User login
The Journal of Clinical Outcomes Management® is an independent, peer-reviewed journal offering evidence-based, practical information for improving the quality, safety, and value of health care.
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
div[contains(@class, 'main-prefix')]
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
New-onset AFib common but unrecognized in the month after cardiac surgery
One in five patients at elevated stroke risk who underwent cardiac surgery with no history of atrial fibrillation preoperatively or at discharge developed postoperative AFib documented on a continuous cardiac rhythm monitoring device within the first 30 days after leaving the hospital in the randomized SEARCH-AF trial.

“Postoperative atrial fibrillation after cardiac surgery is not confined to the hospitalization period per se. We believe that these data should help inform on clinical practice guidelines on monitoring for postoperative atrial fibrillation in such patients,” said Subodh Verma, MD, PhD, reporting the results at the virtual American Heart Association scientific sessions.
“Guidelines provide little or no direction on optimal monitoring post cardiac surgery, particularly if patients are in sinus rhythm at discharge,” the surgeon noted.
SEARCH-AF was an open-label, multicenter study that included 336 patients at elevated stroke risk with an average CHA2DS2-VASc score of 4, no history of preoperative AFib, and none more than briefly with resolution during hospitalization. They were randomized to 30 days of postdischarge continuous cardiac rhythm monitoring with Medtronic’s SEEQ device, to Icentia’s CardioSTAT device, or to usual care, with Holter monitoring at the discretion of the treating physicians.
The primary result was a cumulative duration of AFib or atrial flutter of 6 minutes or longer during that 30-day period. This outcome occurred in 19.6% of the enhanced cardiac monitoring group and 1.7% of usual-care controls. Thus, there is an ongoing persistent occult risk of AFib that typically goes unrecognized. This 10-fold difference in the incidence of postoperative AFib translated into an absolute 17.9% between-group difference and a number-needed-to-treat of 6.
The secondary outcome of a cumulative atrial fib/flutter burden of 6 hours or more during 30 days occurred in 8.6% of the continuously monitored group and none of the controls. A cumulative AFib/flutter burden of 24 hours or greater occurred in 3.1% of the enhanced cardiac monitoring group and zero controls. These are AFib burdens that in other studies have been linked to increased risks of stroke and death, said Dr. Verma, professor of cardiovascular surgery at the University of Toronto.
“From a clinical standpoint, what this trial tells me is for my patients being discharged home tomorrow from the hospital, where they haven’t had AFib and I haven’t initiated anticoagulation, I have a low threshold to monitor these patients and to watch for periods of sustained unrecognized atrial fibrillation,” the surgeon added.
Experts: Results won’t change guidelines
Discussant Ben Freedman, MBBS, PhD, noted that the U.S. Preventive Services Task Force has stated that there are insufficient data available to recommend ECG screening for AFib to prevent stroke. Before the task force can be convinced to recommend it and for payers to cover it, a number of key questions need to be answered. And the SEARCH-AF trial doesn’t provide those answers, said Dr. Freedman, professor of cardiology and deputy director of the Heart Research Institute at the University of Sydney.
First off, it’ll be necessary to know if the risk posed by screen-detected AFib, including postoperative AFib, is similar to that of clinical AFib. Next, it must be shown that this screen-detected postoperative AFib is actionable; that is, that a screening strategy to detect postoperative AFib arising after discharge and then treat with oral anticoagulants will actually prevent more strokes than with usual care. There are large studies underway addressing that question, including HEARTLINE, STROKESTOP, and SAFERGUARD-AF, he observed.
In an interview, Rod S. Passman, MD, who gave a state-of-the-art talk on AFib detection at the meeting and wasn’t involved in SEARCH-AF, said he doesn’t consider the results practice-changing.
“It’s not guideline-changing because you’ve only shown that more intensive monitoring finds more AFib. Guideline-changing would be that finding that AFib and doing something about it impacts hard outcomes, and we don’t have that data yet,” said Dr. Passman, an electrophysiologist who is director of the Center for Arrhythmia Research and professor of medicine and preventive medicine at Northwestern University, Chicago.
The SEARCH-AF trial was funded by the Heart and Stroke Foundation of Canada, Bristol Myers Squibb, Pfizer, and Boehringer Ingelheim. Dr. Verma reported having received speaker’s fees and/or research support from those and other pharmaceutical companies. Dr. Freedman disclosed having no financial conflicts.
One in five patients at elevated stroke risk who underwent cardiac surgery with no history of atrial fibrillation preoperatively or at discharge developed postoperative AFib documented on a continuous cardiac rhythm monitoring device within the first 30 days after leaving the hospital in the randomized SEARCH-AF trial.
“Postoperative atrial fibrillation after cardiac surgery is not confined to the hospitalization period per se. We believe that these data should help inform on clinical practice guidelines on monitoring for postoperative atrial fibrillation in such patients,” said Subodh Verma, MD, PhD, reporting the results at the virtual American Heart Association scientific sessions.
“Guidelines provide little or no direction on optimal monitoring post cardiac surgery, particularly if patients are in sinus rhythm at discharge,” the surgeon noted.
SEARCH-AF was an open-label, multicenter study that included 336 patients at elevated stroke risk with an average CHA2DS2-VASc score of 4, no history of preoperative AFib, and none more than briefly with resolution during hospitalization. They were randomized to 30 days of postdischarge continuous cardiac rhythm monitoring with Medtronic’s SEEQ device, to Icentia’s CardioSTAT device, or to usual care, with Holter monitoring at the discretion of the treating physicians.
The primary result was a cumulative duration of AFib or atrial flutter of 6 minutes or longer during that 30-day period. This outcome occurred in 19.6% of the enhanced cardiac monitoring group and 1.7% of usual-care controls. Thus, there is an ongoing persistent occult risk of AFib that typically goes unrecognized. This 10-fold difference in the incidence of postoperative AFib translated into an absolute 17.9% between-group difference and a number-needed-to-treat of 6.
The secondary outcome of a cumulative atrial fib/flutter burden of 6 hours or more during 30 days occurred in 8.6% of the continuously monitored group and none of the controls. A cumulative AFib/flutter burden of 24 hours or greater occurred in 3.1% of the enhanced cardiac monitoring group and zero controls. These are AFib burdens that in other studies have been linked to increased risks of stroke and death, said Dr. Verma, professor of cardiovascular surgery at the University of Toronto.
“From a clinical standpoint, what this trial tells me is for my patients being discharged home tomorrow from the hospital, where they haven’t had AFib and I haven’t initiated anticoagulation, I have a low threshold to monitor these patients and to watch for periods of sustained unrecognized atrial fibrillation,” the surgeon added.
Experts: Results won’t change guidelines
Discussant Ben Freedman, MBBS, PhD, noted that the U.S. Preventive Services Task Force has stated that there are insufficient data available to recommend ECG screening for AFib to prevent stroke. Before the task force can be convinced to recommend it and for payers to cover it, a number of key questions need to be answered. And the SEARCH-AF trial doesn’t provide those answers, said Dr. Freedman, professor of cardiology and deputy director of the Heart Research Institute at the University of Sydney.
First off, it’ll be necessary to know if the risk posed by screen-detected AFib, including postoperative AFib, is similar to that of clinical AFib. Next, it must be shown that this screen-detected postoperative AFib is actionable; that is, that a screening strategy to detect postoperative AFib arising after discharge and then treat with oral anticoagulants will actually prevent more strokes than with usual care. There are large studies underway addressing that question, including HEARTLINE, STROKESTOP, and SAFERGUARD-AF, he observed.
In an interview, Rod S. Passman, MD, who gave a state-of-the-art talk on AFib detection at the meeting and wasn’t involved in SEARCH-AF, said he doesn’t consider the results practice-changing.
“It’s not guideline-changing because you’ve only shown that more intensive monitoring finds more AFib. Guideline-changing would be that finding that AFib and doing something about it impacts hard outcomes, and we don’t have that data yet,” said Dr. Passman, an electrophysiologist who is director of the Center for Arrhythmia Research and professor of medicine and preventive medicine at Northwestern University, Chicago.
The SEARCH-AF trial was funded by the Heart and Stroke Foundation of Canada, Bristol Myers Squibb, Pfizer, and Boehringer Ingelheim. Dr. Verma reported having received speaker’s fees and/or research support from those and other pharmaceutical companies. Dr. Freedman disclosed having no financial conflicts.
One in five patients at elevated stroke risk who underwent cardiac surgery with no history of atrial fibrillation preoperatively or at discharge developed postoperative AFib documented on a continuous cardiac rhythm monitoring device within the first 30 days after leaving the hospital in the randomized SEARCH-AF trial.
“Postoperative atrial fibrillation after cardiac surgery is not confined to the hospitalization period per se. We believe that these data should help inform on clinical practice guidelines on monitoring for postoperative atrial fibrillation in such patients,” said Subodh Verma, MD, PhD, reporting the results at the virtual American Heart Association scientific sessions.
“Guidelines provide little or no direction on optimal monitoring post cardiac surgery, particularly if patients are in sinus rhythm at discharge,” the surgeon noted.
SEARCH-AF was an open-label, multicenter study that included 336 patients at elevated stroke risk with an average CHA2DS2-VASc score of 4, no history of preoperative AFib, and none more than briefly with resolution during hospitalization. They were randomized to 30 days of postdischarge continuous cardiac rhythm monitoring with Medtronic’s SEEQ device, to Icentia’s CardioSTAT device, or to usual care, with Holter monitoring at the discretion of the treating physicians.
The primary result was a cumulative duration of AFib or atrial flutter of 6 minutes or longer during that 30-day period. This outcome occurred in 19.6% of the enhanced cardiac monitoring group and 1.7% of usual-care controls. Thus, there is an ongoing persistent occult risk of AFib that typically goes unrecognized. This 10-fold difference in the incidence of postoperative AFib translated into an absolute 17.9% between-group difference and a number-needed-to-treat of 6.
The secondary outcome of a cumulative atrial fib/flutter burden of 6 hours or more during 30 days occurred in 8.6% of the continuously monitored group and none of the controls. A cumulative AFib/flutter burden of 24 hours or greater occurred in 3.1% of the enhanced cardiac monitoring group and zero controls. These are AFib burdens that in other studies have been linked to increased risks of stroke and death, said Dr. Verma, professor of cardiovascular surgery at the University of Toronto.
“From a clinical standpoint, what this trial tells me is for my patients being discharged home tomorrow from the hospital, where they haven’t had AFib and I haven’t initiated anticoagulation, I have a low threshold to monitor these patients and to watch for periods of sustained unrecognized atrial fibrillation,” the surgeon added.
Experts: Results won’t change guidelines
Discussant Ben Freedman, MBBS, PhD, noted that the U.S. Preventive Services Task Force has stated that there are insufficient data available to recommend ECG screening for AFib to prevent stroke. Before the task force can be convinced to recommend it and for payers to cover it, a number of key questions need to be answered. And the SEARCH-AF trial doesn’t provide those answers, said Dr. Freedman, professor of cardiology and deputy director of the Heart Research Institute at the University of Sydney.
First off, it’ll be necessary to know if the risk posed by screen-detected AFib, including postoperative AFib, is similar to that of clinical AFib. Next, it must be shown that this screen-detected postoperative AFib is actionable; that is, that a screening strategy to detect postoperative AFib arising after discharge and then treat with oral anticoagulants will actually prevent more strokes than with usual care. There are large studies underway addressing that question, including HEARTLINE, STROKESTOP, and SAFERGUARD-AF, he observed.
In an interview, Rod S. Passman, MD, who gave a state-of-the-art talk on AFib detection at the meeting and wasn’t involved in SEARCH-AF, said he doesn’t consider the results practice-changing.
“It’s not guideline-changing because you’ve only shown that more intensive monitoring finds more AFib. Guideline-changing would be that finding that AFib and doing something about it impacts hard outcomes, and we don’t have that data yet,” said Dr. Passman, an electrophysiologist who is director of the Center for Arrhythmia Research and professor of medicine and preventive medicine at Northwestern University, Chicago.
The SEARCH-AF trial was funded by the Heart and Stroke Foundation of Canada, Bristol Myers Squibb, Pfizer, and Boehringer Ingelheim. Dr. Verma reported having received speaker’s fees and/or research support from those and other pharmaceutical companies. Dr. Freedman disclosed having no financial conflicts.
FROM AHA 2020
VTEs tied to immune checkpoint inhibitor cancer treatment
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.

“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.
“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
Cancer patients who receive an immune checkpoint inhibitor have more than a doubled rate of venous thromboembolism during the subsequent 2 years, compared with their rate during the 2 years before treatment, according to a retrospective analysis of more than 2,800 patients treated at a single U.S. center.
The study focused on cancer patients treated with an immune checkpoint inhibitor (ICI) at Massachusetts General Hospital in Boston. It showed that during the 2 years prior to treatment with any type of ICI, the incidence of venous thromboembolic events (VTE) was 4.85/100 patient-years that then jumped to 11.75/100 patient-years during the 2 years following treatment. This translated into an incidence rate ratio of 2.43 during posttreatment follow-up, compared with pretreatment, Jingyi Gong, MD, said at the virtual American Heart Association scientific sessions.
The increased VTE rate resulted from rises in both the rate of deep vein thrombosis, which had an IRR of 3.23 during the posttreatment period, and for pulmonary embolism, which showed an IRR of 2.24, said Dr. Gong, a physician at Brigham and Women’s Hospital in Boston. She hypothesized that this effect may result from a procoagulant effect of the immune activation and inflammation triggered by ICIs.
Hypothesis-generating results
Cardiologists cautioned that these findings should only be considered hypothesis generating, but raise an important alert for clinicians to have heightened awareness of the potential for VTE following ICI treatment.
“A clear message is to be aware that there is this signal, and be vigilant for patients who might present with VTE following ICI treatment,” commented Richard J. Kovacs, MD, a cardiologist and professor at Indiana University, Indianapolis. The data that Dr. Gong reported are “moderately convincing,” he added in an interview.
“Awareness that patients who receive ICI may be at increased VTE risk is very important,” agreed Umberto Campia, MD, a cardiologist, vascular specialist, and member of the cardio-oncology group at Brigham and Women’s Hospital, who was not involved in the new study.
