User login
Vitamin D deficiency in older adults
Low vitamin D levels can impact cognitive functioning in older adults.1 As vitamin D levels decrease, cognitive impairment increases.
Vitamin D deficiency can occur because few foods contain this nutrient2 and patients have limited exposure to sunlight—vitamin D is produced when sunlight strikes the skin.2 In addition to rickets, low levels of vitamin D have been linked to slower information processing in middle age and older men, cognitive decline, mood disorders, and altered brain development and function resulting in neurodegenerative diseases and other medical disorders.3
One study suggested that one-half of adults age >60 do not get sufficient vitamin D, with an even higher rate among women with Alzheimer’s disease.4 Patients in dementia units typically are not tested for vitamin D levels. These patients rarely leave the unit, which leaves them deprived of the vitamin D provided by sunlight. Even patients exposed to sunlight may receive minimal vitamin D because they use sunscreen.
The following protocol can help patients who may benefit from vitamin D supplementation and increased sun exposure.
Obtain and assess vitamin D levels. Evaluate your patient’s level in the context of physical or cognitive symptoms and other lab values:
- deficient: <12 ng/mL
- inadequate: 12 to 20 ng/mL
- adequate: ≥20 ng/mL.2
Order dietary assessment to identify foods that may increase vitamin D levels. The best sources are fish—salmon, tuna, and mackerel—fish oils, beef, liver, cheese, and egg yolks.2 Several food products, including milk and orange juice, are fortified with vitamin D.
Suggest a daily vitamin D supplement ranging from 400 IU/d to 1,000 IU/d. The Institute of Medicine suggests 600 IU/d for patients age 60 to 70 and 800 IU/d for those age ≥71. For vitamin D deficient patients, recommend >1,000 IU/d.1
Recommend 15 minutes per day in the sun without sunscreen from spring to autumn; late summer to fall is ideal because vitamin D’s half-life is 30 days. Midday is the best time to produce vitamin D.5
Recheck the patient’s Mini-Mental State Examination score every 4 months. Vitamin D supplementation is correlated with cognitive functioning.6
Disclosure
Dr. LaFerney reports no financial, relationship with any company whose, products are mentioned in this article, or with manufacturers of competing, products.
1. Mayo Clinic. Vitamin D. http://www.mayoclinic.com/health/vitamin-d/NS_patient-vitamind/DSECTION=dosing. Updated October 1 2011. Accessed September 26, 2012.
2. National Institutes of Health. Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional. Accessed September 26, 2012.
3. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
4. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
5. Webb AR, Engelsen O. Calculated ultraviolet exposure levels for a healthy vitamin D status. Photochem Photobiol. 2006;82(6):1697-1703.
6. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
Low vitamin D levels can impact cognitive functioning in older adults.1 As vitamin D levels decrease, cognitive impairment increases.
Vitamin D deficiency can occur because few foods contain this nutrient2 and patients have limited exposure to sunlight—vitamin D is produced when sunlight strikes the skin.2 In addition to rickets, low levels of vitamin D have been linked to slower information processing in middle age and older men, cognitive decline, mood disorders, and altered brain development and function resulting in neurodegenerative diseases and other medical disorders.3
One study suggested that one-half of adults age >60 do not get sufficient vitamin D, with an even higher rate among women with Alzheimer’s disease.4 Patients in dementia units typically are not tested for vitamin D levels. These patients rarely leave the unit, which leaves them deprived of the vitamin D provided by sunlight. Even patients exposed to sunlight may receive minimal vitamin D because they use sunscreen.
The following protocol can help patients who may benefit from vitamin D supplementation and increased sun exposure.
Obtain and assess vitamin D levels. Evaluate your patient’s level in the context of physical or cognitive symptoms and other lab values:
- deficient: <12 ng/mL
- inadequate: 12 to 20 ng/mL
- adequate: ≥20 ng/mL.2
Order dietary assessment to identify foods that may increase vitamin D levels. The best sources are fish—salmon, tuna, and mackerel—fish oils, beef, liver, cheese, and egg yolks.2 Several food products, including milk and orange juice, are fortified with vitamin D.
Suggest a daily vitamin D supplement ranging from 400 IU/d to 1,000 IU/d. The Institute of Medicine suggests 600 IU/d for patients age 60 to 70 and 800 IU/d for those age ≥71. For vitamin D deficient patients, recommend >1,000 IU/d.1
Recommend 15 minutes per day in the sun without sunscreen from spring to autumn; late summer to fall is ideal because vitamin D’s half-life is 30 days. Midday is the best time to produce vitamin D.5
Recheck the patient’s Mini-Mental State Examination score every 4 months. Vitamin D supplementation is correlated with cognitive functioning.6
Disclosure
Dr. LaFerney reports no financial, relationship with any company whose, products are mentioned in this article, or with manufacturers of competing, products.
Low vitamin D levels can impact cognitive functioning in older adults.1 As vitamin D levels decrease, cognitive impairment increases.
Vitamin D deficiency can occur because few foods contain this nutrient2 and patients have limited exposure to sunlight—vitamin D is produced when sunlight strikes the skin.2 In addition to rickets, low levels of vitamin D have been linked to slower information processing in middle age and older men, cognitive decline, mood disorders, and altered brain development and function resulting in neurodegenerative diseases and other medical disorders.3
One study suggested that one-half of adults age >60 do not get sufficient vitamin D, with an even higher rate among women with Alzheimer’s disease.4 Patients in dementia units typically are not tested for vitamin D levels. These patients rarely leave the unit, which leaves them deprived of the vitamin D provided by sunlight. Even patients exposed to sunlight may receive minimal vitamin D because they use sunscreen.
The following protocol can help patients who may benefit from vitamin D supplementation and increased sun exposure.
Obtain and assess vitamin D levels. Evaluate your patient’s level in the context of physical or cognitive symptoms and other lab values:
- deficient: <12 ng/mL
- inadequate: 12 to 20 ng/mL
- adequate: ≥20 ng/mL.2
Order dietary assessment to identify foods that may increase vitamin D levels. The best sources are fish—salmon, tuna, and mackerel—fish oils, beef, liver, cheese, and egg yolks.2 Several food products, including milk and orange juice, are fortified with vitamin D.
Suggest a daily vitamin D supplement ranging from 400 IU/d to 1,000 IU/d. The Institute of Medicine suggests 600 IU/d for patients age 60 to 70 and 800 IU/d for those age ≥71. For vitamin D deficient patients, recommend >1,000 IU/d.1
Recommend 15 minutes per day in the sun without sunscreen from spring to autumn; late summer to fall is ideal because vitamin D’s half-life is 30 days. Midday is the best time to produce vitamin D.5
Recheck the patient’s Mini-Mental State Examination score every 4 months. Vitamin D supplementation is correlated with cognitive functioning.6
Disclosure
Dr. LaFerney reports no financial, relationship with any company whose, products are mentioned in this article, or with manufacturers of competing, products.
1. Mayo Clinic. Vitamin D. http://www.mayoclinic.com/health/vitamin-d/NS_patient-vitamind/DSECTION=dosing. Updated October 1 2011. Accessed September 26, 2012.
2. National Institutes of Health. Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional. Accessed September 26, 2012.
3. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
4. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
5. Webb AR, Engelsen O. Calculated ultraviolet exposure levels for a healthy vitamin D status. Photochem Photobiol. 2006;82(6):1697-1703.
6. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
1. Mayo Clinic. Vitamin D. http://www.mayoclinic.com/health/vitamin-d/NS_patient-vitamind/DSECTION=dosing. Updated October 1 2011. Accessed September 26, 2012.
2. National Institutes of Health. Office of Dietary Supplements. Dietary supplement fact sheet: vitamin D. http://ods.od.nih.gov/factsheets/VitaminD-HealthProfessional. Accessed September 26, 2012.
3. Lee DM, Tajar A, Ulubaev A, et al. Association between 25-hydroxyvitamin D levels and cognitive performance in middle-aged and older European men. J Neurol Neurosurg Psychiatry. 2009;80(7):722-729.
4. Wilkins CH, Sheline YI, Roe CM, et al. Vitamin D deficiency is associated with low mood and worse cognitive performance in older adults. Am J Geriatr Psychiatry. 2006;14(12):1032-1040.
5. Webb AR, Engelsen O. Calculated ultraviolet exposure levels for a healthy vitamin D status. Photochem Photobiol. 2006;82(6):1697-1703.
6. Przybelski RJ, Binkley NC. Is vitamin D important for preserving cognition? A positive correlation of serum 25-hydroxyvitamin D concentration with cognitive function. Arch Biochem Biophys. 2007;460(2):202-205.
An open-label trial of escitalopram for PPD: Considerations for research
Challenges in recruiting women to postpartum depression (PPD) antidepressant treatment trials, which we encountered when conducting a trial of escitalopram, contribute to the limited body of knowledge about PPD treatment. Here we discuss results from a preliminary trial of escitalopram for PPD, and challenges of research in this area.
Escitalopram, the S-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with high selectivity and potency that is FDA-approved for treating major depressive disorder (MDD) and generalized anxiety disorder. An agent with antidepressant and anxiolytic effects is particularly desirable for PPD because anxiety is more common in postpartum major depressive episodes than non-postpartum MDD.1 Anxiety and depressive disorders commonly are comorbid in postpartum women.2
We conducted an open-label trial of escitalopram for women with PPD and anxiety. We initially attempted to recruit 20 women.
Methods
Patients received 8 weeks of treatment with escitalopram, 10 to 20 mg/d (flexible dose). After completing the initial phone screen, patients had 5 follow-up visits, once every 2 weeks for 8 weeks. The institutional review board at Massachusetts General Hospital approved this study and we obtained written informed consent from all patients at the first visit. Twelve patients completed the phone screen and 7 eligible patients were enrolled in the study over 32 months. Reasons for ineligibility included having a history of psychosis, onset of symptoms >3 months postpartum, or presenting >6 months after onset. Others declined to participate because of concern about the time commitment or because they pursued nonpharmacologic treatments after the evaluation visit. One patient was lost to follow-up. Three patients completed the study. The study was halted because of the slow pace of recruitment.
Patient selection. Patients were screened for a major depressive episode with postpartum onset within 3 months of childbirth; depressive symptoms may have developed during pregnancy and worsened postpartum to meet criteria for MDD. Women were eligible for the study if they:
- were age 18 to 45
- experienced a major depressive episode with symptoms developing within 3 months of childbirth
- presented within 6 months of childbirth
- had a Montgomery-Åsberg Depression Rating Scale (MADRS) score >15
- had a Beck Anxiety Inventory (BAI) score >10.
Patients who were pregnant or breast-feeding were excluded from the study per an agreement with the sponsor. In addition, women were excluded if they had taken any psychotropic medication within 2 weeks of enrollment; had active suicidal ideation, homicidal ideation, or presence of psychotic symptoms; had chronic depression or dysthymia; had chronic or treatment-resistant anxiety disorders; had a history of mania or hypomania; or had active alcohol or substance abuse within the past year.
Treatment. Patients received escitalopram, 10 mg/d, after the baseline visit. At the investigator’s discretion, the dose could be increased to 20 mg/d or lowered to 5 mg/d if side effects occurred.
Measures. At the first visit, patients were assessed with the Mini-International Neuropsychiatric Interview to verify MDD and exclude diagnoses that would determine ineligibility. MADRS and Edinburgh Postnatal Depression Scale (EPDS) were used at each visit to measure depressive symptoms.3,4 The BAI was completed at each visit to measure anxiety symptoms. Obsessions and compulsions were measured with the Yale-Brown Obsessive Compulsive Scale (Y-BOCS)5 at baseline, and at all following visits if the patient scored >8 at baseline. The Clinical Global Impression Scales for severity and improvement were completed at each visit.6
Results
Of 7 patients enrolled, 3 completed the study, 2 were ineligible after the baseline visit, and 2 did not participate after the baseline visit (1 selected to pursue psychotherapy, and 1 was lost to follow-up).
Two of 3 patients responded to escitalopram (≥50% decrease on MADRS), and both were remitters (MADRS score <7). All 3 patients were responders on EPDS and BAI. One patient had Y-BOCS >8 at baseline (Total Y-BOCS score of 9, and final Y-BOCS score of 8) (Table).
Table
Symptom rating scale scores at baseline and study end
| Baseline (Visit 1) | Final (Visit 5) | |||||
|---|---|---|---|---|---|---|
| Patient | MADRS | BAI | EPDS | MADRS | BAI | EPDS |
| Ms. A | 21 | 18 | 22 | 12 | 0 | 0 |
| Ms. B | 28 | 28 | 19 | 4 | 5 | 2 |
| Ms. C | 37 | 6 | 19 | 6 | 2 | 0 |
| BAI: Beck Anxiety Inventory; EPDS: Edinburgh Postnatal Depression Scale; MADRS: Montgomery-Åsberg Depression Rating Scale | ||||||
Discussion
Patients who stayed in treatment improved during the course of this study. Recruitment was difficult; we were able to recruit only 7 patients out of a projected 20 for the screening visit. We solicited feedback from local obstetrics health care providers and social workers on recruitment and attractiveness of the study as part of our routine collaboration with obstetrical services that screen for PPD. Primary reasons patients were not referred were that they were breast-feeding or they stated they would prefer to receive treatment from their primary care doctor. Recruitment difficulty in this study was in stark contrast to other recent studies completed at our center. For example, we have successfully recruited for menopausal depression and premenstrual dysphoric disorder treatment studies, and have completed large naturalistic studies of women with unipolar depression and bipolar disorder across pregnancy and postpartum. We suspect that many patients who were eligible for the study preferred to seek care from an obstetrician or primary care doctor with whom they already had a therapeutic alliance, and we also suspect that many women with PPD do not seek treatment at all, which is consistent with findings from other research groups.
Lessons learned from PPD research include:
- Including women who are breast-feeding is important because many women choose to breast-feed and suffer from PPD. Because antidepressant use during breast-feeding has been closely studied, it is appropriate to include potential research participants who are breast-feeding as long as they receive adequate information and are able to provide informed consent.
- Participants in PPD studies may require accommodations that take into account their role as a new mother, such as on-site childcare, home visits, or other strategies.
- Because of recruitment challenges in postpartum patients, multisite trials may be required to include adequate numbers of participants.
Related Resource
- Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-21.
Drug Brand Names
- Citalopram • Celexa
- Escitalopram • Lexapro
Disclosures
Dr. Freeman has received grant or research support from Eli Lilly and Company, Forest Laboratories, and GlaxoSmithKline, is on the advisory boards of Otsuka and Takeda/Lundbeck, and is a consultant for PamLab LLC.
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
This study was funded as an investigator-initiated trial by Forest Pharmaceuticals.
