoul, Korea, and Yun Soo Hong, MD, Johns Hopkins Bloomberg School of Public Health, Baltimore — explored the link between different degrees of physical activity and the progression of CAC scores in healthy adults.
“While physical activity improves a wide array of cardiovascular and metabolic biomarkers, endurance athletes were more likely to have a coronary artery calcium (CAC) score >300 Agatston units or coronary plaques compared with sedentary men with a similar risk profile. It is not clear if exercise may itself be associated with calcification of the arteries,” the authors write.
The researchers studied 25,485 participants (22,741 men and 2,744 women) who were part of the Kangbuk Samsung Health Study. All were free of cardiovascular disease at study entry and underwent comprehensive health screening exams at one of two major health centers in Seoul and Suwon, South Korea, between March 1, 2011, and December 31, 2017.
At each exam, participants filled out a questionnaire that included questions on medical and family history, smoking habits, alcohol intake, and education level.
Participants were also quizzed at baseline about their physical activity, using the Korean version of the International Physical Activity Questionnaire Short Form (IPAQ-SF).
On the basis of that, they were categorized into one of three categories: inactive; moderately active, defined as at least 3 days of vigorous-intensity activity for at least 20 min/day or at least 5 days of moderate-intensity activity or walking for at least 30 min/ day or at least 5 days of any combination of walking and moderate- or vigorous-intensity activities, attaining at least 600 MET-min/week; or health-enhancing physically active (HEPA), defined as at least 3 days of vigorous-intensity activity, attaining at least 1,500 MET-min/week or 7 days of any combination of walking or moderate- or vigorous-intensity activities, attaining at least 3000 MET-min/week.
Of the study participants, 47% were classified as inactive, 38% as moderately active, and 15% as HEPA.
Those who were more physically active tended to be older and less likely to smoke than less physically active participants. They also had lower total cholesterol, more hypertension, and existing evidence of calcium deposits in their coronary arteries.
A graded association between physical activity level and the prevalence and progression of coronary artery calcification was seen, irrespective of CAC scores at the start of monitoring.
At baseline, the estimated adjusted average baseline CAC scores in inactive participants was 9.45 (95% CI, 8.76 - 10.14), in moderately active participants was 10.20 (95% CI, 9.40 - 11.00), and in HEPA participants was 12.04 (95% CI, 10.81 - 13.26).
Compared with the least active participants, the estimated adjusted 5-year average increases in CAC was 3.20 (95% CI, 0.72 - 5.69) in moderately active participants and 8.16 (95% CI, 4.80 - 11.53) in HEPA participants.
A higher level of physical activity was associated with faster progression of CAC scores, both in participants with CAC score of 0 at baseline and in those with prevalent CAC.
The authors note there are several limitations to consider when interpreting their findings. These include the absence of an objective assessment of physical activity, the inability to evaluate the association between physical activity and CAC levels with incident cardiovascular events because of a lack of data, and the lack of information on incident myocardial infarction, stroke, CAC density, or volume.
Physical activity might increase coronary atherosclerosis through mechanical stress and vessel wall injury of coronary arteries; physiologic responses during exercise, such as increased blood pressure; increased parathyroid hormone levels; and changes in coronary hemodynamics and inflammation. “In addition, other factors, such as diet, vitamins, and minerals, may change with physical activity,” the authors write.
“The second possibility is that physical activity may increase CAC scores without increasing cardiovascular disease risk,” they write.
“The cardiovascular benefits of physical activity are unquestionable,” the authors emphasize, adding that the national guidelines recommend at least 150 to 300 minutes per week of moderate-intensity or 75 to 150 minutes per week of vigorous-intensity aerobic physical activity.
“Patients and physicians, however, need to consider that engaging in physical activity may accelerate the progression of coronary calcium, possibly due to plaque healing, stabilization and calcification,” they conclude.
Dr. Guallar added: “We would like to link our research to clinical outcomes, so that we can really be sure that the increase in coronary calcium scores does not imply an increase in risk.”
“Do these findings mean that we should stop using coronary artery calcium scores to assess coronary artery disease?” ask Gaurav Gulsin, MD, and Alastair James Moss, MD, University of Leicester, United Kingdom, in an accompanying editorial.
The study highlights the complexity of interpreting CAC scores in patients who have implemented recommendations for physical activity or started statin therapy, they note.
“While proponents would argue that it is an effective tool to screen for subclinical atherosclerosis in asymptomatic individuals, clinicians should be cautious regarding the overuse of this test in otherwise healthy individuals. The coronary artery calcium paradox should not result in paradoxical care for our patients,” Dr. Gulsin and Dr. Moss conclude.
Dr. Sung, Dr. Hong, and the other study authors report no relevant financial relationships. The British Heart Foundation provides funding support for Dr. Gulsin and Dr. Moss.
A version of this article first appeared on Medscape.com.
Lifestyle interventions improve resistant hypertension
A 4-month, structured diet and exercise intervention lowered blood pressure in adults with resistant blood pressure, according to results from a randomized, clinical trial. The program also led to improvements in baroreflex sensitivity, heart rate variability, and flow-mediated dilation, compared with individuals who received only a single education session.

The intervention included instruction from a nutritionist on how to follow the Dietary Approaches to Stop Hypertension (DASH) diet, as well as restricting calories and sodium to less than 2,300 mg/day. It included weekly, 45-minute group counseling sessions, run by a clinical psychiatrist, focusing on eating behaviors. The exercise component included 30- to 45-minute sessions at 70%-85% of initial heart rate reserve, carried out three times per week at a cardiac rehabilitation facility.
“While some individuals can make the lifestyle changes on their own, a structured program of supervised exercise and dietary modification conducted by a multidisciplinary team of physicians, psychologists, nutritionists, and physical therapists/exercise physiologists found in cardiac rehabilitation programs throughout the country is likely to be more effective. There are many cardiac rehabilitation programs throughout the country that are accessible to most patients,” said lead author James Blumenthal, PhD, the J.P. Gibbons Professor in Psychiatry and Behavioral Sciences at Duke University, Durham, N.C.
The study, called Treating Resistant Hypertension Using Lifestyle Modification to Promote Health (TRIUMPH), was published in Circulation. It is one of few that have examined lifestyle interventions in resistant hypertension, defined as systolic blood pressure ≥ 130 mm Hg or diastolic blood pressure ≥ 80 mm Hg after adherence to three or more optimally-dosed antihypertensive medications of three different classes, including one diuretic.

“This is a nice study [that] emphasizes what we often forget: Lifestyle factors, especially salt intake, are important drivers of resistant hypertension. Our own studies have shown that this is predominantly a salt retaining state, and one would expect dietary salt restriction to be particularly effective in this group of patients and that is what this study showed,” said Bryan Williams, MD, who was asked to comment on the study. Dr. Williams is chair of medicine at University College London.
The results should also be reassuring to some who worried that exercise might lead to worsened blood pressure. “This study showed that in patients with resistant hypertension, though not out of control blood pressures, exercise was not only safe, but effective in lowering their blood pressure,” said Deepak L. Bhatt, MD, MPH, who was asked to comment. He is executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, and professor of medicine at Harvard Medical School, both in Boston.

The new research isn’t unique. In August, researchers in Portugal and Brazil showed that a 12-week exercise-only intervention reduced 24-hour and daytime ambulatory systolic and diastolic blood pressure. The two studies communicate the same message. “Lifestyle changes can work for resistant hypertension. So, two independent, modest-sized studies with essentially the same, positive, actionable conclusion,” said Dr. Bhatt, adding that the next step should be a larger, multicenter trial.
In the new study, 90 patients were assigned to the diet and exercise intervention and 50 to the control group. They had a mean age of 63 years, and 48% were women. Participants attended 94% of DASH diet classes and 89% of exercise sessions, and both groups had excellent adherence to medications.
The treatment group had a greater reduction in clinic systolic BP (–12.5 versus –7.1 mm Hg; P = .005) and diastolic BP (–5.9 versus –3.7 mm Hg; P = .034), as well as 24-hour ambulatory systolic BP (–7.0 versus –0.3 mm Hg; P = .001). The treatment group also had more improvement in resting baroreflex sensitivity (2.3 versus –1.1 ms/mm Hg; P = .003), high-frequency heart rate variability (0.4 versus –0.2 ln ms2; P = .025, and flow-mediated dilation (0.3% versus –1.4%, P = .022). The two groups had similar outcomes with respect to pulse wave velocity and left ventricular mass.
