User login
-
JAMA podcast on racism in medicine faces backlash
Published on Feb. 23, the episode is hosted on JAMA’s learning platform for doctors and is available for continuing medical education credits.
“No physician is racist, so how can there be structural racism in health care? An explanation of the idea by doctors for doctors in this user-friendly podcast,” JAMA wrote in a Twitter post to promote the episode. That tweet has since been deleted.
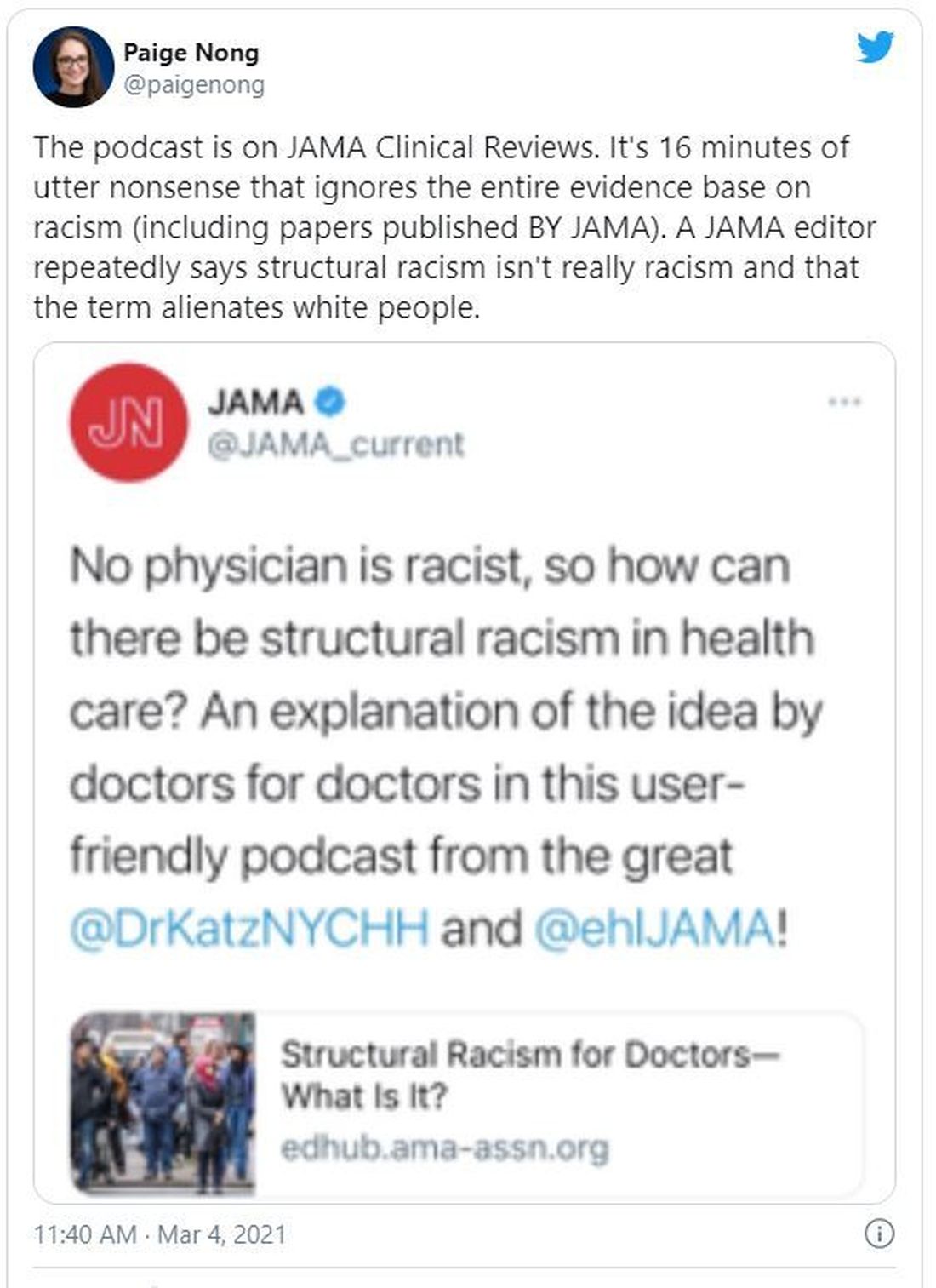
The episode features host Ed Livingston, MD, deputy editor for clinical reviews and education at JAMA, and guest Mitchell Katz, MD, president and CEO for NYC Health + Hospitals and deputy editor for JAMA Internal Medicine. Dr. Livingston approaches the episode as “structural racism for skeptics,” and Dr. Katz tries to explain how structural racism deepens health disparities and what health systems can do about it.
“Many physicians are skeptical of structural racism, the idea that economic, educational, and other societal systems preferentially disadvantage Black Americans and other communities of color,” the episode description says.
In the podcast, Dr. Livingston and Dr. Katz speak about health care disparities and racial inequality. Dr. Livingston, who says he “didn’t understand the concept” going into the episode, suggests that racism was made illegal in the 1960s and that the discussion of “structural racism” should shift away from the term “racism” and focus on socioeconomic status instead.
“What you’re talking about isn’t so much racism ... it isn’t their race, it isn’t their color, it’s their socioeconomic status,” Dr. Livingston says. “Is that a fair statement?”
But Dr. Katz says that “acknowledging structural racism can be helpful to us. Structural racism refers to a system in which policies or practices or how we look at people perpetuates racial inequality.”
Dr. Katz points to the creation of a hospital in San Francisco in the 1880s to treat patients of Chinese ethnicity separately. Outside of health care, he talks about environmental racism between neighborhoods with inequalities in hospitals, schools, and social services.
“All of those things have an impact on that minority person,” Dr. Katz says. “The big thing we can all do is move away from trying to interrogate each other’s opinions and move to a place where we are looking at the policies of our institutions and making sure that they promote equality.”
Dr. Livingston concludes the episode by reemphasizing that “racism” should be taken out of the conversation and it should instead focus on the “structural” aspect of socioeconomics.
“Minorities ... aren’t [in those neighborhoods] because they’re not allowed to buy houses or they can’t get a job because they’re Black or Hispanic. That would be illegal,” Dr. Livingston says. “But disproportionality does exist.”
Efforts to reach Dr. Livingston were unsuccessful. Dr. Katz distanced himself from Dr. Livingston in a statement released on March 4.
“Systemic and interpersonal racism both still exist in our country — they must be rooted out. I do not share the JAMA host’s belief of doing away with the word ‘racism’ will help us be more successful in ending inequities that exists across racial and ethnic lines,” Dr. Katz said. “Further, I believe that we will only produce an equitable society when social and political structures do not continue to produce and perpetuate disparate results based on social race and ethnicity.”
Dr. Katz reiterated that both interpersonal and structural racism continue to exist in the United States, “and it is woefully naive to say that no physician is a racist just because the Civil Rights Act of 1964 forbade it.”
He also recommended JAMA use this controversy “as a learning opportunity for continued dialogue and create another podcast series as an open conversation that invites diverse experts in the field to have an open discussion about structural racism in healthcare.”
The podcast and JAMA’s tweet promoting it were widely criticized on Twitter. In interviews with WebMD, many doctors expressed disbelief that such a respected journal would lend its name to this podcast episode.
B. Bobby Chiong, MD, a radiologist in New York, said although JAMA’s effort to engage with its audience about racism is laudable, it missed the mark.
“I think the backlash comes from how they tried to make a podcast about the subject and somehow made themselves an example of unconscious bias and unfamiliarity with just how embedded in our system is structural racism,” he said.
Perhaps the podcast’s worst offense was its failure to address the painful history of racial bias in this country that still permeates the medical community, says Tamara Saint-Surin, MD, assistant professor at the University of North Carolina at Chapel Hill.
“For physicians in leadership to have the belief that structural racism does not exist in medicine, they don’t really appreciate what affects their patients and what their patients were dealing with,” Dr. Saint-Surin said in an interview. “It was a very harmful podcast and goes to show we still have so much work to do.”
Along with a flawed premise, she says, the podcast was not nearly long enough to address such a nuanced issue. And Dr. Livingston focused on interpersonal racism rather than structural racism, she said, failing to address widespread problems such as higher rates of asthma among Black populations living in areas with poor air quality.
The number of Black doctors remains low and the lack of representation adds to an environment already rife with racism, according to many medical professionals.
Shirlene Obuobi, MD, an internal medicine doctor in Chicago, said JAMA failed to live up to its own standards by publishing material that lacked research and expertise.
“I can’t submit a clinical trial to JAMA without them combing through methods with a fine-tooth comb,” Dr. Obuobi said. “They didn’t uphold the standards they normally apply to anyone else.”
Both the editor of JAMA and the head of the American Medical Association issued statements criticizing the episode and the tweet that promoted it.
JAMA Editor-in-Chief Howard Bauchner, MD, said, “The language of the tweet, and some portions of the podcast, do not reflect my commitment as editorial leader of JAMA and JAMA Network to call out and discuss the adverse effects of injustice, inequity, and racism in society and medicine as JAMA has done for many years.” He said JAMA will schedule a future podcast to address the concerns raised about the recent episode.
AMA CEO James L. Madara, MD, said, “The AMA’s House of Delegates passed policy stating that racism is structural, systemic, cultural, and interpersonal, and we are deeply disturbed – and angered – by a recent JAMA podcast that questioned the existence of structural racism and the affiliated tweet that promoted the podcast and stated ‘no physician is racist, so how can there be structural racism in health care?’ ”
He continued: “JAMA has editorial independence from AMA, but this tweet and podcast are inconsistent with the policies and views of AMA, and I’m concerned about and acknowledge the harms they have caused. Structural racism in health care and our society exists, and it is incumbent on all of us to fix it.”
This article was updated 3/5/21.
A version of this article first appeared on WebMD.com.
Published on Feb. 23, the episode is hosted on JAMA’s learning platform for doctors and is available for continuing medical education credits.
“No physician is racist, so how can there be structural racism in health care? An explanation of the idea by doctors for doctors in this user-friendly podcast,” JAMA wrote in a Twitter post to promote the episode. That tweet has since been deleted.
The episode features host Ed Livingston, MD, deputy editor for clinical reviews and education at JAMA, and guest Mitchell Katz, MD, president and CEO for NYC Health + Hospitals and deputy editor for JAMA Internal Medicine. Dr. Livingston approaches the episode as “structural racism for skeptics,” and Dr. Katz tries to explain how structural racism deepens health disparities and what health systems can do about it.
“Many physicians are skeptical of structural racism, the idea that economic, educational, and other societal systems preferentially disadvantage Black Americans and other communities of color,” the episode description says.
In the podcast, Dr. Livingston and Dr. Katz speak about health care disparities and racial inequality. Dr. Livingston, who says he “didn’t understand the concept” going into the episode, suggests that racism was made illegal in the 1960s and that the discussion of “structural racism” should shift away from the term “racism” and focus on socioeconomic status instead.
“What you’re talking about isn’t so much racism ... it isn’t their race, it isn’t their color, it’s their socioeconomic status,” Dr. Livingston says. “Is that a fair statement?”
But Dr. Katz says that “acknowledging structural racism can be helpful to us. Structural racism refers to a system in which policies or practices or how we look at people perpetuates racial inequality.”
Dr. Katz points to the creation of a hospital in San Francisco in the 1880s to treat patients of Chinese ethnicity separately. Outside of health care, he talks about environmental racism between neighborhoods with inequalities in hospitals, schools, and social services.
“All of those things have an impact on that minority person,” Dr. Katz says. “The big thing we can all do is move away from trying to interrogate each other’s opinions and move to a place where we are looking at the policies of our institutions and making sure that they promote equality.”
Dr. Livingston concludes the episode by reemphasizing that “racism” should be taken out of the conversation and it should instead focus on the “structural” aspect of socioeconomics.
“Minorities ... aren’t [in those neighborhoods] because they’re not allowed to buy houses or they can’t get a job because they’re Black or Hispanic. That would be illegal,” Dr. Livingston says. “But disproportionality does exist.”
Efforts to reach Dr. Livingston were unsuccessful. Dr. Katz distanced himself from Dr. Livingston in a statement released on March 4.
“Systemic and interpersonal racism both still exist in our country — they must be rooted out. I do not share the JAMA host’s belief of doing away with the word ‘racism’ will help us be more successful in ending inequities that exists across racial and ethnic lines,” Dr. Katz said. “Further, I believe that we will only produce an equitable society when social and political structures do not continue to produce and perpetuate disparate results based on social race and ethnicity.”
Dr. Katz reiterated that both interpersonal and structural racism continue to exist in the United States, “and it is woefully naive to say that no physician is a racist just because the Civil Rights Act of 1964 forbade it.”
He also recommended JAMA use this controversy “as a learning opportunity for continued dialogue and create another podcast series as an open conversation that invites diverse experts in the field to have an open discussion about structural racism in healthcare.”
The podcast and JAMA’s tweet promoting it were widely criticized on Twitter. In interviews with WebMD, many doctors expressed disbelief that such a respected journal would lend its name to this podcast episode.
B. Bobby Chiong, MD, a radiologist in New York, said although JAMA’s effort to engage with its audience about racism is laudable, it missed the mark.
“I think the backlash comes from how they tried to make a podcast about the subject and somehow made themselves an example of unconscious bias and unfamiliarity with just how embedded in our system is structural racism,” he said.
Perhaps the podcast’s worst offense was its failure to address the painful history of racial bias in this country that still permeates the medical community, says Tamara Saint-Surin, MD, assistant professor at the University of North Carolina at Chapel Hill.
“For physicians in leadership to have the belief that structural racism does not exist in medicine, they don’t really appreciate what affects their patients and what their patients were dealing with,” Dr. Saint-Surin said in an interview. “It was a very harmful podcast and goes to show we still have so much work to do.”
Along with a flawed premise, she says, the podcast was not nearly long enough to address such a nuanced issue. And Dr. Livingston focused on interpersonal racism rather than structural racism, she said, failing to address widespread problems such as higher rates of asthma among Black populations living in areas with poor air quality.
The number of Black doctors remains low and the lack of representation adds to an environment already rife with racism, according to many medical professionals.
Shirlene Obuobi, MD, an internal medicine doctor in Chicago, said JAMA failed to live up to its own standards by publishing material that lacked research and expertise.
“I can’t submit a clinical trial to JAMA without them combing through methods with a fine-tooth comb,” Dr. Obuobi said. “They didn’t uphold the standards they normally apply to anyone else.”
Both the editor of JAMA and the head of the American Medical Association issued statements criticizing the episode and the tweet that promoted it.
JAMA Editor-in-Chief Howard Bauchner, MD, said, “The language of the tweet, and some portions of the podcast, do not reflect my commitment as editorial leader of JAMA and JAMA Network to call out and discuss the adverse effects of injustice, inequity, and racism in society and medicine as JAMA has done for many years.” He said JAMA will schedule a future podcast to address the concerns raised about the recent episode.
AMA CEO James L. Madara, MD, said, “The AMA’s House of Delegates passed policy stating that racism is structural, systemic, cultural, and interpersonal, and we are deeply disturbed – and angered – by a recent JAMA podcast that questioned the existence of structural racism and the affiliated tweet that promoted the podcast and stated ‘no physician is racist, so how can there be structural racism in health care?’ ”
He continued: “JAMA has editorial independence from AMA, but this tweet and podcast are inconsistent with the policies and views of AMA, and I’m concerned about and acknowledge the harms they have caused. Structural racism in health care and our society exists, and it is incumbent on all of us to fix it.”
This article was updated 3/5/21.
A version of this article first appeared on WebMD.com.
