User login
-
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
Study: COVID cases have been ‘severely undercounted’
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
Large numbers of COVID-19 cases have been undetected and unreported, which has resulted in severe undercounting of the total number of people who have been infected during the pandemic, according to a new study published Monday in the journal PLOS ONE.
In the United States, the number of COVID-19 cases is likely three times that of reported cases. According to the study, more than 71 million Americans have contracted the virus during the pandemic, and 7 million were infected or potentially contagious last week.
Public health officials rely on case counts to guide decisions, so the undercounting should be considered while trying to end the pandemic.
“The estimates of actual infections reveal for the first time the true severity of COVID-19 across the U.S. and in countries worldwide,” Jungsik Noh, PhD, a bioinformatics professor at the University of Texas Southwestern Medical Center, said in a statement.
Dr. Noh and colleague Gaudenz Danuser created a computational model that uses machine-learning strategies to estimate the actual number of daily cases in the United States and the 50 most-infected countries.
The model pulls data from the Johns Hopkins University database and the COVID Tracking Project, as well as large-scale surveys conducted by the CDC and several states. The algorithm uses the number of reported deaths, which is thought to be more accurate than the number of lab-confirmed cases, as the basis for calculations.
In 25 of the 50 countries, the “actual” cumulative cases were estimated to be 5-20 times greater than the confirmed cases. In the United States, Belgium, and Brazil, about 10% of the population has contracted the coronavirus, according to the model. At the beginning of February, about 11% of the population in Pennsylvania had current infections, which was the highest rate of any state. About 0.15% of residents in Minnesota had infections, and about 2.5% of residents in New York and Texas had infections.
“Knowing the true severity in different regions will help us effectively fight against the virus spreading,” Dr. Noh said. “The currently infected population is the cause of future infections and deaths. Its actual size in a region is a crucial variable required when determining the severity of COVID-19 and building strategies against regional outbreaks.”
A version of this article first appeared on WebMD.com.
COVID-19: Peginterferon lambda may prevent clinical deterioration, shorten viral shedding
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.

Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
and shorten the duration of viral shedding, according to results of a double-blind, placebo-controlled trial (NCT04354259).
Reductions in severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) RNA were greater with peginterferon lambda than with placebo from day 3 onward in the phase 2 study led by Jordan J. Feld, MD, of the Toronto Centre for Liver Disease. The findings were reported in The Lancet Respiratory Medicine.
Fewer side effects
To date in randomized clinical trials, efficacy in treatment of COVID-19 has been shown only for remdesivir and dexamethasone in hospitalized patients, and in an interim analysis of accelerated viral clearance for a monoclonal antibody infusion in outpatients.
Activity against respiratory pathogens has been demonstrated for interferon lambda-1, a type III interferon shown to be involved in innate antiviral responses. Interferons, Dr. Feld and coauthors stated, drive induction of genes with antiviral, antiproliferative and immunoregulatory properties, and early treatment with interferons might halt clinical progression and shorten the duration of viral shedding with reduced onward transmission. In addition, interferon lambdas (type III) use a distinct receptor complex with high expression levels limited to epithelial cells in the lung, liver, and intestine, leading to fewer side effects than other interferons, including avoiding risk of promoting cytokine storm syndrome.
The researchers investigated peginterferon lambda safety and efficacy in treatment of patients with laboratory-confirmed, mild to moderate COVID-19. Sixty patients (median age 46 years, about 60% female, about 50% White) were recruited from outpatient testing centers at six institutions in Toronto, and referred to a single ambulatory site. Patients were randomly assigned 1:1 to a single subcutaneous injection of peginterferon lambda 180 mcg or placebo within 7 days of symptom onset or, if asymptomatic, of their first positive swab. Mean time from symptom onset to injection was about 4.5 days, and about 18.5% were asymptomatic. The primary outcome was the proportion of patients negative for SARS-CoV-2 RNA on day 7 after the injection.
Greater benefit with higher baseline load
A higher baseline SARS-CoV-2 RNA concentration found in the peginterferon lambda group was found to be significantly associated with day 7 clearance (odds ratio [OR] 0.69 [95% confidence interval 0.51-0.87]; P = ·001). In the peginterferon lambda group, also, the mean decline in SARS-CoV-2 RNA was significantly larger than in the placebo group across all time points (days 3, 5, 7, and14). While viral load decline was 0.81 log greater in the treatment group (P = .14) by day 3, viral load decline increased to 1.67 log copies per mL by day 5 (P = .013) and 2.42 log copies per mL by day 7 (P = .0041). At day 14, the viral decline was 1.77 log copies per mL larger in the peginterferon lambda group (P = .048). The investigators pointed out that the difference in viral load decline between groups was greater in patients with high baseline viral load (at or above 106 copies per mL). In the peginterferon lambda high baseline viral load group, the reduction was 7.17 log copies per mL, versus 4.92 log copies per mL in the placebo group (P = .004).
More patients SARS-CoV-2 RNA negative
By day 7, 80% of patients in the peginterferon lambda group were negative for SARS-CoV-2 RNA, compared with 63% in the placebo group (P = .15). After baseline load adjustment, however, the peginterferon lambda treatment was significantly associated with day 7 clearance (OR 4·12 [95% CI 1·15-16·73]; P = .029).
Respiratory symptoms improved faster
Most symptoms in both groups were mild to moderate, without difference in frequency or severity. While symptom improvement was generally similar over time for both groups, respiratory symptoms improved faster with peginterferon lambda, with the effect more pronounced in the high baseline viral load group (OR 5·88 (0·81-42·46; P =. 079).
Laboratory adverse events, similar for both groups, were mild.
“Peginterferon lambda has potential to prevent clinical deterioration and shorten duration of viral shedding,” the investigators concluded.
