User login
The Journal of Clinical Outcomes Management® is an independent, peer-reviewed journal offering evidence-based, practical information for improving the quality, safety, and value of health care.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
Replacement meals boost nutrient intake by pregnant women with obesity
LAS VEGAS – Pregnant women with overweight or obesity who replaced two meals a day with bars or shakes starting at their second trimester not only had a significantly reduced rate of gestational weight gain but also benefited from significant improvements in their intake of several micronutrients, in a randomized study of 211 women who completed the regimen.

Further research needs “to examine the generalizability and effectiveness of this prenatal lifestyle modification program in improving micronutrient sufficiency in other populations and settings,” Suzanne Phelan, PhD, said at a meeting presented by The Obesity Society and the American Society for Metabolic and Bariatric Surgery. The study she presented ran at two U.S. sites, in California and Rhode Island, and enrolled a population that was 42% Hispanic/Latina. Despite uncertainty about the applicability of the findings to other populations, the results suggested that partial meal replacement is a way to better control gestational weight gain in women with overweight or obesity while simultaneously increasing micronutrient intake, said Dr. Phelan, a clinical psychologist and professor of kinesiology and public health at the California Polytechnic State University in San Luis Obispo.
She reported data from the Healthy Beginnings/Comienzos Saludables (Preventing Excessive Gestational Weight Gain in Obese Women) study, which enrolled 257 women with overweight or obesity (body mass index of at least 25 kg/m2) at week 9-16 of pregnancy and randomized them to either a multifactorial behavioral lifestyle intervention that included two daily meal replacements, or to “enhanced” usual care. About 80% of participants in both arms, a total of 211 women, completed the study with final follow-up at 35-36 weeks’ gestational age, after enrolling at an average gestational age of just under 14 weeks. In addition to eating nutrition bars or drinking nutrition shakes as the replacement meal options, participants also ate one conventional meal daily as well as 2-4 healthy snacks. The enrolled women included 41% with overweight and 59% with obesity.
The study’s primary endpoint was the rate of gestational weight gain per week, which was 0.33 kg in the intervention group and 0.39 kg in the controls, a statistically significant difference. The proportion of women who exceeded the Institute of Medicine’s recommended maximum gestational weight gain maximum was 41% among those in the intervention group and 54% among the controls, also a statistically significant difference (Am J Clin Nutr. 2018 Feb;107[2]:183-94).
The secondary micronutrient analysis that Dr. Phelan reported documented the high prevalence of micronutrient deficiencies among the study participants at baseline. More than 90% had deficient intake of vitamin D and fiber, more than 80% had inadequate dietary levels of iron, vitamin E, and choline, and more than half had too little dietary magnesium, vitamin K, and folate. There were additional deficiencies for other micronutrients in lesser proportions of study participants.
The analysis also showed how the behavioral and diet intervention through the end of the third trimester normalized many of these deficiencies, compared with the placebo arm. For example, the prevalence of a magnesium dietary deficiency in the intervention arm dropped from 69% at baseline to 37% at follow-up, compared with hardly any change in the control arm, so that women in the intervention group had a 64% reduced rate of magnesium deficiency compared with the controls, a statistically significant difference.
Other micronutrients that had significant drops in deficiency rate included calcium, with a 63% relative reduction in the deficiency prevalence, vitamin A with a 61% cut, vitamin E with an 83% relative reduction, and vitamin K with a 51% relative drop. Other micronutrient intake levels that showed statistically significant increases during the study compared with controls included vitamin D and copper, but choline showed an inexplicable drop in consumption in the intervention group, a “potential concern,” Dr. Phelan said. The intervention also significantly reduced sodium intake. Dr. Phelan and her associates published these findings (Nutrients. 2019 May 14;11[5]:1071; doi: 10.3390/nu11051071).

“The diet quality of many of the pregnant women we have studied was poor, often eating less than half the recommended amounts of fruits and vegetables,” said Leanne M. Redman, PhD, a professor at Louisiana State University and director of the Reproductive Endocrinology and Women’s Health Laboratory at the university’s Pennington Biomedical Research Center in Baton Rouge. “Meal replacement with bars and shakes will be really important for future efforts at improving diet quality” in pregnant women with obesity, predicted Dr. Redman, who did not collaborate on the study Dr. Phelan reported.
SOURCE: Phelan S et al. Obesity Week 2019. Abstract T-OR-2081.
LAS VEGAS – Pregnant women with overweight or obesity who replaced two meals a day with bars or shakes starting at their second trimester not only had a significantly reduced rate of gestational weight gain but also benefited from significant improvements in their intake of several micronutrients, in a randomized study of 211 women who completed the regimen.
Further research needs “to examine the generalizability and effectiveness of this prenatal lifestyle modification program in improving micronutrient sufficiency in other populations and settings,” Suzanne Phelan, PhD, said at a meeting presented by The Obesity Society and the American Society for Metabolic and Bariatric Surgery. The study she presented ran at two U.S. sites, in California and Rhode Island, and enrolled a population that was 42% Hispanic/Latina. Despite uncertainty about the applicability of the findings to other populations, the results suggested that partial meal replacement is a way to better control gestational weight gain in women with overweight or obesity while simultaneously increasing micronutrient intake, said Dr. Phelan, a clinical psychologist and professor of kinesiology and public health at the California Polytechnic State University in San Luis Obispo.
She reported data from the Healthy Beginnings/Comienzos Saludables (Preventing Excessive Gestational Weight Gain in Obese Women) study, which enrolled 257 women with overweight or obesity (body mass index of at least 25 kg/m2) at week 9-16 of pregnancy and randomized them to either a multifactorial behavioral lifestyle intervention that included two daily meal replacements, or to “enhanced” usual care. About 80% of participants in both arms, a total of 211 women, completed the study with final follow-up at 35-36 weeks’ gestational age, after enrolling at an average gestational age of just under 14 weeks. In addition to eating nutrition bars or drinking nutrition shakes as the replacement meal options, participants also ate one conventional meal daily as well as 2-4 healthy snacks. The enrolled women included 41% with overweight and 59% with obesity.
The study’s primary endpoint was the rate of gestational weight gain per week, which was 0.33 kg in the intervention group and 0.39 kg in the controls, a statistically significant difference. The proportion of women who exceeded the Institute of Medicine’s recommended maximum gestational weight gain maximum was 41% among those in the intervention group and 54% among the controls, also a statistically significant difference (Am J Clin Nutr. 2018 Feb;107[2]:183-94).
The secondary micronutrient analysis that Dr. Phelan reported documented the high prevalence of micronutrient deficiencies among the study participants at baseline. More than 90% had deficient intake of vitamin D and fiber, more than 80% had inadequate dietary levels of iron, vitamin E, and choline, and more than half had too little dietary magnesium, vitamin K, and folate. There were additional deficiencies for other micronutrients in lesser proportions of study participants.
The analysis also showed how the behavioral and diet intervention through the end of the third trimester normalized many of these deficiencies, compared with the placebo arm. For example, the prevalence of a magnesium dietary deficiency in the intervention arm dropped from 69% at baseline to 37% at follow-up, compared with hardly any change in the control arm, so that women in the intervention group had a 64% reduced rate of magnesium deficiency compared with the controls, a statistically significant difference.
Other micronutrients that had significant drops in deficiency rate included calcium, with a 63% relative reduction in the deficiency prevalence, vitamin A with a 61% cut, vitamin E with an 83% relative reduction, and vitamin K with a 51% relative drop. Other micronutrient intake levels that showed statistically significant increases during the study compared with controls included vitamin D and copper, but choline showed an inexplicable drop in consumption in the intervention group, a “potential concern,” Dr. Phelan said. The intervention also significantly reduced sodium intake. Dr. Phelan and her associates published these findings (Nutrients. 2019 May 14;11[5]:1071; doi: 10.3390/nu11051071).
“The diet quality of many of the pregnant women we have studied was poor, often eating less than half the recommended amounts of fruits and vegetables,” said Leanne M. Redman, PhD, a professor at Louisiana State University and director of the Reproductive Endocrinology and Women’s Health Laboratory at the university’s Pennington Biomedical Research Center in Baton Rouge. “Meal replacement with bars and shakes will be really important for future efforts at improving diet quality” in pregnant women with obesity, predicted Dr. Redman, who did not collaborate on the study Dr. Phelan reported.
SOURCE: Phelan S et al. Obesity Week 2019. Abstract T-OR-2081.
LAS VEGAS – Pregnant women with overweight or obesity who replaced two meals a day with bars or shakes starting at their second trimester not only had a significantly reduced rate of gestational weight gain but also benefited from significant improvements in their intake of several micronutrients, in a randomized study of 211 women who completed the regimen.