Published on Feb. 23, the episode is hosted on JAMA’s learning platform for doctors and is available for continuing medical education credits.
“No physician is racist, so how can there be structural racism in health care? An explanation of the idea by doctors for doctors in this user-friendly podcast,” JAMA wrote in a Twitter post to promote the episode. That tweet has since been deleted.
The episode features host Ed Livingston, MD, deputy editor for clinical reviews and education at JAMA, and guest Mitchell Katz, MD, president and CEO for NYC Health + Hospitals and deputy editor for JAMA Internal Medicine. Dr. Livingston approaches the episode as “structural racism for skeptics,” and Dr. Katz tries to explain how structural racism deepens health disparities and what health systems can do about it.
“Many physicians are skeptical of structural racism, the idea that economic, educational, and other societal systems preferentially disadvantage Black Americans and other communities of color,” the episode description says.
In the podcast, Dr. Livingston and Dr. Katz speak about health care disparities and racial inequality. Dr. Livingston, who says he “didn’t understand the concept” going into the episode, suggests that racism was made illegal in the 1960s and that the discussion of “structural racism” should shift away from the term “racism” and focus on socioeconomic status instead.
“What you’re talking about isn’t so much racism ... it isn’t their race, it isn’t their color, it’s their socioeconomic status,” Dr. Livingston says. “Is that a fair statement?”
But Dr. Katz says that “acknowledging structural racism can be helpful to us. Structural racism refers to a system in which policies or practices or how we look at people perpetuates racial inequality.”
Dr. Katz points to the creation of a hospital in San Francisco in the 1880s to treat patients of Chinese ethnicity separately. Outside of health care, he talks about environmental racism between neighborhoods with inequalities in hospitals, schools, and social services.
“All of those things have an impact on that minority person,” Dr. Katz says. “The big thing we can all do is move away from trying to interrogate each other’s opinions and move to a place where we are looking at the policies of our institutions and making sure that they promote equality.”
Dr. Livingston concludes the episode by reemphasizing that “racism” should be taken out of the conversation and it should instead focus on the “structural” aspect of socioeconomics.
“Minorities ... aren’t [in those neighborhoods] because they’re not allowed to buy houses or they can’t get a job because they’re Black or Hispanic. That would be illegal,” Dr. Livingston says. “But disproportionality does exist.”
Efforts to reach Dr. Livingston were unsuccessful. Dr. Katz distanced himself from Dr. Livingston in a statement released on March 4.
“Systemic and interpersonal racism both still exist in our country — they must be rooted out. I do not share the JAMA host’s belief of doing away with the word ‘racism’ will help us be more successful in ending inequities that exists across racial and ethnic lines,” Dr. Katz said. “Further, I believe that we will only produce an equitable society when social and political structures do not continue to produce and perpetuate disparate results based on social race and ethnicity.”
Dr. Katz reiterated that both interpersonal and structural racism continue to exist in the United States, “and it is woefully naive to say that no physician is a racist just because the Civil Rights Act of 1964 forbade it.”
He also recommended JAMA use this controversy “as a learning opportunity for continued dialogue and create another podcast series as an open conversation that invites diverse experts in the field to have an open discussion about structural racism in healthcare.”
The podcast and JAMA’s tweet promoting it were widely criticized on Twitter. In interviews with WebMD, many doctors expressed disbelief that such a respected journal would lend its name to this podcast episode.
B. Bobby Chiong, MD, a radiologist in New York, said although JAMA’s effort to engage with its audience about racism is laudable, it missed the mark.
“I think the backlash comes from how they tried to make a podcast about the subject and somehow made themselves an example of unconscious bias and unfamiliarity with just how embedded in our system is structural racism,” he said.
Perhaps the podcast’s worst offense was its failure to address the painful history of racial bias in this country that still permeates the medical community, says Tamara Saint-Surin, MD, assistant professor at the University of North Carolina at Chapel Hill.
“For physicians in leadership to have the belief that structural racism does not exist in medicine, they don’t really appreciate what affects their patients and what their patients were dealing with,” Dr. Saint-Surin said in an interview. “It was a very harmful podcast and goes to show we still have so much work to do.”
Along with a flawed premise, she says, the podcast was not nearly long enough to address such a nuanced issue. And Dr. Livingston focused on interpersonal racism rather than structural racism, she said, failing to address widespread problems such as higher rates of asthma among Black populations living in areas with poor air quality.
The number of Black doctors remains low and the lack of representation adds to an environment already rife with racism, according to many medical professionals.
Shirlene Obuobi, MD, an internal medicine doctor in Chicago, said JAMA failed to live up to its own standards by publishing material that lacked research and expertise.
“I can’t submit a clinical trial to JAMA without them combing through methods with a fine-tooth comb,” Dr. Obuobi said. “They didn’t uphold the standards they normally apply to anyone else.”
Both the editor of JAMA and the head of the American Medical Association issued statements criticizing the episode and the tweet that promoted it.
JAMA Editor-in-Chief Howard Bauchner, MD, said, “The language of the tweet, and some portions of the podcast, do not reflect my commitment as editorial leader of JAMA and JAMA Network to call out and discuss the adverse effects of injustice, inequity, and racism in society and medicine as JAMA has done for many years.” He said JAMA will schedule a future podcast to address the concerns raised about the recent episode.
AMA CEO James L. Madara, MD, said, “The AMA’s House of Delegates passed policy stating that racism is structural, systemic, cultural, and interpersonal, and we are deeply disturbed – and angered – by a recent JAMA podcast that questioned the existence of structural racism and the affiliated tweet that promoted the podcast and stated ‘no physician is racist, so how can there be structural racism in health care?’ ”
He continued: “JAMA has editorial independence from AMA, but this tweet and podcast are inconsistent with the policies and views of AMA, and I’m concerned about and acknowledge the harms they have caused. Structural racism in health care and our society exists, and it is incumbent on all of us to fix it.”
This article was updated 3/5/21.
A version of this article first appeared on WebMD.com.
Docs become dog groomers and warehouse workers after COVID-19 work loss
One of the biggest conundrums of the COVID-19 pandemic has been the simultaneous panic-hiring of medical professionals in hot spots and significant downsizing of staff across the country. From huge hospital systems to private practices, the stoppage of breast reductions and knee replacements, not to mention the drops in motor vehicle accidents and bar fights, have quieted operating rooms and emergency departments and put doctors’ jobs on the chopping block. A widely cited survey suggests that 21% of doctors have had a work reduction due to COVID-19.
For many American doctors, this is their first extended period of unemployment. Unlike engineers or those with MBAs who might see their fortunes rise and fall with the whims of recessions and boom times, physicians are not exactly accustomed to being laid off. However, doctors were already smarting for years due to falling salaries and decreased autonomy, punctuated by endless clicks on electronic medical records software.
Stephanie Eschenbach Morgan, MD, a breast radiologist in North Carolina, trained for 10 years after college before earning a true physician’s salary.
“Being furloughed was awful. Initially, it was only going to be 2 weeks, and then it turned into 2 months with no pay,” she reflected.
Dr. Eschenbach Morgan and her surgeon husband, who lost a full quarter’s salary, had to ask for grace periods on their credit card and mortgage payments because they had paid a large tax bill right before the pandemic began. “We couldn’t get any stimulus help, so that added insult to injury,” she said.
With her time spent waiting in a holding pattern, Dr. Eschenbach Morgan homeschooled her two young children and started putting a home gym together. She went on a home organizing spree, started a garden, and, perhaps most impressively, caught up with 5 years of photo albums.
A bonus she noted: “I didn’t set an alarm for 2 months.”
Shella Farooki, MD, a radiologist in California, was also focused on homeschooling, itself a demanding job, and veered toward retirement. When one of her work contracts furloughed her (“at one point, I made $30K a month for [their business]”), she started saving money at home, teaching the kids, and applied for a Paycheck Protection Program loan. Her husband, a hospitalist, had had his shifts cut. Dr. Farooki tried a radiology artificial intelligence firm but backed out when she was asked to read 9,200 studies for them for $2,000 per month.
Now, she thinks about leaving medicine “every day.”
Some doctors are questioning whether they should be in medicine in the first place. Family medicine physician Jonathan Polak, MD, faced with his own pink slip, turned to pink T-shirts instead. His girlfriend manages an outlet of the teen fashion retailer Justice. Dr. Polak, who finished his residency just 2 years ago, didn’t hesitate to take a $10-an-hour gig as a stock doc, once even finding himself delivering a shelving unit from the shuttering store to a physician fleeing the city for rural New Hampshire to “escape.”
There’s no escape for him – yet. Saddled with “astronomical” student loans, he had considered grocery store work as well. Dr. Polak knows he can’t work part time or go into teaching long term, as he might like.
Even so, he’s doing everything he can to not be in patient care for the long haul – it’s just not what he thought it would be.
“The culture of medicine, bureaucracy, endless paperwork and charting, and threat of litigation sucks a lot of the joy out of it to the point that I don’t see myself doing it forever when imagining myself 5-10 years into it.”
Still, he recently took an 18-month hospital contract that will force him to move to Florida, but he’s also been turning himself into a veritable Renaissance man; composing music, training for an ultramarathon, studying the latest medical findings, roadtripping, and launching a podcast about dog grooming with a master groomer. “We found parallels between medicine and dog grooming,” he says, somewhat convincingly.
Also working the ruff life is Jen Tserng, MD, a former forensic pathologist who landed on news websites in recent years for becoming a professional dogwalker and housesitter without a permanent home. Dr. Tserng knows doctors were restless and unhappy before COVID-19, their thoughts wandering where the grass might be greener.
As her profile grew, she found her inbox gathering messages from disaffected medical minions: students with a fear of failing or staring down residency application season and employed doctors sick of the constant grind. As she recounted those de facto life coach conversations (“What do you really enjoy?” “Do you really like dogs?”) by phone from New York, she said matter-of-factly, “They don’t call because of COVID. They call because they hate their lives.”
Michelle Mudge-Riley, MD, a physician in Texas, has been seeing this shift for some time as well. She recently held a virtual version of her Physicians Helping Physicians conference, where doctors hear from their peers working successfully in fields like pharmaceuticals and real estate investing.
When COVID-19 hit, Dr. Mudge-Riley quickly pivoted to a virtual platform, where the MDs and DOs huddled in breakout rooms having honest chats about their fears and tentative hopes about their new careers.
“There has been increased interest in nonclinical exploration into full- and part-time careers, as well as side hustles, since COVID began,” she said. “Many physicians have had their hours or pay cut, and some have been laid off. Others are furloughed. Some just want out of an environment where they don’t feel safe.”
An ear, nose, and throat surgeon, Maansi Doshi, MD, from central California, didn’t feel safe – so she left. She had returned from India sick with a mystery virus right as the pandemic began (she said her COVID-19 tests were all negative) and was waiting to get well enough to go back to her private practice job. However, she said she clashed with Trump-supporting colleagues she feared might not be taking the pandemic seriously enough.
Finally getting over a relapse of her mystery virus, Dr. Doshi emailed her resignation in May. Her husband, family practice doctor Mark Mangiapane, MD, gave his job notice weeks later in solidarity because he worked in the same building. Together, they have embraced gardening, a Peloton splurge, and learning business skills to open private practices – solo primary care for him; ENT with a focus on her favorite surgery, rhinoplasty, for her.
Dr. Mangiapane had considered editing medical brochures and also tried to apply for a job as a county public health officer in rural California, but he received his own shock when he learned the county intended to open schools in the midst of the pandemic despite advisement to the contrary by the former health officer.
He retreated from job listings altogether after hearing his would-be peers were getting death threats – targeting their children.
Both doctors felt COVID-19 pushed them beyond their comfort zones. “If COVID hadn’t happened, I would be working. ... Be ‘owned.’ In a weird way, COVID made me more independent and take a risk with my career.”
Obstetrician Kwandaa Roberts, MD, certainly did; she took a budding interest in decorating dollhouses straight to Instagram and national news fame, and she is now a TV-show expert on “Sell This House.”
Like Dr. Doshi and Dr. Mangiapane, Dr. Polak wants to be more in control of his future – even if selling T-shirts at a mall means a certain loss of status along the way.
“Aside from my passion to learn and to have that connection with people, I went into medicine ... because of the job security I thought existed,” he said. “I would say that my getting furloughed has changed my view of the United States in a dramatic way. I do not feel as confident in the U.S. economy and general way of life as I did a year ago. And I am taking a number of steps to put myself in a more fluid, adaptable position in case another crisis like this occurs or if the current state of things worsens.”
A version of this article first appeared on Medscape.com.
One of the biggest conundrums of the COVID-19 pandemic has been the simultaneous panic-hiring of medical professionals in hot spots and significant downsizing of staff across the country. From huge hospital systems to private practices, the stoppage of breast reductions and knee replacements, not to mention the drops in motor vehicle accidents and bar fights, have quieted operating rooms and emergency departments and put doctors’ jobs on the chopping block. A widely cited survey suggests that 21% of doctors have had a work reduction due to COVID-19.
For many American doctors, this is their first extended period of unemployment. Unlike engineers or those with MBAs who might see their fortunes rise and fall with the whims of recessions and boom times, physicians are not exactly accustomed to being laid off. However, doctors were already smarting for years due to falling salaries and decreased autonomy, punctuated by endless clicks on electronic medical records software.
Stephanie Eschenbach Morgan, MD, a breast radiologist in North Carolina, trained for 10 years after college before earning a true physician’s salary.
“Being furloughed was awful. Initially, it was only going to be 2 weeks, and then it turned into 2 months with no pay,” she reflected.
Dr. Eschenbach Morgan and her surgeon husband, who lost a full quarter’s salary, had to ask for grace periods on their credit card and mortgage payments because they had paid a large tax bill right before the pandemic began. “We couldn’t get any stimulus help, so that added insult to injury,” she said.
With her time spent waiting in a holding pattern, Dr. Eschenbach Morgan homeschooled her two young children and started putting a home gym together. She went on a home organizing spree, started a garden, and, perhaps most impressively, caught up with 5 years of photo albums.
A bonus she noted: “I didn’t set an alarm for 2 months.”
Shella Farooki, MD, a radiologist in California, was also focused on homeschooling, itself a demanding job, and veered toward retirement. When one of her work contracts furloughed her (“at one point, I made $30K a month for [their business]”), she started saving money at home, teaching the kids, and applied for a Paycheck Protection Program loan. Her husband, a hospitalist, had had his shifts cut. Dr. Farooki tried a radiology artificial intelligence firm but backed out when she was asked to read 9,200 studies for them for $2,000 per month.
Now, she thinks about leaving medicine “every day.”
Some doctors are questioning whether they should be in medicine in the first place. Family medicine physician Jonathan Polak, MD, faced with his own pink slip, turned to pink T-shirts instead. His girlfriend manages an outlet of the teen fashion retailer Justice. Dr. Polak, who finished his residency just 2 years ago, didn’t hesitate to take a $10-an-hour gig as a stock doc, once even finding himself delivering a shelving unit from the shuttering store to a physician fleeing the city for rural New Hampshire to “escape.”
There’s no escape for him – yet. Saddled with “astronomical” student loans, he had considered grocery store work as well. Dr. Polak knows he can’t work part time or go into teaching long term, as he might like.
Even so, he’s doing everything he can to not be in patient care for the long haul – it’s just not what he thought it would be.
“The culture of medicine, bureaucracy, endless paperwork and charting, and threat of litigation sucks a lot of the joy out of it to the point that I don’t see myself doing it forever when imagining myself 5-10 years into it.”
Still, he recently took an 18-month hospital contract that will force him to move to Florida, but he’s also been turning himself into a veritable Renaissance man; composing music, training for an ultramarathon, studying the latest medical findings, roadtripping, and launching a podcast about dog grooming with a master groomer. “We found parallels between medicine and dog grooming,” he says, somewhat convincingly.
