User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Muckle-Wells Syndrome in the Setting of Basal Cell Nevus Syndrome
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
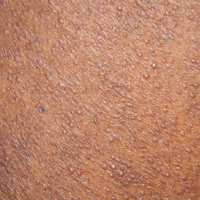
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
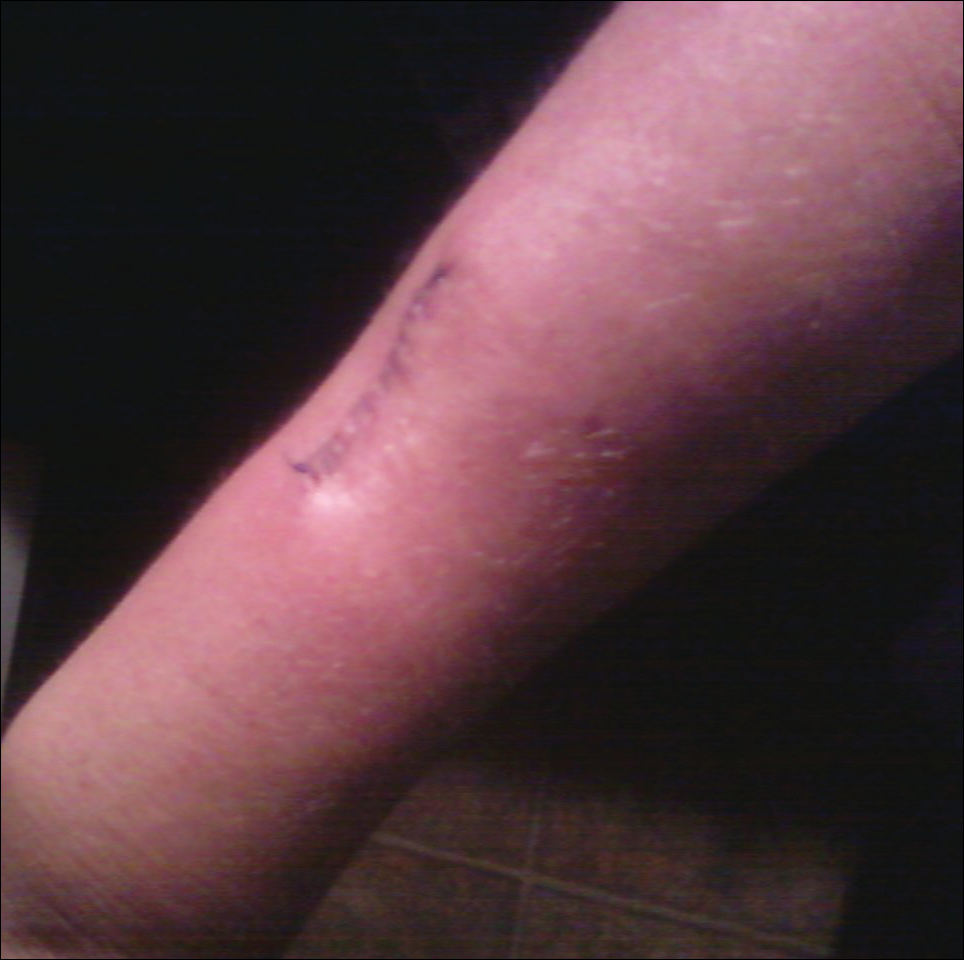

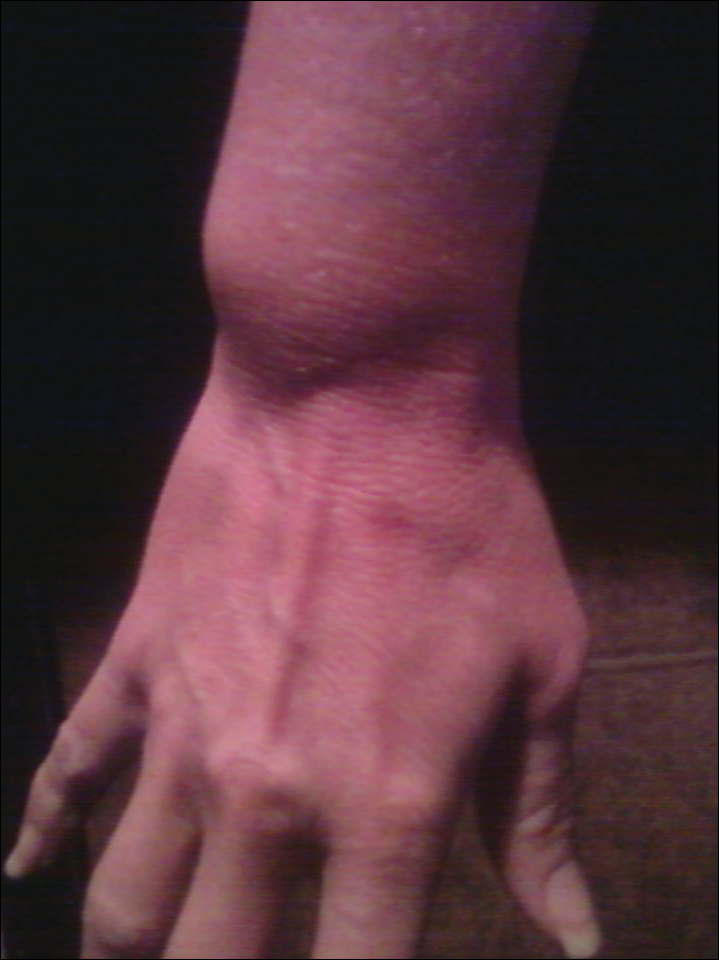
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
Muckle-Wells syndrome (MWS) was first described in 1962 and is part of a broad category of hereditary periodic fever syndromes that include the autoinflammatory syndromes and the cryopyrin-associated periodic syndromes (CAPSs). Unlike autoimmune diseases, autoinflammatory syndromes are not associated with antigen-specific T-cell responses or high titers of autoantibodies but are related to disorders of the innate immune system. Basal cell nevus syndrome (BCNS), or Gorlin syndrome, is a rare genodermatosis inherited in an autosomal-dominant fashion that is characterized by a broad range of anomalies. Most notable is the early and strong predisposition to develop several to hundreds of basal cell carcinomas (BCCs). Classic clinical features of MWS and a thorough history and physical examination can assist in the diagnosis of this rare entity.
Case Report
A 35-year-old woman with a history of BCNS, which had been diagnosed at 24 years of age based on the presence of more than 2 BCCs and a family history of BCNS in her mother, presented with intermittent pruritic urticaria on the chest and back, episodic fevers, associated joint pain and swelling that worsened several hours after exercise, headache, conjunctivitis, blurred vision, and severe debilitating fatigue that had been present since childhood. The symptoms had progressively worsened with age and symptom-free intervals became shorter. She was diagnosed by her rheumatologist with biopsy-proven MWS and a positive NLRP3 (NLR family pyrin domain containing 3) gene mutation at 29 years of age. She was treated unsuccessfully with prednisone and antihistamines and entered a trial with anakinra. She showed improvement for 2 weeks but developed severe swelling and erythema at the injection sites at week 3, along with large leathery patches on the legs and difficulty ambulating.
The patient subsequently underwent excision of her BCCs and reported each site became erythematous, edematous, warm, and painful 6 hours after excision, which lasted for hours to days (Figures 1–3). After the first excision on the right forearm, she was seen in the emergency department, started on intravenous antibiotics and prednisone, and kept overnight in the hospital. She was discharged the following day and the edema in the right forearm subsided over several days. Bacterial culture and laboratory evaluation for infection were negative after the first excision on the right forearm. Because of the symptoms she experienced following this excision, she was referred to the plastic surgery department for excision followed by postoperative monitoring in the hospital. The patient continued to undergo excisions for BCCs and developed more severe symptoms including erythema, edema, warmth, and tenderness at the surrounding sites. Once again, the excision sites were cultured and laboratory work to rule out infection was ordered with a negative result. After several excisions and subsequent clinical findings, the patients’ symptoms were deemed consistent with MWS and not a result of infectious etiology. A diagnosis of MWS and BCNS with exacerbation of MWS with surgical procedures was made.
The patient has continued therapy with rilonacept for MWS, which is managed by her rheumatologist. She has tolerated rilonacept without adverse effects and has experienced a reduction in symptoms that has enhanced her quality of life and allows for further treatment of her BCNS. Her dermatologist (J.W.L.) has been treating her BCCs with vismodegib, but treatment has been sporadic due to muscle cramping after 7 days of therapy. She reported subjective improvement to her dermatologist and has tried alternating 7 days on and 7 days off vismodegib. The muscle cramping still has limited her treatment with this regimen, and she is currently on a trial of 3 days on, 4 days off per week.
Comment
Classification and Clinical Presentation
The hereditary periodic fever syndromes include the autoinflammatory syndromes and the CAPSs. The autoinflammatory syndromes include familial Mediterranean fever, hyperimmunoglobulinemia D with periodic fever syndrome, and tumor necrosis factor receptor–associated periodic syndrome. The CAPSs are similar but distinct and include familial cold autoinflammatory syndrome, neonatal-onset multisystem inflammatory disease (also known as chronic infantile neurologic cutaneous and articular syndrome, or cutaneous articular syndrome) and MWS.1,2
Cryopyrin-associated periodic syndromes are rare inherited diseases that result from mutations in the NLRP3 gene. There is a gain-of-function mutation on the NLRP3 gene located on the long arm of chromosome 1 at position 44, which codes for cryopyrin. An NLRP3 gene mutation causes cryopyrin to become hyperactive, leading to the formation of an inflammasome, which is a group of cryopyrin molecules. Inflammasomes, along with other proteins, activate caspase 1 to produce excess IL-1β, leading to persistent inflammatory symptoms.3 IL-1β is one of the key mediators of the body’s response to microbial invasion, inflammation, immunologic reactions, and tissue injury. It affects a large range of cells and organs. Although IL-1β production is critical for the control of pathogenic infections, excessive cytokine production is harmful to the host and can even be fatal.3,4
Cryopyrin-associated periodic syndromes encompass a disease continuum. The 3 distinct entities share many overlapping features as well as unique and distinguishing characteristics. Familial cold autoinflammatory syndrome is the mildest phenotype and is inherited in an autosomal-dominant fashion. It is characterized by a chronic urticarial eruption that starts early in infancy or childhood. The distribution of the cutaneous eruption is widespread and favors the arms and legs over the face and trunk. A low-grade fever often is seen along with musculoskeletal concerns of arthralgia and pain. Other commonly reported symptoms include conjunctivitis, myalgia, fatigue, and headache. Neurologic symptoms can include headaches. Symptoms usually begin 1 to 2 hours after cold exposure and last less than 24 hours.5-8
Neonatal-onset multisystem inflammatory disease is the most severe phenotype and occurs sporadically. Continuous symptoms and flares are characteristic and the length of the flare can vary from minutes to days. The cutaneous eruption favors the face, trunk, arms, and legs, and varies in intensity, beginning in infancy or childhood. Fever may be intermittent, mild, or absent. Rheumatologic manifestations include arthralgia and swelling, with approximately one-third of patients experiencing severe disabling arthropathy that causes gross joint deformity. Ocular findings include conjunctivitis, uveitis, papilledema, and even blindness. Neurologic sequelae include headaches, sensorineural hearing loss, and aseptic meningitis. Amyloidosis has been seen as a late complication.5,8
Muckle-Wells syndrome is a rare hereditary inflammatory disorder. It has no ethnic predisposition and is mostly inherited in an autosomal-dominant fashion. Classically, the condition is characterized by recurrent urticaria beginning at birth with intermittent episodic fever and malaise. The eruption has a predilection for the face, trunk, arms, and legs, which is similar to neonatal-onset multisystem inflammatory disease. Associated myalgia and arthralgia are common as well as ocular findings of conjunctivitis and episcleritis. Neurologic manifestations include headache and progressive sensorineural hearing loss in 60% to 70% of patients.6 Abdominal pain may be seen along with rare serositis in MWS but is rare in the other CAPSs. Amyloidosis caused by chronic inflammation is the most serious complication of MWS and is seen in approximately one-third of patients, manifesting as proteinuria followed by renal impairment. Symptoms of MWS may occur daily but vary individually, are broad in intensity and duration, and can last 1 to 2 days before resolving spontaneously. The symptoms can result from metabolic stressors including cold, stress, and exercise, as well as microbial pathogens. Leukocytosis and increased acute-phase reactants are observed during episodes of inflammation.4,6,8
Histopathology
Mild phenotypic variability exists between individuals, and many of the symptoms overlap in CAPSs. Although CAPSs display several distinguishing clinical characteristics, interestingly they share the same histopathological features regardless of the syndrome. The typical histopathological finding is a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. Vasodilation and dermal edema also may be seen. These histopathological findings contrast with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria. Similar histopathologic findings have been seen in other neutrophilic urticarial dermatoses such as Schnitzler syndrome.4,6
Differential
The differential diagnoses for CAPSs include Schnitzler syndrome, cold urticaria, systemic-onset juvenile idiopathic arthritis/adult-onset Still disease, and deficiency in IL-1ra. It is important to consider these differential diagnoses for management and treatment options.
Management
The discovery of the NLRP3 gene mutation as well as an understanding of IL-1 biology has led to targeted therapy for these syndromes. Cryopyrin-associated periodic syndromes are mediated by IL-1β with an in vivo rate 5 times higher than in healthy patients.4 The blockade of IL-1β results in complete resolution of symptoms.
In the last several years, anakinra, rilonacept, and canakinumab have shown efficacy in targeting IL-1β as receptor antagonists. Anakinra is a short-acting recombinant IL-1ra with a half-life of 4 to 6 hours. This short half-life requires daily injections and the most common adverse events included injection-site reaction and upper respiratory tract infection.2,4 Rilonacept is a dimeric fusion protein that contains binding regions for the type 1 receptor and the IL-1 receptor accessory protein and is fused to the fragment, crystallizable (Fc) portion of human IgG1. Rilonacept is long acting with a circulating half-life of 8.6 days and offers patients ease of dosing with weekly subcutaneous injections. Rilonacept generally is well tolerated, with the most frequent adverse effects being injection-site reaction, upper respiratory tract infection, headache, arthralgia, and diarrhea.2,7
The newest of the treatments for patients with CAPS is canakinumab. It is a fully human IL-1β monoclonal antibody that is specific for IL-1β and not other members of the IL-1 family. It has a mean half-life of 26 days and is dosed subcutaneously once every 8 weeks. The most common adverse effects include nasopharyngitis, rhinitis, nausea, diarrhea, and vertigo.4 In one study, most patients did not report injection-site reactions.7 Studies also are underway on VX-765, a caspace-1 targeted therapy that acts upstream in the IL-1β pathway. Treatment with anakinra, rilonacept, and canakinumab generally offers rapid and sustained remission in the majority of MWS patients and helps prevent the development of systemic amyloidosis and lessens the potential for end organ damage.2,7
MWS and BCNS
Our patient had an unusual presentation of MWS complicated by BCNS, another rare autosomal-dominant inherited genodermatosis. In an extensive review of PubMed articles indexed for MEDLINE using the search terms Muckle-Wells syndrome and basal cell nevus syndrome, no association was identified between MWS and BCNS. Basal cell nevus syndrome is linked to PTCH1 (patched 1) gene mutation with an incidence of 1:150,000 in the United States and Europe and is characterized by a broad range of anomalies including skeletal abnormalities, ectopic calcification, odontogenic keratocysts, facial dysmorphism with macrocephaly, palmoplantar pits, and numerous tumors. Most notable is the early and strong predisposition to develop several to hundreds of BCCs.9
Conclusion
Muckle-Wells syndrome may go undiagnosed for many years or may be misdiagnosed as refractory urticaria, as in our patient. It is important to include periodic fever syndromes in the differential diagnosis of refractory urticaria with episodic fever to diagnose these cases of MWS earlier.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
- Kagami S, Saeki H, Kuwano Y, et al. A probable case of Muckle-Wells syndrome. J Dermatol. 2006;2:118-121.
- Kanazawa N, Furukawa F. Autoinflammatory syndromes with a dermatological perspective. J Dermatol. 2007;34:601-618.
- Martinon F, Tschopp J. Inflammatory caspases: linking an intracellular innate immune system to autoinflammatory diseases. Cell. 2004;117:561-574.
- Mueller SM, Itin P, Haeusermann P. Muckle-Wells syndrome effectively treated with canakinumab: is the recommended dosing schedule mandatory? Dermatology. 2011;223:113-118.
- Neven B, Prieur A, Quartier dit Maire P. Cryopyrinopathies: update on pathogenesis and treatment. Nat Clin Pract Rheumatol. 2008;4:481-489.
- Newell L, August S, Foria V, et al. Lifelong urticaria and multiple unexplained systemic symptoms. Clin Exp Dermatol. 2011;36:431-433.
- Yu JR, Kieron KS. Cryopyrin-associated periodic syndrome: an update on diagnosis and treatment response. Curr Allergy Asthma Rep. 2011;11:12-20.
- Bolognia JL, Jorizzo JL, Rapini RP, et al, eds. Dermatology. 2nd ed. Barcelona, Spain: Mosby Elsevier; 2008. 9. Göppner D, Leverkus M. Basal cell carcinoma: from the molecular understanding of the pathogenesis to targeted therapy of progressive disease. J Skin Cancer. 2011;2011:650258.
Practice Points
- An urticarial rash occurring in childhood with symptoms of fever, joint pain, and swelling along with visual symptoms should prompt consideration of a cryopyrin-associated periodic syndrome.
- Histopathology shows a dermal neutrophilic infiltrate that tends to be perivascular and also may be perieccrine. This atypical urticaria contrasts with the typical lymphocytic and eosinophilic infiltrate seen in classic urticaria.
Magnification for the Dermatologic Surgeon
Dermatologic surgeons are susceptible to work-related ailments given the nature of their working posture, the most common of which are pain and stiffness in the neck, shoulders, and lower back, as well as headaches.1,2 Awkward posture and positioning, for the sake of getting a better view of the task at hand, puts the surgeon in ergonomically disagreeable positions. Because the prime working years for a dermatologic surgeon tend to coincide with the age of presbyopia onset, magnification may help reduce and thwart musculoskeletal problems and eye strain. Indeed, a multitude of surgical specialties and dentists use intraoperative magnification.3 Knowledge and use of available magnification options can be a key addition to the dermatologic surgeon’s armamentarium. We discuss the need for magnification and review magnification devices that are readily available to the dermatologic surgeon. Table 1 presents a summary of all magnification options discussed.
