User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Cyanosis of the Foot
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.

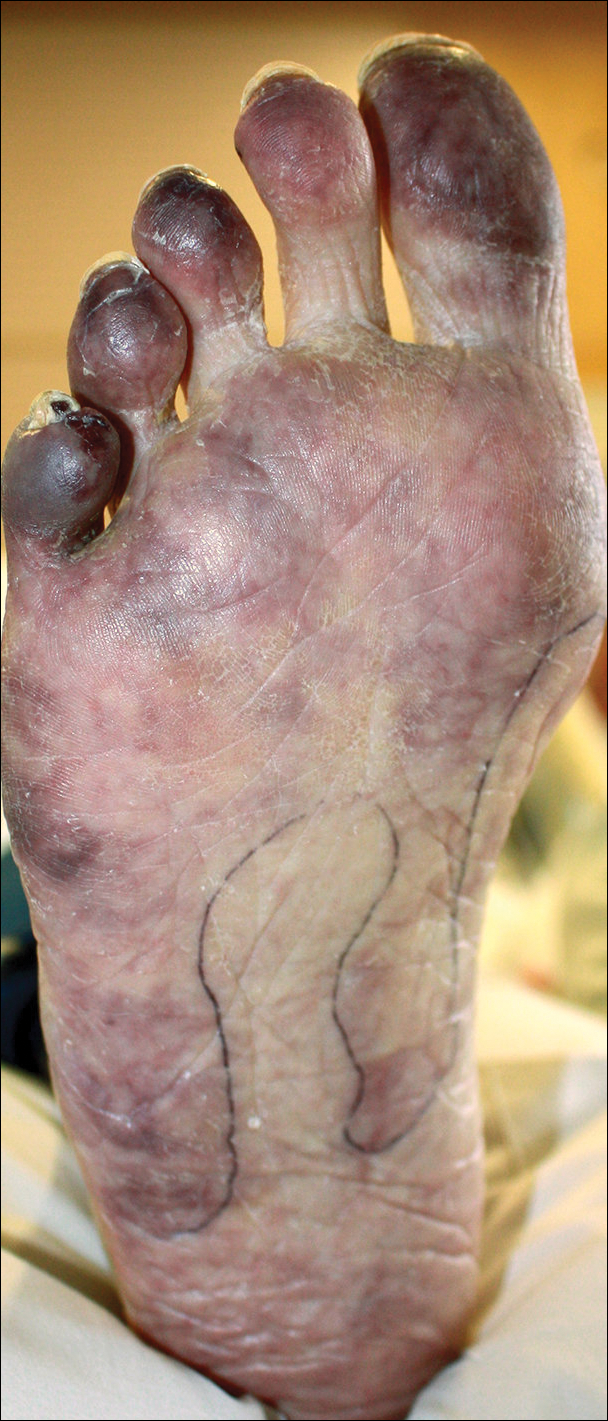
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
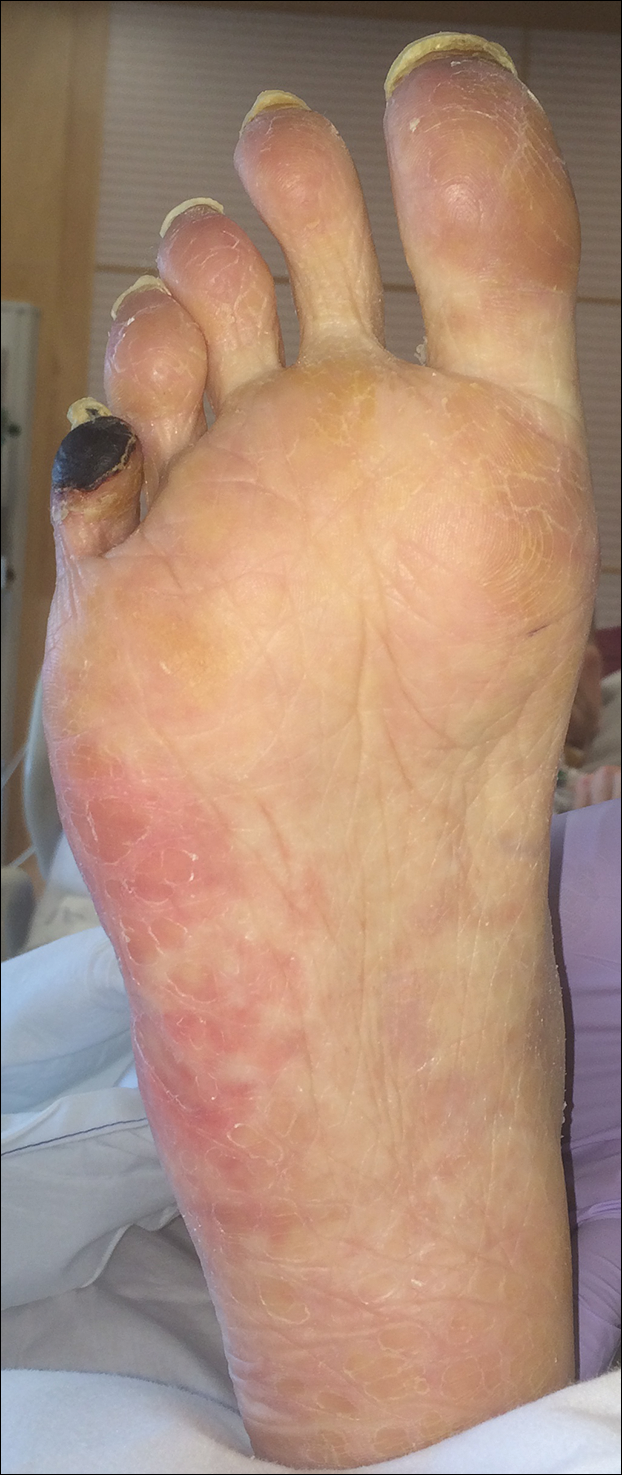
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
The Diagnosis: Antiphospholipid Antibody Syndrome
A biopsy demonstrated scattered intravascular thrombi in the dermis and subcutis, intact vascular walls, and scant lymphocytic inflammation in a background of stasis (Figure 1). A periodic acid-Schiff stain was negative for fungal elements and highlighted the intravascular thrombi. Histologic findings were consistent with thrombotic vasculopathy. On further laboratory workup, lupus anticoagulant studies, including a mixing study, diluted Russell viper venom test, and hexagonal phase phospholipid neutralization test, were abnormal. Titers of anticardiolipin and β2-glycoprotein I antibodies were elevated (anticardiolipin IgG, 137.7 calculated units [normal, <15 calculated units]; β2-glycoprotein I IgG, 256.4 calculated units [normal, <20 calculated units]). Tissue cultures showed no growth of microorganisms and studies for cryoglobulinemia were negative.
The patient was diagnosed with primary antiphospholipid syndrome (APS). He remained on anticoagulation therapy with fondaparinux as an inpatient and was treated with pulse-dose intravenous (IV) corticosteroids followed by a slow oral taper, daily plasmapheresis for 1 week, IV immunoglobulin (0.5 g/kg) for 3 doses, and 4 weekly doses of rituximab (375 mg/m2). His cutaneous findings slowly improved over the next several weeks (Figure 2).
Antiphospholipid syndrome is an autoimmune disorder characterized by thrombotic events and the presence of autoantibodies. The syndrome is defined by 2 major criteria: (1) the occurrence of at least 1 clinical feature of either an episode of vascular thrombosis or pregnancy morbidity such as unexplained fetal death beyond 10 weeks of gestation or recurrent unexplained pregnancy losses; and (2) the presence of at least 1 type of autoantibody, including lupus anticoagulant, anticardiolipin, or β2-glycoprotein antibodies, on 2 separate occasions at least 12 weeks apart.1 Antiphospholipid syndrome can either be primary with no identifiable associated rheumatologic disease or secondary to another autoimmune disease such as systemic lupus erythematosus. Cutaneous manifestations are common and frequently are the first sign of disease in 30% to 40% of patients.2 The most common skin finding is persistent livedo reticularis, which can be seen in 20% to 25% of patients. Patients also may develop skin necrosis, ulcerations, digital gangrene, splinter hemorrhages, and livedoid vasculopathy.2 Systemic manifestations of APS include thrombocytopenia, nephropathy, cognitive dysfunction, and cardiac valve abnormalities.
The exact pathogenesis of APS remains unknown. It is thought to be due to the combination of an inflammatory stimulus that has yet to be characterized in conjunction with autoantibodies that affect multiple target cells including monocytes, platelets, and endothelial cells, which results in activation of the complement system and clotting cascade.3 In rare cases, the disorder can progress to catastrophic antiphospholipid syndrome (CAPS), which requires fulfillment of 4 criteria: (1) evidence of involvement of 3 organs, tissues, or systems; (2) development of manifestations simultaneously or in less than 1 week; (3) laboratory confirmation of the presence of antiphospholipid antibodies; and (4) confirmation by histopathology of small vessel occlusion.4 Probable CAPS is diagnosed when 3 of 4 criteria are present. Our patient met criteria for probable CAPS, as his antibody titers remained elevated 15 weeks after initial presentation. Precipitating factors that can lead to CAPS are thought to include infection, surgical procedures, medications, or discontinuation of anticoagulation drugs.2 Although the mainstay of management of APS is anticoagulation therapy with warfarin and antiplatelet agents such as aspirin, first-line treatment of CAPS involves high-dose systemic glucocorticoids and plasma exchange. Intravenous immunoglobulin also may be employed in treatment. Data from the CAPS registry demonstrate a role for rituximab, an anti-CD20 antibody, at 375 mg/m2 weekly for 4 weeks (the regimen described in our case) or 1 g every 14 days for 2 sessions.5 A majority of the registry patients treated with rituximab recovered (75% [15/20]) and had no recurrent thrombosis (87% [13/15]) at follow-up.5 Data also are emerging on the role of eculizumab, an anti-C5 antibody that inhibits the terminal complement cascade, as a therapy in difficult-to-treat or refractory CAPS.6-8 The prognosis for CAPS patients without treatment is poor, and mortality has been reported in up to 44% of patients. However, with intervention mortality is reduced by more than 2-fold.9,10
It is important to recognize that acral cyanosis with persistent livedo reticularis and digital gangrene can be a presenting manifestation of APS. These cutaneous manifestations should prompt histologic evaluation for thrombotic vasculopathy in addition to serologic tests for APS autoantibodies. Although APS may be treated with anticoagulants and antiplatelet agents, CAPS may require more aggressive therapy with systemic steroids, plasma exchange, IV immunoglobulin, rituximab, and/or eculizumab.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
- Wilson WA, Gharavi AE, Koike T, et al. International consensus statement on preliminary classification criteria for definite antiphospholipid syndrome: report of an international workshop. Arthritis Rheum. 1999;42:1309-1311.
- Pinto-Almeida T, Caetano M, Sanches M, et al. Cutaneous manifestations of antiphospholipid syndrome: a review of the clinical features, diagnosis and management. Acta Reumatol Port. 2013;38:10-18.
- Meroni PL, Chighizola CB, Rovelli F, et al. Antiphospholipid syndrome in 2014: more clinical manifestations, novel pathogenic players and emerging biomarkers. Arthritis Res Ther. 2014;16:209.
- Asherson RA, Cervera R, de Grott PG, et al; Catastrophic Antiphospholipid Syndrome Registry Project Group. Catastrophic antiphospholipid syndrome: international consensus statement on classification criteria and treatment guidelines. Lupus. 2003;12:530-534.
- Berman H, Rodríguez-Pintó I, Cervera R, et al. Rituximab use in the catastrophic antiphospholipid syndrome: descriptive analysis of the CAPS registry patients receiving rituximab [published online June 15, 2013]. Autoimmun Rev. 2013;12:1085-1090.
- Shapira I, Andrade D, Allen SL, et al. Brief report: induction of sustained remission in recurrent catastrophic antiphospholipid syndrome via inhibition of terminal complement with eculizumab. Arthritis Rheum. 2012;64:2719-2723.
- Strakhan M, Hurtado-Sbordoni M, Galeas N, et al. 36-year-old female with catastrophic antiphospholipid syndrome treated with eculizumab: a case report and review of literature. Case Rep Hematol. 2014;2014:704371.
- Lonze BE, Zachary AA, Magro CM, et al. Eculizumab prevents recurrent antiphospholipid antibody syndrome and enables successful renal transplantation. Am J Transplant. 2014;14:459-465.
- Bucciarelli S, Espinosa G, Cervera R, et al. Mortality in the catastrophic antiphospholipid syndrome: causes of death and prognostic factors in a series of 250 patients. Arthritis Rheum. 2006;54:2568-2576.
- Asherson RA, Cervera R, Piette JC, et al. Catastrophic antiphospholipid syndrome. clinical and laboratory features of 50 patients. Medicine (Baltimore). 1998;77:195-207.
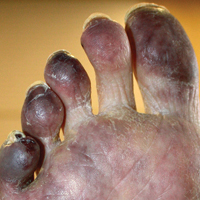
A man in his 50s with a medical history of arterial thrombosis of the right arm, multiple deep vein thromboses (DVTs) of the legs on long-term warfarin, ischemic stroke, atrial fibrillation, and peripheral arterial disease presented with discoloration of the right foot and increasing tenderness of 1 month's duration. There was no history of trauma or recent change in outpatient medications. A family history was notable for an aunt and 2 cousins with DVTs and protein S deficiency. Physical examination revealed livedo reticularis on the sole and lateral aspect of the right foot. There was violaceous discoloration of the volar aspects of all 5 toes and a focal area of ulceration on the fifth toe. Pulses were palpable bilaterally. Initial laboratory evaluation was notable for thrombocytopenia, and preliminary blood cultures revealed no growth of bacterial or fungal organisms. Imaging studies revealed increased arterial stenosis of the right leg as well as DVT of the right great saphenous vein. A punch biopsy of the right medial foot was performed for hematoxylin and eosin stain as well as tissue culture.
Assessing the Effectiveness of Knowledge-Based Interventions in Increasing Skin Cancer Awareness, Knowledge, and Protective Behaviors in Skin of Color Populations
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
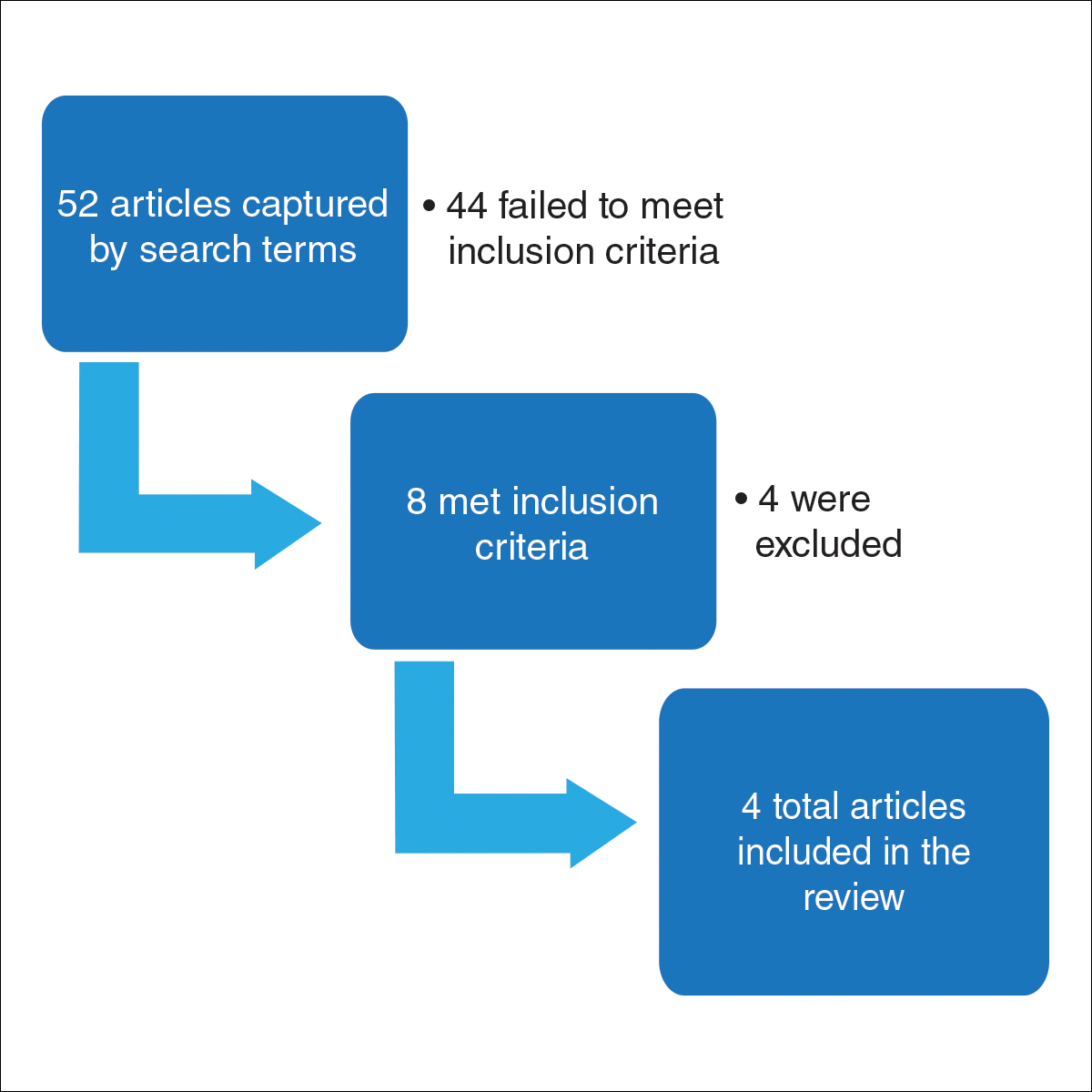
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
Results
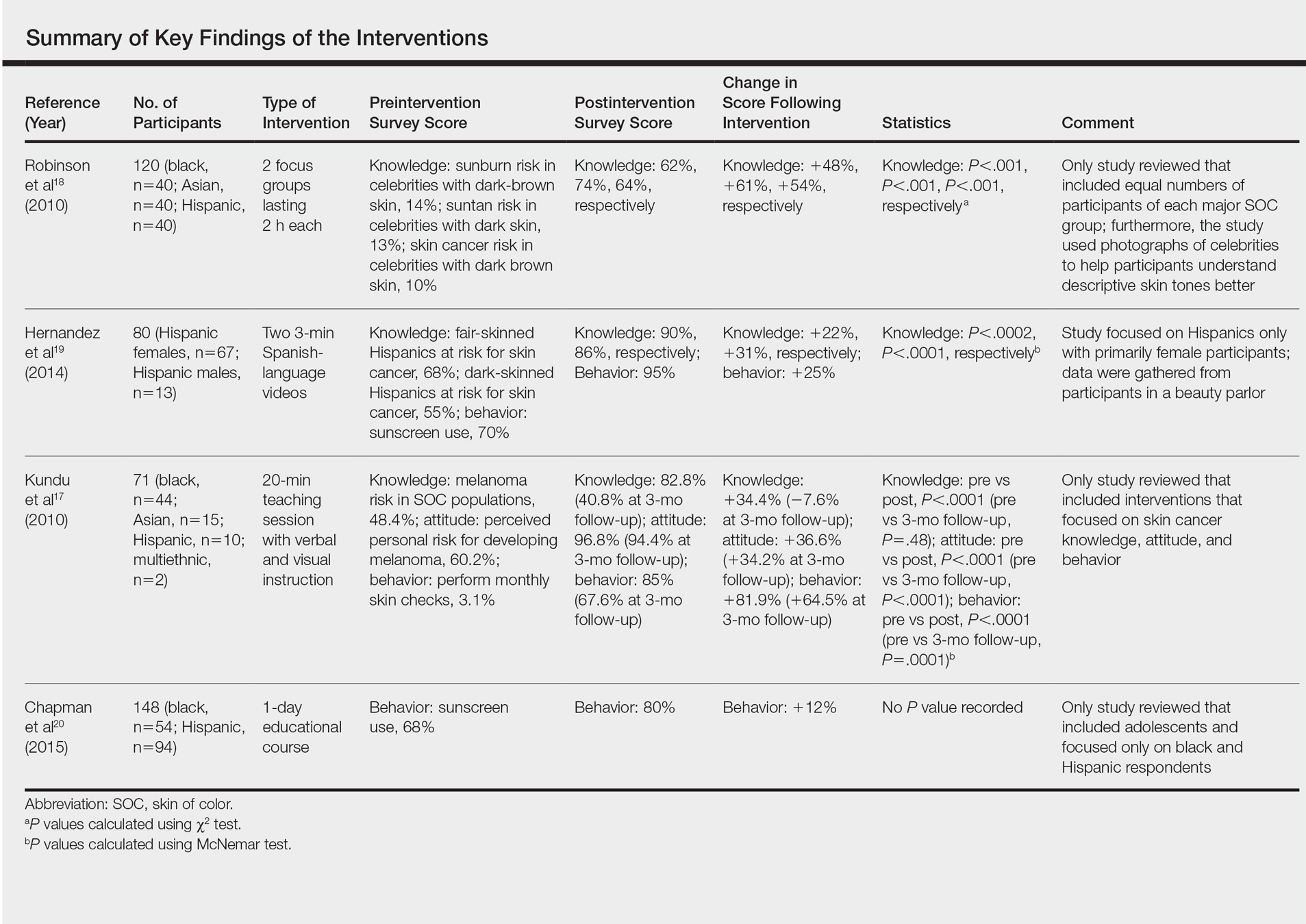
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
Results
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
Malignant melanoma, basal cell carcinoma, and squamous cell carcinoma account for approximately 40% of all neoplasms among the white population in the United States. Skin cancer is the most common malignancy in the United States.1 However, despite this occurrence, there are limited data regarding skin cancer in individuals with skin of color (SOC). The 5-year survival rates for melanoma are 58.2% for black individuals, 69.7% for Hispanics, and 70.9% for Asians compared to 79.8% for white individuals in the United States.2 Even though SOC populations have lower incidences of skin cancer—melanoma, basal cell carcinoma, and squamous cell carcinoma—they exhibit higher death rates.3-7 Nonetheless, no specific guidelines exist to address sun exposure and safety habits in SOC populations.6,8 Furthermore, current demographics suggest that by the year 2050, approximately half of the US population will be nonwhite.4 Paradoxically, despite having increased sun protection from greater amounts of melanin in their skin, black individuals are more likely to present with advanced-stage melanoma (eg, stage III/IV) compared to white individuals.8-12 Furthermore, those of nonwhite populations are more likely to present with more advanced stages of acral lentiginous melanomas than white individuals.13,14 Hispanics also face an increasing incidence of more invasive acral lentiginous melanomas.15 Overall, SOC patients have the poorest skin cancer prognosis, and the data suggest that the reason for this paradox is delayed diagnosis.1
Although skin cancer is largely a preventable condition, the literature suggests that lack of awareness of melanoma among ethnic minorities is one of the main reasons for their poor skin cancer prognosis.16 This lack of awareness decreases the likelihood that an SOC patient would be alert to early detection of cancerous changes.17 Because educating at-risk SOC populations is key to decreasing skin cancer risk, this study focused on determining the efficacy of major knowledge-based interventions conducted to date.1 Overall, we sought to answer the question, do knowledge-based interventions increase skin cancer awareness, knowledge, and protective behavior among people of color?