Also working the ruff life is Jen Tserng, MD, a former forensic pathologist who landed on news websites in recent years for becoming a professional dogwalker and housesitter without a permanent home. Dr. Tserng knows doctors were restless and unhappy before COVID-19, their thoughts wandering where the grass might be greener.
As her profile grew, she found her inbox gathering messages from disaffected medical minions: students with a fear of failing or staring down residency application season and employed doctors sick of the constant grind. As she recounted those de facto life coach conversations (“What do you really enjoy?” “Do you really like dogs?”) by phone from New York, she said matter-of-factly, “They don’t call because of COVID. They call because they hate their lives.”
Michelle Mudge-Riley, MD, a physician in Texas, has been seeing this shift for some time as well. She recently held a virtual version of her Physicians Helping Physicians conference, where doctors hear from their peers working successfully in fields like pharmaceuticals and real estate investing.
When COVID-19 hit, Dr. Mudge-Riley quickly pivoted to a virtual platform, where the MDs and DOs huddled in breakout rooms having honest chats about their fears and tentative hopes about their new careers.
“There has been increased interest in nonclinical exploration into full- and part-time careers, as well as side hustles, since COVID began,” she said. “Many physicians have had their hours or pay cut, and some have been laid off. Others are furloughed. Some just want out of an environment where they don’t feel safe.”
An ear, nose, and throat surgeon, Maansi Doshi, MD, from central California, didn’t feel safe – so she left. She had returned from India sick with a mystery virus right as the pandemic began (she said her COVID-19 tests were all negative) and was waiting to get well enough to go back to her private practice job. However, she said she clashed with Trump-supporting colleagues she feared might not be taking the pandemic seriously enough.
Finally getting over a relapse of her mystery virus, Dr. Doshi emailed her resignation in May. Her husband, family practice doctor Mark Mangiapane, MD, gave his job notice weeks later in solidarity because he worked in the same building. Together, they have embraced gardening, a Peloton splurge, and learning business skills to open private practices – solo primary care for him; ENT with a focus on her favorite surgery, rhinoplasty, for her.
Dr. Mangiapane had considered editing medical brochures and also tried to apply for a job as a county public health officer in rural California, but he received his own shock when he learned the county intended to open schools in the midst of the pandemic despite advisement to the contrary by the former health officer.
He retreated from job listings altogether after hearing his would-be peers were getting death threats – targeting their children.
Both doctors felt COVID-19 pushed them beyond their comfort zones. “If COVID hadn’t happened, I would be working. ... Be ‘owned.’ In a weird way, COVID made me more independent and take a risk with my career.”
Obstetrician Kwandaa Roberts, MD, certainly did; she took a budding interest in decorating dollhouses straight to Instagram and national news fame, and she is now a TV-show expert on “Sell This House.”
Like Dr. Doshi and Dr. Mangiapane, Dr. Polak wants to be more in control of his future – even if selling T-shirts at a mall means a certain loss of status along the way.
“Aside from my passion to learn and to have that connection with people, I went into medicine ... because of the job security I thought existed,” he said. “I would say that my getting furloughed has changed my view of the United States in a dramatic way. I do not feel as confident in the U.S. economy and general way of life as I did a year ago. And I am taking a number of steps to put myself in a more fluid, adaptable position in case another crisis like this occurs or if the current state of things worsens.”
A version of this article first appeared on Medscape.com.
One of the biggest conundrums of the COVID-19 pandemic has been the simultaneous panic-hiring of medical professionals in hot spots and significant downsizing of staff across the country. From huge hospital systems to private practices, the stoppage of breast reductions and knee replacements, not to mention the drops in motor vehicle accidents and bar fights, have quieted operating rooms and emergency departments and put doctors’ jobs on the chopping block. A widely cited survey suggests that 21% of doctors have had a work reduction due to COVID-19.
For many American doctors, this is their first extended period of unemployment. Unlike engineers or those with MBAs who might see their fortunes rise and fall with the whims of recessions and boom times, physicians are not exactly accustomed to being laid off. However, doctors were already smarting for years due to falling salaries and decreased autonomy, punctuated by endless clicks on electronic medical records software.
Stephanie Eschenbach Morgan, MD, a breast radiologist in North Carolina, trained for 10 years after college before earning a true physician’s salary.
“Being furloughed was awful. Initially, it was only going to be 2 weeks, and then it turned into 2 months with no pay,” she reflected.
Dr. Eschenbach Morgan and her surgeon husband, who lost a full quarter’s salary, had to ask for grace periods on their credit card and mortgage payments because they had paid a large tax bill right before the pandemic began. “We couldn’t get any stimulus help, so that added insult to injury,” she said.
With her time spent waiting in a holding pattern, Dr. Eschenbach Morgan homeschooled her two young children and started putting a home gym together. She went on a home organizing spree, started a garden, and, perhaps most impressively, caught up with 5 years of photo albums.
A bonus she noted: “I didn’t set an alarm for 2 months.”
Shella Farooki, MD, a radiologist in California, was also focused on homeschooling, itself a demanding job, and veered toward retirement. When one of her work contracts furloughed her (“at one point, I made $30K a month for [their business]”), she started saving money at home, teaching the kids, and applied for a Paycheck Protection Program loan. Her husband, a hospitalist, had had his shifts cut. Dr. Farooki tried a radiology artificial intelligence firm but backed out when she was asked to read 9,200 studies for them for $2,000 per month.
Now, she thinks about leaving medicine “every day.”
Some doctors are questioning whether they should be in medicine in the first place. Family medicine physician Jonathan Polak, MD, faced with his own pink slip, turned to pink T-shirts instead. His girlfriend manages an outlet of the teen fashion retailer Justice. Dr. Polak, who finished his residency just 2 years ago, didn’t hesitate to take a $10-an-hour gig as a stock doc, once even finding himself delivering a shelving unit from the shuttering store to a physician fleeing the city for rural New Hampshire to “escape.”
There’s no escape for him – yet. Saddled with “astronomical” student loans, he had considered grocery store work as well. Dr. Polak knows he can’t work part time or go into teaching long term, as he might like.
Even so, he’s doing everything he can to not be in patient care for the long haul – it’s just not what he thought it would be.
“The culture of medicine, bureaucracy, endless paperwork and charting, and threat of litigation sucks a lot of the joy out of it to the point that I don’t see myself doing it forever when imagining myself 5-10 years into it.”
Still, he recently took an 18-month hospital contract that will force him to move to Florida, but he’s also been turning himself into a veritable Renaissance man; composing music, training for an ultramarathon, studying the latest medical findings, roadtripping, and launching a podcast about dog grooming with a master groomer. “We found parallels between medicine and dog grooming,” he says, somewhat convincingly.
Also working the ruff life is Jen Tserng, MD, a former forensic pathologist who landed on news websites in recent years for becoming a professional dogwalker and housesitter without a permanent home. Dr. Tserng knows doctors were restless and unhappy before COVID-19, their thoughts wandering where the grass might be greener.
As her profile grew, she found her inbox gathering messages from disaffected medical minions: students with a fear of failing or staring down residency application season and employed doctors sick of the constant grind. As she recounted those de facto life coach conversations (“What do you really enjoy?” “Do you really like dogs?”) by phone from New York, she said matter-of-factly, “They don’t call because of COVID. They call because they hate their lives.”
Michelle Mudge-Riley, MD, a physician in Texas, has been seeing this shift for some time as well. She recently held a virtual version of her Physicians Helping Physicians conference, where doctors hear from their peers working successfully in fields like pharmaceuticals and real estate investing.
When COVID-19 hit, Dr. Mudge-Riley quickly pivoted to a virtual platform, where the MDs and DOs huddled in breakout rooms having honest chats about their fears and tentative hopes about their new careers.
“There has been increased interest in nonclinical exploration into full- and part-time careers, as well as side hustles, since COVID began,” she said. “Many physicians have had their hours or pay cut, and some have been laid off. Others are furloughed. Some just want out of an environment where they don’t feel safe.”
An ear, nose, and throat surgeon, Maansi Doshi, MD, from central California, didn’t feel safe – so she left. She had returned from India sick with a mystery virus right as the pandemic began (she said her COVID-19 tests were all negative) and was waiting to get well enough to go back to her private practice job. However, she said she clashed with Trump-supporting colleagues she feared might not be taking the pandemic seriously enough.
Finally getting over a relapse of her mystery virus, Dr. Doshi emailed her resignation in May. Her husband, family practice doctor Mark Mangiapane, MD, gave his job notice weeks later in solidarity because he worked in the same building. Together, they have embraced gardening, a Peloton splurge, and learning business skills to open private practices – solo primary care for him; ENT with a focus on her favorite surgery, rhinoplasty, for her.
Dr. Mangiapane had considered editing medical brochures and also tried to apply for a job as a county public health officer in rural California, but he received his own shock when he learned the county intended to open schools in the midst of the pandemic despite advisement to the contrary by the former health officer.
He retreated from job listings altogether after hearing his would-be peers were getting death threats – targeting their children.
Both doctors felt COVID-19 pushed them beyond their comfort zones. “If COVID hadn’t happened, I would be working. ... Be ‘owned.’ In a weird way, COVID made me more independent and take a risk with my career.”
Obstetrician Kwandaa Roberts, MD, certainly did; she took a budding interest in decorating dollhouses straight to Instagram and national news fame, and she is now a TV-show expert on “Sell This House.”
Like Dr. Doshi and Dr. Mangiapane, Dr. Polak wants to be more in control of his future – even if selling T-shirts at a mall means a certain loss of status along the way.
“Aside from my passion to learn and to have that connection with people, I went into medicine ... because of the job security I thought existed,” he said. “I would say that my getting furloughed has changed my view of the United States in a dramatic way. I do not feel as confident in the U.S. economy and general way of life as I did a year ago. And I am taking a number of steps to put myself in a more fluid, adaptable position in case another crisis like this occurs or if the current state of things worsens.”
A version of this article first appeared on Medscape.com.
‘Phenomenal’ results with CAR T cells in R/R multiple myeloma
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
Patients with multiple myeloma that has continued to progress despite many lines of therapy have shown deep and durable responses to a new chimeric antigen receptor (CAR) T-cell therapy, idecabtagene vicleucel (ide-cel, under development by Bristol-Myers Squibb and Bluebird Bio).
An expert not involved in the trial described the results as “phenomenal.”
Krina Patel, MD, an associate professor in the department of lymphoma/myeloma at the University of Texas MD Anderson Cancer Center, Houston, said that “the response rate of 73% in a patient population with a median of six lines of therapy, and with one-third of those patients achieving a deep response of complete response or better, is phenomenal.”
“We are very excited as a myeloma community for this study of idecabtagene vicleucel for relapsed/refractory patients,” Dr. Patel said.
The new data on ide-cell, from a trial in 128 patients, were published Feb. 25 in the New England Journal of Medicine.
Lead investigator of the study Nikhil Munshi, MD, of Dana-Farber Cancer Institute, Boston, said: “The results of this trial represent a true turning point in the treatment of this disease. In my 30 years of treating myeloma, I have not seen any other therapy as effective in this group of patients.”
Both experts highlighted the poor prognosis for this population of relapsed/refractory patients. Recent decades have seen a flurry of new agents for myeloma, and there are now three main classes of agents: immunomodulatory agents, proteasome inhibitors, and anti-CD38 antibodies. Nevertheless, in some patients, the disease continues to progress. For patients who have failed all three classes of drugs, the median progression-free survival is about 3-4 months, with a median overall survival of 8-9 months.
Product is awaiting approval
Ide-cel is currently awaiting FDA approval, with a decision date slated for March 27.
Several CAR T-cell products are already marketed for use in certain leukemias and lymphomas, and there is another for use in multiple myeloma, ciltacabtagene autoleucel (cilta-cel, under development by Janssen), that is awaiting approval in Europe.
Strong and sustained responses
The trial involved 128 patients treated with ide-cel infusions. At the time of data cutoff for this report (Jan. 14, 2020), 62 patients remained in the primary study. Of the 128 treated patients, the median age was 61 years and the median time since diagnosis was 6 years. About half (51%) had a high tumor burden (≥50% bone marrow plasma cells), 39% had extramedullary disease, 16% had stage III disease, and 35% had a high-risk cytogenetic abnormality, defined as del(17p), t(4;14), or t(14;16).
Patients in the cohort had received a median of six previous antimyeloma regimens (range, 3-16), and most of the patients (120, 94%) had undergone autologous hematopoietic stem cell transplants. In addition, the majority of patients (84%) had disease that was triple refractory (to an immunomodulatory agent, a proteasome inhibitor, and an anti-CD38 antibody), 60% had disease that was penta exposed (to bortezomib, carfilzomib, lenalidomide, pomalidomide, and daratumumab), and 26% had disease that was penta refractory.
At a median follow-up of 13.3 months, 94 of 128 patients (73%) showed a response to therapy (P < .001), with 42 (33%) showing a complete or stringent complete response, and 67 patients (52%) showing a “very good partial response or better.”
Overall median progression-free survival was 8.8 months at the 450×106 dose but more than double that (20.2 months) for patients who achieved a complete or stringent complete response. Estimated median overall survival was 19.4 months, with an overall survival of 78% at 12 months. The authors noted that overall survival data are not yet mature.
After experiencing disease progression, 28 patients were retreated with ide-cel, with 6 patients showing a second response. The durations of response ranged from 1.9 to 6.8 months.
All patients in the cohort experienced adverse events, primarily grade 3 or 4 events that occurred in 127 patients (99%). The most common events reported were hematologic toxicities, including neutropenia in 114 patients (89%), anemia in 77 (60%), and thrombocytopenia in 67 (52%), and were at least partially related to the lymphodepleting chemotherapy administered before ide-cel infusion, the authors note. Cytokine-release syndrome occurred in 107 patients (84%), primarily grade 1 or 2.
“Results of the KarMMa study support substantial antitumor activity for ide-cel across a target dose range of 150×106 to 450×106 CAR+ T cells,” the authors conclude. “The 450×106 dose appeared to be somewhat more effective than the other doses.”
New option?
“What this study further highlights is that higher cell dose tends to increase cell expansion, which correlates to improved response and duration of response,” said Dr. Patel.
Importantly, multiple vulnerable subgroups experienced impressive outcomes, such as those who are older or with high risk or extramedullary disease, she noted.
“My patients who have undergone this therapy, albeit on other clinical trials, all say that their quality of life during this time of remission is priceless,” Dr. Patel added. “The is the first therapy in the relapsed/refractory setting that allows patients to have a significant chemo-free period. We need to find more ways to do this for our patients.”
The study was supported by Bluebird Bio and Bristol-Myers Squibb. Dr. Patel has served on the advisory board for Janssen and Bristol-Myers Squibb. She also reports a speaking engagement with Oncopeptides. Dr. Munshi acts as a consultant for several pharmaceutical companies, and many coauthors also have relationships with industry, as listed in the original article.
A version of this article first appeared on Medscape.com.
Maribavir seen as superior to other antivirals for CMV clearance post transplant
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
Maribavir, an investigational antiviral agent with a novel mechanism of action, was superior to other antiviral strategies at clearing cytomegalovirus (CMV) viremia and controlling symptoms in hematopoietic cell or solid-organ transplant recipients, results of a phase 3 clinical trial showed.
CMV viremia clearance at study week 8 was seen in 55.7% of all patients randomized to receive maribavir, compared with 23.9% for patients assigned to receive investigator-assigned therapy (IAT), Francisco Marty, MD, from the Dana-Farber Cancer Institute in Boston reported at the Transplant & Cellular Therapies Meetings.