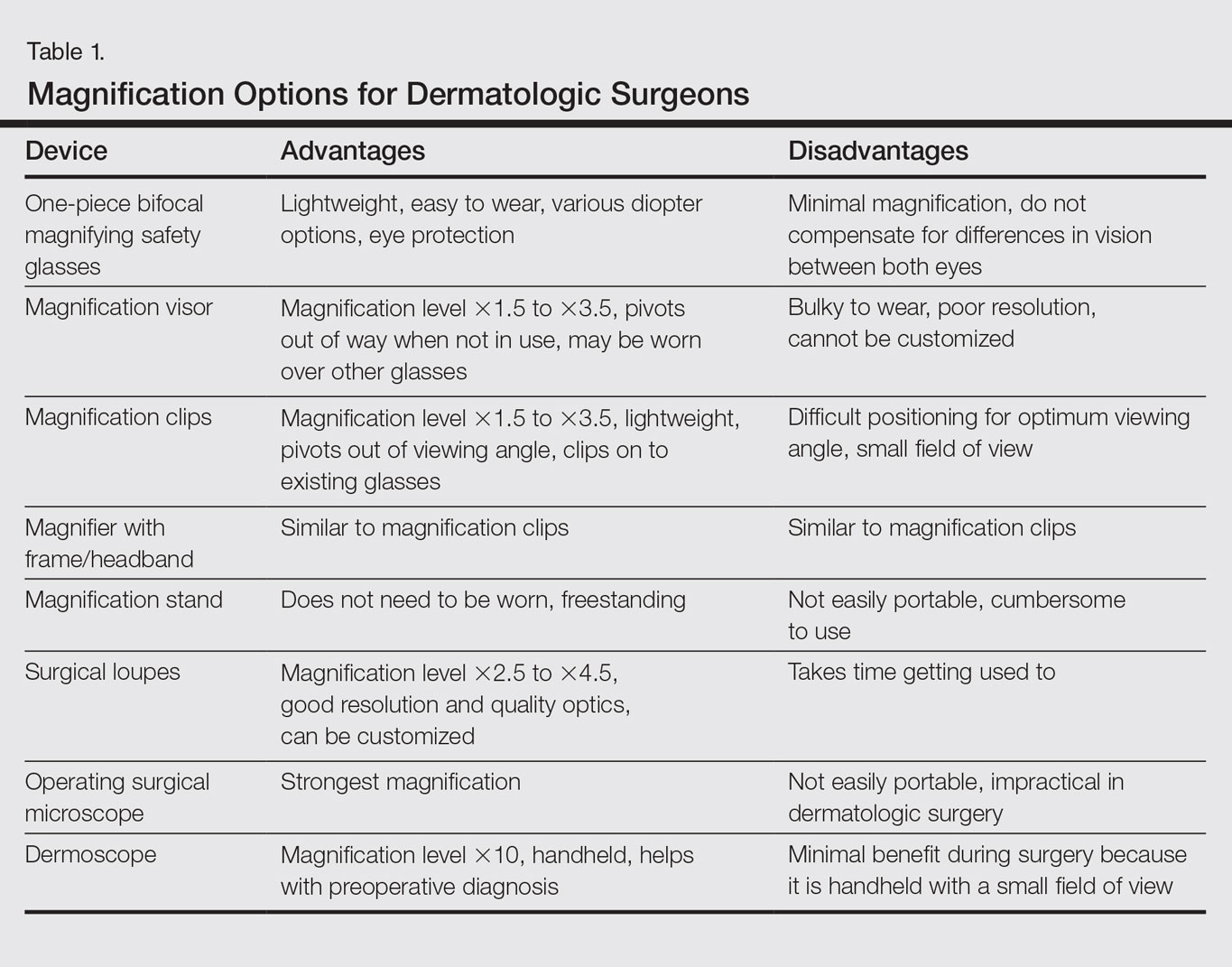
Need for Magnification
Presbyopia is a condition of aging in which one loses the ability to accommodate and focus at near distances. The estimated prevalence of presbyopia in North America is 83%, typically with onset by 45 years of age.4 Individuals with presbyopia often hold objects farther away from their eyes to bring them into focus, causing eye strain, headaches, and musculoskeletal injury.
Use of intraoperative magnification allows for enhanced visualization of fine anatomic details and precise suture placement for the surgeon with or without presbyopia. Higher magnification produces a larger image; however, it also reduces field of view and depth of field (ie, the amount of depth that stays in focus without repositioning). The resolution and quality of the image are dependent on the optical properties of the lens system. The ideal optic system is surgeon dependent and involves a combination of magnification level that will not result in dramatic loss of view and depth of field, while maintaining crispness and quality of image.
Intraoperative magnification yields ergonomic benefits by promoting a safer neck flexion angle by increasing the working distance to a more ideal position (Figure). In doing so, it improves posture and minimizes eye and musculoskeletal strain secondary to awkward positioning and presbyopia.1,5 Stationary working position and neck flexion and rotation with precise and repetitive tasks are risk factors for strain and injuries that dermatologic surgeons often encounter.1 Magnification devices are tools that the dermatologic surgeon can utilize for a more ergonomically sound practice. Indeed, magnification has been shown to improve posture in the dental literature, a specialty with similar occupational risk factors to dermatologic surgery.6-8 Ergonomic practice reduces occupational injuries and improves work quality and productivity, thereby having a favorable effect on both the patient and the physician.
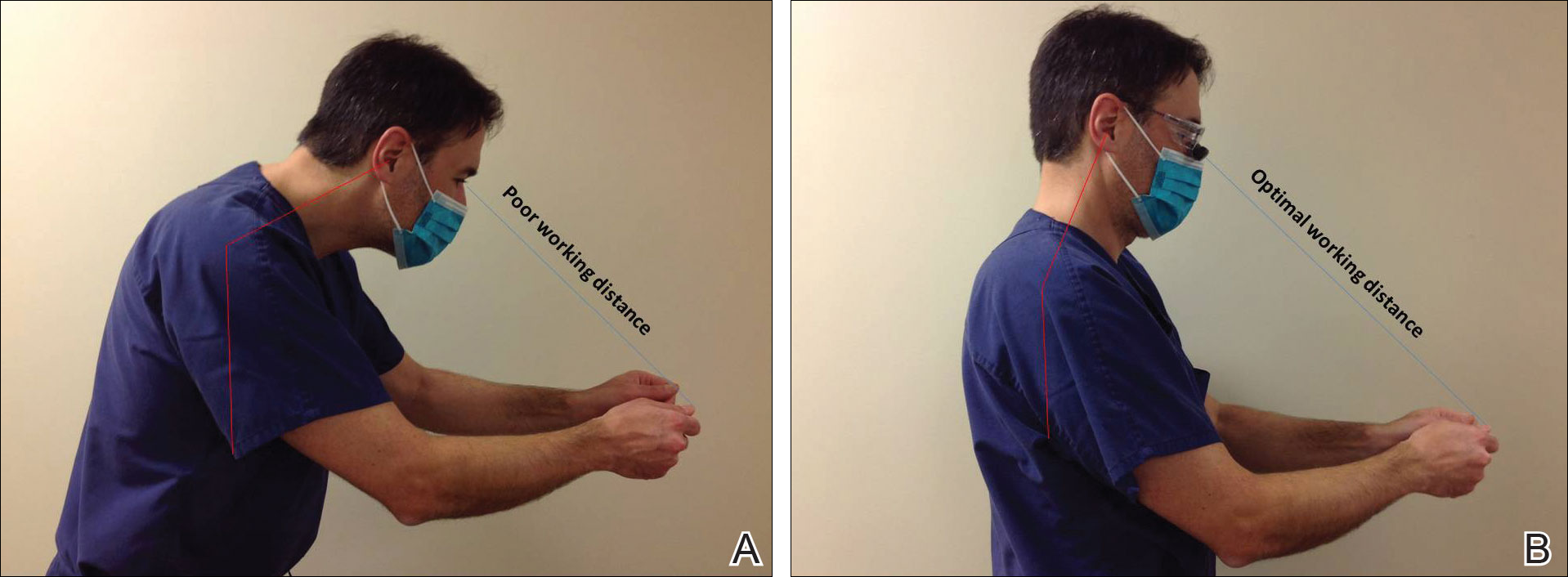
Improved Outcomes With Magnification
There are many examples of improved surgical quality and outcomes with magnification in other specialties. Hart and Hall5 illustrated the advantage of magnification in laceration repairs in the emergency department. In one study, increased magnification resulted in a substantial decrease in positive surgical margin rates in open radical retropubic prostatectomy.9 Schoeffl et al10 demonstrated that the microsurgical success of fine surgical procedures was directly related to optical magnification strength when comparing the unaided eye, surgical loupes, and the operating microscope. The dental literature also has numerous examples of magnification producing improved quality dentistry.11-13 Although magnification is not a novel concept to dermatologic surgery, little has been written about its use in the dermatologic surgery literature.
Magnification Options
One-Piece Bifocal Magnifying Safety Glasses
Bifocal magnifying safety glasses are polycarbonate safety glasses made with lenses in which the lower half is a magnifying lens. They are available in +1.5, +2.0, +2.5, and +3.0 diopter strengths. The total magnification power is calculated as follows: (diopter/4) + 1. The glasses are lightweight, easy to wear, inexpensive, and protect the eyes; however, they provide minimal magnification and do not compensate for differences in vision between both eyes.
Magnification Visor
The magnification visor is a headband visor with magnification lenses. It comes in various levels of magnification ranging from ×1.5 to ×3.5. It can be worn over prescription or safety glasses, may be pivoted out of the way when not in use, and is inexpensive. Conversely, it may be bulky to wear, cannot be customized, and does not offer the best resolution.
Magnification Clips
Magnification clips are hard-coated magnifying lens plates that fasten to eyeglass frames and range in level of magnification from ×1.5 to ×3.5. They can be pivoted out of the viewing angle, are lightweight, and are inexpensive; however, positioning may be difficult for ideal working distance and viewing angle.
Magnifier With Frame/Headband
The magnifier with frame is similar to magnification clips, but the magnification lens plate comes with a frame. It can be used with or without glasses and comes in magnification levels of ×1.5 to ×3.5. It is light, inexpensive, and may be pivoted out of sight, but similar to magnification clips, positioning for the right viewing angle and working distance may be difficult.
The magnifier with headband is essentially the same as the magnifier with frame. The only difference is the magnification plate is attached to a headband as opposed to a frame. It has similar benefits and limitations as the magnifier with frame.
Magnification Stand
The magnification stand comes as a large magnification lens with a flexible arm attached to a stand. It is a basic magnification tool and does not need to be worn; however, the stand is not easily portable and may be cumbersome to use.
Surgical Loupes
Surgical loupes are a robust magnification choice and the mainstay in magnification for the dermatologic surgeon. Loupes have proven to have comparable results in some procedures to the powerful operating surgical microscope.14-17 Factors to consider with loupes include brand, design, lens, magnification, resolution, optimal working distance, field depth, and declination angle.18
The 2 surgical loupe designs—flip-up loupes and through-the-lens loupes—differ in the mounting of the optic lenses on safety glasses. Flip-up loupes have the optics mounted to the bridge of the frame, whereas through-the-lens loupes are fixed in the lenses.
There are 3 different optical systems for surgical loupe magnification: simple, compound, and prismatic. Simple lenses consist of one pair of positive meniscus lenses similar to reading glasses. Compound lenses are made of 2 magnification lenses. Prismatic lenses magnify using a prism that folds and lengthens the light path.19,20
Loupes range in magnification level from ×2.5 to ×4.5. Compared to other magnification modalities, they can be customized and offer better resolution with quality magnification. Additionally, loupes can be fitted with a light source; however, they are expensive and surgeons need time to get used to the increased magnification as well as wearing the loupes.
There are advantages and disadvantages to the different loupe designs (Table 2). Flip-up loupes are more versatile, allowing for use on various safety glasses. They can be flipped out of view, and the declination angle may be altered; however, flip-up loupes have a narrower field of view and are heavier and bulkier than through-the-lens loupes. Through-the-lens loupes are lighter and have a larger field of view, as the optics are closer to the eye. They are customized to the declination angle and working distance of the surgeon. Conversely, through-the-lens loupes are more expensive and cannot be adjusted or moved from the line of vision.

Operating Surgical Microscope
The operating surgical microscope is not practical in the dermatologic surgeon’s practice. It is expensive and provides unnecessarily powerful magnification for dermatologic surgery. This tool usually is used in the operating room for suturing nerves and vessels with sutures sized 8-0 and smaller. Most skin procedures require size 6-0 and larger sutures.
Dermoscope
Dermoscopy, also known as epiluminescence microscopy, is a technique utilizing a handheld device made up of polarized light and a ×10 magnifying lens to evaluate skin lesions. In skilled hands, dermoscopy allows for the examination of characteristic patterns and morphologic features of skin lesions to enhance the clinician’s diagnostic accuracy.21 It may aid the dermatologic surgeon in identifying the surgical margins of difficult-to-define skin cancers. It is small and mobile; however, it has minimal benefit to the dermatologic surgeon during surgery because it is handheld and has a small field of view.
Conclusion
Good ergonomic practices facilitate a healthier and prolonged career for the dermatologic surgeon. When used properly, magnification devices can be a beneficial adjunct to the dermatologic surgeon by promoting better posture, preventing eyestrain, and providing enhanced visualization of the operating field and instruments. Use of magnification devices has been demonstrated to improve patient outcomes in other specialties. There are opportunities for further research specific to magnification improving dermatologic surgery outcomes given the high level of precision and accuracy needed for Mohs micrographic surgery, wound reconstruction, nail surgery, and hair transplantation.
- Liang CA, Levine VJ, Dusza SW, et al. Musculoskeletal disorders and ergonomics in dermatologic surgery: a survey of Mohs surgeons in 2010. Dermatol Surg. 2012;38:240-248.
- Esser AC, Koshy JG, Randle HW. Ergonomics in office-based surgery: a survey-guided observational study. Dermatol Surg. 2007;33:1304-1313; discussion, 1313-1314.
- Jarrett PM. Intraoperative magnification: who uses it? Microsurgery. 2004;24:420-422.
- Holden BA, Fricke TR, Ho SM, et al. Global vision impairment due to uncorrected presbyopia. Arch Ophthalmol. 2008;126:1731-1739.
- Hart RG, Hall J. The value of loupe magnification: an underused tool in emergency medicine. Am J Emerg Med. 2007;25:704-707.
- Branson BG, Bray KK, Gadbury-Amyot C, et al. Effect of magnification lenses on student operator posture. J Dent Educ. 2004;68:384-389.
- Maillet JP, Millar AM, Burke JM, et al. Effect of magnification loupes on dental hygiene student posture. J Dent Educ. 2008;72:33-44.
- Branson BG, Black MA, Simmer-Beck M. Changes in posture: a case study of a dental hygienist’s use of magnification loupes. Work. 2010;35:467-476.
- Magera JS Jr, Inman BA, Slezak JM, et al. Increased optical magnification from 2.5× to 4.3× with technical modification lowers the positive margin rate in open radical retropubic prostatectomy [published online November 13, 2007].J Urol. 2008;179:130-135.
- Schoeffl H, Lazzeri D, Schnelzer R, et al. Optical magnification should be mandatory for microsurgery: scientific basis and clinical data contributing to quality assurance. Arch Plast Surg. 2013;40:104-108.
- Taschieri S, Del Fabbro M, Testori T, et al. Endodontic surgery using 2 different magnification devices: preliminary results of a randomized controlled study. J Oral Maxillofac Surg. 2006;64:235-242.
- Christensen GJ. Magnification in dentistry: useful tool or another gimmick? J Am Dent Assoc. 2003;134:1647-1650.
- Syme SE, Fried JL, Strassler HE. Enhanced visualization using magnification systems. J Dent Hyg. 1997;71:202-206.
- Pieptu D, Luchian S. Loupes-only microsurgery. Microsurgery. 2003;23:181-188.
- Shenaq SM, Klebuc MJ, Vargo D. Free-tissue transfer with the aid of loupe magnification: experience with 251 procedures. Plast Reconstr Surg. 1995;95:261-269.
- Serletti JM, Deuber MA, Guidera PM, et al. Comparison of the operating microscope and loupes for free microvascular tissue transfer. Plast Reconstr Surg. 1995;95:270-276.
- Ross DA, Ariyan S, Restifo R, et al. Use of the operating microscope and loupes for head and neck free microvascular tissue transfer: a retrospective comparison. Arch Otolaryngol Head Neck Surg. 2003;129:189-193.
- Mungadi IA. Refinement on surgical technique: role of magnification. J Surg Tech Case Rep. 2010;2:1-2.
- Stanbury SJ, Elfar J. The use of surgical loupes in microsurgery. J Hand Surg Am. 2011;36:154-156.
- Baker JM, Meals RA. A practical guide to surgical loupes. J Hand Surg Am. 1997;22:967-974.
- Campos-do-Carmo G, Ramos-e-Silva M. Dermoscopy: basic concepts. Int J Dermatol. 2008;47:712-719.
Dermatologic surgeons are susceptible to work-related ailments given the nature of their working posture, the most common of which are pain and stiffness in the neck, shoulders, and lower back, as well as headaches.1,2 Awkward posture and positioning, for the sake of getting a better view of the task at hand, puts the surgeon in ergonomically disagreeable positions. Because the prime working years for a dermatologic surgeon tend to coincide with the age of presbyopia onset, magnification may help reduce and thwart musculoskeletal problems and eye strain. Indeed, a multitude of surgical specialties and dentists use intraoperative magnification.3 Knowledge and use of available magnification options can be a key addition to the dermatologic surgeon’s armamentarium. We discuss the need for magnification and review magnification devices that are readily available to the dermatologic surgeon. Table 1 presents a summary of all magnification options discussed.
Need for Magnification
Presbyopia is a condition of aging in which one loses the ability to accommodate and focus at near distances. The estimated prevalence of presbyopia in North America is 83%, typically with onset by 45 years of age.4 Individuals with presbyopia often hold objects farther away from their eyes to bring them into focus, causing eye strain, headaches, and musculoskeletal injury.