The potential impact of ICI treatment on VTE risk is slowly emerging, added Dr. Campia. Until recently, the literature primarily was case reports, but recently another retrospective, single-center study came out that reported a 13% incidence of VTE in cancer patients following ICI treatment. On the other hand, a recently published meta-analysis of more than 20,000 patients from 68 ICI studies failed to find a suggestion of increased VTE incidence following ICI interventions.
Attempting to assess the impact of treatment on VTE risk in cancer patients is challenging because cancer itself boosts the risk. Recommendations on the use of VTE prophylaxis in cancer patients most recently came out in 2014 from the American Society of Clinical Oncology, which said that VTE prophylaxis for ambulatory cancer patients “may be considered for highly select high-risk patients.” The impact of cancer therapy on VTE risk and the need for prophylaxis is usually assessed by applying the Khorana score, Dr. Campia said in an interview.
VTE spikes acutely after ICI treatment
Dr. Gong analyzed VTE incidence rates by time during the total 4-year period studied, and found that the rate gradually and steadily rose with time throughout the 2 years preceding treatment, spiked immediately following ICI treatment, and then gradually and steadily fell back to roughly the rate seen just before treatment, reaching that level about a year after treatment. She ran a sensitivity analysis that excluded patients who died during the first year following their ICI treatment, and in this calculation an acute spike in VTE following ICI treatment still occurred but with reduced magnitude.
She also reported the results of several subgroup analyses. The IRRs remained consistent among women and men, among patients who were aged over or under 65 years, and regardless of cancer type or treatment with corticosteroids. But the subgroup analyses identified two parameters that seemed to clearly split VTE rates.
Among patients on treatment with an anticoagulant agent at the time of their ICI treatment, roughly 10% of the patients, the IRR was 0.56, compared with a ratio of 3.86 among the other patients, suggesting possible protection. A second factor that seemed linked with VTE incidence was the number of ICI treatment cycles a patient received. Those who received more than five cycles had a risk ratio of 3.95, while those who received five or fewer cycles had a RR of 1.66.
Her analysis included 2,842 cancer patients who received treatment with an ICI at Massachusetts General Hospital. Patients averaged 64 years of age, slightly more than half were men, and 13% had a prior history of VTE. Patients received an average of 5 ICI treatment cycles, but a quarter of the patients received more than 10 cycles.
During the 2-year follow-up, 244 patients (9%) developed VTE. The patients who developed VTE were significantly younger than those who did not, with an average age of 63 years, compared with 65. And the patients who eventually developed VTE had a significantly higher prevalence of prior VTE at 18%, compared with 12% among the patients who stayed VTE free.
The cancer types patients had were non–small cell lung, 29%; melanoma, 28%; head and neck, 12%; renal genitourinary, 6%; and other, 25%. ICIs have been available for routine U.S. practice since 2011. The class includes agents such as pembrolizumab (Keytruda) and durvalumab (Imfinzi).
Researchers would need to perform a prospective, randomized study to determine whether anticoagulant prophylaxis is clearly beneficial for patients receiving ICI treatment, Dr. Gong said. But both Dr. Kovacs and Dr. Campia said that more data on this topic are first needed.
“We need to confirm that treatment with ICI is associated with VTEs. Retrospective data are not definitive,” said Dr. Campia. “We would need to prospectively assess the impact of ICI,” which will not be easy, as it’s quickly become a cornerstone for treating many cancers. “We need to become more familiar with the adverse effects of these drugs. We are still learning about their toxicities.”
The study had no commercial funding. Dr. Gong, Dr. Kovacs, and Dr. Campia had no disclosures.
FROM AHA 2020
Using telehealth to deliver palliative care to cancer patients
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.

Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.
Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
Traditional delivery of palliative care to outpatients with cancer is associated with many challenges.
Telehealth can eliminate some of these challenges but comes with issues of its own, according to results of the REACH PC trial.
Jennifer S. Temel, MD, of Massachusetts General Hospital in Boston, discussed the use of telemedicine in palliative care, including results from REACH PC, during an educational session at the ASCO Virtual Quality Care Symposium 2020.
Dr. Temel noted that, for cancer patients, an in-person visit with a palliative care specialist can cost time, induce fatigue, and increase financial burden from transportation and parking expenses.
For caregivers and family, an in-person visit may necessitate absence from family and/or work, require complex scheduling to coordinate with other office visits, and result in additional transportation and/or parking expenses.
For health care systems, to have a dedicated palliative care clinic requires precious space and financial expenditures for office personnel and other resources.
These issues make it attractive to consider whether telehealth could be used for palliative care services.
Scarcity of palliative care specialists
In the United States, there is roughly 1 palliative care physician for every 20,000 older adults with a life-limiting illness, according to research published in Annual Review of Public Health in 2014.
In its 2019 state-by-state report card, the Center to Advance Palliative Care noted that only 72% of U.S. hospitals with 50 or more beds have a palliative care team.
For patients with serious illnesses and those who are socioeconomically or geographically disadvantaged, palliative care is often inaccessible.
Inefficiencies in the current system are an additional impediment. Palliative care specialists frequently see patients during a portion of the patient’s routine visit to subspecialty or primary care clinics. This limits the palliative care specialist’s ability to perform comprehensive assessments and provide patient-centered care efficiently.
Special considerations regarding telehealth for palliative care
As a specialty, palliative care involves interactions that could make the use of telehealth problematic. For example, conveyance of interest, warmth, and touch are challenging or impossible in a video format.
Palliative care specialists engage with patients regarding relatively serious topics such as prognosis and end-of-life preferences. There is uncertainty about how those discussions would be received by patients and their caregivers via video.
Furthermore, there are logistical impediments such as prescribing opioids with video or across state lines.
Despite these concerns, the ENABLE study showed that supplementing usual oncology care with weekly (transitioning to monthly) telephone-based educational palliative care produced higher quality of life and mood than did usual oncology care alone. These results were published in JAMA in 2009.
REACH PC study demonstrates feasibility of telehealth model
Dr. Temel described the ongoing REACH PC trial in which palliative care is delivered via video visits and compared with in-person palliative care for patients with advanced non–small cell lung cancer.
The primary aim of REACH PC is to determine whether telehealth palliative care is equivalent to traditional palliative care in improving quality of life as a supplement to routine oncology care.
Currently, REACH PC has enrolled 581 patients at its 20 sites, spanning a geographically diverse area. Just over half of patients approached about REACH PC agreed to enroll in it. Ultimately, 1,250 enrollees are sought.
Among patients who declined to participate, 7.6% indicated “discomfort with technology” as the reason. Most refusals were due to lack of interest in research (35.1%) and/or palliative care (22.9%).
Older adults were prominent among enrollees. More than 60% were older than 60 years of age, and more than one-third were older than 70 years.
Among patients who began the trial, there were slightly more withdrawals in the telehealth participants, in comparison with in-person participants (13.6% versus 9.1%).
When palliative care clinicians were queried about video visits, 64.3% said there were no challenges. This is comparable to the 65.5% of clinicians who had no challenges with in-person visits.
When problems occurred with video visits, they were most frequently technical (19.1%). Only 1.4% of clinicians reported difficulty addressing topics that felt uncomfortable over video, and 1.5% reported difficulty establishing rapport.
The success rates of video and in-person visits were similar. About 80% of visits accomplished planned goals.
‘Webside’ manner
Strategies such as reflective listening and summarizing what patients say (to verify an accurate understanding of the patient’s perspective) are key to successful palliative care visits, regardless of the setting.
For telehealth visits, Dr. Temel described techniques she defined as “webside manner,” to compensate for the inability of the clinician to touch a patient. These techniques include leaning in toward the camera, nodding, and pausing to be certain the patient has finished speaking before the clinician speaks again.
Is telehealth the future of palliative care?
I include myself among those oncologists who have voiced concern about moving from face-to-face to remote visits for complicated consultations such as those required for palliative care. Nonetheless, from the preliminary results of the REACH PC trial, it appears that telehealth could be a valuable tool.
To minimize differences between in-person and remote delivery of palliative care, practical strategies for ensuring rapport and facilitating a trusting relationship should be defined further and disseminated.
In addition, we need to be vigilant for widening inequities of care from rapid movement to the use of technology (i.e., an equity gap). In their telehealth experience during the COVID-19 pandemic, investigators at Houston Methodist Cancer Center found that patients declining virtual visits tended to be older, lower-income, and less likely to have commercial insurance. These results were recently published in JCO Oncology Practice.
For the foregoing reasons, hybrid systems for palliative care services will probably always be needed.
Going forward, we should heed the advice of Alvin Toffler in his book Future Shock. Mr. Toffler said, “The illiterate of the 21st century will not be those who cannot read and write, but those who cannot learn, unlearn, and relearn.”
The traditional model for delivering palliative care will almost certainly need to be reimagined and relearned.
Dr. Temel disclosed institutional research funding from Pfizer.
Dr. Lyss was a community-based medical oncologist and clinical researcher for more than 35 years before his recent retirement. His clinical and research interests were focused on breast and lung cancers, as well as expanding clinical trial access to medically underserved populations. He is based in St. Louis. He has no conflicts of interest.
FROM ASCO QUALITY CARE SYMPOSIUM 2020
Open enrollment 2021: A big start for HealthCare.gov
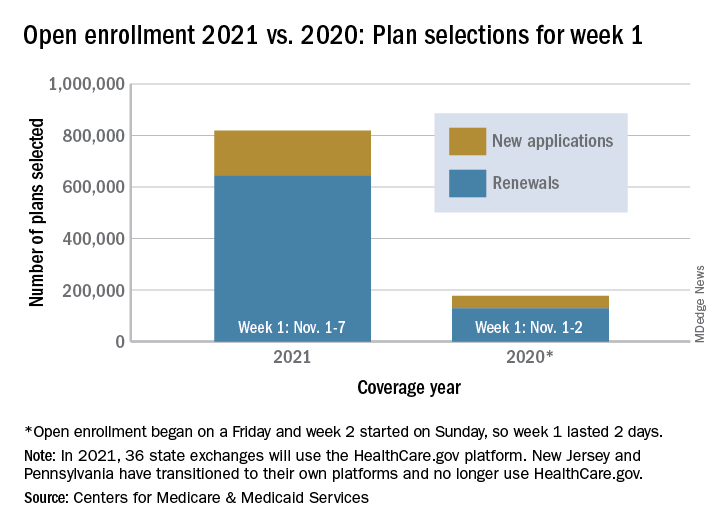
Over 818,000 plans were selected for the 2021 coverage year during the first week, Nov.1-7, of this year’s open enrollment on the federal health insurance exchange, according to the Centers for Medicare & Medicaid Services.
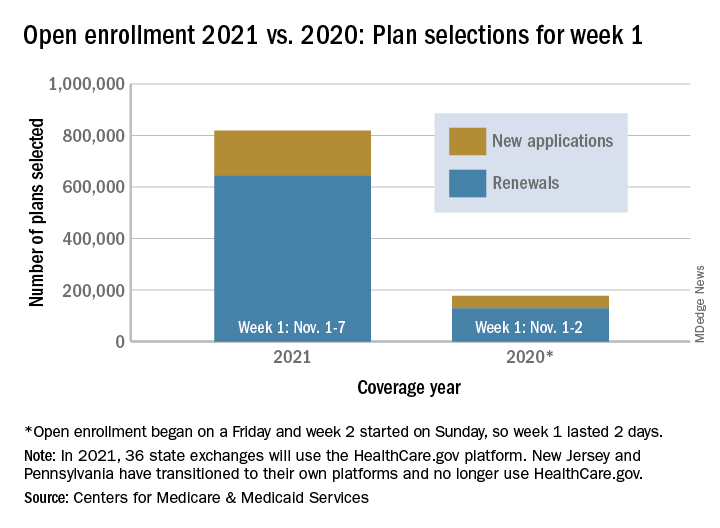
The bulk of those plans, nearly 79%, were renewals by consumers who had coverage through the federal exchange this year. The balance covers new plans selected by individuals who were not covered through HealthCare.gov this year, the CMS noted in a written statement.
The total enrollment for week 1 marks a considerable increase over last year’s first week of open enrollment, which saw approximately 177,000 plans selected, but Nov. 1 fell on a Friday in 2019, so that total represents only 2 days since weeks are tracked as running from Sunday to Saturday, the CMS explained.
For the 2021 benefit year, the HealthCare.gov platform covers 36 states, down from 38 for the 2020 benefit year, because New Jersey and Pennsylvania have “transitioned to their own state-based exchange platforms,” the CMS noted, adding that the two accounted for 7% of all plans selected last year.
“The final number of plan selections associated with enrollment activity during a reporting period may change due to plan modifications or cancellations,” CMS said, and its weekly snapshot “does not report the number of consumers who have paid premiums to effectuate their enrollment.”
This year’s open-enrollment period on HealthCare.gov is scheduled to conclude Dec. 15.
Over 818,000 plans were selected for the 2021 coverage year during the first week, Nov.1-7, of this year’s open enrollment on the federal health insurance exchange, according to the Centers for Medicare & Medicaid Services.
The bulk of those plans, nearly 79%, were renewals by consumers who had coverage through the federal exchange this year. The balance covers new plans selected by individuals who were not covered through HealthCare.gov this year, the CMS noted in a written statement.
The total enrollment for week 1 marks a considerable increase over last year’s first week of open enrollment, which saw approximately 177,000 plans selected, but Nov. 1 fell on a Friday in 2019, so that total represents only 2 days since weeks are tracked as running from Sunday to Saturday, the CMS explained.
For the 2021 benefit year, the HealthCare.gov platform covers 36 states, down from 38 for the 2020 benefit year, because New Jersey and Pennsylvania have “transitioned to their own state-based exchange platforms,” the CMS noted, adding that the two accounted for 7% of all plans selected last year.
“The final number of plan selections associated with enrollment activity during a reporting period may change due to plan modifications or cancellations,” CMS said, and its weekly snapshot “does not report the number of consumers who have paid premiums to effectuate their enrollment.”
