User login
Ocular manifestations of small-vessel vasculitis
We have long understood that vasculitic conditions have various clinical manifestations. The Chapel Hill Consensus Conference classification of systemic vasculitis in 19941 contributed significantly to our understanding of the spectrum of vasculitides and their manifestations, enhancing our diagnostic ability and the likelihood of appropriate treatment.
The ophthalmic manifestations of vasculitis are protean and nonspecific, and should be considered in the overall context of the disease. Patients should be evaluated with the following questions in mind:
- Are the manifestations related to the vasculitis itself?
- Are the manifestations a result or complication of therapy?
- Are the manifestations signs of a completely unrelated and superimposed condition?
This article reviews the three areas of ocular inflammation related to vasculitis and comments on the role of tissue biopsy in the management of these patients.
THREE AREAS OF OCULAR INFLAMMATION
Orbital inflammation
Orbital disease can affect the lacrimal gland (inflammatory dacryoadenitis), extraocular muscles (orbital myositis), and the orbital soft tissues (inflammatory orbital pseudotumor). Orbital inflammation is characterized by relatively sudden onset (within days) of pain, erythema, and proptosis. Diplopia and visual loss from either compression or inflammation of the optic nerve or nerve sheath may be present. Depending upon the structures involved and the degree of involvement, orbital inflammation can be sight-threatening.
Either computed tomography or magnetic resonance imaging should be performed to assess orbital or extraorbital involvement. The orbital structures are particularly amenable to biopsy, which, in this author’s opinion, should be performed whenever possible. The biopsy may need to be interpreted within the context of previous or concurrent immunosuppressive therapy, which can alter the histologic picture, minimize inflammation, and make detection of vasculitis difficult. In addition to identifying inflammation, biopsy helps to identify fungal infection or lymphoma that can follow prolonged immunosuppressive therapy.
Treatment of orbital inflammation requires corticosteroid therapy or some other type of systemic immunosuppression.
Ocular, or globe, inflammation
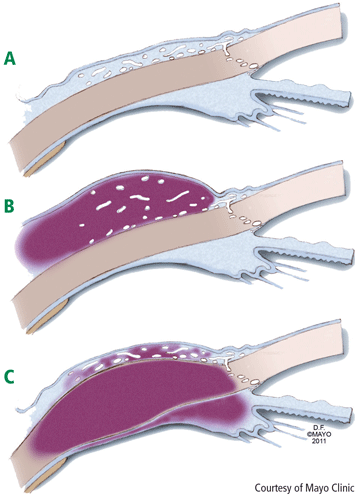
Episcleritis: observation or topical therapy. Episcleritis usually manifests as an otherwise asymptomatic red eye with typical sector-shaped inflammation. Pain is generally not an issue, although patients often report that the eye does not feel normal. Vision is unaffected and there is no potential threat to sight.
The slit-lamp examination shows dilated vessels in the episcleral tissues that blanch after instillation of a drop of 10% phenylephrine. Simple observation may be the best management course, but topical nonsteroidal anti-inflammatory drugs (NSAIDs) or topical corticosteroids may help some patients who have discomfort. There is probably a spectrum of disease in that some patients may have either severe episcleritis or mild scleritis (Figure 1B). At times it can be difficult to differentiate between severe episcleritis and mild scleritis. Although scleritis generally requires systemic therapy, topical therapy is justified for mild scleritis. Episcleritis is associated with systemic disease in approximately 36% of patients.2–4
Scleritis: may be sight-threatening; requires systemic therapy. Scleritis characteristically presents with intense pain and a red eye.3,5–7 Patients may be sensitive to light and their vision may be compromised. Cataracts and glaucoma can complicate the course of scleritis.
With slit-lamp examination, the redness does not blanch upon instillation of topical 10% phenylephrine as it does with episcleritis. The adjacent cornea may also be affected (Figure 1C). Healed scleritis leaves an area of thinned sclera that appears as a visible blue spot, so if the patient’s history includes red eye with pain and a blue area is visible, the clinician can be confident that a prior episode of scleritis occurred.
Scleritis can be anterior or posterior, and the implications are slightly different for each type. Anterior scleritis can be subclassified as diffuse, nodular, or necrotizing. The necrotizing type can be characterized by painful inflammation or, in the case of scleromalacia perforans, no inflammation and no pain. Posterior scleritis may have minimal pain.
Akpek et al5 reported on a group of 243 patients with scleritis (average age, 52 years; range, 5 to 93 years) who were followed for an average of 1.7 years (range, 0 to 16.6 years). An associated medical condition was present in 107 (44%) patients. Rheumatologic conditions accounted for 37%, with rheumatoid arthritis being most common; infectious disease, with herpes zoster ophthalmicus being most common, accounted for 7%. Of those with an associated medical condition, 78% had been diagnosed previously; the remaining 22% were diagnosed at presentation or the condition developed during follow-up.
Treatment typically requires systemic therapy with NSAIDs, but more often oral or intravenous corticosteroids or even methotrexate, mycophenolate mofetil, cyclophosphamide, or rituximab may be required. Patients with antineutrophil cytoplasmic antibody (ANCA)–positive disease may require more intensive therapy than those with ANCA-negative disease.
Keratitis: may be sight-threatening. Patients with keratitis should be evaluated in the same spirit as patients with scleritis (Figure 1C). Although many patients may have superficial keratitis, which is often related to a dry eye and has no prognostic significance, deep or peripheral ulcerative keratitis is not only consistent with systemic vasculitis but also sight-threatening. Symptoms similar to those observed with scleritis typically include severe pain and photophobia and, as with scleritis, treatment usually involves systemic therapy.
Intraocular inflammation
There is no specific treatment for the eye other than treating the underlying condition. Vascular occlusions can sometimes give rise to neovascularization and patients should be followed for this possibility. As with a central nervous system ischemic event, recovery can be variable.
Uveitis. The term “uvea,” derived from the Greek word for grape, describes the shape of the iris, ciliary body, and choroid. Uveitis is a generic term for intraocular inflammation affecting any or all of these structures.
Iritis, or anterior uveitis, is a frequent accompaniment of keratitis or scleritis. Primarily uveitic involvement with retinal vessel vasculitis involving both arteries and veins is uncommon in general but typical of Behçet disease, especially if a hypopyon uveitis is present.
Anterior uveitis can be treated with topical corticosteroids and cycloplegic drugs, but middle and posterior uveitis almost always requires systemic therapy. Most recently, use of anti–tumor necrosis factor-α drugs has been effective in treating Behçet uveitis.8 The visual prognosis with Behçet disease remains guarded.
GRANULOMATOSIS WITH POLYANGIITIS: EYE INVOLVEMENT IS COMMON
In terms of specific small-vessel vasculitic diseases that affect the eye, granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is the quintessential condition. In data obtained from the Wegener Granulomatosis Support Group,9 eye involvement was noted at presentation in 211 of 701 patients (30%), and during the course of their disease an additional 147 patients developed eye involvement. From the time of initial presentation through the course of follow-up, 359 of the 701 patients (51%) eventually had some type of eye involvement.
In a series of patients seen at the Mayo Clinic,10 orbital inflammatory disease and scleritis were the two most frequent manifestations of eye involvement with GPA. Orbital involvement typically presents with pain, erythema, swelling, and proptosis. Varying degrees of ptosis, diplopia, or visual loss may also be present. Imaging may show an infiltrate that is usually adjacent to the maxillary or ethmoid sinus. This same process can affect the superior temporal orbital quadrant, an area apart from any sinus, and involve the lacrimal gland.
BIOPSY IS ADVISED
Biopsy, either incisional, at times to include debulking, or excisional if possible, is recommended to establish a diagnosis or aid in the selection of therapy. Orbital disease has been observed to progress in patients who are receiving maintenance therapy with methotrexate and have no evidence of systemic disease activity. Acute and chronic inflammation with evidence of active vasculitis is usually seen histologically. Personal observations suggest that intraorbital corticosteroid injection followed by rituximab has been effective therapy for this limited subset of patients. Diagnostic biopsies often must be interpreted in light of partial treatment, making histopathologic diagnosis challenging at times. Biopsy is important for exclusion of lymphoproliferative disease or fungal infection.
CONCLUSION
Underlying vasculitis might play a role in patients with nonspecific ocular presentations. It is essential that the ophthalmologist collaborate with a specialist in vasculitis (and vice versa) for evaluation and subsequent therapy, which often involves some form of immunosuppression.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an International Consensus Conference. Arthritis Rheum 1994; 37:187–192.
- Pavesio CE, Meier FM. Systemic disorders associated with episcleritis and scleritis. Curr Opin Ophthalmol 2001; 12:471–478.
- Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol 2000; 130:469–476.
- Akpek EK, Uy HS, Christen W, Gurdal C, Foster CS. Severity of episcleritis and systemic disease association. Ophthalmology 1999; 106:729–731.
- Akpek EK, Thorne JE, Qazi FA, Do DV, Jabs DA. Evaluation of patients with scleritis for systemic disease. Ophthalmology 2004; 111:501–506.
- McCluskey PJ, Watson PG, Lightman S, Haybittle J, Restori M, Branley M. Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology 1999; 106:2380–2386.
- Riono WP, Hidayat AA, Rao NA. Scleritis: a clinicopathologic study of 55 cases. Ophthalmology 1999; 106:1328–1333.
- Tabbara KF, Al-Hemidan AI. Infliximab effects compared to conventional therapy in the management of retinal vasculitis in Behçet disease [published online ahead of print October 17, 2008]. Am J Ophthalmol 2008; 146:845–850. doi: 10.1016/j.ajo.2008.09.010
- Abdou NI, Kullman GJ, Hoffman GS, et al. Wegener’s granulomatosis— survey of 701 patients in North America: changes in outcome in the 1990s. J Rheumatol 2002; 29:309–316.
- Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegener’s granulomatosis. Ophthalmology 1983; 90:279–290.
We have long understood that vasculitic conditions have various clinical manifestations. The Chapel Hill Consensus Conference classification of systemic vasculitis in 19941 contributed significantly to our understanding of the spectrum of vasculitides and their manifestations, enhancing our diagnostic ability and the likelihood of appropriate treatment.
The ophthalmic manifestations of vasculitis are protean and nonspecific, and should be considered in the overall context of the disease. Patients should be evaluated with the following questions in mind:
- Are the manifestations related to the vasculitis itself?
- Are the manifestations a result or complication of therapy?
- Are the manifestations signs of a completely unrelated and superimposed condition?
This article reviews the three areas of ocular inflammation related to vasculitis and comments on the role of tissue biopsy in the management of these patients.
THREE AREAS OF OCULAR INFLAMMATION
Orbital inflammation
Orbital disease can affect the lacrimal gland (inflammatory dacryoadenitis), extraocular muscles (orbital myositis), and the orbital soft tissues (inflammatory orbital pseudotumor). Orbital inflammation is characterized by relatively sudden onset (within days) of pain, erythema, and proptosis. Diplopia and visual loss from either compression or inflammation of the optic nerve or nerve sheath may be present. Depending upon the structures involved and the degree of involvement, orbital inflammation can be sight-threatening.
Either computed tomography or magnetic resonance imaging should be performed to assess orbital or extraorbital involvement. The orbital structures are particularly amenable to biopsy, which, in this author’s opinion, should be performed whenever possible. The biopsy may need to be interpreted within the context of previous or concurrent immunosuppressive therapy, which can alter the histologic picture, minimize inflammation, and make detection of vasculitis difficult. In addition to identifying inflammation, biopsy helps to identify fungal infection or lymphoma that can follow prolonged immunosuppressive therapy.
Treatment of orbital inflammation requires corticosteroid therapy or some other type of systemic immunosuppression.
Ocular, or globe, inflammation
Episcleritis: observation or topical therapy. Episcleritis usually manifests as an otherwise asymptomatic red eye with typical sector-shaped inflammation. Pain is generally not an issue, although patients often report that the eye does not feel normal. Vision is unaffected and there is no potential threat to sight.
The slit-lamp examination shows dilated vessels in the episcleral tissues that blanch after instillation of a drop of 10% phenylephrine. Simple observation may be the best management course, but topical nonsteroidal anti-inflammatory drugs (NSAIDs) or topical corticosteroids may help some patients who have discomfort. There is probably a spectrum of disease in that some patients may have either severe episcleritis or mild scleritis (Figure 1B). At times it can be difficult to differentiate between severe episcleritis and mild scleritis. Although scleritis generally requires systemic therapy, topical therapy is justified for mild scleritis. Episcleritis is associated with systemic disease in approximately 36% of patients.2–4
Scleritis: may be sight-threatening; requires systemic therapy. Scleritis characteristically presents with intense pain and a red eye.3,5–7 Patients may be sensitive to light and their vision may be compromised. Cataracts and glaucoma can complicate the course of scleritis.
With slit-lamp examination, the redness does not blanch upon instillation of topical 10% phenylephrine as it does with episcleritis. The adjacent cornea may also be affected (Figure 1C). Healed scleritis leaves an area of thinned sclera that appears as a visible blue spot, so if the patient’s history includes red eye with pain and a blue area is visible, the clinician can be confident that a prior episode of scleritis occurred.
Scleritis can be anterior or posterior, and the implications are slightly different for each type. Anterior scleritis can be subclassified as diffuse, nodular, or necrotizing. The necrotizing type can be characterized by painful inflammation or, in the case of scleromalacia perforans, no inflammation and no pain. Posterior scleritis may have minimal pain.
Akpek et al5 reported on a group of 243 patients with scleritis (average age, 52 years; range, 5 to 93 years) who were followed for an average of 1.7 years (range, 0 to 16.6 years). An associated medical condition was present in 107 (44%) patients. Rheumatologic conditions accounted for 37%, with rheumatoid arthritis being most common; infectious disease, with herpes zoster ophthalmicus being most common, accounted for 7%. Of those with an associated medical condition, 78% had been diagnosed previously; the remaining 22% were diagnosed at presentation or the condition developed during follow-up.
Treatment typically requires systemic therapy with NSAIDs, but more often oral or intravenous corticosteroids or even methotrexate, mycophenolate mofetil, cyclophosphamide, or rituximab may be required. Patients with antineutrophil cytoplasmic antibody (ANCA)–positive disease may require more intensive therapy than those with ANCA-negative disease.
Keratitis: may be sight-threatening. Patients with keratitis should be evaluated in the same spirit as patients with scleritis (Figure 1C). Although many patients may have superficial keratitis, which is often related to a dry eye and has no prognostic significance, deep or peripheral ulcerative keratitis is not only consistent with systemic vasculitis but also sight-threatening. Symptoms similar to those observed with scleritis typically include severe pain and photophobia and, as with scleritis, treatment usually involves systemic therapy.
Intraocular inflammation
There is no specific treatment for the eye other than treating the underlying condition. Vascular occlusions can sometimes give rise to neovascularization and patients should be followed for this possibility. As with a central nervous system ischemic event, recovery can be variable.
Uveitis. The term “uvea,” derived from the Greek word for grape, describes the shape of the iris, ciliary body, and choroid. Uveitis is a generic term for intraocular inflammation affecting any or all of these structures.
Iritis, or anterior uveitis, is a frequent accompaniment of keratitis or scleritis. Primarily uveitic involvement with retinal vessel vasculitis involving both arteries and veins is uncommon in general but typical of Behçet disease, especially if a hypopyon uveitis is present.
Anterior uveitis can be treated with topical corticosteroids and cycloplegic drugs, but middle and posterior uveitis almost always requires systemic therapy. Most recently, use of anti–tumor necrosis factor-α drugs has been effective in treating Behçet uveitis.8 The visual prognosis with Behçet disease remains guarded.
GRANULOMATOSIS WITH POLYANGIITIS: EYE INVOLVEMENT IS COMMON
In terms of specific small-vessel vasculitic diseases that affect the eye, granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is the quintessential condition. In data obtained from the Wegener Granulomatosis Support Group,9 eye involvement was noted at presentation in 211 of 701 patients (30%), and during the course of their disease an additional 147 patients developed eye involvement. From the time of initial presentation through the course of follow-up, 359 of the 701 patients (51%) eventually had some type of eye involvement.
In a series of patients seen at the Mayo Clinic,10 orbital inflammatory disease and scleritis were the two most frequent manifestations of eye involvement with GPA. Orbital involvement typically presents with pain, erythema, swelling, and proptosis. Varying degrees of ptosis, diplopia, or visual loss may also be present. Imaging may show an infiltrate that is usually adjacent to the maxillary or ethmoid sinus. This same process can affect the superior temporal orbital quadrant, an area apart from any sinus, and involve the lacrimal gland.
BIOPSY IS ADVISED
Biopsy, either incisional, at times to include debulking, or excisional if possible, is recommended to establish a diagnosis or aid in the selection of therapy. Orbital disease has been observed to progress in patients who are receiving maintenance therapy with methotrexate and have no evidence of systemic disease activity. Acute and chronic inflammation with evidence of active vasculitis is usually seen histologically. Personal observations suggest that intraorbital corticosteroid injection followed by rituximab has been effective therapy for this limited subset of patients. Diagnostic biopsies often must be interpreted in light of partial treatment, making histopathologic diagnosis challenging at times. Biopsy is important for exclusion of lymphoproliferative disease or fungal infection.
CONCLUSION
Underlying vasculitis might play a role in patients with nonspecific ocular presentations. It is essential that the ophthalmologist collaborate with a specialist in vasculitis (and vice versa) for evaluation and subsequent therapy, which often involves some form of immunosuppression.
We have long understood that vasculitic conditions have various clinical manifestations. The Chapel Hill Consensus Conference classification of systemic vasculitis in 19941 contributed significantly to our understanding of the spectrum of vasculitides and their manifestations, enhancing our diagnostic ability and the likelihood of appropriate treatment.
The ophthalmic manifestations of vasculitis are protean and nonspecific, and should be considered in the overall context of the disease. Patients should be evaluated with the following questions in mind:
- Are the manifestations related to the vasculitis itself?
- Are the manifestations a result or complication of therapy?
- Are the manifestations signs of a completely unrelated and superimposed condition?
This article reviews the three areas of ocular inflammation related to vasculitis and comments on the role of tissue biopsy in the management of these patients.
THREE AREAS OF OCULAR INFLAMMATION
Orbital inflammation
Orbital disease can affect the lacrimal gland (inflammatory dacryoadenitis), extraocular muscles (orbital myositis), and the orbital soft tissues (inflammatory orbital pseudotumor). Orbital inflammation is characterized by relatively sudden onset (within days) of pain, erythema, and proptosis. Diplopia and visual loss from either compression or inflammation of the optic nerve or nerve sheath may be present. Depending upon the structures involved and the degree of involvement, orbital inflammation can be sight-threatening.
Either computed tomography or magnetic resonance imaging should be performed to assess orbital or extraorbital involvement. The orbital structures are particularly amenable to biopsy, which, in this author’s opinion, should be performed whenever possible. The biopsy may need to be interpreted within the context of previous or concurrent immunosuppressive therapy, which can alter the histologic picture, minimize inflammation, and make detection of vasculitis difficult. In addition to identifying inflammation, biopsy helps to identify fungal infection or lymphoma that can follow prolonged immunosuppressive therapy.
Treatment of orbital inflammation requires corticosteroid therapy or some other type of systemic immunosuppression.
Ocular, or globe, inflammation
Episcleritis: observation or topical therapy. Episcleritis usually manifests as an otherwise asymptomatic red eye with typical sector-shaped inflammation. Pain is generally not an issue, although patients often report that the eye does not feel normal. Vision is unaffected and there is no potential threat to sight.
The slit-lamp examination shows dilated vessels in the episcleral tissues that blanch after instillation of a drop of 10% phenylephrine. Simple observation may be the best management course, but topical nonsteroidal anti-inflammatory drugs (NSAIDs) or topical corticosteroids may help some patients who have discomfort. There is probably a spectrum of disease in that some patients may have either severe episcleritis or mild scleritis (Figure 1B). At times it can be difficult to differentiate between severe episcleritis and mild scleritis. Although scleritis generally requires systemic therapy, topical therapy is justified for mild scleritis. Episcleritis is associated with systemic disease in approximately 36% of patients.2–4
Scleritis: may be sight-threatening; requires systemic therapy. Scleritis characteristically presents with intense pain and a red eye.3,5–7 Patients may be sensitive to light and their vision may be compromised. Cataracts and glaucoma can complicate the course of scleritis.
With slit-lamp examination, the redness does not blanch upon instillation of topical 10% phenylephrine as it does with episcleritis. The adjacent cornea may also be affected (Figure 1C). Healed scleritis leaves an area of thinned sclera that appears as a visible blue spot, so if the patient’s history includes red eye with pain and a blue area is visible, the clinician can be confident that a prior episode of scleritis occurred.
Scleritis can be anterior or posterior, and the implications are slightly different for each type. Anterior scleritis can be subclassified as diffuse, nodular, or necrotizing. The necrotizing type can be characterized by painful inflammation or, in the case of scleromalacia perforans, no inflammation and no pain. Posterior scleritis may have minimal pain.
Akpek et al5 reported on a group of 243 patients with scleritis (average age, 52 years; range, 5 to 93 years) who were followed for an average of 1.7 years (range, 0 to 16.6 years). An associated medical condition was present in 107 (44%) patients. Rheumatologic conditions accounted for 37%, with rheumatoid arthritis being most common; infectious disease, with herpes zoster ophthalmicus being most common, accounted for 7%. Of those with an associated medical condition, 78% had been diagnosed previously; the remaining 22% were diagnosed at presentation or the condition developed during follow-up.
Treatment typically requires systemic therapy with NSAIDs, but more often oral or intravenous corticosteroids or even methotrexate, mycophenolate mofetil, cyclophosphamide, or rituximab may be required. Patients with antineutrophil cytoplasmic antibody (ANCA)–positive disease may require more intensive therapy than those with ANCA-negative disease.
Keratitis: may be sight-threatening. Patients with keratitis should be evaluated in the same spirit as patients with scleritis (Figure 1C). Although many patients may have superficial keratitis, which is often related to a dry eye and has no prognostic significance, deep or peripheral ulcerative keratitis is not only consistent with systemic vasculitis but also sight-threatening. Symptoms similar to those observed with scleritis typically include severe pain and photophobia and, as with scleritis, treatment usually involves systemic therapy.
Intraocular inflammation
There is no specific treatment for the eye other than treating the underlying condition. Vascular occlusions can sometimes give rise to neovascularization and patients should be followed for this possibility. As with a central nervous system ischemic event, recovery can be variable.
Uveitis. The term “uvea,” derived from the Greek word for grape, describes the shape of the iris, ciliary body, and choroid. Uveitis is a generic term for intraocular inflammation affecting any or all of these structures.
Iritis, or anterior uveitis, is a frequent accompaniment of keratitis or scleritis. Primarily uveitic involvement with retinal vessel vasculitis involving both arteries and veins is uncommon in general but typical of Behçet disease, especially if a hypopyon uveitis is present.
Anterior uveitis can be treated with topical corticosteroids and cycloplegic drugs, but middle and posterior uveitis almost always requires systemic therapy. Most recently, use of anti–tumor necrosis factor-α drugs has been effective in treating Behçet uveitis.8 The visual prognosis with Behçet disease remains guarded.
GRANULOMATOSIS WITH POLYANGIITIS: EYE INVOLVEMENT IS COMMON
In terms of specific small-vessel vasculitic diseases that affect the eye, granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is the quintessential condition. In data obtained from the Wegener Granulomatosis Support Group,9 eye involvement was noted at presentation in 211 of 701 patients (30%), and during the course of their disease an additional 147 patients developed eye involvement. From the time of initial presentation through the course of follow-up, 359 of the 701 patients (51%) eventually had some type of eye involvement.
In a series of patients seen at the Mayo Clinic,10 orbital inflammatory disease and scleritis were the two most frequent manifestations of eye involvement with GPA. Orbital involvement typically presents with pain, erythema, swelling, and proptosis. Varying degrees of ptosis, diplopia, or visual loss may also be present. Imaging may show an infiltrate that is usually adjacent to the maxillary or ethmoid sinus. This same process can affect the superior temporal orbital quadrant, an area apart from any sinus, and involve the lacrimal gland.
BIOPSY IS ADVISED
Biopsy, either incisional, at times to include debulking, or excisional if possible, is recommended to establish a diagnosis or aid in the selection of therapy. Orbital disease has been observed to progress in patients who are receiving maintenance therapy with methotrexate and have no evidence of systemic disease activity. Acute and chronic inflammation with evidence of active vasculitis is usually seen histologically. Personal observations suggest that intraorbital corticosteroid injection followed by rituximab has been effective therapy for this limited subset of patients. Diagnostic biopsies often must be interpreted in light of partial treatment, making histopathologic diagnosis challenging at times. Biopsy is important for exclusion of lymphoproliferative disease or fungal infection.
CONCLUSION
Underlying vasculitis might play a role in patients with nonspecific ocular presentations. It is essential that the ophthalmologist collaborate with a specialist in vasculitis (and vice versa) for evaluation and subsequent therapy, which often involves some form of immunosuppression.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an International Consensus Conference. Arthritis Rheum 1994; 37:187–192.
- Pavesio CE, Meier FM. Systemic disorders associated with episcleritis and scleritis. Curr Opin Ophthalmol 2001; 12:471–478.
- Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol 2000; 130:469–476.
- Akpek EK, Uy HS, Christen W, Gurdal C, Foster CS. Severity of episcleritis and systemic disease association. Ophthalmology 1999; 106:729–731.
- Akpek EK, Thorne JE, Qazi FA, Do DV, Jabs DA. Evaluation of patients with scleritis for systemic disease. Ophthalmology 2004; 111:501–506.
- McCluskey PJ, Watson PG, Lightman S, Haybittle J, Restori M, Branley M. Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology 1999; 106:2380–2386.
- Riono WP, Hidayat AA, Rao NA. Scleritis: a clinicopathologic study of 55 cases. Ophthalmology 1999; 106:1328–1333.
- Tabbara KF, Al-Hemidan AI. Infliximab effects compared to conventional therapy in the management of retinal vasculitis in Behçet disease [published online ahead of print October 17, 2008]. Am J Ophthalmol 2008; 146:845–850. doi: 10.1016/j.ajo.2008.09.010
- Abdou NI, Kullman GJ, Hoffman GS, et al. Wegener’s granulomatosis— survey of 701 patients in North America: changes in outcome in the 1990s. J Rheumatol 2002; 29:309–316.
- Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegener’s granulomatosis. Ophthalmology 1983; 90:279–290.
- Jennette JC, Falk RJ, Andrassy K, et al. Nomenclature of systemic vasculitides: proposal of an International Consensus Conference. Arthritis Rheum 1994; 37:187–192.
- Pavesio CE, Meier FM. Systemic disorders associated with episcleritis and scleritis. Curr Opin Ophthalmol 2001; 12:471–478.
- Jabs DA, Mudun A, Dunn JP, Marsh MJ. Episcleritis and scleritis: clinical features and treatment results. Am J Ophthalmol 2000; 130:469–476.
- Akpek EK, Uy HS, Christen W, Gurdal C, Foster CS. Severity of episcleritis and systemic disease association. Ophthalmology 1999; 106:729–731.
- Akpek EK, Thorne JE, Qazi FA, Do DV, Jabs DA. Evaluation of patients with scleritis for systemic disease. Ophthalmology 2004; 111:501–506.
- McCluskey PJ, Watson PG, Lightman S, Haybittle J, Restori M, Branley M. Posterior scleritis: clinical features, systemic associations, and outcome in a large series of patients. Ophthalmology 1999; 106:2380–2386.
- Riono WP, Hidayat AA, Rao NA. Scleritis: a clinicopathologic study of 55 cases. Ophthalmology 1999; 106:1328–1333.
- Tabbara KF, Al-Hemidan AI. Infliximab effects compared to conventional therapy in the management of retinal vasculitis in Behçet disease [published online ahead of print October 17, 2008]. Am J Ophthalmol 2008; 146:845–850. doi: 10.1016/j.ajo.2008.09.010
- Abdou NI, Kullman GJ, Hoffman GS, et al. Wegener’s granulomatosis— survey of 701 patients in North America: changes in outcome in the 1990s. J Rheumatol 2002; 29:309–316.
- Bullen CL, Liesegang TJ, McDonald TJ, DeRemee RA. Ocular complications of Wegener’s granulomatosis. Ophthalmology 1983; 90:279–290.
Monitoring patients with vasculitis
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.

Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
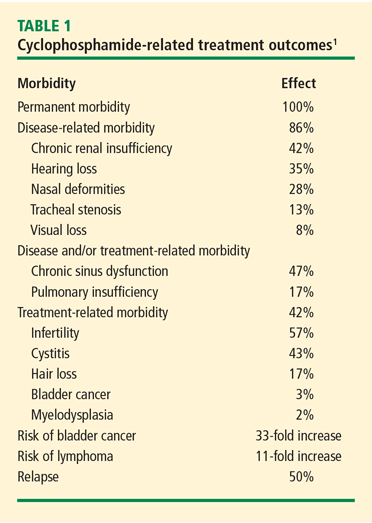
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.

Therapy-related complications
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.

Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.

Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.
Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.
Therapy-related complications
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.
Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.
Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
Granulomatosis with polyangiitis (GPA), is one of the most common types of small-vessel vasculitis, with an estimated prevalence in the United States of 3 per 100,000 people. It is distinguished from other necrotizing vasculitides by its tendency to affect the upper and lower respiratory system and the kidneys. Despite the success of induction and maintenance treatments with cyclophosphamide (CYC), glucocorticoids, and less toxic immunosuppressive alternative therapies in improving the disease course, significant treatment-related toxicities and frequent disease relapses demand stringent patient-specific monitoring in order to provide early treatment of relapses and prevent or decrease morbidity.
SMALL-VESSEL VASCULITIS MANAGEMENT OVERVIEW
Granulomatosis with polyangiitis (formerly Wegener’s granulomatosis, or WG) is an antineutrophil cytoplasmic antibody (ANCA)–associated vasculitis that often affects the respiratory system and kidneys across a broad spectrum of clinical presentations, from mild through life-threatening disease. Patients with severe disease present with significant multisystem manifestations, which, in addition to the respiratory system and kidneys, may involve the joints, eyes, and other organs.
Managing patients diagnosed with systemic small-vessel vasculitides such as GPA and microscopic polyangiitis (MPA) is an inexact science. The goals of treatment are to increase survival, induce and maintain remission, reduce relapses, and minimize treatment-related toxicity. Inducing and maintaining remission have become realistic goals because of the availability of medications that prolong life. On the other hand, extended periods of treatment associated with prolonged life increase the risk of treatment-related toxicity in patients who are inadequately monitored.
MONITORING CONSIDERATIONS
Achieving treatment goals requires long-term monitoring of both disease activity and treatment-related toxicities, with constant adjustments to meet the needs of the individual patient and address the often rapidly changing disease and treatment course. The monitoring protocol consists of regularly scheduled follow-up office visits, urine sediment analyses at every office visit whether or not the patient has relapse symptoms, laboratory tests at regular intervals as indicated by the patient’s medication plan and disease presentation, additional tests such as lung computed tomography (CT), and patient education regarding new symptoms and the frequency of office visits. A consistent monitoring strategy will help detect a relapse before it can produce more severe morbidity, identify treatment-related complications, and—equally important—identify the achievement of remission. An example of the consequences of inconsistent monitoring is presented in “Relapse in a nonadherent patient.”
Because there is no definitive cure for small-vessel vasculitis, relapse is always a possibility. The early diagnosis and treatment of relapse may prevent or decrease morbidity from disease, but strict monitoring is needed to identify relapse and initiate treatment before morbidity occurs (see “Relapse in a patient with new symptoms”). Repeat induction therapy following a relapse introduces risk of drug toxicity and requires careful monitoring, as does long-term maintenance therapy.
In addition to induction and maintenance therapy, several other situations, including prior therapeutic complications, serum creatinine levels, and risk of cardiovascular disease, require special monitoring attention.
Induction therapy: monitor response
Response to treatment during induction must be monitored to identify whether remission is achieved. Induction monitoring requires complete assessment of organ-system involvement at every visit with tools such as the Birmingham Vasculitis Activity Score (BVAS) and, when appropriate, the BVAS/WG. If new or worsening symptoms develop during induction therapy, then the patient needs assessment for continued disease activity as well as treatment complications such as infections related to immunosuppressive therapy.
During induction therapy with daily oral CYC, monitoring should include weekly complete blood cell counts to ensure early identification of leukopenia and other cytopenias. The risk of morbidities increases with the cumulative dose, so a stable blood count for 2 months does not obviate the risk of leukopenia. If persistent hematuria is present without cellular casts, cystoscopy is indicated to look for signs of hemorrhagic cystitis. Prophylaxis against Pneumocystis jirovecii is recommended in all patients who receive immunosuppressive therapy. Finally, bone density measurements should be done at baseline.
Maintenance therapy: frequency can be extended
Monitoring during maintenance therapy is similar to induction monitoring; however, when the dosage of methotrexate or azathioprine is stabilized, the frequency of some tests can be extended to monthly rather than weekly. For example, a complete blood cell count, comprehensive metabolic panel, sedimentation rate, C-reactive protein measurement, and urinalysis should be performed monthly. Follow-up visits should include urine sediment analyses and monitoring for cardiovascular disease risk factors. Medication monitoring should include cystoscopy for persistent hematuria without cellular casts, bone density measurements, and ophthalmologic examinations as frequently as indicated for each individual’s needs. P jirovecii prophylaxis should continue as long as the patient receives immunosuppressive medication.
Therapy-related complications
Bladder complications. In a retrospective analysis of 145 patients with GPA treated with CYC and followed for 0.5 to 27 years (median 8.5 years), nonglomerular hematuria developed in 50% of the patients and bladder carcinoma in 5%.2 The cumulative CYC dose (19 to 251 g) in this group was much higher than what is currently used. Cytologic examination of the urine showed 43% sensitivity for dysplasia (specificity 100%) and 29% sensitivity for atypia (specificity 89%). In contrast, in a retrospective outcomes analysis involving newly diagnosed patients with GPA treated with CYC or methotrexate, 82 patients followed for up to 12 years had no incidents of cystitis or bladder cancer.3 Patients in this study were treated with CYC for only 3 to 6 months and therefore received a lower cumulative dose.
To prevent cystitis during treatment with CYC, the patient should be well hydrated, especially in the morning when CYC should be taken. The bladder should be emptied frequently. The addition of mesna when administering intravenous CYC decreases the risk of cystitis. Serial cystoscopy and urine cytology should be used only in patients with nonglomerular hematuria.
Infertility. Preservation of ovarian function is a concern with CYC therapy in women of childbearing age. The cumulative dose threshold for gonadal failure is unknown, because data from cancer studies4 demonstrating gonadal failure involve higher cumulative CYC doses than are typical for vasculitis treatment. It is also unknown whether duration of amenorrhea predicts the recovery of menses or fertility. The primary option for preservation of ovarian function is the use of gonadotropin-releasing hormone agonists. Oral contraceptives also may be used, but the best prevention is to avoid CYC in these patients if possible.
Osteoporosis. At glucocorticoid dosages of 5 mg/day or greater, bone mineral density begins a rapid decline within the first 3 months and peaks at 6 months.5 The American College of Rheumatology has provided recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis.5 Table 2 presents recommendations for postmenopausal women and men aged 50 years and older who will use glucocorticoids for 3 months or more.5 Recommendations are also available for premenopausal women and men younger than 50 years of age who have a history of fragility fracture.
Leukopenia. Leukopenia should be avoided during CYC treatment. The target white blood cell count should be within the normal range. During treatment with daily oral CYC, the patient should be monitored with a weekly complete blood cell count and medication should be adjusted to maintain the target white blood cell count.
Upon completion of induction therapy, after 3 to 6 months, the patient is switched to maintenance therapy with an alternative immunosuppressive agent such as azathioprine or methotrexate, depending on the serum creatinine concentration and other factors. This transition, characterized by full-dose immunosuppressive therapy when the bone marrow has been previously suppressed by CYC treatment, may induce pancytopenia. Monitoring with weekly complete blood counts for at least 4 weeks after initiating maintenance therapy can help ensure stability during the transition period.
Monitor serum creatinine and adjust dosages
The serum creatinine concentration may increase as CYC treatment progresses; in some cases, the serum creatinine concentration increases before a response to treatment is seen. The CYC dosages should be adjusted as necessary in response to serum creatinine changes. Careful monitoring of serum creatinine is necessary during methotrexate therapy, as methotrexate treatment in the setting of renal insufficiency increases the risk of bone marrow suppression.
Cardiovascular disease in GPA and MPA
Premature atherosclerosis has been well described in patients with GPA.6 Within 5 years of diagnosis of GPA or MPA, a cardiovascular event will occur in 14% of patients.7 In the absence of specific guidelines for prevention of cardiovascular disease in patients with vasculitis, it is essential to monitor patients and treat modifiable traditional risk factors aggressively, especially in younger patients. Suppiah et al found that independent determinants of cardiovascular outcome included older age, diastolic hypertension, and positive proteinase-3–ANCA status in patients without prior cardiovascular disease.7
In the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study, Merkel et al showed an increased incidence of thrombosis in patients with active GPA8 (see “Relapse presenting as thrombosis,” left). As with cardiovascular disease, there are no specific guidelines for monitoring asymptomatic patients for thrombosis or for duration of anticoagulation in patients with GPA. It is recommended that patients be evaluated for active GPA or relapse in the setting of acute thrombosis whether or not symptoms of active GPA are present.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Talar-Williams C, Hijazi YM, Walther MM, et al. Cyclophosphamide-induced cystitis and bladder cancer in patients with Wegener granulomatosis. Ann Intern Med 1996; 124:477–484.
- Villa-Forte A, Clark TM, Gomes M, et al. Substitution of methotrexate for cyclophosphamide in Wegener granulomatosis: a 12-year single-practice experience. Medicine 2007; 86:269–277.
- Harel S, Fermé C, Poirot C. Management of fertility in patients treated for Hodgkin’s lymphoma [published online ahead of print August 9, 2011]. Haematologica 2011; 96:1692–1699. doi: 10.3324/haematol.2011.045856
- Grossman JM, Gordon R, Ranganath VK, et al. American College of Rheumatology 2010 recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis [published online ahead of print July 26, 2010]. Arthritis Care Res (Hoboken) 2010; 62:1515–1526. doi: 10.1002/acr.20295
- Faurschou M, Mellemkjaer L, Sorensen IJ, Svalgaard Thomsen B, Dreyer L, Baslund B. Increased morbidity from ischemic heart disease in patients with Wegener’s granulomatosis. Arthritis Rheum 2009; 60:1187–1192.
- Suppiah R, Judge A, Batra R, et al. A model to predict cardiovascular events in patients with newly diagnosed Wegener’s granulomatosis and microscopic polyangiitis. Arthritis Care Res (Hoboken) 2011; 63:588–596.