“Results of the TRIUMPH study suggest that policymakers should consider resistant hypertension as a new indication for cardiac rehabilitation with appropriate coverage by governmental agencies and private insurers,” Dr. Blumenthal said.
“This is an important new, evidence-based intervention for resistant hypertension. It is safe and relatively inexpensive. It should now be something physicians routinely offer these patients. Hopefully in the future, insurers will cover cardiac rehabilitation for patients with resistant hypertension,” Dr. Bhatt said.
The study also pointed towards an effective approach for patients who may be unable to sustain lifestyle changes. “Of course, not every patient will be able to maintain a healthy diet and an exercise program on their own, thus a cardiac rehabilitation program can be an excellent way to increase the likelihood of successful lifestyle modification,” Dr. Bhatt said.
TRIUMPH was sponsored by grants from the National Heart, Lung, and Blood Institute. None of the authors had disclosures to report. Dr. Bhatt disclosed having financial relationships with more than 40 pharmaceutical companies. Dr. Williams reported having no relevant financial disclosures.
A 4-month, structured diet and exercise intervention lowered blood pressure in adults with resistant blood pressure, according to results from a randomized, clinical trial. The program also led to improvements in baroreflex sensitivity, heart rate variability, and flow-mediated dilation, compared with individuals who received only a single education session.
The intervention included instruction from a nutritionist on how to follow the Dietary Approaches to Stop Hypertension (DASH) diet, as well as restricting calories and sodium to less than 2,300 mg/day. It included weekly, 45-minute group counseling sessions, run by a clinical psychiatrist, focusing on eating behaviors. The exercise component included 30- to 45-minute sessions at 70%-85% of initial heart rate reserve, carried out three times per week at a cardiac rehabilitation facility.
“While some individuals can make the lifestyle changes on their own, a structured program of supervised exercise and dietary modification conducted by a multidisciplinary team of physicians, psychologists, nutritionists, and physical therapists/exercise physiologists found in cardiac rehabilitation programs throughout the country is likely to be more effective. There are many cardiac rehabilitation programs throughout the country that are accessible to most patients,” said lead author James Blumenthal, PhD, the J.P. Gibbons Professor in Psychiatry and Behavioral Sciences at Duke University, Durham, N.C.
The study, called Treating Resistant Hypertension Using Lifestyle Modification to Promote Health (TRIUMPH), was published in Circulation. It is one of few that have examined lifestyle interventions in resistant hypertension, defined as systolic blood pressure ≥ 130 mm Hg or diastolic blood pressure ≥ 80 mm Hg after adherence to three or more optimally-dosed antihypertensive medications of three different classes, including one diuretic.
“This is a nice study [that] emphasizes what we often forget: Lifestyle factors, especially salt intake, are important drivers of resistant hypertension. Our own studies have shown that this is predominantly a salt retaining state, and one would expect dietary salt restriction to be particularly effective in this group of patients and that is what this study showed,” said Bryan Williams, MD, who was asked to comment on the study. Dr. Williams is chair of medicine at University College London.
The results should also be reassuring to some who worried that exercise might lead to worsened blood pressure. “This study showed that in patients with resistant hypertension, though not out of control blood pressures, exercise was not only safe, but effective in lowering their blood pressure,” said Deepak L. Bhatt, MD, MPH, who was asked to comment. He is executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, and professor of medicine at Harvard Medical School, both in Boston.
The new research isn’t unique. In August, researchers in Portugal and Brazil showed that a 12-week exercise-only intervention reduced 24-hour and daytime ambulatory systolic and diastolic blood pressure. The two studies communicate the same message. “Lifestyle changes can work for resistant hypertension. So, two independent, modest-sized studies with essentially the same, positive, actionable conclusion,” said Dr. Bhatt, adding that the next step should be a larger, multicenter trial.
In the new study, 90 patients were assigned to the diet and exercise intervention and 50 to the control group. They had a mean age of 63 years, and 48% were women. Participants attended 94% of DASH diet classes and 89% of exercise sessions, and both groups had excellent adherence to medications.
The treatment group had a greater reduction in clinic systolic BP (–12.5 versus –7.1 mm Hg; P = .005) and diastolic BP (–5.9 versus –3.7 mm Hg; P = .034), as well as 24-hour ambulatory systolic BP (–7.0 versus –0.3 mm Hg; P = .001). The treatment group also had more improvement in resting baroreflex sensitivity (2.3 versus –1.1 ms/mm Hg; P = .003), high-frequency heart rate variability (0.4 versus –0.2 ln ms2; P = .025, and flow-mediated dilation (0.3% versus –1.4%, P = .022). The two groups had similar outcomes with respect to pulse wave velocity and left ventricular mass.
“Results of the TRIUMPH study suggest that policymakers should consider resistant hypertension as a new indication for cardiac rehabilitation with appropriate coverage by governmental agencies and private insurers,” Dr. Blumenthal said.
“This is an important new, evidence-based intervention for resistant hypertension. It is safe and relatively inexpensive. It should now be something physicians routinely offer these patients. Hopefully in the future, insurers will cover cardiac rehabilitation for patients with resistant hypertension,” Dr. Bhatt said.
The study also pointed towards an effective approach for patients who may be unable to sustain lifestyle changes. “Of course, not every patient will be able to maintain a healthy diet and an exercise program on their own, thus a cardiac rehabilitation program can be an excellent way to increase the likelihood of successful lifestyle modification,” Dr. Bhatt said.
TRIUMPH was sponsored by grants from the National Heart, Lung, and Blood Institute. None of the authors had disclosures to report. Dr. Bhatt disclosed having financial relationships with more than 40 pharmaceutical companies. Dr. Williams reported having no relevant financial disclosures.
A 4-month, structured diet and exercise intervention lowered blood pressure in adults with resistant blood pressure, according to results from a randomized, clinical trial. The program also led to improvements in baroreflex sensitivity, heart rate variability, and flow-mediated dilation, compared with individuals who received only a single education session.
The intervention included instruction from a nutritionist on how to follow the Dietary Approaches to Stop Hypertension (DASH) diet, as well as restricting calories and sodium to less than 2,300 mg/day. It included weekly, 45-minute group counseling sessions, run by a clinical psychiatrist, focusing on eating behaviors. The exercise component included 30- to 45-minute sessions at 70%-85% of initial heart rate reserve, carried out three times per week at a cardiac rehabilitation facility.
“While some individuals can make the lifestyle changes on their own, a structured program of supervised exercise and dietary modification conducted by a multidisciplinary team of physicians, psychologists, nutritionists, and physical therapists/exercise physiologists found in cardiac rehabilitation programs throughout the country is likely to be more effective. There are many cardiac rehabilitation programs throughout the country that are accessible to most patients,” said lead author James Blumenthal, PhD, the J.P. Gibbons Professor in Psychiatry and Behavioral Sciences at Duke University, Durham, N.C.
The study, called Treating Resistant Hypertension Using Lifestyle Modification to Promote Health (TRIUMPH), was published in Circulation. It is one of few that have examined lifestyle interventions in resistant hypertension, defined as systolic blood pressure ≥ 130 mm Hg or diastolic blood pressure ≥ 80 mm Hg after adherence to three or more optimally-dosed antihypertensive medications of three different classes, including one diuretic.
“This is a nice study [that] emphasizes what we often forget: Lifestyle factors, especially salt intake, are important drivers of resistant hypertension. Our own studies have shown that this is predominantly a salt retaining state, and one would expect dietary salt restriction to be particularly effective in this group of patients and that is what this study showed,” said Bryan Williams, MD, who was asked to comment on the study. Dr. Williams is chair of medicine at University College London.
The results should also be reassuring to some who worried that exercise might lead to worsened blood pressure. “This study showed that in patients with resistant hypertension, though not out of control blood pressures, exercise was not only safe, but effective in lowering their blood pressure,” said Deepak L. Bhatt, MD, MPH, who was asked to comment. He is executive director of interventional cardiovascular programs at Brigham and Women’s Hospital, and professor of medicine at Harvard Medical School, both in Boston.
The new research isn’t unique. In August, researchers in Portugal and Brazil showed that a 12-week exercise-only intervention reduced 24-hour and daytime ambulatory systolic and diastolic blood pressure. The two studies communicate the same message. “Lifestyle changes can work for resistant hypertension. So, two independent, modest-sized studies with essentially the same, positive, actionable conclusion,” said Dr. Bhatt, adding that the next step should be a larger, multicenter trial.