“Maribavir’s benefit was driven by lower incidence of treatment-limiting toxicities, compared with IAT,” he said a late-breaking abstract session during the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
“Available anti-CMV antivirals are limited by development of resistance and toxicities, particularly myelosuppression with the use of valganciclovir and nephrotoxicity with the use of foscarnet and cidofovir. Alternative treatment options are required to address this unmet medical need,” he said.
Maribavir inhibits the CMV UL97 protein kinase and is thought to affect several critical processes in CMV replication, including viral DNA synthesis, viral gene expression, encapsidation, and egress of mature capsids from the nucleus.
Details of trial
In the phase 3 SHP620-30e trial (NCT02931539), Dr. Marty and colleagues enrolled patients with relapsed or refractory CMV infections after hematopoietic cell transplant (HCT) or solid-organ transplant (SOT) and after stratification by transplant type and screening CMV DNA level randomly assigned them on a 2:1 basis to receive either maribavir 400 mg twice daily (235 patients) or IAT (117 patients), consisting of either ganciclovir/valganciclovir, foscarnet, cidofovir, or combined foscarnet and val/ganciclovir.
The primary endpoint of viremia clearance at 8 weeks was defined as plasma CMV DNA less than 137 IU/mL in two consecutive tests at a central laboratory at least 5 days apart beginning at the end of week 8.
The trial met its primary endpoint, with a viremia clearance rate of 55.7% with maribavir versus 23.9% with IAT.
The viremia clearance rates were similar in each of the transplant groups: 55.9% versus 20.8%, respectively, in patients who underwent HCT, and 55.6% versus 26.1% in patients who underwent SOT (P < .001).
Clearance rates among patients with CMV DNA below 9,100 IU/mL at baseline were 62.1% with maribavir versus 24.7% with IAT. Among patients with baseline CMV DNA of 9100 IU/mL or above, the respective rates were 43.9% versus 21.9%.
CMV viremia clearance continued from week 8 to week 16 in 18.7% of patients assigned to maribavir and to 10.3% of patients randomized to IAT (P < .013).
The median time to first CMV viremia clearance as 22 days with maribavir versus 27 days with IAT (P = .039).
All-cause mortality was similar between the groups, at 11.5% versus 11.1%, respectively.
The incidences of serious and severe treatment-emergent adverse events (TEAE) were 38.5% and 32.1%, respectively, in the maribavir group, and 37.1% and 37.9% in the IAT group.
Any TEAE leading to study drug discontinuation was less common with maribavir, occurring in 13.2% of patients, compared with 31.9% of patients on IAT. Serious TEAEs leading to drug discontinuation occurred in 8.5% versus 14.7%, respectively.
Serious TEAEs leading to death occurred in 6.8% of patients on maribavir versus 5.2% of those on IAT.
Role of letermovir
In the question-and-answer session following the presentation, comoderator Monalisa Ghosh, MD, from the University of Michigan, Ann Arbor, asked whether any patients in the study were currently on letermovir (Prevymis) prophylaxis, and whether any patients had previously been treated with letermovir but had CMV reactivation and were then treated on study.
Dr. Marty noted that the trial was designed before letermovir was approved for CMV prophylaxis in adults who have undergone an allogeneic HCT.
“Nobody was on letermovir at the beginning of the trial,” he replied, but noted that some patients who were enrolled and had infections that were refractory or resistant to valganciclovir, foscarnet, or a combination of the two received letermovir as secondary prophylaxis.
“I haven’t got the data to tell you how often [letermovir] was used; I think part of the lack of mortality benefit [with maribavir] may be due to the fact that people jumped into secondary prophylaxis with letermovir to minimize the toxicities that we saw,” he said.
Although maribavir has not as of this writing received Food and Drug Administration approval, the drug may be available to some patients through a compassionate-use program from Takeda, Dr. Marty noted.
The study was funded by Shire ViroPharma. Dr. Marty disclosed research funding from Shire and from others. Dr. Ghosh had no relevant disclosures.
FROM TCT 2021
CAR-T in children branching out to solid tumors
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
Although the only pediatric indication for chimeric antigen receptor T-cell therapy currently approved by the Food and Drug Administration is B-lineage acute lymphoblastic leukemia (ALL) that is refractory to at least two frontline induction attempts or is in second or later relapse, clinical trials of CAR-T therapy for pediatric solid tumors are also currently in progress, said Gregory Yanik, MD, from the CS Mott Children’s Hospital at the University of Michigan, Ann Arbor, at the Transplant & Cellular Therapies Meetings.
In his presentation, Dr. Yanik discussed progress in solid tumor studies as well as some issues involving the current use of CAR-T therapy for ALL.
Solid tumor studies
Malignancies such as sarcomas, brain tumors, and neuroblastomas pose unique challenges, “In contrast to hematologic malignancies, the protein we’re targeting may not be present on the cell surface of all the tumor cells. There are lower-expression profiles, and this is a problem. In fact, many people have postulated that with CAR-T for pediatric solid tumors we’ll have to do repeated cycles, almost like we do with chemotherapy,” he said at the meeting held by the American Society for Blood and Marrow Transplantation and the Center for International Blood and Marrow Transplant Research.
There are currently 14 studies of CAR-T for central nervous system tumors in children, targeting either epidermal growth factor receptor (EGFR) in glioblastoma multiforme and high-grade gliomas, HER2 in a variety of CNS tumors, the GD2 antigen on pontine gliomas, and the checkpoint molecular B7H3 in medulloblastomas and pontine gliomas.
“In sarcomas in kids there are currently 12 trials in progress. Most of the targeting epitopes are targeting either HER2 or GD2. Repetitive CAR-T infusions are being used in several of these trials in sarcomas.
For neuroblastomas there are currently 13 studies in progress, nearly all of which target GD2. Some of the trials include combining CAR-T with immune checkpoint inhibitors or C7R, an engineered cytokine driver designed to prevent T-cell exhaustion.
In addition, several trials of tumor pulsed dendritic cell vaccines are underway for treatment of children with Wilms tumor, Dr. Yanik noted.
Unresolved procedural questions
It’s still early days in CAR-T therapy, and there are several still unanswered questions regarding optimal therapy for and management of patients undergoing CAR-T procedures, Dr. Yanik said.
For example, the optimal time to collect T cells during apheresis is still unclear, he said. Collecting prior to reinduction therapy raises the risk of transducing leukemic cells, while collecting after reinduction may result in inadequate quantity or quality of cells. Regardless of when cells are collected, apheresis should be performed only when the absolute lymphocyte count is above 500/mcL or the CD3 count is above 150/mcL at the time of apheresis.
In the case tisagenlecleucel (Kymriah), his center typically collects 1x109 CD3 cells regardless of age or weight.
The number of CAR T-cells infused also appears to matter, as responses are improved at CAR-T doses above 1.5x106/kg, while risk for higher-grade cytokine release syndrome (CRS) occurs at higher infusion doses.
Blinatumomab or inotuzumab?
Along with CAR-T, two other agents, the bispecific T-cell engager blinatumomab (Blincyto) and the antibody conjugate inotuzumab ozogamicin (Besponsa) are also approved for the treatment of patients with relapsed/refractory B-cell ALL.
Like CAR-T therapy, the primary toxicities associated with blinatumomab are CRS and neurologic adverse events, whereas at inotuzumab is largely associated with hematologic and hepatic toxicities.
The logistics of therapy differ widely, with a 28-day infusion required for blinatumomab, compared with weekly dosing of inotuzumab, and the multiple visits for apheresis and infusion required for CAR-T.
Blinatumomab is approved for both children and adults with relapsed/refractory ALL, but inotuzumab is approved only for adults, and CAR-T with tisagenlecleucel is approved only for children in this indication.
CD-19 expression
There is evidence to suggest that CD19 expression prior to CAR-T has an effect on outcomes, Dr. Yanik said.
“Does blinatumomab pre–CAR-T impact outcome? The answer is probably yes,” he said.
He referred to a study by investigators at the Children’s Hospital of Philadelphia showing that, “if you’re giving blinatumomab prior to CAR-T therapy, you’re potentially reducing the cell-surface expression of CD19 on your leukemic blasts, and now while you’re bringing these patients in for CAR-T therapy, you’re getting a much higher population of dim CD19 expressers, and this is associated with a higher relapse rate and lower remission rate.”
Predicting relapse
Dr. Yanik referred to a study, currently unpublished, which will show that next-generation sequencing (NGS) is more sensitive than flow cytometry for detection of minimal residual disease (MRD), and that MRD analysis of marrow was more sensitive than analysis of peripheral blood.
“Poor outcomes were seen post CAR-T for patients who were in morphologic remission on day 28 or day 100, but had positive MRD. This especially held true if it was next-gen sequencing MRD-positive at day 100, for which relapse rates were over 95%,” he said.
The absence of B-cells is a surrogate marker for the persistence of CAR-T, and conversely, the recovery of CD19-positive B cells may be a predictor for relapse, especially if the B-cell recovery occurs within the first 6 months following CAR-T infusion.
Transplant after CAR-T?
Bone marrow transplant after CAR-T is recommend for patients with high risk of relapse, including those with B-cell recovery within the first 6 months after CAR-T, patients with MRD positivity at days 28 or 100, and patients with mixed lineage leukemia.
“Should we transplant good-risk patients, meaning, if you have NGS-MRD negative patients, is there a role for transplant? You have to look at the risk versus benefit there. These patients may have a cure rate that’s in the 80%-plus range, could we potentially optimize that even more if we consolidate them with an allo[geneic] transplant,” Dr. Yank said.
Move CAR-T up front?
A Children’s Oncology Group study is currently examining whether giving CAR-T therapy to patients with MRD of 0.01% or greater following first consolidation could result in lower tumor burden, fewer relapse, and less CRS with CAR-T.
Dr. Yanik reported that he had no conflicts of interest to disclose.
FROM TCT 2021
New ASH guidelines: VTE prevention and treatment in cancer patients
New guidelines from the American Society of Hematology “strongly recommend” using no thromboprophylaxis over using parenteral thromboprophylaxis in ambulatory patients receiving cancer chemotherapy who have low venous thromboembolism (VTE) risk, and using no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists in those at any VTE risk level.
The evidence-based guidelines for the prevention and treatment of VTE in patient with cancer, published online in Blood Advances, also include a “conditional recommendation” for using either thromboprophylaxis with the direct oral anticoagulants (DOACs) apixaban or rivaroxaban or using no thromboprophylaxis in ambulatory patients with intermediate risk and using the DOACs over no thromboprophylaxis in those with high VTE risk.
The purpose of the guidelines, which also address VTE prophylaxis in hospitalized patients with cancer and the use of anticoagulation for VTE treatment in patients with cancer, is to provide clinical decision support for shared decision-making by patients and clinicians, Gary H. Lyman, MD, of Fred Hutchinson Cancer Research Center, Seattle and Marc Carrier, MD, of the University of Ottawa, and their colleagues from the multidisciplinary guidelines panel explained.
“The recommendations take into consideration the strength of the evidence, risks of mortality, VTE, and bleeding, as well as quality of life, acceptability, and cost considerations,” they wrote, noting that VTE is a common complication in patients with cancer, who are at markedly increased risk for morbidity and mortality from VTE.
Levels of evidence
The panel members relied on updated and original systematic evidence reviews. Conditional recommendations, as opposed to strong recommendations, are defined by the panel as “suggestions,” and all 33 recommendations that make up the guidelines include a statement on the strength of the relevant evidence.
For example, the thromboprophylaxis recommendations for low, intermediate, and high VTE risk are made based on “moderate certainty in the evidence of effects,” and the recommendation for no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists is a strong recommendation based on “very low certainty in the evidence of benefits, but high certainty about the harms.”
The guidelines panel also strongly recommends, based on moderate certainty in the evidence of effects, using low-molecular-weight heparin over unfractionated heparin for the initial treatment of VTE in patients with cancer, and suggests, based on “very low certainty in the evidence of effects,” using LMWH over fondaparinux in this setting.
In addition to primary prophylaxis in ambulatory and hospitalized patients and initial VTE treatment, they also address primary prophylaxis for patients with cancer who have a central venous catheter, VTE treatment in surgical patients with cancer, short-term VTE treatment, and long-term VTE treatment.
For example, the guidelines panel conditionally recommends:
- Not using parenteral or oral thromboprophylaxis in patients with cancer and a central venous catheter
- Using LMWH or fondaparinux for surgical patients with cancer
- Using DOACS for the short-term treatment of VTE, and LMWH or DOACs for the long-term treatment of VTE in patients with cancer.
The perils of VTE
VTE in patients with cancer can interfere with treatment, increase mortality risk, and increase costs, the authors noted, adding that VTE can also adversely affect cancer patients’ quality of life.
“Some have even reported the experience of VTE to be more upsetting than that of the cancer,” they wrote. “More than 50% of thrombotic events occur within 3 months of the cancer diagnosis, a time when most cancer treatments will be underway. Patients, who are still coming to terms with a recent cancer diagnosis, often view the occurrence of VTE as a further threat to life, confirmation of the severity of their condition, and a poor prognostic sign.”
Therefore, the new guidelines aim to reduce VTE frequency, risk of bleeding complications, morbidity, and costs, thereby improving quality of life and the patient experience, the authors said, noting that three other recent guidelines on VTEs in patients with cancer have been published: the 2019 American Society of Clinical Oncology guidelines, the 2019 International Initiative on Thrombosis and Cancer guidelines, and the 2020 National Comprehensive Cancer Network guidelines.
The ASH guidelines are similar in many ways to the other guidelines, but differ in some ways, as well. An example is the timing of initiation of pharmacological thromboprophylaxis in patients undergoing cancer-related major abdominal surgery. The ASCO and ITAC guidelines advise starting thromboprophylaxis preoperatively, whereas the ASH guidelines recommend initiating thromboprophylaxis postoperatively, citing “the limited advantages to initiating thromboprophylaxis preoperatively, in addition to the potential bleeding and logistical considerations associated with neuraxial anesthesia.”
These differences highlight a lack of data in that setting and the need for additional studies, the authors said.
ASH vs. ASCO
James Douketis, MD, a practicing clinician and professor of medicine at McMaster University, Hamilton, Ont., highlighted another difference between the ASH and ASCO guidelines.
“For the treatment of [cancer-associated thrombosis], ASCO gives a strong recommendation to use LMWH or DOACs (with some caveats), which is easy to follow. ASH, on the other hand, suggests LMWH or a DOAC for the first 7-10 days, DOACs for the first 3-6 months, and back to LMWH or DOACs after 6 months,” he said in an interview.
The recommendation is “very evidence based but ambiguous and not helpful for the practicing clinician,” added Dr. Douketis, who helped develop the ITAC guidelines, but was not part of the ASH or ASCO guideline panels.
ASCO also provides a clear recommendation for giving VTE prophylaxis for 4 weeks after cancer surgery in patients with high VTE risk, whereas ASH gives “a somewhat vague recommendation” for thromboprophylaxis after hospital discharge.
The guidelines are “pretty well aligned” with respect to recommendations on VTE prophylaxis in medical cancer patients receiving chemotherapy, and although the “extremely academic” ASH guidelines were developed by “a superb team using the same evidence and excellent methodology,” they are interpreted in slightly different ways and fall short when it comes to being clinician friendly, Dr. Douketis said.
“At the end of day, for practicing clinicians, the ASH guidelines don’t provide a message that’s easy to digest,” he added.
ASH has, however, provided a resource page that includes tools and information for implementing the guidelines in clinical practice, and will maintain the guidelines “through surveillance for new evidence, ongoing review by experts, and regular revisions,” the authors said.