1. Bernstein IH, Rush AJ, Yonkers K, et al. Symptom features of postpartum depression: are they distinct? Depress Anxiety. 2008;25(1):20-26.
2. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
3. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
4. Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
5. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.
6. Guy W. ECDEU assessment manual for psychopharmacology. Rockville MD: US Department of Health and Human Services; 1976. Department of Health, Education, and Welfare Publication (ADM) 76–338.
Challenges in recruiting women to postpartum depression (PPD) antidepressant treatment trials, which we encountered when conducting a trial of escitalopram, contribute to the limited body of knowledge about PPD treatment. Here we discuss results from a preliminary trial of escitalopram for PPD, and challenges of research in this area.
Escitalopram, the S-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with high selectivity and potency that is FDA-approved for treating major depressive disorder (MDD) and generalized anxiety disorder. An agent with antidepressant and anxiolytic effects is particularly desirable for PPD because anxiety is more common in postpartum major depressive episodes than non-postpartum MDD.1 Anxiety and depressive disorders commonly are comorbid in postpartum women.2
We conducted an open-label trial of escitalopram for women with PPD and anxiety. We initially attempted to recruit 20 women.
Methods
Patients received 8 weeks of treatment with escitalopram, 10 to 20 mg/d (flexible dose). After completing the initial phone screen, patients had 5 follow-up visits, once every 2 weeks for 8 weeks. The institutional review board at Massachusetts General Hospital approved this study and we obtained written informed consent from all patients at the first visit. Twelve patients completed the phone screen and 7 eligible patients were enrolled in the study over 32 months. Reasons for ineligibility included having a history of psychosis, onset of symptoms >3 months postpartum, or presenting >6 months after onset. Others declined to participate because of concern about the time commitment or because they pursued nonpharmacologic treatments after the evaluation visit. One patient was lost to follow-up. Three patients completed the study. The study was halted because of the slow pace of recruitment.
Patient selection. Patients were screened for a major depressive episode with postpartum onset within 3 months of childbirth; depressive symptoms may have developed during pregnancy and worsened postpartum to meet criteria for MDD. Women were eligible for the study if they:
- were age 18 to 45
- experienced a major depressive episode with symptoms developing within 3 months of childbirth
- presented within 6 months of childbirth
- had a Montgomery-Åsberg Depression Rating Scale (MADRS) score >15
- had a Beck Anxiety Inventory (BAI) score >10.
Patients who were pregnant or breast-feeding were excluded from the study per an agreement with the sponsor. In addition, women were excluded if they had taken any psychotropic medication within 2 weeks of enrollment; had active suicidal ideation, homicidal ideation, or presence of psychotic symptoms; had chronic depression or dysthymia; had chronic or treatment-resistant anxiety disorders; had a history of mania or hypomania; or had active alcohol or substance abuse within the past year.
Treatment. Patients received escitalopram, 10 mg/d, after the baseline visit. At the investigator’s discretion, the dose could be increased to 20 mg/d or lowered to 5 mg/d if side effects occurred.
Measures. At the first visit, patients were assessed with the Mini-International Neuropsychiatric Interview to verify MDD and exclude diagnoses that would determine ineligibility. MADRS and Edinburgh Postnatal Depression Scale (EPDS) were used at each visit to measure depressive symptoms.3,4 The BAI was completed at each visit to measure anxiety symptoms. Obsessions and compulsions were measured with the Yale-Brown Obsessive Compulsive Scale (Y-BOCS)5 at baseline, and at all following visits if the patient scored >8 at baseline. The Clinical Global Impression Scales for severity and improvement were completed at each visit.6
Results
Of 7 patients enrolled, 3 completed the study, 2 were ineligible after the baseline visit, and 2 did not participate after the baseline visit (1 selected to pursue psychotherapy, and 1 was lost to follow-up).
Two of 3 patients responded to escitalopram (≥50% decrease on MADRS), and both were remitters (MADRS score <7). All 3 patients were responders on EPDS and BAI. One patient had Y-BOCS >8 at baseline (Total Y-BOCS score of 9, and final Y-BOCS score of 8) (Table).
Table
Symptom rating scale scores at baseline and study end
| Baseline (Visit 1) | Final (Visit 5) | |||||
|---|---|---|---|---|---|---|
| Patient | MADRS | BAI | EPDS | MADRS | BAI | EPDS |
| Ms. A | 21 | 18 | 22 | 12 | 0 | 0 |
| Ms. B | 28 | 28 | 19 | 4 | 5 | 2 |
| Ms. C | 37 | 6 | 19 | 6 | 2 | 0 |
| BAI: Beck Anxiety Inventory; EPDS: Edinburgh Postnatal Depression Scale; MADRS: Montgomery-Åsberg Depression Rating Scale | ||||||
Discussion
Patients who stayed in treatment improved during the course of this study. Recruitment was difficult; we were able to recruit only 7 patients out of a projected 20 for the screening visit. We solicited feedback from local obstetrics health care providers and social workers on recruitment and attractiveness of the study as part of our routine collaboration with obstetrical services that screen for PPD. Primary reasons patients were not referred were that they were breast-feeding or they stated they would prefer to receive treatment from their primary care doctor. Recruitment difficulty in this study was in stark contrast to other recent studies completed at our center. For example, we have successfully recruited for menopausal depression and premenstrual dysphoric disorder treatment studies, and have completed large naturalistic studies of women with unipolar depression and bipolar disorder across pregnancy and postpartum. We suspect that many patients who were eligible for the study preferred to seek care from an obstetrician or primary care doctor with whom they already had a therapeutic alliance, and we also suspect that many women with PPD do not seek treatment at all, which is consistent with findings from other research groups.
Lessons learned from PPD research include:
- Including women who are breast-feeding is important because many women choose to breast-feed and suffer from PPD. Because antidepressant use during breast-feeding has been closely studied, it is appropriate to include potential research participants who are breast-feeding as long as they receive adequate information and are able to provide informed consent.
- Participants in PPD studies may require accommodations that take into account their role as a new mother, such as on-site childcare, home visits, or other strategies.
- Because of recruitment challenges in postpartum patients, multisite trials may be required to include adequate numbers of participants.
Related Resource
- Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-21.
Drug Brand Names
- Citalopram • Celexa
- Escitalopram • Lexapro
Disclosures
Dr. Freeman has received grant or research support from Eli Lilly and Company, Forest Laboratories, and GlaxoSmithKline, is on the advisory boards of Otsuka and Takeda/Lundbeck, and is a consultant for PamLab LLC.
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
This study was funded as an investigator-initiated trial by Forest Pharmaceuticals.
Challenges in recruiting women to postpartum depression (PPD) antidepressant treatment trials, which we encountered when conducting a trial of escitalopram, contribute to the limited body of knowledge about PPD treatment. Here we discuss results from a preliminary trial of escitalopram for PPD, and challenges of research in this area.
Escitalopram, the S-enantiomer of citalopram, is a selective serotonin reuptake inhibitor with high selectivity and potency that is FDA-approved for treating major depressive disorder (MDD) and generalized anxiety disorder. An agent with antidepressant and anxiolytic effects is particularly desirable for PPD because anxiety is more common in postpartum major depressive episodes than non-postpartum MDD.1 Anxiety and depressive disorders commonly are comorbid in postpartum women.2
We conducted an open-label trial of escitalopram for women with PPD and anxiety. We initially attempted to recruit 20 women.
Methods
Patients received 8 weeks of treatment with escitalopram, 10 to 20 mg/d (flexible dose). After completing the initial phone screen, patients had 5 follow-up visits, once every 2 weeks for 8 weeks. The institutional review board at Massachusetts General Hospital approved this study and we obtained written informed consent from all patients at the first visit. Twelve patients completed the phone screen and 7 eligible patients were enrolled in the study over 32 months. Reasons for ineligibility included having a history of psychosis, onset of symptoms >3 months postpartum, or presenting >6 months after onset. Others declined to participate because of concern about the time commitment or because they pursued nonpharmacologic treatments after the evaluation visit. One patient was lost to follow-up. Three patients completed the study. The study was halted because of the slow pace of recruitment.
Patient selection. Patients were screened for a major depressive episode with postpartum onset within 3 months of childbirth; depressive symptoms may have developed during pregnancy and worsened postpartum to meet criteria for MDD. Women were eligible for the study if they:
- were age 18 to 45
- experienced a major depressive episode with symptoms developing within 3 months of childbirth
- presented within 6 months of childbirth
- had a Montgomery-Åsberg Depression Rating Scale (MADRS) score >15
- had a Beck Anxiety Inventory (BAI) score >10.
Patients who were pregnant or breast-feeding were excluded from the study per an agreement with the sponsor. In addition, women were excluded if they had taken any psychotropic medication within 2 weeks of enrollment; had active suicidal ideation, homicidal ideation, or presence of psychotic symptoms; had chronic depression or dysthymia; had chronic or treatment-resistant anxiety disorders; had a history of mania or hypomania; or had active alcohol or substance abuse within the past year.
Treatment. Patients received escitalopram, 10 mg/d, after the baseline visit. At the investigator’s discretion, the dose could be increased to 20 mg/d or lowered to 5 mg/d if side effects occurred.
Measures. At the first visit, patients were assessed with the Mini-International Neuropsychiatric Interview to verify MDD and exclude diagnoses that would determine ineligibility. MADRS and Edinburgh Postnatal Depression Scale (EPDS) were used at each visit to measure depressive symptoms.3,4 The BAI was completed at each visit to measure anxiety symptoms. Obsessions and compulsions were measured with the Yale-Brown Obsessive Compulsive Scale (Y-BOCS)5 at baseline, and at all following visits if the patient scored >8 at baseline. The Clinical Global Impression Scales for severity and improvement were completed at each visit.6
Results
Of 7 patients enrolled, 3 completed the study, 2 were ineligible after the baseline visit, and 2 did not participate after the baseline visit (1 selected to pursue psychotherapy, and 1 was lost to follow-up).
Two of 3 patients responded to escitalopram (≥50% decrease on MADRS), and both were remitters (MADRS score <7). All 3 patients were responders on EPDS and BAI. One patient had Y-BOCS >8 at baseline (Total Y-BOCS score of 9, and final Y-BOCS score of 8) (Table).
Table
Symptom rating scale scores at baseline and study end
| Baseline (Visit 1) | Final (Visit 5) | |||||
|---|---|---|---|---|---|---|
| Patient | MADRS | BAI | EPDS | MADRS | BAI | EPDS |
| Ms. A | 21 | 18 | 22 | 12 | 0 | 0 |
| Ms. B | 28 | 28 | 19 | 4 | 5 | 2 |
| Ms. C | 37 | 6 | 19 | 6 | 2 | 0 |
| BAI: Beck Anxiety Inventory; EPDS: Edinburgh Postnatal Depression Scale; MADRS: Montgomery-Åsberg Depression Rating Scale | ||||||
Discussion
Patients who stayed in treatment improved during the course of this study. Recruitment was difficult; we were able to recruit only 7 patients out of a projected 20 for the screening visit. We solicited feedback from local obstetrics health care providers and social workers on recruitment and attractiveness of the study as part of our routine collaboration with obstetrical services that screen for PPD. Primary reasons patients were not referred were that they were breast-feeding or they stated they would prefer to receive treatment from their primary care doctor. Recruitment difficulty in this study was in stark contrast to other recent studies completed at our center. For example, we have successfully recruited for menopausal depression and premenstrual dysphoric disorder treatment studies, and have completed large naturalistic studies of women with unipolar depression and bipolar disorder across pregnancy and postpartum. We suspect that many patients who were eligible for the study preferred to seek care from an obstetrician or primary care doctor with whom they already had a therapeutic alliance, and we also suspect that many women with PPD do not seek treatment at all, which is consistent with findings from other research groups.
Lessons learned from PPD research include:
- Including women who are breast-feeding is important because many women choose to breast-feed and suffer from PPD. Because antidepressant use during breast-feeding has been closely studied, it is appropriate to include potential research participants who are breast-feeding as long as they receive adequate information and are able to provide informed consent.
- Participants in PPD studies may require accommodations that take into account their role as a new mother, such as on-site childcare, home visits, or other strategies.
- Because of recruitment challenges in postpartum patients, multisite trials may be required to include adequate numbers of participants.
Related Resource
- Freeman MP, Joffe H, Cohen LS. Postpartum depression: Help patients find the right treatment. Current Psychiatry. 2012;11(11):14-21.
Drug Brand Names
- Citalopram • Celexa
- Escitalopram • Lexapro
Disclosures
Dr. Freeman has received grant or research support from Eli Lilly and Company, Forest Laboratories, and GlaxoSmithKline, is on the advisory boards of Otsuka and Takeda/Lundbeck, and is a consultant for PamLab LLC.
Dr. Joffe has received grant or research support from Cephalon/Teva, and is a consultant to Noven and Sunovion.
Dr. Cohen has received research support from AstraZeneca, Bayer HealthCare Pharmaceuticals, Bristol-Myers Squibb, Forest Laboratories, GlaxoSmithKline, National Institute of Mental Health, National Institute on Aging, National Institutes of Health, Ortho-McNeil Janssen, and Pfizer and has served on an advisory board for PamLab LLC.
This study was funded as an investigator-initiated trial by Forest Pharmaceuticals.
1. Bernstein IH, Rush AJ, Yonkers K, et al. Symptom features of postpartum depression: are they distinct? Depress Anxiety. 2008;25(1):20-26.
2. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
3. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
4. Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
5. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.
6. Guy W. ECDEU assessment manual for psychopharmacology. Rockville MD: US Department of Health and Human Services; 1976. Department of Health, Education, and Welfare Publication (ADM) 76–338.
1. Bernstein IH, Rush AJ, Yonkers K, et al. Symptom features of postpartum depression: are they distinct? Depress Anxiety. 2008;25(1):20-26.
2. Wenzel A, Haugen EN, Jackson LC, et al. Anxiety symptoms and disorders at eight weeks postpartum. J Anxiety Disord. 2005;19(3):295-311.
3. Cox JL, Holden JM, Sagovsky R. Detection of postnatal depression. Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987;150:782-786.
4. Montgomery SA, Åsberg M. A new depression scale designed to be sensitive to change. Br J Psychiatry. 1979;134:382-389.
5. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.
6. Guy W. ECDEU assessment manual for psychopharmacology. Rockville MD: US Department of Health and Human Services; 1976. Department of Health, Education, and Welfare Publication (ADM) 76–338.