In the new study, 90 patients were assigned to the diet and exercise intervention and 50 to the control group. They had a mean age of 63 years, and 48% were women. Participants attended 94% of DASH diet classes and 89% of exercise sessions, and both groups had excellent adherence to medications.
The treatment group had a greater reduction in clinic systolic BP (–12.5 versus –7.1 mm Hg; P = .005) and diastolic BP (–5.9 versus –3.7 mm Hg; P = .034), as well as 24-hour ambulatory systolic BP (–7.0 versus –0.3 mm Hg; P = .001). The treatment group also had more improvement in resting baroreflex sensitivity (2.3 versus –1.1 ms/mm Hg; P = .003), high-frequency heart rate variability (0.4 versus –0.2 ln ms2; P = .025, and flow-mediated dilation (0.3% versus –1.4%, P = .022). The two groups had similar outcomes with respect to pulse wave velocity and left ventricular mass.
“Results of the TRIUMPH study suggest that policymakers should consider resistant hypertension as a new indication for cardiac rehabilitation with appropriate coverage by governmental agencies and private insurers,” Dr. Blumenthal said.
“This is an important new, evidence-based intervention for resistant hypertension. It is safe and relatively inexpensive. It should now be something physicians routinely offer these patients. Hopefully in the future, insurers will cover cardiac rehabilitation for patients with resistant hypertension,” Dr. Bhatt said.
The study also pointed towards an effective approach for patients who may be unable to sustain lifestyle changes. “Of course, not every patient will be able to maintain a healthy diet and an exercise program on their own, thus a cardiac rehabilitation program can be an excellent way to increase the likelihood of successful lifestyle modification,” Dr. Bhatt said.
TRIUMPH was sponsored by grants from the National Heart, Lung, and Blood Institute. None of the authors had disclosures to report. Dr. Bhatt disclosed having financial relationships with more than 40 pharmaceutical companies. Dr. Williams reported having no relevant financial disclosures.
FROM CIRCULATION
Apple devices identify early Parkinson’s disease
, new research shows. Results from the WATCH-PD study show clear differences in a finger-tapping task in the Parkinson’s disease versus control group. The finger-tapping task also correlated with “traditional measures,” such as the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), investigators reported.
“And then the smartphone and smartwatch also showed differences in gait between groups,” said lead investigator Jamie Adams, MD, University of Rochester, New York.
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
WATCH-PD
The 12-month WATCH-PD study included 132 individuals at 17 Parkinson’s Study Group sites, 82 with Parkinson’s disease and 50 controls.
Participants with Parkinson’s disease were untreated, were no more than 2 years out from diagnosis (mean disease duration, 10.0 ±7.3 months), and were in Hoehn and Yahr stage 1 or 2.
Apple Watches and iPhones were provided to participants, all of whom underwent in-clinic assessments at baseline and at months 1, 3, 6, 9, and 12. The assessments included motor and cognitive tasks using the devices, which contained motion sensors.
The phone also contained an app that could assess verbal, cognitive, and other abilities. Participants wore a set of inertial sensors (APDM Mobility Lab) while performing the MDS-UPDRS Part III motor examination.
In addition, there were biweekly at-home tasks. Questions and tests on the watch assessed symptoms of mood, fatigue, cognition, and falls as well as cognitive performance involving perceptual, verbal, visual spatial, and fine motor abilities. Both the watch and iPhone were used to gauge gait, balance, and tremor.
Ages of the participants were approximately the same in the Parkinson’s disease and control groups (63.3 years vs. 60.2 years, respectively), but male to female ratios differed between the groups. There were more men in the Parkinson’s disease cohort (56% men vs. 44% women) and more women in the control cohort (36% vs. 64%; P =.03).
Between-group differences
Results showed that MDS-UPDRS total scores and on all individual parts of the rating scale were significantly better for the control group (lower scores are better), as shown in the following table.
Similarly, the control group performed better than the Parkinson’s disease group on the Montreal Cognitive Assessment (MoCA), with higher scores showing better performance on the 0 to 30 scale (28.1 vs. 27.6, respectively).
Touchscreen assessments on the phone also showed group differences in a finger-tapping task, with more taps by the control group than by the Parkinson’s disease group. The difference was more pronounced when the dominant hand was used.
The median numbers of taps in 20 seconds for the dominant hand were 103.7 for the Parkinson’s disease cohort versus 131.9 for control cohort (P < .005); and for the nondominant hand the numbers of taps were 106.6 versus 122.1 (P < .05), respectively. The control group also scored better on tests of hand fine-motor control (P < .01) and on the mobile digit symbols modalities test (P < .05)
Measures of gait in a 1-minute walk test also showed group differences.
“The five gait measures that differed most were cadence, which is steps per minute, double support, arm swing amplitude, arm swing variation, and turn duration,” Dr. Adams said.
‘Tremendous interest’
Commenting on the findings, Ludy Shih, MD, MMSc, of Boston University, noted that in the future, devices such as the ones used in this study may help clinicians remotely monitor their patients’ Parkinson’s disease conditions and response to therapy.
That would “eliminate some of the transportation barrier for people with Parkinson’s disease,” said Dr. Shih, who was not involved with the research.
The devices can give objective measurements, reducing inter-rater variability in assessment of movements, she noted.
“I think there’s tremendous interest in using digital measures to pick up on subtle disease phenotypes earlier than a clinical diagnosis can be made,” Dr. Shih said.
She also referred to literature “going back a few decades” showing that finger tapping can be used as a pharmacodynamic measure of how well a patient’s dopaminergic medications are working, so the devices may be a way to remotely assess treatment efficacy and decide when it is time to make adjustments.
Dr. Shih said she thinks regulatory agencies are now open “to consider these as part of the totality of evidence that a therapeutic [device] might be working.”
Whether these would need to be professional grade and approved as medical devices or if patients could just buy smartwatches and smartphones to generate useful data is still a question, she said. Already, there are several Parkinson’s apps that the public can download to track symptoms, improve voice, provide exercises, find support groups or research studies, and more.
Dr. Shih predicted that the biweekly at-home tasks, as in the current protocol, could be a burden to some people. If only a segment of the population were willing to comply, it could call into question how generalizable the results were, she added.
“There’s even a prior publication showing that compliance rate really dropped like a rock,” she noted. However, for those people willing to perform the tasks on a regular schedule, the results could be valuable, Dr. Shih said.
Dr. Adams concurred, saying that she had received feedback from some of her study participants that the biweekly tasks were a bit much.
The study was supported by Biogen and Takeda Pharmaceuticals. Dr. Adams receives research support from Biogen. Dr. Shih has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research shows. Results from the WATCH-PD study show clear differences in a finger-tapping task in the Parkinson’s disease versus control group. The finger-tapping task also correlated with “traditional measures,” such as the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), investigators reported.
“And then the smartphone and smartwatch also showed differences in gait between groups,” said lead investigator Jamie Adams, MD, University of Rochester, New York.
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
WATCH-PD
The 12-month WATCH-PD study included 132 individuals at 17 Parkinson’s Study Group sites, 82 with Parkinson’s disease and 50 controls.
Participants with Parkinson’s disease were untreated, were no more than 2 years out from diagnosis (mean disease duration, 10.0 ±7.3 months), and were in Hoehn and Yahr stage 1 or 2.
Apple Watches and iPhones were provided to participants, all of whom underwent in-clinic assessments at baseline and at months 1, 3, 6, 9, and 12. The assessments included motor and cognitive tasks using the devices, which contained motion sensors.
The phone also contained an app that could assess verbal, cognitive, and other abilities. Participants wore a set of inertial sensors (APDM Mobility Lab) while performing the MDS-UPDRS Part III motor examination.
In addition, there were biweekly at-home tasks. Questions and tests on the watch assessed symptoms of mood, fatigue, cognition, and falls as well as cognitive performance involving perceptual, verbal, visual spatial, and fine motor abilities. Both the watch and iPhone were used to gauge gait, balance, and tremor.
Ages of the participants were approximately the same in the Parkinson’s disease and control groups (63.3 years vs. 60.2 years, respectively), but male to female ratios differed between the groups. There were more men in the Parkinson’s disease cohort (56% men vs. 44% women) and more women in the control cohort (36% vs. 64%; P =.03).
Between-group differences
Results showed that MDS-UPDRS total scores and on all individual parts of the rating scale were significantly better for the control group (lower scores are better), as shown in the following table.
