User login
Welcome to Current Psychiatry, a leading source of information, online and in print, for practitioners of psychiatry and its related subspecialties, including addiction psychiatry, child and adolescent psychiatry, and geriatric psychiatry. This Web site contains evidence-based reviews of the prevention, diagnosis, and treatment of mental illness and psychological disorders; case reports; updates on psychopharmacology; news about the specialty of psychiatry; pearls for practice; and other topics of interest and use to this audience.
Dear Drupal User: You're seeing this because you're logged in to Drupal, and not redirected to MDedge.com/psychiatry.
Depression
adolescent depression
adolescent major depressive disorder
adolescent schizophrenia
adolescent with major depressive disorder
animals
autism
baby
brexpiprazole
child
child bipolar
child depression
child schizophrenia
children with bipolar disorder
children with depression
children with major depressive disorder
compulsive behaviors
cure
elderly bipolar
elderly depression
elderly major depressive disorder
elderly schizophrenia
elderly with dementia
first break
first episode
gambling
gaming
geriatric depression
geriatric major depressive disorder
geriatric schizophrenia
infant
kid
major depressive disorder
major depressive disorder in adolescents
major depressive disorder in children
parenting
pediatric
pediatric bipolar
pediatric depression
pediatric major depressive disorder
pediatric schizophrenia
pregnancy
pregnant
rexulti
skin care
teen
wine
section[contains(@class, 'nav-hidden')]
footer[@id='footer']
div[contains(@class, 'pane-pub-article-current-psychiatry')]
div[contains(@class, 'pane-pub-home-current-psychiatry')]
div[contains(@class, 'pane-pub-topic-current-psychiatry')]
div[contains(@class, 'panel-panel-inner')]
div[contains(@class, 'pane-node-field-article-topics')]
section[contains(@class, 'footer-nav-section-wrapper')]
Does your patient have postpartum OCD?
Childbirth is a trigger for first-onse
In one prospective study of 461 women who recently gave birth, researchers found the prevalence of OCD symptoms was 11% at 2 weeks postpartum.1 Mothers with OCD may have time-consuming or functionally impairing obsessions and/or compulsions that can include:
- anticipatory anxiety of contamination (eg, germs, illness)
- thoughts of accidental or intentional harm to their infant
- compulsions comprised of cleaning and checking behaviors
- avoidance of situations
- thought suppression.
Because both clinicians and patients may not be aware of the effects of childbirth on women with OCD, postpartum OCD may go underdiagnosed or be misdiagnosed as major depressive disorder (MDD) or an anxiety disorder. Additionally, women with OCD who lack insight or have delusional beliefs might be misdiagnosed with postpartum psychosis.
Here I offer methods to help effectively identify OCD in postpartum women, and suggest how to implement an individualized treatment approach.
Keys to identification and diagnosis
Mothers who present with postpartum anxiety or depression may have obsessions and compulsions. It is important to specifically screen for these symptoms because some mothers may be reluctant to discuss the content of their thoughts or behaviors.
Screen women who present with postpartum anxiety or depression for obsessions and compulsions by using questions based on DSM-5 criteria,2 such as:
- Do you have unpleasant thoughts, urges, or images that repeatedly enter your mind?
- Do you feel driven to perform certain behaviors or mental acts over and over again?
A validated scale, such as the Yale-Brown Obsessive Compulsive Scale (Y-BOCS),3 can also be used to screen for obsessive/compulsive symptoms in these patients.
Continue to: Evaluate women who endorse...
Evaluate women who endorse obsessions or compulsions for OCD. Women who meet diagnostic criteria for OCD should also be assessed for common psychiatric comorbidities, including MDD, anxiety disorders, or bipolar disorder. Obsessive-compulsive disorder with absent insight and delusional beliefs should be differentiated from postpartum psychosis, which is often a manifestation of bipolar disorder.
Treatment: What to consider
When selecting a treatment, consider factors such as symptom severity, psychiatric comorbidities, the patient’s insight into her OCD symptoms, patient preference, and breastfeeding status. Cognitive-behavioral therapy with exposure response prevention is indicated for patients with mild to moderate OCD. Pharmacotherapy should be reserved for individuals with severe OCD. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay pharmacologic treatment of postpartum OCD; however, there are currently no randomized controlled trials of SSRIs for women with postpartum OCD. Augmentation with quetiapine should be considered for women who have an inadequate response to SSRIs.
Acknowledgment
The author thanks Christine Baczynski for her help with the preparation of this article.
1. Miller ES, Chu C, Gollan J, et al. Obsessive-compulsive symptoms during the postpartum period. A prospective cohort. J Reprod Med. 2013;58(3-4):115-122.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.
Childbirth is a trigger for first-onse
In one prospective study of 461 women who recently gave birth, researchers found the prevalence of OCD symptoms was 11% at 2 weeks postpartum.1 Mothers with OCD may have time-consuming or functionally impairing obsessions and/or compulsions that can include:
- anticipatory anxiety of contamination (eg, germs, illness)
- thoughts of accidental or intentional harm to their infant
- compulsions comprised of cleaning and checking behaviors
- avoidance of situations
- thought suppression.
Because both clinicians and patients may not be aware of the effects of childbirth on women with OCD, postpartum OCD may go underdiagnosed or be misdiagnosed as major depressive disorder (MDD) or an anxiety disorder. Additionally, women with OCD who lack insight or have delusional beliefs might be misdiagnosed with postpartum psychosis.
Here I offer methods to help effectively identify OCD in postpartum women, and suggest how to implement an individualized treatment approach.
Keys to identification and diagnosis
Mothers who present with postpartum anxiety or depression may have obsessions and compulsions. It is important to specifically screen for these symptoms because some mothers may be reluctant to discuss the content of their thoughts or behaviors.
Screen women who present with postpartum anxiety or depression for obsessions and compulsions by using questions based on DSM-5 criteria,2 such as:
- Do you have unpleasant thoughts, urges, or images that repeatedly enter your mind?
- Do you feel driven to perform certain behaviors or mental acts over and over again?
A validated scale, such as the Yale-Brown Obsessive Compulsive Scale (Y-BOCS),3 can also be used to screen for obsessive/compulsive symptoms in these patients.
Continue to: Evaluate women who endorse...
Evaluate women who endorse obsessions or compulsions for OCD. Women who meet diagnostic criteria for OCD should also be assessed for common psychiatric comorbidities, including MDD, anxiety disorders, or bipolar disorder. Obsessive-compulsive disorder with absent insight and delusional beliefs should be differentiated from postpartum psychosis, which is often a manifestation of bipolar disorder.
Treatment: What to consider
When selecting a treatment, consider factors such as symptom severity, psychiatric comorbidities, the patient’s insight into her OCD symptoms, patient preference, and breastfeeding status. Cognitive-behavioral therapy with exposure response prevention is indicated for patients with mild to moderate OCD. Pharmacotherapy should be reserved for individuals with severe OCD. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay pharmacologic treatment of postpartum OCD; however, there are currently no randomized controlled trials of SSRIs for women with postpartum OCD. Augmentation with quetiapine should be considered for women who have an inadequate response to SSRIs.
Acknowledgment
The author thanks Christine Baczynski for her help with the preparation of this article.
Childbirth is a trigger for first-onse
In one prospective study of 461 women who recently gave birth, researchers found the prevalence of OCD symptoms was 11% at 2 weeks postpartum.1 Mothers with OCD may have time-consuming or functionally impairing obsessions and/or compulsions that can include:
- anticipatory anxiety of contamination (eg, germs, illness)
- thoughts of accidental or intentional harm to their infant
- compulsions comprised of cleaning and checking behaviors
- avoidance of situations
- thought suppression.
Because both clinicians and patients may not be aware of the effects of childbirth on women with OCD, postpartum OCD may go underdiagnosed or be misdiagnosed as major depressive disorder (MDD) or an anxiety disorder. Additionally, women with OCD who lack insight or have delusional beliefs might be misdiagnosed with postpartum psychosis.
Here I offer methods to help effectively identify OCD in postpartum women, and suggest how to implement an individualized treatment approach.
Keys to identification and diagnosis
Mothers who present with postpartum anxiety or depression may have obsessions and compulsions. It is important to specifically screen for these symptoms because some mothers may be reluctant to discuss the content of their thoughts or behaviors.
Screen women who present with postpartum anxiety or depression for obsessions and compulsions by using questions based on DSM-5 criteria,2 such as:
- Do you have unpleasant thoughts, urges, or images that repeatedly enter your mind?
- Do you feel driven to perform certain behaviors or mental acts over and over again?
A validated scale, such as the Yale-Brown Obsessive Compulsive Scale (Y-BOCS),3 can also be used to screen for obsessive/compulsive symptoms in these patients.
Continue to: Evaluate women who endorse...
Evaluate women who endorse obsessions or compulsions for OCD. Women who meet diagnostic criteria for OCD should also be assessed for common psychiatric comorbidities, including MDD, anxiety disorders, or bipolar disorder. Obsessive-compulsive disorder with absent insight and delusional beliefs should be differentiated from postpartum psychosis, which is often a manifestation of bipolar disorder.
Treatment: What to consider
When selecting a treatment, consider factors such as symptom severity, psychiatric comorbidities, the patient’s insight into her OCD symptoms, patient preference, and breastfeeding status. Cognitive-behavioral therapy with exposure response prevention is indicated for patients with mild to moderate OCD. Pharmacotherapy should be reserved for individuals with severe OCD. Selective serotonin reuptake inhibitors (SSRIs) are the mainstay pharmacologic treatment of postpartum OCD; however, there are currently no randomized controlled trials of SSRIs for women with postpartum OCD. Augmentation with quetiapine should be considered for women who have an inadequate response to SSRIs.
Acknowledgment
The author thanks Christine Baczynski for her help with the preparation of this article.
1. Miller ES, Chu C, Gollan J, et al. Obsessive-compulsive symptoms during the postpartum period. A prospective cohort. J Reprod Med. 2013;58(3-4):115-122.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.
1. Miller ES, Chu C, Gollan J, et al. Obsessive-compulsive symptoms during the postpartum period. A prospective cohort. J Reprod Med. 2013;58(3-4):115-122.
2. American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders, 5th ed. Washington, DC: American Psychiatric Association; 2013.
3. Goodman WK, Price LH, Rasmussen SA, et al. The Yale-Brown Obsessive Compulsive Scale. I. Development, use, and reliability. Arch Gen Psychiatry. 1989;46(11):1006-1011.

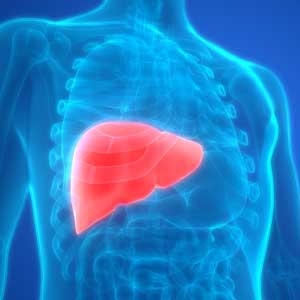
Young, angry, and in need of a liver transplant
CASE Rash, fever, extreme lethargy; multiple hospital visits
Ms. L, age 21, a single woman with a history of major depressive disorder (MDD), is directly admitted from an outside community hospital to our tertiary care academic hospital with acute liver failure.
One month earlier, Ms. L had an argument with her family and punched a wall, fracturing her hand. Following the episode, Ms. L’s primary care physician (PCP) prescribed valproic acid, 500 mg/d, to address “mood swings,” which included angry outbursts and irritability. According to her PCP, no baseline laboratory tests were ordered for Ms. L when she started valproic acid because she was young and otherwise healthy.
After Ms. L had been taking valproic acid for approximately 2 weeks, her mother noticed she became extremely lethargic and took her to the emergency department (ED) of a community hospital (Visit 1) (Table 1). At this time, her laboratory results were notable for an aspartate aminotransferase (AST) level of 303 IU/L (reference range: 8 to 40 IU/L) and an alanine aminotransferase (ALT) level of 241 IU/L (reference range: 20 to 60 IU/L). She also underwent a liver ultrasound, urine toxicology screen, blood alcohol level, and acetaminophen level; the results of all of these tests were unremarkable. Her valproic acid level was within therapeutic limits, consistent with patient adherence; her ammonia level was within normal limits. At Visit 1, Ms. L’s transaminitis was presumed to be secondary to valproic acid. The ED clinicians told her to stop taking valproic acid and discharged her. Her PCP did not give her any follow-up instructions for further laboratory tests or any other recommendations.

During the next week, even though she stopped taking the valproic acid as instructed, Ms. L developed a rash and fever, and continued to have lethargy and general malaise. When she returned to the ED (Visit 2) (Table 1), she was febrile, tachycardic, and hypotensive, with an elevated white blood cell count, eosinophilia, low platelets, and elevated liver function tests. At Visit 2, she was alert and oriented to person, place, time, and situation. Ms. L insisted that she had not overdosed on any medications, or used illicit drugs or alcohol. A test for hepatitis C was negative. Her ammonia level was 58 µmol/L (reference range: 11 to 32 µmol/L). Ms. L received N-acetylcysteine (NAC), prednisone, diphenhydramine, famotidine, and ibuprofen before she was transferred to our tertiary care hospital.
When she arrives at our facility (Visit 3) (Table 1), Ms. L is admitted with acute liver failure. She has an ALT level of 4,091 IU/L, and an AST level of 2,049 IU/L. Ms. L’s mother says that her daughter had been taking sertraline for depression for “some time” with no adverse effects, although she is not clear on the dose or frequency. Her mother says that Ms. L generally likes to spend most of her time at home, and does not believe her daughter is a danger to herself or others. Ms. L’s mother could not describe any episodes of mania or recurrent, dangerous anger episodes. Ms. L has no other medical history and has otherwise been healthy.
On hospital Day 2, Ms. L’s ammonia level is 72 µmol/L, which is slightly elevated. The hepatology team confirms that Ms. L may require a liver transplantation. The primary team consults the inpatient psychiatry consultation-liaison (C-L) team for a pre-transplant psychiatric evaluation.
[polldaddy:10307646]
The authors’ observations
The differential diagnosis for Ms. L was broad and included both accidental and intentional medication overdose. The primary team consulted the inpatient psychiatry C-L team not only for a pre-transplant evaluation, but also to assess for possible overdose.
Continue to: A review of the records...
A review of the records from Visit 1 and Visit 2 at the outside hospital found no acetaminophen in Ms. L’s system and verified that there was no evidence of a current valproic acid overdose. Ms. L had stated that she had not overdosed on any other medications or used any illicit drugs or alcohol. Ms. L’s complex symptoms—namely fever, acute liver failure, and rash—were more consistent with an adverse effect of valproic acid or possibly an inherent autoimmune process.

Liver damage from valproic acid
Valproic acid is FDA-approved for treating bipolar disorder, epilepsy, and migraine headaches1 (Table 21). Common adverse effects include nausea, vomiting, sleepiness, and dry mouth. Rarely, valproic acid can impair liver function. While receiving valproic acid, 5% to 10% of patients develop elevated ALT levels, but most are asymptomatic and resolve with time, even if the patient continues taking valproic acid.2 Valproic acid hepatotoxicity resulting in liver transplantation for a healthy patient is extremely rare (Table 31). Liver failure, both fatal and non-fatal, is more prevalent in patients concurrently taking other medications, such as antiepileptics, benzodiazepines, and antipsychotics, as compared with patients receiving only valproic acid.3

There are 3 clinically distinguishable forms of hepatotoxicity due to valproic acid2:
- hyperammonemia
- acute liver failure and jaundice
- Myriad ProReye-like syndrome, which is generally seen in children.
In case reports of hyperammonemia due to valproic acid, previously healthy patients experience confusion, lethargy, and eventual coma in the context of elevated serum ammonia levels; these symptoms resolved upon discontinuing valproic acid.4,5 Liver function remained normal, with normal to near-normal liver enzymes and bilirubin.3 Hyperammonemia and resulting encephalopathy generally occurred within 1 to 3 weeks after initiation of valproate therapy, with resolution of hyperammonemia and resulting symptoms within a few days after stopping valproic acid.2-4
At Visit 2, Ms. L’s presentation was not initially consistent with hepatic encephalopathy. She was alert and oriented to person, place, time, and situation. Additionally, Ms. L’s presenting problem was elevated liver function tests, not elevated ammonia levels. At Visit 2, her ammonia level was 58 µmol/L; on Day 2 (Visit 3) of her hospital stay, her ammonia level was 72 µmol/L (slightly elevated).
Continue to: At Visit 2 in the ED...
At Visit 2 in the ED, Ms. L was started on NAC because the team suspected she was experiencing drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. This syndrome is characterized by extensive rash, fever, and involvement of at least 1 internal organ. It is a variation of a drug-induced hypersensitivity syndrome. Ms. L’s unremarkable valproic acid levels combined with the psychiatry assessment ruled out valproic hepatotoxicity due to overdose, either intentional or accidental.
In case reports, patients who developed acute liver failure due to valproic acid typically had a hepatitis-like syndrome consisting of moderate elevation in liver enzymes, jaundice, and liver failure necessitating transplantation after at least 1 month of treatment with valproic acid.2 In addition to the typical hepatitis-like syndrome resulting from valproic acid, case reports have also linked treatment with valproic acid to DRESS syndrome.2 This syndrome is known to occur with anticonvulsants such as phenobarbital, lamotrigine, and phenytoin, but there are only a few reported cases of DRESS syndrome due to valproic acid therapy alone.6 Drug rash with eosinophilia and systemic symptoms syndrome differs from other acute liver failure cases in that patients also develop lymphadenopathy, fever, and rash.2,6,7 Patients with DRESS syndrome typically respond to corticosteroid therapy and discontinuation of valproic acid, and the liver damage resolves after several weeks, without a need for transplantation.2,6,7
Ms. L seemed to have similarities to DRESS syndrome. However, the severity of her liver damage, which might require transplantation even after only 2 weeks of valproic acid therapy, initially led the hepatology and C-L teams to consider her presentation similar to severe hepatitis-like cases.
EVALUATION Consent for transplantation
As an inpatient, Ms. L undergoes further laboratory testing. Her hepatic function panel demonstrates a total protein level of 4.8 g/dL, an albumin level of 2.0 g/dL, total bilirubin level of 12.2 mg/dL, and alkaline phosphatase of 166 IU/L. Her laboratory results indicate a prothrombin time (PT) of 77.4 seconds, partial thromboplastin time of 61.5 seconds, and PT international normalized ratio (INR) of 9.6. Ms. L’s basic metabolic panel is within normal limits except for a blood urea nitrogen level of 6 mg/dL, glucose level of 136 mg/dL, and calcium level of 7.0 mg/dL. Her complete blood count indicates a white blood cell count of 12.1, hemoglobin of 10.3 g/dL, hematocrit of 30.4%, mean corpuscular volume of 85.9 fL, and platelet count of 84. Her lipase level is normal at 49 U/L. Her serum acetaminophen concentration is <3.0 mcg/mL, valproic acid level was <2 µg/mL, and she is negative for hepatitis A, B, and C. A urine toxicology screen and testing for herpes simplex, rapid plasma reagin, and human immunodeficiency virus are all negative. Results from several auto-antibodies tests are negative and within normal limits, except filamentous actin (F-actin) antibody, which is slightly higher than normal at 21.4 ELISA units. Based on these results, Ms. L’s liver failure seemed most likely secondary to a reaction to valproic acid.
During her pre-transplant psychiatric evaluation, Ms. L is found to be a poor historian with minimal speech production, flat affect, and clouded sensorium. She denies overdosing on her prescribed valproic acid or sertraline, reports no current suicidal ideation, and does not want to die. She accurately recalls her correct daily dosing of each medication, and verifies that she stopped taking valproic acid 2 weeks ago after being advised to do so by the ED clinicians at Visit 2. She continued to take sertraline until Visit 2. She denied any past or present episodes consistent with mania, which was consistent with her mother’s report.
Continue to: Ms. L becomes agitated...
Ms. L becomes agitated upon further questioning, and requests immediate discharge so that she can return to her family. The evaluation is postponed briefly.
When they reconvene, the C-L team performs a decision-making capacity evaluation, which reveals that Ms. L’s mood and affect are consistent with fear of her impending liver transplant and being alone and approximately 2 hours from her family. This is likely complicated by delirium due to hepatotoxicity. Further discussion between Ms. L and the multidisciplinary team focuses on the risks, benefits, adverse effects of, and alternatives to her current treatment; the possibility of needing a liver transplantation; and how to help her family with transportation to the hospital. Following the discussion, Ms. L is fully cooperative with further treatment, and the pre-transplant psychiatric evaluation is completed.
On physical examination, Ms. L is noted to have a widespread morbilliform rash covering 50% to 60% of her body.
[polldaddy:10307651]
The authors’ observations
L-carnitine supplementation
Multiple studies have shown that supplementation with L-carnitine may increase survival from severe hepatotoxicity due to valproic acid.8,9 Valproic acid may contribute to carnitine deficiency due to its inhibition of carnitine biosynthesis via a decrease in alpha-ketoglutarate concentration.8 Hepatotoxicity or hyperammonemia due to valproic acid may be potentiated by a carnitine deficiency, either pre-existing or resulting from valproic acid.8 L-carnitine supplementation has hastened the decrease of valproic acid–induced ammonemia in a dose-dependent manner,10 and it is currently recommended in cases of valproic acid toxicity, especially in children.8 Children at high risk for developing carnitine deficiency who need to receive valproic acid can be given carnitine supplementation.11 It is not known whether L-carnitine is clinically effective in protecting the liver or hastening liver recovery,8 but it is believed that it might prevent adverse effects of hepatotoxicity and hyperammonemia, especially in patients who receive long-term valproic acid therapy.12
TREATMENT Decompensation and transplantation
Ms. L’s treatment regimen includes NAC, lactulose, and L-carnitine supplementation. During the course of Ms. L’s hospital stay, her liver enzymes begin to trend downward, but her INR and PT remain elevated.
Continue to: On hospital Day 6...
On hospital Day 6, she develops more severe symptoms of hepatic encephalopathy, with significant altered mental status and inattention. Ms. L is transferred to the ICU, intubated, and placed on the liver transplant list.
On hospital Day 9, she undergoes a liver transplantation.
[polldaddy:10307652]
The authors’ observations
Baseline laboratory testing should have been conducted prior to initiating valproic acid. As Ms. L’s symptoms worsened, better communication with her PCP and closer monitoring after starting valproic acid might have resulted in more immediate care. Early recognition of her symptoms and decompensation may have triggered earlier inpatient admission and/or transfer to a tertiary care facility for observation and treatment. Additionally, repeat laboratory testing and instructions on when to return to the ED should have been provided at Visit 1.

This case demonstrates the need for all clinicians who prescribe valproic acid to remain diligent about the accurate diagnosis of mood and behavioral symptoms, knowing when psychotropic medications are indicated, and carefully considering and discussing even rare, potentially life-threatening adverse effects of all medications with patients.