- Merkel PA, Lo GH, Holbrook JT, et al; for Wegener’s Granulomatosis Etanercept Trial Research Group. Brief communication: high incidence of venous thrombotic events among patients with Wegener granulomatosis: the Wegener’s Clinical Occurrence of Thrombosis (WeCLOT) study. Ann Intern Med 2005; 142:620–626.
Safety issues in vasculitis: Infections and immunizations in the immunosuppressed host
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
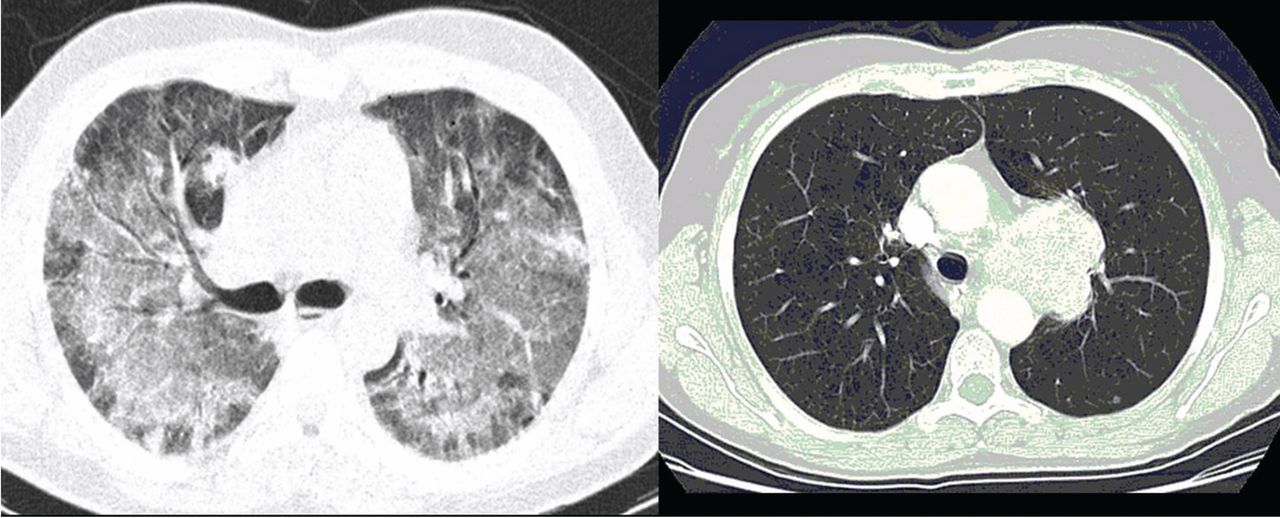
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
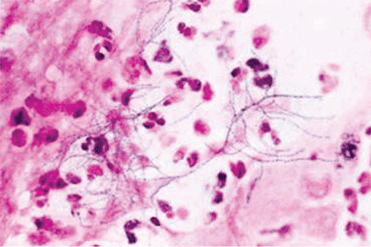
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
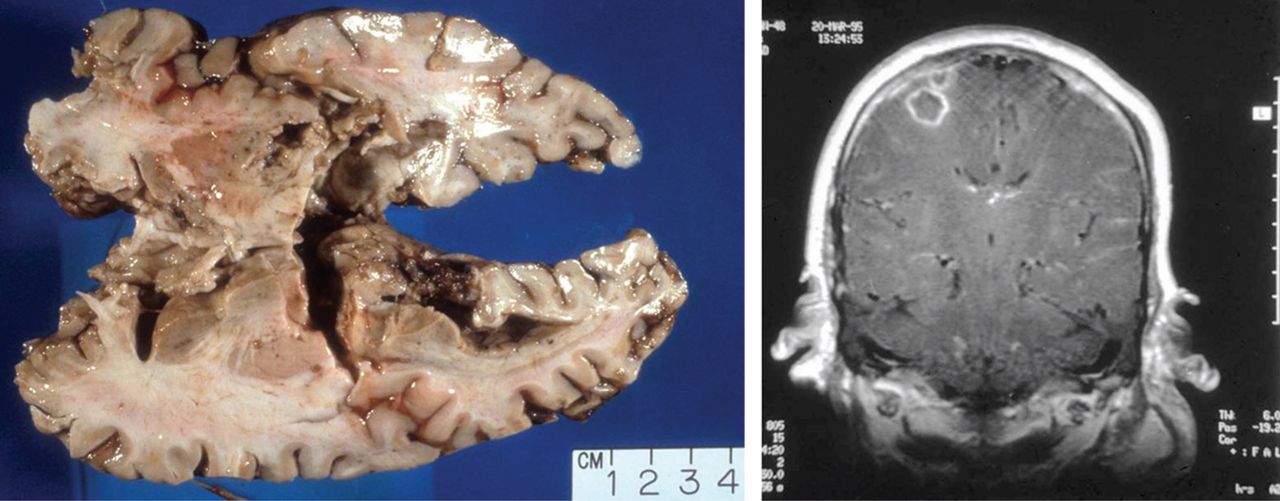
Varicella zoster
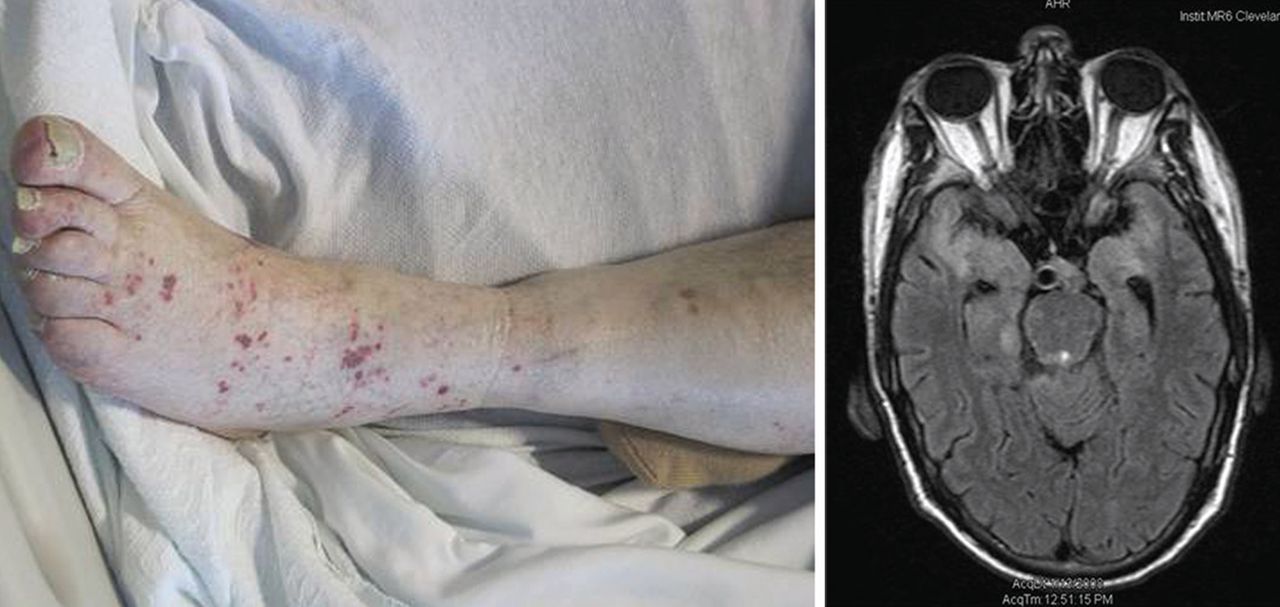
JC virus
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
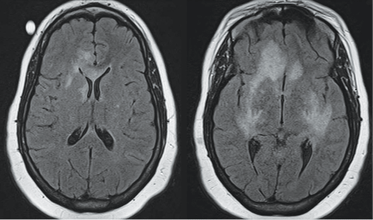
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
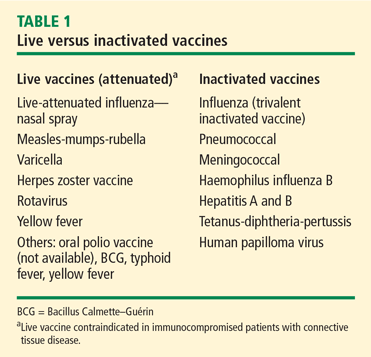
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
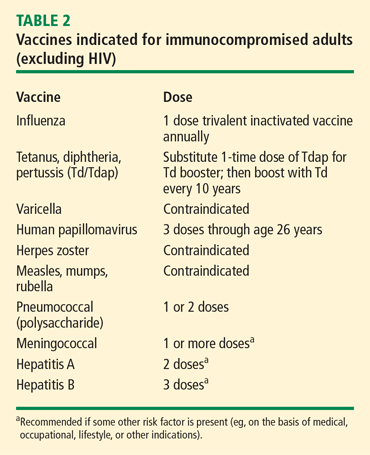
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
Varicella zoster
JC virus
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
In 2007, Falagas et al1 provided a systematic review of studies focusing on infection-related morbidity and mortality in patients with connective tissue diseases. Many of the studies reviewed were published prior to the introduction of biologic agents for the treatment of rheumatologic disorders. In 39 studies focusing on infection incidence, patient outcomes, or both in patients with systemic lupus erythematosus (SLE), rheumatoid arthritis (RA), polymyositis/dermatomyositis, granulomatosis with polyangiitis (GPA, [Wegener’s granulomatosis]), and systemic sclerosis, serious infection developed in 29% of patients and 24% of these died due to the infection with a median attributable mortality of 5.2%. Most of the reported infections were common bacterial syndromes such as pneumonia or bacteremia, and opportunistic fungal (Pneumocystis) infections.
Similarly, in 2006 Alarcón2 reported that 25% to 50% of patients with SLE had significant morbidity primarily from common bacterial infections, with viral, fungal, and parasitic infection less common. Staphylococcus aureus was a common cause of soft tissue infection, septic arthritis, and bacteremia. Streptococcus pneumoniae typically caused respiratory infections, although meningitis and sepsis were reported with SLE. Gram-negative bacteria such as Escherichia coli, Klebsiella species, and Pseudomonas species usually caused urinary tract infections and nosocomial pneumonia. Other bacterial infections included Nocardia species, Mycobacterium tuberculosis, and, rarely, Listeria monocytogenes. The most common viral infection was herpes zoster. Fungal infections included Pneumocystis jirovecii (formerly known as Pneumocystis carinii) and Candida species.
In scleroderma, another connective tissue disease evaluated in the literature by Alarcón,2 reports of bacterial, viral, and fungal infections are limited to case reports. In scleroderma patients, viral infections with cytomegalovirus (CMV), parvovirus B19, and P jirovecii were similar to pathogens observed with SLE.
In polymyositis/dermatomyositis, gram-positive pneumonia affected 15% to 20% of patients and S aureus occurred frequently in the juvenile form of the disease. Herpes zoster was commonly observed, but CMV was relatively rare. Other viral infections included Coxsackie virus, parvovirus B19, and hepatitis C in polymyositis/dermatomyositis. Infection with P jirovecii is frequently fatal in these patients. Other fungal infections seen in polymyositis/dermatomyositis include candidiasis and histoplasmosis.2
Since the approval of antitumor necrosis factor (anti-TNF) agents for RA in the late 1990s, as well as other more recent biologic agents, there has been heightened awareness of infectious complications in rheumatologic patients. A major concern with the anti-TNF agents is the risk of granulomatous infection, particularly mycobacterial disease and dimorphic fungal infections such as histoplasmosis and coccidioidomycosis. Formation of granulomas is the major host defense against mycobacterial infection and is mediated in large part by TNF-alpha. The precise risk of infection associated with each of the various biologic agents is still under study, and rates from randomized trials have differed from postmarketing surveillance studies. Important pathogens associated with biologic agents include Nocardia, CMV, Listeria, Aspergillus, and JC virus (JCV).3,4 Delays in the diagnosis of these infections in immunocompromised patients have led to poor outcomes.
KEY PATHOGENS IN INFECTIONS OF IMMUNOCOMPROMISED HOSTS
Pneumocystis jirovecii
For many decades, P jirovecii was classified as a protozoan but, based on gene sequencing, the organism has been reclassified as a fungus. P jirovecii is a low-virulence, unicellular organism that is the causative agent of Pneumocystis pneumonia (PCP). Epidemiologically, primary infection most likely occurs in infants and children. Colonization may be transient, entering the airways and then resolving over a period of weeks or months. Alternatively, the organism may enter a latent state similar to tuberculosis with reactivation occurring during times of intense immunosuppression. However, molecular epidemiology studies show that new cases of PCP are likely environmentally acquired through multiple exposures rather than reactivation of latent infection.5,6 Transmission is thought to be airborne from person to person. Pathogenically, the trophic form of the organism attaches to type 1 alveolar cells and remains in the extracellular compartment of the alveoli. This colonization evokes an influx of inflammatory cells (CD8 cells, neutrophils, and macrophages). However, not all colonizations result in pneumonia—even in advanced human immunodeficiency virus (HIV) infection. While there is an innate immunity through alveolar macrophages and pulmonary surfactant, alveolar macrophage response is impaired in HIV when the CD4 count is low. Cell-mediated immunity is the main defense against progression to pneumonia with assistance from costimulatory molecules (such as CD28 and CD2) as well as B cells.
Laboratory diagnosis. P jirovecii cannot be grown in culture for clinical purposes, and it is extremely difficult to culture even in the research setting. Cytologic stains such as the Wright-Giemsa and methamine silver stains are the mainstay of laboratory diagnosis. The yield for P jirovecii from routine expectorated sputum is very low and some laboratories discourage this approach. The sensitivity of nebulized sputum using hypertonic saline ranges from 50% to 90%.9
In patients with acquired immune deficiency syndrome (AIDS), bronchoscopy provides 90% to 98% sensitivity by BAL. Transbronchial biopsy may provide some additional yield over BAL in a few situations, such as patients who have been receiving partial P jirovecii prophylaxis. Immunofluorescence techniques using monoclonal antibodies to P jirovecii are commercially available and are first-line diagnostic tools in some laboratories. Recently, polymerase chain reaction (PCR) assay has been introduced into clinical practice as a reproducible test with high sensitivity.
Primary therapy. Primary therapy for PCP consists of trimethoprim-sulfamethoxazole (TMP-SMX) or pentamidine. TMP-SMX is considered the drug of choice and is usually administered intravenously for 21 days in HIV patients and 14 days for non-HIV patients. The oral form may be used in patients with less severe PCP with a functioning gastrointestinal tract. Common adverse reactions to TMP-SMX include rash, Stevens-Johnson syndrome, neutropenia, changes in pulmonary function, and nausea/vomiting/diarrhea.10 Pentamidine is as effective as TMP-SMX, but is associated with renal toxicity, hypotension, severe hypoglycemia, cardiac arrhythmias, and diabetes.11 It is generally reserved for severe cases of PCP in patients who are allergic to or otherwise intolerant of sulfa. Other treatments include atovaquone and trimethoprim-dapsone. Adjunctive corticosteroids have been shown to be beneficial in moderate to severe PCP in HIV patients to reduce the local host inflammatory response to dead or dying organisms. Recent guidelines have recommended corticosteroids for HIV patients with PCP who have an arterial oxygen pressure of 70 mm Hg or less on room air, or an alveolar-arterial (A-a) gradient of oxygen 35 mm Hg or greater.12 Little is known about the role of adjunctive corticosteroids in non-HIV patients, given a lack of clinical studies.
Prevention. Recent estimates of disease burden from a meta-analysis of 11,900 patients with connective tissue diseases found PCP in 12% of patients with GPA, in 6% of those with polydermatomyositis, in 5% of those with SLE, and in 1% of those with RA.1 Mortality due to PCP is higher in patients with rheumatic diseases, ranging from 30% in RA to 63% in GPA, than in those with HIV (10% to 20%).13 One key risk factor predisposing patients with connective tissue diseases to infection with P jirovecii is recent corticosteroid use. Among patients with connective tissue disease, more than 90% of those infected with P jirovecii have recently received steroid therapy.14 Additionally, in almost all patients with P jirovecii, lymphopenia with absolute lymphocyte counts less than 1,000/mm3 is present.15
In patients with HIV, prophylaxis is initiated at a CD4 level of 200/mm3.13 However, the cutoff is less clear for non-HIV rheumatic patients. A cutoff of less than 300 cells/mm3 has been proposed for prophylaxis of PCP. However, at that range, approximately 50% of patients with connective tissue disease would remain above the threshold.13 One possible solution is to screen by PCR and treat colonization. Other algorithms have been proposed, but there is no general consensus on treatment of non-HIV rheumatic patients.13,16 Generally, prophylaxis should be considered in patients at the highest risk for PCP. These include patients taking prednisone at doses greater than 20 mg/day for 1 month plus a cytotoxic agent, a TNF inhibitor plus glucocorticoids, and methotrexate plus glucocorticoids in GPA.13
Nocardia asteroides and Nocardia species
Classically, Nocardia infection results in abscess formation with infiltrates of polymorphonuclear cells, debris, and thin-walled abscesses. The most frequent site of primary infection is pulmonary. Characteristically, multiple pulmonary nodules or cavities are seen, and Nocardia should be considered in the differential diagnosis of an immunocompromised patient with nodular pneumonia. The nodules can also be masslike in appearance (greater than 2 cm). The presentation of new cavitary lung opacities with systemic symptoms may be mistaken for GPA.22Nocardia may disseminate to the central nervous system (CNS), skin, joints, and spine, usually causing suppurative infection at these sites. Nocardia has a very strong tropism for neural tissue. In the CNS, Nocardia can cause single or multiple brain abscesses that may be asymptomatic; patients with pulmonary nocardiosis require imaging to rule out occult CNS involvement.
Nocardia species are resistant to several antibiotics. The treatment of choice for Nocardia species is TMPSMX, but imipenem, amikacin, third-generation cephalosporins, and other options such as minocycline and linezolid may be considered depending on the species and the antimicrobial susceptibility pattern.
Histoplasma capsulatum
Histoplasma capsulatum is a dimorphic fungus that causes disease in both healthy and immunocompromised hosts. The organism differs from other pathogenic fungi in that it is an intracellular organism, mainly involving the reticuloendothelial system, and is rarely in the extracellular space. In the United States, infections are clustered endemically in areas such as the Mississippi and Ohio River Valleys, but infections are common worldwide. The fungus is found in soil, mulch, bird excrement, and bat guano. Asymptomatic or mild infections are common in healthy persons residing in endemic areas and occur on a sporadic basis. Epidemics can occur when contaminated material is aerosolized. Histoplasmosis is also an opportunistic infection in patients with impaired T-cell immunity such as persons with AIDS, organ transplant recipients, hematologic malignancies, and corticosteroid use. Clinically significant cases of histoplasmosis have been described in patients with RA while receiving methotrexate alone, corticosteroids alone, and combinations of disease-modifying agents.23 Histoplasmosis was recently identified in 240 patients in association with TNF inhibitors, translating to 17 per 100,000 patients treated with infliximab.21,24
Pathogenesis. Infection initially occurs through inhalation of contaminated material from the environment, primarily causing pulmonary infection. The organism converts from a mold form in the environment to a pathogenic yeast form in the host. Once inhaled, the mediastinal lymph nodes provide the first line of defense. Following draining of the lymph nodes, the organism enters the bloodstream in both immunocompetent and immunosuppressed patients. It is spread hematogenously into the spleen, liver, and reticulo-endothelial system, where it is eventually cleared. In immunocompetent patients, cellular immunity limits infection within 7 to 14 days and humoral immunity is not protective.25 Granuloma formation is the hallmark of host defense.
Spectrum of illness. Histoplasmosis is associated with a wide spectrum of illness, with presentation ranging from asymptomatic to mild pulmonary illness to overwhelming pneumonia. Symptomatic pulmonary histoplasmosis typically presents with fever, flulike symptoms, and cough, often with retrosternal chest pain. X-rays show patchy or nodular infiltrates, with hilar or mediastinal lymphadenopathy. In some cases the lung parenchyma is clear and the main feature is fever and bilateral hilar adenopathy. Pulmonary histoplasmosis may be difficult to distinguish from sarcoidosis and tuberculosis. Extrapulmonary disease can present as hepatitis, infective endocarditis, and chronic meningitis. In immunocompromised patients, histoplasmosis can present as a progressive disseminated disease which can be acute, subacute, or chronic. Chronic disseminated histoplasmosis is characterized by cough, persistent fever, wasting, hepatosplenomegaly, oral ulcerations, and progressive cytopenias. Acute disseminated histoplasmosis has a much more fulminant course characterized by respiratory insufficiency, hypotension, multisystem organ failure, coagulopathies, and encephalopathy. Histoplasmosis is primarily a pulmonary disease, but in disseminated disease more than 50% of patients have no pulmonary symptoms and 30% may have normal chest x-rays.26 In one series of infliximab-related cases (n = 10), all came from an endemic area 1 week to 6 months after the first dose of infliximab. Patients presented with cough, fever, and shortness of breath.27 The pathogenesis of histoplasmosis in patients receiving TNF inhibitors is not entirely clear; such patients may be suffering a new primary infection, a reinfection, or, least likely, reactivation of latent infection.
Definitive diagnosis requires culture confirmation from appropriate body fluids or identification of characteristic yeast forms from histopathologic sections of tissue biopsies. Serologic tests may also be used to confirm the diagnosis. Detection of H and M precipitins or bands by immunodiffusion is a routine test in many laboratories. M bands are present in 50% of acute cases but their presence does not distinguish acute from remote infection. H bands are present in only 10% of all acute cases, but their presence is very specific for acute histoplasmosis.28
When looking at complement fixation antibodies to yeast (HY) and mycelial (HMy) forms in pulmonary histoplasmosis, a fourfold rise in titer establishes the diagnosis retrospectively, and a single titer greater than 1:32 is strongly suggestive of active infection. However, in progressive disseminated histoplasmosis, the complement fixation antibodies are frequently negative.29 Detection of antigen in urine and serum by enzyme immunoassay has become a mainstay of diagnosis, with a sensitivity of approximately 90% in progressive disseminated disease.30 Of note, most cases of histoplasmosis associated with biologic agents have detectable urinary antigen tests.
Treatment. Acute pulmonary histoplasmosis is usually self-limited, requiring no treatment. The 2007 Infective Diseases Society of America (IDSA) guidelines recommend observation alone in most cases of mild to moderate pulmonary histoplasmosis unless symptoms persist longer than 1 month. For moderately severe or severe acute pulmonary histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 to 5.0 mg/kg/day) or deosycholate amphotericin B (0.7 to 1.0 mg/kg/day) for 1 to 2 weeks followed by itraconazole 200 mg twice daily for a total of 12 weeks. Methylprednisolone at a dose of 0.5 to 1.0 mg/kg/day intravenously for 1 to 2 weeks is also recommended. For moderately severe to severe disseminated histoplasmosis, the IDSA recommends lipid formulations of amphotericin B (3.0 mg/kg/day) for 1 to 2 weeks followed by oral itraconazole 200 mg three times daily for 3 days and then 200 mg twice daily for a total of at least 12 months.31 Commonly, the immunosuppressive agent is held during treatment.
Aspergillus species
Another emerging pathogen is Aspergillus species—a ubiquitous mold spread by aerosols of spores. There are many different species of Aspergillus, but the most common human pathogens include A fumigates, A niger, and A flavus. To date, 39 cases of Aspergillus infection associated with infliximab and etanercept have been reported in the Adverse Event Reporting System, translating to 9 to 12 cases per 100,000 patients.21
Varicella zoster
JC virus
More than 80% of adults are seropositive for JCV, a DNA virus of the genus Polyomavirus that causes lytic infection of oligodendrocytes.34 In immunocompromised hosts, JCV causes progressive multifocal leukoencephalopathy (PML), a rare but devastating demyelinating disease. PML was first described in malignancy, leukemia, and various other immunocompromised states, prior to its strong association with AIDS in the 1980s. More recently, JCV has been associated with natalizumab for multiple sclerosis and Crohn disease, rituximab for oncology patients, efalizumab for psoriasis,35 and mycophenolate mofetil for transplant recipients.36
In 2006 the US Food and Drug Administration issued a safety alert regarding PML in two patients with SLE treated with rituximab and other immunosuppressives.37 In a review of PML in rheumatic disease, 36 cases were identified in patients who had not previously received a biologic agent. Most of these patients (60%) had SLE.38 Of these, many had little or no immunosuppression over the 6 months prior to the diagnosis of PML, suggesting that SLE itself may predispose to PML. Interestingly, PML is rarely associated with TNF inhibitors.
Classic presentation of PML includes motor weakness, aphasia, dysarthria, vision loss, and cognitive loss. Atypical presentation includes seizures, headaches, and brainstem involvement. PML usually spares the optic nerves, spinal cord, peripheral nerves, and muscles. In persons with underlying rheumatic diseases, PML can be difficult to distinguish from neuropsychiatric SLE or CNS vasculitis.
Treatment. In clinical trials no antiviral agent has been effective in the treatment of PML. In HIV patients who develop PML, highly active antiretroviral therapy should be initiated (if antiretroviral-naïve) or existing antiviral regimens optimized. Antiretroviral therapy in this situation may stabilize disease and possibly increase survival.42 For HIV-negative patients who develop PML, the cornerstone of management is immediate decrease or discontinuation of immunosuppression.43 Several adjunctive measures have been reported mainly in natalizumab-associated PML, including corticosteroids, mirtazapine, plasma exchange, and others.
VACCINES
Vaccination is important in the prevention of infectious disease in immunocompromised patients with connective tissue diseases. Because live vaccines are contraindicated in immunocompromised patients, inactivated or component vaccines should be used. It is recommended that patients who will start immunosuppressive therapy be vaccinated 2 to 4 weeks before beginning therapy. If this is not possible, vaccination should be administered during disease remission, 3 months after immunosuppression and 1 to 3 months after administration of high-dose corticosteroids.
- Short-term (less than 14 days)
- At a dose of less than 20 mg/day of prednisone or equivalent
- Long-term on alternate days with short-acting preparations
- At a physiologic dose of prednisone
- Topical, inhaled, intra-articular, bursal, or via tendon.44
Until definitive guidelines are developed, practitioners must evaluate and treat each patient individually to maximize the efficacy of disease treatments while preventing infection morbidity and mortality in their patients with connective tissue diseases.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.
- Falagas ME, Manta KG, Betsi GI, Pappas G. Infection-related morbidity and mortality in patients with connective tissue diseases: a systematic review. Clin Rheumatol 2007; 26:663–670.
- Alarcón GS. Infections in systemic connective tissue diseases: systemic lupus erythematosus, scleroderma, and polymyositis/dermatomyositis. Infect Dis Clin North Am 2006; 20:849–875.
- Crum NF, Lederman ER, Wallace MR. Infections associated with tumor necrosis factor-alpha antagonists. Medicine (Baltimore) 2005; 84:291–302.
- Rychly DJ, DiPiro JT. Infections associated with tumor necrosis factor-alpha antagonists. Pharmacotherapy 2005; 25:1181–1192.
- Wakefield AE, Lindley AR, Ambrose HE, Denis CM, Miller RF. Limited asymptomatic carriage of Pneumocystis jiroveci in human immunodeficiency virus-infected patients [published online ahead of print March 6, 2003]. J Infect Dis 2003; 187:901–908. doi: 10.1086/368165
- Beard CB, Carter JL, Keely SP, et al. Genetic variation in Pneumocystis carinii isolates from different geographic regions: implications for transmission. Emerg Infect Dis 2000; 6:265–272.
- Walzer PD, Smulian AG. Pneumocystis species. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Hartman TE, Primack SL, Müller NL, Staples CA. Diagnosis of thoracic complications in AIDS: accuracy of CT. Am J Roentgenol 1994; 162:547–553.
- Shelhamer JH, Gill VJ, Quinn TC, et al. The laboratory evaluation of opportunistic pulmonary infections. Ann Intern Med 1996; 124:585–599.
- Wharton JM, Coleman DL, Wofsy CB, et al. Trimethoprim-sulfamethoxazole or pentamidine for Pneumocystis carinii pneumonia in the acquired immunodeficiency syndrome. Ann Intern Med 1986; 105:37–44.
- Stein DS, Stevens RC. Treatment-associated toxicities: incidence and mechanisms. In:Sattler FR, Walzer PD, eds. Pneumocystis carinii. London: Bailliere Tindall; 1995:505–530.
- Consensus statement on the use of corticosteroids as adjunctive therapy for Pneumocystis pneumonia in the acquired immunodeficiency syndrome. The National Institutes of Health-University of California Expert Panel for Corticosteroids as Adjunctive Therapy for Pneumocystis Pneumonia. N Engl J Med 1990; 323:1500–1504.
- Stamp LK, Hurst M. Is there a role for consensus guidelines for P. jiroveci pneumonia prophylaxis in immunosuppressed patients with rheumatic diseases? J Rheumatol 2010; 37:686–688.
- Yale S, Limper A. Pneumocystis carinii pneumonia in patients without acquired immunodeficiency syndrome: associated illnesses and prior corticosteroid therapy. Mayo Clin Proc 1996; 71:5–13.
- Sowden E, Carmichael A. Autoimmune inflammatory disorders, systemic corticosteroids and Pneumocystis pneumonia: a strategy for prevention [published online October 16, 2004]. BMC Infect Dis 2004; 4:42. doi: 10.1186/1471-2334-4-42
- Cettomai D, Gelber AC, Christopher-Stine L. A survey of rheumatologists’ practice for prescribing Pneumocystis prophylaxis. J Rheumatol 2010; 37:792–799.
- Keegan JM, Byrd JW. Nocardiosis associated with low dose methotrexate for rheumatoid arthritis. J Rheumatol 1988; 15:1585–1586.
- Gruberg L, Thaler M, Rozenman J, et al. Nocardia asteroides infection complicating rheumatoid arthritis. J Rheumatol 1991; 18:459–461.
- Corneliessen JJ, Bakker LJ, van der Veen MJ, et al. Nocardia asteroides pneumonia complicating low dose methotrexate treatment of refractory rheumatoid arthritis. Ann Rheum Dis 1991; 50;642–644.
- Silva C, Faccioli LH. Tumor necrosis factor and macrophage activation are important in clearance of Nocardia brasiliensis from the livers and spleens of mice. Infect Immun 1992; 60:3566–3570.
- Wallis RS, Broder MS, Wong JY, Hanson ME, Beenhouwer DO. Granulomatous infectious diseases associated with tumor necrosis factor antagonists. Clin Infect Dis 2004; 38:1261–1265.
- Gibb W, Williams A. Nocardiosis mimicking Wegener’s granulomatosis. Scand J Infect Dis 1986; 18:583–585.
- Olson TC, Bongartz T, Crowson CS, Roberts GD, Orenstein R, Matteson EI. Histoplasmosis infection in patients with rheumatoid arthritis, 1998–2009 [published online May 23, 2011]. BMC Infectious Diseases 2011; 11:145. doi: 10.1186/1471-2334-11-145
- Information for Healthcare Professionals: Cimzia (certolizumab pegol), Enbrel etanercept), Humira (adalimumab), and Remicade (infliximab). U.S. Food and Drug Administration Web site. http://www.fda.gov/Drugs/DrugSafety/PostmarketDrugSafetyInformationforPatientsandProviders/ucm124185.htm. Updated January 25, 2010. Accessed September 27, 2012.
- Paya CV, Roberts GD, Cockerill FR. Transient fungemia in acute pulmonary histoplasmosis: detection by new blood-culturing techniques. J Infect Dis 1987; 156:313–315.
- Goodwin RA, Shapiro JL, Thurman GH, Thurman SS, Des Prez RM. Disseminated histoplasmosis: clinical and pathologic correlations. Medicine (Baltimore) 1980; 59:1–33.
- Lee JH, Slifman NR, Gershon SK, et al. Life-threatening histoplasmosis complicating immunotherapy with tumor necrosis factor alpha antagonists infliximab and etanercept. Arthritis Rheum 2002; 46:2565–2570.
- Picardi JL, Kauffman CA, Schwarz J, Phair JP. Detection of precipitating antibodies to Histoplasma capsulatum by counterimmunoelectrophoresis. Am Rev Respir Dis 1976; 114:171–176.
- Deepe GS. Histoplasma capsulatum. In: Mandell GL, Bennett JE, Dolin R, eds. Principles and Practice of Infectious Diseases. 7th ed. Philadelphia, Pa: Elsevier Churchill Livingstone; 2009.
- Kauffman CA. Histoplasmosis: a clinical and laboratory update. Clin Microbiol Rev 2007; 20:115–132.
- Wheat LJ, Freifeld AG, Kleiman MB, et al; Infectious Diseases Society of America. Clinical practice guidelines for the management of patients with histoplasmosis: 2007 update by the Infectious Diseases Society of America [published online ahead of print August 27, 2007]. Clin Infect Dis 2007; 45:807–825. doi: 10.1086/521259
- Johnson RW. Herpes zoster and postherpetic neuralgia. Expert Rev Vaccines 2010; 9( 3 suppl):21–26.
- Wolfe F, Michaud K, Chakravarty EF. Rates and predictors of herpes zoster in patients with rheumatoid arthritis and non-inflammatory musculoskeletal disorders. Rheumatology 2006; 45:1370–1375.
- Weber T, Trebst C, Frye S, et al. Analysis of the systemic and intrathecal humoral immune response in progressive multifocal leukoencephalopathy. J Infect Dis 1997; 176:250–254.
- Kothary N, Diak IL, Brinker A, Bezabeh S, Avigan M, Dal Pan G. Progressive multifocal leukoencephalopathy associated with efalizumab use in psoriasis patients. J Am Acad Dermatol 2011; 65:546–551.
- Neff RT, Hurst FP, Falta EM, et al. Progressive multifocal leukoencephalopathy and use of mycophenolate mofetil after kidney transplantation. Transplantation 2008; 86:1474–1478.
- Rituxan warning. FDA Consum 2007; 41:3.
- Calabrese LH, Molloy ES, Huang D, Ransohoff RM. Progressive multifocal leukoencephalopathy in rheumatic diseases. Arthritis Rheum 2007; 56:2116–2128.
- Cinque P, Scarpellini P, Vago L, Linde A, Lazzarin A. Diagnosis of central nervous system complications in HIV-infected patients: cerebrospinal fluid analysis by the polymerase chain reaction. AIDS 1997; 11:1–17.
- Ryschkewitsch C, Jensen P, Hou J, Fahle G, Fischer S, Major EO. Comparison of PCR-southern hybridization and quantitative real-time PCR for the detection of JC and BK viral nucleotide sequences in urine and cerebrospinal fluid. J Virol Methods 2004; 121:217–221.
- Major EO. History and current concepts in the pathogenesis of PML. Cleve Clin J Med 2011; 78( suppl 2):S3–S7.
- Antinori A, Ammassari A, Giancola ML, et al. Epidemiology and prognosis of AIDS-associated progressive multifocal leukoencephalopathy in the HAART era. J Neurovirol 2001; 7:323–328.
- Calabrese L. A rational approach to PML for the clinician. Cleve Clin J Med 2011; 78 (suppl 2):S38–S41.
- Kroger AT, Sumaya CV, Pickering LK, Atkinson WL. General recommendations on immunization. Recommendations of the Advisory Committee on Immunization Practices (ACIP). MMWR Morb Mortal Wkly Rep 2011; 60:1–60.
Treating vasculitis with conventional immunosuppressive agents
In 1958, shortly after the first descriptions of granulomatosis with polyangiitis, or GPA (Wegener’s granulomatosis), the 1-year mortality was 18%,1 mainly due to renal failure. Physicians tried to combat the disease using various immunosuppressive drugs (nitrogen mustard and, in later years, azathioprine and methotrexate), but measurable success came only after investigators introduced cyclophosphamide (CYC) in combination with the glucocorticoid prednisone.2
A key 1992 study showed that the CYC/prednisone combination markedly improved the disease status in 91% of patients,3 with 75% achieving complete remission. The treatment came at a price, however, with almost all patients suffering serious morbidity or side effects. The results also highlighted concerns about potential malignancies caused by prolonged use of CYC and glucocorticoids. Those concerns motivated the European Vasculitis Study Group in the late 1980s and early 1990s to design and validate testing for antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAV) and pursue consensus regarding treatment.4
ALTERNATIVES TO STANDARD THERAPY
The accepted therapeutic strategy for GPA is to first induce remission using high doses of CYC and then prevent relapse with longer-term, less toxic therapeutic alternatives. These less toxic therapies include newer agents as well as new methods of delivery, particularly for patients with nonsevere forms of disease.
Methotrexate—effective for early treatment
Methotrexate showed early promise in several nonrandomized trials of patients with nonsevere disease. In one such study, de Groot et al subclassified 100 patients at diagnosis according to the extent and severity of the disease.5 Patients were then randomized to receive either standard oral CYC or methotrexate, each combined with prednisolone. Remission rates (90% to 94%) were comparable regardless of whether patients received CYC or methotrexate, although patients with more severe disease who were taking methotrexate took longer to achieve remission. At the same time, relapse rates were higher for methotrexate-taking patients (70%) compared with the CYC group (47%). Thus, while methotrexate could replace CYC for initial treatment of early AAV, CYC had a greater influence on subsequent relapse rates, particularly in patients with more severe forms of disease.
Pulse cyclophosphamide—a new method
Investigators tested pulse delivery of CYC compared with oral daily administration as a means of reducing the CYC dose. An analysis of 14 relatively small studies showed that pulse CYC had the same survival and renal failure rates as continuous therapy.6
One such trial, the CYC Daily Oral Versus Pulsed (CYCLOPS) trial, involved 149 patients with generalized disease (nephritis, GPA, and microscopic polyangiitis [MPA]) who were administered either an intravenous (IV) pulse or a daily oral CYC regimen.7 The pulse CYC neither shortened patients’ time to remission nor increased the proportion of patients who achieved it. Patients receiving pulse CYC suffered one-third the rate of leukopenia experienced by patients who received the oral regimen. Since infection is a source of mortality in vasculitis, this finding is an important consideration when balancing the benefits of day-to-day control offered by oral administration against the safety of at-risk patients such as the elderly.
This treatment strategy may be relevant for patients with renal impairment. It was once thought that patients with renal failure after receiving CYC had more aggressive disease and therefore needed higher dosages. Investigators who studied the impact of renal insufficiency and hemodialysis on the pharmacokinetics of CYC found that clearance of CYC is impaired in patients with reduced renal function.8 Thus, when renal function is suppressed, the CYC dosage should be reduced rather than increased.