Similarly, the control group performed better than the Parkinson’s disease group on the Montreal Cognitive Assessment (MoCA), with higher scores showing better performance on the 0 to 30 scale (28.1 vs. 27.6, respectively).
Touchscreen assessments on the phone also showed group differences in a finger-tapping task, with more taps by the control group than by the Parkinson’s disease group. The difference was more pronounced when the dominant hand was used.
The median numbers of taps in 20 seconds for the dominant hand were 103.7 for the Parkinson’s disease cohort versus 131.9 for control cohort (P < .005); and for the nondominant hand the numbers of taps were 106.6 versus 122.1 (P < .05), respectively. The control group also scored better on tests of hand fine-motor control (P < .01) and on the mobile digit symbols modalities test (P < .05)
Measures of gait in a 1-minute walk test also showed group differences.
“The five gait measures that differed most were cadence, which is steps per minute, double support, arm swing amplitude, arm swing variation, and turn duration,” Dr. Adams said.
‘Tremendous interest’
Commenting on the findings, Ludy Shih, MD, MMSc, of Boston University, noted that in the future, devices such as the ones used in this study may help clinicians remotely monitor their patients’ Parkinson’s disease conditions and response to therapy.
That would “eliminate some of the transportation barrier for people with Parkinson’s disease,” said Dr. Shih, who was not involved with the research.
The devices can give objective measurements, reducing inter-rater variability in assessment of movements, she noted.
“I think there’s tremendous interest in using digital measures to pick up on subtle disease phenotypes earlier than a clinical diagnosis can be made,” Dr. Shih said.
She also referred to literature “going back a few decades” showing that finger tapping can be used as a pharmacodynamic measure of how well a patient’s dopaminergic medications are working, so the devices may be a way to remotely assess treatment efficacy and decide when it is time to make adjustments.
Dr. Shih said she thinks regulatory agencies are now open “to consider these as part of the totality of evidence that a therapeutic [device] might be working.”
Whether these would need to be professional grade and approved as medical devices or if patients could just buy smartwatches and smartphones to generate useful data is still a question, she said. Already, there are several Parkinson’s apps that the public can download to track symptoms, improve voice, provide exercises, find support groups or research studies, and more.
Dr. Shih predicted that the biweekly at-home tasks, as in the current protocol, could be a burden to some people. If only a segment of the population were willing to comply, it could call into question how generalizable the results were, she added.
“There’s even a prior publication showing that compliance rate really dropped like a rock,” she noted. However, for those people willing to perform the tasks on a regular schedule, the results could be valuable, Dr. Shih said.
Dr. Adams concurred, saying that she had received feedback from some of her study participants that the biweekly tasks were a bit much.
The study was supported by Biogen and Takeda Pharmaceuticals. Dr. Adams receives research support from Biogen. Dr. Shih has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
, new research shows. Results from the WATCH-PD study show clear differences in a finger-tapping task in the Parkinson’s disease versus control group. The finger-tapping task also correlated with “traditional measures,” such as the Movement Disorder Society–Unified Parkinson’s Disease Rating Scale (MDS-UPDRS), investigators reported.
“And then the smartphone and smartwatch also showed differences in gait between groups,” said lead investigator Jamie Adams, MD, University of Rochester, New York.
The findings were presented at the International Congress of Parkinson’s Disease and Movement Disorders.
WATCH-PD
The 12-month WATCH-PD study included 132 individuals at 17 Parkinson’s Study Group sites, 82 with Parkinson’s disease and 50 controls.
Participants with Parkinson’s disease were untreated, were no more than 2 years out from diagnosis (mean disease duration, 10.0 ±7.3 months), and were in Hoehn and Yahr stage 1 or 2.
Apple Watches and iPhones were provided to participants, all of whom underwent in-clinic assessments at baseline and at months 1, 3, 6, 9, and 12. The assessments included motor and cognitive tasks using the devices, which contained motion sensors.
The phone also contained an app that could assess verbal, cognitive, and other abilities. Participants wore a set of inertial sensors (APDM Mobility Lab) while performing the MDS-UPDRS Part III motor examination.
In addition, there were biweekly at-home tasks. Questions and tests on the watch assessed symptoms of mood, fatigue, cognition, and falls as well as cognitive performance involving perceptual, verbal, visual spatial, and fine motor abilities. Both the watch and iPhone were used to gauge gait, balance, and tremor.
Ages of the participants were approximately the same in the Parkinson’s disease and control groups (63.3 years vs. 60.2 years, respectively), but male to female ratios differed between the groups. There were more men in the Parkinson’s disease cohort (56% men vs. 44% women) and more women in the control cohort (36% vs. 64%; P =.03).
Between-group differences
Results showed that MDS-UPDRS total scores and on all individual parts of the rating scale were significantly better for the control group (lower scores are better), as shown in the following table.
Similarly, the control group performed better than the Parkinson’s disease group on the Montreal Cognitive Assessment (MoCA), with higher scores showing better performance on the 0 to 30 scale (28.1 vs. 27.6, respectively).
Touchscreen assessments on the phone also showed group differences in a finger-tapping task, with more taps by the control group than by the Parkinson’s disease group. The difference was more pronounced when the dominant hand was used.
The median numbers of taps in 20 seconds for the dominant hand were 103.7 for the Parkinson’s disease cohort versus 131.9 for control cohort (P < .005); and for the nondominant hand the numbers of taps were 106.6 versus 122.1 (P < .05), respectively. The control group also scored better on tests of hand fine-motor control (P < .01) and on the mobile digit symbols modalities test (P < .05)
Measures of gait in a 1-minute walk test also showed group differences.
“The five gait measures that differed most were cadence, which is steps per minute, double support, arm swing amplitude, arm swing variation, and turn duration,” Dr. Adams said.
‘Tremendous interest’
Commenting on the findings, Ludy Shih, MD, MMSc, of Boston University, noted that in the future, devices such as the ones used in this study may help clinicians remotely monitor their patients’ Parkinson’s disease conditions and response to therapy.
That would “eliminate some of the transportation barrier for people with Parkinson’s disease,” said Dr. Shih, who was not involved with the research.
The devices can give objective measurements, reducing inter-rater variability in assessment of movements, she noted.
“I think there’s tremendous interest in using digital measures to pick up on subtle disease phenotypes earlier than a clinical diagnosis can be made,” Dr. Shih said.
She also referred to literature “going back a few decades” showing that finger tapping can be used as a pharmacodynamic measure of how well a patient’s dopaminergic medications are working, so the devices may be a way to remotely assess treatment efficacy and decide when it is time to make adjustments.
Dr. Shih said she thinks regulatory agencies are now open “to consider these as part of the totality of evidence that a therapeutic [device] might be working.”
Whether these would need to be professional grade and approved as medical devices or if patients could just buy smartwatches and smartphones to generate useful data is still a question, she said. Already, there are several Parkinson’s apps that the public can download to track symptoms, improve voice, provide exercises, find support groups or research studies, and more.
Dr. Shih predicted that the biweekly at-home tasks, as in the current protocol, could be a burden to some people. If only a segment of the population were willing to comply, it could call into question how generalizable the results were, she added.
“There’s even a prior publication showing that compliance rate really dropped like a rock,” she noted. However, for those people willing to perform the tasks on a regular schedule, the results could be valuable, Dr. Shih said.
Dr. Adams concurred, saying that she had received feedback from some of her study participants that the biweekly tasks were a bit much.
The study was supported by Biogen and Takeda Pharmaceuticals. Dr. Adams receives research support from Biogen. Dr. Shih has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM MDS VIRTUAL CONGRESS 2021
The soccer punch
Soccer is the most popular sport on Earth.
A recent JAMA Neurology article noted the incidence of neurodegenerative disease in retired professional soccer players. It found that, not surprisingly, the frequency of such was higher than in the general population, highest amongst defenders and lowest in goalkeepers (presumably because the latter can use their hands).

The point here shouldn’t surprise anyone: Repeatedly hitting your head on solid objects is a bad idea.
Somewhere, a long time ago, early vertebrates developed a bony case to protect their centralized nervous system. Its success is shown by the fact that skulls and spinal cords among vertebrates have more similarities than differences: They work. It’s true that some ungulates use their heads to fight, but their skulls are adapted for such, being thicker and having horns and antlers to lessen the impacts.