Use of intraoperative magnification allows for enhanced visualization of fine anatomic details and precise suture placement for the surgeon with or without presbyopia. Higher magnification produces a larger image; however, it also reduces field of view and depth of field (ie, the amount of depth that stays in focus without repositioning). The resolution and quality of the image are dependent on the optical properties of the lens system. The ideal optic system is surgeon dependent and involves a combination of magnification level that will not result in dramatic loss of view and depth of field, while maintaining crispness and quality of image.
Intraoperative magnification yields ergonomic benefits by promoting a safer neck flexion angle by increasing the working distance to a more ideal position (Figure). In doing so, it improves posture and minimizes eye and musculoskeletal strain secondary to awkward positioning and presbyopia.1,5 Stationary working position and neck flexion and rotation with precise and repetitive tasks are risk factors for strain and injuries that dermatologic surgeons often encounter.1 Magnification devices are tools that the dermatologic surgeon can utilize for a more ergonomically sound practice. Indeed, magnification has been shown to improve posture in the dental literature, a specialty with similar occupational risk factors to dermatologic surgery.6-8 Ergonomic practice reduces occupational injuries and improves work quality and productivity, thereby having a favorable effect on both the patient and the physician.
Improved Outcomes With Magnification
There are many examples of improved surgical quality and outcomes with magnification in other specialties. Hart and Hall5 illustrated the advantage of magnification in laceration repairs in the emergency department. In one study, increased magnification resulted in a substantial decrease in positive surgical margin rates in open radical retropubic prostatectomy.9 Schoeffl et al10 demonstrated that the microsurgical success of fine surgical procedures was directly related to optical magnification strength when comparing the unaided eye, surgical loupes, and the operating microscope. The dental literature also has numerous examples of magnification producing improved quality dentistry.11-13 Although magnification is not a novel concept to dermatologic surgery, little has been written about its use in the dermatologic surgery literature.
Magnification Options
One-Piece Bifocal Magnifying Safety Glasses
Bifocal magnifying safety glasses are polycarbonate safety glasses made with lenses in which the lower half is a magnifying lens. They are available in +1.5, +2.0, +2.5, and +3.0 diopter strengths. The total magnification power is calculated as follows: (diopter/4) + 1. The glasses are lightweight, easy to wear, inexpensive, and protect the eyes; however, they provide minimal magnification and do not compensate for differences in vision between both eyes.
Magnification Visor
The magnification visor is a headband visor with magnification lenses. It comes in various levels of magnification ranging from ×1.5 to ×3.5. It can be worn over prescription or safety glasses, may be pivoted out of the way when not in use, and is inexpensive. Conversely, it may be bulky to wear, cannot be customized, and does not offer the best resolution.
Magnification Clips
Magnification clips are hard-coated magnifying lens plates that fasten to eyeglass frames and range in level of magnification from ×1.5 to ×3.5. They can be pivoted out of the viewing angle, are lightweight, and are inexpensive; however, positioning may be difficult for ideal working distance and viewing angle.
Magnifier With Frame/Headband
The magnifier with frame is similar to magnification clips, but the magnification lens plate comes with a frame. It can be used with or without glasses and comes in magnification levels of ×1.5 to ×3.5. It is light, inexpensive, and may be pivoted out of sight, but similar to magnification clips, positioning for the right viewing angle and working distance may be difficult.
The magnifier with headband is essentially the same as the magnifier with frame. The only difference is the magnification plate is attached to a headband as opposed to a frame. It has similar benefits and limitations as the magnifier with frame.
Magnification Stand
The magnification stand comes as a large magnification lens with a flexible arm attached to a stand. It is a basic magnification tool and does not need to be worn; however, the stand is not easily portable and may be cumbersome to use.
Surgical Loupes
Surgical loupes are a robust magnification choice and the mainstay in magnification for the dermatologic surgeon. Loupes have proven to have comparable results in some procedures to the powerful operating surgical microscope.14-17 Factors to consider with loupes include brand, design, lens, magnification, resolution, optimal working distance, field depth, and declination angle.18
The 2 surgical loupe designs—flip-up loupes and through-the-lens loupes—differ in the mounting of the optic lenses on safety glasses. Flip-up loupes have the optics mounted to the bridge of the frame, whereas through-the-lens loupes are fixed in the lenses.
There are 3 different optical systems for surgical loupe magnification: simple, compound, and prismatic. Simple lenses consist of one pair of positive meniscus lenses similar to reading glasses. Compound lenses are made of 2 magnification lenses. Prismatic lenses magnify using a prism that folds and lengthens the light path.19,20
Loupes range in magnification level from ×2.5 to ×4.5. Compared to other magnification modalities, they can be customized and offer better resolution with quality magnification. Additionally, loupes can be fitted with a light source; however, they are expensive and surgeons need time to get used to the increased magnification as well as wearing the loupes.
There are advantages and disadvantages to the different loupe designs (Table 2). Flip-up loupes are more versatile, allowing for use on various safety glasses. They can be flipped out of view, and the declination angle may be altered; however, flip-up loupes have a narrower field of view and are heavier and bulkier than through-the-lens loupes. Through-the-lens loupes are lighter and have a larger field of view, as the optics are closer to the eye. They are customized to the declination angle and working distance of the surgeon. Conversely, through-the-lens loupes are more expensive and cannot be adjusted or moved from the line of vision.
Operating Surgical Microscope
The operating surgical microscope is not practical in the dermatologic surgeon’s practice. It is expensive and provides unnecessarily powerful magnification for dermatologic surgery. This tool usually is used in the operating room for suturing nerves and vessels with sutures sized 8-0 and smaller. Most skin procedures require size 6-0 and larger sutures.
Dermoscope
Dermoscopy, also known as epiluminescence microscopy, is a technique utilizing a handheld device made up of polarized light and a ×10 magnifying lens to evaluate skin lesions. In skilled hands, dermoscopy allows for the examination of characteristic patterns and morphologic features of skin lesions to enhance the clinician’s diagnostic accuracy.21 It may aid the dermatologic surgeon in identifying the surgical margins of difficult-to-define skin cancers. It is small and mobile; however, it has minimal benefit to the dermatologic surgeon during surgery because it is handheld and has a small field of view.
Conclusion
Good ergonomic practices facilitate a healthier and prolonged career for the dermatologic surgeon. When used properly, magnification devices can be a beneficial adjunct to the dermatologic surgeon by promoting better posture, preventing eyestrain, and providing enhanced visualization of the operating field and instruments. Use of magnification devices has been demonstrated to improve patient outcomes in other specialties. There are opportunities for further research specific to magnification improving dermatologic surgery outcomes given the high level of precision and accuracy needed for Mohs micrographic surgery, wound reconstruction, nail surgery, and hair transplantation.
Dermatologic surgeons are susceptible to work-related ailments given the nature of their working posture, the most common of which are pain and stiffness in the neck, shoulders, and lower back, as well as headaches.1,2 Awkward posture and positioning, for the sake of getting a better view of the task at hand, puts the surgeon in ergonomically disagreeable positions. Because the prime working years for a dermatologic surgeon tend to coincide with the age of presbyopia onset, magnification may help reduce and thwart musculoskeletal problems and eye strain. Indeed, a multitude of surgical specialties and dentists use intraoperative magnification.3 Knowledge and use of available magnification options can be a key addition to the dermatologic surgeon’s armamentarium. We discuss the need for magnification and review magnification devices that are readily available to the dermatologic surgeon. Table 1 presents a summary of all magnification options discussed.
Need for Magnification
Presbyopia is a condition of aging in which one loses the ability to accommodate and focus at near distances. The estimated prevalence of presbyopia in North America is 83%, typically with onset by 45 years of age.4 Individuals with presbyopia often hold objects farther away from their eyes to bring them into focus, causing eye strain, headaches, and musculoskeletal injury.
Use of intraoperative magnification allows for enhanced visualization of fine anatomic details and precise suture placement for the surgeon with or without presbyopia. Higher magnification produces a larger image; however, it also reduces field of view and depth of field (ie, the amount of depth that stays in focus without repositioning). The resolution and quality of the image are dependent on the optical properties of the lens system. The ideal optic system is surgeon dependent and involves a combination of magnification level that will not result in dramatic loss of view and depth of field, while maintaining crispness and quality of image.
Intraoperative magnification yields ergonomic benefits by promoting a safer neck flexion angle by increasing the working distance to a more ideal position (Figure). In doing so, it improves posture and minimizes eye and musculoskeletal strain secondary to awkward positioning and presbyopia.1,5 Stationary working position and neck flexion and rotation with precise and repetitive tasks are risk factors for strain and injuries that dermatologic surgeons often encounter.1 Magnification devices are tools that the dermatologic surgeon can utilize for a more ergonomically sound practice. Indeed, magnification has been shown to improve posture in the dental literature, a specialty with similar occupational risk factors to dermatologic surgery.6-8 Ergonomic practice reduces occupational injuries and improves work quality and productivity, thereby having a favorable effect on both the patient and the physician.
Improved Outcomes With Magnification
There are many examples of improved surgical quality and outcomes with magnification in other specialties. Hart and Hall5 illustrated the advantage of magnification in laceration repairs in the emergency department. In one study, increased magnification resulted in a substantial decrease in positive surgical margin rates in open radical retropubic prostatectomy.9 Schoeffl et al10 demonstrated that the microsurgical success of fine surgical procedures was directly related to optical magnification strength when comparing the unaided eye, surgical loupes, and the operating microscope. The dental literature also has numerous examples of magnification producing improved quality dentistry.11-13 Although magnification is not a novel concept to dermatologic surgery, little has been written about its use in the dermatologic surgery literature.
Magnification Options
One-Piece Bifocal Magnifying Safety Glasses
Bifocal magnifying safety glasses are polycarbonate safety glasses made with lenses in which the lower half is a magnifying lens. They are available in +1.5, +2.0, +2.5, and +3.0 diopter strengths. The total magnification power is calculated as follows: (diopter/4) + 1. The glasses are lightweight, easy to wear, inexpensive, and protect the eyes; however, they provide minimal magnification and do not compensate for differences in vision between both eyes.
Magnification Visor
The magnification visor is a headband visor with magnification lenses. It comes in various levels of magnification ranging from ×1.5 to ×3.5. It can be worn over prescription or safety glasses, may be pivoted out of the way when not in use, and is inexpensive. Conversely, it may be bulky to wear, cannot be customized, and does not offer the best resolution.
Magnification Clips
Magnification clips are hard-coated magnifying lens plates that fasten to eyeglass frames and range in level of magnification from ×1.5 to ×3.5. They can be pivoted out of the viewing angle, are lightweight, and are inexpensive; however, positioning may be difficult for ideal working distance and viewing angle.
Magnifier With Frame/Headband
The magnifier with frame is similar to magnification clips, but the magnification lens plate comes with a frame. It can be used with or without glasses and comes in magnification levels of ×1.5 to ×3.5. It is light, inexpensive, and may be pivoted out of sight, but similar to magnification clips, positioning for the right viewing angle and working distance may be difficult.
The magnifier with headband is essentially the same as the magnifier with frame. The only difference is the magnification plate is attached to a headband as opposed to a frame. It has similar benefits and limitations as the magnifier with frame.
Magnification Stand
The magnification stand comes as a large magnification lens with a flexible arm attached to a stand. It is a basic magnification tool and does not need to be worn; however, the stand is not easily portable and may be cumbersome to use.
Surgical Loupes
Surgical loupes are a robust magnification choice and the mainstay in magnification for the dermatologic surgeon. Loupes have proven to have comparable results in some procedures to the powerful operating surgical microscope.14-17 Factors to consider with loupes include brand, design, lens, magnification, resolution, optimal working distance, field depth, and declination angle.18
The 2 surgical loupe designs—flip-up loupes and through-the-lens loupes—differ in the mounting of the optic lenses on safety glasses. Flip-up loupes have the optics mounted to the bridge of the frame, whereas through-the-lens loupes are fixed in the lenses.
There are 3 different optical systems for surgical loupe magnification: simple, compound, and prismatic. Simple lenses consist of one pair of positive meniscus lenses similar to reading glasses. Compound lenses are made of 2 magnification lenses. Prismatic lenses magnify using a prism that folds and lengthens the light path.19,20
Loupes range in magnification level from ×2.5 to ×4.5. Compared to other magnification modalities, they can be customized and offer better resolution with quality magnification. Additionally, loupes can be fitted with a light source; however, they are expensive and surgeons need time to get used to the increased magnification as well as wearing the loupes.
There are advantages and disadvantages to the different loupe designs (Table 2). Flip-up loupes are more versatile, allowing for use on various safety glasses. They can be flipped out of view, and the declination angle may be altered; however, flip-up loupes have a narrower field of view and are heavier and bulkier than through-the-lens loupes. Through-the-lens loupes are lighter and have a larger field of view, as the optics are closer to the eye. They are customized to the declination angle and working distance of the surgeon. Conversely, through-the-lens loupes are more expensive and cannot be adjusted or moved from the line of vision.
Operating Surgical Microscope
The operating surgical microscope is not practical in the dermatologic surgeon’s practice. It is expensive and provides unnecessarily powerful magnification for dermatologic surgery. This tool usually is used in the operating room for suturing nerves and vessels with sutures sized 8-0 and smaller. Most skin procedures require size 6-0 and larger sutures.
Dermoscope
Dermoscopy, also known as epiluminescence microscopy, is a technique utilizing a handheld device made up of polarized light and a ×10 magnifying lens to evaluate skin lesions. In skilled hands, dermoscopy allows for the examination of characteristic patterns and morphologic features of skin lesions to enhance the clinician’s diagnostic accuracy.21 It may aid the dermatologic surgeon in identifying the surgical margins of difficult-to-define skin cancers. It is small and mobile; however, it has minimal benefit to the dermatologic surgeon during surgery because it is handheld and has a small field of view.
Conclusion
Good ergonomic practices facilitate a healthier and prolonged career for the dermatologic surgeon. When used properly, magnification devices can be a beneficial adjunct to the dermatologic surgeon by promoting better posture, preventing eyestrain, and providing enhanced visualization of the operating field and instruments. Use of magnification devices has been demonstrated to improve patient outcomes in other specialties. There are opportunities for further research specific to magnification improving dermatologic surgery outcomes given the high level of precision and accuracy needed for Mohs micrographic surgery, wound reconstruction, nail surgery, and hair transplantation.
- Liang CA, Levine VJ, Dusza SW, et al. Musculoskeletal disorders and ergonomics in dermatologic surgery: a survey of Mohs surgeons in 2010. Dermatol Surg. 2012;38:240-248.
- Esser AC, Koshy JG, Randle HW. Ergonomics in office-based surgery: a survey-guided observational study. Dermatol Surg. 2007;33:1304-1313; discussion, 1313-1314.
- Jarrett PM. Intraoperative magnification: who uses it? Microsurgery. 2004;24:420-422.
- Holden BA, Fricke TR, Ho SM, et al. Global vision impairment due to uncorrected presbyopia. Arch Ophthalmol. 2008;126:1731-1739.
- Hart RG, Hall J. The value of loupe magnification: an underused tool in emergency medicine. Am J Emerg Med. 2007;25:704-707.
- Branson BG, Bray KK, Gadbury-Amyot C, et al. Effect of magnification lenses on student operator posture. J Dent Educ. 2004;68:384-389.
- Maillet JP, Millar AM, Burke JM, et al. Effect of magnification loupes on dental hygiene student posture. J Dent Educ. 2008;72:33-44.
- Branson BG, Black MA, Simmer-Beck M. Changes in posture: a case study of a dental hygienist’s use of magnification loupes. Work. 2010;35:467-476.
- Magera JS Jr, Inman BA, Slezak JM, et al. Increased optical magnification from 2.5× to 4.3× with technical modification lowers the positive margin rate in open radical retropubic prostatectomy [published online November 13, 2007].J Urol. 2008;179:130-135.
- Schoeffl H, Lazzeri D, Schnelzer R, et al. Optical magnification should be mandatory for microsurgery: scientific basis and clinical data contributing to quality assurance. Arch Plast Surg. 2013;40:104-108.
- Taschieri S, Del Fabbro M, Testori T, et al. Endodontic surgery using 2 different magnification devices: preliminary results of a randomized controlled study. J Oral Maxillofac Surg. 2006;64:235-242.
- Christensen GJ. Magnification in dentistry: useful tool or another gimmick? J Am Dent Assoc. 2003;134:1647-1650.
- Syme SE, Fried JL, Strassler HE. Enhanced visualization using magnification systems. J Dent Hyg. 1997;71:202-206.
- Pieptu D, Luchian S. Loupes-only microsurgery. Microsurgery. 2003;23:181-188.
- Shenaq SM, Klebuc MJ, Vargo D. Free-tissue transfer with the aid of loupe magnification: experience with 251 procedures. Plast Reconstr Surg. 1995;95:261-269.
- Serletti JM, Deuber MA, Guidera PM, et al. Comparison of the operating microscope and loupes for free microvascular tissue transfer. Plast Reconstr Surg. 1995;95:270-276.
- Ross DA, Ariyan S, Restifo R, et al. Use of the operating microscope and loupes for head and neck free microvascular tissue transfer: a retrospective comparison. Arch Otolaryngol Head Neck Surg. 2003;129:189-193.
- Mungadi IA. Refinement on surgical technique: role of magnification. J Surg Tech Case Rep. 2010;2:1-2.
- Stanbury SJ, Elfar J. The use of surgical loupes in microsurgery. J Hand Surg Am. 2011;36:154-156.
- Baker JM, Meals RA. A practical guide to surgical loupes. J Hand Surg Am. 1997;22:967-974.
- Campos-do-Carmo G, Ramos-e-Silva M. Dermoscopy: basic concepts. Int J Dermatol. 2008;47:712-719.
- Liang CA, Levine VJ, Dusza SW, et al. Musculoskeletal disorders and ergonomics in dermatologic surgery: a survey of Mohs surgeons in 2010. Dermatol Surg. 2012;38:240-248.
- Esser AC, Koshy JG, Randle HW. Ergonomics in office-based surgery: a survey-guided observational study. Dermatol Surg. 2007;33:1304-1313; discussion, 1313-1314.
- Jarrett PM. Intraoperative magnification: who uses it? Microsurgery. 2004;24:420-422.
