User login
Cutis is a peer-reviewed clinical journal for the dermatologist, allergist, and general practitioner published monthly since 1965. Concise clinical articles present the practical side of dermatology, helping physicians to improve patient care. Cutis is referenced in Index Medicus/MEDLINE and is written and edited by industry leaders.
ass lick
assault rifle
balls
ballsac
black jack
bleach
Boko Haram
bondage
causas
cheap
child abuse
cocaine
compulsive behaviors
cost of miracles
cunt
Daech
display network stats
drug paraphernalia
explosion
fart
fda and death
fda AND warn
fda AND warning
fda AND warns
feom
fuck
gambling
gfc
gun
human trafficking
humira AND expensive
illegal
ISIL
ISIS
Islamic caliphate
Islamic state
madvocate
masturbation
mixed martial arts
MMA
molestation
national rifle association
NRA
nsfw
nuccitelli
pedophile
pedophilia
poker
porn
porn
pornography
psychedelic drug
recreational drug
sex slave rings
shit
slot machine
snort
substance abuse
terrorism
terrorist
texarkana
Texas hold 'em
UFC
section[contains(@class, 'nav-hidden')]
section[contains(@class, 'nav-hidden active')
A peer-reviewed, indexed journal for dermatologists with original research, image quizzes, cases and reviews, and columns.
Red-Brown Plaque on the Leg
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.

Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
The Diagnosis: Wells Syndrome
A punch biopsy taken from the perimeter of the lesion demonstrated mild spongiosis overlying a dense nodular to diffuse infiltrate of lymphocytes, neutrophils, and numerous eosinophils, some involving underlying fat lobules (Figure, A and B). In some areas, eosinophilic degeneration of collagen bundles surrounded by a rim of histiocytes, "flame features," were observed (Figure C). The clinical and histological features were consistent with Wells syndrome (WS), also known as eosinophilic cellulitis. Given the localized mild nature of the disease, the patient was started on a midpotency topical corticosteroid.
Wells syndrome is a rare inflammatory condition characterized by clinical polymorphism, suggestive histologic findings, and a recurrent course.1,2 This condition is especially rare in children.3,4 Caputo et al1 described 7 variants in their case series of 19 patients: classic plaque-type variant (the most common clinical presentation in children); annular granuloma-like (the most common clinical presentation in adults); urticarialike; bullous; papulonodular; papulovesicular; and fixed drug eruption-like. Wells syndrome is thought to result from excess production of IL-5 in response to a hypersensitivity reaction to an exogenous or endogenous circulating antigen.3,4 Increased levels of IL-5 enhance eosinophil accumulation in the skin, degranulation, and subsequent tissue destruction.3,4 Reported triggers include insect bites, viral and bacterial infections, drug eruptions, recent vaccination, and paraphenylenediamine in henna tattoos.3-7 Additionally, WS has been reported in the setting of gastrointestinal pathologies, such as celiac disease and ulcerative colitis, and with asthma exacerbations.8,9 However, in half of pediatric cases, no trigger can be identified.7
Clinically, WS presents with pruritic, mildly tender plaques.7 Lesions may be localized or diffuse and range from mild annular or circinate plaques with infiltrated borders to cellulitic-appearing lesions that are occasionally associated with bullae.5,6 Patients often report prodromal symptoms of burning and pruritus.5,6 Lesions rapidly progress over 2 to 3 days, pass through a blue grayish discoloration phase, and gradually resolve over 2 to 8 weeks.5,6,10 Although patients generally heal without scarring, WS lesions have been described to resolve with atrophy and hyperpigmentation resembling morphea.5-7 Additionally, patients typically experience a relapsing remitting course over months to years with eventual spontaneous resolution.1,5 Patients also may experience systemic symptoms including fever, lymphadenopathy, and arthralgia, though they do not develop more widespread systemic manifestations.2,3,7
Diagnosis of WS is based on clinicopathologic correlation. Histopathology of WS lesions demonstrates 3 phases. The acute phase demonstrates edema of the superficial and mid dermis with a dense dermal eosinophilic infiltrate.1,6,10 The subacute granulomatous phase demonstrates flame figures in the dermis.1,2,6,7,10 Flame figures consist of palisading groups of eosinophils and histiocytes around a core of degenerating basophilic collagen bundles associated with major basic protein.1,2,6,7,10 Finally, in the resolution phase, eosinophils gradually disappear while histiocytes and giant cells persist, forming microgranulomas.1,2,10 Notably, no vasculitis is observed and direct immunofluorescence is negative.3,7 Although flame figures are suggestive of WS, they are not pathognomonic and are observed in other conditions including Churg-Strauss syndrome, parasitic and fungal infections, herpes gestationis, bullous pemphigoid, and follicular mucinosis.2,5
Wells syndrome is a self-resolving and benign condition.1,10 Physicians are recommended to gather a complete history including review of medications and vaccinations; a history of insect bites, infections, and asthma; laboratory workup consisting of a complete blood cell count with differential and stool samples for ova and parasites; and a skin biopsy if the diagnosis is unclear.7 Identification and treatment of underlying causes often results in resolution.6 Systemic corticosteroids frequently are used in both adult and pediatric patients, though practitioners should consider alternative treatments when recurrences occur to avoid steroid side effects.3,6 Midpotency topical corticosteroids present a safe alternative to systemic corticosteroids in the pediatric population, especially in cases of localized WS without systemic symptoms.3 Other medications reported in the literature include cyclosporine, dapsone, antimalarial medications, and azathioprine.6 Despite appropriate therapy, patients and physicians should anticipate recurrence over months to years.1,6
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
- Caputo R, Marzano AV, Vezzoli P, et al. Wells syndrome in adults and children: a report of 19 cases. Arch Dermatol. 2006;142:1157-1161.
- Smith SM, Kiracofe EA, Clark LN, et al. Idiopathic hypereosinophilic syndrome with cutaneous manifestations and flame figures: a spectrum of eosinophilic dermatoses whose features overlap with Wells' syndrome. Am J Dermatopathol. 2015;37:910-914.
- Gilliam AE, Bruckner AL, Howard RM, et al. Bullous "cellulitis" with eosinophilia: case report and review of Wells' syndrome in childhood. Pediatrics. 2005;116:E149-E155.
- Nacaroglu HT, Celegen M, Karkıner CS, et al. Eosinophilic cellulitis (Wells' syndrome) caused by a temporary henna tattoo. Postepy Dermatol Alergol. 2014;31:322-324.
- Heelan K, Ryan JF, Shear NH, et al. Wells syndrome (eosinophilic cellulitis): proposed diagnostic criteria and a literature review of the drug-induced variant. J Dermatol Case Rep. 2013;7:113-120.
- Sinno H, Lacroix JP, Lee J, et al. Diagnosis and management of eosinophilic cellulitis (Wells' syndrome): a case series and literature review. Can J Plast Surg. 2012;20:91-97.