But humans? Nope. The skull can support up to nine tons of (slowly-applied) weight (don’t try this at home) but repeated impacts aren’t good for its contents.
There is no degree of external protection that will prevent this, either. We talk about helmets, but the reality is that, while they definitely reduce exterior injuries, they do very little to prevent the effects of rapid acceleration/deceleration on the brain inside. This is what results in concussions, coup & contra-coup injuries, and the shearing effects of diffuse axonal injury.
I’m not saying we should ban soccer, or football, or any of the other activities that clearly have a high risk of repeated head trauma. They’re ingrained into the cultures of our societies.
At this point it’s pretty much impossible for participants and their family members to not be aware of the risks posed by these sports. The popular press has covered it in great detail.
At some point there’s only so much you can warn people about. Like tobacco smoking or riding without a helmet, you accept the risks, fully aware of the serious potential consequences. For those who wish to participate, it’s their decision.
But it’s also time to stop blinding ourselves to the simple facts. Minimizing them, pointing out their delayed onset, and turning a blind eye won’t change that.
If we’re going to continue enjoying contact sports, we have to accept that someone is going to pay the price for it, even if they’ve been forewarned. And no amount of protective gear, at today’s technology, is going to change that.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Soccer is the most popular sport on Earth.
A recent JAMA Neurology article noted the incidence of neurodegenerative disease in retired professional soccer players. It found that, not surprisingly, the frequency of such was higher than in the general population, highest amongst defenders and lowest in goalkeepers (presumably because the latter can use their hands).
The point here shouldn’t surprise anyone: Repeatedly hitting your head on solid objects is a bad idea.
Somewhere, a long time ago, early vertebrates developed a bony case to protect their centralized nervous system. Its success is shown by the fact that skulls and spinal cords among vertebrates have more similarities than differences: They work. It’s true that some ungulates use their heads to fight, but their skulls are adapted for such, being thicker and having horns and antlers to lessen the impacts.
But humans? Nope. The skull can support up to nine tons of (slowly-applied) weight (don’t try this at home) but repeated impacts aren’t good for its contents.
There is no degree of external protection that will prevent this, either. We talk about helmets, but the reality is that, while they definitely reduce exterior injuries, they do very little to prevent the effects of rapid acceleration/deceleration on the brain inside. This is what results in concussions, coup & contra-coup injuries, and the shearing effects of diffuse axonal injury.
I’m not saying we should ban soccer, or football, or any of the other activities that clearly have a high risk of repeated head trauma. They’re ingrained into the cultures of our societies.
At this point it’s pretty much impossible for participants and their family members to not be aware of the risks posed by these sports. The popular press has covered it in great detail.
At some point there’s only so much you can warn people about. Like tobacco smoking or riding without a helmet, you accept the risks, fully aware of the serious potential consequences. For those who wish to participate, it’s their decision.
But it’s also time to stop blinding ourselves to the simple facts. Minimizing them, pointing out their delayed onset, and turning a blind eye won’t change that.
If we’re going to continue enjoying contact sports, we have to accept that someone is going to pay the price for it, even if they’ve been forewarned. And no amount of protective gear, at today’s technology, is going to change that.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Soccer is the most popular sport on Earth.
A recent JAMA Neurology article noted the incidence of neurodegenerative disease in retired professional soccer players. It found that, not surprisingly, the frequency of such was higher than in the general population, highest amongst defenders and lowest in goalkeepers (presumably because the latter can use their hands).
The point here shouldn’t surprise anyone: Repeatedly hitting your head on solid objects is a bad idea.
Somewhere, a long time ago, early vertebrates developed a bony case to protect their centralized nervous system. Its success is shown by the fact that skulls and spinal cords among vertebrates have more similarities than differences: They work. It’s true that some ungulates use their heads to fight, but their skulls are adapted for such, being thicker and having horns and antlers to lessen the impacts.
But humans? Nope. The skull can support up to nine tons of (slowly-applied) weight (don’t try this at home) but repeated impacts aren’t good for its contents.
There is no degree of external protection that will prevent this, either. We talk about helmets, but the reality is that, while they definitely reduce exterior injuries, they do very little to prevent the effects of rapid acceleration/deceleration on the brain inside. This is what results in concussions, coup & contra-coup injuries, and the shearing effects of diffuse axonal injury.
I’m not saying we should ban soccer, or football, or any of the other activities that clearly have a high risk of repeated head trauma. They’re ingrained into the cultures of our societies.
At this point it’s pretty much impossible for participants and their family members to not be aware of the risks posed by these sports. The popular press has covered it in great detail.
At some point there’s only so much you can warn people about. Like tobacco smoking or riding without a helmet, you accept the risks, fully aware of the serious potential consequences. For those who wish to participate, it’s their decision.
But it’s also time to stop blinding ourselves to the simple facts. Minimizing them, pointing out their delayed onset, and turning a blind eye won’t change that.
If we’re going to continue enjoying contact sports, we have to accept that someone is going to pay the price for it, even if they’ve been forewarned. And no amount of protective gear, at today’s technology, is going to change that.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Updates to CDC’s STI guidelines relevant to midlife women too
Sexually transmitted infection rates have not increased as dramatically in older women as they have in women in their teens and 20s, but rates of chlamydia and gonorrhea in women over age 35 have seen a steady incline over the past decade, and syphilis rates have climbed steeply, according to data from the Centers for Disease Control and Prevention.
That makes the STI treatment guidelines released by the CDC in July even timelier for practitioners of menopause medicine, according to Michael S. Policar, MD, MPH, a professor emeritus of ob.gyn. and reproductive sciences at the University of California, San Francisco.
Dr. Policar discussed what clinicians need to know about STIs in midlife women at the hybrid annual meeting of the North American Menopause Society. Even the nomenclature change in the guidelines from “sexually transmitted diseases” to “sexually transmitted infections” is important “because they want to acknowledge the fact that a lot of the sexually transmitted infections that we’re treating are asymptomatic, are colonizations, and are not yet diseases,” Dr. Policar said. “We’re trying to be much more expansive in thinking about finding these infections before they actually start causing morbidity in the form of a disease.”
Sexual history
The primary guidelines update for taking sexual history is the recommendation to ask patients about their intentions regarding pregnancy. The “5 Ps” of sexual history are now Partners, Practices, Protection from STIs, Past history of STIs, and Pregnancy intention.
“There should be a sixth P that has to do with pleasure questions,” Policar added. “We ask all the time for patients that we see in the context of perimenopausal and menopausal services, ‘Are you satisfied with your sexual relationship with your partner?’ Hopefully that will make it into the CDC guidelines as the sixth P at some point, but for now, that’s aspirational.”
In asking about partners, instead of asking patients whether they have sex with men, women, or both, clinicians should ask first if the patient is having sex of any kind – oral, vaginal, or anal – with anyone. From there, providers should ask how many sex partners the patient has had, the gender(s) of the partners, and whether they or their partners have other sex partners, using more gender-inclusive language.
When asking about practices, in addition to asking about the type of sexual contact patients have had, additional questions include whether the patient met their partners online or through apps, whether they or any of their partners use drugs, and whether the patient has exchanged sex for any needs, such as money, housing, or drugs. The additional questions can identify those at higher risk for STIs.
After reviewing the CDC’s list of risk factors for gonorrhea and chlamydia screening, Dr. Policar shared the screening list from the California Department of Public Health, which he finds more helpful:
- History of gonorrhea, chlamydia, or pelvic inflammatory disease (PID) in the past 2 years.
- More than 1 sexual partner in the past year.
- New sexual partner within 90 days.
- Reason to believe that a sex partner has had other partners in the past year.
- Exchanging sex for drugs or money within the past year.
- Other factors identified locally, including prevalence of infection in the community.
STI screening guidelines
For those with a positive gonorrhea/chlamydia (GC/CT) screen, a nucleic acid amplification test (NAAT) vaginal swab is the preferred specimen source, and self-collection is fine for women of any age, Dr. Policar said. In addition, cis-women who received anal intercourse in the preceding year should consider undergoing a rectal GC/CT NAAT, and those who performed oral sex should consider a pharyngeal GC/CT NAAT, based on shared clinical decision-making. A rectal swab requires an insertion of 3-4 cm and a 360-degree twirl of the wrist, not the swab, to ensure you get a sample from the entire circumference. Pharyngeal samples require swabbing both tonsillar pillars while taking care for those who may gag.