Methods
For this review, the Cochrane method of analysis was used to conduct a thorough search of PubMed articles indexed for MEDLINE (1994-2016), as well as a search of CINAHL (1997-2016), PsycINFO (1999-2016), and Web of Science (1965-2016), using a combination of more than 100 search terms including but not limited to skin cancer, skin of color, intervention, and ethnic skin. The search yielded a total of 52 articles (Figure). Following review, only 8 articles met inclusion criteria, which were as follows: (1) study was related to skin cancer in SOC patients, which included an intervention to increase skin cancer awareness and knowledge; (2) study included adult participants or adolescents aged 12 to 18 years; (3) study was written in English; and (4) study was published in a peer-reviewed journal. Of the remaining 8 articles, 4 were excluded due to the following criteria: (1) study failed to provide both preintervention and postintervention data, (2) study failed to provide quantitative data, and (3) study included participants who worked as health care professionals or ancillary staff. As a result, a total of 4 articles were analyzed and discussed in this review (Table).
Results
Robinson et al18 conducted 12 focus groups with 120 total participants (40 black, 40 Asian, and 40 Hispanic patients). Participants engaged in a 2-hour tape-recorded focus group with a moderator guide on melanoma and skin cancer. Furthermore, they also were asked to assess skin cancer risk in 5 celebrities with different skin tones. The statistically significant preintervention results of the study (χ2=4.6, P<.001) were as follows: only 2%, 4%, and 14% correctly reported that celebrities with a very fair skin type, a fair skin type, and very dark skin type, respectively, could get sunburn, compared to 75%, 76%, and 62% post-intervention. Additionally, prior to intervention, 14% of the study population believed that dark brown skin type could get sunburn compared to 62% of the same group postintervention. This study demonstrated that the intervention helped SOC patients better identify their ability to get sunburn and identify their skin cancer risk.18
Hernandez et al19 used a video-based intervention in a Hispanic community, which was in contrast to the multiracial focus group intervention conducted by Robinson et al.18 Eighty Hispanic individuals were recruited from beauty salons to participate in the study. Participants watched two 3-minute videos in Spanish and completed a preintervention and postintervention survey. The first video emphasized the photoaging benefits of sun protection, while the second focused on skin cancer prevention. Preintervention surveys indicated that only 54 (68%) participants believed that fair-skinned Hispanics were at risk for skin cancer, which improved to 72 (90%) participants postintervention. Furthermore, initially only 44 (55%) participants thought those with darker skin types could develop skin cancer, but this number increased to 69 (86%) postintervention. For both questions regarding fair and dark skin, the agreement proportion was significantly different between the preeducation and posteducation videos (P<.0002 for the fair skin question and P<.0001 for the dark skin question). This study greatly increased awareness of skin cancer risk among Hispanics,19 similar to the Robinson et al18 study.
In contrast to 2-hour focus groups or 3-minute video–based interventions, a study by Kundu et al17 employed a 20-minute educational class-based intervention with both verbal and visual instruction. This study assessed the efficacy of an educational tutorial on improving awareness and early detection of melanoma in SOC individuals. Photographs were used to help participants recognize the ABCDEs of melanoma and to show examples of acral lentiginous melanomas in white individuals. A total of 71 participants completed a preintervention questionnaire, participated in a 20-minute class, and completed a postintervention questionnaire immediately after and 3 months following the class. The study population included 44 black, 15 Asian, 10 Hispanic, and 2 multiethnic participants. Knowledge that melanoma is a skin cancer increased from 83.9% to 100% immediately postintervention (P=.0001) and 97.2% at 3 months postintervention (P=.0075). Additionally, knowledge that people of color are at risk for melanoma increased from 48.4% preintervention to 82.8% immediately postintervention (P<.0001). However, only 40.8% of participants retained this knowledge at 3 months postintervention. Because only 1 participant reported a family history of skin cancer, the authors hypothesized that the reason for this loss of knowledge was that most participants were not personally affected by friends or family members with melanoma. A future study with an appropriate control group would be needed to support this claim. This study shed light on the potential of class-based interventions to increase both awareness and knowledge of skin cancer in SOC populations.17
A study by Chapman et al20 examined the effects of a sun protection educational program on increasing awareness of skin cancer in Hispanic and black middle school students in southern Los Angeles, California. It was the only study we reviewed that focused primarily on adolescents. Furthermore, it included the largest sample size (N=148) analyzed here. Students were given a preintervention questionnaire to evaluate their awareness of skin cancer and current sun-protection practices. Based on these results, the investigators devised a set of learning goals and incorporated them into an educational pamphlet. The intervention, called “Skin Teaching Day,” was a 1-day program discussing skin cancer and the importance of sun protection. Prior to the intervention, 68% of participants reported that they used sunscreen. Three months after completing the program, 80% of participants reported sunscreen use, an increase of 12% prior to the intervention. The results of this study demonstrated the unique effectiveness and potential of pamphlets in increasing sunscreen use.20
Comment
Overall, various methods of interventions such as focus groups, videos, pamphlets, and lectures improved knowledge of skin cancer risk and sun-protection behaviors in SOC populations. Furthermore, the unique differences of each study provided important insights into the successful design of an intervention.
An important characteristic of the Robinson et al18 study was the addition of photographs, which allowed participants not only to visualize different skin tones but also provided them with the opportunity to relate themselves to the photographs; by doing so, participants could effectively pick out the skin tone that best suited them. Written SOC scales are limited to mere descriptions and thus make it more difficult for participants to accurately identify the tone that best fits them. Kundu et al17 used photographs to teach skin self-examination and ABCDEs for detection of melanoma. Additionally, both studies used photographs to demonstrate examples of skin cancer.17,18 Recent evidence suggests the use of visuals can be efficacious for improving skin cancer knowledge and awareness; a study in 16 SOC kidney transplant recipients found that the addition of photographs of squamous cell carcinoma in various skin tones to a sun-protection educational pamphlet was more effective than the original pamphlet without photographs.21
In contrast to the Robinson et al18 study and Hernandez et al19 study, the Kundu et al17 study showed photographs of acral lentiginous melanomas in white patients rather than SOC patients. However, SOC populations may be less likely to relate to or identify skin changes in skin types that are different from their own. This technique was still beneficial, as acral lentiginous melanoma is the most common type of melanoma in SOC populations. Another benefit of the study was that it was the only study reviewed that included a follow-up postintervention questionnaire. Such data is useful, as it demonstrates how muchinformation is retained by participants and may be more likely to predict compliance with skin cancer protective behaviors.17
The Hernandez et al19 study is unique in that it was the only one to include an educational intervention entirely in Spanish, which is important to consider, as language may be a hindrance to participants’ understanding in the other studies, particularly Hispanics, possibly leading to a lack of information retention regarding sun-protective behaviors. Furthermore, it also was the only study to utilize videos as a method for interventions. The 3-minute videos demonstrated that interventions could be efficient as compared to the 2-hour in-class intervention used by Robinson et al18 and the 20-minute intervention used by Kundu et al.17 Additionally, videos also could be more cost-effective, as incentives for large focus groups would no longer be needed. Furthermore, in the Hernandez et al19 study, there was minimal to no disruption in the participants’ daily routine, as the participants were getting cosmetic services while watching the videos, perhaps allowing them to be more attentive. In contrast, both the Robinson et al18 and Kundu et al17 studies required time out from the participants’ daily schedules. In addition, these studies were notably longer than the Hernandez et al19 study. The 8-hour intervention in the Chapman et al20 study also may not be feasible for the general population because of its excessive length. However, the intervention was successful among the adolescent participants, which suggested that shorter durations are effective in the adult population and longer interventions may be more appropriate for adolescents because they benefit from peer activity.
Despite the success of the educational interventions as outlined in the 4 studies described here, a major epidemiologic flaw is that these interventions included only a small percentage of the target population. The largest total number of adults surveyed and undergoing an intervention in any of the populations was only 120.17 By failing to reach a substantial proportion of the population at risk, the number of preventable deaths likely will not decrease. The authors believe a larger-scale intervention would provide meaningful change. Australia’s SunSmart campaign to increase skin cancer awareness in the Australian population is an example of one such large-scale national intervention. The campaign focused on massive television advertisements in the summer to educate participants about the dangers of skin cancer and the importance of protective behaviors. Telephone surveys conducted from 1987 to 2011 demonstrated that more exposure to the advertisements in the SunSmart campaign meant that individuals were more likely to use sunscreen and avoid sun exposure.22 In the United States, a similar intervention would be of great benefit in educating SOC populations regarding skin cancer risk. Additionally, dermatology residents need to be adequately trained to educate patients of color about the risk for skin cancer, as survey data indicated more than 80% of Australian dermatologists desired more SOC teaching during their training and 50% indicated that they would have time to learn it during their training if offered.23 Furthermore, one study suggested that future interventions must include primary-, secondary-, and tertiary-prevention methods to effectively reduce skin cancer risk among patients of color.24 Primary prevention involves sun avoidance, secondary prevention involves detecting cancerous lesions, and tertiary prevention involves undergoing treatment of skin malignancies. However, increased knowledge does not necessarily mean increased preventative action will be employed (eg, sunscreen use, wearing sun-protective clothing and sunglasses, avoiding tanning beds and excessive sun exposure). Additional studies that demonstrate a notable increase in sun-protective behaviors related to increased knowledge are needed.
Because retention of skin cancer knowledge decreased in several postintervention surveys, there also is a dire need for continuing skin cancer education in patients of color, which may be accomplished through a combination effort of television advertisement campaigns, pamphlets, social media, community health departments, or even community members. For example, a pilot program found that Hispanic lay health workers who are educated about skin cancer may serve as a bridge between medical providers and the Hispanic community by encouraging individuals in this population to get regular skin examinations from a physician.25 Overall, there are currently gaps in the understanding and treatment of skin cancer in people of color.26 Identifying the advantages and disadvantages of all relevant skin cancer interventions conducted in the SOC population will hopefully guide future studies to help close these gaps by allowing others to design the best possible intervention. By doing so, researchers can generate an intervention that is precise, well-informed, and effective in decreasing mortality rates from skin cancer among SOC populations.
Conclusion
All of the studies reviewed demonstrated that instructional and educational interventions are promising methods for improving either knowledge, awareness, or safe skin practices and sun-protective behaviors in SOC populations to differing degrees (Table). Although each of the 4 interventions employed their own methods, they all increased 1 or more of the 3 aforementioned concepts—knowledge, awareness, or safe skin practices and sun-protective behaviors—when comparing postsurvey to presurvey data. However, the critically important message derived from this research is that there is a tremendous need for a substantial large-scale educational intervention to increase knowledge regarding skin cancer in SOC populations.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
- Agbai ON, Buster K, Sanchez M, et al. Skin cancer and photoprotection in people of color: a review and recommendations for physicians and the public. J Am Acad Dermatol. 2014;70:748-762.
- Cormier JN, Xing Y, Ding M, et al. Ethnic differences among patients with cutaneous melanoma. Arch Intern Med. 2006;166:1907-1914.
- Gloster HM Jr, Neal K. Skin cancer in skin of color. J Am Acad Dermatol. 2006;55:741-760.
- Dawes SM, Tsai S, Gittleman H, et al. Racial disparities in melanoma survival. J Am Acad Dermatol. 2016;75:983-991.
- Byrd KM, Wilson DC, Hoyler SS, et al. Advanced presentation of melanoma in African Americans. J Am Acad Dermatol. 2004;50:21-24.
- Hu S, Parmet Y, Allen G, et al. Disparity in melanoma: a trend analysis of melanoma incidence and stage at diagnosis among whites, Hispanics, and blacks in Florida. Arch Dermatol. 2009;145:1369-1374.
- Wu XC, Eide MJ, King J, et al. Racial and ethnic variations in incidence and survival of cutaneous melanoma in the United States, 1999-2006. J Am Acad Dermatol. 2011;65(5, suppl 1):S26-S37.
- Byrd-Miles K, Toombs EL, Peck GL. Skin cancer in individuals of African, Asian, Latin-American, and American-Indian descent: differences in incidence, clinical presentation, and survival compared to Caucasians. J Drugs Dermatol. 2007;6:10-16.
- Hu S, Soza-Vento RM, Parker DF, et al. Comparison of stage at diagnosis of melanoma among Hispanic, black, and white patients in Miami-Dade County, Florida. Arch Dermatol. 2006;142:704-708.
- Hu S, Parker DF, Thomas AG, et al. Advanced presentation of melanoma in African Americans: the Miami-Dade County experience. J Am Acad Dermatol. 2004;5:1031-1032.
- Bellows CF, Belafsky P, Fortgang IS, et al. Melanoma in African-Americans: trends in biological behavior and clinical characteristics over two decades. J Surg Oncol. 2001;78:10-16.
- Pritchett EN, Doyle A, Shaver CM, et al. Nonmelanoma skin cancer in nonwhite organ transplant recipients. JAMA Dermatol. 2016;152:1348-1353.
- Shin S, Palis BE, Phillips JL, et al. Cutaneous melanoma in Asian-Americans. J Surg Oncol. 2009;99:114-118.
- Stubblefield J, Kelly B. Melanoma in non-caucasian populations. Surg Clin North Am. 2014;94:1115-1126.
- Bradford PT, Goldstein AM, McMaster ML, et al. Acral lentiginous melanoma: incidence and survival patterns in the United States, 1986-2005. Arch Dermatol. 2009;145:427-434.
- Pichon LC, Corral I, Landrine H, et al. Perceived skin cancer risk and sunscreen use among African American adults. J Health Psychol. 2010;15:1181-1189.
- Kundu RV, Kamaria M, Ortiz S, et al. Effectiveness of a knowledge-based intervention for melanoma among those with ethnic skin. J Am Acad Dermatol. 2010;62:777-784.
- Robinson JK, Joshi KM, Ortiz S, et al. Melanoma knowledge, perception, and awareness in ethnic minorities in Chicago: recommendations regarding education. Psychooncology. 2010;20:313-320.
- Hernandez C, Wang S, Abraham I, et al. Evaluation of educational videos to increase skin cancer risk awareness and sun safe behaviors among adult Hispanics. J Cancer Educ. 2014;29:563-569.
- Chapman LW, Ochoa A, Tenconi F, et al. Dermatologic health literacy in underserved communities: a case report of south Los Angeles middle schools. Dermatol Online J. 2015;21. pii:13030/qt8671p40n.
- Yanina G, Gaber R, Clayman ML, et al. Sun protection education for diverse audiences: need for skin cancer pictures. J Cancer Educ. 2015;30:187-189.
- Dobbinson SJ, Volkov A, Wakefield MA. Continued impact of sunsmart advertising on youth and adults’ behaviors. Am J Prev Med. 2015;49:20-28.
- Rodrigues MA, Ross AL, Gilmore S, et al. Australian dermatologists’ perspective on skin of colour: results of a national survey [published online December 9, 2016]. Australas J Dermatol. doi:10.1111/ajd.12556.
- Jacobsen A, Galvan A, Lachapelle CC, et al. Defining the need for skin cancer prevention education in uninsured, minority, and immigrant communities. JAMA Dermatol. 2016;152:1342-1347.
- Hernandez C, Kim H, Mauleon G, et al. A pilot program in collaboration with community centers to increase awareness and participation in skin cancer screening among Latinos in Chicago. J Cancer Educ. 2013;28:342-345.
- Kailas A, Solomon JA, Mostow EN, et al. Gaps in the understanding and treatment of skin cancer in people of color. J Am Acad Dermatol. 2016;74:144-149.
Practice Points
- Patients of color should be informed that they are at risk for skin cancer including melanoma.
- Patients of color should be taught to identify suspicious skin lesions including the ABCDEs of melanoma.
- Patients of color should be instructed to perform self-body skin examinations, especially of the palms and soles, for any evolving skin lesions. Patients should be instructed on the importance of visiting a physician for an evolving or suspicious mole or lesion.
Cosmetic Corner: Dermatologists Weigh in on Postprocedural Makeup
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.
To improve patient care and outcomes, leading dermatologists offered their recommendations on postprocedural makeup. Consideration must be given to:
- Dual Action Redness Relief
PCA Skin
“This product is great immediately after laser treatment or filler/botulinum toxin injections to reduce postprocedural redness.”— Gary Goldenberg, MD, New York, New York
- Isdinceutics Skin Drops
ISDIN
“This product is great to reduce or camouflage postprocedural bruising or redness.”—Gary Goldenberg, MD, New York, New York
- Oxygenating Foundation
Oxygenetix
“This is my favorite postprocedural makeup. Originally designed for burn victims, this makeup has botanicals, SPF, and is water resistant and soothing.”—Jeannette Graf, MD, Great Neck, New York
- Quick-Fix Concealer Stick
Dermablend
“This product is customized to match your patient’s skin type. It’s great at covering up purpura postprocedure.”—Shari Lipner, MD, PhD, New York, New York
“I love Dermablend because it can essentially camouflage anything postprocedure, getting patients back to work or their social activities.”— Jerome Potozkin, MD, Danville, California
Cutis invites readers to send us their recommendations. Pigment corrector, lip plumper, moisturizers for men, and wet skin moisturizers will be featured in upcoming editions of Cosmetic Corner. Please e-mail your recommendation(s) to the Editorial Office.
Disclaimer: Opinions expressed herein do not necessarily reflect those of Cutis or Frontline Medical Communications Inc. and shall not be used for product endorsement purposes. Any reference made to a specific commercial product does not indicate or imply that Cutis or Frontline Medical Communications Inc. endorses, recommends, or favors the product mentioned. No guarantee is given to the effects of recommended products.