Although rare, after starting valproic acid, a patient may experience a rapid decompensation and life-threatening illness. Ideally, clinicians should closely monitor patients after initiating valproic acid (Table 41). Clinicians must have a clear knowledge of the recommended monitoring and indications for hospitalization and treatment when they note adverse effects such as elevated liver enzymes or transaminitis (Table 513,14). Even after stopping valproic acid, patients who have experienced adverse events should be closely monitored to ensure complete resolution.
Continue to: Consider patient-specific factors
Consider patient-specific factors
Consider the mental state, intellectual capacity, and social support of each patient before initiating valproic acid. Its use as a mood stabilizer for “mood swings” outside of the context of bipolar disorder is questionable. Valproic acid is FDA-approved for treating bipolar disorder and seizures, but not for anger outbursts/irritability. Prior to starting valproic acid, Ms. L may have benefited from alternative nonpharmacologic treatments, such as psychotherapy, for her anger outbursts and poor coping skills. Therapeutic techniques that focused on helping her acquire better coping mechanisms may have been useful, especially because her mood symptoms did not meet criteria for bipolar disorder, and her depression had long been controlled with sertraline monotherapy.
OUTCOME Discharged after 20 days
Ms. L stays in the hospital for 10 days after receiving her liver transplant. She has low appetite and some difficulty with sleep after the transplant; therefore, the C-L team recommends mirtazapine, 15 mg/d. She has no behavioral problems during her stay, and is set up with home health, case management, and psychiatry follow-up. On hospital Day 20, she is discharged.
Bottom Line
Use caution when prescribing valproic acid, even in young, otherwise healthy patients. Although rare, some patients may experience a rapid decompensation and life-threatening illness after starting valproic acid. When prescribing valproic acid, ensure close follow-up after initiation, including mental status examinations, physical examinations, and laboratory testing.
Related Resource
- Doroudgar S, Chou TI. How to modify psychotropic therapy for patients who have liver dysfunction. Current Psychiatry. 2014;13(12):46-49.
Drug Brand Names
Diphenhydramine • Benadryl
Famotidine • Fluxid, Pepcid
Lamotrigine • Lamictal
Mirtazapine • Remeron
N-acetylcysteine • Mucomyst
Phenobarbital • Luminal
Phenytoin • Dilantin
Prednisone • Cortan, Deltasone
Sertraline • Zoloft
Valproic acid • Depakene
1. Depakote [package insert]. North Chicago, IL: AbbVie, Inc.; 2019.
2. National Institutes of Health. U.S. Department of Health and Human Services. Drug Record: Valproate. https://livertox.nlm.nih.gov/Valproate.htm. Updated October 30, 2018. Accessed March 21, 2019.
3. Schmid MM, Freudenmann RW, Keller F, et al. Non-fatal and fatal liver failure associated with valproic acid. Pharmacopsychiatry. 2013;46(2):63-68.
4. Patel N, Landry KB, Fargason RE, et al. Reversible encephalopathy due to valproic acid induced hyperammonemia in a patient with Bipolar I disorder: a cautionary report. Psychopharmacol Bull. 2017;47(1):40-44.
5. Eze E, Workman M, Donley B. Hyperammonemia and coma developed by a woman treated with valproic acid for affective disorder. Psychiatr Serv. 1998;49(10):1358-1359.
6. Darban M and Bagheri B. Drug reaction with eosinophilia and systemic symptoms induced by valproic acid: a case report. Iran Red Crescent Med J. 2016;18(9): e35825.
7. van Zoelen MA, de Graaf M, van Dijk MR, et al. Valproic acid-induced DRESS syndrome with acute liver failure. Neth J Med. 2012;70(3):155.
8. Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila). 2009;47(2):101-111.
9. Bohan TP, Helton E, McDonald I, et al. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology. 2001;56(10):1405-1409.
10. Böhles H, Sewell AC, Wenzel D. The effect of carnitine supplementation in valproate-induced hyperammonaemia. Acta Paediatr. 1996;85(4):446-449.
11. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638.
12. Romero-Falcón A, de la Santa-Belda E, García-Contreras R, et al. A case of valproate-associated hepatotoxicity treated with L-carnitine. Eur J Intern Med. 2003;14(5):338-340.
13. National Institute for Health and Clinical Excellence. Bipolar disorder: the management of bipolar disorder in adults, children, and adolescents, in primary and secondary care. https://www.nice.org.uk/guidance/cg185. Updated April 2018. Accessed March 21, 2019.
14 . Hirschfeld RMA, Bowden CL, Gitlin MJ, et al. Practice guideline for the treatment of patients with biopolar disorder: second edition. American Psychiatric Association. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/bipolar.pdf. Published 2002. Accessed March 21, 2019.
CASE Rash, fever, extreme lethargy; multiple hospital visits
Ms. L, age 21, a single woman with a history of major depressive disorder (MDD), is directly admitted from an outside community hospital to our tertiary care academic hospital with acute liver failure.
One month earlier, Ms. L had an argument with her family and punched a wall, fracturing her hand. Following the episode, Ms. L’s primary care physician (PCP) prescribed valproic acid, 500 mg/d, to address “mood swings,” which included angry outbursts and irritability. According to her PCP, no baseline laboratory tests were ordered for Ms. L when she started valproic acid because she was young and otherwise healthy.
After Ms. L had been taking valproic acid for approximately 2 weeks, her mother noticed she became extremely lethargic and took her to the emergency department (ED) of a community hospital (Visit 1) (Table 1). At this time, her laboratory results were notable for an aspartate aminotransferase (AST) level of 303 IU/L (reference range: 8 to 40 IU/L) and an alanine aminotransferase (ALT) level of 241 IU/L (reference range: 20 to 60 IU/L). She also underwent a liver ultrasound, urine toxicology screen, blood alcohol level, and acetaminophen level; the results of all of these tests were unremarkable. Her valproic acid level was within therapeutic limits, consistent with patient adherence; her ammonia level was within normal limits. At Visit 1, Ms. L’s transaminitis was presumed to be secondary to valproic acid. The ED clinicians told her to stop taking valproic acid and discharged her. Her PCP did not give her any follow-up instructions for further laboratory tests or any other recommendations.
During the next week, even though she stopped taking the valproic acid as instructed, Ms. L developed a rash and fever, and continued to have lethargy and general malaise. When she returned to the ED (Visit 2) (Table 1), she was febrile, tachycardic, and hypotensive, with an elevated white blood cell count, eosinophilia, low platelets, and elevated liver function tests. At Visit 2, she was alert and oriented to person, place, time, and situation. Ms. L insisted that she had not overdosed on any medications, or used illicit drugs or alcohol. A test for hepatitis C was negative. Her ammonia level was 58 µmol/L (reference range: 11 to 32 µmol/L). Ms. L received N-acetylcysteine (NAC), prednisone, diphenhydramine, famotidine, and ibuprofen before she was transferred to our tertiary care hospital.
When she arrives at our facility (Visit 3) (Table 1), Ms. L is admitted with acute liver failure. She has an ALT level of 4,091 IU/L, and an AST level of 2,049 IU/L. Ms. L’s mother says that her daughter had been taking sertraline for depression for “some time” with no adverse effects, although she is not clear on the dose or frequency. Her mother says that Ms. L generally likes to spend most of her time at home, and does not believe her daughter is a danger to herself or others. Ms. L’s mother could not describe any episodes of mania or recurrent, dangerous anger episodes. Ms. L has no other medical history and has otherwise been healthy.
On hospital Day 2, Ms. L’s ammonia level is 72 µmol/L, which is slightly elevated. The hepatology team confirms that Ms. L may require a liver transplantation. The primary team consults the inpatient psychiatry consultation-liaison (C-L) team for a pre-transplant psychiatric evaluation.
[polldaddy:10307646]
The authors’ observations
The differential diagnosis for Ms. L was broad and included both accidental and intentional medication overdose. The primary team consulted the inpatient psychiatry C-L team not only for a pre-transplant evaluation, but also to assess for possible overdose.
Continue to: A review of the records...
A review of the records from Visit 1 and Visit 2 at the outside hospital found no acetaminophen in Ms. L’s system and verified that there was no evidence of a current valproic acid overdose. Ms. L had stated that she had not overdosed on any other medications or used any illicit drugs or alcohol. Ms. L’s complex symptoms—namely fever, acute liver failure, and rash—were more consistent with an adverse effect of valproic acid or possibly an inherent autoimmune process.
Liver damage from valproic acid
Valproic acid is FDA-approved for treating bipolar disorder, epilepsy, and migraine headaches1 (Table 21). Common adverse effects include nausea, vomiting, sleepiness, and dry mouth. Rarely, valproic acid can impair liver function. While receiving valproic acid, 5% to 10% of patients develop elevated ALT levels, but most are asymptomatic and resolve with time, even if the patient continues taking valproic acid.2 Valproic acid hepatotoxicity resulting in liver transplantation for a healthy patient is extremely rare (Table 31). Liver failure, both fatal and non-fatal, is more prevalent in patients concurrently taking other medications, such as antiepileptics, benzodiazepines, and antipsychotics, as compared with patients receiving only valproic acid.3
There are 3 clinically distinguishable forms of hepatotoxicity due to valproic acid2:
- hyperammonemia
- acute liver failure and jaundice
- Myriad ProReye-like syndrome, which is generally seen in children.
In case reports of hyperammonemia due to valproic acid, previously healthy patients experience confusion, lethargy, and eventual coma in the context of elevated serum ammonia levels; these symptoms resolved upon discontinuing valproic acid.4,5 Liver function remained normal, with normal to near-normal liver enzymes and bilirubin.3 Hyperammonemia and resulting encephalopathy generally occurred within 1 to 3 weeks after initiation of valproate therapy, with resolution of hyperammonemia and resulting symptoms within a few days after stopping valproic acid.2-4
At Visit 2, Ms. L’s presentation was not initially consistent with hepatic encephalopathy. She was alert and oriented to person, place, time, and situation. Additionally, Ms. L’s presenting problem was elevated liver function tests, not elevated ammonia levels. At Visit 2, her ammonia level was 58 µmol/L; on Day 2 (Visit 3) of her hospital stay, her ammonia level was 72 µmol/L (slightly elevated).
Continue to: At Visit 2 in the ED...
At Visit 2 in the ED, Ms. L was started on NAC because the team suspected she was experiencing drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. This syndrome is characterized by extensive rash, fever, and involvement of at least 1 internal organ. It is a variation of a drug-induced hypersensitivity syndrome. Ms. L’s unremarkable valproic acid levels combined with the psychiatry assessment ruled out valproic hepatotoxicity due to overdose, either intentional or accidental.
In case reports, patients who developed acute liver failure due to valproic acid typically had a hepatitis-like syndrome consisting of moderate elevation in liver enzymes, jaundice, and liver failure necessitating transplantation after at least 1 month of treatment with valproic acid.2 In addition to the typical hepatitis-like syndrome resulting from valproic acid, case reports have also linked treatment with valproic acid to DRESS syndrome.2 This syndrome is known to occur with anticonvulsants such as phenobarbital, lamotrigine, and phenytoin, but there are only a few reported cases of DRESS syndrome due to valproic acid therapy alone.6 Drug rash with eosinophilia and systemic symptoms syndrome differs from other acute liver failure cases in that patients also develop lymphadenopathy, fever, and rash.2,6,7 Patients with DRESS syndrome typically respond to corticosteroid therapy and discontinuation of valproic acid, and the liver damage resolves after several weeks, without a need for transplantation.2,6,7
Ms. L seemed to have similarities to DRESS syndrome. However, the severity of her liver damage, which might require transplantation even after only 2 weeks of valproic acid therapy, initially led the hepatology and C-L teams to consider her presentation similar to severe hepatitis-like cases.
EVALUATION Consent for transplantation
As an inpatient, Ms. L undergoes further laboratory testing. Her hepatic function panel demonstrates a total protein level of 4.8 g/dL, an albumin level of 2.0 g/dL, total bilirubin level of 12.2 mg/dL, and alkaline phosphatase of 166 IU/L. Her laboratory results indicate a prothrombin time (PT) of 77.4 seconds, partial thromboplastin time of 61.5 seconds, and PT international normalized ratio (INR) of 9.6. Ms. L’s basic metabolic panel is within normal limits except for a blood urea nitrogen level of 6 mg/dL, glucose level of 136 mg/dL, and calcium level of 7.0 mg/dL. Her complete blood count indicates a white blood cell count of 12.1, hemoglobin of 10.3 g/dL, hematocrit of 30.4%, mean corpuscular volume of 85.9 fL, and platelet count of 84. Her lipase level is normal at 49 U/L. Her serum acetaminophen concentration is <3.0 mcg/mL, valproic acid level was <2 µg/mL, and she is negative for hepatitis A, B, and C. A urine toxicology screen and testing for herpes simplex, rapid plasma reagin, and human immunodeficiency virus are all negative. Results from several auto-antibodies tests are negative and within normal limits, except filamentous actin (F-actin) antibody, which is slightly higher than normal at 21.4 ELISA units. Based on these results, Ms. L’s liver failure seemed most likely secondary to a reaction to valproic acid.
During her pre-transplant psychiatric evaluation, Ms. L is found to be a poor historian with minimal speech production, flat affect, and clouded sensorium. She denies overdosing on her prescribed valproic acid or sertraline, reports no current suicidal ideation, and does not want to die. She accurately recalls her correct daily dosing of each medication, and verifies that she stopped taking valproic acid 2 weeks ago after being advised to do so by the ED clinicians at Visit 2. She continued to take sertraline until Visit 2. She denied any past or present episodes consistent with mania, which was consistent with her mother’s report.
Continue to: Ms. L becomes agitated...
Ms. L becomes agitated upon further questioning, and requests immediate discharge so that she can return to her family. The evaluation is postponed briefly.
When they reconvene, the C-L team performs a decision-making capacity evaluation, which reveals that Ms. L’s mood and affect are consistent with fear of her impending liver transplant and being alone and approximately 2 hours from her family. This is likely complicated by delirium due to hepatotoxicity. Further discussion between Ms. L and the multidisciplinary team focuses on the risks, benefits, adverse effects of, and alternatives to her current treatment; the possibility of needing a liver transplantation; and how to help her family with transportation to the hospital. Following the discussion, Ms. L is fully cooperative with further treatment, and the pre-transplant psychiatric evaluation is completed.
On physical examination, Ms. L is noted to have a widespread morbilliform rash covering 50% to 60% of her body.
[polldaddy:10307651]
The authors’ observations
L-carnitine supplementation
Multiple studies have shown that supplementation with L-carnitine may increase survival from severe hepatotoxicity due to valproic acid.8,9 Valproic acid may contribute to carnitine deficiency due to its inhibition of carnitine biosynthesis via a decrease in alpha-ketoglutarate concentration.8 Hepatotoxicity or hyperammonemia due to valproic acid may be potentiated by a carnitine deficiency, either pre-existing or resulting from valproic acid.8 L-carnitine supplementation has hastened the decrease of valproic acid–induced ammonemia in a dose-dependent manner,10 and it is currently recommended in cases of valproic acid toxicity, especially in children.8 Children at high risk for developing carnitine deficiency who need to receive valproic acid can be given carnitine supplementation.11 It is not known whether L-carnitine is clinically effective in protecting the liver or hastening liver recovery,8 but it is believed that it might prevent adverse effects of hepatotoxicity and hyperammonemia, especially in patients who receive long-term valproic acid therapy.12
TREATMENT Decompensation and transplantation
Ms. L’s treatment regimen includes NAC, lactulose, and L-carnitine supplementation. During the course of Ms. L’s hospital stay, her liver enzymes begin to trend downward, but her INR and PT remain elevated.
Continue to: On hospital Day 6...
On hospital Day 6, she develops more severe symptoms of hepatic encephalopathy, with significant altered mental status and inattention. Ms. L is transferred to the ICU, intubated, and placed on the liver transplant list.
On hospital Day 9, she undergoes a liver transplantation.
[polldaddy:10307652]
The authors’ observations
Baseline laboratory testing should have been conducted prior to initiating valproic acid. As Ms. L’s symptoms worsened, better communication with her PCP and closer monitoring after starting valproic acid might have resulted in more immediate care. Early recognition of her symptoms and decompensation may have triggered earlier inpatient admission and/or transfer to a tertiary care facility for observation and treatment. Additionally, repeat laboratory testing and instructions on when to return to the ED should have been provided at Visit 1.
This case demonstrates the need for all clinicians who prescribe valproic acid to remain diligent about the accurate diagnosis of mood and behavioral symptoms, knowing when psychotropic medications are indicated, and carefully considering and discussing even rare, potentially life-threatening adverse effects of all medications with patients.
Although rare, after starting valproic acid, a patient may experience a rapid decompensation and life-threatening illness. Ideally, clinicians should closely monitor patients after initiating valproic acid (Table 41). Clinicians must have a clear knowledge of the recommended monitoring and indications for hospitalization and treatment when they note adverse effects such as elevated liver enzymes or transaminitis (Table 513,14). Even after stopping valproic acid, patients who have experienced adverse events should be closely monitored to ensure complete resolution.
Continue to: Consider patient-specific factors
Consider patient-specific factors
Consider the mental state, intellectual capacity, and social support of each patient before initiating valproic acid. Its use as a mood stabilizer for “mood swings” outside of the context of bipolar disorder is questionable. Valproic acid is FDA-approved for treating bipolar disorder and seizures, but not for anger outbursts/irritability. Prior to starting valproic acid, Ms. L may have benefited from alternative nonpharmacologic treatments, such as psychotherapy, for her anger outbursts and poor coping skills. Therapeutic techniques that focused on helping her acquire better coping mechanisms may have been useful, especially because her mood symptoms did not meet criteria for bipolar disorder, and her depression had long been controlled with sertraline monotherapy.
OUTCOME Discharged after 20 days
Ms. L stays in the hospital for 10 days after receiving her liver transplant. She has low appetite and some difficulty with sleep after the transplant; therefore, the C-L team recommends mirtazapine, 15 mg/d. She has no behavioral problems during her stay, and is set up with home health, case management, and psychiatry follow-up. On hospital Day 20, she is discharged.
Bottom Line
Use caution when prescribing valproic acid, even in young, otherwise healthy patients. Although rare, some patients may experience a rapid decompensation and life-threatening illness after starting valproic acid. When prescribing valproic acid, ensure close follow-up after initiation, including mental status examinations, physical examinations, and laboratory testing.
Related Resource
- Doroudgar S, Chou TI. How to modify psychotropic therapy for patients who have liver dysfunction. Current Psychiatry. 2014;13(12):46-49.
Drug Brand Names
Diphenhydramine • Benadryl
Famotidine • Fluxid, Pepcid
Lamotrigine • Lamictal
Mirtazapine • Remeron
N-acetylcysteine • Mucomyst
Phenobarbital • Luminal
Phenytoin • Dilantin
Prednisone • Cortan, Deltasone
Sertraline • Zoloft
Valproic acid • Depakene
CASE Rash, fever, extreme lethargy; multiple hospital visits
Ms. L, age 21, a single woman with a history of major depressive disorder (MDD), is directly admitted from an outside community hospital to our tertiary care academic hospital with acute liver failure.
One month earlier, Ms. L had an argument with her family and punched a wall, fracturing her hand. Following the episode, Ms. L’s primary care physician (PCP) prescribed valproic acid, 500 mg/d, to address “mood swings,” which included angry outbursts and irritability. According to her PCP, no baseline laboratory tests were ordered for Ms. L when she started valproic acid because she was young and otherwise healthy.
After Ms. L had been taking valproic acid for approximately 2 weeks, her mother noticed she became extremely lethargic and took her to the emergency department (ED) of a community hospital (Visit 1) (Table 1). At this time, her laboratory results were notable for an aspartate aminotransferase (AST) level of 303 IU/L (reference range: 8 to 40 IU/L) and an alanine aminotransferase (ALT) level of 241 IU/L (reference range: 20 to 60 IU/L). She also underwent a liver ultrasound, urine toxicology screen, blood alcohol level, and acetaminophen level; the results of all of these tests were unremarkable. Her valproic acid level was within therapeutic limits, consistent with patient adherence; her ammonia level was within normal limits. At Visit 1, Ms. L’s transaminitis was presumed to be secondary to valproic acid. The ED clinicians told her to stop taking valproic acid and discharged her. Her PCP did not give her any follow-up instructions for further laboratory tests or any other recommendations.
During the next week, even though she stopped taking the valproic acid as instructed, Ms. L developed a rash and fever, and continued to have lethargy and general malaise. When she returned to the ED (Visit 2) (Table 1), she was febrile, tachycardic, and hypotensive, with an elevated white blood cell count, eosinophilia, low platelets, and elevated liver function tests. At Visit 2, she was alert and oriented to person, place, time, and situation. Ms. L insisted that she had not overdosed on any medications, or used illicit drugs or alcohol. A test for hepatitis C was negative. Her ammonia level was 58 µmol/L (reference range: 11 to 32 µmol/L). Ms. L received N-acetylcysteine (NAC), prednisone, diphenhydramine, famotidine, and ibuprofen before she was transferred to our tertiary care hospital.
When she arrives at our facility (Visit 3) (Table 1), Ms. L is admitted with acute liver failure. She has an ALT level of 4,091 IU/L, and an AST level of 2,049 IU/L. Ms. L’s mother says that her daughter had been taking sertraline for depression for “some time” with no adverse effects, although she is not clear on the dose or frequency. Her mother says that Ms. L generally likes to spend most of her time at home, and does not believe her daughter is a danger to herself or others. Ms. L’s mother could not describe any episodes of mania or recurrent, dangerous anger episodes. Ms. L has no other medical history and has otherwise been healthy.
On hospital Day 2, Ms. L’s ammonia level is 72 µmol/L, which is slightly elevated. The hepatology team confirms that Ms. L may require a liver transplantation. The primary team consults the inpatient psychiatry consultation-liaison (C-L) team for a pre-transplant psychiatric evaluation.
[polldaddy:10307646]
The authors’ observations
The differential diagnosis for Ms. L was broad and included both accidental and intentional medication overdose. The primary team consulted the inpatient psychiatry C-L team not only for a pre-transplant evaluation, but also to assess for possible overdose.
Continue to: A review of the records...
A review of the records from Visit 1 and Visit 2 at the outside hospital found no acetaminophen in Ms. L’s system and verified that there was no evidence of a current valproic acid overdose. Ms. L had stated that she had not overdosed on any other medications or used any illicit drugs or alcohol. Ms. L’s complex symptoms—namely fever, acute liver failure, and rash—were more consistent with an adverse effect of valproic acid or possibly an inherent autoimmune process.
Liver damage from valproic acid
Valproic acid is FDA-approved for treating bipolar disorder, epilepsy, and migraine headaches1 (Table 21). Common adverse effects include nausea, vomiting, sleepiness, and dry mouth. Rarely, valproic acid can impair liver function. While receiving valproic acid, 5% to 10% of patients develop elevated ALT levels, but most are asymptomatic and resolve with time, even if the patient continues taking valproic acid.2 Valproic acid hepatotoxicity resulting in liver transplantation for a healthy patient is extremely rare (Table 31). Liver failure, both fatal and non-fatal, is more prevalent in patients concurrently taking other medications, such as antiepileptics, benzodiazepines, and antipsychotics, as compared with patients receiving only valproic acid.3
There are 3 clinically distinguishable forms of hepatotoxicity due to valproic acid2:
- hyperammonemia
- acute liver failure and jaundice
- Myriad ProReye-like syndrome, which is generally seen in children.
In case reports of hyperammonemia due to valproic acid, previously healthy patients experience confusion, lethargy, and eventual coma in the context of elevated serum ammonia levels; these symptoms resolved upon discontinuing valproic acid.4,5 Liver function remained normal, with normal to near-normal liver enzymes and bilirubin.3 Hyperammonemia and resulting encephalopathy generally occurred within 1 to 3 weeks after initiation of valproate therapy, with resolution of hyperammonemia and resulting symptoms within a few days after stopping valproic acid.2-4
At Visit 2, Ms. L’s presentation was not initially consistent with hepatic encephalopathy. She was alert and oriented to person, place, time, and situation. Additionally, Ms. L’s presenting problem was elevated liver function tests, not elevated ammonia levels. At Visit 2, her ammonia level was 58 µmol/L; on Day 2 (Visit 3) of her hospital stay, her ammonia level was 72 µmol/L (slightly elevated).
Continue to: At Visit 2 in the ED...
At Visit 2 in the ED, Ms. L was started on NAC because the team suspected she was experiencing drug rash with eosinophilia and systemic symptoms (DRESS) syndrome. This syndrome is characterized by extensive rash, fever, and involvement of at least 1 internal organ. It is a variation of a drug-induced hypersensitivity syndrome. Ms. L’s unremarkable valproic acid levels combined with the psychiatry assessment ruled out valproic hepatotoxicity due to overdose, either intentional or accidental.
In case reports, patients who developed acute liver failure due to valproic acid typically had a hepatitis-like syndrome consisting of moderate elevation in liver enzymes, jaundice, and liver failure necessitating transplantation after at least 1 month of treatment with valproic acid.2 In addition to the typical hepatitis-like syndrome resulting from valproic acid, case reports have also linked treatment with valproic acid to DRESS syndrome.2 This syndrome is known to occur with anticonvulsants such as phenobarbital, lamotrigine, and phenytoin, but there are only a few reported cases of DRESS syndrome due to valproic acid therapy alone.6 Drug rash with eosinophilia and systemic symptoms syndrome differs from other acute liver failure cases in that patients also develop lymphadenopathy, fever, and rash.2,6,7 Patients with DRESS syndrome typically respond to corticosteroid therapy and discontinuation of valproic acid, and the liver damage resolves after several weeks, without a need for transplantation.2,6,7
Ms. L seemed to have similarities to DRESS syndrome. However, the severity of her liver damage, which might require transplantation even after only 2 weeks of valproic acid therapy, initially led the hepatology and C-L teams to consider her presentation similar to severe hepatitis-like cases.
EVALUATION Consent for transplantation
As an inpatient, Ms. L undergoes further laboratory testing. Her hepatic function panel demonstrates a total protein level of 4.8 g/dL, an albumin level of 2.0 g/dL, total bilirubin level of 12.2 mg/dL, and alkaline phosphatase of 166 IU/L. Her laboratory results indicate a prothrombin time (PT) of 77.4 seconds, partial thromboplastin time of 61.5 seconds, and PT international normalized ratio (INR) of 9.6. Ms. L’s basic metabolic panel is within normal limits except for a blood urea nitrogen level of 6 mg/dL, glucose level of 136 mg/dL, and calcium level of 7.0 mg/dL. Her complete blood count indicates a white blood cell count of 12.1, hemoglobin of 10.3 g/dL, hematocrit of 30.4%, mean corpuscular volume of 85.9 fL, and platelet count of 84. Her lipase level is normal at 49 U/L. Her serum acetaminophen concentration is <3.0 mcg/mL, valproic acid level was <2 µg/mL, and she is negative for hepatitis A, B, and C. A urine toxicology screen and testing for herpes simplex, rapid plasma reagin, and human immunodeficiency virus are all negative. Results from several auto-antibodies tests are negative and within normal limits, except filamentous actin (F-actin) antibody, which is slightly higher than normal at 21.4 ELISA units. Based on these results, Ms. L’s liver failure seemed most likely secondary to a reaction to valproic acid.
During her pre-transplant psychiatric evaluation, Ms. L is found to be a poor historian with minimal speech production, flat affect, and clouded sensorium. She denies overdosing on her prescribed valproic acid or sertraline, reports no current suicidal ideation, and does not want to die. She accurately recalls her correct daily dosing of each medication, and verifies that she stopped taking valproic acid 2 weeks ago after being advised to do so by the ED clinicians at Visit 2. She continued to take sertraline until Visit 2. She denied any past or present episodes consistent with mania, which was consistent with her mother’s report.
Continue to: Ms. L becomes agitated...
Ms. L becomes agitated upon further questioning, and requests immediate discharge so that she can return to her family. The evaluation is postponed briefly.
When they reconvene, the C-L team performs a decision-making capacity evaluation, which reveals that Ms. L’s mood and affect are consistent with fear of her impending liver transplant and being alone and approximately 2 hours from her family. This is likely complicated by delirium due to hepatotoxicity. Further discussion between Ms. L and the multidisciplinary team focuses on the risks, benefits, adverse effects of, and alternatives to her current treatment; the possibility of needing a liver transplantation; and how to help her family with transportation to the hospital. Following the discussion, Ms. L is fully cooperative with further treatment, and the pre-transplant psychiatric evaluation is completed.
On physical examination, Ms. L is noted to have a widespread morbilliform rash covering 50% to 60% of her body.
[polldaddy:10307651]
The authors’ observations
L-carnitine supplementation
Multiple studies have shown that supplementation with L-carnitine may increase survival from severe hepatotoxicity due to valproic acid.8,9 Valproic acid may contribute to carnitine deficiency due to its inhibition of carnitine biosynthesis via a decrease in alpha-ketoglutarate concentration.8 Hepatotoxicity or hyperammonemia due to valproic acid may be potentiated by a carnitine deficiency, either pre-existing or resulting from valproic acid.8 L-carnitine supplementation has hastened the decrease of valproic acid–induced ammonemia in a dose-dependent manner,10 and it is currently recommended in cases of valproic acid toxicity, especially in children.8 Children at high risk for developing carnitine deficiency who need to receive valproic acid can be given carnitine supplementation.11 It is not known whether L-carnitine is clinically effective in protecting the liver or hastening liver recovery,8 but it is believed that it might prevent adverse effects of hepatotoxicity and hyperammonemia, especially in patients who receive long-term valproic acid therapy.12
TREATMENT Decompensation and transplantation
Ms. L’s treatment regimen includes NAC, lactulose, and L-carnitine supplementation. During the course of Ms. L’s hospital stay, her liver enzymes begin to trend downward, but her INR and PT remain elevated.
Continue to: On hospital Day 6...
On hospital Day 6, she develops more severe symptoms of hepatic encephalopathy, with significant altered mental status and inattention. Ms. L is transferred to the ICU, intubated, and placed on the liver transplant list.
On hospital Day 9, she undergoes a liver transplantation.
[polldaddy:10307652]
The authors’ observations
Baseline laboratory testing should have been conducted prior to initiating valproic acid. As Ms. L’s symptoms worsened, better communication with her PCP and closer monitoring after starting valproic acid might have resulted in more immediate care. Early recognition of her symptoms and decompensation may have triggered earlier inpatient admission and/or transfer to a tertiary care facility for observation and treatment. Additionally, repeat laboratory testing and instructions on when to return to the ED should have been provided at Visit 1.
This case demonstrates the need for all clinicians who prescribe valproic acid to remain diligent about the accurate diagnosis of mood and behavioral symptoms, knowing when psychotropic medications are indicated, and carefully considering and discussing even rare, potentially life-threatening adverse effects of all medications with patients.
Although rare, after starting valproic acid, a patient may experience a rapid decompensation and life-threatening illness. Ideally, clinicians should closely monitor patients after initiating valproic acid (Table 41). Clinicians must have a clear knowledge of the recommended monitoring and indications for hospitalization and treatment when they note adverse effects such as elevated liver enzymes or transaminitis (Table 513,14). Even after stopping valproic acid, patients who have experienced adverse events should be closely monitored to ensure complete resolution.
Continue to: Consider patient-specific factors
Consider patient-specific factors
Consider the mental state, intellectual capacity, and social support of each patient before initiating valproic acid. Its use as a mood stabilizer for “mood swings” outside of the context of bipolar disorder is questionable. Valproic acid is FDA-approved for treating bipolar disorder and seizures, but not for anger outbursts/irritability. Prior to starting valproic acid, Ms. L may have benefited from alternative nonpharmacologic treatments, such as psychotherapy, for her anger outbursts and poor coping skills. Therapeutic techniques that focused on helping her acquire better coping mechanisms may have been useful, especially because her mood symptoms did not meet criteria for bipolar disorder, and her depression had long been controlled with sertraline monotherapy.
OUTCOME Discharged after 20 days
Ms. L stays in the hospital for 10 days after receiving her liver transplant. She has low appetite and some difficulty with sleep after the transplant; therefore, the C-L team recommends mirtazapine, 15 mg/d. She has no behavioral problems during her stay, and is set up with home health, case management, and psychiatry follow-up. On hospital Day 20, she is discharged.
Bottom Line
Use caution when prescribing valproic acid, even in young, otherwise healthy patients. Although rare, some patients may experience a rapid decompensation and life-threatening illness after starting valproic acid. When prescribing valproic acid, ensure close follow-up after initiation, including mental status examinations, physical examinations, and laboratory testing.
Related Resource
- Doroudgar S, Chou TI. How to modify psychotropic therapy for patients who have liver dysfunction. Current Psychiatry. 2014;13(12):46-49.
Drug Brand Names
Diphenhydramine • Benadryl
Famotidine • Fluxid, Pepcid
Lamotrigine • Lamictal
Mirtazapine • Remeron
N-acetylcysteine • Mucomyst
Phenobarbital • Luminal
Phenytoin • Dilantin
Prednisone • Cortan, Deltasone
Sertraline • Zoloft
Valproic acid • Depakene
1. Depakote [package insert]. North Chicago, IL: AbbVie, Inc.; 2019.
2. National Institutes of Health. U.S. Department of Health and Human Services. Drug Record: Valproate. https://livertox.nlm.nih.gov/Valproate.htm. Updated October 30, 2018. Accessed March 21, 2019.
3. Schmid MM, Freudenmann RW, Keller F, et al. Non-fatal and fatal liver failure associated with valproic acid. Pharmacopsychiatry. 2013;46(2):63-68.
4. Patel N, Landry KB, Fargason RE, et al. Reversible encephalopathy due to valproic acid induced hyperammonemia in a patient with Bipolar I disorder: a cautionary report. Psychopharmacol Bull. 2017;47(1):40-44.
5. Eze E, Workman M, Donley B. Hyperammonemia and coma developed by a woman treated with valproic acid for affective disorder. Psychiatr Serv. 1998;49(10):1358-1359.
6. Darban M and Bagheri B. Drug reaction with eosinophilia and systemic symptoms induced by valproic acid: a case report. Iran Red Crescent Med J. 2016;18(9): e35825.
7. van Zoelen MA, de Graaf M, van Dijk MR, et al. Valproic acid-induced DRESS syndrome with acute liver failure. Neth J Med. 2012;70(3):155.
8. Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila). 2009;47(2):101-111.
9. Bohan TP, Helton E, McDonald I, et al. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology. 2001;56(10):1405-1409.
10. Böhles H, Sewell AC, Wenzel D. The effect of carnitine supplementation in valproate-induced hyperammonaemia. Acta Paediatr. 1996;85(4):446-449.
11. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638.
12. Romero-Falcón A, de la Santa-Belda E, García-Contreras R, et al. A case of valproate-associated hepatotoxicity treated with L-carnitine. Eur J Intern Med. 2003;14(5):338-340.
13. National Institute for Health and Clinical Excellence. Bipolar disorder: the management of bipolar disorder in adults, children, and adolescents, in primary and secondary care. https://www.nice.org.uk/guidance/cg185. Updated April 2018. Accessed March 21, 2019.
14 . Hirschfeld RMA, Bowden CL, Gitlin MJ, et al. Practice guideline for the treatment of patients with biopolar disorder: second edition. American Psychiatric Association. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/bipolar.pdf. Published 2002. Accessed March 21, 2019.
1. Depakote [package insert]. North Chicago, IL: AbbVie, Inc.; 2019.
2. National Institutes of Health. U.S. Department of Health and Human Services. Drug Record: Valproate. https://livertox.nlm.nih.gov/Valproate.htm. Updated October 30, 2018. Accessed March 21, 2019.
3. Schmid MM, Freudenmann RW, Keller F, et al. Non-fatal and fatal liver failure associated with valproic acid. Pharmacopsychiatry. 2013;46(2):63-68.
4. Patel N, Landry KB, Fargason RE, et al. Reversible encephalopathy due to valproic acid induced hyperammonemia in a patient with Bipolar I disorder: a cautionary report. Psychopharmacol Bull. 2017;47(1):40-44.
5. Eze E, Workman M, Donley B. Hyperammonemia and coma developed by a woman treated with valproic acid for affective disorder. Psychiatr Serv. 1998;49(10):1358-1359.
6. Darban M and Bagheri B. Drug reaction with eosinophilia and systemic symptoms induced by valproic acid: a case report. Iran Red Crescent Med J. 2016;18(9): e35825.
7. van Zoelen MA, de Graaf M, van Dijk MR, et al. Valproic acid-induced DRESS syndrome with acute liver failure. Neth J Med. 2012;70(3):155.
8. Lheureux PE, Hantson P. Carnitine in the treatment of valproic acid-induced toxicity. Clin Toxicol (Phila). 2009;47(2):101-111.
9. Bohan TP, Helton E, McDonald I, et al. Effect of L-carnitine treatment for valproate-induced hepatotoxicity. Neurology. 2001;56(10):1405-1409.
10. Böhles H, Sewell AC, Wenzel D. The effect of carnitine supplementation in valproate-induced hyperammonaemia. Acta Paediatr. 1996;85(4):446-449.
11. Raskind JY, El-Chaar GM. The role of carnitine supplementation during valproic acid therapy. Ann Pharmacother. 2000;34(5):630-638.
12. Romero-Falcón A, de la Santa-Belda E, García-Contreras R, et al. A case of valproate-associated hepatotoxicity treated with L-carnitine. Eur J Intern Med. 2003;14(5):338-340.
13. National Institute for Health and Clinical Excellence. Bipolar disorder: the management of bipolar disorder in adults, children, and adolescents, in primary and secondary care. https://www.nice.org.uk/guidance/cg185. Updated April 2018. Accessed March 21, 2019.
14 . Hirschfeld RMA, Bowden CL, Gitlin MJ, et al. Practice guideline for the treatment of patients with biopolar disorder: second edition. American Psychiatric Association. https://psychiatryonline.org/pb/assets/raw/sitewide/practice_guidelines/guidelines/bipolar.pdf. Published 2002. Accessed March 21, 2019.
Gut microbiota and its implications for psychiatry: A review of 3 studies
The “human microbiota” describes all microorganisms within the human body, including bacteria, viruses, and eukaryotes. The related term “microbiome” refers to the complete catalog of these microbes and their genes.1 There is a growing awareness that the human microbiota plays an important role in maintaining mental health, and that a disruption in its composition can contribute to manifestations of psychiatric disorders. A growing body of evidence has also linked mental health outcomes to the gut microbiome, suggesting that the gut microbiota can modulate the gut-brain axis.2
Numerous neurotransmitters, including dopamine, serotonin, gamma-aminobutyric acid, and acetylcholine, are produced in the gastrointestinal (GI) tract, and our diet is vital in sustaining and replenishing them. At the same time, our brain regulates our GI tract by secretion of hormones such as oxytocin, leptin, ghrelin, neuropeptide Y, corticotrophin-releasing factor, and a plethora of others. Dysregulation of this microbiome can lead to both physical and mental illnesses. Symptoms of psychiatric disorders, such as depression, psychosis, anxiety, and autism, can be a consequence of this dysregulation.2
Our diet can also modify the gut microorganisms and therefore many of its metabolic pathways. More attention has been given to pre- and probiotics and their effects on DNA by epigenetic changes. One can quickly start to appreciate how this intricate crosstalk can lead to a variety of pathologic and psychiatric problems that have an adverse effect on autoimmune, inflammatory, metabolic, cognitive, and behavioral processes.2,3
Thus far, links have mostly been reported in animal models, and human studies are limited.4 Researchers are just beginning to elucidate how the microbiota affect gut-brain signaling in humans. Such mechanisms may include alterations in microbial composition, immune activation, vagus nerve signaling, alterations in tryptophan metabolism, production of specific microbial neuroactive metabolites, and bacterial cell wall sugars.5 The microbiota-gut-brain axis plays a part in regulating/programming the hypothalamic-pituitary-adrenal (HPA) axis throughout the life span.3 The interactions between the gut microbiome, the immune system, and the CNS are regulated through pathways that involve endocrine functions (HPA axis), the immune system, and metabolic factors.3,4 Recent research focusing on the gut microbiome has also given rise to international projects such as the Human Microbiome Project (Human Microbiome Project Consortium, 2012).3
Several studies have looked into psychiatry and inflammatory/immune pathways. Here we review 3 recent studies that have focused on the gut-brain axis (Table6-8).

1. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
The aim of this study was to evaluate immunoglobulin G (IgG) response against 40 food products in patients with depression vs those in a control group, along with changes in inflammatory markers, psychological stress, and dietary variables.6
Study design
- N = 63, IgG levels against 44 food products, cortisol levels, tumor necrosis factor (TNF)-alpha, interleukin 6 (IL-6), and IL-1 beta levels were recorded. The psychological parameters of 34 participants with depression and 29 controls were compared using the Hamilton Depression Rating scale, (HAM-D-17), Perceived Stress scale, and Symptom Checklist scale. The study was conducted in Poland.
Continue to: Outcomes
Outcomes
- Patients who were depressed had lower IgG levels against dairy products compared to controls when there was high dairy consumption. However, there was no overall difference between patients and controls in mean IgG concentration against food products.
- Patients who were depressed had higher levels of cortisol. Levels of cortisol had a positive correlation with HAM-D-17 score. Patients with depression had lower levels of TNF-alpha.
Conclusion
- Patients with depression had lower levels of IgG against dairy protein. Patients with depression had high cortisol levels but decreased levels of TNF-alpha, which could explain an immune suppression of IgG in these patients. There were no differences in IL-6 or IL-1beta levels.
Hypercortisolemia is present in approximately 60% of patients with depression. Elevated cortisol levels have a negative effect on lymphocyte function. B-lymphocytes (CD 10+ and CD 19+) are sensitive to glucocorticoids. Studies in mice have demonstrated that elevated glucocorticoid levels are associated with a 50% decrease in serum B-lymphocytes, and this can be explained by downregulation of c-myc protein, which plays a role in cell proliferation and cell survival. Glucocorticoids also decrease levels of protein kinases that are vital for the cell cycle to continue, and they upregulate p27 and p21, which are cell cycle inhibitors. Therefore, if high cortisol suppresses B-lymphocyte production, this can explain how patients with depression have low IgG levels, since B-lymphocytes differentiate into plasma cells that will produce antibodies.6
Depression can trigger an inflammatory response by increasing levels of inflammatory cytokines, acute phase reactants, and oxidative molecules. The inflammatory response can lead to intestinal wall disruption, and therefore bacteria can migrate across the GI barrier, along with food antigens, which could then lead to food antigen hypersensitivity.6
The significance of diet
Many studies have looked into specific types of diets, such as the Mediterranean diet, the ketogenic diet, and the addition of supplements such as probiotics, omega-3 fatty acids, zinc, and multivitamins.7 The Mediterranean diet is high in fiber, nuts, legumes, and fish.7 The ketogenic diet includes a controlled amount of fat, but is low in protein and carbohydrates.7 The main point is that a balanced diet can have a positive effect on mental health.7 The Mediterranean diet has shown to decrease the incidence of cardiovascular disease and lower the risk of depression.7 In animal studies, the ketogenic diet has improved anxiety, depression, and autism.7 Diet clearly affects gut microbiota and, as a consequence, the body’s level of inflammation.7
Continue to: The following review...
The following review highlighted the significance of diet on gut microbiome and mental health.7
2. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut- brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17: 1-9.
Study design
- These researchers provided a narrative review of the significance of a healthy diet and nutritional supplements on the gut microbiome and the treatment of patients with psychiatric illness.
Outcomes
- This review suggested dietary coaching as a nonpharmacologic treatment for patients with psychiatric illness.
Conclusion
- The utilization of nutritional advice, along with medication management, therapy, and physical activity, can provide a holistic approach to the biopsychosocial treatment of patients with psychiatric illness.
This review also emphasized the poor dietary trends of Westernized countries, which include calorie-dense, genetically altered, processed meals. As Mörkl et al7 noted, we are overfed but undernourished. Mörkl et al7 reviewed studies that involve dietary coaching as part of the treatment plan of patients with mental illness. In one of these studies, patients who received nutritional advice and coaching over 6 weeks had a 40% to 50% decrease in depressive symptoms. These effects persisted for 2 more years. Mörkl et al7 also reviewed an Italian study that found that providing nutritional advice in patients with affective disorders and psychosis helped improve symptom severity and sleep.7
Continue to: Mörkl et al...
Mörkl et al7 also reviewed dietary supplements. Some studies have linked use of omega-3 fatty acids with improvement in affective disorders, Alzheimer’s disease, and posttraumatic stress disorder, as well as cardiovascular conditions. Omega-3 fatty acids may exert beneficial effects by enhancing brain-derived neurotrophic factor and neurogenesis as well as by decreasing inflammation.7
Zinc supplementation can also improve depression, as it has been linked to cytokine variation and hippocampal neuronal growth. Vitamin B9 deficiency and vitamin D deficiency also have been associated with depression. Mörkl et al7 emphasized that a balanced diet that incorporates a variety of nutrients is more beneficial than supplementation of any individual vitamin alone.
Researchers have long emphasized the importance of a healthy balanced diet when treating patients with medical conditions such as cardiovascular or cerebrovascular diseases. Based on the studies Mörkl et al7 reviewed, the same emphasis should be communicated to our patients who suffer from psychiatric conditions.
The gut and anxiety
The gut microbiome has also been an area of research when studying generalized anxiety disorder (GAD).8
3. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
The aim of the study was to determine if there were changes in the composition of the gut microbiome in patients with GAD compared with healthy controls.8
Continue to: Study design
Study design
- A cross-sectional study of 76 patients in Zhejiang, China. Forty patients with GAD in the active state and 36 healthy controls were compared in terms of composition of GI microbacterial flora.
- Researchers also examined a subgroup of 12 patients who were treatment-naïve and 17 controls. Stool samples were collected from the 12 patients who were treatment-naïve before initiating medication.
- Researchers also conducted a prospective study in a subgroup of 9 patients with GAD in both the active state and remissive state. Two stool samples were collected from each patient—one during the active state of GAD and one during the remissive state—for a total of 18 samples. Stool samples analyzed with the use of polymerase chain reaction and microbial analysis.
- Patients completed the Hamilton Anxiety Rating (HAM-A) scale and were classified into groups. Those with HAM-A scores >14 were classified as being in the active state of GAD, and those with scores <7 were classified as being in the remissive state.
Outcomes
- Among the samples collected, 8 bacterial taxa were found in different amounts in patients with GAD and healthy controls. Bacteroidetes, Ruminococcus gnavus, and Fusobacterium were increased in patients with GAD compared with controls, while Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus were increased in healthy controls.
- Bacterial variety was notably lower in the 12 patients who were treatment-naïve compared with the control group.
- There was no notable difference in microbial composition between patients in the active vs remissive state.
Conclusion
- Patients with GAD had less short chain fatty acid–producing bacteria (Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus) compared with controls. Decreased formation of short chain fatty acids could lead to GI barrier disruption. Fusobacterium and Ruminococcus were increased in patients with GAD. Fusobacterium can cause disease and be invasive when it disseminates within the body. The inflammatory characteristics of Fusobacterium contribute to the immunologic activation in GAD. Ruminococcus breaks down mucin, which could then increase GI permeability by mucous degradation of the GI lumen.
Changes in food processing and manufacturing have led to changes in our diets. Changes in our normal GI microbacterial flora could lead to increased gut permeability, bacterial dissemination, and subsequent systemic inflammation. Research has shown that the composition of the microbiota changes across the life span.9 A balanced intake of nutrients is important for both our physical and mental health and safeguards the basis of gut microbiome regulation. A well-regulated gut microbiome ensures low levels of inflammation in the brain and body. Lifestyle modifications and dietary coaching could be practical interventions for patients with psychiatric conditions.5 Current advances in technology now offer precise analyses of thousands of metabolites, enabling metabolomics to offer the promise of discovering new drug targets and biomarkers that may help pave a way to precision medicine.
1. Dave M, Higgins PD, Middha S, et al. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res. 2012;160:246-257.
2. Nasrallah HA. It takes guts to be mentally ill: microbiota and psychopathology. Current Psychiatry. 2018;17(9):4-6.
3. Malan-Muller S, Valles-Colomer M, Raes J, et al. The gut microbiome and mental health: implications for anxiety-and trauma-related disorders. OMICS. 2018;22(2):90-107.
4. Du Toit A. The gut microbiome and mental health. Nat Rev Microbiol. 2019;17(4):196.
5. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701-712.
6. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
7. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut-brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17:1-9.
8. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
9. Douglas-Escobar M, Elliott E, Neu J. Effect of intestinal microbial ecology on the developing brain. JAMA Pediatr. 2013;167(4):374-379.
The “human microbiota” describes all microorganisms within the human body, including bacteria, viruses, and eukaryotes. The related term “microbiome” refers to the complete catalog of these microbes and their genes.1 There is a growing awareness that the human microbiota plays an important role in maintaining mental health, and that a disruption in its composition can contribute to manifestations of psychiatric disorders. A growing body of evidence has also linked mental health outcomes to the gut microbiome, suggesting that the gut microbiota can modulate the gut-brain axis.2
Numerous neurotransmitters, including dopamine, serotonin, gamma-aminobutyric acid, and acetylcholine, are produced in the gastrointestinal (GI) tract, and our diet is vital in sustaining and replenishing them. At the same time, our brain regulates our GI tract by secretion of hormones such as oxytocin, leptin, ghrelin, neuropeptide Y, corticotrophin-releasing factor, and a plethora of others. Dysregulation of this microbiome can lead to both physical and mental illnesses. Symptoms of psychiatric disorders, such as depression, psychosis, anxiety, and autism, can be a consequence of this dysregulation.2
Our diet can also modify the gut microorganisms and therefore many of its metabolic pathways. More attention has been given to pre- and probiotics and their effects on DNA by epigenetic changes. One can quickly start to appreciate how this intricate crosstalk can lead to a variety of pathologic and psychiatric problems that have an adverse effect on autoimmune, inflammatory, metabolic, cognitive, and behavioral processes.2,3
Thus far, links have mostly been reported in animal models, and human studies are limited.4 Researchers are just beginning to elucidate how the microbiota affect gut-brain signaling in humans. Such mechanisms may include alterations in microbial composition, immune activation, vagus nerve signaling, alterations in tryptophan metabolism, production of specific microbial neuroactive metabolites, and bacterial cell wall sugars.5 The microbiota-gut-brain axis plays a part in regulating/programming the hypothalamic-pituitary-adrenal (HPA) axis throughout the life span.3 The interactions between the gut microbiome, the immune system, and the CNS are regulated through pathways that involve endocrine functions (HPA axis), the immune system, and metabolic factors.3,4 Recent research focusing on the gut microbiome has also given rise to international projects such as the Human Microbiome Project (Human Microbiome Project Consortium, 2012).3
Several studies have looked into psychiatry and inflammatory/immune pathways. Here we review 3 recent studies that have focused on the gut-brain axis (Table6-8).
1. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
The aim of this study was to evaluate immunoglobulin G (IgG) response against 40 food products in patients with depression vs those in a control group, along with changes in inflammatory markers, psychological stress, and dietary variables.6
Study design
- N = 63, IgG levels against 44 food products, cortisol levels, tumor necrosis factor (TNF)-alpha, interleukin 6 (IL-6), and IL-1 beta levels were recorded. The psychological parameters of 34 participants with depression and 29 controls were compared using the Hamilton Depression Rating scale, (HAM-D-17), Perceived Stress scale, and Symptom Checklist scale. The study was conducted in Poland.
Continue to: Outcomes
Outcomes
- Patients who were depressed had lower IgG levels against dairy products compared to controls when there was high dairy consumption. However, there was no overall difference between patients and controls in mean IgG concentration against food products.
- Patients who were depressed had higher levels of cortisol. Levels of cortisol had a positive correlation with HAM-D-17 score. Patients with depression had lower levels of TNF-alpha.
Conclusion
- Patients with depression had lower levels of IgG against dairy protein. Patients with depression had high cortisol levels but decreased levels of TNF-alpha, which could explain an immune suppression of IgG in these patients. There were no differences in IL-6 or IL-1beta levels.
Hypercortisolemia is present in approximately 60% of patients with depression. Elevated cortisol levels have a negative effect on lymphocyte function. B-lymphocytes (CD 10+ and CD 19+) are sensitive to glucocorticoids. Studies in mice have demonstrated that elevated glucocorticoid levels are associated with a 50% decrease in serum B-lymphocytes, and this can be explained by downregulation of c-myc protein, which plays a role in cell proliferation and cell survival. Glucocorticoids also decrease levels of protein kinases that are vital for the cell cycle to continue, and they upregulate p27 and p21, which are cell cycle inhibitors. Therefore, if high cortisol suppresses B-lymphocyte production, this can explain how patients with depression have low IgG levels, since B-lymphocytes differentiate into plasma cells that will produce antibodies.6
Depression can trigger an inflammatory response by increasing levels of inflammatory cytokines, acute phase reactants, and oxidative molecules. The inflammatory response can lead to intestinal wall disruption, and therefore bacteria can migrate across the GI barrier, along with food antigens, which could then lead to food antigen hypersensitivity.6
The significance of diet
Many studies have looked into specific types of diets, such as the Mediterranean diet, the ketogenic diet, and the addition of supplements such as probiotics, omega-3 fatty acids, zinc, and multivitamins.7 The Mediterranean diet is high in fiber, nuts, legumes, and fish.7 The ketogenic diet includes a controlled amount of fat, but is low in protein and carbohydrates.7 The main point is that a balanced diet can have a positive effect on mental health.7 The Mediterranean diet has shown to decrease the incidence of cardiovascular disease and lower the risk of depression.7 In animal studies, the ketogenic diet has improved anxiety, depression, and autism.7 Diet clearly affects gut microbiota and, as a consequence, the body’s level of inflammation.7
Continue to: The following review...
The following review highlighted the significance of diet on gut microbiome and mental health.7
2. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut- brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17: 1-9.
Study design
- These researchers provided a narrative review of the significance of a healthy diet and nutritional supplements on the gut microbiome and the treatment of patients with psychiatric illness.
Outcomes
- This review suggested dietary coaching as a nonpharmacologic treatment for patients with psychiatric illness.
Conclusion
- The utilization of nutritional advice, along with medication management, therapy, and physical activity, can provide a holistic approach to the biopsychosocial treatment of patients with psychiatric illness.
This review also emphasized the poor dietary trends of Westernized countries, which include calorie-dense, genetically altered, processed meals. As Mörkl et al7 noted, we are overfed but undernourished. Mörkl et al7 reviewed studies that involve dietary coaching as part of the treatment plan of patients with mental illness. In one of these studies, patients who received nutritional advice and coaching over 6 weeks had a 40% to 50% decrease in depressive symptoms. These effects persisted for 2 more years. Mörkl et al7 also reviewed an Italian study that found that providing nutritional advice in patients with affective disorders and psychosis helped improve symptom severity and sleep.7
Continue to: Mörkl et al...
Mörkl et al7 also reviewed dietary supplements. Some studies have linked use of omega-3 fatty acids with improvement in affective disorders, Alzheimer’s disease, and posttraumatic stress disorder, as well as cardiovascular conditions. Omega-3 fatty acids may exert beneficial effects by enhancing brain-derived neurotrophic factor and neurogenesis as well as by decreasing inflammation.7
Zinc supplementation can also improve depression, as it has been linked to cytokine variation and hippocampal neuronal growth. Vitamin B9 deficiency and vitamin D deficiency also have been associated with depression. Mörkl et al7 emphasized that a balanced diet that incorporates a variety of nutrients is more beneficial than supplementation of any individual vitamin alone.
Researchers have long emphasized the importance of a healthy balanced diet when treating patients with medical conditions such as cardiovascular or cerebrovascular diseases. Based on the studies Mörkl et al7 reviewed, the same emphasis should be communicated to our patients who suffer from psychiatric conditions.
The gut and anxiety
The gut microbiome has also been an area of research when studying generalized anxiety disorder (GAD).8
3. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
The aim of the study was to determine if there were changes in the composition of the gut microbiome in patients with GAD compared with healthy controls.8
Continue to: Study design
Study design
- A cross-sectional study of 76 patients in Zhejiang, China. Forty patients with GAD in the active state and 36 healthy controls were compared in terms of composition of GI microbacterial flora.
- Researchers also examined a subgroup of 12 patients who were treatment-naïve and 17 controls. Stool samples were collected from the 12 patients who were treatment-naïve before initiating medication.
- Researchers also conducted a prospective study in a subgroup of 9 patients with GAD in both the active state and remissive state. Two stool samples were collected from each patient—one during the active state of GAD and one during the remissive state—for a total of 18 samples. Stool samples analyzed with the use of polymerase chain reaction and microbial analysis.
- Patients completed the Hamilton Anxiety Rating (HAM-A) scale and were classified into groups. Those with HAM-A scores >14 were classified as being in the active state of GAD, and those with scores <7 were classified as being in the remissive state.
Outcomes
- Among the samples collected, 8 bacterial taxa were found in different amounts in patients with GAD and healthy controls. Bacteroidetes, Ruminococcus gnavus, and Fusobacterium were increased in patients with GAD compared with controls, while Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus were increased in healthy controls.
- Bacterial variety was notably lower in the 12 patients who were treatment-naïve compared with the control group.
- There was no notable difference in microbial composition between patients in the active vs remissive state.
Conclusion
- Patients with GAD had less short chain fatty acid–producing bacteria (Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus) compared with controls. Decreased formation of short chain fatty acids could lead to GI barrier disruption. Fusobacterium and Ruminococcus were increased in patients with GAD. Fusobacterium can cause disease and be invasive when it disseminates within the body. The inflammatory characteristics of Fusobacterium contribute to the immunologic activation in GAD. Ruminococcus breaks down mucin, which could then increase GI permeability by mucous degradation of the GI lumen.
Changes in food processing and manufacturing have led to changes in our diets. Changes in our normal GI microbacterial flora could lead to increased gut permeability, bacterial dissemination, and subsequent systemic inflammation. Research has shown that the composition of the microbiota changes across the life span.9 A balanced intake of nutrients is important for both our physical and mental health and safeguards the basis of gut microbiome regulation. A well-regulated gut microbiome ensures low levels of inflammation in the brain and body. Lifestyle modifications and dietary coaching could be practical interventions for patients with psychiatric conditions.5 Current advances in technology now offer precise analyses of thousands of metabolites, enabling metabolomics to offer the promise of discovering new drug targets and biomarkers that may help pave a way to precision medicine.
The “human microbiota” describes all microorganisms within the human body, including bacteria, viruses, and eukaryotes. The related term “microbiome” refers to the complete catalog of these microbes and their genes.1 There is a growing awareness that the human microbiota plays an important role in maintaining mental health, and that a disruption in its composition can contribute to manifestations of psychiatric disorders. A growing body of evidence has also linked mental health outcomes to the gut microbiome, suggesting that the gut microbiota can modulate the gut-brain axis.2
Numerous neurotransmitters, including dopamine, serotonin, gamma-aminobutyric acid, and acetylcholine, are produced in the gastrointestinal (GI) tract, and our diet is vital in sustaining and replenishing them. At the same time, our brain regulates our GI tract by secretion of hormones such as oxytocin, leptin, ghrelin, neuropeptide Y, corticotrophin-releasing factor, and a plethora of others. Dysregulation of this microbiome can lead to both physical and mental illnesses. Symptoms of psychiatric disorders, such as depression, psychosis, anxiety, and autism, can be a consequence of this dysregulation.2
Our diet can also modify the gut microorganisms and therefore many of its metabolic pathways. More attention has been given to pre- and probiotics and their effects on DNA by epigenetic changes. One can quickly start to appreciate how this intricate crosstalk can lead to a variety of pathologic and psychiatric problems that have an adverse effect on autoimmune, inflammatory, metabolic, cognitive, and behavioral processes.2,3
Thus far, links have mostly been reported in animal models, and human studies are limited.4 Researchers are just beginning to elucidate how the microbiota affect gut-brain signaling in humans. Such mechanisms may include alterations in microbial composition, immune activation, vagus nerve signaling, alterations in tryptophan metabolism, production of specific microbial neuroactive metabolites, and bacterial cell wall sugars.5 The microbiota-gut-brain axis plays a part in regulating/programming the hypothalamic-pituitary-adrenal (HPA) axis throughout the life span.3 The interactions between the gut microbiome, the immune system, and the CNS are regulated through pathways that involve endocrine functions (HPA axis), the immune system, and metabolic factors.3,4 Recent research focusing on the gut microbiome has also given rise to international projects such as the Human Microbiome Project (Human Microbiome Project Consortium, 2012).3
Several studies have looked into psychiatry and inflammatory/immune pathways. Here we review 3 recent studies that have focused on the gut-brain axis (Table6-8).
1. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
The aim of this study was to evaluate immunoglobulin G (IgG) response against 40 food products in patients with depression vs those in a control group, along with changes in inflammatory markers, psychological stress, and dietary variables.6
Study design
- N = 63, IgG levels against 44 food products, cortisol levels, tumor necrosis factor (TNF)-alpha, interleukin 6 (IL-6), and IL-1 beta levels were recorded. The psychological parameters of 34 participants with depression and 29 controls were compared using the Hamilton Depression Rating scale, (HAM-D-17), Perceived Stress scale, and Symptom Checklist scale. The study was conducted in Poland.
Continue to: Outcomes
Outcomes
- Patients who were depressed had lower IgG levels against dairy products compared to controls when there was high dairy consumption. However, there was no overall difference between patients and controls in mean IgG concentration against food products.
- Patients who were depressed had higher levels of cortisol. Levels of cortisol had a positive correlation with HAM-D-17 score. Patients with depression had lower levels of TNF-alpha.
Conclusion
- Patients with depression had lower levels of IgG against dairy protein. Patients with depression had high cortisol levels but decreased levels of TNF-alpha, which could explain an immune suppression of IgG in these patients. There were no differences in IL-6 or IL-1beta levels.
Hypercortisolemia is present in approximately 60% of patients with depression. Elevated cortisol levels have a negative effect on lymphocyte function. B-lymphocytes (CD 10+ and CD 19+) are sensitive to glucocorticoids. Studies in mice have demonstrated that elevated glucocorticoid levels are associated with a 50% decrease in serum B-lymphocytes, and this can be explained by downregulation of c-myc protein, which plays a role in cell proliferation and cell survival. Glucocorticoids also decrease levels of protein kinases that are vital for the cell cycle to continue, and they upregulate p27 and p21, which are cell cycle inhibitors. Therefore, if high cortisol suppresses B-lymphocyte production, this can explain how patients with depression have low IgG levels, since B-lymphocytes differentiate into plasma cells that will produce antibodies.6
Depression can trigger an inflammatory response by increasing levels of inflammatory cytokines, acute phase reactants, and oxidative molecules. The inflammatory response can lead to intestinal wall disruption, and therefore bacteria can migrate across the GI barrier, along with food antigens, which could then lead to food antigen hypersensitivity.6
The significance of diet
Many studies have looked into specific types of diets, such as the Mediterranean diet, the ketogenic diet, and the addition of supplements such as probiotics, omega-3 fatty acids, zinc, and multivitamins.7 The Mediterranean diet is high in fiber, nuts, legumes, and fish.7 The ketogenic diet includes a controlled amount of fat, but is low in protein and carbohydrates.7 The main point is that a balanced diet can have a positive effect on mental health.7 The Mediterranean diet has shown to decrease the incidence of cardiovascular disease and lower the risk of depression.7 In animal studies, the ketogenic diet has improved anxiety, depression, and autism.7 Diet clearly affects gut microbiota and, as a consequence, the body’s level of inflammation.7
Continue to: The following review...
The following review highlighted the significance of diet on gut microbiome and mental health.7
2. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut- brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17: 1-9.
Study design
- These researchers provided a narrative review of the significance of a healthy diet and nutritional supplements on the gut microbiome and the treatment of patients with psychiatric illness.
Outcomes
- This review suggested dietary coaching as a nonpharmacologic treatment for patients with psychiatric illness.
Conclusion
- The utilization of nutritional advice, along with medication management, therapy, and physical activity, can provide a holistic approach to the biopsychosocial treatment of patients with psychiatric illness.
This review also emphasized the poor dietary trends of Westernized countries, which include calorie-dense, genetically altered, processed meals. As Mörkl et al7 noted, we are overfed but undernourished. Mörkl et al7 reviewed studies that involve dietary coaching as part of the treatment plan of patients with mental illness. In one of these studies, patients who received nutritional advice and coaching over 6 weeks had a 40% to 50% decrease in depressive symptoms. These effects persisted for 2 more years. Mörkl et al7 also reviewed an Italian study that found that providing nutritional advice in patients with affective disorders and psychosis helped improve symptom severity and sleep.7
Continue to: Mörkl et al...
Mörkl et al7 also reviewed dietary supplements. Some studies have linked use of omega-3 fatty acids with improvement in affective disorders, Alzheimer’s disease, and posttraumatic stress disorder, as well as cardiovascular conditions. Omega-3 fatty acids may exert beneficial effects by enhancing brain-derived neurotrophic factor and neurogenesis as well as by decreasing inflammation.7
Zinc supplementation can also improve depression, as it has been linked to cytokine variation and hippocampal neuronal growth. Vitamin B9 deficiency and vitamin D deficiency also have been associated with depression. Mörkl et al7 emphasized that a balanced diet that incorporates a variety of nutrients is more beneficial than supplementation of any individual vitamin alone.
Researchers have long emphasized the importance of a healthy balanced diet when treating patients with medical conditions such as cardiovascular or cerebrovascular diseases. Based on the studies Mörkl et al7 reviewed, the same emphasis should be communicated to our patients who suffer from psychiatric conditions.
The gut and anxiety
The gut microbiome has also been an area of research when studying generalized anxiety disorder (GAD).8
3. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
The aim of the study was to determine if there were changes in the composition of the gut microbiome in patients with GAD compared with healthy controls.8
Continue to: Study design
Study design
- A cross-sectional study of 76 patients in Zhejiang, China. Forty patients with GAD in the active state and 36 healthy controls were compared in terms of composition of GI microbacterial flora.
- Researchers also examined a subgroup of 12 patients who were treatment-naïve and 17 controls. Stool samples were collected from the 12 patients who were treatment-naïve before initiating medication.
- Researchers also conducted a prospective study in a subgroup of 9 patients with GAD in both the active state and remissive state. Two stool samples were collected from each patient—one during the active state of GAD and one during the remissive state—for a total of 18 samples. Stool samples analyzed with the use of polymerase chain reaction and microbial analysis.
- Patients completed the Hamilton Anxiety Rating (HAM-A) scale and were classified into groups. Those with HAM-A scores >14 were classified as being in the active state of GAD, and those with scores <7 were classified as being in the remissive state.
Outcomes
- Among the samples collected, 8 bacterial taxa were found in different amounts in patients with GAD and healthy controls. Bacteroidetes, Ruminococcus gnavus, and Fusobacterium were increased in patients with GAD compared with controls, while Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus were increased in healthy controls.
- Bacterial variety was notably lower in the 12 patients who were treatment-naïve compared with the control group.
- There was no notable difference in microbial composition between patients in the active vs remissive state.
Conclusion
- Patients with GAD had less short chain fatty acid–producing bacteria (Faecalibacterium, Eubacterium rectale, Sutterella, Lachnospira, and Butyricicoccus) compared with controls. Decreased formation of short chain fatty acids could lead to GI barrier disruption. Fusobacterium and Ruminococcus were increased in patients with GAD. Fusobacterium can cause disease and be invasive when it disseminates within the body. The inflammatory characteristics of Fusobacterium contribute to the immunologic activation in GAD. Ruminococcus breaks down mucin, which could then increase GI permeability by mucous degradation of the GI lumen.
Changes in food processing and manufacturing have led to changes in our diets. Changes in our normal GI microbacterial flora could lead to increased gut permeability, bacterial dissemination, and subsequent systemic inflammation. Research has shown that the composition of the microbiota changes across the life span.9 A balanced intake of nutrients is important for both our physical and mental health and safeguards the basis of gut microbiome regulation. A well-regulated gut microbiome ensures low levels of inflammation in the brain and body. Lifestyle modifications and dietary coaching could be practical interventions for patients with psychiatric conditions.5 Current advances in technology now offer precise analyses of thousands of metabolites, enabling metabolomics to offer the promise of discovering new drug targets and biomarkers that may help pave a way to precision medicine.
1. Dave M, Higgins PD, Middha S, et al. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res. 2012;160:246-257.
2. Nasrallah HA. It takes guts to be mentally ill: microbiota and psychopathology. Current Psychiatry. 2018;17(9):4-6.
3. Malan-Muller S, Valles-Colomer M, Raes J, et al. The gut microbiome and mental health: implications for anxiety-and trauma-related disorders. OMICS. 2018;22(2):90-107.
4. Du Toit A. The gut microbiome and mental health. Nat Rev Microbiol. 2019;17(4):196.
5. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701-712.
6. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
7. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut-brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17:1-9.
8. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
9. Douglas-Escobar M, Elliott E, Neu J. Effect of intestinal microbial ecology on the developing brain. JAMA Pediatr. 2013;167(4):374-379.
1. Dave M, Higgins PD, Middha S, et al. The human gut microbiome: current knowledge, challenges, and future directions. Transl Res. 2012;160:246-257.
2. Nasrallah HA. It takes guts to be mentally ill: microbiota and psychopathology. Current Psychiatry. 2018;17(9):4-6.
3. Malan-Muller S, Valles-Colomer M, Raes J, et al. The gut microbiome and mental health: implications for anxiety-and trauma-related disorders. OMICS. 2018;22(2):90-107.
4. Du Toit A. The gut microbiome and mental health. Nat Rev Microbiol. 2019;17(4):196.
5. Cryan JF, Dinan TG. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 2012;13(10):701-712.
6. Rudzki L, Pawlak D, Pawlak K, et al. Immune suppression of IgG response against dairy proteins in major depression. BMC Psychiatry. 2017;17(1):268.
7. Mörkl S, Wagner-Skacel J, Lahousen T, et al. The role of nutrition and the gut-brain axis in psychiatry: a review of the literature. Neuropsychobiology. 2018;17:1-9.
8. Jiang HY, Zhang X, Yu ZH, et al. Altered gut microbiota profile in patients with generalized anxiety disorder. J Psychiatr Res. 2018;104:130-136.
9. Douglas-Escobar M, Elliott E, Neu J. Effect of intestinal microbial ecology on the developing brain. JAMA Pediatr. 2013;167(4):374-379.