This year’s open-enrollment period on HealthCare.gov is scheduled to conclude Dec. 15.
Over 818,000 plans were selected for the 2021 coverage year during the first week, Nov.1-7, of this year’s open enrollment on the federal health insurance exchange, according to the Centers for Medicare & Medicaid Services.
The bulk of those plans, nearly 79%, were renewals by consumers who had coverage through the federal exchange this year. The balance covers new plans selected by individuals who were not covered through HealthCare.gov this year, the CMS noted in a written statement.
The total enrollment for week 1 marks a considerable increase over last year’s first week of open enrollment, which saw approximately 177,000 plans selected, but Nov. 1 fell on a Friday in 2019, so that total represents only 2 days since weeks are tracked as running from Sunday to Saturday, the CMS explained.
For the 2021 benefit year, the HealthCare.gov platform covers 36 states, down from 38 for the 2020 benefit year, because New Jersey and Pennsylvania have “transitioned to their own state-based exchange platforms,” the CMS noted, adding that the two accounted for 7% of all plans selected last year.
“The final number of plan selections associated with enrollment activity during a reporting period may change due to plan modifications or cancellations,” CMS said, and its weekly snapshot “does not report the number of consumers who have paid premiums to effectuate their enrollment.”
This year’s open-enrollment period on HealthCare.gov is scheduled to conclude Dec. 15.
Moderna: Interim data show 94.5% efficacy for COVID-19 vaccine, will seek FDA EUA
The Moderna mRNA-1273 vaccine, in development to prevent COVID-19, yielded 94.5% efficacy in early results and is generally well tolerated, the company announced early Monday. The product can be stored at refrigeration temperatures common to many physician offices, pharmacies, and hospitals.
The first interim results of the phase 3 COVE trial included 95 participants with confirmed COVID-19. An independent data safety monitoring board, which was appointed by the National Institutes of Health, informed Moderna that 90 of the patients who were positive for COVID-19 were in a placebo group and that 5 patients were in the mRNA-1273 vaccine group, resulting in a vaccine efficacy of 94.5% (P < .0001).
Interim data included 11 patients with severe COVID-19, all of whom were in the placebo group.
“This positive interim analysis from our phase 3 study has given us the first clinical validation that our vaccine can prevent COVID-19 disease, including severe disease,” said Stéphane Bancel, CEO of Moderna, said in a statement.
The vaccine met its primary study endpoint, which was based on adjudicated data that were collected starting 2 weeks after the second dose of mRNA-1273. The interim study population included people who could be at higher risk for COVID-19, including 15 adults aged 65 years and older and 20 participants from diverse communities.
Safety data
The DSMB also reviewed safety data for the COVE study interim results. The vaccine was generally safe and well tolerated, as determined on the basis of solicited adverse events. Most adverse events were mild to moderate and were generally short-lived, according to a company news release.
Injection-site pain was reported in 2.7% of participants after the first dose. After the second dose, 9.7% of participants reported fatigue, 8.9% reported myalgia, 5.2% reported arthralgia, 4.5% reported headache, 4.1% reported pain, and 2.0% reported erythema or redness at the injection site.
Moderna plans to request emergency-use authorization (EUA) from the Food and Drug Administration in the coming weeks. The company expects that the EUA will be based on more data from the COVE study, including a final analysis of 151 patients with a median follow-up of more than 2 months. Moderna also plans to seek authorizations from global regulatory agencies.
The company expects to have approximately 20 million doses of mRNA-1273 ready to ship in the United States by the end of the year. In addition, the company says it remains on track to manufacture between 500 million and 1 billion doses globally in 2021.
Moderna is developing distribution plans in conjunction with the Centers for Disease Control and Prevention, the federal government’s Operation Warp Speed, and McKesson, a COVID-19 vaccine distributor contracted by the U.S. government.
Refrigeration requirements
The mRNA-1273 vaccine can be shipped and stored for up to 6 months at –20° C (about –4° F), a temperature maintained in most home or medical freezers, according to Moderna. The company expects that, after the product thaws, it will remain stable at standard refrigerator temperatures of 2°-8° C (36°-46° F) for up to 30 days within the 6-month shelf life.
Because the mRNA-1273 vaccine is stable at these refrigerator temperatures, it can be stored at most physicians’ offices, pharmacies, and hospitals, the company noted. In contrast, the similar Pfizer BTN162b2 vaccine – early results for which showed a 90% efficacy rate – requires shipment and storage at “deep-freeze” conditions of –70° C or –80° C, which is more challenging from a logistic point of view.
Moderna’s mRNA-1273 can be kept at room temperature for up to 12 hours after removal from a refrigerator for patient administration. The vaccine will not require dilution prior to use.
More than 30,000 people aged older than 18 years in the United States are enrolled in the COVE study. The research is being conducted in collaboration with the National Institute of Allergy and Infectious Diseases and the Biomedical Advanced Research and Development Authority, part of the Office of the Assistant Secretary for Preparedness and Response at the Department of Health & Human Services.
A version of this article originally appeared on Medscape.com.
The Moderna mRNA-1273 vaccine, in development to prevent COVID-19, yielded 94.5% efficacy in early results and is generally well tolerated, the company announced early Monday. The product can be stored at refrigeration temperatures common to many physician offices, pharmacies, and hospitals.
The first interim results of the phase 3 COVE trial included 95 participants with confirmed COVID-19. An independent data safety monitoring board, which was appointed by the National Institutes of Health, informed Moderna that 90 of the patients who were positive for COVID-19 were in a placebo group and that 5 patients were in the mRNA-1273 vaccine group, resulting in a vaccine efficacy of 94.5% (P < .0001).
Interim data included 11 patients with severe COVID-19, all of whom were in the placebo group.
“This positive interim analysis from our phase 3 study has given us the first clinical validation that our vaccine can prevent COVID-19 disease, including severe disease,” said Stéphane Bancel, CEO of Moderna, said in a statement.
The vaccine met its primary study endpoint, which was based on adjudicated data that were collected starting 2 weeks after the second dose of mRNA-1273. The interim study population included people who could be at higher risk for COVID-19, including 15 adults aged 65 years and older and 20 participants from diverse communities.
Safety data
The DSMB also reviewed safety data for the COVE study interim results. The vaccine was generally safe and well tolerated, as determined on the basis of solicited adverse events. Most adverse events were mild to moderate and were generally short-lived, according to a company news release.
Injection-site pain was reported in 2.7% of participants after the first dose. After the second dose, 9.7% of participants reported fatigue, 8.9% reported myalgia, 5.2% reported arthralgia, 4.5% reported headache, 4.1% reported pain, and 2.0% reported erythema or redness at the injection site.
Moderna plans to request emergency-use authorization (EUA) from the Food and Drug Administration in the coming weeks. The company expects that the EUA will be based on more data from the COVE study, including a final analysis of 151 patients with a median follow-up of more than 2 months. Moderna also plans to seek authorizations from global regulatory agencies.
The company expects to have approximately 20 million doses of mRNA-1273 ready to ship in the United States by the end of the year. In addition, the company says it remains on track to manufacture between 500 million and 1 billion doses globally in 2021.
Moderna is developing distribution plans in conjunction with the Centers for Disease Control and Prevention, the federal government’s Operation Warp Speed, and McKesson, a COVID-19 vaccine distributor contracted by the U.S. government.
Refrigeration requirements
The mRNA-1273 vaccine can be shipped and stored for up to 6 months at –20° C (about –4° F), a temperature maintained in most home or medical freezers, according to Moderna. The company expects that, after the product thaws, it will remain stable at standard refrigerator temperatures of 2°-8° C (36°-46° F) for up to 30 days within the 6-month shelf life.
Because the mRNA-1273 vaccine is stable at these refrigerator temperatures, it can be stored at most physicians’ offices, pharmacies, and hospitals, the company noted. In contrast, the similar Pfizer BTN162b2 vaccine – early results for which showed a 90% efficacy rate – requires shipment and storage at “deep-freeze” conditions of –70° C or –80° C, which is more challenging from a logistic point of view.
Moderna’s mRNA-1273 can be kept at room temperature for up to 12 hours after removal from a refrigerator for patient administration. The vaccine will not require dilution prior to use.
More than 30,000 people aged older than 18 years in the United States are enrolled in the COVE study. The research is being conducted in collaboration with the National Institute of Allergy and Infectious Diseases and the Biomedical Advanced Research and Development Authority, part of the Office of the Assistant Secretary for Preparedness and Response at the Department of Health & Human Services.
A version of this article originally appeared on Medscape.com.
The Moderna mRNA-1273 vaccine, in development to prevent COVID-19, yielded 94.5% efficacy in early results and is generally well tolerated, the company announced early Monday. The product can be stored at refrigeration temperatures common to many physician offices, pharmacies, and hospitals.
The first interim results of the phase 3 COVE trial included 95 participants with confirmed COVID-19. An independent data safety monitoring board, which was appointed by the National Institutes of Health, informed Moderna that 90 of the patients who were positive for COVID-19 were in a placebo group and that 5 patients were in the mRNA-1273 vaccine group, resulting in a vaccine efficacy of 94.5% (P < .0001).
Interim data included 11 patients with severe COVID-19, all of whom were in the placebo group.
“This positive interim analysis from our phase 3 study has given us the first clinical validation that our vaccine can prevent COVID-19 disease, including severe disease,” said Stéphane Bancel, CEO of Moderna, said in a statement.
The vaccine met its primary study endpoint, which was based on adjudicated data that were collected starting 2 weeks after the second dose of mRNA-1273. The interim study population included people who could be at higher risk for COVID-19, including 15 adults aged 65 years and older and 20 participants from diverse communities.
Safety data
The DSMB also reviewed safety data for the COVE study interim results. The vaccine was generally safe and well tolerated, as determined on the basis of solicited adverse events. Most adverse events were mild to moderate and were generally short-lived, according to a company news release.
Injection-site pain was reported in 2.7% of participants after the first dose. After the second dose, 9.7% of participants reported fatigue, 8.9% reported myalgia, 5.2% reported arthralgia, 4.5% reported headache, 4.1% reported pain, and 2.0% reported erythema or redness at the injection site.
Moderna plans to request emergency-use authorization (EUA) from the Food and Drug Administration in the coming weeks. The company expects that the EUA will be based on more data from the COVE study, including a final analysis of 151 patients with a median follow-up of more than 2 months. Moderna also plans to seek authorizations from global regulatory agencies.
The company expects to have approximately 20 million doses of mRNA-1273 ready to ship in the United States by the end of the year. In addition, the company says it remains on track to manufacture between 500 million and 1 billion doses globally in 2021.
Moderna is developing distribution plans in conjunction with the Centers for Disease Control and Prevention, the federal government’s Operation Warp Speed, and McKesson, a COVID-19 vaccine distributor contracted by the U.S. government.
Refrigeration requirements
The mRNA-1273 vaccine can be shipped and stored for up to 6 months at –20° C (about –4° F), a temperature maintained in most home or medical freezers, according to Moderna. The company expects that, after the product thaws, it will remain stable at standard refrigerator temperatures of 2°-8° C (36°-46° F) for up to 30 days within the 6-month shelf life.
Because the mRNA-1273 vaccine is stable at these refrigerator temperatures, it can be stored at most physicians’ offices, pharmacies, and hospitals, the company noted. In contrast, the similar Pfizer BTN162b2 vaccine – early results for which showed a 90% efficacy rate – requires shipment and storage at “deep-freeze” conditions of –70° C or –80° C, which is more challenging from a logistic point of view.
Moderna’s mRNA-1273 can be kept at room temperature for up to 12 hours after removal from a refrigerator for patient administration. The vaccine will not require dilution prior to use.
More than 30,000 people aged older than 18 years in the United States are enrolled in the COVE study. The research is being conducted in collaboration with the National Institute of Allergy and Infectious Diseases and the Biomedical Advanced Research and Development Authority, part of the Office of the Assistant Secretary for Preparedness and Response at the Department of Health & Human Services.
A version of this article originally appeared on Medscape.com.
STRENGTH trial questions CV benefit of high-dose omega-3s
Questions about the cardiovascular benefit of omega-3 fatty acids and the high-dose eicosapentaenoic acid (EPA) product, icosapent ethyl (Vascepa, Amarin), have resurfaced with the presentation and publication of the STRENGTH trial using a combined high-dose omega-3 fatty acid product.
The STRENGTH trial showed no benefit on cardiovascular event rates of a high-dose combination of EPA and docosahexaenoic acid (DHA) in a new branded product (Epanova, AstraZeneca).
It was announced in January that the trial was being stopped because of a low likelihood of showing any benefit.
Full results were presented Nov. 15 at the virtual American Heart Association scientific sessions and simultaneously published online in JAMA.
These results showed similar cardiovascular event rates with the high-dose EPA/DHA product and placebo, with a hazard ratio for the primary endpoint of 0.99. In addition to no benefit, more adverse effects occurred in the active treatment arm, with a higher rate of gastrointestinal adverse events and atrial fibrillation.
“We found no benefit of a high-dose combination of EPA and DHA. Despite a 270% to 300% increase in EPA, the hazard curves for the active and placebo groups were superimposable,” STRENGTH investigator A. Michael Lincoff, MD, of the Cleveland Clinic, said at the AHA meeting.
The big question is how the negative results of the STRENGTH trial can be reconciled with the very positive results of the REDUCE-IT trial, which showed an impressive 25% relative risk reduction in major adverse cardiovascular events with the high-dose purified EPA product, icosapent ethyl.
Dr. Lincoff proposed several possible explanations for the different results between these two trials, although he cautioned that all explanations were speculative.
The one explanation that Dr. Lincoff highlighted in particular was the different placebos used in the two trials. REDUCE-IT used a placebo of mineral oil, which Dr. Lincoff noted increases LDL, apolipoprotein B, and high-sensitivity C-reactive protein, whereas the corn oil placebo used in STRENGTH “is truly neutral on a broad range lipid and cardiovascular biomarkers,” he said.
“It must therefore be considered that at least part of the benefit in REDUCE-IT is due to an increase in adverse cardiovascular event rate in the control arm, and our results from STRENGTH cast uncertainly on the net benefit or harm of any omega-3 fatty acid preparation,” Dr. Lincoff said.