Sweet Syndrome Induced by Oral Acetaminophen-Codeine Following Repair of a Facial Fracture
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
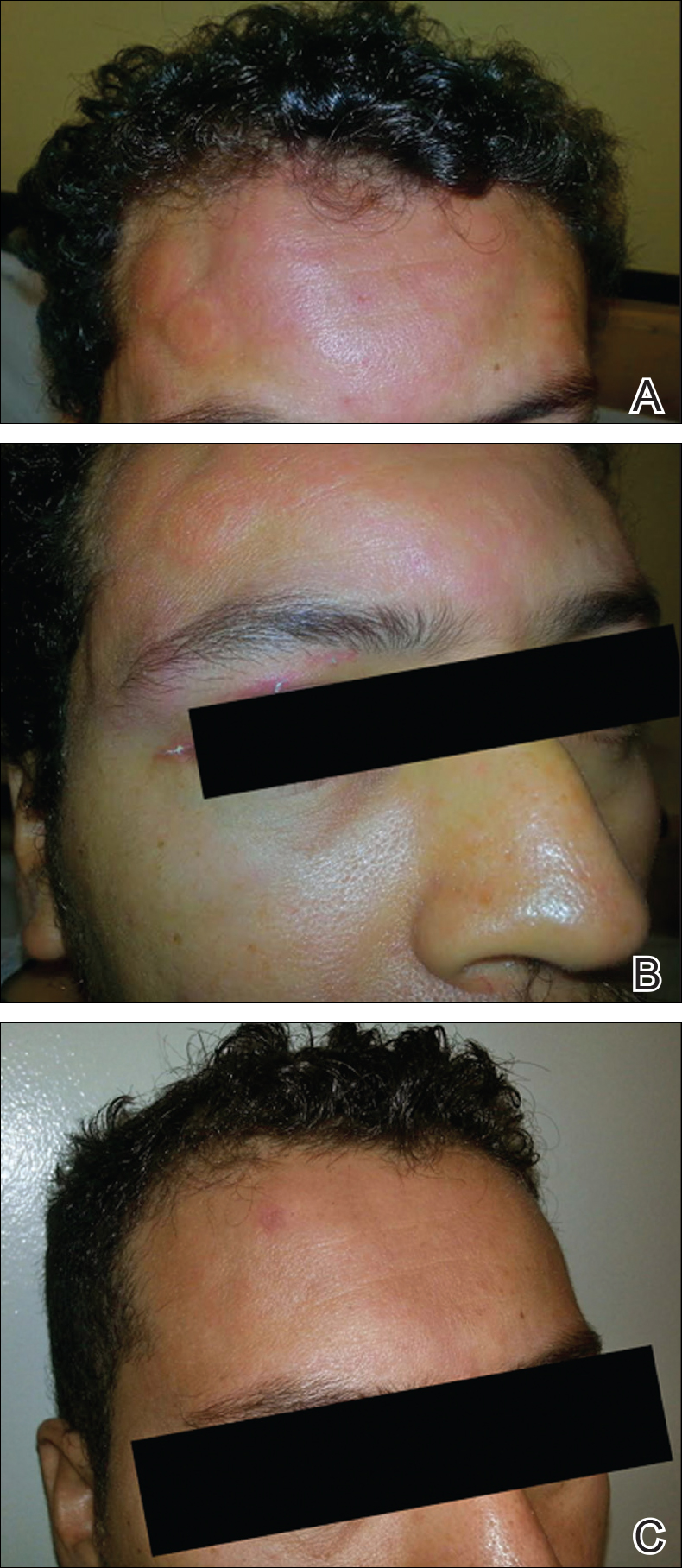
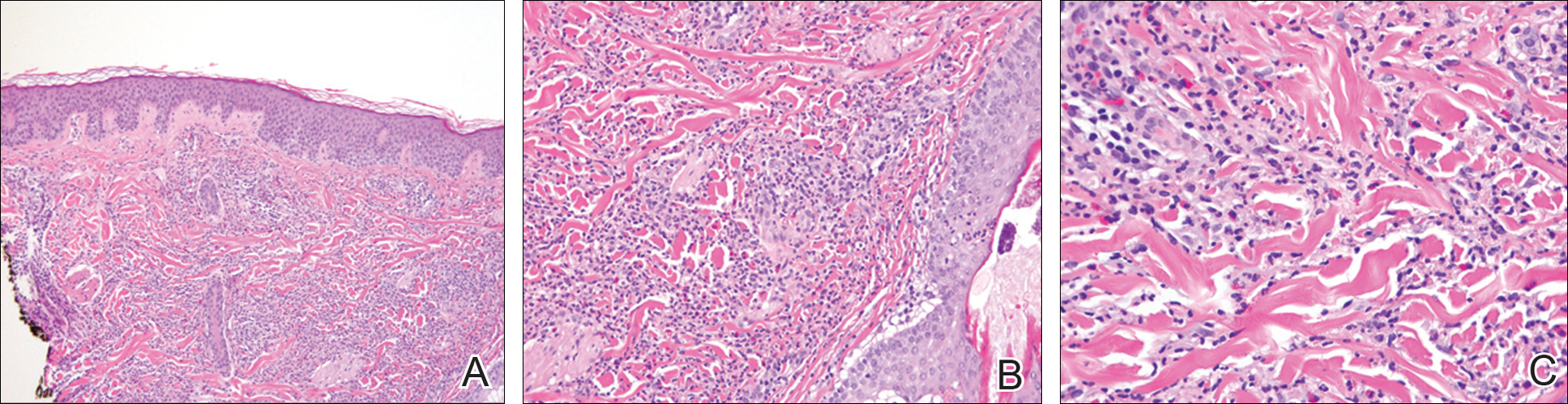
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
In 1964, Sweet1 described 8 women with acute onset of fever and erythematous plaques associated with a nonspecific infection of the respiratory or gastrointestinal tract. The lesions were histologically characterized by a neutrophilic infiltrate, and the author named the constellation of findings acute febrile neutrophilic dermatosis.1 In 1968, Whittle et al2 reported on similar cases and coined the term Sweet syndrome (SS).
Although the etiology and pathogenesis of SS remain unknown, several theories have been proposed. Because SS often is preceded by a respiratory or gastrointestinal tract infection, it has been postulated that it may represent a hypersensitivity reaction or may be related to local or systemic dysregulation of cytokine secretion.3,4 In addition to respiratory or gastrointestinal tract infections, SS has been reported in association with malignancies, autoimmune diseases, drugs, vaccines, pregnancy, inflammatory bowel disease, and chemotherapy. It also may be idiopathic.5
The eruption of SS manifests as erythematous, indurated, and sharply demarcated plaques or nodules that typically favor the head, neck, and arms, with a particularly strong predilection for the dorsal aspects of the hands.6 Plaques and nodules are histologically characterized by a diffuse dermal neutrophilic infiltrate, papillary dermal edema, neutrophilic spongiosis, subcorneal pustules, and leukocytoclasia. Vasculitic features are not seen.7 The eruption typically resolves spontaneously in 5 to 12 weeks but recurs in approximately 30% of cases.8 Relatively common extracutaneous findings include ocular involvement, arthralgia, myalgia, and arthritis.4,9 Both cutaneous and extracutaneous findings typically are responsive to prednisone at a dosage of 0.5 to 1 mg/kg daily for 4 to 6 weeks. Prolonged low-dose prednisone for 2 to 3 additional months may be necessary to suppress recurrence.8 Potassium iodide at 900 mg daily may be used as an alternative regimen.3,8
Sweet syndrome is divided into 5 subcategories based on the underlying etiology: (1) classic or idiopathic, (2) paraneoplastic, (3) inflammatory and/or autoimmune disease related, (4) pregnancy related, and (5) drug induced.3 Although drug-induced SS comprises the minority of total cases (<5%), its reported incidence has been rising in recent years and has been associated with an escalating number of medications.10 We report a rare case of SS induced by administration of oral acetaminophen-codeine.
Case Report
A 32-year-old man with a history of diabetes mellitus underwent postoperative repair of a facial fracture. The patient was administered an oral acetaminophen-codeine suspension for postoperative pain control. One week later, he developed a painful eruption on the forehead and presented to the emergency department. He was prescribed acetaminophen-codeine 300/30-mg tablets every 6 hours in addition to hydrocortisone cream 1% applied every 6 hours. After this reintroduction of oral acetaminophen-codeine, he experienced intermittent fevers and an exacerbation of the initial cutaneous eruption. The patient presented for a second time 2 days after being seen in the emergency department and a dermatology consultation was obtained.
At the time of consultation, the patient was noted to have injected conjunctiva and erythematous, well-demarcated, and indurated plaques on the forehead with associated pain and burning (Figures 1A and 1B). Additional erythematous annular plaques were found on the palms, arms, and right knee. Laboratory workup revealed only mild anemia on complete blood cell count with a white blood cell count of 10.1×109/L (reference range, 4.5–11.0×109/L), hemoglobin of 12.9 g/dL (reference range, 14.0–17.4 g/dL), and hematocrit of 37.3% (reference range, 41%–50%). The platelet count was 284×103/µL (reference range, 150–350×103/µL). Basic metabolic panel was notable for an elevated glucose level of 418 mg/dL (reference range, 70–110 mg/dL). The most recent hemoglobin A1C (several months prior) was notable at 14.7% of total hemoglobin (reference range, 4%–7% of total hemoglobin). A 4-mm punch biopsy of the right side of the forehead demonstrated minimal to mild papillary dermal edema and a diffuse dermal neutrophilic infiltrate spanning the upper, middle, and lower dermis with evidence of mild leukocytoclasia and no evidence of leukocytoclastic vasculitis (Figure 2). These histologic features together with the clinical presentation were consistent with a diagnosis of SS.
After an initial dose of intravenous methylprednisolone sodium succinate 125 mg in the emergency department, the patient was admitted for additional intravenous steroid administration in the context of uncontrolled hyperglycemia and history of poor glucose control. Upon admission, acetaminophen-codeine was discontinued and the patient was transitioned to intravenous methylprednisolone sodium succinate 60 mg every 8 hours. The patient also was given intravenous diphenhydramine 25 mg every 6 hours and desonide ointment 0.05% was applied to facial lesions. The inpatient medication regimen resulted in notable improvement of
Comment
Although SS itself is relatively rare, there has been an increasing incidence of the drug-induced subtype, most often in association with use of granulocyte colony-stimulating factor and granulocyte monocyte-stimulating factor. There also have been reported associations with a growing number of medications that include antibiotics, antiepileptic drugs, furosemide, hydralazine, and all-trans retinoic acid.11-19 Moghim
Several therapies for advanced melanoma also have been reviewed in the literature, including ipilimumab and vemurafenib,27-30 as have several medications for the treatment of myelodysplastic syndrome including azacitidine.31,32 A seve
Additional medications more recently involved in the pathogenesis of drug-induced SS include the chemotherapeutic agents topetecan, mitoxantrone, gemcitabine, and vorinostat.34-37 The antimalarial medication chloroquine also has been implicated, as have selective cyclooxygenase-2 inhibitors, hypomethylating agents, the tumor necrosis factor inhibitor adalimumab, IL-2 therapies, aripiprazole, and several other medications.38-49
Despite drug-induced SS being reported in association with an increasing number of medications, there had been a lack of appropriate diagnostic criteria. To tha
Conclusion
The number of cases of drug-induced SS in the literature continues to climb; however, the association with acetaminophen-codeine is unique. The importance of this case lies in educating both physicians and pharmacists alike regarding a newly recognized adverse effect of acetaminophen-codeine. Because acetaminophen-codeine often is used for its analgesic properties, and the predominant symptom of the cutaneous eruption of SS is pain, the therapeutic value of acetaminophen-codeine is substantially diminished in acetaminophen-codeine–induced SS. Accordingly, in these cases, the medication may be discontinued or substituted upon recognition of this adverse reaction to reduce patient morbidity.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
- Sweet RD. An acute febrile neutrophilic dermatosis. Br J Dermatol. 1964;76:349-356.
- Whittle CH, Back GA, Champion RH. Recurrent neutrophilic dermatosis of the face—a variant of Sweet’s syndrome. Br J Dermatol. 1968;80:806-810.
- Von den Driesch P. Sweet’s syndrome (acute febrile neutrophilic dermatosis). J Am Acad Dermatol. 1994;31:535-536.
- Honigsmann H, Cohen PR, Wolff K. Acute febrile neutrophilic dermatosis (Sweet’s syndrome). Wien Klin Wochenschr. 1979;91:842-847.
- Limdiwala PG, Parikh SJ, Shah JS. Sweet’s Syndrome. Indian J Dent Res. 2014;25:401-405.
- Walling HW, Snipes CJ, Gerami P, et al. The relationship between neutrophilic dermatosis of the dorsal hands and sweet syndrome: report of 9 cases and comparison to atypical pyoderma gangrenosum. Arch Dermatol. 2006;142:57-63.
- Ratzinger G, Burgdorf W, Zelger BG, et al. Acute febrile neutrophilic dermatosis: a histopathologic study of 31 cases with review of literature. Am J Dermatopathol. 2007;29:125-133.
- Moschella SL, Davis MDP. Neutrophilic dermatoses. In: Bolognia J, Jorizzo J, Rapini R, eds. Dermatology. 2nd ed. Philadelphia, PA: Elsevier; 2012:423-428.
- Fett DL, Gibson LE, Su WP. Sweet’s syndrome: signs and symptoms and associated disorders. Mayo Clinic Proc. 1995;70:234-240.
- Carvalho R, Fernandes C, Afonso A, et al. Drug-induced Sweet’s syndrome by alclofenac. Cutan Ocul Toxicol. 2011;30:315-316.
- Moghimi J, Pahlevan D, Azizzadeh M, et al. Isotretinoin-associated Sweet’s syndrome: a case report. Daru. 2014;22:69.
- Cholongitas E, Pipili C, Dasenaki M, et al. Piperacillin/tazobactam-induced Sweet syndrome in a patient with chronic lymphocytic leukemia and autoimmune cholangitis. Am J Dermatopathol. 2008;30:203-204.
- Kandula S, Burke WS, Goldfarb JN. Clindamycin-induced Sweet syndrome. J Am Acad Dermatol. 2010;62:898-900.
- Jamet A, Lagarce L, Le Clec’h C, et al. Doxycycline-induced Sweet’s syndrome. Eur J Dermatol. 2008;18:595-596.
- Cartee TV, Chen SC. Sweet syndrome associated with hydralazine-induced lupus erythematosus. Cutis. 2012;89:121-124.
- Baybay H, Elhatimi A, Idrissi R, et al. Sweet’s syndrome following oral ciprofloxacin therapy. Ann Dermatol Venereol. 2011;138:606-607.
- Khaled A, Kharfi M, Fazaa B, et al. A first case of trimethoprim-sulfamethoxazole induced Sweet’s syndrome in a child. Pediatr Dermatol. 2009;26:744-746.
- Calixto R, Menezes Y, Ostronoff M, et al. Favorable outcome of severe, extensive, granulocyte colony-stimulating factor-induced, corticosteroid-resistant Sweet’s syndrome treated with high-dose intravenous immunoglobulin. J Clin Oncol. 2014;32:E1-E2.
- Margaretten ME, Ruben BS, Fye K. Systemic sulfa-induced Sweet’s syndrome. Arthritis Rheum. 2008;59:1044-1046.
- Tanguy-Schmidt A, Avenel-Audran M, Croué A, et al. Bortezomib-induced acute neutrophilic dermatosis. Ann Dermatol Venereol. 2009;136:443-446.
- Choonhakarn C, Chaowattanapanit S. Azathioprine-induced Sweet’s syndrome and published work review. J Dermatol. 2013;40:267-271.
- Cyrus N, Stavert R, Mason AR, et al. Neutrophilic dermatosis after azathioprine exposure. JAMA Dermatol. 2013;149:592-597.
- Hurtado-Garcia R, Escribano-Stablé JC, Pascual JC, et al. Neutrophilic dermatosis caused by azathioprine hypersensitivity. Int J Dermatol. 2012;51:1522-1525.
- Valentine MC, Walsh JS. Neutrophilic dermatosis caused by azathioprine. Skinmed. 2011;9:386-388.
- Kim JS, Roh HS, Lee JW, et al. Distinct variant of Sweet’s syndrome: bortezomib-induced histiocytoid Sweet’s syndrome in a patient with multiple myeloma. Int J Dermatol. 2012;51:1491-1493.
- Ozlem C, Deram B, Mustafa S, et al. Propylthiouracil-induced anti-neutrophil cytoplasmic antibodies and agranulocytosis together with granulocyte colony-stimulating factor induced Sweet’s syndrome in a patient with Graves’ disease. Intern Med. 2011;50:1973-1976.
- Kyllo RL, Parker MK, Rosman I, et al. Ipilimumab-associated Sweet syndrome in a patient with high-risk melanoma. J Am Acad Dermatol. 2014;70:E85-E86.
- Pintova S, Sidhu H, Friedlander PA, et al. Sweet’s syndrome in a patient with metastatic melanoma after ipilimumab therapy. Melanoma Res. 2013;23:498-501.
- Yorio JT, Mays SR, Ciurea AM, et al. Case of vemurafenib-induced Sweet’s syndrome. J Dermatol. 2014;41:817-820.
- Pattanaprichakul P, Tetzlaff MT, Lapolla WJ, et al. Sweet syndrome following vemurafenib therapy for recurrent cholangiocarcinoma. J Cutan Pathol. 2014;41:326-328.
- Trickett HB, Cumpston A, Craig M. Azacitidine-associated Sweet’s syndrome. Am J Health Syst Pharm. 2012;69:869-871.
- Tintle S, Patel V, Ruskin A, et al. Azacitidine: a new medication associated with Sweet syndrome. J Am Acad Dermatol. 2011;64:E77-E79.
- Thieu KP, Rosenbach M, Xu X, et al. Neutrophilic dermatosis complicating lenalidomide therapy. J Am Acad Dermatol. 2009;61:709-710.
- Dickson EL, Bakhru A, Chan MP. Topotecan-induced Sweet’s syndrome: a case report. Gynecol Oncol Case Rep. 2013;4:50-52.
- Kümpfel T, Gerdes LA, Flaig M, et al. Drug-induced Sweet’s syndrome after mitoxantrone therapy in a patient with multiple sclerosis. Mult Scler. 2011;17:495-497.
- Martorell-Calatayud A, Requena C, Sanmartin O, et al. Gemcitabine-associated sweet syndrome-like eruption. J Am Acad Dermatol. 2011;65:1236-1238.
- Pang A, Tan KB, Aw D, et al. A case of Sweet’s syndrome due to 5-azacytidine and vorinostat in a patient with NK/T cell lymphoma. Cutan Ocul Toxicol. 2012;31:64-66.
- El Moutaoui L, Zouhair K, Benchikhi H. Sweet syndrome induced by chloroquine. Ann Dermatol Venereol. 2009;136:56-57.
- Rosmaninho A, Lobo I, Selores M. Sweet’s syndrome associated with the intake of a selective cyclooxygenase-2 (COX-2) inhibitor. Cutan Ocul Toxicol. 2011;30:298-301.
- Alencar C, Abramowtiz M, Parekh S, et al. Atypical presentations of Sweet’s syndrome in patients with MDS/AML receiving combinations of hypomethylating agents with histone deacetylase inhibitors. Am J Hematol. 2009;84:688-689.
- Keidel S, McColl A, Edmonds S. Sweet’s syndrome after adalimumab therapy for refractory relapsing polychondritis. BMJ Case Rep. 2011;2011.
- Rondina A, Watson AC. Bullous Sweet’s syndrome and pseudolymphoma precipitated by IL-2 therapy. Cutis. 2010;85:206-213.
- Gheorghe L, Cotruta B, Trifu V, et al. Drug-induced Sweet’s syndrome secondary to hepatitis C antiviral therapy. Int J Dermatol. 2008;47:957-959.
- Zobniw CM, Saad SA, Kostoff D, et al. Bortezomib-induced Sweet’s syndrome confirmed by rechallenge. Pharmacotherapy. 2014;34:E18-E21.
- Kolb-Mäurer A, Kneitz H, Goebeler M. Sweet-like syndrome induced by bortezomib. J Dtsch Dermatol Ges. 2013;11:1200-1202.
- Thuillier D, Lenglet A, Chaby G, et al. Bortezomib-induced eruption: Sweet syndrome? two case reports [in French]. Ann Dermatol Venereol. 2009;136:427-430.
- Kim MJ, Jang KT, Choe YH. Azathioprine hypersensitivity presenting as sweet syndrome in a child with ulcerative colitis. Indian Pediatr. 2011;48:969-971.
- Truchuelo M, Bagazgoitia L, Alcántara J, et al. Sweet-like lesions induced by bortezomib: a review of the literature and a report of 2 cases. Actas Dermosifiliogr. 2012;103:829-831.
- Hoelt P, Fattouh K, Villani AP. Dermpath & clinic: drug-induced Sweet syndrome. Eur J Dermatol. 2016;26:641-642.
- Walker DC, Cohen PR. Trimethoprim-sulfamethoxazole-associated acute febrile neutrophilic dermatosis: case report and review of drug-induced Sweet’s syndrome. J Am Acad Dermatol. 1996;34:918-923.
- Thompson DF, Montarella KE. Drug-induced Sweet’s syndrome. Ann Pharmacother. 2007;41:802-811.
Practice Points
- The rate of medication-induced Sweet syndrome is on the rise.
- Oral acetaminophen-codeine may induce Sweet syndrome.