For contact testing – asymptomatic people who have had a high-risk sexual exposure – providers should test for gonorrhea, chlamydia, HIV, and syphilis but not for herpes, high-risk HPV, hepatitis B, hepatitis C, or bacterial vaginosis. “Maybe we’ll do a screen for trichomoniasis, and maybe we’ll offer herpes type 2 serology or antibody screening,” Dr. Policar said. Providers should also ask patients requesting contact testing if they have been vaccinated for hepatitis B. If not, “the conversation should be how can we get you vaccinated for hepatitis B,” Dr. Policar said.
HIV screening only needs to occur once between the ages of 15 and 65 for low-risk people and then once annually (or more often if necessary) for those who have a sex partner with HIV, use injectable drugs, engage in commercial sex work, have a new sex partner with unknown HIV status, received care at an STD or TB clinic, or were in a correctional facility or homeless shelter.
Those at increased risk for syphilis include men who have sex with men, men under age 29, and anyone living with HIV or who has a history of incarceration or a history of commercial sex work. In addition, African Americans have the greatest risk for syphilis of racial/ethnic groups, followed by Hispanics. Most adults only require hepatitis C screening with anti-hep C antibody testing once in their lifetime. Periodic hepatitis C screening should occur for people who inject drugs. If the screening is positive, providers should conduct an RNA polymerase chain reaction (PCR) test to determine whether a chronic infection is present.
Trichomoniasis screening should occur annually in women living with HIV or in correctional facilities. Others to consider screening include people with new or multiple sex partners, a history of STIs, inconsistent condom use, a history of sex work, and intravenous drug use. Dr. Policar also noted that several new assays, including NAAT, PCR, and a rapid test, are available for trichomoniasis.
STI treatment guidelines
For women with mucoprurulent cervicitis, the cause could be chlamydia, gonorrhea, herpes, trichomonas, mycoplasma, or even progesterone from pregnancy or contraception, Dr. Policar said. The new preferred treatment is 100 mg of doxycycline. The alternative, albeit less preferred, treatment is 1 g azithromycin.
The preferred treatment for chlamydia is now 100 mg oral doxycycline twice daily, or doxycycline 200 mg delayed-release once daily, for 7 days. Alternative regimens include 1 g oral azithromycin in a single dose or 500 mg oral levofloxacin once daily for 7 days. The switch to recommending doxycycline over azithromycin is based on recent evidence showing that doxycycline has a slightly higher efficacy for urogenital chlamydia and a substantially higher efficacy for rectal chlamydia. In addition, an increasing proportion of gonorrheal infections have shown resistance to azithromycin, particularly beginning in 2014.
Preferred treatment of new, uncomplicated gonorrhea infections of the cervix, urethra, rectum, and pharynx is one 500-mg dose of ceftriaxone for those weighing under 150 kg and 1 g for those weighing 150 kg or more. If ceftriaxone is unavailable, the new alternative recommended treatment for gonorrhea is 800 mg cefixime. For pharyngeal gonorrhea only, the CDC recommends a test-of-cure 7-14 days after treatment.
For gonorrheal infections, the CDC also recommends treatment with doxycycline if chlamydia has not been excluded, but the agency no longer recommends dual therapy with azithromycin unless it’s used in place of doxycycline for those who are pregnant, have an allergy, or may not be compliant with a 7-day doxycycline regimen.
The preferred treatment for bacterial vaginosis has not changed. The new recommended regimen for trichomoniasis is 500 mg oral metronidazole for 7 days, with the alternative being a single 2-g dose of tinidazole. Male partners should receive 2 g oral metronidazole. The CDC also notes that patients taking metronidazole no longer need to abstain from alcohol during treatment.
”Another area where the guidelines changed is in their description of expedited partner therapy, which means that, when we find an index case who has gonorrhea or chlamydia, we always have a discussion with her about getting her partners treated,” Dr. Policar said. “The CDC was quite clear that the responsibility for discussing partner treatment rests with us as the diagnosing provider” since city and county health departments don’t have the time or resources for contact tracing these STIs.
The two main ways to treat partners are to have the patient bring their partner(s) to the appointment with them or to do patient-delivered partner therapy. Ideally, clinicians who dispense their own medications can give the patient enough drugs to give her partner(s) a complete dose as well. Otherwise, providers can prescribe extra doses in the index patients’ name or write prescriptions in the partner’s name.
“In every state of the union now, it is legal for you to to prescribe antibiotics for partners sight unseen, Dr. Policar said.
Margaret Sullivan, MD, an ob.gyn. from rural western North Carolina, noted during the Q&A that an obstacle to partner therapy at her practice has been cost, particularly since many of the men don’t have insurance.
“I have not heard before of prescribing the extra doses for partners under the patient’s name,” Dr. Sullivan said. “I’ve thought about doing it, but [was worried about] it potentially being fraudulent if that patient has Medicaid and we’re prescribing extra doses under her name, so how do you work around that?”
Dr. Policar acknowledged that barrier and recommended that patients use the website/app Goodrx.com to find discounts for out-of-pocket generic medications. He also noted the occasional obstacle of pharmacists balking at filling a double or triple dose.
“What we’ve been suggesting in that circumstance is to literally copy that part of the CDC guidelines, which explains expedited partner therapy or patient-delivered partner therapy and send that off to the pharmacist so they can see that it’s a national recommendation of the CDC,” Dr. Policar said.
Claudia Rodriguez, MD, an ob.gyn. who works at Sherman Hospital in Elgin, Ill., asked about the CDC recommendations for HPV vaccination in older women. Although the CDC permits women over age 26 to receive the HPV vaccine, the agency does not “make a solid recommendation to have this done, which oftentimes makes a big difference in whether or not health insurance will actually pay for vaccination in that circumstance,” Dr. Policar said.
Patients are welcome to request the vaccine after shared decision-making, but “we should never present this as something which is routine,” he said. For women in their 50s, for example, “there’s virtually no data about any additional degree of protection that you would get” from HPV vaccination, Dr. Policar said in response to a similar question from Tara Allmen, MD, an ob.gyn. in New York City. “If you ask me for my personal clinical opinion about it, I would say it’s not going to be worth it,” he said.
Dr Policar had no disclosures. Disclosures were unavailable for attendees who spoke.
Sexually transmitted infection rates have not increased as dramatically in older women as they have in women in their teens and 20s, but rates of chlamydia and gonorrhea in women over age 35 have seen a steady incline over the past decade, and syphilis rates have climbed steeply, according to data from the Centers for Disease Control and Prevention.
That makes the STI treatment guidelines released by the CDC in July even timelier for practitioners of menopause medicine, according to Michael S. Policar, MD, MPH, a professor emeritus of ob.gyn. and reproductive sciences at the University of California, San Francisco.
Dr. Policar discussed what clinicians need to know about STIs in midlife women at the hybrid annual meeting of the North American Menopause Society. Even the nomenclature change in the guidelines from “sexually transmitted diseases” to “sexually transmitted infections” is important “because they want to acknowledge the fact that a lot of the sexually transmitted infections that we’re treating are asymptomatic, are colonizations, and are not yet diseases,” Dr. Policar said. “We’re trying to be much more expansive in thinking about finding these infections before they actually start causing morbidity in the form of a disease.”
Sexual history
The primary guidelines update for taking sexual history is the recommendation to ask patients about their intentions regarding pregnancy. The “5 Ps” of sexual history are now Partners, Practices, Protection from STIs, Past history of STIs, and Pregnancy intention.
“There should be a sixth P that has to do with pleasure questions,” Policar added. “We ask all the time for patients that we see in the context of perimenopausal and menopausal services, ‘Are you satisfied with your sexual relationship with your partner?’ Hopefully that will make it into the CDC guidelines as the sixth P at some point, but for now, that’s aspirational.”
In asking about partners, instead of asking patients whether they have sex with men, women, or both, clinicians should ask first if the patient is having sex of any kind – oral, vaginal, or anal – with anyone. From there, providers should ask how many sex partners the patient has had, the gender(s) of the partners, and whether they or their partners have other sex partners, using more gender-inclusive language.
When asking about practices, in addition to asking about the type of sexual contact patients have had, additional questions include whether the patient met their partners online or through apps, whether they or any of their partners use drugs, and whether the patient has exchanged sex for any needs, such as money, housing, or drugs. The additional questions can identify those at higher risk for STIs.