Mycophenolate mofetil—efficacy not yet confirmed
Another alternative to CYC, mycophenolate mofetil (MMF), has gained much attention, although its effectiveness is not yet certain. Pilot data show that 13 of 17 patients with MPA achieved remission after 6 months of treatment with MMF.9 Meanwhile, the so-called MYCYC trial, in which patients with newly diagnosed AAV receive either the CYCLOPS regimen or MMF, is under way.10
Deoxyspergualin—remission not sustained
A nonstandard drug that warrants attention is deoxyspergualin (now called gusperimus), licensed in Japan for 15 years. In a prospective, open-label trial of 45 patients with relapsing or refractory GPA, investigators showed that 95% achieved partial remission and 45% full remission, although remission was not sustained when therapy was stopped.11 Because the drug must be administered daily for 21 days by subcutaneous injection, deoxyspergualin is not easy to use. It may represent an alternative, however, because it permitted prednisolone dosage reduction.
EVALUATING RISK AND CHOOSING THERAPIES
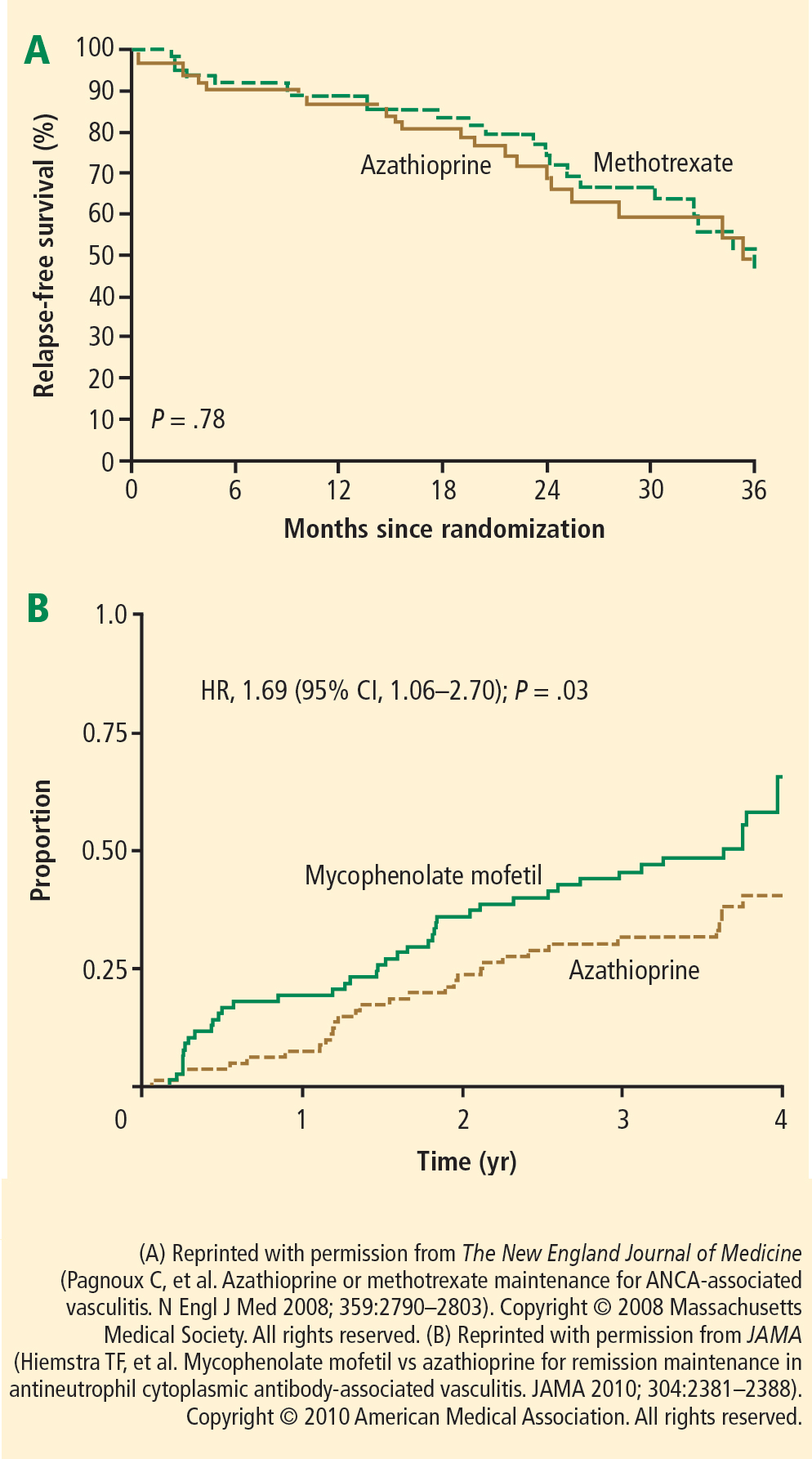
CONSIDERATIONS IN CHOOSING REMISSION THERAPY
Overall, when planning remission therapy and its duration, clinicians must balance the efficacy of CYC and glucocorticoids against their toxicity. Close monitoring and the patient’s capacity to adhere to instructions are two critical issues. Other important considerations include the risk and consequences of relapse, which vary in different circumstances, and the association of cancer with CYC therapy.
Relapse risk is variable
Certain patients are at higher risk of relapse than others. Patients with GPA or proteinase-3-ANCA–positive disease are at higher relapse risk than those who have MPA. ANCA-positive disease in remission or rising ANCA markers both increase the risk of relapse. Ear, nose, throat, and lung diseases increase the likelihood of relapse. Patients with GPA who are Staphylococcus aureus carriers have increased risk. Serum creatinine levels of 2.0 to 3.0 mg/dL at the end of induction therapy should arouse concern about renal relapse.
Most relapses affect the ear, nose, and throat system and do not threaten vital organs. Relapse does not increase the risk of end-stage renal disease or death.
Consider mortality and cancer data
Although the strongest predictor of early death is infection, advanced age and renal impairment also predict death. Chronic kidney disease stage at entry and glomerular filtration rate significantly predict mortality.21 More than 36 g CYC (equivalent to 9 to 12 months of standard oral therapy) increases the risk of bladder cancer 10-fold and myeloid leukemia 60-fold, but the cancer risk is time-dependent; malignancy requires 12 years on average to emerge.22
CONCLUSION
Cyclophosphamide in combination with glucocorticoids remains the standard therapy for GPA and related vasculitides, despite the risk of significant treatment-related comorbidities. Several strategies can be employed to reduce exposure, such as sequential withdrawal of CYC and IV administration. The optimization of glucocorticoid dosing will be a major research focus in the next decade. Newer agents may improve the maintenance of remission; for example, azathioprine and methotrexate show equal efficacy and safety, while MMF is less effective. When planning remission maintenance therapy, the relapse risk should be considered carefully because it varies among clinical scenarios. Other factors in the decision include the consequences for the patient, monitoring requirements, and the patient’s ability to understand and adhere to instructions.
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Novack SN, Pearson CM. Cyclophosphamide therapy in Wegener’s granulomatosis. N Engl J Med 1971; 284:938–942.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Rasmussen N, Jayne DRW, Abramowicz D, et al. European therapeutic trials in ANCA-associated systemic vasculitis: disease scoring, consensus regimens and proposed clinical trials. Clin Exp Immunol 1995; 101 (suppl 1):29–34.
- de Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:2461–2469.
- de Groot K, Adu D, Savage COS; for EUVAS (European Vasculitis Study Group). The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant 2001; 16:2018–2027.
- de Groot K, Harper L, Jayne DRW, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009; 150:670–680.
- Haubitz M, Bohnenstengel F, Brunkhorst R, Schwab M, Hofmann U, Busse D. Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney Int 2002; 61:1495–1501.
- Silva F, Specks U, Kalra S, et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement: a prospective, open-label pilot trial [published online ahead of print January 21, 2010]. Clin J Am Soc Nephrol 2010; 5:445–453. doi: 10.2215/CJN.06010809
- MYCYC clinical trial protocol. The European Vasculitis Society Web site. http://www.vasculitis.org. Updated April 12, 2011. Accessed June 13, 2012.
- Flossmann O, Baslund B, Bruchfeld A, et al. Deoxyspergualin in relapsing and refractory Wegener’s granulomatosis [published online ahead of print August 19, 2008]. Ann Rheum Dis 2009; 68:1125–1130. doi: 10.1136/ard.2008.092429
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of primary small and medium vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:310–317. doi: 10.1136/ard.2008.088096
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:318–323. doi: 10.1136/ard.2008.088351
- Hellmich B, Flossmann O, Gross WL, et al; on behalf of the European Vasculitis Study Group. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis [published online ahead of print December 14, 2006]. Ann Rheum Dis 2007; 66:605–617. doi: 10.1136/ard.2006.062711
- Jayne D, Rasmussen N, Andrassy K, et al; for the European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Pagnoux C, Mahr A, Hamidou MA, et al; for the French Vasculitis Study Group. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008; 359:2790–2803.
- Hiemstra TF, Walsh M, Mahr A, et al; for the European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibodyassociated vasculitis: a randomized controlled trial [published online ahead of print November 8, 2010]. JAMA 2010; 304:2381–2388. doi: 10.1001/jama.2010.1658
- Metzler C, Miehle N, Manger K, et al; for the German Network of Rheumatic Diseases. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis [published online ahead of print May 22, 2007]. Rheumatology 2007; 46:1087–1091. doi: 10.1093/rheumatology/kem029
- Walsh M, Merkel PA, Mahr A, Jayne D. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Arthritis Care Res (Hoboken) 2010; 62:1166–1173.
- Vanková Z, Ríhová Z, Jancová E, Rysavá R, Merta M, Tesar V. Optimizing the therapeutic strategies in ANCA-associated vasculitis— single centre experience with international randomized trials. Prague Med Rep 2006; 107:199–212.
- Flossmann O, Berden A, de Groot K, et al; for the European Vasculitis Study Group. Long-term patient survival in ANCA-associated vasculitis [published online ahead of print November 24, 2010]. Ann Rheum Dis 2011; 70:488–494. doi: 10.1136/ard.2010.137778
- Faurschou M, Sorensen IJ, Mellemkjaer L, et al. Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients [published online ahead of print October 15, 2007]. J Rheumatol 2008; 35:100–105.
In 1958, shortly after the first descriptions of granulomatosis with polyangiitis, or GPA (Wegener’s granulomatosis), the 1-year mortality was 18%,1 mainly due to renal failure. Physicians tried to combat the disease using various immunosuppressive drugs (nitrogen mustard and, in later years, azathioprine and methotrexate), but measurable success came only after investigators introduced cyclophosphamide (CYC) in combination with the glucocorticoid prednisone.2
A key 1992 study showed that the CYC/prednisone combination markedly improved the disease status in 91% of patients,3 with 75% achieving complete remission. The treatment came at a price, however, with almost all patients suffering serious morbidity or side effects. The results also highlighted concerns about potential malignancies caused by prolonged use of CYC and glucocorticoids. Those concerns motivated the European Vasculitis Study Group in the late 1980s and early 1990s to design and validate testing for antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAV) and pursue consensus regarding treatment.4
ALTERNATIVES TO STANDARD THERAPY
The accepted therapeutic strategy for GPA is to first induce remission using high doses of CYC and then prevent relapse with longer-term, less toxic therapeutic alternatives. These less toxic therapies include newer agents as well as new methods of delivery, particularly for patients with nonsevere forms of disease.
Methotrexate—effective for early treatment
Methotrexate showed early promise in several nonrandomized trials of patients with nonsevere disease. In one such study, de Groot et al subclassified 100 patients at diagnosis according to the extent and severity of the disease.5 Patients were then randomized to receive either standard oral CYC or methotrexate, each combined with prednisolone. Remission rates (90% to 94%) were comparable regardless of whether patients received CYC or methotrexate, although patients with more severe disease who were taking methotrexate took longer to achieve remission. At the same time, relapse rates were higher for methotrexate-taking patients (70%) compared with the CYC group (47%). Thus, while methotrexate could replace CYC for initial treatment of early AAV, CYC had a greater influence on subsequent relapse rates, particularly in patients with more severe forms of disease.
Pulse cyclophosphamide—a new method
Investigators tested pulse delivery of CYC compared with oral daily administration as a means of reducing the CYC dose. An analysis of 14 relatively small studies showed that pulse CYC had the same survival and renal failure rates as continuous therapy.6
One such trial, the CYC Daily Oral Versus Pulsed (CYCLOPS) trial, involved 149 patients with generalized disease (nephritis, GPA, and microscopic polyangiitis [MPA]) who were administered either an intravenous (IV) pulse or a daily oral CYC regimen.7 The pulse CYC neither shortened patients’ time to remission nor increased the proportion of patients who achieved it. Patients receiving pulse CYC suffered one-third the rate of leukopenia experienced by patients who received the oral regimen. Since infection is a source of mortality in vasculitis, this finding is an important consideration when balancing the benefits of day-to-day control offered by oral administration against the safety of at-risk patients such as the elderly.
This treatment strategy may be relevant for patients with renal impairment. It was once thought that patients with renal failure after receiving CYC had more aggressive disease and therefore needed higher dosages. Investigators who studied the impact of renal insufficiency and hemodialysis on the pharmacokinetics of CYC found that clearance of CYC is impaired in patients with reduced renal function.8 Thus, when renal function is suppressed, the CYC dosage should be reduced rather than increased.
Mycophenolate mofetil—efficacy not yet confirmed
Another alternative to CYC, mycophenolate mofetil (MMF), has gained much attention, although its effectiveness is not yet certain. Pilot data show that 13 of 17 patients with MPA achieved remission after 6 months of treatment with MMF.9 Meanwhile, the so-called MYCYC trial, in which patients with newly diagnosed AAV receive either the CYCLOPS regimen or MMF, is under way.10
Deoxyspergualin—remission not sustained
A nonstandard drug that warrants attention is deoxyspergualin (now called gusperimus), licensed in Japan for 15 years. In a prospective, open-label trial of 45 patients with relapsing or refractory GPA, investigators showed that 95% achieved partial remission and 45% full remission, although remission was not sustained when therapy was stopped.11 Because the drug must be administered daily for 21 days by subcutaneous injection, deoxyspergualin is not easy to use. It may represent an alternative, however, because it permitted prednisolone dosage reduction.
EVALUATING RISK AND CHOOSING THERAPIES
CONSIDERATIONS IN CHOOSING REMISSION THERAPY
Overall, when planning remission therapy and its duration, clinicians must balance the efficacy of CYC and glucocorticoids against their toxicity. Close monitoring and the patient’s capacity to adhere to instructions are two critical issues. Other important considerations include the risk and consequences of relapse, which vary in different circumstances, and the association of cancer with CYC therapy.
Relapse risk is variable
Certain patients are at higher risk of relapse than others. Patients with GPA or proteinase-3-ANCA–positive disease are at higher relapse risk than those who have MPA. ANCA-positive disease in remission or rising ANCA markers both increase the risk of relapse. Ear, nose, throat, and lung diseases increase the likelihood of relapse. Patients with GPA who are Staphylococcus aureus carriers have increased risk. Serum creatinine levels of 2.0 to 3.0 mg/dL at the end of induction therapy should arouse concern about renal relapse.
Most relapses affect the ear, nose, and throat system and do not threaten vital organs. Relapse does not increase the risk of end-stage renal disease or death.
Consider mortality and cancer data
Although the strongest predictor of early death is infection, advanced age and renal impairment also predict death. Chronic kidney disease stage at entry and glomerular filtration rate significantly predict mortality.21 More than 36 g CYC (equivalent to 9 to 12 months of standard oral therapy) increases the risk of bladder cancer 10-fold and myeloid leukemia 60-fold, but the cancer risk is time-dependent; malignancy requires 12 years on average to emerge.22
CONCLUSION
Cyclophosphamide in combination with glucocorticoids remains the standard therapy for GPA and related vasculitides, despite the risk of significant treatment-related comorbidities. Several strategies can be employed to reduce exposure, such as sequential withdrawal of CYC and IV administration. The optimization of glucocorticoid dosing will be a major research focus in the next decade. Newer agents may improve the maintenance of remission; for example, azathioprine and methotrexate show equal efficacy and safety, while MMF is less effective. When planning remission maintenance therapy, the relapse risk should be considered carefully because it varies among clinical scenarios. Other factors in the decision include the consequences for the patient, monitoring requirements, and the patient’s ability to understand and adhere to instructions.
In 1958, shortly after the first descriptions of granulomatosis with polyangiitis, or GPA (Wegener’s granulomatosis), the 1-year mortality was 18%,1 mainly due to renal failure. Physicians tried to combat the disease using various immunosuppressive drugs (nitrogen mustard and, in later years, azathioprine and methotrexate), but measurable success came only after investigators introduced cyclophosphamide (CYC) in combination with the glucocorticoid prednisone.2
A key 1992 study showed that the CYC/prednisone combination markedly improved the disease status in 91% of patients,3 with 75% achieving complete remission. The treatment came at a price, however, with almost all patients suffering serious morbidity or side effects. The results also highlighted concerns about potential malignancies caused by prolonged use of CYC and glucocorticoids. Those concerns motivated the European Vasculitis Study Group in the late 1980s and early 1990s to design and validate testing for antineutrophil cytoplasmic antibody (ANCA)–associated vasculitides (AAV) and pursue consensus regarding treatment.4
ALTERNATIVES TO STANDARD THERAPY
The accepted therapeutic strategy for GPA is to first induce remission using high doses of CYC and then prevent relapse with longer-term, less toxic therapeutic alternatives. These less toxic therapies include newer agents as well as new methods of delivery, particularly for patients with nonsevere forms of disease.
Methotrexate—effective for early treatment
Methotrexate showed early promise in several nonrandomized trials of patients with nonsevere disease. In one such study, de Groot et al subclassified 100 patients at diagnosis according to the extent and severity of the disease.5 Patients were then randomized to receive either standard oral CYC or methotrexate, each combined with prednisolone. Remission rates (90% to 94%) were comparable regardless of whether patients received CYC or methotrexate, although patients with more severe disease who were taking methotrexate took longer to achieve remission. At the same time, relapse rates were higher for methotrexate-taking patients (70%) compared with the CYC group (47%). Thus, while methotrexate could replace CYC for initial treatment of early AAV, CYC had a greater influence on subsequent relapse rates, particularly in patients with more severe forms of disease.
Pulse cyclophosphamide—a new method
Investigators tested pulse delivery of CYC compared with oral daily administration as a means of reducing the CYC dose. An analysis of 14 relatively small studies showed that pulse CYC had the same survival and renal failure rates as continuous therapy.6
One such trial, the CYC Daily Oral Versus Pulsed (CYCLOPS) trial, involved 149 patients with generalized disease (nephritis, GPA, and microscopic polyangiitis [MPA]) who were administered either an intravenous (IV) pulse or a daily oral CYC regimen.7 The pulse CYC neither shortened patients’ time to remission nor increased the proportion of patients who achieved it. Patients receiving pulse CYC suffered one-third the rate of leukopenia experienced by patients who received the oral regimen. Since infection is a source of mortality in vasculitis, this finding is an important consideration when balancing the benefits of day-to-day control offered by oral administration against the safety of at-risk patients such as the elderly.
This treatment strategy may be relevant for patients with renal impairment. It was once thought that patients with renal failure after receiving CYC had more aggressive disease and therefore needed higher dosages. Investigators who studied the impact of renal insufficiency and hemodialysis on the pharmacokinetics of CYC found that clearance of CYC is impaired in patients with reduced renal function.8 Thus, when renal function is suppressed, the CYC dosage should be reduced rather than increased.
Mycophenolate mofetil—efficacy not yet confirmed
Another alternative to CYC, mycophenolate mofetil (MMF), has gained much attention, although its effectiveness is not yet certain. Pilot data show that 13 of 17 patients with MPA achieved remission after 6 months of treatment with MMF.9 Meanwhile, the so-called MYCYC trial, in which patients with newly diagnosed AAV receive either the CYCLOPS regimen or MMF, is under way.10
Deoxyspergualin—remission not sustained
A nonstandard drug that warrants attention is deoxyspergualin (now called gusperimus), licensed in Japan for 15 years. In a prospective, open-label trial of 45 patients with relapsing or refractory GPA, investigators showed that 95% achieved partial remission and 45% full remission, although remission was not sustained when therapy was stopped.11 Because the drug must be administered daily for 21 days by subcutaneous injection, deoxyspergualin is not easy to use. It may represent an alternative, however, because it permitted prednisolone dosage reduction.
EVALUATING RISK AND CHOOSING THERAPIES
CONSIDERATIONS IN CHOOSING REMISSION THERAPY
Overall, when planning remission therapy and its duration, clinicians must balance the efficacy of CYC and glucocorticoids against their toxicity. Close monitoring and the patient’s capacity to adhere to instructions are two critical issues. Other important considerations include the risk and consequences of relapse, which vary in different circumstances, and the association of cancer with CYC therapy.
Relapse risk is variable
Certain patients are at higher risk of relapse than others. Patients with GPA or proteinase-3-ANCA–positive disease are at higher relapse risk than those who have MPA. ANCA-positive disease in remission or rising ANCA markers both increase the risk of relapse. Ear, nose, throat, and lung diseases increase the likelihood of relapse. Patients with GPA who are Staphylococcus aureus carriers have increased risk. Serum creatinine levels of 2.0 to 3.0 mg/dL at the end of induction therapy should arouse concern about renal relapse.
Most relapses affect the ear, nose, and throat system and do not threaten vital organs. Relapse does not increase the risk of end-stage renal disease or death.
Consider mortality and cancer data
Although the strongest predictor of early death is infection, advanced age and renal impairment also predict death. Chronic kidney disease stage at entry and glomerular filtration rate significantly predict mortality.21 More than 36 g CYC (equivalent to 9 to 12 months of standard oral therapy) increases the risk of bladder cancer 10-fold and myeloid leukemia 60-fold, but the cancer risk is time-dependent; malignancy requires 12 years on average to emerge.22
CONCLUSION
Cyclophosphamide in combination with glucocorticoids remains the standard therapy for GPA and related vasculitides, despite the risk of significant treatment-related comorbidities. Several strategies can be employed to reduce exposure, such as sequential withdrawal of CYC and IV administration. The optimization of glucocorticoid dosing will be a major research focus in the next decade. Newer agents may improve the maintenance of remission; for example, azathioprine and methotrexate show equal efficacy and safety, while MMF is less effective. When planning remission maintenance therapy, the relapse risk should be considered carefully because it varies among clinical scenarios. Other factors in the decision include the consequences for the patient, monitoring requirements, and the patient’s ability to understand and adhere to instructions.
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Novack SN, Pearson CM. Cyclophosphamide therapy in Wegener’s granulomatosis. N Engl J Med 1971; 284:938–942.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Rasmussen N, Jayne DRW, Abramowicz D, et al. European therapeutic trials in ANCA-associated systemic vasculitis: disease scoring, consensus regimens and proposed clinical trials. Clin Exp Immunol 1995; 101 (suppl 1):29–34.
- de Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:2461–2469.
- de Groot K, Adu D, Savage COS; for EUVAS (European Vasculitis Study Group). The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant 2001; 16:2018–2027.
- de Groot K, Harper L, Jayne DRW, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009; 150:670–680.
- Haubitz M, Bohnenstengel F, Brunkhorst R, Schwab M, Hofmann U, Busse D. Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney Int 2002; 61:1495–1501.
- Silva F, Specks U, Kalra S, et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement: a prospective, open-label pilot trial [published online ahead of print January 21, 2010]. Clin J Am Soc Nephrol 2010; 5:445–453. doi: 10.2215/CJN.06010809
- MYCYC clinical trial protocol. The European Vasculitis Society Web site. http://www.vasculitis.org. Updated April 12, 2011. Accessed June 13, 2012.
- Flossmann O, Baslund B, Bruchfeld A, et al. Deoxyspergualin in relapsing and refractory Wegener’s granulomatosis [published online ahead of print August 19, 2008]. Ann Rheum Dis 2009; 68:1125–1130. doi: 10.1136/ard.2008.092429
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of primary small and medium vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:310–317. doi: 10.1136/ard.2008.088096
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:318–323. doi: 10.1136/ard.2008.088351
- Hellmich B, Flossmann O, Gross WL, et al; on behalf of the European Vasculitis Study Group. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis [published online ahead of print December 14, 2006]. Ann Rheum Dis 2007; 66:605–617. doi: 10.1136/ard.2006.062711
- Jayne D, Rasmussen N, Andrassy K, et al; for the European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Pagnoux C, Mahr A, Hamidou MA, et al; for the French Vasculitis Study Group. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008; 359:2790–2803.
- Hiemstra TF, Walsh M, Mahr A, et al; for the European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibodyassociated vasculitis: a randomized controlled trial [published online ahead of print November 8, 2010]. JAMA 2010; 304:2381–2388. doi: 10.1001/jama.2010.1658
- Metzler C, Miehle N, Manger K, et al; for the German Network of Rheumatic Diseases. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis [published online ahead of print May 22, 2007]. Rheumatology 2007; 46:1087–1091. doi: 10.1093/rheumatology/kem029
- Walsh M, Merkel PA, Mahr A, Jayne D. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Arthritis Care Res (Hoboken) 2010; 62:1166–1173.
- Vanková Z, Ríhová Z, Jancová E, Rysavá R, Merta M, Tesar V. Optimizing the therapeutic strategies in ANCA-associated vasculitis— single centre experience with international randomized trials. Prague Med Rep 2006; 107:199–212.
- Flossmann O, Berden A, de Groot K, et al; for the European Vasculitis Study Group. Long-term patient survival in ANCA-associated vasculitis [published online ahead of print November 24, 2010]. Ann Rheum Dis 2011; 70:488–494. doi: 10.1136/ard.2010.137778
- Faurschou M, Sorensen IJ, Mellemkjaer L, et al. Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients [published online ahead of print October 15, 2007]. J Rheumatol 2008; 35:100–105.
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Novack SN, Pearson CM. Cyclophosphamide therapy in Wegener’s granulomatosis. N Engl J Med 1971; 284:938–942.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Rasmussen N, Jayne DRW, Abramowicz D, et al. European therapeutic trials in ANCA-associated systemic vasculitis: disease scoring, consensus regimens and proposed clinical trials. Clin Exp Immunol 1995; 101 (suppl 1):29–34.
- de Groot K, Rasmussen N, Bacon PA, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:2461–2469.
- de Groot K, Adu D, Savage COS; for EUVAS (European Vasculitis Study Group). The value of pulse cyclophosphamide in ANCA-associated vasculitis: meta-analysis and critical review. Nephrol Dial Transplant 2001; 16:2018–2027.
- de Groot K, Harper L, Jayne DRW, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Ann Intern Med 2009; 150:670–680.
- Haubitz M, Bohnenstengel F, Brunkhorst R, Schwab M, Hofmann U, Busse D. Cyclophosphamide pharmacokinetics and dose requirements in patients with renal insufficiency. Kidney Int 2002; 61:1495–1501.
- Silva F, Specks U, Kalra S, et al. Mycophenolate mofetil for induction and maintenance of remission in microscopic polyangiitis with mild to moderate renal involvement: a prospective, open-label pilot trial [published online ahead of print January 21, 2010]. Clin J Am Soc Nephrol 2010; 5:445–453. doi: 10.2215/CJN.06010809
- MYCYC clinical trial protocol. The European Vasculitis Society Web site. http://www.vasculitis.org. Updated April 12, 2011. Accessed June 13, 2012.
- Flossmann O, Baslund B, Bruchfeld A, et al. Deoxyspergualin in relapsing and refractory Wegener’s granulomatosis [published online ahead of print August 19, 2008]. Ann Rheum Dis 2009; 68:1125–1130. doi: 10.1136/ard.2008.092429
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of primary small and medium vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:310–317. doi: 10.1136/ard.2008.088096
- Mukhtyar C, Guillevin L, Cid MC, et al; for the European Vasculitis Study Group. EULAR recommendations for the management of large vessel vasculitis [published online ahead of print April 15, 2008]. Ann Rheum Dis 2009; 68:318–323. doi: 10.1136/ard.2008.088351
- Hellmich B, Flossmann O, Gross WL, et al; on behalf of the European Vasculitis Study Group. EULAR recommendations for conducting clinical studies and/or clinical trials in systemic vasculitis: focus on anti-neutrophil cytoplasm antibody-associated vasculitis [published online ahead of print December 14, 2006]. Ann Rheum Dis 2007; 66:605–617. doi: 10.1136/ard.2006.062711
- Jayne D, Rasmussen N, Andrassy K, et al; for the European Vasculitis Study Group. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Pagnoux C, Mahr A, Hamidou MA, et al; for the French Vasculitis Study Group. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med 2008; 359:2790–2803.
- Hiemstra TF, Walsh M, Mahr A, et al; for the European Vasculitis Study Group (EUVAS). Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibodyassociated vasculitis: a randomized controlled trial [published online ahead of print November 8, 2010]. JAMA 2010; 304:2381–2388. doi: 10.1001/jama.2010.1658
- Metzler C, Miehle N, Manger K, et al; for the German Network of Rheumatic Diseases. Elevated relapse rate under oral methotrexate versus leflunomide for maintenance of remission in Wegener’s granulomatosis [published online ahead of print May 22, 2007]. Rheumatology 2007; 46:1087–1091. doi: 10.1093/rheumatology/kem029
- Walsh M, Merkel PA, Mahr A, Jayne D. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: a meta-analysis. Arthritis Care Res (Hoboken) 2010; 62:1166–1173.
- Vanková Z, Ríhová Z, Jancová E, Rysavá R, Merta M, Tesar V. Optimizing the therapeutic strategies in ANCA-associated vasculitis— single centre experience with international randomized trials. Prague Med Rep 2006; 107:199–212.
- Flossmann O, Berden A, de Groot K, et al; for the European Vasculitis Study Group. Long-term patient survival in ANCA-associated vasculitis [published online ahead of print November 24, 2010]. Ann Rheum Dis 2011; 70:488–494. doi: 10.1136/ard.2010.137778
- Faurschou M, Sorensen IJ, Mellemkjaer L, et al. Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients [published online ahead of print October 15, 2007]. J Rheumatol 2008; 35:100–105.
Biologic agents in the treatment of granulomatosis with polyangiitis
Granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is a vasculitis that affects the renal and respiratory systems. Remission can be induced in most patients with the combination of glucocorticoids and cyclophosphamide. Unfortunately, patients often suffer disease relapses requiring re-treatment and exposure to the cumulative toxicities of repeated cyclophosphamide use. In recent years, improved understanding of the mechanisms of action of cyclophosphamide has led to investigation of treatment strategies that target the role of B cells more specifically in the pathogenesis of the disease.
This article reviews the results of recent studies involving the use of biologic therapy in the treatment of GPA, with a brief examination of historic events that influenced the design of recent trials.
HISTORICAL PERSPECTIVE
The natural history of GPA was characterized in 19581 in a retrospective study showing that 50% of those afflicted died within 6 months, and 80% died by 18 months. Prednisone and cyclophosphamide changed this dismal outcome. The combination markedly improved the status of 91% to 93% of patients,2,3 with most achieving complete remission. Treatment came with a price, however. Almost all patients suffered serious morbidity or side effects, including chronic renal insufficiency (11% requiring dialysis), recurrent infections, hearing loss, infertility, and diabetes. In addition, most patients (99 of 155 in one study) suffered relapse and a significant number (19 of 155) died because of the disease or its treatment.
Investigators’ pursuit of treatment alternatives included foregoing cyclophosphamide in patients who had limited or early systemic GPA and reducing the duration of treatment for patients with severe disease.4 Studies conducted in the late 1990s defined what eventually became standard therapy for GPA: remission induction with glucocorticoids and methotrexate for limited GPA and with glucocorticoids and cyclophosphamide for severe disease. Following remission induction, after 3 to 6 months cyclophosphamide is replaced by azathioprine or methotrexate for remission maintenance. While helpful, these alternatives still fell short of achieving safe, long-term remission.
THERAPY WITH BIOLOGICS
Targeting tumor necrosis factor
The first randomized placebo-controlled trial of a biologic agent in GPA, the Wegener’s Granulomatosis Etanercept Trial (WGET),4 evaluated whether etanercept, a soluble inhibitor of tumor necrosis factor (TNF), would be an effective adjunct to standard therapy. The results showed that etanercept did not confer any beneficial effect and, in fact, if combined with exposure to cyclophosphamide, etanercept increased the risk for solid tumors. Thus, anti-TNF therapy has a limited or no role in the management of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV).
Targeting B cells
The mechanisms of cyclophosphamide effects on disease activity were not clearly understood. In the late 1970s, however, National Institutes of Health investigators found that cyclophosphamide, at the doses administered for GPA, had a profound effect on B-cell function.5 Later investigations showed that disease activity of GPA was clearly related to the frequency of activated B cells detectable in the peripheral blood, while abnormally activated T cells were also detectable in patients in remission.6 These findings suggested that activated B cells might be responsible for disease activity, whereas persistently activated T cells might explain the chronically relapsing nature of the disease.6
B cells are the precursors of short-lived plasma cells, which are thought to be the primary source of autoantibodies, including ANCA. Based on clinical observations as well as in vitro and some animal model experiments, investigators have ascribed pathogenic roles to ANCA. Consequently, targeting the cells that produce these autoantibodies (short-lived plasma cells of B-cell origin) might form the basis of a novel treatment. Why not target cells of the B-cell lineage, thereby eliminating the short-lived plasma cells that would otherwise produce autoantibodies? This might be achieved with rituximab, a monoclonal antibody directed against the CD20 molecule found on pre-B and mature B cells.7 Our group first successfully deployed this strategy in the early 2000s, followed by an open-label pilot study.8–10
The RAVE trial
The Rituximab in ANCA-Associated Vasculitis (RAVE) trial was a multicenter, randomized, placebo-controlled trial that compared rituximab for remission induction and maintenance with standard therapy consisting of cyclophosphamide followed by azathioprine in patients with severe AAV.11 The results of a pilot trial in 200610 set the stage for the RAVE trial, which hypothesized that treatment with rituximab plus glucocorticoids would not be inferior to daily cyclophosphamide plus glucocorticoids. Both would induce remission and permit discontinuation of prednisone after 6 months.
Nine centers enrolled a total of 197 patients with severe GPA or microscopic polyangiitis (MPA), all positive for ANCA, with active disease severe enough to warrant treatment with prednisone and cyclophosphamide. All participants received 1 to 3 g of methyl-prednisolone intravenously followed by prednisone (1 mg/kg per day). The treatment group received rituximab (375 mg/m2 once weekly for 4 weeks) and the control group received standard therapy with cyclophosphamide (2 mg/kg per day) followed by azathioprine (2 mg/kg per day) after 3 to 6 months when remission was achieved.
The primary end point was complete remission, defined as a Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG) of 0 and successful tapering of prednisone by 6 months. Secondary end points included rates of disease flares, cumulative glucocorticoid doses, rates of adverse events, and Medical Outcomes Study 36-item short-form health survey (SF-36, a measure of quality of life) scores. Among patients receiving rituximab, 64% reached the primary end point compared with 53% of patients in the control group. Rituximab was judged not inferior to standard therapy.
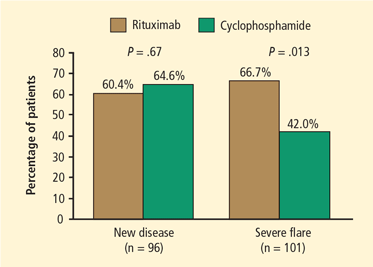
Results were similar for the secondary end point of disease remission while taking less than 10 mg/d of prednisone, with 71% of rituximab patients and 62% of control-group patients achieving remission. Rituximab was also as effective as cyclophosphamide in the treatment of patients with major renal disease or alveolar hemorrhage. Most strikingly, rituximab proved superior to the cyclophosphamide-based regimen for inducing remission in patients who entered the trial with relapsing disease (67% rituximab versus 42% cyclophosphamide) (Figure 1). Those who entered the trial with a new diagnosis did not show the same difference in efficacy.
Rituximab also proved significantly more effective than cyclophosphamide for patients who had proteinase-3 (PR3) ANCA, whereas the efficacy of both agents was equivalent among patients who had myeloperoxidase ANCA. Patients in the cyclophosphamide arm experienced more leukopenia compared with the rituximab arm, but this did not lead to more infections.
In summary, the RAVE trial showed that rituximab matched the efficacy of cyclophosphamide (standard therapy) in inducing remission in patients with severe AAV. The results held true for subsets of patients with major renal disease and those with alveolar hemorrhage. Most strikingly, among patients who entered the trial with a severe relapse, those who received rituximab responded better than those treated with cyclophosphamide. There were no significant differences in flare rates by 6 months and no difference in the rate of severe adverse events. However, participants receiving cyclophosphamide experienced more selected adverse events, particularly leukopenias.
Clinically speaking, rituximab represents the first proven alternative to cyclophosphamide for remission induction in this patient population. The treatment presents the preferred option for patients interested in preserving fertility or who need to be re-treated for a severe disease flare. Based on these data, the US Food and Drug Administration recently extended the labeling of rituximab for treatment of GPA and MPA.
The RITUXVAS trial
The European Vasculitis Study Group (EUVAS) launched another trial comparing the efficacy of rituximab with cyclophosphamide for remission induction.12 The trial design differed from that of the RAVE trial in that investigators did not discontinue prednisone in all patients, followed patients for 12 months, and assessed sustained remission as the primary end point. In this trial, patients in the rituximab arm also received two single intravenous cyclophosphamide infusions, and cyclophosphamide in the control arm was given intravenously. All 44 patients enrolled in the trial and randomized 3:1 to the rituximab versus the cyclophosphamide control arm were ANCA-positive and had active renal disease. The patient population overall was older and had more severe renal disease than the patients enrolled in the RAVE trial. Overall, one course of rituximab achieved the same results as 6 months of intravenous pulse cyclophosphamide followed by oral azathioprine in terms of rate of sustained remission at 12 months, time to relapse, improvement of renal function, and rate of adverse events.
Mayo Clinic cohort study
Our group at Mayo Clinic evaluated the safety and effectiveness of rituximab when used repeatedly in order to maintain long-term remission.13 The study involved 53 patients who had a long-term (10 years, on average) diagnosis of refractory AAV. The patients received, on average, four courses of rituximab. All of these patients had GPA and all but one were PR3-ANCA–positive.
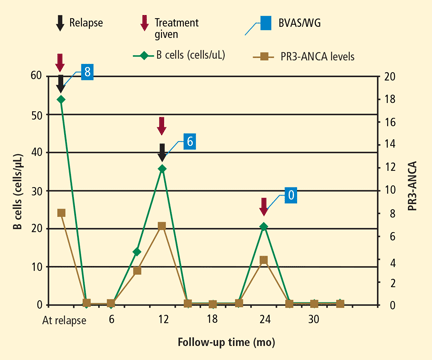
In this cohort, rituximab was effective and safe for induction and maintenance of remission in patients with relapsing GPA. The study showed that B-cell depletion effectively maintains remission in these patients, supporting the notion that B cells play an important role in GPA. Because rituximab works by depleting B cells and ANCA, timing of re-treatment can be individualized based on B-cell counts and ANCA levels. Thus, rituximab represents a promising alternative to standard therapy and a means for long-term patient management, particularly for those in whom other agents have failed to achieve or maintain remission in the past.