Panic disorder: Break the fear circuit

Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
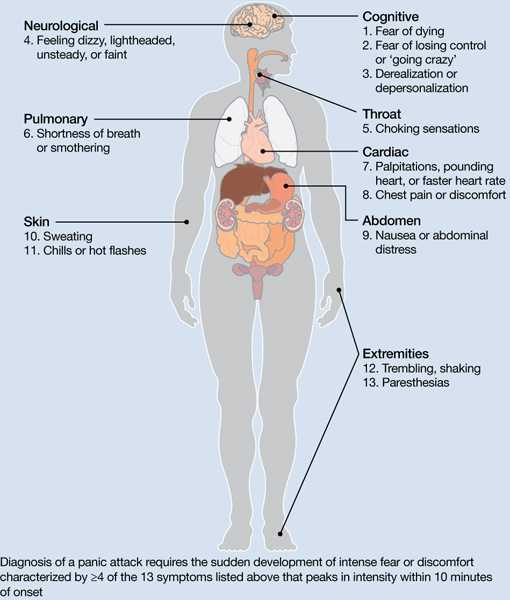
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.

Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.
Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Ms. K, a 24-year-old waitress who lives with her boyfriend, was referred by her primary care physician for evaluation of panic attacks that began “out of nowhere” at work approximately 6 months ago. The unpredictable attacks occur multiple times per week, causing her to leave work and cancel shifts.
Ms. K reports that before the panic attacks began, she felt happy in her relationship, enjoyed hobbies, and was hopeful about the future. However, she has become concerned that a potentially catastrophic illness is causing her panic attacks. She researches her symptoms on the Internet, and is preoccupied with the possibility of sudden death due to an undiagnosed heart condition. Multiple visits to the emergency room have not identified any physical abnormalities. Her primary care doctor prescribed alprazolam, 0.5 mg as needed for panic attacks, which she reports is helpful, “but only in the moment of the attacks.” Ms. K avoids alcohol and illicit substances and limits her caffeine intake. She is not willing to accept that her life “feels so limited.” Her dream of earning a nursing degree and eventually starting a family now seems unattainable.
Panic disorder (PD) occurs in 3% to 5% of adults, with women affected at roughly twice the rate of men.1 Causing a broad range of distress and varying degrees of impairment, PD commonly occurs with other psychiatric disorders. For most patients, treatment is effective, but those who do not respond to initial approaches require a thoughtful, stepped approach to care. Key considerations include establishing an accurate diagnosis, clarifying comorbid illnesses, ascertaining patient beliefs and expectations, and providing appropriately dosed and maintained treatments.
Panic attacks vs PD
Panic attacks consist of rapid onset of intense anxiety, with prominent somatic symptoms, that peaks within 10 minutes (Figure).2 Attacks in which <4 of the listed symptoms occur are considered limited-symptom panic attacks.
Figure: Body locations of panic attack symptoms
Diagnosis of a panic attack requires the sudden development of intense fear or discomfort characterized by ≥4 of the 13 symptoms listed above that peaks in intensity within 10 minutes of onset
Source: Reference 2
Panic attacks can occur with various disorders, including other anxiety disorders, mood disorders, and substance intoxication or withdrawal. Because serious medical conditions can present with panic-like symptoms, the initial occurrence of such symptoms warrants consideration of physiological causes. For a Box2 that describes the differential diagnosis of panic attacks, see this article at CurrentPsychiatry.com.
To meet diagnostic criteria for panic disorder, panic attacks must initially occur “out of the blue,” meaning no specific object or situation induced the attack. The differential diagnosis of panic attacks includes assessing for other psychiatric disorders that may involve panic attacks. Evaluation requires considering the context in which the panic attacks occur, including their start date, pattern of attacks, instigating situations, and associated thoughts.
Social phobia. Attacks occur only during or immediately before a social interaction in which the patient fears embarrassing himself or herself.
Obsessive-compulsive disorder (OCD). Attacks occur when the patient cannot avoid exposure to an obsessional fear or is prevented from performing a ritual that diffuses obsessional anxiety.
Posttraumatic stress disorder (PTSD). Attacks occur when confronted by a trauma-related memory or trigger.
Specific phobia. Attacks occur only when the patient encounters a specifically feared object, place, or situation, unrelated to social phobia, OCD, or PTSD.
Medical conditions. Conditions to consider include—but are not limited to—hyperthyroidism, pulmonary embolism, myocardial infarction, cardiac dysrhythmias, hypoglycemia, asthma, partial complex seizures, and pheochromocytoma.
Source: Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington, DC: American Psychiatric Association; 2000
A PD diagnosis requires that repeated panic attacks initially must occur from “out of the blue,” meaning no specific object or situation induced the attack. In addition, the diagnosis requires 1 of 3 types of psychological or behavioral changes as a result of the attacks (Table 1).2 Agoraphobia is diagnosed if 1 of the behavioral changes is avoidance of places or situations from which escape might be embarrassing or difficult should an attack occur. A patient can be diagnosed as having PD with agoraphobia, PD without agoraphobia, or agoraphobia without PD (ie, experiences only limited symptom panic attacks, but avoids situations or stimuli associated with them).
Table 1
Definitions of panic disorder and agoraphobia
| Panic disorder |
|---|
|
| Agoraphobia |
| Anxiety about, or avoidance of, being in places or situations from which escape might be difficult or embarrassing, or in which help may not be available in the event of having an unexpected or situationally predisposed panic attack or panic-like symptoms. Agoraphobic fears typically involve characteristic clusters of situations that include being outside the home alone, being in a crowd, standing in a line, being on a bridge, or traveling in a bus, train, or automobile |
| Source: Reference 2 |
Comorbidities are common in patients with PD and predict greater difficulty achieving remission (Box).1,3-6
The most common psychiatric conditions that co-occur with panic disorder (PD) are other anxiety disorders, mood disorders, personality disorders, and substance use disorders.1 Carefully assess the severity and degree of impairment or distress arising from each condition to prioritize treatment goals. For example, treating panic attacks would be a lower priority in a patient with untreated bipolar disorder.
Assessing comorbid substance abuse is important in selecting PD treatments. Benzodiazepines should almost always be avoided in patients with a history of drug abuse—illicit or prescribed. Although complete abstinence should not be a prerequisite for beginning PD treatment, detoxification and concomitant substance abuse treatment are essential.3
Comorbid mood disorders also affect the course of PD treatment. Antidepressants are effective for treating depression and PD, whereas benzodiazepines are not effective for depression.4 Antidepressants in patients with bipolar disorder are controversial because these medications might induce mixed or elevated mood states or rapid cycling. In these complicated patients, consider antidepressants lower in the treatment algorithm.5
Other conditions to consider before beginning treatment include pregnancy or the possibility of becoming pregnant in the near future and suicidal ideation. PD is associated with increased risk for suicidal ideation and progression to suicide attempts, particularly in patients with a comorbid mood or psychotic disorder.6 In addition, consider the potential impact of medications on comorbid medical conditions.
Treatment begins with education
The goal of treatment is remission of symptoms, ideally including an absence of panic attacks, agoraphobic avoidance, and anticipatory anxiety.1 The Panic Disorder Severity Scale self-report is a validated measure of panic symptoms that may be useful in clinical practice.7
The first step in treatment is educating patients about panic attacks, framing them as an overreactive fear circuit in the brain that produces physical symptoms that are not dangerous. Using a brain model that shows the location of the amygdala, hippocampus, and prefrontal cortex—which play crucial roles in generating and controlling anxiety and fear—can make this discussion more concrete.8 Although highly simplified, such models allow clinicians to demonstrate that excessive reactivity of limbic regions can be reduced by both top-down (cortico-limbic connections via cognitive-behavioral therapy [CBT]) and bottom-up (pharmacotherapy directly acting on limbic structures) approaches. Such discussions lead to treatment recommendations for CBT, pharmacotherapy, or their combination.
No single treatment has emerged as the definitive “best” for PD, and no reliable predictors can guide specific treatment for an individual.3 Combining CBT with pharmacotherapy produces higher short-term response rates than either treatment alone, but in the long term, combination treatment does not appear to be superior to CBT alone.9 Base the initial treatment selection for PD on patient preference, treatment availability and cost, and comorbid medical and psychiatric conditions. For an Algorithm to guide treatment decisions, see this article at CurrentPsychiatry.com.
Algorithm: Treatment for panic disorder: A suggested algorithm
aPoor response to an SSRI should lead to a switch to venlafaxine extended-release, and vice versa
bBenzodiazepines are relatively contraindicated in geriatric patients and patients with a history of substance abuse or dependence
CBT: cognitive-behavioral therapy; MAOI: monoamine oxidase inhibitor; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; Ven XR: venlafaxine extended-release
First-line treatments
Psychotherapy. CBT is the most efficacious psychotherapy for PD. Twelve to 15 sessions of CBT has demonstrated efficacy for PD, with additional effects on comorbid anxiety and depressive symptoms.10 No large clinical trials of CBT have used cognitive restructuring alone; all have included at least some component of exposure that requires the patient to confront feared physical sensations. Gains during treatment may be steady and gradual or sudden and uneven, with rapid improvement in some but not all symptoms. CBT and pharmacotherapy have demonstrated similar levels of benefit in short-term trials, but CBT has proven superior in most9 but not all11 trials evaluating long-term outcomes, particularly compared with pharmacotherapy that is discontinued during follow-up. Although less studied, group CBT also may be considered if a patient cannot afford individual CBT.
Pharmacotherapy. Evidence supports selective serotonin reuptake inhibitors (SSRIs), venlafaxine extended-release (XR), benzodiazepines, and tricyclic antidepressants (TCAs) as effective treatments for PD.3 No class of medication has demonstrated superiority over others in short-term treatment.3,12 Because of the medical risks associated with benzodiazepines and TCAs, an SSRI or venlafaxine XR should be the first medication option for most patients. Fluoxetine, paroxetine, sertraline, and venlafaxine XR are FDA-approved for PD. Paroxetine is associated with weight gain and may increase the risk for panic recurrence upon discontinuation more than sertraline, making it a less favorable option for many patients.13 Start doses at half the normal starting dose used for treating major depressive disorder and continue for 4 to 7 days, then increase to the minimal effective dose. For a Table3 that lists dosing recommendations for antidepressants to treat PD, see this article at CurrentPsychiatry.com. If there is no improvement by 4 weeks, increase the dose every 2 to 4 weeks until remission is achieved or side effects prevent further dose increases.
Table
Recommended doses for antidepressants used to treat panic disorder
| Medication | Starting dose (mg/d) | Therapeutic range (mg/d) |
|---|---|---|
| SSRIs | ||
| Citalopram | 10 | 20 to 40 |
| Escitalopram | 5 | 10 to 40 |
| Fluoxetine | 5 to 10 | 20 to 80 |
| Fluvoxamine | 25 | 100 to 300 |
| Paroxetine | 10 | 20 to 80 |
| Paroxetine CR | 12.5 | 25 to 50 |
| Sertraline | 25 | 100 to 200 |
| SNRIs | ||
| Duloxetine | 20 to 30 | 60 to 120 |
| Venlafaxine XR | 37.5 | 150 to 225 |
| TCAs | ||
| Clomipramine | 10 to 25 | 100 to 300 |
| Imipramine | 10 | 100 to 300 |
| MAOI | ||
| Phenelzine | 15 | 45 to 90 |
| CR: controlled release; MAOI: monoamine oxidase inhibitor; SNRIs: serotonin-norepinephrine reuptake inhibitors; SSRIs: selective serotonin reuptake inhibitors; TCAs: tricyclic antidepressants; XR: extended release Source: American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington, DC: American Psychiatric Association; 2009 | ||
Treatment nonresponse. True non-response needs to be distinguished from poor response caused by inadequate treatment delivery, eg, patients not completing homework assignments in CBT or not adhering to pharmacotherapy. Asking patients about adverse effects or personal and family beliefs about treatment may reveal reasons for nonadherence.
Second-line treatments
Little data are available to guide next-step treatment options in patients who don’t achieve remission from their initial treatment. Patients who benefit from an SSRI, venlafaxine XR, or CBT but still have symptoms should be started on combination treatment. For a patient who experiences complete non-response to the initial treatment, discontinue the first treatment and switch to the other modality. In general, completely ineffective treatments should be discontinued when another treatment is added, but when partial improvement (>30%) occurs, continue the original treatment and augment it with another approach.
For patients pursuing pharmacotherapy, poor response to an adequate SSRI trial usually should lead to a switch to venlafaxine XR, and vice versa. Failure to respond to both of these medication classes should prompt a switch to a benzodiazepine or TCA.
Benzodiazepines are a fast-acting, effective treatment for PD, with efficacy similar to SSRIs in acute and long-term treatment.14 Benzodiazepines may be prescribed with antidepressants at the beginning of treatment to improve response speed.15 Clonazepam and alprazolam are FDA-approved for treating PD. A high-potency, long-acting agent, clonazepam is the preferred initial benzodiazepine, dosed 0.5 to 4 mg/d on a fixed schedule. Although substantial data support using alprazolam for PD, it requires more frequent dosing and has a greater risk of rebound anxiety and abuse potential because of its more rapid onset of action. Compared with immediate-release alprazolam, alprazolam XR has a slower absorption rate and longer steady state in the blood, but this formulation does not have lower abuse potential or greater efficacy. Although not FDA-approved for PD, diazepam and lorazepam also have proven efficacy for PD.3
Benzodiazepines should be considered contraindicated in patients with a history of substance abuse, except in select cases.4 Benzodiazepines generally should be avoided in older patients because of increased risk for falls, cognitive impairment, and motor vehicle accidents. Table 2 lists situations in which benzodiazepines may be used to treat PD.