After reviewing the CDC’s list of risk factors for gonorrhea and chlamydia screening, Dr. Policar shared the screening list from the California Department of Public Health, which he finds more helpful:
- History of gonorrhea, chlamydia, or pelvic inflammatory disease (PID) in the past 2 years.
- More than 1 sexual partner in the past year.
- New sexual partner within 90 days.
- Reason to believe that a sex partner has had other partners in the past year.
- Exchanging sex for drugs or money within the past year.
- Other factors identified locally, including prevalence of infection in the community.
STI screening guidelines
For those with a positive gonorrhea/chlamydia (GC/CT) screen, a nucleic acid amplification test (NAAT) vaginal swab is the preferred specimen source, and self-collection is fine for women of any age, Dr. Policar said. In addition, cis-women who received anal intercourse in the preceding year should consider undergoing a rectal GC/CT NAAT, and those who performed oral sex should consider a pharyngeal GC/CT NAAT, based on shared clinical decision-making. A rectal swab requires an insertion of 3-4 cm and a 360-degree twirl of the wrist, not the swab, to ensure you get a sample from the entire circumference. Pharyngeal samples require swabbing both tonsillar pillars while taking care for those who may gag.
For contact testing – asymptomatic people who have had a high-risk sexual exposure – providers should test for gonorrhea, chlamydia, HIV, and syphilis but not for herpes, high-risk HPV, hepatitis B, hepatitis C, or bacterial vaginosis. “Maybe we’ll do a screen for trichomoniasis, and maybe we’ll offer herpes type 2 serology or antibody screening,” Dr. Policar said. Providers should also ask patients requesting contact testing if they have been vaccinated for hepatitis B. If not, “the conversation should be how can we get you vaccinated for hepatitis B,” Dr. Policar said.
HIV screening only needs to occur once between the ages of 15 and 65 for low-risk people and then once annually (or more often if necessary) for those who have a sex partner with HIV, use injectable drugs, engage in commercial sex work, have a new sex partner with unknown HIV status, received care at an STD or TB clinic, or were in a correctional facility or homeless shelter.
Those at increased risk for syphilis include men who have sex with men, men under age 29, and anyone living with HIV or who has a history of incarceration or a history of commercial sex work. In addition, African Americans have the greatest risk for syphilis of racial/ethnic groups, followed by Hispanics. Most adults only require hepatitis C screening with anti-hep C antibody testing once in their lifetime. Periodic hepatitis C screening should occur for people who inject drugs. If the screening is positive, providers should conduct an RNA polymerase chain reaction (PCR) test to determine whether a chronic infection is present.
Trichomoniasis screening should occur annually in women living with HIV or in correctional facilities. Others to consider screening include people with new or multiple sex partners, a history of STIs, inconsistent condom use, a history of sex work, and intravenous drug use. Dr. Policar also noted that several new assays, including NAAT, PCR, and a rapid test, are available for trichomoniasis.
STI treatment guidelines
For women with mucoprurulent cervicitis, the cause could be chlamydia, gonorrhea, herpes, trichomonas, mycoplasma, or even progesterone from pregnancy or contraception, Dr. Policar said. The new preferred treatment is 100 mg of doxycycline. The alternative, albeit less preferred, treatment is 1 g azithromycin.
The preferred treatment for chlamydia is now 100 mg oral doxycycline twice daily, or doxycycline 200 mg delayed-release once daily, for 7 days. Alternative regimens include 1 g oral azithromycin in a single dose or 500 mg oral levofloxacin once daily for 7 days. The switch to recommending doxycycline over azithromycin is based on recent evidence showing that doxycycline has a slightly higher efficacy for urogenital chlamydia and a substantially higher efficacy for rectal chlamydia. In addition, an increasing proportion of gonorrheal infections have shown resistance to azithromycin, particularly beginning in 2014.
Preferred treatment of new, uncomplicated gonorrhea infections of the cervix, urethra, rectum, and pharynx is one 500-mg dose of ceftriaxone for those weighing under 150 kg and 1 g for those weighing 150 kg or more. If ceftriaxone is unavailable, the new alternative recommended treatment for gonorrhea is 800 mg cefixime. For pharyngeal gonorrhea only, the CDC recommends a test-of-cure 7-14 days after treatment.
For gonorrheal infections, the CDC also recommends treatment with doxycycline if chlamydia has not been excluded, but the agency no longer recommends dual therapy with azithromycin unless it’s used in place of doxycycline for those who are pregnant, have an allergy, or may not be compliant with a 7-day doxycycline regimen.
The preferred treatment for bacterial vaginosis has not changed. The new recommended regimen for trichomoniasis is 500 mg oral metronidazole for 7 days, with the alternative being a single 2-g dose of tinidazole. Male partners should receive 2 g oral metronidazole. The CDC also notes that patients taking metronidazole no longer need to abstain from alcohol during treatment.
”Another area where the guidelines changed is in their description of expedited partner therapy, which means that, when we find an index case who has gonorrhea or chlamydia, we always have a discussion with her about getting her partners treated,” Dr. Policar said. “The CDC was quite clear that the responsibility for discussing partner treatment rests with us as the diagnosing provider” since city and county health departments don’t have the time or resources for contact tracing these STIs.
The two main ways to treat partners are to have the patient bring their partner(s) to the appointment with them or to do patient-delivered partner therapy. Ideally, clinicians who dispense their own medications can give the patient enough drugs to give her partner(s) a complete dose as well. Otherwise, providers can prescribe extra doses in the index patients’ name or write prescriptions in the partner’s name.
“In every state of the union now, it is legal for you to to prescribe antibiotics for partners sight unseen, Dr. Policar said.
Margaret Sullivan, MD, an ob.gyn. from rural western North Carolina, noted during the Q&A that an obstacle to partner therapy at her practice has been cost, particularly since many of the men don’t have insurance.
“I have not heard before of prescribing the extra doses for partners under the patient’s name,” Dr. Sullivan said. “I’ve thought about doing it, but [was worried about] it potentially being fraudulent if that patient has Medicaid and we’re prescribing extra doses under her name, so how do you work around that?”
Dr. Policar acknowledged that barrier and recommended that patients use the website/app Goodrx.com to find discounts for out-of-pocket generic medications. He also noted the occasional obstacle of pharmacists balking at filling a double or triple dose.
“What we’ve been suggesting in that circumstance is to literally copy that part of the CDC guidelines, which explains expedited partner therapy or patient-delivered partner therapy and send that off to the pharmacist so they can see that it’s a national recommendation of the CDC,” Dr. Policar said.
Claudia Rodriguez, MD, an ob.gyn. who works at Sherman Hospital in Elgin, Ill., asked about the CDC recommendations for HPV vaccination in older women. Although the CDC permits women over age 26 to receive the HPV vaccine, the agency does not “make a solid recommendation to have this done, which oftentimes makes a big difference in whether or not health insurance will actually pay for vaccination in that circumstance,” Dr. Policar said.
Patients are welcome to request the vaccine after shared decision-making, but “we should never present this as something which is routine,” he said. For women in their 50s, for example, “there’s virtually no data about any additional degree of protection that you would get” from HPV vaccination, Dr. Policar said in response to a similar question from Tara Allmen, MD, an ob.gyn. in New York City. “If you ask me for my personal clinical opinion about it, I would say it’s not going to be worth it,” he said.
Dr Policar had no disclosures. Disclosures were unavailable for attendees who spoke.
Sexually transmitted infection rates have not increased as dramatically in older women as they have in women in their teens and 20s, but rates of chlamydia and gonorrhea in women over age 35 have seen a steady incline over the past decade, and syphilis rates have climbed steeply, according to data from the Centers for Disease Control and Prevention.
That makes the STI treatment guidelines released by the CDC in July even timelier for practitioners of menopause medicine, according to Michael S. Policar, MD, MPH, a professor emeritus of ob.gyn. and reproductive sciences at the University of California, San Francisco.
Dr. Policar discussed what clinicians need to know about STIs in midlife women at the hybrid annual meeting of the North American Menopause Society. Even the nomenclature change in the guidelines from “sexually transmitted diseases” to “sexually transmitted infections” is important “because they want to acknowledge the fact that a lot of the sexually transmitted infections that we’re treating are asymptomatic, are colonizations, and are not yet diseases,” Dr. Policar said. “We’re trying to be much more expansive in thinking about finding these infections before they actually start causing morbidity in the form of a disease.”
Sexual history
The primary guidelines update for taking sexual history is the recommendation to ask patients about their intentions regarding pregnancy. The “5 Ps” of sexual history are now Partners, Practices, Protection from STIs, Past history of STIs, and Pregnancy intention.