“This clinical trial is important” because it suggests that a single intravenous dose of peginterferon lambda administered to outpatients with a positive SARS-CoV-2 nasopharyngeal swab speeds reduction of SARS-CoV-2 viral load, David L. Bowton, MD, FCCP, professor emeritus, Wake Forest Baptist Health, Winston-Salem, N.C., said in an interview. He observed that the smaller viral load difference observed at day 14 likely reflects host immune responses.
Dr. Bowton also noted that two placebo group baseline characteristics (five placebo group patients with anti-SARS-CoV-2 S protein IgG antibodies; two times more undetectable SARS-CoV-2 RNA at baseline assessment) would tend to reduce differences between the peginterferon lambda and placebo groups. He added that the study findings were concordant with another phase 2 trial of hospitalized COVID-19 patients receiving inhaled interferon beta-1a.
“Thus, interferons may find a place in the treatment of COVID-19 and perhaps other severe viral illnesses,” Dr. Bowton said.
The study was funded by the Toronto COVID-19 Action Initiative, University of Toronto, and the Ontario First COVID-19 Rapid Research Fund, Toronto General & Western Hospital Foundation.
Dr. Bowton had no disclosures. Disclosures for Dr. Feld and coauthors are listed on the Lancet Respiratory Medicine website.
FROM THE LANCET RESPIRATORY MEDICINE
Teenagers get in the queue for COVID-19 vaccines
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
The vaccinations can’t come soon enough for parents like Stacy Hillenburg, a developmental therapist in Aurora, Ill., whose 9-year-old son takes immunosuppressants because he had a heart transplant when he was 7 weeks old. Although school-age children aren’t yet included in clinical trials, if her 12- and 13-year-old daughters could get vaccinated, along with both parents, then the family could relax some of the protocols they currently follow to prevent infection.
Whenever they are around other people, even masked and socially distanced, they come home and immediately shower and change their clothes. So far, no one in the family has been infected with COVID, but the anxiety is ever-present. “I can’t wait for it to come out,” Ms. Hillenburg said of a pediatric COVID vaccine. “It will ease my mind so much.”
She isn’t alone in that anticipation. In the fall, the American Academy of Pediatrics and other pediatric vaccine experts urged faster action on pediatric vaccine trials and worried that children would be left behind as adults gained protection from COVID. But recent developments have eased those concerns.
“Over the next couple of months, we will be doing trials in an age-deescalation manner,” with studies moving gradually to younger children, Anthony S. Fauci, MD, chief medical adviser on COVID-19 to the president, said in a coronavirus response team briefing on Jan. 29. “So that hopefully, as we get to the late spring and summer, we will have children being able to be vaccinated.”
Pfizer completed enrollment of 2,259 teens aged 12-15 years in late January and expects to move forward with a separate pediatric trial of children aged 5-11 years by this spring, Keanna Ghazvini, senior associate for global media relations at Pfizer, said in an interview.
Enrollment in Moderna’s TeenCove study of adolescents ages 12-17 years began slowly in late December, but the pace has since picked up, said company spokesperson Colleen Hussey. “We continue to bring clinical trial sites online, and we are on track to provide updated data around mid-year 2021.” A trial extension in children 11 years and younger is expected to begin later in 2021.
Johnson & Johnson and AstraZeneca said they expect to begin adolescent trials in early 2021, according to data shared by the Advisory Committee on Immunization Practices. An interim analysis of J&J’s Janssen COVID-19 vaccine trial data, released on Jan. 29, showed it was 72% effective in US participants aged 18 years or older. AstraZeneca’s U.S. trial in adults is ongoing.
Easing the burden
Vaccination could lessen children’s risk of severe disease as well as the social and emotional burdens of the pandemic, says James Campbell, MD, a pediatric infectious disease specialist at the University of Maryland’s Center for Vaccine Development in Baltimore, which was involved in the Moderna and early-phase Pfizer trials. He coauthored a September 2020 article in Clinical Infectious Diseases titled: “Warp Speed for COVID-19 vaccines: Why are children stuck in neutral?”
The adolescent trials are a vital step to ensure timely vaccine access for teens and younger children. “It is reasonable, when you have limited vaccine, that your rollout goes to the highest priority and then moves to lower and lower priorities. In adults, we’re just saying: ‘Wait your turn,’ ” he said of the current vaccination effort. “If we didn’t have the [vaccine trial] data in children, we’d be saying: ‘You don’t have a turn.’ ”
As the pandemic has worn on, the burden on children has grown. As of Tuesday, 269 children had died of COVID-19. That is well above the highest annual death toll recorded during a regular flu season – 188 flu deaths among children and adolescents under 18 in the 2019-2020 and 2017-2018 flu seasons.
Children are less likely to transmit COVID-19 in their household than adults, according to a meta-analysis of 54 studies published in JAMA Network Open. But that does not necessarily mean children are less infectious, the authors said, noting that unmeasured factors could have affected the spread of infection among adults.
Moreover, children and adolescents need protection from COVID infection – and from the potential for severe disease or lingering effects – and, given that there are 74 million children and teens in the United States, their vaccination is an important part of stopping the pandemic, said Grace Lee, MD, professor of pediatrics at Stanford (Calif.) University, and cochair of ACIP’s COVID-19 Vaccine Safety Technical Subgroup.
“In order to interrupt transmission, I don’t see how we’re going to do that without vaccinating children and adolescents,” she said.
Dr. Lee said her 16-year-old daughter misses the normal teenage social life and is excited about getting the vaccine when she is eligible. (Adolescents without high-risk conditions are in the lowest vaccination tier, according to ACIP recommendations.) “There is truly individual protection to be gained,” Dr. Lee said.
She noted that researchers continue to assess the immune responses to the adult vaccines – even looking at immune characteristics of the small percentage of people who aren’t protected from infection – and that information helps in the evaluation of the pediatric immune responses. As the trials expand to younger children and infants, dosing will be a major focus. “How many doses do they need they need to receive the same immunity? Safety considerations will be critically important,” she said.