- Cherng E, McClung AA, Rosenthal HM, et al. Wells' syndrome associated with parvovirus in a 5-year-old boy. Pediatr Dermatol. 2012;29:762-764.
- Eren M, Açikalin M. A case report of Wells' syndrome in a celiac patient. Turk J Gastroenterol. 2010;21:172-174.
- Cruz MJ, Mota A, Baudrier T, et al. Recurrent Wells' syndrome associated with allergic asthma exacerbation. Cutan Ocul Toxicol. 2012;31:154-156.
- Van der Straaten S, Wojciechowski M, Salgado R, et al. Eosinophilic cellulitis or Wells' syndrome in a 6-year-old child. Eur J Pediatr. 2006;165:197-198.
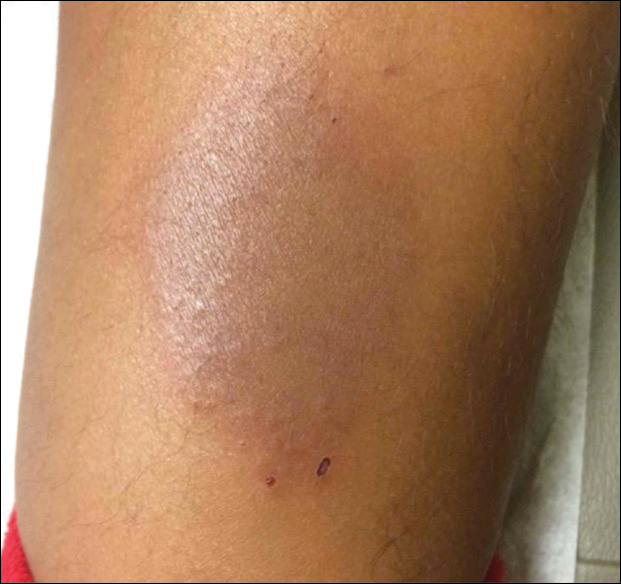
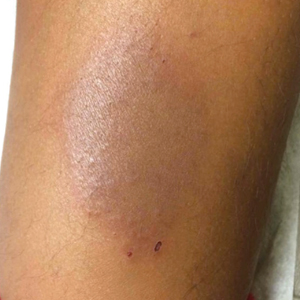
A healthy 7-year-old boy presented with an enlarging hyperpigmented plaque on the anterior aspect of the lower left leg of 2 months' duration. His mother reported onset following a mosquito bite. Clotrimazole was used without improvement. His mother denied recent travel, similar lesions in close contacts, fever, asthma, and arthralgia. Physical examination revealed a 5.2 ×3-cm nonscaly, red-brown, ovoid, thin plaque with a slightly raised border.
A Peek at Our June 2018 Issue
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel
The video associated with this article is no longer available on this site. Please view all of our videos on the MDedge YouTube channel

What’s Eating You? Clothes Moths (Tineola Species)
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
 larva.")
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
Clothes moths are common pests found inside buildings such as homes, stores, and museums. The most common species of importance include the webbing clothes moth (Tineola bisselliella)(Figure) and the casemaking clothes moth (Tineola pellionella). Both species target textiles such as wool, rugs, feathers, felts, hair, furs, and even grains.1,2 They avoid synthetics and plant materials such as cottons.1
Characteristics
Adult clothes moths extend 7 to 8 mm and are a golden (T bisselliella) to brown (T pellionella) color with fringed wings and a tuft on their heads.1,3 Adults do not eat; females die within a few days of laying eggs, while males live approximately 1 month. Once laid, eggs hatch within 4 to 10 days.1,3 The larvae (caterpillars) incur damage to clothes and other household goods. Fully mature larvae are 12- to 13-mm long, and the Tineola species have white- to cream-colored bodies with brown heads. The webbing clothes moth larva lacks ocelli (eyes), while the casemaking moth larva has a singular ocellus.1
Transmission
An infestation is evidenced by woolen items that have furrows or holes in them. Pheromone traps also can expose an active infestation.3 The webbing moth larvae can be found beneath a self-spun silken mat on the food source that offers the insect protection and camouflage while it eats; the mat collects frass (feces) and clothes particles.1,3,4 The casemaking moth larvae drag around a portable silken bag that takes on the color of the fabric being eaten and serves as a refuge when disturbed.1,3,4 Both adult and larval stages prefer low light conditions. The total time of development from caterpillar to adult varies depending on the temperature and humidity of the environment, but most clothes moths complete their life cycle within 1 to 3 months.1
Management of an Infestation
Multiple infestation treatment options exist should a patient present with a clothes moth infestation. Infested clothing articles or small blankets and rugs can be dry-cleaned or laundered. Any items not in use should be laundered before being sealed in airtight storage containers. Mothball vapor at appropriate concentrations is lethal to the moths, and when possible, clothing should be stored with mothballs or flakes at the concentration recommended by the manufacturer.4 Individuals should avoid application of household insecticides to clothing or bedding, which may be poisonous to people.1,4 Freezing, heating, and dry ice fumigation techniques also can be used to treat infested products.3 Cedarwood usually is insufficient to deter an infestation, as the oil vapor rarely reaches an effective concentration to repel or harm the insects.3,4 Strict housekeeping with attention to vacuuming carpets, baseboards, closets, and laundering all linens and furniture covers can further reduce an infestation.4 Clothes items can be set in the sunlight and brushed to help loosen the pests, as they dislike direct light and may fall from the garments.3 Dust insecticides also can be used per the manufacturer label to treat crevices and baseboards in an active area of infestation that may otherwise be difficult to clean.3 If an extensive infestation exists or larger items are infested, then a professional pest control agency should be employed for proper eradication.
Conclusion
Understanding the life cycle and basic biology of clothes moths and other common household pests will help the clinician identify an infestation and counsel patients if an insect is a true ectoparasite. Clothes moth larvae are not parasites but can be found on clothing and can be confused with myiasis or true parasites.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
- Jacobs S. Clothes moth. Penn State Extension website. http://ento.psu.edu/extension/factsheets/clothes-moth. Updated January 2013. Accessed May 14, 2018.
- Querner P. Insect pests and integrated pest management in museums, libraries and historic buildings. Insects. 2015;6:595-607.
- Choe D-H. Pest notes: clothes moths (publication 7435). University of California Agriculture & Natural Resources website. http://ipm.ucanr.edu/PMG/PESTNOTES/pn7435.html. Updated March 2013. Accessed May 14, 2018.
- Potter M. Entfact-609: clothes moths. Entomology at the University of Kentucky website. https://entomology.ca.uky.edu/ef609. Updated October 2001. Accessed May 14, 2018.
Practice Points
- Clothes moth larvae are common household pests that may be misidentified as a parasitic infection such as myiasis when found on a person.
- Understanding the basic biology of clothes moths will help the clinician identify an infestation and appropriately counsel patients that clothes moths do not pose a considerable health risk.