Further research needs “to examine the generalizability and effectiveness of this prenatal lifestyle modification program in improving micronutrient sufficiency in other populations and settings,” Suzanne Phelan, PhD, said at a meeting presented by The Obesity Society and the American Society for Metabolic and Bariatric Surgery. The study she presented ran at two U.S. sites, in California and Rhode Island, and enrolled a population that was 42% Hispanic/Latina. Despite uncertainty about the applicability of the findings to other populations, the results suggested that partial meal replacement is a way to better control gestational weight gain in women with overweight or obesity while simultaneously increasing micronutrient intake, said Dr. Phelan, a clinical psychologist and professor of kinesiology and public health at the California Polytechnic State University in San Luis Obispo.
She reported data from the Healthy Beginnings/Comienzos Saludables (Preventing Excessive Gestational Weight Gain in Obese Women) study, which enrolled 257 women with overweight or obesity (body mass index of at least 25 kg/m2) at week 9-16 of pregnancy and randomized them to either a multifactorial behavioral lifestyle intervention that included two daily meal replacements, or to “enhanced” usual care. About 80% of participants in both arms, a total of 211 women, completed the study with final follow-up at 35-36 weeks’ gestational age, after enrolling at an average gestational age of just under 14 weeks. In addition to eating nutrition bars or drinking nutrition shakes as the replacement meal options, participants also ate one conventional meal daily as well as 2-4 healthy snacks. The enrolled women included 41% with overweight and 59% with obesity.
The study’s primary endpoint was the rate of gestational weight gain per week, which was 0.33 kg in the intervention group and 0.39 kg in the controls, a statistically significant difference. The proportion of women who exceeded the Institute of Medicine’s recommended maximum gestational weight gain maximum was 41% among those in the intervention group and 54% among the controls, also a statistically significant difference (Am J Clin Nutr. 2018 Feb;107[2]:183-94).
The secondary micronutrient analysis that Dr. Phelan reported documented the high prevalence of micronutrient deficiencies among the study participants at baseline. More than 90% had deficient intake of vitamin D and fiber, more than 80% had inadequate dietary levels of iron, vitamin E, and choline, and more than half had too little dietary magnesium, vitamin K, and folate. There were additional deficiencies for other micronutrients in lesser proportions of study participants.
The analysis also showed how the behavioral and diet intervention through the end of the third trimester normalized many of these deficiencies, compared with the placebo arm. For example, the prevalence of a magnesium dietary deficiency in the intervention arm dropped from 69% at baseline to 37% at follow-up, compared with hardly any change in the control arm, so that women in the intervention group had a 64% reduced rate of magnesium deficiency compared with the controls, a statistically significant difference.
Other micronutrients that had significant drops in deficiency rate included calcium, with a 63% relative reduction in the deficiency prevalence, vitamin A with a 61% cut, vitamin E with an 83% relative reduction, and vitamin K with a 51% relative drop. Other micronutrient intake levels that showed statistically significant increases during the study compared with controls included vitamin D and copper, but choline showed an inexplicable drop in consumption in the intervention group, a “potential concern,” Dr. Phelan said. The intervention also significantly reduced sodium intake. Dr. Phelan and her associates published these findings (Nutrients. 2019 May 14;11[5]:1071; doi: 10.3390/nu11051071).
“The diet quality of many of the pregnant women we have studied was poor, often eating less than half the recommended amounts of fruits and vegetables,” said Leanne M. Redman, PhD, a professor at Louisiana State University and director of the Reproductive Endocrinology and Women’s Health Laboratory at the university’s Pennington Biomedical Research Center in Baton Rouge. “Meal replacement with bars and shakes will be really important for future efforts at improving diet quality” in pregnant women with obesity, predicted Dr. Redman, who did not collaborate on the study Dr. Phelan reported.
SOURCE: Phelan S et al. Obesity Week 2019. Abstract T-OR-2081.
REPORTING FROM OBESITY WEEK 2019
New cystic fibrosis therapy raises hopes among specialists and patients
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”

The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.

Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”
The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.
Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
A newly approved triple-combination modulator to treat cystic fibrosis (CF) has raised expectations of a treatment turning point among patients and specialists. If the early results are sustained, elexacaftor/ivacaftor/tezacaftor (Trikafta) could prove to be the rare case of a much-touted new medicine that meets high expectations.
“CF even in infants causes inflammation, so we know that lung damage can start early and progress,” said Susan Millard, MD, FCCP, of Helen DeVos Children’s Hospital in Grand Rapids, Mich., and the local clinical research director for the pediatric pulmonary and sleep medicine section. “This oral drug therapy is actually treating the underlying problem, as opposed to many of the therapies we have that take hours to nebulize and only work locally in the airways.”
Dr. Millard is the recent past pediatric editor for Chest Physician and has been a local principal investigator at Helen DeVos Children’s Hospital for many Vertex-sponsored clinical studies.
The pivotal studies
The Food and Drug Administration approval of Trikafta rested on two pivotal phase 3, placebo-controlled studies, one in patients with two copies of the most common CF mutations, F508del, and the second in patients with one copy of F508del and a second mutation that was called a “minimal-function” mutation. The findings have ignited the hopes of many people with CF and their physicians. The drug was approved in October 2019 for patients aged 12 years and older who have at least one F508del mutation of the cystic fibrosis transmembrane conductance regulator gene. About 90% of patients in the United States have at least one copy of F508del. In the study looking at patients with one copy of F508del, the mean predicted forced expiratory volume in 1 second increased 13.8% in patients taking the drug versus placebo (N Engl J Med. 2019 Oct 31. doi: 10.1056/NEJMoa1908639). The number of pulmonary exacerbations decreased by 63% in the Trikafta group, compared with placebo. Pulmonary exacerbations were described as a change in specific symptoms that required treatment with a new oral, intravenous, or inhaled antibiotic. Serious adverse drug reactions that occurred more frequently in patients receiving Trikafta, compared with placebo, were rash and influenza events.
In the study that included patients with two copies of F508del, on average, the lung function increased 10% versus patients on ivacaftor/tezacaftor at 4 weeks. In addition, there was a 45.1 mmol/L on average decrease in the sweat chloride level in the Trikafta group, compared with ivacaftor/tezacaftor.
A hopeful start
Robert Giusti, MD, a pediatric pulmonologist at New York University Langone Health, is also hopeful. “This could be the kind of treatment that will make a revolution in terms of [cystic fibrosis] care if it can be started very early in life shortly after diagnosis. We anticipate that patients will be disease free for a longer period of time.”
The Cystic Fibrosis Foundation’s (CFF) “venture philanthropy” initiative played an important role in the development of the drug by Vertex Pharmaceuticals. The CFF has invested many millions of dollars in research by drug companies since the 1980s and was an early backer of Vertex. According to a statement on the CFF website, the Foundation sold its royalty rights for treatments developed by Vertex for $3.3 billion in 2014. The drug has a list price of about $311,000 a year. Payment issues may arise in the future, but for now, Vertex has stated that insurers and some Medicaid programs have begun paying claims for Trikafta
Specialists who treat CF now are watching to see how well patients tolerate this highly anticipated drug – and how well it meets expectations. The Therapeutic Development Network, the clinical research division of the CFF, is enrolling patients taking Trikafta in an observational study to follow for long-term follow-up.
Meeting expectations
“[Long-term efficacy is] something that we’re always concerned about. When the drug comes to market, is it going to be as effective as we thought it might be?” said Ryan Thomas, MD, director of the Cystic Fibrosis Center at Michigan State University, East Lansing. The MSU Cystic Fibrosis Center receives funding from the Cystic Fibrosis Foundation.
Almost one in five patients could not tolerate treatment with Orkambi, most often because of adverse breathing events, according to a French study published in the American Journal of Respiratory and Critical Care Medicine. The investigators wrote: “Among the 845 patients (292 adolescents, 553 adults) who initiated lumacaftor/ivacaftor, 18.2% (154 patients) discontinued treatment, often due to respiratory (48.1%, 74 patients) or nonrespiratory (27.9%, 43 patients) adverse events” and that the discontinuation rate was considerably higher than previously reported in clinical trials.
“We thought [Orkambi] was going to be something that could have a big effect,” Dr. Thomas said. “It turned out that it was harder for people to tolerate than we thought and the improvements weren’t as sustained as we thought they might be. I really don’t think this will end up being the case with Trikafta.”
Longer-term data are starting to emerge, which may ease some of the concerns inherent in working with a newer medicine. “These [data] suggest that this is going to be a game changer,” Dr. Thomas said. “If Trikafta is this efficacious, well, we’re talking about having people with CF who will live full lifespans without a lung transplant, and that is so rare.”
The decrease in hospitalizations, improved CT scans, and lower rates of lung function decline suggest it could be “the Holy Grail,” Dr. Thomas said.
A different disease
Trikafta is the latest in a series of improvements of CF treatment in recent decades, recalled Dr. Giusti, who has been in this field for about 3 decades. “It used to be that I attended many funerals for children with CF. Now with patients living longer and healthier lives I am invited to attend their weddings and even their children’s baptisms and bris ceremonies. It is a very different disease than it used to be.”