Intranasal esketamine
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal

How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.

Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).

Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc

A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).

Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.

Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal
How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.
Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).
Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc
A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).
Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.
Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
Treatment-resistant depression (TRD) is a common clinical struggle that practicing clinicians address on a daily basis. Major depressive disorder affects nearly 1 in 5 Americans at some point in their life and, by definition, impairs social and occupational functioning. Historic treatments have focused on the monoamine theories of depression—modulating the monoamines serotonin, norepinephrine, and/or dopamine. Limitations of currently available antidepressants include delayed onset of effect and low remission rates. To further complicate the matter, numerous studies have shown that with each subsequent antidepressant trial, patients have a decreasing likelihood of responding to subsequent antidepressant treatment options. For example, in the classic STAR*D trial, by the time a patient had not responded to the first 2 antidepressant options, the chance that they would respond to a third or fourth antidepressant had decreased to approximately 15% per antidepressant treatment course.1
To address the need for new treatments for patients with TRD, on March 5, 2019 the FDA-approved intranasal
How it works
Modern research has looked beyond the monoamine system to explore the neuro-modulatory effects of glutamate and gamma-aminobutyric acid (GABA).3 The yin and yang of glutamate and GABA revolves around neural excitation vs neural inhibition at a local synaptic level. The primary effects of the glutamate and GABA systems (Table 2) can be broken down into several key areas of understanding.
Glutamate modulates ionotropic N-methyl-
Esketamine, the S-enantiomer of ketamine, has a higher affinity for the NMDA receptor than the R-enantiomer and has been developed as an intranasal adjunctive treatment for TRD. Esketamine blocks NMDA receptors on GABA interneurons. This allows for increased pulsatile release of glutamate into the synapse. Intrasynaptic glutamate then stimulates postsynaptic AMPA receptors. Glutamate stimulation of postsynaptic AMPA receptors results in an intracellular cascade that activates the enzymes tropomyosin receptor kinase B (TrkB) and mammalian target of rapamycin (mTOR). TrkB stimulation results in increased production and release of BDNF. mTor stimulation increases neuronal membrane protein formation with subsequent increased neural plasticity. Taken together, preclinical models show that esketamine’s inhibition of the NMDA receptor on the GABA interneuron results in a cascade of increased BDNF release and synaptogenesis with increased neuroplasticity (Table 3).
Clinical implications
Treatment-resistant depression affects nearly one-third of patients currently receiving standard antidepressant treatment. Major depressive disorder is currently the second leading cause of disability for working adults within the United States and one of the largest causes of disability worldwide. The esketamine nasal spray could be beneficial for patients who have experienced TRD with standard monoamine antidepressants.
Supporting evidence
Clinical trials examining intranasal esketamine include both short- and long-term studies of patients with TRD.
Continue to: Esketamine was evaluated...
Esketamine was evaluated in a randomized, placebo-controlled, double-blind, multicenter, short-term (4-week) phase III study in adult patients age 18 to 65 with TRD (they had not responded to at least 2 different antidepressants of adequate dose and duration).4 After discontinuing prior antidepressant treatments, all patients were started on a newly initiated antidepressant and were also randomized to concomitant intranasal esketamine or intranasal placebo as follows:
- 114 patients were randomized to the intranasal esketamine plus newly initiated oral antidepressant arm
- 109 patients were randomized to the placebo nasal spray plus newly initiated oral antidepressant arm
- The mean baseline Montgomery-Åsberg Depression Rating Scale (MADRS) score for each group was 37 (ie, moderately to severely depressed).
Newly started antidepressants included esc
A long-term, double-blind multicenter maintenance-of-effect trial examined adults age 18 to 65 with TRD.5-6 Patients in this study were responders in 1 of 2 short-term studies or in an open-label direct enrollment study. Stable remission was defined as a MADRS total score <12 for at least 3 of the last 4 weeks of the study, and stable response was defined as a MADRS reduction of >50% but not in remission. After 16 weeks of intranasal esketamine plus an oral antidepressant, stable remitters and stable responders were then randomized separately to continue intranasal esketamine or switch to placebo nasal spray, with both groups continuing on their concomitant oral antidepressant. The primary study endpoint was time to relapse. Relapse was defined as a MADRS total score >22 for more than 2 consecutive weeks, hospitalization for worsening of depression, or any other clinically relevant event. The median age was 48, 66% were female, 90% were White and 4% were black. Patients in stable response or stable remission experienced a significantly longer time to relapse compared with patients who continued their oral antidepressant but were switched to placebo intranasal spray. In this remission response study, patients could receive intranasal treatment weekly or bi-weekly based on symptom severity (Figure 22).
Impact on driving. Two studies examined the impact of esketamine on driving performance. One examined adults with major depressive disorder and the other examined healthy participants. The effects of a single 84-mg dose of esketamine nasal spray on a patient’s ability to drive was assessed in 23 healthy adults. In this study, mirt
A second study evaluated the effects of repeated esketamine administration on driving performance in 25 adults with major depressive disorder. In this study, an ethanol-containing beverage was used as an active control. After administration of a single 84-mg dose of intranasal esketamine, driving performance was the same as a placebo at 18 hours. In the multiple dose phase, standard driving performance was similar for esketamine nasal spray and placebo at 6 hours postdose on Days 11, 18, and 25.
Continue to: Pharmacologic profile
Pharmacologic profile
Adverse events. The most common adverse events in patients treated with esketamine nasal spray were dissociation (41%), dizziness (29%), nausea (28%), sedation (23%), and vertigo (23%).2 The majority of these effects were short-term and resolved during the 2-hour observation period.
In addition to spontaneously reported events, sedation and dissociation were further monitored with specific scales. Sedation was measured with the Modified Observer’s Alertness and Sedation Scale. Using this scale, 50% of patients receiving 56 mg and 61% of patients receiving 84 mg of esketamine met criteria for sedation.
Similarly, dissociation/perceptional changes were measured with spontaneously reported events and also with the Clinician Administered Dissociative State Scale. On this scale, 61% of patients receiving the 56-mg dose, and 69% of patients receiving the 84-mg dose met criteria for dissociation/perceptional changes after dose administration.
Increases in blod pressure. Esketamine intranasal spray was associated with a 7 to 9 mm Hg increase in systolic blood pressure and a 4 to 6 mm Hg increase in diastolic blood pressure, both of which peaked 40 minutes post-dose.
Nausea and vomiting. Intranasal esketamine was associated with a 27% rate of nausea at 56 mg, and 32% at 84 mg, with a 6% rate of vomiting at 56 mg and 12% at 84 mg.
Continue to: Pharmacokinetics
Pharmacokinetics
Esketamine exposure increases from 28 to 84 mg in a fairly dose-proportional range. No accumulation of esketamine was observed in the plasma following twice-weekly administration. Bioavailability is approximately 48% following nasal administration. The Tmax for esketamine plasma concentration is 20 to 40 minutes after the last nasal spray. Protein binding of esketamine is approximately 43% to 45%. The brain-to-plasma ratio of noresketamine is 4 to 6 times lower than that of esketamine. The half-life of esketamine ranged from 7 to 12 hours. The mean half-life of nore
Potential drug interactions
Central nervous system depressants. Concomitant use of esketamine and other CNS depressants (ie, benzodiazepines, opioids, alcohol) may increase sedation. Patients receiving esketamine with concomitant use of other CNS depressants should be closely monitored for sedation.
Psychostimulants. Concomitant use of esketamine and psychostimulants (ie, amphetamines, methylphenidates, moda
Monoamine oxidase inhibitors. Concomitant use of esketamine with monoamine oxidase inhibitors may increase blood pressure. Closely monitor blood pressure with concomitant use of esketamine and monoamine oxidase inhibitors.
Use in special populations. Because of concerns of increased sedation, intranasal esketamine should be administered cautiously in patients receiving other CNS depressants, such as benzodiazepines. In patients with psychosis or a prior history of psychosis, esketamine should be used with increased caution and the risk/benefit ratio should be carefully considered.
Continue to: Because of potential teratogenicity...
Because of potential teratogenicity, esketamine is not recommended in women who are pregnant, may become pregnant, or who are currently nursing.
Intranasal esketamine was examined in a phase III trial of 194 patients age ≥65. At the end of 4 weeks, there was no statistically significant difference in groups on the MADRS, the primary efficacy endpoint. There were no overall differences in the safety profile in patients >65 years compared with younger patients; however, the mean esketamine Cmax and area under the curve were higher in older patients compared with younger adults. The mean esketamine half-life was longer in patients with moderate hepatic impairment.
Abuse liability
Esketamine is a CIII controlled substance and concerns about abuse, misuse, and diversion have been taken into account within the REMS drug safety program.2 Patients with a prior history of substance abuse or misuse should be considered with regard to the risk/benefit ratio.
The REMS drug safety program
Due to the nature of its usually transient adverse effects, including sedation, dissociation, hypertension, and nausea, intranasal esketamine will be administered through a REMS drug safety program at certified REMS treatment centers. Certified REMS treatment centers will receive training on how to safely and effectively counsel and monitor patients. Prior to treatment, patients will receive blood pressure monitoring and anticipated adverse effects will be discussed. Patients will be instructed to not eat solid food for 2 hours pre-dose and to not drink anything for 30 minutes prior.
A treatment session consists of nasal administration and a minimum 2-hour post-administration observation period. Blood pressure must be assessed prior to administration and if elevated, (ie, systolic blood pressure >140 mm Hg, diastolic >90 mm Hg), clinicians should consider the risk of short-term increases in blood pressure that may occur. Do not administer if increases in blood pressure or intracranial pressure pose a serious risk.
Continue to: After each intranasal...
After each intranasal administration the patient will be observed for 5 minutes before the second nasal inhaler is utilized and for another 5 minutes when the patient is receiving 84 mg (ie, each inhaler equals 28 mg). After administering, blood pressure should be reassessed at approximately 40 minutes, which corresponds to the Cmax of intranasal esketamine, and periodically thereafter as warranted.
The patient will then be monitored in a quiet environment for a minimum of 2 hours to make sure that dissociative phenomenon, sedation, and hypertensive reactions have normalized prior to discharge from a certified REMS treatment center.
Dosing and administration
Each intranasal device is primed for 2 infusions (1 in each nostril) for a total dose of 28 mg of esketamine. Combinations of devices can be used to adjust the dose as appropriate for individual patients. The recommended starting dose is 56 mg (ie, 2 devices, with a 5-minute gap between devices). The dose can be increased to 84 mg (ie, 3 intranasal devices spaced at 5-minute intervals) by the second dose based on clinical judgment.
The patient will be instructed to recline the head to a 45° angle, clear his or her nostrils prior to the first treatment, and then self-administer a dose to each nostril while holding the reciprocal nostril closed and inhaling. This process is then repeated every 5 minutes for each subsequent device, with a maximum total dose of 3 devices, or 84 mg (Figure 32). The patient will then be monitored for blood pressure, heart rate, and signs of psychologic or physiologic changes for the next 2 hours. Patients may not drive a car or operate any type of motor equipment until the following day after receiving a normal night’s sleep. Patients will be released from the REMS treatment center after 2 hours if both psychological and physical adverse effects have normalized.
Missed treatment sessions. If a patient misses a treatment session and there is worsening of depressive symptoms, consider returning the patient to the previous dosing schedule (ie, every 2 weeks to once weekly, or weekly to twice weekly).
Continue to: Contraindications for...
Contraindications for intranasal esketamine include:
- aneurysmal vascular disease, including thoracic and abdominal aortic, intracranial, and peripheral arterial vessels, or arterial venous malformations
- history of intracerebral hemorrhage
- hypersensitivity to esketamine, ketamine, or any of the excipients.
Clinical considerations
Intranasal esketamine represents a unique delivery system for the first glutamatergic treatment approved for patients with TRD.
Why Rx? Treatment-resistant depression is found in nearly 1 out of 3 patients with currently available monoaminergic antidepressant treatment options. Patients with TRD are at increased risk of physical and psychological impairment, subsequent worsening of their condition, and social and occupational disability.
Bottom Line
Intranasal esketamine is the first glutamatergic treatment option FDA-approved for patients with treatment-resistant depression who have not responded to standard antidepressant treatment options. In short-term trials, intranasal esketamine significantly improved depressive symptoms as quickly as 24 hours after treatment, with significant improvement maintained through 4 weeks of ongoing administration. In addition, intranasal esketamine was shown to significantly decrease time to relapse for patients who had achieved stable remission or stable response.
Related Resource
- Sullivan MG. FDA approves intranasal esketamine for refractory major depressive disorder. Clinical Psychiatry News. https://www.mdedge.com/psychiatry/article/195712/depression/fda-approves-intranasal-esketamine-refractory-major-depressive. Published March 5, 2019.
Drug Brand Names
Armodafinil • Nuvigil
Duloxetine • Cymbalta
Escitalopram • Lexapro
Esketamine • Spravato
Mirtazapine • Remeron
Modafinil • Provigil
Sertraline • Zoloft
Venlafaxine • Effexor
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
1. Rush AG, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR D Report. Am J Psychiatry. 2006;163(11):1905-1917.
2. Spravato [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2019.
3. Duman RS, Aghajanian GK, Sanacora G, et al. Synaptic plasticity and depression: new insights from stress and rapid-acting anti-depression. Nat Med. 2016;22(3):238-249.
4. Daly EJ, Singh JB, Fedgchin M, et al. Efficacy and safety of intranasal esketamine adjunctive to oral antidepressant therapy in treatment-resistant depression: a randomized clinical trial. JAMA Psychiatry. 2018;75(2):139-148.
5. Daly EJ, Trivedi M, Janik A, et al. A randomized withdrawal, double-blind, multicenter study of esketamine nasal spray plus an oral antidepressant for relapse prevention in treatment-resistant depression. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.
6. Wajs E, Aluisio L, Morrison R, et al. Long-term safety of esketamine nasal spray plus oral antidepressants in patients with treatment-resistant depression: phase III open-label safety and efficacy study. Poster presented at the 2018 American Society of Clinical Psychopharmacology Annual Meeting; May 2018; Miami, Florida.

The importance of engaging with local mental health organizations
“Hi Dr. Burke, thanks for coming in today. My daughter struggles with depression and I feel like every time I try to reach out, I hit a dead end with her. How do I connect with someone, who by the nature of their disease, is hard to reach?”
The answer? I’m not quite sure. I stood in front of a classroom of parents, siblings, and persons struggling with mental health issues, lecturing about depression. I can tell you about the complex interplay of biologic, psychological, and social factors that can lead one to become depressed. I can tell you the prevalence of depression in today’s society, and how it is rising among all age groups. I can tell you a myriad of different treatments, from pharmacologic to therapeutic to procedural, for depression. But how, from a parent’s perspective, can you connect with your child struggling with depression when they do not want your help? That I cannot tell you, at least not yet, anyways.
I had connected with the National Alliance on Mental Illness (NAMI) in the Fall of 2018, when a patient of mine was discharged from hospitalization and told by a faith-based substance use treatment program that he would not be allowed to use any “mind-altering” medications when he returned to their program. Concerned about my patient, whom I had just stabilized with the use of medications, I did my best to work through that organization’s resistance to psychotropic medications. When that failed, I reached out to NAMI for help in advocating for persons with mental illness. My involvement escalated to giving a lecture on “Living with Depression” to our local chapter of approximately 25 individuals that night. I had expected to lecture to an engaged crowd about what I thought was my immense knowledge of depression, from diagnosis to development to treatment. What I had not expected, however, was to have a learning experience of my own.
I stood at the front of the room, listening to story after story of persons with depression and their family members discussing their experiences. Throughout the 90-minute lecture, my emotions ranged from being impressed to shocked, scared, and, ultimately, proud. For the past year and 7 months, I had been spending time with persons with mental illness on what was likely the worst days of their lives. I had seen a variety of severe presentations, from grossly psychotic to acutely manic to majorly depressed to highly agitated. With that wealth of experience, I had thought I was becoming an expert; however, at the front of that classroom that night, I realized how little I actually knew. Yes, I had contemplated before how much severe mental illness and hospitalization could affect a person and their loved ones. However, it was a different level of understanding to hear first-hand accounts of the loss of relationships, the struggle to connect, and the fall-out from intensive inpatient treatment.
In residency, we spend what seems like an immeasurable amount of time on inpatient psychiatric units, in outpatient clinics, and everywhere in between. We see so many patients on a daily, weekly, monthly, and yearly basis that it becomes easy to lose the individuality of each patient. We start associating patients with their disorder, rather than with who they are. However, if we take a step back and allow a larger perspective—one that considers not only the patient but their families and communities—we likely would be able to provide greater and more comprehensive care.
My experience at NAMI was one that I will treasure forever. It opened my eyes to struggles that had I failed to even notice, and for that, and many other connections I made, I am grateful to have been blessed with this experience. My greatest recommendation to my fellow residents would be to engage with your local community organizations in the hope that you, too, can have an eye-opening experience that will strengthen your practice.
“Hi Dr. Burke, thanks for coming in today. My daughter struggles with depression and I feel like every time I try to reach out, I hit a dead end with her. How do I connect with someone, who by the nature of their disease, is hard to reach?”
The answer? I’m not quite sure. I stood in front of a classroom of parents, siblings, and persons struggling with mental health issues, lecturing about depression. I can tell you about the complex interplay of biologic, psychological, and social factors that can lead one to become depressed. I can tell you the prevalence of depression in today’s society, and how it is rising among all age groups. I can tell you a myriad of different treatments, from pharmacologic to therapeutic to procedural, for depression. But how, from a parent’s perspective, can you connect with your child struggling with depression when they do not want your help? That I cannot tell you, at least not yet, anyways.
I had connected with the National Alliance on Mental Illness (NAMI) in the Fall of 2018, when a patient of mine was discharged from hospitalization and told by a faith-based substance use treatment program that he would not be allowed to use any “mind-altering” medications when he returned to their program. Concerned about my patient, whom I had just stabilized with the use of medications, I did my best to work through that organization’s resistance to psychotropic medications. When that failed, I reached out to NAMI for help in advocating for persons with mental illness. My involvement escalated to giving a lecture on “Living with Depression” to our local chapter of approximately 25 individuals that night. I had expected to lecture to an engaged crowd about what I thought was my immense knowledge of depression, from diagnosis to development to treatment. What I had not expected, however, was to have a learning experience of my own.
I stood at the front of the room, listening to story after story of persons with depression and their family members discussing their experiences. Throughout the 90-minute lecture, my emotions ranged from being impressed to shocked, scared, and, ultimately, proud. For the past year and 7 months, I had been spending time with persons with mental illness on what was likely the worst days of their lives. I had seen a variety of severe presentations, from grossly psychotic to acutely manic to majorly depressed to highly agitated. With that wealth of experience, I had thought I was becoming an expert; however, at the front of that classroom that night, I realized how little I actually knew. Yes, I had contemplated before how much severe mental illness and hospitalization could affect a person and their loved ones. However, it was a different level of understanding to hear first-hand accounts of the loss of relationships, the struggle to connect, and the fall-out from intensive inpatient treatment.
In residency, we spend what seems like an immeasurable amount of time on inpatient psychiatric units, in outpatient clinics, and everywhere in between. We see so many patients on a daily, weekly, monthly, and yearly basis that it becomes easy to lose the individuality of each patient. We start associating patients with their disorder, rather than with who they are. However, if we take a step back and allow a larger perspective—one that considers not only the patient but their families and communities—we likely would be able to provide greater and more comprehensive care.
My experience at NAMI was one that I will treasure forever. It opened my eyes to struggles that had I failed to even notice, and for that, and many other connections I made, I am grateful to have been blessed with this experience. My greatest recommendation to my fellow residents would be to engage with your local community organizations in the hope that you, too, can have an eye-opening experience that will strengthen your practice.
“Hi Dr. Burke, thanks for coming in today. My daughter struggles with depression and I feel like every time I try to reach out, I hit a dead end with her. How do I connect with someone, who by the nature of their disease, is hard to reach?”
The answer? I’m not quite sure. I stood in front of a classroom of parents, siblings, and persons struggling with mental health issues, lecturing about depression. I can tell you about the complex interplay of biologic, psychological, and social factors that can lead one to become depressed. I can tell you the prevalence of depression in today’s society, and how it is rising among all age groups. I can tell you a myriad of different treatments, from pharmacologic to therapeutic to procedural, for depression. But how, from a parent’s perspective, can you connect with your child struggling with depression when they do not want your help? That I cannot tell you, at least not yet, anyways.
I had connected with the National Alliance on Mental Illness (NAMI) in the Fall of 2018, when a patient of mine was discharged from hospitalization and told by a faith-based substance use treatment program that he would not be allowed to use any “mind-altering” medications when he returned to their program. Concerned about my patient, whom I had just stabilized with the use of medications, I did my best to work through that organization’s resistance to psychotropic medications. When that failed, I reached out to NAMI for help in advocating for persons with mental illness. My involvement escalated to giving a lecture on “Living with Depression” to our local chapter of approximately 25 individuals that night. I had expected to lecture to an engaged crowd about what I thought was my immense knowledge of depression, from diagnosis to development to treatment. What I had not expected, however, was to have a learning experience of my own.
I stood at the front of the room, listening to story after story of persons with depression and their family members discussing their experiences. Throughout the 90-minute lecture, my emotions ranged from being impressed to shocked, scared, and, ultimately, proud. For the past year and 7 months, I had been spending time with persons with mental illness on what was likely the worst days of their lives. I had seen a variety of severe presentations, from grossly psychotic to acutely manic to majorly depressed to highly agitated. With that wealth of experience, I had thought I was becoming an expert; however, at the front of that classroom that night, I realized how little I actually knew. Yes, I had contemplated before how much severe mental illness and hospitalization could affect a person and their loved ones. However, it was a different level of understanding to hear first-hand accounts of the loss of relationships, the struggle to connect, and the fall-out from intensive inpatient treatment.
In residency, we spend what seems like an immeasurable amount of time on inpatient psychiatric units, in outpatient clinics, and everywhere in between. We see so many patients on a daily, weekly, monthly, and yearly basis that it becomes easy to lose the individuality of each patient. We start associating patients with their disorder, rather than with who they are. However, if we take a step back and allow a larger perspective—one that considers not only the patient but their families and communities—we likely would be able to provide greater and more comprehensive care.
My experience at NAMI was one that I will treasure forever. It opened my eyes to struggles that had I failed to even notice, and for that, and many other connections I made, I am grateful to have been blessed with this experience. My greatest recommendation to my fellow residents would be to engage with your local community organizations in the hope that you, too, can have an eye-opening experience that will strengthen your practice.