Modifier -25 Victory, But the Battle Is Not Over
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
On February 23, 2018, Anthem Insurance Companies, Inc, announced the reversal of its proposed policy to reduce reimbursement for evaluation and management (E/M) services billed using modifier -25.1 This win for physicians was the result of a broad-based, multipronged advocacy campaign, and the American Academy of Dermatology Association (AADA) was critical to this victory.
Dermatology Took the Lead in Opposing Policy That Would Reduce Reimbursement
Dermatology was the first to trumpet the dangers of modifier -25 reduction policies and explain why other specialty societies should care. The AADA took the lead in assembling a coalition of physician groups to oppose the proposed policy by sharing language for opposition letters and meeting talking points with many societies outside of dermatology as well as producing the first draft for the American Medical Association (AMA) House of Delegates’ resolution that spurred action against Anthem’s proposed policy.2 Members of the AADA attended numerous conference calls and in-person meetings with health insurance officials to urge them to reverse the policy and helped coordinate opposition from state and national specialty societies. The influence and advocacy of the AMA was critical in reversing the proposed policy, but dermatology started the opposition and organized the players to bring the fight against Anthem.
AADA Continues to Fight
Despite the victory against Anthem, challenges to fair reimbursement of modifier -25 claims are ongoing. Two insurers recently announced implementation of new modifier -25 reduction policies.3,4 Moreover, 4 other insurers have existing modifier -25 reduction policies in place.5-8 The AADA has engaged, and will continue engaging, each of these insurers with the message that the ability to perform procedures and distinct E/M services on the same day is integral to efficient, patient-centered care of dermatologic diseases. The AADA argues that insurers’ rationale (overlapping indirect practice expenses) for payment reduction is improper and that appropriately documented modifier -25 claims should be reimbursed at 100% of allowable charges.
In addition to the existing insurer policies affecting modifier -25, Anthem has announced that it plans to conduct audits of modifier -25 claims with recoupments of inappropriate charges.1 Some Medicare Administrative Contractors also have cited modifier -25 as an area of concern and issued guidelines for reporting modifier -25, which frequently precede audits.9 The AADA is concerned that if Anthem follows through with its aggressive audits to recoup money, which can result in substantial take-backs, and finds a high error rate in modifier -25 claims, it also may consider revisiting its reduction policy. Moreover, it is likely that other insurers will use failed modifier -25 audits as an excuse to continue, expand, or adopt modifier -25 reduction policies. It is clear that blanket reduction in payment is much easier and less expensive for insurers than auditing medical records and not paying those who abuse modifier -25. As long as insurers are under financial pressure, modifier -25 will be a tempting target for reducing health care costs.
How to Use Modifier -25 Correctly
In addition to opposing inappropriate reimbursement of modifier -25, the AADA is committed to educating its members and insurers on the correct use and documentation of modifier -25. In December 2017, Mollie A. MacCormack, MD (Nashua, New Hampshire), and I led an educational webinar on modifier -25 for more than 1100 attendees.10 We discussed the performance standards and documentation requirements of modifier -25. It was clear that dermatologists were interested not only in the correct coding of modifier -25 claims but also avoiding the consequences of audits.
My peers frequently ask, “What can we [dermatologists] do to prepare for potential modifier -25 audits?” My advice is always, “Physician audit thyself.” I recommend self-auditing to make sure you and your practice are in compliance with modifier -25 documentation requirements. In a March 2017 Cutis column on modifier -25, I discussed what constitutes separate and distinct E/M services and what is included in a procedure’s global surgical package.11 A self-audit is as simple as pulling 10 to 20 medical records in which a same-day procedure and E/M service were billed. Cross out any information in the note included in the procedure’s global surgical package including history associated with establishing the diagnosis, physical examination of the procedure area(s), and discussion of treatment options. If complete documentation for an E/M is still present after removing the procedure and associated evaluation, you have passed the self-audit. If not, consider changing your coding to better reflect the documentation in your records. In my experience, insurance auditors are not physicians and often are not even medical professionals. Clear documentation and clear existence of a separate, distinct, and medically necessary E/M service is needed to succeed in a modifier -25 audit.
Final Thoughts
Dermatologists should rejoice that Anthem decided to rescind its modifier -25 policy. If this policy had gone into effect, the modifier -25 reduction would likely have spread to most other insurers as industry standard. This victory certainly shows what can be accomplished when organized medicine works together. Your state dermatology and medical societies, national societies, and the AMA collaborated on a critical existential threat to cost-effective and efficient patient care and won. These organizations deserve your membership and support as well as your thanks; however, our celebration must be short-lived, as there still are other insurers with modifier -25 reductions in place, and audits have been promised. We must continue to focus on proper use and documentation of modifier -25. This effort will not only help dermatologists decrease the risk of large recoupments from audits but also help the AADA continue to oppose inappropriate payment policies.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
- Anthem Insurance Companies, Inc. Network Update. April 2018. https://www11.anthem.com/provider/ct/f5/s1/t0/pw_g334020.pdf?refer=ahpprovider&state=ct. Accessed May 25, 2018.
- American Academy of Dermatology, American Society for Dermatologic Surgery Association, American College of Mohs Surgery, American Society of Dermatopathology, Society for Investigative Dermatology. Opposition to reduced payment for the 25 modifier. https://www.ama-assn.org/sites/default/files/media-browser/public/hod/i17-808.pdf. Accessed May 14, 2018.
- We’re changing the payment policy when evaluation and management services are billed with surgery. Blue Cross Blue Shield of Michigan website. https://www.bcbsm.com/newsletter/therecord/2018/record_0418/Record_0418u.shtml. Published April 2018. Accessed May 14, 2018.
- Centene Corporation. Payment policy: problem oriented visits billed with surgical procedures. https://www.superiorhealthplan.com/content/dam/centene/Superior/Provider/PDFs/Problem%20Oriented%20Visits%20Billed
%20with%20Surgical%20Procedures%20(Ambetter%20MMP
%20and%20Medicare%20Advantage%20Only).pdf. Accessed May 14, 2018. - Blue Cross Blue Shield of Rhode Island. Payment Policy: Coding and Payment Guidelines. August 16, 2016. https://www.bcbsri.com/sites/default/files/polices/Coding-and-Payment-Guidelines-Oct2016.pdf. Accessed May 9, 2018.
- Modifier payment policy. Tufts Health Plan website. https://tuftshealthplan.com/documents/providers/payment-policies/modifier-payment-policy. Updated November 2017. Accessed May 14, 2018.