Complications of Cosmetic Eye Whitening
First introduced in 2008 as a surgical treatment of chronic conjunctival injection, cosmetic eye whitening became popularized in South Kore
The procedure involves performing a localized conjunctivectomy with or without removal of the Tenon capsule.4 Brimonidine tartrate is given for vascular constriction. When conjunctivectomy is performed in the right eye, the medial conjunctiva is incised from the 2-o’clock to 5-o’clock positions and the lateral conjunctiva is incised from the 10-o’clock to 7-o’clock positions. After the conjunctiva and Tenon capsule are excised, hemostasis is achieved with electrocauterization. Postoperative management may consist of topical mitomycin C (MMC) 0.02% 4 times daily for 2 to 5 days along with topical steroids. The addition of bevacizumab 1.25 mg/mL also has been described.5
In this report, we provide a comprehensive review of the complications of cosmetic eye whitening based on a review of the literature. Clinicians in both aesthetic practice and ophthalmology should be aware of the potential complications to accurately educate their patients about the possible risks and benefits of this procedure.
Methods
A review of PubMed articles indexed for MEDLINE (January 2009 to July 2017) using the search terms cosmetic eye whitening, cosmetic wide conjunctivectomy, I-Brite, and chronic hyperemic conjuctiva was conducted to evaluate the number of reports of complications from cosmetic eye whitening. A total of 10 articles were included in the study based on a review of abstracts. Non–English-language abstracts were not reviewed.
Results
Based on a review of 10 articles commenting on the complications of cosmetic eye whitening, a total of 2400 patients had undergone a cosmetic conjunctivectomy with various postoperative complications and recurrences (Table).4-13 The most commonly recurring complications based on the reported frequencies in the articles included chronic conjunctival epithelial defects, scleral thinning, calcific plaques, dry eye syndrome, diplopia (sometimes requiring strabismus surgery), and elevated intraocular pressure.
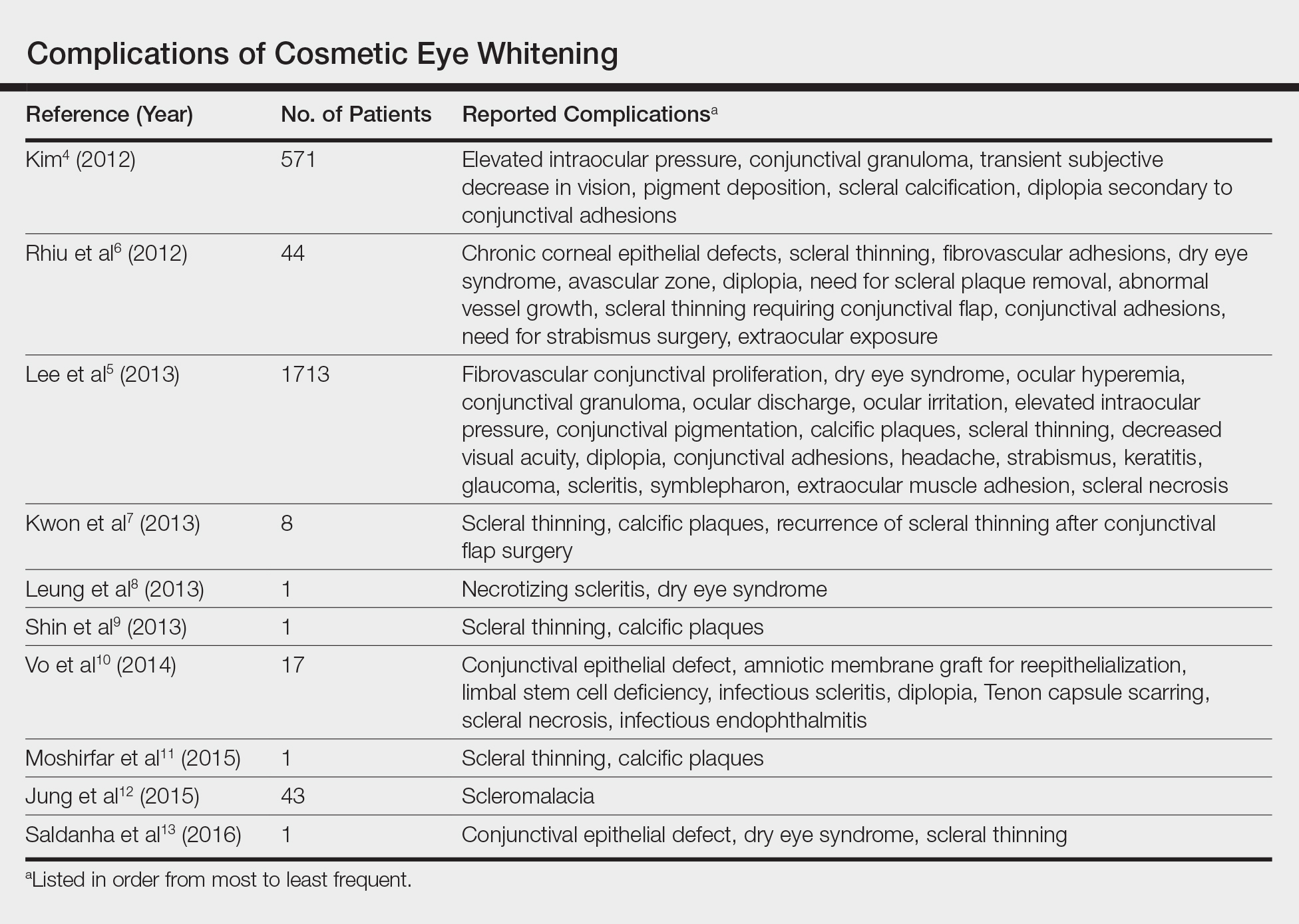
Kim4 was the first to report this surgical technique for irreversible hyperemic conjunctiva (N=1815). The reported success rate in South Korea was overwhelmingly high at 94.6%. In a mean (SD) follow-up time of 12.9 (7.8) months (range, 2–27 months), less than 20% of patients required surgical revision. During this time, the most common postoperative complications included elevation in intraocular pressure (17.2%), conjunctival granuloma (8.4%), transient vision decrease (7.5%), pigment deposition (5.3%), scleral calcifications (3.9%), and diplopia secondary to conjunctival adhesions (1.6%). No permanent defects were reported, and complications improved with surgical and medical management.4
Contrary to the findings of Kim,4 a large number of complications were seen; thus, on March 4, 2011, the Korean Ministry of Health & Welfare issued a declaration to discontinue the procedure under Article 49 of the Medical Service Act. Medical records from the single clinic in Korea from November 2007 to May 2010 were reviewed.5 One of the largest reviews of cosmetic eye whitening complications reviewed 1713 patients who underwent conjunctivectomy plus topical MMC with or without bevacizumab injection. Pterygium and chronic conjunctival hyperemia were the most common diagnoses that prompted patients to undergo treatment. Over an average follow-up period of 10.9 months, the overall complication rate was 82.9%, with severe complications being fibrovascular conjunctival proliferation (43.8%), recurrent hyperemic conjunctiva (28.1%), intraocular pressure (13.1%), scleral thinning with calcified plaques (6.2%), scleral thinning (4.4%), and diplopia (3.6%). A total of 56.9% of patients reported being satisfied with the cosmetic outcome of the surgery.5
In some of the smaller case series and case reports we reviewed, more vision-threatening complications have been described. Infectious endophthalmitis, infectious scleritis, and necrotizing scleritis have all been reported as complications of cosmetic eye whitening.8,10
Comment
The pathophysiology of the complications of cosmetic eye whitening stem from the disruption of the normal conjunctiva, destruction of the vascularization to the sclera, and loss of limbal stem cells. Mitomycin C is a topical antimetabolite antibiotic agent that inhibits DNA synthesis. This relatively safe and inexpensive product has decreased the recurrence rate in pterygium surgery as early as 1963.14,15 Complications of MMC in pterygium surgery include infectious scleritis, necrotizing scleritis, calcium formation, and even scleromalacia, occurring at incidence rates as low as 1.4%.16 These risks are balanced against the medical necessity of using MMC. Given the elective nature of cosmetic eye whitening, these complications in a cosmetic setting may not be justified.
The debate of the use of this procedure continues to occur in ophthalmologic societies. Both the Korean Ministry of Health & Welfare and the American Society of Cataract Refractive Surgery do not condone the use of regional conjunctivectomy for cosmetic eye whitening.5,17 Evidence shows that complications from cosmetic conjunctivectomy can be devastating and unnecessary given its elective nature. Although some complications (eg, dry eye syndrome, pain, discomfort) may be considered mild, the number of potentially serious complications brings the usefulness of the procedure into question.
This review is a launchpad to inform the medical community of the potential downside to conjunctivectomy for cosmetic eye whitening with the hope that it can initiate meaningful risk-benefit discussions between providers and physicians.
- Kim BH. Cosmetic eye whitening. Poster presented at: American Society of Cataract and Refractive Surgery; April 4-9, 2008; Chicago, IL.
- Kim BH. Cosmetic eye whitening by regional conjunctivectomy. Poster presented at: European Society of Cataract & Refractive Surgeons; September 13-17, 2008; Berlin, Germany.
- Raiskup F, Solomon A, Landau D, et al. Mitomycin C for pterygium: long term evaluation. Br J Ophthalmol. 2004;88:1425-1428.
- Kim BH. Regional conjunctivectomy with postoperative mitomycin C to treat chronic hyperemic conjunctiva. Cornea. 2012;31:236-244.
- Lee S, Go J, Rhiu S, et al. Cosmetic regional conjunctivectomy with postoperative mitomycin C application with or without bevacizumab injection [published online April 6, 2013]. Am J Ophthalmol. 2013;156:616-622.
- Rhiu S, Shim J, Kim EK, et al. Complications of cosmetic wide conjunctivectomy combined with postsurgical mitomycin C application. Cornea. 2012;31:245-252.
- Kwon HJ, Nam SM, Lee SY, et al. Conjunctival flap surgery for calcified scleromalacia after cosmetic conjunctivectomy. Cornea. 2013;32:821-825.
- Leung TG, Dunn JP, Akpek EK, et al. Necrotizing scleritis as a complication of cosmetic eye whitening procedure. J Ophthalmic Inflamm Infect. 2013;3:39.
- Shin HY, Kim MS, Chung SK. The development of scleromalacia after regional conjunctivectomy with the postoperative application of mitomycin C as an adjuvant therapy. Korean J Ophthalmol. 2013;27:208-210.
- Vo RC, Stafeeva K, Aldave AJ, et al. Complications related to a cosmetic eye-whitening procedure. Am J Ophthalmol. 2014;158:967-973.
- Moshirfar M, McCaughey MV, Fenzl CR, et al. Delayed manifestation of bilateral scleral thinning after I-BRITE® procedure and review of literature for cosmetic eye-whitening procedures. Clin Ophthalmol. 2015;9:445-451.
- Jung JW, Kwon KY, Choi DL, et al. Long-term clinical outcomes of conjunctival flap surgery for calcified scleromalacia after periocular surgery. Cornea. 2015;34:308-312.
- Saldanha MJ, Yang PT, Chan CC. Scleral thinning after I-BRITE procedure treated with amniotic membrane graft. Can J Ophthalmol. 2016;51:e115-e116.
- Seiler T, Schnelle B, Wollensak J. Pterygium excision using 193-nm excimer laser smoothing and topical mitomycin C. Ger J Ophthalmol. 1992;1:429-431.
- Singh G, Wilson MR, Foster CS. Long-term follow-up study of mitomycin eye drops as adjunctive treatment of pterygia and its comparison with conjunctival autograft transplantation. Cornea. 1990;9:331-334.
- Lam DS, Wong AK, Fan DS, et al. Intraoperative mitomycin C to prevent recurrence of pterygium after excision: a 30-month follow-up study. Ophthalmology. 1998;105:901-904; discussion 904-905.
- ASCRS Cornea Clinical Committee. Clinical alert: eye-whitening procedure: regional conjunctivectomy with mitomycin-C application [press release]. Fairfax, VA: American Society of Cataract and Refractive Surgery. http://www.ascrs.org/node/1352. Accessed January 22, 2015.
First introduced in 2008 as a surgical treatment of chronic conjunctival injection, cosmetic eye whitening became popularized in South Kore
The procedure involves performing a localized conjunctivectomy with or without removal of the Tenon capsule.4 Brimonidine tartrate is given for vascular constriction. When conjunctivectomy is performed in the right eye, the medial conjunctiva is incised from the 2-o’clock to 5-o’clock positions and the lateral conjunctiva is incised from the 10-o’clock to 7-o’clock positions. After the conjunctiva and Tenon capsule are excised, hemostasis is achieved with electrocauterization. Postoperative management may consist of topical mitomycin C (MMC) 0.02% 4 times daily for 2 to 5 days along with topical steroids. The addition of bevacizumab 1.25 mg/mL also has been described.5
In this report, we provide a comprehensive review of the complications of cosmetic eye whitening based on a review of the literature. Clinicians in both aesthetic practice and ophthalmology should be aware of the potential complications to accurately educate their patients about the possible risks and benefits of this procedure.
Methods
A review of PubMed articles indexed for MEDLINE (January 2009 to July 2017) using the search terms cosmetic eye whitening, cosmetic wide conjunctivectomy, I-Brite, and chronic hyperemic conjuctiva was conducted to evaluate the number of reports of complications from cosmetic eye whitening. A total of 10 articles were included in the study based on a review of abstracts. Non–English-language abstracts were not reviewed.
Results
Based on a review of 10 articles commenting on the complications of cosmetic eye whitening, a total of 2400 patients had undergone a cosmetic conjunctivectomy with various postoperative complications and recurrences (Table).4-13 The most commonly recurring complications based on the reported frequencies in the articles included chronic conjunctival epithelial defects, scleral thinning, calcific plaques, dry eye syndrome, diplopia (sometimes requiring strabismus surgery), and elevated intraocular pressure.
Kim4 was the first to report this surgical technique for irreversible hyperemic conjunctiva (N=1815). The reported success rate in South Korea was overwhelmingly high at 94.6%. In a mean (SD) follow-up time of 12.9 (7.8) months (range, 2–27 months), less than 20% of patients required surgical revision. During this time, the most common postoperative complications included elevation in intraocular pressure (17.2%), conjunctival granuloma (8.4%), transient vision decrease (7.5%), pigment deposition (5.3%), scleral calcifications (3.9%), and diplopia secondary to conjunctival adhesions (1.6%). No permanent defects were reported, and complications improved with surgical and medical management.4
Contrary to the findings of Kim,4 a large number of complications were seen; thus, on March 4, 2011, the Korean Ministry of Health & Welfare issued a declaration to discontinue the procedure under Article 49 of the Medical Service Act. Medical records from the single clinic in Korea from November 2007 to May 2010 were reviewed.5 One of the largest reviews of cosmetic eye whitening complications reviewed 1713 patients who underwent conjunctivectomy plus topical MMC with or without bevacizumab injection. Pterygium and chronic conjunctival hyperemia were the most common diagnoses that prompted patients to undergo treatment. Over an average follow-up period of 10.9 months, the overall complication rate was 82.9%, with severe complications being fibrovascular conjunctival proliferation (43.8%), recurrent hyperemic conjunctiva (28.1%), intraocular pressure (13.1%), scleral thinning with calcified plaques (6.2%), scleral thinning (4.4%), and diplopia (3.6%). A total of 56.9% of patients reported being satisfied with the cosmetic outcome of the surgery.5
In some of the smaller case series and case reports we reviewed, more vision-threatening complications have been described. Infectious endophthalmitis, infectious scleritis, and necrotizing scleritis have all been reported as complications of cosmetic eye whitening.8,10
Comment
The pathophysiology of the complications of cosmetic eye whitening stem from the disruption of the normal conjunctiva, destruction of the vascularization to the sclera, and loss of limbal stem cells. Mitomycin C is a topical antimetabolite antibiotic agent that inhibits DNA synthesis. This relatively safe and inexpensive product has decreased the recurrence rate in pterygium surgery as early as 1963.14,15 Complications of MMC in pterygium surgery include infectious scleritis, necrotizing scleritis, calcium formation, and even scleromalacia, occurring at incidence rates as low as 1.4%.16 These risks are balanced against the medical necessity of using MMC. Given the elective nature of cosmetic eye whitening, these complications in a cosmetic setting may not be justified.
The debate of the use of this procedure continues to occur in ophthalmologic societies. Both the Korean Ministry of Health & Welfare and the American Society of Cataract Refractive Surgery do not condone the use of regional conjunctivectomy for cosmetic eye whitening.5,17 Evidence shows that complications from cosmetic conjunctivectomy can be devastating and unnecessary given its elective nature. Although some complications (eg, dry eye syndrome, pain, discomfort) may be considered mild, the number of potentially serious complications brings the usefulness of the procedure into question.
This review is a launchpad to inform the medical community of the potential downside to conjunctivectomy for cosmetic eye whitening with the hope that it can initiate meaningful risk-benefit discussions between providers and physicians.
First introduced in 2008 as a surgical treatment of chronic conjunctival injection, cosmetic eye whitening became popularized in South Kore
The procedure involves performing a localized conjunctivectomy with or without removal of the Tenon capsule.4 Brimonidine tartrate is given for vascular constriction. When conjunctivectomy is performed in the right eye, the medial conjunctiva is incised from the 2-o’clock to 5-o’clock positions and the lateral conjunctiva is incised from the 10-o’clock to 7-o’clock positions. After the conjunctiva and Tenon capsule are excised, hemostasis is achieved with electrocauterization. Postoperative management may consist of topical mitomycin C (MMC) 0.02% 4 times daily for 2 to 5 days along with topical steroids. The addition of bevacizumab 1.25 mg/mL also has been described.5
In this report, we provide a comprehensive review of the complications of cosmetic eye whitening based on a review of the literature. Clinicians in both aesthetic practice and ophthalmology should be aware of the potential complications to accurately educate their patients about the possible risks and benefits of this procedure.
Methods
A review of PubMed articles indexed for MEDLINE (January 2009 to July 2017) using the search terms cosmetic eye whitening, cosmetic wide conjunctivectomy, I-Brite, and chronic hyperemic conjuctiva was conducted to evaluate the number of reports of complications from cosmetic eye whitening. A total of 10 articles were included in the study based on a review of abstracts. Non–English-language abstracts were not reviewed.
Results
Based on a review of 10 articles commenting on the complications of cosmetic eye whitening, a total of 2400 patients had undergone a cosmetic conjunctivectomy with various postoperative complications and recurrences (Table).4-13 The most commonly recurring complications based on the reported frequencies in the articles included chronic conjunctival epithelial defects, scleral thinning, calcific plaques, dry eye syndrome, diplopia (sometimes requiring strabismus surgery), and elevated intraocular pressure.
Kim4 was the first to report this surgical technique for irreversible hyperemic conjunctiva (N=1815). The reported success rate in South Korea was overwhelmingly high at 94.6%. In a mean (SD) follow-up time of 12.9 (7.8) months (range, 2–27 months), less than 20% of patients required surgical revision. During this time, the most common postoperative complications included elevation in intraocular pressure (17.2%), conjunctival granuloma (8.4%), transient vision decrease (7.5%), pigment deposition (5.3%), scleral calcifications (3.9%), and diplopia secondary to conjunctival adhesions (1.6%). No permanent defects were reported, and complications improved with surgical and medical management.4
Contrary to the findings of Kim,4 a large number of complications were seen; thus, on March 4, 2011, the Korean Ministry of Health & Welfare issued a declaration to discontinue the procedure under Article 49 of the Medical Service Act. Medical records from the single clinic in Korea from November 2007 to May 2010 were reviewed.5 One of the largest reviews of cosmetic eye whitening complications reviewed 1713 patients who underwent conjunctivectomy plus topical MMC with or without bevacizumab injection. Pterygium and chronic conjunctival hyperemia were the most common diagnoses that prompted patients to undergo treatment. Over an average follow-up period of 10.9 months, the overall complication rate was 82.9%, with severe complications being fibrovascular conjunctival proliferation (43.8%), recurrent hyperemic conjunctiva (28.1%), intraocular pressure (13.1%), scleral thinning with calcified plaques (6.2%), scleral thinning (4.4%), and diplopia (3.6%). A total of 56.9% of patients reported being satisfied with the cosmetic outcome of the surgery.5
In some of the smaller case series and case reports we reviewed, more vision-threatening complications have been described. Infectious endophthalmitis, infectious scleritis, and necrotizing scleritis have all been reported as complications of cosmetic eye whitening.8,10
Comment
The pathophysiology of the complications of cosmetic eye whitening stem from the disruption of the normal conjunctiva, destruction of the vascularization to the sclera, and loss of limbal stem cells. Mitomycin C is a topical antimetabolite antibiotic agent that inhibits DNA synthesis. This relatively safe and inexpensive product has decreased the recurrence rate in pterygium surgery as early as 1963.14,15 Complications of MMC in pterygium surgery include infectious scleritis, necrotizing scleritis, calcium formation, and even scleromalacia, occurring at incidence rates as low as 1.4%.16 These risks are balanced against the medical necessity of using MMC. Given the elective nature of cosmetic eye whitening, these complications in a cosmetic setting may not be justified.