- Holden BA, Fricke TR, Ho SM, et al. Global vision impairment due to uncorrected presbyopia. Arch Ophthalmol. 2008;126:1731-1739.
- Hart RG, Hall J. The value of loupe magnification: an underused tool in emergency medicine. Am J Emerg Med. 2007;25:704-707.
- Branson BG, Bray KK, Gadbury-Amyot C, et al. Effect of magnification lenses on student operator posture. J Dent Educ. 2004;68:384-389.
- Maillet JP, Millar AM, Burke JM, et al. Effect of magnification loupes on dental hygiene student posture. J Dent Educ. 2008;72:33-44.
- Branson BG, Black MA, Simmer-Beck M. Changes in posture: a case study of a dental hygienist’s use of magnification loupes. Work. 2010;35:467-476.
- Magera JS Jr, Inman BA, Slezak JM, et al. Increased optical magnification from 2.5× to 4.3× with technical modification lowers the positive margin rate in open radical retropubic prostatectomy [published online November 13, 2007].J Urol. 2008;179:130-135.
- Schoeffl H, Lazzeri D, Schnelzer R, et al. Optical magnification should be mandatory for microsurgery: scientific basis and clinical data contributing to quality assurance. Arch Plast Surg. 2013;40:104-108.
- Taschieri S, Del Fabbro M, Testori T, et al. Endodontic surgery using 2 different magnification devices: preliminary results of a randomized controlled study. J Oral Maxillofac Surg. 2006;64:235-242.
- Christensen GJ. Magnification in dentistry: useful tool or another gimmick? J Am Dent Assoc. 2003;134:1647-1650.
- Syme SE, Fried JL, Strassler HE. Enhanced visualization using magnification systems. J Dent Hyg. 1997;71:202-206.
- Pieptu D, Luchian S. Loupes-only microsurgery. Microsurgery. 2003;23:181-188.
- Shenaq SM, Klebuc MJ, Vargo D. Free-tissue transfer with the aid of loupe magnification: experience with 251 procedures. Plast Reconstr Surg. 1995;95:261-269.
- Serletti JM, Deuber MA, Guidera PM, et al. Comparison of the operating microscope and loupes for free microvascular tissue transfer. Plast Reconstr Surg. 1995;95:270-276.
- Ross DA, Ariyan S, Restifo R, et al. Use of the operating microscope and loupes for head and neck free microvascular tissue transfer: a retrospective comparison. Arch Otolaryngol Head Neck Surg. 2003;129:189-193.
- Mungadi IA. Refinement on surgical technique: role of magnification. J Surg Tech Case Rep. 2010;2:1-2.
- Stanbury SJ, Elfar J. The use of surgical loupes in microsurgery. J Hand Surg Am. 2011;36:154-156.
- Baker JM, Meals RA. A practical guide to surgical loupes. J Hand Surg Am. 1997;22:967-974.
- Campos-do-Carmo G, Ramos-e-Silva M. Dermoscopy: basic concepts. Int J Dermatol. 2008;47:712-719.
Practice Points
- Ergonomic practice is paramount in preserving the longevity and productivity of the dermatologic surgeon.
- A magnification device may be a helpful addition for a dermatologic surgeon to achieve a healthier and more productive practice.
Program for Maintenance of Certification by the American Board of Dermatology
Maintenance of Certification (MOC) was adopted by the 24 certifying boards constituting the American Board of Medical Specialties (ABMS) in 2000. The American Board of Dermatology (ABD) granted its first time-limited certificates in 1991 with the first cohort of diplomates entering MOC in 2006. The rationale for MOC centered on 2 propositions: First, continuing medical education (CME) alone was insufficient to assure the public that physicians were remaining up-to-date with an expanding knowledge base and offered little opportunity to engage in meaningful self-assessment and practice improvement. Second, parties external to the medical profession were focusing increased attention on physician error and quality assurance in medical practice. Maintenance of Certification, therefore, provided a mechanism of physician self-regulation in meeting public scrutiny.1,2
The basic framework of MOC remains unchanged since its inception, though notable effort has been expended in simplifying the tools available. All MOC components offered directly by ABD including the MOC examination are covered by the $150 annual fee.
Professional Standing
Diplomates attest to the status of all state medical licenses and level of clinical activity. All licenses must be unrestricted. “Clinically active” is defined as any patient care delivered within the prior 12 months. Having a restricted license or being clinically inactive does not automatically trigger loss of certification but does result in an ABD review.
Self-assessment
Diplomates complete 300 credits (1 question=1 credit) over 10 years and complete, or attest to prior completion of, a foundational course in patient safety. Self-assessment questions are widely available from various sources, including the Question of the Week offered by the American Academy of Dermatology, Clinicopathologic Correlation and CME-designated articles offered by JAMA Dermatology, and Photo Challenges and Dermatopathology Diagnosis quizzes offered by Cutis. The ABD recognizes patient safety education satisfied as part of medical school and residency as well as various other venues. Online courses offering CME and MOC credit also are available. Credit is accrued whether the item is answered correctly or not.
Cognitive Expertise
Dermatologists take a general dermatology module and choose one subspecialty module composed of questions directed to the clinical practitioner. The general module consists of 100 image items, most of which ask for a diagnosis. The list of entities potentially included on the assessment is made available in advance for self-study. The subspecialty module consists of 50 questions targeting the specific content area selected: medical dermatology, surgical dermatology, pediatric dermatology, and dermatopathology. The actual questions also are made available in advance for self-study. Board-certified pediatric dermatologists and dermatopathologists are offered a second 50-question set of items in their specialty to allow maintenance of the second certificate. Venues include Pearson VUE testing centers and at-home or in-office tests by remote proctoring.
The ABD is considering participation in the longitudinal assessment program developed by the ABMS. If adopted, it will offer questions distributed over a many-year span in small packets, on mobile devices, and on personal computers. Diplomates will have the ability to select content and pace, including opt-out periods as life events dictate. A minimum number of correctly answered items over time will form the basis for summative assessment.
Practice Improvement
A critical element of MOC, practice improvement affords the physician the opportunity to study how patients receive care in a wide range of settings. Beginning in 2015, the ABD developed focused practice improvement modules, now totaling 21, with many more coming in the future. The free modules are offered on an online platform (https://secure.dataharborsolutions.com/ABDermOrg/Default.aspx) and target narrow content areas. The broad range of offerings allows diplomates to choose an area of specific interest. The participant is asked to read an overview and rationale for the module, consider reading selected references that provide the evidence base, and perform 5 chart abstractions consisting of yes or no answers to no more than 5 questions narrowly focused on the chosen topic. If a first round shows no room for improvement, the participant is finished. If a deficiency is identified, the diplomate can reflect on and implement any necessary changes in process of care and pursue a second round. These modules have been very well received, with typical diplomates’ comments expressing appreciation for the ease of use and relevance to practice. Unedited and unselected reviews can be found online (https://www.abderm.org/diplomates/fulfilling-moc-requirements/resource-vendor-list/practice-improvement/american-board-of-dermatology-focused-pi-modules-free.aspx).
Future Direction
The ABD continuously communicates with diplomates about changes and new opportunities in its MOC program with a goal of maximizing value and minimizing cost in terms of dollars and time.3 The directors of the ABD continue to seek feedback about the MOC program and are committed to further refinements to achieve this goal. A critical feature of the redesigned website (http://www.abderm.org/) allows diplomates to submit and read anonymous reviews of all tools available to fulfill MOC requirements. This thoughtful diplomate feedback informs MOC developmental efforts.
All directors and executive staff of the ABD, regardless of certificate status, pay the annual fee and participate in MOC. Active participation in MOC is made public on the ABMS website. This acknowledgment is an assurance to patients that the physician’s professional standing is sound, that the physician periodically self-assesses what he/she knows, that this knowledge meets psychometrically valid standards set by dermatologists, and that physicians explore the quality of care delivered in specific practice settings. It’s the right thing to do!
- Kohn LT, Corrigan JM, Donaldson MS, eds. To Err is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
- Institute of Medicine. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press; 2001:337.
- American Board of Dermatology. We are simplifying maintenance of certification. here’s how. http://eepurl.com/bLd9vz. Published January 3, 2016. Accessed May 11, 2017.
Maintenance of Certification (MOC) was adopted by the 24 certifying boards constituting the American Board of Medical Specialties (ABMS) in 2000. The American Board of Dermatology (ABD) granted its first time-limited certificates in 1991 with the first cohort of diplomates entering MOC in 2006. The rationale for MOC centered on 2 propositions: First, continuing medical education (CME) alone was insufficient to assure the public that physicians were remaining up-to-date with an expanding knowledge base and offered little opportunity to engage in meaningful self-assessment and practice improvement. Second, parties external to the medical profession were focusing increased attention on physician error and quality assurance in medical practice. Maintenance of Certification, therefore, provided a mechanism of physician self-regulation in meeting public scrutiny.1,2
The basic framework of MOC remains unchanged since its inception, though notable effort has been expended in simplifying the tools available. All MOC components offered directly by ABD including the MOC examination are covered by the $150 annual fee.
Professional Standing
Diplomates attest to the status of all state medical licenses and level of clinical activity. All licenses must be unrestricted. “Clinically active” is defined as any patient care delivered within the prior 12 months. Having a restricted license or being clinically inactive does not automatically trigger loss of certification but does result in an ABD review.
Self-assessment
Diplomates complete 300 credits (1 question=1 credit) over 10 years and complete, or attest to prior completion of, a foundational course in patient safety. Self-assessment questions are widely available from various sources, including the Question of the Week offered by the American Academy of Dermatology, Clinicopathologic Correlation and CME-designated articles offered by JAMA Dermatology, and Photo Challenges and Dermatopathology Diagnosis quizzes offered by Cutis. The ABD recognizes patient safety education satisfied as part of medical school and residency as well as various other venues. Online courses offering CME and MOC credit also are available. Credit is accrued whether the item is answered correctly or not.
Cognitive Expertise
Dermatologists take a general dermatology module and choose one subspecialty module composed of questions directed to the clinical practitioner. The general module consists of 100 image items, most of which ask for a diagnosis. The list of entities potentially included on the assessment is made available in advance for self-study. The subspecialty module consists of 50 questions targeting the specific content area selected: medical dermatology, surgical dermatology, pediatric dermatology, and dermatopathology. The actual questions also are made available in advance for self-study. Board-certified pediatric dermatologists and dermatopathologists are offered a second 50-question set of items in their specialty to allow maintenance of the second certificate. Venues include Pearson VUE testing centers and at-home or in-office tests by remote proctoring.
The ABD is considering participation in the longitudinal assessment program developed by the ABMS. If adopted, it will offer questions distributed over a many-year span in small packets, on mobile devices, and on personal computers. Diplomates will have the ability to select content and pace, including opt-out periods as life events dictate. A minimum number of correctly answered items over time will form the basis for summative assessment.
Practice Improvement
A critical element of MOC, practice improvement affords the physician the opportunity to study how patients receive care in a wide range of settings. Beginning in 2015, the ABD developed focused practice improvement modules, now totaling 21, with many more coming in the future. The free modules are offered on an online platform (https://secure.dataharborsolutions.com/ABDermOrg/Default.aspx) and target narrow content areas. The broad range of offerings allows diplomates to choose an area of specific interest. The participant is asked to read an overview and rationale for the module, consider reading selected references that provide the evidence base, and perform 5 chart abstractions consisting of yes or no answers to no more than 5 questions narrowly focused on the chosen topic. If a first round shows no room for improvement, the participant is finished. If a deficiency is identified, the diplomate can reflect on and implement any necessary changes in process of care and pursue a second round. These modules have been very well received, with typical diplomates’ comments expressing appreciation for the ease of use and relevance to practice. Unedited and unselected reviews can be found online (https://www.abderm.org/diplomates/fulfilling-moc-requirements/resource-vendor-list/practice-improvement/american-board-of-dermatology-focused-pi-modules-free.aspx).
Future Direction
The ABD continuously communicates with diplomates about changes and new opportunities in its MOC program with a goal of maximizing value and minimizing cost in terms of dollars and time.3 The directors of the ABD continue to seek feedback about the MOC program and are committed to further refinements to achieve this goal. A critical feature of the redesigned website (http://www.abderm.org/) allows diplomates to submit and read anonymous reviews of all tools available to fulfill MOC requirements. This thoughtful diplomate feedback informs MOC developmental efforts.
All directors and executive staff of the ABD, regardless of certificate status, pay the annual fee and participate in MOC. Active participation in MOC is made public on the ABMS website. This acknowledgment is an assurance to patients that the physician’s professional standing is sound, that the physician periodically self-assesses what he/she knows, that this knowledge meets psychometrically valid standards set by dermatologists, and that physicians explore the quality of care delivered in specific practice settings. It’s the right thing to do!
Maintenance of Certification (MOC) was adopted by the 24 certifying boards constituting the American Board of Medical Specialties (ABMS) in 2000. The American Board of Dermatology (ABD) granted its first time-limited certificates in 1991 with the first cohort of diplomates entering MOC in 2006. The rationale for MOC centered on 2 propositions: First, continuing medical education (CME) alone was insufficient to assure the public that physicians were remaining up-to-date with an expanding knowledge base and offered little opportunity to engage in meaningful self-assessment and practice improvement. Second, parties external to the medical profession were focusing increased attention on physician error and quality assurance in medical practice. Maintenance of Certification, therefore, provided a mechanism of physician self-regulation in meeting public scrutiny.1,2
The basic framework of MOC remains unchanged since its inception, though notable effort has been expended in simplifying the tools available. All MOC components offered directly by ABD including the MOC examination are covered by the $150 annual fee.
Professional Standing
Diplomates attest to the status of all state medical licenses and level of clinical activity. All licenses must be unrestricted. “Clinically active” is defined as any patient care delivered within the prior 12 months. Having a restricted license or being clinically inactive does not automatically trigger loss of certification but does result in an ABD review.
Self-assessment
Diplomates complete 300 credits (1 question=1 credit) over 10 years and complete, or attest to prior completion of, a foundational course in patient safety. Self-assessment questions are widely available from various sources, including the Question of the Week offered by the American Academy of Dermatology, Clinicopathologic Correlation and CME-designated articles offered by JAMA Dermatology, and Photo Challenges and Dermatopathology Diagnosis quizzes offered by Cutis. The ABD recognizes patient safety education satisfied as part of medical school and residency as well as various other venues. Online courses offering CME and MOC credit also are available. Credit is accrued whether the item is answered correctly or not.
Cognitive Expertise
Dermatologists take a general dermatology module and choose one subspecialty module composed of questions directed to the clinical practitioner. The general module consists of 100 image items, most of which ask for a diagnosis. The list of entities potentially included on the assessment is made available in advance for self-study. The subspecialty module consists of 50 questions targeting the specific content area selected: medical dermatology, surgical dermatology, pediatric dermatology, and dermatopathology. The actual questions also are made available in advance for self-study. Board-certified pediatric dermatologists and dermatopathologists are offered a second 50-question set of items in their specialty to allow maintenance of the second certificate. Venues include Pearson VUE testing centers and at-home or in-office tests by remote proctoring.
The ABD is considering participation in the longitudinal assessment program developed by the ABMS. If adopted, it will offer questions distributed over a many-year span in small packets, on mobile devices, and on personal computers. Diplomates will have the ability to select content and pace, including opt-out periods as life events dictate. A minimum number of correctly answered items over time will form the basis for summative assessment.
Practice Improvement
A critical element of MOC, practice improvement affords the physician the opportunity to study how patients receive care in a wide range of settings. Beginning in 2015, the ABD developed focused practice improvement modules, now totaling 21, with many more coming in the future. The free modules are offered on an online platform (https://secure.dataharborsolutions.com/ABDermOrg/Default.aspx) and target narrow content areas. The broad range of offerings allows diplomates to choose an area of specific interest. The participant is asked to read an overview and rationale for the module, consider reading selected references that provide the evidence base, and perform 5 chart abstractions consisting of yes or no answers to no more than 5 questions narrowly focused on the chosen topic. If a first round shows no room for improvement, the participant is finished. If a deficiency is identified, the diplomate can reflect on and implement any necessary changes in process of care and pursue a second round. These modules have been very well received, with typical diplomates’ comments expressing appreciation for the ease of use and relevance to practice. Unedited and unselected reviews can be found online (https://www.abderm.org/diplomates/fulfilling-moc-requirements/resource-vendor-list/practice-improvement/american-board-of-dermatology-focused-pi-modules-free.aspx).
Future Direction
The ABD continuously communicates with diplomates about changes and new opportunities in its MOC program with a goal of maximizing value and minimizing cost in terms of dollars and time.3 The directors of the ABD continue to seek feedback about the MOC program and are committed to further refinements to achieve this goal. A critical feature of the redesigned website (http://www.abderm.org/) allows diplomates to submit and read anonymous reviews of all tools available to fulfill MOC requirements. This thoughtful diplomate feedback informs MOC developmental efforts.
All directors and executive staff of the ABD, regardless of certificate status, pay the annual fee and participate in MOC. Active participation in MOC is made public on the ABMS website. This acknowledgment is an assurance to patients that the physician’s professional standing is sound, that the physician periodically self-assesses what he/she knows, that this knowledge meets psychometrically valid standards set by dermatologists, and that physicians explore the quality of care delivered in specific practice settings. It’s the right thing to do!
- Kohn LT, Corrigan JM, Donaldson MS, eds. To Err is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
- Institute of Medicine. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press; 2001:337.
- American Board of Dermatology. We are simplifying maintenance of certification. here’s how. http://eepurl.com/bLd9vz. Published January 3, 2016. Accessed May 11, 2017.
- Kohn LT, Corrigan JM, Donaldson MS, eds. To Err is Human: Building a Safer Health System. Washington, DC: National Academy Press; 2000.
- Institute of Medicine. Crossing the Quality Chasm: A New Health System for the 21st Century. Washington, DC: National Academy Press; 2001:337.
- American Board of Dermatology. We are simplifying maintenance of certification. here’s how. http://eepurl.com/bLd9vz. Published January 3, 2016. Accessed May 11, 2017.