- Harvard Pilgrim Health Care. Payment Policies: Evaluation and Management. February 2018. https://www.harvardpilgrim.org/pls/portal/docs/PAGE/PROVIDERS/MANUALS/PAYMENT%20POLICIES/H-2%20EVALUATION-MGT_02
0118.PDF. Accessed May 25, 2018. - Updates to the policies on modifier 25 reporting and reimbursement. Independence Blue Cross website. http://provcomm.ibx.com/ProvComm/ProvComm.nsf/4bcc623b93e226638525792c00575962/bbe9e72728cc01e285258167005c629d!OpenDocument. Published July 24, 2017. Accessed May 14, 2018.
- Centers for Medicare & Medicaid Services. Payment for evaluation and management services provided during global period of surgery. MLN Matters. May 19, 2006. https://www.cms.gov/Outreach-and-Education/Medicare-Learning-Network-MLN/MLNMattersArticles/downloads/MM5025.pdf. Accessed May 14, 2018.
- Audits on modifier 25 are coming—complimentary live webinar. American Academy of Dermatology website. https://store.aad.org/products/11928. Accessed May 9, 2018.
- Rogers HW. One diagnosis and modifier -25: appropriate or audit target? Cutis. 2017;99:165-166.
Practice Points
- Insurers are increasingly targeting modifier -25 for audits and payment reductions.
- Physicians should understand modifier -25 documentation requirements and self-audit to ensure compliance.

Updated Guidelines on Peanut Allergy Prevention in Infants With Atopic Dermatitis
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
It has been said that “extraordinary claims require extraordinary evidence.”1 In the pursuit of evidence-based medicine, we are encouraged to follow a similar standard, with an emphasis on waiting for multiple studies with good-quality data and high levels of agreement before changing any aspect of our clinical practice. The ostensible purpose is that studies can be flawed, conclusions can be incorrect, or biases can be overlooked. In such cases, acting on questionable results could imperil patients. It is for this reason that so many review articles sometimes frustratingly seem to conclude that further evidence is needed.2
Based on this standard, recently published addendum guidelines from the National Institute of Allergy and Infectious Diseases for prevention of peanut allergy in the United States3 are somewhat striking in that they make fairly bold recommendations based on results from the 2015 Learning Early about Peanut Allergy (LEAP) study,4 a randomized trial evaluating early peanut introduction as a preventive strategy for peanut allergy. Of note, this study was not placebo controlled, was conducted at only 1 site in the United Kingdom, and only included 640 children, though the number of participants was admittedly large for this type of study.4 Arguably, the LEAP study alone does not provide enough evidence upon which to base what essentially amounts to an about-face in the official recommendations for prevention of peanut and other food allergies, which emphasized delayed introduction of high-risk foods, especially in high-risk individuals.5-7 To better understand this shift, we need to briefly explore the context of the addendum guidelines.
As many as one-third of pediatric patients with atopic dermatitis (AD) have food allergies, thus diet often is invoked by patients and providers alike as an underlying cause of the disease.8 Many patients in my practice are so focused on potential food allergies that actual treatment of the affected skin is marginalized and often dismissed as a stopgap that does not address the root of the problem. A 2004 study of 100 children with AD found that diet was manipulated by the parents in 75% of patients in an attempt to manage the disease.9
Patients are not the only ones who consider food allergies to be a driving force in AD. The medical literature indicates that this theory has existed for centuries; for instance, with regard to the relationship between diet and AD, the author of an article from 1830 quipped, “There is probably no subject in which more deeply rooted convictions have been held . . . than the connection between diet and disease, both as regards the causation and treatment of the latter . . .”10 More apropos perhaps is a statement from the 2010 National Institute of Allergy and Infectious Diseases guidelines on food allergy management, which noted that while the expert group “does not mean to imply that AD results from [food allergies], the role of [food allergies] in the pathogenesis and severity of this condition remains controversial.”11
Prior to the LEAP study, food allergy recommendations for clinical practice in the United Kingdom in 199812 and the United States in 200013 recommended excluding allergenic foods (eg, peanuts, tree nuts, soy, milk, eggs) from the diet in infants with a family history of atopy until 3 years of age. However, those recommendations did not seem to be working, when in fact just the opposite was happening. From 1997 until the LEAP study was conducted in 2015, the prevalence of peanut allergy more than quadrupled and became the leading cause of anaphylaxis and death related to food allergy.14 Additionally, study after study concluded that elimination diets did not seem to help most patients with AD.15 As is required in good scientific thinking, when a hypothesis is proven false, other approaches must be considered.
The idea arose that perhaps delaying introduction of allergenic foods was the opposite of the answer.4 The LEAP study tested the notion that peanut allergies are rare in countries where peanuts are introduced early and if telling families to delay introduction of peanuts in infants might actually be causing development of a peanut allergy, and the tests bore fruit. It was found that giving infants peanut-containing foods resulted in a more than 80% reduction in peanut allergy at 5 years of age (P<.001).4 What was perhaps even more interesting was the connection between AD and peanut allergy. An important idea articulated in the LEAP study is in some ways revolutionary: Rather than foods causing AD, it could be that “early environmental exposure (through the skin) to peanut may account for early sensitization, whereas early oral exposure may lead to immune tolerance.”4 This concept—that impaired eczematous skin may actually lead to the development of food allergies—turns the whole thing upside down.
What do these updated guidelines actually suggest? The first guideline focuses on infants with severe AD, egg allergy, or both, who therefore are thought to be at the highest risk for developing peanut allergy.3 Because of the higher baseline risk in this subgroup, measurement of the peanut-specific IgE (peanut sIgE) level, skin prick testing (SPT), or both is strongly recommended before introducing peanut protein into the diet. This testing can be performed by qualified providers as a screening measure, but if positive (≥0.35 kUA/L for peanut sIgE or >2 mm on the peanut SPT), referral to an allergy specialist is warranted. If these studies are negative, it is thought the likelihood of peanut allergy is low, and it is recommended that caregivers introduce age-appropriate peanut-containing foods (eg, peanut butter snack puffs, diluted peanut butter) as early as 4 to 6 months of age. The second guideline recommends that peanut-containing foods should be introduced into the diets of infants with mild or moderate AD at approximately 6 months of age without the need for prior screening via peanut sIgE or SPT. Lastly, the third guideline recommends that caregivers freely introduce peanut-containing foods together with other solid foods in infants without AD or food allergies in accordance with family preference.3
The results of the LEAP study are certainly exciting, and although the theoretical basis makes good scientific sense and the updated guidelines truly address an important and growing problem, there are several issues with this update that are worth considering. Given the constraints of the LEAP study, it certainly seems possible that the results will not be applicable to all populations or foods. More research is needed to ensure that this robust finding applies to other children and to explore the introduction of other allergenic foods, which the LEAP study investigators also emphasized.4
In fairness, the updated guidelines clearly state the quality of evidence of their recommendations and make it clear that expert opinion is playing a large role.3 For the first guideline regarding recommendations for those with severe AD and/or egg allergy, the quality of evidence is deemed moderate, while the contribution of expert opinion is listed as significant. For the second and third guidelines regarding recommendations for mild to moderate AD and those without AD, respectively, the quality of evidence is low and expert opinion is again listed as significant.3
Importantly, delineating severe AD from moderate disease—which is necessary because only severe AD warrants evaluation with peanut sIgE and/or SPT—can be difficult, as the distinction relies on a degree of subjectivity that may vary between specialists. Indeed, 2 publications suggest extending the definition of severe AD to include infants with early-onset AD (<3 months of age) and those with moderate AD not responding to treatment.16,17
Despite these reservations, the updated guidelines represent a breakthrough in understanding in an area truly in need of advancement. Although the evidence may not be exactly extraordinary, the context for these developments and our deeper understanding suggest that we do indeed live in extraordinary times.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.