On a cautionary note, rituximab is an immunosuppressive agent. Risk of infection during treatment seems similar to that associated with carefully monitored cyclophosphamide followed by azathioprine. To avoid complications, physicians should also maintain Pneumocystis prophylaxis for at least the duration of B-cell depletion.
CONCLUSION
Enhanced understanding of the mechanism of action of cyclophosphamide has led to investigation of the role of B cells in the development of AAV and, from there, to the potential for treatment with biologics such as rituximab. Rituximab is equivalent in efficacy to cyclophosphamide for remission induction in AAV. It effectively restores remission and prevents relapse, and it is a better option than cyclophosphamide for PR3-ANCA–associated relapsing vasculitis. Future investigations should further address how to best prevent relapses after B-cell reconstitution.
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Cupps TR, Edgar LC, Fauci AS. Suppression of human B lymphocyte function by cyclophosphamide. J Immunol 1982; 128:2453–2457.
- Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B- and T-cell activation in Wegener’s granulomatosis. J Allergy Clin Immunol 1999; 103:885–894.
- Salama AD, Pusey CD. Drug insight: rituximab in renal disease and transplantation. Nat Clin Pract Nephrol 2006; 2:221–230.
- Specks U, Fervenza FC, McDonald TJ, Hogan MC. Response of Wegener’s granulomatosis to anti-CD20 chimeric monoclonal antibody therapy. Arthritis Rheum 2001; 44:2836–2840.
- Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:262–268.
- Keogh KA, Ytterberg SR, Fervenza FC, Carlson KA, Schroeder DR, Specks U. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial [published online ahead of print October 13, 2005]. Am J Respir Crit Care Med 2006; 173:180–187. doi: 10.1164/rccm.200507-1144OC
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010; 363:211–220.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
Granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is a vasculitis that affects the renal and respiratory systems. Remission can be induced in most patients with the combination of glucocorticoids and cyclophosphamide. Unfortunately, patients often suffer disease relapses requiring re-treatment and exposure to the cumulative toxicities of repeated cyclophosphamide use. In recent years, improved understanding of the mechanisms of action of cyclophosphamide has led to investigation of treatment strategies that target the role of B cells more specifically in the pathogenesis of the disease.
This article reviews the results of recent studies involving the use of biologic therapy in the treatment of GPA, with a brief examination of historic events that influenced the design of recent trials.
HISTORICAL PERSPECTIVE
The natural history of GPA was characterized in 19581 in a retrospective study showing that 50% of those afflicted died within 6 months, and 80% died by 18 months. Prednisone and cyclophosphamide changed this dismal outcome. The combination markedly improved the status of 91% to 93% of patients,2,3 with most achieving complete remission. Treatment came with a price, however. Almost all patients suffered serious morbidity or side effects, including chronic renal insufficiency (11% requiring dialysis), recurrent infections, hearing loss, infertility, and diabetes. In addition, most patients (99 of 155 in one study) suffered relapse and a significant number (19 of 155) died because of the disease or its treatment.
Investigators’ pursuit of treatment alternatives included foregoing cyclophosphamide in patients who had limited or early systemic GPA and reducing the duration of treatment for patients with severe disease.4 Studies conducted in the late 1990s defined what eventually became standard therapy for GPA: remission induction with glucocorticoids and methotrexate for limited GPA and with glucocorticoids and cyclophosphamide for severe disease. Following remission induction, after 3 to 6 months cyclophosphamide is replaced by azathioprine or methotrexate for remission maintenance. While helpful, these alternatives still fell short of achieving safe, long-term remission.
THERAPY WITH BIOLOGICS
Targeting tumor necrosis factor
The first randomized placebo-controlled trial of a biologic agent in GPA, the Wegener’s Granulomatosis Etanercept Trial (WGET),4 evaluated whether etanercept, a soluble inhibitor of tumor necrosis factor (TNF), would be an effective adjunct to standard therapy. The results showed that etanercept did not confer any beneficial effect and, in fact, if combined with exposure to cyclophosphamide, etanercept increased the risk for solid tumors. Thus, anti-TNF therapy has a limited or no role in the management of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV).
Targeting B cells
The mechanisms of cyclophosphamide effects on disease activity were not clearly understood. In the late 1970s, however, National Institutes of Health investigators found that cyclophosphamide, at the doses administered for GPA, had a profound effect on B-cell function.5 Later investigations showed that disease activity of GPA was clearly related to the frequency of activated B cells detectable in the peripheral blood, while abnormally activated T cells were also detectable in patients in remission.6 These findings suggested that activated B cells might be responsible for disease activity, whereas persistently activated T cells might explain the chronically relapsing nature of the disease.6
B cells are the precursors of short-lived plasma cells, which are thought to be the primary source of autoantibodies, including ANCA. Based on clinical observations as well as in vitro and some animal model experiments, investigators have ascribed pathogenic roles to ANCA. Consequently, targeting the cells that produce these autoantibodies (short-lived plasma cells of B-cell origin) might form the basis of a novel treatment. Why not target cells of the B-cell lineage, thereby eliminating the short-lived plasma cells that would otherwise produce autoantibodies? This might be achieved with rituximab, a monoclonal antibody directed against the CD20 molecule found on pre-B and mature B cells.7 Our group first successfully deployed this strategy in the early 2000s, followed by an open-label pilot study.8–10
The RAVE trial
The Rituximab in ANCA-Associated Vasculitis (RAVE) trial was a multicenter, randomized, placebo-controlled trial that compared rituximab for remission induction and maintenance with standard therapy consisting of cyclophosphamide followed by azathioprine in patients with severe AAV.11 The results of a pilot trial in 200610 set the stage for the RAVE trial, which hypothesized that treatment with rituximab plus glucocorticoids would not be inferior to daily cyclophosphamide plus glucocorticoids. Both would induce remission and permit discontinuation of prednisone after 6 months.
Nine centers enrolled a total of 197 patients with severe GPA or microscopic polyangiitis (MPA), all positive for ANCA, with active disease severe enough to warrant treatment with prednisone and cyclophosphamide. All participants received 1 to 3 g of methyl-prednisolone intravenously followed by prednisone (1 mg/kg per day). The treatment group received rituximab (375 mg/m2 once weekly for 4 weeks) and the control group received standard therapy with cyclophosphamide (2 mg/kg per day) followed by azathioprine (2 mg/kg per day) after 3 to 6 months when remission was achieved.
The primary end point was complete remission, defined as a Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG) of 0 and successful tapering of prednisone by 6 months. Secondary end points included rates of disease flares, cumulative glucocorticoid doses, rates of adverse events, and Medical Outcomes Study 36-item short-form health survey (SF-36, a measure of quality of life) scores. Among patients receiving rituximab, 64% reached the primary end point compared with 53% of patients in the control group. Rituximab was judged not inferior to standard therapy.
Results were similar for the secondary end point of disease remission while taking less than 10 mg/d of prednisone, with 71% of rituximab patients and 62% of control-group patients achieving remission. Rituximab was also as effective as cyclophosphamide in the treatment of patients with major renal disease or alveolar hemorrhage. Most strikingly, rituximab proved superior to the cyclophosphamide-based regimen for inducing remission in patients who entered the trial with relapsing disease (67% rituximab versus 42% cyclophosphamide) (Figure 1). Those who entered the trial with a new diagnosis did not show the same difference in efficacy.
Rituximab also proved significantly more effective than cyclophosphamide for patients who had proteinase-3 (PR3) ANCA, whereas the efficacy of both agents was equivalent among patients who had myeloperoxidase ANCA. Patients in the cyclophosphamide arm experienced more leukopenia compared with the rituximab arm, but this did not lead to more infections.
In summary, the RAVE trial showed that rituximab matched the efficacy of cyclophosphamide (standard therapy) in inducing remission in patients with severe AAV. The results held true for subsets of patients with major renal disease and those with alveolar hemorrhage. Most strikingly, among patients who entered the trial with a severe relapse, those who received rituximab responded better than those treated with cyclophosphamide. There were no significant differences in flare rates by 6 months and no difference in the rate of severe adverse events. However, participants receiving cyclophosphamide experienced more selected adverse events, particularly leukopenias.
Clinically speaking, rituximab represents the first proven alternative to cyclophosphamide for remission induction in this patient population. The treatment presents the preferred option for patients interested in preserving fertility or who need to be re-treated for a severe disease flare. Based on these data, the US Food and Drug Administration recently extended the labeling of rituximab for treatment of GPA and MPA.
The RITUXVAS trial
The European Vasculitis Study Group (EUVAS) launched another trial comparing the efficacy of rituximab with cyclophosphamide for remission induction.12 The trial design differed from that of the RAVE trial in that investigators did not discontinue prednisone in all patients, followed patients for 12 months, and assessed sustained remission as the primary end point. In this trial, patients in the rituximab arm also received two single intravenous cyclophosphamide infusions, and cyclophosphamide in the control arm was given intravenously. All 44 patients enrolled in the trial and randomized 3:1 to the rituximab versus the cyclophosphamide control arm were ANCA-positive and had active renal disease. The patient population overall was older and had more severe renal disease than the patients enrolled in the RAVE trial. Overall, one course of rituximab achieved the same results as 6 months of intravenous pulse cyclophosphamide followed by oral azathioprine in terms of rate of sustained remission at 12 months, time to relapse, improvement of renal function, and rate of adverse events.
Mayo Clinic cohort study
Our group at Mayo Clinic evaluated the safety and effectiveness of rituximab when used repeatedly in order to maintain long-term remission.13 The study involved 53 patients who had a long-term (10 years, on average) diagnosis of refractory AAV. The patients received, on average, four courses of rituximab. All of these patients had GPA and all but one were PR3-ANCA–positive.
In this cohort, rituximab was effective and safe for induction and maintenance of remission in patients with relapsing GPA. The study showed that B-cell depletion effectively maintains remission in these patients, supporting the notion that B cells play an important role in GPA. Because rituximab works by depleting B cells and ANCA, timing of re-treatment can be individualized based on B-cell counts and ANCA levels. Thus, rituximab represents a promising alternative to standard therapy and a means for long-term patient management, particularly for those in whom other agents have failed to achieve or maintain remission in the past.
On a cautionary note, rituximab is an immunosuppressive agent. Risk of infection during treatment seems similar to that associated with carefully monitored cyclophosphamide followed by azathioprine. To avoid complications, physicians should also maintain Pneumocystis prophylaxis for at least the duration of B-cell depletion.
CONCLUSION
Enhanced understanding of the mechanism of action of cyclophosphamide has led to investigation of the role of B cells in the development of AAV and, from there, to the potential for treatment with biologics such as rituximab. Rituximab is equivalent in efficacy to cyclophosphamide for remission induction in AAV. It effectively restores remission and prevents relapse, and it is a better option than cyclophosphamide for PR3-ANCA–associated relapsing vasculitis. Future investigations should further address how to best prevent relapses after B-cell reconstitution.
Granulomatosis with polyangiitis (GPA [Wegener’s granulomatosis]) is a vasculitis that affects the renal and respiratory systems. Remission can be induced in most patients with the combination of glucocorticoids and cyclophosphamide. Unfortunately, patients often suffer disease relapses requiring re-treatment and exposure to the cumulative toxicities of repeated cyclophosphamide use. In recent years, improved understanding of the mechanisms of action of cyclophosphamide has led to investigation of treatment strategies that target the role of B cells more specifically in the pathogenesis of the disease.
This article reviews the results of recent studies involving the use of biologic therapy in the treatment of GPA, with a brief examination of historic events that influenced the design of recent trials.
HISTORICAL PERSPECTIVE
The natural history of GPA was characterized in 19581 in a retrospective study showing that 50% of those afflicted died within 6 months, and 80% died by 18 months. Prednisone and cyclophosphamide changed this dismal outcome. The combination markedly improved the status of 91% to 93% of patients,2,3 with most achieving complete remission. Treatment came with a price, however. Almost all patients suffered serious morbidity or side effects, including chronic renal insufficiency (11% requiring dialysis), recurrent infections, hearing loss, infertility, and diabetes. In addition, most patients (99 of 155 in one study) suffered relapse and a significant number (19 of 155) died because of the disease or its treatment.
Investigators’ pursuit of treatment alternatives included foregoing cyclophosphamide in patients who had limited or early systemic GPA and reducing the duration of treatment for patients with severe disease.4 Studies conducted in the late 1990s defined what eventually became standard therapy for GPA: remission induction with glucocorticoids and methotrexate for limited GPA and with glucocorticoids and cyclophosphamide for severe disease. Following remission induction, after 3 to 6 months cyclophosphamide is replaced by azathioprine or methotrexate for remission maintenance. While helpful, these alternatives still fell short of achieving safe, long-term remission.
THERAPY WITH BIOLOGICS
Targeting tumor necrosis factor
The first randomized placebo-controlled trial of a biologic agent in GPA, the Wegener’s Granulomatosis Etanercept Trial (WGET),4 evaluated whether etanercept, a soluble inhibitor of tumor necrosis factor (TNF), would be an effective adjunct to standard therapy. The results showed that etanercept did not confer any beneficial effect and, in fact, if combined with exposure to cyclophosphamide, etanercept increased the risk for solid tumors. Thus, anti-TNF therapy has a limited or no role in the management of antineutrophil cytoplasmic antibody (ANCA)-associated vasculitis (AAV).
Targeting B cells
The mechanisms of cyclophosphamide effects on disease activity were not clearly understood. In the late 1970s, however, National Institutes of Health investigators found that cyclophosphamide, at the doses administered for GPA, had a profound effect on B-cell function.5 Later investigations showed that disease activity of GPA was clearly related to the frequency of activated B cells detectable in the peripheral blood, while abnormally activated T cells were also detectable in patients in remission.6 These findings suggested that activated B cells might be responsible for disease activity, whereas persistently activated T cells might explain the chronically relapsing nature of the disease.6
B cells are the precursors of short-lived plasma cells, which are thought to be the primary source of autoantibodies, including ANCA. Based on clinical observations as well as in vitro and some animal model experiments, investigators have ascribed pathogenic roles to ANCA. Consequently, targeting the cells that produce these autoantibodies (short-lived plasma cells of B-cell origin) might form the basis of a novel treatment. Why not target cells of the B-cell lineage, thereby eliminating the short-lived plasma cells that would otherwise produce autoantibodies? This might be achieved with rituximab, a monoclonal antibody directed against the CD20 molecule found on pre-B and mature B cells.7 Our group first successfully deployed this strategy in the early 2000s, followed by an open-label pilot study.8–10
The RAVE trial
The Rituximab in ANCA-Associated Vasculitis (RAVE) trial was a multicenter, randomized, placebo-controlled trial that compared rituximab for remission induction and maintenance with standard therapy consisting of cyclophosphamide followed by azathioprine in patients with severe AAV.11 The results of a pilot trial in 200610 set the stage for the RAVE trial, which hypothesized that treatment with rituximab plus glucocorticoids would not be inferior to daily cyclophosphamide plus glucocorticoids. Both would induce remission and permit discontinuation of prednisone after 6 months.
Nine centers enrolled a total of 197 patients with severe GPA or microscopic polyangiitis (MPA), all positive for ANCA, with active disease severe enough to warrant treatment with prednisone and cyclophosphamide. All participants received 1 to 3 g of methyl-prednisolone intravenously followed by prednisone (1 mg/kg per day). The treatment group received rituximab (375 mg/m2 once weekly for 4 weeks) and the control group received standard therapy with cyclophosphamide (2 mg/kg per day) followed by azathioprine (2 mg/kg per day) after 3 to 6 months when remission was achieved.
The primary end point was complete remission, defined as a Birmingham Vasculitis Activity Score for Wegener’s Granulomatosis (BVAS/WG) of 0 and successful tapering of prednisone by 6 months. Secondary end points included rates of disease flares, cumulative glucocorticoid doses, rates of adverse events, and Medical Outcomes Study 36-item short-form health survey (SF-36, a measure of quality of life) scores. Among patients receiving rituximab, 64% reached the primary end point compared with 53% of patients in the control group. Rituximab was judged not inferior to standard therapy.
Results were similar for the secondary end point of disease remission while taking less than 10 mg/d of prednisone, with 71% of rituximab patients and 62% of control-group patients achieving remission. Rituximab was also as effective as cyclophosphamide in the treatment of patients with major renal disease or alveolar hemorrhage. Most strikingly, rituximab proved superior to the cyclophosphamide-based regimen for inducing remission in patients who entered the trial with relapsing disease (67% rituximab versus 42% cyclophosphamide) (Figure 1). Those who entered the trial with a new diagnosis did not show the same difference in efficacy.
Rituximab also proved significantly more effective than cyclophosphamide for patients who had proteinase-3 (PR3) ANCA, whereas the efficacy of both agents was equivalent among patients who had myeloperoxidase ANCA. Patients in the cyclophosphamide arm experienced more leukopenia compared with the rituximab arm, but this did not lead to more infections.
In summary, the RAVE trial showed that rituximab matched the efficacy of cyclophosphamide (standard therapy) in inducing remission in patients with severe AAV. The results held true for subsets of patients with major renal disease and those with alveolar hemorrhage. Most strikingly, among patients who entered the trial with a severe relapse, those who received rituximab responded better than those treated with cyclophosphamide. There were no significant differences in flare rates by 6 months and no difference in the rate of severe adverse events. However, participants receiving cyclophosphamide experienced more selected adverse events, particularly leukopenias.
Clinically speaking, rituximab represents the first proven alternative to cyclophosphamide for remission induction in this patient population. The treatment presents the preferred option for patients interested in preserving fertility or who need to be re-treated for a severe disease flare. Based on these data, the US Food and Drug Administration recently extended the labeling of rituximab for treatment of GPA and MPA.
The RITUXVAS trial
The European Vasculitis Study Group (EUVAS) launched another trial comparing the efficacy of rituximab with cyclophosphamide for remission induction.12 The trial design differed from that of the RAVE trial in that investigators did not discontinue prednisone in all patients, followed patients for 12 months, and assessed sustained remission as the primary end point. In this trial, patients in the rituximab arm also received two single intravenous cyclophosphamide infusions, and cyclophosphamide in the control arm was given intravenously. All 44 patients enrolled in the trial and randomized 3:1 to the rituximab versus the cyclophosphamide control arm were ANCA-positive and had active renal disease. The patient population overall was older and had more severe renal disease than the patients enrolled in the RAVE trial. Overall, one course of rituximab achieved the same results as 6 months of intravenous pulse cyclophosphamide followed by oral azathioprine in terms of rate of sustained remission at 12 months, time to relapse, improvement of renal function, and rate of adverse events.
Mayo Clinic cohort study
Our group at Mayo Clinic evaluated the safety and effectiveness of rituximab when used repeatedly in order to maintain long-term remission.13 The study involved 53 patients who had a long-term (10 years, on average) diagnosis of refractory AAV. The patients received, on average, four courses of rituximab. All of these patients had GPA and all but one were PR3-ANCA–positive.
In this cohort, rituximab was effective and safe for induction and maintenance of remission in patients with relapsing GPA. The study showed that B-cell depletion effectively maintains remission in these patients, supporting the notion that B cells play an important role in GPA. Because rituximab works by depleting B cells and ANCA, timing of re-treatment can be individualized based on B-cell counts and ANCA levels. Thus, rituximab represents a promising alternative to standard therapy and a means for long-term patient management, particularly for those in whom other agents have failed to achieve or maintain remission in the past.
On a cautionary note, rituximab is an immunosuppressive agent. Risk of infection during treatment seems similar to that associated with carefully monitored cyclophosphamide followed by azathioprine. To avoid complications, physicians should also maintain Pneumocystis prophylaxis for at least the duration of B-cell depletion.
CONCLUSION
Enhanced understanding of the mechanism of action of cyclophosphamide has led to investigation of the role of B cells in the development of AAV and, from there, to the potential for treatment with biologics such as rituximab. Rituximab is equivalent in efficacy to cyclophosphamide for remission induction in AAV. It effectively restores remission and prevents relapse, and it is a better option than cyclophosphamide for PR3-ANCA–associated relapsing vasculitis. Future investigations should further address how to best prevent relapses after B-cell reconstitution.
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Cupps TR, Edgar LC, Fauci AS. Suppression of human B lymphocyte function by cyclophosphamide. J Immunol 1982; 128:2453–2457.
- Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B- and T-cell activation in Wegener’s granulomatosis. J Allergy Clin Immunol 1999; 103:885–894.
- Salama AD, Pusey CD. Drug insight: rituximab in renal disease and transplantation. Nat Clin Pract Nephrol 2006; 2:221–230.
- Specks U, Fervenza FC, McDonald TJ, Hogan MC. Response of Wegener’s granulomatosis to anti-CD20 chimeric monoclonal antibody therapy. Arthritis Rheum 2001; 44:2836–2840.
- Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:262–268.
- Keogh KA, Ytterberg SR, Fervenza FC, Carlson KA, Schroeder DR, Specks U. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial [published online ahead of print October 13, 2005]. Am J Respir Crit Care Med 2006; 173:180–187. doi: 10.1164/rccm.200507-1144OC
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010; 363:211–220.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
- Walton EW. Giant-cell granuloma of the respiratory tract (Wegener’s granulomatosis). Br Med J 1958; 2:265–270.
- Hoffman GS, Kerr GS, Leavitt RY, et al. Wegener granulomatosis: an analysis of 158 patients. Ann Intern Med 1992; 116:488–498.
- Reinhold-Keller E, Beuge N, Latza U, et al. An interdisciplinary approach to the care of patients with Wegener’s granulomatosis: long-term outcome in 155 patients. Arthritis Rheum 2000; 43:1021–1032.
- Jayne D, Rasmussen N, Andrassy K, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med 2003; 349:36–44.
- Cupps TR, Edgar LC, Fauci AS. Suppression of human B lymphocyte function by cyclophosphamide. J Immunol 1982; 128:2453–2457.
- Popa ER, Stegeman CA, Bos NA, Kallenberg CG, Tervaert JW. Differential B- and T-cell activation in Wegener’s granulomatosis. J Allergy Clin Immunol 1999; 103:885–894.
- Salama AD, Pusey CD. Drug insight: rituximab in renal disease and transplantation. Nat Clin Pract Nephrol 2006; 2:221–230.
- Specks U, Fervenza FC, McDonald TJ, Hogan MC. Response of Wegener’s granulomatosis to anti-CD20 chimeric monoclonal antibody therapy. Arthritis Rheum 2001; 44:2836–2840.
- Keogh KA, Wylam ME, Stone JH, Specks U. Induction of remission by B lymphocyte depletion in eleven patients with refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis Rheum 2005; 52:262–268.
- Keogh KA, Ytterberg SR, Fervenza FC, Carlson KA, Schroeder DR, Specks U. Rituximab for refractory Wegener’s granulomatosis: report of a prospective, open-label pilot trial [published online ahead of print October 13, 2005]. Am J Respir Crit Care Med 2006; 173:180–187. doi: 10.1164/rccm.200507-1144OC
- Stone JH, Merkel PA, Spiera R, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med 2010; 363:221–232.
- Jones RB, Tervaert JW, Hauser T, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med 2010; 363:211–220.
- Cartin-Ceba R, Golbin J, Keogh KA, et al. Rituximab for remission induction and maintenance in granulomatosis with polyangiitis (Wegener’s): a single-center ten-year experience [published online ahead of print June 21, 2012]. Arthritis Rheum. doi: 10.1002/art.34584
History of vasculitis: The life and work of Adolf Kussmaul
Adolf Kussmaul, who lived and practiced medicine in the 19th century, is known for his clinical skills, his scientific acumen, his gift for teaching, and his mastery of diverse areas of knowledge. He was a contemporary of such luminaries as pathologist Rudolf Virchow. In the rheumatology community, he is best known for describing the first case of polyarteritis nodosa (PAN).
FIRST CASE
In the first volume of the first edition of German Archive for Clinical Medicine, Kussmaul, along with his pathology associate Rudolf Maier, reported the case of Carl Seufarth, a 27-year-old tailor’s journeyman. Seufarth arrived at the University of Freiburg internal medicine clinic on May 4, 1865, at 10 am. Kussmaul was at that time head of medicine at Freiburg. Seufarth’s journeyman’s log recorded that he had been healthy when he left his hometown of Gernsbach in southwest Germany on January 30, 1865. His entry indicated that he was 5 feet 2 inches tall, was of strong build, and had healthy facial color.
Kussmaul’s 1866 description of Seufarth upon his arrival at the clinic is among the most memorable passages in medical literature:
Despite his frail appearance, Seufarth was able to walk into the hospital and climb the two flights of stairs to the internal medicine clinic without assistance. He had had a cold followed by a productive cough in the autumn of 1864, but felt well afterward. In the 8 days prior to admission to the University of Freiburg, he developed diarrhea and frequent chills with fevers and sweats. He had felt unwell for the preceding 2 to 3 weeks, during which he was hospitalized briefly for scabies, wandered from one place to another, and eventually arrived in Freiburg. Freiburg police imprisoned him on May 2 for begging and brought him to the internal medicine department on May 4 because of weakness.
Over the next several days, Seufarth experienced rapidly developing weakness, numbness in the left hand and eventually other extremities, and paralysis of the arm and hand muscles. He was closely monitored at the clinic with his temperature recorded every morning and evening. On the 28th day of hospitalization, pea-sized nodules were discovered in the subcutaneous skin of the abdomen and chest. By June 2, the patient was in a state of extreme weakness. He died on June 3, 1865, at 2 am.
This description is what we understand today as typical of vascular involvement in PAN. Maier also examined the tissue microscopically. In his report, he described the aneurysmal dilatations, narrowings, and inflammation occurring at the branches of the arteries. His sketch of involved organs depicted neutrophilic infiltration into the walls of the vessels.
When consulted by Kussmaul for a second opinion, pathologist Rudolf Virchow said he had not observed patients with disease similar to that of Seufarth. In his archives, however, he later found a specimen of an aneurysm in a branch of the superior mesenteric artery.
Kussmaul and Maier published the case under the title “On a previously undescribed peculiar arterial disease (periarteritis nodosa) accompanied by Bright’s disease and rapidly progressive general muscle weakness.” “Periarteritis nodosa” was later termed “polyarteritis nodosa” to better describe the inflammation of multiple medium-and small-vessel arteries rather than inflammation around the arteries as Maier had initially envisioned it.
BIOGRAPHICAL NOTES
The son of a German army surgeon, Kussmaul was born in 1822 in Graben near Karlsruhe, a small town in the Black Forest of southwestern Germany. Kussmaul began his medical studies at the University of Heidelberg in 1840. That same year, he constructed the first ophthalmoscope. The device did not function as intended because he had not discovered the light orientation needed to prevent the iris from contracting. But, as he later said, “It was the best ophthalmoscope of the time. Its only drawback was that it did not work.”
After graduating from the University of Heidelberg, Kussmaul went into private practice in Wiesloch. He returned to the University a year later, after having developed pericarditis, where he served as an assistant in 1846 and 1847 and engaged not only in medicine and medical discovery, but also poetry, publishing, and social movements. He founded a magazine that published short stories, poetry, and spoofs on the government; and he coined the term “Biedermeier,” which refers to a furniture style as well as a German social movement.
With plans to further his medical education, Kussmaul and his friend, Edward Bronner, traveled to Vienna and Prague in 1847 and 1848. In Vienna, they met the anatomic pathologist Karl Rokitansky. Although the young men hoped to study with the renowned scientist, they were soon dissuaded by Rokitansky’s clear dislike of working with students. He also had little use for patients, holding that the best patient was a dead patient because of all that one could learn by doing an autopsy.
Kussmaul and Bronner returned to Germany, Kussmaul having been called to serve as a physician in the Baden battalion during the German-Danish war. There, he contributed significantly to the health of the army by insisting that wounded soldiers not be bled—a common treatment at that time that actually accelerated the deaths of many soldiers in the field.
ACADEMICIAN, SCIENTIST, AND CLINICIAN
Shortly after his 1850 marriage to Luise Amanda Wolf, the daughter of a famous surgeon, Kussmaul developed an ascending polyradiculopathy, which at one time was called Landry-Kussmaul paralysis and later Guillain-Barré syndrome. This condition, along with his previous history of pericarditis, stimulated Kussmaul’s pursuit of medical knowledge for better understanding of his own afflictions as well as medicine in general.
He completed his doctoral dissertation at the University of Würzburg in 1853. There, he worked with pathology professor Virchow, who is known as the father of the theory of coagulation and the cellular theory of disease. It is perhaps less well known that in a treatise on histopathology in 1847, Virchow proposed that vasculitis actually might occur in blood vessels and originate in the adventitia. This profound insight was lost at the time because of inadequate understanding of vasculitic disorders.
Returning to the University of Heidelberg in 1854, Kussmaul earned the rank of assistant professor of medicine and, by 1857, professor of medicine. Two years later, he relocated to the University of Erlangen as a professor of medicine. His inaugural lecture at the University of Erlangen was the presentation of two cases of Landry-Kussmaul paralysis. Kussmaul’s research at Erlangen focused on differentiating the symptoms of mercurialism from syphilis (mercury was used for the treatment of syphilis).
Kussmaul was then called to the University of Freiburg in 1863 as head of the department of medicine. Among Kussmaul’s achievements at the University of Freiburg in the 1860s were the description of paradoxical pulse in obstructive pericarditis that we know as the Kussmaul pulse, and the description of the breathing characteristic of diabetic acidotic coma that we know as Kussmaul respiration. There he also performed the first gastroscopy on a sword-swallowing circus performer using a derivation of a laryngoscope; unfortunately, again his invention was thwarted by lack of an adequate light source. He also studied peptic ulcer disease and described a technique for dilating a stenosed peptic ulcer lesion with a balloon device. He later worked with Czerny and Billroth to develop the surgical procedure used routinely for nearly 100 years to relieve peptic ulcer disease prior to the introduction of drugs such as ranitidine.
RHEUMATOLOGY “WORMS”
Kussmaul and Maier initially published the Seufarth case in abstract form and called it “human worm aneurysm,” because they thought that the vascular pea-shaped or pea-sized structures represented worm and nematode infiltration. When they examined the specimens microscopically, however, they realized that they were viewing an inflammatory disease process.
Ironically, vessel disease of the PAN type was described in 1852 by Rokitansky. Rokitansky reported finding mesenteric aneurysms in the branch points of the arteries; however, because he eschewed technology, he did not examine the specimen microscopically and failed to recognize the inflammatory process. His student, Hans Eppinger, revisited the specimen some 30 years later and, under microscopic examination, clearly defined the aneurysmal dilatations and inflammatory infiltrates (Figure 2).
A final rheumatology worm episode occurred late in Kussmaul’s career in Strasburg, where he had become head of the department of medicine in 1878. Kussmaul asked his assistant and biographer, Albert Kahn, to administer naphthalene to a patient to eradicate intestinal worms. Strangely, the worms survived, but the fever resolved. Due to a pharmacy error, acetanilide, an anti-inflammatory marketed by Bayer, had been dispensed rather than naphthalene. Bayer subsequently marketed the product as Antifebrin.
REMEMBERED AND COMMEMORATED
Kussmaul was a much-loved teacher and a well-respected physician. After he retired in 1888, he returned to Heidelberg as emeritus professor of medicine. He died in 1902 at age 80. His desire to understand disease, his clinical observations, his teaching abilities, and his ability to apply medical technology to the bedside all played roles in his contributions to clinical medicine. One of several Kussmaul commemoration sites is a lunette in Lenox Hill Hospital, New York, New York, where his portrait plaque is displayed alongside those of Ismar Boas and Carl Anton Ewald, the founders of modern gastroenterology.
Adolf Kussmaul, who lived and practiced medicine in the 19th century, is known for his clinical skills, his scientific acumen, his gift for teaching, and his mastery of diverse areas of knowledge. He was a contemporary of such luminaries as pathologist Rudolf Virchow. In the rheumatology community, he is best known for describing the first case of polyarteritis nodosa (PAN).
FIRST CASE
In the first volume of the first edition of German Archive for Clinical Medicine, Kussmaul, along with his pathology associate Rudolf Maier, reported the case of Carl Seufarth, a 27-year-old tailor’s journeyman. Seufarth arrived at the University of Freiburg internal medicine clinic on May 4, 1865, at 10 am. Kussmaul was at that time head of medicine at Freiburg. Seufarth’s journeyman’s log recorded that he had been healthy when he left his hometown of Gernsbach in southwest Germany on January 30, 1865. His entry indicated that he was 5 feet 2 inches tall, was of strong build, and had healthy facial color.
Kussmaul’s 1866 description of Seufarth upon his arrival at the clinic is among the most memorable passages in medical literature:
Despite his frail appearance, Seufarth was able to walk into the hospital and climb the two flights of stairs to the internal medicine clinic without assistance. He had had a cold followed by a productive cough in the autumn of 1864, but felt well afterward. In the 8 days prior to admission to the University of Freiburg, he developed diarrhea and frequent chills with fevers and sweats. He had felt unwell for the preceding 2 to 3 weeks, during which he was hospitalized briefly for scabies, wandered from one place to another, and eventually arrived in Freiburg. Freiburg police imprisoned him on May 2 for begging and brought him to the internal medicine department on May 4 because of weakness.
Over the next several days, Seufarth experienced rapidly developing weakness, numbness in the left hand and eventually other extremities, and paralysis of the arm and hand muscles. He was closely monitored at the clinic with his temperature recorded every morning and evening. On the 28th day of hospitalization, pea-sized nodules were discovered in the subcutaneous skin of the abdomen and chest. By June 2, the patient was in a state of extreme weakness. He died on June 3, 1865, at 2 am.
This description is what we understand today as typical of vascular involvement in PAN. Maier also examined the tissue microscopically. In his report, he described the aneurysmal dilatations, narrowings, and inflammation occurring at the branches of the arteries. His sketch of involved organs depicted neutrophilic infiltration into the walls of the vessels.
When consulted by Kussmaul for a second opinion, pathologist Rudolf Virchow said he had not observed patients with disease similar to that of Seufarth. In his archives, however, he later found a specimen of an aneurysm in a branch of the superior mesenteric artery.
Kussmaul and Maier published the case under the title “On a previously undescribed peculiar arterial disease (periarteritis nodosa) accompanied by Bright’s disease and rapidly progressive general muscle weakness.” “Periarteritis nodosa” was later termed “polyarteritis nodosa” to better describe the inflammation of multiple medium-and small-vessel arteries rather than inflammation around the arteries as Maier had initially envisioned it.
BIOGRAPHICAL NOTES
The son of a German army surgeon, Kussmaul was born in 1822 in Graben near Karlsruhe, a small town in the Black Forest of southwestern Germany. Kussmaul began his medical studies at the University of Heidelberg in 1840. That same year, he constructed the first ophthalmoscope. The device did not function as intended because he had not discovered the light orientation needed to prevent the iris from contracting. But, as he later said, “It was the best ophthalmoscope of the time. Its only drawback was that it did not work.”
After graduating from the University of Heidelberg, Kussmaul went into private practice in Wiesloch. He returned to the University a year later, after having developed pericarditis, where he served as an assistant in 1846 and 1847 and engaged not only in medicine and medical discovery, but also poetry, publishing, and social movements. He founded a magazine that published short stories, poetry, and spoofs on the government; and he coined the term “Biedermeier,” which refers to a furniture style as well as a German social movement.
With plans to further his medical education, Kussmaul and his friend, Edward Bronner, traveled to Vienna and Prague in 1847 and 1848. In Vienna, they met the anatomic pathologist Karl Rokitansky. Although the young men hoped to study with the renowned scientist, they were soon dissuaded by Rokitansky’s clear dislike of working with students. He also had little use for patients, holding that the best patient was a dead patient because of all that one could learn by doing an autopsy.
Kussmaul and Bronner returned to Germany, Kussmaul having been called to serve as a physician in the Baden battalion during the German-Danish war. There, he contributed significantly to the health of the army by insisting that wounded soldiers not be bled—a common treatment at that time that actually accelerated the deaths of many soldiers in the field.
ACADEMICIAN, SCIENTIST, AND CLINICIAN
Shortly after his 1850 marriage to Luise Amanda Wolf, the daughter of a famous surgeon, Kussmaul developed an ascending polyradiculopathy, which at one time was called Landry-Kussmaul paralysis and later Guillain-Barré syndrome. This condition, along with his previous history of pericarditis, stimulated Kussmaul’s pursuit of medical knowledge for better understanding of his own afflictions as well as medicine in general.
He completed his doctoral dissertation at the University of Würzburg in 1853. There, he worked with pathology professor Virchow, who is known as the father of the theory of coagulation and the cellular theory of disease. It is perhaps less well known that in a treatise on histopathology in 1847, Virchow proposed that vasculitis actually might occur in blood vessels and originate in the adventitia. This profound insight was lost at the time because of inadequate understanding of vasculitic disorders.
Returning to the University of Heidelberg in 1854, Kussmaul earned the rank of assistant professor of medicine and, by 1857, professor of medicine. Two years later, he relocated to the University of Erlangen as a professor of medicine. His inaugural lecture at the University of Erlangen was the presentation of two cases of Landry-Kussmaul paralysis. Kussmaul’s research at Erlangen focused on differentiating the symptoms of mercurialism from syphilis (mercury was used for the treatment of syphilis).
Kussmaul was then called to the University of Freiburg in 1863 as head of the department of medicine. Among Kussmaul’s achievements at the University of Freiburg in the 1860s were the description of paradoxical pulse in obstructive pericarditis that we know as the Kussmaul pulse, and the description of the breathing characteristic of diabetic acidotic coma that we know as Kussmaul respiration. There he also performed the first gastroscopy on a sword-swallowing circus performer using a derivation of a laryngoscope; unfortunately, again his invention was thwarted by lack of an adequate light source. He also studied peptic ulcer disease and described a technique for dilating a stenosed peptic ulcer lesion with a balloon device. He later worked with Czerny and Billroth to develop the surgical procedure used routinely for nearly 100 years to relieve peptic ulcer disease prior to the introduction of drugs such as ranitidine.
RHEUMATOLOGY “WORMS”
Kussmaul and Maier initially published the Seufarth case in abstract form and called it “human worm aneurysm,” because they thought that the vascular pea-shaped or pea-sized structures represented worm and nematode infiltration. When they examined the specimens microscopically, however, they realized that they were viewing an inflammatory disease process.
Ironically, vessel disease of the PAN type was described in 1852 by Rokitansky. Rokitansky reported finding mesenteric aneurysms in the branch points of the arteries; however, because he eschewed technology, he did not examine the specimen microscopically and failed to recognize the inflammatory process. His student, Hans Eppinger, revisited the specimen some 30 years later and, under microscopic examination, clearly defined the aneurysmal dilatations and inflammatory infiltrates (Figure 2).