Asked whether he used omega-3 fatty acids in his practice, Dr. Lincoff replied, “Aside from patients with triglycerides greater than 500 – for which there is other evidence of benefit – I do not routinely prescribe omega-3 fatty acids. For the reasons discussed, I think there are questions about whether the risks and benefits have a favorable ratio.”
Asked at an AHA press conference what advice he would give to other physicians on the use of Vascepa, Dr. Lincoff said, “On the one hand, we could take the REDUCE-IT study results at face value, but there are potential concerns on the construct of that trial and whether the effects were exaggerated. That having been said, the [Food and Drug Administration] has approved that initial indication, so this is not a straightforward issue of whether or not that trial result is valid.
“What I would like to see is a trial in future with a clearly neutral comparator. It’s hard to recommend taking your patients off Vascepa now, but I have a high threshold at this point to start patients on it because of these concerns,” he added.
A “manufactured controversy”
The lead investigator of the REDUCE-IT trial, Deepak L. Bhatt, MD, professor of medicine, Harvard Medical School, Boston, described Dr. Lincoff’s comments as “absurd.”

In an interview, he said the Japanese JELIS trial, while having some limitations, also showed a benefit of icosapent ethyl, which “in the context of this manufactured controversy about the mineral oil placebo in REDUCE-IT, completely rebuts concerns about the placebo in REDUCE-IT being toxic.”
Dr. Bhatt also suggested that DHA may counter some of the benefits of EPA. “It appears that the STRENGTH trial leadership is trying to stir up controversy, rather than just reporting objectively that they have a negative trial,” he added.
Dr. Lincoff outlined other possible explanations for the difference between the two trials.
He noted that icosapent ethyl increased levels of EPA by 45% in REDUCE-IT more than did the combined product used in STRENGTH. “But this moderate difference seems insufficient to account for the markedly different results of the two trials,” Dr. Lincoff added, “and both trials showed a 19% reduction in triglycerides, suggesting they have similar biochemical effects.”
There is also the possibility of an adverse effect of DHA, he noted, “but this has never been seen in previous studies.”
Another explanation could be differences in trial populations, with REDUCE-IT including more patients with established cardiovascular disease. “But the results were no different in this group compared to the patients without established cardiovascular disease, so this explanation is unlikely,” Dr. Lincoff suggested.
STRENGTH trial
The STRENGTH trial included 13,078 statin-treated participants with high cardiovascular risk, hypertriglyceridemia, and low levels of HDL cholesterol from 22 countries.
They were randomly assigned to a 4 g per day of carboxylic acid formulation of EPA and DHA or to corn oil as an inert comparator.
The trial was halted when 1,384 patients had experienced a primary endpoint event (of a planned 1,600 events), based on an interim analysis that indicated a low probability of clinical benefit of the active treatment.
At this point, the primary efficacy endpoint – a composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization – occurred in 12.0% of patients treated with the omega-3 product vs. 12.2% of those who received corn oil (hazard ratio [HR], 0.99; P = .84).
A greater rate of gastrointestinal adverse events was observed in the omega-3 group (24.7%) than in corn oil–treated patients (14.7%). An increased rate of new-onset atrial fibrillation was also observed in the omega-3 group (2.2% vs 1.3%; HR, 1.69).
Uncertainty prevails
The moderator of an AHA press conference at which the STRENGTH trial was discussed, Amit Khera, MD, professor of medicine and director of preventive cardiology at UT Southwestern Medical Center, Dallas, said in an interview that questions about how Vascepa brought about the benefits shown in REDUCE-IT have been ongoing since that trial was published.
“I think for now, we have to accept the REDUCE-IT results as a positive finding. However, the STRENGTH trial did amplify these questions a bit since there was no signal at all for benefit, and this heightens the call for additional trials of high-dose EPA formulations, including icosapent ethyl, versus corn oil or another neutral comparator,” he said.
Discussant of the STRENGTH trial at the AHA late-breaker session, Alberico Catapano, MD, PhD, University of Milan, noted that DHA may have less biological activity than EPA.
“We don’t know for certain, but there are studies suggesting that EPA may have more effect on stabilizing plaque membranes,” Dr. Catapano said. “Certainly, the dose of EPA was different in the two studies, and in my view this could be part of the explanation. But we are still in place where we need more evidence.”
In an editorial accompanying the JAMA publication of STRENGTH, Garima Sharma, MD, Seth S. Martin, MD, and Roger S. Blumenthal, MD, Johns Hopkins University, Baltimore, said the trial’s findings “may invigorate further investigation regarding IPE [icosapent ethyl], generate additional constructive debate around the optimal placebo control, and should prompt reconsideration of over-the-counter mixed omega-3 fatty acid products for ASCVD [atherosclerotic cardiovascular disease] prevention.
“This latter point is especially important given the lack of evidence for benefit, and the potential for harm due to increased AF [atrial fibrillation],” they noted.
“The reasons the findings from the REDUCE-IT trial were positive and the STRENGTH trial were not, and that EPA levels correlated with outcomes in REDUCE-IT but did not in STRENGTH, remain uncertain,” they concluded. “The importance of the specific omega-3 formulation in achieving ASCVD risk reduction and the degree to which the placebo (i.e., mineral oil vs corn oil) may have affected outcomes remain unresolved.”
The STRENGTH trial was sponsored by AstraZeneca. Dr. Lincoff reported receiving grants from AstraZeneca during the conduct of the study. Dr. Catapano has received honoraria, lecture fees, or research grants from Sigma-Tau, Manarini, Kowa Pharmaceuticals, Recordati, Eli Lilly, AstraZeneca, Mediolanum, Pfizer, Merck, Sanofi, Aegerion, Amgen, Genzyme, Bayer, Sanofi, Regeneron Daiichi Sankyo, and Amarin. Dr. Martin reports receiving consulting fees from AstraZeneca, Amgen, Esperion, and REGENXBIO, and has a patent pending for a system of LDL-C estimation filed by Johns Hopkins University. Dr. Bhatt reports serving as principal investigator for REDUCE-IT and that Brigham and Women’s Hospital has received research funding from Amarin.
A version of this article originally appeared on Medscape.com.
Questions about the cardiovascular benefit of omega-3 fatty acids and the high-dose eicosapentaenoic acid (EPA) product, icosapent ethyl (Vascepa, Amarin), have resurfaced with the presentation and publication of the STRENGTH trial using a combined high-dose omega-3 fatty acid product.
The STRENGTH trial showed no benefit on cardiovascular event rates of a high-dose combination of EPA and docosahexaenoic acid (DHA) in a new branded product (Epanova, AstraZeneca).
It was announced in January that the trial was being stopped because of a low likelihood of showing any benefit.
Full results were presented Nov. 15 at the virtual American Heart Association scientific sessions and simultaneously published online in JAMA.
These results showed similar cardiovascular event rates with the high-dose EPA/DHA product and placebo, with a hazard ratio for the primary endpoint of 0.99. In addition to no benefit, more adverse effects occurred in the active treatment arm, with a higher rate of gastrointestinal adverse events and atrial fibrillation.
“We found no benefit of a high-dose combination of EPA and DHA. Despite a 270% to 300% increase in EPA, the hazard curves for the active and placebo groups were superimposable,” STRENGTH investigator A. Michael Lincoff, MD, of the Cleveland Clinic, said at the AHA meeting.
The big question is how the negative results of the STRENGTH trial can be reconciled with the very positive results of the REDUCE-IT trial, which showed an impressive 25% relative risk reduction in major adverse cardiovascular events with the high-dose purified EPA product, icosapent ethyl.
Dr. Lincoff proposed several possible explanations for the different results between these two trials, although he cautioned that all explanations were speculative.
The one explanation that Dr. Lincoff highlighted in particular was the different placebos used in the two trials. REDUCE-IT used a placebo of mineral oil, which Dr. Lincoff noted increases LDL, apolipoprotein B, and high-sensitivity C-reactive protein, whereas the corn oil placebo used in STRENGTH “is truly neutral on a broad range lipid and cardiovascular biomarkers,” he said.
“It must therefore be considered that at least part of the benefit in REDUCE-IT is due to an increase in adverse cardiovascular event rate in the control arm, and our results from STRENGTH cast uncertainly on the net benefit or harm of any omega-3 fatty acid preparation,” Dr. Lincoff said.
Asked whether he used omega-3 fatty acids in his practice, Dr. Lincoff replied, “Aside from patients with triglycerides greater than 500 – for which there is other evidence of benefit – I do not routinely prescribe omega-3 fatty acids. For the reasons discussed, I think there are questions about whether the risks and benefits have a favorable ratio.”
Asked at an AHA press conference what advice he would give to other physicians on the use of Vascepa, Dr. Lincoff said, “On the one hand, we could take the REDUCE-IT study results at face value, but there are potential concerns on the construct of that trial and whether the effects were exaggerated. That having been said, the [Food and Drug Administration] has approved that initial indication, so this is not a straightforward issue of whether or not that trial result is valid.
“What I would like to see is a trial in future with a clearly neutral comparator. It’s hard to recommend taking your patients off Vascepa now, but I have a high threshold at this point to start patients on it because of these concerns,” he added.
A “manufactured controversy”
The lead investigator of the REDUCE-IT trial, Deepak L. Bhatt, MD, professor of medicine, Harvard Medical School, Boston, described Dr. Lincoff’s comments as “absurd.”
In an interview, he said the Japanese JELIS trial, while having some limitations, also showed a benefit of icosapent ethyl, which “in the context of this manufactured controversy about the mineral oil placebo in REDUCE-IT, completely rebuts concerns about the placebo in REDUCE-IT being toxic.”
Dr. Bhatt also suggested that DHA may counter some of the benefits of EPA. “It appears that the STRENGTH trial leadership is trying to stir up controversy, rather than just reporting objectively that they have a negative trial,” he added.
Dr. Lincoff outlined other possible explanations for the difference between the two trials.
He noted that icosapent ethyl increased levels of EPA by 45% in REDUCE-IT more than did the combined product used in STRENGTH. “But this moderate difference seems insufficient to account for the markedly different results of the two trials,” Dr. Lincoff added, “and both trials showed a 19% reduction in triglycerides, suggesting they have similar biochemical effects.”
There is also the possibility of an adverse effect of DHA, he noted, “but this has never been seen in previous studies.”
Another explanation could be differences in trial populations, with REDUCE-IT including more patients with established cardiovascular disease. “But the results were no different in this group compared to the patients without established cardiovascular disease, so this explanation is unlikely,” Dr. Lincoff suggested.
STRENGTH trial
The STRENGTH trial included 13,078 statin-treated participants with high cardiovascular risk, hypertriglyceridemia, and low levels of HDL cholesterol from 22 countries.
They were randomly assigned to a 4 g per day of carboxylic acid formulation of EPA and DHA or to corn oil as an inert comparator.
The trial was halted when 1,384 patients had experienced a primary endpoint event (of a planned 1,600 events), based on an interim analysis that indicated a low probability of clinical benefit of the active treatment.
At this point, the primary efficacy endpoint – a composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization – occurred in 12.0% of patients treated with the omega-3 product vs. 12.2% of those who received corn oil (hazard ratio [HR], 0.99; P = .84).
A greater rate of gastrointestinal adverse events was observed in the omega-3 group (24.7%) than in corn oil–treated patients (14.7%). An increased rate of new-onset atrial fibrillation was also observed in the omega-3 group (2.2% vs 1.3%; HR, 1.69).
Uncertainty prevails
The moderator of an AHA press conference at which the STRENGTH trial was discussed, Amit Khera, MD, professor of medicine and director of preventive cardiology at UT Southwestern Medical Center, Dallas, said in an interview that questions about how Vascepa brought about the benefits shown in REDUCE-IT have been ongoing since that trial was published.
“I think for now, we have to accept the REDUCE-IT results as a positive finding. However, the STRENGTH trial did amplify these questions a bit since there was no signal at all for benefit, and this heightens the call for additional trials of high-dose EPA formulations, including icosapent ethyl, versus corn oil or another neutral comparator,” he said.
Discussant of the STRENGTH trial at the AHA late-breaker session, Alberico Catapano, MD, PhD, University of Milan, noted that DHA may have less biological activity than EPA.
“We don’t know for certain, but there are studies suggesting that EPA may have more effect on stabilizing plaque membranes,” Dr. Catapano said. “Certainly, the dose of EPA was different in the two studies, and in my view this could be part of the explanation. But we are still in place where we need more evidence.”
In an editorial accompanying the JAMA publication of STRENGTH, Garima Sharma, MD, Seth S. Martin, MD, and Roger S. Blumenthal, MD, Johns Hopkins University, Baltimore, said the trial’s findings “may invigorate further investigation regarding IPE [icosapent ethyl], generate additional constructive debate around the optimal placebo control, and should prompt reconsideration of over-the-counter mixed omega-3 fatty acid products for ASCVD [atherosclerotic cardiovascular disease] prevention.
“This latter point is especially important given the lack of evidence for benefit, and the potential for harm due to increased AF [atrial fibrillation],” they noted.
“The reasons the findings from the REDUCE-IT trial were positive and the STRENGTH trial were not, and that EPA levels correlated with outcomes in REDUCE-IT but did not in STRENGTH, remain uncertain,” they concluded. “The importance of the specific omega-3 formulation in achieving ASCVD risk reduction and the degree to which the placebo (i.e., mineral oil vs corn oil) may have affected outcomes remain unresolved.”
The STRENGTH trial was sponsored by AstraZeneca. Dr. Lincoff reported receiving grants from AstraZeneca during the conduct of the study. Dr. Catapano has received honoraria, lecture fees, or research grants from Sigma-Tau, Manarini, Kowa Pharmaceuticals, Recordati, Eli Lilly, AstraZeneca, Mediolanum, Pfizer, Merck, Sanofi, Aegerion, Amgen, Genzyme, Bayer, Sanofi, Regeneron Daiichi Sankyo, and Amarin. Dr. Martin reports receiving consulting fees from AstraZeneca, Amgen, Esperion, and REGENXBIO, and has a patent pending for a system of LDL-C estimation filed by Johns Hopkins University. Dr. Bhatt reports serving as principal investigator for REDUCE-IT and that Brigham and Women’s Hospital has received research funding from Amarin.