The debate of the use of this procedure continues to occur in ophthalmologic societies. Both the Korean Ministry of Health & Welfare and the American Society of Cataract Refractive Surgery do not condone the use of regional conjunctivectomy for cosmetic eye whitening.5,17 Evidence shows that complications from cosmetic conjunctivectomy can be devastating and unnecessary given its elective nature. Although some complications (eg, dry eye syndrome, pain, discomfort) may be considered mild, the number of potentially serious complications brings the usefulness of the procedure into question.
This review is a launchpad to inform the medical community of the potential downside to conjunctivectomy for cosmetic eye whitening with the hope that it can initiate meaningful risk-benefit discussions between providers and physicians.
- Kim BH. Cosmetic eye whitening. Poster presented at: American Society of Cataract and Refractive Surgery; April 4-9, 2008; Chicago, IL.
- Kim BH. Cosmetic eye whitening by regional conjunctivectomy. Poster presented at: European Society of Cataract & Refractive Surgeons; September 13-17, 2008; Berlin, Germany.
- Raiskup F, Solomon A, Landau D, et al. Mitomycin C for pterygium: long term evaluation. Br J Ophthalmol. 2004;88:1425-1428.
- Kim BH. Regional conjunctivectomy with postoperative mitomycin C to treat chronic hyperemic conjunctiva. Cornea. 2012;31:236-244.
- Lee S, Go J, Rhiu S, et al. Cosmetic regional conjunctivectomy with postoperative mitomycin C application with or without bevacizumab injection [published online April 6, 2013]. Am J Ophthalmol. 2013;156:616-622.
- Rhiu S, Shim J, Kim EK, et al. Complications of cosmetic wide conjunctivectomy combined with postsurgical mitomycin C application. Cornea. 2012;31:245-252.
- Kwon HJ, Nam SM, Lee SY, et al. Conjunctival flap surgery for calcified scleromalacia after cosmetic conjunctivectomy. Cornea. 2013;32:821-825.
- Leung TG, Dunn JP, Akpek EK, et al. Necrotizing scleritis as a complication of cosmetic eye whitening procedure. J Ophthalmic Inflamm Infect. 2013;3:39.
- Shin HY, Kim MS, Chung SK. The development of scleromalacia after regional conjunctivectomy with the postoperative application of mitomycin C as an adjuvant therapy. Korean J Ophthalmol. 2013;27:208-210.
- Vo RC, Stafeeva K, Aldave AJ, et al. Complications related to a cosmetic eye-whitening procedure. Am J Ophthalmol. 2014;158:967-973.
- Moshirfar M, McCaughey MV, Fenzl CR, et al. Delayed manifestation of bilateral scleral thinning after I-BRITE® procedure and review of literature for cosmetic eye-whitening procedures. Clin Ophthalmol. 2015;9:445-451.
- Jung JW, Kwon KY, Choi DL, et al. Long-term clinical outcomes of conjunctival flap surgery for calcified scleromalacia after periocular surgery. Cornea. 2015;34:308-312.
- Saldanha MJ, Yang PT, Chan CC. Scleral thinning after I-BRITE procedure treated with amniotic membrane graft. Can J Ophthalmol. 2016;51:e115-e116.
- Seiler T, Schnelle B, Wollensak J. Pterygium excision using 193-nm excimer laser smoothing and topical mitomycin C. Ger J Ophthalmol. 1992;1:429-431.
- Singh G, Wilson MR, Foster CS. Long-term follow-up study of mitomycin eye drops as adjunctive treatment of pterygia and its comparison with conjunctival autograft transplantation. Cornea. 1990;9:331-334.
- Lam DS, Wong AK, Fan DS, et al. Intraoperative mitomycin C to prevent recurrence of pterygium after excision: a 30-month follow-up study. Ophthalmology. 1998;105:901-904; discussion 904-905.
- ASCRS Cornea Clinical Committee. Clinical alert: eye-whitening procedure: regional conjunctivectomy with mitomycin-C application [press release]. Fairfax, VA: American Society of Cataract and Refractive Surgery. http://www.ascrs.org/node/1352. Accessed January 22, 2015.
- Kim BH. Cosmetic eye whitening. Poster presented at: American Society of Cataract and Refractive Surgery; April 4-9, 2008; Chicago, IL.
- Kim BH. Cosmetic eye whitening by regional conjunctivectomy. Poster presented at: European Society of Cataract & Refractive Surgeons; September 13-17, 2008; Berlin, Germany.
- Raiskup F, Solomon A, Landau D, et al. Mitomycin C for pterygium: long term evaluation. Br J Ophthalmol. 2004;88:1425-1428.
- Kim BH. Regional conjunctivectomy with postoperative mitomycin C to treat chronic hyperemic conjunctiva. Cornea. 2012;31:236-244.
- Lee S, Go J, Rhiu S, et al. Cosmetic regional conjunctivectomy with postoperative mitomycin C application with or without bevacizumab injection [published online April 6, 2013]. Am J Ophthalmol. 2013;156:616-622.
- Rhiu S, Shim J, Kim EK, et al. Complications of cosmetic wide conjunctivectomy combined with postsurgical mitomycin C application. Cornea. 2012;31:245-252.
- Kwon HJ, Nam SM, Lee SY, et al. Conjunctival flap surgery for calcified scleromalacia after cosmetic conjunctivectomy. Cornea. 2013;32:821-825.
- Leung TG, Dunn JP, Akpek EK, et al. Necrotizing scleritis as a complication of cosmetic eye whitening procedure. J Ophthalmic Inflamm Infect. 2013;3:39.
- Shin HY, Kim MS, Chung SK. The development of scleromalacia after regional conjunctivectomy with the postoperative application of mitomycin C as an adjuvant therapy. Korean J Ophthalmol. 2013;27:208-210.
- Vo RC, Stafeeva K, Aldave AJ, et al. Complications related to a cosmetic eye-whitening procedure. Am J Ophthalmol. 2014;158:967-973.
- Moshirfar M, McCaughey MV, Fenzl CR, et al. Delayed manifestation of bilateral scleral thinning after I-BRITE® procedure and review of literature for cosmetic eye-whitening procedures. Clin Ophthalmol. 2015;9:445-451.
- Jung JW, Kwon KY, Choi DL, et al. Long-term clinical outcomes of conjunctival flap surgery for calcified scleromalacia after periocular surgery. Cornea. 2015;34:308-312.
- Saldanha MJ, Yang PT, Chan CC. Scleral thinning after I-BRITE procedure treated with amniotic membrane graft. Can J Ophthalmol. 2016;51:e115-e116.
- Seiler T, Schnelle B, Wollensak J. Pterygium excision using 193-nm excimer laser smoothing and topical mitomycin C. Ger J Ophthalmol. 1992;1:429-431.
- Singh G, Wilson MR, Foster CS. Long-term follow-up study of mitomycin eye drops as adjunctive treatment of pterygia and its comparison with conjunctival autograft transplantation. Cornea. 1990;9:331-334.
- Lam DS, Wong AK, Fan DS, et al. Intraoperative mitomycin C to prevent recurrence of pterygium after excision: a 30-month follow-up study. Ophthalmology. 1998;105:901-904; discussion 904-905.
- ASCRS Cornea Clinical Committee. Clinical alert: eye-whitening procedure: regional conjunctivectomy with mitomycin-C application [press release]. Fairfax, VA: American Society of Cataract and Refractive Surgery. http://www.ascrs.org/node/1352. Accessed January 22, 2015.
Resident Pearl
- Cosmetic eye whitening has severe and vision-threatening complications that should be aware to all cosmetic surgeons.

Concurrent Sturge-Weber Syndrome, Facial Infantile Hemangioma, and Cutis Marmorata Telangiectatica Congenita
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
Sturge-Weber syndrome (SWS) is a disease of dermatologic, neurologic, and ocular significance.1 The most distinctive manifestation is facial capillary malformation, commonly referred to as a port-wine stain or nevus flammeus. The dysregulated angiogenesis, caused by somatic mutations of the G protein subunit alpha Q gene, GNAQ, also affects the central nervous system.2 Seizures, intellectual disability, and glaucoma are common consequences.1 Not all port-wine stains are associated with SWS.3 Distribution in the ophthalmic dermatome is associated with increased risk for SWS, with 8% of patients with port-wine stains also having SWS.4 The disease is more serious when bilateral lesions are present.5 Diagnosis is clinical based on dermatologic, nervous system, and ophthalmologic findings.6 The disease is nonheritable because the mutation is found only in the somatic cell lines.2 The possibility of epigenetic influence on disease development has to be investigated. The treatment is aimed at managing complications, as there is no cure.7
Infantile hemangioma (IH) likewise represents a disruption in the process of vascular development but without the widespread consequences of SWS. The pathogenesis of hemangioma development has not been fully elucidated, with presence of GLUT1 (glucose transporter 1) protein implicated in lesions.4 Facial infantile hemangiomas have an incidence of approximately 5 in every 100 births, and the prevalence decreases with age. Most hemangiomas undergo growth followed by an involution process, with most lesions vanishing by 5 years of age.4 They typically are seen at 2 to 3 weeks of age, growing rapidly for the first 6 months, which is a contrast to the static nature of nevus flammeus. Infantile hemangiomas are regarded as sporadic, though autosomal-dominant inheritance patterns have been observed.4 Our patient demonstrated facial IH at birth, which is a rare and interesting finding suggesting that some epigenetic factors influenced this modification of the disease course in this patient.
Cutis marmorata telangiectatica congenita (CMTC) is a rare cutaneous vascular condition found in newborns. Its extraordinary infrequency is reflected in the fact that only 300 cases have been reported worldwide.8 At birth, CMTC manifests as a pinkish reticulated pattern all over the body mimicking cutis marmorata; however, unlike cutis marmorata, the lesions do not improve with warming.9 The lesions of CMTC gradually lighten as the patient ages.8 Limb asymmetry is the most common extravascular complication of CMTC and, similar to SWS, glaucoma also can occur.10 Cutis marmorata telangiectatica congenita has been known to occur simultaneously with SWS or IH, but the combination of all 3 conditions in our patient is unique. Due to the scarcity of cases, the pathophysiology and treatment is poorly understood, with appropriate monitoring for sequelae recommended.9
Case Report
The patient was born at 39 weeks’ gestation following an uncomplicated pregnancy and delivery. She weighed 2950 g, her length was 19 in, and her head circumference was 13.25 in, correlating to the 10th, 50th, and 25th percentiles, respectively. Her Apgar score was 8/9 at 1 and 5 minutes. Her parents were nonconsanguineous and in good health. The patient’s family lived in poverty, which led us to conjecture about the role that toxins played in the epigenetics of the patient and her family. It was the mother’s third pregnancy; both prior pregnancies resulted in healthy children. The patient was breastfed. No family history of heritable vascular disorders was reported.
On the first day of life during the newborn examination, dark red pigment changes were noticed under the nose and erythematous pigmentation over the whole body was observed (Figure). On examination, 2-toned reticular lesions identified as extensive nevus flammeus were found bilaterally over the distribution of the ophthalmic division of the trigeminal nerve. A separate erythematous plaque over the maxilla also was recognized. The pediatrician suspected SWS and facial IH. The patient was discharged after 3 days with a referral to pediatric dermatology, and appropriate follow-up with a pediatrician was scheduled. The patient returned for these appointments and the significance of SWS was explained to her parents. Consultation with pediatric dermatology at 2 weeks of age confirmed the diagnosis of SWS as well as facial IH.
Upon further follow-up with pediatric dermatology at 2 months of age, the patient received an additional diagnosis of CMTC. These exceedingly rare lesions were located over the back, trunk, arms, and legs. The patient’s parents were counseled about the management of these conditions and appropriate follow-up.
Comment
This case describes 3 different vascular malformations in the same patient. Cutis marmorata telangiectatica congenita is rare and yet is described in this patient along with 2 other notable endothelial abnormalities. The clinical interest of this case is heightened by the presence of CMTC.
The causative factor of SWS is a well-documented mutation of the GNAQ gene, but there is considerable variability in how it affects the patient. Unlike in SWS, no single factor can be attributed to the development of IH. This case shows that these 3 diseases are not mutually exclusive and can present with unusually severe features when they occur concomitantly. The embryologic basis of SWS traces its roots back to the first trimester during vascular development, wher
The severity of the SWS in our patient was highlighted by the extensive nevus flammeus. These lesions occurred over the face, trunk, arms, and legs. The port-wine stain with dermatomal distribution of the ophthalmic nerve was the most concerning feature regarding the development of neurologic complications in this patient. Although the developmental delays associated with SWS can be serious, early intervention is important and can improve long-term outcomes. The facial IH arising at birth was contrary to the typical presentation. All of these factors will be kept in mind as the patient progresses and patient-centered care is provided. Because this patient’s presentation differed from other patients with IH, we will be more vigilant in providing close follow-up and monitoring for other medical problems involving other organs (eg, the brain); for instance, we will monitor for seizures and developmental delay.
Conclusion
In our patient, a unique pattern of SWS, facial IH, and CMTC are described in a pediatric patient. Many disciplines are involved in the treatment. In the patient’s first days of life, extensive collaboration between pediatrics and dermatologists was pivotal, with ophthalmology, pathology, and radiology consultations at hand. This case highlights that several vascular malformations of different origins can occur in the same patient. Epigenetic along with genetic factors likely contributed to this fascinating presentation. The importance of parental education and maintaining appropriate follow-up for this patient is crucial for a favorable outcome.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
- Sinawat S, Auvichayapat N, Auvichayapat P, et al. 12-year retrospective study of Sturge-weber syndrome and literature review. J Med Assoc Thail. 2014;97:742-750.
- Shirley MD, Tang H, Gallione CJ, et al. Sturge-Weber syndrome and port-wine stains caused by somatic mutation in GNAQ. N Engl J Med. 2013;368:1971-1979.
- Sudarsanam A, Ardern-Holmes SL. Sturge-Weber syndrome: from the past to the present [published online November 7, 2013]. Eur J Paediat Neurol. 2014;18:257-266.
- Paller AS, Mancini AJ. Hurwitz Clinical Pediatric Dermatology: A Textbook of Skin Disorders of Childhood and Adolescence. Philadelphia, PA: Elsevier Saunders; 2011.
- Sujansky E, Conradi S. Sturge-Weber syndrome: age of onset of seizures and glaucoma and the prognosis for affected children. J Child Neurol. 1995;10:49-58.
- Lo W, Marchuk DA, Ball KL, et al. Updates and future horizons on the understanding, diagnosis, and treatment of Sturge-Weber syndrome brain involvement. Dev Med Child Neurol. 2012;54:214-223.
- Comi AM. Update on Sturge-Weber syndrome: diagnosis, treatment, quantitative measures, and controversies. Lymphat Res Biol. 2007;5:257-264.
- Resende CI, Araujo C, Vieira AP, et al. Cutis marmorata telangiectatica congenital [published online October 17, 2013]. BMJ Case Rep. doi:10.1136/bcr-2013-200056.
- Levy R, Lam JM. Cutis marmorata telangiectatica congenita: a mimicker of a common disorder. CMAJ. 2011;183:E249-E251.
- Kienast AK, Hoeger PH. Cutis marmorata telangiectatica congenita: a prospective study of 27 cases and review of the literature with proposal of diagnostic criteria. Clin Exp Dermatol. 2009;34:319-323.
- Comi AM. Topical review: pathophysiology of Sturge-Weber syndrome. J Child Neurol. 2003;18:509-516.
Practice Points
- This case highlights that several vascular malformations of different origins can occur in the same patient.
- Epigenetic factors along with genetic factors can lead to development of complex vascular conditions.
- Close collaborations of different medical specialties is necessary to make an accurate diagnosis and to follow up to achieve optimal long-term outcomes for patients with complex medical conditions.
Ideals of Facial Beauty
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4

The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4
The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
Several concepts of ideal aesthetic measurements can be traced back to ancient Greek and European Renaissance art. In examining canons of beauty, these classical ideals often are compared to modern-day standards, allowing clinicians to delineate the parameters of an attractive facial appearance and facilitate the planning of cosmetic procedures.
Given the growing number of available cosmetic interventions, dermatologists have a powerful ability to modify facial proportions; however, changes to individual structures should be made with a mindful approach to improving overall facial harmony. This article reviews the established parameters of facial beauty to assist the clinician in enhancing cosmetic outcomes.
Canons of Facial Aesthetics
Horizontal Thirds
In his writings on human anatomy, Leonardo da Vinci described dividing the face into equal thirds (Figure 1). The upper third measures from the trichion (the midline point of the normal hairline) to the glabella (the smooth prominence between the eyebrows). The middle third measures from the glabella to the subnasale (the midline point where the nasal septum meets the upper lip). The lower third measures from the subnasale to the menton (the most inferior point of the chin).1
Although the validity of the canon is intended to apply across race and gender, these proportions may vary by ethnicity (Table). In white individuals, the middle third of the face tends to be shorter than the upper and lower thirds.2 This same relationship has been observed in black males.3 In Chinese females, the upper third commonly is shorter than the middle and lower thirds, correlating with a less prominent forehead. In contrast, black females tend to have a relatively longer upper third.4
The relationship between modern perceptions of attractiveness and the neoclassical norm of equal thirds remains a topic of interest. Milutinovic et al1 examined facial thirds in white female celebrities from beauty and fashion magazines and compared them to a group of anonymous white females from the general population. The group of anonymous females showed statistically significant (P<.05) differences between the sizes of the 3 facial segments, whereas the group of celebrity faces demonstrated uniformity between the facial thirds.1
The lower face can itself be divided into thirds, with the upper third measured from the subnasale to the stomion (the midline point of the oral fissure when the lips are closed), and the lower two-thirds measured from the stomion to the menton (Figure 1). Mommaerts and Moerenhout5 examined photographs of 105 attractive celebrity faces and compared their proportions to those of classical sculptures of gods and goddesses (antique faces). The authors identified an upper one-third to lower two-thirds ratio of 69.8% in celebrity females and 69.1% in celebrity males; these ratios were not significantly different from the 72.4% seen in antique females and 73.1% in antique males. The authors concluded that a 30% upper lip to 70% lower lip-chin proportion may be the most appropriate to describe contemporary standards.5
Vertical Fifths
In the vertical dimension, the neoclassical canon of facial proportions divides the face into equal fifths (Figure 2).6 The 2 most lateral fifths are measured from the lateral helix of each ear to the exocanthus of each eye. The eye fissure lengths (measured between the endocanthion and exocanthion of each eye) represent one-fifth. The middle fifth is measured between the medial canthi of both eyes (endocanthion to endocanthion). This distance is equal to the width of the nose, as measured between both alae. Finally, the width of the mouth represents 1.5-times the width of the nose. These ratios of the vertical fifths apply to both males and females.6
Anthropometric studies have examined deviations from the neoclassical canon according to ethnicity. Wang et al7 compared the measurements of North American white and Han Chinese patients to these standards. White patients demonstrated a greater ratio of mouth width to nose width relative to the canon. In contrast, Han Chinese patients demonstrated a relatively wider nose and narrower mouth.7
In black individuals, it has been observed that the dimensions of most facial segments correspond to the neoclassical standards; however, nose width is relatively wider in black individuals relative to the canon as well as relative to white individuals.8
Milutinovic et al1 also compared vertical fifths between white celebrities and anonymous females. In the anonymous female group, statistically significant (P<.05) variations were found between the sizes of the different facial components. In contrast, the celebrity female group showed balance between the widths of vertical fifths.1
Lips
In the lower facial third, the lips represent a key element of attractiveness. Recently, lip augmentation, aimed at creating fuller and plumper lips, has dominated the popular culture and social media landscape.9 Although the aesthetic ideal of lips continues to evolve over time, recent studies have aimed at quantifying modern notions of attractive lip appearance.
Popenko et al10 examined lip measurements using computer-generated images of white women with different variations of lip sizes and lower face proportions. Computer-generated faces were graded on attractiveness by more than 400 individuals from focus groups. An upper lip to lower lip ratio of 1:2 was judged to be the most attractive, while a ratio of 2:1 was judged to be the least attractive. Results also showed that the surface area of the most attractive lips comprised roughly 10% of the lower third of the face.10
Penna et al11 analyzed various parameters of the lips and lower facial third using photographs of 176 white males and females that were judged on attractiveness by 250 volunteer evaluators. Faces were graded on a scale from 1 (absolutely attractive) to 7 (absolutely unattractive). Attractive males and females (grades 1 and 2) both demonstrated an average ratio of upper vermilion height to nose-mouth distance (measured from the subnasalae to the lower edge of the upper vermilion border) of 0.28, which was significantly greater than the average ratio observed in less attractive individuals (grades 6 or 7)(P<.05). In addition, attractive males and females demonstrated a ratio of upper vermilion height to nose-chin distance (measured from the subnasalae to the menton) of 0.09, which again was larger than the average ratio seen in less attractive individuals. Figure 3 demonstrates an aesthetic ideal of the lips derived from these 2 studies, though consideration should be given to the fact that these studies were based in white populations.