A final rheumatology worm episode occurred late in Kussmaul’s career in Strasburg, where he had become head of the department of medicine in 1878. Kussmaul asked his assistant and biographer, Albert Kahn, to administer naphthalene to a patient to eradicate intestinal worms. Strangely, the worms survived, but the fever resolved. Due to a pharmacy error, acetanilide, an anti-inflammatory marketed by Bayer, had been dispensed rather than naphthalene. Bayer subsequently marketed the product as Antifebrin.
REMEMBERED AND COMMEMORATED
Kussmaul was a much-loved teacher and a well-respected physician. After he retired in 1888, he returned to Heidelberg as emeritus professor of medicine. He died in 1902 at age 80. His desire to understand disease, his clinical observations, his teaching abilities, and his ability to apply medical technology to the bedside all played roles in his contributions to clinical medicine. One of several Kussmaul commemoration sites is a lunette in Lenox Hill Hospital, New York, New York, where his portrait plaque is displayed alongside those of Ismar Boas and Carl Anton Ewald, the founders of modern gastroenterology.
Adolf Kussmaul, who lived and practiced medicine in the 19th century, is known for his clinical skills, his scientific acumen, his gift for teaching, and his mastery of diverse areas of knowledge. He was a contemporary of such luminaries as pathologist Rudolf Virchow. In the rheumatology community, he is best known for describing the first case of polyarteritis nodosa (PAN).
FIRST CASE
In the first volume of the first edition of German Archive for Clinical Medicine, Kussmaul, along with his pathology associate Rudolf Maier, reported the case of Carl Seufarth, a 27-year-old tailor’s journeyman. Seufarth arrived at the University of Freiburg internal medicine clinic on May 4, 1865, at 10 am. Kussmaul was at that time head of medicine at Freiburg. Seufarth’s journeyman’s log recorded that he had been healthy when he left his hometown of Gernsbach in southwest Germany on January 30, 1865. His entry indicated that he was 5 feet 2 inches tall, was of strong build, and had healthy facial color.
Kussmaul’s 1866 description of Seufarth upon his arrival at the clinic is among the most memorable passages in medical literature:
Despite his frail appearance, Seufarth was able to walk into the hospital and climb the two flights of stairs to the internal medicine clinic without assistance. He had had a cold followed by a productive cough in the autumn of 1864, but felt well afterward. In the 8 days prior to admission to the University of Freiburg, he developed diarrhea and frequent chills with fevers and sweats. He had felt unwell for the preceding 2 to 3 weeks, during which he was hospitalized briefly for scabies, wandered from one place to another, and eventually arrived in Freiburg. Freiburg police imprisoned him on May 2 for begging and brought him to the internal medicine department on May 4 because of weakness.
Over the next several days, Seufarth experienced rapidly developing weakness, numbness in the left hand and eventually other extremities, and paralysis of the arm and hand muscles. He was closely monitored at the clinic with his temperature recorded every morning and evening. On the 28th day of hospitalization, pea-sized nodules were discovered in the subcutaneous skin of the abdomen and chest. By June 2, the patient was in a state of extreme weakness. He died on June 3, 1865, at 2 am.
This description is what we understand today as typical of vascular involvement in PAN. Maier also examined the tissue microscopically. In his report, he described the aneurysmal dilatations, narrowings, and inflammation occurring at the branches of the arteries. His sketch of involved organs depicted neutrophilic infiltration into the walls of the vessels.
When consulted by Kussmaul for a second opinion, pathologist Rudolf Virchow said he had not observed patients with disease similar to that of Seufarth. In his archives, however, he later found a specimen of an aneurysm in a branch of the superior mesenteric artery.
Kussmaul and Maier published the case under the title “On a previously undescribed peculiar arterial disease (periarteritis nodosa) accompanied by Bright’s disease and rapidly progressive general muscle weakness.” “Periarteritis nodosa” was later termed “polyarteritis nodosa” to better describe the inflammation of multiple medium-and small-vessel arteries rather than inflammation around the arteries as Maier had initially envisioned it.
BIOGRAPHICAL NOTES
The son of a German army surgeon, Kussmaul was born in 1822 in Graben near Karlsruhe, a small town in the Black Forest of southwestern Germany. Kussmaul began his medical studies at the University of Heidelberg in 1840. That same year, he constructed the first ophthalmoscope. The device did not function as intended because he had not discovered the light orientation needed to prevent the iris from contracting. But, as he later said, “It was the best ophthalmoscope of the time. Its only drawback was that it did not work.”
After graduating from the University of Heidelberg, Kussmaul went into private practice in Wiesloch. He returned to the University a year later, after having developed pericarditis, where he served as an assistant in 1846 and 1847 and engaged not only in medicine and medical discovery, but also poetry, publishing, and social movements. He founded a magazine that published short stories, poetry, and spoofs on the government; and he coined the term “Biedermeier,” which refers to a furniture style as well as a German social movement.
With plans to further his medical education, Kussmaul and his friend, Edward Bronner, traveled to Vienna and Prague in 1847 and 1848. In Vienna, they met the anatomic pathologist Karl Rokitansky. Although the young men hoped to study with the renowned scientist, they were soon dissuaded by Rokitansky’s clear dislike of working with students. He also had little use for patients, holding that the best patient was a dead patient because of all that one could learn by doing an autopsy.
Kussmaul and Bronner returned to Germany, Kussmaul having been called to serve as a physician in the Baden battalion during the German-Danish war. There, he contributed significantly to the health of the army by insisting that wounded soldiers not be bled—a common treatment at that time that actually accelerated the deaths of many soldiers in the field.
ACADEMICIAN, SCIENTIST, AND CLINICIAN
Shortly after his 1850 marriage to Luise Amanda Wolf, the daughter of a famous surgeon, Kussmaul developed an ascending polyradiculopathy, which at one time was called Landry-Kussmaul paralysis and later Guillain-Barré syndrome. This condition, along with his previous history of pericarditis, stimulated Kussmaul’s pursuit of medical knowledge for better understanding of his own afflictions as well as medicine in general.
He completed his doctoral dissertation at the University of Würzburg in 1853. There, he worked with pathology professor Virchow, who is known as the father of the theory of coagulation and the cellular theory of disease. It is perhaps less well known that in a treatise on histopathology in 1847, Virchow proposed that vasculitis actually might occur in blood vessels and originate in the adventitia. This profound insight was lost at the time because of inadequate understanding of vasculitic disorders.
Returning to the University of Heidelberg in 1854, Kussmaul earned the rank of assistant professor of medicine and, by 1857, professor of medicine. Two years later, he relocated to the University of Erlangen as a professor of medicine. His inaugural lecture at the University of Erlangen was the presentation of two cases of Landry-Kussmaul paralysis. Kussmaul’s research at Erlangen focused on differentiating the symptoms of mercurialism from syphilis (mercury was used for the treatment of syphilis).
Kussmaul was then called to the University of Freiburg in 1863 as head of the department of medicine. Among Kussmaul’s achievements at the University of Freiburg in the 1860s were the description of paradoxical pulse in obstructive pericarditis that we know as the Kussmaul pulse, and the description of the breathing characteristic of diabetic acidotic coma that we know as Kussmaul respiration. There he also performed the first gastroscopy on a sword-swallowing circus performer using a derivation of a laryngoscope; unfortunately, again his invention was thwarted by lack of an adequate light source. He also studied peptic ulcer disease and described a technique for dilating a stenosed peptic ulcer lesion with a balloon device. He later worked with Czerny and Billroth to develop the surgical procedure used routinely for nearly 100 years to relieve peptic ulcer disease prior to the introduction of drugs such as ranitidine.
RHEUMATOLOGY “WORMS”
Kussmaul and Maier initially published the Seufarth case in abstract form and called it “human worm aneurysm,” because they thought that the vascular pea-shaped or pea-sized structures represented worm and nematode infiltration. When they examined the specimens microscopically, however, they realized that they were viewing an inflammatory disease process.
Ironically, vessel disease of the PAN type was described in 1852 by Rokitansky. Rokitansky reported finding mesenteric aneurysms in the branch points of the arteries; however, because he eschewed technology, he did not examine the specimen microscopically and failed to recognize the inflammatory process. His student, Hans Eppinger, revisited the specimen some 30 years later and, under microscopic examination, clearly defined the aneurysmal dilatations and inflammatory infiltrates (Figure 2).
A final rheumatology worm episode occurred late in Kussmaul’s career in Strasburg, where he had become head of the department of medicine in 1878. Kussmaul asked his assistant and biographer, Albert Kahn, to administer naphthalene to a patient to eradicate intestinal worms. Strangely, the worms survived, but the fever resolved. Due to a pharmacy error, acetanilide, an anti-inflammatory marketed by Bayer, had been dispensed rather than naphthalene. Bayer subsequently marketed the product as Antifebrin.
REMEMBERED AND COMMEMORATED
Kussmaul was a much-loved teacher and a well-respected physician. After he retired in 1888, he returned to Heidelberg as emeritus professor of medicine. He died in 1902 at age 80. His desire to understand disease, his clinical observations, his teaching abilities, and his ability to apply medical technology to the bedside all played roles in his contributions to clinical medicine. One of several Kussmaul commemoration sites is a lunette in Lenox Hill Hospital, New York, New York, where his portrait plaque is displayed alongside those of Ismar Boas and Carl Anton Ewald, the founders of modern gastroenterology.
Analysis Details the GI Disease Burden in U.S.
Clostridium difficile contributes mightily to the overall burden of gastrointestinal disease in the United States and was associated with a 237% increase in hospitalizations in the last decade.
Researchers who examined the latest data on the nationwide toll of GI and liver disease also found a 314% rise in hospitalizations related to morbid obesity and a continuing national health burden exacted by reflux symptoms, Barrett’s esophagus, and colorectal cancer.
Video from the American Gastroenterological Association (http://www.youtube.com/amergastroassn)
"We compiled the most recently available statistics on GI symptoms, quality of life, outpatient diagnoses, hospitalizations, costs, mortality, and endoscopic utilization from a variety of publicly and privately held databases," Dr. Anne F. Peery of the University of North Carolina, Chapel Hill, and her colleagues reported in the November issue of Gastroenterology (doi:10.1053/j.gastro.2012.08.002).
"Payers, policy makers, clinicians, and others interested in resource utilization may use these statistics to better understand evolving disease trends, and the best way to meet the challenge of these diseases."
The findings are based on data for 2009, the most recent year for which complete information was available, from the National Ambulatory Medical Care Survey, sponsored by the U.S. Centers for Disease Control and Prevention; the United States National Health and Wellness Survey, sponsored by the private company Kantar Health; the Nationwide Inpatient Sample, sponsored by the Agency for Healthcare Research and Quality; the Surveillance, Epidemiology, and End Results database of the National Cancer Institute; the National Vital Statistics System, sponsored by the National Center for Health Statistics and the CDC; and the Thomson Reuters MarketScan’s databases of commercial, Medicare, and Medicaid records.
Among the findings:
• C. difficile hospitalizations have increased 237% since 2000 and were associated with 4% in-hospital mortality. Now the ninth leading GI cause of mortality, with an absolute increase of 230% in the number of C. difficile–related deaths since 2002, the infection also markedly impairs quality of life and the capacity for work and other activities.
• Hospitalizations related to obesity remained relatively stable since 2000, but those associated with morbid obesity rose by 314%, and many were likely caused by the marked increase in bariatric surgery.
• Gastroesophageal reflux remains the most common GI-associated diagnosis in primary care, accounting for 9 million outpatient visits in 2009, and the most common GI-associated discharge diagnosis, with 4.4 million such diagnoses in 2009. Obesity was associated with 1.7 million discharge diagnoses and constipation with 1 million.
• Barrett’s esophagus accounted for almost half a million outpatient visits in 2009, when an estimated 3.3 million Americans had this diagnosis. Given that endoscopic surveillance is recommended every 3-5 years, Barrett’s contributes substantially to resource utilization.
• Colorectal cancer, with an estimated 147,000 patients diagnosed in 2008, accounts for more than half of all GI cancer diagnoses and continues to be the primary cause of GI-associated mortality. Pancreatic and hepatobiliary cancers are the next most frequently diagnosed GI cancers.
• Of the approximately 2.5 million deaths in the United States in 2009, 10% were attributed to an underlying GI cause. Chronic liver disease and cirrhosis are the 12th leading causes of death in the country.
• The total outpatient cost for GI endoscopy in 2009 was estimated to be $32.4 billion, which is higher than previously published estimates. An estimated 6.9 million upper endoscopies, 11.5 million lower endoscopies, and 228,000 biliary endoscopies were performed in the United States in 2009.
• Chronic liver disease and viral hepatitis were associated with 6% mortality and cost an estimated $1.8 billion per year in inpatient cost.
• Hospitalizations for nonalcoholic fatty liver disease increased 97% since 2000.
This study was supported in part by the National Institutes of Health. No financial conflicts of interest were reported.
Digestive (GI and liver) diseases constitute a substantial and growing burden in the United States. As detailed in the report by Dr. Peery and colleagues, there were over 46 million outpatient encounters associated with the top 20 digestive disease diagnoses in 2009, with approximately 10% of deaths nationwide with an underlying digestive disease cause. The observed increased prevalence of hospitalizations for many diagnoses (e.g., a 14% increase with principal discharge diagnosis of chronic liver disease with viral hepatitis) and procedures (e.g., a 17% increase in lower GI endoscopies among commercially insured patients) between 2000 and 2009 is expected given population growth and aging. In addition, dramatic increases in hospitalizations associated with C. difficile and morbid obesity were also noted.
The report provides crucial information for diverse constituencies, including healthcare planners, clinicians and researchers. However, as acknowledged by the authors, there are some important caveats with respect to coverage or quality for some data sources to bear in mind when interpreting these results. This is particularly relevant for non-alcoholic fatty liver disease, which is likely underestimated because of well-known problems in diagnostic code specificity and use.

Several factors suggest that the prevalence and costs of digestive diseases will increase substantially during the next decade. These include: an aging population with the number of people aged 65 years or older projected to be greater than 54 million by 2020;the estimated tens of millions of individuals with newly available healthcare coverage as of 2014 as part of the Affordable Care Act; continued increases in obesity rates; and the recent CDC recommendation that all baby-boomers be screened for hepatitis C. This report will facilitate timely planning and also serve as benchmark to help measure the impact of these forces on the scope and burden of digestive diseases and their clinical management.
DONNA L. WHITE, PH.D., MPH, is an investigator in the Clinical Epidemiology and Outcomes Program in the Houston VA Health Services Research and Development Center of Excellence at the Michael E. DeBakey VA Medical Center, Houston. She also is an assistant professor in the department of medicine at Baylor College of Medicine, Houston.
Digestive (GI and liver) diseases constitute a substantial and growing burden in the United States. As detailed in the report by Dr. Peery and colleagues, there were over 46 million outpatient encounters associated with the top 20 digestive disease diagnoses in 2009, with approximately 10% of deaths nationwide with an underlying digestive disease cause. The observed increased prevalence of hospitalizations for many diagnoses (e.g., a 14% increase with principal discharge diagnosis of chronic liver disease with viral hepatitis) and procedures (e.g., a 17% increase in lower GI endoscopies among commercially insured patients) between 2000 and 2009 is expected given population growth and aging. In addition, dramatic increases in hospitalizations associated with C. difficile and morbid obesity were also noted.
The report provides crucial information for diverse constituencies, including healthcare planners, clinicians and researchers. However, as acknowledged by the authors, there are some important caveats with respect to coverage or quality for some data sources to bear in mind when interpreting these results. This is particularly relevant for non-alcoholic fatty liver disease, which is likely underestimated because of well-known problems in diagnostic code specificity and use.
Several factors suggest that the prevalence and costs of digestive diseases will increase substantially during the next decade. These include: an aging population with the number of people aged 65 years or older projected to be greater than 54 million by 2020;the estimated tens of millions of individuals with newly available healthcare coverage as of 2014 as part of the Affordable Care Act; continued increases in obesity rates; and the recent CDC recommendation that all baby-boomers be screened for hepatitis C. This report will facilitate timely planning and also serve as benchmark to help measure the impact of these forces on the scope and burden of digestive diseases and their clinical management.
DONNA L. WHITE, PH.D., MPH, is an investigator in the Clinical Epidemiology and Outcomes Program in the Houston VA Health Services Research and Development Center of Excellence at the Michael E. DeBakey VA Medical Center, Houston. She also is an assistant professor in the department of medicine at Baylor College of Medicine, Houston.
Digestive (GI and liver) diseases constitute a substantial and growing burden in the United States. As detailed in the report by Dr. Peery and colleagues, there were over 46 million outpatient encounters associated with the top 20 digestive disease diagnoses in 2009, with approximately 10% of deaths nationwide with an underlying digestive disease cause. The observed increased prevalence of hospitalizations for many diagnoses (e.g., a 14% increase with principal discharge diagnosis of chronic liver disease with viral hepatitis) and procedures (e.g., a 17% increase in lower GI endoscopies among commercially insured patients) between 2000 and 2009 is expected given population growth and aging. In addition, dramatic increases in hospitalizations associated with C. difficile and morbid obesity were also noted.
The report provides crucial information for diverse constituencies, including healthcare planners, clinicians and researchers. However, as acknowledged by the authors, there are some important caveats with respect to coverage or quality for some data sources to bear in mind when interpreting these results. This is particularly relevant for non-alcoholic fatty liver disease, which is likely underestimated because of well-known problems in diagnostic code specificity and use.
Several factors suggest that the prevalence and costs of digestive diseases will increase substantially during the next decade. These include: an aging population with the number of people aged 65 years or older projected to be greater than 54 million by 2020;the estimated tens of millions of individuals with newly available healthcare coverage as of 2014 as part of the Affordable Care Act; continued increases in obesity rates; and the recent CDC recommendation that all baby-boomers be screened for hepatitis C. This report will facilitate timely planning and also serve as benchmark to help measure the impact of these forces on the scope and burden of digestive diseases and their clinical management.
DONNA L. WHITE, PH.D., MPH, is an investigator in the Clinical Epidemiology and Outcomes Program in the Houston VA Health Services Research and Development Center of Excellence at the Michael E. DeBakey VA Medical Center, Houston. She also is an assistant professor in the department of medicine at Baylor College of Medicine, Houston.
Clostridium difficile contributes mightily to the overall burden of gastrointestinal disease in the United States and was associated with a 237% increase in hospitalizations in the last decade.
Researchers who examined the latest data on the nationwide toll of GI and liver disease also found a 314% rise in hospitalizations related to morbid obesity and a continuing national health burden exacted by reflux symptoms, Barrett’s esophagus, and colorectal cancer.
Video from the American Gastroenterological Association (http://www.youtube.com/amergastroassn)
"We compiled the most recently available statistics on GI symptoms, quality of life, outpatient diagnoses, hospitalizations, costs, mortality, and endoscopic utilization from a variety of publicly and privately held databases," Dr. Anne F. Peery of the University of North Carolina, Chapel Hill, and her colleagues reported in the November issue of Gastroenterology (doi:10.1053/j.gastro.2012.08.002).
"Payers, policy makers, clinicians, and others interested in resource utilization may use these statistics to better understand evolving disease trends, and the best way to meet the challenge of these diseases."
The findings are based on data for 2009, the most recent year for which complete information was available, from the National Ambulatory Medical Care Survey, sponsored by the U.S. Centers for Disease Control and Prevention; the United States National Health and Wellness Survey, sponsored by the private company Kantar Health; the Nationwide Inpatient Sample, sponsored by the Agency for Healthcare Research and Quality; the Surveillance, Epidemiology, and End Results database of the National Cancer Institute; the National Vital Statistics System, sponsored by the National Center for Health Statistics and the CDC; and the Thomson Reuters MarketScan’s databases of commercial, Medicare, and Medicaid records.
Among the findings:
• C. difficile hospitalizations have increased 237% since 2000 and were associated with 4% in-hospital mortality. Now the ninth leading GI cause of mortality, with an absolute increase of 230% in the number of C. difficile–related deaths since 2002, the infection also markedly impairs quality of life and the capacity for work and other activities.
• Hospitalizations related to obesity remained relatively stable since 2000, but those associated with morbid obesity rose by 314%, and many were likely caused by the marked increase in bariatric surgery.
• Gastroesophageal reflux remains the most common GI-associated diagnosis in primary care, accounting for 9 million outpatient visits in 2009, and the most common GI-associated discharge diagnosis, with 4.4 million such diagnoses in 2009. Obesity was associated with 1.7 million discharge diagnoses and constipation with 1 million.
• Barrett’s esophagus accounted for almost half a million outpatient visits in 2009, when an estimated 3.3 million Americans had this diagnosis. Given that endoscopic surveillance is recommended every 3-5 years, Barrett’s contributes substantially to resource utilization.
• Colorectal cancer, with an estimated 147,000 patients diagnosed in 2008, accounts for more than half of all GI cancer diagnoses and continues to be the primary cause of GI-associated mortality. Pancreatic and hepatobiliary cancers are the next most frequently diagnosed GI cancers.
• Of the approximately 2.5 million deaths in the United States in 2009, 10% were attributed to an underlying GI cause. Chronic liver disease and cirrhosis are the 12th leading causes of death in the country.
• The total outpatient cost for GI endoscopy in 2009 was estimated to be $32.4 billion, which is higher than previously published estimates. An estimated 6.9 million upper endoscopies, 11.5 million lower endoscopies, and 228,000 biliary endoscopies were performed in the United States in 2009.
• Chronic liver disease and viral hepatitis were associated with 6% mortality and cost an estimated $1.8 billion per year in inpatient cost.
• Hospitalizations for nonalcoholic fatty liver disease increased 97% since 2000.
This study was supported in part by the National Institutes of Health. No financial conflicts of interest were reported.
Clostridium difficile contributes mightily to the overall burden of gastrointestinal disease in the United States and was associated with a 237% increase in hospitalizations in the last decade.
Researchers who examined the latest data on the nationwide toll of GI and liver disease also found a 314% rise in hospitalizations related to morbid obesity and a continuing national health burden exacted by reflux symptoms, Barrett’s esophagus, and colorectal cancer.
Video from the American Gastroenterological Association (http://www.youtube.com/amergastroassn)
"We compiled the most recently available statistics on GI symptoms, quality of life, outpatient diagnoses, hospitalizations, costs, mortality, and endoscopic utilization from a variety of publicly and privately held databases," Dr. Anne F. Peery of the University of North Carolina, Chapel Hill, and her colleagues reported in the November issue of Gastroenterology (doi:10.1053/j.gastro.2012.08.002).
"Payers, policy makers, clinicians, and others interested in resource utilization may use these statistics to better understand evolving disease trends, and the best way to meet the challenge of these diseases."
The findings are based on data for 2009, the most recent year for which complete information was available, from the National Ambulatory Medical Care Survey, sponsored by the U.S. Centers for Disease Control and Prevention; the United States National Health and Wellness Survey, sponsored by the private company Kantar Health; the Nationwide Inpatient Sample, sponsored by the Agency for Healthcare Research and Quality; the Surveillance, Epidemiology, and End Results database of the National Cancer Institute; the National Vital Statistics System, sponsored by the National Center for Health Statistics and the CDC; and the Thomson Reuters MarketScan’s databases of commercial, Medicare, and Medicaid records.
Among the findings:
• C. difficile hospitalizations have increased 237% since 2000 and were associated with 4% in-hospital mortality. Now the ninth leading GI cause of mortality, with an absolute increase of 230% in the number of C. difficile–related deaths since 2002, the infection also markedly impairs quality of life and the capacity for work and other activities.
• Hospitalizations related to obesity remained relatively stable since 2000, but those associated with morbid obesity rose by 314%, and many were likely caused by the marked increase in bariatric surgery.
• Gastroesophageal reflux remains the most common GI-associated diagnosis in primary care, accounting for 9 million outpatient visits in 2009, and the most common GI-associated discharge diagnosis, with 4.4 million such diagnoses in 2009. Obesity was associated with 1.7 million discharge diagnoses and constipation with 1 million.
• Barrett’s esophagus accounted for almost half a million outpatient visits in 2009, when an estimated 3.3 million Americans had this diagnosis. Given that endoscopic surveillance is recommended every 3-5 years, Barrett’s contributes substantially to resource utilization.
• Colorectal cancer, with an estimated 147,000 patients diagnosed in 2008, accounts for more than half of all GI cancer diagnoses and continues to be the primary cause of GI-associated mortality. Pancreatic and hepatobiliary cancers are the next most frequently diagnosed GI cancers.
• Of the approximately 2.5 million deaths in the United States in 2009, 10% were attributed to an underlying GI cause. Chronic liver disease and cirrhosis are the 12th leading causes of death in the country.
• The total outpatient cost for GI endoscopy in 2009 was estimated to be $32.4 billion, which is higher than previously published estimates. An estimated 6.9 million upper endoscopies, 11.5 million lower endoscopies, and 228,000 biliary endoscopies were performed in the United States in 2009.
• Chronic liver disease and viral hepatitis were associated with 6% mortality and cost an estimated $1.8 billion per year in inpatient cost.
• Hospitalizations for nonalcoholic fatty liver disease increased 97% since 2000.
This study was supported in part by the National Institutes of Health. No financial conflicts of interest were reported.
FROM GASTROENTEROLOGY
Liver Candidates Decline Many Organ Offers
Eighty-four percent of candidates on the wait-list for liver transplant who either died or were removed from the list before they were able to undergo transplantation declined at least one offer of a donor liver, Dr. Jennifer Cindy Lai of the University of California, San Francisco, and her colleagues reported in the November issue of Gastroenterology.
Even more surprising, most of these candidates declined "not just one or two but a median of six liver offers during their time on the wait-list."
The "declined" donor organs were then successfully transplanted into lower-priority recipients.
These findings suggest that mortality among wait-listed patients "is not simply a result of not having the opportunity for transplantation, as many of us assume. Rather, wait-list mortality appears to result from opportunities for transplantation that were declined," Dr. Lai and her associates wrote.
The reasons that so many viable donor livers were initially declined are not yet clear. General, somewhat vague reasons were listed but not fully explained in the records the researchers analyzed for this study, which they obtained from the United Network for Organ Sharing/Organ Procurement Transplantation Network database.
The investigators assessed organ offers to 33,389 liver transplant candidates aged 18 years and older who were wait-listed across the United States between 2005 and 2010.
The reasons that proffered organs were declined, as listed in the medical records, fit into six broad categories: unfavorable donor age or quality of organ; unfavorable donor organ size/weight; other unfavorable donor factors, such as ABO blood transfusion incompatibility, "social history," "positive serologic tests," or "organ anatomical damage or defect"; unreadiness of the recipient, usually because he or she was ill, unavailable, refused the organ, or required multiple organ transplants at the same time; problems with the transplant program itself, such as a "heavy workload" or unavailability of a surgeon or operating room at the recipient’s medical center, failure to respond to the offer in a timely way, or excessive distance to ship the organ.
A total of 20% of the study population (6,737 patients) died or were removed from the wait-list because they became too sick before they could undergo transplantation. A total of 5,680 (84%) of those patients had been offered one or more donor livers before they died or were taken off the list.
Offers of donor livers were declined most often (68%) because of "unfavorable donor age or quality of organ," whereas 9% were declined because of unfavorable organ size, 15% because of "other donor factors," 4% because the recipient wasn’t ready, and 4% because of transplant program or miscellaneous other factors.
However, the dominant use of the "donor quality or age" refusal code in the database almost certainly "does not accurately or fully capture the true refusal reason," Dr. Lai and her associates said.
Even livers judged to be of high quality according to standard criteria were declined because of supposed "unfavorable donor age or quality of organ." But the investigators found no difference in the risk of graft failure between such high-quality livers that were declined and other high-quality livers that were accepted on the first offer.
Other reasons must be playing an important role in this high rate refusal, but "the nuances of these refusals cannot be determined" without more individualized data, they said.
Dr. Lai and her colleagues suggested that to cut down on refusals of apparently viable organs, the transplant community should "reduce the stigma associated with non–ideal livers, and set realistic expectations for wait-listed candidates" so that they’re less likely to pass up a suitable donation while assuming that a better offer will come along.
Patients also should be educated about the unpredictability of death or of sudden worsening of liver disease while on the wait-list. They should be advised that there is a survival benefit associated with the transplantation of any graft, compared with continuing on the wait-list.
In addition, the current regulatory environment focuses on transplant centers’ outcomes, which may influence some centers to discourage the acceptance of less than optimal donor organs. "This may be especially relevant for low-volume transplant centers, for whom even a small number of poor outcomes ... may make a relatively large difference in the centers’ perceived performance," the researchers wrote.
Finally, wait-list candidates should be encouraged to complete their transplant work-ups as expeditiously as possible to avoid having to refuse a donor offer simply because they have not yet undergone the necessary cardiac testing or cancer screening.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of California, San Francisco. No financial conflicts of interest were reported.
Eighty-four percent of candidates on the wait-list for liver transplant who either died or were removed from the list before they were able to undergo transplantation declined at least one offer of a donor liver, Dr. Jennifer Cindy Lai of the University of California, San Francisco, and her colleagues reported in the November issue of Gastroenterology.
Even more surprising, most of these candidates declined "not just one or two but a median of six liver offers during their time on the wait-list."
The "declined" donor organs were then successfully transplanted into lower-priority recipients.
These findings suggest that mortality among wait-listed patients "is not simply a result of not having the opportunity for transplantation, as many of us assume. Rather, wait-list mortality appears to result from opportunities for transplantation that were declined," Dr. Lai and her associates wrote.
The reasons that so many viable donor livers were initially declined are not yet clear. General, somewhat vague reasons were listed but not fully explained in the records the researchers analyzed for this study, which they obtained from the United Network for Organ Sharing/Organ Procurement Transplantation Network database.
The investigators assessed organ offers to 33,389 liver transplant candidates aged 18 years and older who were wait-listed across the United States between 2005 and 2010.
The reasons that proffered organs were declined, as listed in the medical records, fit into six broad categories: unfavorable donor age or quality of organ; unfavorable donor organ size/weight; other unfavorable donor factors, such as ABO blood transfusion incompatibility, "social history," "positive serologic tests," or "organ anatomical damage or defect"; unreadiness of the recipient, usually because he or she was ill, unavailable, refused the organ, or required multiple organ transplants at the same time; problems with the transplant program itself, such as a "heavy workload" or unavailability of a surgeon or operating room at the recipient’s medical center, failure to respond to the offer in a timely way, or excessive distance to ship the organ.
A total of 20% of the study population (6,737 patients) died or were removed from the wait-list because they became too sick before they could undergo transplantation. A total of 5,680 (84%) of those patients had been offered one or more donor livers before they died or were taken off the list.
Offers of donor livers were declined most often (68%) because of "unfavorable donor age or quality of organ," whereas 9% were declined because of unfavorable organ size, 15% because of "other donor factors," 4% because the recipient wasn’t ready, and 4% because of transplant program or miscellaneous other factors.
However, the dominant use of the "donor quality or age" refusal code in the database almost certainly "does not accurately or fully capture the true refusal reason," Dr. Lai and her associates said.
Even livers judged to be of high quality according to standard criteria were declined because of supposed "unfavorable donor age or quality of organ." But the investigators found no difference in the risk of graft failure between such high-quality livers that were declined and other high-quality livers that were accepted on the first offer.
Other reasons must be playing an important role in this high rate refusal, but "the nuances of these refusals cannot be determined" without more individualized data, they said.
Dr. Lai and her colleagues suggested that to cut down on refusals of apparently viable organs, the transplant community should "reduce the stigma associated with non–ideal livers, and set realistic expectations for wait-listed candidates" so that they’re less likely to pass up a suitable donation while assuming that a better offer will come along.
Patients also should be educated about the unpredictability of death or of sudden worsening of liver disease while on the wait-list. They should be advised that there is a survival benefit associated with the transplantation of any graft, compared with continuing on the wait-list.
In addition, the current regulatory environment focuses on transplant centers’ outcomes, which may influence some centers to discourage the acceptance of less than optimal donor organs. "This may be especially relevant for low-volume transplant centers, for whom even a small number of poor outcomes ... may make a relatively large difference in the centers’ perceived performance," the researchers wrote.
Finally, wait-list candidates should be encouraged to complete their transplant work-ups as expeditiously as possible to avoid having to refuse a donor offer simply because they have not yet undergone the necessary cardiac testing or cancer screening.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of California, San Francisco. No financial conflicts of interest were reported.
Eighty-four percent of candidates on the wait-list for liver transplant who either died or were removed from the list before they were able to undergo transplantation declined at least one offer of a donor liver, Dr. Jennifer Cindy Lai of the University of California, San Francisco, and her colleagues reported in the November issue of Gastroenterology.
Even more surprising, most of these candidates declined "not just one or two but a median of six liver offers during their time on the wait-list."
The "declined" donor organs were then successfully transplanted into lower-priority recipients.
These findings suggest that mortality among wait-listed patients "is not simply a result of not having the opportunity for transplantation, as many of us assume. Rather, wait-list mortality appears to result from opportunities for transplantation that were declined," Dr. Lai and her associates wrote.
The reasons that so many viable donor livers were initially declined are not yet clear. General, somewhat vague reasons were listed but not fully explained in the records the researchers analyzed for this study, which they obtained from the United Network for Organ Sharing/Organ Procurement Transplantation Network database.
The investigators assessed organ offers to 33,389 liver transplant candidates aged 18 years and older who were wait-listed across the United States between 2005 and 2010.
The reasons that proffered organs were declined, as listed in the medical records, fit into six broad categories: unfavorable donor age or quality of organ; unfavorable donor organ size/weight; other unfavorable donor factors, such as ABO blood transfusion incompatibility, "social history," "positive serologic tests," or "organ anatomical damage or defect"; unreadiness of the recipient, usually because he or she was ill, unavailable, refused the organ, or required multiple organ transplants at the same time; problems with the transplant program itself, such as a "heavy workload" or unavailability of a surgeon or operating room at the recipient’s medical center, failure to respond to the offer in a timely way, or excessive distance to ship the organ.
A total of 20% of the study population (6,737 patients) died or were removed from the wait-list because they became too sick before they could undergo transplantation. A total of 5,680 (84%) of those patients had been offered one or more donor livers before they died or were taken off the list.
Offers of donor livers were declined most often (68%) because of "unfavorable donor age or quality of organ," whereas 9% were declined because of unfavorable organ size, 15% because of "other donor factors," 4% because the recipient wasn’t ready, and 4% because of transplant program or miscellaneous other factors.
However, the dominant use of the "donor quality or age" refusal code in the database almost certainly "does not accurately or fully capture the true refusal reason," Dr. Lai and her associates said.
Even livers judged to be of high quality according to standard criteria were declined because of supposed "unfavorable donor age or quality of organ." But the investigators found no difference in the risk of graft failure between such high-quality livers that were declined and other high-quality livers that were accepted on the first offer.
Other reasons must be playing an important role in this high rate refusal, but "the nuances of these refusals cannot be determined" without more individualized data, they said.
Dr. Lai and her colleagues suggested that to cut down on refusals of apparently viable organs, the transplant community should "reduce the stigma associated with non–ideal livers, and set realistic expectations for wait-listed candidates" so that they’re less likely to pass up a suitable donation while assuming that a better offer will come along.
Patients also should be educated about the unpredictability of death or of sudden worsening of liver disease while on the wait-list. They should be advised that there is a survival benefit associated with the transplantation of any graft, compared with continuing on the wait-list.
In addition, the current regulatory environment focuses on transplant centers’ outcomes, which may influence some centers to discourage the acceptance of less than optimal donor organs. "This may be especially relevant for low-volume transplant centers, for whom even a small number of poor outcomes ... may make a relatively large difference in the centers’ perceived performance," the researchers wrote.
Finally, wait-list candidates should be encouraged to complete their transplant work-ups as expeditiously as possible to avoid having to refuse a donor offer simply because they have not yet undergone the necessary cardiac testing or cancer screening.
This study was supported by the National Institute of Diabetes and Digestive and Kidney Diseases and the University of California, San Francisco. No financial conflicts of interest were reported.
FROM GASTROENTEROLOGY
Pretreatment Care Predicts HCV Outcomes
Patients with hepatitis C infections are more likely to initiate appropriate antiviral therapy and achieve a sustained virologic response if they receive high quality health care, Dr. Fasiha Kanwal of the Michael E. DeBakey Veterans Affairs Medical Center, Houston, and her colleagues reported in the November issue of Clinical Gastroenterology and Hepatology.
In their study of nearly 35,000 adults with HCV, the odds of initiating antiviral therapy were threefold higher in patients who received optimal care from the moment HCV infection was diagnosed than in those who did not. And among patients who initiated antiviral therapy, the quality of care they received before that therapy even began strongly predicted whether they would complete antiviral therapy and achieve a sustained virologic response, the investigators said.
The study showed, however, that only 11% of patients received all of the appropriate initial care.
Process-of-care measures are frequently used to assess the quality of care for HCV, but until now no study has assessed whether these measures actually correlate with better outcomes. To address this issue, Dr. Kanwal and her associates "evaluated the relationship between adherence to a broad set of process-based measures in HCV and 3 subsequent HCV-specific endpoints: receipt of antiviral treatment, completion of antiviral treatment, and the clinical outcome associated with improved survival: sustained virologic response."
The investigators used data from the VA registry on HCV clinical care, which covers patient demographics, lab tests, pharmacy information, and data on inpatient and outpatient care for approximately 300,000 patients across the country. For this study, they included data on 34,749 adults.
The mean subject age was 53 years, and 97% were men. Approximately half the study population was white and 26% was black; ethnicity was not reported for the others.
The researchers assessed seven process-of-care measures: confirmation of HCV viremia, evaluation by HCV specialists, HCV genotype testing, liver biopsy for those found to have genotype 1 HCV, and the ruling out of liver diseases related to hepatitis B, autoimmunity, or iron overload.
They also assessed seven process-of-care measures related to the prevention and management of comorbid conditions: HIV testing; hepatitis A and B serology testing, hepatitis A and B vaccination if serology results proved negative; treatment of depression; and treatment of substance abuse disorder.
Finally, they assessed six process-of-care measures related to monitoring of antiviral therapy’s effects: testing of viral load before antivirals were initiated and again at weeks 12, 24, and 48; reduction of ribavirin dose if anemia developed during treatment; and avoidance of prescribing growth-stimulating factors for leukopenia during antiviral therapy.
Overall, only 11% of the study subjects received all of the appropriate initial care, and 8% received all the appropriate care related to prevention and management of comorbid conditions. Moreover, of the study subjects who received antiviral therapy, just 37% received all the appropriate monitoring of treatment effects Dr. Kanwal and her associates said.
In patients who received optimal care before a definitive diagnosis was made, the odds of receiving antiviral therapy were 3.2 times higher than in patients who did not receive optimal care before diagnosis, they reported.