BSA75, BSA90, and BSA100: New Clinical Tools for Measuring Improvement in Psoriasis
Currently, there is no widely accepted tool for assessing the severity of psoriasis in the clinical setting.1-5 Moreover, there is still a need for a simple assessment tool to assist in evaluating a patient’s response to therapy in clinical practice.6
The body surface area (BSA) is a familiar and widely used measurement by clinicians. It is easily calculated by the rule of nines or with the patient’s open palm and thumb approximating 1% of the BSA.7 Body surface area is an uncomplicated concept for patients to understand and interpret. It also promotes patient empowerment and self-care by allowing patients to monitor short-term and long-term response to therapy.
The National Psoriasis Foundation Medical Board published treatment targets for plaque psoriasis. One of the conclusions states, “The acceptable response at 3 months postinitiation was either BSA 3% or less or BSA improvement 75% or more from baseline.”8
We propose a new nomenclature that a 75% improvement in BSA be recognized as BSA75, a 90% improvement in BSA as BSA90, and a 100% improvement in BSA as BSA100. These classifications would be analogous to corresponding improvements in the following psoriasis area and severity index (PASI) scores: PASI 75, PASI 90, PASI 100.9 A loss of BSA goals/milestones (ie, BSA75) could encourage and facilitate physician-patient conversations and further direct modifications to disease management and treatment therapy.
A potential drawback to the implementation of this novel categorization system is that other notable aspects of psoriasis would not be assessed, such as erythema, induration, or scale; subjective measurements; patient quality of life; patient symptoms; areas of involvement (eg, palms, soles of feet); and disease course. Nevertheless, the BSA75, BSA90, and BSA100 classifications can serve as practical, objective, and straightforward tools to monitor disease progression and treatment response in psoriasis patients, which may potentially promote improved patient outcomes in clinical practice.
- van de Kerkhof PC. The Psoriasis Area and Severity Index and alternative approaches for the assessment of severity: persisting areas of confusion. Br J Dermatol. 1997;137:661-662.
- Langley RG, Ellis CN. Evaluating psoriasis with Psoriasis Area and Severity Index, Psoriasis Global Assessment, and Lattice System Physician’s Global Assessment. J Am Acad Dermatol. 2004;51:563-569.
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
- Ashcroft DM, Wan Po AL, Williams HC, et al. Clinical measures of disease severity and outcome in psoriasis: a critical appraisal of their quality. Br J Dermatol. 1999;141:185-191.
- Gottlieb AB, Chaudhari U, Baker DG, et al. The National Psoriasis Foundation Psoriasis Score (NPF-PS) system versus the Psoriasis Area Severity Index (PASI) and Physician’s Global Assessment (PGA): a comparison. J Drugs Dermatol. 2003;2:260-266.
- Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica. 1978;157:238-244.
- Sheridan RL, Petras L, Basha G, et al. Planimetry study of the percent of body surface represented by the hand and palm: sizing irregular burns is more accurately done with the palm. J Burn Care Rehabil. 1995;16:605-606.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Manalo IF, Gilbert KE, Wu JJ. Time to raise the bar to Psoriasis Area Severity Index 90 and 100. J Drugs Dermatol. 2015;14:1086-1088.
Currently, there is no widely accepted tool for assessing the severity of psoriasis in the clinical setting.1-5 Moreover, there is still a need for a simple assessment tool to assist in evaluating a patient’s response to therapy in clinical practice.6
The body surface area (BSA) is a familiar and widely used measurement by clinicians. It is easily calculated by the rule of nines or with the patient’s open palm and thumb approximating 1% of the BSA.7 Body surface area is an uncomplicated concept for patients to understand and interpret. It also promotes patient empowerment and self-care by allowing patients to monitor short-term and long-term response to therapy.
The National Psoriasis Foundation Medical Board published treatment targets for plaque psoriasis. One of the conclusions states, “The acceptable response at 3 months postinitiation was either BSA 3% or less or BSA improvement 75% or more from baseline.”8
We propose a new nomenclature that a 75% improvement in BSA be recognized as BSA75, a 90% improvement in BSA as BSA90, and a 100% improvement in BSA as BSA100. These classifications would be analogous to corresponding improvements in the following psoriasis area and severity index (PASI) scores: PASI 75, PASI 90, PASI 100.9 A loss of BSA goals/milestones (ie, BSA75) could encourage and facilitate physician-patient conversations and further direct modifications to disease management and treatment therapy.
A potential drawback to the implementation of this novel categorization system is that other notable aspects of psoriasis would not be assessed, such as erythema, induration, or scale; subjective measurements; patient quality of life; patient symptoms; areas of involvement (eg, palms, soles of feet); and disease course. Nevertheless, the BSA75, BSA90, and BSA100 classifications can serve as practical, objective, and straightforward tools to monitor disease progression and treatment response in psoriasis patients, which may potentially promote improved patient outcomes in clinical practice.
Currently, there is no widely accepted tool for assessing the severity of psoriasis in the clinical setting.1-5 Moreover, there is still a need for a simple assessment tool to assist in evaluating a patient’s response to therapy in clinical practice.6
The body surface area (BSA) is a familiar and widely used measurement by clinicians. It is easily calculated by the rule of nines or with the patient’s open palm and thumb approximating 1% of the BSA.7 Body surface area is an uncomplicated concept for patients to understand and interpret. It also promotes patient empowerment and self-care by allowing patients to monitor short-term and long-term response to therapy.
The National Psoriasis Foundation Medical Board published treatment targets for plaque psoriasis. One of the conclusions states, “The acceptable response at 3 months postinitiation was either BSA 3% or less or BSA improvement 75% or more from baseline.”8
We propose a new nomenclature that a 75% improvement in BSA be recognized as BSA75, a 90% improvement in BSA as BSA90, and a 100% improvement in BSA as BSA100. These classifications would be analogous to corresponding improvements in the following psoriasis area and severity index (PASI) scores: PASI 75, PASI 90, PASI 100.9 A loss of BSA goals/milestones (ie, BSA75) could encourage and facilitate physician-patient conversations and further direct modifications to disease management and treatment therapy.
A potential drawback to the implementation of this novel categorization system is that other notable aspects of psoriasis would not be assessed, such as erythema, induration, or scale; subjective measurements; patient quality of life; patient symptoms; areas of involvement (eg, palms, soles of feet); and disease course. Nevertheless, the BSA75, BSA90, and BSA100 classifications can serve as practical, objective, and straightforward tools to monitor disease progression and treatment response in psoriasis patients, which may potentially promote improved patient outcomes in clinical practice.
- van de Kerkhof PC. The Psoriasis Area and Severity Index and alternative approaches for the assessment of severity: persisting areas of confusion. Br J Dermatol. 1997;137:661-662.
- Langley RG, Ellis CN. Evaluating psoriasis with Psoriasis Area and Severity Index, Psoriasis Global Assessment, and Lattice System Physician’s Global Assessment. J Am Acad Dermatol. 2004;51:563-569.
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
- Ashcroft DM, Wan Po AL, Williams HC, et al. Clinical measures of disease severity and outcome in psoriasis: a critical appraisal of their quality. Br J Dermatol. 1999;141:185-191.
- Gottlieb AB, Chaudhari U, Baker DG, et al. The National Psoriasis Foundation Psoriasis Score (NPF-PS) system versus the Psoriasis Area Severity Index (PASI) and Physician’s Global Assessment (PGA): a comparison. J Drugs Dermatol. 2003;2:260-266.
- Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica. 1978;157:238-244.
- Sheridan RL, Petras L, Basha G, et al. Planimetry study of the percent of body surface represented by the hand and palm: sizing irregular burns is more accurately done with the palm. J Burn Care Rehabil. 1995;16:605-606.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Manalo IF, Gilbert KE, Wu JJ. Time to raise the bar to Psoriasis Area Severity Index 90 and 100. J Drugs Dermatol. 2015;14:1086-1088.
- van de Kerkhof PC. The Psoriasis Area and Severity Index and alternative approaches for the assessment of severity: persisting areas of confusion. Br J Dermatol. 1997;137:661-662.
- Langley RG, Ellis CN. Evaluating psoriasis with Psoriasis Area and Severity Index, Psoriasis Global Assessment, and Lattice System Physician’s Global Assessment. J Am Acad Dermatol. 2004;51:563-569.
- Finlay AY, Khan GK. Dermatology Life Quality Index (DLQI)—a simple practical measure for routine clinical use. Clin Exp Dermatol. 1994;19:210-216.
- Ashcroft DM, Wan Po AL, Williams HC, et al. Clinical measures of disease severity and outcome in psoriasis: a critical appraisal of their quality. Br J Dermatol. 1999;141:185-191.
- Gottlieb AB, Chaudhari U, Baker DG, et al. The National Psoriasis Foundation Psoriasis Score (NPF-PS) system versus the Psoriasis Area Severity Index (PASI) and Physician’s Global Assessment (PGA): a comparison. J Drugs Dermatol. 2003;2:260-266.
- Fredriksson T, Pettersson U. Severe psoriasis—oral therapy with a new retinoid. Dermatologica. 1978;157:238-244.
- Sheridan RL, Petras L, Basha G, et al. Planimetry study of the percent of body surface represented by the hand and palm: sizing irregular burns is more accurately done with the palm. J Burn Care Rehabil. 1995;16:605-606.
- Armstrong AW, Siegel MP, Bagel J, et al. From the Medical Board of the National Psoriasis Foundation: treatment targets for plaque psoriasis. J Am Acad Dermatol. 2017;76:290-298.
- Manalo IF, Gilbert KE, Wu JJ. Time to raise the bar to Psoriasis Area Severity Index 90 and 100. J Drugs Dermatol. 2015;14:1086-1088.

Cosmetic Corner: Dermatologists Weigh in on Face Scrubs
To improve patient care and outcomes, leading dermatologists offered their recommendations on face scrubs. Consideration must be given to:
- Crystal Peel Microdermabrasion Exfoliating Face Crème
Formulary for Physicians, Inc
“This product is a highly effective facial scrub for patients with thick skin. Its exfoliating ingredient is corundum, another name for aluminum oxide, the crystal used by most microabrasion machines.”— Mark G. Rubin, MD, Beverly Hills, California
- Facial Fuel Energizing Scrub
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Olay Regenerist Regenerating Cream Cleanser
Procter & Gamble
“Oxygenated beads in the creamy formula help to gently exfoliate the skin without overdrying and stripping the skin’s outer layer, leaving the skin soft and fresh.”—Jeannette Graf, MD, New York, New York
- PRESCRIBEDsolutions: Starting Up/Face, Surface Improvement
Biopelle, Inc
“I use Starting Up/Face as my daily cleanser, as it contains salicylic acid and helps improve the overall texture plus minimize bumps from shaving, and Surface Improvement about every other day on my face in the shower.”—Joel L. Cohen, MD, Greenwood Village, Colorado
- St. Ives Apricot Blemish Control Scrub
Unilever
“It exfoliates and has salicylic acid. After exfoliating, I recommend allowing it to sit on the skin for 5 minutes before washing off.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and redness-reducing products will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on face scrubs. Consideration must be given to:
- Crystal Peel Microdermabrasion Exfoliating Face Crème
Formulary for Physicians, Inc
“This product is a highly effective facial scrub for patients with thick skin. Its exfoliating ingredient is corundum, another name for aluminum oxide, the crystal used by most microabrasion machines.”— Mark G. Rubin, MD, Beverly Hills, California
- Facial Fuel Energizing Scrub
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Olay Regenerist Regenerating Cream Cleanser
Procter & Gamble
“Oxygenated beads in the creamy formula help to gently exfoliate the skin without overdrying and stripping the skin’s outer layer, leaving the skin soft and fresh.”—Jeannette Graf, MD, New York, New York
- PRESCRIBEDsolutions: Starting Up/Face, Surface Improvement
Biopelle, Inc
“I use Starting Up/Face as my daily cleanser, as it contains salicylic acid and helps improve the overall texture plus minimize bumps from shaving, and Surface Improvement about every other day on my face in the shower.”—Joel L. Cohen, MD, Greenwood Village, Colorado
- St. Ives Apricot Blemish Control Scrub
Unilever
“It exfoliates and has salicylic acid. After exfoliating, I recommend allowing it to sit on the skin for 5 minutes before washing off.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and redness-reducing products will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on face scrubs. Consideration must be given to:
- Crystal Peel Microdermabrasion Exfoliating Face Crème
Formulary for Physicians, Inc
“This product is a highly effective facial scrub for patients with thick skin. Its exfoliating ingredient is corundum, another name for aluminum oxide, the crystal used by most microabrasion machines.”— Mark G. Rubin, MD, Beverly Hills, California
- Facial Fuel Energizing Scrub
Kiehl’s
Recommended by Gary Goldenberg, MD, New York, New York
- Olay Regenerist Regenerating Cream Cleanser
Procter & Gamble
“Oxygenated beads in the creamy formula help to gently exfoliate the skin without overdrying and stripping the skin’s outer layer, leaving the skin soft and fresh.”—Jeannette Graf, MD, New York, New York
- PRESCRIBEDsolutions: Starting Up/Face, Surface Improvement
Biopelle, Inc
“I use Starting Up/Face as my daily cleanser, as it contains salicylic acid and helps improve the overall texture plus minimize bumps from shaving, and Surface Improvement about every other day on my face in the shower.”—Joel L. Cohen, MD, Greenwood Village, Colorado
- St. Ives Apricot Blemish Control Scrub
Unilever
“It exfoliates and has salicylic acid. After exfoliating, I recommend allowing it to sit on the skin for 5 minutes before washing off.”—Anthony M. Rossi, MD, New York, New York
Cutis invites readers to send us their recommendations. Athlete’s foot treatments, cleansing devices, and redness-reducing products will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Management of Poorly Controlled Indolent Systemic Mastocytosis Using Narrowband UVB Phototherapy
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.

The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012.
- Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
- Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
- Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
- Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
- Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
- Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
- Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
- Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
- Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
- Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
- Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
- Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
- Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.
The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
Systemic mastocytosis is a heterogeneous disorder of stem cell origin defined by abnormal hyperplasia and accumulation of mast cells (MCs) in one or more tissues.1,2 The most commonly affected tissues are the bone marrow, gastrointestinal tract, and skin. Based on a number of major and minor criteria defined by the World Health Organization (WHO), the mastocytoses are subdivided into 7 variants that range from isolated cutaneous involvement to widespread systemic disease.1-4 The most frequently diagnosed subtype is indolent systemic mastocytosis (ISM), a chronic disorder characterized by diffuse cutaneous macules and papules as well as bone marrow involvement in the form of multifocal dense infiltrates of MCs that frequently are phenotypically positive for c-KIT and tryptase. Serum tryptase levels are nearly invariably elevated in patients with this condition.1,2
Symptoms of ISM are determined by the intermittent release of histamine and leukotrienes from hyperproliferating MCs as well as IL-6 and eosinophil chemotactic factors. As the burden of MC secretory products increases, patients experience worsening pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.1,2,5 The mainstay of treatment of this condition involves symptom control through the inhibition of MC mediators.1 The majority of patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of MC degranulation.1,2,5
Unfortunately, some patients are unable to achieve adequate symptom control through the use of mediator-targeting treatments alone. In these cases, physicians often are faced with the following treatment dilemma: Either attempt to use therapies such as interferon alfa, which is cytoreductive to MCs, or 2-chlorodeoxyadenosine to reduce the overall MC burden, or turn to newer nonimmunosuppressive second-line options. We present the case of a patient with chronic ISM with progressive cutaneous lesions and poorly controlled pruritus that was previously managed with topical corticosteroids and antihistamines who responded favorably to treatment with narrowband UVB (NB-UVB) phototherapy.
Case Report
A 57-year-old woman presented with a 10-year history of widespread red-brown macules and papules on the trunk and upper and lower extremities. The lesions were intermittently pruritic, a symptom that was exacerbated on sun and heat exposure. A skin biopsy performed by an outside dermatologist 9 years prior confirmed the presence of mastocytosis. The patient was originally treated with triamcinolone cream and oral antihistamines, which controlled her symptoms successfully for nearly a decade.
At the current presentation, the patient reported increasingly severe pruritus and lesional spread to the neck and face of 15 months’ duration. She denied any symptoms of flushing, diarrhea, syncopal episodes, or lightheadedness. Physical examination revealed a well-appearing middle-aged woman with multiple 3- to 8-mm, red-brown, blanchable macules and papules with areas coalescing into plaques that primarily involved the legs (Figure 1A); arms; back; and to a lesser extent the abdomen, neck, and face. There was no palpable lymphadenopathy.
Laboratory results revealed a complete blood cell count and basic metabolic profile within reference range; however, the serum tryptase level was elevated at 65 ng/mL (reference range, <11.4 ng/mL). A positron emission tomography–computed tomography scan was negative, as well as a c-KIT mutation analysis. A review of the skin biopsy from 9 years prior demonstrated slight acanthosis with dermal proliferation of mononuclear cells (Figure 2A), some of which had abundant cytoplasm and oval-shaped nuclei. There were few eosinophils and marked dermal telangiectasias. Giemsa stain revealed increased numbers of MCs in the upper dermis (Figure 2B). A bone marrow biopsy performed 9 years later showed multifocal lesions composed of MCs with associated lymphoid aggregates without notable myelodyspoiesis (or myeloproliferative neoplasm). These features were all consistent with WHO criteria for ISM. Based on the most current clinical, laboratory, and histopathologic findings, the patient was diagnosed with category IB ISM.
The patient’s symptoms had remained stable for 9 years with a regimen of triamcinolone cream 0.1% twice daily, doxepin cream 5% daily as needed, and oral fexofenadine 180 mg once daily. The patient continues to use topical steroids and oral antihistamines. Due to inadequate symptom control, breakthrough pruritus, and the development of new skin lesions on the head and neck, she was started on NB-UVB treatment 2 months after presentation. The patient’s symptoms and the extent of cutaneous maculopapular lesions improved after 20 light treatments (Figure 1B), with even more dramatic results after 40 cycles of therapy (Figure 1C). Overall, the lower legs have proved most recalcitrant to this treatment modality. She is currently continuing to receive NB-UVB treatment twice weekly.