- Encyclopaedia Galactica [television transcript]. Cosmos: A Personal Voyage. Public Broadcasting Service. December 14, 1980.
- Ezzo J, Bausell B, Moerman DE, et al. Reviewing the reviews: how strong is the evidence? how clear are the conclusions? Int J Technol Assess Health Care. 2001;17:457-466.
- Togias A, Cooper SF, Acebal ML, et al. Addendum guidelines for the prevention of peanut allergy in the United States: report of the National Institute of Allergy and Infectious Diseases–sponsored expert panel.J Allergy Clin Immunol. 2017;139:29-44.
- Du Toit G, Roberts G, Sayre PH, et al. Randomized trial of peanut consumption in infants at risk for peanut allergy. N Engl J Med. 2015;372:803-813.
- Høst A, Koletzko B, Dreborg S, et al. Dietary products used in infants for treatment and prevention of food allergy. joint statement of the European Society for Paediatric Allergology and Clinical Immunology (ESPACI) Committee on Hypoallergenic Formulas and the European Society for Paediatric Gastroenterology, Hepatology and Nutrition (ESPGHAN) Committee on Nutrition. Arch Dis Child. 1999;81:80-84.
- American Academy of Pediatrics. Committee on Nutrition. hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Fiocchi A, Assa’ad A, Bahna S; Adverse Reactions to Foods Committee; American College of Allergy, Asthma and Immunology. Food allergy and the introduction of solid foods to infants: a consensus document. Ann Allergy Asthma Immunol. 2006;97:10-20.
- Thompson MM, Hanifin JM. Effective therapy of childhood atopic dermatitis allays food allergy concerns. J Am Acad Dermatol. 2005;53(2 suppl 2):S214-S219.
- Johnston GA, Bilbao RM, Graham-Brown RA. The use of dietary manipulation by parents of children with atopic dermatitis. Br J Dermatol. 2004;150:1186-1189.
- Mackenzie S. The inaugural address on the advantages to be derived from the study of dermatology. BMJ. 1830;1:193-197.
- Boyce JA, Assa’ad A, Burks AW, et al; NIAID-Sponsored Expert Panel. Guidelines for the diagnosis and management of food allergy in the United States: report of the NIAID-sponsored expert panel. J Allergy Clin Immunol. 2010;126(6 suppl):S1-S58.
- Committee on Toxicity of Chemicals in Food, Consumer Products and the Environment. Peanut Allergy. London, England: Department of Health; 1998.
- American Academy of Pediatrics Committee on Nutrition. Hypoallergenic infant formulas. Pediatrics. 2000;106(2, pt 1):346-349.
- Gruchalla RS, Sampson HA. Preventing peanut allergy through early consumption—ready for prime time? N Engl J Med. 2015;372:875-877.
- Lim NR, Lohman ME, Lio PA. The role of elimination diets in atopic dermatitis: a comprehensive review. Pediatr Dermatol. 2017;34:516-527.
- Wong CC, Allen KJ, Orchard D. Changes to infant feeding guidelines: relevance to dermatologists. Australas J Dermatol. 2017;58:e171-e175.
- Martin PE, Eckert JK, Koplin JJ, et al. Which infants with eczema are at risk of food allergy? results from a population-based cohort. Clin Exp Allergy. 2015;45:255-264.

Beltlike Lichen Planus Pigmentosus Complicated With Focal Amyloidosis
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.

Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.

.")
.")
.")
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
To the Editor:
A 68-year-old man presented with slightly itchy macules on the waist and abdomen of approximately 2 years’ duration. He reported that the initial lesions were dark red and subsequently coalesced to form a beltlike pigmentation on the abdomen. He denied any prior treatment, and the lesions did not spontaneously resolve. The patient was taking escitalopram oxalate, telmisartan, and aspirin for depression and cardiovascular disease that was diagnosed 3 years prior. He reported no exposure to UV radiation or a heat source. He denied use of any cosmetics on the body as well as a family history of similar symptoms.
Physical examination showed reticulate brown-purple macules with slight scale on the surface that had become confluent, forming a beltlike pigmentation on the waist and abdomen (Figure 1). Wickham striae were not seen. The oral mucosa and nails were not affected. Microscopic examination for fungal infections was negative.
Systematic physical and laboratory examinations revealed no abnormalities. A skin biopsy from a macule on the abdomen showed hyperkeratosis, thinned out stratum spinosum with flattening of rete ridges, hypergranulosis with vacuolar alteration of the basal cell layer, and bandlike infiltration of lymphocytes and melanophages with incontinence of pigment (Figure 2). Focalized purplish homogeneous deposits were observed in the upper dermis (Figure 3), of which positive crystal violet staining indicated amyloidosis (Figure 4). Congo red stain revealed amyloid deposition (Figure 5). Thus, the diagnosis of lichen planus pigmentosus (LPP) complicated with focal amyloidosis was made. The patient was treated with topical corticosteroids and tretinoin, and no notable therapeutic effects were observed at 3-month follow-up.
Lichen planus pigmentosus, a variant of lichen planus, is a condition of unknown etiology exhibiting dark brown macules and/or papules and a long clinical course. The face, neck, trunk, arms, and legs are the most common areas of presentation, whereas involvement of the scalp, nails, or oral mucosa is relatively rare.
The first clinicohistopathological study with a large sample size was documented by Bhutani et al1 in 1974 who termed the currently recognized entity lichen planus pigmentosus. Lichen planus pigmentosus is a frequently encountered hyperpigmentation disorder in Indians, whereas sporadic cases also are reported in other regions and ethnicities.2 In cases of LPP, the pigmentation is symmetrical, and its pattern most often is diffuse, then reticular, blotchy, and perifollicular.3 Two unique patterns of LPP have been documented, including linear/blaschkoid LPP and zosteriform LPP.4,5 Our patient showed a unique beltlike distribution pattern.
The pathogenesis of LPP still is unclear, and several inciting factors such as mustard oil, gold therapy,6 and hepatitis C virus infection have been cited.7 Mancuso and Berdondini8 reported a case of LPP flaring immediately after relapse of nephrotic syndrome. It also has been considered as a paraneoplastic phenomen.9 No exact cause was found in our patient after a series of relative examinations.