Table 2
Clinical scenarios in which to consider using benzodiazepines
| Coadministration for 2 to 4 weeks when initiating treatment with an SSRI or venlafaxine XR to achieve more rapid relief and mitigate potential antidepressant-induced anxiety |
| For patients who wish to avoid antidepressants because of concern about sexual dysfunction |
| For patients who need chronic aspirin or an NSAID, which may increase the risk for upper gastrointestinal bleeding when taken in combination with an SSRI |
| For patients with comorbid bipolar disorder or epilepsy |
| Next-step monotherapy or augmentation in patients who respond poorly to an SSRI, venlafaxine XR, TCA, or CBT |
| CBT: cognitive-behavioral therapy; NSAID: nonsteroidal anti-inflammatory drug; SSRI: selective serotonin reuptake inhibitor; TCA: tricyclic antidepressant; XR: extended release |
TCAs are effective as monotherapy for PD. Most support comes from studies of imipramine or clomipramine.12 Similar to SSRIs and venlafaxine XR, use a low initial dose and gradually increase until the patient remits or side effects prevent further increases. SSRI and TCA combinations rarely are used unless the TCA is a relatively specific norepinephrine reuptake inhibitor (eg, desipramine, nortriptyline). Because TCAs are metabolized via the cytochrome P450 2D6 system and some SSRIs—particularly fluoxetine and paroxetine—strongly inhibit 2D6, combinations of TCAs with these agents may lead to dangerously high plasma TCA levels, placing patients at risk for cardiac dysrhythmias and other side effects.16
Monoamine oxidase inhibitors (MAOIs)—particularly phenelzine—are underused for PD. They have the strongest efficacy data for any class of medications outside the first- and second-line agents and have a unique mechanism of action. In patients who can comply with the dietary and medication limitations, an MAOI generally should be the next step after nonresponse to other treatments.3
Alternative treatments
For patients who do not respond to any of the treatments described above, data from uncontrolled studies support mirtazapine, levetiracetam, and the serotonin-norepinephrine reuptake inhibitors duloxetine and milnacipran as monotherapy for PD.17 Pindolol—a beta blocker and 5-HT1A receptor antagonist—proved superior to placebo as an adjunctive agent to SSRIs in treatment-resistant PD in 1 of 2 trials.17 Minimal evidence supports the atypical antipsychotics risperidone and olanzapine in treatment-resistant PD, although a placebo-controlled trial of quetiapine SR coadministered with SSRIs recently was completed (NCT00619892; results pending). Atypical antipsychotics are best reserved for patients with a primary psychotic disorder or bipolar disorder who experience panic attacks.5
Panic-focused psychodynamic psychotherapy, a 12-week (approximately 24 sessions) form of psychotherapy, has demonstrated superiority vs applied relaxation therapy.18 This treatment could be considered for patients who do not respond to standard first-line treatments, but few community therapists are familiar with this method.
For many patients with PD, complementary and alternative medicine (CAM) approaches are appealing. See this article at CurrentPsychiatry.com for a Box that discusses CAM for PD.
Although no complementary and alternative medicine treatments have strong evidence of efficacy as monotherapy for panic disorder (PD), several have data that suggest benefit with little evidence of risk. These include bibliotherapy, yoga, aerobic exercise, and the dietary supplements kava and inositol.a Exercise as a treatment poses a challenge because it can induce symptoms that the patient fears, such as tachycardia and shortness of breath. In addition to any direct physiologic benefit from aerobic exercise, there is also an exposure component that can be harnessed by gradually increasing the exertion level.
Another approach undergoing extensive evaluation is Internet-provided cognitive-behavioral therapy (CBT). Using guided CBT modules with or without therapist support, Internet-provided CBT provides an option for motivated patients unable to complete in-person CBT because of logistical factors.b A helpful resource that reviews Internet self-help and psychotherapy guided programs for PD and other psychiatric conditions is http://beacon.anu.edu.au.
References
a. Antonacci DJ, Davis E, Bloch RM, et al. CAM for your anxious patient: what the evidence says. Current Psychiatry. 2010;9(10):42-52.
b. Johnston L, Titov N, Andrews G, et al. A RCT of a transdiagnostic internet-delivered treatment for three anxiety disorders: examination of support roles and disorder-specific outcomes. PLoS One. 2011;6(11):e28079.
Maintenance treatment
Patients who complete a course of CBT for PD often follow up with several “booster sessions” at monthly or longer intervals that focus on relapse prevention techniques. Few controlled trials have evaluated pharmacotherapy discontinuation in PD. Most guidelines recommend continuing treatment for ≥1 year after achieving remission to minimize the risk of relapse.3 Researchers are focusing on whether medication dosage can be reduced during maintenance without loss of efficacy.
Treatment discontinuation
In the absence of urgent medical need, taper medications for PD gradually over several months. PD patients are highly sensitive to unusual physical sensations, which can occur while discontinuing antidepressants or benzodiazepines. If a benzodiazepine is used in conjunction with an antidepressant, the benzodiazepine should be discontinued first, so that the antidepressant can help ease benzodiazepine-associated discontinuation symptoms. A brief course of CBT during pharmacotherapy discontinuation may increase the likelihood of successful tapering.19
CASE CONTINUED: A successful switch
Ms. K has to discontinue sequential trials of fluoxetine, 40 mg/d, and venlafaxine XR, 225 mg/d because of side effects, and she does not reduce the frequency of her alprazolam use. She agrees to switch from alprazolam to clonazepam, 0.5 mg every morning and 1 mg at bedtime, and to start CBT. Clonazepam reduces her anxiety sufficiently so she can address her symptoms in therapy. Through CBT she becomes motivated to monitor her thoughts and treat them as guesses rather than facts, reviewing the evidence for her thoughts and generating rational responses. She participates in exposure exercises, which she practices between sessions, and grows to tolerate uncomfortable sensations until they no longer signal danger. After 12 CBT sessions, she is panic-free. Despite some trepidation, she agrees to a slow taper off clonazepam, reducing the dose by 0.25 mg every 2 weeks. She continues booster sessions with her therapist to manage any re-emerging anxiety. After an additional 12 weeks, she successfully discontinues clonazepam and remains panic-free.
Related Resources
- American Psychiatric Association. Panic disorder. http://healthyminds.org/Main-Topic/Panic-Disorder.aspx.
- Anxiety and Depression Association of America. Panic disorder & agoraphobia. http://adaa.org/understanding-anxiety/panic-disorder-agoraphobia.
- Mayo Clinic. Panic attacks and panic disorder. www.mayoclinic.com/health/panic-attacks/DS00338.
- National Health Service Self-Help Guides. www.ntw.nhs.uk/pic/selfhelp.
- National Institute of Mental Health. Panic disorder. www.nimh.nih.gov/health/topics/panic-disorder/index.shtml.
Drug Brand Names
- Alprazolam • Xanax
- Alprazolam XR • Xanax XR
- Citalopram • Celexa
- Clomipramine • Anafranil
- Clonazepam • Klonopin
- Desipramine • Norpramin
- Diazepam • Valium
- Duloxetine • Cymbalta
- Escitalopram • Lexapro
- Fluoxetine • Prozac
- Fluvoxamine • Luvox
- Imipramine • Tofranil
- Levetiracetam • Keppra
- Lorazepam • Ativan
- Milnacipran • Savella
- Mirtazapine • Remeron
- Nortriptyline • Aventyl, Pamelor
- Olanzapine • Zyprexa
- Paroxetine • Paxil
- Paroxetine CR • Paxil CR
- Phenelzine • Nardil
- Pindolol • Visken
- Quetiapine SR • Seroquel SR
- Risperidone • Risperdal
- Sertraline • Zoloft
- Venlafaxine XR • Effexor XR
Disclosures
Dr. Dunlop receives research support from Bristol-Myers Squibb, GlaxoSmithKline, and the National Institute of Mental Health. He serves as a consultant to MedAvante and Roche.
Ms. Schneider and Dr. Gerardi report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
1. Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet. 2006;368(9540):1023-1032.
2. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
3. American Psychiatric Association. Practice guideline for the treatment of patients with panic disorder. 2nd ed. Washington DC: American Psychiatric Association; 2009.
4. Dunlop BW, Davis PG. Combination treatment with benzodiazepines and SSRIs for comorbid anxiety and depression: a review. Prim Care Companion J Clin Psychiatry. 2008;10(3):222-228.
5. Rakofsky JJ, Dunlop BW. Treating nonspecific anxiety and anxiety disorders in patients with bipolar disorder: a review. J Clin Psychiatry. 2011;72(1):81-90.
6. Sareen J, Cox BJ, Afifi TO, et al. Anxiety disorders and risk for suicidal ideation and suicide attempts: a population-based longitudinal study of adults. Arch Gen Psychiatry. 2005;62(11):1249-1257.
7. Houck PR, Spiegel DA, Shear MK, et al. Reliability of the self-report version of the panic disorder severity scale. Depress Anxiety. 2002;15(4):183-185.
8. Ninan PT, Dunlop BW. Neurobiology and etiology of panic disorder. J Clin Psychiatry. 2005;66(suppl 4):3-7.
9. Furukawa TA, Watanabe N, Churchill R. Psychotherapy plus antidepressant for panic disorder with or without agoraphobia: systematic review. Br J Psychiatry. 2006;188:305-312.
10. Barlow DH, Gorman JM, Shear MK, et al. Cognitive-behavioral therapy, imipramine, or their combination for panic disorder: a randomized controlled trial. JAMA. 2000;283(19):2529-2536.
11. van Apeldoorn FJ, Timmerman ME, Mersch PP, et al. A randomized trial of cognitive-behavioral therapy or selective serotonin reuptake inhibitor or both combined for panic disorder with or without agoraphobia: treatment results through 1-year follow-up. J Clin Psychiatry. 2010;71(5):574-586.
12. Bakker A, van Balkom AJ, Spinhoven P. SSRIs vs. TCAs in the treatment of panic disorder: a meta-analysis. Acta Psychiatr Scand. 2002;106(3):163-167.
13. Bandelow B, Behnke K, Lenoir S, et al. Sertraline versus paroxetine in the treatment of panic disorder: an acute, double-blind noninferiority comparison. J Clin Psychiatry. 2004;65(3):405-413.
14. Nardi AE, Freire RC, Mochcovitch MD, et al. A randomized, naturalistic, parallel-group study for the long-term treatment of panic disorder with clonazepam or paroxetine. J Clin Psychopharmacol. 2012;32(1):120-126.
15. Goddard AW, Brouette T, Almai A, et al. Early coadministration of clonazepam with sertraline for panic disorder. Arch Gen Psychiatry. 2001;58(7):681-686.
16. Preskorn SH, Shah R, Neff M, et al. The potential for clinically significant drug-drug interactions involving the CYP 2D6 system: effects with fluoxetine and paroxetine versus sertraline. J Psychiatr Pract. 2007;13(1):5-12.
17. Perna G, Guerriero G, Caldirola D. Emerging drugs for panic disorder. Expert Opin Emerg Drugs. 2011;16(4):631-645.
18. Milrod B, Leon AC, Busch F, et al. A randomized controlled clinical trial of psychoanalytic psychotherapy for panic disorder. Am J Psychiatry. 2007;164(2):265-272.
19. Otto MW, Pollack MH, Sachs GS, et al. Discontinuation of benzodiazepine treatment: efficacy of cognitive-behavioral therapy for patients with panic disorder. Am J Psychiatry. 1993;150(10):1485-1490.
Differentiating Alzheimer’s disease from dementia with Lewy bodies
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
Discuss this article at www.facebook.com/CurrentPsychiatry
Alzheimer’s disease (AD) and dementia with Lewy bodies (DLB) are the first and second most common causes of neurodegenerative dementia, respectively.“New Alzheimer’s disease guidelines: Implications for clinicians,” Current Psychiatry, March 2012, p. 15-20; http://bit.ly/UNYikk.
The 2005 report of the DLB Consortium5 recognizes central, core, suggestive, and supportive features of DLB (Table 1).5,10 These features are considered in the context of other confounding clinical conditions and the timing of cognitive and motor symptoms. The revised DLB criteria5 require a central feature of progressive cognitive decline. “Probable DLB” is when a patient presents with 2 core features or 1 core feature and ≥1 suggestive features. A diagnosis of “possible DLB” requires 1 core feature or 1 suggestive feature in the presence of progressive cognitive decline.
Table 1
Diagnostic criteria for AD and DLB
| NIA-AA criteria for AD (2011)10 |
| Possible AD: Clinical and cognitive criteria (DSM-IV-TR) for AD are met and there is an absence of biomarkers to support the diagnosis or there is evidence of a secondary disorder that can cause dementia |
| Probable AD: Clinical and cognitive criteria for AD are met and there is documented progressive cognitive decline or abnormal biomarker(s) suggestive of AD or evidence of proven AD autosomal dominant genetic mutation (presenilin-1, presenilin-2, amyloid-β precursor protein) |
| Definite AD: Clinical criteria for probable AD are met and there is histopathologic evidence of the disorder |
| Revised clinical diagnostic criteria for DLB (2005)5 |
| Core features: Fluctuating cognition, recurrent visual hallucinations, soft motor features of parkinsonism |
| Suggestive features: REM sleep behavior disorder, severe antipsychotic sensitivity, decreased tracer uptake in striatum on SPECT dopamine transporter imaging or on myocardial scintigraphy with MIBG |
| Supportive features (common but lacking diagnostic specificity): repeated falls and syncope; transient, unexplained loss of consciousness; systematized delusions; hallucinations other than visual; relative preservation of medial temporal lobe on CT or MRI scan; decreased tracer uptake on SPECT or PET imaging in occipital regions; prominent slow waves on EEG with temporal lobe transient sharp waves |
| AD: Alzheimer’s disease; DLB: dementia with Lewy bodies; MIBG: metaiodobenzylguanidine; NIA-AA: National Institute on Aging and the Alzheimer’s Association; PET: positron emission tomography; REM: rapid eye movement; SPECT: single photon emission computed tomography |
Biomarkers for AD, but not DLB
The 2011 diagnostic criteria for AD incorporate biomarkers that can be measured in vivo and reflect speci?c features of disease-related pathophysiologic processes. Biomarkers for AD are divided into 2 categories:11
- amyloid-beta (Aβ) accumulation: abnormal tracer retention on amyloid positron emission topography (PET) imaging and low cerebrospinal fluid (CSF) Aβ42
- neuronal degeneration or injury: elevated CSF tau (total and phosphorylated tau), decreased ?uorodeoxyglucose uptake on PET in temporo-parietal cortices, and atrophy on structural MRI in the hippocampal and temporo-parietal regions.
No clinically applicable genotypic or CSF markers exist to support a DLB diagnosis, but there are many promising candidates, including elevated levels of CSF p-tau 181, CSF levels of alpha- and beta-synuclein,12 and CSF beta-glucocerebrosidase levels.13 PET mapping of brain acetylcholinesterase activity,14 123I-2β-carbomethoxy-3β- (4-iodophenyl)-N-(3-fluoropropyl)nortropane single photon emission computed tomography (SPECT) dopamine transporter (DaT) imaging15 and metaiodobenzylguanidine (MIBG) scintigraphy also are promising methods. DaT scan SPECT is FDA-approved for detecting loss of functional dopaminergic neuron terminals in the striatum and can differentiate between AD and DLB with a sensitivity and specificity of 78% to 88% and 94% to 100%, respectively.16 This test is covered by Medicare for differentiating AD and DLB.
Differences in presentation
Cognitive impairment. Contrary to the early memory impairment that characterizes AD, memory deficits in DLB usually appear later in the disease course.5 Patients with DLB manifest greater attentional, visuospatial, and executive impairments than those with AD, whereas AD causes more profound episodic (declarative) memory impairment than DLB. DLB patients show more preserved consolidation and storage of verbal information than AD patients because of less neuroanatomical and cholinergic compromise in the medial temporal lobe. There is no evidence of significant differences in remote memory, semantic memory, and language (naming and fluency).