Comment
Systemic mastocytosis is a heterogeneous disorder characterized by the proliferation and accumulation of atypical MCs in tissues, principally in the bone marrow and skin, though involvement of the gastrointestinal tract, liver, spleen, and lymphatic system also have been reported.1,2,6 The WHO classification of mastocytosis divides this condition into 7 subtypes.4 Indolent systemic mastocytosis is the most common variant.2,6 The etiology of ISM is not fully understood, but there is evidence suggesting that an activating mutation of KIT proto-oncogene receptor tyrosine kinase, KIT (usually D816V), present in the MCs of nearly 80% of patients with ISM may be involved.1,3-5,7 Patients occasionally present with predominantly cutaneous findings but typically seek medical attention due to the recurrent systemic symptoms of the disease (eg, pruritus, flushing, syncope, palpitations, headache, dyspepsia, vomiting, diarrhea), which are related to the release of MC mediators.1,2
The management of ISM is complex and based primarily on symptom reduction without alteration of disease course.1,2,5,7 Patients should avoid symptom triggers such as heat, humidity, emotional and physical stress, alcohol, and certain medications (ie, aspirin, opioids, radiocontrast agents).7 Patients are initially treated with histamine H1- and H2-receptor antagonists to alleviate MC mediator release symptoms.1,2,8 Although H1 blockers are most effective in mitigating cutaneous symptoms and limiting pruritus, H2 blockers are used to control gastric hypersecretion and dyspepsia.2 Proton pump inhibitors are useful in patients with peptic ulcer disease who are unresponsive to H2-receptor antagonist therapy.2,7 Cromolyn sodium and ketotifen fumarate are MC stabilizers that help prevent degranulation, which is helpful in relieving most major ISM symptoms. Leukotriene antagonists, such as zafirlukast, montelukast sodium, or zileuton, also may be employed to target the proinflammatory and pruritogenic leukotrienes, also products of the MC protein.2,7 Imatinib mesylate and masitinib mesylate, both tyrosine kinase inhibitors, have been shown to improve symptoms and reduce MC mediator levels in ISM; however, most patients harbor the resistant KIT D816V mutation, which limits the utility of this medication.Patients with sensitive KIT mutations or those who have the wild-type KIT D816 mutation may be more appropriate candidates for imatinib or masitinib therapy, which can ameliorate symptoms of flushing, pruritus, and depression.7-10 Treatment with omalizumab, a humanized murine anti-IgE monoclonal antibody, can be effective in treating recurrent, treatment-refractory anaphylaxis in ISM patients.5,7
Symptoms unresponsive to these therapies can be effectively treated with a short course of oral corticosteroids,6,7 while MC cytoreductive therapies such as interferon alfa or 2-chlorodeoxyadenosine (cladribine/2-CdA) are reserved for refractory cases.2,7 Alternative therapies such as NB-UVB2 or psoralen plus UVA phototherapy11 also have demonstrated success in treating ISM symptoms. In the past, NB-UVB has shown efficacy in controlling pruriginous conditions ranging from chronic urticaria12,13 to atopic dermatitis14 to psoriasis.15 This evidence has spurred studies to evaluate if NB-UVB has a role in the management of uncontrolled cases of cutaneous and ISM.2,13,16,17 To date, the evidence has been promising. The majority of patients treated with this regimen report subjective reduction in pruritus in addition to clinical cutaneous disease burden.2,11 Also, laboratory analysis demonstrates decreased levels of tryptase in patients utilizing NB-UVB phototherapy.2 Thus far, the use of NB-UVB phototherapy in the treatment of pruriginous disorders such as ISM has not been associated with any severe side effects such as increased rates of anaphylaxis, though some research has suggested that this therapy may lower the threshold for patients to develop symptomatic dermographism.12 Overall, patients treated with NB-UVB phototherapy report improved quality of life related to more effective symptom control.16
Although ISM is currently considered an incurable chronic condition,6 this case illustrates that symptomatic management is possible, even in cases of long-standing, severe disease. Patients should still be encouraged to avoid triggering factors and be vigilant in preventing potential anaphylaxis. However, NB-UVB phototherapy provides a supplemental or alternative treatment choice when other therapies have failed. We hope that the success of NB-UVB demonstrated in this case provides further evidence that this light-based therapy is a valuable treatment option in mastocytosis patients with unremitting or poorly controlled symptoms.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012.
- Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
- Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
- Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
- Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
- Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
- Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
- Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
- Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
- Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
- Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
- Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
- Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
- Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
- Bolognia J, Jorizzo JL, Schaffer JV, eds. Dermatology. 3rd ed. St. Louis, MO: Mosby/Elsevier; 2012.
- Brazzelli V, Grasso V, Manna G, et al. Indolent systemic mastocytosis treated with narrow-band UVB phototherapy: study of five cases [published online May 13, 2011]. J Eur Acad Dermatol Venereol. 2012;26:465-469.
- Pardanani A, Lim KH, Lasho TL, et al. WHO subvariants of indolent mastocytosis: clinical details and prognostic evaluation in 159 consecutive adults. Blood. 2010;115:150-151.
- Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes [published online April 8, 2009]. Blood. 2009;114:937-951.
- Wolff K, Komar M, Petzelbauer P. Clinical and histopathological aspects of cutaneous mastocytosis. Leuk Res. 2001;25:519-528.
- Marone G, Spadaro G, Granata F, et al. Treatment of mastocytosis: pharmacologic basis and current concepts. Leuk Res. 2001;25:583-594.
- Pardanani A. How I treat patients with indolent and smoldering mastocytosis (rare conditions but difficult to manage)[published online February 20, 2013]. Blood. 2013;121:3085-3094.
- Hartmann K, Henz BM. Mastocytosis: recent advances in defining the disease. Br J Dermatol. 2001;144:682-695.
- Vega-Ruiz A, Cortes JE, Sever M, et al. Phase II study of imatinib mesylate as therapy for patients with systemic mastocytosis. Leuk Res. 2009;33:1481-1484.
- Lortholary O, Chandesris MO, Bulai Livideanu C, et al. Masitinib for treatment of severely symptomatic indolent systemic mastocytosis: a randomised, placebo-controlled, phase 3 study. Lancet. 2017;389:612-620.
- Godt O, Proksch E, Streit V, et al. Short-and long-term effectiveness of oral and bath PUVA therapy in urticaria pigmentosa and systemic mastocytosis. Dermatology. 1997;1:35-39.
- Berroeta L, Clark C, Ibbotson SH, et al. Narrow-band (TL-01) ultraviolet B phototherapy for chronic urticaria. Clin Exp Dermatol. 2004;29:91-99.
- Engin B, Ozdemir M, Balevi A, et al. Treatment of chronic urticaria with narrowband ultraviolet B phototherapy: a randomized controlled trial. Acta Derm Venereol. 2008;3:247-251.
- Meduri NB, Vandergriff T, Rasmussen H, et al. Phototherapy in the management of atopic dermatitis: a systemic review. Photodermatol Photoimmunol Photomed. 2007;23:106-112.
- Nguyen T, Gattu S, Pugashetti R, et al. Practice of phototherapy in the treatment of moderate-to severe psoriasis. Curr Probl Dermatol. 2009;38:59-78.
- Brazzelli V, Grassi S, Merante S, et al. Narrow-band UVB phototherapy and psoralen-ultraviolet A photochemotherapy in the treatment of cutaneous mastocytosis: a study in 20 patients. Photodermatol Photoimmunol Photomed. 2016;32:238-246.
- Prignano F, Troiano M, Lotti T. Cutaneous mastocytosis: successful treatment with narrowband ultraviolet B phototherapy. Clin Exp Dermatol. 2010;35:914-915.
Practice Points
- Patients with cutaneous lesions and symptoms consistent with mastocytosis should be worked up for potential systemic involvement.
- Symptoms of indolent systemic mastocytosis (ISM) include pruritus, flushing, palpitations, vomiting, and anaphylaxis in severe instances.
- Most patients respond well to antihistamines, antileukotriene agents, and oral corticosteroids during severe episodes of mast cell degranulation.
- Narrowband UVB is a safe, effective, and well-tolerated treatment option for symptom control in refractory ISM cases.

Hydralazine-Associated Cutaneous Vasculitis Presenting With Aerodigestive Tract Involvement
Hydralazine-induced antineutrophil cytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
RELATED ARTICLE: Sweet Syndrome Associated With Hydralazine-Induced Lupus Erythematosus
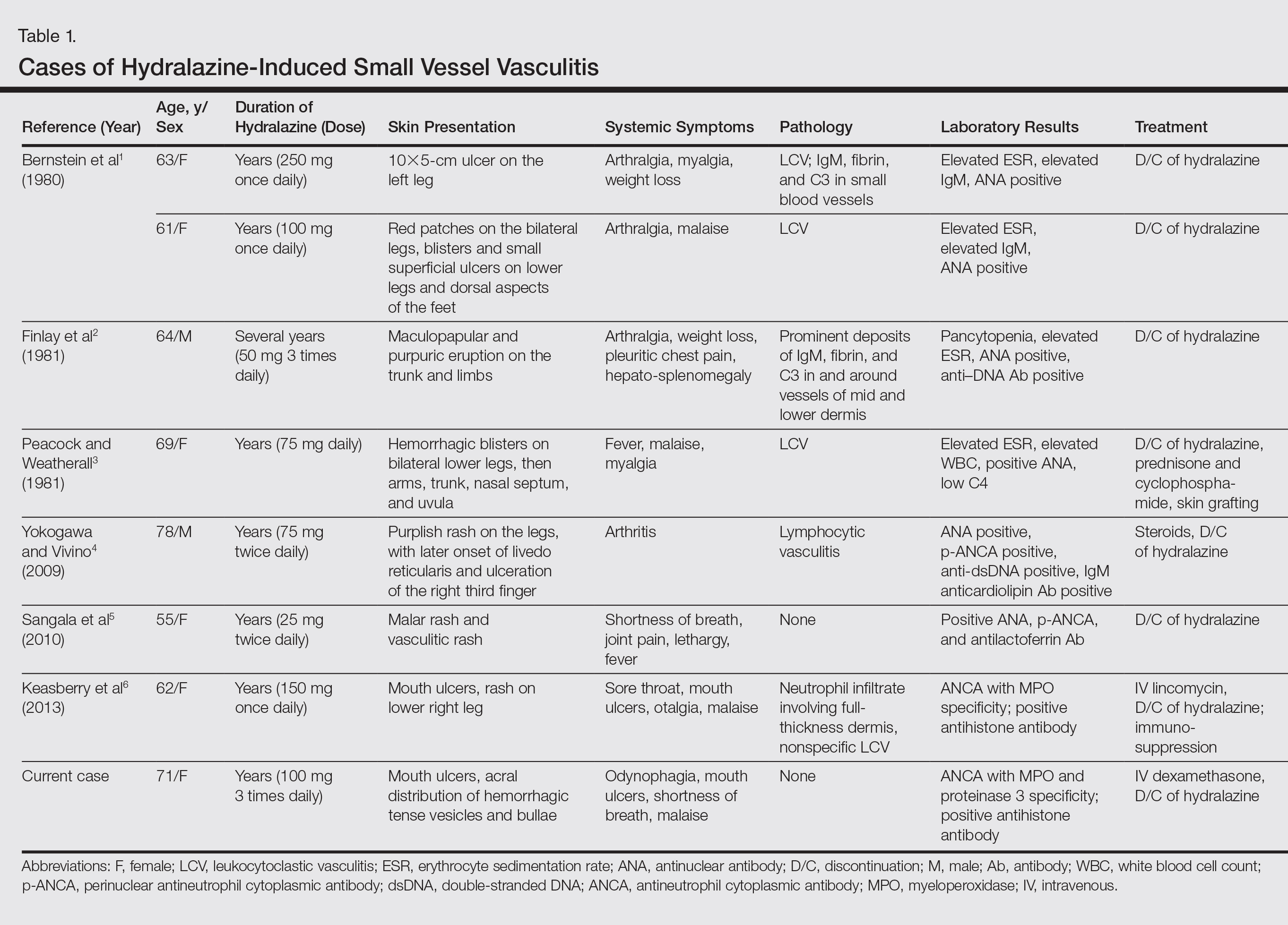
Case Report
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
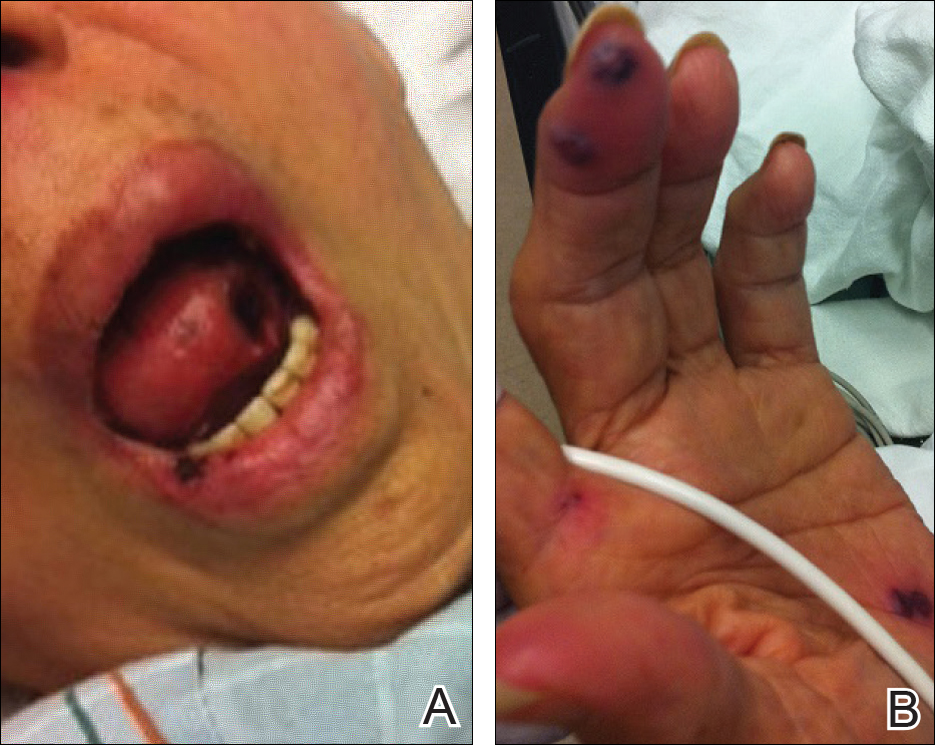
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virus screening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.

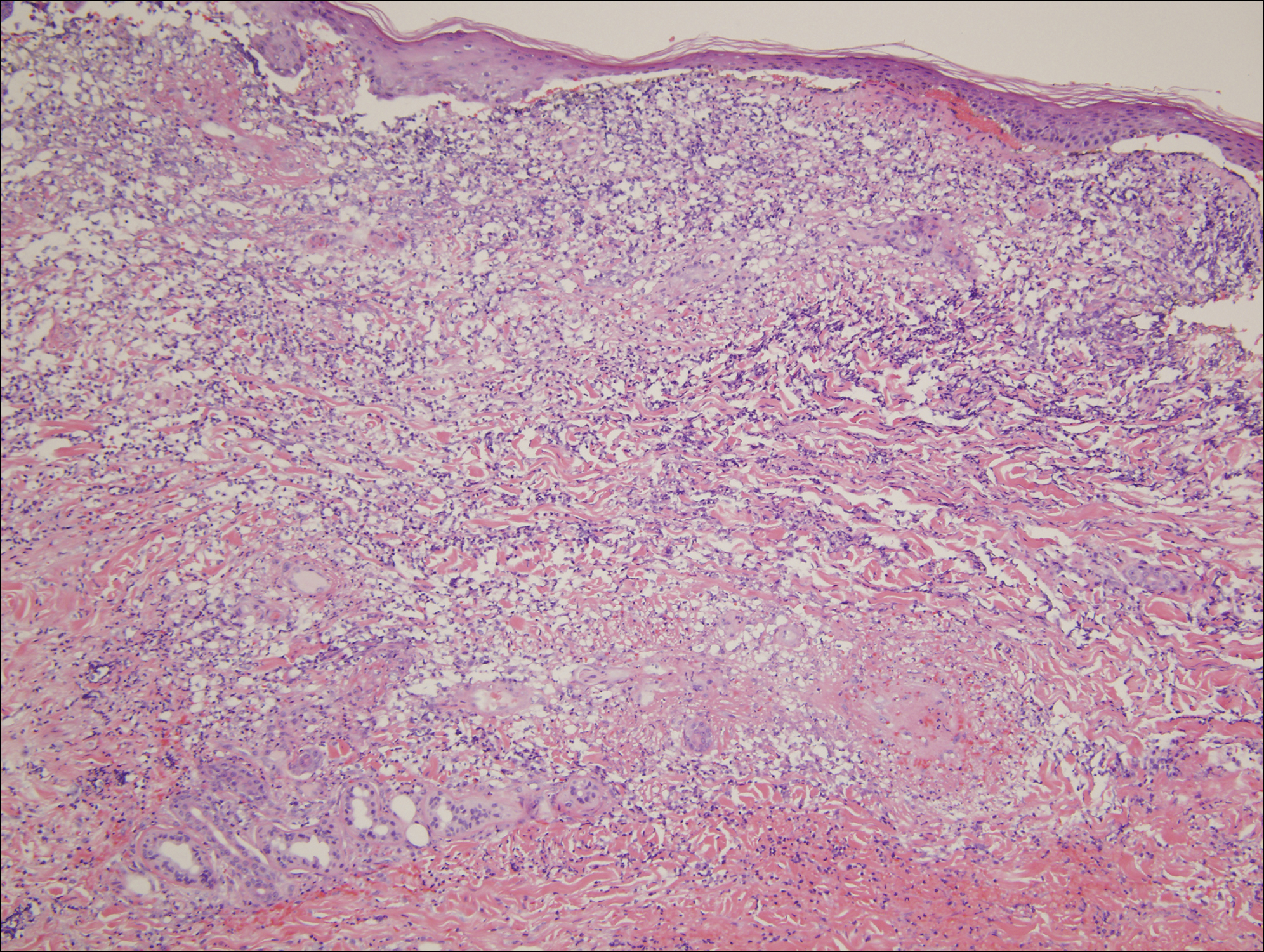
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
- Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
- Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
- Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1121-1122.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
- Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
- Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
- Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
Hydralazine-induced antineutrophil cytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
RELATED ARTICLE: Sweet Syndrome Associated With Hydralazine-Induced Lupus Erythematosus
Case Report
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virus screening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
Hydralazine-induced antineutrophil cytoplasmic antibody (ANCA)–positive vasculitis is a complex entity characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement. Although it is an established entity, a PubMed search of articles indexed for MEDLINE using the terms hydralazine vasculitis, ANCA positive vasculitis, and hydralazine associated vasculitis revealed a limited number of cases reported in the dermatologic literature (Table 1).1-6 We report a rare case of hydralazine-induced vasculitis associated with airway compromise and severe gastrointestinal tract bleeding.