The histopathologic changes associated with LPP consist of atrophic epidermis; bandlike lymphocytic infiltrate with vacuolar degeneration of the basal layer in the epidermis; and prominent melanin incontinence in the upper dermis, which can be diverse depending on different sites of skin biopsy and the phase of LPP. Histopathologic findings in our patient were consistent with LPP. The differential diagnosis for the reticulate pattern of pigmentation seen in our patient included confluent and reticulated papillomatosis and poikilodermalike cutaneous amyloidosis, both easily excluded with histopathologic confirmation.
Local amyloidosis also was confirmed by crystal violet staining in our case and its etiology was uncertain. Generalized and local amyloidosis has been reported in association with lichen planus. The diagnosis of lichen planus was followed by the diagnosis of amyloidosis, and the typical skin lesions of these 2 conditions were able to be differentiated in these reported cases.10,11 However, beltlike pigmentation was the only manifestation for our patient and we could not separate the 2 conditions with the naked eye.
Chronic irritation to the skin resulting in excessive production of degenerate keratins and their subsequent conversion into amyloid deposits has been proposed to be an etiologic factor of amyloidosis.11 Because of the distribution pattern in our case, we believe focal amyloidosis could be attributed to chronic friction and scratching.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
- Bhutani LK, Bedi TR, Pandhi RK, et al. Lichen planus pigmentosus. Dermatologica. 1974;149:43-50.
- Kanwar AJ, Kaur S. Lichen planus pigmentosus. J Am Acad Dermatol. 1989;21(4, pt 1):815.
- Kanwar AJ, Dogra S, Handa S, et al. A study of 124 Indian patients with lichen planus pigmentosus. Clin Exp Dermatol. 2003;28:481-485.
- Akarsu S, Ilknur T, Özer E, et al. Lichen planus pigmentosus distributed along the lines of Blaschko. Int J Dermatol. 2013;52:253-254.
- Cho S, Whang KK. Lichen planus pigmentosus presenting in zosteriform pattern. J Dermatol. 1997;24:193-197.
- Ingber A, Weissmann-Katzenelson V, David M, et al. Lichen planus and lichen planus pigmentosus following gold therapy—case reports and review of the literature [in German]. Z Hautkr. 1986;61:315-319.
- Al-Mutairi N, El-Khalawany M. Clinicopathological characteristics of lichen planus pigmentosus and its response to tacrolimus ointment: an open label, non-randomized, prospective study. J Eur Acad Dermatol Venereol. 2010;24:535-540.
- Mancuso G, Berdondini RM. Coexistence of lichen planus pigmentosus and minimal change nephrotic syndrome. Eur J Dermatol. 2009;19:389-390.
- Sassolas B, Zagnoli A, Leroy JP, et al. Lichen planus pigmentosus associated with acrokeratosis of Bazex. Clin Exp Dermatol. 1994;19:70-73.
- Maeda H, Ohta S, Saito Y, et al. Epidermal origin of the amyloid in localized cutaneous amyloidosis. Br J Dermatol. 1982;106:345-351.
- Hongcharu W, Baldassano M, Gonzalez E. Generalized lichen amyloidosis associated with chronic lichen planus. J Am Acad Dermatol. 2000;43:346-348.
Practice Points
- Lichen planus pigmentosus can present in a unique beltlike distribution pattern.
- Focal amyloidosis due to chronic friction and scratching cannot be excluded from the differential diagnosis.
Acute Generalized Exanthematous Pustulosis Caused by Pantoprazole
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.

Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
To the Editor:
A 34-year-old woman presented with a generalized pustular eruption with subjective fevers, chills, night sweats, and light-headedness. Ten days prior to admission she developed a generalized erythematous and pruritic rash; she had started pantoprazole for reflux 4 days prior to the rash. On admission, skin examination revealed facial edema and diffuse erythema covering 80% of the total body surface area with multiple 1- to 4-mm pustules coalescing into lakes of pus on the trunk as well as bilateral upper and lower arms and legs sparing the palms and soles. Desquamation and serous drainage with crust were observed on the skin of the head, upper trunk, and thighs (Figure 1). Vital signs were notable for hypotension. Laboratory tests on admission were remarkable for leukocytosis (white blood cell count: 22.5×103/μL [reference range, 4.5–11×103/μL]) with absolute eosinophilia but no neutrophilia. C-reactive protein (CRP) was elevated (237.9 mg/L [reference range, 5.0–9.9 mg/L]). Renal and hepatic functions were normal. Blood cultures grew methicillin-sensitive Staphylococcus aureus (MSSA). Further infectious disease workup for viral and fungal pathogens was negative.
Skin biopsy from the left thigh revealed subcorneal, pustular, acute spongiotic dermatitis with marked intraepidermal spongiosis and papillary edema; exocytosis of eosinophils; and single cell necrosis of keratinocytes (Figure 2). These findings were consistent with acute generalized exanthematous pustulosis (AGEP). Pantoprazole was discontinued, and cardiovascular support and antibiotic therapy for MSSA bacteremia were initiated. Respiratory, kidney, and liver functions remained normal throughout the 11-day hospitalization, and the pustular dermatitis, MSSA bacteremia, and cardiovascular symptoms resolved within 10 days.
Acute generalized exanthematous pustulosis is an uncommon, self-limited, generalized sterile pustular eruption notable for the usual absence of systemic symptoms and extracutaneous organ involvement. Hotz et al1 found that mean peripheral neutrophil counts (mean, 21.5×103/μL) and CRP levels (mean, 241.6 mg/L) were notably elevated in patients with systemic (ie, hepatic, pulmonary, renal, bone marrow) involvement. In our patient, only the CRP approached the elevated value reported by Hotz et al.1 However, the patient exhibited only cardiovascular instability in the context of secondary bacteremia and no other systemic symptoms. The combination of highly elevated neutrophilia and CRP may be a better marker for AGEP-precipitated extracutaneous organ involvement.
Although infectious pathogens such as Epstein-Barr virus and cytomegalovirus have been implicated, the majority of AGEP cases are adverse reactions (ARs) to medications, such as β-lactam antibiotics. In our patient, the widely prescribed proton pump inhibitor (PPI) pantoprazole was the most likely cause. Acute generalized exanthematous pustulosis was reported in a patient taking another PPI, omeprazole.2 However, PPIs are recognized to cause many cutaneous and other organ ARs, though prevalence of ARs is still low. In Thailand, Chularojanamontri et al3 reported 13.8 per 100,000 individuals developed a cutaneous AR to PPIs, and the ARs most frequently were attributed to omeprazole. They found that drug exanthems were the most common cutaneous ARs.3 However, more severe hypersensitivity reactions have been reported, including Stevens-Johnson syndrome, toxic epidermal necrolysis, and autoimmune eruptions such as cutaneous lupus erythematosus.3,4 Other systemic reactions to PPIs include increased risks for urticaria, pneumonia, Clostridium difficile infections, and acute interstitial nephritis.4,5
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
- Hotz C, Valeyrie-Allanore L, Haddad C, et al. Systemic involvement of acute generalized exanthematous pustulosis: a retrospective study on 58 patients. Br J Dermatol. 2013;169:1223-1232.