New guidelines from the American Society of Hematology “strongly recommend” using no thromboprophylaxis over using parenteral thromboprophylaxis in ambulatory patients receiving cancer chemotherapy who have low venous thromboembolism (VTE) risk, and using no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists in those at any VTE risk level.
The evidence-based guidelines for the prevention and treatment of VTE in patient with cancer, published online in Blood Advances, also include a “conditional recommendation” for using either thromboprophylaxis with the direct oral anticoagulants (DOACs) apixaban or rivaroxaban or using no thromboprophylaxis in ambulatory patients with intermediate risk and using the DOACs over no thromboprophylaxis in those with high VTE risk.
The purpose of the guidelines, which also address VTE prophylaxis in hospitalized patients with cancer and the use of anticoagulation for VTE treatment in patients with cancer, is to provide clinical decision support for shared decision-making by patients and clinicians, Gary H. Lyman, MD, of Fred Hutchinson Cancer Research Center, Seattle and Marc Carrier, MD, of the University of Ottawa, and their colleagues from the multidisciplinary guidelines panel explained.
“The recommendations take into consideration the strength of the evidence, risks of mortality, VTE, and bleeding, as well as quality of life, acceptability, and cost considerations,” they wrote, noting that VTE is a common complication in patients with cancer, who are at markedly increased risk for morbidity and mortality from VTE.
Levels of evidence
The panel members relied on updated and original systematic evidence reviews. Conditional recommendations, as opposed to strong recommendations, are defined by the panel as “suggestions,” and all 33 recommendations that make up the guidelines include a statement on the strength of the relevant evidence.
For example, the thromboprophylaxis recommendations for low, intermediate, and high VTE risk are made based on “moderate certainty in the evidence of effects,” and the recommendation for no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists is a strong recommendation based on “very low certainty in the evidence of benefits, but high certainty about the harms.”
The guidelines panel also strongly recommends, based on moderate certainty in the evidence of effects, using low-molecular-weight heparin over unfractionated heparin for the initial treatment of VTE in patients with cancer, and suggests, based on “very low certainty in the evidence of effects,” using LMWH over fondaparinux in this setting.
In addition to primary prophylaxis in ambulatory and hospitalized patients and initial VTE treatment, they also address primary prophylaxis for patients with cancer who have a central venous catheter, VTE treatment in surgical patients with cancer, short-term VTE treatment, and long-term VTE treatment.
For example, the guidelines panel conditionally recommends:
- Not using parenteral or oral thromboprophylaxis in patients with cancer and a central venous catheter
- Using LMWH or fondaparinux for surgical patients with cancer
- Using DOACS for the short-term treatment of VTE, and LMWH or DOACs for the long-term treatment of VTE in patients with cancer.
The perils of VTE
VTE in patients with cancer can interfere with treatment, increase mortality risk, and increase costs, the authors noted, adding that VTE can also adversely affect cancer patients’ quality of life.
“Some have even reported the experience of VTE to be more upsetting than that of the cancer,” they wrote. “More than 50% of thrombotic events occur within 3 months of the cancer diagnosis, a time when most cancer treatments will be underway. Patients, who are still coming to terms with a recent cancer diagnosis, often view the occurrence of VTE as a further threat to life, confirmation of the severity of their condition, and a poor prognostic sign.”
Therefore, the new guidelines aim to reduce VTE frequency, risk of bleeding complications, morbidity, and costs, thereby improving quality of life and the patient experience, the authors said, noting that three other recent guidelines on VTEs in patients with cancer have been published: the 2019 American Society of Clinical Oncology guidelines, the 2019 International Initiative on Thrombosis and Cancer guidelines, and the 2020 National Comprehensive Cancer Network guidelines.
The ASH guidelines are similar in many ways to the other guidelines, but differ in some ways, as well. An example is the timing of initiation of pharmacological thromboprophylaxis in patients undergoing cancer-related major abdominal surgery. The ASCO and ITAC guidelines advise starting thromboprophylaxis preoperatively, whereas the ASH guidelines recommend initiating thromboprophylaxis postoperatively, citing “the limited advantages to initiating thromboprophylaxis preoperatively, in addition to the potential bleeding and logistical considerations associated with neuraxial anesthesia.”
These differences highlight a lack of data in that setting and the need for additional studies, the authors said.
ASH vs. ASCO
James Douketis, MD, a practicing clinician and professor of medicine at McMaster University, Hamilton, Ont., highlighted another difference between the ASH and ASCO guidelines.
“For the treatment of [cancer-associated thrombosis], ASCO gives a strong recommendation to use LMWH or DOACs (with some caveats), which is easy to follow. ASH, on the other hand, suggests LMWH or a DOAC for the first 7-10 days, DOACs for the first 3-6 months, and back to LMWH or DOACs after 6 months,” he said in an interview.
The recommendation is “very evidence based but ambiguous and not helpful for the practicing clinician,” added Dr. Douketis, who helped develop the ITAC guidelines, but was not part of the ASH or ASCO guideline panels.
ASCO also provides a clear recommendation for giving VTE prophylaxis for 4 weeks after cancer surgery in patients with high VTE risk, whereas ASH gives “a somewhat vague recommendation” for thromboprophylaxis after hospital discharge.
The guidelines are “pretty well aligned” with respect to recommendations on VTE prophylaxis in medical cancer patients receiving chemotherapy, and although the “extremely academic” ASH guidelines were developed by “a superb team using the same evidence and excellent methodology,” they are interpreted in slightly different ways and fall short when it comes to being clinician friendly, Dr. Douketis said.
“At the end of day, for practicing clinicians, the ASH guidelines don’t provide a message that’s easy to digest,” he added.
ASH has, however, provided a resource page that includes tools and information for implementing the guidelines in clinical practice, and will maintain the guidelines “through surveillance for new evidence, ongoing review by experts, and regular revisions,” the authors said.
New guidelines from the American Society of Hematology “strongly recommend” using no thromboprophylaxis over using parenteral thromboprophylaxis in ambulatory patients receiving cancer chemotherapy who have low venous thromboembolism (VTE) risk, and using no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists in those at any VTE risk level.
The evidence-based guidelines for the prevention and treatment of VTE in patient with cancer, published online in Blood Advances, also include a “conditional recommendation” for using either thromboprophylaxis with the direct oral anticoagulants (DOACs) apixaban or rivaroxaban or using no thromboprophylaxis in ambulatory patients with intermediate risk and using the DOACs over no thromboprophylaxis in those with high VTE risk.
The purpose of the guidelines, which also address VTE prophylaxis in hospitalized patients with cancer and the use of anticoagulation for VTE treatment in patients with cancer, is to provide clinical decision support for shared decision-making by patients and clinicians, Gary H. Lyman, MD, of Fred Hutchinson Cancer Research Center, Seattle and Marc Carrier, MD, of the University of Ottawa, and their colleagues from the multidisciplinary guidelines panel explained.
“The recommendations take into consideration the strength of the evidence, risks of mortality, VTE, and bleeding, as well as quality of life, acceptability, and cost considerations,” they wrote, noting that VTE is a common complication in patients with cancer, who are at markedly increased risk for morbidity and mortality from VTE.
Levels of evidence
The panel members relied on updated and original systematic evidence reviews. Conditional recommendations, as opposed to strong recommendations, are defined by the panel as “suggestions,” and all 33 recommendations that make up the guidelines include a statement on the strength of the relevant evidence.
For example, the thromboprophylaxis recommendations for low, intermediate, and high VTE risk are made based on “moderate certainty in the evidence of effects,” and the recommendation for no thromboprophylaxis over oral thromboprophylaxis with vitamin K antagonists is a strong recommendation based on “very low certainty in the evidence of benefits, but high certainty about the harms.”
The guidelines panel also strongly recommends, based on moderate certainty in the evidence of effects, using low-molecular-weight heparin over unfractionated heparin for the initial treatment of VTE in patients with cancer, and suggests, based on “very low certainty in the evidence of effects,” using LMWH over fondaparinux in this setting.
In addition to primary prophylaxis in ambulatory and hospitalized patients and initial VTE treatment, they also address primary prophylaxis for patients with cancer who have a central venous catheter, VTE treatment in surgical patients with cancer, short-term VTE treatment, and long-term VTE treatment.
For example, the guidelines panel conditionally recommends:
- Not using parenteral or oral thromboprophylaxis in patients with cancer and a central venous catheter
- Using LMWH or fondaparinux for surgical patients with cancer
- Using DOACS for the short-term treatment of VTE, and LMWH or DOACs for the long-term treatment of VTE in patients with cancer.
The perils of VTE
VTE in patients with cancer can interfere with treatment, increase mortality risk, and increase costs, the authors noted, adding that VTE can also adversely affect cancer patients’ quality of life.
“Some have even reported the experience of VTE to be more upsetting than that of the cancer,” they wrote. “More than 50% of thrombotic events occur within 3 months of the cancer diagnosis, a time when most cancer treatments will be underway. Patients, who are still coming to terms with a recent cancer diagnosis, often view the occurrence of VTE as a further threat to life, confirmation of the severity of their condition, and a poor prognostic sign.”
Therefore, the new guidelines aim to reduce VTE frequency, risk of bleeding complications, morbidity, and costs, thereby improving quality of life and the patient experience, the authors said, noting that three other recent guidelines on VTEs in patients with cancer have been published: the 2019 American Society of Clinical Oncology guidelines, the 2019 International Initiative on Thrombosis and Cancer guidelines, and the 2020 National Comprehensive Cancer Network guidelines.
The ASH guidelines are similar in many ways to the other guidelines, but differ in some ways, as well. An example is the timing of initiation of pharmacological thromboprophylaxis in patients undergoing cancer-related major abdominal surgery. The ASCO and ITAC guidelines advise starting thromboprophylaxis preoperatively, whereas the ASH guidelines recommend initiating thromboprophylaxis postoperatively, citing “the limited advantages to initiating thromboprophylaxis preoperatively, in addition to the potential bleeding and logistical considerations associated with neuraxial anesthesia.”
These differences highlight a lack of data in that setting and the need for additional studies, the authors said.
ASH vs. ASCO
James Douketis, MD, a practicing clinician and professor of medicine at McMaster University, Hamilton, Ont., highlighted another difference between the ASH and ASCO guidelines.
“For the treatment of [cancer-associated thrombosis], ASCO gives a strong recommendation to use LMWH or DOACs (with some caveats), which is easy to follow. ASH, on the other hand, suggests LMWH or a DOAC for the first 7-10 days, DOACs for the first 3-6 months, and back to LMWH or DOACs after 6 months,” he said in an interview.
The recommendation is “very evidence based but ambiguous and not helpful for the practicing clinician,” added Dr. Douketis, who helped develop the ITAC guidelines, but was not part of the ASH or ASCO guideline panels.
ASCO also provides a clear recommendation for giving VTE prophylaxis for 4 weeks after cancer surgery in patients with high VTE risk, whereas ASH gives “a somewhat vague recommendation” for thromboprophylaxis after hospital discharge.
The guidelines are “pretty well aligned” with respect to recommendations on VTE prophylaxis in medical cancer patients receiving chemotherapy, and although the “extremely academic” ASH guidelines were developed by “a superb team using the same evidence and excellent methodology,” they are interpreted in slightly different ways and fall short when it comes to being clinician friendly, Dr. Douketis said.
“At the end of day, for practicing clinicians, the ASH guidelines don’t provide a message that’s easy to digest,” he added.
ASH has, however, provided a resource page that includes tools and information for implementing the guidelines in clinical practice, and will maintain the guidelines “through surveillance for new evidence, ongoing review by experts, and regular revisions,” the authors said.
FROM BLOOD ADVANCES
FDA places clinical hold on sickle cell gene therapy
The Food and Drug Administration placed a clinical hold yesterday on two gene therapy trials for sickle cell disease (SCD) after two recent complications: one participant developed acute myeloid leukemia (AML) and another developed myelodysplastic syndrome (MDS). The sponsoring company, bluebird bio, suspended the trials last week upon learning of the cases.
The company has also put the brakes on a treatment for beta thalassemia already approved in the European Union and the United Kingdom, betibeglogene autotemcel (Zynteglo). The treatment hasn’t been associated with problems but uses the same gene delivery vector, a lentivirus, as that used in the SCD trials.
Overall, the company has enrolled 47 SCD patients and 63 with beta thalassemia in trials.
The gene therapy “space” is one with spectacular successes – for a form of retinal blindness and spinal muscular atrophy – rising against a backdrop of recent setbacks and failures – for Duchenne muscular dystrophy, lipoprotein lipase deficiency, and myotubular myopathy.
A lentiviral vector
The retooled lentivirus used in the SCD trials, LentiGlobin, delivers a beta-globin gene with one amino acid replacement to hematopoietic stem cells outside the patient’s body. The modified cells are then infused back into the patient. The gene therapy reshapes red blood cells, enabling them to circulate through narrow blood vessels without sickling and adhering into painful logjams.
What is worrisome is that in the patient who developed AML, and who received the gene therapy more than 5 years ago, the cancer cells contained the vector. Those test results aren’t yet available for the participant who has MDS.
The finding raises suspicion that the gene therapy had a role in the cancer, but is only correlative.
Lentiviral vectors have a good track record, but the two cases evoke memories of 2 decades ago. In 2001, five children being treated for an inherited immunodeficiency (SCID-X1) with a gamma retroviral vector developed leukemia and one died. Those viruses inserted into an oncogene. Happening 2 years after the death of 18-year-old Jesse Gelsinger in another gene therapy trial, the SCID trial had a chilling effect on the field.
Since then, lentiviral vectors have been reinvented to be “self-inactivating,” minimizing the risk for inserting willy-nilly into a genome. “Lentiviral vectors have been expressly designed to avoid insertional oncogenesis, based on prior experience with the gamma retroviruses. We don’t have evidence that the vector is causative, but our studies will shed some light on whether that’s true in these cases,” said bluebird bio chief scientific officer Philip Gregory, DPhil, on a conference call Feb. 16.
Lentiviral vectors have been successful as the backbone of chimeric antigen receptor T-cell (CAR-T) therapy, which directs modified T cells to certain blood cancers. “Among the hundreds to thousands of patients treated with CAR-T cell therapy, lentivirus vector hasn’t been associated with any malignancies,” said bluebird’s chief medical officer, Dave Davidson, MD.
Jeanne Loring, PhD, director of the Center for Regenerative Medicine at Scripps Research, agreed. “Gene therapy is having some extreme highs and lows these days. Most studies use [adeno-associated viral] vectors, which don’t integrate into the genome. But some people have antibodies to AAV vectors, and AAV is diluted out when cells divide. That’s why lentivirus, which integrates into the genome, is used for blood stem cells and T cells in CAR-T therapy.”
Pinpointing causality
At bluebird bio, investigation into the possible “genetic gymnastics” of the lentivirus vector is focusing on where it integrates into the genome – whether it harpoons an oncogene like the gamma retroviral vectors, or affects genome stability, Dr. Gregory explained. To be causative, the affected gene must be a “driver” of the cancer, and not just a “passenger.”
Another suspect is busulfan, a drug used to “condition” the recipient’s bone marrow, making room for modified stem cells. “It’s possible that busulfan is the main problem, as it is a carcinogen unto itself,” said Paul Knoepfler, PhD, a stem cell researcher at the University of California, Davis.
In addition to the two more recent reports of complications, a third trial participant, who had participated in a phase 1/2 trial, developed MDS in 2018 and died of AML in July 2020. The cancer cells from that patient did not contain viral vectors and the MDS was attributed to busulfan conditioning.