RELATED ARTICLE: Sweet Syndrome Associated With Hydralazine-Induced Lupus Erythematosus
Case Report
A 71-year-old woman with a history of end-stage renal disease treated with hemodialysis, as well as hypertension, diabetes mellitus, and ischemic cardiomyopathy, presented to our emergency department with odynophagia, muscle weakness, shortness of breath, and a distinctive mucocutaneous eruption on the left eyelid, lips, and tongue of 2 days’ duration. Physical examination revealed an ill-appearing, afebrile, dyspneic woman with swelling of the left upper eyelid, conjunctival injection, ulcerations on the lips and tongue, and tense hemorrhagic vesicles, as well as vesiculopustules on the elbows, palms, fingers, lower legs, and toes (Figure 1). Given her dyspnea, flexible laryngoscopy was performed and revealed ulceration and edema involving the epiglottis, aryepiglottic folds, and arytenoids. The patient was intubated for airway protection and started on intravenous dexamethasone.
An extensive diagnostic workup commenced. Bacterial, viral, and fungal cultures of blood, skin tissue, and respiratory secretions, as well as human immunodeficiency virus screening, were all negative. Specifically, a tissue culture was performed on skin from the left thigh, viral culture and direct fluorescent antibody were performed on a vesicle on the right knee for herpes simplex virus and herpes zoster, and a superficial wound culture was taken from the left arm, all showing no growth. The patient’s home medications were reviewed and revealed she was currently taking hydralazine (100 mg 3 times daily), which was started approximately 2 years prior. Laboratory results revealed a positive antinuclear antibody titer of 1:320 (diffuse pattern), positive antihistone antibody, and positive ANCA with cytoplasmic and perinuclear accentuation (Table 2). Enzyme-linked immunosorbent assays showed IgG antibodies to myeloperoxidase (MPO) and proteinase 3 (Table 2). Skin biopsies from the right lower leg and right upper arm were compatible with necrotizing leukocytoclastic vasculitis characterized by mural and luminal fibrin deposition involving capillaries and venules of the superficial and deep dermis (Figure 2). The vessel walls were infiltrated by neutrophils with concomitant leukocytoclasia. Vessels in the mid dermis were occluded by cellular fibrin thrombi. Foci of neutrophilic interface dermatitis with subepidermal bulla formation were observed. Infectious stains were negative. On direct immunofluorescence, striking homogeneous mantles of staining of IgG were present within the cutaneous vasculature.
Because the infectious workup was negative and there was no other known instigating factor of vasculitis, concern for a drug-induced process prompted thorough review of the patient’s home medications and discontinuation of hydralazine. A diagnosis of hydralazine-associated cutaneous vasculitis was made when laboratory workup confirmed no underlying infectious process or rheumatologic condition and the medication known to cause her symptoms was on her medication list. The dexamethasone dose was increased, leading to rapid improvement of her mucocutaneous findings; however, on initiation of a steroid taper, she developed substantial gastrointestinal tract bleeding. An esophageal biopsy revealed a neutrophil-rich necrotizing process that essentially mirrored the cutaneous biopsy consistent with vasculitic involvement of the gastrointestinal tract. Steroids were again increased with resolution in gastrointestinal tract bleeding.
Comment
Our case highlights a distinct clinical presentation of hydralazine-induced ANCA-positive cutaneous vasculitis associated with severe involvement of the aerodigestive tract with gastrointestinal tract bleeding and airway compromise requiring intubation. Although discontinuation of hydralazine and in certain cases the addition of immunosuppressive agents may be adequate for resolution of symptoms, some cases progress despite treatment, leading to skin grafting, amputation, and death.3,4 Therefore, early recognition of hydralazine-induced cutaneous vasculitis and discontinuation of hydralazine are of paramount importance.
Reporting hydralazine-induced vasculitis is valuable because of its unique cutaneous, extracutaneous, and serologic findings. In our case, the cutaneous vasculitis presented clinically with acral hemorrhagic vesiculopustules and necrotic ulcerations resembling septic emboli, as opposed to classic lesions of palpable purpura typical of drug-induced leukocytoclastic vasculitis. Similar cutaneous findings have been described in other cases of hydralazine-induced vasculitis, indicating that this pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration is characteristic of this entity.1,3,6 In addition, involvement of the oral cavity, larynx, and gastrointestinal tract have been reported in cases of hydralazine-induced vasculitis, indicating mucosal involvement is an important feature of this disease.3,6 Although involvement of the oral mucosa, larynx, and acral sites appears to be characteristic, the exact basis for this site localization remains elusive. A precedent has been established for a similar pattern of intraoral and laryngeal involvement in other ANCA-positive vasculitic syndromes, most notably Wegener granulomatosis.7 Similarly, there are certain occlusive vasculitic syndromes that show acral localization including chronic septic vasculitis and vasculitis of collagen vascular disease.
Serologic trends can aid in diagnosing hydralazine-induced vasculitis. In theory, the nonspecific cutaneous findings, often in association with joint pain and positive antinuclear antibodies, may lead clinicians to the misdiagnosis of a connective tissue disease, such as systemic lupus erythematosus (SLE). However, unlike SLE, hydralazine-induced vasculitis is associated with positive ANCAs, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.8,9 Our patient had both positive perinuclear ANCA with cytoplasmic ANCA as well as a positive antihistone antibodies, a combination highly suggestive of a drug-induced process.
Despite the often acute presentation of hydralazine-induced ANCA-positive vasculitis, afflicted patients have characteristically been on the drug for a long period of time. Our patient is exemplary of most reported cases, as the time from initiation of hydralazine to onset of vasculitis was 2 years.4
The mechanism by which hydralazine causes this reaction is still a matter of debate. It seems clear that there are certain at-risk populations, such as slow acetylators and patients with an underlying hypercoagulable state. There are several theories by which hydralazine induces autoantibody formation. The first involves hydralazine metabolization by MPO released from activated neutrophils to form reactive intermediate metabolites. Such metabolites can be cytotoxic and may cause abnormal degradation of chromatin in susceptible individuals, leading to an autoimmune response against histone-DNA complexes. Alternatively, hydralazine may act as a hapten and bind to MPO, inducing an immune response against the hydralazine-MPO complex, with resultant formation of anti-MPO antibodies in susceptible individuals.10
Conclusion
Hydralazine-induced ANCA-positive vasculitis is a syndromic complex characterized by a distinctive clinical presentation comprising acral hemorrhagic vesiculopustules and necrotic ulcerations, at times with severe mucosal involvement along with a characteristic ANCA-positive serologic profile. Drug withdrawal is the cornerstone of therapy, and depending on the severity of symptoms, additional immunosuppressive treatment such as corticosteroids may be necessary. Older age of onset, female gender, and underlying autoimmune diatheses likely define important risk factors. With more recognition and reporting of this disease, further trends in both clinical and serological presentation will emerge.
- Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
- Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
- Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1121-1122.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
- Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
- Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
- Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
- Bernstein RM, Egerton-Vernon J, Webster J. Hydrallazine-induced cutaneous vasculitis. Br Med J. 1980;280:156-157.
- Finlay AY, Statham B, Knight AG. Hydrallazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1703-1704.
- Peacock A, Weatherall D. Hydralazine-induced necrotising vasculitis. Br Med J (Clin Res Ed). 1981;282:1121-1122.
- Yokogawa N, Vivino FB. Hydralazine-induced autoimmune disease: comparison to idiopathic lupus and ANCA-positive vasculitis. Mod Rheumatol. 2009;19:338-347.
- Sangala N, Lee RW, Horsfield C, et al. Combined ANCA-associated vasculitis and lupus syndrome following prolonged use of hydralazine: a timely reminder of an old foe. Int Urol Nephrol. 2010;42:503-506.
- Keasberry J, Frazier J, Isbel NM, et al. Hydralazine-induced anti-neutrophil cytoplasmic antibody-positive renal vasculitis presenting with a vasculitic syndrome, acute nephritis and a puzzling skin rash: a case report. J Med Case Rep. 2013;7:20.
- Wojciechowska J, Krajewski W, Krajewski P, et al. Granulomatosis with polyangiitis in otolaryngologist practice: a review of current knowledge. Clin Exp Otorhinolaryngol. 2016;9:8-13.
- Short AK, Lockwood CM. Antigen specificity in hydralazine associated ANCA positive systemic vasculitis. QJM. 1995;88:775-783.
- Nässberger L, Hultquist R, Sturfelt G. Occurrence of anti-lactoferrin antibodies in patients with systemic lupus erythematosus, hydralazine-induced lupus, and rheumatoid arthritis. Scand J Rheumatol. 1994;23:206-210.
- Cambridge G, Wallace H, Bernstein RM, et al. Autoantibodies to myeloperoxidase in idiopathic and drug-induced systemic lupus erythematosus and vasculitis. Br J Rheumatol. 1994;33:109-114.
Practice Points
- Hydralazine-induced small vessel vasculitis has a characteristic pattern of acral pseudoembolic vesiculopustules with necrosis and ulceration, along with involvement of the aerodigestive tract.
- Unlike systemic lupus erythematosus (SLE), hydralazine-induced vasculitis is associated with positive antineutrophil cytoplasmic antibodies, while antibodies against double-stranded DNA, a highly specific antibody for SLE, are uncommon.
- Increased recognition of the clinical and serological features of hydralazine-induced small vessel vasculitis may lead to earlier recognition of this disease and decreased time to discontinuation of hydralazine when appropriate.
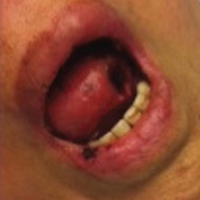
Systemic Interferon Alfa Injections for the Treatment of a Giant Orf
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
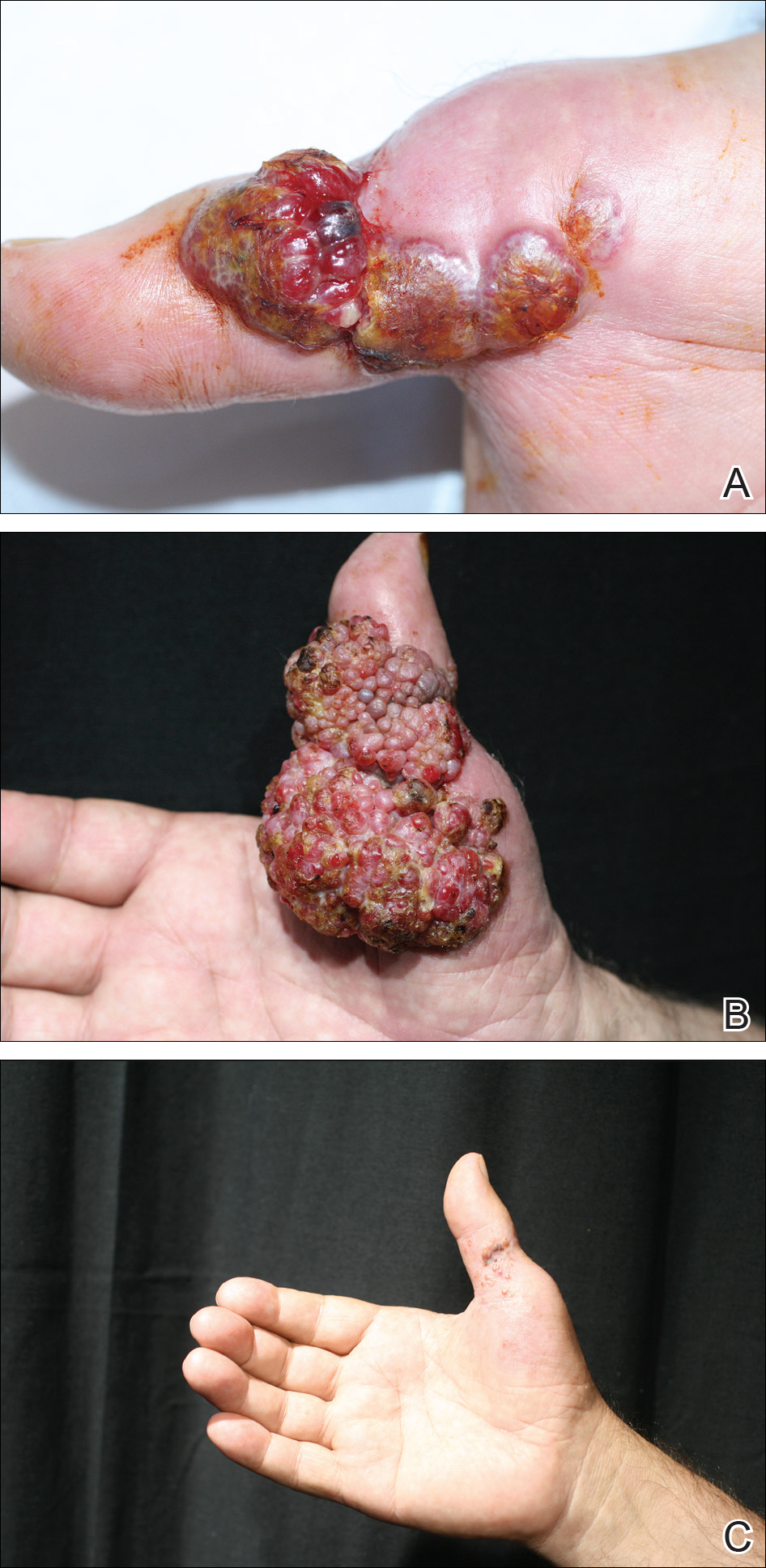
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
Orf, also known as ecthyma contagiosum, is a common viral zoonotic infection caused by a parapoxvirus. It is widespread among small ruminants such as sheep and goats, and it can be transmitted to humans by close contact with infected animals or contaminated fomites. It usually manifests as vesiculoulcerative lesions or nodules on the inoculation sites, mostly on the hands, but other sites such as the head and scalp occasionally may be involved.1 We report the case of an orf that proliferated dramatically and became giant after total excision. It was successfully treated with systemic interferon alfa-2a injections and imiquimod cream.
Case Report
A 68-year-old man presented with a rapidly enlarging mass on the left hand that developed 4 weeks prior after close contact with a freshly slaughtered sheep during an Islamic holiday in Turkey. His medical history was remarkable for chronic lymphocytic leukemia (CLL), which was diagnosed one year prior. The patient had been treated with systemic prednisolone and cyclophosphamide therapies, but his disease was in remission at the current presentation and he currently was not receiving any treatment. On physical examination, a 2-cm, exophytic, pinkish gray, weeping nodule was observed on the proximal aspect of the right thumb. Based on the clinical findings and typical anamnesis, a diagnosis of an orf was concluded. It was decided to monitor the patient without any intervention; however, because the lesion did not resolve and remained stable, he was referred to a plastic surgeon for surgical removal after 6 weeks of follow-up.
Histopathologic examination of the excision specimen revealed pseudoepitheliomatous hyperplasia, massive capillary proliferation, and viral cytopathic changes in keratinocytes characterized by ballooning degeneration and eosinophilic cytoplasmic inclusions, which was consistent with the clinical diagnosis of an orf. Unfortunately, the lesion relapsed rapidly following excision (Figure, A). Treatment with oral valacyclovir (1 g 3 times daily) and imiquimod cream 5% (3 times weekly) was initiated. However, this treatment was unsuccessful and was discontinued after 6 weeks, as the lesion kept growing, reaching a diameter of approximately 5 cm and becoming lobulated on the surface (Figure, B). Combination therapy was started with imiquimod cream 5% (3 times weekly) and intralesional interferon alfa-2a injections (3 million IU twice weekly). The injections were so painful that the patient refused further therapy after only 2 injections. The therapy was switched from intralesional to systemic subcutaneous injections of interferon alfa-2a (3 million IU twice weekly) with concomitant imiquimod cream 5% 3 times weekly. This treatment was well tolerated by our patient with no notable side effects, except for mild fever on the night of each injection. Three weeks after the commencement of systemic injections, remarkable healing of the lesions with reduced size and exudation was noted. The frequency of injections was decreased to once weekly, which was then discontinued after 6 weeks when the lesion totally resolved (Figure, C). At 12 months’ follow-up, there were no signs of relapse.
Comment
Orf is an occupational disease that usually develops in farmers, butchers, and veterinarians; however, epidemic outbreaks of human orf are commonly observed in Turkey after the feast of sacrifice, as many individuals have close contact with the animals during sacrification.2 In Turkey, orf is well recognized by dermatologists, and clinical diagnosis usually is not difficult.
Human orf has a self-limited course in which lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, such as our patient with CLL, orf lesions may be persistent, atypical, and giant, requiring early and effective treatment. Treatment options for giant orf tumors in immunocompromised individuals include surgical excision,3 cryotherapy, topical imiquimod,4,5 topical or intralesional cidofovir,6 and intralesional interferon alfa injections.7 According to our clinical observations, surgical interventions for treatment of orfs usually cause a delay in the natural healing process; however, because surgical excision is a recommended treatment option for exophytic and recalcitrant orfs, we decided to treat our patient with surgical excision, which resulted in rapid recurrence and massive proliferation. A similar case of giant orf that was aggravated after surgery has been reported.8 In light of these cases, it is our opinion that treatment options other than surgery may be reasonable.
Chronic lymphocytic leukemia may show features of both humoral and cell-mediated deficiency. Patients are known to be prone to viral infections such as varicella-zoster virus, herpes simplex virus, cytomegalovirus, and human papillomavirus. A giant orf infection on the background of CLL also has been described.9
Interferons were first discovered in 1957 and named after their ability to interfere with viral replication. They represent a family of cytokines that has an essential role in the innate immune response to virus infections. Because of their antiviral properties, recombinant forms of interferon alfa are widely used with success in the treatment of chronic hepatitis B and hepatitis C virus infections. A few other antiviral clinical applications of interferon alfa include infections caused by human herpesvirus 8 (the etiological agent in Kaposi sarcoma) and human papillomatosis virus (the etiological agent in juvenile laryngeal papillomatosis and condyloma acuminatum).10
In a report by Ran et al,7 intralesional interferon alfa injections were successfully used for treatment of giant orf lesions in an immunocompromised patient. As a result, we started treating the patient with intralesional interferon alfa-2a, but it was not well tolerated by our patient, as it was quite painful. We then decided to continue the therapy with systemic interferon alfa-2a injections, as we believed that it was a good option due to its antiviral, antiproliferative, and antiangiogenic properties. With the experimental combined therapy of systemic interferon alfa-2a and topical imiquimod, our patient achieved a complete response in 9 weeks (3 weeks of twice weekly injections and then 6 weeks of once weekly injections) and had no relapses during 12 months of follow-up.