- Nantes Castillejo O, Zozaya Urmeneta JM, Valcayo Peñalba A, et al. Acute generalized exanthematous pustulosis induced by omeprazole [in Spanish]. Gastroenterol Hepatol. 2008;31:295-298.
- Chularojanamontri L, Jiamton S, Manapajon A, et al. Cutaneous reactions to proton pump inhibitors: a case-control study. J Drugs Dermatol. 2012;11:E43-E47.
- Chang YS. Hypersensitivity reactions to proton pump inhibitors. Curr Opin Allergy Clin Immunol. 2012;12:348-353.
- Wilhelm SM, Rjater RG, Kale-Pradhan PB. Perils and pitfalls of long-term effects of proton pump inhibitors. Expert Rev Clin Pharmacol. 2013;6:443-551.
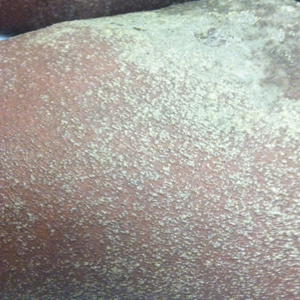
Interstitial Granulomatous Dermatitis and Palisaded Neutrophilic Granulomatous Dermatitis
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.

.")
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.

.")
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
To the Editor:
Palisaded neutrophilic granulomatous dermatitis (PNGD) is a rare disorder that often is associated with systemic disease. It has been shown to manifest in the presence of systemic lupus erythematosus; rheumatoid arthritis; Wegener granulomatosis; and other diseases, mainly autoimmune conditions. Interstitial granulomatous dermatitis (IGD) associated with arthritis was first described by Ackerman et al1 in 1993. In 1994, IGD was placed among the spectrum of PNGD by Chu et al.2 The disease entities included in the spectrum of PNGD of the immune complex disease are Churg-Strauss granuloma, cutaneous extravascular necrotizing granuloma, rheumatoid papules, superficial ulcerating rheumatoid necrobiosis, and IGD with arthritis.2 It has been suggested that IGD has a distinct clinical presentation with associated histopathology, while others suggest it still is part of the PNGD spectrum.2,3 We present 2 cases of granulomatous dermatitis and their findings related to IGD and PNGD.
A 58-year-old woman presented with recurrent painful lesions on the trunk, arms, and legs of 2 years’ duration. The lesions spontaneously resolved without scarring or hyperpigmentation but would recur in different areas on the trunk. She was diagnosed with rheumatoid arthritis following a recent autoimmune workup. At presentation, physical examination revealed tender erythematous edematous plaques on the bilateral upper back (Figure 1) and erythematous nodules on the bilateral upper arms. The patient previously had an antinuclear antibody titer of 1:320 with a speckled pattern. A repeat antinuclear antibody titer taken 1 year later was negative. Her rheumatoid factor initially was positive and remained positive upon repeat testing. Punch biopsies were performed for histologic evaluation of the lesions and immunofluorescence. Biopsies examined with hematoxylin and eosin stain revealed perivascular and interstitial mixed (lymphocytic, neutrophilic, eosinophilic) bottom-heavy inflammation with nuclear dust and basophilic degeneration of collagen (Figure 2). Immunofluorescence studies were negative. The patient deferred treatment.
A 74-year-old man presented with a rash on the flank and back with associated pruritus and occasional pain of 2 months’ duration. His primary care physician prescribed a course of cephalexin, but the rash did not improve. Review of systems was positive for intermittent swelling of the hands, feet, and lips, and negative for arthritis. His medical history included 2 episodes of rheumatic fever, one complicated by pneumonia. His medications included finasteride, simvastatin, bisoprolol-hydrochlorothiazide, aspirin, tiotropium, vitamin D, and fish oil. At presentation, physical examination revealed tender violaceous plaques with induration and central clearing distributed on the left side of the back, left side of the flank, and left axilla. The lesion on the axilla measured 30.0×3.5 cm and the lesions on the left side of the back measured 30.0×9.0 cm. The rims of the lesions were elevated and consistent with the rope sign (Figure 3). A punch biopsy of the lesion on the left axilla showed perivascular and interstitial infiltrate of lymphocytes, neutrophils, histiocytes, and eosinophils. There was no evidence of fibrin deposition in the blood vessels. Small areas of necrobiotic collagen surrounded by multinucleated giant cells and lymphocytes were noted (Figure 4). The rash improved spontaneously at the time of suture removal. No treatment was initiated.
Granulomatous dermatitis in the presence of an autoimmune disorder can present as IGD or PNGD. Both forms of granulomatous dermatitis are rare conditions and considered to be part of the same clinicopathological spectrum. These conditions can be difficult to distinguish clinically but are histologically unique.
Interstitial granulomatous dermatitis and PNGD can have a variable clinical expression. Palisaded neutrophilic granulomatous dermatitis generally presents as flesh-colored to erythematous papules or plaques, most commonly located on the upper arms. The lesions may have a central umbilication with perforation and ulceration.4 Interstitial granulomatous dermatitis most commonly presents as erythematous plaques and papules. The lesions are symmetric and asymptomatic. They most commonly appear on the trunk, axillae, buttocks, thighs, and groin. Subcutaneous linear cords (the rope sign) is a characteristic associated with IGD.3,5 However, the rope sign also has been reported in a patient with PNGD with systemic lupus,6 which further demonstrates the overlapping spectrum of clinical expression seen in these 2 forms of granulomatous dermatitis. Therefore, a diagnosis cannot be made by clinical expression alone; histologic findings are needed for confirmation.
When differentiating IGD and PNGD histologically, it is important to keep in mind that these features exist on a spectrum and depend on the age of the lesion. Deposition of the immune complex around the dermal blood vessel initiates the pathogenesis. Early lesions of PNGD show a neutrophilic infiltrate, focal leukocytoclastic vasculitis, and dense nuclear dust. Developed lesions show zones of basophilic degenerated collagen surrounded by palisades of histiocytes, neutrophils, and nuclear debris.2 The histologic pattern of IGD features smaller areas of palisading histiocytes surrounding foci of degenerated collagen. Neutrophils and eosinophils are seen among the degenerated collagen. There is no evidence of vasculitis and dermal mucin usually is absent.7
Palisaded neutrophilic granulomatous dermatitis has been reported to improve with systemic steroids and dapsone.8 Th
Some authors have disputed the spectrum that Chu et al2 had determined in their study and proposed IGD is a separate entity from the PNGD spectrum. Verneuil et al9 stated that the clinical presentations in Chu et al’s2 study (symmetric papules of the extremities) had not been reported in a patient with IGD. However, in a study of IGD by Peroni et al,3 7 of 12 patients presented with symmetrical papules of the extremities. We believe that the spectrum proposed by Chu et al2 still holds true.