The promise of Trikafta leaves behind the minority of patients for whom the drug won’t work. This is for the 10% of patients that have rare mutations. That can lead to difficult conversations with parents about why this new option is not a choice for their child, Dr. Millard said. “It just crushes you, but the Cystic Fibrosis Foundation is committing a lot of new research in that direction. Their mantra is ‘until it is done.’ ”
Realistic expectations
William (Randy) Hunt, MD, FAAP, FACP, assistant professor of medicine in the Division of Pulmonary, Allergy, Critical Care and Sleep, Emory University School of Medicine, Atlanta, agrees that Trikafta is an exciting development in CF treatment. He noted, “Starting this medication early in life may very well significantly attenuate the disease, but it is not a cure. For individuals who already have significant disease, we may not see the same level of improvements in lung function as what we saw in the studies. The studies generally excluded individuals with ppFEV1 < 40%. Nevertheless, I remain optimistic and have been prescribing it to nearly everyone that qualifies after a discussion.”
Dr. Hunt added, “Patients are asking if they can stop their current chronic CF therapies once they start Trikafta. The answer is “no, at least not right now.” While all the relatively short-term data around Trikafta are very promising, we do not yet know how sustained the long-term benefits will be. Still, safely removing therapeutic burden from our patient population is a real interest. There are plans underway by the CFF and other institutions to systematically research whether discontinuing chronic CF therapies is safe in the setting of Trikafta.”
He concluded that 10% of individuals with CF mutations still do not respond to the modulators currently available. “We will not leave that population behind, but treating these remaining mutations is going to take continued efforts and likely modulators that are therapeutically differently from the mechanism of actions of those that are currently available,” he said.
Therese Borden contributed to this article.
1/2/2020 - This story was updated.
FDA okays first generics for Eliquis
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.

The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
The Food and Drug Administration has approved two applications for first generic versions of apixaban (Eliquis, Bristol-Myers Squibb/Pfizer) tablets to reduce the risk for stroke and systemic embolism in patients with nonvalvular atrial fibrillation.
The FDA gave the go-ahead to market generic versions of apixaban to Micro Labs Limited and Mylan Pharmaceuticals.
“Today’s approvals of the first generics of apixaban are an example of how the FDA’s generic drug program improves access to lower-cost, safe, and high-quality medicines,” Janet Woodcock, MD, director of the FDA’s Center for Drug Evaluation and Research, said in a statement today. “These approvals mark the first generic approvals of a direct oral anticoagulant.”
It is estimated that between 2.7 and 6.1 million people in the United States have atrial fibrillation. Many of these individuals use anticoagulants or anticlotting drugs to reduce that risk. Direct oral anticoagulants, however, do not require repeated blood testing.
Apixaban was approved by the FDA in December 2012 for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. Additional indications in the United States are to treat and prevent the recurrence of deep vein thrombosis (DVT) and pulmonary embolism (PE) and as DVT/PE prophylaxis in adults who have undergone hip or knee replacement surgery.
The FDA reminds providers that, as with brand name apixaban, generic versions must be dispensed with a medication guide that provides important instructions on the drug’s uses and risks. Healthcare professionals should counsel patients on signs and symptoms of possible bleeding.
As with other FDA-approved anticlotting drugs, bleeding, including life-threatening and fatal bleeding, is the most serious risk with apixaban.
Full prescribing information for the drug also warns about the increased risk for stroke in patients who discontinue use of the drug without taking some other form of anticoagulation. Epidural or spinal hematoma, which may cause long-term or permanent paralysis, may occur in patients treated with apixaban who are undergoing spinal epidural anesthesia or spinal puncture.
This story first appeared on Medscape.com.
FDA okays ubrogepant for acute migraine treatment
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.

Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
The Food and Drug Administration has approved ubrogepant (Ubrelvy, Allergan) for the acute treatment of migraine with or without aura in adults.
Ubrogepant is the first drug in the class of oral calcitonin gene–related peptide receptor antagonists approved for the acute treatment of migraine. It is approved in two dose strengths (50 mg and 100 mg).
The drug is not indicated, however, for the preventive treatment of migraine.
“Migraine is an often disabling condition that affects an estimated 37 million people in the U.S.,” Billy Dunn, MD, acting director of the office of neuroscience in the FDA’s Center for Drug Evaluation and Research, said in an FDA news release.
Ubrogepant represents “an important new option for the acute treatment of migraine in adults, as it is the first drug in its class approved for this indication. The FDA is pleased to approve a novel treatment for patients suffering from migraine and will continue to work with stakeholders to promote the development of new safe and effective migraine therapies,” added Dr. Dunn.
The safety and efficacy of ubrogepant for the acute treatment of migraine was demonstrated in two randomized, double-blind, placebo-controlled trials (ACHIEVE I and ACHIEVE II). In total, 1,439 adults with a history of migraine, with and without aura, received ubrogepant to treat an ongoing migraine.
“Both 50-mg and 100-mg dose strengths demonstrated significantly greater rates of pain freedom and freedom from the most bothersome migraine-associated symptom at 2 hours, compared with placebo,” Allergan said in a news release announcing approval.
The most common side effects reported by patients in the clinical trials were nausea, tiredness, and dry mouth. Ubrogepant is contraindicated for coadministration with strong CYP3A4 inhibitors.
The company expects to have ubrogepant available in the first quarter of 2020.
A version of this story originally appeared on Medscape.com.
Can insulin plus metformin improve pregnancy outcomes in women with type 2 diabetes?
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
WASHINGTON – Insulin is the preferred agent for type 2 diabetes in pregnant women, yet about a third of pregnancies still have an adverse outcome, according Kim Boggess, MD, who spoke at the biennial meeting of the Diabetes in Pregnancy Study Group of North America.
“We are not where we need to be,” said Dr. Boggess, who is leading a trial that brings metformin, the first-line agent for type 2 diabetes outside of pregnancy, back into the picture for pregnant women – as an add-on to insulin.
It is an interesting twist, because pregnant women taking metformin for preexisting type 2 or gestational diabetes have been shown in some studies to require supplemental insulin, more than occasionally, to achieve target glycemic control.
This was the case in a small, randomized, controlled trial at Dr. Boggess’ institution, the University of North Carolina at Chapel Hill, in which 43% of pregnant women with type 2 diabetes who were assigned to metformin required supplemental insulin (Am J Perinatol. 2013;30[6]:483-90). (0% vs. 36%, respectively) and fewer reports of glucose values less than 60 mg/dL (7.1% vs. 50%).
“I don’t consider this [need for supplemental insulin] ‘metformin failure,’ because studies that use metformin as monotherapy and that [show some patients] ultimately requiring insulin support ... also show that these women need less insulin,” she said. “What’s the risk of insulin alone? Hypoglycemia. So using less insulin could be a good thing.”
Other research suggests there may be less maternal weight gain, less neonatal hypoglycemia, fewer neonatal complications, and improved maternal glycemic control in patients treated with metformin, alone or with add-on insulin, than with insulin alone. “We’re starting to get a sense in the literature that, at least in the [pregnant] population with type 2 diabetes, there may be a role for metformin,” said Dr. Boggess, professor and program director for maternal-fetal medicine at the university.
Currently, the multisite MOMPOD trial (Medical Optimization of Management of T2DM Complicating Pregnancy) is randomizing 950 women to insulin plus 1,000 mg metformin twice daily or insulin plus placebo. The primary outcome of the trial is a composite of pregnancy loss, preterm birth, birth injury, neonatal hypoglycemia, or hyperbilirubinemia. Infant fat mass (within 72 hours of birth) is a secondary outcome, along with maternal safety and maternal side effects.
The MiTy (Metformin in Women with T2DM in Pregnancy) trial in Canada, with similar randomization arms and outcomes measures, is completed and undergoing analysis. “Hopefully we’ll [soon] be able to say whether the addition of adjuvant metformin to insulin to treat type 2 diabetes brings the perinatal adverse outcome rate down from 30%,” said Dr. Boggess.
Metformin is the recommended first-line agent for type 2 diabetes in nonpregnant adults. But during pregnancy, insulin, which does not cross the placenta, is the preferred agent, according to recommendations of the American Diabetes Association and the American College of Obstetricians and Gynecologists, she noted. Lingering in the background is the fact that the long-term effects of in utero metformin exposure on offspring – and of exposure to any oral hypoglycemic agent – are unknown, she said*
A majority of the adverse pregnancy outcomes that occur in the context of type 2 diabetes involve macrosomia. “It’s a big deal,” Dr. Boggess said, that results in numerous maternal and infant risks and complications. “We also know that the in utero environment that contributes to, or causes, macrosomia predisposes to childhood obesity and obesity later on.”
Diabetes is the “leading risk factor” for adverse pregnancy outcomes today, said E. Albert Reece, MD, PhD, MBA, executive vice president for medical affairs at the University of Maryland, Baltimore, and the John Z. and Akiko K. Bowers distinguished professor and dean of the University of Maryland School of Medicine. In the United States, 11% of women aged 20 years and older have diabetes, and the disease affects more than 1% of all pregnancies, he said.