Similarly, patients who received optimal preventive and comorbid-condition care showed rates of antiviral therapy that were 36% higher than those of patients who received suboptimal preventive and comorbid-condition care.
The strong association between fulfillment of these process-of-care measures and appropriate antiviral therapy remained robust in a series of sensitivity analyses, which means it’s likely that meeting process-of-care goals directly leads to better HCV outcomes, Dr. Kanwal and her colleagues said.
The investigators could not, however, rule out the possibility that meeting these goals is simply a marker of more compliant patients, which in turn produces better outcomes.
The study findings imply that the effectiveness of the two new direct-acting antiviral agents that recently became available for HCV may hinge on the quality of care patients are receiving before they even start taking these drugs, rather than simply on the effectiveness of the drugs alone, Dr. Kanwal and her associates said.
This study was supported by the U.S. Department of Veterans Affairs Health Services Research and Development Service. No financial conflicts of interest were reported.
Financial reimbursement in medicine has long been driven by volume rather than quality. This incentive structure is changing, and in the near future practitioners will experience increased scrutiny of the quality of care we provide.

|
|
Quality can be divided into structure (having the proper equipment to clean endoscopes), process (testing for latent tuberculosis before beginning anti–tumor necrosis factor therapy), and outcomes (perforation rate during colonoscopy). The latter is, of course, the most important, but it is also the most difficult to measure because of low event rates and inadequate risk adjustment. Therefore, most quality measures are based on processes of care. For quality measurement to yield any true value to the patient, however, it is important that these processes are clearly linked to better patient outcomes.
Hepatitis C, which affects 1.3% of Americans, has recently been a target disease for measuring and improving quality of care. Dr. Kanwal and her colleagues found that patients receiving optimum process-related quality care were more likely to undergo antiviral therapy. Such patients also were more likely to complete treatment once started and to achieve sustained virologic response if treatment was completed. Since these findings persisted despite adjustment for numerous potential confounders such as comorbidities, it appears that higher quality of care (as measured by processes) may truly lead to better patient outcomes.
What does this mean for practitioners? Particularly in the era of triple therapy, hepatitis C virus (HCV) infection cannot be managed like any other disease. Protocols and tracking systems need to be developed to ensure that quality measures are met. Many practices find it helpful to funnel HCV patients to a single person for case management, such as a nurse or midlevel provider. Hopefully, these efforts, in conjunction with newer antivirals, will soon eradicate hepatitis C altogether.
Michael Volk, M.D., is assistant professor of hepatology at the University of Michigan, Ann Arbor.
Financial reimbursement in medicine has long been driven by volume rather than quality. This incentive structure is changing, and in the near future practitioners will experience increased scrutiny of the quality of care we provide.
|
|
|
Quality can be divided into structure (having the proper equipment to clean endoscopes), process (testing for latent tuberculosis before beginning anti–tumor necrosis factor therapy), and outcomes (perforation rate during colonoscopy). The latter is, of course, the most important, but it is also the most difficult to measure because of low event rates and inadequate risk adjustment. Therefore, most quality measures are based on processes of care. For quality measurement to yield any true value to the patient, however, it is important that these processes are clearly linked to better patient outcomes.
Hepatitis C, which affects 1.3% of Americans, has recently been a target disease for measuring and improving quality of care. Dr. Kanwal and her colleagues found that patients receiving optimum process-related quality care were more likely to undergo antiviral therapy. Such patients also were more likely to complete treatment once started and to achieve sustained virologic response if treatment was completed. Since these findings persisted despite adjustment for numerous potential confounders such as comorbidities, it appears that higher quality of care (as measured by processes) may truly lead to better patient outcomes.
What does this mean for practitioners? Particularly in the era of triple therapy, hepatitis C virus (HCV) infection cannot be managed like any other disease. Protocols and tracking systems need to be developed to ensure that quality measures are met. Many practices find it helpful to funnel HCV patients to a single person for case management, such as a nurse or midlevel provider. Hopefully, these efforts, in conjunction with newer antivirals, will soon eradicate hepatitis C altogether.
Michael Volk, M.D., is assistant professor of hepatology at the University of Michigan, Ann Arbor.
Financial reimbursement in medicine has long been driven by volume rather than quality. This incentive structure is changing, and in the near future practitioners will experience increased scrutiny of the quality of care we provide.
|
|
|
Quality can be divided into structure (having the proper equipment to clean endoscopes), process (testing for latent tuberculosis before beginning anti–tumor necrosis factor therapy), and outcomes (perforation rate during colonoscopy). The latter is, of course, the most important, but it is also the most difficult to measure because of low event rates and inadequate risk adjustment. Therefore, most quality measures are based on processes of care. For quality measurement to yield any true value to the patient, however, it is important that these processes are clearly linked to better patient outcomes.
Hepatitis C, which affects 1.3% of Americans, has recently been a target disease for measuring and improving quality of care. Dr. Kanwal and her colleagues found that patients receiving optimum process-related quality care were more likely to undergo antiviral therapy. Such patients also were more likely to complete treatment once started and to achieve sustained virologic response if treatment was completed. Since these findings persisted despite adjustment for numerous potential confounders such as comorbidities, it appears that higher quality of care (as measured by processes) may truly lead to better patient outcomes.
What does this mean for practitioners? Particularly in the era of triple therapy, hepatitis C virus (HCV) infection cannot be managed like any other disease. Protocols and tracking systems need to be developed to ensure that quality measures are met. Many practices find it helpful to funnel HCV patients to a single person for case management, such as a nurse or midlevel provider. Hopefully, these efforts, in conjunction with newer antivirals, will soon eradicate hepatitis C altogether.
Michael Volk, M.D., is assistant professor of hepatology at the University of Michigan, Ann Arbor.
Patients with hepatitis C infections are more likely to initiate appropriate antiviral therapy and achieve a sustained virologic response if they receive high quality health care, Dr. Fasiha Kanwal of the Michael E. DeBakey Veterans Affairs Medical Center, Houston, and her colleagues reported in the November issue of Clinical Gastroenterology and Hepatology.
In their study of nearly 35,000 adults with HCV, the odds of initiating antiviral therapy were threefold higher in patients who received optimal care from the moment HCV infection was diagnosed than in those who did not. And among patients who initiated antiviral therapy, the quality of care they received before that therapy even began strongly predicted whether they would complete antiviral therapy and achieve a sustained virologic response, the investigators said.
The study showed, however, that only 11% of patients received all of the appropriate initial care.
Process-of-care measures are frequently used to assess the quality of care for HCV, but until now no study has assessed whether these measures actually correlate with better outcomes. To address this issue, Dr. Kanwal and her associates "evaluated the relationship between adherence to a broad set of process-based measures in HCV and 3 subsequent HCV-specific endpoints: receipt of antiviral treatment, completion of antiviral treatment, and the clinical outcome associated with improved survival: sustained virologic response."
The investigators used data from the VA registry on HCV clinical care, which covers patient demographics, lab tests, pharmacy information, and data on inpatient and outpatient care for approximately 300,000 patients across the country. For this study, they included data on 34,749 adults.
The mean subject age was 53 years, and 97% were men. Approximately half the study population was white and 26% was black; ethnicity was not reported for the others.
The researchers assessed seven process-of-care measures: confirmation of HCV viremia, evaluation by HCV specialists, HCV genotype testing, liver biopsy for those found to have genotype 1 HCV, and the ruling out of liver diseases related to hepatitis B, autoimmunity, or iron overload.
They also assessed seven process-of-care measures related to the prevention and management of comorbid conditions: HIV testing; hepatitis A and B serology testing, hepatitis A and B vaccination if serology results proved negative; treatment of depression; and treatment of substance abuse disorder.
Finally, they assessed six process-of-care measures related to monitoring of antiviral therapy’s effects: testing of viral load before antivirals were initiated and again at weeks 12, 24, and 48; reduction of ribavirin dose if anemia developed during treatment; and avoidance of prescribing growth-stimulating factors for leukopenia during antiviral therapy.
Overall, only 11% of the study subjects received all of the appropriate initial care, and 8% received all the appropriate care related to prevention and management of comorbid conditions. Moreover, of the study subjects who received antiviral therapy, just 37% received all the appropriate monitoring of treatment effects Dr. Kanwal and her associates said.
In patients who received optimal care before a definitive diagnosis was made, the odds of receiving antiviral therapy were 3.2 times higher than in patients who did not receive optimal care before diagnosis, they reported.
Similarly, patients who received optimal preventive and comorbid-condition care showed rates of antiviral therapy that were 36% higher than those of patients who received suboptimal preventive and comorbid-condition care.
The strong association between fulfillment of these process-of-care measures and appropriate antiviral therapy remained robust in a series of sensitivity analyses, which means it’s likely that meeting process-of-care goals directly leads to better HCV outcomes, Dr. Kanwal and her colleagues said.
The investigators could not, however, rule out the possibility that meeting these goals is simply a marker of more compliant patients, which in turn produces better outcomes.
The study findings imply that the effectiveness of the two new direct-acting antiviral agents that recently became available for HCV may hinge on the quality of care patients are receiving before they even start taking these drugs, rather than simply on the effectiveness of the drugs alone, Dr. Kanwal and her associates said.
This study was supported by the U.S. Department of Veterans Affairs Health Services Research and Development Service. No financial conflicts of interest were reported.
Patients with hepatitis C infections are more likely to initiate appropriate antiviral therapy and achieve a sustained virologic response if they receive high quality health care, Dr. Fasiha Kanwal of the Michael E. DeBakey Veterans Affairs Medical Center, Houston, and her colleagues reported in the November issue of Clinical Gastroenterology and Hepatology.
In their study of nearly 35,000 adults with HCV, the odds of initiating antiviral therapy were threefold higher in patients who received optimal care from the moment HCV infection was diagnosed than in those who did not. And among patients who initiated antiviral therapy, the quality of care they received before that therapy even began strongly predicted whether they would complete antiviral therapy and achieve a sustained virologic response, the investigators said.
The study showed, however, that only 11% of patients received all of the appropriate initial care.
Process-of-care measures are frequently used to assess the quality of care for HCV, but until now no study has assessed whether these measures actually correlate with better outcomes. To address this issue, Dr. Kanwal and her associates "evaluated the relationship between adherence to a broad set of process-based measures in HCV and 3 subsequent HCV-specific endpoints: receipt of antiviral treatment, completion of antiviral treatment, and the clinical outcome associated with improved survival: sustained virologic response."
The investigators used data from the VA registry on HCV clinical care, which covers patient demographics, lab tests, pharmacy information, and data on inpatient and outpatient care for approximately 300,000 patients across the country. For this study, they included data on 34,749 adults.
The mean subject age was 53 years, and 97% were men. Approximately half the study population was white and 26% was black; ethnicity was not reported for the others.
The researchers assessed seven process-of-care measures: confirmation of HCV viremia, evaluation by HCV specialists, HCV genotype testing, liver biopsy for those found to have genotype 1 HCV, and the ruling out of liver diseases related to hepatitis B, autoimmunity, or iron overload.
They also assessed seven process-of-care measures related to the prevention and management of comorbid conditions: HIV testing; hepatitis A and B serology testing, hepatitis A and B vaccination if serology results proved negative; treatment of depression; and treatment of substance abuse disorder.
Finally, they assessed six process-of-care measures related to monitoring of antiviral therapy’s effects: testing of viral load before antivirals were initiated and again at weeks 12, 24, and 48; reduction of ribavirin dose if anemia developed during treatment; and avoidance of prescribing growth-stimulating factors for leukopenia during antiviral therapy.
Overall, only 11% of the study subjects received all of the appropriate initial care, and 8% received all the appropriate care related to prevention and management of comorbid conditions. Moreover, of the study subjects who received antiviral therapy, just 37% received all the appropriate monitoring of treatment effects Dr. Kanwal and her associates said.
In patients who received optimal care before a definitive diagnosis was made, the odds of receiving antiviral therapy were 3.2 times higher than in patients who did not receive optimal care before diagnosis, they reported.
Similarly, patients who received optimal preventive and comorbid-condition care showed rates of antiviral therapy that were 36% higher than those of patients who received suboptimal preventive and comorbid-condition care.
The strong association between fulfillment of these process-of-care measures and appropriate antiviral therapy remained robust in a series of sensitivity analyses, which means it’s likely that meeting process-of-care goals directly leads to better HCV outcomes, Dr. Kanwal and her colleagues said.
The investigators could not, however, rule out the possibility that meeting these goals is simply a marker of more compliant patients, which in turn produces better outcomes.
The study findings imply that the effectiveness of the two new direct-acting antiviral agents that recently became available for HCV may hinge on the quality of care patients are receiving before they even start taking these drugs, rather than simply on the effectiveness of the drugs alone, Dr. Kanwal and her associates said.
This study was supported by the U.S. Department of Veterans Affairs Health Services Research and Development Service. No financial conflicts of interest were reported.
FROM CLINICAL GASTROENTEROLOGY AND HEPATOLOGY
Androgen deficiency in older men: Indications, advantages, and pitfalls of testosterone replacement therapy
Editor’s note: This is the second of two articles on hypogonadism in men and focuses on the appropriate use of testosterone therapy. The first article, published last month, focused in more detail on the differential diagnosis of hypogonadism.
As men age, testosterone production gradually decreases. In our increasingly aged population, clinicians will continue to see an increase in the number of men with seemingly nonspecific symptoms of aging that are possibly due to low serum testosterone (eg, low energy level, depressive symptoms, erectile dysfunction, decreased libido). These clinical symptoms, coupled with low serum testosterone, may adversely affect quality of life and life expectancy. Testosterone replacement therapy (TRT) may improve symptoms and quality of life. Given the nonspecific nature of these symptoms, accurate diagnosis and treatment of clinically significant low testosterone with a goal of symptom and quality of life improvement can prove challenging.
These challenges in diagnosis and treatment result in a lack of standardized nomenclature. The terms male menopause and andropause, although popular, are the least helpful, as they have few correlates with the better-defined female menopause. Late-onset hypogonadism implies a well-defined, later age of decline, which is inaccurate since the decline in serum testosterone in men begins in middle age and is gradual. Testosterone deficiency syndrome implies a set of specific and well-defined symptoms. Androgen deficiency in the aging male (ADAM) and Androgen deficiency in the older male are common terms specifying an age cohort (> 40 years old) and an abnormal laboratory value without mention of symptoms. While all these terms have their limitations, we will primarily use ADAM in this discussion.
PREVALENCE OF LOW TESTOSTERONE
Serum testosterone levels begin to decline in men in their mid-40s, with an approximately 1% to 2% decline annually and a marked decline after age 60.1
Araujo and colleagues2 studied the prevalence of androgen-deficient men, with androgen deficiency defined as at least three signs or symptoms and either a total testosterone less than 200 ng/dL or a total testosterone 200 ng/dL to 400 ng/dL with a free testosterone less than 8.91 ng/dL. The overall prevalence of low testosterone on initial measurement was 6%, which doubled to 12% with repeat measurement.
Serial measures are important: one study that followed untreated men over 15 years found normal testosterone on serial measures in 50%.3 In a multicenter cross-sectional study, 11.8% of men had low testosterone and low or normal luteinizing hormone (LH) levels (secondary hypogonadism/hypothalamic-pituitary failure), with 2% of patients with low testosterone and elevated LH (primary hypogonadism/testicular failure).4
CLINICAL PRESENTATION AND DIAGNOSIS
A biochemical diagnosis of low testosterone is dependent on accurate measurement. Testosterone release is diurnal, with the highest levels in the early morning, and often has week-to-week variability. Thus, it is important to collect blood in the early morning and to confirm a diagnosis of low testosterone with at least one repeat measurement several days later, including LH assessment. LH levels will help differentiate primary hypogonadism from secondary hypogonadism, which may alter diagnosis and treatment in certain patients, with secondary hypogonadism associated with pituitary dysfunction, and primary hypogonadism associated with aging.4
Testosterone binds in the bloodstream to sex hormone-binding globulin (SHBG), and this bound form is generally considered biologically inactive, although there are in vitro and animal studies suggesting SHBG-bound androgen may indeed have biological activity. 5,6 “Bioavailable” testosterone is active and includes both free testosterone and testosterone bound to albumin.
There is no general agreement on the acceptable normal range of testosterone, with variability within the literature and between laboratories. “Normal” total testosterone levels have ranged from more than 280 ng/dL to more than 350 ng/dL (12 nmol/L).7,8 Similarly, there is no generally accepted lower limit of normal, although some studies report a threshold level of testosterone less than 230 ng/dL (8 nmol/L) as “abnormal.” Values between these two upper and lower limits are considered “borderline.”7,8 These intermediate or borderline values coupled with clinical symptoms of testosterone deficiency syndrome or ADAM should be considered abnormal.
When total testosterone is borderline, measurement of free or bioavailable testosterone (free plus albumin-bound) should be considered. Total testosterone is typically measured using automated immunoassay platforms, with method-related differences leading to significant variability in measurement accuracy and precision. This variability is seen most dramatically in those with low total testosterone.9 However, the variability of total testosterone measurements is substantially smaller among mass spectrometry assays than among immunoassays. 10
The gold standards for free testosterone measurement are centrifugal ultrafiltration and equilibrium dialysis.9 However, these techniques are laborious and usually unavailable in local laboratories. Calculated free testosterone values using total testosterone and SHBG are most commonly used and are sufficiently accurate for clinical practice.11
Free testosterone levels can be diagnostic when total testosterone levels do not correspond with clinical presentation. However, the clinical utility of free testosterone is difficult to assess due to the variability among laboratory assays and a lack of consensus on threshold parameters. A threshold free testosterone level of more than 225 pmol/L (65 pg/mL) is generally considered normal.7,8 Before starting a patient on TRT, measurement of hemoglobin and prostate-specific antigen (PSA) and digital rectal examination of the prostate (if age is > 39) are essential.
Prolactin levels are recommended when low testosterone is confirmed, especially in patients at high clinical risk for hyperprolactinemia. Once hyperprolactinemia is identified, Endocrine Society guidelines recommend excluding medication use, renal failure, hypothyroidism, and parasellar tumors as possible causes of elevated prolactin levels.12
Low testosterone values should be treated only in patients with clinically significant symptoms that are likely to be caused by the low testosterone itself. Symptoms associated with age-related decline in testosterone that may improve with TRT include low libido,13,14 low energy,14 depressed mood,15–17 low muscle mass, osteoporosis, and hot flashes. Men with erectile dysfunction have also shown a significant improvement with TRT compared with placebo, but with a variable overall response independent of normalization of testosterone. 18,19 This is likely due to the multifactorial nature of erectile dysfunction, including vascular, neurologic, psychogenic, and endocrinologic causes.
Screening questionnaires have been developed for symptoms of low testosterone, but their clinical utility is unclear. The ADAM questionnaire is used as a screening tool for low testosterone but not to monitor response to TRT, and it is highly nonspecific.20 The Aging Male Symptom Scale questionnaire includes psychological, somatovegetative, and sexual components and is used both to screen for low testosterone and to measure outcomes.21 However, a recent observational study comparing the ability of these questionnaires to assess clinical symptoms revealed a low sensitivity and a low specificity to detect androgen deficiency in men with a total testosterone level less than 300 ng/dL.22 Overall, the current data do not conclusively support the use of hypogonadism questionnaires for screening.

The patient history when evaluating for ADAM should include evaluation of sexual and constitutional symptoms as described above and in Table 1. In addition, a history of traumatic, medical, or surgical events that could affect testosterone production should be obtained, including cryptorchidism, scrotal, inguinal, or abdominal surgery, pituitary surgery or radiation, prior issues with infertility, timing of puberty, history of renal or hepatic failure, chemotherapy (for cancer or autoimmune diseases), and prior use of anabolic steroids or opiates.
A complete physical examination should include assessment of virilization, gynecomastia, and the genitalia, including the size, position, and volume of the testes. The size and consistency of the prostate should be assessed on digital rectal examination.
LOW TESTOSTERONE AND ASSOCIATED COMORBIDITIES
Low testosterone is associated with many comorbidities, including metabolic syndrome, depression, type 2 diabetes mellitus, and cardiovascular disease, as discussed later in this section. Low testosterone has also shown associations with osteoporosis, cognitive impairment, hypertension, hyperlipidemia, decreased physical performance, end-stage renal disease, and treatment with steroids or opiates.23–26 However, the studies that found these associations included men younger than 40 years and may not be fully applicable to the ADAM population.
The association of metabolic syndrome and type 2 diabetes mellitus with low testosterone is well established in multiple studies. Grossman and colleagues27 investigated the association of type 2 diabetes mellitus and low testosterone, with low total testosterone defined as below 10 nmol/L and low calculated free testosterone less than 0.23 nmol/L. The prevalence of low total testosterone was 43%, and the prevalence of low free testosterone was 57%. In addition, a recent meta-analysis comparing total testosterone of men with and without metabolic syndrome revealed an association between a baseline decrease in mean total and free testosterone levels in men with metabolic syndrome compared with controls. This study found a total testosterone mean difference of –2.64 nmol/L (95% confidence interval [CI] –2.95 to –2.32) and a free testosterone mean difference of –0.26 pmol/L (95% CI –0.39 to –0.13), respectively, when comparing men with metabolic syndrome against those without.28
Testosterone has also been suggested to be protective against type 2 diabetes mellitus, with 42% lower risk of type 2 diabetes mellitus in men with testosterone levels ranging from 450 ng/dL to 605 ng/dL.29
Obesity has been specifically linked with secondary hypogonadism.4,23,24 A prospective cohort of 58 men with an average age of 46 years and a body mass index ranging from 30 to 45 kg/m2 were monitored on a low-calorie diet for 9 weeks. Afterward, biochemical analysis revealed an increase in free testosterone from 185 pmol/L ± 66 to 208 ± 70 pmol/L (P = .002) with a mean weight loss of 16.3 kg ± 4.5 kg.30 This emphasizes the importance of lifestyle changes in the management of hypogonadal men.
LOW TESTOSTERONE AND THE OVERALL MORTALITY RATE
Low testosterone is associated unfavorably with the rate of all-cause mortality. A retrospective study in male veterans over age 40 with repeated testosterone levels over a 5-year period found that the risk of death from all causes in men with normal testosterone (> 250 ng/dL or free testosterone > 0.75 ng/dL) was 20% (95% CI 16.2%–241%) vs 35% (95% CI 28.5%–41.4%) in men with low testosterone (< 250 ng/dL or free testosterone < 0.75 ng/dL). In multivariate analysis, men with testosterone less than 250 ng/dL (< 8.7 nmol/L) or free testosterone less than 0.75 ng/dL (< 0.03 nmol/L) had up to an 88% higher death rate than men with normal testosterone levels.31
Low testosterone has also been associated with other end-organ, disease-specific mortality. In men with end-stage renal disease, low testosterone was an independent predictor of death from all causes and from cardiovascular disease.32 A prospective European health study revealed an association between low testosterone and increased risk of death from cardiovascular disease and cancer.33 A recent meta-analysis of population-based studies confirmed this association, despite significant interstudy heterogeneity. 34 Although multiple studies show an independent association of low testosterone and increased mortality rate, causality remains unconfirmed. This may be difficult to prove, given the available study designs and the nonspecific nature of symptoms related to low testosterone and potentially associated comorbidities.
TRT: INDICATIONS AND CONTRAINDICATIONS
The indications, benefits, and risks of TRT are controversial, with current data lacking long-term follow-up and consistent biochemical target values. Treatment of low testosterone is not indicated at the present time in the absence of clinical symptoms.
According to recently published guidelines, TRT is recommended for symptomatic men with low or borderline total testosterone or free testosterone (< 350 ng/dL or < 65 pg/mL).7,8 Patients with borderline biochemical values (total testosterone 200–350 ng/dL, free testosterone 40–65 pg/mL) and possible related symptoms should be treated with TRT for at least 3 months and then reevaluated to verify improved testosterone levels and to assess for symptom amelioration or resolution.35 Dose escalation is recommended in patients with subtherapeutic testosterone levels and limited clinical improvement after 3 months of treatment.
Target maintenance testosterone levels have not been defined, with mid to lower young adult male serum testosterone levels recommended at this time.8 Given that the current literature does not specify a target testosterone replacement range, we recommend monitoring the clinical response along with total testosterone to decide adjustments in TRT. Ultimately, treatment goals of TRT should be the resolution of signs and symptoms, including improvement of sexual function, libido, and preservation of bone mineral density.7,8
Contraindications
TRT is not recommended in men with the following:
- Breast cancer
- Polycythemia (hematocrit > 50%)
- Untreated obstructive sleep apnea
- Lower urinary tract symptoms caused by an enlarged prostate; International Prostate Symptom Score > 19
- Poorly controlled heart failure
- Desire for fertility.
The role of TRT in prostate cancer remains controversial (see below) and remains contraindicated in recent Endocrine Society clinical practice guidelines.7 Guidelines recommend urologic consultation prior to initiation of TRT in patients at increased risk of prostate cancer,7 based on age, race, family history, PSA, PSA velocity, and history of prostate biopsy.
One prominent historic concern about androgen replacement therapy regards the potential for de novo development of prostate cancer. Numerous studies have failed to find elevated risk of new diagnosis, progression, or recurrence of prostate cancer in patients on TRT.36,37 Nevertheless, patients who develop elevated PSA, increased PSA velocity, or an abnormal digital rectal examination while on TRT should undergo prostate biopsy.
TRT FORMULATIONS AND TREATMENT OPTIONS
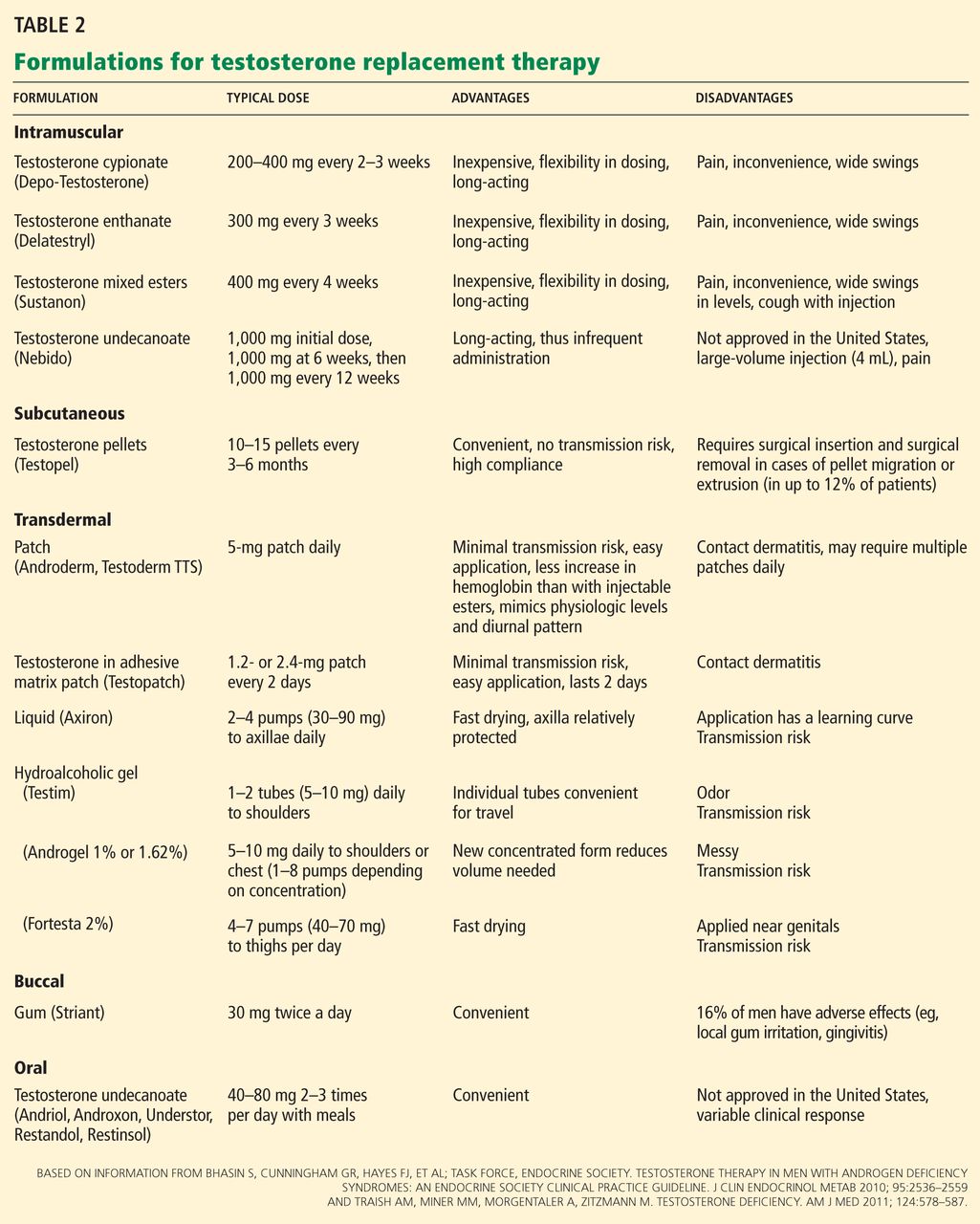
A number of effective formulations of TRT are available (Table 2). Transdermal and parenteral formulations are most commonly used. Enteric testosterone formulations are not available in the United States and are associated with hepatotoxicity. While buccal testosterone therapy is available, it often leads to local gingival irritation and has not gained widespread popularity.
Parenteral TRT can be administered intramuscularly (IM) or subcutaneously (SQ). Testosterone cypionate (Depo-Testosterone) is the only IM form available in the United States and is given every 2 to 3 weeks. It is the least expensive form of TRT, but it requires frequent administration (by either the clinical practitioner or the patient himself). Testosterone cypionate injections lead to markedly wide swings of testosterone levels, ranging from supraphysiologic levels for a few days after administration to hypogonadal levels before the next injection. This may be mitigated by more-frequent injections. The longer-acting form testosterone undecanoate is available outside the United States and is given every 12 weeks when stable levels are reached.
The other parenteral option is SQ slow-release pellets (Testopel). These pellets have 75 mg of testosterone. Typically 8 to 14 pellets are placed subcutaneously in the buttock area, which will provide coverage for 3 to 6 months.38 The insertion procedure is simple with a short learning curve, limited compliance issues, and elimination of risk of transdermal transmission of drug to others. Disadvantages include wound infection and pellet extrusion, seen in 0.3% to 12% of patients in various studies.38
Another route of TRT is transdermal, including patches, liquids, and gels. Patches are applied daily and are rotated to different sites with minimal risk for skin transmission to others, although use may be limited by site dermatitis. Three hydro-alcoholic gel formulations are currently available in the United States: Androgel (1% or 1.62%), which is applied to the chest or the shoulders; Testim 1%, which is applied to the shoulders; and Fortesta (2%), which is applied to the thighs. A liquid preparation, Axiron, is applied to the axillae. Because secondary transfer to women and children is possible, it is important to thoroughly wash hands after application and to cover the treated skin with clothing. In 3 to 4 hours, all the medication is absorbed, and the area should then be washed before direct skin contact with others (Table 2).
MONITORING PATIENTS ON TRT
Patients starting TRT will require clinical and biochemical monitoring to evaluate response to therapy as well as possible side effects. The first set of laboratory values should be obtained 6 to 12 weeks after initiation of therapy and then typically quarterly for 1 year, every 6 months for the second year, and annually thereafter. Laboratory values monitored should include total testosterone, PSA, and hematocrit.
Men on daily therapy (patch, gel, liquid) should have testosterone drawn approximately 2 hours after application. Current TRT regimen data lack an appropriate target testosterone value, and guidelines suggest a mid to lower young adult male testosterone level.8 Since this is not clearly delineated in the current literature, the authors recommend monitoring clinical symptoms along with testosterone levels when adjusting TRT. It is important to document that serum testosterone was actually increased to the normal range in treated men without clinical improvement.
A rise in PSA of up to 24% would be an acceptable response in a benign prostate gland, but a higher increase or increase above 4.0 ng/dL should prompt consideration of prostate biopsy. 39 Similarly, hemoglobin and hematocrit typically increase, but a hematocrit greater than 55% should prompt dose reduction or cessation.7 Transaminases do not need routine monitoring during parenteral or transdermal therapy. Bone mineral density should be monitored every 1 to 2 years.7,8
CLINICAL BENEFITS OF TRT
There are promising data regarding the clinical benefits of TRT in patients with metabolic syndrome and type 2 diabetes mellitus. A recent meta-analysis investigating the effect of TRT on metabolic syndrome revealed an improvement in fasting plasma glucose, homeostatic model assessment index, triglycerides, treadmill duration, high-density lipoprotein cholesterol, and waist circumference.40,41 TRT also decreased insulin resistance and improved glycemic control in type 2 diabetic hypogonadal men.42 Results from a randomized controlled trial comparing 12 weeks of intramuscular testosterone treatment vs placebo in men with metabolic syndrome revealed an improvement in mean waist circumference from 108 cm ± 8 cm to 105.5 cm ± 7.7 cm. Sixty percent of men initially diagnosed with metabolic syndrome and treated with testosterone no longer met diagnostic criteria for metabolic syndrome according to the National Cholesterol Education Program–Third Adult Treatment Panel (NCEP-ATP III) and the International Diabetes Federation (IDF) guidelines.43
Depression has also been associated with low testosterone, with free testosterone levels below 170 pmol/L associated with frank depressive symptoms and levels below 220 pmol/L predictive of future onset of depressive symptoms.15 Testosterone replacement therapy has been shown to improve depressive symptoms in hypogonadal men.16,17 Shores et al16 conducted a randomized placebo-controlled study of testosterone replacement in men older than 50 years with dysthymia or minor depression. Men treated with testosterone gel for 12 weeks showed an improvement of baseline total testosterone levels from 291 ng/dL to 449 ng/dL. Men treated with testosterone also had a 53% rate of depression remission compared with 19% in the placebo group.16
The evidence supporting improved sexual function with TRT is variable. Some studies indicate limited or transient improvement of sexual function after TRT in men with erectile dysfunction,18,19 while others report an improvement in sexual function after 3 months of TRT.44 Because of the multifactorial nature of erectile dysfunction, men with erectile dysfunction and ADAM may require TRT and a phosphodiesterase type 5 (PDE5) inhibitor, as TRT alone may be insufficient. In a prospective observational study of men with erectile dysfunction and an initial testosterone lower than 300 ng/dL, testosterone gel was administered for at least 1 year, and improvement in sexual function was seen. Results revealed a correlation between improvement in sexual function and concurrent therapy with a PDE5 inhibitor.45 In a recent multicenter placebo-controlled study of PDE5 inhibitor nonresponders, the addition of a testosterone gel to tadalafil (Cialis) improved sexual function, again suggesting a synergistic effect when treating erectile dysfunction with both TRT and a PDE5 inhibitor.46
ADVERSE EVENTS RELATED TO TRT
Despite the aforementioned benefits, it must be emphasized that TRT should be used for specific target symptoms related to hypogonadism in older men and that the general health benefits and safety of TRT in an asymptomatic man with a low measured testosterone alone remains unproven.
Cardiovascular events. In a recent study of 209 elderly men with low testosterone and limited mobility associated with other chronic illnesses, 6 months of TRT resulted in the development of cardiovascular-related adverse events in 23 patients compared with 5 men in the placebo group.47 This may have been related to how adverse events were reported, with cumulative adverse events reviewed every 6 months, ranging from peripheral edema, hypertension, arrhythmias, and electrocardiographic changes. Serious adverse events were reviewed as they occurred, including stroke and acute myocardial events.
Other studies41,43 have revealed a favorable effect of TRT on cardiovascular disease and its surrogate markers but have lacked detailed reports and close monitoring of adverse events. Thus, variation of outcome measurement and reporting may obfuscate the detection of adverse cardiovascular events. Outcomes may also depend on the testosterone formulation and the target serum concentration.43
Larger, long-term placebo-controlled trials are needed to elucidate cardiovascular risk as a primary outcome in older androgen-deficient men undergoing TRT.
Other adverse effects related to TRT include erythrocytosis, seen in 3% to 18% of patients with transdermal administration,48,49 and up to 44% of patients undergoing IM therapy.48 Gynecomastia can occur and is more likely to resolve after treatment cessation of transdermal testosterone treatment than IM injections.48 Other potential clinical side effects that should prompt dose-reduction or discontinuation are irritability, bothersome acne, fluid retention, testicular atrophy, worsening of lower urinary tract symptoms from an enlarged prostate, and new or worsening heart failure. Infrequently, obstructive sleep apnea may be worsened by TRT, although currently the data linking sleep apnea and TRT are limited.50
TRT AND PROSTATE CANCER
The relationship between prostate cancer growth and testosterone is well established, with androgen ablation remaining the cornerstone of treatment for metastatic disease. Since androgen deprivation leads to the regression of prostate cancer, there has been concern that TRT may lead to growth or de novo development of prostate cancer. TRT has thus been strongly prohibited in patients with prostate cancer.7 However, recent data challenge this paradigm.
In a retrospective study of 81 men (mean age 56.8 years) treated with TRT, only 4 men (4.9%) developed prostate cancer over a 5-year period.51 This is less than the estimated 16.7% probability of developing prostate cancer in the general US population.52
Recent accumulating data support the concept of testosterone reaching a saturation level when binding androgen receptors within the prostate at extremely low levels. Increases above this level with TRT as with ADAM do not increase the risk of development or progression of prostate cancer.53 In addition, large doses of dihydrotestosterone do not seem to alter PSA, prostate volume, or International Prostate Symptom Score.54 These findings may have implications in future androgen therapies in hypogonadal older men.
Pathologic studies suggest low testosterone is associated with a higher Gleason grade of prostate cancer,55 although this association remains unconfirmed.56
In men with erectile dysfunction after prostate cancer treatment, TRT appears safe after brachytherapy57 or radical prostatectomy.58 A small study of 15 hypogonadal men with castrate-resistant prostate cancer and minimal or no metastatic disease showed only 1 patient had symptomatic progression.59 Moreover, a recent small study of 13 men with known prostate cancer on active surveillance showed that TRT did not lead to local progression or metastatic disease in any of the patients.60
While these data are provocative, it should still be emphasized that the standard of care for prostate cancer screening should be followed in age-appropriate men with ADAM. In addition, hypogonadal men with prostate cancer should only be treated with testosterone in conjunction with careful counseling and ongoing monitoring.
TRT SHOULD NOT REPLACE HEALTHY LIFESTYLE CHANGES
There has been a dramatic increase in TRT initiation for nonspecific symptoms of low testosterone in older androgen-deficient men. With this increase in initiation of TRT, there is a significant risk of overtreating. While there are many encouraging associations between treatment of androgen deficiency and improvement in rates of of morbidity and mortality, much remains unknown about the overall long-term risks and benefits of TRT. It is important to emphasize that TRT should not replace healthy lifestyle changes including regular exercise, weight loss, and diet modifications, which may provide the patient symptom resolution. Thoughtful dialogue with the patient is critical prior to TRT initiation, including thorough disclosure of the risks and benefits of treatment, and the limitations of the data as it evolves.