Teen trials underway
Pfizer/BioNTech extended its adult trial to 16- and 17-year-olds in October, which enabled older teens to be included in its emergency-use authorization. They and younger teens, ages 12-15, receive the same dose as adults.
The ongoing trials with Pfizer and Moderna vaccines are immunobridging trials, designed to study safety and immunogenicity. Investigators will compare the teens’ immune response with the findings from the larger adult trials. When the trials expand to school-age children (6-12 years), protocols call for testing the safety and immunogenicity of a half-dose vaccine as well as the full dose.
Children ages 2-5 years and infants and toddlers will be enrolled in future trials, studying safety and immunogenicity of full, half, or even quarter dosages. The Pediatric Research Equity Act of 2003 requires licensed vaccines to be tested for safety and efficacy in children, unless they are not appropriate for a pediatric population.
Demand for the teen trials has been strong. At Cincinnati Children’s Hospital Medical Center, 259 teenagers joined the Pfizer/BioNTech trial, but some teenagers were turned away when the trial’s national enrollment closed in late January.
“Many of the children are having no side effects, and if they are, they’re having the same [effects] as the young adults – local redness or pain, fatigue, and headaches,” said Robert Frenck, MD, director of the Cincinnati Children’s Gamble Program for Clinical Studies.
Parents may share some of the vaccine hesitancy that has affected adult vaccination. But that is balanced by the hope that vaccines will end the pandemic and usher in a new normal. “If it looks like [vaccines] will increase the likelihood of children returning to school safely, that may be a motivating factor,” Dr. Frenck said.
Cody Meissner, MD, chief of the pediatric infectious disease service at Tufts Medical Center, Boston, was initially cautious about the extension of vaccination to adolescents. A member of the Vaccine and Related Biological Products Advisory Committee, which evaluates data and makes recommendations to the Food and Drug Administration, Dr. Meissner initially abstained in the vote on the Pfizer/BioNTech emergency-use authorization for people 16 and older.
He noted that, at the time the committee reviewed the Pfizer vaccine, the company had data available for just 134 teenagers, half of whom received a placebo. But the vaccination of 34 million adults has provided robust data about the vaccine’s safety, and the trial expansion into adolescents is important.
“I’m comfortable with the way these trials are going now,” he said. “This is the way I was hoping they would go.”
Ms. Hillenburg is on the parent advisory board of Voices for Vaccines, an organization of parents supporting vaccination that is affiliated with the Task Force for Global Health, an Atlanta-based independent public health organization. Dr. Campbell’s institution has received funds to conduct clinical trials from the National Institutes of Health and several companies, including Merck, GlaxoSmithKline, Sanofi, Pfizer, and Moderna. He has served pro bono on many safety and data monitoring committees. Dr. Frenck’s institution is receiving funds to conduct the Pfizer trial. In the past 5 years, he has also participated in clinical trials for GlaxoSmithKline, Merck, and Meissa vaccines. Dr. Lee and Dr. Meissner disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Model could reduce some disparities in lung cancer screening
New research suggests that proposed lung cancer screening guidelines could inadvertently increase racial and ethnic disparities, but adding in a risk prediction model could reduce some of these disparities by identifying people with high predicted benefit, regardless of race or ethnicity.
The draft United States Preventive Services Task Force (USPSTF) 2020 guidelines recommend annual lung cancer screening for individuals aged 50-80 who currently smoke or quit in the last 15 years, and who have a smoking history equivalent to at least one pack of cigarettes per day for 20 years or more.
This expands the age range and smoking history requirement compared to the 2013 USPSTF recommendations in an attempt to partially ameliorate racial disparities in screening eligibility. The 2013 guidelines recommend screening ever-smokers aged 55-80 with 30 or more pack-years and 15 or fewer quit-years.
However, neither the 2013 nor the 2020 USPSTF recommendations consider the higher risk of lung cancer and younger ages at diagnosis among African Americans, despite their smoking less than Whites, according to Rebecca Landy, PhD, of the National Cancer Institute in Bethesda, Md.
“For the same age and smoking history as Whites, minorities have substantially different lung cancer risk,” Dr. Landy said. “Incorporating individualized prediction models into USPSTF guidelines may reduce racial/ethnic disparities in lung cancer screening eligibility.”
Dr. Landy and colleagues set out to test that theory, and she presented the results at the 2020 World Congress on Lung Cancer (Abstract 3564), which was rescheduled for January 2021. The results were published in the Journal of the National Cancer Institute.
Study details
Dr. Landy and colleagues modeled the performance of National Lung Screening Trial–like screening (three annual CT screens, 5 years of follow-up) among three cohorts of ever-smokers aged 50-80 using the 2015 National Health Interview Survey.
One group was eligible by USPSTF 2013 guidelines, another by draft USPSTF 2020 guidelines, and yet another by augmenting the USPSTF 2020 guidelines using risk prediction to include individuals with 12 or more days of life gained according to the Life-Years From Screening–CT (LYFS-CT) model.
“Among each race/ethnicity, we calculated the number eligible for screening, proportion of preventable lung cancer deaths prevented, proportion of gainable life-years gained, and screening effectiveness, as well as the relative disparities in lung cancer deaths prevented and life-years gained,” Dr. Landy said.
Results
Under the 2013 guidelines, 8 million ever-smokers were eligible. The disparities in lung cancer death sensitivity, compared to Whites, were 15% for African Americans, 15% for Asian Americans, and 24% for Hispanic Americans. Disparities for life-year gained sensitivity were 15%, 13%, and 24%, respectively.
Under the 2020 draft guidelines, 14.5 million ever-smokers were eligible, but racial/ethnic disparities persisted. Disparities in lung cancer death sensitivity were 13% for African Americans, 19% for Asian Americans, and 27% for Hispanic Americans. Disparities for life-year gained sensitivity were 16%, 19%, and 27%, respectively.