The MOMPOD trial is sponsored by the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr. Boggess reported no conflicts of interest.
* This article was updated 1/2/2020.
REPORTING FROM DPSG-NA 2019
Vitamin E acetate confirmed as likely source of EVALI
Vitamin E acetate was found in fluid from the lungs of 94% of patients with electronic cigarette, or vaping, product use–associated lung injury, data from a convenience sample of 51 patients indicate. The findings were published in the New England Journal of Medicine.
Cases of electronic cigarette, or vaping, product use–associated lung injury (EVALI) were reported to the Centers for Disease Control and Prevention starting in early 2019, and numbers rose throughout the year, “which suggests new or increased exposure to one or more toxicants from the use of e-cigarette products,” wrote Benjamin C. Blount, PhD, of the National Center for Environmental Health at the CDC, and colleagues.
To further investigate potential toxins in patients with EVALI, the researchers examined bronchoalveolar-lavage (BAL) fluid from 51 EVALI patients and 99 healthy controls.
After the researchers used isotope dilution mass spectrometry on the samples, 48 of the 51 patients (94%) showed vitamin E acetate in their BAL samples. No other potential toxins – including plant oils, medium-chain triglyceride oil, petroleum distillates, and diluent terpenes – were identified. The samples of one patient each showed coconut oil and limonene.
A total of 47 of 51 patients for whom complete laboratory data were available either reported vaping tetrahydrocannabinol products within 90 days of becoming ill, or showed tetrahydrocannabinol or its metabolites in their BAL fluid. In addition, 30 of 47 patients showed nicotine or nicotine metabolites in their BAL fluid.
The average age of the patients was 23 years, 69% were male. Overall, 25 were confirmed EVALI cases and 26 were probable cases, and probable cases included the three patients who showed no vitamin E acetate.
The safety of inhaling vitamin E acetate, which is a common ingredient in dietary supplements and skin care creams, has not been well studied. It could contribute to lung injury when heated in e-cigarette products by splitting the acetate to create the reactive compound and potential lung irritant ketene, the researchers said.
The study findings were limited by several factors including the possibility that vitamin E acetate is a marker for exposure to other toxicants, a lack of data on the impact of heating vitamin e acetate, and the inability to assess the timing of the vitamin E acetate exposure compared to BAL sample collection, the researchers noted.
However, the results suggest that vitamin E acetate may play a role in EVALI because of the high detection rate in patients from across the United States, the biologically possible potential for lung injury from vitamin e acetate, and the timing of the rise of EVALI and the use of vitamin E acetate in vaping products, they concluded.
The research was supported by the National Cancer Institute, the FDA Center for Tobacco Products, and The Ohio State University Pelotonia intramural research program. The authors had no financial conflicts to disclose.
SOURCE: Blount BC et al. N Engl J Med. 2019 Dec 20. doi: 10.1056/NEJMoa1916433.
Vitamin E acetate was found in fluid from the lungs of 94% of patients with electronic cigarette, or vaping, product use–associated lung injury, data from a convenience sample of 51 patients indicate. The findings were published in the New England Journal of Medicine.
Cases of electronic cigarette, or vaping, product use–associated lung injury (EVALI) were reported to the Centers for Disease Control and Prevention starting in early 2019, and numbers rose throughout the year, “which suggests new or increased exposure to one or more toxicants from the use of e-cigarette products,” wrote Benjamin C. Blount, PhD, of the National Center for Environmental Health at the CDC, and colleagues.
To further investigate potential toxins in patients with EVALI, the researchers examined bronchoalveolar-lavage (BAL) fluid from 51 EVALI patients and 99 healthy controls.
After the researchers used isotope dilution mass spectrometry on the samples, 48 of the 51 patients (94%) showed vitamin E acetate in their BAL samples. No other potential toxins – including plant oils, medium-chain triglyceride oil, petroleum distillates, and diluent terpenes – were identified. The samples of one patient each showed coconut oil and limonene.
A total of 47 of 51 patients for whom complete laboratory data were available either reported vaping tetrahydrocannabinol products within 90 days of becoming ill, or showed tetrahydrocannabinol or its metabolites in their BAL fluid. In addition, 30 of 47 patients showed nicotine or nicotine metabolites in their BAL fluid.
The average age of the patients was 23 years, 69% were male. Overall, 25 were confirmed EVALI cases and 26 were probable cases, and probable cases included the three patients who showed no vitamin E acetate.
The safety of inhaling vitamin E acetate, which is a common ingredient in dietary supplements and skin care creams, has not been well studied. It could contribute to lung injury when heated in e-cigarette products by splitting the acetate to create the reactive compound and potential lung irritant ketene, the researchers said.
The study findings were limited by several factors including the possibility that vitamin E acetate is a marker for exposure to other toxicants, a lack of data on the impact of heating vitamin e acetate, and the inability to assess the timing of the vitamin E acetate exposure compared to BAL sample collection, the researchers noted.
However, the results suggest that vitamin E acetate may play a role in EVALI because of the high detection rate in patients from across the United States, the biologically possible potential for lung injury from vitamin e acetate, and the timing of the rise of EVALI and the use of vitamin E acetate in vaping products, they concluded.
The research was supported by the National Cancer Institute, the FDA Center for Tobacco Products, and The Ohio State University Pelotonia intramural research program. The authors had no financial conflicts to disclose.
SOURCE: Blount BC et al. N Engl J Med. 2019 Dec 20. doi: 10.1056/NEJMoa1916433.
Vitamin E acetate was found in fluid from the lungs of 94% of patients with electronic cigarette, or vaping, product use–associated lung injury, data from a convenience sample of 51 patients indicate. The findings were published in the New England Journal of Medicine.
Cases of electronic cigarette, or vaping, product use–associated lung injury (EVALI) were reported to the Centers for Disease Control and Prevention starting in early 2019, and numbers rose throughout the year, “which suggests new or increased exposure to one or more toxicants from the use of e-cigarette products,” wrote Benjamin C. Blount, PhD, of the National Center for Environmental Health at the CDC, and colleagues.
To further investigate potential toxins in patients with EVALI, the researchers examined bronchoalveolar-lavage (BAL) fluid from 51 EVALI patients and 99 healthy controls.
After the researchers used isotope dilution mass spectrometry on the samples, 48 of the 51 patients (94%) showed vitamin E acetate in their BAL samples. No other potential toxins – including plant oils, medium-chain triglyceride oil, petroleum distillates, and diluent terpenes – were identified. The samples of one patient each showed coconut oil and limonene.
A total of 47 of 51 patients for whom complete laboratory data were available either reported vaping tetrahydrocannabinol products within 90 days of becoming ill, or showed tetrahydrocannabinol or its metabolites in their BAL fluid. In addition, 30 of 47 patients showed nicotine or nicotine metabolites in their BAL fluid.
The average age of the patients was 23 years, 69% were male. Overall, 25 were confirmed EVALI cases and 26 were probable cases, and probable cases included the three patients who showed no vitamin E acetate.
The safety of inhaling vitamin E acetate, which is a common ingredient in dietary supplements and skin care creams, has not been well studied. It could contribute to lung injury when heated in e-cigarette products by splitting the acetate to create the reactive compound and potential lung irritant ketene, the researchers said.
The study findings were limited by several factors including the possibility that vitamin E acetate is a marker for exposure to other toxicants, a lack of data on the impact of heating vitamin e acetate, and the inability to assess the timing of the vitamin E acetate exposure compared to BAL sample collection, the researchers noted.
However, the results suggest that vitamin E acetate may play a role in EVALI because of the high detection rate in patients from across the United States, the biologically possible potential for lung injury from vitamin e acetate, and the timing of the rise of EVALI and the use of vitamin E acetate in vaping products, they concluded.
The research was supported by the National Cancer Institute, the FDA Center for Tobacco Products, and The Ohio State University Pelotonia intramural research program. The authors had no financial conflicts to disclose.
SOURCE: Blount BC et al. N Engl J Med. 2019 Dec 20. doi: 10.1056/NEJMoa1916433.