Nick Leschly, chief of bluebird, pointed out the importance of clinical context in implicating the vector. Because SCD itself stresses the bone marrow, patients already face an increased risk of developing blood cancer, he said. “Now layer on other risks of the gene therapy. It’s challenging because we’re dealing with patients who have life expectancy in the mid 40s.” Previous treatments, such the antisickling drug hydroxyurea, may also contribute to patient vulnerability.
A patient’s view
SCD affects more than 100,000 people in the United States, and about 20 million globally. Charles Hough is one of them. He can attest to the severity of the disease as well as the promise of gene therapy
Mr. Hough was diagnosed at age 2, and endured the profound fatigue, pain crises, and even coma characteristic of severe cases. He cited his “rebirth” as Sept. 25, 2018, when he received his first modified stem cells at the National Institutes of Health. Mr. Hough told his story a year ago in a webinar for the National Organization for Rare Disorders. This news organization caught up with him in light of the clinical trial hold.
Although the preparative regimens for the gene therapy were tough, his sickle cell symptoms vanished after gene therapy. Even hearing about the current hold on the clinical trial, Mr. Hough doesn’t regret his participation.
“I had a lot of friends who passed because of the complications from sickle cell. I was always worried that I wouldn’t live to see the next day. Now I don’t have that stress hanging over my head and I feel like I can live a normal life. Becoming sickle cell free was my dream.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration placed a clinical hold yesterday on two gene therapy trials for sickle cell disease (SCD) after two recent complications: one participant developed acute myeloid leukemia (AML) and another developed myelodysplastic syndrome (MDS). The sponsoring company, bluebird bio, suspended the trials last week upon learning of the cases.
The company has also put the brakes on a treatment for beta thalassemia already approved in the European Union and the United Kingdom, betibeglogene autotemcel (Zynteglo). The treatment hasn’t been associated with problems but uses the same gene delivery vector, a lentivirus, as that used in the SCD trials.
Overall, the company has enrolled 47 SCD patients and 63 with beta thalassemia in trials.
The gene therapy “space” is one with spectacular successes – for a form of retinal blindness and spinal muscular atrophy – rising against a backdrop of recent setbacks and failures – for Duchenne muscular dystrophy, lipoprotein lipase deficiency, and myotubular myopathy.
A lentiviral vector
The retooled lentivirus used in the SCD trials, LentiGlobin, delivers a beta-globin gene with one amino acid replacement to hematopoietic stem cells outside the patient’s body. The modified cells are then infused back into the patient. The gene therapy reshapes red blood cells, enabling them to circulate through narrow blood vessels without sickling and adhering into painful logjams.
What is worrisome is that in the patient who developed AML, and who received the gene therapy more than 5 years ago, the cancer cells contained the vector. Those test results aren’t yet available for the participant who has MDS.
The finding raises suspicion that the gene therapy had a role in the cancer, but is only correlative.
Lentiviral vectors have a good track record, but the two cases evoke memories of 2 decades ago. In 2001, five children being treated for an inherited immunodeficiency (SCID-X1) with a gamma retroviral vector developed leukemia and one died. Those viruses inserted into an oncogene. Happening 2 years after the death of 18-year-old Jesse Gelsinger in another gene therapy trial, the SCID trial had a chilling effect on the field.
Since then, lentiviral vectors have been reinvented to be “self-inactivating,” minimizing the risk for inserting willy-nilly into a genome. “Lentiviral vectors have been expressly designed to avoid insertional oncogenesis, based on prior experience with the gamma retroviruses. We don’t have evidence that the vector is causative, but our studies will shed some light on whether that’s true in these cases,” said bluebird bio chief scientific officer Philip Gregory, DPhil, on a conference call Feb. 16.
Lentiviral vectors have been successful as the backbone of chimeric antigen receptor T-cell (CAR-T) therapy, which directs modified T cells to certain blood cancers. “Among the hundreds to thousands of patients treated with CAR-T cell therapy, lentivirus vector hasn’t been associated with any malignancies,” said bluebird’s chief medical officer, Dave Davidson, MD.
Jeanne Loring, PhD, director of the Center for Regenerative Medicine at Scripps Research, agreed. “Gene therapy is having some extreme highs and lows these days. Most studies use [adeno-associated viral] vectors, which don’t integrate into the genome. But some people have antibodies to AAV vectors, and AAV is diluted out when cells divide. That’s why lentivirus, which integrates into the genome, is used for blood stem cells and T cells in CAR-T therapy.”
Pinpointing causality
At bluebird bio, investigation into the possible “genetic gymnastics” of the lentivirus vector is focusing on where it integrates into the genome – whether it harpoons an oncogene like the gamma retroviral vectors, or affects genome stability, Dr. Gregory explained. To be causative, the affected gene must be a “driver” of the cancer, and not just a “passenger.”
Another suspect is busulfan, a drug used to “condition” the recipient’s bone marrow, making room for modified stem cells. “It’s possible that busulfan is the main problem, as it is a carcinogen unto itself,” said Paul Knoepfler, PhD, a stem cell researcher at the University of California, Davis.
In addition to the two more recent reports of complications, a third trial participant, who had participated in a phase 1/2 trial, developed MDS in 2018 and died of AML in July 2020. The cancer cells from that patient did not contain viral vectors and the MDS was attributed to busulfan conditioning.
Nick Leschly, chief of bluebird, pointed out the importance of clinical context in implicating the vector. Because SCD itself stresses the bone marrow, patients already face an increased risk of developing blood cancer, he said. “Now layer on other risks of the gene therapy. It’s challenging because we’re dealing with patients who have life expectancy in the mid 40s.” Previous treatments, such the antisickling drug hydroxyurea, may also contribute to patient vulnerability.
A patient’s view
SCD affects more than 100,000 people in the United States, and about 20 million globally. Charles Hough is one of them. He can attest to the severity of the disease as well as the promise of gene therapy
Mr. Hough was diagnosed at age 2, and endured the profound fatigue, pain crises, and even coma characteristic of severe cases. He cited his “rebirth” as Sept. 25, 2018, when he received his first modified stem cells at the National Institutes of Health. Mr. Hough told his story a year ago in a webinar for the National Organization for Rare Disorders. This news organization caught up with him in light of the clinical trial hold.
Although the preparative regimens for the gene therapy were tough, his sickle cell symptoms vanished after gene therapy. Even hearing about the current hold on the clinical trial, Mr. Hough doesn’t regret his participation.
“I had a lot of friends who passed because of the complications from sickle cell. I was always worried that I wouldn’t live to see the next day. Now I don’t have that stress hanging over my head and I feel like I can live a normal life. Becoming sickle cell free was my dream.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration placed a clinical hold yesterday on two gene therapy trials for sickle cell disease (SCD) after two recent complications: one participant developed acute myeloid leukemia (AML) and another developed myelodysplastic syndrome (MDS). The sponsoring company, bluebird bio, suspended the trials last week upon learning of the cases.
The company has also put the brakes on a treatment for beta thalassemia already approved in the European Union and the United Kingdom, betibeglogene autotemcel (Zynteglo). The treatment hasn’t been associated with problems but uses the same gene delivery vector, a lentivirus, as that used in the SCD trials.
Overall, the company has enrolled 47 SCD patients and 63 with beta thalassemia in trials.
The gene therapy “space” is one with spectacular successes – for a form of retinal blindness and spinal muscular atrophy – rising against a backdrop of recent setbacks and failures – for Duchenne muscular dystrophy, lipoprotein lipase deficiency, and myotubular myopathy.
A lentiviral vector
The retooled lentivirus used in the SCD trials, LentiGlobin, delivers a beta-globin gene with one amino acid replacement to hematopoietic stem cells outside the patient’s body. The modified cells are then infused back into the patient. The gene therapy reshapes red blood cells, enabling them to circulate through narrow blood vessels without sickling and adhering into painful logjams.
What is worrisome is that in the patient who developed AML, and who received the gene therapy more than 5 years ago, the cancer cells contained the vector. Those test results aren’t yet available for the participant who has MDS.
The finding raises suspicion that the gene therapy had a role in the cancer, but is only correlative.
Lentiviral vectors have a good track record, but the two cases evoke memories of 2 decades ago. In 2001, five children being treated for an inherited immunodeficiency (SCID-X1) with a gamma retroviral vector developed leukemia and one died. Those viruses inserted into an oncogene. Happening 2 years after the death of 18-year-old Jesse Gelsinger in another gene therapy trial, the SCID trial had a chilling effect on the field.
Since then, lentiviral vectors have been reinvented to be “self-inactivating,” minimizing the risk for inserting willy-nilly into a genome. “Lentiviral vectors have been expressly designed to avoid insertional oncogenesis, based on prior experience with the gamma retroviruses. We don’t have evidence that the vector is causative, but our studies will shed some light on whether that’s true in these cases,” said bluebird bio chief scientific officer Philip Gregory, DPhil, on a conference call Feb. 16.
Lentiviral vectors have been successful as the backbone of chimeric antigen receptor T-cell (CAR-T) therapy, which directs modified T cells to certain blood cancers. “Among the hundreds to thousands of patients treated with CAR-T cell therapy, lentivirus vector hasn’t been associated with any malignancies,” said bluebird’s chief medical officer, Dave Davidson, MD.
Jeanne Loring, PhD, director of the Center for Regenerative Medicine at Scripps Research, agreed. “Gene therapy is having some extreme highs and lows these days. Most studies use [adeno-associated viral] vectors, which don’t integrate into the genome. But some people have antibodies to AAV vectors, and AAV is diluted out when cells divide. That’s why lentivirus, which integrates into the genome, is used for blood stem cells and T cells in CAR-T therapy.”
Pinpointing causality
At bluebird bio, investigation into the possible “genetic gymnastics” of the lentivirus vector is focusing on where it integrates into the genome – whether it harpoons an oncogene like the gamma retroviral vectors, or affects genome stability, Dr. Gregory explained. To be causative, the affected gene must be a “driver” of the cancer, and not just a “passenger.”
Another suspect is busulfan, a drug used to “condition” the recipient’s bone marrow, making room for modified stem cells. “It’s possible that busulfan is the main problem, as it is a carcinogen unto itself,” said Paul Knoepfler, PhD, a stem cell researcher at the University of California, Davis.
In addition to the two more recent reports of complications, a third trial participant, who had participated in a phase 1/2 trial, developed MDS in 2018 and died of AML in July 2020. The cancer cells from that patient did not contain viral vectors and the MDS was attributed to busulfan conditioning.
Nick Leschly, chief of bluebird, pointed out the importance of clinical context in implicating the vector. Because SCD itself stresses the bone marrow, patients already face an increased risk of developing blood cancer, he said. “Now layer on other risks of the gene therapy. It’s challenging because we’re dealing with patients who have life expectancy in the mid 40s.” Previous treatments, such the antisickling drug hydroxyurea, may also contribute to patient vulnerability.
A patient’s view
SCD affects more than 100,000 people in the United States, and about 20 million globally. Charles Hough is one of them. He can attest to the severity of the disease as well as the promise of gene therapy
Mr. Hough was diagnosed at age 2, and endured the profound fatigue, pain crises, and even coma characteristic of severe cases. He cited his “rebirth” as Sept. 25, 2018, when he received his first modified stem cells at the National Institutes of Health. Mr. Hough told his story a year ago in a webinar for the National Organization for Rare Disorders. This news organization caught up with him in light of the clinical trial hold.
Although the preparative regimens for the gene therapy were tough, his sickle cell symptoms vanished after gene therapy. Even hearing about the current hold on the clinical trial, Mr. Hough doesn’t regret his participation.
“I had a lot of friends who passed because of the complications from sickle cell. I was always worried that I wouldn’t live to see the next day. Now I don’t have that stress hanging over my head and I feel like I can live a normal life. Becoming sickle cell free was my dream.”
A version of this article first appeared on Medscape.com.
Fired for good judgment a sign of physicians’ lost respect
What happened to Hasan Gokal, MD, should stick painfully in the craws of all physicians. It should serve as a call to action, because Dr. Gokal is sitting at home today without a job and under threat of further legal action while we continue about our day.

Dr. Gokal’s “crime” is that he vaccinated 10 strangers and acquaintances with soon-to-expire doses of the Moderna COVID-19 vaccine. He drove to the homes of some in the dark of night and injected others on his Sugar Land, Texas, lawn. He spent hours in a frantic search for willing recipients to beat the expiration clock. With minutes to spare, he gave the last dose to his at-risk wife, who has symptomatic pulmonary sarcoidosis, but whose age meant she did not fall into a vaccine priority tier.
According to the New York Times, Dr. Gokal’s wife was hesitant, afraid he might get into trouble. But why would she be hesitant? He wasn’t doing anything immoral. Perhaps she knew how far physicians have fallen and how bitterly they both could suffer.
In Barren County, Ky., where I live, a state of emergency was declared by our judge executive because of inclement weather. This directive allows our emergency management to “waive procedures and formalities otherwise required by the law.” It’s too bad that the same courtesy was not afforded to Dr. Gokal in Texas. It’s a shame that ice and snow didn’t drive his actions. Perhaps that would have protected him against the harsh criticism. Rather, it was his oath to patients and dedication to his fellow humans that motivated him, and for that, he was made to suffer.
Dr. Gokal was right to think that pouring the last 10 vaccine doses down the toilet would be an egregious act. But he was wrong in thinking his decision to find takers for the vaccine would be viewed as expedient. Instead, he was accused of graft and even nepotism. And there is the rub. That he was fired and charged with the theft of $137 worth of vaccines says everything about how physicians are treated in the year 2021. Dr. Gokal’s lawyer says the charge carried a maximum penalty of 1 year in prison and a fine of nearly $4,000.
Thank God a sage judge threw out the case and “rebuked” the office of District Attorney Kim Ogg. That hasn’t stopped her from threatening to bring the case to a grand jury. That threat invites anyone faced with the same scenario to flush the extra vaccine doses into the septic system. It encourages us to choose the toilet handle to avoid a mug shot.
And we can’t ignore the racial slant to this story. The Times reported that Dr. Gokal asked the officials, “Are you suggesting that there were too many Indian names in this group?”
“Exactly” was the answer. Let that sink in.
None of this would have happened 20 years ago. Back then, no one would have questioned the wisdom a physician gains from all our years of training and residency. In an age when anyone who conducts an office visit is now called “doctor,” respect for the letters “MD” has been leveled. We physicians have lost our autonomy and been cowed into submission.
But whatever his profession, Hasan Gokal was fired for being a good human. Today, the sun rose on 10 individuals who now enjoy better protection against a deadly pandemic. They include a bed-bound nonagenarian. A woman in her 80s with dementia. A mother with a child who uses a ventilator. All now have antibodies against SARS-CoV2 because of the tireless actions of Dr. Gokal.
Yet Dr. Gokal’s future is uncertain. Will we help him, or will we leave him to the wolves? In an email exchange with his lawyer’s office, I learned that Dr. Gokal has received offers of employment but is unable to entertain them because the actions by the Harris County District Attorney triggered an automatic review by the Texas Medical Board. A GoFundMe page was launched, but an appreciative Dr. Gokal stated publicly that he’d rather the money go to a needy charity.
In the last paragraph of the Times article, Dr. Gokal asks, “How can I take it back?” referencing stories about “the Pakistani doctor in Houston who stole all those vaccines.”
Let’s help him take back his story. In helping him, perhaps we can take back a little control. We could start with letters of support that could be mailed to his lawyer, Paul Doyle, Esq., of Houston, or tweet, respectfully of course, to the district attorney @Kimoggforda.
We can also let the Harris County Public Health Department in Houston know what we think of their actions.
On Martin Luther King Day, Kim Ogg, the district attorney who charged Dr. Gokal, tweeted MLK’s famous quote: “Injustice anywhere is a threat to justice everywhere.”
Let that motivate us to action.