Using the LYFS-CT predictive-risk model added an additional 3.5 million people and “nearly eliminated” disparities for African Americans, Dr. Landy noted. However, disparities persisted for Asian Americans and Hispanic Americans.
Disparities in lung cancer death sensitivity were 0% for African Americans, 19% for Asian Americans, and 23% for Hispanic Americans. Disparities for life-year gained sensitivity were 1%, 19%, and 24%, respectively.
More and widening disparity
The results showed that augmenting USPSTF criteria to include high-benefit people selected significantly more African Americans than Whites and could therefore reduce or even eliminate disparities between Whites and African Americans.
“The 2020 USPSTF draft recommendations would make 6.5 million more people eligible to be screened, in addition to the 8 million from the 2013 criteria,” said Gerard Silvestri, MD, of the Medical University of South Carolina, Charleston, who was not involved in this study.
“But there will be more White people than African American people added, and the disparity between them may widen. Using the risk prediction model outlined in this well-researched study could close the gap in disparity. It’s important to identify individual risk and life expectancy.”
Dr. Silvestri pointed out that, compared to Whites, African Americans develop lung cancer at an earlier age with fewer pack-years history of smoking and have worse outcomes.
“We can’t just focus on one aspect of disparity,” he said. “African Americans are much less likely to be insured or to identify a primary care provider for integrated care. We know that screening works. The 2020 USPSTF draft recommendations will enlarge the pool of eligible African Americans and reduce disparities if the other part of the equation holds; that is, they get access to care and screening.”
This study was funded by the National Institutes of Health/National Cancer Institute. Dr. Landy and Dr. Silvestri have no disclosures.
New research suggests that proposed lung cancer screening guidelines could inadvertently increase racial and ethnic disparities, but adding in a risk prediction model could reduce some of these disparities by identifying people with high predicted benefit, regardless of race or ethnicity.
The draft United States Preventive Services Task Force (USPSTF) 2020 guidelines recommend annual lung cancer screening for individuals aged 50-80 who currently smoke or quit in the last 15 years, and who have a smoking history equivalent to at least one pack of cigarettes per day for 20 years or more.
This expands the age range and smoking history requirement compared to the 2013 USPSTF recommendations in an attempt to partially ameliorate racial disparities in screening eligibility. The 2013 guidelines recommend screening ever-smokers aged 55-80 with 30 or more pack-years and 15 or fewer quit-years.
However, neither the 2013 nor the 2020 USPSTF recommendations consider the higher risk of lung cancer and younger ages at diagnosis among African Americans, despite their smoking less than Whites, according to Rebecca Landy, PhD, of the National Cancer Institute in Bethesda, Md.
“For the same age and smoking history as Whites, minorities have substantially different lung cancer risk,” Dr. Landy said. “Incorporating individualized prediction models into USPSTF guidelines may reduce racial/ethnic disparities in lung cancer screening eligibility.”
Dr. Landy and colleagues set out to test that theory, and she presented the results at the 2020 World Congress on Lung Cancer (Abstract 3564), which was rescheduled for January 2021. The results were published in the Journal of the National Cancer Institute.
Study details
Dr. Landy and colleagues modeled the performance of National Lung Screening Trial–like screening (three annual CT screens, 5 years of follow-up) among three cohorts of ever-smokers aged 50-80 using the 2015 National Health Interview Survey.
One group was eligible by USPSTF 2013 guidelines, another by draft USPSTF 2020 guidelines, and yet another by augmenting the USPSTF 2020 guidelines using risk prediction to include individuals with 12 or more days of life gained according to the Life-Years From Screening–CT (LYFS-CT) model.
“Among each race/ethnicity, we calculated the number eligible for screening, proportion of preventable lung cancer deaths prevented, proportion of gainable life-years gained, and screening effectiveness, as well as the relative disparities in lung cancer deaths prevented and life-years gained,” Dr. Landy said.
Results
Under the 2013 guidelines, 8 million ever-smokers were eligible. The disparities in lung cancer death sensitivity, compared to Whites, were 15% for African Americans, 15% for Asian Americans, and 24% for Hispanic Americans. Disparities for life-year gained sensitivity were 15%, 13%, and 24%, respectively.
Under the 2020 draft guidelines, 14.5 million ever-smokers were eligible, but racial/ethnic disparities persisted. Disparities in lung cancer death sensitivity were 13% for African Americans, 19% for Asian Americans, and 27% for Hispanic Americans. Disparities for life-year gained sensitivity were 16%, 19%, and 27%, respectively.
Using the LYFS-CT predictive-risk model added an additional 3.5 million people and “nearly eliminated” disparities for African Americans, Dr. Landy noted. However, disparities persisted for Asian Americans and Hispanic Americans.
Disparities in lung cancer death sensitivity were 0% for African Americans, 19% for Asian Americans, and 23% for Hispanic Americans. Disparities for life-year gained sensitivity were 1%, 19%, and 24%, respectively.
More and widening disparity
The results showed that augmenting USPSTF criteria to include high-benefit people selected significantly more African Americans than Whites and could therefore reduce or even eliminate disparities between Whites and African Americans.
“The 2020 USPSTF draft recommendations would make 6.5 million more people eligible to be screened, in addition to the 8 million from the 2013 criteria,” said Gerard Silvestri, MD, of the Medical University of South Carolina, Charleston, who was not involved in this study.
“But there will be more White people than African American people added, and the disparity between them may widen. Using the risk prediction model outlined in this well-researched study could close the gap in disparity. It’s important to identify individual risk and life expectancy.”
Dr. Silvestri pointed out that, compared to Whites, African Americans develop lung cancer at an earlier age with fewer pack-years history of smoking and have worse outcomes.
“We can’t just focus on one aspect of disparity,” he said. “African Americans are much less likely to be insured or to identify a primary care provider for integrated care. We know that screening works. The 2020 USPSTF draft recommendations will enlarge the pool of eligible African Americans and reduce disparities if the other part of the equation holds; that is, they get access to care and screening.”