FROM NEW ENGLAND JOURNAL OF MEDICINE
Influenza activity continues to be unusually high
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
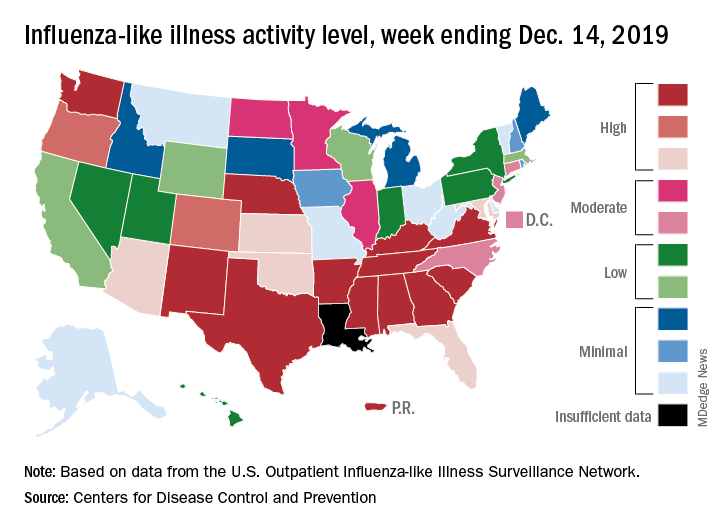
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
The 2019-2020 flu season continues its unusually early rise in activity, with the Centers for Disease Control and Prevention estimating that 3.7 million cases have occurred through Dec. 14.
which is up from 3.2% the previous week and is the sixth consecutive week that the United States has been at or above the national baseline of 2.4%, the CDC reported Dec. 20. This year’s 3.9% is the highest mid-December rate recorded since 2003, when it reached almost 7.4%.
Most of the influenza activity so far this season is being driven by influenza B/Victoria viruses. Nationwide testing puts influenza B prevalence at 68.5% of all positive specimens, exactly the same as last week, but A(H1N1) viruses “are increasing in proportion relative to other influenza viruses in some regions,” the CDC’s influenza division said.
A look at this week’s activity map shows that 21 states, compared with 12 last week, were in the “high” range of activity – that’s levels 8-10 on the CDC’s 1-10 scale. Twelve of those states, along with Puerto Rico, were at level 10, which was up from nine a week earlier, the CDC said.
The overall hospitalization rate through the week of Dec. 8-14 (5.5 per 100,000 population) “is similar to what has been seen at this time during recent seasons,” the CDC noted. The highest rates are occurring among adults over age 65 years (12.7 per 100,000) and children aged 0-4 years (10.9 per 100,000).
Three ILI-related deaths among children that occurred last week were reported, which brings the total for the 2019-2020 season to 19, the CDC said.
Survival data reported from largest CAR T trial in B-cell lymphoma
ORLANDO – Updated results from the TRANSCEND NHL trial include survival data with lisocabtagene maraleucel (liso-cel), an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, in patients with relapsed/refractory B-cell lymphomas.

The median progression-free survival (PFS) was 6.8 months, and the median overall survival was 21.1 months. PFS results were best among complete responders and among patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma.
Jeremy S. Abramson, MD, of Massachusetts General Hospital in Boston, presented these results at the annual meeting of the American Society of Hematology.
“TRANSCEND NHL is the largest clinical study to date of CD19-directed CAR T cells in patients with relapsed/refractory aggressive B-cell lymphoma,” Dr. Abramson said.
The phase 1 trial (NCT02631044) includes 269 patients who received liso-cel. They were diagnosed with transformed follicular lymphoma (22%) or other indolent lymphoma (7%), high-grade B-cell lymphoma (13%), primary mediastinal large B-cell lymphoma (6%), grade 3B follicular lymphoma (1%), or diffuse large B-cell lymphoma not otherwise specified (51%).
At baseline, patients had received a median of three prior systemic therapies (range, one to eight). Some patients had received autologous (33%) or allogeneic (3%) transplant. Many patients were chemotherapy refractory (67%) or had never achieved a complete response to prior therapy (44%).
More than half of patients (59%) received bridging therapy during liso-cel manufacturing. All patients received lymphodepletion with fludarabine and cyclophosphamide, followed by liso-cel at 50 x 106 CAR T cells, 100 x 106 CAR T cells, or 150 x 106 CAR T cells.
Response and survival
The median follow-up was 12.0 months. The overall response rate was 73%, and the complete response rate was 53%.
“Remissions were rapid, with a median of 1 month from CAR T-cell infusion, and durable, with a median duration of response that has not been reached and 55% of patients remaining in response at 1 year,” Dr. Abramson said.
The median PFS was 6.8 months overall, not reached for patients who achieved a complete response, 2.8 months for patients with a partial response, and 1.1 months for patients with stable disease or progressive disease.
The median PFS was not reached for patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma, 5.0 months for high-grade B-cell lymphoma, 3.0 months for diffuse large B-cell lymphoma not otherwise specified, and 2.9 months in transformed indolent non-Hodgkin lymphoma.
The median overall survival was 21.1 months overall, not reached for patients who achieved a complete response, 9.0 months for patients who had a partial response, and 5.1 months for patients with stable disease or progressive disease.
Safety
Common treatment-emergent adverse events were neutropenia (63%), anemia (48%), fatigue (44%), nausea (33%), thrombocytopenia (31%), headache (30%), decreased appetite (28%), and diarrhea (26%).
Cytokine release syndrome (CRS) occurred in 42% of patients, and neurologic events occurred in 30%. Grade 3-4 CRS occurred in 2% of patients, and grade 3-4 neurologic events occurred in 10%. There were no cases of grade 5 CRS or neurologic events.
The median time to CRS onset was 5 days, and the median time to onset of neurologic events was 9 days. The median time to resolution of CRS and neurologic events was 5 days and 11 days, respectively.
“The low incidence of severe CRS and neurologic events and their late time of onset support using this product in a large range of patients and in the outpatient setting,” Dr. Abramson said.
There were seven grade 5 treatment-related adverse events, including diffuse alveolar damage, pulmonary hemorrhage, multiple organ dysfunction syndrome, cardiomyopathy, fludarabine leukoencephalopathy, septic shock, and progressive multifocal leukoencephalopathy.
This trial is sponsored by Bristol-Myers Squibb. Dr. Abramson reported relationships with Juno Therapeutics and Celgene, now owned by Bristol-Myers Squibb, and a range of other companies.
SOURCE: Abramson JS et al. ASH 2019, Abstract 241.
ORLANDO – Updated results from the TRANSCEND NHL trial include survival data with lisocabtagene maraleucel (liso-cel), an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, in patients with relapsed/refractory B-cell lymphomas.
The median progression-free survival (PFS) was 6.8 months, and the median overall survival was 21.1 months. PFS results were best among complete responders and among patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma.
Jeremy S. Abramson, MD, of Massachusetts General Hospital in Boston, presented these results at the annual meeting of the American Society of Hematology.
“TRANSCEND NHL is the largest clinical study to date of CD19-directed CAR T cells in patients with relapsed/refractory aggressive B-cell lymphoma,” Dr. Abramson said.
The phase 1 trial (NCT02631044) includes 269 patients who received liso-cel. They were diagnosed with transformed follicular lymphoma (22%) or other indolent lymphoma (7%), high-grade B-cell lymphoma (13%), primary mediastinal large B-cell lymphoma (6%), grade 3B follicular lymphoma (1%), or diffuse large B-cell lymphoma not otherwise specified (51%).
At baseline, patients had received a median of three prior systemic therapies (range, one to eight). Some patients had received autologous (33%) or allogeneic (3%) transplant. Many patients were chemotherapy refractory (67%) or had never achieved a complete response to prior therapy (44%).
More than half of patients (59%) received bridging therapy during liso-cel manufacturing. All patients received lymphodepletion with fludarabine and cyclophosphamide, followed by liso-cel at 50 x 106 CAR T cells, 100 x 106 CAR T cells, or 150 x 106 CAR T cells.
Response and survival
The median follow-up was 12.0 months. The overall response rate was 73%, and the complete response rate was 53%.
“Remissions were rapid, with a median of 1 month from CAR T-cell infusion, and durable, with a median duration of response that has not been reached and 55% of patients remaining in response at 1 year,” Dr. Abramson said.
The median PFS was 6.8 months overall, not reached for patients who achieved a complete response, 2.8 months for patients with a partial response, and 1.1 months for patients with stable disease or progressive disease.
The median PFS was not reached for patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma, 5.0 months for high-grade B-cell lymphoma, 3.0 months for diffuse large B-cell lymphoma not otherwise specified, and 2.9 months in transformed indolent non-Hodgkin lymphoma.
The median overall survival was 21.1 months overall, not reached for patients who achieved a complete response, 9.0 months for patients who had a partial response, and 5.1 months for patients with stable disease or progressive disease.
Safety
Common treatment-emergent adverse events were neutropenia (63%), anemia (48%), fatigue (44%), nausea (33%), thrombocytopenia (31%), headache (30%), decreased appetite (28%), and diarrhea (26%).
Cytokine release syndrome (CRS) occurred in 42% of patients, and neurologic events occurred in 30%. Grade 3-4 CRS occurred in 2% of patients, and grade 3-4 neurologic events occurred in 10%. There were no cases of grade 5 CRS or neurologic events.
The median time to CRS onset was 5 days, and the median time to onset of neurologic events was 9 days. The median time to resolution of CRS and neurologic events was 5 days and 11 days, respectively.
“The low incidence of severe CRS and neurologic events and their late time of onset support using this product in a large range of patients and in the outpatient setting,” Dr. Abramson said.