Paternalism vs autonomy: Why watching our words is important
Two patients were admitted to our unit at the same time: Mr. P, age 27, an architect with unspecified personality disorder, and Mr. D, age 62, a bank manager who has had bipolar disorder for 40 years and was experiencing a moderate depressive episode. Mr. P’s discomfort with the treatment team informing him of his treatment plan was evident, and he discussed at length his terms and stipulations for management. Mr. D, on the other hand, was loath to shoulder the burden of any decision-making, even in minor matters such as what time he should take his daily walk.
Patient autonomy is a central factor in the present-day doctor–patient equation. In psychiatry, this is sometimes further complicated by a patient’s impaired judgment and lowered decision-making capacity (DMC). In our clinical practice, we often notice that younger patients (ie, millennials) prefer to have autonomy rather than being given instructions, which they may find patronizing, whereas the older generation relies more on the doctor for decision-making.
What the decision-making process entails
The decision-making process involves 3 steps:
- information gathering
- deliberation
- implementation.
Decision-making preferences fall on a spectrum that ranges from paternalism at one end to autonomy on the other, with many intervening components, characterized by varying amounts of responsibility shared between doctor and patient.1 This typically comes into play when there is more than one treatment option with similar outcomes.2 Paternalism is defined as an action performed with the intent of promoting another’s good but occurring against the other’s will, or without consent.3 Here, the patient is not privy to the deliberation process, and no explanations are provided.1
Two other decision-making constructs are shared decision-making (SDM) and informed decision-making (IDM). In SDM, the deliberation process involves participation of both patient and doctor, with active discussion and a final decision after both parties reach an agreement. In IDM, the deliberation is conducted solely by the patient, after he or she receives all information. Shared decision-making and IDM are frequently used interchangeably, but in the latter, the doctor has no role other than to provide information.1,5
Before choosing SDM or IDM, it is necessary to assess the patient’s DMC—the ability to understand information about choices, make a judgment that respects personal values, understand potential outcomes, and freely communicate his or her wishes.6
Benefits and risks
The progression from paternalism to autonomy began in the mid-20th century as a consequence of the Nuremberg Trials, from which the concept of “informed consent” first came into existence.7 The Indian value system has always regarded the medical profession and its practitioners with high esteem, as evidenced by the Sanskrit quote “Vaidyo Narayano Harihi,” which translates to “The doctor is God.” A significant chunk of the Indian population still considers the doctor’s word to be law, and they hand over health-related decisions to medical professionals. Here, the expectation of a paternalistic attitude is decidedly unequivocal.
Continue to: Of course...
Of course, there are pros and cons to every approach. Making patients’ independence a priority is the highest virtue of autonomy, but in such cases a patient may have difficulty comprehending medical consequences, and therefore may miss out on the benefits of a sound professional perspective. Paternalism may be superior medically, but the doctor may not be aware of all patient-specific factors, and it would not be prudent to make a decision for a patient without being privy to the entire picture.
The 21st century has witnessed a change in attitudes regarding medical care. With an increasing interest in patient autonomy, it is time for us to adopt these changes and move towards the patient-centred end of the spectrum. However, this should occur only after the patient improves enough symptomatically to regain DMC; autonomy is unlikely to be appropriate for patients with serious mental illness. Ideally, SDM includes the best of both worlds, and results in optimal outcomes. However, when SDM breaks down, a selective, soft paternalistic attitude would be most beneficial, without impinging on the patient’s basic personal rights.
1. Charles C, Gafni A, Whelan T. Decision-making in the physician-patient encounter: revisiting the shared treatment decision-making model. Soc Sci Med. 1999;49(5):651-661.
2. Barry MJ, Edgman-Levitan S. Shared decision making—pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781.
3. Sartorius RE. Paternalism. Minneapolis, MN: University of Minnesota Press; 1983.
4. Dong R. Paternalism in medical decision making. Duke University. https://dukespace.lib.duke.edu/dspace/bitstream/handle/10161/3958/Dong_Thesis.pdf. Published 2011. Accessed April 17, 2019.
5. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
6. Beauchamp TL, Childress JF. Principles of biomedical ethics. 5th ed. New York, NY: Oxford University Press; 2001:57-112.
7. Weindling P. The origins of informed consent: the International Scientific Commission on Medical War Crimes, and the Nuremberg Code. Bull Hist Med. 2001;75(1):37-71.
Two patients were admitted to our unit at the same time: Mr. P, age 27, an architect with unspecified personality disorder, and Mr. D, age 62, a bank manager who has had bipolar disorder for 40 years and was experiencing a moderate depressive episode. Mr. P’s discomfort with the treatment team informing him of his treatment plan was evident, and he discussed at length his terms and stipulations for management. Mr. D, on the other hand, was loath to shoulder the burden of any decision-making, even in minor matters such as what time he should take his daily walk.
Patient autonomy is a central factor in the present-day doctor–patient equation. In psychiatry, this is sometimes further complicated by a patient’s impaired judgment and lowered decision-making capacity (DMC). In our clinical practice, we often notice that younger patients (ie, millennials) prefer to have autonomy rather than being given instructions, which they may find patronizing, whereas the older generation relies more on the doctor for decision-making.
What the decision-making process entails
The decision-making process involves 3 steps:
- information gathering
- deliberation
- implementation.
Decision-making preferences fall on a spectrum that ranges from paternalism at one end to autonomy on the other, with many intervening components, characterized by varying amounts of responsibility shared between doctor and patient.1 This typically comes into play when there is more than one treatment option with similar outcomes.2 Paternalism is defined as an action performed with the intent of promoting another’s good but occurring against the other’s will, or without consent.3 Here, the patient is not privy to the deliberation process, and no explanations are provided.1
Two other decision-making constructs are shared decision-making (SDM) and informed decision-making (IDM). In SDM, the deliberation process involves participation of both patient and doctor, with active discussion and a final decision after both parties reach an agreement. In IDM, the deliberation is conducted solely by the patient, after he or she receives all information. Shared decision-making and IDM are frequently used interchangeably, but in the latter, the doctor has no role other than to provide information.1,5
Before choosing SDM or IDM, it is necessary to assess the patient’s DMC—the ability to understand information about choices, make a judgment that respects personal values, understand potential outcomes, and freely communicate his or her wishes.6
Benefits and risks
The progression from paternalism to autonomy began in the mid-20th century as a consequence of the Nuremberg Trials, from which the concept of “informed consent” first came into existence.7 The Indian value system has always regarded the medical profession and its practitioners with high esteem, as evidenced by the Sanskrit quote “Vaidyo Narayano Harihi,” which translates to “The doctor is God.” A significant chunk of the Indian population still considers the doctor’s word to be law, and they hand over health-related decisions to medical professionals. Here, the expectation of a paternalistic attitude is decidedly unequivocal.
Continue to: Of course...
Of course, there are pros and cons to every approach. Making patients’ independence a priority is the highest virtue of autonomy, but in such cases a patient may have difficulty comprehending medical consequences, and therefore may miss out on the benefits of a sound professional perspective. Paternalism may be superior medically, but the doctor may not be aware of all patient-specific factors, and it would not be prudent to make a decision for a patient without being privy to the entire picture.
The 21st century has witnessed a change in attitudes regarding medical care. With an increasing interest in patient autonomy, it is time for us to adopt these changes and move towards the patient-centred end of the spectrum. However, this should occur only after the patient improves enough symptomatically to regain DMC; autonomy is unlikely to be appropriate for patients with serious mental illness. Ideally, SDM includes the best of both worlds, and results in optimal outcomes. However, when SDM breaks down, a selective, soft paternalistic attitude would be most beneficial, without impinging on the patient’s basic personal rights.
Two patients were admitted to our unit at the same time: Mr. P, age 27, an architect with unspecified personality disorder, and Mr. D, age 62, a bank manager who has had bipolar disorder for 40 years and was experiencing a moderate depressive episode. Mr. P’s discomfort with the treatment team informing him of his treatment plan was evident, and he discussed at length his terms and stipulations for management. Mr. D, on the other hand, was loath to shoulder the burden of any decision-making, even in minor matters such as what time he should take his daily walk.
Patient autonomy is a central factor in the present-day doctor–patient equation. In psychiatry, this is sometimes further complicated by a patient’s impaired judgment and lowered decision-making capacity (DMC). In our clinical practice, we often notice that younger patients (ie, millennials) prefer to have autonomy rather than being given instructions, which they may find patronizing, whereas the older generation relies more on the doctor for decision-making.
What the decision-making process entails
The decision-making process involves 3 steps:
- information gathering
- deliberation
- implementation.
Decision-making preferences fall on a spectrum that ranges from paternalism at one end to autonomy on the other, with many intervening components, characterized by varying amounts of responsibility shared between doctor and patient.1 This typically comes into play when there is more than one treatment option with similar outcomes.2 Paternalism is defined as an action performed with the intent of promoting another’s good but occurring against the other’s will, or without consent.3 Here, the patient is not privy to the deliberation process, and no explanations are provided.1
Two other decision-making constructs are shared decision-making (SDM) and informed decision-making (IDM). In SDM, the deliberation process involves participation of both patient and doctor, with active discussion and a final decision after both parties reach an agreement. In IDM, the deliberation is conducted solely by the patient, after he or she receives all information. Shared decision-making and IDM are frequently used interchangeably, but in the latter, the doctor has no role other than to provide information.1,5
Before choosing SDM or IDM, it is necessary to assess the patient’s DMC—the ability to understand information about choices, make a judgment that respects personal values, understand potential outcomes, and freely communicate his or her wishes.6
Benefits and risks
The progression from paternalism to autonomy began in the mid-20th century as a consequence of the Nuremberg Trials, from which the concept of “informed consent” first came into existence.7 The Indian value system has always regarded the medical profession and its practitioners with high esteem, as evidenced by the Sanskrit quote “Vaidyo Narayano Harihi,” which translates to “The doctor is God.” A significant chunk of the Indian population still considers the doctor’s word to be law, and they hand over health-related decisions to medical professionals. Here, the expectation of a paternalistic attitude is decidedly unequivocal.
Continue to: Of course...
Of course, there are pros and cons to every approach. Making patients’ independence a priority is the highest virtue of autonomy, but in such cases a patient may have difficulty comprehending medical consequences, and therefore may miss out on the benefits of a sound professional perspective. Paternalism may be superior medically, but the doctor may not be aware of all patient-specific factors, and it would not be prudent to make a decision for a patient without being privy to the entire picture.
The 21st century has witnessed a change in attitudes regarding medical care. With an increasing interest in patient autonomy, it is time for us to adopt these changes and move towards the patient-centred end of the spectrum. However, this should occur only after the patient improves enough symptomatically to regain DMC; autonomy is unlikely to be appropriate for patients with serious mental illness. Ideally, SDM includes the best of both worlds, and results in optimal outcomes. However, when SDM breaks down, a selective, soft paternalistic attitude would be most beneficial, without impinging on the patient’s basic personal rights.
1. Charles C, Gafni A, Whelan T. Decision-making in the physician-patient encounter: revisiting the shared treatment decision-making model. Soc Sci Med. 1999;49(5):651-661.
2. Barry MJ, Edgman-Levitan S. Shared decision making—pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781.
3. Sartorius RE. Paternalism. Minneapolis, MN: University of Minnesota Press; 1983.
4. Dong R. Paternalism in medical decision making. Duke University. https://dukespace.lib.duke.edu/dspace/bitstream/handle/10161/3958/Dong_Thesis.pdf. Published 2011. Accessed April 17, 2019.
5. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
6. Beauchamp TL, Childress JF. Principles of biomedical ethics. 5th ed. New York, NY: Oxford University Press; 2001:57-112.
7. Weindling P. The origins of informed consent: the International Scientific Commission on Medical War Crimes, and the Nuremberg Code. Bull Hist Med. 2001;75(1):37-71.
1. Charles C, Gafni A, Whelan T. Decision-making in the physician-patient encounter: revisiting the shared treatment decision-making model. Soc Sci Med. 1999;49(5):651-661.
2. Barry MJ, Edgman-Levitan S. Shared decision making—pinnacle of patient-centered care. N Engl J Med. 2012;366(9):780-781.
3. Sartorius RE. Paternalism. Minneapolis, MN: University of Minnesota Press; 1983.
4. Dong R. Paternalism in medical decision making. Duke University. https://dukespace.lib.duke.edu/dspace/bitstream/handle/10161/3958/Dong_Thesis.pdf. Published 2011. Accessed April 17, 2019.
5. Charles C, Gafni A, Whelan T. Shared decision-making in the medical encounter: what does it mean? (or it takes at least two to tango). Soc Sci Med. 1997;44(5):681-692.
6. Beauchamp TL, Childress JF. Principles of biomedical ethics. 5th ed. New York, NY: Oxford University Press; 2001:57-112.
7. Weindling P. The origins of informed consent: the International Scientific Commission on Medical War Crimes, and the Nuremberg Code. Bull Hist Med. 2001;75(1):37-71.

Your patient’s brain is different at every visit
Unlike other organs in the human body, the brain is constantly changing. The main driver for this ongoing re-engineering across various neural circuits is “experiential neuroplasticity,” which creates billions of new synapses and dendrite spines as well as new connections. And as the brain reinvents itself from day to day, the mind evolves as well.
The neurobiologic re-sculpting of the brain’s complex innards continuously encodes memories of what we learn and experience during waking hours, including all that we see, hear, feel, think, contemplate, plan, and decide. However, in addition to the ongoing intrinsic neuroplasticity that records life’s experiences within neural circuits, there are many extrinsic factors that can further modify the brain and the “psyche” it generates via electrical, neurochemical, and physiological mechanisms. That’s why every patient a psychiatrist sees at follow-up visits will have a brain that will be different from the previous encounter.
Consider the following factors that can modify a patient’s brain (for better or worse) between sessions:
- Psychotherapy that the patient received at the last session will biologically modify his or her brain. Creating new insights and understanding of one’s behavior and “connecting the dots” of the past and present emotions and reactions are all associated with neuroplastic changes within the brain.
- Mood or psychotic episodes. Depressive, manic, or psychotic episodes are associated with neuroinflammation, oxidative stress, and apoptotic effects, which can disrupt the brain’s cytoarchitecture. That’s why psychiatrists must inquire about such episodes between visits and document the possible effects on the patient’s mental status.
- Psychotropic medications all bind to one or more brain receptors to exert therapeutic or adverse effects, both of which are associated with changes in neurotransmitter pathways. A key component of every follow-up visit is to gauge the risks and benefits of the pharmacotherapy prescribed at the prior visit.
- Nonpsychiatric prescription medications are often associated with iatrogenic effects on the brain apart from their intended target organs. These iatrogenic effects include anxiety, depression, mania, psychosis, and cognitive changes. That’s why during each visit, the physician or nurse practitioner must review all prescription medications and consider their potential effects on the patient’s mental status.
- Over-the-counter drugs and supplements may exert neurologic effects via histaminergic, muscarinic, glutamatergic, adrenergic, or serotonergic effects—all of which can alter brain chemistry and contribute to mental status changes. They can also inhibit or induce cytochrome enzymes and induce adverse effects or loss of efficacy of the primary psychotropic medication the patient takes.
- Medical illness, even as simple as an upper respiratory viral infection, can alter brain function due to illness-induced physiological aberrations, including pain and peripheral inflammation, with neurologic consequences. Common metabolic disorders such as diabetes, hyperlipidemia, and hypertension can exert mental status changes.
- Alcohol and drugs of abuse alter brain structure and function and can induce psychological and cognitive changes. Inquiring about the amount and frequency of alcohol and recreational drug use must be done in detail at every visit.
- Stressful events. It is almost impossible for a psychiatric patient not to encounter stressful life events between visits. Coping with any mental disorder can be quite stressful and challenging due to its social, vocational, or personal consequences. Stress increases cortisol, which is associated with deleterious inflammatory effects on the brain. Persistent stress can lead to hippocampal atrophy because of the abundance of glucocorticoid receptors in the hippocampus. Inquiry about stressors must be part of every psychiatric follow-up visit. Multiple psychological, physiological, and behavioral effects are well known to be generated by stress, especially in individuals already impaired by mental illness.
- Diet. What a patient eats (or avoids eating) can affect the brain. High-fat diets can be inflammatory, while a diet rich in fruits, vegetables, and nuts can be neuroprotective. The microbiota and the enteric brain—both in the gastrointestinal tract—have been reported to influence mood and behavior. (For more on this, see “Gut microbiota and its implications for psychiatry: A review of 3 studies” on page 40 and “It takes guts to be mentally ill: Microbiota and psychopathology,” From the Editor,
Current Psychiatry , September 2018, p. 4-6.) - Obesity is associated with brain atrophy as well as depression. Weight should be assessed at every visit and coupled with counseling about diet and exercise.
- Exercise, or the lack of it, can alter the brain in good or bad ways. Many studies have shown that regular exercise can induce hippocampal neurogenesis and sharpen memory and cognition. On the other hand, a sedentary lifestyle can be detrimental to the heart, bones, and brain, with an elevation in cerebrovascular and cardiovascular risks, both of which can progressively alter brain structure and function.
- Concussion, contusions, and traumatic brain injury obviously can activate the microglia and trigger neurologic sequelae and mental repercussions. At every visit, patients should be asked if they have experienced a mild or severe head injury, whether it is accidental or sports-related.
- Dehydration, especially on the day of the visit, can alter mental status in subtle ways. Cerebral ventricular volume has been shown to change with dehydration. Asking a patient about daily fluid intake should be a standard question, especially for older patients, who may experience hypotension and mental status changes due to hypovolemia.
- Sleep, whether too much or too little, is associated with brain effects and can impact cognition and behavior. Asking patients about sleep is important because it can affect the brain, and also can be a symptom of unresolved psychiatric disorders. Chronic sleep disorders are associated with neuroinflammation.
- Menstrual cycle. Various neurotransmitters fluctuate during a woman’s menstrual cycle. Her cognition becomes sharper around ovulation, and that may influence her mental status and perhaps the neuroplasticity of her brain.
- Pregnancy and its major hormone changes can change brain structure and function. Estrogen, progesterone, and prolactin have different structural effects on the brain that can help the future mother care for her dependent baby. Asking about missed periods and pregnancy during childbearing years can be useful during psychiatric encounters.
Continue to: In summary...
In summary, numerous variables can affect the patient’s brain between visits, influencing his or her mental status. The ever-changing brain can be challenging to assess, especially in brief 15- to 20-minute follow-up sessions that have become more common in psychiatry. Perhaps patients should help their psychiatrists or nurse practitioners by completing a checklist with all the above variables, either online on the day of their appointment or on a form in the waiting room immediately prior to the visit. This might also increase patients’ awareness of the importance of participating in monitoring themselves.
And finally, let’s not forget that the psychiatrist’s brain also changes continuously due to his or her own daily experiences, stresses, diet, lifestyle, medical illness, or medications. Thus, at every psychiatric session, the brains of both patient and psychiatrist are very different from the previous encounter!
To comment on this editorial or other topics of interest: [email protected].
Unlike other organs in the human body, the brain is constantly changing. The main driver for this ongoing re-engineering across various neural circuits is “experiential neuroplasticity,” which creates billions of new synapses and dendrite spines as well as new connections. And as the brain reinvents itself from day to day, the mind evolves as well.
The neurobiologic re-sculpting of the brain’s complex innards continuously encodes memories of what we learn and experience during waking hours, including all that we see, hear, feel, think, contemplate, plan, and decide. However, in addition to the ongoing intrinsic neuroplasticity that records life’s experiences within neural circuits, there are many extrinsic factors that can further modify the brain and the “psyche” it generates via electrical, neurochemical, and physiological mechanisms. That’s why every patient a psychiatrist sees at follow-up visits will have a brain that will be different from the previous encounter.
Consider the following factors that can modify a patient’s brain (for better or worse) between sessions:
- Psychotherapy that the patient received at the last session will biologically modify his or her brain. Creating new insights and understanding of one’s behavior and “connecting the dots” of the past and present emotions and reactions are all associated with neuroplastic changes within the brain.
- Mood or psychotic episodes. Depressive, manic, or psychotic episodes are associated with neuroinflammation, oxidative stress, and apoptotic effects, which can disrupt the brain’s cytoarchitecture. That’s why psychiatrists must inquire about such episodes between visits and document the possible effects on the patient’s mental status.
- Psychotropic medications all bind to one or more brain receptors to exert therapeutic or adverse effects, both of which are associated with changes in neurotransmitter pathways. A key component of every follow-up visit is to gauge the risks and benefits of the pharmacotherapy prescribed at the prior visit.
- Nonpsychiatric prescription medications are often associated with iatrogenic effects on the brain apart from their intended target organs. These iatrogenic effects include anxiety, depression, mania, psychosis, and cognitive changes. That’s why during each visit, the physician or nurse practitioner must review all prescription medications and consider their potential effects on the patient’s mental status.
- Over-the-counter drugs and supplements may exert neurologic effects via histaminergic, muscarinic, glutamatergic, adrenergic, or serotonergic effects—all of which can alter brain chemistry and contribute to mental status changes. They can also inhibit or induce cytochrome enzymes and induce adverse effects or loss of efficacy of the primary psychotropic medication the patient takes.
- Medical illness, even as simple as an upper respiratory viral infection, can alter brain function due to illness-induced physiological aberrations, including pain and peripheral inflammation, with neurologic consequences. Common metabolic disorders such as diabetes, hyperlipidemia, and hypertension can exert mental status changes.
- Alcohol and drugs of abuse alter brain structure and function and can induce psychological and cognitive changes. Inquiring about the amount and frequency of alcohol and recreational drug use must be done in detail at every visit.
- Stressful events. It is almost impossible for a psychiatric patient not to encounter stressful life events between visits. Coping with any mental disorder can be quite stressful and challenging due to its social, vocational, or personal consequences. Stress increases cortisol, which is associated with deleterious inflammatory effects on the brain. Persistent stress can lead to hippocampal atrophy because of the abundance of glucocorticoid receptors in the hippocampus. Inquiry about stressors must be part of every psychiatric follow-up visit. Multiple psychological, physiological, and behavioral effects are well known to be generated by stress, especially in individuals already impaired by mental illness.
- Diet. What a patient eats (or avoids eating) can affect the brain. High-fat diets can be inflammatory, while a diet rich in fruits, vegetables, and nuts can be neuroprotective. The microbiota and the enteric brain—both in the gastrointestinal tract—have been reported to influence mood and behavior. (For more on this, see “Gut microbiota and its implications for psychiatry: A review of 3 studies” on page 40 and “It takes guts to be mentally ill: Microbiota and psychopathology,” From the Editor,
Current Psychiatry , September 2018, p. 4-6.) - Obesity is associated with brain atrophy as well as depression. Weight should be assessed at every visit and coupled with counseling about diet and exercise.
- Exercise, or the lack of it, can alter the brain in good or bad ways. Many studies have shown that regular exercise can induce hippocampal neurogenesis and sharpen memory and cognition. On the other hand, a sedentary lifestyle can be detrimental to the heart, bones, and brain, with an elevation in cerebrovascular and cardiovascular risks, both of which can progressively alter brain structure and function.
- Concussion, contusions, and traumatic brain injury obviously can activate the microglia and trigger neurologic sequelae and mental repercussions. At every visit, patients should be asked if they have experienced a mild or severe head injury, whether it is accidental or sports-related.
- Dehydration, especially on the day of the visit, can alter mental status in subtle ways. Cerebral ventricular volume has been shown to change with dehydration. Asking a patient about daily fluid intake should be a standard question, especially for older patients, who may experience hypotension and mental status changes due to hypovolemia.
- Sleep, whether too much or too little, is associated with brain effects and can impact cognition and behavior. Asking patients about sleep is important because it can affect the brain, and also can be a symptom of unresolved psychiatric disorders. Chronic sleep disorders are associated with neuroinflammation.
- Menstrual cycle. Various neurotransmitters fluctuate during a woman’s menstrual cycle. Her cognition becomes sharper around ovulation, and that may influence her mental status and perhaps the neuroplasticity of her brain.
- Pregnancy and its major hormone changes can change brain structure and function. Estrogen, progesterone, and prolactin have different structural effects on the brain that can help the future mother care for her dependent baby. Asking about missed periods and pregnancy during childbearing years can be useful during psychiatric encounters.
Continue to: In summary...
In summary, numerous variables can affect the patient’s brain between visits, influencing his or her mental status. The ever-changing brain can be challenging to assess, especially in brief 15- to 20-minute follow-up sessions that have become more common in psychiatry. Perhaps patients should help their psychiatrists or nurse practitioners by completing a checklist with all the above variables, either online on the day of their appointment or on a form in the waiting room immediately prior to the visit. This might also increase patients’ awareness of the importance of participating in monitoring themselves.
And finally, let’s not forget that the psychiatrist’s brain also changes continuously due to his or her own daily experiences, stresses, diet, lifestyle, medical illness, or medications. Thus, at every psychiatric session, the brains of both patient and psychiatrist are very different from the previous encounter!
To comment on this editorial or other topics of interest: [email protected].
Unlike other organs in the human body, the brain is constantly changing. The main driver for this ongoing re-engineering across various neural circuits is “experiential neuroplasticity,” which creates billions of new synapses and dendrite spines as well as new connections. And as the brain reinvents itself from day to day, the mind evolves as well.
The neurobiologic re-sculpting of the brain’s complex innards continuously encodes memories of what we learn and experience during waking hours, including all that we see, hear, feel, think, contemplate, plan, and decide. However, in addition to the ongoing intrinsic neuroplasticity that records life’s experiences within neural circuits, there are many extrinsic factors that can further modify the brain and the “psyche” it generates via electrical, neurochemical, and physiological mechanisms. That’s why every patient a psychiatrist sees at follow-up visits will have a brain that will be different from the previous encounter.
Consider the following factors that can modify a patient’s brain (for better or worse) between sessions:
- Psychotherapy that the patient received at the last session will biologically modify his or her brain. Creating new insights and understanding of one’s behavior and “connecting the dots” of the past and present emotions and reactions are all associated with neuroplastic changes within the brain.
- Mood or psychotic episodes. Depressive, manic, or psychotic episodes are associated with neuroinflammation, oxidative stress, and apoptotic effects, which can disrupt the brain’s cytoarchitecture. That’s why psychiatrists must inquire about such episodes between visits and document the possible effects on the patient’s mental status.
- Psychotropic medications all bind to one or more brain receptors to exert therapeutic or adverse effects, both of which are associated with changes in neurotransmitter pathways. A key component of every follow-up visit is to gauge the risks and benefits of the pharmacotherapy prescribed at the prior visit.
- Nonpsychiatric prescription medications are often associated with iatrogenic effects on the brain apart from their intended target organs. These iatrogenic effects include anxiety, depression, mania, psychosis, and cognitive changes. That’s why during each visit, the physician or nurse practitioner must review all prescription medications and consider their potential effects on the patient’s mental status.
- Over-the-counter drugs and supplements may exert neurologic effects via histaminergic, muscarinic, glutamatergic, adrenergic, or serotonergic effects—all of which can alter brain chemistry and contribute to mental status changes. They can also inhibit or induce cytochrome enzymes and induce adverse effects or loss of efficacy of the primary psychotropic medication the patient takes.
- Medical illness, even as simple as an upper respiratory viral infection, can alter brain function due to illness-induced physiological aberrations, including pain and peripheral inflammation, with neurologic consequences. Common metabolic disorders such as diabetes, hyperlipidemia, and hypertension can exert mental status changes.
- Alcohol and drugs of abuse alter brain structure and function and can induce psychological and cognitive changes. Inquiring about the amount and frequency of alcohol and recreational drug use must be done in detail at every visit.
- Stressful events. It is almost impossible for a psychiatric patient not to encounter stressful life events between visits. Coping with any mental disorder can be quite stressful and challenging due to its social, vocational, or personal consequences. Stress increases cortisol, which is associated with deleterious inflammatory effects on the brain. Persistent stress can lead to hippocampal atrophy because of the abundance of glucocorticoid receptors in the hippocampus. Inquiry about stressors must be part of every psychiatric follow-up visit. Multiple psychological, physiological, and behavioral effects are well known to be generated by stress, especially in individuals already impaired by mental illness.
- Diet. What a patient eats (or avoids eating) can affect the brain. High-fat diets can be inflammatory, while a diet rich in fruits, vegetables, and nuts can be neuroprotective. The microbiota and the enteric brain—both in the gastrointestinal tract—have been reported to influence mood and behavior. (For more on this, see “Gut microbiota and its implications for psychiatry: A review of 3 studies” on page 40 and “It takes guts to be mentally ill: Microbiota and psychopathology,” From the Editor,
Current Psychiatry , September 2018, p. 4-6.) - Obesity is associated with brain atrophy as well as depression. Weight should be assessed at every visit and coupled with counseling about diet and exercise.
- Exercise, or the lack of it, can alter the brain in good or bad ways. Many studies have shown that regular exercise can induce hippocampal neurogenesis and sharpen memory and cognition. On the other hand, a sedentary lifestyle can be detrimental to the heart, bones, and brain, with an elevation in cerebrovascular and cardiovascular risks, both of which can progressively alter brain structure and function.
- Concussion, contusions, and traumatic brain injury obviously can activate the microglia and trigger neurologic sequelae and mental repercussions. At every visit, patients should be asked if they have experienced a mild or severe head injury, whether it is accidental or sports-related.
- Dehydration, especially on the day of the visit, can alter mental status in subtle ways. Cerebral ventricular volume has been shown to change with dehydration. Asking a patient about daily fluid intake should be a standard question, especially for older patients, who may experience hypotension and mental status changes due to hypovolemia.
- Sleep, whether too much or too little, is associated with brain effects and can impact cognition and behavior. Asking patients about sleep is important because it can affect the brain, and also can be a symptom of unresolved psychiatric disorders. Chronic sleep disorders are associated with neuroinflammation.
- Menstrual cycle. Various neurotransmitters fluctuate during a woman’s menstrual cycle. Her cognition becomes sharper around ovulation, and that may influence her mental status and perhaps the neuroplasticity of her brain.
- Pregnancy and its major hormone changes can change brain structure and function. Estrogen, progesterone, and prolactin have different structural effects on the brain that can help the future mother care for her dependent baby. Asking about missed periods and pregnancy during childbearing years can be useful during psychiatric encounters.
Continue to: In summary...
In summary, numerous variables can affect the patient’s brain between visits, influencing his or her mental status. The ever-changing brain can be challenging to assess, especially in brief 15- to 20-minute follow-up sessions that have become more common in psychiatry. Perhaps patients should help their psychiatrists or nurse practitioners by completing a checklist with all the above variables, either online on the day of their appointment or on a form in the waiting room immediately prior to the visit. This might also increase patients’ awareness of the importance of participating in monitoring themselves.
And finally, let’s not forget that the psychiatrist’s brain also changes continuously due to his or her own daily experiences, stresses, diet, lifestyle, medical illness, or medications. Thus, at every psychiatric session, the brains of both patient and psychiatrist are very different from the previous encounter!
To comment on this editorial or other topics of interest: [email protected].

When a disaster disrupts access to psychiatric medications
In recent decades, disasters such as storms, earthquakes, and terrorism have occurred with increasing frequency. Disaster planners assess the needs and vulnerabilities of communities in order to save lives during these events. They focus on providing electricity and clean water and addressing other public health measures. What is not adequately planned for, in our opinion, is a disruption in the pharmaceutical supply chain, particularly supplies of psychiatric medications.
There is now a rich literature on disaster psychiatry.1-4 However, there’s been a lack of information about disrupted access to psychiatric medications. Disruptive behavior after Hurricanes Katrina, Maria, Rita, and others were a consequence of a lack of medications or difficulty obtaining medications following these disasters.5-7
This article discusses the pharmaceutical supply chain, the lack of stockpiles of psychiatric medications, and how clinicians can prepare themselves and their patients in the event a disaster strikes.
Supply chains
Each day, nearly 12 million prescriptions are filled in the United States, with gratifying swiftness, efficiency, and accuracy. Our confidence in the nation’s pharmaceutical dependability, however, rests squarely upon the strength and resilience of vast, interconnected supply chains that involve the myriad aspects of private industry—from manufacturing to shipping and transport to last-mile delivery from pharmacy to patient. The failure of any one of the links in any of these supply chains can result in the instant unavailability of critical medications.
Supply chains are fundamental to modern life and must fluctuate to address disruptions; however, common supplemental and gap-filling functions that address minor changes may be insufficient to mitigate supply chain disruptions during a disaster. While supply chains can be extremely complex and can vary significantly from product to product, all supply chains can generally be presented through the components found in Table 1.

All components within a supply chain, such as the transportation mechanisms between nodes, facilities, people, and communication networks, can affect a supply chain’s resilience. For a supply chain to be resilient, key players—in this case, psychiatrists and associated medical professionals—must be acutely aware of the supply chain elements within their vision and reasonable anticipation: known nodes and links, their potential vulnerabilities, and ways and means to mitigate expected disruption.
Recent natural disasters, especially Hurricanes Katrina, Sandy, Harvey, and Maria, have given both government emergency management (at all levels) and clinicians the opportunity to understand the full effects of broken pharmaceutical supply chains under varying and extreme circumstances.
Continue to: As stated in a...
As stated in a recent Department of Homeland Security health care supply chain report, “Pharmaceuticals are one of the top concerns for healthcare providers in terms of supply chain disruptions. They are prone to various supply chain problems, including limited sources, lack of alternatives, time sensitivity, frequent shortages, and minimal on-site inventories. Each stakeholder along the pharmaceutical supply chain faces challenges with understanding and planning for possible disruptions emerging further up the chain. The rapidly expanding use of just-in-time inventory practices by distributors and healthcare customers is creating an increasingly fragile supply-demand balance that could be highly disrupted by a major event either further up the supply chain or within the last mile of delivery.”8,9
No national stockpiles of psychiatric medications
The CDC maintains stockpiles of emergency medications, but these supplies focus on medications to combat infection. In these caches, there are no psychiatric medications other than diazepam, which is stocked for its ability to combat the effects of nerve agents.
In major storm-related events, such as Hurricane Katrina in New Orleans in 2005, the disruptions in all supply chains included psychiatric medications. In the aftermath, many people with addictions and/or severe mental illnesses did not receive either their drugs of choice and/or antimanic and antipsychotic medications. As a result, disruptive behavior became common, especially in the shelters.5-7
During a widespread public emergency, police and emergency services are often stretched very thin. In calmer times, police or emergency services may take a person with disruptive and aggressive behavior to a local emergency department. However, in times of chaos, such as during Hurricane Katrina, patients with aggressive or disruptive behaviors were forcefully incapacitated (ie, “tased”) or shot.
Withdrawal from antidepressants, opiates, alcohol, and benzodiazepines has its own risks. Withdrawal from alcohol or benzodiazepines can be life-threatening. Therefore, it is critically important that clinicians think about how to ensure their patients have a supply of their medications. This may imply stockpiling on a personal or community basis.
Continue to: What to consider before disruption
What to consider before disruption
Many psychiatrists, especially those who have not practiced through a local disaster, may have never contemplated how they would support their patients during a disruptive event. Psychiatrists should carefully consider the questions outlined in Table 2 before a disaster strikes.