This study was funded by the National Institutes of Health/National Cancer Institute. Dr. Landy and Dr. Silvestri have no disclosures.
New research suggests that proposed lung cancer screening guidelines could inadvertently increase racial and ethnic disparities, but adding in a risk prediction model could reduce some of these disparities by identifying people with high predicted benefit, regardless of race or ethnicity.
The draft United States Preventive Services Task Force (USPSTF) 2020 guidelines recommend annual lung cancer screening for individuals aged 50-80 who currently smoke or quit in the last 15 years, and who have a smoking history equivalent to at least one pack of cigarettes per day for 20 years or more.
This expands the age range and smoking history requirement compared to the 2013 USPSTF recommendations in an attempt to partially ameliorate racial disparities in screening eligibility. The 2013 guidelines recommend screening ever-smokers aged 55-80 with 30 or more pack-years and 15 or fewer quit-years.
However, neither the 2013 nor the 2020 USPSTF recommendations consider the higher risk of lung cancer and younger ages at diagnosis among African Americans, despite their smoking less than Whites, according to Rebecca Landy, PhD, of the National Cancer Institute in Bethesda, Md.
“For the same age and smoking history as Whites, minorities have substantially different lung cancer risk,” Dr. Landy said. “Incorporating individualized prediction models into USPSTF guidelines may reduce racial/ethnic disparities in lung cancer screening eligibility.”
Dr. Landy and colleagues set out to test that theory, and she presented the results at the 2020 World Congress on Lung Cancer (Abstract 3564), which was rescheduled for January 2021. The results were published in the Journal of the National Cancer Institute.
Study details
Dr. Landy and colleagues modeled the performance of National Lung Screening Trial–like screening (three annual CT screens, 5 years of follow-up) among three cohorts of ever-smokers aged 50-80 using the 2015 National Health Interview Survey.
One group was eligible by USPSTF 2013 guidelines, another by draft USPSTF 2020 guidelines, and yet another by augmenting the USPSTF 2020 guidelines using risk prediction to include individuals with 12 or more days of life gained according to the Life-Years From Screening–CT (LYFS-CT) model.
“Among each race/ethnicity, we calculated the number eligible for screening, proportion of preventable lung cancer deaths prevented, proportion of gainable life-years gained, and screening effectiveness, as well as the relative disparities in lung cancer deaths prevented and life-years gained,” Dr. Landy said.
Results
Under the 2013 guidelines, 8 million ever-smokers were eligible. The disparities in lung cancer death sensitivity, compared to Whites, were 15% for African Americans, 15% for Asian Americans, and 24% for Hispanic Americans. Disparities for life-year gained sensitivity were 15%, 13%, and 24%, respectively.
Under the 2020 draft guidelines, 14.5 million ever-smokers were eligible, but racial/ethnic disparities persisted. Disparities in lung cancer death sensitivity were 13% for African Americans, 19% for Asian Americans, and 27% for Hispanic Americans. Disparities for life-year gained sensitivity were 16%, 19%, and 27%, respectively.
Using the LYFS-CT predictive-risk model added an additional 3.5 million people and “nearly eliminated” disparities for African Americans, Dr. Landy noted. However, disparities persisted for Asian Americans and Hispanic Americans.
Disparities in lung cancer death sensitivity were 0% for African Americans, 19% for Asian Americans, and 23% for Hispanic Americans. Disparities for life-year gained sensitivity were 1%, 19%, and 24%, respectively.
More and widening disparity
The results showed that augmenting USPSTF criteria to include high-benefit people selected significantly more African Americans than Whites and could therefore reduce or even eliminate disparities between Whites and African Americans.
“The 2020 USPSTF draft recommendations would make 6.5 million more people eligible to be screened, in addition to the 8 million from the 2013 criteria,” said Gerard Silvestri, MD, of the Medical University of South Carolina, Charleston, who was not involved in this study.
“But there will be more White people than African American people added, and the disparity between them may widen. Using the risk prediction model outlined in this well-researched study could close the gap in disparity. It’s important to identify individual risk and life expectancy.”
Dr. Silvestri pointed out that, compared to Whites, African Americans develop lung cancer at an earlier age with fewer pack-years history of smoking and have worse outcomes.
“We can’t just focus on one aspect of disparity,” he said. “African Americans are much less likely to be insured or to identify a primary care provider for integrated care. We know that screening works. The 2020 USPSTF draft recommendations will enlarge the pool of eligible African Americans and reduce disparities if the other part of the equation holds; that is, they get access to care and screening.”
This study was funded by the National Institutes of Health/National Cancer Institute. Dr. Landy and Dr. Silvestri have no disclosures.
FROM WCLC 2020
Some COVID-19 vaccine reactions could be pseudoallergic, experts say
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.
On Jan. 13, 2 days after a drive-through vaccination “superstation” opened in San Diego, six people were treated for anaphylaxis after they received the Moderna vaccine, leading the California state epidemiologist to recommend pausing the administration of that particular lot.
A group of allergy and immunology experts and public health officials reviewed the cases, as well as an incident that occurred the day before, and concluded that at least some of the responses were angioedema, or swelling — a serious allergic reaction — but none were actually anaphylaxis. No similar clusters had occurred with the same vaccine lot in other states, and California resumed using the doses.
Yet questions remain about the reactions and the mechanisms for them. Some might have been triggered by an allergy to a vaccine component, most likely the polyethylene glycol (PEG) that stabilizes the lipid surrounding the mRNA, the key vaccine component in both the Moderna and Pfizer vaccines. Another possible explanation is that some could be pseudoallergic reactions to a blood protein known as complement, a little-understood process that resembles an antigen-based reaction but doesn’t leave an immune memory and might not recur.
Cases of complement-activation-related pseudoallergy look like a severe allergic reaction but occur through a different mechanism and don’t require previous exposure to an allergen.