Golden Ratio
The golden ratio, also known as Phi, can be observed in nature, art, and architecture. Approximately equal to 1.618, the golden ratio also has been identified as a possible marker of beauty in the human face and has garnered attention in the lay press. The ratio has been applied to several proportions and structures in the face, such as the ratio of mouth width to nose width or the ratio of tooth height to tooth width, with investigation providing varying levels of validation about whether these ratios truly correlate with perceptions of beauty.12 Swift and Remington13 advocated for application of the golden ratio toward a comprehensive set of facial proportions. Marquardt14 used the golden ratio to create a 3-dimensional representation of an idealized face, known as the golden decagon mask. Although the golden ratio and the golden decagon mask have been proposed as analytic tools, their utility in clinical practice may be limited. Firstly, due to its popularity in the lay press, the golden ratio has been inconsistently applied to a wide range of facial ratios, which may undermine confidence in its representation as truth rather than coincidence. Secondly, although some authors have found validity of the golden decagon mask in representing unified ratios of attractiveness, others have asserted that it characterizes a masculinized white female and fails to account for ethnic differences.15-19
Age-Related Changes
In addition to the facial proportions guided by genetics, several changes occur with increased age. Over the course of a lifetime, predictable patterns emerge in the dimensions of the skin, soft tissue, and bone. These alterations in structural proportions may ultimately lead to an unevenness in facial aesthetics.
In skeletal structure, gradual bone resorption and expansion causes a reduction in facial height as well as an increase in facial width and depth.20 Fat atrophy and hypertrophy affect soft tissue proportions, visualized as hollowing at the temples, cheeks, and around the eyes, along with fullness in the submental region and jowls.21 Finally, decreases in skin elasticity and collagen exacerbate the appearance of rhytides and sagging. In older patients who desire a more youthful appearance, various applications of dermal fillers, fat grafting, liposuction, and skin tightening techniques can help to mitigate these changes.
Conclusion
Improving facial aesthetics relies on an understanding of the norms of facial proportions. Although cosmetic interventions commonly are advertised or described based on a single anatomical unit, it is important to appreciate the relationships between facial structures. Most notably, clinicians should be mindful of facial ratios when considering the introduction of filler materials or implants. Augmentation procedures at the temples, zygomatic arch, jaw, chin, and lips all have the possibility to alter facial ratios. Changes should therefore be considered in the context of improving overall facial harmony, with the clinician remaining cognizant of the ideal vertical and horizontal divisions of the face. Understanding such concepts and communicating them to patients can help in appropriately addressing all target areas, thereby leading to greater patient satisfaction.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
- Milutinovic J, Zelic K, Nedeljkovic N. Evaluation of facial beauty using anthropometric proportions. ScientificWorldJournal. 2014;2014:428250. doi:10.1155/2014/428250.
- Farkas LG, Hreczko TA, Kolar JC, et al. Vertical and horizontal proportions of the face in young-adult North-American Caucasians: revision of neoclassical canons. Plast Reconstr Surg. 1985;75:328-338.
- Porter JP. The average African American male face: an anthropometric analysis. Arch Facial Plast Surg. 2004;6:78-81.
- Porter JP, Olson KL. Anthropometric facial analysis of the African American woman. Arch Facial Plast Surg. 2001;3:191-197.
- Mommaerts MY, Moerenhout BA. Ideal proportions in full face front view, contemporary versus antique. J Craniomaxillofac Surg. 2011;39:107-110.
- Vegter F, Hage JJ. Clinical anthropometry and canons of the face in historical perspective. Plast Reconstr Surg. 2000;106:1090-1096.
- Wang D, Qian G, Zhang M, et al. Differences in horizontal, neoclassical facial canons in Chinese (Han) and North American Caucasian populations. Aesthetic Plast Surg. 1997;21:265-269.
- Farkas LG, Forrest CR, Litsas L. Revision of neoclassical facial canons in young adult Afro-Americans. Aesthetic Plast Surg. 2000;24:179-184.
- Coleman GG, Lindauer SJ, Tüfekçi E, et al. Influence of chin prominence on esthetic lip profile preferences. Am J Orthod Dentofacial Orthop. 2007;132:36-42.
- Popenko NA, Tripathi PB, Devcic Z, et al. A quantitative approach to determining the ideal female lip aesthetic and its effect on facial attractiveness. JAMA Facial Plast Surg. 2017;19:261-267.
- Penna V, Fricke A, Iblher N, et al. The attractive lip: a photomorphometric analysis. J Plast Reconstr Aesthet Surg. 2015;68:920-929.
- Prokopakis EP, Vlastos IM, Picavet VA, et al. The golden ratio in facial symmetry. Rhinology. 2013;51:18-21.
- Swift A, Remington K. BeautiPHIcationTM: a global approach to facial beauty. Clin Plast Surg. 2011;38:247-277.
- Marquardt SR. Dr. Stephen R. Marquardt on the Golden Decagon and human facial beauty. interview by Dr. Gottlieb. J Clin Orthod. 2002;36:339-347.
- Veerala G, Gandikota CS, Yadagiri PK, et al. Marquardt’s facial Golden Decagon mask and its fitness with South Indian facial traits. J Clin Diagn Res. 2016;10:ZC49-ZC52.
- Holland E. Marquardt’s Phi mask: pitfalls of relying on fashion models and the golden ratio to describe a beautiful face. Aesthetic Plast Surg. 2008;32:200-208.
- Alam MK, Mohd Noor NF, Basri R, et al. Multiracial facial golden ratio and evaluation of facial appearance. PLoS One. 2015;10:e0142914.
- Kim YH. Easy facial analysis using the facial golden mask. J Craniofac Surg. 2007;18:643-649.
- Bashour M. An objective system for measuring facial attractiveness. Plast Reconstr Surg. 2006;118:757-774; discussion 775-776.
- Bartlett SP, Grossman R, Whitaker LA. Age-related changes of the craniofacial skeleton: an anthropometric and histologic analysis. Plast Reconstr Surg. 1992;90:592-600.
- Donofrio LM. Fat distribution: a morphologic study of the aging face. Dermatol Surg. 2000;26:1107-1112.
Practice Points
- Canons of ideal facial dimensions have existed since antiquity and remain relevant in modern times.
- Horizontal and vertical anatomical ratios can provide a useful framework for cosmetic interventions.
- To maximize aesthetic results, alterations to individual cosmetic units should be made with thoughtful consideration of overall facial harmony.
Do Infants Fed Rice and Rice Products Have an Increased Risk for Skin Cancer?
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
To the Editor:
Rice and rice products, such as rice cereal and rice snacks, contain inorganic arsenic. Exposure to arsenicin utero and during early life may be associated with adverse fetal growth, adverse infant and child immune response, and adverse neurodevelopmental outcomes. Therefore, the World Health Organization, the Food and Agriculture Organization of the United Nations, the European Union, and the US Food and Drug Administration have suggested maximum arsenic ingestion recommendations for infants: 100 ng/g for inorganic arsenic in products geared toward infants. However, infants consuming only a few servings of rice products may exceed the weekly tolerable intake of arsenic.
Karagas et al1 obtained dietary data on 759 infants who were enrolled in the New Hampshire Birth Cohort Study from 2011 to 2014. They noted that 80% of the infants had been introduced to rice cereal during the first year. Additional data on diet and total urinary arsenic at 12 months was available for 129 infants: 32.6% of these infants were fed rice snacks. In addition, the total urinary arsenic concentration was higher among infants who ate rice cereal or rice snacks as compared to infants who did not eat rice or rice products.
Chronic arsenic exposure can result in patchy dark brown hyperpigmentation with scattered pale spots referred to as “raindrops on a dusty road.” The axilla, eyelids, groin, neck, nipples, and temples often are affected. However, the hyperpigmentation can extend across the chest, abdomen, and back in severe cases.
Horizontal white lines across the nails (Mees lines) may develop. Keratoses, often on the palms (arsenic keratoses), may appear; they persist and may progress to skin cancers. In addition, patients with arsenic exposure are more susceptible to developing nonmelanoma skin cancers.2
It is unknown if exposure to inorganic arsenic in infancy predisposes these individuals to skin cancer when they become adults. Long-term longitudinal follow-up of the participants in this study may provide additional insight. Perhaps infants should not receive rice cereals and rice snacks or their parents should more carefully monitor the amount of rice and rice products that they ingest.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
- Karagas MR, Punshon T, Sayarath V, et al. Association of rice and rice-product consumption with arsenic exposure early in life. JAMA Pediatr. 2016;170:609-616.
- Mayer JE, Goldman RH. Arsenic and skin cancer in the USA: the current evidence regarding arsenic-contaminated drinking water. Int J Dermatol. 2016;55;e585-e591.
Clinical Trial Designs for Topical Antifungal Treatments of Onychomycosis and Implications on Clinical Practice
Onychomycosis is a fungal nail infection primarily caused by dermatophytes.1 If left untreated, the infection can cause nail destruction and deformities,1 resulting in pain and discomfort,2 impaired foot mobility,3 and an overall reduced quality of life.1 Onychomycosis is a chronic condition that requires long treatment periods due to the slow growth rates of toenails.1 To successfully cure the condition, fungal eradication must be achieved.
Prior to the US Food and Drug Administration (FDA) approval of tavaborole and efinaconazole, ciclopirox was the only approved topical treatment for onychomycosis.4 The recent approval of tavaborole and efinaconazole has increased treatment options available to patients and has started to pave the way for future topical treatments. This article discusses the 3 approved topical treatments for onychomycosis and focuses on the design of the phase 3 clinical trials that led to their approval.
Topical Agents Used to Treat Onychomycosis
Tavaborole, efinaconazole, and ciclopirox have undergone extensive clinical investigation to receive FDA approval. Results from pivotal phase 3 studies establishing the efficacy and safety of each agent formed the basis for regulatory submission. Although it may seem intuitive to compare the relative performance of these agents based on their respective phase 3 clinical trial data, there are important differences in study methodology, conduct, and populations that prevent direct comparisons. The FDA provides limited guidance to the pharmaceutical industry on how to conduct clinical trials for potential onychomycosis treatments. Comparative efficacy and safety claims are limited based on cross-study comparisons. The details of the phase 3 trial designs are summarized in the Table.
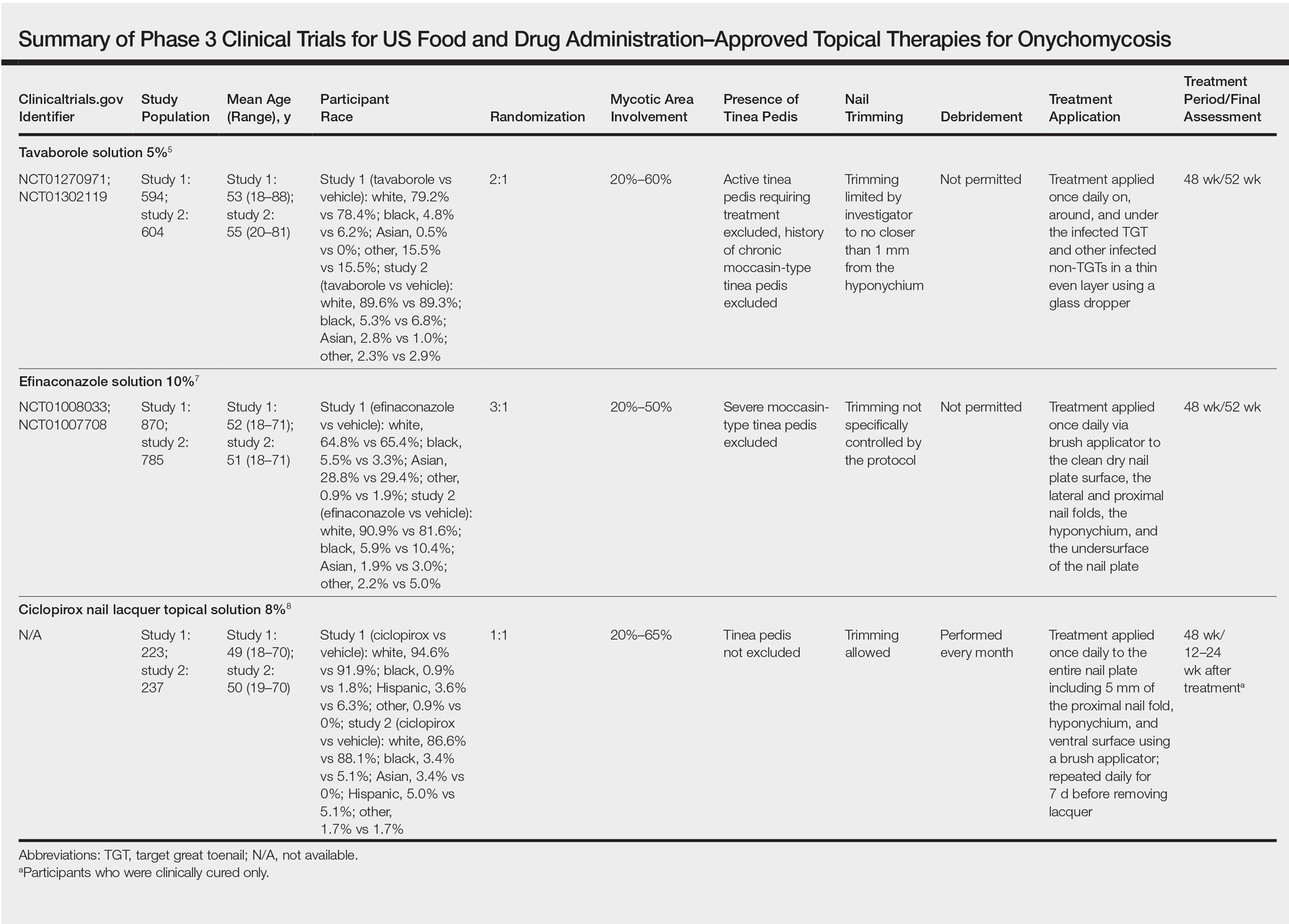
Tavaborole
Tavaborole is a boron-based treatment with a novel mechanism of action.5 Tavaborole binds to the editing domain of leucyl–transfer ribonucleic acid synthetase via an integrated boron atom and inhibits fungal protein synthesis.6 Two identical randomized, double-blind, vehicle-controlled, parallel-group, phase 3 clinical trials evaluating tavaborole were performed.5 The first study (registered at www.clinicaltrials.gov with the identifier NCT01270971) included 594 participants from27 sites in the United States and Mexico and was conducted between December 2010 and November 2012. The second study (NCT01302119) included 604 participants from 32 sites in the United States and Canada and was conducted between February 2011 and January 2013.
Eligible participants 18 years and older had distal subungual onychomycosis (DSO) of the toenails affecting 20% to 60% of 1 or more target great toenails (TGTs), tested positive for fungus using potassium hydroxide (KOH) wet mounts and positive for Trichophyton rubrum and Trichophyton mentagrophytes on fungal culture diagnostic tests, had distal TGT thickness of 3 mm or less, and had 3 mm or more of clear nail between the proximal nail fold and the most proximal visible mycotic border.5 Those with active tinea pedis requiring treatment or with a history of chronic moccasin-type tinea pedis were excluded. Participants were randomized to receive either tavaborole or vehicle (2:1). Treatments were applied once daily to all infected toenails for a total of 48 weeks, and nail debridement (defined as partial or complete removal of the toenail) was not permitted. Notably, controlled trimming of the nail was allowed to 1 mm of the leading nail edge. Regular assessments of each toenail for disease involvement, onycholysis, and subungual hyperkeratosis were made at screening, baseline, week 2, week 6, and every 6 weeks thereafter until week 52. Subungual TGT samples were taken at screening and every 12 weeks during the study for examination at a mycology laboratory, which performed KOH and fungal culture tests. A follow-up assessment was made at week 52.5
The primary end point was complete cure of the TGT at week 52, with secondary end points of completely or almost clear TGT nail (≤10% dystrophic nail), completely or almost clear TGT nail (≤10% dystrophic nail) plus negative mycology, and negative mycology of TGT.5 Examples of TGTs in participants who achieved complete cure and almost clear nails with negative mycology before and after treatment with tavaborole are shown in Figure 1. An example of a patient considered to have treatment failure is shown in Figure 2. This patient showed marked improvement in nail appearance and had a negative culture result but had a positive KOH test, which demonstrates the stringency in which topical agents are judged in onychomycosis trials.5
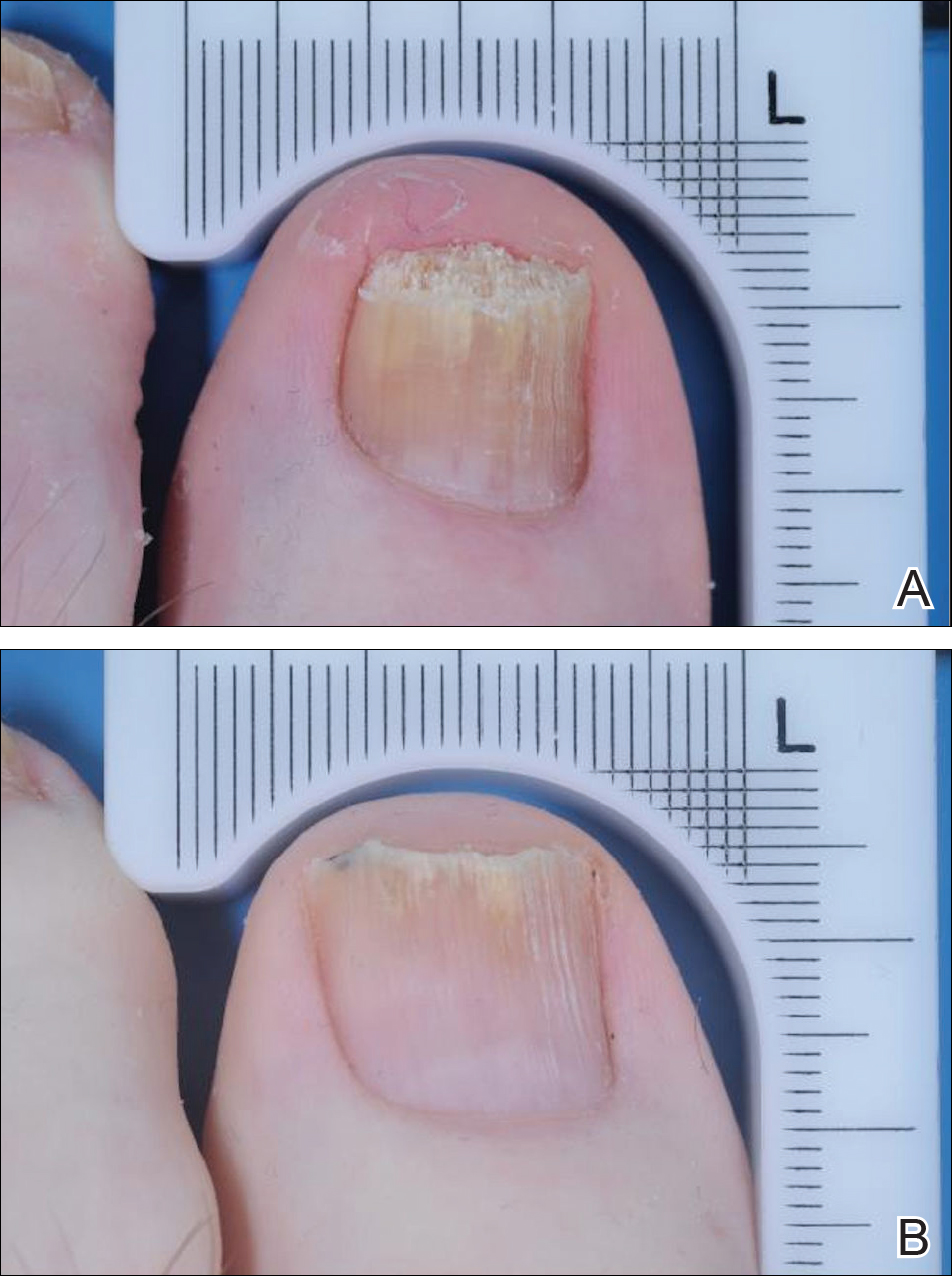
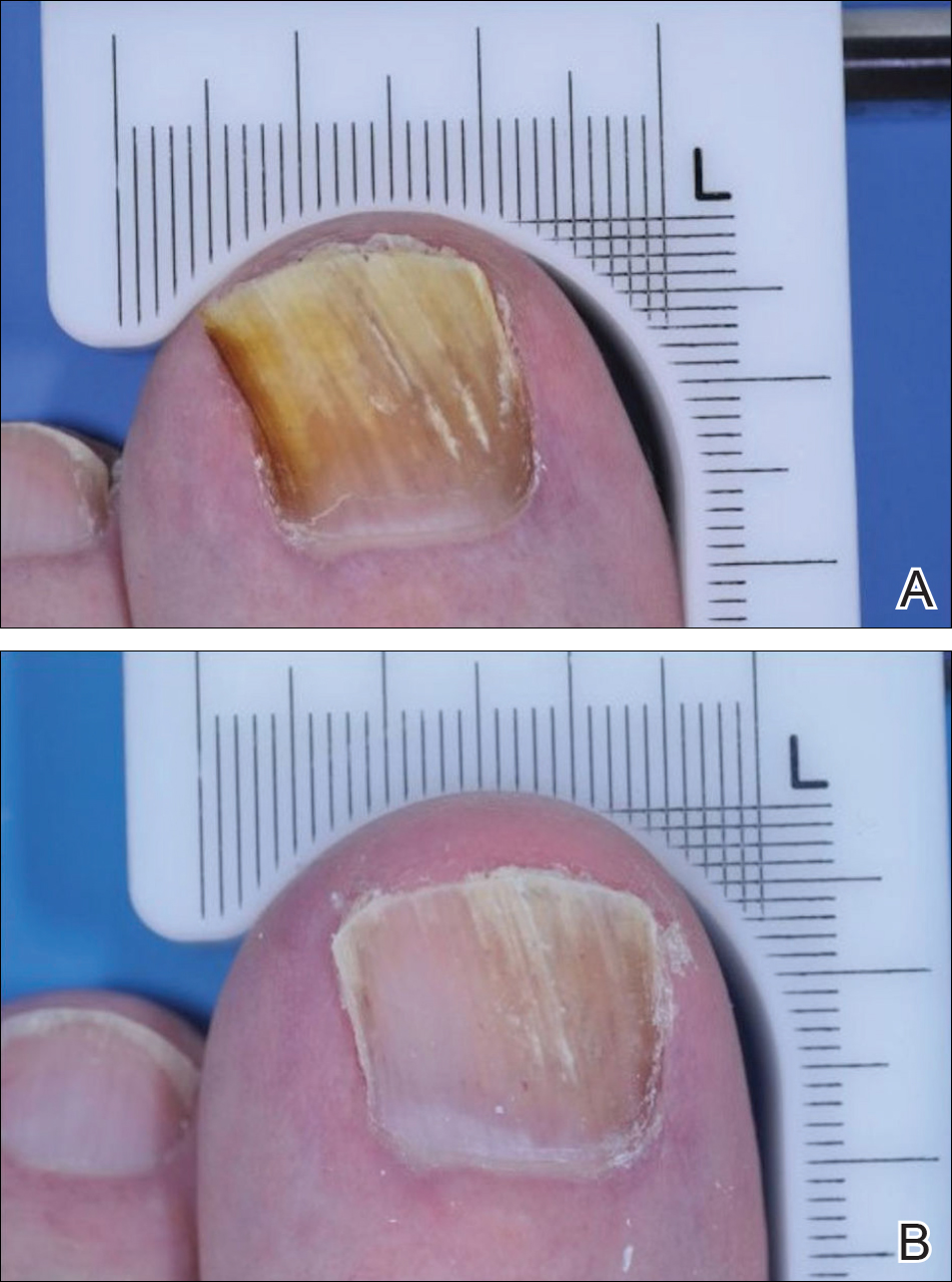
Efinaconazole
Efinaconazole is a topical triazole antifungal specifically indicated to treat onychomycosis. Two identical randomized, vehicle-controlled, double-blind, multicenter trials were performed to assess the safety and efficacy of efinaconazole solution 10%.7 The first study (NCT01008033) involved 870 participants and was conducted at a total of 74 sites in Japan (33 sites), Canada (7 sites), and the United States (34 sites) between December 2009 and September 2011. The second study (NCT01007708) had 785 participants and was conducted at 44 sites in Canada (8 sites) and the United States (36 sites) between December 2009 and October 2011.