There were seven grade 5 treatment-related adverse events, including diffuse alveolar damage, pulmonary hemorrhage, multiple organ dysfunction syndrome, cardiomyopathy, fludarabine leukoencephalopathy, septic shock, and progressive multifocal leukoencephalopathy.
This trial is sponsored by Bristol-Myers Squibb. Dr. Abramson reported relationships with Juno Therapeutics and Celgene, now owned by Bristol-Myers Squibb, and a range of other companies.
SOURCE: Abramson JS et al. ASH 2019, Abstract 241.
ORLANDO – Updated results from the TRANSCEND NHL trial include survival data with lisocabtagene maraleucel (liso-cel), an anti-CD19 chimeric antigen receptor (CAR) T-cell therapy, in patients with relapsed/refractory B-cell lymphomas.
The median progression-free survival (PFS) was 6.8 months, and the median overall survival was 21.1 months. PFS results were best among complete responders and among patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma.
Jeremy S. Abramson, MD, of Massachusetts General Hospital in Boston, presented these results at the annual meeting of the American Society of Hematology.
“TRANSCEND NHL is the largest clinical study to date of CD19-directed CAR T cells in patients with relapsed/refractory aggressive B-cell lymphoma,” Dr. Abramson said.
The phase 1 trial (NCT02631044) includes 269 patients who received liso-cel. They were diagnosed with transformed follicular lymphoma (22%) or other indolent lymphoma (7%), high-grade B-cell lymphoma (13%), primary mediastinal large B-cell lymphoma (6%), grade 3B follicular lymphoma (1%), or diffuse large B-cell lymphoma not otherwise specified (51%).
At baseline, patients had received a median of three prior systemic therapies (range, one to eight). Some patients had received autologous (33%) or allogeneic (3%) transplant. Many patients were chemotherapy refractory (67%) or had never achieved a complete response to prior therapy (44%).
More than half of patients (59%) received bridging therapy during liso-cel manufacturing. All patients received lymphodepletion with fludarabine and cyclophosphamide, followed by liso-cel at 50 x 106 CAR T cells, 100 x 106 CAR T cells, or 150 x 106 CAR T cells.
Response and survival
The median follow-up was 12.0 months. The overall response rate was 73%, and the complete response rate was 53%.
“Remissions were rapid, with a median of 1 month from CAR T-cell infusion, and durable, with a median duration of response that has not been reached and 55% of patients remaining in response at 1 year,” Dr. Abramson said.
The median PFS was 6.8 months overall, not reached for patients who achieved a complete response, 2.8 months for patients with a partial response, and 1.1 months for patients with stable disease or progressive disease.
The median PFS was not reached for patients with primary mediastinal large B-cell lymphoma or transformed follicular lymphoma, 5.0 months for high-grade B-cell lymphoma, 3.0 months for diffuse large B-cell lymphoma not otherwise specified, and 2.9 months in transformed indolent non-Hodgkin lymphoma.
The median overall survival was 21.1 months overall, not reached for patients who achieved a complete response, 9.0 months for patients who had a partial response, and 5.1 months for patients with stable disease or progressive disease.
Safety
Common treatment-emergent adverse events were neutropenia (63%), anemia (48%), fatigue (44%), nausea (33%), thrombocytopenia (31%), headache (30%), decreased appetite (28%), and diarrhea (26%).
Cytokine release syndrome (CRS) occurred in 42% of patients, and neurologic events occurred in 30%. Grade 3-4 CRS occurred in 2% of patients, and grade 3-4 neurologic events occurred in 10%. There were no cases of grade 5 CRS or neurologic events.
The median time to CRS onset was 5 days, and the median time to onset of neurologic events was 9 days. The median time to resolution of CRS and neurologic events was 5 days and 11 days, respectively.
“The low incidence of severe CRS and neurologic events and their late time of onset support using this product in a large range of patients and in the outpatient setting,” Dr. Abramson said.
There were seven grade 5 treatment-related adverse events, including diffuse alveolar damage, pulmonary hemorrhage, multiple organ dysfunction syndrome, cardiomyopathy, fludarabine leukoencephalopathy, septic shock, and progressive multifocal leukoencephalopathy.
This trial is sponsored by Bristol-Myers Squibb. Dr. Abramson reported relationships with Juno Therapeutics and Celgene, now owned by Bristol-Myers Squibb, and a range of other companies.
SOURCE: Abramson JS et al. ASH 2019, Abstract 241.
REPORTING FROM ASH 2019
Vitamin D alone does not reduce fracture risk
Vitamin D supplementation alone does not appear to reduce the risk of fracture, but a combination of vitamin D and calcium may, according to a systematic review and meta-analysis published in JAMA Network Open.
Pang Yao, PhD, from the Nuffield Department of Population Health at the University of Oxford (England) and coauthors wrote that, while randomized, controlled trials (RCTs) of vitamin D supplements – either alone or in combination with calcium supplementation – have found conflicting results, most only had limited power to detect differences in the risk of fracture.
Dr. Yao and associates performed a meta-analysis of 11 observational studies with 39,141 participants, 11 RCTs of vitamin D supplementation alone in 34,243 participants, and 6 RCTs of calcium plus vitamin D involving 49,282 participants.
The analysis of the observational studies revealed that each 10.0-ng/mL increase in blood 25-hydroxyvitamin D concentrations was associated with a 7% lower risk of any fracture. However the authors noted significant heterogeneity between individual studies.
The meta-analysis of the 11 trials of vitamin D alone found that supplementation was not associated with significant change in the risk for any fracture or for hip fracture. Even subgroup analyses looking at age, residential status, location, study design, daily supplementation, or duration of supplementation failed to find any effect. However, there was a median difference in blood 25-hydroxyvitamin D concentrations of 8.4 ng/mL with vitamin D supplementation.
In the meta-analysis of the six vitamin D plus calcium trials, there was a significant 6% reduction in the rate of any fracture and a 16% reduction in hip fracture rate with supplementation. Overall, there was a 1% reduction in the risk of any fracture for each 0.4-ng/mL difference in blood 25-hydroxyvitamin D concentration and 2% reduction in the risk of hip fracture.
However, the authors judged five of those six vitamin D plus calcium trials to be at high risk of bias, with two having open-label designs, although there was little heterogeneity among the studies. All the trials used either 800 or 400 IU/day of vitamin D and 1,200 or 800 mg/day of calcium, and the mean duration of treatment was 5.9 years.
Participants aged 80 years or older living in institutions showed greater reductions in the risk of any fracture with calcium plus vitamin D supplementation, compared with those younger than 80 years who were living in the community.
“In this systematic review and meta-analysis, the available evidence from completed RCTs provided no support for the effects of vitamin D alone on prevention of fracture, but most of these RCTs were constrained by methodological problems,” they wrote. “Meta-analyses of ongoing RCTs assessing the effects of higher daily doses of vitamin D on fracture risk are needed before making recommendations on the use of vitamin D for prevention of fracture.”
One author was supported by a Sino-British Fellowship Trust scholarship, and another received grants from the U.K. Medical Research Council. No conflicts of interest were declared.
SOURCE: Yao P et al. JAMA Netw Open. 2019. doi: 10.1001/jamanetworkopen.2019.17789.
Vitamin D supplementation alone does not appear to reduce the risk of fracture, but a combination of vitamin D and calcium may, according to a systematic review and meta-analysis published in JAMA Network Open.
Pang Yao, PhD, from the Nuffield Department of Population Health at the University of Oxford (England) and coauthors wrote that, while randomized, controlled trials (RCTs) of vitamin D supplements – either alone or in combination with calcium supplementation – have found conflicting results, most only had limited power to detect differences in the risk of fracture.
Dr. Yao and associates performed a meta-analysis of 11 observational studies with 39,141 participants, 11 RCTs of vitamin D supplementation alone in 34,243 participants, and 6 RCTs of calcium plus vitamin D involving 49,282 participants.
The analysis of the observational studies revealed that each 10.0-ng/mL increase in blood 25-hydroxyvitamin D concentrations was associated with a 7% lower risk of any fracture. However the authors noted significant heterogeneity between individual studies.
The meta-analysis of the 11 trials of vitamin D alone found that supplementation was not associated with significant change in the risk for any fracture or for hip fracture. Even subgroup analyses looking at age, residential status, location, study design, daily supplementation, or duration of supplementation failed to find any effect. However, there was a median difference in blood 25-hydroxyvitamin D concentrations of 8.4 ng/mL with vitamin D supplementation.
In the meta-analysis of the six vitamin D plus calcium trials, there was a significant 6% reduction in the rate of any fracture and a 16% reduction in hip fracture rate with supplementation. Overall, there was a 1% reduction in the risk of any fracture for each 0.4-ng/mL difference in blood 25-hydroxyvitamin D concentration and 2% reduction in the risk of hip fracture.