Melissa Walton-Shirley, MD, is a native Kentuckian who retired from full-time invasive cardiology. She enjoys locums work in Montana and is a champion of physician rights and patient safety. In addition to opinion writing, she enjoys spending time with her husband, daughters and parents, and sidelines as a backing vocalist for local rock bands. A version of this article first appeared on Medscape.com.
What happened to Hasan Gokal, MD, should stick painfully in the craws of all physicians. It should serve as a call to action, because Dr. Gokal is sitting at home today without a job and under threat of further legal action while we continue about our day.
Dr. Gokal’s “crime” is that he vaccinated 10 strangers and acquaintances with soon-to-expire doses of the Moderna COVID-19 vaccine. He drove to the homes of some in the dark of night and injected others on his Sugar Land, Texas, lawn. He spent hours in a frantic search for willing recipients to beat the expiration clock. With minutes to spare, he gave the last dose to his at-risk wife, who has symptomatic pulmonary sarcoidosis, but whose age meant she did not fall into a vaccine priority tier.
According to the New York Times, Dr. Gokal’s wife was hesitant, afraid he might get into trouble. But why would she be hesitant? He wasn’t doing anything immoral. Perhaps she knew how far physicians have fallen and how bitterly they both could suffer.
In Barren County, Ky., where I live, a state of emergency was declared by our judge executive because of inclement weather. This directive allows our emergency management to “waive procedures and formalities otherwise required by the law.” It’s too bad that the same courtesy was not afforded to Dr. Gokal in Texas. It’s a shame that ice and snow didn’t drive his actions. Perhaps that would have protected him against the harsh criticism. Rather, it was his oath to patients and dedication to his fellow humans that motivated him, and for that, he was made to suffer.
Dr. Gokal was right to think that pouring the last 10 vaccine doses down the toilet would be an egregious act. But he was wrong in thinking his decision to find takers for the vaccine would be viewed as expedient. Instead, he was accused of graft and even nepotism. And there is the rub. That he was fired and charged with the theft of $137 worth of vaccines says everything about how physicians are treated in the year 2021. Dr. Gokal’s lawyer says the charge carried a maximum penalty of 1 year in prison and a fine of nearly $4,000.
Thank God a sage judge threw out the case and “rebuked” the office of District Attorney Kim Ogg. That hasn’t stopped her from threatening to bring the case to a grand jury. That threat invites anyone faced with the same scenario to flush the extra vaccine doses into the septic system. It encourages us to choose the toilet handle to avoid a mug shot.
And we can’t ignore the racial slant to this story. The Times reported that Dr. Gokal asked the officials, “Are you suggesting that there were too many Indian names in this group?”
“Exactly” was the answer. Let that sink in.
None of this would have happened 20 years ago. Back then, no one would have questioned the wisdom a physician gains from all our years of training and residency. In an age when anyone who conducts an office visit is now called “doctor,” respect for the letters “MD” has been leveled. We physicians have lost our autonomy and been cowed into submission.
But whatever his profession, Hasan Gokal was fired for being a good human. Today, the sun rose on 10 individuals who now enjoy better protection against a deadly pandemic. They include a bed-bound nonagenarian. A woman in her 80s with dementia. A mother with a child who uses a ventilator. All now have antibodies against SARS-CoV2 because of the tireless actions of Dr. Gokal.
Yet Dr. Gokal’s future is uncertain. Will we help him, or will we leave him to the wolves? In an email exchange with his lawyer’s office, I learned that Dr. Gokal has received offers of employment but is unable to entertain them because the actions by the Harris County District Attorney triggered an automatic review by the Texas Medical Board. A GoFundMe page was launched, but an appreciative Dr. Gokal stated publicly that he’d rather the money go to a needy charity.
In the last paragraph of the Times article, Dr. Gokal asks, “How can I take it back?” referencing stories about “the Pakistani doctor in Houston who stole all those vaccines.”
Let’s help him take back his story. In helping him, perhaps we can take back a little control. We could start with letters of support that could be mailed to his lawyer, Paul Doyle, Esq., of Houston, or tweet, respectfully of course, to the district attorney @Kimoggforda.
We can also let the Harris County Public Health Department in Houston know what we think of their actions.
On Martin Luther King Day, Kim Ogg, the district attorney who charged Dr. Gokal, tweeted MLK’s famous quote: “Injustice anywhere is a threat to justice everywhere.”
Let that motivate us to action.
Melissa Walton-Shirley, MD, is a native Kentuckian who retired from full-time invasive cardiology. She enjoys locums work in Montana and is a champion of physician rights and patient safety. In addition to opinion writing, she enjoys spending time with her husband, daughters and parents, and sidelines as a backing vocalist for local rock bands. A version of this article first appeared on Medscape.com.
What happened to Hasan Gokal, MD, should stick painfully in the craws of all physicians. It should serve as a call to action, because Dr. Gokal is sitting at home today without a job and under threat of further legal action while we continue about our day.
Dr. Gokal’s “crime” is that he vaccinated 10 strangers and acquaintances with soon-to-expire doses of the Moderna COVID-19 vaccine. He drove to the homes of some in the dark of night and injected others on his Sugar Land, Texas, lawn. He spent hours in a frantic search for willing recipients to beat the expiration clock. With minutes to spare, he gave the last dose to his at-risk wife, who has symptomatic pulmonary sarcoidosis, but whose age meant she did not fall into a vaccine priority tier.
According to the New York Times, Dr. Gokal’s wife was hesitant, afraid he might get into trouble. But why would she be hesitant? He wasn’t doing anything immoral. Perhaps she knew how far physicians have fallen and how bitterly they both could suffer.
In Barren County, Ky., where I live, a state of emergency was declared by our judge executive because of inclement weather. This directive allows our emergency management to “waive procedures and formalities otherwise required by the law.” It’s too bad that the same courtesy was not afforded to Dr. Gokal in Texas. It’s a shame that ice and snow didn’t drive his actions. Perhaps that would have protected him against the harsh criticism. Rather, it was his oath to patients and dedication to his fellow humans that motivated him, and for that, he was made to suffer.
Dr. Gokal was right to think that pouring the last 10 vaccine doses down the toilet would be an egregious act. But he was wrong in thinking his decision to find takers for the vaccine would be viewed as expedient. Instead, he was accused of graft and even nepotism. And there is the rub. That he was fired and charged with the theft of $137 worth of vaccines says everything about how physicians are treated in the year 2021. Dr. Gokal’s lawyer says the charge carried a maximum penalty of 1 year in prison and a fine of nearly $4,000.
Thank God a sage judge threw out the case and “rebuked” the office of District Attorney Kim Ogg. That hasn’t stopped her from threatening to bring the case to a grand jury. That threat invites anyone faced with the same scenario to flush the extra vaccine doses into the septic system. It encourages us to choose the toilet handle to avoid a mug shot.
And we can’t ignore the racial slant to this story. The Times reported that Dr. Gokal asked the officials, “Are you suggesting that there were too many Indian names in this group?”
“Exactly” was the answer. Let that sink in.
None of this would have happened 20 years ago. Back then, no one would have questioned the wisdom a physician gains from all our years of training and residency. In an age when anyone who conducts an office visit is now called “doctor,” respect for the letters “MD” has been leveled. We physicians have lost our autonomy and been cowed into submission.
But whatever his profession, Hasan Gokal was fired for being a good human. Today, the sun rose on 10 individuals who now enjoy better protection against a deadly pandemic. They include a bed-bound nonagenarian. A woman in her 80s with dementia. A mother with a child who uses a ventilator. All now have antibodies against SARS-CoV2 because of the tireless actions of Dr. Gokal.
Yet Dr. Gokal’s future is uncertain. Will we help him, or will we leave him to the wolves? In an email exchange with his lawyer’s office, I learned that Dr. Gokal has received offers of employment but is unable to entertain them because the actions by the Harris County District Attorney triggered an automatic review by the Texas Medical Board. A GoFundMe page was launched, but an appreciative Dr. Gokal stated publicly that he’d rather the money go to a needy charity.
In the last paragraph of the Times article, Dr. Gokal asks, “How can I take it back?” referencing stories about “the Pakistani doctor in Houston who stole all those vaccines.”
Let’s help him take back his story. In helping him, perhaps we can take back a little control. We could start with letters of support that could be mailed to his lawyer, Paul Doyle, Esq., of Houston, or tweet, respectfully of course, to the district attorney @Kimoggforda.
We can also let the Harris County Public Health Department in Houston know what we think of their actions.
On Martin Luther King Day, Kim Ogg, the district attorney who charged Dr. Gokal, tweeted MLK’s famous quote: “Injustice anywhere is a threat to justice everywhere.”
Let that motivate us to action.
Melissa Walton-Shirley, MD, is a native Kentuckian who retired from full-time invasive cardiology. She enjoys locums work in Montana and is a champion of physician rights and patient safety. In addition to opinion writing, she enjoys spending time with her husband, daughters and parents, and sidelines as a backing vocalist for local rock bands. A version of this article first appeared on Medscape.com.
FDA grants emergency use authorization to Johnson & Johnson COVID-19 vaccine
And then there were three.

More vaccine availability at a time of high demand and limited supply could help officials vaccinate more Americans, more quickly. In addition, the J&J vaccine offers one-dose convenience and storage at conventional refrigeration temperatures.
Initial reactions to the EUA for the J&J vaccine have been positive.
“The advantages of having a third vaccine, especially one that is a single shot and can be stored without special refrigeration requirements, will be a major contribution in getting the general public vaccinated sooner, both in the U.S. and around the world,” Phyllis Tien, MD, professor of medicine in the division of infectious diseases at the University of California, San Francisco, told Medscape Medical News.
“It’s great news. We have yet a third vaccine that is highly effective at preventing COVID, and even more effective at preventing severe COVID,” said Paul Goepfert, MD. It’s a “tremendous boon for our country and other countries as well.”
“This vaccine has also been shown to be effective against the B.1.351 strain that was first described in South Africa,” added Dr. Goepfert, director of the Alabama Vaccine Research Clinic and infectious disease specialist at the University of Alabama at Birmingham.
The EUA “is indeed exciting news,” Colleen Kraft, MD, associate chief medical officer at Emory University Hospital and associate professor at Emory University School of Medicine in Atlanta, said during a February 25 media briefing.
One recent concern centers on people aged 60 years and older. Documents the FDA released earlier this week suggest a lower efficacy, 42%, for the J&J immunization among people in this age group with certain relevant comorbidities. In contrast, without underlying conditions like heart disease or diabetes, efficacy in this cohort was 72%.
The more the merrier
The scope and urgency of the COVID-19 pandemic necessitates as many protective measures as possible, said Raj Shah, MD, geriatrician, and associate professor of family medicine and codirector of the Center for Community Health Equity at Rush University in Chicago.
“Trying to vaccinate as many individuals living in the United States to prevent the spread of COVID is such a big project that no one company or one vaccine was going to be able to ramp up fast enough on its own,” Dr. Shah told Medscape Medical News.“This has been the hope for us,” he added, “to get to multiple vaccines with slightly different properties that will provide more options.”
Experience with the J&J vaccine so far suggests reactions are less severe. “The nice thing about the Johnson and Johnson [vaccine] is that it definitely has less side effects,” Dr. Kraft said.
On the other hand, low-grade fever, chills, or fatigue after vaccination can be considered a positive because they can reflect how well the immune system is responding, she added.
One and done?
Single-dose administration could be more than a convenience — it could also help clinicians vaccinate members of underserved communities and rural locations, where returning for a second dose could be more difficult for some people.
“In a controlled setting, in a clinical trial, we do a lot to make sure people get all the treatment they need,” Dr. Shah said. “We’re not seeing it right now, but we’re always worried when we have more than one dose that has to be administered, that some people will drop off and not come back for the second vaccine.”
This group could include the needle-phobic, he added. “For them, having it done once alleviates a lot of the anxiety.”
Looking beyond the numbers
The phase 3 ENSEMBLE study of the J&J vaccine revealed a 72% efficacy for preventing moderate-to-severe COVID-19 among U.S. participants. In contrast, researchers reported 94% to 95% efficacy for the Pfizer/BioNTech and Moderna vaccines.
However, experts agreed that focusing solely on these numbers can miss more important points. For example, no participants who received the J&J vaccine in the phase 3 trial died from COVID-19-related illness. There were five such deaths in the placebo cohort.
“One of the things that these vaccines do very well is they minimize severe disease,” Dr. Kraft said. “As somebody that has spent an inordinate time in the hospital taking care of patients with severe disease from COVID, this is very much a welcome addition to our armamentarium to fight this virus.”
“If you can give something that prevents people from dying, that is a true path to normalcy,” Dr. Goepfert added.
More work to do
“The demand is strong from all groups right now. We just have to work on getting more vaccines out there,” Dr. Shah said.
“We are at a point in this country where we are getting better with the distribution of the vaccine,” he added, “but we are nowhere close to achieving that distribution of vaccines to get to everybody.”
Dr. Goepfert, Dr. Shah, and Dr. Kraft disclosed no relevant financial relationships. Dr. Tien received support from Johnson & Johnson to conduct the J&J COVID-19 vaccine trial in the San Francisco VA Health Care System.
A version of this article first appeared on Medscape.com.
And then there were three.
More vaccine availability at a time of high demand and limited supply could help officials vaccinate more Americans, more quickly. In addition, the J&J vaccine offers one-dose convenience and storage at conventional refrigeration temperatures.
Initial reactions to the EUA for the J&J vaccine have been positive.
“The advantages of having a third vaccine, especially one that is a single shot and can be stored without special refrigeration requirements, will be a major contribution in getting the general public vaccinated sooner, both in the U.S. and around the world,” Phyllis Tien, MD, professor of medicine in the division of infectious diseases at the University of California, San Francisco, told Medscape Medical News.
“It’s great news. We have yet a third vaccine that is highly effective at preventing COVID, and even more effective at preventing severe COVID,” said Paul Goepfert, MD. It’s a “tremendous boon for our country and other countries as well.”
“This vaccine has also been shown to be effective against the B.1.351 strain that was first described in South Africa,” added Dr. Goepfert, director of the Alabama Vaccine Research Clinic and infectious disease specialist at the University of Alabama at Birmingham.
The EUA “is indeed exciting news,” Colleen Kraft, MD, associate chief medical officer at Emory University Hospital and associate professor at Emory University School of Medicine in Atlanta, said during a February 25 media briefing.
One recent concern centers on people aged 60 years and older. Documents the FDA released earlier this week suggest a lower efficacy, 42%, for the J&J immunization among people in this age group with certain relevant comorbidities. In contrast, without underlying conditions like heart disease or diabetes, efficacy in this cohort was 72%.
The more the merrier
The scope and urgency of the COVID-19 pandemic necessitates as many protective measures as possible, said Raj Shah, MD, geriatrician, and associate professor of family medicine and codirector of the Center for Community Health Equity at Rush University in Chicago.
“Trying to vaccinate as many individuals living in the United States to prevent the spread of COVID is such a big project that no one company or one vaccine was going to be able to ramp up fast enough on its own,” Dr. Shah told Medscape Medical News.“This has been the hope for us,” he added, “to get to multiple vaccines with slightly different properties that will provide more options.”
Experience with the J&J vaccine so far suggests reactions are less severe. “The nice thing about the Johnson and Johnson [vaccine] is that it definitely has less side effects,” Dr. Kraft said.
On the other hand, low-grade fever, chills, or fatigue after vaccination can be considered a positive because they can reflect how well the immune system is responding, she added.
One and done?
Single-dose administration could be more than a convenience — it could also help clinicians vaccinate members of underserved communities and rural locations, where returning for a second dose could be more difficult for some people.