A version of this article originally appeared on Medscape.com.
Questions about the cardiovascular benefit of omega-3 fatty acids and the high-dose eicosapentaenoic acid (EPA) product, icosapent ethyl (Vascepa, Amarin), have resurfaced with the presentation and publication of the STRENGTH trial using a combined high-dose omega-3 fatty acid product.
The STRENGTH trial showed no benefit on cardiovascular event rates of a high-dose combination of EPA and docosahexaenoic acid (DHA) in a new branded product (Epanova, AstraZeneca).
It was announced in January that the trial was being stopped because of a low likelihood of showing any benefit.
Full results were presented Nov. 15 at the virtual American Heart Association scientific sessions and simultaneously published online in JAMA.
These results showed similar cardiovascular event rates with the high-dose EPA/DHA product and placebo, with a hazard ratio for the primary endpoint of 0.99. In addition to no benefit, more adverse effects occurred in the active treatment arm, with a higher rate of gastrointestinal adverse events and atrial fibrillation.
“We found no benefit of a high-dose combination of EPA and DHA. Despite a 270% to 300% increase in EPA, the hazard curves for the active and placebo groups were superimposable,” STRENGTH investigator A. Michael Lincoff, MD, of the Cleveland Clinic, said at the AHA meeting.
The big question is how the negative results of the STRENGTH trial can be reconciled with the very positive results of the REDUCE-IT trial, which showed an impressive 25% relative risk reduction in major adverse cardiovascular events with the high-dose purified EPA product, icosapent ethyl.
Dr. Lincoff proposed several possible explanations for the different results between these two trials, although he cautioned that all explanations were speculative.
The one explanation that Dr. Lincoff highlighted in particular was the different placebos used in the two trials. REDUCE-IT used a placebo of mineral oil, which Dr. Lincoff noted increases LDL, apolipoprotein B, and high-sensitivity C-reactive protein, whereas the corn oil placebo used in STRENGTH “is truly neutral on a broad range lipid and cardiovascular biomarkers,” he said.
“It must therefore be considered that at least part of the benefit in REDUCE-IT is due to an increase in adverse cardiovascular event rate in the control arm, and our results from STRENGTH cast uncertainly on the net benefit or harm of any omega-3 fatty acid preparation,” Dr. Lincoff said.
Asked whether he used omega-3 fatty acids in his practice, Dr. Lincoff replied, “Aside from patients with triglycerides greater than 500 – for which there is other evidence of benefit – I do not routinely prescribe omega-3 fatty acids. For the reasons discussed, I think there are questions about whether the risks and benefits have a favorable ratio.”
Asked at an AHA press conference what advice he would give to other physicians on the use of Vascepa, Dr. Lincoff said, “On the one hand, we could take the REDUCE-IT study results at face value, but there are potential concerns on the construct of that trial and whether the effects were exaggerated. That having been said, the [Food and Drug Administration] has approved that initial indication, so this is not a straightforward issue of whether or not that trial result is valid.
“What I would like to see is a trial in future with a clearly neutral comparator. It’s hard to recommend taking your patients off Vascepa now, but I have a high threshold at this point to start patients on it because of these concerns,” he added.
A “manufactured controversy”
The lead investigator of the REDUCE-IT trial, Deepak L. Bhatt, MD, professor of medicine, Harvard Medical School, Boston, described Dr. Lincoff’s comments as “absurd.”
In an interview, he said the Japanese JELIS trial, while having some limitations, also showed a benefit of icosapent ethyl, which “in the context of this manufactured controversy about the mineral oil placebo in REDUCE-IT, completely rebuts concerns about the placebo in REDUCE-IT being toxic.”
Dr. Bhatt also suggested that DHA may counter some of the benefits of EPA. “It appears that the STRENGTH trial leadership is trying to stir up controversy, rather than just reporting objectively that they have a negative trial,” he added.
Dr. Lincoff outlined other possible explanations for the difference between the two trials.
He noted that icosapent ethyl increased levels of EPA by 45% in REDUCE-IT more than did the combined product used in STRENGTH. “But this moderate difference seems insufficient to account for the markedly different results of the two trials,” Dr. Lincoff added, “and both trials showed a 19% reduction in triglycerides, suggesting they have similar biochemical effects.”
There is also the possibility of an adverse effect of DHA, he noted, “but this has never been seen in previous studies.”
Another explanation could be differences in trial populations, with REDUCE-IT including more patients with established cardiovascular disease. “But the results were no different in this group compared to the patients without established cardiovascular disease, so this explanation is unlikely,” Dr. Lincoff suggested.
STRENGTH trial
The STRENGTH trial included 13,078 statin-treated participants with high cardiovascular risk, hypertriglyceridemia, and low levels of HDL cholesterol from 22 countries.
They were randomly assigned to a 4 g per day of carboxylic acid formulation of EPA and DHA or to corn oil as an inert comparator.
The trial was halted when 1,384 patients had experienced a primary endpoint event (of a planned 1,600 events), based on an interim analysis that indicated a low probability of clinical benefit of the active treatment.
At this point, the primary efficacy endpoint – a composite of cardiovascular death, nonfatal myocardial infarction, nonfatal stroke, coronary revascularization, or unstable angina requiring hospitalization – occurred in 12.0% of patients treated with the omega-3 product vs. 12.2% of those who received corn oil (hazard ratio [HR], 0.99; P = .84).
A greater rate of gastrointestinal adverse events was observed in the omega-3 group (24.7%) than in corn oil–treated patients (14.7%). An increased rate of new-onset atrial fibrillation was also observed in the omega-3 group (2.2% vs 1.3%; HR, 1.69).
Uncertainty prevails
The moderator of an AHA press conference at which the STRENGTH trial was discussed, Amit Khera, MD, professor of medicine and director of preventive cardiology at UT Southwestern Medical Center, Dallas, said in an interview that questions about how Vascepa brought about the benefits shown in REDUCE-IT have been ongoing since that trial was published.
“I think for now, we have to accept the REDUCE-IT results as a positive finding. However, the STRENGTH trial did amplify these questions a bit since there was no signal at all for benefit, and this heightens the call for additional trials of high-dose EPA formulations, including icosapent ethyl, versus corn oil or another neutral comparator,” he said.
Discussant of the STRENGTH trial at the AHA late-breaker session, Alberico Catapano, MD, PhD, University of Milan, noted that DHA may have less biological activity than EPA.
“We don’t know for certain, but there are studies suggesting that EPA may have more effect on stabilizing plaque membranes,” Dr. Catapano said. “Certainly, the dose of EPA was different in the two studies, and in my view this could be part of the explanation. But we are still in place where we need more evidence.”
In an editorial accompanying the JAMA publication of STRENGTH, Garima Sharma, MD, Seth S. Martin, MD, and Roger S. Blumenthal, MD, Johns Hopkins University, Baltimore, said the trial’s findings “may invigorate further investigation regarding IPE [icosapent ethyl], generate additional constructive debate around the optimal placebo control, and should prompt reconsideration of over-the-counter mixed omega-3 fatty acid products for ASCVD [atherosclerotic cardiovascular disease] prevention.
“This latter point is especially important given the lack of evidence for benefit, and the potential for harm due to increased AF [atrial fibrillation],” they noted.
“The reasons the findings from the REDUCE-IT trial were positive and the STRENGTH trial were not, and that EPA levels correlated with outcomes in REDUCE-IT but did not in STRENGTH, remain uncertain,” they concluded. “The importance of the specific omega-3 formulation in achieving ASCVD risk reduction and the degree to which the placebo (i.e., mineral oil vs corn oil) may have affected outcomes remain unresolved.”
The STRENGTH trial was sponsored by AstraZeneca. Dr. Lincoff reported receiving grants from AstraZeneca during the conduct of the study. Dr. Catapano has received honoraria, lecture fees, or research grants from Sigma-Tau, Manarini, Kowa Pharmaceuticals, Recordati, Eli Lilly, AstraZeneca, Mediolanum, Pfizer, Merck, Sanofi, Aegerion, Amgen, Genzyme, Bayer, Sanofi, Regeneron Daiichi Sankyo, and Amarin. Dr. Martin reports receiving consulting fees from AstraZeneca, Amgen, Esperion, and REGENXBIO, and has a patent pending for a system of LDL-C estimation filed by Johns Hopkins University. Dr. Bhatt reports serving as principal investigator for REDUCE-IT and that Brigham and Women’s Hospital has received research funding from Amarin.
A version of this article originally appeared on Medscape.com.
Pembrolizumab approved for triple-negative breast cancer
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
The Food and Drug Administration has granted accelerated approval for pembrolizumab (Keytruda) in combination with chemotherapy to treat locally recurrent unresectable or metastatic triple-negative breast cancer (TNBC) that expresses PD-L1, as determined by a combined positive score of 10 or greater on an FDA-approved assay.
The FDA also approved a PD-L1 assay for selecting TNBC patients for pembrolizumab, the PD-L1 IHC 22C3 pharmDx.
Pembrolizumab is approved for numerous indications in the United States, but the new approval is its first breast cancer indication.
The accelerated approval for pembrolizumab in TNBC was based on progression-free survival (PFS) in the KEYNOTE-355 trial. The FDA noted that continued approval of pembrolizumab in TNBC “may be contingent upon verification and description of clinical benefit in the confirmatory trials.”
KEYNOTE-355 enrolled patients with locally recurrent unresectable or metastatic TNBC who had not received chemotherapy in the metastatic setting. Patients were randomized to chemotherapy (nab-paclitaxel, paclitaxel, or gemcitabine plus carboplatin) plus placebo (n = 281) or chemotherapy plus pembrolizumab at 200 mg on day 1 every 3 weeks (n = 562).
Among PD-L1-positive patients (n = 323), the median PFS was 5.6 months in the placebo arm and 9.7 months in the pembrolizumab arm (hazard ratio, 0.65; P = .0012).
The recommended pembrolizumab dose in TNBC is 200 mg every 3 weeks or 400 mg every 6 weeks administered prior to chemotherapy until disease progression, unacceptable toxicity, or up to 24 months.
Pembrolizumab can cause immune-mediated adverse reactions that may be severe or fatal, according to Merck, the manufacturer of pembrolizumab. These adverse reactions include pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and complications of allogeneic hematopoietic stem cell transplant.
“Based on the severity of the adverse reaction, [pembrolizumab] should be withheld or discontinued and corticosteroids administered if appropriate,” the company noted.
For more details on pembrolizumab, see the full prescribing information.
FROM THE FOOD AND DRUG ADMINISTRATION
SCAPIS: Simple questionnaire can identify silent atherosclerosis
Individuals in the general population with high levels of silent coronary atherosclerosis can be successfully identified with a simple questionnaire that they can complete themselves at home, a new study suggests.
The Swedish CardioPulmonary BioImage Study (SCAPIS) found that 40% of middle-aged adults without known heart disease had evidence of coronary atherosclerosis on coronary CT angiography (CCTA), and 13% had extensive atherosclerotic disease.
The authors found that the screening questionnaire could identify individuals who had extensive coronary atherosclerosis with a reasonably high predictive value.
Initial results from the study were presented today at the virtual American Heart Association (AHA) Scientific Sessions 2020.
“Our study is looking to see if we can estimate how many people in the general population have significant coronary atherosclerosis and therefore could benefit from preventative treatment,” lead author, Göran Bergström, MD, explained to Medscape Medical News.
Bergström, who is professor and lead physician at Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden, said there are no good data on this as yet. “There are studies of atherosclerosis burden in patients who have had a cardiovascular event, but our study was conducted in a random selection of the middle-aged general population who did not have symptoms of heart disease.”
“Our study also suggests that in future we may be able to identify these people with an online questionnaire, and those that reached a certain score could be referred for an imaging test,” he added.
SCAPIS included more than 30,000 men and women, age 50 to 64 years, who had no history of cardiovascular events or cardiac intervention. They were asked questions about sex, age, lifestyle, smoking, body measurements, cholesterol medication, and blood pressure to predict their risk for coronary artery disease.
Researchers then used CCTA images to examine patients’ arteries for the presence of plaque. More than 25,000 individuals from the original sample were successfully imaged.
Results showed that 40% of the middle-aged population had some coronary atherosclerosis and 5% had severe atherosclerosis, defined as the presence of a stenosis blocking 50% or more of blood flow in one of the coronary arteries.
A second aim of the study was to use data from the questionnaire to develop a prediction model to identify people with widespread atherosclerosis — those with any type of stenosis in four different segments of their coronary arteries, who made up 13% of the population.
The questionnaire included data on 120 different variables. Of these variables, around 100 could be assessed by the patients themselves and another 20 measurements could be performed in the clinic, such as blood pressure and cholesterol levels.
The researchers then used artificial intelligence to assess which variables were associated with widespread atherosclerosis. This had an area under the curve (AUC, a measure of the predictive value) of 0.8.
“An AUC of 1.0 would show a perfect prediction, and a value of 0.5 shows no value. A result of 0.8 shows reasonable predictive potential. This is an encouraging result and suggests this strategy could work,” Bergström said.
“We know silent atherosclerosis is a big problem and causes sudden cardiac events in people who have not shown symptoms,” he said.
The goal is to identify these patients before they have an event and offer them preventive treatments. “At present we try and identify patients at high risk of cardiovascular events by using cholesterol and blood pressure measurements and cardiovascular risk scores such as Framingham. But this is not so effective,” Bergström explained.
“Using imaging such as CCTA, where you can actually see atherosclerotic plaque, could be better for prediction, but we can’t image everyone. So, we wanted to see whether we could narrow down the population who should receive imaging with a detailed questionnaire, and it looks like we can.”
The study found that including clinical measurements such as blood pressure and cholesterol did not add much to the predictive value for identifying people with extensive coronary atherosclerosis, a result that Bergström said was surprising.
Which population to target?