However, the authors judged five of those six vitamin D plus calcium trials to be at high risk of bias, with two having open-label designs, although there was little heterogeneity among the studies. All the trials used either 800 or 400 IU/day of vitamin D and 1,200 or 800 mg/day of calcium, and the mean duration of treatment was 5.9 years.
Participants aged 80 years or older living in institutions showed greater reductions in the risk of any fracture with calcium plus vitamin D supplementation, compared with those younger than 80 years who were living in the community.
“In this systematic review and meta-analysis, the available evidence from completed RCTs provided no support for the effects of vitamin D alone on prevention of fracture, but most of these RCTs were constrained by methodological problems,” they wrote. “Meta-analyses of ongoing RCTs assessing the effects of higher daily doses of vitamin D on fracture risk are needed before making recommendations on the use of vitamin D for prevention of fracture.”
One author was supported by a Sino-British Fellowship Trust scholarship, and another received grants from the U.K. Medical Research Council. No conflicts of interest were declared.
SOURCE: Yao P et al. JAMA Netw Open. 2019. doi: 10.1001/jamanetworkopen.2019.17789.
Vitamin D supplementation alone does not appear to reduce the risk of fracture, but a combination of vitamin D and calcium may, according to a systematic review and meta-analysis published in JAMA Network Open.
Pang Yao, PhD, from the Nuffield Department of Population Health at the University of Oxford (England) and coauthors wrote that, while randomized, controlled trials (RCTs) of vitamin D supplements – either alone or in combination with calcium supplementation – have found conflicting results, most only had limited power to detect differences in the risk of fracture.
Dr. Yao and associates performed a meta-analysis of 11 observational studies with 39,141 participants, 11 RCTs of vitamin D supplementation alone in 34,243 participants, and 6 RCTs of calcium plus vitamin D involving 49,282 participants.
The analysis of the observational studies revealed that each 10.0-ng/mL increase in blood 25-hydroxyvitamin D concentrations was associated with a 7% lower risk of any fracture. However the authors noted significant heterogeneity between individual studies.
The meta-analysis of the 11 trials of vitamin D alone found that supplementation was not associated with significant change in the risk for any fracture or for hip fracture. Even subgroup analyses looking at age, residential status, location, study design, daily supplementation, or duration of supplementation failed to find any effect. However, there was a median difference in blood 25-hydroxyvitamin D concentrations of 8.4 ng/mL with vitamin D supplementation.
In the meta-analysis of the six vitamin D plus calcium trials, there was a significant 6% reduction in the rate of any fracture and a 16% reduction in hip fracture rate with supplementation. Overall, there was a 1% reduction in the risk of any fracture for each 0.4-ng/mL difference in blood 25-hydroxyvitamin D concentration and 2% reduction in the risk of hip fracture.
However, the authors judged five of those six vitamin D plus calcium trials to be at high risk of bias, with two having open-label designs, although there was little heterogeneity among the studies. All the trials used either 800 or 400 IU/day of vitamin D and 1,200 or 800 mg/day of calcium, and the mean duration of treatment was 5.9 years.
Participants aged 80 years or older living in institutions showed greater reductions in the risk of any fracture with calcium plus vitamin D supplementation, compared with those younger than 80 years who were living in the community.
“In this systematic review and meta-analysis, the available evidence from completed RCTs provided no support for the effects of vitamin D alone on prevention of fracture, but most of these RCTs were constrained by methodological problems,” they wrote. “Meta-analyses of ongoing RCTs assessing the effects of higher daily doses of vitamin D on fracture risk are needed before making recommendations on the use of vitamin D for prevention of fracture.”
One author was supported by a Sino-British Fellowship Trust scholarship, and another received grants from the U.K. Medical Research Council. No conflicts of interest were declared.
SOURCE: Yao P et al. JAMA Netw Open. 2019. doi: 10.1001/jamanetworkopen.2019.17789.
FROM JAMA NETWORK OPEN
Chemotherapy better for metastatic breast cancer maintenance than durvalumab
SAN ANTONIO – Maintenance therapy with the immune checkpoint inhibitor durvalumab (Imfinzi) did not improve outcomes compared with chemotherapy for patients with metastatic breast cancer, although there was a trend toward benefit for patients with triple-negative breast cancer and those whose tumors were positive for programmed death ligand 1 (PD-L1), a substudy of SAFIR-02 Breast.

Median progression-free survival for 131 patients with metastatic or locally advanced, HER2-negative, hormone receptor–negative or endocrine-resistant breast cancer who received durvalumab maintenance following chemotherapy in the study was 2.7 months, compared with 4.4 months for 68 patients who received maintenance chemotherapy, reported Florence Dalenc, MD, of Institut Claudius Regaud, Toulouse, France.
“For hormone receptor–positive metastatic breast cancer, exploratory analyses suggest that anti-PD-L1 as a single agent is less effective than chemotherapy as maintenance,” she said at the San Antonio Breast Cancer Symposium.
The rationale for investigating an anti-PD-L1 agent for maintenance in patients with metastatic breast cancer is that the tumor is (presumably) not growing when immunotherapy is introduced and disease burden, a mechanism of immune suppression, has been reduced by chemotherapy. In addition, chemotherapy attracts tumor infiltrating lymphocytes (TILs) to the tumor bed, and response to chemotherapy may be a marker for immunogenic tumors, she said.
Durvalumab is a human immunoglobulin G1 kappa monoclonal antibody that blocks the interaction of the programmed death protein 1 with PD-L1. In the GeparNuevo trial, the addition of durvalumab to anthracycline-/taxane-based neoadjuvant chemotherapy increased the pathologic complete response (pCR) rate particularly in patients treated with durvalumab alone before the start of chemotherapy.
To see whether durvalumab could offer a benefit over chemotherapy in the maintenance setting, Dr. Dalenc and colleagues studied patients who were enrolled in SAFIR-02 Breast, which was designed to assess the efficacy of genomic analysis as a therapeutic decision tool in patients with metastatic breast cancer.
In the trial, patients with HER2-negative metastatic breast cancer who have a complete or partial response or stable disease following six to eight chemotherapy cycles (or 4 cycles if chemotherapy was stopped for toxicities) are randomized based on the presence or absence of targetable molecular alterations. Patients with identified alteration are then randomized to either targeted therapy matched to genomics, or to maintenance chemotherapy.
Patients without molecular alterations – the population included in the SAFIR02-IMMUNO substudy reported by Dr. Dalenc and colleagues – are stratified by one or two prior lines of chemotherapy and by response type, and then stratified on a 2:1 basis to maintenance with either durvalumab 10 mg/kg every 14 days or standard maintenance chemotherapy for up to 2 years.
In each arm, the median patient age was 56, and approximately 58% of patients had Eastern Cooperative Oncology Group (ECOG) performance status of 0. Approximately 43% of patients in each arm had three or more metastatic sites, and roughly half had liver metastases, while slightly more than one-fourth had metastases to lung.
As noted before, median PFS in the overall substudy population, the primary endpoint, was 4.6 months for patients on maintenance chemotherapy, compared with 2.7 months with durvalumab. This difference translated into an adjusted hazard ratio for progression with durvalumab of 1.40 (P = .047).
Also as noted, there were nonsignificant trends toward benefit with durvalumab among patients with triple-negative breast cancer or PD-L1-positive tumors. For all other subgroups, however, maintenance chemotherapy was favored, with significant benefit seen among patients with disease that progressed after second-line chemotherapy, patients with a clinical response at the time of re-randomization, those 50 years and older, patients with non-triple-negative disease, patients with liver metastases, and those whose tumors were negative for PD-L1.
Median overall survival (OS) was numerically longer with durvalumab (21.7 vs. 17.9 months), but this difference was not statistically significant.
In exploratory analyses, median OS for patients with triple-negative disease was 21 vs. 14 months, respectively, with an unadjusted hazard ratio favoring durvalumab of 0.54 (P = .0377). Among patients with PD-L1 positive tumors, the median OS was 26 months with durvalumab vs. 12 months with chemotherapy, with an unadjusted hazard ratio of 0.42, which just missed statistical significance (P = .0552).
Dr. Dalenc said that the study generates the hypothesis that single-agent durvalumab could improve outcomes in metastatic triple-negative breast cancer, but also raises questions about which disease parameters limit the response to anti-PD-L1 therapy.
The study was sponsored by UNICANCER with support from Fondation ARC and AstraZeneca. Dr. Dalenc reported research grants, travel, and/or advisory board activity for AstraZeneca and others.
SOURCE: Dalenc F et al. SABCS 2019. Abstract GS3-02.
SAN ANTONIO – Maintenance therapy with the immune checkpoint inhibitor durvalumab (Imfinzi) did not improve outcomes compared with chemotherapy for patients with metastatic breast cancer, although there was a trend toward benefit for patients with triple-negative breast cancer and those whose tumors were positive for programmed death ligand 1 (PD-L1), a substudy of SAFIR-02 Breast.