“There should be a sixth P that has to do with pleasure questions,” Policar added. “We ask all the time for patients that we see in the context of perimenopausal and menopausal services, ‘Are you satisfied with your sexual relationship with your partner?’ Hopefully that will make it into the CDC guidelines as the sixth P at some point, but for now, that’s aspirational.”
In asking about partners, instead of asking patients whether they have sex with men, women, or both, clinicians should ask first if the patient is having sex of any kind – oral, vaginal, or anal – with anyone. From there, providers should ask how many sex partners the patient has had, the gender(s) of the partners, and whether they or their partners have other sex partners, using more gender-inclusive language.
When asking about practices, in addition to asking about the type of sexual contact patients have had, additional questions include whether the patient met their partners online or through apps, whether they or any of their partners use drugs, and whether the patient has exchanged sex for any needs, such as money, housing, or drugs. The additional questions can identify those at higher risk for STIs.
After reviewing the CDC’s list of risk factors for gonorrhea and chlamydia screening, Dr. Policar shared the screening list from the California Department of Public Health, which he finds more helpful:
- History of gonorrhea, chlamydia, or pelvic inflammatory disease (PID) in the past 2 years.
- More than 1 sexual partner in the past year.
- New sexual partner within 90 days.
- Reason to believe that a sex partner has had other partners in the past year.
- Exchanging sex for drugs or money within the past year.
- Other factors identified locally, including prevalence of infection in the community.
STI screening guidelines
For those with a positive gonorrhea/chlamydia (GC/CT) screen, a nucleic acid amplification test (NAAT) vaginal swab is the preferred specimen source, and self-collection is fine for women of any age, Dr. Policar said. In addition, cis-women who received anal intercourse in the preceding year should consider undergoing a rectal GC/CT NAAT, and those who performed oral sex should consider a pharyngeal GC/CT NAAT, based on shared clinical decision-making. A rectal swab requires an insertion of 3-4 cm and a 360-degree twirl of the wrist, not the swab, to ensure you get a sample from the entire circumference. Pharyngeal samples require swabbing both tonsillar pillars while taking care for those who may gag.
For contact testing – asymptomatic people who have had a high-risk sexual exposure – providers should test for gonorrhea, chlamydia, HIV, and syphilis but not for herpes, high-risk HPV, hepatitis B, hepatitis C, or bacterial vaginosis. “Maybe we’ll do a screen for trichomoniasis, and maybe we’ll offer herpes type 2 serology or antibody screening,” Dr. Policar said. Providers should also ask patients requesting contact testing if they have been vaccinated for hepatitis B. If not, “the conversation should be how can we get you vaccinated for hepatitis B,” Dr. Policar said.
HIV screening only needs to occur once between the ages of 15 and 65 for low-risk people and then once annually (or more often if necessary) for those who have a sex partner with HIV, use injectable drugs, engage in commercial sex work, have a new sex partner with unknown HIV status, received care at an STD or TB clinic, or were in a correctional facility or homeless shelter.
Those at increased risk for syphilis include men who have sex with men, men under age 29, and anyone living with HIV or who has a history of incarceration or a history of commercial sex work. In addition, African Americans have the greatest risk for syphilis of racial/ethnic groups, followed by Hispanics. Most adults only require hepatitis C screening with anti-hep C antibody testing once in their lifetime. Periodic hepatitis C screening should occur for people who inject drugs. If the screening is positive, providers should conduct an RNA polymerase chain reaction (PCR) test to determine whether a chronic infection is present.
Trichomoniasis screening should occur annually in women living with HIV or in correctional facilities. Others to consider screening include people with new or multiple sex partners, a history of STIs, inconsistent condom use, a history of sex work, and intravenous drug use. Dr. Policar also noted that several new assays, including NAAT, PCR, and a rapid test, are available for trichomoniasis.
STI treatment guidelines
For women with mucoprurulent cervicitis, the cause could be chlamydia, gonorrhea, herpes, trichomonas, mycoplasma, or even progesterone from pregnancy or contraception, Dr. Policar said. The new preferred treatment is 100 mg of doxycycline. The alternative, albeit less preferred, treatment is 1 g azithromycin.
The preferred treatment for chlamydia is now 100 mg oral doxycycline twice daily, or doxycycline 200 mg delayed-release once daily, for 7 days. Alternative regimens include 1 g oral azithromycin in a single dose or 500 mg oral levofloxacin once daily for 7 days. The switch to recommending doxycycline over azithromycin is based on recent evidence showing that doxycycline has a slightly higher efficacy for urogenital chlamydia and a substantially higher efficacy for rectal chlamydia. In addition, an increasing proportion of gonorrheal infections have shown resistance to azithromycin, particularly beginning in 2014.
Preferred treatment of new, uncomplicated gonorrhea infections of the cervix, urethra, rectum, and pharynx is one 500-mg dose of ceftriaxone for those weighing under 150 kg and 1 g for those weighing 150 kg or more. If ceftriaxone is unavailable, the new alternative recommended treatment for gonorrhea is 800 mg cefixime. For pharyngeal gonorrhea only, the CDC recommends a test-of-cure 7-14 days after treatment.
For gonorrheal infections, the CDC also recommends treatment with doxycycline if chlamydia has not been excluded, but the agency no longer recommends dual therapy with azithromycin unless it’s used in place of doxycycline for those who are pregnant, have an allergy, or may not be compliant with a 7-day doxycycline regimen.
The preferred treatment for bacterial vaginosis has not changed. The new recommended regimen for trichomoniasis is 500 mg oral metronidazole for 7 days, with the alternative being a single 2-g dose of tinidazole. Male partners should receive 2 g oral metronidazole. The CDC also notes that patients taking metronidazole no longer need to abstain from alcohol during treatment.
”Another area where the guidelines changed is in their description of expedited partner therapy, which means that, when we find an index case who has gonorrhea or chlamydia, we always have a discussion with her about getting her partners treated,” Dr. Policar said. “The CDC was quite clear that the responsibility for discussing partner treatment rests with us as the diagnosing provider” since city and county health departments don’t have the time or resources for contact tracing these STIs.
The two main ways to treat partners are to have the patient bring their partner(s) to the appointment with them or to do patient-delivered partner therapy. Ideally, clinicians who dispense their own medications can give the patient enough drugs to give her partner(s) a complete dose as well. Otherwise, providers can prescribe extra doses in the index patients’ name or write prescriptions in the partner’s name.
“In every state of the union now, it is legal for you to to prescribe antibiotics for partners sight unseen, Dr. Policar said.
Margaret Sullivan, MD, an ob.gyn. from rural western North Carolina, noted during the Q&A that an obstacle to partner therapy at her practice has been cost, particularly since many of the men don’t have insurance.
“I have not heard before of prescribing the extra doses for partners under the patient’s name,” Dr. Sullivan said. “I’ve thought about doing it, but [was worried about] it potentially being fraudulent if that patient has Medicaid and we’re prescribing extra doses under her name, so how do you work around that?”
Dr. Policar acknowledged that barrier and recommended that patients use the website/app Goodrx.com to find discounts for out-of-pocket generic medications. He also noted the occasional obstacle of pharmacists balking at filling a double or triple dose.
“What we’ve been suggesting in that circumstance is to literally copy that part of the CDC guidelines, which explains expedited partner therapy or patient-delivered partner therapy and send that off to the pharmacist so they can see that it’s a national recommendation of the CDC,” Dr. Policar said.
Claudia Rodriguez, MD, an ob.gyn. who works at Sherman Hospital in Elgin, Ill., asked about the CDC recommendations for HPV vaccination in older women. Although the CDC permits women over age 26 to receive the HPV vaccine, the agency does not “make a solid recommendation to have this done, which oftentimes makes a big difference in whether or not health insurance will actually pay for vaccination in that circumstance,” Dr. Policar said.
Patients are welcome to request the vaccine after shared decision-making, but “we should never present this as something which is routine,” he said. For women in their 50s, for example, “there’s virtually no data about any additional degree of protection that you would get” from HPV vaccination, Dr. Policar said in response to a similar question from Tara Allmen, MD, an ob.gyn. in New York City. “If you ask me for my personal clinical opinion about it, I would say it’s not going to be worth it,” he said.
Dr Policar had no disclosures. Disclosures were unavailable for attendees who spoke.
FROM NAMS 2021