Compromised attention in DLB may be the basis for fluctuating cognition, a characteristic of the disease. The greater attentional impairment and reaction time variability in DLB compared with AD is evident during complex tasks for attention and may be a function of the executive and visuospatial demands of the tasks.17
Executive functions critical to adaptive, goal-directed behavior are more impaired in DLB than AD. DLB patients are more susceptible to distraction and have difficulty engaging in a task and shifting from 1 task to another. This, together with a tendency for confabulation and perseveration, are signs of executive dysfunction.
Neuropsychiatric features. DLB patients are more likely than AD patients to exhibit psychiatric symptoms and have more functional impairment.18 In an analysis of autopsy-confirmed cases, hallucinations and delusions were more frequent with Lewy body pathology (75%) than AD (21%) at initial clinical evaluation.18 By the end stages of both illnesses, the degree of psychotic symptoms is comparable.19 Depression is common in DLB; whether base rates of depressed mood and major depression differ between DLB and AD is uncertain.20
Psychosis in AD can be induced by medication or delirium, or triggered by poor sensory perceptions. Psychotic symptoms occur more frequently during the moderate and advanced stages of AD, when patients present with visual hallucinations, delusions, or delusional misidentifications. As many as 10% to 20% of patients with AD experience hallucinations, typically visual. Delusions occur in 30% to 50% of AD patients, usually in the later stages of the disease. The most common delusional themes are infidelity, theft, and paranoia. Female sex is a risk factor for psychosis in AD. Delusions co-occur with aggression, anxiety, and aberrant motor behavior.
Visual hallucinations—mostly vivid, well-formed, false perceptions of insects, animals, or people—are the defining feature of DLB.21 Many patients recognize that they are experiencing visual hallucinations and can ignore them. DLB patients also may experience visual illusions, such as misperceiving household objects as living beings. Delusions—typically paranoid—are common among DLB patients, as are depression and anxiety.1 Agitation or aggressive behavior tends to occur late in the illness, if at all.
The causes of psychotic symptoms in DLB are not fully understood, but dopamine dysfunction likely is involved in hallucinations, delusions, and agitation, and serotonin dysfunction may be associated with depression and anxiety. Rapid eye movement (REM) sleep/wakefulness dysregulation, in which the dream imagery of REM sleep may occur during wakefulness, also has been proposed as a mechanism for visual hallucinations in DLB.22 In DLB, psychotic symptoms occur early and are a hallmark of this illness, whereas in AD they usually occur in the middle to late stages of the disease.
Motor symptoms. In AD, extrapyramidal symptoms (EPS) are common later in the disease, are strongly correlated with disease severity, and are a strong, independent predictor of depression severity.23 EPS are more common in DLB than in AD24 and DLB patients are at higher risk of developing EPS even with low doses of typical antipsychotics, compared with AD patients.25
Other symptoms. REM sleep behavior disorder (RBD) is characterized by enacting dreams—often violent—during REM sleep. RBD is common in DLB and many patients also have excessive daytime somnolence. Other sleep disorders in DLB include insomnia, obstructive sleep apnea, central sleep apnea, restless legs syndrome, and periodic limb movements during sleep.
In AD patients, common sleep behaviors include confusion in the early evening (“sundowning”) and frequent nighttime awakenings, often accompanied by wandering.26 Orthostatic hypotension, impotence, urinary incontinence, and constipation are common in DLB. Lack of insight concerning personal cognitive, mood, and behavioral state is highly prevalent in AD patients and more common than in DLB.
Diagnostic evaluation
Because there are no definitive clinical markers for DLB, diagnosis is based on a detailed clinical and family history from the patient and a reliable informant, as well as a physical, neurologic, and mental status examination that looks for associated noncognitive symptoms, and neuropsychological evaluation. Reasons DLB may be misdiagnosed include:
- Some “core” clinical features of DLB may not appear or may overlap with AD.
- Presence and severity of concurrent AD pathology in DLB may modify the clinical presentation, with decreased rates of hallucinations and parkinsonism and increased neurofibrillary tangles.
- Failure to reliably identify fluctuations—variations in cognition and arousal, such as periods of unresponsiveness while awake (“zoning out”), excessive daytime somnolence, and disorganized speech.27
Detecting and characterizing cognitive deficits in dementia patients using neuropsychological testing is important in establishing a clinical diagnosis, determining baseline levels of impairment, forming a prognosis, and initiating disease-specific treatments. Differences in neuropsychological findings in AD and DLB are outlined in Table 2.16,28-33 Several studies have suggested using these measures to differentiate patients with DLB from those with AD.20
Table 2
Diagnostic testing for Alzheimer’s disease and dementia with Lewy bodies
| Alzheimer’s disease | Dementia with Lewy bodies |
|---|---|
| Neuropsychological testing findings | |
| Relatively more impairment on verbal memory tasks, delayed recall, delayed recognition, and encoding and storing information.28 Dysfunction of episodic memory function | Relatively more impairment on attention or concentration, verbal fluency, visuoperceptual, visuoconstructive, visual memory tests, and frontal executive functions.28 Relatively preserved confrontation naming and verbal memory |
| MRI findings | |
| Diffuse cortical atrophy, relatively greater volume loss in hippocampus and medial temporal lobe structures (strong correlation with severity)29 | Mild generalized cerebral cortical atrophy with minimal hippocampal atrophy and relative preservation of medial temporal lobe structures30 |
| [18F]FDG PET | |
| Widespread metabolic deficits in neocortical association areas, with sparing of the basal ganglia, thalamus, cerebellum, primary sensory motor cortex, and visual cortex | Widespread cortical hypometabolism, more marked in primary visual and occipital association areas, and less severe in parietal, frontal, and anterior cingulate cortices.31 Severe cholinergic deafferentation of the neocortex, particularly in posterior cortical regions32 |
| Single photon emission computed tomography | |
| Parietotemporal hypoperfusion | Occipital hypoperfusion |
| 123I-FP-CIT SPECT (DaT scan) | |
| No significant loss of DaT | Low nigrostriatal terminal density of DaT caused by severe nigrostriatal degeneration16 |
| Myocardial scintigraphy with MIBG | |
| No significant change in MIBG uptake | Decreased MIBG uptake33 |
| 123I-FP-CIT: 123I-2β-carbomethoxy-3β-(4-iodophenyl)-N-(3-fluoropropyl)nortropane; DaT: dopamine transporter; FDG PET: [18F]-fluoro-d-glucose positron emission tomography; MIBG: metaiodobenzylguanidine; SPECT: single photon emission computed tomography | |
Evidence is insufficient to support using electroencephalographic and polysomnographic studies when initially evaluating patients with dementia. Brain CT or MRI are recommended as part of the initial evaluation of dementia patients to exclude treatable causes of dementia and help clarify the differential diagnosis. Occipital hypometabolism and hypoperfusion demonstrated on PET and SPECT imaging have high sensitivity and specificity for differentiating AD from DLB.
To diagnose DLB more consistently, look for core features of the disease, RBD, antipsychotic hypersensitivity, and decreased striatal binding at presynaptic DaT sites.15 Abnormal (low binding) DaT activity is the most reliable diagnostic marker for DLB.34 Myocardial scintigraphy with MIBG is sensitive to pathologic changes of DLB before clinical expression and could overcome the difficulties of using clinical criteria alone to identify patients with DLB.35 MIBG scintigraphy may be preferred to DaT scan because it is less expensive and its sensitivity and specificity to DLB are independent of the presence of parkinsonism.35
For an overview of pharmacotherapy options for patients with AD or DLB, see Box 2.
Pharmacotherapy options for patients with Alzheimer’s disease (AD) or dementia with Lewy bodies (DLB) include cholinesterase inhibitors, memantine, antipsychotics, and other agents.
Cholinesterase inhibitors. Donepezil, rivastigmine, and galantamine are FDA-approved for treating AD. Their efficacy appears to be similar, so the choice of agent is based largely on cost, patient tolerability, and physician experience.
No medications are FDA-approved for treating DLB. Neocortical cholinergic activity assessed by choline acetyltransferase levels is more severely depleted in DLB than in AD, and this deficit is correlated with the presence of visual hallucinations and global severity of cognitive impairment.a Therefore, drugs that enhance central cholinergic function offer a therapeutic approach for DLB; cognitive and hallucinatory symptoms are the anticipated targets. Multiple anecdotal reports, open-label studies,b,c and 1 randomized, placebo-controlled triald suggest that cholinesterase inhibitors are efficacious in DLB, with reported benefits in cognition, fluctuations, psychotic symptoms, and parkinsonian symptoms. A 20-week randomized, double-blind, placebo-controlled multicenter studyd of patients with DLB found rivastigmine, 6 to 12 mg/d, was superior to placebo. Patients receiving rivastigmine exhibited significantly reduced anxiety, delusions, and hallucinations and significantly better performance on a computerized battery of neuropsychological tests, especially tasks that required sustained attention. Differences between rivastigmine and placebo disappeared after drug discontinuation.
Memantine is a noncompetitive antagonist of N-methyl-d-aspartate receptors that is effective in AD.e The possible involvement of glutamate in DLB has provided a rationale for treating DLB with memantine. Two randomized controlled trials in DLB found that patients treated with memantine for 24 weeks performed better on Clinical Global Impression of Change, but not on most other secondary outcome measures.f,g In both studies, memantine was well tolerated. However, other studies have noted worsening of delusions and hallucinations with memantine in DLB patients.h
Antipsychotics. Agitation is common in moderate and advanced AD. Atypical antipsychotics have been used with variable efficacy to treat agitation, but their use is associated with excess mortality. DLB patients pose a considerable therapeutic challenge because antipsychotics—the mainstay of treatment of psychosis and behavioral problems in most other disorders—can provoke severe, irreversible, and often fatal sensitivity reactions in this type of dementia.i A 2- to 3-fold increased mortality risk associated with antipsychotic sensitivity reactions in DLB is partly mediated via acute blockade of postsynaptic dopamine D2 receptors in the striatum. For severe and disabling psychosis, a trial of a cholinesterase inhibitor and/or lowering the dose of antiparkinsonian medication should be considered first. In urgent situations, small doses of an atypical antipsychotic that is least associated with parkinsonism side effects—such as quetiapine or aripiprazole—should be used.
Other treatments. Treatment of parkinsonian symptoms in DLB patients is similar to that for Parkinson’s disease, but the risk of psychotic symptoms in DLB warrants a conservative approach. Levodopa seems to be more effective than dopamine agonists and produces fewer side effects.j Rapid eye movement sleep behavior disorder often responds to low doses of clonazepam (0.25 to 1.5 mg). Depression and anxiety disorders are common in AD at all stages and their treatment is not fundamentally different than in geriatric patients without dementia. Selective serotonin reuptake inhibitors and electroconvulsive therapy have been used successfully in depressed patients with AD or DLB.k,l
Disease-modifying treatments. Researchers are evaluating an array of antiamyloid and neuroprotective therapeutic approaches for AD based on the hypothesis that amyloid-beta protein plays a pivotal role in disease onset and progression. Interventions that reduce amyloid production, limit aggregation, or increase clearance may block the cascade of events comprising AD pathogenesis. Reducing tau hyperphosphorylation, limiting oxidation and excitotoxicity, and controlling inflammation also may be beneficial strategies. Potentially neuroprotective and restorative treatments such as neurotrophins, neurotrophic factor enhancers, and stem cell-related approaches also are being investigated.
There are no large-scale studies of disease-modifying treatments for DLB. Potential areas of research include the relationship between proteasome function and a-synuclein pathology, the role of beta-synuclein, and the impact of alterations to alpha-synuclein on its propensity to aggregate.
References
a. Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931-1934.
b. Beversdorf DQ, Warner JL, Davis RA, et al. Donepezil in the treatment of dementia with lewy bodies. Am J Geriatr Psychiatry. 2004;12(5):542-544.
c. Edwards K, Royall D, Hershey L, et al. Efficacy and safety of galantamine in patients with dementia with Lewy bodies: a 24-week open-label study. Dement Geriatr Cogn Disord. 2007;23(6):401-405.
d. McKeith I, Del Ser T, Spano P, et al. Efficacy of rivastigmine in dementia with Lewy bodies: a randomised, double-blind, placebo-controlled international study. Lancet. 2000;356(9247):2031-2036.
e. Tariot PN, Farlow MR, Grossberg GT, et al. Memantine treatment in patients with moderate to severe Alzheimer disease already receiving donepezil: a randomized controlled trial. JAMA. 2004;291(3):317-324.
f. Aarsland D, Ballard C, Walker Z, et al. Memantine in patients with Parkinson’s disease dementia or dementia with Lewy bodies: a double-blind, placebo-controlled, multicentre trial. Lancet Neurol. 2009;8(7):613-618.
g. Emre M, Tsolaki M, Bonuccelli U, et al. Memantine for patients with Parkinson’s disease dementia or dementia with Lewy bodies: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2010;9(10):969-977.
h. Ridha BH, Josephs KA, Rossor MN. Delusions and hallucinations in dementia with Lewy bodies: worsening with memantine. Neurology. 2005;65(3):481-482.
i. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
j. Fernandez HH, Wu CK, Ott BR. Pharmacotherapy of dementia with Lewy bodies. Expert Opin Pharmacother. 2003;4(11):2027-2037.
k. Swartz M, Barak Y, Mirecki I, et al. Treating depression in Alzheimer’s disease: integration of differing guidelines. Int Psychogeriatr. 2000;12(3):353-358.
l. Takahashi S, Mizukami K, Yasuno F, et al. Depression associated with dementia with Lewy bodies (DLB) and the effect of somatotherapy. Psychogeriatrics. 2009;9(2):56-61.
Related Resources
- Hanyu H, Sato T, Hirao K, et al. Differences in clinical course between dementia with Lewy bodies and Alzheimer’s disease. Eur J Neurol. 2009;16(2):212-217.
- Walker Z, McKeith I, Rodda J, et al. Comparison of cognitive decline between dementia with Lewy bodies and Alzheimer’s disease: a cohort study. BMJ Open. 2012;2:e000380.
Drug Brand Names
- Aripiprazole • Abilify
- Clonazepam • Klonopin
- Donepezil • Aricept
- Galantamine • Razadyne, Reminyl
- Levodopa • Dopar, Larodopa
- Memantine • Namenda
- Quetiapine • Seroquel
- Rivastigmine • Exelon
Disclosure
Drs. Bishnoi and Manepalli report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Grossberg serves as a consultant to Forest, Janssen, Novartis, and Pfizer. His department receives research funding from Novartis, Janssen, and Pfizer.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
1. McKeith IG, Galasko D, Kosaka K, et al. Consensus guidelines for the clinical and pathologic diagnosis of dementia with Lewy bodies (DLB): report of the consortium on DLB international workshop. Neurology. 1996;47(5):1113-1124.