Discussant of the study, Pamela Douglas, MD, professor of research in cardiovascular diseases at Duke University, Durham, North Carolina, congratulated the SCAPIS investigators on creating “a very rich data set for current and future study.”

“The SCAPIS study has already yielded novel data on the prevalence of coronary artery disease in the general population, and will address many critical questions over the long term,” she said.
But Douglas suggested that individuals with extensive coronary atherosclerosis were not the most appropriate target population to identify.
“The rationale for choosing this cutpoint is unclear as clinical risk/mortality is higher in all nonobstructive coronary artery disease, starting at one-vessel involvement,” she noted. “Therefore, effective preventive strategies likely need to start with detection and treatment of patients with even minimal plaque.”
Responding to Medscape Medical News, Bergström said this was a valid argument. “We plan to reanalyze our results with different populations as the target — that is something that we can do in the future.
But targeting everyone with just one coronary plaque is going to identify a large group — it was 40% of the population in our study. This will be too many people in whom to perform confirmatory CCTA imaging. It would be impractical to try and conduct cardiac imaging on that many people.”
Bergström noted that more data are needed on the danger of various levels of coronary atherosclerosis in this population who have not had any symptoms.
“We don’t have this information at present, but we are continuing to follow our population and we will have data on cardiac events in a few years’ time. Then we will know which level of atherosclerosis we need to target. It will probably be somewhere in between the extensive levels we used in this first analysis (which occurred in 13% of people) and the 40% of people who showed just one area of plaque.”
This study is the first report from SCAPIS, a collaborative project between six Swedish universities with the following vision statement: to “reduce the risk of cardiovascular and respiratory diseases for generations to come.”
The SCAPIS project is funded by the Swedish Heart and Lung Foundation. Bergström reports no disclosures.
This article first appeared on Medscape.com.
Individuals in the general population with high levels of silent coronary atherosclerosis can be successfully identified with a simple questionnaire that they can complete themselves at home, a new study suggests.
The Swedish CardioPulmonary BioImage Study (SCAPIS) found that 40% of middle-aged adults without known heart disease had evidence of coronary atherosclerosis on coronary CT angiography (CCTA), and 13% had extensive atherosclerotic disease.
The authors found that the screening questionnaire could identify individuals who had extensive coronary atherosclerosis with a reasonably high predictive value.
Initial results from the study were presented today at the virtual American Heart Association (AHA) Scientific Sessions 2020.
“Our study is looking to see if we can estimate how many people in the general population have significant coronary atherosclerosis and therefore could benefit from preventative treatment,” lead author, Göran Bergström, MD, explained to Medscape Medical News.
Bergström, who is professor and lead physician at Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden, said there are no good data on this as yet. “There are studies of atherosclerosis burden in patients who have had a cardiovascular event, but our study was conducted in a random selection of the middle-aged general population who did not have symptoms of heart disease.”
“Our study also suggests that in future we may be able to identify these people with an online questionnaire, and those that reached a certain score could be referred for an imaging test,” he added.
SCAPIS included more than 30,000 men and women, age 50 to 64 years, who had no history of cardiovascular events or cardiac intervention. They were asked questions about sex, age, lifestyle, smoking, body measurements, cholesterol medication, and blood pressure to predict their risk for coronary artery disease.
Researchers then used CCTA images to examine patients’ arteries for the presence of plaque. More than 25,000 individuals from the original sample were successfully imaged.
Results showed that 40% of the middle-aged population had some coronary atherosclerosis and 5% had severe atherosclerosis, defined as the presence of a stenosis blocking 50% or more of blood flow in one of the coronary arteries.
A second aim of the study was to use data from the questionnaire to develop a prediction model to identify people with widespread atherosclerosis — those with any type of stenosis in four different segments of their coronary arteries, who made up 13% of the population.
The questionnaire included data on 120 different variables. Of these variables, around 100 could be assessed by the patients themselves and another 20 measurements could be performed in the clinic, such as blood pressure and cholesterol levels.
The researchers then used artificial intelligence to assess which variables were associated with widespread atherosclerosis. This had an area under the curve (AUC, a measure of the predictive value) of 0.8.
“An AUC of 1.0 would show a perfect prediction, and a value of 0.5 shows no value. A result of 0.8 shows reasonable predictive potential. This is an encouraging result and suggests this strategy could work,” Bergström said.
“We know silent atherosclerosis is a big problem and causes sudden cardiac events in people who have not shown symptoms,” he said.
The goal is to identify these patients before they have an event and offer them preventive treatments. “At present we try and identify patients at high risk of cardiovascular events by using cholesterol and blood pressure measurements and cardiovascular risk scores such as Framingham. But this is not so effective,” Bergström explained.
“Using imaging such as CCTA, where you can actually see atherosclerotic plaque, could be better for prediction, but we can’t image everyone. So, we wanted to see whether we could narrow down the population who should receive imaging with a detailed questionnaire, and it looks like we can.”
The study found that including clinical measurements such as blood pressure and cholesterol did not add much to the predictive value for identifying people with extensive coronary atherosclerosis, a result that Bergström said was surprising.
Which population to target?
Discussant of the study, Pamela Douglas, MD, professor of research in cardiovascular diseases at Duke University, Durham, North Carolina, congratulated the SCAPIS investigators on creating “a very rich data set for current and future study.”
“The SCAPIS study has already yielded novel data on the prevalence of coronary artery disease in the general population, and will address many critical questions over the long term,” she said.
But Douglas suggested that individuals with extensive coronary atherosclerosis were not the most appropriate target population to identify.
“The rationale for choosing this cutpoint is unclear as clinical risk/mortality is higher in all nonobstructive coronary artery disease, starting at one-vessel involvement,” she noted. “Therefore, effective preventive strategies likely need to start with detection and treatment of patients with even minimal plaque.”
Responding to Medscape Medical News, Bergström said this was a valid argument. “We plan to reanalyze our results with different populations as the target — that is something that we can do in the future.
But targeting everyone with just one coronary plaque is going to identify a large group — it was 40% of the population in our study. This will be too many people in whom to perform confirmatory CCTA imaging. It would be impractical to try and conduct cardiac imaging on that many people.”
Bergström noted that more data are needed on the danger of various levels of coronary atherosclerosis in this population who have not had any symptoms.
“We don’t have this information at present, but we are continuing to follow our population and we will have data on cardiac events in a few years’ time. Then we will know which level of atherosclerosis we need to target. It will probably be somewhere in between the extensive levels we used in this first analysis (which occurred in 13% of people) and the 40% of people who showed just one area of plaque.”
This study is the first report from SCAPIS, a collaborative project between six Swedish universities with the following vision statement: to “reduce the risk of cardiovascular and respiratory diseases for generations to come.”
The SCAPIS project is funded by the Swedish Heart and Lung Foundation. Bergström reports no disclosures.
This article first appeared on Medscape.com.
Individuals in the general population with high levels of silent coronary atherosclerosis can be successfully identified with a simple questionnaire that they can complete themselves at home, a new study suggests.
The Swedish CardioPulmonary BioImage Study (SCAPIS) found that 40% of middle-aged adults without known heart disease had evidence of coronary atherosclerosis on coronary CT angiography (CCTA), and 13% had extensive atherosclerotic disease.
The authors found that the screening questionnaire could identify individuals who had extensive coronary atherosclerosis with a reasonably high predictive value.
Initial results from the study were presented today at the virtual American Heart Association (AHA) Scientific Sessions 2020.
“Our study is looking to see if we can estimate how many people in the general population have significant coronary atherosclerosis and therefore could benefit from preventative treatment,” lead author, Göran Bergström, MD, explained to Medscape Medical News.
Bergström, who is professor and lead physician at Sahlgrenska Academy, Gothenburg University, Gothenburg, Sweden, said there are no good data on this as yet. “There are studies of atherosclerosis burden in patients who have had a cardiovascular event, but our study was conducted in a random selection of the middle-aged general population who did not have symptoms of heart disease.”
“Our study also suggests that in future we may be able to identify these people with an online questionnaire, and those that reached a certain score could be referred for an imaging test,” he added.
SCAPIS included more than 30,000 men and women, age 50 to 64 years, who had no history of cardiovascular events or cardiac intervention. They were asked questions about sex, age, lifestyle, smoking, body measurements, cholesterol medication, and blood pressure to predict their risk for coronary artery disease.
Researchers then used CCTA images to examine patients’ arteries for the presence of plaque. More than 25,000 individuals from the original sample were successfully imaged.
Results showed that 40% of the middle-aged population had some coronary atherosclerosis and 5% had severe atherosclerosis, defined as the presence of a stenosis blocking 50% or more of blood flow in one of the coronary arteries.
A second aim of the study was to use data from the questionnaire to develop a prediction model to identify people with widespread atherosclerosis — those with any type of stenosis in four different segments of their coronary arteries, who made up 13% of the population.
The questionnaire included data on 120 different variables. Of these variables, around 100 could be assessed by the patients themselves and another 20 measurements could be performed in the clinic, such as blood pressure and cholesterol levels.
The researchers then used artificial intelligence to assess which variables were associated with widespread atherosclerosis. This had an area under the curve (AUC, a measure of the predictive value) of 0.8.
“An AUC of 1.0 would show a perfect prediction, and a value of 0.5 shows no value. A result of 0.8 shows reasonable predictive potential. This is an encouraging result and suggests this strategy could work,” Bergström said.
“We know silent atherosclerosis is a big problem and causes sudden cardiac events in people who have not shown symptoms,” he said.
The goal is to identify these patients before they have an event and offer them preventive treatments. “At present we try and identify patients at high risk of cardiovascular events by using cholesterol and blood pressure measurements and cardiovascular risk scores such as Framingham. But this is not so effective,” Bergström explained.
“Using imaging such as CCTA, where you can actually see atherosclerotic plaque, could be better for prediction, but we can’t image everyone. So, we wanted to see whether we could narrow down the population who should receive imaging with a detailed questionnaire, and it looks like we can.”
The study found that including clinical measurements such as blood pressure and cholesterol did not add much to the predictive value for identifying people with extensive coronary atherosclerosis, a result that Bergström said was surprising.
Which population to target?
Discussant of the study, Pamela Douglas, MD, professor of research in cardiovascular diseases at Duke University, Durham, North Carolina, congratulated the SCAPIS investigators on creating “a very rich data set for current and future study.”
“The SCAPIS study has already yielded novel data on the prevalence of coronary artery disease in the general population, and will address many critical questions over the long term,” she said.
But Douglas suggested that individuals with extensive coronary atherosclerosis were not the most appropriate target population to identify.
“The rationale for choosing this cutpoint is unclear as clinical risk/mortality is higher in all nonobstructive coronary artery disease, starting at one-vessel involvement,” she noted. “Therefore, effective preventive strategies likely need to start with detection and treatment of patients with even minimal plaque.”
Responding to Medscape Medical News, Bergström said this was a valid argument. “We plan to reanalyze our results with different populations as the target — that is something that we can do in the future.
But targeting everyone with just one coronary plaque is going to identify a large group — it was 40% of the population in our study. This will be too many people in whom to perform confirmatory CCTA imaging. It would be impractical to try and conduct cardiac imaging on that many people.”
Bergström noted that more data are needed on the danger of various levels of coronary atherosclerosis in this population who have not had any symptoms.
“We don’t have this information at present, but we are continuing to follow our population and we will have data on cardiac events in a few years’ time. Then we will know which level of atherosclerosis we need to target. It will probably be somewhere in between the extensive levels we used in this first analysis (which occurred in 13% of people) and the 40% of people who showed just one area of plaque.”
This study is the first report from SCAPIS, a collaborative project between six Swedish universities with the following vision statement: to “reduce the risk of cardiovascular and respiratory diseases for generations to come.”
The SCAPIS project is funded by the Swedish Heart and Lung Foundation. Bergström reports no disclosures.
This article first appeared on Medscape.com.

Omega-3 caps, vitamin D both fail for atrial fib primary prevention: VITAL-Rhythm
Clinical trials of omega-3 fatty acid or vitamin D supplements have followed a long and winding road in search of benefits in cardiovascular (CV) disease, with wildly mixed results. But the journey may be in vain in one of cardiology’s frontier research areas, primary prevention of atrial fibrillation (AF), suggest primary results of the VITAL-Rhythm trial, presented Nov. 13 during the American Heart Association (AHA) Scientific Sessions 2020 virtual meeting.

Neither marine-oil caps nor the vitamin D3 supplements made a difference to risk for incident AF, whether paroxysmal or persistent, over more than 5 years in the study, with more than 25,000 adults in the community. Nor did they seem to cause harm.
“To our knowledge, this is the first large-scale, long-term, randomized placebo-controlled trial to test the effect of any intervention on incident AF,” Christine M. Albert, MD, MPH, Cedars-Sinai Medical Center in Los Angeles, said at a media briefing on VITAL-Rhythm before her formal presentation of the trial during the conference.
Its findings, she said, don’t support the use of marine-oil caps or vitamin D3 for primary prevention of incident AF. “Fortunately, they also do not show any increased risk in atrial fibrillation for patients who are using these supplements for other indications.”
Both agents are widely taken without physician supervision for their perceived benefits, and marine-oil caps in particular – often in special prescription formulations – may be used for reducing elevated triglyceride levels and, based on the results of REDUCE-IT, cutting cardiovascular risk.
“It’s pretty clear that there’s no evidence to suggest that either of these supplements is helpful for preventing atrial fibrillation. And I think that’s clear from the evidence these investigators presented,” said Jonathan P. Piccini, MD, MHS, Duke University, Durham, N.C., who wasn’t part of the study.
“It’s also a little disappointing because atrial fibrillation is such a huge problem, and the inability to identify preventative strategies is a repeated theme,” he said in an interview.
VITAL-Rhythm is an ancillary study within the VITAL trial, which showed no benefit from either supplement regarding risk for incident cancer or CV events, as reported at the AHA sessions 2 years ago. In fact, their effects seem sweepingly negative throughout the trial; in another ancillary study, VITAL-DKD, neither supplement helped preserve renal function over 5 years in patients with type 2 diabetes.