Participants aged 18 to 70 years with a clinical diagnosis of DSO affecting 1 or more TGT were eligible to participate.7 Other eligibility criteria included an uninfected toenail length 3 mm or more from the proximal nail fold, a maximum toenail thickness of 3 mm, positive KOH wet mounts, and positive dermatophyte or mixed dermatophyte/candida cultures. Dermatophytes included T rubrum and T mentagrophytes. Those with severe moccasin-type tinea pedis were excluded. Participants were randomized to receive efinaconazole or vehicle (3:1). Once-daily treatments were self-applied to nails for 48 weeks. Clinical assessments were made at baseline and every 12 weeks until week 48, with a follow-up assessment at week 52. No nail trimming protocol was provided.7
The primary end point of the efinaconazole phase 3 trials was complete cure at week 52, with secondary end points including mycologic cure, treatment success (≤5% mycotic nail), and complete or almost complete cure (negative culture and KOH, ≤5% mycotic nail). An example of a complete cure from baseline to week 52 is shown in Figure 3.7
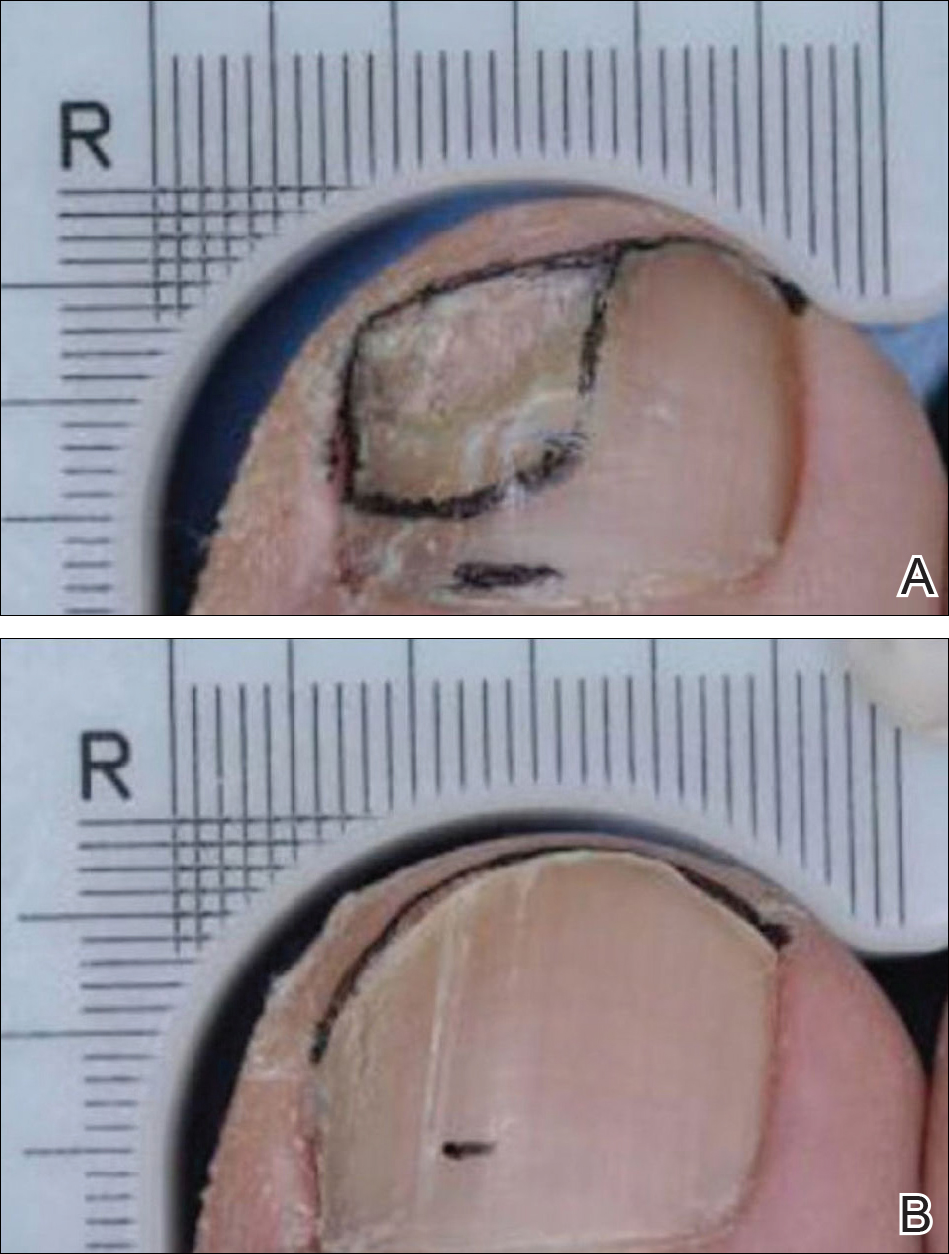
Ciclopirox
Ciclopirox was the first topical therapy to be approved for the treatment of onychomycosis. Ciclopirox is a broad-spectrum antifungal agent that inhibits metal-dependent enzymes, which are responsible for the degradation of toxic peroxides in fungal cells. The safety and efficacy of ciclopirox nail lacquer topical solution 8% also was investigated in 2 identical phase 3 clinical trials.8 The first study was conducted at 9 sites in the United States between June 1994 and June 1996 and included 223 participants. The second study was conducted at 9 sites in the United States between July 1994 and April 1996 and included 237 participants.
Eligible participants were required to have DSO in at least one TGT, positive KOH wet mount with positive dermatophyte culture, and 20% to 65% nail involvement.8 Those with tinea pedis were not excluded. Participants were randomized to receive once-daily treatment with ciclopirox or vehicle (1:1)(applied to all toenails and affected fingernails) for 48 weeks. The product was to be removed by the patient with alcohol on a weekly basis. Trimming was allowed as necessary, and mechanical debridement by the physician could be performed monthly. Assessments were made every 4 weeks, and mycologic examinations were performed every 12 weeks. Participants who were clinically cured were assessed further in a 12- to 24-week posttreatment follow-up period.8
The primary end point of complete cure and secondary end points of treatment success (negative culture and KOH, ≤10% mycotic nail), mycologic cure, and negative mycologic culture were assessed at week 48.8
Phase 3 Clinical Trial Similarities and Differences
The phase 3 clinical trials used to investigate the safety and efficacy of tavaborole,5 efinaconazole,7 and ciclopirox8 were similar in their overall design. All trials were randomized, double-blind, vehicle-controlled studies in patients with DSO. Each agent was assessed using a once-daily application for a treatment period of 48 weeks.
Primary differences among study designs included the age range of participants, the range of mycotic nail involvement, the presence/absence of tinea pedis, and the nail trimming/debridement protocols used. Differences were observed in the patient eligibility criteria of these trials. Both mycotic area and participant age range were inconsistent for each agent (eTable). Participants with larger mycotic areas usually have a poorer prognosis, as they tend to have a greater fungal load.9 A baseline mycotic area of 20% to 60%,5 20% to 50%,7 and 20% to 65%8 at baseline was required for the tavaborole, efinaconazole, and ciclopirox trials, respectively. Variations in mycotic area between trials can affect treatment efficacy, as clinical cures can be reached quicker by patients with smaller areas of infection. Of note, the average mycotic area of involvement was not reported in the tavaborole studies but was 36% and 40% for the efinaconazole and ciclopirox studies, respectively.5,8 It also is more difficult to achieve complete cure in older patients, as they have poor circulation and reduced nail growth rates.1,10 The participant age range was 18 to 88 years in the tavaborole trials, with 8% of the participants older than 70 years,5 compared to 18 to 71 years in both the efinaconazole and ciclopirox trials.7,8 The average age of participants in each study was approximately 54, 51, and 50 years for tavaborole, efinaconazole, and ciclopirox, respectively. Because factors impacting treatment failure can increase with age, efficacy results can be confounded by differing age distributions across different studies.
Another important feature that differed between the clinical trials was the approach to nail trimming—defined as shortening of the free edge of the nail distal to the hyponychium—which varies from debridement in that the nail plate is removed or reduced in thickness proximal to the hyponychium. In the tavaborole trials, trimming was controlled to within 1 mm of the free edge of the nail,5 whereas the protocol used for the ciclopirox trials allowed nail trimming as necessary as well as moderate debridement before treatment application and on a monthly basis.8 Debridement is an important component in all ciclopirox trials, as it is used to reduce fungal load.11 No trimming control was provided during the efinaconazole trials; however, debridement was prohibited.7 These differences can dramatically affect the study results, as residual fungal elements and portions of infected nails are removed during the trimming process in an uncontrolled manner, which can affect mycologic testing results as well as the clinical efficacy results determined through investigator evaluation. Discrepancies regarding nail trimming approach inevitably makes the trial results difficult to compare, as mycologic cure is not translatable between studies.
Furthermore, somewhat unusually, complete cure rate variations were observed between different study centers in the efinaconazole trials. Japanese centers in the first efinaconazole study (NCT01008033) had higher complete cure rates in both the efinaconazole and vehicle treatment arms, which is notable because approximately 29% of participants in this study were Asian, mostly hailing from 33 Japanese centers. The reason for these confounding results is unknown and requires further analysis.
Lastly, the presence or absence of tinea pedis can affect the response to onychomycosis treatment. In the tavaborole trials, patients with active interdigital tinea pedis or exclusively plantar tinea pedis or chronic moccasin-type tinea pedis requiring treatment were excluded from the studies.5 In contrast, only patients with severe moccasin-type tinea pedis were excluded in efinaconazole trials.7 The ciclopirox studies had no exclusions based on presence of tinea pedis.8 These differences are noteworthy, as tinea pedis can serve as a reservoir for fungal infection if not treated and can lead to recurrence of onychomycosis.12
Conclusion
In recent years, disappointing efficacy has resulted in the failure of several topical agents for onychomycosis during their development; however, there are several aspects to consider when examining efficacy data in onychomycosis studies. Obtaining a complete cure in onychomycosis is difficult. Because patients applying treatments at home are unlikely to undergo mycologic testing to confirm complete cure, visual inspections are helpful to determine treatment efficacy.
Despite similar overall designs, notable differences in the study designs of the phase 3 clinical trials investigating tavaborole, efinaconazole, and ciclopirox are likely to have had an effect on the reported results, making the efficacy of the agents difficult to compare. It is particularly tempting to compare the primary end point results of each trial, especially considering tavaborole and efinaconazole had primary end points with the same parameters; however, there are several other factors (eg, age range of study population, extent of infection, nail trimming, patient demographics) that may have affected the outcomes of the studies and precluded a direct comparison of any end points. Without head-to-head investigations, there is room for prescribing clinicians to interpret results differently.
Acknowledgment
Writing and editorial assistance was provided by ApotheCom Associates, LLC, Yardley, Pennsylvania, and was supported by Sandoz, a Novartis division.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Scher RK. Onychomycosis: a significant medical disorder. J Am Acad Dermatol. 1996;35(3, pt 2):S2-S5.
- Del Rosso JQ. The role of topical antifungal therapy for onychomycosis and the emergence of newer agents. J Clin Aesthet Dermatol. 2014;7:10-18.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Gupta AK, Joseph WS. Ciclopirox 8% nail lacquer in the treatment of onychomycosis of the toenails in the United States. J Am Pod Med Assoc. 2000;90:495-501.
- Carney C, Tosti A, Daniel R, et al. A new classification system for grading the severity of onychomycosis: Onychomycosis Severity Index. Arch Dermatol. 2011;147:1277-1282.
- Gupta AK. Onychomycosis in the elderly. Drugs Aging. 2000;16:397-407.
- Gupta AK, Malkin KF. Ciclopirox nail lacquer and podiatric practice. J Am Podiatr Med Assoc. 2000;90:502-507.
- Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
Onychomycosis is a fungal nail infection primarily caused by dermatophytes.1 If left untreated, the infection can cause nail destruction and deformities,1 resulting in pain and discomfort,2 impaired foot mobility,3 and an overall reduced quality of life.1 Onychomycosis is a chronic condition that requires long treatment periods due to the slow growth rates of toenails.1 To successfully cure the condition, fungal eradication must be achieved.
Prior to the US Food and Drug Administration (FDA) approval of tavaborole and efinaconazole, ciclopirox was the only approved topical treatment for onychomycosis.4 The recent approval of tavaborole and efinaconazole has increased treatment options available to patients and has started to pave the way for future topical treatments. This article discusses the 3 approved topical treatments for onychomycosis and focuses on the design of the phase 3 clinical trials that led to their approval.
Topical Agents Used to Treat Onychomycosis
Tavaborole, efinaconazole, and ciclopirox have undergone extensive clinical investigation to receive FDA approval. Results from pivotal phase 3 studies establishing the efficacy and safety of each agent formed the basis for regulatory submission. Although it may seem intuitive to compare the relative performance of these agents based on their respective phase 3 clinical trial data, there are important differences in study methodology, conduct, and populations that prevent direct comparisons. The FDA provides limited guidance to the pharmaceutical industry on how to conduct clinical trials for potential onychomycosis treatments. Comparative efficacy and safety claims are limited based on cross-study comparisons. The details of the phase 3 trial designs are summarized in the Table.
Tavaborole
Tavaborole is a boron-based treatment with a novel mechanism of action.5 Tavaborole binds to the editing domain of leucyl–transfer ribonucleic acid synthetase via an integrated boron atom and inhibits fungal protein synthesis.6 Two identical randomized, double-blind, vehicle-controlled, parallel-group, phase 3 clinical trials evaluating tavaborole were performed.5 The first study (registered at www.clinicaltrials.gov with the identifier NCT01270971) included 594 participants from27 sites in the United States and Mexico and was conducted between December 2010 and November 2012. The second study (NCT01302119) included 604 participants from 32 sites in the United States and Canada and was conducted between February 2011 and January 2013.
Eligible participants 18 years and older had distal subungual onychomycosis (DSO) of the toenails affecting 20% to 60% of 1 or more target great toenails (TGTs), tested positive for fungus using potassium hydroxide (KOH) wet mounts and positive for Trichophyton rubrum and Trichophyton mentagrophytes on fungal culture diagnostic tests, had distal TGT thickness of 3 mm or less, and had 3 mm or more of clear nail between the proximal nail fold and the most proximal visible mycotic border.5 Those with active tinea pedis requiring treatment or with a history of chronic moccasin-type tinea pedis were excluded. Participants were randomized to receive either tavaborole or vehicle (2:1). Treatments were applied once daily to all infected toenails for a total of 48 weeks, and nail debridement (defined as partial or complete removal of the toenail) was not permitted. Notably, controlled trimming of the nail was allowed to 1 mm of the leading nail edge. Regular assessments of each toenail for disease involvement, onycholysis, and subungual hyperkeratosis were made at screening, baseline, week 2, week 6, and every 6 weeks thereafter until week 52. Subungual TGT samples were taken at screening and every 12 weeks during the study for examination at a mycology laboratory, which performed KOH and fungal culture tests. A follow-up assessment was made at week 52.5
The primary end point was complete cure of the TGT at week 52, with secondary end points of completely or almost clear TGT nail (≤10% dystrophic nail), completely or almost clear TGT nail (≤10% dystrophic nail) plus negative mycology, and negative mycology of TGT.5 Examples of TGTs in participants who achieved complete cure and almost clear nails with negative mycology before and after treatment with tavaborole are shown in Figure 1. An example of a patient considered to have treatment failure is shown in Figure 2. This patient showed marked improvement in nail appearance and had a negative culture result but had a positive KOH test, which demonstrates the stringency in which topical agents are judged in onychomycosis trials.5
Efinaconazole
Efinaconazole is a topical triazole antifungal specifically indicated to treat onychomycosis. Two identical randomized, vehicle-controlled, double-blind, multicenter trials were performed to assess the safety and efficacy of efinaconazole solution 10%.7 The first study (NCT01008033) involved 870 participants and was conducted at a total of 74 sites in Japan (33 sites), Canada (7 sites), and the United States (34 sites) between December 2009 and September 2011. The second study (NCT01007708) had 785 participants and was conducted at 44 sites in Canada (8 sites) and the United States (36 sites) between December 2009 and October 2011.
Participants aged 18 to 70 years with a clinical diagnosis of DSO affecting 1 or more TGT were eligible to participate.7 Other eligibility criteria included an uninfected toenail length 3 mm or more from the proximal nail fold, a maximum toenail thickness of 3 mm, positive KOH wet mounts, and positive dermatophyte or mixed dermatophyte/candida cultures. Dermatophytes included T rubrum and T mentagrophytes. Those with severe moccasin-type tinea pedis were excluded. Participants were randomized to receive efinaconazole or vehicle (3:1). Once-daily treatments were self-applied to nails for 48 weeks. Clinical assessments were made at baseline and every 12 weeks until week 48, with a follow-up assessment at week 52. No nail trimming protocol was provided.7
The primary end point of the efinaconazole phase 3 trials was complete cure at week 52, with secondary end points including mycologic cure, treatment success (≤5% mycotic nail), and complete or almost complete cure (negative culture and KOH, ≤5% mycotic nail). An example of a complete cure from baseline to week 52 is shown in Figure 3.7
Ciclopirox
Ciclopirox was the first topical therapy to be approved for the treatment of onychomycosis. Ciclopirox is a broad-spectrum antifungal agent that inhibits metal-dependent enzymes, which are responsible for the degradation of toxic peroxides in fungal cells. The safety and efficacy of ciclopirox nail lacquer topical solution 8% also was investigated in 2 identical phase 3 clinical trials.8 The first study was conducted at 9 sites in the United States between June 1994 and June 1996 and included 223 participants. The second study was conducted at 9 sites in the United States between July 1994 and April 1996 and included 237 participants.