2. Buracchio T, Arvanitakis Z, Gorbien M. Dementia with Lewy bodies: current concepts. Dement Geriatr Cogn Disord. 2005;20(5):306-320.
3. Fujishiro H, Iseki E, Higashi S, et al. Distribution of cerebral amyloid deposition and its relevance to clinical phenotype in Lewy body dementia. Neurosci Lett. 2010;486(1):19-23.
4. Kosaka K. Diffuse Lewy body disease. Neuropathology. 2000;20(suppl):S73-S78.
5. McKeith IG, Dickson DW, Lowe J, et al. Consortium on DLB. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology. 2005;65(12):1863-1872.
6. Cummings JL, Cole G. Alzheimer disease. JAMA. 2002;287(18):2335-2338.
7. Zaccai J, McCracken C, Brayne C. A systematic review of prevalence and incidence studies of dementia with Lewy bodies. Age Ageing. 2005;34(6):561-566.
8. Bradshaw J, Saling M, Hopwood M, et al. Fluctuating cognition in dementia with Lewy bodies and Alzheimer’s disease is qualitatively distinct. J Neurol Neurosurg Psychiatry. 2004;75(3):382-387.
9. Singleton AB, Wharton A, O’Brien KK, et al. Clinical and neuropathological correlates of apolipoprotein E genotype in dementia with Lewy bodies. Dement Geriatr Cogn Disord. 2002;14(4):167-175.
10. McKhann GM, Knopman DS, Chertkow H, et al. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263-269.
11. Jack CR, Jr, Albert MS, Knopman DS, et al. Introduction to the recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):257-262.
12. Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF alpha-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Exp Neurol. 2008;213(2):315-325.
13. Parnetti L, Balducci C, Pierguidi L, et al. Cerebrospinal fluid beta-glucocerebrosidase activity is reduced in dementia with Lewy bodies. Neurobiol Dis. 2009;34(3):484-486.
14. Shimada H, Hirano S, Shinotoh H, et al. Mapping of brain acetylcholinesterase alterations in Lewy body disease by PET. Neurology. 2009;73(4):273-278.
15. McKeith I, O’Brien J, Walker Z, et al. Sensitivity and specificity of dopamine transporter imaging with 123I-FP-CIT SPECT in dementia with Lewy bodies: a phase III, multicentre study. Lancet Neurol. 2007;6(4):305-313.
16. Walker Z, Jaros E, Walker RW, et al. Dementia with Lewy bodies: a comparison of clinical diagnosis, FP-CIT single photon emission computed tomography imaging and autopsy. J Neurol Neurosurg Psychiatry. 2007;78(11):1176-1181.
17. Bradshaw JM, Saling M, Anderson V, et al. Higher cortical deficits influence attentional processing in dementia with Lewy bodies, relative to patients with dementia of the Alzheimer’s type and controls. J Neurol Neurosurg Psychiatry. 2006;77(10):1129-1135.
18. Weiner MF, Hynan LS, Parikh B, et al. Can Alzheimer’s disease and dementias with Lewy bodies be distinguished clinically? J Geriatr Psychiatry Neurol. 2003;16(4):245-250.
19. Stavitsky K, Brickman AM, Scarmeas N, et al. The progression of cognition, psychiatric symptoms, and functional abilities in dementia with Lewy bodies and Alzheimer disease. Arch Neurol. 2006;63(10):1450-1456.
20. Ferman TJ, Smith GE, Boeve BF, et al. Neuropsychological differentiation of dementia with Lewy bodies from normal aging and Alzheimer’s disease. Clin Neuropsychol. 2006;20(4):623-636.
21. McKeith IG, Perry EK, Perry RH. Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology. 1999;53(5):902-905.
22. Boeve BF, Silber MH, Ferman TJ, et al. Association of REM sleep behavior disorder and neurodegenerative disease may reflect an underlying synucleinopathy. Mov Disord. 2001;16(4):622-630.
23. Portet F, Scarmeas N, Cosentino S, et al. Extrapyramidal signs before and after diagnosis of incident Alzheimer disease in a prospective population study. Arch Neurol. 2009;66(9):1120-1126.
24. McKeith I, Fairbairn A, Perry R, et al. Neuroleptic sensitivity in patients with senile dementia of Lewy body type. BMJ. 1992;305(6855):673-678.
25. Tarawneh R, Galvin JE. Distinguishing Lewy body dementias from Alzheimer’s disease. Expert Rev Neurother. 2007;7(11):1499-1516.
26. Ancoli-Israel S, Klauber MR, Gillin JC, et al. Sleep in non-institutionalized Alzheimer’s disease patients. Aging (Milano). 1994;6(6):451-458.
27. Ferman TJ, Smith GE, Boeve BF, et al. DLB fluctuations: specific features that reliably differentiate DLB from AD and normal aging. Neurology. 2004;62(2):181-187.
28. Salmon DP, Galasko D, Hansen LA, et al. Neuropsychological deficits associated with diffuse Lewy body disease. Brain Cogn. 1996;31(2):148-165.
29. Jack CR, Jr, Petersen RC, Xu Y, et al. Rates of hippocampal atrophy correlate with change in clinical status in aging and AD. Neurology. 2000;55(4):484-489.
30. Burton EJ, Barber R, Mukaetova-Ladinska EB, et al. Medial temporal lobe atrophy on MRI differentiates Alzheimer’s disease from dementia with Lewy bodies and vascular cognitive impairment: a prospective study with pathological verification of diagnosis. Brain. 2009;132(pt 1):195-203.
31. Ishii K, Soma T, Kono AK, et al. Comparison of regional brain volume and glucose metabolism between patients with mild dementia with lewy bodies and those with mild Alzheimer’s disease. J Nucl Med. 2007;48(5):704-711.
32. Klein JC, Eggers C, Kalbe E, et al. Neurotransmitter changes in dementia with Lewy bodies and Parkinson disease dementia in vivo. Neurology. 2010;74(11):885-892.
33. Fujishiro H, Nakamura S, Kitazawa M, et al. Early detection of dementia with Lewy bodies in patients with amnestic mild cognitive impairment using 123I-MIBG cardiac scintigraphy. J Neurol Sci. 2012;315(1-2):115-119.
34. O’Brien JT, McKeith IG, Walker Z, et al. Diagnostic accuracy of 123I-FP-CIT SPECT in possible dementia with Lewy bodies. Br J Psychiatry. 2009;194:34-39.
35. Yoshita M, Taki J, Yokoyama K, et al. Value of 123I-MIBG radioactivity in the differential diagnosis of DLB from AD. Neurology. 2006;66(12):1850-1854.
Why do cancer patients smoke and what can providers do about it?
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Despite the widespread dissemination of information about the health risks associated with smoking, many cancer patients continue to smoke, which results in a decreased quality of life, an increased probability of cancer recurrence, and a decreased survival time. Efficacious interventions are available to assist cancer patients to quit smoking, yet smoking cessation interventions are often not implemented. This review describes how clinicians, administrators, insurers, and purchasers can encourage a culture of health care in which tobacco cessation interventions are implemented consistent with evidence-based standards of care. Implementing efficacious tobacco cessation interventions can reduce morbidity and mortality among cancer patients...
*Click on the link to the left of this introduction for a PDF of the full article.
Dasatinib in the first-line treatment of chronic myeloid leukemia
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Dasatinib has been approved for first-line treatment of chronic-phase chronic myeloid leukemia by the Food and Drug Administration and is recommended as a first-line treatment option by the National Comprehensive Cancer Network. Based on in vitro data, dasatinib seems to be less susceptible to the resistance mechanisms that affect imatinib. Dasatinib is an effective second-line treatment in patients who are resistant to imatinib. First-line clinical data show that dasatinib provides more rapid and deeper degrees of response than does imatinib, which may correlate with improvements in long-term patient outcome. Grade 1 or 2 cytopenias are the most common adverse events of first-line dasatinib treatment. In a phase 3 comparison with imatinib, several types of nonhematologic adverse events were less frequent in the dasatinib arm; frequencies of grade 3 and 4 events were 2%. Among patients with a minimum follow-up of 24 months, grade 1 or 2 pleural effusion was reported in 14% of dasatinib-treated patients and was manageable in almost all cases; no grade 3 or 4 pleural effusion occurred. Prompt and effective monitoring and management of dasatinib toxicities is essential to minimize intolerance and nonadherence to therapy. Patient education is important to increase the likelihood of prompt management and provide reassurance. Recommendations for patient monitoring, management, and education are provided.
*For a PDF of the full article, click on the link to the left of this introduction.
Hospitalists Urged to Watch for Fungal Meningitis Cases in Midst of National Outbreak
A national outbreak of fungal meningitis tied to contaminated steroids in epidural injections should make hospitalists vigilant with patients who present potential symptoms, says an infectious-disease (ID) specialist.
Earlier this month, health officials linked the outbreak to tainted batches of steroids used in spinal injections, and they say it could be weeks, or even months, before they know whether the incubation period for the disease is over, according to The New York Times.
"The key in my mind is that hospitalists ought to have a high index of suspicion for this right now," says hospitalist and ID expert James Pile, MD, FACP, SFHM, of the Cleveland Clinic. "If you encounter a patient you think may have meningitis, may have a brain stem stroke, may have an epidural abscess or vertebral osteomyelitis...at least think and ask the patient, or their family member, 'Did you receive an epidural steroid injection recently?'"
The answer to that question will help determine the best care delivery for hospitalists, and physicians should not rely on patients to relay the information without being asked for it, Dr. Pile says.
The outbreak has been traced to three contaminated batches of methylprednisolone produced by the New England Compounding Center in Framingham, Mass. The company, which is under criminal investigation, has been linked to at least 25 deaths and more than 317 infected patients. Although 14,000 people might have been injected with the contaminated compound, CDC officials say the likelihood of infection remains relatively low.
Dr. Pile says that while hospitalists might see only a handful of fungal meningitis cases in their careers, they still need to keep the possibility in mind when examining patients. It's a safe approach to take, particularly as the CDC continues to investigate the extent of the outbreak. The CDC has advised against antifungal prophylaxis or presumptive treatment of exposed asymptomatic patients without a diagnosed case of meningitis.
"This is just unfolding so quickly, it's a moving target," Dr. Pile adds. "How big it ends up being and what kinds of new or unusual manifestations present remain to be seen."
Visit our website for more information about infectious disease and hospital medicine.
A national outbreak of fungal meningitis tied to contaminated steroids in epidural injections should make hospitalists vigilant with patients who present potential symptoms, says an infectious-disease (ID) specialist.
Earlier this month, health officials linked the outbreak to tainted batches of steroids used in spinal injections, and they say it could be weeks, or even months, before they know whether the incubation period for the disease is over, according to The New York Times.
"The key in my mind is that hospitalists ought to have a high index of suspicion for this right now," says hospitalist and ID expert James Pile, MD, FACP, SFHM, of the Cleveland Clinic. "If you encounter a patient you think may have meningitis, may have a brain stem stroke, may have an epidural abscess or vertebral osteomyelitis...at least think and ask the patient, or their family member, 'Did you receive an epidural steroid injection recently?'"
The answer to that question will help determine the best care delivery for hospitalists, and physicians should not rely on patients to relay the information without being asked for it, Dr. Pile says.
The outbreak has been traced to three contaminated batches of methylprednisolone produced by the New England Compounding Center in Framingham, Mass. The company, which is under criminal investigation, has been linked to at least 25 deaths and more than 317 infected patients. Although 14,000 people might have been injected with the contaminated compound, CDC officials say the likelihood of infection remains relatively low.
Dr. Pile says that while hospitalists might see only a handful of fungal meningitis cases in their careers, they still need to keep the possibility in mind when examining patients. It's a safe approach to take, particularly as the CDC continues to investigate the extent of the outbreak. The CDC has advised against antifungal prophylaxis or presumptive treatment of exposed asymptomatic patients without a diagnosed case of meningitis.
"This is just unfolding so quickly, it's a moving target," Dr. Pile adds. "How big it ends up being and what kinds of new or unusual manifestations present remain to be seen."
Visit our website for more information about infectious disease and hospital medicine.
A national outbreak of fungal meningitis tied to contaminated steroids in epidural injections should make hospitalists vigilant with patients who present potential symptoms, says an infectious-disease (ID) specialist.
Earlier this month, health officials linked the outbreak to tainted batches of steroids used in spinal injections, and they say it could be weeks, or even months, before they know whether the incubation period for the disease is over, according to The New York Times.
"The key in my mind is that hospitalists ought to have a high index of suspicion for this right now," says hospitalist and ID expert James Pile, MD, FACP, SFHM, of the Cleveland Clinic. "If you encounter a patient you think may have meningitis, may have a brain stem stroke, may have an epidural abscess or vertebral osteomyelitis...at least think and ask the patient, or their family member, 'Did you receive an epidural steroid injection recently?'"
The answer to that question will help determine the best care delivery for hospitalists, and physicians should not rely on patients to relay the information without being asked for it, Dr. Pile says.
The outbreak has been traced to three contaminated batches of methylprednisolone produced by the New England Compounding Center in Framingham, Mass. The company, which is under criminal investigation, has been linked to at least 25 deaths and more than 317 infected patients. Although 14,000 people might have been injected with the contaminated compound, CDC officials say the likelihood of infection remains relatively low.
Dr. Pile says that while hospitalists might see only a handful of fungal meningitis cases in their careers, they still need to keep the possibility in mind when examining patients. It's a safe approach to take, particularly as the CDC continues to investigate the extent of the outbreak. The CDC has advised against antifungal prophylaxis or presumptive treatment of exposed asymptomatic patients without a diagnosed case of meningitis.
"This is just unfolding so quickly, it's a moving target," Dr. Pile adds. "How big it ends up being and what kinds of new or unusual manifestations present remain to be seen."
Visit our website for more information about infectious disease and hospital medicine.
Guidelines Help Slash CLABSI Rate by 40% in the ICU
The largest effort to date to tackle central-line-associated bloodstream infections (CLABSIs) has reduced infection rates in ICUs nationwide by 40%, according to preliminary findings from the federal Agency for Healthcare Research and Quality (AHRQ).
AHRQ attributes the decrease to a CLABSI safety checklist from the Comprehensive Unit-Based Safety Program (CUSP) that encourages hospital staff to wash their hands prior to inserting central lines, avoid the femoral site, remove lines when they are no longer needed, and use the antimicrobial agent chlorhexidine to clean the patient's insertion site.
The checklist was developed by Peter Pronovost, MD, PhD, FCCM, and colleagues at Johns Hopkins University in Baltimore, and originally implemented in ICUs statewide in Michigan as the Keystone Project. Since 2009, CUSP has recruited more than 1,000 participating hospitals in 44 states. CUSP collectively reported a decrease to 1.25 from 1.87 CLABSIs per 1,000 central-line days 10-12 months after implementing the program, according to AHRQ [PDF].