Conclusion
We present a rare case of a giant orf treated with systemic interferon alfa-2a injections. Because intralesional injections are quite painful, systemic subcutaneous injections of interferon might be a good and safe alternative for recalcitrant orf lesions in immunocompromised patients. However, more studies and reports are needed to confirm its effectiveness and safety.
The 9th Cosmetic Surgery Forum will be held November 29-December 2, 2017, in Las Vegas, Nevada. Get more information at www.cosmeticsurgeryforum.com.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
- Gurel MS, Ozardali I, Bitiren M, et al. Giant orf on the nose. Eur J Dermatol. 2002;12:183-185.
- Uzel M, Sasmaz S, Bakaris S, et al. A viral infection of the hand commonly seen after the feast of sacrifice: human orf (orf of the hand). Epidemiol Infect. 2005;133:653-657.
- Ballanger F, Barbarot S, Mollat C, et al. Two giant orf lesions in a heart/lung transplant patient. Eur J Dermatol. 2006;16:284-286.
- Zaharia D, Kanitakis J, Pouteil-Noble C, et al. Rapidly growing orf in a renal transplant recipient: favourable outcome with reduction of immunosuppression and imiquimod. Transpl Int. 2010;23:E62-E64.
- Lederman ER, Green GM, DeGroot HE, et al. Progressive ORF virus infection in a patient with lymphoma: successful treatment using imiquimod. Clin Infect Dis. 2007;44:e100-e103.
- Geerinck K, Lukito G, Snoeck R, et al. A case of human orf in an immunocompromised patient treated successfully with cidofovir cream. J Med Virol. 2001;64:543-549.
- Ran M, Lee M, Gong J, et al. Oral acyclovir and intralesional interferon injections for treatment of giant pyogenic granuloma–like lesions in an immunocompromised patient with human orf. JAMA Dermatol. 2015;151:1032-1034.
- Key SJ, Catania J, Mustafa SF, et al. Unusual presentation of human giant orf (ecthyma contagiosum). J Craniofac Surg. 2007;18:1076-1078.
- Hunskaar S. Giant orf in a patient with chronic lymphocytic leukaemia. Br J Dermatol. 1986;114:631-634.
- Friedman RM. Clinical uses of interferons. Br J Clin Pharmacol. 2008;65:158-162.
Resident Pearl
- Human orf lesions spontaneously resolve in 4 to 8 weeks; however, in immunocompromised patients, orf lesions may be persistent, atypical, and giant. We observed that surgical interventions for treatment of orfs cause a delay in the natural healing process, and other treatment options such as subcutaneous interferon alfa-2a may be used.
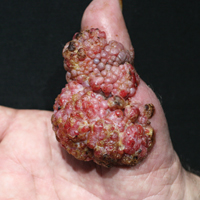
Perifollicular Papules on the Trunk
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
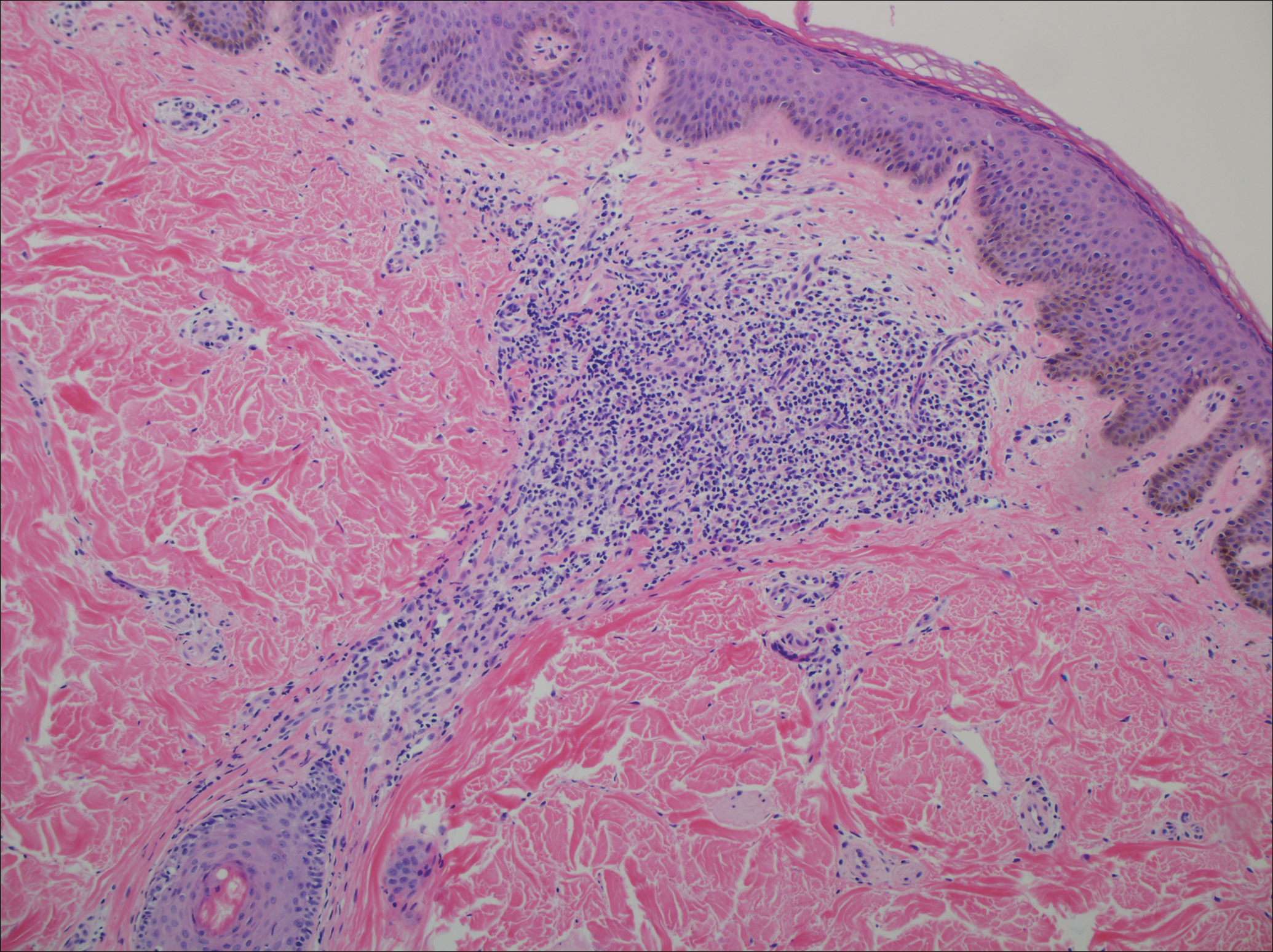
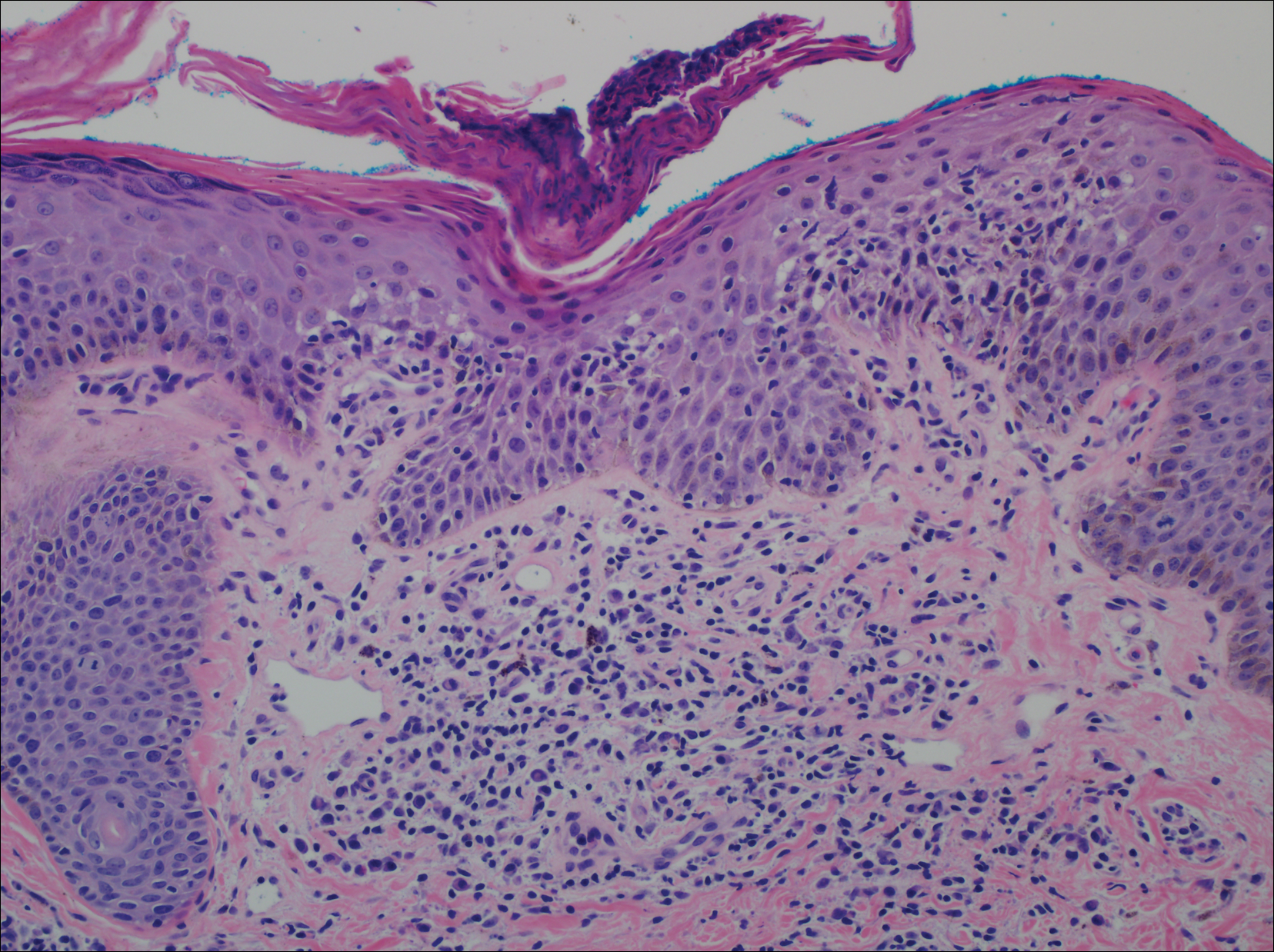
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
The Diagnosis: Disseminate and Recurrent Infundibulofolliculitis
A punch biopsy of a representative lesion on the trunk was performed. Histopathologic examination revealed a chronic lymphohistiocytic proliferation, focal spongiosis, and lymphocytic exocytosis primarily involving the isthmus of the hair follicle (Figure 1). At the follicular opening there was associated parakeratosis of the adjacent epidermis (Figure 2). Given these clinical and histopathological findings, a diagnosis of disseminate and recurrent infundibulofolliculitis (DRIF) was made.
Disseminate and recurrent infundibulofolliculitis was first described by Hitch and Lund1 in 1968 in a healthy 27-year-old black man as a widespread recurrent follicular eruption. Disseminate and recurrent infundibulofolliculitis usually affects young adult males with darkly pigmented skin.2,3 It has less commonly been described in children, females, and white individuals.3,4 Associations with atopy, systemic diseases, or medications are unknown.3-6 The onset usually is sudden and the disease course may be characterized by intermittent recurrences. Pruritus usually is reported but may be mild.5
Histopathology is characterized by spongiosis centered on the infundibulum of the hair follicle and a primarily lymphocytic inflammatory infiltrate. Neutrophils also may be identified.3 Disseminate and recurrent infundibulofolliculitis can be differentiated histologically from clinically similar entities such as keratosis pilaris, which has a keratin plug filling the infundibulum; lichen nitidus, which is characterized by a clawlike downgrowth of the rete ridges surrounding a central foci of inflammation; or folliculitis, which is characterized by perifollicular suppurative inflammation.
Treatment of DRIF is anecdotal and limited to case reports. Vitamin A alone or in combination with vitamin E has been reported to lead to some improvement.5 Tetracycline-class antibiotics, keratolytics, antihistamines, and topical retinoids have not been successful, and mixed results have been seen with topical steroids.5-7 There is a reported case of improvement with a 3-week regimen of psoralen plus UVA followed by twice-weekly maintenance.8 Promising results in the treatment of DRIF have been shown with oral isotretinoin once daily.3-5 Finally, DRIF may resolve independently6; therefore, treatment of DRIF should be addressed on a case-by-case basis.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis: report of a case. Arch Dermatol. 1968;97:432-435.
- Hitch JM, Lund HZ. Disseminate and recurrent infundibulo-folliculitis. Arch Dermatol. 1972;105:580-583.
- Calka O, Metin A, Ozen S. A case of disseminated and recurrent infundibulofolliculitis responsive to treatment with systemic isotretinoin. J Dermatol. 2002;29:431-434.
- Aroni K, Grapsa A, Agapitos E. Disseminate and recurrent infundibulofolliculitis: response to isotretinoin. J Drugs Dermatol. 2004;3:434-435.
- Aroni K, Aivaliotis M, Davaris P. Disseminated and recurrent infundibular folliculitis (D.R.I.F.): report of a case successfully treated with isotretinoin. J Dermatol. 1998;25:51-53.
- Owen WR, Wood C. Disseminate and recurrent infundibulofolliculitis. Arch Dermatol. 1979;115:174-175.
- Hinds GA, Heald PW. A case of disseminate and recurrent infundibulofolliculitis responsive to treatment with topical steroids. Dermatol Online J. 2008;14:11.
- Goihman-Yahr M. Disseminate and recurrent infundibulofolliculitis: response to psoralen plus UVA therapy. Int J Dermatol. 1999;38:75-78.
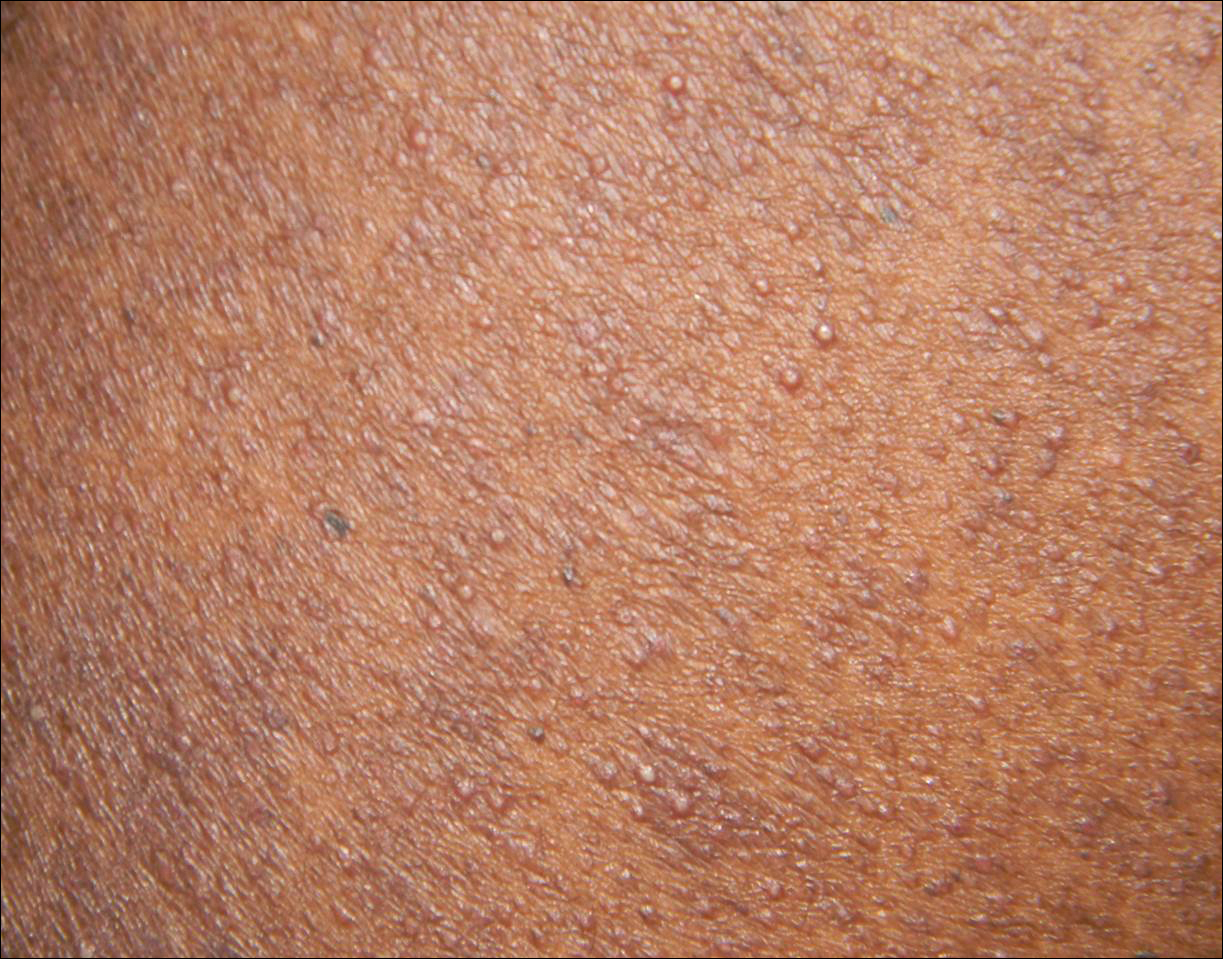
A 40-year-old black man presented with numerous perifollicular flesh-colored papules on the back, chest, abdomen, and proximal aspect of the arms of 6 years' duration. He described these lesions as persistent, nonpainful, and nonpruritic. He previously was treated with an unknown cream without any benefit. These lesions were cosmetically bothersome.