“It has the same signs and symptoms and is treated the same way, but it occurs through a different pathway,” explained Neal Halsey, MD, director emeritus of the Institute for Vaccine Safety and emeritus professor at the Johns Hopkins Bloomberg School of Public Health in Baltimore.
Pseudoallergies are not well understood, but they have been associated with reactions to the contrast media used in imaging, such as with MRI. “If people have had an anaphylaxis-type reaction following the injection of contrast-dye material, that is a strong signal that it might be a complement-activation-related pseudoallergy,” said Dr. Halsey, a member of the Clinical Immunization Safety Assessment Network. “Those are the people who definitely need to consider seeing an allergist before getting the COVID vaccines.”
When Aleena Banerji, MD, clinical director of the allergy and clinical immunology unit at Massachusetts General Hospital in Boston, talks to patients about vaccine reactions, she addresses the risk for COVID-19 infection. All of the people who developed allergies after the Pfizer and Moderna vaccines recovered, but more than 445,000 Americans have died from COVID-19.
Most people with common allergies, such as to food or oral medications, don’t need to worry about reactions, said Dr. Banerji, lead author of a review that assessed the risk for allergic reactions to the Pfizer and Moderna vaccines.
Investigating reactions
As investigators search for the answers to what causes reactions, transparency is crucial to trust, said Kathryn Edwards, MD, principal investigator of the Clinical Immunization Safety Assessment Project, a vaccine safety network funded by the Centers for Disease Control and Prevention.
“Unless the public knows that we’re really investigating and we’re taking this seriously, then I think the vaccine hesitancy is going to increase,” said Dr. Edwards, professor of pediatrics at Vanderbilt University Medical Center and scientific director of the Vanderbilt Vaccine Research Program in Nashville, Tenn.
First reports of anaphylaxis came quickly after COVID-19 vaccinations began. In the 2 weeks before the holidays, almost 2 million health care workers received the Pfizer vaccine, and 21 of them developed anaphylaxis, according to CDC researchers who reviewed case reports from the Vaccine Adverse Event Reporting System (VAERS). That rate of about 1 in 100,000 is 10 times higher than the occurrence with other vaccines. No deaths from anaphylaxis were reported.
As the vaccinations ramped up, the rate declined. As of Jan. 18, 50 cases of anaphylaxis were reported to VAERS after the administration of 9,943,247 Pfizer doses, for a rate of 5.0 per million, according to data presented at the Jan. 27 meeting of the CDC Advisory Committee on Immunization Practices. And 21 cases of anaphylaxis were reported to VAERS after the administration of 7,581,429 Moderna doses, for a rate of 2.8 per million.
The anaphylaxis occurred almost exclusively in women; only three of the VAERS anaphylaxis reports were from men. Only 24% had a history of anaphylaxis.
The earlier CDC report explored the potential link to allergies. One person with anaphylaxis had a history of allergy to iodinated contrast media, and others had allergies to various medications, vaccines, foods, and animals. The researchers reported 86 nonanaphylaxis allergic reactions and 61 nonallergic adverse events among the 175 case reports they reviewed as possible cases of severe allergic reaction.
Of 1,266 reports that VAERS received from Dec. 21 to Jan. 10, the CDC identified 108 possible cases of severe allergic reaction after the Moderna vaccine. Only 10 met the case definition of anaphylaxis put forward by the Brighton Collaboration, a vaccine safety organization. All but one case involved a history of allergies or allergic reactions; only five had a previously experienced anaphylaxis.
There were 47 nonanaphylaxis allergic reactions.
The San Diego cluster also met the Brighton case definition for anaphylaxis, Dr. Edwards reported. This discrepancy highlights the difficulties in characterizing vaccine reactions.
Measuring a pseudoallergic reaction is a challenge. It requires that a blood sample be drawn soon after the incident and then frozen to protect heat-sensitive blood markers, Dr. Edwards explained.
And as vaccinations rise, so do adverse-event reports. But unlike in clinical trials, there is no control group for comparison. That is why vaccine safety experts urge caution when evaluating events and, where possible, advise looking at background rates.
“A major way to determine whether the adverse event is causally related is to assess the incidence of the adverse event in vaccines versus nonvaccines,” said Walter Orenstein, MD, who directed the U.S. Immunization Program from 1988 to 2004 and is now associate director of the Emory Vaccine Center and professor of infectious diseases at Emory University in Atlanta. Public health officials could then identify vaccine risk factors, he said.
When a reaction occurs almost immediately after vaccination, vaccine safety investigators look for probable triggers. If allergy to PEG is the culprit in anaphylactic reactions, then the individuals would have had a previous exposure, perhaps from injectable medications, Dr. Edwards said.
It might be feasible to perform a skin test for allergy to PEG. “If the skin testing is negative, that doesn’t completely rule out allergy, but it can be used in the decision-making about giving the first or second vaccine dose,” Dr. Banerji said.
Other vaccines, such as childhood vaccines, contain polysorbate as a stabilizer, which has a similar chemical structure, and it’s not clear why someone would react to PEG but not to polysorbate, Dr. Edwards said.
Meanwhile, other illnesses and even deaths sometimes occur in the days after vaccination, but that doesn’t mean the vaccine caused them, cautioned Steve Black, MD, emeritus professor of pediatrics at Cincinnati Children’s Hospital and cofounder of the Global Vaccine Data Network, an international vaccine safety collaboration.
“Different events and clusters of events will occur by chance alone, as these events can occur without vaccines. We need to not immediately assume that they’re due to the vaccine,” he said. “You don’t want to undermine the whole vaccine program every time something comes up and assume that it’s associated with the vaccine.”
The CDC only has three contraindications for the vaccines:
- Severe allergic reaction (such as anaphylaxis) after a previous dose of an mRNA COVID-19 vaccine or any of its components.
- Immediate allergic reaction of any severity to a previous dose of an mRNA COVID-19 vaccine or any of its components (including PEG).