Median progression-free survival for 131 patients with metastatic or locally advanced, HER2-negative, hormone receptor–negative or endocrine-resistant breast cancer who received durvalumab maintenance following chemotherapy in the study was 2.7 months, compared with 4.4 months for 68 patients who received maintenance chemotherapy, reported Florence Dalenc, MD, of Institut Claudius Regaud, Toulouse, France.
“For hormone receptor–positive metastatic breast cancer, exploratory analyses suggest that anti-PD-L1 as a single agent is less effective than chemotherapy as maintenance,” she said at the San Antonio Breast Cancer Symposium.
The rationale for investigating an anti-PD-L1 agent for maintenance in patients with metastatic breast cancer is that the tumor is (presumably) not growing when immunotherapy is introduced and disease burden, a mechanism of immune suppression, has been reduced by chemotherapy. In addition, chemotherapy attracts tumor infiltrating lymphocytes (TILs) to the tumor bed, and response to chemotherapy may be a marker for immunogenic tumors, she said.
Durvalumab is a human immunoglobulin G1 kappa monoclonal antibody that blocks the interaction of the programmed death protein 1 with PD-L1. In the GeparNuevo trial, the addition of durvalumab to anthracycline-/taxane-based neoadjuvant chemotherapy increased the pathologic complete response (pCR) rate particularly in patients treated with durvalumab alone before the start of chemotherapy.
To see whether durvalumab could offer a benefit over chemotherapy in the maintenance setting, Dr. Dalenc and colleagues studied patients who were enrolled in SAFIR-02 Breast, which was designed to assess the efficacy of genomic analysis as a therapeutic decision tool in patients with metastatic breast cancer.
In the trial, patients with HER2-negative metastatic breast cancer who have a complete or partial response or stable disease following six to eight chemotherapy cycles (or 4 cycles if chemotherapy was stopped for toxicities) are randomized based on the presence or absence of targetable molecular alterations. Patients with identified alteration are then randomized to either targeted therapy matched to genomics, or to maintenance chemotherapy.
Patients without molecular alterations – the population included in the SAFIR02-IMMUNO substudy reported by Dr. Dalenc and colleagues – are stratified by one or two prior lines of chemotherapy and by response type, and then stratified on a 2:1 basis to maintenance with either durvalumab 10 mg/kg every 14 days or standard maintenance chemotherapy for up to 2 years.
In each arm, the median patient age was 56, and approximately 58% of patients had Eastern Cooperative Oncology Group (ECOG) performance status of 0. Approximately 43% of patients in each arm had three or more metastatic sites, and roughly half had liver metastases, while slightly more than one-fourth had metastases to lung.
As noted before, median PFS in the overall substudy population, the primary endpoint, was 4.6 months for patients on maintenance chemotherapy, compared with 2.7 months with durvalumab. This difference translated into an adjusted hazard ratio for progression with durvalumab of 1.40 (P = .047).
Also as noted, there were nonsignificant trends toward benefit with durvalumab among patients with triple-negative breast cancer or PD-L1-positive tumors. For all other subgroups, however, maintenance chemotherapy was favored, with significant benefit seen among patients with disease that progressed after second-line chemotherapy, patients with a clinical response at the time of re-randomization, those 50 years and older, patients with non-triple-negative disease, patients with liver metastases, and those whose tumors were negative for PD-L1.
Median overall survival (OS) was numerically longer with durvalumab (21.7 vs. 17.9 months), but this difference was not statistically significant.
In exploratory analyses, median OS for patients with triple-negative disease was 21 vs. 14 months, respectively, with an unadjusted hazard ratio favoring durvalumab of 0.54 (P = .0377). Among patients with PD-L1 positive tumors, the median OS was 26 months with durvalumab vs. 12 months with chemotherapy, with an unadjusted hazard ratio of 0.42, which just missed statistical significance (P = .0552).
Dr. Dalenc said that the study generates the hypothesis that single-agent durvalumab could improve outcomes in metastatic triple-negative breast cancer, but also raises questions about which disease parameters limit the response to anti-PD-L1 therapy.
The study was sponsored by UNICANCER with support from Fondation ARC and AstraZeneca. Dr. Dalenc reported research grants, travel, and/or advisory board activity for AstraZeneca and others.
SOURCE: Dalenc F et al. SABCS 2019. Abstract GS3-02.
SAN ANTONIO – Maintenance therapy with the immune checkpoint inhibitor durvalumab (Imfinzi) did not improve outcomes compared with chemotherapy for patients with metastatic breast cancer, although there was a trend toward benefit for patients with triple-negative breast cancer and those whose tumors were positive for programmed death ligand 1 (PD-L1), a substudy of SAFIR-02 Breast.
Median progression-free survival for 131 patients with metastatic or locally advanced, HER2-negative, hormone receptor–negative or endocrine-resistant breast cancer who received durvalumab maintenance following chemotherapy in the study was 2.7 months, compared with 4.4 months for 68 patients who received maintenance chemotherapy, reported Florence Dalenc, MD, of Institut Claudius Regaud, Toulouse, France.
“For hormone receptor–positive metastatic breast cancer, exploratory analyses suggest that anti-PD-L1 as a single agent is less effective than chemotherapy as maintenance,” she said at the San Antonio Breast Cancer Symposium.
The rationale for investigating an anti-PD-L1 agent for maintenance in patients with metastatic breast cancer is that the tumor is (presumably) not growing when immunotherapy is introduced and disease burden, a mechanism of immune suppression, has been reduced by chemotherapy. In addition, chemotherapy attracts tumor infiltrating lymphocytes (TILs) to the tumor bed, and response to chemotherapy may be a marker for immunogenic tumors, she said.
Durvalumab is a human immunoglobulin G1 kappa monoclonal antibody that blocks the interaction of the programmed death protein 1 with PD-L1. In the GeparNuevo trial, the addition of durvalumab to anthracycline-/taxane-based neoadjuvant chemotherapy increased the pathologic complete response (pCR) rate particularly in patients treated with durvalumab alone before the start of chemotherapy.
To see whether durvalumab could offer a benefit over chemotherapy in the maintenance setting, Dr. Dalenc and colleagues studied patients who were enrolled in SAFIR-02 Breast, which was designed to assess the efficacy of genomic analysis as a therapeutic decision tool in patients with metastatic breast cancer.
In the trial, patients with HER2-negative metastatic breast cancer who have a complete or partial response or stable disease following six to eight chemotherapy cycles (or 4 cycles if chemotherapy was stopped for toxicities) are randomized based on the presence or absence of targetable molecular alterations. Patients with identified alteration are then randomized to either targeted therapy matched to genomics, or to maintenance chemotherapy.
Patients without molecular alterations – the population included in the SAFIR02-IMMUNO substudy reported by Dr. Dalenc and colleagues – are stratified by one or two prior lines of chemotherapy and by response type, and then stratified on a 2:1 basis to maintenance with either durvalumab 10 mg/kg every 14 days or standard maintenance chemotherapy for up to 2 years.
In each arm, the median patient age was 56, and approximately 58% of patients had Eastern Cooperative Oncology Group (ECOG) performance status of 0. Approximately 43% of patients in each arm had three or more metastatic sites, and roughly half had liver metastases, while slightly more than one-fourth had metastases to lung.
As noted before, median PFS in the overall substudy population, the primary endpoint, was 4.6 months for patients on maintenance chemotherapy, compared with 2.7 months with durvalumab. This difference translated into an adjusted hazard ratio for progression with durvalumab of 1.40 (P = .047).
Also as noted, there were nonsignificant trends toward benefit with durvalumab among patients with triple-negative breast cancer or PD-L1-positive tumors. For all other subgroups, however, maintenance chemotherapy was favored, with significant benefit seen among patients with disease that progressed after second-line chemotherapy, patients with a clinical response at the time of re-randomization, those 50 years and older, patients with non-triple-negative disease, patients with liver metastases, and those whose tumors were negative for PD-L1.
Median overall survival (OS) was numerically longer with durvalumab (21.7 vs. 17.9 months), but this difference was not statistically significant.
In exploratory analyses, median OS for patients with triple-negative disease was 21 vs. 14 months, respectively, with an unadjusted hazard ratio favoring durvalumab of 0.54 (P = .0377). Among patients with PD-L1 positive tumors, the median OS was 26 months with durvalumab vs. 12 months with chemotherapy, with an unadjusted hazard ratio of 0.42, which just missed statistical significance (P = .0552).
Dr. Dalenc said that the study generates the hypothesis that single-agent durvalumab could improve outcomes in metastatic triple-negative breast cancer, but also raises questions about which disease parameters limit the response to anti-PD-L1 therapy.
The study was sponsored by UNICANCER with support from Fondation ARC and AstraZeneca. Dr. Dalenc reported research grants, travel, and/or advisory board activity for AstraZeneca and others.
SOURCE: Dalenc F et al. SABCS 2019. Abstract GS3-02.
REPORTING FROM SABCS 2019