Medication-specific issues
During major disasters, patients may not have access to their medications, or the medications may not be able to be fed into the health care system for dispersion. Other issues include closed pharmacies, expired medications as a result of limited refrigeration service, inability to deliver medications to an affected area, and the inability of manufacturing plants to produce medications. For example, after Hurricane Maria, sterile water was in short supply.
After a major disaster, clinicians often leave their communities because they cannot support themselves or their practices. Thus, clinicians may not be available to prescribe needed medications. Available clinicians—often primary care physicians—may not be aware of a patient’s medication history, or they may be uncomfortable prescribing psychiatric medications, especially antipsychotics.
Abrupt discontinuation of psychiatric medications can have severe consequences. Patients may experience withdrawal symptoms, worsening psychiatric symptoms, new-onset psychiatric symptoms, thoughts of harm to self or others, psychosis, or cravings. These issues may be particularly problematic for patients receiving antidepressants, antipsychotics, benzodiazepines, or medication-assisted treatment for opioid use disorder.
Antidepressants. Patients experiencing antidepressant withdrawal, particularly withdrawal from selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors, may exhibit severe symptoms. In addition to the potential recurrence of depressive or anxiety symptoms and suicidal thoughts, patients may experience irritability, insomnia, headache, nausea, and electric shock–like sensations. Prescribing an antidepressant with a longer half-life could potentially prevent an abrupt withdrawal in the event a disaster occurs.
Continue to: Antipsychotics
Antipsychotics. Rapid or abrupt withdrawal of antipsychotics could lead to an increase in psychosis, paranoia, hallucinations, or delusions. Withdrawal of antipsychotics could also lead to agitation, restlessness, insomnia, paresthesia, and anxiety. If a known disaster is likely to occur, such as in the case of a hurricane forecast, clinicians may consider switching a patient a long-acting injectable antipsychotic to minimize the risk of withdrawal and symptom exacerbation.
Benzodiazepines. The abrupt withdrawal of benzodiazepines could result in symptoms that include rebound anxiety, insomnia, restlessness, muscle tension, irritability, nausea, malaise, blurred vision, diaphoresis, nightmares, and seizures. Additionally, many people use benzodiazepines recreationally, and their illicit supply may run out during disasters, which could lead to untreated withdrawal and violence in the community.
Clinicians need to develop action plans for any patients who are receiving scheduled benzodiazepine dosing in order to prevent abrupt withdrawal if a disaster occurs.
Opioids. Opioid cravings and withdrawal are also a major concern during times of disrupted supply. Patients receiving chronic opioid therapy may not be able to receive their maintenance medications, which could lead to withdrawal. Additionally, patients taking illicit opioids may also be at risk of withdrawal.
Early symptoms of opioid withdrawal include watery eyes, runny nose, sweating, anxiety and irritability, poor sleep, and muscle pain. Later symptoms could include cramping, diarrhea, vomiting, increased heart rate and blood pressure, restlessness, shakiness, chills, sweating, and dilated pupils.
Continue to: Contingency planning...
Contingency planning should be a part of the treatment plan for every patient receiving chronic opioid therapy who lives in an area where major disasters are likely to occur.
Medication-assisted treatment for opioid use disorder. Patients receiving treatment for opioid use disorder may be prescribed the partial opioid agonist buprenorphine, either by itself or in combination with the opioid antagonist naloxone. This could be particularly problematic to continue in a major disaster due to the lack of credentialed clinicians, limited supplies, and patients only receiving small amounts of the medication at a time due to the risk of diversion.
Symptoms of buprenorphine withdrawal are similar to those associated with opioid withdrawal. Developing a thoughtful plan in case of a disaster should be part of all buprenorphine prescribing. Patients should be aware of withdrawal symptoms and what to do if they run out of medication.
Additionally, emergency clinicians should have access to buprenorphine and buprenorphine/naloxone and the ability to prescribe them in disaster situations. As with all aspects of disaster response, it is wise to work out issues in advance.
Help your patients get ready
Advise your patients to prepare emergency kits that contain their psychiatric medications that they could quickly grab and go if needed. Because there may be times when it is not possible to gather all necessary medications, having even a small supply ready to go at a moment’s notice would be beneficial. If permitted, patients should also consider keeping medications in multiple locations, including at their place of work, home, or a family member’s home.
Continue to: Additionally, instruct patients...
Additionally, instruct patients to always carry a list of all medications they currently take. Ideally, this list should also include past medications and responses, allergies, and provider contact information. During a disaster, this information could prove vital to an emergency clinician. At a minimum, verify that your patient maintains a list of current medications.
Clinicians should develop emergency plans for all psychiatric medications they prescribe. Document and discuss with your patients any necessary considerations for patients who take medications that require more intensive monitoring, such as lithium or clozapine.
Clinicians, patients, emergency responders, and health care workers need to work together to prepare for major disasters to avoid withdrawal and other consequences of disrupted access to psychiatric medications.
Bottom Line
Consult with local public health officials to determine and develop contingency plans to provide psychiatric medications to your patients in the event of a disaster. Discuss treatment plans and contingency planning with patients, particularly those in regions most likely to be affected by a disaster. Instruct patients to refill medications prior to a foreseeable disaster and to maintain a personal stockpile of medications when appropriate.
Related Resources
- Ochi S, Hodgson S, Landeg O, et al. Disaster-driven evacuation and medication loss: A systematic literature review. PLoS Curr. 2014;6.b. doi: 10.1371/currents.dis.fa417630b566a0c7dfdbf945910edd96.
- Pate JE, Fisher JW. Disaster ethics: What are the ground rules? Current Psychiatry. 2007;6(6):69-78.
Drug Brand Names
Buprenorphine • Subutex
Buprenorphine/naloxone • Suboxone
Clozapine • Clozaril
Diazepam • Valium
Lithium • Eskalith, Lithobid
1. National Institute of Mental Health. Mental health and mass violence: evidence based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. https://cpa.ca/docs/File/Emergencies/massviolence.pdf. Published 2002. Accessed March 11, 2019.
2. Ritchie EC, Friedman M, Watson P. Interventions following mass violence and disasters: strategies for mental health practice. New York, NY: Guilford Press; 2006.
3. Ritchie EC, O’Brien K, Grant M, et al. Disaster psychiatry. In: Stern TA, Rosenbaum JF, Fava M, et al. The Massachusetts General Hospital textbook of comprehensive clinical psychiatry, 2nd edition. Philadelphia, PA: Mosby/Elsevier; 2016:968-974.
4. Ritchie EC, Hamilton S. Early interventions and risk assessment following disaster. Psychiatric Annals. 2004;34(8):605-610.
5. Kessler RC, Galea S, Gruber MJ, et al. Trends in mental illness and suicidality after Hurricane Katrina. Mol Psychiatry. 2008;13(4):374-384.
6. Weisler RH, Barbee JG IV, Townsend MH. Mental health and recovery in the Gulf Coast after Hurricanes Katrina and Rita. JAMA. 2006;296(5):585-588.
7. Galea S, Brewin CR, Gruber M, et al. Exposure to hurricane-related stressors and mental illness after Hurricane Katrina. Arch Gen Psychiatry. 2007;64(12).1427-1434.
8. Federal Emergency Management Agency. Supply Chain Resilience Guide Department of Homeland Security. https://www.fema.gov/media-library-data/1544795397837-767851ba177c7097bf8672aadf8a93c9/NE_DRAFT_Supply_Chain_Resilience.pdf. Published December 17, 2018. Accessed January 2, 2019.
9. Durkin J, Telab M, Fitzmaurice P, et al. Only as strong as its weakest link: resilience of the healthcare supply chain in New York. https://www.hstoday.us/subject-matter-areas/emergency-preparedness/only-as-strong-as-its-weakest-link-the-resilience-of-the-healthcare-supply-chain-in-new-york/. Published October 26, 2018. Accessed February 14, 2019.
In recent decades, disasters such as storms, earthquakes, and terrorism have occurred with increasing frequency. Disaster planners assess the needs and vulnerabilities of communities in order to save lives during these events. They focus on providing electricity and clean water and addressing other public health measures. What is not adequately planned for, in our opinion, is a disruption in the pharmaceutical supply chain, particularly supplies of psychiatric medications.
There is now a rich literature on disaster psychiatry.1-4 However, there’s been a lack of information about disrupted access to psychiatric medications. Disruptive behavior after Hurricanes Katrina, Maria, Rita, and others were a consequence of a lack of medications or difficulty obtaining medications following these disasters.5-7
This article discusses the pharmaceutical supply chain, the lack of stockpiles of psychiatric medications, and how clinicians can prepare themselves and their patients in the event a disaster strikes.
Supply chains
Each day, nearly 12 million prescriptions are filled in the United States, with gratifying swiftness, efficiency, and accuracy. Our confidence in the nation’s pharmaceutical dependability, however, rests squarely upon the strength and resilience of vast, interconnected supply chains that involve the myriad aspects of private industry—from manufacturing to shipping and transport to last-mile delivery from pharmacy to patient. The failure of any one of the links in any of these supply chains can result in the instant unavailability of critical medications.
Supply chains are fundamental to modern life and must fluctuate to address disruptions; however, common supplemental and gap-filling functions that address minor changes may be insufficient to mitigate supply chain disruptions during a disaster. While supply chains can be extremely complex and can vary significantly from product to product, all supply chains can generally be presented through the components found in Table 1.
All components within a supply chain, such as the transportation mechanisms between nodes, facilities, people, and communication networks, can affect a supply chain’s resilience. For a supply chain to be resilient, key players—in this case, psychiatrists and associated medical professionals—must be acutely aware of the supply chain elements within their vision and reasonable anticipation: known nodes and links, their potential vulnerabilities, and ways and means to mitigate expected disruption.
Recent natural disasters, especially Hurricanes Katrina, Sandy, Harvey, and Maria, have given both government emergency management (at all levels) and clinicians the opportunity to understand the full effects of broken pharmaceutical supply chains under varying and extreme circumstances.
Continue to: As stated in a...
As stated in a recent Department of Homeland Security health care supply chain report, “Pharmaceuticals are one of the top concerns for healthcare providers in terms of supply chain disruptions. They are prone to various supply chain problems, including limited sources, lack of alternatives, time sensitivity, frequent shortages, and minimal on-site inventories. Each stakeholder along the pharmaceutical supply chain faces challenges with understanding and planning for possible disruptions emerging further up the chain. The rapidly expanding use of just-in-time inventory practices by distributors and healthcare customers is creating an increasingly fragile supply-demand balance that could be highly disrupted by a major event either further up the supply chain or within the last mile of delivery.”8,9
No national stockpiles of psychiatric medications
The CDC maintains stockpiles of emergency medications, but these supplies focus on medications to combat infection. In these caches, there are no psychiatric medications other than diazepam, which is stocked for its ability to combat the effects of nerve agents.
In major storm-related events, such as Hurricane Katrina in New Orleans in 2005, the disruptions in all supply chains included psychiatric medications. In the aftermath, many people with addictions and/or severe mental illnesses did not receive either their drugs of choice and/or antimanic and antipsychotic medications. As a result, disruptive behavior became common, especially in the shelters.5-7
During a widespread public emergency, police and emergency services are often stretched very thin. In calmer times, police or emergency services may take a person with disruptive and aggressive behavior to a local emergency department. However, in times of chaos, such as during Hurricane Katrina, patients with aggressive or disruptive behaviors were forcefully incapacitated (ie, “tased”) or shot.
Withdrawal from antidepressants, opiates, alcohol, and benzodiazepines has its own risks. Withdrawal from alcohol or benzodiazepines can be life-threatening. Therefore, it is critically important that clinicians think about how to ensure their patients have a supply of their medications. This may imply stockpiling on a personal or community basis.
Continue to: What to consider before disruption
What to consider before disruption
Many psychiatrists, especially those who have not practiced through a local disaster, may have never contemplated how they would support their patients during a disruptive event. Psychiatrists should carefully consider the questions outlined in Table 2 before a disaster strikes.
Medication-specific issues
During major disasters, patients may not have access to their medications, or the medications may not be able to be fed into the health care system for dispersion. Other issues include closed pharmacies, expired medications as a result of limited refrigeration service, inability to deliver medications to an affected area, and the inability of manufacturing plants to produce medications. For example, after Hurricane Maria, sterile water was in short supply.
After a major disaster, clinicians often leave their communities because they cannot support themselves or their practices. Thus, clinicians may not be available to prescribe needed medications. Available clinicians—often primary care physicians—may not be aware of a patient’s medication history, or they may be uncomfortable prescribing psychiatric medications, especially antipsychotics.
Abrupt discontinuation of psychiatric medications can have severe consequences. Patients may experience withdrawal symptoms, worsening psychiatric symptoms, new-onset psychiatric symptoms, thoughts of harm to self or others, psychosis, or cravings. These issues may be particularly problematic for patients receiving antidepressants, antipsychotics, benzodiazepines, or medication-assisted treatment for opioid use disorder.
Antidepressants. Patients experiencing antidepressant withdrawal, particularly withdrawal from selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors, may exhibit severe symptoms. In addition to the potential recurrence of depressive or anxiety symptoms and suicidal thoughts, patients may experience irritability, insomnia, headache, nausea, and electric shock–like sensations. Prescribing an antidepressant with a longer half-life could potentially prevent an abrupt withdrawal in the event a disaster occurs.
Continue to: Antipsychotics
Antipsychotics. Rapid or abrupt withdrawal of antipsychotics could lead to an increase in psychosis, paranoia, hallucinations, or delusions. Withdrawal of antipsychotics could also lead to agitation, restlessness, insomnia, paresthesia, and anxiety. If a known disaster is likely to occur, such as in the case of a hurricane forecast, clinicians may consider switching a patient a long-acting injectable antipsychotic to minimize the risk of withdrawal and symptom exacerbation.
Benzodiazepines. The abrupt withdrawal of benzodiazepines could result in symptoms that include rebound anxiety, insomnia, restlessness, muscle tension, irritability, nausea, malaise, blurred vision, diaphoresis, nightmares, and seizures. Additionally, many people use benzodiazepines recreationally, and their illicit supply may run out during disasters, which could lead to untreated withdrawal and violence in the community.
Clinicians need to develop action plans for any patients who are receiving scheduled benzodiazepine dosing in order to prevent abrupt withdrawal if a disaster occurs.
Opioids. Opioid cravings and withdrawal are also a major concern during times of disrupted supply. Patients receiving chronic opioid therapy may not be able to receive their maintenance medications, which could lead to withdrawal. Additionally, patients taking illicit opioids may also be at risk of withdrawal.
Early symptoms of opioid withdrawal include watery eyes, runny nose, sweating, anxiety and irritability, poor sleep, and muscle pain. Later symptoms could include cramping, diarrhea, vomiting, increased heart rate and blood pressure, restlessness, shakiness, chills, sweating, and dilated pupils.
Continue to: Contingency planning...
Contingency planning should be a part of the treatment plan for every patient receiving chronic opioid therapy who lives in an area where major disasters are likely to occur.
Medication-assisted treatment for opioid use disorder. Patients receiving treatment for opioid use disorder may be prescribed the partial opioid agonist buprenorphine, either by itself or in combination with the opioid antagonist naloxone. This could be particularly problematic to continue in a major disaster due to the lack of credentialed clinicians, limited supplies, and patients only receiving small amounts of the medication at a time due to the risk of diversion.
Symptoms of buprenorphine withdrawal are similar to those associated with opioid withdrawal. Developing a thoughtful plan in case of a disaster should be part of all buprenorphine prescribing. Patients should be aware of withdrawal symptoms and what to do if they run out of medication.
Additionally, emergency clinicians should have access to buprenorphine and buprenorphine/naloxone and the ability to prescribe them in disaster situations. As with all aspects of disaster response, it is wise to work out issues in advance.
Help your patients get ready
Advise your patients to prepare emergency kits that contain their psychiatric medications that they could quickly grab and go if needed. Because there may be times when it is not possible to gather all necessary medications, having even a small supply ready to go at a moment’s notice would be beneficial. If permitted, patients should also consider keeping medications in multiple locations, including at their place of work, home, or a family member’s home.
Continue to: Additionally, instruct patients...
Additionally, instruct patients to always carry a list of all medications they currently take. Ideally, this list should also include past medications and responses, allergies, and provider contact information. During a disaster, this information could prove vital to an emergency clinician. At a minimum, verify that your patient maintains a list of current medications.
Clinicians should develop emergency plans for all psychiatric medications they prescribe. Document and discuss with your patients any necessary considerations for patients who take medications that require more intensive monitoring, such as lithium or clozapine.
Clinicians, patients, emergency responders, and health care workers need to work together to prepare for major disasters to avoid withdrawal and other consequences of disrupted access to psychiatric medications.
Bottom Line
Consult with local public health officials to determine and develop contingency plans to provide psychiatric medications to your patients in the event of a disaster. Discuss treatment plans and contingency planning with patients, particularly those in regions most likely to be affected by a disaster. Instruct patients to refill medications prior to a foreseeable disaster and to maintain a personal stockpile of medications when appropriate.
Related Resources
- Ochi S, Hodgson S, Landeg O, et al. Disaster-driven evacuation and medication loss: A systematic literature review. PLoS Curr. 2014;6.b. doi: 10.1371/currents.dis.fa417630b566a0c7dfdbf945910edd96.
- Pate JE, Fisher JW. Disaster ethics: What are the ground rules? Current Psychiatry. 2007;6(6):69-78.
Drug Brand Names
Buprenorphine • Subutex
Buprenorphine/naloxone • Suboxone
Clozapine • Clozaril
Diazepam • Valium
Lithium • Eskalith, Lithobid
In recent decades, disasters such as storms, earthquakes, and terrorism have occurred with increasing frequency. Disaster planners assess the needs and vulnerabilities of communities in order to save lives during these events. They focus on providing electricity and clean water and addressing other public health measures. What is not adequately planned for, in our opinion, is a disruption in the pharmaceutical supply chain, particularly supplies of psychiatric medications.
There is now a rich literature on disaster psychiatry.1-4 However, there’s been a lack of information about disrupted access to psychiatric medications. Disruptive behavior after Hurricanes Katrina, Maria, Rita, and others were a consequence of a lack of medications or difficulty obtaining medications following these disasters.5-7
This article discusses the pharmaceutical supply chain, the lack of stockpiles of psychiatric medications, and how clinicians can prepare themselves and their patients in the event a disaster strikes.
Supply chains
Each day, nearly 12 million prescriptions are filled in the United States, with gratifying swiftness, efficiency, and accuracy. Our confidence in the nation’s pharmaceutical dependability, however, rests squarely upon the strength and resilience of vast, interconnected supply chains that involve the myriad aspects of private industry—from manufacturing to shipping and transport to last-mile delivery from pharmacy to patient. The failure of any one of the links in any of these supply chains can result in the instant unavailability of critical medications.
Supply chains are fundamental to modern life and must fluctuate to address disruptions; however, common supplemental and gap-filling functions that address minor changes may be insufficient to mitigate supply chain disruptions during a disaster. While supply chains can be extremely complex and can vary significantly from product to product, all supply chains can generally be presented through the components found in Table 1.
All components within a supply chain, such as the transportation mechanisms between nodes, facilities, people, and communication networks, can affect a supply chain’s resilience. For a supply chain to be resilient, key players—in this case, psychiatrists and associated medical professionals—must be acutely aware of the supply chain elements within their vision and reasonable anticipation: known nodes and links, their potential vulnerabilities, and ways and means to mitigate expected disruption.
Recent natural disasters, especially Hurricanes Katrina, Sandy, Harvey, and Maria, have given both government emergency management (at all levels) and clinicians the opportunity to understand the full effects of broken pharmaceutical supply chains under varying and extreme circumstances.
Continue to: As stated in a...
As stated in a recent Department of Homeland Security health care supply chain report, “Pharmaceuticals are one of the top concerns for healthcare providers in terms of supply chain disruptions. They are prone to various supply chain problems, including limited sources, lack of alternatives, time sensitivity, frequent shortages, and minimal on-site inventories. Each stakeholder along the pharmaceutical supply chain faces challenges with understanding and planning for possible disruptions emerging further up the chain. The rapidly expanding use of just-in-time inventory practices by distributors and healthcare customers is creating an increasingly fragile supply-demand balance that could be highly disrupted by a major event either further up the supply chain or within the last mile of delivery.”8,9
No national stockpiles of psychiatric medications
The CDC maintains stockpiles of emergency medications, but these supplies focus on medications to combat infection. In these caches, there are no psychiatric medications other than diazepam, which is stocked for its ability to combat the effects of nerve agents.
In major storm-related events, such as Hurricane Katrina in New Orleans in 2005, the disruptions in all supply chains included psychiatric medications. In the aftermath, many people with addictions and/or severe mental illnesses did not receive either their drugs of choice and/or antimanic and antipsychotic medications. As a result, disruptive behavior became common, especially in the shelters.5-7
During a widespread public emergency, police and emergency services are often stretched very thin. In calmer times, police or emergency services may take a person with disruptive and aggressive behavior to a local emergency department. However, in times of chaos, such as during Hurricane Katrina, patients with aggressive or disruptive behaviors were forcefully incapacitated (ie, “tased”) or shot.
Withdrawal from antidepressants, opiates, alcohol, and benzodiazepines has its own risks. Withdrawal from alcohol or benzodiazepines can be life-threatening. Therefore, it is critically important that clinicians think about how to ensure their patients have a supply of their medications. This may imply stockpiling on a personal or community basis.
Continue to: What to consider before disruption
What to consider before disruption
Many psychiatrists, especially those who have not practiced through a local disaster, may have never contemplated how they would support their patients during a disruptive event. Psychiatrists should carefully consider the questions outlined in Table 2 before a disaster strikes.
Medication-specific issues
During major disasters, patients may not have access to their medications, or the medications may not be able to be fed into the health care system for dispersion. Other issues include closed pharmacies, expired medications as a result of limited refrigeration service, inability to deliver medications to an affected area, and the inability of manufacturing plants to produce medications. For example, after Hurricane Maria, sterile water was in short supply.
After a major disaster, clinicians often leave their communities because they cannot support themselves or their practices. Thus, clinicians may not be available to prescribe needed medications. Available clinicians—often primary care physicians—may not be aware of a patient’s medication history, or they may be uncomfortable prescribing psychiatric medications, especially antipsychotics.
Abrupt discontinuation of psychiatric medications can have severe consequences. Patients may experience withdrawal symptoms, worsening psychiatric symptoms, new-onset psychiatric symptoms, thoughts of harm to self or others, psychosis, or cravings. These issues may be particularly problematic for patients receiving antidepressants, antipsychotics, benzodiazepines, or medication-assisted treatment for opioid use disorder.
Antidepressants. Patients experiencing antidepressant withdrawal, particularly withdrawal from selective serotonin reuptake inhibitors or serotonin-norepinephrine reuptake inhibitors, may exhibit severe symptoms. In addition to the potential recurrence of depressive or anxiety symptoms and suicidal thoughts, patients may experience irritability, insomnia, headache, nausea, and electric shock–like sensations. Prescribing an antidepressant with a longer half-life could potentially prevent an abrupt withdrawal in the event a disaster occurs.
Continue to: Antipsychotics
Antipsychotics. Rapid or abrupt withdrawal of antipsychotics could lead to an increase in psychosis, paranoia, hallucinations, or delusions. Withdrawal of antipsychotics could also lead to agitation, restlessness, insomnia, paresthesia, and anxiety. If a known disaster is likely to occur, such as in the case of a hurricane forecast, clinicians may consider switching a patient a long-acting injectable antipsychotic to minimize the risk of withdrawal and symptom exacerbation.
Benzodiazepines. The abrupt withdrawal of benzodiazepines could result in symptoms that include rebound anxiety, insomnia, restlessness, muscle tension, irritability, nausea, malaise, blurred vision, diaphoresis, nightmares, and seizures. Additionally, many people use benzodiazepines recreationally, and their illicit supply may run out during disasters, which could lead to untreated withdrawal and violence in the community.
Clinicians need to develop action plans for any patients who are receiving scheduled benzodiazepine dosing in order to prevent abrupt withdrawal if a disaster occurs.
Opioids. Opioid cravings and withdrawal are also a major concern during times of disrupted supply. Patients receiving chronic opioid therapy may not be able to receive their maintenance medications, which could lead to withdrawal. Additionally, patients taking illicit opioids may also be at risk of withdrawal.
Early symptoms of opioid withdrawal include watery eyes, runny nose, sweating, anxiety and irritability, poor sleep, and muscle pain. Later symptoms could include cramping, diarrhea, vomiting, increased heart rate and blood pressure, restlessness, shakiness, chills, sweating, and dilated pupils.
Continue to: Contingency planning...
Contingency planning should be a part of the treatment plan for every patient receiving chronic opioid therapy who lives in an area where major disasters are likely to occur.
Medication-assisted treatment for opioid use disorder. Patients receiving treatment for opioid use disorder may be prescribed the partial opioid agonist buprenorphine, either by itself or in combination with the opioid antagonist naloxone. This could be particularly problematic to continue in a major disaster due to the lack of credentialed clinicians, limited supplies, and patients only receiving small amounts of the medication at a time due to the risk of diversion.
Symptoms of buprenorphine withdrawal are similar to those associated with opioid withdrawal. Developing a thoughtful plan in case of a disaster should be part of all buprenorphine prescribing. Patients should be aware of withdrawal symptoms and what to do if they run out of medication.
Additionally, emergency clinicians should have access to buprenorphine and buprenorphine/naloxone and the ability to prescribe them in disaster situations. As with all aspects of disaster response, it is wise to work out issues in advance.
Help your patients get ready
Advise your patients to prepare emergency kits that contain their psychiatric medications that they could quickly grab and go if needed. Because there may be times when it is not possible to gather all necessary medications, having even a small supply ready to go at a moment’s notice would be beneficial. If permitted, patients should also consider keeping medications in multiple locations, including at their place of work, home, or a family member’s home.
Continue to: Additionally, instruct patients...
Additionally, instruct patients to always carry a list of all medications they currently take. Ideally, this list should also include past medications and responses, allergies, and provider contact information. During a disaster, this information could prove vital to an emergency clinician. At a minimum, verify that your patient maintains a list of current medications.
Clinicians should develop emergency plans for all psychiatric medications they prescribe. Document and discuss with your patients any necessary considerations for patients who take medications that require more intensive monitoring, such as lithium or clozapine.
Clinicians, patients, emergency responders, and health care workers need to work together to prepare for major disasters to avoid withdrawal and other consequences of disrupted access to psychiatric medications.
Bottom Line
Consult with local public health officials to determine and develop contingency plans to provide psychiatric medications to your patients in the event of a disaster. Discuss treatment plans and contingency planning with patients, particularly those in regions most likely to be affected by a disaster. Instruct patients to refill medications prior to a foreseeable disaster and to maintain a personal stockpile of medications when appropriate.
Related Resources
- Ochi S, Hodgson S, Landeg O, et al. Disaster-driven evacuation and medication loss: A systematic literature review. PLoS Curr. 2014;6.b. doi: 10.1371/currents.dis.fa417630b566a0c7dfdbf945910edd96.
- Pate JE, Fisher JW. Disaster ethics: What are the ground rules? Current Psychiatry. 2007;6(6):69-78.
Drug Brand Names
Buprenorphine • Subutex
Buprenorphine/naloxone • Suboxone
Clozapine • Clozaril
Diazepam • Valium
Lithium • Eskalith, Lithobid
1. National Institute of Mental Health. Mental health and mass violence: evidence based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. https://cpa.ca/docs/File/Emergencies/massviolence.pdf. Published 2002. Accessed March 11, 2019.
2. Ritchie EC, Friedman M, Watson P. Interventions following mass violence and disasters: strategies for mental health practice. New York, NY: Guilford Press; 2006.
3. Ritchie EC, O’Brien K, Grant M, et al. Disaster psychiatry. In: Stern TA, Rosenbaum JF, Fava M, et al. The Massachusetts General Hospital textbook of comprehensive clinical psychiatry, 2nd edition. Philadelphia, PA: Mosby/Elsevier; 2016:968-974.
4. Ritchie EC, Hamilton S. Early interventions and risk assessment following disaster. Psychiatric Annals. 2004;34(8):605-610.
5. Kessler RC, Galea S, Gruber MJ, et al. Trends in mental illness and suicidality after Hurricane Katrina. Mol Psychiatry. 2008;13(4):374-384.
6. Weisler RH, Barbee JG IV, Townsend MH. Mental health and recovery in the Gulf Coast after Hurricanes Katrina and Rita. JAMA. 2006;296(5):585-588.
7. Galea S, Brewin CR, Gruber M, et al. Exposure to hurricane-related stressors and mental illness after Hurricane Katrina. Arch Gen Psychiatry. 2007;64(12).1427-1434.
8. Federal Emergency Management Agency. Supply Chain Resilience Guide Department of Homeland Security. https://www.fema.gov/media-library-data/1544795397837-767851ba177c7097bf8672aadf8a93c9/NE_DRAFT_Supply_Chain_Resilience.pdf. Published December 17, 2018. Accessed January 2, 2019.
9. Durkin J, Telab M, Fitzmaurice P, et al. Only as strong as its weakest link: resilience of the healthcare supply chain in New York. https://www.hstoday.us/subject-matter-areas/emergency-preparedness/only-as-strong-as-its-weakest-link-the-resilience-of-the-healthcare-supply-chain-in-new-york/. Published October 26, 2018. Accessed February 14, 2019.
1. National Institute of Mental Health. Mental health and mass violence: evidence based early psychological intervention for victims/survivors of mass violence. A workshop to reach consensus on best practices. https://cpa.ca/docs/File/Emergencies/massviolence.pdf. Published 2002. Accessed March 11, 2019.
2. Ritchie EC, Friedman M, Watson P. Interventions following mass violence and disasters: strategies for mental health practice. New York, NY: Guilford Press; 2006.
3. Ritchie EC, O’Brien K, Grant M, et al. Disaster psychiatry. In: Stern TA, Rosenbaum JF, Fava M, et al. The Massachusetts General Hospital textbook of comprehensive clinical psychiatry, 2nd edition. Philadelphia, PA: Mosby/Elsevier; 2016:968-974.
4. Ritchie EC, Hamilton S. Early interventions and risk assessment following disaster. Psychiatric Annals. 2004;34(8):605-610.
5. Kessler RC, Galea S, Gruber MJ, et al. Trends in mental illness and suicidality after Hurricane Katrina. Mol Psychiatry. 2008;13(4):374-384.
6. Weisler RH, Barbee JG IV, Townsend MH. Mental health and recovery in the Gulf Coast after Hurricanes Katrina and Rita. JAMA. 2006;296(5):585-588.
7. Galea S, Brewin CR, Gruber M, et al. Exposure to hurricane-related stressors and mental illness after Hurricane Katrina. Arch Gen Psychiatry. 2007;64(12).1427-1434.
8. Federal Emergency Management Agency. Supply Chain Resilience Guide Department of Homeland Security. https://www.fema.gov/media-library-data/1544795397837-767851ba177c7097bf8672aadf8a93c9/NE_DRAFT_Supply_Chain_Resilience.pdf. Published December 17, 2018. Accessed January 2, 2019.
9. Durkin J, Telab M, Fitzmaurice P, et al. Only as strong as its weakest link: resilience of the healthcare supply chain in New York. https://www.hstoday.us/subject-matter-areas/emergency-preparedness/only-as-strong-as-its-weakest-link-the-resilience-of-the-healthcare-supply-chain-in-new-york/. Published October 26, 2018. Accessed February 14, 2019.