- Immediate allergic reaction of any severity to polysorbate (due to potential cross-reactive hypersensitivity with PEG).
People who have had an immediate allergic reaction to other vaccines or injectable therapies should consider consulting with an allergist or immunologist before getting the Pfizer or Moderna vaccines, the CDC advises.
The CDC also says that people with a history of anaphylaxis from any cause should be observed for 30 minutes after vaccination. Vaccination protocol calls for everyone else to wait on site for 15 minutes after vaccination.
A version of this article first appeared on Medscape.com.
Inhaled hyaluronan may bring sigh of relief to COPD patients
(COPD), findings of a new study suggest.

HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.

“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.

Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
(COPD), findings of a new study suggest.
HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.
“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.
Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
(COPD), findings of a new study suggest.
HMW-HA was associated with a significantly shorter duration of noninvasive positive-pressure ventilation (NIPPV), lower systemic inflammatory markers, and lower measured peak airway pressure, compared with placebo, reported lead author Flavia Galdi, MD, of Campus Bio-Medico University Hospital, Rome, and colleagues.
“HMW-HA is a naturally occurring sugar that is abundant in the extracellular matrix, including in the lung,” the investigators wrote in Respiratory Research. “[It] has been used routinely, together with hypertonic saline, in cystic fibrosis patients [for several years] with no reported side effects; rather, it improves tolerability and decreases the need for bronchodilators in these patients.”
According to Robert A. Sandhaus, MD, PhD, FCCP, of National Jewish Health, Denver, the role of hyaluronan in lung disease was first recognized decades ago.
“Data stretching back into the 1970s has identified decreases in hyaluronan content in emphysematous lung tissue, protection of lung connective tissue from proteolysis by hyaluronan, and potential therapeutic roles for hyaluronan in a variety of disease, especially of the lungs,” he said in an interview.
For patients with COPD, treatment with HMW-HA may provide benefit by counteracting an imbalance in diseased lung tissue, wrote Dr. Galdi and colleagues.
“Emerging evidence suggests that imbalance between declining HMW-HA levels, and increasing smaller fragments of hyaluronan may contribute to chronic airway disease pathogenesis,” they wrote. “This has led to the hypothesis that exogenous supplementation of HMW-HA may restore hyaluronan homeostasis in favor of undegraded molecules, inhibit inflammation and loss of lung function, and ameliorate COPD progression.”
To test this hypothesis, the investigators screened 44 patients with a history of acute exacerbations of COPD necessitating NIPPV, ultimately excluding 3 patients because of heart failure. Following 1:1 randomization, 20 patients received HMW-HA while 21 received placebo, each twice daily, in conjunction with NIPPV and standard medical therapy. Treatment continued until NIPPV failure or liberation from NIPPV. Most patients received NIPPV in the hospital; however, home/chronic NIPPV was given to four patients in the placebo group and three patients in the HMW-HA group.
The primary outcome was duration of NIPPV. Secondary outcomes included markers of systemic inflammation associated with acute exacerbations of COPD and respiratory physiology parameters. Adverse events were also reported.
Results showed that patients treated with HMW-HA were liberated sooner from NIPPV than were those who received placebo (mean, 5.2 vs 6.4 days; P < .037). Similarly, patients in the HMW-HA group had significantly shorter hospital stay, on average, than those in the placebo group (mean, 7.2 vs 10.2 days; P = .039). Median values followed a similar pattern.
“These data suggest that HMW-HA shortened the duration of acute respiratory failure, need for NIPPV and, consequently, hospital length of stay in these patients,” the investigators wrote.
Secondary outcomes further supported these therapeutic benefits. Compared with placebo, HMW-HA was associated with significantly lower peak pressure and greater improvements in both pCO2/FiO2 ratio and inflammatory markers. No adverse events were reported.
Further analyses involving human bronchial epithelial cell cultures offered some mechanistic insight. Using micro-optical coherence tomography imaging, the investigators found that HMW-HA treatment was associated with “a prominent effect on mucociliary transport” in cell cultures derived from COPD patients and in healthy nonsmoker cell cultures exposed to cigarette smoke extract.
“Our study shows for the first time the therapeutic potential of an extracellular matrix molecule in acute exacerbation of human lung disease,” the investigators concluded, noting a “clinically meaningful salutary effect” on duration of NIPPV.
Dr. Galdi and colleagues went on to predict that benefits in a real-world patient population could be even more meaningful.
“Since the serum samples were collected at the end of NIPPV, HMW-HA–treated patients were on average sampled a day earlier than placebo-treated patients (because they were liberated from NIPPV a day earlier on average),” the investigators wrote. “Thus, HMW-HA treatment effects may have been underestimated in our study.”
According to Dr. Sandhaus, “The current report, while a relatively small single-center study, is well controlled and the results suggest that inhaled hyaluronan decreased time on noninvasive ventilation, decreased hospital stay duration, and decreased some mediators of inflammation.”
He also suggested that HMW-HA may have a role in the prophylactic setting.
“The limitations of this pilot study are appropriately explored by the authors but do not dampen the exciting possibility that this therapeutic approach may hold promise not only in severe exacerbations of COPD but potentially for the prevention of such exacerbations,” Dr. Sandhaus said.
Jerome O. Cantor, MD, FCCP, of St. John’s University, New York, who previously conducted a pilot study for using lower molecular weight hyaluronan in COPD and published a review on the subject, said that more studies are necessary.
“Further clinical trials are needed to better determine the role of hyaluronan as an adjunct to existing therapies for COPD exacerbations,” he said.
The study was supported by the National Institutes of Health. The investigators and Dr. Sandhaus declared no conflicts of interest. Dr. Cantor disclosed a relationship with MatRx Therapeutics.