Eligible participants were required to have DSO in at least one TGT, positive KOH wet mount with positive dermatophyte culture, and 20% to 65% nail involvement.8 Those with tinea pedis were not excluded. Participants were randomized to receive once-daily treatment with ciclopirox or vehicle (1:1)(applied to all toenails and affected fingernails) for 48 weeks. The product was to be removed by the patient with alcohol on a weekly basis. Trimming was allowed as necessary, and mechanical debridement by the physician could be performed monthly. Assessments were made every 4 weeks, and mycologic examinations were performed every 12 weeks. Participants who were clinically cured were assessed further in a 12- to 24-week posttreatment follow-up period.8
The primary end point of complete cure and secondary end points of treatment success (negative culture and KOH, ≤10% mycotic nail), mycologic cure, and negative mycologic culture were assessed at week 48.8
Phase 3 Clinical Trial Similarities and Differences
The phase 3 clinical trials used to investigate the safety and efficacy of tavaborole,5 efinaconazole,7 and ciclopirox8 were similar in their overall design. All trials were randomized, double-blind, vehicle-controlled studies in patients with DSO. Each agent was assessed using a once-daily application for a treatment period of 48 weeks.
Primary differences among study designs included the age range of participants, the range of mycotic nail involvement, the presence/absence of tinea pedis, and the nail trimming/debridement protocols used. Differences were observed in the patient eligibility criteria of these trials. Both mycotic area and participant age range were inconsistent for each agent (eTable). Participants with larger mycotic areas usually have a poorer prognosis, as they tend to have a greater fungal load.9 A baseline mycotic area of 20% to 60%,5 20% to 50%,7 and 20% to 65%8 at baseline was required for the tavaborole, efinaconazole, and ciclopirox trials, respectively. Variations in mycotic area between trials can affect treatment efficacy, as clinical cures can be reached quicker by patients with smaller areas of infection. Of note, the average mycotic area of involvement was not reported in the tavaborole studies but was 36% and 40% for the efinaconazole and ciclopirox studies, respectively.5,8 It also is more difficult to achieve complete cure in older patients, as they have poor circulation and reduced nail growth rates.1,10 The participant age range was 18 to 88 years in the tavaborole trials, with 8% of the participants older than 70 years,5 compared to 18 to 71 years in both the efinaconazole and ciclopirox trials.7,8 The average age of participants in each study was approximately 54, 51, and 50 years for tavaborole, efinaconazole, and ciclopirox, respectively. Because factors impacting treatment failure can increase with age, efficacy results can be confounded by differing age distributions across different studies.
Another important feature that differed between the clinical trials was the approach to nail trimming—defined as shortening of the free edge of the nail distal to the hyponychium—which varies from debridement in that the nail plate is removed or reduced in thickness proximal to the hyponychium. In the tavaborole trials, trimming was controlled to within 1 mm of the free edge of the nail,5 whereas the protocol used for the ciclopirox trials allowed nail trimming as necessary as well as moderate debridement before treatment application and on a monthly basis.8 Debridement is an important component in all ciclopirox trials, as it is used to reduce fungal load.11 No trimming control was provided during the efinaconazole trials; however, debridement was prohibited.7 These differences can dramatically affect the study results, as residual fungal elements and portions of infected nails are removed during the trimming process in an uncontrolled manner, which can affect mycologic testing results as well as the clinical efficacy results determined through investigator evaluation. Discrepancies regarding nail trimming approach inevitably makes the trial results difficult to compare, as mycologic cure is not translatable between studies.
Furthermore, somewhat unusually, complete cure rate variations were observed between different study centers in the efinaconazole trials. Japanese centers in the first efinaconazole study (NCT01008033) had higher complete cure rates in both the efinaconazole and vehicle treatment arms, which is notable because approximately 29% of participants in this study were Asian, mostly hailing from 33 Japanese centers. The reason for these confounding results is unknown and requires further analysis.
Lastly, the presence or absence of tinea pedis can affect the response to onychomycosis treatment. In the tavaborole trials, patients with active interdigital tinea pedis or exclusively plantar tinea pedis or chronic moccasin-type tinea pedis requiring treatment were excluded from the studies.5 In contrast, only patients with severe moccasin-type tinea pedis were excluded in efinaconazole trials.7 The ciclopirox studies had no exclusions based on presence of tinea pedis.8 These differences are noteworthy, as tinea pedis can serve as a reservoir for fungal infection if not treated and can lead to recurrence of onychomycosis.12
Conclusion
In recent years, disappointing efficacy has resulted in the failure of several topical agents for onychomycosis during their development; however, there are several aspects to consider when examining efficacy data in onychomycosis studies. Obtaining a complete cure in onychomycosis is difficult. Because patients applying treatments at home are unlikely to undergo mycologic testing to confirm complete cure, visual inspections are helpful to determine treatment efficacy.
Despite similar overall designs, notable differences in the study designs of the phase 3 clinical trials investigating tavaborole, efinaconazole, and ciclopirox are likely to have had an effect on the reported results, making the efficacy of the agents difficult to compare. It is particularly tempting to compare the primary end point results of each trial, especially considering tavaborole and efinaconazole had primary end points with the same parameters; however, there are several other factors (eg, age range of study population, extent of infection, nail trimming, patient demographics) that may have affected the outcomes of the studies and precluded a direct comparison of any end points. Without head-to-head investigations, there is room for prescribing clinicians to interpret results differently.
Acknowledgment
Writing and editorial assistance was provided by ApotheCom Associates, LLC, Yardley, Pennsylvania, and was supported by Sandoz, a Novartis division.
Onychomycosis is a fungal nail infection primarily caused by dermatophytes.1 If left untreated, the infection can cause nail destruction and deformities,1 resulting in pain and discomfort,2 impaired foot mobility,3 and an overall reduced quality of life.1 Onychomycosis is a chronic condition that requires long treatment periods due to the slow growth rates of toenails.1 To successfully cure the condition, fungal eradication must be achieved.
Prior to the US Food and Drug Administration (FDA) approval of tavaborole and efinaconazole, ciclopirox was the only approved topical treatment for onychomycosis.4 The recent approval of tavaborole and efinaconazole has increased treatment options available to patients and has started to pave the way for future topical treatments. This article discusses the 3 approved topical treatments for onychomycosis and focuses on the design of the phase 3 clinical trials that led to their approval.
Topical Agents Used to Treat Onychomycosis
Tavaborole, efinaconazole, and ciclopirox have undergone extensive clinical investigation to receive FDA approval. Results from pivotal phase 3 studies establishing the efficacy and safety of each agent formed the basis for regulatory submission. Although it may seem intuitive to compare the relative performance of these agents based on their respective phase 3 clinical trial data, there are important differences in study methodology, conduct, and populations that prevent direct comparisons. The FDA provides limited guidance to the pharmaceutical industry on how to conduct clinical trials for potential onychomycosis treatments. Comparative efficacy and safety claims are limited based on cross-study comparisons. The details of the phase 3 trial designs are summarized in the Table.
Tavaborole
Tavaborole is a boron-based treatment with a novel mechanism of action.5 Tavaborole binds to the editing domain of leucyl–transfer ribonucleic acid synthetase via an integrated boron atom and inhibits fungal protein synthesis.6 Two identical randomized, double-blind, vehicle-controlled, parallel-group, phase 3 clinical trials evaluating tavaborole were performed.5 The first study (registered at www.clinicaltrials.gov with the identifier NCT01270971) included 594 participants from27 sites in the United States and Mexico and was conducted between December 2010 and November 2012. The second study (NCT01302119) included 604 participants from 32 sites in the United States and Canada and was conducted between February 2011 and January 2013.
Eligible participants 18 years and older had distal subungual onychomycosis (DSO) of the toenails affecting 20% to 60% of 1 or more target great toenails (TGTs), tested positive for fungus using potassium hydroxide (KOH) wet mounts and positive for Trichophyton rubrum and Trichophyton mentagrophytes on fungal culture diagnostic tests, had distal TGT thickness of 3 mm or less, and had 3 mm or more of clear nail between the proximal nail fold and the most proximal visible mycotic border.5 Those with active tinea pedis requiring treatment or with a history of chronic moccasin-type tinea pedis were excluded. Participants were randomized to receive either tavaborole or vehicle (2:1). Treatments were applied once daily to all infected toenails for a total of 48 weeks, and nail debridement (defined as partial or complete removal of the toenail) was not permitted. Notably, controlled trimming of the nail was allowed to 1 mm of the leading nail edge. Regular assessments of each toenail for disease involvement, onycholysis, and subungual hyperkeratosis were made at screening, baseline, week 2, week 6, and every 6 weeks thereafter until week 52. Subungual TGT samples were taken at screening and every 12 weeks during the study for examination at a mycology laboratory, which performed KOH and fungal culture tests. A follow-up assessment was made at week 52.5
The primary end point was complete cure of the TGT at week 52, with secondary end points of completely or almost clear TGT nail (≤10% dystrophic nail), completely or almost clear TGT nail (≤10% dystrophic nail) plus negative mycology, and negative mycology of TGT.5 Examples of TGTs in participants who achieved complete cure and almost clear nails with negative mycology before and after treatment with tavaborole are shown in Figure 1. An example of a patient considered to have treatment failure is shown in Figure 2. This patient showed marked improvement in nail appearance and had a negative culture result but had a positive KOH test, which demonstrates the stringency in which topical agents are judged in onychomycosis trials.5
Efinaconazole
Efinaconazole is a topical triazole antifungal specifically indicated to treat onychomycosis. Two identical randomized, vehicle-controlled, double-blind, multicenter trials were performed to assess the safety and efficacy of efinaconazole solution 10%.7 The first study (NCT01008033) involved 870 participants and was conducted at a total of 74 sites in Japan (33 sites), Canada (7 sites), and the United States (34 sites) between December 2009 and September 2011. The second study (NCT01007708) had 785 participants and was conducted at 44 sites in Canada (8 sites) and the United States (36 sites) between December 2009 and October 2011.
Participants aged 18 to 70 years with a clinical diagnosis of DSO affecting 1 or more TGT were eligible to participate.7 Other eligibility criteria included an uninfected toenail length 3 mm or more from the proximal nail fold, a maximum toenail thickness of 3 mm, positive KOH wet mounts, and positive dermatophyte or mixed dermatophyte/candida cultures. Dermatophytes included T rubrum and T mentagrophytes. Those with severe moccasin-type tinea pedis were excluded. Participants were randomized to receive efinaconazole or vehicle (3:1). Once-daily treatments were self-applied to nails for 48 weeks. Clinical assessments were made at baseline and every 12 weeks until week 48, with a follow-up assessment at week 52. No nail trimming protocol was provided.7
The primary end point of the efinaconazole phase 3 trials was complete cure at week 52, with secondary end points including mycologic cure, treatment success (≤5% mycotic nail), and complete or almost complete cure (negative culture and KOH, ≤5% mycotic nail). An example of a complete cure from baseline to week 52 is shown in Figure 3.7
Ciclopirox
Ciclopirox was the first topical therapy to be approved for the treatment of onychomycosis. Ciclopirox is a broad-spectrum antifungal agent that inhibits metal-dependent enzymes, which are responsible for the degradation of toxic peroxides in fungal cells. The safety and efficacy of ciclopirox nail lacquer topical solution 8% also was investigated in 2 identical phase 3 clinical trials.8 The first study was conducted at 9 sites in the United States between June 1994 and June 1996 and included 223 participants. The second study was conducted at 9 sites in the United States between July 1994 and April 1996 and included 237 participants.
Eligible participants were required to have DSO in at least one TGT, positive KOH wet mount with positive dermatophyte culture, and 20% to 65% nail involvement.8 Those with tinea pedis were not excluded. Participants were randomized to receive once-daily treatment with ciclopirox or vehicle (1:1)(applied to all toenails and affected fingernails) for 48 weeks. The product was to be removed by the patient with alcohol on a weekly basis. Trimming was allowed as necessary, and mechanical debridement by the physician could be performed monthly. Assessments were made every 4 weeks, and mycologic examinations were performed every 12 weeks. Participants who were clinically cured were assessed further in a 12- to 24-week posttreatment follow-up period.8
The primary end point of complete cure and secondary end points of treatment success (negative culture and KOH, ≤10% mycotic nail), mycologic cure, and negative mycologic culture were assessed at week 48.8
Phase 3 Clinical Trial Similarities and Differences
The phase 3 clinical trials used to investigate the safety and efficacy of tavaborole,5 efinaconazole,7 and ciclopirox8 were similar in their overall design. All trials were randomized, double-blind, vehicle-controlled studies in patients with DSO. Each agent was assessed using a once-daily application for a treatment period of 48 weeks.
Primary differences among study designs included the age range of participants, the range of mycotic nail involvement, the presence/absence of tinea pedis, and the nail trimming/debridement protocols used. Differences were observed in the patient eligibility criteria of these trials. Both mycotic area and participant age range were inconsistent for each agent (eTable). Participants with larger mycotic areas usually have a poorer prognosis, as they tend to have a greater fungal load.9 A baseline mycotic area of 20% to 60%,5 20% to 50%,7 and 20% to 65%8 at baseline was required for the tavaborole, efinaconazole, and ciclopirox trials, respectively. Variations in mycotic area between trials can affect treatment efficacy, as clinical cures can be reached quicker by patients with smaller areas of infection. Of note, the average mycotic area of involvement was not reported in the tavaborole studies but was 36% and 40% for the efinaconazole and ciclopirox studies, respectively.5,8 It also is more difficult to achieve complete cure in older patients, as they have poor circulation and reduced nail growth rates.1,10 The participant age range was 18 to 88 years in the tavaborole trials, with 8% of the participants older than 70 years,5 compared to 18 to 71 years in both the efinaconazole and ciclopirox trials.7,8 The average age of participants in each study was approximately 54, 51, and 50 years for tavaborole, efinaconazole, and ciclopirox, respectively. Because factors impacting treatment failure can increase with age, efficacy results can be confounded by differing age distributions across different studies.
Another important feature that differed between the clinical trials was the approach to nail trimming—defined as shortening of the free edge of the nail distal to the hyponychium—which varies from debridement in that the nail plate is removed or reduced in thickness proximal to the hyponychium. In the tavaborole trials, trimming was controlled to within 1 mm of the free edge of the nail,5 whereas the protocol used for the ciclopirox trials allowed nail trimming as necessary as well as moderate debridement before treatment application and on a monthly basis.8 Debridement is an important component in all ciclopirox trials, as it is used to reduce fungal load.11 No trimming control was provided during the efinaconazole trials; however, debridement was prohibited.7 These differences can dramatically affect the study results, as residual fungal elements and portions of infected nails are removed during the trimming process in an uncontrolled manner, which can affect mycologic testing results as well as the clinical efficacy results determined through investigator evaluation. Discrepancies regarding nail trimming approach inevitably makes the trial results difficult to compare, as mycologic cure is not translatable between studies.
Furthermore, somewhat unusually, complete cure rate variations were observed between different study centers in the efinaconazole trials. Japanese centers in the first efinaconazole study (NCT01008033) had higher complete cure rates in both the efinaconazole and vehicle treatment arms, which is notable because approximately 29% of participants in this study were Asian, mostly hailing from 33 Japanese centers. The reason for these confounding results is unknown and requires further analysis.
Lastly, the presence or absence of tinea pedis can affect the response to onychomycosis treatment. In the tavaborole trials, patients with active interdigital tinea pedis or exclusively plantar tinea pedis or chronic moccasin-type tinea pedis requiring treatment were excluded from the studies.5 In contrast, only patients with severe moccasin-type tinea pedis were excluded in efinaconazole trials.7 The ciclopirox studies had no exclusions based on presence of tinea pedis.8 These differences are noteworthy, as tinea pedis can serve as a reservoir for fungal infection if not treated and can lead to recurrence of onychomycosis.12
Conclusion
In recent years, disappointing efficacy has resulted in the failure of several topical agents for onychomycosis during their development; however, there are several aspects to consider when examining efficacy data in onychomycosis studies. Obtaining a complete cure in onychomycosis is difficult. Because patients applying treatments at home are unlikely to undergo mycologic testing to confirm complete cure, visual inspections are helpful to determine treatment efficacy.
Despite similar overall designs, notable differences in the study designs of the phase 3 clinical trials investigating tavaborole, efinaconazole, and ciclopirox are likely to have had an effect on the reported results, making the efficacy of the agents difficult to compare. It is particularly tempting to compare the primary end point results of each trial, especially considering tavaborole and efinaconazole had primary end points with the same parameters; however, there are several other factors (eg, age range of study population, extent of infection, nail trimming, patient demographics) that may have affected the outcomes of the studies and precluded a direct comparison of any end points. Without head-to-head investigations, there is room for prescribing clinicians to interpret results differently.
Acknowledgment
Writing and editorial assistance was provided by ApotheCom Associates, LLC, Yardley, Pennsylvania, and was supported by Sandoz, a Novartis division.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Scher RK. Onychomycosis: a significant medical disorder. J Am Acad Dermatol. 1996;35(3, pt 2):S2-S5.
- Del Rosso JQ. The role of topical antifungal therapy for onychomycosis and the emergence of newer agents. J Clin Aesthet Dermatol. 2014;7:10-18.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Gupta AK, Joseph WS. Ciclopirox 8% nail lacquer in the treatment of onychomycosis of the toenails in the United States. J Am Pod Med Assoc. 2000;90:495-501.
- Carney C, Tosti A, Daniel R, et al. A new classification system for grading the severity of onychomycosis: Onychomycosis Severity Index. Arch Dermatol. 2011;147:1277-1282.
- Gupta AK. Onychomycosis in the elderly. Drugs Aging. 2000;16:397-407.
- Gupta AK, Malkin KF. Ciclopirox nail lacquer and podiatric practice. J Am Podiatr Med Assoc. 2000;90:502-507.
- Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
- Elewski BE. Onychomycosis: pathogenesis, diagnosis, and management. Clin Microbiol Rev. 1998;11:415-429.
- Thomas J, Jacobson GA, Narkowicz CK, et al. Toenail onychomycosis: an important global disease burden. J Clin Pharm Ther. 2010;35:497-519.
- Scher RK. Onychomycosis: a significant medical disorder. J Am Acad Dermatol. 1996;35(3, pt 2):S2-S5.
- Del Rosso JQ. The role of topical antifungal therapy for onychomycosis and the emergence of newer agents. J Clin Aesthet Dermatol. 2014;7:10-18.
- Elewski BE, Aly R, Baldwin SL, et al. Efficacy and safety of tavaborole topical solution, 5%, a novel boron-based antifungal agent, for the treatment of toenail onychomycosis: results from 2 randomized phase-III studies. J Am Acad Dermatol. 2015;73:62-69.
- Rock FL, Mao W, Yaremchuk A, et al. An antifungal agent inhibits an aminoacyl-tRNA synthetase by trapping tRNA in the editing site. Science. 2007;316:1759-1761.
- Elewski BE, Rich P, Pollak R, et al. Efinaconazole 10% solution in the treatment of toenail onychomycosis: two phase III multicenter, randomized, double-blind studies. J Am Acad Dermatol. 2013;68:600-608.
- Gupta AK, Joseph WS. Ciclopirox 8% nail lacquer in the treatment of onychomycosis of the toenails in the United States. J Am Pod Med Assoc. 2000;90:495-501.
- Carney C, Tosti A, Daniel R, et al. A new classification system for grading the severity of onychomycosis: Onychomycosis Severity Index. Arch Dermatol. 2011;147:1277-1282.
- Gupta AK. Onychomycosis in the elderly. Drugs Aging. 2000;16:397-407.
- Gupta AK, Malkin KF. Ciclopirox nail lacquer and podiatric practice. J Am Podiatr Med Assoc. 2000;90:502-507.
- Scher RK, Baran R. Onychomycosis in clinical practice: factors contributing to recurrence. Br J Dermatol. 2003;149(suppl 65):5-9.
Practice Points
- Despite similar overall designs, notable differences in the study designs of phase 3 clinical trials investigating tavaborole, efinaconazole, and ciclopirox for the treatment of onychomycosis are likely to have had an effect on the reported results, making the efficacy of these agents difficult to compare.
- The primary difference between studies for tavaborole, efinaconazole, and ciclopirox include the age range of participants, the range of mycotic nail involvement, the presence/absence of tinea pedis, and the nail trimming/debridement protocols used.
- Without head-to-head investigations, there is room for prescribing clinicians to interpret study results for these agents differently.