- Feldman HA, Longcope C, Derby CA, et al. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab 2002; 87:589–598.
- Araujo AB, O’Donnell AB, Brambilla DJ, et al. Prevalence and incidence of androgen deficiency in middle-aged and older men: estimates from the Massachusetts Male Aging Study. J Clin Endocrinol Metab 2004; 89:5920–5926.
- Travison TG, Shackelton R, Araujo AB, et al. The natural history of symptomatic androgen deficiency in men: onset, progression, and spontaneous remission. J Am Geriatr Soc 2008; 56:831–839.
- Tajar A, Forti G, O’Neill TW, et al; EMAS Group. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 2010; 95:1810–1818.
- Hammes A, Andreassen TK, Spoelgen R, et al. Role of endocytosis in cellular uptake of sex steroids. Cell 2005; 122:751–762.
- Rosner W, Hryb DJ, Kahn SM, Nakhla AM, Romas NA. Interactions of sex hormone-binding globulin with target cells. Mol Cell Endocrinol 2010; 316:79–85.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wang C, Nieschlag E, Swerdloff R, et al; International Society of Andrology (ISA). Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. J Androl 2009; 30:1–9.
- Morley JE, Patrick P, Perry HM. Evaluation of assays available to measure free testosterone. Metabolism 2002; 51:554–559.
- Vesper HW, Bhasin S, Wang C, et al. Interlaboratory comparison study of serum total testosterone [corrected] measurements performed by mass spectrometry methods. Steroids 2009; 74:498–503.
- Ly LP, Sartorius G, Hull L, et al. Accuracy of calculated free testosterone formulae in men. Clin Endocrinol (Oxf) 2010; 73:382–388.
- Melmed S, Casanueva FF, Hoffman AR, et al; Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011; 96:273–288.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Kelleher S, Conway AJ, Handelsman DJ. Blood testosterone threshold for androgen deficiency symptoms. J Clin Endocrinol Metab 2004; 89:3813–3817.
- Joshi D, van Schoor NM, de Ronde W, et al. Low free testosterone levels are associated with prevalence and incidence of depressive symptoms in older men. Clin Endocrinol (Oxf) 2010; 72:232–240.
- Shores MM, Kivlahan DR, Sadak TI, Li EJ, Matsumoto AM. A randomized, double-blind, placebo-controlled study of testosterone treatment in hypogonadal older men with subthreshold depression (dysthymia or minor depression). J Clin Psychiatry 2009; 70:1009–1016.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. The effect of testosterone supplementation on depression symptoms in hypogonadal men from the Testim Registry in the US (TRiUS). Aging Male 2012; 15:14–21.
- Jain P, Rademaker AW, McVary KT. Testosterone supplementation for erectile dysfunction: results of a meta-analysis. J Urol 2000; 164:371–375.
- Mulhall JP, Valenzuela R, Aviv N, Parker M. Effect of testosterone supplementation on sexual function in hypogonadal men with erectile dysfunction. Urology 2004; 63:348–352.
- Morley JE, Charlton E, Patrick P, et al. Validation of a screening questionnaire for androgen deficiency in aging males. Metabolism 2000; 49:1239–1242.
- Moore C, Huebler D, Zimmermann T, Heinemann LA, Saad F, Thai DM. The Aging Males’ Symptoms scale (AMS) as outcome measure for treatment of androgen deficiency. Eur Urol 2004; 46:80–87.
- Chueh KS, Huang SP, Lee YC, et al. The Comparison of the Aging Male Symptoms (AMS) Scale and Androgen Deficiency in the Aging Male (ADAM) Questionnaire to Detect Androgen Deficiency in Middle-Aged Men. J Androl 2012[Epub ahead of print]
- Mulligan T, Frick MF, Zuraw QC, Stemhagen A, McWhirter C. Prevalence of hypogonadism in males aged at least 45 years: the HIM study. Int J Clin Pract 2006; 60:762–769.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Krasnoff JB, Basaria S, Pencina MJ, et al. Free testosterone levels are associated with mobility limitation and physical performance in community-dwelling men: the Framingham Offspring Study. J Clin Endocrinol Metab 2010; 95:2790–2799.
- Carrero JJ, Qureshi AR, Nakashima A, et al. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol Dial Transplant 2011; 26:184–190.
- Grossmann M, Thomas MC, Panagiotopoulos S, et al. Low testosterone levels are common and associated with insulin resistance in men with diabetes. J Clin Endocrinol Metab 2008; 93:1834–1840.
- Brand JS, van der Tweel I, Grobbee DE, Emmelot-Vonk MH, van der Schouw YT. Testosterone, sex hormone-binding globulin and the metabolic syndrome: a systematic review and meta-analysis of observational studies. Int J Epidemiol 2011; 40:189–207.
- Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 2006; 295:1288–1299.
- Niskanen L, Laaksonen DE, Punnonen K, Mustajoki P, Kaukua J, Rissanen A. Changes in sex hormone-binding globulin and testosterone during weight loss and weight maintenance in abdominally obese men with the metabolic syndrome. Diabetes Obes Metab 2004; 6:208–215.
- Shores MM, Matsumoto AM, Sloan KL, Kivlahan DR. Low serum testosterone and mortality in male veterans. Arch Intern Med 2006; 166:1660–1665.
- Carrero JJ, Qureshi AR, Parini P, et al. Low serum testosterone increases mortality risk among male dialysis patients. J Am Soc Nephrol 2009; 20:613–620.
- Haring R, Völzke H, Steveling A, et al. Low serum testosterone levels are associated with increased risk of mortality in a population-based cohort of men aged 20–79. Eur Heart J 2010; 31:1494–1501.
- Araujo AB, Dixon JM, Suarez EA, Murad MH, Guey LT, Wittert GA. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab 2011; 96:3007–3019.
- Rhoden EL, Morgentaler A. Risks of testosterone-replacement therapy and recommendations for monitoring. N Engl J Med 2004; 350:482–492.
- Isbarn H, Pinthus JH, Marks LS, et al. Testosterone and prostate cancer: revisiting old paradigms. Eur Urol 2009; 56:48–56.
- Traish AM, Miner MM, Morgentaler A, Zitzmann M. Testosterone deficiency. Am J Med 2011; 124:578–587.
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med 2009; 6:3177–3192.
- Gerstenbluth RE, Maniam PN, Corty EW, Seftel AD. Prostate-specific antigen changes in hypogonadal men treated with testosterone replacement. J Androl 2002; 23:922–926.
- Corona G, Monami M, Rastrelli G, et al. Testosterone and metabolic syndrome: a meta-analysis study. J Sex Med 2011; 8:272–283.
- Corona G, Rastrelli G, Monami M, et al. Hypogonadism as a risk factor for cardiovascular mortality in men: a meta-analytic study. Eur J Endocrinol 2011; 165:687–701.
- Kapoor D, Goodwin E, Channer KS, Jones TH. Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur J Endocrinol 2006; 154:899–906.
- Aversa A, Bruzziches R, Francomano D, et al. Effects of testosterone undecanoate on cardiovascular risk factors and atherosclerosis in middle-aged men with late-onset hypogonadism and metabolic syndrome: results from a 24-month, randomized, double-blind, placebo-controlled study. J Sex Med 2010; 7:3495–3503.
- Rhoden EL, Morgentaler A. Symptomatic response rates to testosterone therapy and the likelihood of completing 12 months of therapy in clinical practice. J Sex Med 2010; 7:277–283.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. Improved sexual function with testosterone replacement therapy in hypogonadal men: real-world data from the Testim Registry in the United States (TRiUS). J Sex Med 2011; 8:3204–3213.
- Buvat J, Montorsi F, Maggi M, et al. Hypogonadal men nonresponders to the PDE5 inhibitor tadalafil benefit from normalization of testosterone levels with a 1% hydroalcoholic testosterone gel in the treatment of erectile dysfunction (TADTEST study). J Sex Med 2011; 8:284–293.
- Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med 2010; 363:109–122.
- Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab 1999; 84:3469–3478.
- Wang C, Swerdloff RS, Iranmanesh A, et al; Testosterone Gel Study Group. Transdermal testosterone gel improves sexual function, mood, muscle strength, and body composition parameters in hypogonadal men. J Clin Endocrinol Metab 2000; 85:2839–2853.
- Hanafy HM. Testosterone therapy and obstructive sleep apnea: is there a real connection? J Sex Med 2007; 4:1241–1246.
- Coward RM, Simhan J, Carson CC. Prostate-specific antigen changes and prostate cancer in hypogonadal men treated with testosterone replacement therapy. BJU Int 2009; 103:1179–1183.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Morgentaler A, Traish AM. Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol 2009; 55:310–320.
- Page ST, Lin DW, Mostaghel EA, et al. Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: a randomized-controlled trial. J Clin Endocrinol Metab 2011; 96:430–437.
- Botto H, Neuzillet Y, Lebret T, Camparo P, Molinie V, Raynaud JP. High incidence of predominant Gleason pattern 4 localized prostate cancer is associated with low serum testosterone. J Urol 2011; 186:1400–1405.
- Salonia A, Gallina A, Briganti A, et al. Preoperative hypogonadism is not an independent predictor of high-risk disease in patients undergoing radical prostatectomy. Cancer 2011; 117:3953–3962.
- Sarosdy MF. Testosterone replacement for hypogonadism after treatment of early prostate cancer with brachytherapy. Cancer 2007; 109:536–541.
- Khera M. Androgens and erectile function: a case for early androgen use in postprostatectomy hypogonadal men. J Sex Med 2009; 6:(suppl 3):234–238.
- Szmulewitz R, Mohile S, Posadas E, et al. A randomized phase 1 study of testosterone replacement for patients with low-risk castration-resistant prostate cancer. Eur Urol 2009; 56:97–103.
- Morgentaler A, Lipshultz LI, Bennett R, Sweeney M, Avila D, Khera M. Testosterone therapy in men with untreated prostate cancer. J Urol 2011; 185:1256–1260.
Editor’s note: This is the second of two articles on hypogonadism in men and focuses on the appropriate use of testosterone therapy. The first article, published last month, focused in more detail on the differential diagnosis of hypogonadism.
As men age, testosterone production gradually decreases. In our increasingly aged population, clinicians will continue to see an increase in the number of men with seemingly nonspecific symptoms of aging that are possibly due to low serum testosterone (eg, low energy level, depressive symptoms, erectile dysfunction, decreased libido). These clinical symptoms, coupled with low serum testosterone, may adversely affect quality of life and life expectancy. Testosterone replacement therapy (TRT) may improve symptoms and quality of life. Given the nonspecific nature of these symptoms, accurate diagnosis and treatment of clinically significant low testosterone with a goal of symptom and quality of life improvement can prove challenging.
These challenges in diagnosis and treatment result in a lack of standardized nomenclature. The terms male menopause and andropause, although popular, are the least helpful, as they have few correlates with the better-defined female menopause. Late-onset hypogonadism implies a well-defined, later age of decline, which is inaccurate since the decline in serum testosterone in men begins in middle age and is gradual. Testosterone deficiency syndrome implies a set of specific and well-defined symptoms. Androgen deficiency in the aging male (ADAM) and Androgen deficiency in the older male are common terms specifying an age cohort (> 40 years old) and an abnormal laboratory value without mention of symptoms. While all these terms have their limitations, we will primarily use ADAM in this discussion.
PREVALENCE OF LOW TESTOSTERONE
Serum testosterone levels begin to decline in men in their mid-40s, with an approximately 1% to 2% decline annually and a marked decline after age 60.1
Araujo and colleagues2 studied the prevalence of androgen-deficient men, with androgen deficiency defined as at least three signs or symptoms and either a total testosterone less than 200 ng/dL or a total testosterone 200 ng/dL to 400 ng/dL with a free testosterone less than 8.91 ng/dL. The overall prevalence of low testosterone on initial measurement was 6%, which doubled to 12% with repeat measurement.
Serial measures are important: one study that followed untreated men over 15 years found normal testosterone on serial measures in 50%.3 In a multicenter cross-sectional study, 11.8% of men had low testosterone and low or normal luteinizing hormone (LH) levels (secondary hypogonadism/hypothalamic-pituitary failure), with 2% of patients with low testosterone and elevated LH (primary hypogonadism/testicular failure).4
CLINICAL PRESENTATION AND DIAGNOSIS
A biochemical diagnosis of low testosterone is dependent on accurate measurement. Testosterone release is diurnal, with the highest levels in the early morning, and often has week-to-week variability. Thus, it is important to collect blood in the early morning and to confirm a diagnosis of low testosterone with at least one repeat measurement several days later, including LH assessment. LH levels will help differentiate primary hypogonadism from secondary hypogonadism, which may alter diagnosis and treatment in certain patients, with secondary hypogonadism associated with pituitary dysfunction, and primary hypogonadism associated with aging.4
Testosterone binds in the bloodstream to sex hormone-binding globulin (SHBG), and this bound form is generally considered biologically inactive, although there are in vitro and animal studies suggesting SHBG-bound androgen may indeed have biological activity. 5,6 “Bioavailable” testosterone is active and includes both free testosterone and testosterone bound to albumin.
There is no general agreement on the acceptable normal range of testosterone, with variability within the literature and between laboratories. “Normal” total testosterone levels have ranged from more than 280 ng/dL to more than 350 ng/dL (12 nmol/L).7,8 Similarly, there is no generally accepted lower limit of normal, although some studies report a threshold level of testosterone less than 230 ng/dL (8 nmol/L) as “abnormal.” Values between these two upper and lower limits are considered “borderline.”7,8 These intermediate or borderline values coupled with clinical symptoms of testosterone deficiency syndrome or ADAM should be considered abnormal.
When total testosterone is borderline, measurement of free or bioavailable testosterone (free plus albumin-bound) should be considered. Total testosterone is typically measured using automated immunoassay platforms, with method-related differences leading to significant variability in measurement accuracy and precision. This variability is seen most dramatically in those with low total testosterone.9 However, the variability of total testosterone measurements is substantially smaller among mass spectrometry assays than among immunoassays. 10
The gold standards for free testosterone measurement are centrifugal ultrafiltration and equilibrium dialysis.9 However, these techniques are laborious and usually unavailable in local laboratories. Calculated free testosterone values using total testosterone and SHBG are most commonly used and are sufficiently accurate for clinical practice.11
Free testosterone levels can be diagnostic when total testosterone levels do not correspond with clinical presentation. However, the clinical utility of free testosterone is difficult to assess due to the variability among laboratory assays and a lack of consensus on threshold parameters. A threshold free testosterone level of more than 225 pmol/L (65 pg/mL) is generally considered normal.7,8 Before starting a patient on TRT, measurement of hemoglobin and prostate-specific antigen (PSA) and digital rectal examination of the prostate (if age is > 39) are essential.
Prolactin levels are recommended when low testosterone is confirmed, especially in patients at high clinical risk for hyperprolactinemia. Once hyperprolactinemia is identified, Endocrine Society guidelines recommend excluding medication use, renal failure, hypothyroidism, and parasellar tumors as possible causes of elevated prolactin levels.12
Low testosterone values should be treated only in patients with clinically significant symptoms that are likely to be caused by the low testosterone itself. Symptoms associated with age-related decline in testosterone that may improve with TRT include low libido,13,14 low energy,14 depressed mood,15–17 low muscle mass, osteoporosis, and hot flashes. Men with erectile dysfunction have also shown a significant improvement with TRT compared with placebo, but with a variable overall response independent of normalization of testosterone. 18,19 This is likely due to the multifactorial nature of erectile dysfunction, including vascular, neurologic, psychogenic, and endocrinologic causes.
Screening questionnaires have been developed for symptoms of low testosterone, but their clinical utility is unclear. The ADAM questionnaire is used as a screening tool for low testosterone but not to monitor response to TRT, and it is highly nonspecific.20 The Aging Male Symptom Scale questionnaire includes psychological, somatovegetative, and sexual components and is used both to screen for low testosterone and to measure outcomes.21 However, a recent observational study comparing the ability of these questionnaires to assess clinical symptoms revealed a low sensitivity and a low specificity to detect androgen deficiency in men with a total testosterone level less than 300 ng/dL.22 Overall, the current data do not conclusively support the use of hypogonadism questionnaires for screening.
The patient history when evaluating for ADAM should include evaluation of sexual and constitutional symptoms as described above and in Table 1. In addition, a history of traumatic, medical, or surgical events that could affect testosterone production should be obtained, including cryptorchidism, scrotal, inguinal, or abdominal surgery, pituitary surgery or radiation, prior issues with infertility, timing of puberty, history of renal or hepatic failure, chemotherapy (for cancer or autoimmune diseases), and prior use of anabolic steroids or opiates.
A complete physical examination should include assessment of virilization, gynecomastia, and the genitalia, including the size, position, and volume of the testes. The size and consistency of the prostate should be assessed on digital rectal examination.
LOW TESTOSTERONE AND ASSOCIATED COMORBIDITIES
Low testosterone is associated with many comorbidities, including metabolic syndrome, depression, type 2 diabetes mellitus, and cardiovascular disease, as discussed later in this section. Low testosterone has also shown associations with osteoporosis, cognitive impairment, hypertension, hyperlipidemia, decreased physical performance, end-stage renal disease, and treatment with steroids or opiates.23–26 However, the studies that found these associations included men younger than 40 years and may not be fully applicable to the ADAM population.
The association of metabolic syndrome and type 2 diabetes mellitus with low testosterone is well established in multiple studies. Grossman and colleagues27 investigated the association of type 2 diabetes mellitus and low testosterone, with low total testosterone defined as below 10 nmol/L and low calculated free testosterone less than 0.23 nmol/L. The prevalence of low total testosterone was 43%, and the prevalence of low free testosterone was 57%. In addition, a recent meta-analysis comparing total testosterone of men with and without metabolic syndrome revealed an association between a baseline decrease in mean total and free testosterone levels in men with metabolic syndrome compared with controls. This study found a total testosterone mean difference of –2.64 nmol/L (95% confidence interval [CI] –2.95 to –2.32) and a free testosterone mean difference of –0.26 pmol/L (95% CI –0.39 to –0.13), respectively, when comparing men with metabolic syndrome against those without.28
Testosterone has also been suggested to be protective against type 2 diabetes mellitus, with 42% lower risk of type 2 diabetes mellitus in men with testosterone levels ranging from 450 ng/dL to 605 ng/dL.29
Obesity has been specifically linked with secondary hypogonadism.4,23,24 A prospective cohort of 58 men with an average age of 46 years and a body mass index ranging from 30 to 45 kg/m2 were monitored on a low-calorie diet for 9 weeks. Afterward, biochemical analysis revealed an increase in free testosterone from 185 pmol/L ± 66 to 208 ± 70 pmol/L (P = .002) with a mean weight loss of 16.3 kg ± 4.5 kg.30 This emphasizes the importance of lifestyle changes in the management of hypogonadal men.
LOW TESTOSTERONE AND THE OVERALL MORTALITY RATE
Low testosterone is associated unfavorably with the rate of all-cause mortality. A retrospective study in male veterans over age 40 with repeated testosterone levels over a 5-year period found that the risk of death from all causes in men with normal testosterone (> 250 ng/dL or free testosterone > 0.75 ng/dL) was 20% (95% CI 16.2%–241%) vs 35% (95% CI 28.5%–41.4%) in men with low testosterone (< 250 ng/dL or free testosterone < 0.75 ng/dL). In multivariate analysis, men with testosterone less than 250 ng/dL (< 8.7 nmol/L) or free testosterone less than 0.75 ng/dL (< 0.03 nmol/L) had up to an 88% higher death rate than men with normal testosterone levels.31
Low testosterone has also been associated with other end-organ, disease-specific mortality. In men with end-stage renal disease, low testosterone was an independent predictor of death from all causes and from cardiovascular disease.32 A prospective European health study revealed an association between low testosterone and increased risk of death from cardiovascular disease and cancer.33 A recent meta-analysis of population-based studies confirmed this association, despite significant interstudy heterogeneity. 34 Although multiple studies show an independent association of low testosterone and increased mortality rate, causality remains unconfirmed. This may be difficult to prove, given the available study designs and the nonspecific nature of symptoms related to low testosterone and potentially associated comorbidities.
TRT: INDICATIONS AND CONTRAINDICATIONS
The indications, benefits, and risks of TRT are controversial, with current data lacking long-term follow-up and consistent biochemical target values. Treatment of low testosterone is not indicated at the present time in the absence of clinical symptoms.
According to recently published guidelines, TRT is recommended for symptomatic men with low or borderline total testosterone or free testosterone (< 350 ng/dL or < 65 pg/mL).7,8 Patients with borderline biochemical values (total testosterone 200–350 ng/dL, free testosterone 40–65 pg/mL) and possible related symptoms should be treated with TRT for at least 3 months and then reevaluated to verify improved testosterone levels and to assess for symptom amelioration or resolution.35 Dose escalation is recommended in patients with subtherapeutic testosterone levels and limited clinical improvement after 3 months of treatment.
Target maintenance testosterone levels have not been defined, with mid to lower young adult male serum testosterone levels recommended at this time.8 Given that the current literature does not specify a target testosterone replacement range, we recommend monitoring the clinical response along with total testosterone to decide adjustments in TRT. Ultimately, treatment goals of TRT should be the resolution of signs and symptoms, including improvement of sexual function, libido, and preservation of bone mineral density.7,8
Contraindications
TRT is not recommended in men with the following:
- Breast cancer
- Polycythemia (hematocrit > 50%)
- Untreated obstructive sleep apnea
- Lower urinary tract symptoms caused by an enlarged prostate; International Prostate Symptom Score > 19
- Poorly controlled heart failure
- Desire for fertility.
The role of TRT in prostate cancer remains controversial (see below) and remains contraindicated in recent Endocrine Society clinical practice guidelines.7 Guidelines recommend urologic consultation prior to initiation of TRT in patients at increased risk of prostate cancer,7 based on age, race, family history, PSA, PSA velocity, and history of prostate biopsy.
One prominent historic concern about androgen replacement therapy regards the potential for de novo development of prostate cancer. Numerous studies have failed to find elevated risk of new diagnosis, progression, or recurrence of prostate cancer in patients on TRT.36,37 Nevertheless, patients who develop elevated PSA, increased PSA velocity, or an abnormal digital rectal examination while on TRT should undergo prostate biopsy.
TRT FORMULATIONS AND TREATMENT OPTIONS
A number of effective formulations of TRT are available (Table 2). Transdermal and parenteral formulations are most commonly used. Enteric testosterone formulations are not available in the United States and are associated with hepatotoxicity. While buccal testosterone therapy is available, it often leads to local gingival irritation and has not gained widespread popularity.
Parenteral TRT can be administered intramuscularly (IM) or subcutaneously (SQ). Testosterone cypionate (Depo-Testosterone) is the only IM form available in the United States and is given every 2 to 3 weeks. It is the least expensive form of TRT, but it requires frequent administration (by either the clinical practitioner or the patient himself). Testosterone cypionate injections lead to markedly wide swings of testosterone levels, ranging from supraphysiologic levels for a few days after administration to hypogonadal levels before the next injection. This may be mitigated by more-frequent injections. The longer-acting form testosterone undecanoate is available outside the United States and is given every 12 weeks when stable levels are reached.
The other parenteral option is SQ slow-release pellets (Testopel). These pellets have 75 mg of testosterone. Typically 8 to 14 pellets are placed subcutaneously in the buttock area, which will provide coverage for 3 to 6 months.38 The insertion procedure is simple with a short learning curve, limited compliance issues, and elimination of risk of transdermal transmission of drug to others. Disadvantages include wound infection and pellet extrusion, seen in 0.3% to 12% of patients in various studies.38
Another route of TRT is transdermal, including patches, liquids, and gels. Patches are applied daily and are rotated to different sites with minimal risk for skin transmission to others, although use may be limited by site dermatitis. Three hydro-alcoholic gel formulations are currently available in the United States: Androgel (1% or 1.62%), which is applied to the chest or the shoulders; Testim 1%, which is applied to the shoulders; and Fortesta (2%), which is applied to the thighs. A liquid preparation, Axiron, is applied to the axillae. Because secondary transfer to women and children is possible, it is important to thoroughly wash hands after application and to cover the treated skin with clothing. In 3 to 4 hours, all the medication is absorbed, and the area should then be washed before direct skin contact with others (Table 2).
MONITORING PATIENTS ON TRT
Patients starting TRT will require clinical and biochemical monitoring to evaluate response to therapy as well as possible side effects. The first set of laboratory values should be obtained 6 to 12 weeks after initiation of therapy and then typically quarterly for 1 year, every 6 months for the second year, and annually thereafter. Laboratory values monitored should include total testosterone, PSA, and hematocrit.
Men on daily therapy (patch, gel, liquid) should have testosterone drawn approximately 2 hours after application. Current TRT regimen data lack an appropriate target testosterone value, and guidelines suggest a mid to lower young adult male testosterone level.8 Since this is not clearly delineated in the current literature, the authors recommend monitoring clinical symptoms along with testosterone levels when adjusting TRT. It is important to document that serum testosterone was actually increased to the normal range in treated men without clinical improvement.
A rise in PSA of up to 24% would be an acceptable response in a benign prostate gland, but a higher increase or increase above 4.0 ng/dL should prompt consideration of prostate biopsy. 39 Similarly, hemoglobin and hematocrit typically increase, but a hematocrit greater than 55% should prompt dose reduction or cessation.7 Transaminases do not need routine monitoring during parenteral or transdermal therapy. Bone mineral density should be monitored every 1 to 2 years.7,8
CLINICAL BENEFITS OF TRT
There are promising data regarding the clinical benefits of TRT in patients with metabolic syndrome and type 2 diabetes mellitus. A recent meta-analysis investigating the effect of TRT on metabolic syndrome revealed an improvement in fasting plasma glucose, homeostatic model assessment index, triglycerides, treadmill duration, high-density lipoprotein cholesterol, and waist circumference.40,41 TRT also decreased insulin resistance and improved glycemic control in type 2 diabetic hypogonadal men.42 Results from a randomized controlled trial comparing 12 weeks of intramuscular testosterone treatment vs placebo in men with metabolic syndrome revealed an improvement in mean waist circumference from 108 cm ± 8 cm to 105.5 cm ± 7.7 cm. Sixty percent of men initially diagnosed with metabolic syndrome and treated with testosterone no longer met diagnostic criteria for metabolic syndrome according to the National Cholesterol Education Program–Third Adult Treatment Panel (NCEP-ATP III) and the International Diabetes Federation (IDF) guidelines.43
Depression has also been associated with low testosterone, with free testosterone levels below 170 pmol/L associated with frank depressive symptoms and levels below 220 pmol/L predictive of future onset of depressive symptoms.15 Testosterone replacement therapy has been shown to improve depressive symptoms in hypogonadal men.16,17 Shores et al16 conducted a randomized placebo-controlled study of testosterone replacement in men older than 50 years with dysthymia or minor depression. Men treated with testosterone gel for 12 weeks showed an improvement of baseline total testosterone levels from 291 ng/dL to 449 ng/dL. Men treated with testosterone also had a 53% rate of depression remission compared with 19% in the placebo group.16
The evidence supporting improved sexual function with TRT is variable. Some studies indicate limited or transient improvement of sexual function after TRT in men with erectile dysfunction,18,19 while others report an improvement in sexual function after 3 months of TRT.44 Because of the multifactorial nature of erectile dysfunction, men with erectile dysfunction and ADAM may require TRT and a phosphodiesterase type 5 (PDE5) inhibitor, as TRT alone may be insufficient. In a prospective observational study of men with erectile dysfunction and an initial testosterone lower than 300 ng/dL, testosterone gel was administered for at least 1 year, and improvement in sexual function was seen. Results revealed a correlation between improvement in sexual function and concurrent therapy with a PDE5 inhibitor.45 In a recent multicenter placebo-controlled study of PDE5 inhibitor nonresponders, the addition of a testosterone gel to tadalafil (Cialis) improved sexual function, again suggesting a synergistic effect when treating erectile dysfunction with both TRT and a PDE5 inhibitor.46
ADVERSE EVENTS RELATED TO TRT
Despite the aforementioned benefits, it must be emphasized that TRT should be used for specific target symptoms related to hypogonadism in older men and that the general health benefits and safety of TRT in an asymptomatic man with a low measured testosterone alone remains unproven.
Cardiovascular events. In a recent study of 209 elderly men with low testosterone and limited mobility associated with other chronic illnesses, 6 months of TRT resulted in the development of cardiovascular-related adverse events in 23 patients compared with 5 men in the placebo group.47 This may have been related to how adverse events were reported, with cumulative adverse events reviewed every 6 months, ranging from peripheral edema, hypertension, arrhythmias, and electrocardiographic changes. Serious adverse events were reviewed as they occurred, including stroke and acute myocardial events.
Other studies41,43 have revealed a favorable effect of TRT on cardiovascular disease and its surrogate markers but have lacked detailed reports and close monitoring of adverse events. Thus, variation of outcome measurement and reporting may obfuscate the detection of adverse cardiovascular events. Outcomes may also depend on the testosterone formulation and the target serum concentration.43
Larger, long-term placebo-controlled trials are needed to elucidate cardiovascular risk as a primary outcome in older androgen-deficient men undergoing TRT.
Other adverse effects related to TRT include erythrocytosis, seen in 3% to 18% of patients with transdermal administration,48,49 and up to 44% of patients undergoing IM therapy.48 Gynecomastia can occur and is more likely to resolve after treatment cessation of transdermal testosterone treatment than IM injections.48 Other potential clinical side effects that should prompt dose-reduction or discontinuation are irritability, bothersome acne, fluid retention, testicular atrophy, worsening of lower urinary tract symptoms from an enlarged prostate, and new or worsening heart failure. Infrequently, obstructive sleep apnea may be worsened by TRT, although currently the data linking sleep apnea and TRT are limited.50
TRT AND PROSTATE CANCER
The relationship between prostate cancer growth and testosterone is well established, with androgen ablation remaining the cornerstone of treatment for metastatic disease. Since androgen deprivation leads to the regression of prostate cancer, there has been concern that TRT may lead to growth or de novo development of prostate cancer. TRT has thus been strongly prohibited in patients with prostate cancer.7 However, recent data challenge this paradigm.
In a retrospective study of 81 men (mean age 56.8 years) treated with TRT, only 4 men (4.9%) developed prostate cancer over a 5-year period.51 This is less than the estimated 16.7% probability of developing prostate cancer in the general US population.52
Recent accumulating data support the concept of testosterone reaching a saturation level when binding androgen receptors within the prostate at extremely low levels. Increases above this level with TRT as with ADAM do not increase the risk of development or progression of prostate cancer.53 In addition, large doses of dihydrotestosterone do not seem to alter PSA, prostate volume, or International Prostate Symptom Score.54 These findings may have implications in future androgen therapies in hypogonadal older men.
Pathologic studies suggest low testosterone is associated with a higher Gleason grade of prostate cancer,55 although this association remains unconfirmed.56
In men with erectile dysfunction after prostate cancer treatment, TRT appears safe after brachytherapy57 or radical prostatectomy.58 A small study of 15 hypogonadal men with castrate-resistant prostate cancer and minimal or no metastatic disease showed only 1 patient had symptomatic progression.59 Moreover, a recent small study of 13 men with known prostate cancer on active surveillance showed that TRT did not lead to local progression or metastatic disease in any of the patients.60
While these data are provocative, it should still be emphasized that the standard of care for prostate cancer screening should be followed in age-appropriate men with ADAM. In addition, hypogonadal men with prostate cancer should only be treated with testosterone in conjunction with careful counseling and ongoing monitoring.
TRT SHOULD NOT REPLACE HEALTHY LIFESTYLE CHANGES
There has been a dramatic increase in TRT initiation for nonspecific symptoms of low testosterone in older androgen-deficient men. With this increase in initiation of TRT, there is a significant risk of overtreating. While there are many encouraging associations between treatment of androgen deficiency and improvement in rates of of morbidity and mortality, much remains unknown about the overall long-term risks and benefits of TRT. It is important to emphasize that TRT should not replace healthy lifestyle changes including regular exercise, weight loss, and diet modifications, which may provide the patient symptom resolution. Thoughtful dialogue with the patient is critical prior to TRT initiation, including thorough disclosure of the risks and benefits of treatment, and the limitations of the data as it evolves.
Editor’s note: This is the second of two articles on hypogonadism in men and focuses on the appropriate use of testosterone therapy. The first article, published last month, focused in more detail on the differential diagnosis of hypogonadism.
As men age, testosterone production gradually decreases. In our increasingly aged population, clinicians will continue to see an increase in the number of men with seemingly nonspecific symptoms of aging that are possibly due to low serum testosterone (eg, low energy level, depressive symptoms, erectile dysfunction, decreased libido). These clinical symptoms, coupled with low serum testosterone, may adversely affect quality of life and life expectancy. Testosterone replacement therapy (TRT) may improve symptoms and quality of life. Given the nonspecific nature of these symptoms, accurate diagnosis and treatment of clinically significant low testosterone with a goal of symptom and quality of life improvement can prove challenging.
These challenges in diagnosis and treatment result in a lack of standardized nomenclature. The terms male menopause and andropause, although popular, are the least helpful, as they have few correlates with the better-defined female menopause. Late-onset hypogonadism implies a well-defined, later age of decline, which is inaccurate since the decline in serum testosterone in men begins in middle age and is gradual. Testosterone deficiency syndrome implies a set of specific and well-defined symptoms. Androgen deficiency in the aging male (ADAM) and Androgen deficiency in the older male are common terms specifying an age cohort (> 40 years old) and an abnormal laboratory value without mention of symptoms. While all these terms have their limitations, we will primarily use ADAM in this discussion.
PREVALENCE OF LOW TESTOSTERONE
Serum testosterone levels begin to decline in men in their mid-40s, with an approximately 1% to 2% decline annually and a marked decline after age 60.1
Araujo and colleagues2 studied the prevalence of androgen-deficient men, with androgen deficiency defined as at least three signs or symptoms and either a total testosterone less than 200 ng/dL or a total testosterone 200 ng/dL to 400 ng/dL with a free testosterone less than 8.91 ng/dL. The overall prevalence of low testosterone on initial measurement was 6%, which doubled to 12% with repeat measurement.
Serial measures are important: one study that followed untreated men over 15 years found normal testosterone on serial measures in 50%.3 In a multicenter cross-sectional study, 11.8% of men had low testosterone and low or normal luteinizing hormone (LH) levels (secondary hypogonadism/hypothalamic-pituitary failure), with 2% of patients with low testosterone and elevated LH (primary hypogonadism/testicular failure).4
CLINICAL PRESENTATION AND DIAGNOSIS
A biochemical diagnosis of low testosterone is dependent on accurate measurement. Testosterone release is diurnal, with the highest levels in the early morning, and often has week-to-week variability. Thus, it is important to collect blood in the early morning and to confirm a diagnosis of low testosterone with at least one repeat measurement several days later, including LH assessment. LH levels will help differentiate primary hypogonadism from secondary hypogonadism, which may alter diagnosis and treatment in certain patients, with secondary hypogonadism associated with pituitary dysfunction, and primary hypogonadism associated with aging.4
Testosterone binds in the bloodstream to sex hormone-binding globulin (SHBG), and this bound form is generally considered biologically inactive, although there are in vitro and animal studies suggesting SHBG-bound androgen may indeed have biological activity. 5,6 “Bioavailable” testosterone is active and includes both free testosterone and testosterone bound to albumin.
There is no general agreement on the acceptable normal range of testosterone, with variability within the literature and between laboratories. “Normal” total testosterone levels have ranged from more than 280 ng/dL to more than 350 ng/dL (12 nmol/L).7,8 Similarly, there is no generally accepted lower limit of normal, although some studies report a threshold level of testosterone less than 230 ng/dL (8 nmol/L) as “abnormal.” Values between these two upper and lower limits are considered “borderline.”7,8 These intermediate or borderline values coupled with clinical symptoms of testosterone deficiency syndrome or ADAM should be considered abnormal.
When total testosterone is borderline, measurement of free or bioavailable testosterone (free plus albumin-bound) should be considered. Total testosterone is typically measured using automated immunoassay platforms, with method-related differences leading to significant variability in measurement accuracy and precision. This variability is seen most dramatically in those with low total testosterone.9 However, the variability of total testosterone measurements is substantially smaller among mass spectrometry assays than among immunoassays. 10
The gold standards for free testosterone measurement are centrifugal ultrafiltration and equilibrium dialysis.9 However, these techniques are laborious and usually unavailable in local laboratories. Calculated free testosterone values using total testosterone and SHBG are most commonly used and are sufficiently accurate for clinical practice.11
Free testosterone levels can be diagnostic when total testosterone levels do not correspond with clinical presentation. However, the clinical utility of free testosterone is difficult to assess due to the variability among laboratory assays and a lack of consensus on threshold parameters. A threshold free testosterone level of more than 225 pmol/L (65 pg/mL) is generally considered normal.7,8 Before starting a patient on TRT, measurement of hemoglobin and prostate-specific antigen (PSA) and digital rectal examination of the prostate (if age is > 39) are essential.
Prolactin levels are recommended when low testosterone is confirmed, especially in patients at high clinical risk for hyperprolactinemia. Once hyperprolactinemia is identified, Endocrine Society guidelines recommend excluding medication use, renal failure, hypothyroidism, and parasellar tumors as possible causes of elevated prolactin levels.12
Low testosterone values should be treated only in patients with clinically significant symptoms that are likely to be caused by the low testosterone itself. Symptoms associated with age-related decline in testosterone that may improve with TRT include low libido,13,14 low energy,14 depressed mood,15–17 low muscle mass, osteoporosis, and hot flashes. Men with erectile dysfunction have also shown a significant improvement with TRT compared with placebo, but with a variable overall response independent of normalization of testosterone. 18,19 This is likely due to the multifactorial nature of erectile dysfunction, including vascular, neurologic, psychogenic, and endocrinologic causes.
Screening questionnaires have been developed for symptoms of low testosterone, but their clinical utility is unclear. The ADAM questionnaire is used as a screening tool for low testosterone but not to monitor response to TRT, and it is highly nonspecific.20 The Aging Male Symptom Scale questionnaire includes psychological, somatovegetative, and sexual components and is used both to screen for low testosterone and to measure outcomes.21 However, a recent observational study comparing the ability of these questionnaires to assess clinical symptoms revealed a low sensitivity and a low specificity to detect androgen deficiency in men with a total testosterone level less than 300 ng/dL.22 Overall, the current data do not conclusively support the use of hypogonadism questionnaires for screening.