These 2 reports demonstrate the diverse presentation of IGD and PNGD. It is important for dermatologists to keep in mind the PNGD spectrum when a patient presents with granulomatous dermatitis in the presence of an autoimmune disorder.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
- Ackerman AB, Guo Y, Vitale P. Clues to diagnosis in dermatopathology. Am Society Clin Pathol. 1993;3:309-312.
- Chu P, Connolly MK, LeBoit PE. The histopathologic spectrum of palisaded neutrophilic and granulomatous dermatitis in patients with collagen vascular disease. Arch Dermatol. 1994;130:1278-1283.
- Peroni A, Colato C, Schena D, et al. Interstitial granulomatous dermatitis: a distinct entity with characteristic histological and clinical pattern. Br J Dermatol. 2012;166:775-783.
- Hantash BM, Chiang D, Kohler S, et al. Palisaded neutrophilic and granulomatous dermatitis associated with limited systemic sclerosis. J Am Acad Dermatol. 2008;58:661-664.
- Garcia-Rabasco A, Esteve-Martinez A, Zaragoza-Ninet V, et al. Interstitial granulomatous dermatitis in a patient with lupus erythematosus. Am J Dermatopathol. 2011;33:871-872.
- Gulati A, Paige D, Yaqoob M, et al. Palisaded neutrophilic granulomatous dermatitis associated with systemic lupus erythematosus presenting with the burning rope sign. J Am Acad Dermatol. 2009;61:711-714.
- Tomasini C, Pippione M. Interstitial granulomatous dermatitis with plaques. J Am Acad Dermatol. 2002;46:892-899.
- Fett N, Kovarik C, Bennett D. Palisaded neutrophilic granulomatous dermatitis without a definable underlying disorder treated with dapsone. J Am Acad Dermatol. 2011;65:E92-E93.
- Verneuil L, Dompmartin A, Comoz F, et al. Interstitial granulomatous dermatitis with cutaneous cords and arthritis: a disorder associated with autoantibodies. J Am Acad Dermatol. 2001;45:286-291.
Practice Points
- The clinical features of interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis exist on a spectrum, and these is considerable overlap between the features of these 2 clinicopathologic entities.
- Interstitial granulomatous dermatitis and palisaded neutrophilic granulomatous dermatitis may respond to systemic steroids or treatment of the underlying systemic disease. Some cases spontaneously resolve.
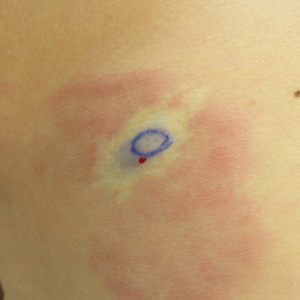
Tinea Incognito in a Tattoo
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.

Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
To the Editor:
Tinea incognito occurs when superficial fungal infections fail to demonstrate typical clinical features in the setting of immune suppression caused by topical or systemic steroids.1,2 A case of tinea corporis obscured by an allergic tattoo reaction is presented.
A 52-year-old man presented for evaluation of a rash overlying a tattoo on the right calf of 3 weeks’ duration (Figure, A). The tattoo was placed 4 years prior to presentation. Within 6 months of the tattoo’s placement, pruritus, scaling, and edema developed in a 2-mm rim around the outer border and in the eyes of the elephant tattoo but not in the lettering portion of the tattoo, which was added by a different tattoo artist with a different red dye. A diagnosis of red dye tattoo allergic reaction was made. Daily treatment with tacrolimus ointment 0.1% and halobetasol propionate cream 0.05% under occlusion for 18 months provided only partial relief of incessant pruritus. Three months prior to presentation the tattoo reaction appeared to become worse with more pruritus and extension outside the bounds of the original tattoo.
Physical examination revealed the red rim of the tattoo was erythematous, edematous, and crusted. In addition, a 5×4-cm well-demarcated, erythematous, scaling patch was present overlying the elephant tattoo on the right calf and extending superiorly and laterally away from the tattoo. Scaling and maceration also were present in the web spaces between the fourth and fifth toes, and the toenails were yellowed, thickened, and dystrophic with signs of distal onycholysis. A potassium hydroxide preparation performed from the plaque on the right calf demonstrated septate fungal hyphae.
The diagnosis of tinea corporis secondary to tinea pedis overlying a red dye tattoo allergic reaction was made. Tacrolimus and halobetasol propionate were discontinued and treatment with ketoconazole cream 2% twice daily and oral terbinafine 250 mg once daily was started. The erythematous patch beyond the borders of the tattoo cleared within weeks, but the patient reported worsening of cracking, itching, and swelling overlying the red dye in the rim of the tattoo following discontinuation of topical anti-inflammatory drugs (Figure, B).
A potassium hydroxide preparation demonstrated that the expansible rash was tinea corporis disguised in its character by the coloration of the tattoo; the erythematous, edematous, pruritic tattoo allergic reaction at its rim; and suppression of the normal inflammatory response by daily use of a topical steroid and a calcineurin inhibitor. The latter effect (an immunocompromised district) impacts the classic exaggerated scaling, inflamed rim, and central clearing of tinea corporis present in individuals with a normal inflammatory response.2 Although tinea incognito is classically described on the ankles and lower legs of patients with stasis dermatitis chronically treated with topical steroids, it could occur anywhere in the setting of immunosuppression.3
An analysis of this case using Occam’s razor suggests that the association of this tattoo and tinea was not a coincidence. This guiding principle (heuristic) suggests that economy and succinctness in the logic of science is most likely to produce a correct medical diagnosis (eg, associated findings can be explained by identifying one underlying cause).4 The topical anti-inflammatory drugs increase the likelihood that the patient’s interdigital tinea would spread to this precise location symmetrically expanding in the outline of the tattoo.2
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
- Gathings RM, Abide JM, Brodell RT. An unusual inflammatory rash. JAMA Pediatr. 2014;168:185-186.
- Ruocco V, Brunetti G, Puca RV, et al. The immunocompromised district: a unifying concept for lymphoedematous, herpes-infected and otherwise damaged sites. J Eur Acad Dermatol Venereol. 2009;23:1364-1373.
- Romano C, Maritati E, Gianni C. Tinea incognito in Italy: a 15-year survey. Mycoses. 2006;49:383-387.
- Jefferys WH, Berger JO. Ockham’s razor and Bayesian analysis. American Scientist. 1992;80:64-72.
Practice Points
- Health care providers should have a low threshold to perform a potassium hydroxide preparation when the possibility of a superficial fungal infection is considered.
- Tinea incognito occurs when a superficial fungal infection has unusual clinical features in the setting of local immune suppression.