Cannabidiol (CBD) for schizophrenia: Promise or pipe dream?
Over the past few decades, it has become increasingly clear that cannabis use can increase the risk of developing a psychotic disorder and worsen the course of existing schizophrenia in a dose-dependent fashion.1-3 Beyond psychosis, although many patients with mental illness use cannabis for recreational purposes or as purported “self-medication,” currently available evidence suggests that marijuana is more likely to represent a harm than a benefit for psychiatric disorders4 (Box4-8). Our current state of knowledge therefore suggests that psychiatrists should caution their patients against using cannabis and prioritize interventions to reduce or discontinue use, especially among those with psychotic disorders.
Box
Data from California in 2006—a decade after the state’s legalization of “medical marijuana”—revealed that 23% of patients in a sample enrolled in medical marijuana clinics were receiving cannabis to treat a mental disorder.5 That was a striking statistic given the dearth of evidence to support a benefit of cannabis for psychiatric conditions at the time, leaving clinicians who provided the necessary recommendations to obtain medical marijuana largely unable to give informed consent about the risks and benefits, much less recommendations about specific products, routes of administration, or dosing. In 2019, we know considerably more about the interaction between cannabinoids and mental health, but research findings thus far warrant more caution than enthusiasm, with one recent review concluding that “whenever an association is observed between cannabis use and psychiatric disorders, the relationship is generally an adverse one.”4
Some critics have argued that the medical marijuana industry represents little more than a front for recreational use. In California and other states that have legalized recreational use, that claim has been rendered all but moot, although the public remains curious about the potential health benefits of cannabinoids and will likely continue to look to clinicians for advice. For those seeking guidance from evidence-based research, the existing state of knowledge can seem like a “Wild West” of anecdotal subjective reports, biased opinions, and uncontrolled clinical studies. Cannabis remains a Schedule I drug at the federal level, and quality clinical research has been limited to a relatively modest number of randomized controlled trials (RCTs), mostly involving FDA-approved cannabinoids rather than smoked cannabis. Randomized controlled trials that have involved smoked marijuana have generally involved low-potency delta-9-tetrahydrocannabinol (THC) cannabis that may not reflect the same therapeutic and adverse effects of the increasingly high potency cannabis now available on the street and in dispensaries.
In psychiatry, a few RCTs are underway exploring cannabis as a viable treatment for mental disorders (eg, posttraumatic stress disorder), but none have yet been completed or published. At best, retrospective studies to date have failed to support a consistent benefit of cannabis for any psychiatric disorder and at worst increasingly suggest a negative impact on psychotic, mood, and anxiety disorders.4,6 Meanwhile, synthetic cannabinoid receptor agonists (eg, “Spice” products) have come to represent a clear public health risk, with both medical and psychiatric toxicity.7
A more cautiously optimistic case for the therapeutic potential of cannabinoids in psychiatry could be made for cannabidiol (CBD), which may possess anxiolytic, antipsychotic, and neuroprotective properties.8 Based on its purported health benefits, it is possible that CBD may even gain widespread popularity as a food supplement. Because a pharmaceutically-manufactured form of CBD was recently FDA-approved for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome, off-label prescribing of CBD for psychiatric disorders can be anticipated. While there is not yet sufficient evidence about risks and benefits to justify CBD being recommended broadly in psychiatry, that same informational vacuum has not stopped eager patients from seeking approval for cannabis, and some physicians from providing it.
Despite that conclusion, because cannabis is classified as a Schedule I drug by the US Drug Enforcement Agency, clinical research investigating the risks and benefits of cannabis has been limited. It therefore remains possible that cannabis, or individual cannabinoids such as cannabidiol (CBD), may yet find a therapeutic niche in psychiatry. This article reviews evidence on CBD for the treatment of schizophrenia.
Cannabinergic drugs as potential antipsychotics
Although the bulk of evidence indicates a harmful effect of cannabis in individuals with or at risk for psychosis, there have been a few published cases of schizophrenia improving with dronabinol, an FDA-approved, synthetic form of delta-9-tetrahydrocannabinol (THC).9,10 THC is the constituent of cannabis that produces euphoric effects. These provocative findings have not been replicated in controlled clinical trials, but suggest at least the theoretical possibility of idiosyncratic benefits from THC for some individuals within the psychotic spectrum.
Still, given that most available evidence supports that THC has a harmful effect on psychosis and psychosis risk, researchers have instead performed randomized controlled trials (RCTs) to investigate a possible therapeutic role for medications that oppose the agonist effects of THC at cannabinoid type 1 (CB1) receptors. To date, 2 RCTs comparing rimonabant, a CB1 inverse agonist, with placebo (PLB) in patients with schizophrenia have failed to demonstrate any benefit for psychotic symptoms or cognitive deficits.11,12 A third trial examining rimonabant for people diagnosed with schizophrenia who were overweight found significant benefits for anxiety and depressive symptoms, but none for positive symptoms or the primary outcome of weight loss.13 While these results are discouraging, the role of THC in precipitating psychosis suggests that novel agents opposing the actions of THC on the cannabinoid system could have antipsychotic properties.14
Cannabidiol: An antipsychotic medication?
In contrast to THC, CBD has minimal euphorigenic properties and has recently been heralded in the popular press as a “miracle drug” with benefits for medical and psychiatric disorders alike.15 It has even been speculated that it could become a popular food supplement.16 In 2018, the FDA gave full approval to a pharmaceutically manufactured form of CBD (brand name: Epidiolex) as a novel treatment for 2 rare and severe forms of pediatric epilepsy, Lennox-Gastaut syndrome and Dravet syndrome,17 based on RCTs supporting its efficacy for these often refractory and life-threatening conditions.18-20
In psychiatry, there have not yet been enough robust clinical studies to support broad therapeutic claims for CBD as a treatment for any mental disorder.21 However, there is growing evidence that CBD has potential as an antipsychotic medication. In 1995, the first case report was published describing the efficacy of CBD, 1,500 mg/d, as standalone therapy in a single individual with schizophrenia.22 In 2006, the same research group followed up with a case series in which only 1 out of 3 patients with treatment-refractory schizophrenia improved with flexible dosing of CBD to a maximum dose of 1,280 mg/d.23
There have been 3 published RCTs exploring the efficacy of CBD in schizophrenia (Table24-26). The first study, published in 2012, included 39 adults with schizophrenia who were randomized to 800 mg/d of CBD or amisulpride (AMS), a second-generation antipsychotic that is popular in Europe but is not available in the United States.24 Over 4 weeks of randomized treatment, CBD resulted in as much improvement in overall symptoms and positive symptoms as AMS, and improvement of negative symptoms was significantly greater with CBD. Compared with patients treated with antipsychotic medication, patients who were treated with CBD had fewer extrapyramidal symptoms, less weight gain, and less prolactin elevation. This initial trial suggests that CBD might be as efficacious in schizophrenia as antipsychotic medication, without its burdensome adverse effects. However, this is the only RCT of CBD monotherapy published to date.