The participants started VITAL without a history of AF, CV disease, or cancer; they were randomly assigned to take about a gram of omega-3 fatty acids, 2000 IU vitamin D3 daily, or their placebos, in a double randomization.
VITAL and its ancillary studies collectively undercut mechanistic theories about how omega-3 fatty acid and vitamin D supplements may affect AF risk, ideas derived from epidemiologic and dietary studies. They were thought perhaps “to have direct antiarrhythmic effects on myocytes through effects on ion channels, electrical remodeling, electrical stabilizing effects, and fluidity of the cell membranes,” observed Renate B. Schnabel, MD, MSc, University Heart Center, Hamburg, Germany, at the briefing. Or such effects might be related to beneficial effects on atherosclerosis, inflammation, or ischemic heart disease, she noted.
Neither idea is likely after VITAL and VITAL-Rhythm, said Dr. Schnabel, who spoke as an invited discussant after Albert’s formal presentation at AHA 2020.
That omega-3 fatty acid supplements may not improve AF incidence or risks has also been evident from many clinical trials and observational studies. Several, including REDUCE-IT, included some evidence for increasing risk for AF with marine-oil supplement intake. That may have happened in VITAL-Rhythm as well.
“While there was no evidence that the omega-3 three fatty acids prevented atrial fibrillation, there was a signal of perhaps more atrial fibrillation in the omega-3 fatty-acids group,” said Dr. Piccini, who directs his center’s electrophysiology clinical trials program.
A sensitivity analysis limited to participants who adhered to their assigned regimens, as opposed to the main intention-to-treat (ITT) analysis, showed a nonsignificant 13% increased hazard ratio for incident AF for the marine-oil supplement group. It reached a P value of .09, which can be interpreted as a trend.
“There are a few studies that have now showed a trend or an increased incidence of arrhythmia in patients treated with omega-3 fatty acids,” Dr. Piccini noted. “I don’t think it’s definitive, but it’s certainly something to keep an eye on.”
VITAL-Rhythm included an electrocardiography (ECG) substudy, yet to be reported, that should yield more insights about any such effects of marine-oil or vitamin D supplements in the trial, Dr. Albert said at the briefing.
The ancillary study assigned its 25,119 patients (mean age, 67 years; 51% women) to take vitamin D3 at 2000 IU/day, marine-oil supplements containing omega-3 fatty acids at 840 mg per day – 460 mg eicosapentaenoic acid (EPA) plus 380 mg docosahexaenoic acid (DHA) (Omacor, Pronova BioPharma) – or their placebos in a 2 x 2 randomization.
Incident cases of AF were identified through annual questionnaires in which the participants self-reported whether they had received a physician diagnosis of the arrhythmia, supplemented by Centers for Medicare & Medicaid Services claims data for AF hospital and clinical visits. Those led to a review of inpatient and outpatient records, from which AF events were adjudicated by an endpoint committee.
An electrocardiogram (72.9%) or physician’s report (27.1%) confirmed the AF diagnosis as the protocol required.
By those standards, 900 incident cases were identified, for a rate of 3.6% over a median of 5.3 years. They were paroxysmal in 58.4%, persistent in 38.4%, and indeterminant in 3.1%, Dr. Albert reported.
Of the 12,542 patients assigned to marine-oil caps by ITT, 469 (3.74%) developed incident AF in the ITT analysis, compared to 431 of 12,577 (3.43%) who received placebo, for an adjusted hazard ratio (HR) of 1.09 (95% CI, 0.96-1.24; P = .19).
The results were similar in two sensitivity analyses, one of which omitted patients with AF who may have had symptoms before randomization and another excluding those whose incident AF was identified solely in CMS data. But in the third “on treatment” sensitivity analysis, the HR for events was 1.13 (95% CI, 0.98-1.30; P = .09).
Outcomes for the vitamin D randomization were nearly the same, for an HR of 1.09 (95% CI, 0.96-1.25; P = .19) by ITT; the results were similar in all three sensitivity analyses.
“It’s not a tremendous signal of risk,” said Piccini of the marine-oil on-treatment analysis. But it, along with consistent evidence from other studies, does give him pause. “If a patient came to me and said,
Doctor, I want to take omega-3 fish oil, because I want to reduce my risk of events, as an arrhythmia doctor I would say, ‘We don’t have great evidence to do that for preventing atrial fibrillation. And there’s actually some evidence that it could mildly increase your risk of developing it.’ ”
For those prescribed evidence-based marine-oil therapy for other indications, he said, “I think the take-home message certainly is, if they report palpitations or other signs or symptoms that could be due to atrial fibrillation, we should be aggressive about screening for atrial fibrillation,” and making the diagnosis as appropriate. If the incident AF resolves after stopping the treatment, “maybe it’s reasonable to refrain from prescribing the medication for that patient.”
VITAL-Rhythm and VITAL are supported by multiple grants from the National Institutes of Health. Albert discloses receiving grant support from St. Jude Medical, Abbott, and Roche. Schnabel reports receiving honoraria from Bristol-Myers Squibb/Pfizer. Piccini previously disclosed receiving research grants from Abbott, the Association for the Advancement of Medical Instrumentation, Bayer, Boston Scientific, and Philips and serving as a consultant to Abbott, Allergan, ARCA Biopharma, Biotronik, Boston Scientific, LivaNova, Medtronic, Milestone, Sanofi, Philips, and UptoDate.
A version of this article originally appeared on Medscape.com.
Clinical trials of omega-3 fatty acid or vitamin D supplements have followed a long and winding road in search of benefits in cardiovascular (CV) disease, with wildly mixed results. But the journey may be in vain in one of cardiology’s frontier research areas, primary prevention of atrial fibrillation (AF), suggest primary results of the VITAL-Rhythm trial, presented Nov. 13 during the American Heart Association (AHA) Scientific Sessions 2020 virtual meeting.
Neither marine-oil caps nor the vitamin D3 supplements made a difference to risk for incident AF, whether paroxysmal or persistent, over more than 5 years in the study, with more than 25,000 adults in the community. Nor did they seem to cause harm.
“To our knowledge, this is the first large-scale, long-term, randomized placebo-controlled trial to test the effect of any intervention on incident AF,” Christine M. Albert, MD, MPH, Cedars-Sinai Medical Center in Los Angeles, said at a media briefing on VITAL-Rhythm before her formal presentation of the trial during the conference.
Its findings, she said, don’t support the use of marine-oil caps or vitamin D3 for primary prevention of incident AF. “Fortunately, they also do not show any increased risk in atrial fibrillation for patients who are using these supplements for other indications.”
Both agents are widely taken without physician supervision for their perceived benefits, and marine-oil caps in particular – often in special prescription formulations – may be used for reducing elevated triglyceride levels and, based on the results of REDUCE-IT, cutting cardiovascular risk.
“It’s pretty clear that there’s no evidence to suggest that either of these supplements is helpful for preventing atrial fibrillation. And I think that’s clear from the evidence these investigators presented,” said Jonathan P. Piccini, MD, MHS, Duke University, Durham, N.C., who wasn’t part of the study.
“It’s also a little disappointing because atrial fibrillation is such a huge problem, and the inability to identify preventative strategies is a repeated theme,” he said in an interview.
VITAL-Rhythm is an ancillary study within the VITAL trial, which showed no benefit from either supplement regarding risk for incident cancer or CV events, as reported at the AHA sessions 2 years ago. In fact, their effects seem sweepingly negative throughout the trial; in another ancillary study, VITAL-DKD, neither supplement helped preserve renal function over 5 years in patients with type 2 diabetes.
The participants started VITAL without a history of AF, CV disease, or cancer; they were randomly assigned to take about a gram of omega-3 fatty acids, 2000 IU vitamin D3 daily, or their placebos, in a double randomization.
VITAL and its ancillary studies collectively undercut mechanistic theories about how omega-3 fatty acid and vitamin D supplements may affect AF risk, ideas derived from epidemiologic and dietary studies. They were thought perhaps “to have direct antiarrhythmic effects on myocytes through effects on ion channels, electrical remodeling, electrical stabilizing effects, and fluidity of the cell membranes,” observed Renate B. Schnabel, MD, MSc, University Heart Center, Hamburg, Germany, at the briefing. Or such effects might be related to beneficial effects on atherosclerosis, inflammation, or ischemic heart disease, she noted.
Neither idea is likely after VITAL and VITAL-Rhythm, said Dr. Schnabel, who spoke as an invited discussant after Albert’s formal presentation at AHA 2020.
That omega-3 fatty acid supplements may not improve AF incidence or risks has also been evident from many clinical trials and observational studies. Several, including REDUCE-IT, included some evidence for increasing risk for AF with marine-oil supplement intake. That may have happened in VITAL-Rhythm as well.
“While there was no evidence that the omega-3 three fatty acids prevented atrial fibrillation, there was a signal of perhaps more atrial fibrillation in the omega-3 fatty-acids group,” said Dr. Piccini, who directs his center’s electrophysiology clinical trials program.
A sensitivity analysis limited to participants who adhered to their assigned regimens, as opposed to the main intention-to-treat (ITT) analysis, showed a nonsignificant 13% increased hazard ratio for incident AF for the marine-oil supplement group. It reached a P value of .09, which can be interpreted as a trend.
“There are a few studies that have now showed a trend or an increased incidence of arrhythmia in patients treated with omega-3 fatty acids,” Dr. Piccini noted. “I don’t think it’s definitive, but it’s certainly something to keep an eye on.”
VITAL-Rhythm included an electrocardiography (ECG) substudy, yet to be reported, that should yield more insights about any such effects of marine-oil or vitamin D supplements in the trial, Dr. Albert said at the briefing.
The ancillary study assigned its 25,119 patients (mean age, 67 years; 51% women) to take vitamin D3 at 2000 IU/day, marine-oil supplements containing omega-3 fatty acids at 840 mg per day – 460 mg eicosapentaenoic acid (EPA) plus 380 mg docosahexaenoic acid (DHA) (Omacor, Pronova BioPharma) – or their placebos in a 2 x 2 randomization.
Incident cases of AF were identified through annual questionnaires in which the participants self-reported whether they had received a physician diagnosis of the arrhythmia, supplemented by Centers for Medicare & Medicaid Services claims data for AF hospital and clinical visits. Those led to a review of inpatient and outpatient records, from which AF events were adjudicated by an endpoint committee.
An electrocardiogram (72.9%) or physician’s report (27.1%) confirmed the AF diagnosis as the protocol required.
By those standards, 900 incident cases were identified, for a rate of 3.6% over a median of 5.3 years. They were paroxysmal in 58.4%, persistent in 38.4%, and indeterminant in 3.1%, Dr. Albert reported.
Of the 12,542 patients assigned to marine-oil caps by ITT, 469 (3.74%) developed incident AF in the ITT analysis, compared to 431 of 12,577 (3.43%) who received placebo, for an adjusted hazard ratio (HR) of 1.09 (95% CI, 0.96-1.24; P = .19).
The results were similar in two sensitivity analyses, one of which omitted patients with AF who may have had symptoms before randomization and another excluding those whose incident AF was identified solely in CMS data. But in the third “on treatment” sensitivity analysis, the HR for events was 1.13 (95% CI, 0.98-1.30; P = .09).
Outcomes for the vitamin D randomization were nearly the same, for an HR of 1.09 (95% CI, 0.96-1.25; P = .19) by ITT; the results were similar in all three sensitivity analyses.
“It’s not a tremendous signal of risk,” said Piccini of the marine-oil on-treatment analysis. But it, along with consistent evidence from other studies, does give him pause. “If a patient came to me and said,
Doctor, I want to take omega-3 fish oil, because I want to reduce my risk of events, as an arrhythmia doctor I would say, ‘We don’t have great evidence to do that for preventing atrial fibrillation. And there’s actually some evidence that it could mildly increase your risk of developing it.’ ”
For those prescribed evidence-based marine-oil therapy for other indications, he said, “I think the take-home message certainly is, if they report palpitations or other signs or symptoms that could be due to atrial fibrillation, we should be aggressive about screening for atrial fibrillation,” and making the diagnosis as appropriate. If the incident AF resolves after stopping the treatment, “maybe it’s reasonable to refrain from prescribing the medication for that patient.”
VITAL-Rhythm and VITAL are supported by multiple grants from the National Institutes of Health. Albert discloses receiving grant support from St. Jude Medical, Abbott, and Roche. Schnabel reports receiving honoraria from Bristol-Myers Squibb/Pfizer. Piccini previously disclosed receiving research grants from Abbott, the Association for the Advancement of Medical Instrumentation, Bayer, Boston Scientific, and Philips and serving as a consultant to Abbott, Allergan, ARCA Biopharma, Biotronik, Boston Scientific, LivaNova, Medtronic, Milestone, Sanofi, Philips, and UptoDate.
A version of this article originally appeared on Medscape.com.
Clinical trials of omega-3 fatty acid or vitamin D supplements have followed a long and winding road in search of benefits in cardiovascular (CV) disease, with wildly mixed results. But the journey may be in vain in one of cardiology’s frontier research areas, primary prevention of atrial fibrillation (AF), suggest primary results of the VITAL-Rhythm trial, presented Nov. 13 during the American Heart Association (AHA) Scientific Sessions 2020 virtual meeting.
Neither marine-oil caps nor the vitamin D3 supplements made a difference to risk for incident AF, whether paroxysmal or persistent, over more than 5 years in the study, with more than 25,000 adults in the community. Nor did they seem to cause harm.
“To our knowledge, this is the first large-scale, long-term, randomized placebo-controlled trial to test the effect of any intervention on incident AF,” Christine M. Albert, MD, MPH, Cedars-Sinai Medical Center in Los Angeles, said at a media briefing on VITAL-Rhythm before her formal presentation of the trial during the conference.
Its findings, she said, don’t support the use of marine-oil caps or vitamin D3 for primary prevention of incident AF. “Fortunately, they also do not show any increased risk in atrial fibrillation for patients who are using these supplements for other indications.”
Both agents are widely taken without physician supervision for their perceived benefits, and marine-oil caps in particular – often in special prescription formulations – may be used for reducing elevated triglyceride levels and, based on the results of REDUCE-IT, cutting cardiovascular risk.