The patient history when evaluating for ADAM should include evaluation of sexual and constitutional symptoms as described above and in Table 1. In addition, a history of traumatic, medical, or surgical events that could affect testosterone production should be obtained, including cryptorchidism, scrotal, inguinal, or abdominal surgery, pituitary surgery or radiation, prior issues with infertility, timing of puberty, history of renal or hepatic failure, chemotherapy (for cancer or autoimmune diseases), and prior use of anabolic steroids or opiates.
A complete physical examination should include assessment of virilization, gynecomastia, and the genitalia, including the size, position, and volume of the testes. The size and consistency of the prostate should be assessed on digital rectal examination.
LOW TESTOSTERONE AND ASSOCIATED COMORBIDITIES
Low testosterone is associated with many comorbidities, including metabolic syndrome, depression, type 2 diabetes mellitus, and cardiovascular disease, as discussed later in this section. Low testosterone has also shown associations with osteoporosis, cognitive impairment, hypertension, hyperlipidemia, decreased physical performance, end-stage renal disease, and treatment with steroids or opiates.23–26 However, the studies that found these associations included men younger than 40 years and may not be fully applicable to the ADAM population.
The association of metabolic syndrome and type 2 diabetes mellitus with low testosterone is well established in multiple studies. Grossman and colleagues27 investigated the association of type 2 diabetes mellitus and low testosterone, with low total testosterone defined as below 10 nmol/L and low calculated free testosterone less than 0.23 nmol/L. The prevalence of low total testosterone was 43%, and the prevalence of low free testosterone was 57%. In addition, a recent meta-analysis comparing total testosterone of men with and without metabolic syndrome revealed an association between a baseline decrease in mean total and free testosterone levels in men with metabolic syndrome compared with controls. This study found a total testosterone mean difference of –2.64 nmol/L (95% confidence interval [CI] –2.95 to –2.32) and a free testosterone mean difference of –0.26 pmol/L (95% CI –0.39 to –0.13), respectively, when comparing men with metabolic syndrome against those without.28
Testosterone has also been suggested to be protective against type 2 diabetes mellitus, with 42% lower risk of type 2 diabetes mellitus in men with testosterone levels ranging from 450 ng/dL to 605 ng/dL.29
Obesity has been specifically linked with secondary hypogonadism.4,23,24 A prospective cohort of 58 men with an average age of 46 years and a body mass index ranging from 30 to 45 kg/m2 were monitored on a low-calorie diet for 9 weeks. Afterward, biochemical analysis revealed an increase in free testosterone from 185 pmol/L ± 66 to 208 ± 70 pmol/L (P = .002) with a mean weight loss of 16.3 kg ± 4.5 kg.30 This emphasizes the importance of lifestyle changes in the management of hypogonadal men.
LOW TESTOSTERONE AND THE OVERALL MORTALITY RATE
Low testosterone is associated unfavorably with the rate of all-cause mortality. A retrospective study in male veterans over age 40 with repeated testosterone levels over a 5-year period found that the risk of death from all causes in men with normal testosterone (> 250 ng/dL or free testosterone > 0.75 ng/dL) was 20% (95% CI 16.2%–241%) vs 35% (95% CI 28.5%–41.4%) in men with low testosterone (< 250 ng/dL or free testosterone < 0.75 ng/dL). In multivariate analysis, men with testosterone less than 250 ng/dL (< 8.7 nmol/L) or free testosterone less than 0.75 ng/dL (< 0.03 nmol/L) had up to an 88% higher death rate than men with normal testosterone levels.31
Low testosterone has also been associated with other end-organ, disease-specific mortality. In men with end-stage renal disease, low testosterone was an independent predictor of death from all causes and from cardiovascular disease.32 A prospective European health study revealed an association between low testosterone and increased risk of death from cardiovascular disease and cancer.33 A recent meta-analysis of population-based studies confirmed this association, despite significant interstudy heterogeneity. 34 Although multiple studies show an independent association of low testosterone and increased mortality rate, causality remains unconfirmed. This may be difficult to prove, given the available study designs and the nonspecific nature of symptoms related to low testosterone and potentially associated comorbidities.
TRT: INDICATIONS AND CONTRAINDICATIONS
The indications, benefits, and risks of TRT are controversial, with current data lacking long-term follow-up and consistent biochemical target values. Treatment of low testosterone is not indicated at the present time in the absence of clinical symptoms.
According to recently published guidelines, TRT is recommended for symptomatic men with low or borderline total testosterone or free testosterone (< 350 ng/dL or < 65 pg/mL).7,8 Patients with borderline biochemical values (total testosterone 200–350 ng/dL, free testosterone 40–65 pg/mL) and possible related symptoms should be treated with TRT for at least 3 months and then reevaluated to verify improved testosterone levels and to assess for symptom amelioration or resolution.35 Dose escalation is recommended in patients with subtherapeutic testosterone levels and limited clinical improvement after 3 months of treatment.
Target maintenance testosterone levels have not been defined, with mid to lower young adult male serum testosterone levels recommended at this time.8 Given that the current literature does not specify a target testosterone replacement range, we recommend monitoring the clinical response along with total testosterone to decide adjustments in TRT. Ultimately, treatment goals of TRT should be the resolution of signs and symptoms, including improvement of sexual function, libido, and preservation of bone mineral density.7,8
Contraindications
TRT is not recommended in men with the following:
- Breast cancer
- Polycythemia (hematocrit > 50%)
- Untreated obstructive sleep apnea
- Lower urinary tract symptoms caused by an enlarged prostate; International Prostate Symptom Score > 19
- Poorly controlled heart failure
- Desire for fertility.
The role of TRT in prostate cancer remains controversial (see below) and remains contraindicated in recent Endocrine Society clinical practice guidelines.7 Guidelines recommend urologic consultation prior to initiation of TRT in patients at increased risk of prostate cancer,7 based on age, race, family history, PSA, PSA velocity, and history of prostate biopsy.
One prominent historic concern about androgen replacement therapy regards the potential for de novo development of prostate cancer. Numerous studies have failed to find elevated risk of new diagnosis, progression, or recurrence of prostate cancer in patients on TRT.36,37 Nevertheless, patients who develop elevated PSA, increased PSA velocity, or an abnormal digital rectal examination while on TRT should undergo prostate biopsy.
TRT FORMULATIONS AND TREATMENT OPTIONS
A number of effective formulations of TRT are available (Table 2). Transdermal and parenteral formulations are most commonly used. Enteric testosterone formulations are not available in the United States and are associated with hepatotoxicity. While buccal testosterone therapy is available, it often leads to local gingival irritation and has not gained widespread popularity.
Parenteral TRT can be administered intramuscularly (IM) or subcutaneously (SQ). Testosterone cypionate (Depo-Testosterone) is the only IM form available in the United States and is given every 2 to 3 weeks. It is the least expensive form of TRT, but it requires frequent administration (by either the clinical practitioner or the patient himself). Testosterone cypionate injections lead to markedly wide swings of testosterone levels, ranging from supraphysiologic levels for a few days after administration to hypogonadal levels before the next injection. This may be mitigated by more-frequent injections. The longer-acting form testosterone undecanoate is available outside the United States and is given every 12 weeks when stable levels are reached.
The other parenteral option is SQ slow-release pellets (Testopel). These pellets have 75 mg of testosterone. Typically 8 to 14 pellets are placed subcutaneously in the buttock area, which will provide coverage for 3 to 6 months.38 The insertion procedure is simple with a short learning curve, limited compliance issues, and elimination of risk of transdermal transmission of drug to others. Disadvantages include wound infection and pellet extrusion, seen in 0.3% to 12% of patients in various studies.38
Another route of TRT is transdermal, including patches, liquids, and gels. Patches are applied daily and are rotated to different sites with minimal risk for skin transmission to others, although use may be limited by site dermatitis. Three hydro-alcoholic gel formulations are currently available in the United States: Androgel (1% or 1.62%), which is applied to the chest or the shoulders; Testim 1%, which is applied to the shoulders; and Fortesta (2%), which is applied to the thighs. A liquid preparation, Axiron, is applied to the axillae. Because secondary transfer to women and children is possible, it is important to thoroughly wash hands after application and to cover the treated skin with clothing. In 3 to 4 hours, all the medication is absorbed, and the area should then be washed before direct skin contact with others (Table 2).
MONITORING PATIENTS ON TRT
Patients starting TRT will require clinical and biochemical monitoring to evaluate response to therapy as well as possible side effects. The first set of laboratory values should be obtained 6 to 12 weeks after initiation of therapy and then typically quarterly for 1 year, every 6 months for the second year, and annually thereafter. Laboratory values monitored should include total testosterone, PSA, and hematocrit.
Men on daily therapy (patch, gel, liquid) should have testosterone drawn approximately 2 hours after application. Current TRT regimen data lack an appropriate target testosterone value, and guidelines suggest a mid to lower young adult male testosterone level.8 Since this is not clearly delineated in the current literature, the authors recommend monitoring clinical symptoms along with testosterone levels when adjusting TRT. It is important to document that serum testosterone was actually increased to the normal range in treated men without clinical improvement.
A rise in PSA of up to 24% would be an acceptable response in a benign prostate gland, but a higher increase or increase above 4.0 ng/dL should prompt consideration of prostate biopsy. 39 Similarly, hemoglobin and hematocrit typically increase, but a hematocrit greater than 55% should prompt dose reduction or cessation.7 Transaminases do not need routine monitoring during parenteral or transdermal therapy. Bone mineral density should be monitored every 1 to 2 years.7,8
CLINICAL BENEFITS OF TRT
There are promising data regarding the clinical benefits of TRT in patients with metabolic syndrome and type 2 diabetes mellitus. A recent meta-analysis investigating the effect of TRT on metabolic syndrome revealed an improvement in fasting plasma glucose, homeostatic model assessment index, triglycerides, treadmill duration, high-density lipoprotein cholesterol, and waist circumference.40,41 TRT also decreased insulin resistance and improved glycemic control in type 2 diabetic hypogonadal men.42 Results from a randomized controlled trial comparing 12 weeks of intramuscular testosterone treatment vs placebo in men with metabolic syndrome revealed an improvement in mean waist circumference from 108 cm ± 8 cm to 105.5 cm ± 7.7 cm. Sixty percent of men initially diagnosed with metabolic syndrome and treated with testosterone no longer met diagnostic criteria for metabolic syndrome according to the National Cholesterol Education Program–Third Adult Treatment Panel (NCEP-ATP III) and the International Diabetes Federation (IDF) guidelines.43
Depression has also been associated with low testosterone, with free testosterone levels below 170 pmol/L associated with frank depressive symptoms and levels below 220 pmol/L predictive of future onset of depressive symptoms.15 Testosterone replacement therapy has been shown to improve depressive symptoms in hypogonadal men.16,17 Shores et al16 conducted a randomized placebo-controlled study of testosterone replacement in men older than 50 years with dysthymia or minor depression. Men treated with testosterone gel for 12 weeks showed an improvement of baseline total testosterone levels from 291 ng/dL to 449 ng/dL. Men treated with testosterone also had a 53% rate of depression remission compared with 19% in the placebo group.16
The evidence supporting improved sexual function with TRT is variable. Some studies indicate limited or transient improvement of sexual function after TRT in men with erectile dysfunction,18,19 while others report an improvement in sexual function after 3 months of TRT.44 Because of the multifactorial nature of erectile dysfunction, men with erectile dysfunction and ADAM may require TRT and a phosphodiesterase type 5 (PDE5) inhibitor, as TRT alone may be insufficient. In a prospective observational study of men with erectile dysfunction and an initial testosterone lower than 300 ng/dL, testosterone gel was administered for at least 1 year, and improvement in sexual function was seen. Results revealed a correlation between improvement in sexual function and concurrent therapy with a PDE5 inhibitor.45 In a recent multicenter placebo-controlled study of PDE5 inhibitor nonresponders, the addition of a testosterone gel to tadalafil (Cialis) improved sexual function, again suggesting a synergistic effect when treating erectile dysfunction with both TRT and a PDE5 inhibitor.46
ADVERSE EVENTS RELATED TO TRT
Despite the aforementioned benefits, it must be emphasized that TRT should be used for specific target symptoms related to hypogonadism in older men and that the general health benefits and safety of TRT in an asymptomatic man with a low measured testosterone alone remains unproven.
Cardiovascular events. In a recent study of 209 elderly men with low testosterone and limited mobility associated with other chronic illnesses, 6 months of TRT resulted in the development of cardiovascular-related adverse events in 23 patients compared with 5 men in the placebo group.47 This may have been related to how adverse events were reported, with cumulative adverse events reviewed every 6 months, ranging from peripheral edema, hypertension, arrhythmias, and electrocardiographic changes. Serious adverse events were reviewed as they occurred, including stroke and acute myocardial events.
Other studies41,43 have revealed a favorable effect of TRT on cardiovascular disease and its surrogate markers but have lacked detailed reports and close monitoring of adverse events. Thus, variation of outcome measurement and reporting may obfuscate the detection of adverse cardiovascular events. Outcomes may also depend on the testosterone formulation and the target serum concentration.43
Larger, long-term placebo-controlled trials are needed to elucidate cardiovascular risk as a primary outcome in older androgen-deficient men undergoing TRT.
Other adverse effects related to TRT include erythrocytosis, seen in 3% to 18% of patients with transdermal administration,48,49 and up to 44% of patients undergoing IM therapy.48 Gynecomastia can occur and is more likely to resolve after treatment cessation of transdermal testosterone treatment than IM injections.48 Other potential clinical side effects that should prompt dose-reduction or discontinuation are irritability, bothersome acne, fluid retention, testicular atrophy, worsening of lower urinary tract symptoms from an enlarged prostate, and new or worsening heart failure. Infrequently, obstructive sleep apnea may be worsened by TRT, although currently the data linking sleep apnea and TRT are limited.50
TRT AND PROSTATE CANCER
The relationship between prostate cancer growth and testosterone is well established, with androgen ablation remaining the cornerstone of treatment for metastatic disease. Since androgen deprivation leads to the regression of prostate cancer, there has been concern that TRT may lead to growth or de novo development of prostate cancer. TRT has thus been strongly prohibited in patients with prostate cancer.7 However, recent data challenge this paradigm.
In a retrospective study of 81 men (mean age 56.8 years) treated with TRT, only 4 men (4.9%) developed prostate cancer over a 5-year period.51 This is less than the estimated 16.7% probability of developing prostate cancer in the general US population.52
Recent accumulating data support the concept of testosterone reaching a saturation level when binding androgen receptors within the prostate at extremely low levels. Increases above this level with TRT as with ADAM do not increase the risk of development or progression of prostate cancer.53 In addition, large doses of dihydrotestosterone do not seem to alter PSA, prostate volume, or International Prostate Symptom Score.54 These findings may have implications in future androgen therapies in hypogonadal older men.
Pathologic studies suggest low testosterone is associated with a higher Gleason grade of prostate cancer,55 although this association remains unconfirmed.56
In men with erectile dysfunction after prostate cancer treatment, TRT appears safe after brachytherapy57 or radical prostatectomy.58 A small study of 15 hypogonadal men with castrate-resistant prostate cancer and minimal or no metastatic disease showed only 1 patient had symptomatic progression.59 Moreover, a recent small study of 13 men with known prostate cancer on active surveillance showed that TRT did not lead to local progression or metastatic disease in any of the patients.60
While these data are provocative, it should still be emphasized that the standard of care for prostate cancer screening should be followed in age-appropriate men with ADAM. In addition, hypogonadal men with prostate cancer should only be treated with testosterone in conjunction with careful counseling and ongoing monitoring.
TRT SHOULD NOT REPLACE HEALTHY LIFESTYLE CHANGES
There has been a dramatic increase in TRT initiation for nonspecific symptoms of low testosterone in older androgen-deficient men. With this increase in initiation of TRT, there is a significant risk of overtreating. While there are many encouraging associations between treatment of androgen deficiency and improvement in rates of of morbidity and mortality, much remains unknown about the overall long-term risks and benefits of TRT. It is important to emphasize that TRT should not replace healthy lifestyle changes including regular exercise, weight loss, and diet modifications, which may provide the patient symptom resolution. Thoughtful dialogue with the patient is critical prior to TRT initiation, including thorough disclosure of the risks and benefits of treatment, and the limitations of the data as it evolves.
- Feldman HA, Longcope C, Derby CA, et al. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab 2002; 87:589–598.
- Araujo AB, O’Donnell AB, Brambilla DJ, et al. Prevalence and incidence of androgen deficiency in middle-aged and older men: estimates from the Massachusetts Male Aging Study. J Clin Endocrinol Metab 2004; 89:5920–5926.
- Travison TG, Shackelton R, Araujo AB, et al. The natural history of symptomatic androgen deficiency in men: onset, progression, and spontaneous remission. J Am Geriatr Soc 2008; 56:831–839.
- Tajar A, Forti G, O’Neill TW, et al; EMAS Group. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 2010; 95:1810–1818.
- Hammes A, Andreassen TK, Spoelgen R, et al. Role of endocytosis in cellular uptake of sex steroids. Cell 2005; 122:751–762.
- Rosner W, Hryb DJ, Kahn SM, Nakhla AM, Romas NA. Interactions of sex hormone-binding globulin with target cells. Mol Cell Endocrinol 2010; 316:79–85.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wang C, Nieschlag E, Swerdloff R, et al; International Society of Andrology (ISA). Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. J Androl 2009; 30:1–9.
- Morley JE, Patrick P, Perry HM. Evaluation of assays available to measure free testosterone. Metabolism 2002; 51:554–559.
- Vesper HW, Bhasin S, Wang C, et al. Interlaboratory comparison study of serum total testosterone [corrected] measurements performed by mass spectrometry methods. Steroids 2009; 74:498–503.
- Ly LP, Sartorius G, Hull L, et al. Accuracy of calculated free testosterone formulae in men. Clin Endocrinol (Oxf) 2010; 73:382–388.
- Melmed S, Casanueva FF, Hoffman AR, et al; Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011; 96:273–288.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Kelleher S, Conway AJ, Handelsman DJ. Blood testosterone threshold for androgen deficiency symptoms. J Clin Endocrinol Metab 2004; 89:3813–3817.
- Joshi D, van Schoor NM, de Ronde W, et al. Low free testosterone levels are associated with prevalence and incidence of depressive symptoms in older men. Clin Endocrinol (Oxf) 2010; 72:232–240.
- Shores MM, Kivlahan DR, Sadak TI, Li EJ, Matsumoto AM. A randomized, double-blind, placebo-controlled study of testosterone treatment in hypogonadal older men with subthreshold depression (dysthymia or minor depression). J Clin Psychiatry 2009; 70:1009–1016.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. The effect of testosterone supplementation on depression symptoms in hypogonadal men from the Testim Registry in the US (TRiUS). Aging Male 2012; 15:14–21.
- Jain P, Rademaker AW, McVary KT. Testosterone supplementation for erectile dysfunction: results of a meta-analysis. J Urol 2000; 164:371–375.
- Mulhall JP, Valenzuela R, Aviv N, Parker M. Effect of testosterone supplementation on sexual function in hypogonadal men with erectile dysfunction. Urology 2004; 63:348–352.
- Morley JE, Charlton E, Patrick P, et al. Validation of a screening questionnaire for androgen deficiency in aging males. Metabolism 2000; 49:1239–1242.
- Moore C, Huebler D, Zimmermann T, Heinemann LA, Saad F, Thai DM. The Aging Males’ Symptoms scale (AMS) as outcome measure for treatment of androgen deficiency. Eur Urol 2004; 46:80–87.
- Chueh KS, Huang SP, Lee YC, et al. The Comparison of the Aging Male Symptoms (AMS) Scale and Androgen Deficiency in the Aging Male (ADAM) Questionnaire to Detect Androgen Deficiency in Middle-Aged Men. J Androl 2012[Epub ahead of print]
- Mulligan T, Frick MF, Zuraw QC, Stemhagen A, McWhirter C. Prevalence of hypogonadism in males aged at least 45 years: the HIM study. Int J Clin Pract 2006; 60:762–769.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Krasnoff JB, Basaria S, Pencina MJ, et al. Free testosterone levels are associated with mobility limitation and physical performance in community-dwelling men: the Framingham Offspring Study. J Clin Endocrinol Metab 2010; 95:2790–2799.
- Carrero JJ, Qureshi AR, Nakashima A, et al. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol Dial Transplant 2011; 26:184–190.
- Grossmann M, Thomas MC, Panagiotopoulos S, et al. Low testosterone levels are common and associated with insulin resistance in men with diabetes. J Clin Endocrinol Metab 2008; 93:1834–1840.
- Brand JS, van der Tweel I, Grobbee DE, Emmelot-Vonk MH, van der Schouw YT. Testosterone, sex hormone-binding globulin and the metabolic syndrome: a systematic review and meta-analysis of observational studies. Int J Epidemiol 2011; 40:189–207.
- Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 2006; 295:1288–1299.
- Niskanen L, Laaksonen DE, Punnonen K, Mustajoki P, Kaukua J, Rissanen A. Changes in sex hormone-binding globulin and testosterone during weight loss and weight maintenance in abdominally obese men with the metabolic syndrome. Diabetes Obes Metab 2004; 6:208–215.
- Shores MM, Matsumoto AM, Sloan KL, Kivlahan DR. Low serum testosterone and mortality in male veterans. Arch Intern Med 2006; 166:1660–1665.
- Carrero JJ, Qureshi AR, Parini P, et al. Low serum testosterone increases mortality risk among male dialysis patients. J Am Soc Nephrol 2009; 20:613–620.
- Haring R, Völzke H, Steveling A, et al. Low serum testosterone levels are associated with increased risk of mortality in a population-based cohort of men aged 20–79. Eur Heart J 2010; 31:1494–1501.
- Araujo AB, Dixon JM, Suarez EA, Murad MH, Guey LT, Wittert GA. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab 2011; 96:3007–3019.
- Rhoden EL, Morgentaler A. Risks of testosterone-replacement therapy and recommendations for monitoring. N Engl J Med 2004; 350:482–492.
- Isbarn H, Pinthus JH, Marks LS, et al. Testosterone and prostate cancer: revisiting old paradigms. Eur Urol 2009; 56:48–56.
- Traish AM, Miner MM, Morgentaler A, Zitzmann M. Testosterone deficiency. Am J Med 2011; 124:578–587.
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med 2009; 6:3177–3192.
- Gerstenbluth RE, Maniam PN, Corty EW, Seftel AD. Prostate-specific antigen changes in hypogonadal men treated with testosterone replacement. J Androl 2002; 23:922–926.
- Corona G, Monami M, Rastrelli G, et al. Testosterone and metabolic syndrome: a meta-analysis study. J Sex Med 2011; 8:272–283.
- Corona G, Rastrelli G, Monami M, et al. Hypogonadism as a risk factor for cardiovascular mortality in men: a meta-analytic study. Eur J Endocrinol 2011; 165:687–701.
- Kapoor D, Goodwin E, Channer KS, Jones TH. Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur J Endocrinol 2006; 154:899–906.
- Aversa A, Bruzziches R, Francomano D, et al. Effects of testosterone undecanoate on cardiovascular risk factors and atherosclerosis in middle-aged men with late-onset hypogonadism and metabolic syndrome: results from a 24-month, randomized, double-blind, placebo-controlled study. J Sex Med 2010; 7:3495–3503.
- Rhoden EL, Morgentaler A. Symptomatic response rates to testosterone therapy and the likelihood of completing 12 months of therapy in clinical practice. J Sex Med 2010; 7:277–283.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. Improved sexual function with testosterone replacement therapy in hypogonadal men: real-world data from the Testim Registry in the United States (TRiUS). J Sex Med 2011; 8:3204–3213.
- Buvat J, Montorsi F, Maggi M, et al. Hypogonadal men nonresponders to the PDE5 inhibitor tadalafil benefit from normalization of testosterone levels with a 1% hydroalcoholic testosterone gel in the treatment of erectile dysfunction (TADTEST study). J Sex Med 2011; 8:284–293.
- Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med 2010; 363:109–122.
- Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab 1999; 84:3469–3478.
- Wang C, Swerdloff RS, Iranmanesh A, et al; Testosterone Gel Study Group. Transdermal testosterone gel improves sexual function, mood, muscle strength, and body composition parameters in hypogonadal men. J Clin Endocrinol Metab 2000; 85:2839–2853.
- Hanafy HM. Testosterone therapy and obstructive sleep apnea: is there a real connection? J Sex Med 2007; 4:1241–1246.
- Coward RM, Simhan J, Carson CC. Prostate-specific antigen changes and prostate cancer in hypogonadal men treated with testosterone replacement therapy. BJU Int 2009; 103:1179–1183.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Morgentaler A, Traish AM. Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol 2009; 55:310–320.
- Page ST, Lin DW, Mostaghel EA, et al. Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: a randomized-controlled trial. J Clin Endocrinol Metab 2011; 96:430–437.
- Botto H, Neuzillet Y, Lebret T, Camparo P, Molinie V, Raynaud JP. High incidence of predominant Gleason pattern 4 localized prostate cancer is associated with low serum testosterone. J Urol 2011; 186:1400–1405.
- Salonia A, Gallina A, Briganti A, et al. Preoperative hypogonadism is not an independent predictor of high-risk disease in patients undergoing radical prostatectomy. Cancer 2011; 117:3953–3962.
- Sarosdy MF. Testosterone replacement for hypogonadism after treatment of early prostate cancer with brachytherapy. Cancer 2007; 109:536–541.
- Khera M. Androgens and erectile function: a case for early androgen use in postprostatectomy hypogonadal men. J Sex Med 2009; 6:(suppl 3):234–238.
- Szmulewitz R, Mohile S, Posadas E, et al. A randomized phase 1 study of testosterone replacement for patients with low-risk castration-resistant prostate cancer. Eur Urol 2009; 56:97–103.
- Morgentaler A, Lipshultz LI, Bennett R, Sweeney M, Avila D, Khera M. Testosterone therapy in men with untreated prostate cancer. J Urol 2011; 185:1256–1260.
- Feldman HA, Longcope C, Derby CA, et al. Age trends in the level of serum testosterone and other hormones in middle-aged men: longitudinal results from the Massachusetts male aging study. J Clin Endocrinol Metab 2002; 87:589–598.
- Araujo AB, O’Donnell AB, Brambilla DJ, et al. Prevalence and incidence of androgen deficiency in middle-aged and older men: estimates from the Massachusetts Male Aging Study. J Clin Endocrinol Metab 2004; 89:5920–5926.
- Travison TG, Shackelton R, Araujo AB, et al. The natural history of symptomatic androgen deficiency in men: onset, progression, and spontaneous remission. J Am Geriatr Soc 2008; 56:831–839.
- Tajar A, Forti G, O’Neill TW, et al; EMAS Group. Characteristics of secondary, primary, and compensated hypogonadism in aging men: evidence from the European Male Ageing Study. J Clin Endocrinol Metab 2010; 95:1810–1818.
- Hammes A, Andreassen TK, Spoelgen R, et al. Role of endocytosis in cellular uptake of sex steroids. Cell 2005; 122:751–762.
- Rosner W, Hryb DJ, Kahn SM, Nakhla AM, Romas NA. Interactions of sex hormone-binding globulin with target cells. Mol Cell Endocrinol 2010; 316:79–85.
- Bhasin S, Cunningham GR, Hayes FJ, et al; Task Force, Endocrine Society. Testosterone therapy in men with androgen deficiency syndromes: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2010; 95:2536–2559.
- Wang C, Nieschlag E, Swerdloff R, et al; International Society of Andrology (ISA). Investigation, treatment, and monitoring of late-onset hypogonadism in males: ISA, ISSAM, EAU, EAA, and ASA recommendations. J Androl 2009; 30:1–9.
- Morley JE, Patrick P, Perry HM. Evaluation of assays available to measure free testosterone. Metabolism 2002; 51:554–559.
- Vesper HW, Bhasin S, Wang C, et al. Interlaboratory comparison study of serum total testosterone [corrected] measurements performed by mass spectrometry methods. Steroids 2009; 74:498–503.
- Ly LP, Sartorius G, Hull L, et al. Accuracy of calculated free testosterone formulae in men. Clin Endocrinol (Oxf) 2010; 73:382–388.
- Melmed S, Casanueva FF, Hoffman AR, et al; Endocrine Society. Diagnosis and treatment of hyperprolactinemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab 2011; 96:273–288.
- Wu FC, Tajar A, Beynon JM, et al; EMAS Group. Identification of late-onset hypogonadism in middle-aged and elderly men. N Engl J Med 2010; 363:123–135.
- Kelleher S, Conway AJ, Handelsman DJ. Blood testosterone threshold for androgen deficiency symptoms. J Clin Endocrinol Metab 2004; 89:3813–3817.
- Joshi D, van Schoor NM, de Ronde W, et al. Low free testosterone levels are associated with prevalence and incidence of depressive symptoms in older men. Clin Endocrinol (Oxf) 2010; 72:232–240.
- Shores MM, Kivlahan DR, Sadak TI, Li EJ, Matsumoto AM. A randomized, double-blind, placebo-controlled study of testosterone treatment in hypogonadal older men with subthreshold depression (dysthymia or minor depression). J Clin Psychiatry 2009; 70:1009–1016.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. The effect of testosterone supplementation on depression symptoms in hypogonadal men from the Testim Registry in the US (TRiUS). Aging Male 2012; 15:14–21.
- Jain P, Rademaker AW, McVary KT. Testosterone supplementation for erectile dysfunction: results of a meta-analysis. J Urol 2000; 164:371–375.
- Mulhall JP, Valenzuela R, Aviv N, Parker M. Effect of testosterone supplementation on sexual function in hypogonadal men with erectile dysfunction. Urology 2004; 63:348–352.
- Morley JE, Charlton E, Patrick P, et al. Validation of a screening questionnaire for androgen deficiency in aging males. Metabolism 2000; 49:1239–1242.
- Moore C, Huebler D, Zimmermann T, Heinemann LA, Saad F, Thai DM. The Aging Males’ Symptoms scale (AMS) as outcome measure for treatment of androgen deficiency. Eur Urol 2004; 46:80–87.
- Chueh KS, Huang SP, Lee YC, et al. The Comparison of the Aging Male Symptoms (AMS) Scale and Androgen Deficiency in the Aging Male (ADAM) Questionnaire to Detect Androgen Deficiency in Middle-Aged Men. J Androl 2012[Epub ahead of print]
- Mulligan T, Frick MF, Zuraw QC, Stemhagen A, McWhirter C. Prevalence of hypogonadism in males aged at least 45 years: the HIM study. Int J Clin Pract 2006; 60:762–769.
- Dhindsa S, Miller MG, McWhirter CL, et al. Testosterone concentrations in diabetic and nondiabetic obese men. Diabetes Care 2010; 33:1186–1192.
- Krasnoff JB, Basaria S, Pencina MJ, et al. Free testosterone levels are associated with mobility limitation and physical performance in community-dwelling men: the Framingham Offspring Study. J Clin Endocrinol Metab 2010; 95:2790–2799.
- Carrero JJ, Qureshi AR, Nakashima A, et al. Prevalence and clinical implications of testosterone deficiency in men with end-stage renal disease. Nephrol Dial Transplant 2011; 26:184–190.
- Grossmann M, Thomas MC, Panagiotopoulos S, et al. Low testosterone levels are common and associated with insulin resistance in men with diabetes. J Clin Endocrinol Metab 2008; 93:1834–1840.
- Brand JS, van der Tweel I, Grobbee DE, Emmelot-Vonk MH, van der Schouw YT. Testosterone, sex hormone-binding globulin and the metabolic syndrome: a systematic review and meta-analysis of observational studies. Int J Epidemiol 2011; 40:189–207.
- Ding EL, Song Y, Malik VS, Liu S. Sex differences of endogenous sex hormones and risk of type 2 diabetes: a systematic review and meta-analysis. JAMA 2006; 295:1288–1299.
- Niskanen L, Laaksonen DE, Punnonen K, Mustajoki P, Kaukua J, Rissanen A. Changes in sex hormone-binding globulin and testosterone during weight loss and weight maintenance in abdominally obese men with the metabolic syndrome. Diabetes Obes Metab 2004; 6:208–215.
- Shores MM, Matsumoto AM, Sloan KL, Kivlahan DR. Low serum testosterone and mortality in male veterans. Arch Intern Med 2006; 166:1660–1665.
- Carrero JJ, Qureshi AR, Parini P, et al. Low serum testosterone increases mortality risk among male dialysis patients. J Am Soc Nephrol 2009; 20:613–620.
- Haring R, Völzke H, Steveling A, et al. Low serum testosterone levels are associated with increased risk of mortality in a population-based cohort of men aged 20–79. Eur Heart J 2010; 31:1494–1501.
- Araujo AB, Dixon JM, Suarez EA, Murad MH, Guey LT, Wittert GA. Clinical review: Endogenous testosterone and mortality in men: a systematic review and meta-analysis. J Clin Endocrinol Metab 2011; 96:3007–3019.
- Rhoden EL, Morgentaler A. Risks of testosterone-replacement therapy and recommendations for monitoring. N Engl J Med 2004; 350:482–492.
- Isbarn H, Pinthus JH, Marks LS, et al. Testosterone and prostate cancer: revisiting old paradigms. Eur Urol 2009; 56:48–56.
- Traish AM, Miner MM, Morgentaler A, Zitzmann M. Testosterone deficiency. Am J Med 2011; 124:578–587.
- Cavender RK, Fairall M. Subcutaneous testosterone pellet implant (Testopel) therapy for men with testosterone deficiency syndrome: a single-site retrospective safety analysis. J Sex Med 2009; 6:3177–3192.
- Gerstenbluth RE, Maniam PN, Corty EW, Seftel AD. Prostate-specific antigen changes in hypogonadal men treated with testosterone replacement. J Androl 2002; 23:922–926.
- Corona G, Monami M, Rastrelli G, et al. Testosterone and metabolic syndrome: a meta-analysis study. J Sex Med 2011; 8:272–283.
- Corona G, Rastrelli G, Monami M, et al. Hypogonadism as a risk factor for cardiovascular mortality in men: a meta-analytic study. Eur J Endocrinol 2011; 165:687–701.
- Kapoor D, Goodwin E, Channer KS, Jones TH. Testosterone replacement therapy improves insulin resistance, glycaemic control, visceral adiposity and hypercholesterolaemia in hypogonadal men with type 2 diabetes. Eur J Endocrinol 2006; 154:899–906.
- Aversa A, Bruzziches R, Francomano D, et al. Effects of testosterone undecanoate on cardiovascular risk factors and atherosclerosis in middle-aged men with late-onset hypogonadism and metabolic syndrome: results from a 24-month, randomized, double-blind, placebo-controlled study. J Sex Med 2010; 7:3495–3503.
- Rhoden EL, Morgentaler A. Symptomatic response rates to testosterone therapy and the likelihood of completing 12 months of therapy in clinical practice. J Sex Med 2010; 7:277–283.
- Khera M, Bhattacharya RK, Blick G, Kushner H, Nguyen D, Miner MM. Improved sexual function with testosterone replacement therapy in hypogonadal men: real-world data from the Testim Registry in the United States (TRiUS). J Sex Med 2011; 8:3204–3213.
- Buvat J, Montorsi F, Maggi M, et al. Hypogonadal men nonresponders to the PDE5 inhibitor tadalafil benefit from normalization of testosterone levels with a 1% hydroalcoholic testosterone gel in the treatment of erectile dysfunction (TADTEST study). J Sex Med 2011; 8:284–293.
- Basaria S, Coviello AD, Travison TG, et al. Adverse events associated with testosterone administration. N Engl J Med 2010; 363:109–122.
- Dobs AS, Meikle AW, Arver S, Sanders SW, Caramelli KE, Mazer NA. Pharmacokinetics, efficacy, and safety of a permeation-enhanced testosterone transdermal system in comparison with bi-weekly injections of testosterone enanthate for the treatment of hypogonadal men. J Clin Endocrinol Metab 1999; 84:3469–3478.
- Wang C, Swerdloff RS, Iranmanesh A, et al; Testosterone Gel Study Group. Transdermal testosterone gel improves sexual function, mood, muscle strength, and body composition parameters in hypogonadal men. J Clin Endocrinol Metab 2000; 85:2839–2853.
- Hanafy HM. Testosterone therapy and obstructive sleep apnea: is there a real connection? J Sex Med 2007; 4:1241–1246.
- Coward RM, Simhan J, Carson CC. Prostate-specific antigen changes and prostate cancer in hypogonadal men treated with testosterone replacement therapy. BJU Int 2009; 103:1179–1183.
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10–29.
- Morgentaler A, Traish AM. Shifting the paradigm of testosterone and prostate cancer: the saturation model and the limits of androgen-dependent growth. Eur Urol 2009; 55:310–320.
- Page ST, Lin DW, Mostaghel EA, et al. Dihydrotestosterone administration does not increase intraprostatic androgen concentrations or alter prostate androgen action in healthy men: a randomized-controlled trial. J Clin Endocrinol Metab 2011; 96:430–437.
- Botto H, Neuzillet Y, Lebret T, Camparo P, Molinie V, Raynaud JP. High incidence of predominant Gleason pattern 4 localized prostate cancer is associated with low serum testosterone. J Urol 2011; 186:1400–1405.
- Salonia A, Gallina A, Briganti A, et al. Preoperative hypogonadism is not an independent predictor of high-risk disease in patients undergoing radical prostatectomy. Cancer 2011; 117:3953–3962.
- Sarosdy MF. Testosterone replacement for hypogonadism after treatment of early prostate cancer with brachytherapy. Cancer 2007; 109:536–541.
- Khera M. Androgens and erectile function: a case for early androgen use in postprostatectomy hypogonadal men. J Sex Med 2009; 6:(suppl 3):234–238.
- Szmulewitz R, Mohile S, Posadas E, et al. A randomized phase 1 study of testosterone replacement for patients with low-risk castration-resistant prostate cancer. Eur Urol 2009; 56:97–103.
- Morgentaler A, Lipshultz LI, Bennett R, Sweeney M, Avila D, Khera M. Testosterone therapy in men with untreated prostate cancer. J Urol 2011; 185:1256–1260.
KEY POINTS
- General health benefits and safety of TRT in asymptomatic patients are not clearly defined by current data.
- Treatment of low testosterone is discouraged in the absence of clinical symptoms.
- A morning serum testosterone should be obtained after ruling out other causes of symptoms. It should also be repeated to confirm androgen deficiency in older men.
- Androgen deficiency in older men is associated with metabolic syndrome, type 2 diabetes mellitus, obesity, osteoporosis, renal failure, anemia, and previous treatment with steroids or opiates.
- TRT in men with a history of prostate cancer remains controversial. The existing limited data suggest that TRT is safe after curative therapy for prostate cancer. Patients treated should be monitored closely and informed of the risks of cancer progression and recurrence while they are on TRT.