Continue to: Two other recently published RCTs...
Two other recently published RCTs compared CBD with PLB as add-on therapy to antipsychotics. McGuire et al25 compared CBD, 1,000 mg/d, to PLB over 6 weeks in 88 patients with schizophrenia. Positive symptom improvement was statistically greater with CBD than with PLB, although the magnitude of clinical change was modest (using the Positive and Negative Syndrome Scale [PANSS] positive symptom subscale: −3.2 points for CBD vs −1.7 points for PLB). Changes in PANSS total score and subscales for general and negative symptoms were not significantly different between treatment groups. There was also no significant difference in overall change in neurocognitive symptoms, although post-hoc analysis revealed significantly greater improvement in motor speed for patients treated with CBD. More than twice the number of patients treated with CBD were rated as “much improved” by the Clinical Global Impressions scale compared with patients treated with PLB, but this was not a statistically significant finding, and most patients experienced only “minimal” or “no improvement.” In terms of adverse events, there were no significant differences between patients in the CBD and PLB groups. Although this study is technically “positive” for CBD and suggests minimal adverse effects, it is not clear whether the statistically significant positive symptom improvements (+1.5 PANSS points for CBD over PLB) were clinically significant.
The most recently published placebo-controlled RCT of CBD as add-on therapy to antipsychotic medication included 36 patients with schizophrenia treated over 6 weeks.26 In this study, there was no benefit of CBD, 600 mg/d, on any PANSS score outcome (total, general, positive, or negative symptoms). For the primary outcome of the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Consensus Cognitive Battery, there were no significant drug × time effects, and post-hoc analyses showed that only patients treated with PLB improved with time. Sedation was more common among patients treated with CBD compared with PLB.
Making sense of the data
There have been mixed results from the few case reports and 3 RCTs of patients with schizophrenia who were treated with CBD. How can we resolve these disparate findings? A few possible interpretations of the data that warrant clarification through additional research include:
Dosing. In the first case report with positive results, CBD was dosed at 1,500 mg/d,22 whereas in the subsequent case series with mixed results, the maximum allowable dose of CBD was 1,280 mg/d.23 Likewise, in the RCTs, positive results were found when CBD was dosed at 800 to 1,000 mg/d,24,25 but not at 600 mg/d.26 The efficacy of CBD for schizophrenia might depend on higher doses.
Treatment resistance. In the second case series in which only 1 out of 3 patients responded to treatment with CBD,23 the patients had demonstrated previous nonresponse to at least 2 first-generation antipsychotics (FGAs) and risperidone, 6 mg/d. In the RCTs, all patients were antipsychotic-responsive.24-26 Cannabidiol may not be as effective for patients with treatment-refractory schizophrenia as it is for patients with schizophrenia who respond to antipsychotics.
Continue to: Clinical stability
Clinical stability. Within the RCTs, the greatest response was observed in the study that enrolled patients who were hospitalized with acute symptoms of schizophrenia.23 In the 2 studies that found either modest or no benefit with CBD, the patients had been stabilized on antipsychotic medications prior to randomization. Cannabidiol may offer limited benefit as add-on therapy to patients who have already responded to antipsychotic treatment, where there is “less room” for additional improvement.
Monotherapy. Both the case reports22,23 and the RCT with the most robust positive findings24 involved treatment with CBD as monotherapy. For some patients with schizophrenia, CBD might be effective as standalone therapy as an alternative to antipsychotics that is better tolerated. Adding CBD to antipsychotic therapy might be redundant and therefore less effective.
Answering questions about CBD
Cannabidiol is becoming increasingly popular for its purported health benefits. The mixed results of the few studies published on CBD for schizophrenia place clinicians in a difficult position when attempting to answer questions about how cannabinoids might fit into treatment of patients with psychosis. Consider the following:
Is cannabis helpful for patients with schizophrenia? No. Aside from the few case reports suggesting that FDA-approved THC (dronabinol) can improve symptoms in some patients,9,10 most of the evidence from anecdotal reports and both experimental and observational studies indicate that cannabis, THC, and synthetic cannabinoids have a harmful effect in patients with or at risk for psychosis.1-3
If you are considering recommending some form of cannabis to patients with schizophrenia, what kind should you recommend? Recommending or encouraging cannabis use for patients with psychosis is ill-advised. Although certain types of cannabis might contain more THC (eg, Cannabis indica vs Cannabis sativa) or variable amounts of CBD, in general the amount of CBD in whole leaf cannabis is minimal, with the ratio of THC to CBD increasingly significantly over the past decade.3,27 Most forms of cannabis should therefore be avoided by individuals with or at risk for psychotic disorders.
Continue to: What about CBD oil and other CBD products sold in dispensaries?
What about CBD oil and other CBD products sold in dispensaries? Cannabidiol is increasingly available in various forms based on its ability to be designated as a legal hemp product (containing <0.3% THC) at the federal level or as a cannabinoid in states where cannabis is legal. However, several studies have now shown that cannabis products sold online or in dispensaries are often labeled inaccurately, with both under- and over-reporting of THC and CBD content.28-30 Some CBD products have been found to have almost no CBD at all.29,30 The unreliability of product labeling makes it difficult to predict the effects of CBD products that are not subject to FDA purity standards for medications or dietary supplements. It also raises questions about the sources of CBD and the reliability of dosing in the studies discussed above.
Why might CBD work as an antipsychotic? Although CBD has minimal affinity for cannabinoid receptors, it appears to act as a partial agonist of dopamine D2 receptors and an agonist at 5-HT1A receptors, with overall effects that decrease mesolimbic dopamine activity.31,32 In addition, CBD increases the availability of the endogenous cannabinoid anandamide, which may have antipsychotic properties.14,33
Now that the FDA has approved CBD manufactured by a pharmaceutical company, should it be prescribed “off-label” for patients with schizophrenia? This is the “million dollar question,” with insufficient evidence to provide a clear answer. It should now be possible to prescribe FDA-approved CBD for off-label purposes, including the treatment of schizophrenia and other psychiatric disorders. No doubt, some clinicians are already doing so. This will predictably yield more anecdotal evidence about efficacy and adverse effects in the future, but there is not yet adequate evidence to support an FDA indication for CBD in schizophrenia. Additional studies of CBD for schizophrenia are ongoing.
Bottom Line
Cannabidiol (CBD) is becoming increasingly popular based on its purported health benefits, but the evidence supporting a therapeutic role in psychiatry is preliminary at best. Although CBD is now available by prescription as an FDA-approved drug for the treatment of 2 rare forms of epilepsy, its benefits in patients with schizophrenia are uncertain based on mixed results in clinical trials.
Related Resources
- Clinicaltrials.gov. Studies of “cannabidiol” and “schizophrenia.” U.S. National Library of Medicine. https://clinicaltrials.gov/ct2/results?cond=Schizophrenia&term=cannabidiol.
- Grinspoon P. Cannabidiol (CBD) – what we know and what we don’t. Harvard Health Blog. https://www.health.harvard.edu/blog/cannabidiol-cbd-what-we-know-and-what-wedont-2018082414476. Published August 24, 2018.
Drug Brand Names
Cannabidiol • Epidiolex
Dronabinol • Marinol
Risperidone • Risperdal
1. Pierre JM. Cannabis, synthetic cannabinoids, and psychosis risk: what the evidence says. Current Psychiatry. 2011;10(9):49-58.
2. Radhakrishan R, Wilkinson ST, D’Souza DC. Gone to pot – a review of the association between cannabis and psychosis. Front Psychiatry. 2014;5:54.
3. Pierre JM. Risks of increasingly potent cannabis: joint effects of potency and frequency. Current Psychiatry. 2016;16(2):14-20.
4. Hanna RC, Perez JM, Ghose S. Cannabis and development of dual diagnoses: a literature review. Am J Drug Alcohol Abuse. 2017;43(4):442-255.
5. Nunberg H, Kilmer B, Pacula RL, et al. An analysis of applicants presenting to a medical marijuana specialty practice in California. J Drug Policy Anal. 2011;4(1):1.
6. Wilkinson ST, Radhakrishnan, D’Souza DC. A systematic review of the evidence for medical marijuana in psychiatric indications. J Clin Psychiatry. 2016;77(8):1050-1064.
7. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: Update 2015. Subst Abus. 2016;38(3):344-366.
8. Crippa JA, Guimarães FS, Campos A, et al. Translational investigation of the therapeutic potential of cannabidiol (CBD): toward a new age. Front Immunol. 2018;9:2009.
9. Schwarz G, Karajgi B. Improvement in refractory psychosis with dronabinol: four case reports. J Clin Psychiatry. 2010;71(11):1552-1553.
10. Schwarz G, Karajgi B, McCarthy R. Synthetic delta-9-tetrahydrocannabinol (dronabinol) can improve the symptoms of schizophrenia. J Clin Psychopharmacol. 2009;29(3):255-258.
11. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry. 2004;161(6):975-984.
12. Boggs DL, Kelly DL, McMahon RP, et al. Rimonabant for neurocognition in schizophrenia: a 16-week double blind placebo controlled trial. Schizophr Res. 2012;134(2-3):207-210.
13. Kelly DL, Gorelick DA, Conley RR, et al. Effects of cannabinoid-1 receptor antagonist rimonabant on psychiatric symptoms in overweight people with schizophrenia: a randomized, double-blind, pilot study. J Clin Psychopharmacol. 2011;31(1):86-91.
14. Leweke FM, Mueller JK, Lange B, et al. Therapeutic potential of cannabinoids in psychosis. Biol Psychiatry. 2016;79(7):604-612.
15. Halperin A. What is CBD? The ‘miracle’ cannabis compound that doesn’t get you high. The Guardian. https://www.theguardian.com/society/2018/may/28/what-is-cbd-cannabidiol-cannabis-medical-uses. Published May 28, 2018. Accessed April 3, 2019.
16. Pierre J. Coca, cola, and cannabis: psychoactive drugs as beverages. Psychology Today (blog) Psych Unseen. https://www.psychologytoday.com/us/blog/psych-unseen/201810/coca-cola-and-cannabis-psychoactive-drugs-beverages. Published October 1, 2018. Accessed April 3, 2019.
17. U.S. Food and Drug Administration. FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy. FDA News Release. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm611046.htm. Published June 25, 2018. Accessed April 3, 2019.
18. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011-2020.
19. Thiele EA, March ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085-1096.
20. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. 2018;378:1888-1897.
21. Khoury JM, Neves MCLD, Rogue MAV, et al. Is there a role of cannabidiol in psychiatry? World J Biol Psychiatry. 2017:1-16.
22. Zuardi AW, Morais SL, Guimares FS, et al. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56(10):485-486.
23. Zuardi AW, Hallak JEC, Dursun SM. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. 2006;20(5):683-686.
24. Leweke FM, Piomelli D, Pahlisch F, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15.
25. McGuire P, Robson P, Cubala WJ, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175(3):225-231.
26. Boggs DL, Surti I, Gupta A, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacol. 2018;235(7):1923-1932.
27. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016; 79(7):613-619.
28. Vandrey R, Raber JC, Raber ME, et al. Cannabinoid dose and label accuracy in edible medical cannabis products. JAMA. 2015;313(24):2491-2492.
29. Ruth AC, Gryniewicz-Ruzicka CM, Trehy ML, et al. Consistency of label claims of internet-purchased hemp oil and cannabis products as determined using IMS and LC-MS: a marketplace study. J Reg Sci. 2016;3:1-6.
30. Bonn-Miller MO, Loflin MJE, Thomas BF, et al. Labeling accuracy of cannabidiol extracts sold online. JAMA. 2017;318(17):1708-1709.
31. Seeman P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl Psychiatry. 2016;6(10):e920. doi: 10.1038/tp.2016.195.
32. Renard J, Norris C, Rushlow W, et al. Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: implications for novel schizophrenia treatments. Neurosci Biobehav Rev. 2017;157-165.
33. Gururajan A, Malone DT. Does cannabidiol have a role in the treatment of schizophrenia? Schizophr Res. 2016;176(2-3):281-290.
Over the past few decades, it has become increasingly clear that cannabis use can increase the risk of developing a psychotic disorder and worsen the course of existing schizophrenia in a dose-dependent fashion.1-3 Beyond psychosis, although many patients with mental illness use cannabis for recreational purposes or as purported “self-medication,” currently available evidence suggests that marijuana is more likely to represent a harm than a benefit for psychiatric disorders4 (Box4-8). Our current state of knowledge therefore suggests that psychiatrists should caution their patients against using cannabis and prioritize interventions to reduce or discontinue use, especially among those with psychotic disorders.
Box
Data from California in 2006—a decade after the state’s legalization of “medical marijuana”—revealed that 23% of patients in a sample enrolled in medical marijuana clinics were receiving cannabis to treat a mental disorder.5 That was a striking statistic given the dearth of evidence to support a benefit of cannabis for psychiatric conditions at the time, leaving clinicians who provided the necessary recommendations to obtain medical marijuana largely unable to give informed consent about the risks and benefits, much less recommendations about specific products, routes of administration, or dosing. In 2019, we know considerably more about the interaction between cannabinoids and mental health, but research findings thus far warrant more caution than enthusiasm, with one recent review concluding that “whenever an association is observed between cannabis use and psychiatric disorders, the relationship is generally an adverse one.”4
Some critics have argued that the medical marijuana industry represents little more than a front for recreational use. In California and other states that have legalized recreational use, that claim has been rendered all but moot, although the public remains curious about the potential health benefits of cannabinoids and will likely continue to look to clinicians for advice. For those seeking guidance from evidence-based research, the existing state of knowledge can seem like a “Wild West” of anecdotal subjective reports, biased opinions, and uncontrolled clinical studies. Cannabis remains a Schedule I drug at the federal level, and quality clinical research has been limited to a relatively modest number of randomized controlled trials (RCTs), mostly involving FDA-approved cannabinoids rather than smoked cannabis. Randomized controlled trials that have involved smoked marijuana have generally involved low-potency delta-9-tetrahydrocannabinol (THC) cannabis that may not reflect the same therapeutic and adverse effects of the increasingly high potency cannabis now available on the street and in dispensaries.
In psychiatry, a few RCTs are underway exploring cannabis as a viable treatment for mental disorders (eg, posttraumatic stress disorder), but none have yet been completed or published. At best, retrospective studies to date have failed to support a consistent benefit of cannabis for any psychiatric disorder and at worst increasingly suggest a negative impact on psychotic, mood, and anxiety disorders.4,6 Meanwhile, synthetic cannabinoid receptor agonists (eg, “Spice” products) have come to represent a clear public health risk, with both medical and psychiatric toxicity.7
A more cautiously optimistic case for the therapeutic potential of cannabinoids in psychiatry could be made for cannabidiol (CBD), which may possess anxiolytic, antipsychotic, and neuroprotective properties.8 Based on its purported health benefits, it is possible that CBD may even gain widespread popularity as a food supplement. Because a pharmaceutically-manufactured form of CBD was recently FDA-approved for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome, off-label prescribing of CBD for psychiatric disorders can be anticipated. While there is not yet sufficient evidence about risks and benefits to justify CBD being recommended broadly in psychiatry, that same informational vacuum has not stopped eager patients from seeking approval for cannabis, and some physicians from providing it.
Despite that conclusion, because cannabis is classified as a Schedule I drug by the US Drug Enforcement Agency, clinical research investigating the risks and benefits of cannabis has been limited. It therefore remains possible that cannabis, or individual cannabinoids such as cannabidiol (CBD), may yet find a therapeutic niche in psychiatry. This article reviews evidence on CBD for the treatment of schizophrenia.
Cannabinergic drugs as potential antipsychotics
Although the bulk of evidence indicates a harmful effect of cannabis in individuals with or at risk for psychosis, there have been a few published cases of schizophrenia improving with dronabinol, an FDA-approved, synthetic form of delta-9-tetrahydrocannabinol (THC).9,10 THC is the constituent of cannabis that produces euphoric effects. These provocative findings have not been replicated in controlled clinical trials, but suggest at least the theoretical possibility of idiosyncratic benefits from THC for some individuals within the psychotic spectrum.
Still, given that most available evidence supports that THC has a harmful effect on psychosis and psychosis risk, researchers have instead performed randomized controlled trials (RCTs) to investigate a possible therapeutic role for medications that oppose the agonist effects of THC at cannabinoid type 1 (CB1) receptors. To date, 2 RCTs comparing rimonabant, a CB1 inverse agonist, with placebo (PLB) in patients with schizophrenia have failed to demonstrate any benefit for psychotic symptoms or cognitive deficits.11,12 A third trial examining rimonabant for people diagnosed with schizophrenia who were overweight found significant benefits for anxiety and depressive symptoms, but none for positive symptoms or the primary outcome of weight loss.13 While these results are discouraging, the role of THC in precipitating psychosis suggests that novel agents opposing the actions of THC on the cannabinoid system could have antipsychotic properties.14
Cannabidiol: An antipsychotic medication?
In contrast to THC, CBD has minimal euphorigenic properties and has recently been heralded in the popular press as a “miracle drug” with benefits for medical and psychiatric disorders alike.15 It has even been speculated that it could become a popular food supplement.16 In 2018, the FDA gave full approval to a pharmaceutically manufactured form of CBD (brand name: Epidiolex) as a novel treatment for 2 rare and severe forms of pediatric epilepsy, Lennox-Gastaut syndrome and Dravet syndrome,17 based on RCTs supporting its efficacy for these often refractory and life-threatening conditions.18-20
In psychiatry, there have not yet been enough robust clinical studies to support broad therapeutic claims for CBD as a treatment for any mental disorder.21 However, there is growing evidence that CBD has potential as an antipsychotic medication. In 1995, the first case report was published describing the efficacy of CBD, 1,500 mg/d, as standalone therapy in a single individual with schizophrenia.22 In 2006, the same research group followed up with a case series in which only 1 out of 3 patients with treatment-refractory schizophrenia improved with flexible dosing of CBD to a maximum dose of 1,280 mg/d.23
There have been 3 published RCTs exploring the efficacy of CBD in schizophrenia (Table24-26). The first study, published in 2012, included 39 adults with schizophrenia who were randomized to 800 mg/d of CBD or amisulpride (AMS), a second-generation antipsychotic that is popular in Europe but is not available in the United States.24 Over 4 weeks of randomized treatment, CBD resulted in as much improvement in overall symptoms and positive symptoms as AMS, and improvement of negative symptoms was significantly greater with CBD. Compared with patients treated with antipsychotic medication, patients who were treated with CBD had fewer extrapyramidal symptoms, less weight gain, and less prolactin elevation. This initial trial suggests that CBD might be as efficacious in schizophrenia as antipsychotic medication, without its burdensome adverse effects. However, this is the only RCT of CBD monotherapy published to date.
Continue to: Two other recently published RCTs...
Two other recently published RCTs compared CBD with PLB as add-on therapy to antipsychotics. McGuire et al25 compared CBD, 1,000 mg/d, to PLB over 6 weeks in 88 patients with schizophrenia. Positive symptom improvement was statistically greater with CBD than with PLB, although the magnitude of clinical change was modest (using the Positive and Negative Syndrome Scale [PANSS] positive symptom subscale: −3.2 points for CBD vs −1.7 points for PLB). Changes in PANSS total score and subscales for general and negative symptoms were not significantly different between treatment groups. There was also no significant difference in overall change in neurocognitive symptoms, although post-hoc analysis revealed significantly greater improvement in motor speed for patients treated with CBD. More than twice the number of patients treated with CBD were rated as “much improved” by the Clinical Global Impressions scale compared with patients treated with PLB, but this was not a statistically significant finding, and most patients experienced only “minimal” or “no improvement.” In terms of adverse events, there were no significant differences between patients in the CBD and PLB groups. Although this study is technically “positive” for CBD and suggests minimal adverse effects, it is not clear whether the statistically significant positive symptom improvements (+1.5 PANSS points for CBD over PLB) were clinically significant.
The most recently published placebo-controlled RCT of CBD as add-on therapy to antipsychotic medication included 36 patients with schizophrenia treated over 6 weeks.26 In this study, there was no benefit of CBD, 600 mg/d, on any PANSS score outcome (total, general, positive, or negative symptoms). For the primary outcome of the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Consensus Cognitive Battery, there were no significant drug × time effects, and post-hoc analyses showed that only patients treated with PLB improved with time. Sedation was more common among patients treated with CBD compared with PLB.
Making sense of the data
There have been mixed results from the few case reports and 3 RCTs of patients with schizophrenia who were treated with CBD. How can we resolve these disparate findings? A few possible interpretations of the data that warrant clarification through additional research include:
Dosing. In the first case report with positive results, CBD was dosed at 1,500 mg/d,22 whereas in the subsequent case series with mixed results, the maximum allowable dose of CBD was 1,280 mg/d.23 Likewise, in the RCTs, positive results were found when CBD was dosed at 800 to 1,000 mg/d,24,25 but not at 600 mg/d.26 The efficacy of CBD for schizophrenia might depend on higher doses.
Treatment resistance. In the second case series in which only 1 out of 3 patients responded to treatment with CBD,23 the patients had demonstrated previous nonresponse to at least 2 first-generation antipsychotics (FGAs) and risperidone, 6 mg/d. In the RCTs, all patients were antipsychotic-responsive.24-26 Cannabidiol may not be as effective for patients with treatment-refractory schizophrenia as it is for patients with schizophrenia who respond to antipsychotics.
Continue to: Clinical stability
Clinical stability. Within the RCTs, the greatest response was observed in the study that enrolled patients who were hospitalized with acute symptoms of schizophrenia.23 In the 2 studies that found either modest or no benefit with CBD, the patients had been stabilized on antipsychotic medications prior to randomization. Cannabidiol may offer limited benefit as add-on therapy to patients who have already responded to antipsychotic treatment, where there is “less room” for additional improvement.
Monotherapy. Both the case reports22,23 and the RCT with the most robust positive findings24 involved treatment with CBD as monotherapy. For some patients with schizophrenia, CBD might be effective as standalone therapy as an alternative to antipsychotics that is better tolerated. Adding CBD to antipsychotic therapy might be redundant and therefore less effective.
Answering questions about CBD
Cannabidiol is becoming increasingly popular for its purported health benefits. The mixed results of the few studies published on CBD for schizophrenia place clinicians in a difficult position when attempting to answer questions about how cannabinoids might fit into treatment of patients with psychosis. Consider the following:
Is cannabis helpful for patients with schizophrenia? No. Aside from the few case reports suggesting that FDA-approved THC (dronabinol) can improve symptoms in some patients,9,10 most of the evidence from anecdotal reports and both experimental and observational studies indicate that cannabis, THC, and synthetic cannabinoids have a harmful effect in patients with or at risk for psychosis.1-3
If you are considering recommending some form of cannabis to patients with schizophrenia, what kind should you recommend? Recommending or encouraging cannabis use for patients with psychosis is ill-advised. Although certain types of cannabis might contain more THC (eg, Cannabis indica vs Cannabis sativa) or variable amounts of CBD, in general the amount of CBD in whole leaf cannabis is minimal, with the ratio of THC to CBD increasingly significantly over the past decade.3,27 Most forms of cannabis should therefore be avoided by individuals with or at risk for psychotic disorders.
Continue to: What about CBD oil and other CBD products sold in dispensaries?
What about CBD oil and other CBD products sold in dispensaries? Cannabidiol is increasingly available in various forms based on its ability to be designated as a legal hemp product (containing <0.3% THC) at the federal level or as a cannabinoid in states where cannabis is legal. However, several studies have now shown that cannabis products sold online or in dispensaries are often labeled inaccurately, with both under- and over-reporting of THC and CBD content.28-30 Some CBD products have been found to have almost no CBD at all.29,30 The unreliability of product labeling makes it difficult to predict the effects of CBD products that are not subject to FDA purity standards for medications or dietary supplements. It also raises questions about the sources of CBD and the reliability of dosing in the studies discussed above.
Why might CBD work as an antipsychotic? Although CBD has minimal affinity for cannabinoid receptors, it appears to act as a partial agonist of dopamine D2 receptors and an agonist at 5-HT1A receptors, with overall effects that decrease mesolimbic dopamine activity.31,32 In addition, CBD increases the availability of the endogenous cannabinoid anandamide, which may have antipsychotic properties.14,33
Now that the FDA has approved CBD manufactured by a pharmaceutical company, should it be prescribed “off-label” for patients with schizophrenia? This is the “million dollar question,” with insufficient evidence to provide a clear answer. It should now be possible to prescribe FDA-approved CBD for off-label purposes, including the treatment of schizophrenia and other psychiatric disorders. No doubt, some clinicians are already doing so. This will predictably yield more anecdotal evidence about efficacy and adverse effects in the future, but there is not yet adequate evidence to support an FDA indication for CBD in schizophrenia. Additional studies of CBD for schizophrenia are ongoing.
Bottom Line
Cannabidiol (CBD) is becoming increasingly popular based on its purported health benefits, but the evidence supporting a therapeutic role in psychiatry is preliminary at best. Although CBD is now available by prescription as an FDA-approved drug for the treatment of 2 rare forms of epilepsy, its benefits in patients with schizophrenia are uncertain based on mixed results in clinical trials.
Related Resources
- Clinicaltrials.gov. Studies of “cannabidiol” and “schizophrenia.” U.S. National Library of Medicine. https://clinicaltrials.gov/ct2/results?cond=Schizophrenia&term=cannabidiol.
- Grinspoon P. Cannabidiol (CBD) – what we know and what we don’t. Harvard Health Blog. https://www.health.harvard.edu/blog/cannabidiol-cbd-what-we-know-and-what-wedont-2018082414476. Published August 24, 2018.
Drug Brand Names
Cannabidiol • Epidiolex
Dronabinol • Marinol
Risperidone • Risperdal
Over the past few decades, it has become increasingly clear that cannabis use can increase the risk of developing a psychotic disorder and worsen the course of existing schizophrenia in a dose-dependent fashion.1-3 Beyond psychosis, although many patients with mental illness use cannabis for recreational purposes or as purported “self-medication,” currently available evidence suggests that marijuana is more likely to represent a harm than a benefit for psychiatric disorders4 (Box4-8). Our current state of knowledge therefore suggests that psychiatrists should caution their patients against using cannabis and prioritize interventions to reduce or discontinue use, especially among those with psychotic disorders.
Box
Data from California in 2006—a decade after the state’s legalization of “medical marijuana”—revealed that 23% of patients in a sample enrolled in medical marijuana clinics were receiving cannabis to treat a mental disorder.5 That was a striking statistic given the dearth of evidence to support a benefit of cannabis for psychiatric conditions at the time, leaving clinicians who provided the necessary recommendations to obtain medical marijuana largely unable to give informed consent about the risks and benefits, much less recommendations about specific products, routes of administration, or dosing. In 2019, we know considerably more about the interaction between cannabinoids and mental health, but research findings thus far warrant more caution than enthusiasm, with one recent review concluding that “whenever an association is observed between cannabis use and psychiatric disorders, the relationship is generally an adverse one.”4
Some critics have argued that the medical marijuana industry represents little more than a front for recreational use. In California and other states that have legalized recreational use, that claim has been rendered all but moot, although the public remains curious about the potential health benefits of cannabinoids and will likely continue to look to clinicians for advice. For those seeking guidance from evidence-based research, the existing state of knowledge can seem like a “Wild West” of anecdotal subjective reports, biased opinions, and uncontrolled clinical studies. Cannabis remains a Schedule I drug at the federal level, and quality clinical research has been limited to a relatively modest number of randomized controlled trials (RCTs), mostly involving FDA-approved cannabinoids rather than smoked cannabis. Randomized controlled trials that have involved smoked marijuana have generally involved low-potency delta-9-tetrahydrocannabinol (THC) cannabis that may not reflect the same therapeutic and adverse effects of the increasingly high potency cannabis now available on the street and in dispensaries.
In psychiatry, a few RCTs are underway exploring cannabis as a viable treatment for mental disorders (eg, posttraumatic stress disorder), but none have yet been completed or published. At best, retrospective studies to date have failed to support a consistent benefit of cannabis for any psychiatric disorder and at worst increasingly suggest a negative impact on psychotic, mood, and anxiety disorders.4,6 Meanwhile, synthetic cannabinoid receptor agonists (eg, “Spice” products) have come to represent a clear public health risk, with both medical and psychiatric toxicity.7
A more cautiously optimistic case for the therapeutic potential of cannabinoids in psychiatry could be made for cannabidiol (CBD), which may possess anxiolytic, antipsychotic, and neuroprotective properties.8 Based on its purported health benefits, it is possible that CBD may even gain widespread popularity as a food supplement. Because a pharmaceutically-manufactured form of CBD was recently FDA-approved for the treatment of seizures associated with Lennox-Gastaut syndrome and Dravet syndrome, off-label prescribing of CBD for psychiatric disorders can be anticipated. While there is not yet sufficient evidence about risks and benefits to justify CBD being recommended broadly in psychiatry, that same informational vacuum has not stopped eager patients from seeking approval for cannabis, and some physicians from providing it.
Despite that conclusion, because cannabis is classified as a Schedule I drug by the US Drug Enforcement Agency, clinical research investigating the risks and benefits of cannabis has been limited. It therefore remains possible that cannabis, or individual cannabinoids such as cannabidiol (CBD), may yet find a therapeutic niche in psychiatry. This article reviews evidence on CBD for the treatment of schizophrenia.
Cannabinergic drugs as potential antipsychotics
Although the bulk of evidence indicates a harmful effect of cannabis in individuals with or at risk for psychosis, there have been a few published cases of schizophrenia improving with dronabinol, an FDA-approved, synthetic form of delta-9-tetrahydrocannabinol (THC).9,10 THC is the constituent of cannabis that produces euphoric effects. These provocative findings have not been replicated in controlled clinical trials, but suggest at least the theoretical possibility of idiosyncratic benefits from THC for some individuals within the psychotic spectrum.
Still, given that most available evidence supports that THC has a harmful effect on psychosis and psychosis risk, researchers have instead performed randomized controlled trials (RCTs) to investigate a possible therapeutic role for medications that oppose the agonist effects of THC at cannabinoid type 1 (CB1) receptors. To date, 2 RCTs comparing rimonabant, a CB1 inverse agonist, with placebo (PLB) in patients with schizophrenia have failed to demonstrate any benefit for psychotic symptoms or cognitive deficits.11,12 A third trial examining rimonabant for people diagnosed with schizophrenia who were overweight found significant benefits for anxiety and depressive symptoms, but none for positive symptoms or the primary outcome of weight loss.13 While these results are discouraging, the role of THC in precipitating psychosis suggests that novel agents opposing the actions of THC on the cannabinoid system could have antipsychotic properties.14
Cannabidiol: An antipsychotic medication?
In contrast to THC, CBD has minimal euphorigenic properties and has recently been heralded in the popular press as a “miracle drug” with benefits for medical and psychiatric disorders alike.15 It has even been speculated that it could become a popular food supplement.16 In 2018, the FDA gave full approval to a pharmaceutically manufactured form of CBD (brand name: Epidiolex) as a novel treatment for 2 rare and severe forms of pediatric epilepsy, Lennox-Gastaut syndrome and Dravet syndrome,17 based on RCTs supporting its efficacy for these often refractory and life-threatening conditions.18-20
In psychiatry, there have not yet been enough robust clinical studies to support broad therapeutic claims for CBD as a treatment for any mental disorder.21 However, there is growing evidence that CBD has potential as an antipsychotic medication. In 1995, the first case report was published describing the efficacy of CBD, 1,500 mg/d, as standalone therapy in a single individual with schizophrenia.22 In 2006, the same research group followed up with a case series in which only 1 out of 3 patients with treatment-refractory schizophrenia improved with flexible dosing of CBD to a maximum dose of 1,280 mg/d.23
There have been 3 published RCTs exploring the efficacy of CBD in schizophrenia (Table24-26). The first study, published in 2012, included 39 adults with schizophrenia who were randomized to 800 mg/d of CBD or amisulpride (AMS), a second-generation antipsychotic that is popular in Europe but is not available in the United States.24 Over 4 weeks of randomized treatment, CBD resulted in as much improvement in overall symptoms and positive symptoms as AMS, and improvement of negative symptoms was significantly greater with CBD. Compared with patients treated with antipsychotic medication, patients who were treated with CBD had fewer extrapyramidal symptoms, less weight gain, and less prolactin elevation. This initial trial suggests that CBD might be as efficacious in schizophrenia as antipsychotic medication, without its burdensome adverse effects. However, this is the only RCT of CBD monotherapy published to date.
Continue to: Two other recently published RCTs...
Two other recently published RCTs compared CBD with PLB as add-on therapy to antipsychotics. McGuire et al25 compared CBD, 1,000 mg/d, to PLB over 6 weeks in 88 patients with schizophrenia. Positive symptom improvement was statistically greater with CBD than with PLB, although the magnitude of clinical change was modest (using the Positive and Negative Syndrome Scale [PANSS] positive symptom subscale: −3.2 points for CBD vs −1.7 points for PLB). Changes in PANSS total score and subscales for general and negative symptoms were not significantly different between treatment groups. There was also no significant difference in overall change in neurocognitive symptoms, although post-hoc analysis revealed significantly greater improvement in motor speed for patients treated with CBD. More than twice the number of patients treated with CBD were rated as “much improved” by the Clinical Global Impressions scale compared with patients treated with PLB, but this was not a statistically significant finding, and most patients experienced only “minimal” or “no improvement.” In terms of adverse events, there were no significant differences between patients in the CBD and PLB groups. Although this study is technically “positive” for CBD and suggests minimal adverse effects, it is not clear whether the statistically significant positive symptom improvements (+1.5 PANSS points for CBD over PLB) were clinically significant.
The most recently published placebo-controlled RCT of CBD as add-on therapy to antipsychotic medication included 36 patients with schizophrenia treated over 6 weeks.26 In this study, there was no benefit of CBD, 600 mg/d, on any PANSS score outcome (total, general, positive, or negative symptoms). For the primary outcome of the Measurement and Treatment Research to Improve Cognition in Schizophrenia (MATRICS) Consensus Cognitive Battery, there were no significant drug × time effects, and post-hoc analyses showed that only patients treated with PLB improved with time. Sedation was more common among patients treated with CBD compared with PLB.
Making sense of the data
There have been mixed results from the few case reports and 3 RCTs of patients with schizophrenia who were treated with CBD. How can we resolve these disparate findings? A few possible interpretations of the data that warrant clarification through additional research include:
Dosing. In the first case report with positive results, CBD was dosed at 1,500 mg/d,22 whereas in the subsequent case series with mixed results, the maximum allowable dose of CBD was 1,280 mg/d.23 Likewise, in the RCTs, positive results were found when CBD was dosed at 800 to 1,000 mg/d,24,25 but not at 600 mg/d.26 The efficacy of CBD for schizophrenia might depend on higher doses.
Treatment resistance. In the second case series in which only 1 out of 3 patients responded to treatment with CBD,23 the patients had demonstrated previous nonresponse to at least 2 first-generation antipsychotics (FGAs) and risperidone, 6 mg/d. In the RCTs, all patients were antipsychotic-responsive.24-26 Cannabidiol may not be as effective for patients with treatment-refractory schizophrenia as it is for patients with schizophrenia who respond to antipsychotics.
Continue to: Clinical stability
Clinical stability. Within the RCTs, the greatest response was observed in the study that enrolled patients who were hospitalized with acute symptoms of schizophrenia.23 In the 2 studies that found either modest or no benefit with CBD, the patients had been stabilized on antipsychotic medications prior to randomization. Cannabidiol may offer limited benefit as add-on therapy to patients who have already responded to antipsychotic treatment, where there is “less room” for additional improvement.
Monotherapy. Both the case reports22,23 and the RCT with the most robust positive findings24 involved treatment with CBD as monotherapy. For some patients with schizophrenia, CBD might be effective as standalone therapy as an alternative to antipsychotics that is better tolerated. Adding CBD to antipsychotic therapy might be redundant and therefore less effective.
Answering questions about CBD
Cannabidiol is becoming increasingly popular for its purported health benefits. The mixed results of the few studies published on CBD for schizophrenia place clinicians in a difficult position when attempting to answer questions about how cannabinoids might fit into treatment of patients with psychosis. Consider the following:
Is cannabis helpful for patients with schizophrenia? No. Aside from the few case reports suggesting that FDA-approved THC (dronabinol) can improve symptoms in some patients,9,10 most of the evidence from anecdotal reports and both experimental and observational studies indicate that cannabis, THC, and synthetic cannabinoids have a harmful effect in patients with or at risk for psychosis.1-3
If you are considering recommending some form of cannabis to patients with schizophrenia, what kind should you recommend? Recommending or encouraging cannabis use for patients with psychosis is ill-advised. Although certain types of cannabis might contain more THC (eg, Cannabis indica vs Cannabis sativa) or variable amounts of CBD, in general the amount of CBD in whole leaf cannabis is minimal, with the ratio of THC to CBD increasingly significantly over the past decade.3,27 Most forms of cannabis should therefore be avoided by individuals with or at risk for psychotic disorders.
Continue to: What about CBD oil and other CBD products sold in dispensaries?
What about CBD oil and other CBD products sold in dispensaries? Cannabidiol is increasingly available in various forms based on its ability to be designated as a legal hemp product (containing <0.3% THC) at the federal level or as a cannabinoid in states where cannabis is legal. However, several studies have now shown that cannabis products sold online or in dispensaries are often labeled inaccurately, with both under- and over-reporting of THC and CBD content.28-30 Some CBD products have been found to have almost no CBD at all.29,30 The unreliability of product labeling makes it difficult to predict the effects of CBD products that are not subject to FDA purity standards for medications or dietary supplements. It also raises questions about the sources of CBD and the reliability of dosing in the studies discussed above.
Why might CBD work as an antipsychotic? Although CBD has minimal affinity for cannabinoid receptors, it appears to act as a partial agonist of dopamine D2 receptors and an agonist at 5-HT1A receptors, with overall effects that decrease mesolimbic dopamine activity.31,32 In addition, CBD increases the availability of the endogenous cannabinoid anandamide, which may have antipsychotic properties.14,33
Now that the FDA has approved CBD manufactured by a pharmaceutical company, should it be prescribed “off-label” for patients with schizophrenia? This is the “million dollar question,” with insufficient evidence to provide a clear answer. It should now be possible to prescribe FDA-approved CBD for off-label purposes, including the treatment of schizophrenia and other psychiatric disorders. No doubt, some clinicians are already doing so. This will predictably yield more anecdotal evidence about efficacy and adverse effects in the future, but there is not yet adequate evidence to support an FDA indication for CBD in schizophrenia. Additional studies of CBD for schizophrenia are ongoing.
Bottom Line
Cannabidiol (CBD) is becoming increasingly popular based on its purported health benefits, but the evidence supporting a therapeutic role in psychiatry is preliminary at best. Although CBD is now available by prescription as an FDA-approved drug for the treatment of 2 rare forms of epilepsy, its benefits in patients with schizophrenia are uncertain based on mixed results in clinical trials.
Related Resources
- Clinicaltrials.gov. Studies of “cannabidiol” and “schizophrenia.” U.S. National Library of Medicine. https://clinicaltrials.gov/ct2/results?cond=Schizophrenia&term=cannabidiol.
- Grinspoon P. Cannabidiol (CBD) – what we know and what we don’t. Harvard Health Blog. https://www.health.harvard.edu/blog/cannabidiol-cbd-what-we-know-and-what-wedont-2018082414476. Published August 24, 2018.
Drug Brand Names
Cannabidiol • Epidiolex
Dronabinol • Marinol
Risperidone • Risperdal
1. Pierre JM. Cannabis, synthetic cannabinoids, and psychosis risk: what the evidence says. Current Psychiatry. 2011;10(9):49-58.
2. Radhakrishan R, Wilkinson ST, D’Souza DC. Gone to pot – a review of the association between cannabis and psychosis. Front Psychiatry. 2014;5:54.
3. Pierre JM. Risks of increasingly potent cannabis: joint effects of potency and frequency. Current Psychiatry. 2016;16(2):14-20.
4. Hanna RC, Perez JM, Ghose S. Cannabis and development of dual diagnoses: a literature review. Am J Drug Alcohol Abuse. 2017;43(4):442-255.
5. Nunberg H, Kilmer B, Pacula RL, et al. An analysis of applicants presenting to a medical marijuana specialty practice in California. J Drug Policy Anal. 2011;4(1):1.
6. Wilkinson ST, Radhakrishnan, D’Souza DC. A systematic review of the evidence for medical marijuana in psychiatric indications. J Clin Psychiatry. 2016;77(8):1050-1064.
7. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: Update 2015. Subst Abus. 2016;38(3):344-366.
8. Crippa JA, Guimarães FS, Campos A, et al. Translational investigation of the therapeutic potential of cannabidiol (CBD): toward a new age. Front Immunol. 2018;9:2009.
9. Schwarz G, Karajgi B. Improvement in refractory psychosis with dronabinol: four case reports. J Clin Psychiatry. 2010;71(11):1552-1553.
10. Schwarz G, Karajgi B, McCarthy R. Synthetic delta-9-tetrahydrocannabinol (dronabinol) can improve the symptoms of schizophrenia. J Clin Psychopharmacol. 2009;29(3):255-258.
11. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry. 2004;161(6):975-984.
12. Boggs DL, Kelly DL, McMahon RP, et al. Rimonabant for neurocognition in schizophrenia: a 16-week double blind placebo controlled trial. Schizophr Res. 2012;134(2-3):207-210.
13. Kelly DL, Gorelick DA, Conley RR, et al. Effects of cannabinoid-1 receptor antagonist rimonabant on psychiatric symptoms in overweight people with schizophrenia: a randomized, double-blind, pilot study. J Clin Psychopharmacol. 2011;31(1):86-91.
14. Leweke FM, Mueller JK, Lange B, et al. Therapeutic potential of cannabinoids in psychosis. Biol Psychiatry. 2016;79(7):604-612.
15. Halperin A. What is CBD? The ‘miracle’ cannabis compound that doesn’t get you high. The Guardian. https://www.theguardian.com/society/2018/may/28/what-is-cbd-cannabidiol-cannabis-medical-uses. Published May 28, 2018. Accessed April 3, 2019.
16. Pierre J. Coca, cola, and cannabis: psychoactive drugs as beverages. Psychology Today (blog) Psych Unseen. https://www.psychologytoday.com/us/blog/psych-unseen/201810/coca-cola-and-cannabis-psychoactive-drugs-beverages. Published October 1, 2018. Accessed April 3, 2019.
17. U.S. Food and Drug Administration. FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy. FDA News Release. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm611046.htm. Published June 25, 2018. Accessed April 3, 2019.
18. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011-2020.
19. Thiele EA, March ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085-1096.
20. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. 2018;378:1888-1897.
21. Khoury JM, Neves MCLD, Rogue MAV, et al. Is there a role of cannabidiol in psychiatry? World J Biol Psychiatry. 2017:1-16.
22. Zuardi AW, Morais SL, Guimares FS, et al. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56(10):485-486.
23. Zuardi AW, Hallak JEC, Dursun SM. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. 2006;20(5):683-686.
24. Leweke FM, Piomelli D, Pahlisch F, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15.
25. McGuire P, Robson P, Cubala WJ, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175(3):225-231.
26. Boggs DL, Surti I, Gupta A, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacol. 2018;235(7):1923-1932.
27. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016; 79(7):613-619.
28. Vandrey R, Raber JC, Raber ME, et al. Cannabinoid dose and label accuracy in edible medical cannabis products. JAMA. 2015;313(24):2491-2492.
29. Ruth AC, Gryniewicz-Ruzicka CM, Trehy ML, et al. Consistency of label claims of internet-purchased hemp oil and cannabis products as determined using IMS and LC-MS: a marketplace study. J Reg Sci. 2016;3:1-6.
30. Bonn-Miller MO, Loflin MJE, Thomas BF, et al. Labeling accuracy of cannabidiol extracts sold online. JAMA. 2017;318(17):1708-1709.
31. Seeman P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl Psychiatry. 2016;6(10):e920. doi: 10.1038/tp.2016.195.
32. Renard J, Norris C, Rushlow W, et al. Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: implications for novel schizophrenia treatments. Neurosci Biobehav Rev. 2017;157-165.
33. Gururajan A, Malone DT. Does cannabidiol have a role in the treatment of schizophrenia? Schizophr Res. 2016;176(2-3):281-290.
1. Pierre JM. Cannabis, synthetic cannabinoids, and psychosis risk: what the evidence says. Current Psychiatry. 2011;10(9):49-58.
2. Radhakrishan R, Wilkinson ST, D’Souza DC. Gone to pot – a review of the association between cannabis and psychosis. Front Psychiatry. 2014;5:54.
3. Pierre JM. Risks of increasingly potent cannabis: joint effects of potency and frequency. Current Psychiatry. 2016;16(2):14-20.
4. Hanna RC, Perez JM, Ghose S. Cannabis and development of dual diagnoses: a literature review. Am J Drug Alcohol Abuse. 2017;43(4):442-255.
5. Nunberg H, Kilmer B, Pacula RL, et al. An analysis of applicants presenting to a medical marijuana specialty practice in California. J Drug Policy Anal. 2011;4(1):1.
6. Wilkinson ST, Radhakrishnan, D’Souza DC. A systematic review of the evidence for medical marijuana in psychiatric indications. J Clin Psychiatry. 2016;77(8):1050-1064.
7. Tournebize J, Gibaja V, Kahn JP. Acute effects of synthetic cannabinoids: Update 2015. Subst Abus. 2016;38(3):344-366.
8. Crippa JA, Guimarães FS, Campos A, et al. Translational investigation of the therapeutic potential of cannabidiol (CBD): toward a new age. Front Immunol. 2018;9:2009.
9. Schwarz G, Karajgi B. Improvement in refractory psychosis with dronabinol: four case reports. J Clin Psychiatry. 2010;71(11):1552-1553.
10. Schwarz G, Karajgi B, McCarthy R. Synthetic delta-9-tetrahydrocannabinol (dronabinol) can improve the symptoms of schizophrenia. J Clin Psychopharmacol. 2009;29(3):255-258.
11. Meltzer HY, Arvanitis L, Bauer D, et al. Placebo-controlled evaluation of four novel compounds for the treatment of schizophrenia and schizoaffective disorder. Am J Psychiatry. 2004;161(6):975-984.
12. Boggs DL, Kelly DL, McMahon RP, et al. Rimonabant for neurocognition in schizophrenia: a 16-week double blind placebo controlled trial. Schizophr Res. 2012;134(2-3):207-210.
13. Kelly DL, Gorelick DA, Conley RR, et al. Effects of cannabinoid-1 receptor antagonist rimonabant on psychiatric symptoms in overweight people with schizophrenia: a randomized, double-blind, pilot study. J Clin Psychopharmacol. 2011;31(1):86-91.
14. Leweke FM, Mueller JK, Lange B, et al. Therapeutic potential of cannabinoids in psychosis. Biol Psychiatry. 2016;79(7):604-612.
15. Halperin A. What is CBD? The ‘miracle’ cannabis compound that doesn’t get you high. The Guardian. https://www.theguardian.com/society/2018/may/28/what-is-cbd-cannabidiol-cannabis-medical-uses. Published May 28, 2018. Accessed April 3, 2019.
16. Pierre J. Coca, cola, and cannabis: psychoactive drugs as beverages. Psychology Today (blog) Psych Unseen. https://www.psychologytoday.com/us/blog/psych-unseen/201810/coca-cola-and-cannabis-psychoactive-drugs-beverages. Published October 1, 2018. Accessed April 3, 2019.
17. U.S. Food and Drug Administration. FDA approves first drug comprised of an active ingredient derived from marijuana to treat rare, severe forms of epilepsy. FDA News Release. https://www.fda.gov/newsevents/newsroom/pressannouncements/ucm611046.htm. Published June 25, 2018. Accessed April 3, 2019.
18. Devinsky O, Cross JH, Laux L, et al. Trial of cannabidiol for drug-resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011-2020.
19. Thiele EA, March ED, French JA, et al. Cannabidiol in patients with seizures associated with Lennox-Gastaut syndrome (GWPCARE4): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet. 2018;391(10125):1085-1096.
20. Devinsky O, Patel AD, Cross JH, et al. Effect of cannabidiol on drop seizures in the Lennox-Gastaut syndrome. N Engl J Med. 2018;378:1888-1897.
21. Khoury JM, Neves MCLD, Rogue MAV, et al. Is there a role of cannabidiol in psychiatry? World J Biol Psychiatry. 2017:1-16.
22. Zuardi AW, Morais SL, Guimares FS, et al. Antipsychotic effect of cannabidiol. J Clin Psychiatry. 1995;56(10):485-486.
23. Zuardi AW, Hallak JEC, Dursun SM. Cannabidiol monotherapy for treatment-resistant schizophrenia. J Psychopharmacol. 2006;20(5):683-686.
24. Leweke FM, Piomelli D, Pahlisch F, et al. Cannabidiol enhances anandamide signaling and alleviates psychotic symptoms of schizophrenia. Transl Psychiatry. 2012;2:e94. doi: 10.1038/tp.2012.15.
25. McGuire P, Robson P, Cubala WJ, et al. Cannabidiol (CBD) as an adjunctive therapy in schizophrenia: a multicenter randomized controlled trial. Am J Psychiatry. 2018;175(3):225-231.
26. Boggs DL, Surti I, Gupta A, et al. The effects of cannabidiol (CBD) on cognition and symptoms in outpatients with chronic schizophrenia a randomized placebo controlled trial. Psychopharmacol. 2018;235(7):1923-1932.
27. ElSohly MA, Mehmedic Z, Foster S, et al. Changes in cannabis potency over the last 2 decades (1995-2014): analysis of current data in the United States. Biol Psychiatry. 2016; 79(7):613-619.
28. Vandrey R, Raber JC, Raber ME, et al. Cannabinoid dose and label accuracy in edible medical cannabis products. JAMA. 2015;313(24):2491-2492.
29. Ruth AC, Gryniewicz-Ruzicka CM, Trehy ML, et al. Consistency of label claims of internet-purchased hemp oil and cannabis products as determined using IMS and LC-MS: a marketplace study. J Reg Sci. 2016;3:1-6.
30. Bonn-Miller MO, Loflin MJE, Thomas BF, et al. Labeling accuracy of cannabidiol extracts sold online. JAMA. 2017;318(17):1708-1709.
31. Seeman P. Cannabidiol is a partial agonist at dopamine D2High receptors, predicting its antipsychotic clinical dose. Transl Psychiatry. 2016;6(10):e920. doi: 10.1038/tp.2016.195.
32. Renard J, Norris C, Rushlow W, et al. Neuronal and molecular effects of cannabidiol on the mesolimbic dopamine system: implications for novel schizophrenia treatments. Neurosci Biobehav Rev. 2017;157-165.
33. Gururajan A, Malone DT. Does cannabidiol have a role in the treatment of schizophrenia? Schizophr Res. 2016;176(2-3):281-290.
 for schizophrenia: Promise or pipe dream?")