The real game-changer for CLABSIs has been the widespread adoption of chlorhexidine as an insertion site disinfectant, says Sanjay Saint, MD, MPH, director of the Veterans Administration at the University of Michigan Patient Safety Enhancement Program in Ann Arbor and professor of medicine at the University of Michigan. Dr. Saint is on the national leadership team of On the CUSP: Stop CAUTI (Catheter-Associated Urinary Tract Infections), an initiative that aims to reduce mean rates of CAUTI infections by 25% in hospitals nationwide.
Although hospitalists don't routinely place central lines, their role in this procedure is growing, both in nonacademic hospitals that lack intensivists and on hospitals' general medicine floors.
"My take-home message for hospitalists: if you are putting in central lines, if you only make one change in practice, is to use chlorhexidine as the site disinfectant," Dr. Saint says.
Visit our website for more information about central-line-associated bloodstream infections.
The largest effort to date to tackle central-line-associated bloodstream infections (CLABSIs) has reduced infection rates in ICUs nationwide by 40%, according to preliminary findings from the federal Agency for Healthcare Research and Quality (AHRQ).
AHRQ attributes the decrease to a CLABSI safety checklist from the Comprehensive Unit-Based Safety Program (CUSP) that encourages hospital staff to wash their hands prior to inserting central lines, avoid the femoral site, remove lines when they are no longer needed, and use the antimicrobial agent chlorhexidine to clean the patient's insertion site.
The checklist was developed by Peter Pronovost, MD, PhD, FCCM, and colleagues at Johns Hopkins University in Baltimore, and originally implemented in ICUs statewide in Michigan as the Keystone Project. Since 2009, CUSP has recruited more than 1,000 participating hospitals in 44 states. CUSP collectively reported a decrease to 1.25 from 1.87 CLABSIs per 1,000 central-line days 10-12 months after implementing the program, according to AHRQ [PDF].
The real game-changer for CLABSIs has been the widespread adoption of chlorhexidine as an insertion site disinfectant, says Sanjay Saint, MD, MPH, director of the Veterans Administration at the University of Michigan Patient Safety Enhancement Program in Ann Arbor and professor of medicine at the University of Michigan. Dr. Saint is on the national leadership team of On the CUSP: Stop CAUTI (Catheter-Associated Urinary Tract Infections), an initiative that aims to reduce mean rates of CAUTI infections by 25% in hospitals nationwide.
Although hospitalists don't routinely place central lines, their role in this procedure is growing, both in nonacademic hospitals that lack intensivists and on hospitals' general medicine floors.
"My take-home message for hospitalists: if you are putting in central lines, if you only make one change in practice, is to use chlorhexidine as the site disinfectant," Dr. Saint says.
Visit our website for more information about central-line-associated bloodstream infections.
The largest effort to date to tackle central-line-associated bloodstream infections (CLABSIs) has reduced infection rates in ICUs nationwide by 40%, according to preliminary findings from the federal Agency for Healthcare Research and Quality (AHRQ).
AHRQ attributes the decrease to a CLABSI safety checklist from the Comprehensive Unit-Based Safety Program (CUSP) that encourages hospital staff to wash their hands prior to inserting central lines, avoid the femoral site, remove lines when they are no longer needed, and use the antimicrobial agent chlorhexidine to clean the patient's insertion site.
The checklist was developed by Peter Pronovost, MD, PhD, FCCM, and colleagues at Johns Hopkins University in Baltimore, and originally implemented in ICUs statewide in Michigan as the Keystone Project. Since 2009, CUSP has recruited more than 1,000 participating hospitals in 44 states. CUSP collectively reported a decrease to 1.25 from 1.87 CLABSIs per 1,000 central-line days 10-12 months after implementing the program, according to AHRQ [PDF].
The real game-changer for CLABSIs has been the widespread adoption of chlorhexidine as an insertion site disinfectant, says Sanjay Saint, MD, MPH, director of the Veterans Administration at the University of Michigan Patient Safety Enhancement Program in Ann Arbor and professor of medicine at the University of Michigan. Dr. Saint is on the national leadership team of On the CUSP: Stop CAUTI (Catheter-Associated Urinary Tract Infections), an initiative that aims to reduce mean rates of CAUTI infections by 25% in hospitals nationwide.
Although hospitalists don't routinely place central lines, their role in this procedure is growing, both in nonacademic hospitals that lack intensivists and on hospitals' general medicine floors.
"My take-home message for hospitalists: if you are putting in central lines, if you only make one change in practice, is to use chlorhexidine as the site disinfectant," Dr. Saint says.
Visit our website for more information about central-line-associated bloodstream infections.
Early Surgery Yields Survival Benefit for Low-Grade Gliomas
Adults in Norway with diffuse low-grade gliomas who were treated at a hospital advocating early surgical resection had better overall survival than those treated at a hospital advocating "watchful waiting," according to a report published online Oct. 30 in JAMA.
This finding significantly strengthens the sparse evidence in support of early resection for newly diagnosed diffuse low-grade gliomas, said Dr. Asgeir S. Jakola of the department of neurosurgery, St. Olav’s University Hospital, Trondheim (Norway) and his associates.
Management of these tumors is one of the major controversies in both neurology and oncology today, largely because the effect of surgery on survival is still unclear. The only evidence available until now was based solely on uncontrolled surgical series; some of these have reported that it is safe to withhold surgery until the lesions progress, while others have reported that immediate resection improves survival and delays the time to malignant transformation.
Both patients and physicians are reluctant to undertake immediate surgery when the evidence supporting that strategy has been so tenuous. They also are concerned that the risk of early and aggressive surgery outweighs the benefit, particularly when most patients are capable of normal activity and have a reasonably long life expectancy at diagnosis, the investigators said.
It is unlikely that a randomized, controlled study comparing the two approaches will ever be performed. Dr. Jakola and his colleagues therefore conducted a retrospective, population-based parallel-cohort study at two neurosurgical centers, each of which preferred one of these strategies over the other. Their "natural experiment" was possible because in Norway, there were two such facilities that were relatively close geographically and served a homogenous population. The nationalized health care system distributes training, resources, and personnel equally throughout the country, so the two hospitals were quite similar in other respects. And patient follow-up is 100%.
The 12-year study involved 153 adults with diffuse, histologically verified supratentorial grade I and II tumors diagnosed in 1998-2009, who were followed until death or until April 2011. The median follow-up was 7 years. Gliomas included astrocytomas, oligodendrogliomas, and oligoastrocytomas.
For patients with newly diagnosed low-grade gliomas, hospital A favored biopsy and watchful waiting. The 66 patients treated there typically were followed with MRI at 3 and 6 months, then yearly thereafter. They usually were offered surgical resection, if the lesions grew or showed signs of malignant transformation.
Hospital B favored immediate maximal safe tumor resection for the 87 patients treated there, with MRI follow-up at 6 and 12 months, then annually thereafter. This strategy was not pursued in some patients, however: notably, those who were elderly or had comorbidities and were likely to die from another cause before malignant transformation would take place, and those who had very widespread tumor infiltration that made resection impractical.
The two study groups were well balanced with regard to patient age and comorbidities, and rates of surgical rescue therapy were the same. There also were no differences between the two groups in complications or acquired neurologic deficits.
At the end of the study period, 34 patients (52%) from hospital A had died, compared with only 28 patients (32%) from hospital B. Median survival was 5.9 years at hospital A, but median survival had not yet been reached at hospital B, the researchers said (JAMA 2012;308: [doi:10.1001/jama.2012.12807]).
This survival advantage increased over time. Expected 3-year survival was 70% at hospital A vs. 80% at hospital B; expected 5-year survival was 60% at hospital A vs. 74% at hospital B; and expected 7-year survival was 44% at hospital A vs. 68% at hospital B.
In a post hoc analysis that attempted to account for differences between the two study groups in prognostic factors, the survival benefit for immediate resection remained robust. It also remained robust in another post hoc analysis that examined the subgroup of patients who had the most common glioma, a grade II astrocytoma. Median survival was 5.6 years at the hospital favoring watchful waiting, compared with 9.7 years at the hospital favoring early resection, in this large subgroup of patients.
Based on these findings, hospital A has changed its preferred strategy from watchful waiting to early resection, Dr. Jakola and his associates said.
"Despite the clear survival advantage seen, clinical judgment is still necessary in individual patients with suspected low-grade glioma since results will depend on patient and disease characteristics together with surgical results in terms of resection grades and complication rates," they added.
One of Dr. Jakola’s associates reported holding stock in Sonowand, manufacturer of the 3-D ultrasound-based imaging system used in one of the study hospitals.
This "natural experiment" may be the best source of evidence supporting early surgical resection that we’re likely to get, but the study by Dr. Jakola and his colleagues did have some limitations, said Dr. James M. Markert.
The confidence intervals around the point estimates for survival in both groups overlapped, which means the patients must be followed for a longer period to ensure that the confidence intervals eventually separate definitively. Also, one potentially important difference between the two study groups was not accounted for: the proportion of oligodendrogliomas, which are highly survivable, was higher at hospital B (19%) than at hospital A (9%).
In addition, radiation therapy was administered more often at the hospital favoring resection (43% of patients) than at the hospital favoring watchful waiting (29%), which may have affected survival rates. And although the authors reported no differences between the two groups in complications or neurologic deficits, "assessment methods were not delineated and the data were insufficient to reach a definitive conclusion," he noted.
Dr. Markert is in the division of neurosurgery at the University of Alabama at Birmingham. He reported ties to Catherex and Tocgen. These remarks were taken from his editorial accompanying Dr. Jakola’s report (JAMA 2012 Oct. 25 [doi:10.1001/jama.2012.14523]).
This "natural experiment" may be the best source of evidence supporting early surgical resection that we’re likely to get, but the study by Dr. Jakola and his colleagues did have some limitations, said Dr. James M. Markert.
The confidence intervals around the point estimates for survival in both groups overlapped, which means the patients must be followed for a longer period to ensure that the confidence intervals eventually separate definitively. Also, one potentially important difference between the two study groups was not accounted for: the proportion of oligodendrogliomas, which are highly survivable, was higher at hospital B (19%) than at hospital A (9%).
In addition, radiation therapy was administered more often at the hospital favoring resection (43% of patients) than at the hospital favoring watchful waiting (29%), which may have affected survival rates. And although the authors reported no differences between the two groups in complications or neurologic deficits, "assessment methods were not delineated and the data were insufficient to reach a definitive conclusion," he noted.
Dr. Markert is in the division of neurosurgery at the University of Alabama at Birmingham. He reported ties to Catherex and Tocgen. These remarks were taken from his editorial accompanying Dr. Jakola’s report (JAMA 2012 Oct. 25 [doi:10.1001/jama.2012.14523]).
This "natural experiment" may be the best source of evidence supporting early surgical resection that we’re likely to get, but the study by Dr. Jakola and his colleagues did have some limitations, said Dr. James M. Markert.
The confidence intervals around the point estimates for survival in both groups overlapped, which means the patients must be followed for a longer period to ensure that the confidence intervals eventually separate definitively. Also, one potentially important difference between the two study groups was not accounted for: the proportion of oligodendrogliomas, which are highly survivable, was higher at hospital B (19%) than at hospital A (9%).
In addition, radiation therapy was administered more often at the hospital favoring resection (43% of patients) than at the hospital favoring watchful waiting (29%), which may have affected survival rates. And although the authors reported no differences between the two groups in complications or neurologic deficits, "assessment methods were not delineated and the data were insufficient to reach a definitive conclusion," he noted.
Dr. Markert is in the division of neurosurgery at the University of Alabama at Birmingham. He reported ties to Catherex and Tocgen. These remarks were taken from his editorial accompanying Dr. Jakola’s report (JAMA 2012 Oct. 25 [doi:10.1001/jama.2012.14523]).
Adults in Norway with diffuse low-grade gliomas who were treated at a hospital advocating early surgical resection had better overall survival than those treated at a hospital advocating "watchful waiting," according to a report published online Oct. 30 in JAMA.
This finding significantly strengthens the sparse evidence in support of early resection for newly diagnosed diffuse low-grade gliomas, said Dr. Asgeir S. Jakola of the department of neurosurgery, St. Olav’s University Hospital, Trondheim (Norway) and his associates.
Management of these tumors is one of the major controversies in both neurology and oncology today, largely because the effect of surgery on survival is still unclear. The only evidence available until now was based solely on uncontrolled surgical series; some of these have reported that it is safe to withhold surgery until the lesions progress, while others have reported that immediate resection improves survival and delays the time to malignant transformation.
Both patients and physicians are reluctant to undertake immediate surgery when the evidence supporting that strategy has been so tenuous. They also are concerned that the risk of early and aggressive surgery outweighs the benefit, particularly when most patients are capable of normal activity and have a reasonably long life expectancy at diagnosis, the investigators said.
It is unlikely that a randomized, controlled study comparing the two approaches will ever be performed. Dr. Jakola and his colleagues therefore conducted a retrospective, population-based parallel-cohort study at two neurosurgical centers, each of which preferred one of these strategies over the other. Their "natural experiment" was possible because in Norway, there were two such facilities that were relatively close geographically and served a homogenous population. The nationalized health care system distributes training, resources, and personnel equally throughout the country, so the two hospitals were quite similar in other respects. And patient follow-up is 100%.
The 12-year study involved 153 adults with diffuse, histologically verified supratentorial grade I and II tumors diagnosed in 1998-2009, who were followed until death or until April 2011. The median follow-up was 7 years. Gliomas included astrocytomas, oligodendrogliomas, and oligoastrocytomas.
For patients with newly diagnosed low-grade gliomas, hospital A favored biopsy and watchful waiting. The 66 patients treated there typically were followed with MRI at 3 and 6 months, then yearly thereafter. They usually were offered surgical resection, if the lesions grew or showed signs of malignant transformation.
Hospital B favored immediate maximal safe tumor resection for the 87 patients treated there, with MRI follow-up at 6 and 12 months, then annually thereafter. This strategy was not pursued in some patients, however: notably, those who were elderly or had comorbidities and were likely to die from another cause before malignant transformation would take place, and those who had very widespread tumor infiltration that made resection impractical.
The two study groups were well balanced with regard to patient age and comorbidities, and rates of surgical rescue therapy were the same. There also were no differences between the two groups in complications or acquired neurologic deficits.
At the end of the study period, 34 patients (52%) from hospital A had died, compared with only 28 patients (32%) from hospital B. Median survival was 5.9 years at hospital A, but median survival had not yet been reached at hospital B, the researchers said (JAMA 2012;308: [doi:10.1001/jama.2012.12807]).