“It’s pretty clear that there’s no evidence to suggest that either of these supplements is helpful for preventing atrial fibrillation. And I think that’s clear from the evidence these investigators presented,” said Jonathan P. Piccini, MD, MHS, Duke University, Durham, N.C., who wasn’t part of the study.
“It’s also a little disappointing because atrial fibrillation is such a huge problem, and the inability to identify preventative strategies is a repeated theme,” he said in an interview.
VITAL-Rhythm is an ancillary study within the VITAL trial, which showed no benefit from either supplement regarding risk for incident cancer or CV events, as reported at the AHA sessions 2 years ago. In fact, their effects seem sweepingly negative throughout the trial; in another ancillary study, VITAL-DKD, neither supplement helped preserve renal function over 5 years in patients with type 2 diabetes.
The participants started VITAL without a history of AF, CV disease, or cancer; they were randomly assigned to take about a gram of omega-3 fatty acids, 2000 IU vitamin D3 daily, or their placebos, in a double randomization.
VITAL and its ancillary studies collectively undercut mechanistic theories about how omega-3 fatty acid and vitamin D supplements may affect AF risk, ideas derived from epidemiologic and dietary studies. They were thought perhaps “to have direct antiarrhythmic effects on myocytes through effects on ion channels, electrical remodeling, electrical stabilizing effects, and fluidity of the cell membranes,” observed Renate B. Schnabel, MD, MSc, University Heart Center, Hamburg, Germany, at the briefing. Or such effects might be related to beneficial effects on atherosclerosis, inflammation, or ischemic heart disease, she noted.
Neither idea is likely after VITAL and VITAL-Rhythm, said Dr. Schnabel, who spoke as an invited discussant after Albert’s formal presentation at AHA 2020.
That omega-3 fatty acid supplements may not improve AF incidence or risks has also been evident from many clinical trials and observational studies. Several, including REDUCE-IT, included some evidence for increasing risk for AF with marine-oil supplement intake. That may have happened in VITAL-Rhythm as well.
“While there was no evidence that the omega-3 three fatty acids prevented atrial fibrillation, there was a signal of perhaps more atrial fibrillation in the omega-3 fatty-acids group,” said Dr. Piccini, who directs his center’s electrophysiology clinical trials program.
A sensitivity analysis limited to participants who adhered to their assigned regimens, as opposed to the main intention-to-treat (ITT) analysis, showed a nonsignificant 13% increased hazard ratio for incident AF for the marine-oil supplement group. It reached a P value of .09, which can be interpreted as a trend.
“There are a few studies that have now showed a trend or an increased incidence of arrhythmia in patients treated with omega-3 fatty acids,” Dr. Piccini noted. “I don’t think it’s definitive, but it’s certainly something to keep an eye on.”
VITAL-Rhythm included an electrocardiography (ECG) substudy, yet to be reported, that should yield more insights about any such effects of marine-oil or vitamin D supplements in the trial, Dr. Albert said at the briefing.
The ancillary study assigned its 25,119 patients (mean age, 67 years; 51% women) to take vitamin D3 at 2000 IU/day, marine-oil supplements containing omega-3 fatty acids at 840 mg per day – 460 mg eicosapentaenoic acid (EPA) plus 380 mg docosahexaenoic acid (DHA) (Omacor, Pronova BioPharma) – or their placebos in a 2 x 2 randomization.
Incident cases of AF were identified through annual questionnaires in which the participants self-reported whether they had received a physician diagnosis of the arrhythmia, supplemented by Centers for Medicare & Medicaid Services claims data for AF hospital and clinical visits. Those led to a review of inpatient and outpatient records, from which AF events were adjudicated by an endpoint committee.
An electrocardiogram (72.9%) or physician’s report (27.1%) confirmed the AF diagnosis as the protocol required.
By those standards, 900 incident cases were identified, for a rate of 3.6% over a median of 5.3 years. They were paroxysmal in 58.4%, persistent in 38.4%, and indeterminant in 3.1%, Dr. Albert reported.
Of the 12,542 patients assigned to marine-oil caps by ITT, 469 (3.74%) developed incident AF in the ITT analysis, compared to 431 of 12,577 (3.43%) who received placebo, for an adjusted hazard ratio (HR) of 1.09 (95% CI, 0.96-1.24; P = .19).
The results were similar in two sensitivity analyses, one of which omitted patients with AF who may have had symptoms before randomization and another excluding those whose incident AF was identified solely in CMS data. But in the third “on treatment” sensitivity analysis, the HR for events was 1.13 (95% CI, 0.98-1.30; P = .09).
Outcomes for the vitamin D randomization were nearly the same, for an HR of 1.09 (95% CI, 0.96-1.25; P = .19) by ITT; the results were similar in all three sensitivity analyses.
“It’s not a tremendous signal of risk,” said Piccini of the marine-oil on-treatment analysis. But it, along with consistent evidence from other studies, does give him pause. “If a patient came to me and said,
Doctor, I want to take omega-3 fish oil, because I want to reduce my risk of events, as an arrhythmia doctor I would say, ‘We don’t have great evidence to do that for preventing atrial fibrillation. And there’s actually some evidence that it could mildly increase your risk of developing it.’ ”
For those prescribed evidence-based marine-oil therapy for other indications, he said, “I think the take-home message certainly is, if they report palpitations or other signs or symptoms that could be due to atrial fibrillation, we should be aggressive about screening for atrial fibrillation,” and making the diagnosis as appropriate. If the incident AF resolves after stopping the treatment, “maybe it’s reasonable to refrain from prescribing the medication for that patient.”
VITAL-Rhythm and VITAL are supported by multiple grants from the National Institutes of Health. Albert discloses receiving grant support from St. Jude Medical, Abbott, and Roche. Schnabel reports receiving honoraria from Bristol-Myers Squibb/Pfizer. Piccini previously disclosed receiving research grants from Abbott, the Association for the Advancement of Medical Instrumentation, Bayer, Boston Scientific, and Philips and serving as a consultant to Abbott, Allergan, ARCA Biopharma, Biotronik, Boston Scientific, LivaNova, Medtronic, Milestone, Sanofi, Philips, and UptoDate.
A version of this article originally appeared on Medscape.com.

Intravenous iron reduces HF readmissions: AFFIRM-AHF
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.

However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.

He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.
However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.
He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
Iron supplementation reduces heart failure (HF) readmissions in iron-deficient patients hospitalized for acute HF, according to results of the AFFIRM-AHF trial.
After 52 weeks, intravenous ferric carboxymaltose (Ferinject) reduced the risk of total HF hospitalizations and cardiovascular (CV) death by 21% compared with placebo (293 vs 372 events; rate ratio [RR] 0.79; 95% CI, 0.62 - 1.01).
Although the composite primary endpoint failed to achieve statistical significance, it was driven by a significant 26% reduction in the risk of total HF hospital readmissions (P = .013) without an effect on CV mortality (P =.809).
Because the management and follow-up of patients was affected by the COVID-19 pandemic, a prespecified sensitivity analysis was performed that censored patients in each country at the date when its first COVID-19 patient was reported, explained principal investigator Piotr Ponikowski, MD, PhD, Wroclaw Medical University, Wroclaw, Poland.
That analysis revealed a significant 30% reduction in total HF readmissions (P = .005) in patients receiving ferric carboxymaltose (FCM), as well as significant benefits on the primary composite and secondary endpoints.
Notably, 80% of patients required only one or two injections and HF hospitalizations were reduced irrespective of anemia status.
“Iron deficiency should be searched in patients hospitalized with acute heart failure — assessed using a simple blood test — and is now an important therapeutic target,” Ponikowski said at the virtual American Heart Association (AHA) Scientific Sessions 2020.
The results were also published simultaneously in The Lancet.
Iron deficiency is present in up to 70% of patients with acute HF and a predictor of poor outcome, independent of anemia and ejection fraction, he noted.
The FAIR-HF, CONFIRM-HF, and EFFECT-HF trials demonstrated that IV iron supplementation improves exercise capacity, symptoms, and quality of life in iron-deficient HF patients.
However, no such benefit was seen with oral IV in the IRONOUT trial. “So it seems if we are to replace iron, it needs to be done using intravenous therapy,” said John McMurray, MD, University of Glasgow, Scotland, who was invited to discuss the results.
He observed that the reduction in HF hospitalizations in AFFIRM-AHF were relatively modest and that the trial was never expected to show a benefit on CV mortality. Also, the COVID-19 sensitivity analysis providing more convincing effects is a valid approach and one recommended by regulators.
Further, the findings are supported by independent evidence in chronic kidney disease, from the PIVOTAL trial, that intravenous iron reduces HF hospitalizations, McMurray said.
“The million-dollar question, of course, is what will the results of this study mean for the guidelines: I think they probably will change the guidelines,” he said. “Certainly, I hope they will change the US guidelines, which have really given a very lukewarm recommendation for intravenous iron and I think that should probably be stronger.”
In a class IIb recommendation, the 2017 American College of Cardiology/AHA/Heart Failure Society of America heart failure guidelines say intravenous iron “might be reasonable” to improve functional status and quality of life in New York Heart Association class II and III patients with iron deficiency.
The 2016 European Society of Cardiology guidelines include a class IIa recommendation that IV iron “should be considered” in iron-deficient patients with symptomatic HF with reduced ejection fraction.
“This is the first large-scale [trial] of IV supplementation that could potentially change the way we approach patients, particularly those with hospitalized heart failure,” past AHA president Clyde Yancy, MD, MSc, Northwestern University Feinberg School of Medicine in Chicago, said during an earlier press briefing.
He pointed out that clinicians have been circumspect about the early IV iron data. “I have to congratulate you because you’ve changed the narrative,” Yancy said. “We have to start thinking about iron deficiency; we have to think about how we incorporate this in treatment protocols.”
Press briefing panelist Marc Pfeffer, MD, PhD, Brigham and Women’s Hospital and Harvard Medical School in Boston, acknowledged he was among those circumspect.
“I’m no longer a skeptic and I want to congratulate them for showing it’s a risk factor,” he said. “It’s one thing to have a risk factor; it’s another to be a modifiable risk factor and I think that’s what’s so exciting about this.”
The double-blind, phase 4 AFFIRM-AHF trial randomly assigned 1132 patients to receive a bolus injection of ferric carboxymaltose or normal saline before hospital discharge for an acute HF episode. Subsequent treatment was given, as needed, up to 24 weeks post-randomization.
At admission, all patients had left ventricular ejection fractions less than 50% and iron deficiency (serum ferritin <100 ng/mL or serum ferritin 100-299 ng/mL if transferrin saturation <20%).
The modified intention-to-treat (mITT) analysis included 558 FCM patients and 550 controls in whom study treatment was started and for whom at least one post-randomization value was available.
Press briefing discussant Nancy Sweitzer, MD, PhD, director of the University of Arizona’s Sarver Heart Center in Tucson, said AFFIRM-AHF is an “important trial likely to change guidelines” and “targeted one of the highest risk populations we have in heart failure.”
Patients with iron deficiency tend to be elderly with more comorbidities, have longer hospital lengths of stay, and higher readmission rates. “So impacting hospitalizations in this population is incredibly impactful,” she said.
“Awareness and assessment of iron deficiency are an important part of inpatient care of patients with ejection fractions less than or equal to 50% and acute decompensated heart failure, and I think all of us in the community need to pay much more attention to this issue.”
As with any new therapy, there are implementation challenges such as how to monitor patients and deliver the therapy in a cost-effective way, Sweitzer said.
The trial focused on the most vulnerable period for HF patients, but these patients should be rechecked every 3 to 4 months for iron deficiency, Ponikowski observed during the briefing.
“This is a modifiable risk factor,” he said. “We only need to remember, we only need to assess it, and we have a very, very simple tool in our hands. We just need to measure two biomarkers, transferrin saturation and ferritin — that’s all.”
Unanswered questions include the mechanism behind the reduction in hospitalization, the relationship of benefit to hemoglobin levels, and whether there is a differential benefit based on age, presence of ischemia, or sex, especially as women tend to be more severely affected by iron deficiency, Sweitzer said.
During the formal presentation, Ponikowski said the primary endpoint was consistent in subgroup analyses across baseline hemoglobin, estimated glomerular filtration rate, and N-terminal pro-brain natriuretic peptide levels, HF etiology, ejection fraction, and whether HF was diagnosed prior to the index hospitalization.
Treatment with FCM was safe, with no significant differences between the FCM and placebo groups in serious adverse events (45% vs 51%) or adverse events leading to study discontinuation (18% vs 17%), he reported. The most common adverse events were cardiac disorders (40.1% vs 44.3%) and infections (18.2% vs 22%).
AFFIRM-AHF is the first of three ongoing mortality and morbidity trials in heart failure with intravenous ferric carboxymaltose; the others are FAIR-HF2 and HEART-FID. Additional insights are also expected next year on intravenous iron isomaltoside from the Scottish-based IRONMAN trial in 1300 HF patients with iron deficiency.
The study was sponsored by Vifor International. Ponikowski has received research grants and personal fees from Vifor Pharma; and personal fees from Amgen, Bayer, Novartis, Abbott Vascular, Boehringer Ingelheim, Merck, Pfizer, Servier, AstraZeneca, Berlin Chemie, Cibiem, Renal Guard Solutions Bristol-Myers Squibb, and Impulse Dynamics.
Pfeffer reported honoraria from AstraZeneca, Corvidia, GlaxoSmithKline, Jazz, MyoKardia, Novartis, Roche, Sanofi, and Servier; other relationships with DalCor and Novo Nordisk; research grants from Novartis; and an ownership interest in DalCor. Sweitzer reported research payments from Merck and Novartis; and consulting fees from Myocardia.
McMurray reported relationships with Amgen, AstraZeneca, Bayer, Boehringer Ingelheim, Cytokinetics, Novartis, and Servier. Yancy reported a relationship with Abbott and JAMA Network.
Lancet. Published online November 13, 2020. Full text
American Heart Association Scientific Sessions 2020: Presented November 13, 2020.
A version of this article originally appeared on Medscape.com.
FROM AHA 2020