“In a controlled setting, in a clinical trial, we do a lot to make sure people get all the treatment they need,” Dr. Shah said. “We’re not seeing it right now, but we’re always worried when we have more than one dose that has to be administered, that some people will drop off and not come back for the second vaccine.”
This group could include the needle-phobic, he added. “For them, having it done once alleviates a lot of the anxiety.”
Looking beyond the numbers
The phase 3 ENSEMBLE study of the J&J vaccine revealed a 72% efficacy for preventing moderate-to-severe COVID-19 among U.S. participants. In contrast, researchers reported 94% to 95% efficacy for the Pfizer/BioNTech and Moderna vaccines.
However, experts agreed that focusing solely on these numbers can miss more important points. For example, no participants who received the J&J vaccine in the phase 3 trial died from COVID-19-related illness. There were five such deaths in the placebo cohort.
“One of the things that these vaccines do very well is they minimize severe disease,” Dr. Kraft said. “As somebody that has spent an inordinate time in the hospital taking care of patients with severe disease from COVID, this is very much a welcome addition to our armamentarium to fight this virus.”
“If you can give something that prevents people from dying, that is a true path to normalcy,” Dr. Goepfert added.
More work to do
“The demand is strong from all groups right now. We just have to work on getting more vaccines out there,” Dr. Shah said.
“We are at a point in this country where we are getting better with the distribution of the vaccine,” he added, “but we are nowhere close to achieving that distribution of vaccines to get to everybody.”
Dr. Goepfert, Dr. Shah, and Dr. Kraft disclosed no relevant financial relationships. Dr. Tien received support from Johnson & Johnson to conduct the J&J COVID-19 vaccine trial in the San Francisco VA Health Care System.
A version of this article first appeared on Medscape.com.
And then there were three.
More vaccine availability at a time of high demand and limited supply could help officials vaccinate more Americans, more quickly. In addition, the J&J vaccine offers one-dose convenience and storage at conventional refrigeration temperatures.
Initial reactions to the EUA for the J&J vaccine have been positive.
“The advantages of having a third vaccine, especially one that is a single shot and can be stored without special refrigeration requirements, will be a major contribution in getting the general public vaccinated sooner, both in the U.S. and around the world,” Phyllis Tien, MD, professor of medicine in the division of infectious diseases at the University of California, San Francisco, told Medscape Medical News.
“It’s great news. We have yet a third vaccine that is highly effective at preventing COVID, and even more effective at preventing severe COVID,” said Paul Goepfert, MD. It’s a “tremendous boon for our country and other countries as well.”
“This vaccine has also been shown to be effective against the B.1.351 strain that was first described in South Africa,” added Dr. Goepfert, director of the Alabama Vaccine Research Clinic and infectious disease specialist at the University of Alabama at Birmingham.
The EUA “is indeed exciting news,” Colleen Kraft, MD, associate chief medical officer at Emory University Hospital and associate professor at Emory University School of Medicine in Atlanta, said during a February 25 media briefing.
One recent concern centers on people aged 60 years and older. Documents the FDA released earlier this week suggest a lower efficacy, 42%, for the J&J immunization among people in this age group with certain relevant comorbidities. In contrast, without underlying conditions like heart disease or diabetes, efficacy in this cohort was 72%.
The more the merrier
The scope and urgency of the COVID-19 pandemic necessitates as many protective measures as possible, said Raj Shah, MD, geriatrician, and associate professor of family medicine and codirector of the Center for Community Health Equity at Rush University in Chicago.
“Trying to vaccinate as many individuals living in the United States to prevent the spread of COVID is such a big project that no one company or one vaccine was going to be able to ramp up fast enough on its own,” Dr. Shah told Medscape Medical News.“This has been the hope for us,” he added, “to get to multiple vaccines with slightly different properties that will provide more options.”
Experience with the J&J vaccine so far suggests reactions are less severe. “The nice thing about the Johnson and Johnson [vaccine] is that it definitely has less side effects,” Dr. Kraft said.
On the other hand, low-grade fever, chills, or fatigue after vaccination can be considered a positive because they can reflect how well the immune system is responding, she added.
One and done?
Single-dose administration could be more than a convenience — it could also help clinicians vaccinate members of underserved communities and rural locations, where returning for a second dose could be more difficult for some people.
“In a controlled setting, in a clinical trial, we do a lot to make sure people get all the treatment they need,” Dr. Shah said. “We’re not seeing it right now, but we’re always worried when we have more than one dose that has to be administered, that some people will drop off and not come back for the second vaccine.”
This group could include the needle-phobic, he added. “For them, having it done once alleviates a lot of the anxiety.”
Looking beyond the numbers
The phase 3 ENSEMBLE study of the J&J vaccine revealed a 72% efficacy for preventing moderate-to-severe COVID-19 among U.S. participants. In contrast, researchers reported 94% to 95% efficacy for the Pfizer/BioNTech and Moderna vaccines.
However, experts agreed that focusing solely on these numbers can miss more important points. For example, no participants who received the J&J vaccine in the phase 3 trial died from COVID-19-related illness. There were five such deaths in the placebo cohort.
“One of the things that these vaccines do very well is they minimize severe disease,” Dr. Kraft said. “As somebody that has spent an inordinate time in the hospital taking care of patients with severe disease from COVID, this is very much a welcome addition to our armamentarium to fight this virus.”
“If you can give something that prevents people from dying, that is a true path to normalcy,” Dr. Goepfert added.
More work to do
“The demand is strong from all groups right now. We just have to work on getting more vaccines out there,” Dr. Shah said.
“We are at a point in this country where we are getting better with the distribution of the vaccine,” he added, “but we are nowhere close to achieving that distribution of vaccines to get to everybody.”
Dr. Goepfert, Dr. Shah, and Dr. Kraft disclosed no relevant financial relationships. Dr. Tien received support from Johnson & Johnson to conduct the J&J COVID-19 vaccine trial in the San Francisco VA Health Care System.
A version of this article first appeared on Medscape.com.
J&J COVID-19 vaccine wins unanimous backing of FDA panel

The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.
The Food and Drug Administration (FDA) is expected to quickly provide an emergency use authorization (EUA) for the vaccine following the recommendation by the panel. The FDA’s Vaccines and Related Biological Products Advisory Committee voted 22-0 on this question: Based on the totality of scientific evidence available, do the benefits of the Johnson & Johnson COVID-19 Vaccine outweigh its risks for use in individuals 18 years of age and older?
The Johnson & Johnson vaccine is expected to offer more convenient dosing and be easier to distribute than the two rival products already available in the United States. Janssen’s vaccine is intended to be given in a single dose. In December, the FDA granted EUAs for the Pfizer/BioNTech and Moderna COVID-19 vaccines, which are each two-dose regimens.
Johnson & Johnson’s vaccine can be stored for at least 3 months at normal refrigerator temperatures of 2°C to 8°C (36°F to 46°F). Its shipping and storage fits into the existing medical supply infrastructure, the company said in its briefing materials for the FDA advisory committee meeting. In contrast, Pfizer’s vaccine is stored in ultracold freezers at temperatures between -80°C and -60°C (-112°F and -76°F), according to the Centers for Disease Control and Prevention. Moderna’s vaccine may be stored in a freezer between -25°C and -15°C (-13°F and 5°F).
But FDA advisers focused more in their deliberations on concerns about Janssen’s vaccine, including emerging reports of allergic reactions.
The advisers also discussed how patients might respond to the widely reported gap between Johnson & Johnson’s topline efficacy rates compared with rivals. The company’s initial unveiling last month of key results for its vaccine caused an initial wave of disappointment, with its overall efficacy against moderate-to-severe COVID-19 28 days postvaccination first reported at about 66% globally. By contrast, results for the Pfizer and Moderna vaccines suggest they have efficacy rates of 95% and 94%.
But in concluding, the advisers spoke of the Janssen vaccine as a much-needed tool to address the COVID-19 pandemic. The death toll in the United States attributed to the virus has reached 501,414, according to the World Health Organization.
“Despite the concerns that were raised during the discussion. I think what we have to keep in mind is that we’re still in the midst of this deadly pandemic,” said FDA adviser Archana Chatterjee, MD, PhD, from Rosalind Franklin University. “There is a shortage of vaccines that are currently authorized, and I think authorization of this vaccine will help meet the needs at the moment.”
The FDA is not bound to accept the recommendations of its advisers, but it often does so.
Anaphylaxis case
FDA advisers raised only a few questions for Johnson & Johnson and FDA staff ahead of their vote. The committee’s deliberations were less contentious and heated than had been during its December reviews of the Pfizer and Moderna vaccines. In those meetings, the panel voted 17-4, with one abstention, in favor of Pfizer’s vaccine and 20-0, with one abstention, on the Moderna vaccine.
“We are very comfortable now with the procedure, as well as the vaccines,” said Arnold Monto, MD, after the Feb. 26 vote on the Janssen vaccine. Dr. Monto, from the University of Michigan School of Public Health in Ann Arbor, has served as the chairman of the FDA panel through its review of all three COVID-19 vaccines.
Among the issues noted in the deliberations was the emergence of a concern about anaphylaxis with the vaccine.
This serious allergic reaction has been seen in people who have taken the Pfizer and Moderna vaccines. Before the week of the panel meeting, though, there had not been reports of anaphylaxis with the Johnson & Johnson vaccine, said Macaya Douoguih, MD, MPH, head of clinical development and medical affairs for Janssen/ Johnson & Johnson’s vaccines division.
However, on February 24, Johnson & Johnson received preliminary reports about two cases of severe allergic reaction from an open-label study in South Africa, with one of these being anaphylaxis, Dr. Douoguih said. The company will continue to closely monitor for these events as outlined in their pharmacovigilance plan, Dr. Douoguih said.
Federal health officials have sought to make clinicians aware of the rare risk for anaphylaxis with COVID vaccines, while reminding the public that this reaction can be managed.
The FDA had Tom Shimabukuro, MD, MPH, MBA, from the CDC, give an update on postmarketing surveillance for the Pfizer and Moderna vaccines as part of the review of the Johnson & Johnson application. Dr. Shimabukuro and CDC colleagues published a report in JAMA on February 14 that looked at an anaphylaxis case reported connected with COVID vaccines between December 14, 2020, and January 18, 2021.
The CDC identified 66 case reports received that met Brighton Collaboration case definition criteria for anaphylaxis (levels 1, 2, or 3): 47 following Pfizer/BioNTech vaccine, for a reporting rate of 4.7 cases/million doses administered, and 19 following Moderna vaccine, for a reporting rate of 2.5 cases/million doses administered, Dr. Shimabukuro and CDC colleagues wrote.
The CDC has published materials to help clinicians prepare for the possibility of this rare event, Dr. Shimabukuro told the FDA advisers.
“The take-home message here is that these are rare events and anaphylaxis, although clinically serious, is treatable,” Dr. Shimabukuro said.
At the conclusion of the meeting, FDA panelist Patrick Moore, MD, MPH, from the University of Pittsburgh in Pennsylvania, stressed the need to convey to the public that the COVID vaccines appear so far to be safe. Many people earlier had doubts about how the FDA could both safely and quickly review the applications for EUAs for these products.
“As of February 26, things are looking good. That could change tomorrow,” Dr. Moore said. But “this whole EUA process does seem to have worked, despite my own personal concerns about it.”
No second-class vaccines
The Johnson & Johnson vaccine, known as Ad26.COV2.S, is composed of a recombinant, replication-incompetent human adenovirus type 26 (Ad26) vector. It’s intended to encode a stabilized form of SARS-CoV-2 spike (S) protein. The Pfizer and Moderna vaccines use a different mechanism. They rely on mRNA.
The FDA advisers also discussed how patients might respond to the widely reported gap between Janssen’s topline efficacy rates compared with rivals. They urged against people parsing study details too finely and seeking to pick and choose their shots.
“It’s important that people do not think that one vaccine is better than another,” said FDA adviser H. Cody Meissner, MD, from Tufts University School of Medicine in Boston.
Dr. Monto agreed, noting that many people in the United States are still waiting for their turn to get COVID vaccines because of the limited early supply.
Trying to game the system to get one vaccine instead of another would not be wise. “In this environment, whatever you can get, get,” Dr. Monto said.
During an open public hearing, Sarah Christopherson, policy advocacy director of the National Women’s Health Network, said that press reports are fueling a damaging impression in the public that there are “first and second-class” vaccines.
“That has the potential to exacerbate existing mistrust” in vaccines, she said. “Public health authorities must address these perceptions head on.”
She urged against attempts to compare the Janssen vaccine to others, noting the potential effects of emerging variants of the virus.
“It’s difficult to make an apples-to-apples comparison between vaccines,” she said.
Johnson & Johnson’s efficacy results, which are lower than those of the mRNA vaccines, may be a reflection of the ways in which SARS-Co-V-2 is mutating and thus becoming more of a threat, according to the company. A key study of the new vaccine, involving about 44,000 people, coincided with the emergence of new SARS-CoV-2 variants, which were emerging in some of the countries where the pivotal COV3001 study was being conducted, the company said.
At least 14 days after vaccination, the Johnson & Johnson COVID vaccine efficacy (95% confidence interval) was 72.0% (58.2, 81.7) in the United States, 68.1% (48.8, 80.7) in Brazil, and 64.0% (41.2, 78.7) in South Africa.
Weakened standards?
Several researchers called on the FDA to maintain a critical attitude when assessing Johnson & Johnson’s application for the EUA, warning of a potential for a permanent erosion of agency rules due to hasty action on COVID vaccines.
They raised concerns about the FDA demanding too little in terms of follow-up studies on COVID vaccines and with persisting murkiness resulting in attempts to determine how well these treatments work beyond the initial study period.
“I worry about FDA lowering its approval standards,” said Peter Doshi, PhD, from The BMJ and a faculty member at the University of Maryland School of Medicine in Baltimore, during an open public hearing at the meeting.
“There’s a real urgency to stand back right now and look at the forest here, as well as the trees, and I urge the committee to consider the effects FDA decisions may have on the entire regulatory approval process,” Dr. Doshi said.
Dr. Doshi asked why Johnson & Johnson did not seek a standard full approval — a biologics license application (BLA) — instead of aiming for the lower bar of an EUA. The FDA already has allowed wide distribution of the Pfizer/BioNTech and Moderna vaccines through EUAs. That removes the sense of urgency that FDA faced last year in his view.
The FDA’s June 2020 guidance on the development of COVID vaccines had asked drugmakers to plan on following participants in COVID vaccine trials for “ideally at least one to two years.” Yet people who got placebo in Moderna and Pfizer trials already are being vaccinated, Dr. Doshi said. And Johnson & Johnson said in its presentation to the FDA that if the Ad26.COV2.S vaccine were granted an EUA, the COV3001 study design would be amended to “facilitate cross-over of placebo participants in all participating countries to receive one dose of active study vaccine as fast as operationally feasible.”
“I’m nervous about the prospect of there never being a COVID vaccine that meets the FDA’s approval standard” for a BLA instead of the more limited EUA, Dr. Doshi said.
Diana Zuckerman, PhD, president of the nonprofit National Center for Health Research, noted that the FDA’s subsequent guidance tailored for EUAs for COVID vaccines “drastically shortened” the follow-up time to a median of 2 months. Dr. Zuckerman said that a crossover design would be “a reasonable compromise, but only if the placebo group has at least 6 months of data.” Dr. Zuckerman opened her remarks in the open public hearing by saying she had inherited Johnson & Johnson stock, so was speaking at the meeting against her own financial interest.
“As soon as a vaccine is authorized, we start losing the placebo group. If FDA lets that happen, that’s a huge loss for public health and a huge loss of information about how we can all stay safe,” Dr. Zuckerman said.
A version of this article first appeared on Medscape.com.