This survival advantage increased over time. Expected 3-year survival was 70% at hospital A vs. 80% at hospital B; expected 5-year survival was 60% at hospital A vs. 74% at hospital B; and expected 7-year survival was 44% at hospital A vs. 68% at hospital B.
In a post hoc analysis that attempted to account for differences between the two study groups in prognostic factors, the survival benefit for immediate resection remained robust. It also remained robust in another post hoc analysis that examined the subgroup of patients who had the most common glioma, a grade II astrocytoma. Median survival was 5.6 years at the hospital favoring watchful waiting, compared with 9.7 years at the hospital favoring early resection, in this large subgroup of patients.
Based on these findings, hospital A has changed its preferred strategy from watchful waiting to early resection, Dr. Jakola and his associates said.
"Despite the clear survival advantage seen, clinical judgment is still necessary in individual patients with suspected low-grade glioma since results will depend on patient and disease characteristics together with surgical results in terms of resection grades and complication rates," they added.
One of Dr. Jakola’s associates reported holding stock in Sonowand, manufacturer of the 3-D ultrasound-based imaging system used in one of the study hospitals.
Adults in Norway with diffuse low-grade gliomas who were treated at a hospital advocating early surgical resection had better overall survival than those treated at a hospital advocating "watchful waiting," according to a report published online Oct. 30 in JAMA.
This finding significantly strengthens the sparse evidence in support of early resection for newly diagnosed diffuse low-grade gliomas, said Dr. Asgeir S. Jakola of the department of neurosurgery, St. Olav’s University Hospital, Trondheim (Norway) and his associates.
Management of these tumors is one of the major controversies in both neurology and oncology today, largely because the effect of surgery on survival is still unclear. The only evidence available until now was based solely on uncontrolled surgical series; some of these have reported that it is safe to withhold surgery until the lesions progress, while others have reported that immediate resection improves survival and delays the time to malignant transformation.
Both patients and physicians are reluctant to undertake immediate surgery when the evidence supporting that strategy has been so tenuous. They also are concerned that the risk of early and aggressive surgery outweighs the benefit, particularly when most patients are capable of normal activity and have a reasonably long life expectancy at diagnosis, the investigators said.
It is unlikely that a randomized, controlled study comparing the two approaches will ever be performed. Dr. Jakola and his colleagues therefore conducted a retrospective, population-based parallel-cohort study at two neurosurgical centers, each of which preferred one of these strategies over the other. Their "natural experiment" was possible because in Norway, there were two such facilities that were relatively close geographically and served a homogenous population. The nationalized health care system distributes training, resources, and personnel equally throughout the country, so the two hospitals were quite similar in other respects. And patient follow-up is 100%.
The 12-year study involved 153 adults with diffuse, histologically verified supratentorial grade I and II tumors diagnosed in 1998-2009, who were followed until death or until April 2011. The median follow-up was 7 years. Gliomas included astrocytomas, oligodendrogliomas, and oligoastrocytomas.
For patients with newly diagnosed low-grade gliomas, hospital A favored biopsy and watchful waiting. The 66 patients treated there typically were followed with MRI at 3 and 6 months, then yearly thereafter. They usually were offered surgical resection, if the lesions grew or showed signs of malignant transformation.
Hospital B favored immediate maximal safe tumor resection for the 87 patients treated there, with MRI follow-up at 6 and 12 months, then annually thereafter. This strategy was not pursued in some patients, however: notably, those who were elderly or had comorbidities and were likely to die from another cause before malignant transformation would take place, and those who had very widespread tumor infiltration that made resection impractical.
The two study groups were well balanced with regard to patient age and comorbidities, and rates of surgical rescue therapy were the same. There also were no differences between the two groups in complications or acquired neurologic deficits.
At the end of the study period, 34 patients (52%) from hospital A had died, compared with only 28 patients (32%) from hospital B. Median survival was 5.9 years at hospital A, but median survival had not yet been reached at hospital B, the researchers said (JAMA 2012;308: [doi:10.1001/jama.2012.12807]).
This survival advantage increased over time. Expected 3-year survival was 70% at hospital A vs. 80% at hospital B; expected 5-year survival was 60% at hospital A vs. 74% at hospital B; and expected 7-year survival was 44% at hospital A vs. 68% at hospital B.
In a post hoc analysis that attempted to account for differences between the two study groups in prognostic factors, the survival benefit for immediate resection remained robust. It also remained robust in another post hoc analysis that examined the subgroup of patients who had the most common glioma, a grade II astrocytoma. Median survival was 5.6 years at the hospital favoring watchful waiting, compared with 9.7 years at the hospital favoring early resection, in this large subgroup of patients.
Based on these findings, hospital A has changed its preferred strategy from watchful waiting to early resection, Dr. Jakola and his associates said.
"Despite the clear survival advantage seen, clinical judgment is still necessary in individual patients with suspected low-grade glioma since results will depend on patient and disease characteristics together with surgical results in terms of resection grades and complication rates," they added.
One of Dr. Jakola’s associates reported holding stock in Sonowand, manufacturer of the 3-D ultrasound-based imaging system used in one of the study hospitals.
FROM JAMA
Major Finding: Overall mortality was 52% with watchful waiting and 32% with early resection; median survival was 5.9 years in the first group but has not yet been reached in the second group.
Data Source: Investigators compared survival rates in one hospital that advocated watchful waiting (66 patients) and another that advocated early resection (87 patients) for low-grade gliomas.
Disclosures: One of Dr. Jakola’s associates reported holding stock in Sonowand, manufacturer of the 3-D ultrasound-based imaging system used in one of the study hospitals.
Memantine Protects Cognitive Function After Whole Brain Irradiation
BOSTON – Memantine, a drug normally prescribed for slowing cognitive decline in Alzheimer’s disease, can help to preserve cognitive function in cancer patients who have undergone whole brain irradiation, a study showed.
In a phase III trial, patients with brain metastases were randomly assigned to take 20 mg memantine (Namenda) or placebo daily for 24 weeks after whole brain radiation therapy (WBRT). The memantine cohort had a 17% relative reduction in cognitive decline compared with patients who got a placebo, Dr. Nadia N. Laack reported at the annual meeting of the American Society for Radiation Oncology.

The finding teetered on the edge of statistical significance (P = .059), however, because only one-third of patients (32%) completed the 24 weeks of drug therapy, due to death (survival was poorer than expected), disease progression, or noncompliance, said Dr. Laack. a radiation oncologist at the Mayo Clinic in Rochester, Minn.
"Overall, we feel that the weight of evidence supports our conclusion that memantine helps to preserve cognitive function after whole brain radiotherapy in patients with brain metastases," Dr. Laack said at a briefing prior to presenting the data at a plenary session.
WBRT is associated with cognitive impairment in a majority of patients who receive it, Dr. Laack said, noting that at 4 months post radiation, 60% of patients will have declines in one or more cognitive domains.
Because the mechanism of decline is similar to that seen with Alzheimer\'s and vascular dementias, and because memantine has been shown to modestly improve mild to moderate cognition in both dementia types, Dr. Laack and his colleagues hypothesized that it might protect brains exposed to therapeutic doses of radiation.
A total of 508 patients were tested at baseline and at 8, 16, 24, and 52 weeks after radiation with 37.5 Gy in 15 fractions. They were evaluated with MRI and cognitive assessment; domains of memory, processing speed, executive function, global function, self-reported cognitive function, and quality of life were evaluated. Median overall follow-up was 12.4 months.
There were no differences between the treatment groups in overall survival at a median of 6 months or in progression-free survival at 5 months.
Among 149 patients available for analysis at 24 weeks, patients who took memantine had a significantly longer time to memory decline than did those on placebo (P = .02), and had a trend toward less decline in the primary end point, the Hopkins Verbal Learning Test–Revised delayed recall instrument (median decline of 0 standard deviation, vs. –2 standard deviations for patients on placebo).
For the secondary objective of cognitive function decline/failure, defined as a change greater than reversible cognitive impairment or 2 standard deviations decline from baseline on any domain of brain function, the hazard ratio for memantine at 24 weeks was 0.784 (P = .01), indicating a significant reduction in the incidence of cognitive dysfunction.
"Although memantine was discontinued at 6 months, the effect on cognitive function was maintained for the duration of the trial, suggesting that memantine may be preventing radiation injury rather than simply treating cognitive dysfunction," Dr. Laack said.
The trial was sponsored by grants from the National Cancer Institute and Forest Pharmaceuticals. Dr. Laack reported no relevant financial disclosures.
BOSTON – Memantine, a drug normally prescribed for slowing cognitive decline in Alzheimer’s disease, can help to preserve cognitive function in cancer patients who have undergone whole brain irradiation, a study showed.
In a phase III trial, patients with brain metastases were randomly assigned to take 20 mg memantine (Namenda) or placebo daily for 24 weeks after whole brain radiation therapy (WBRT). The memantine cohort had a 17% relative reduction in cognitive decline compared with patients who got a placebo, Dr. Nadia N. Laack reported at the annual meeting of the American Society for Radiation Oncology.
The finding teetered on the edge of statistical significance (P = .059), however, because only one-third of patients (32%) completed the 24 weeks of drug therapy, due to death (survival was poorer than expected), disease progression, or noncompliance, said Dr. Laack. a radiation oncologist at the Mayo Clinic in Rochester, Minn.
"Overall, we feel that the weight of evidence supports our conclusion that memantine helps to preserve cognitive function after whole brain radiotherapy in patients with brain metastases," Dr. Laack said at a briefing prior to presenting the data at a plenary session.
WBRT is associated with cognitive impairment in a majority of patients who receive it, Dr. Laack said, noting that at 4 months post radiation, 60% of patients will have declines in one or more cognitive domains.
Because the mechanism of decline is similar to that seen with Alzheimer\'s and vascular dementias, and because memantine has been shown to modestly improve mild to moderate cognition in both dementia types, Dr. Laack and his colleagues hypothesized that it might protect brains exposed to therapeutic doses of radiation.
A total of 508 patients were tested at baseline and at 8, 16, 24, and 52 weeks after radiation with 37.5 Gy in 15 fractions. They were evaluated with MRI and cognitive assessment; domains of memory, processing speed, executive function, global function, self-reported cognitive function, and quality of life were evaluated. Median overall follow-up was 12.4 months.
There were no differences between the treatment groups in overall survival at a median of 6 months or in progression-free survival at 5 months.
Among 149 patients available for analysis at 24 weeks, patients who took memantine had a significantly longer time to memory decline than did those on placebo (P = .02), and had a trend toward less decline in the primary end point, the Hopkins Verbal Learning Test–Revised delayed recall instrument (median decline of 0 standard deviation, vs. –2 standard deviations for patients on placebo).
For the secondary objective of cognitive function decline/failure, defined as a change greater than reversible cognitive impairment or 2 standard deviations decline from baseline on any domain of brain function, the hazard ratio for memantine at 24 weeks was 0.784 (P = .01), indicating a significant reduction in the incidence of cognitive dysfunction.
"Although memantine was discontinued at 6 months, the effect on cognitive function was maintained for the duration of the trial, suggesting that memantine may be preventing radiation injury rather than simply treating cognitive dysfunction," Dr. Laack said.
The trial was sponsored by grants from the National Cancer Institute and Forest Pharmaceuticals. Dr. Laack reported no relevant financial disclosures.
BOSTON – Memantine, a drug normally prescribed for slowing cognitive decline in Alzheimer’s disease, can help to preserve cognitive function in cancer patients who have undergone whole brain irradiation, a study showed.
In a phase III trial, patients with brain metastases were randomly assigned to take 20 mg memantine (Namenda) or placebo daily for 24 weeks after whole brain radiation therapy (WBRT). The memantine cohort had a 17% relative reduction in cognitive decline compared with patients who got a placebo, Dr. Nadia N. Laack reported at the annual meeting of the American Society for Radiation Oncology.
The finding teetered on the edge of statistical significance (P = .059), however, because only one-third of patients (32%) completed the 24 weeks of drug therapy, due to death (survival was poorer than expected), disease progression, or noncompliance, said Dr. Laack. a radiation oncologist at the Mayo Clinic in Rochester, Minn.
"Overall, we feel that the weight of evidence supports our conclusion that memantine helps to preserve cognitive function after whole brain radiotherapy in patients with brain metastases," Dr. Laack said at a briefing prior to presenting the data at a plenary session.
WBRT is associated with cognitive impairment in a majority of patients who receive it, Dr. Laack said, noting that at 4 months post radiation, 60% of patients will have declines in one or more cognitive domains.
Because the mechanism of decline is similar to that seen with Alzheimer\'s and vascular dementias, and because memantine has been shown to modestly improve mild to moderate cognition in both dementia types, Dr. Laack and his colleagues hypothesized that it might protect brains exposed to therapeutic doses of radiation.
A total of 508 patients were tested at baseline and at 8, 16, 24, and 52 weeks after radiation with 37.5 Gy in 15 fractions. They were evaluated with MRI and cognitive assessment; domains of memory, processing speed, executive function, global function, self-reported cognitive function, and quality of life were evaluated. Median overall follow-up was 12.4 months.
There were no differences between the treatment groups in overall survival at a median of 6 months or in progression-free survival at 5 months.
Among 149 patients available for analysis at 24 weeks, patients who took memantine had a significantly longer time to memory decline than did those on placebo (P = .02), and had a trend toward less decline in the primary end point, the Hopkins Verbal Learning Test–Revised delayed recall instrument (median decline of 0 standard deviation, vs. –2 standard deviations for patients on placebo).
For the secondary objective of cognitive function decline/failure, defined as a change greater than reversible cognitive impairment or 2 standard deviations decline from baseline on any domain of brain function, the hazard ratio for memantine at 24 weeks was 0.784 (P = .01), indicating a significant reduction in the incidence of cognitive dysfunction.
"Although memantine was discontinued at 6 months, the effect on cognitive function was maintained for the duration of the trial, suggesting that memantine may be preventing radiation injury rather than simply treating cognitive dysfunction," Dr. Laack said.
The trial was sponsored by grants from the National Cancer Institute and Forest Pharmaceuticals. Dr. Laack reported no relevant financial disclosures.
AT THE ANNUAL MEETING OF THE AMERICAN SOCIETY FOR RADIATION ONCOLOGY
Major Finding: Cancer patients with brain metastases had a 17% relative reduction in cognitive decline after whole brain radiation if they took memantine vs. placebo for 24 weeks.
Data Source: Investigators randomized 508 patients in a placebo-controlled clinical trial.
Disclosures: The trial was sponsored by grants from the National Cancer Institute and Forest Pharmaceuticals. Dr. Laack reported no relevant financial disclosures.