FROM RESPIRATORY RESEARCH
COVID-19 in children: New cases down for third straight week
New COVID-19 cases in children dropped for the third consecutive week, even as children continue to make up a larger share of all cases, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
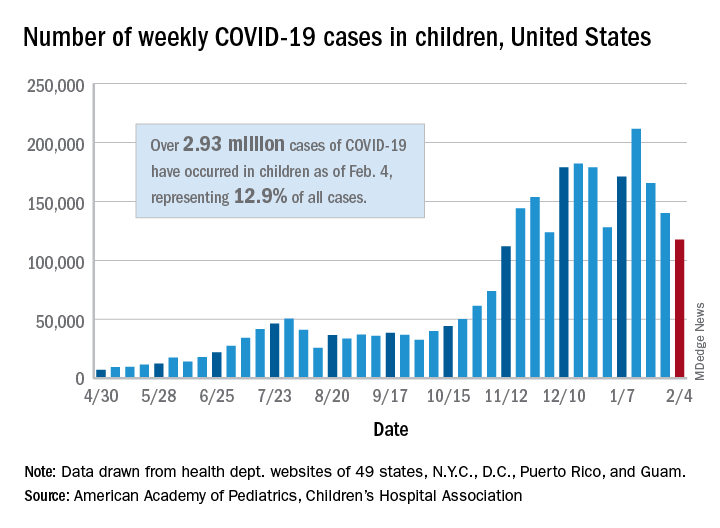
New child cases totaled almost 118,000 for the week of Jan. 29-Feb. 4, continuing the decline that began right after the United States topped 200,000 cases for the only time Jan. 8-14, the AAP and the CHA said in their weekly COVID-19 report.
For the latest week, however, children represented 16.0% of all new COVID-19 cases, continuing a 5-week increase that began in early December 2020, after the proportion had dropped to 12.6%, based on data collected from the health departments of 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. During the week of Sept. 11-17, children made up 16.9% of all cases, the highest level seen during the pandemic.
The 2.93 million cases that have been reported in children make up 12.9% of all cases since the pandemic began, and the overall rate of pediatric coronavirus infection is 3,899 cases per 100,000 children in the population. Taking a step down from the national level, 30 states are above that rate and 18 are below it, along with D.C., New York City, Puerto Rico, and Guam (New York and Texas are excluded), the AAP and CHA reported.
There were 12 new COVID-19–related child deaths in the 43 states, along with New York City and Guam, that are reporting such data, bringing the total to 227. Nationally, 0.06% of all deaths have occurred in children, with rates ranging from 0.00% (11 states) to 0.26% (Nebraska) in the 45 jurisdictions, the AAP/CHA report shows.
Child hospitalizations rose to 1.9% of all hospitalizations after holding at 1.8% since mid-November in 25 reporting jurisdictions (24 states and New York City), but the hospitalization rate among children with COVID held at 0.8%, where it has been for the last 4 weeks. Hospitalization rates as high as 3.8% were recorded early in the pandemic, the AAP and CHA noted.
New COVID-19 cases in children dropped for the third consecutive week, even as children continue to make up a larger share of all cases, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
New child cases totaled almost 118,000 for the week of Jan. 29-Feb. 4, continuing the decline that began right after the United States topped 200,000 cases for the only time Jan. 8-14, the AAP and the CHA said in their weekly COVID-19 report.
For the latest week, however, children represented 16.0% of all new COVID-19 cases, continuing a 5-week increase that began in early December 2020, after the proportion had dropped to 12.6%, based on data collected from the health departments of 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. During the week of Sept. 11-17, children made up 16.9% of all cases, the highest level seen during the pandemic.
The 2.93 million cases that have been reported in children make up 12.9% of all cases since the pandemic began, and the overall rate of pediatric coronavirus infection is 3,899 cases per 100,000 children in the population. Taking a step down from the national level, 30 states are above that rate and 18 are below it, along with D.C., New York City, Puerto Rico, and Guam (New York and Texas are excluded), the AAP and CHA reported.
There were 12 new COVID-19–related child deaths in the 43 states, along with New York City and Guam, that are reporting such data, bringing the total to 227. Nationally, 0.06% of all deaths have occurred in children, with rates ranging from 0.00% (11 states) to 0.26% (Nebraska) in the 45 jurisdictions, the AAP/CHA report shows.
Child hospitalizations rose to 1.9% of all hospitalizations after holding at 1.8% since mid-November in 25 reporting jurisdictions (24 states and New York City), but the hospitalization rate among children with COVID held at 0.8%, where it has been for the last 4 weeks. Hospitalization rates as high as 3.8% were recorded early in the pandemic, the AAP and CHA noted.
New COVID-19 cases in children dropped for the third consecutive week, even as children continue to make up a larger share of all cases, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
New child cases totaled almost 118,000 for the week of Jan. 29-Feb. 4, continuing the decline that began right after the United States topped 200,000 cases for the only time Jan. 8-14, the AAP and the CHA said in their weekly COVID-19 report.
For the latest week, however, children represented 16.0% of all new COVID-19 cases, continuing a 5-week increase that began in early December 2020, after the proportion had dropped to 12.6%, based on data collected from the health departments of 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. During the week of Sept. 11-17, children made up 16.9% of all cases, the highest level seen during the pandemic.
The 2.93 million cases that have been reported in children make up 12.9% of all cases since the pandemic began, and the overall rate of pediatric coronavirus infection is 3,899 cases per 100,000 children in the population. Taking a step down from the national level, 30 states are above that rate and 18 are below it, along with D.C., New York City, Puerto Rico, and Guam (New York and Texas are excluded), the AAP and CHA reported.
There were 12 new COVID-19–related child deaths in the 43 states, along with New York City and Guam, that are reporting such data, bringing the total to 227. Nationally, 0.06% of all deaths have occurred in children, with rates ranging from 0.00% (11 states) to 0.26% (Nebraska) in the 45 jurisdictions, the AAP/CHA report shows.
Child hospitalizations rose to 1.9% of all hospitalizations after holding at 1.8% since mid-November in 25 reporting jurisdictions (24 states and New York City), but the hospitalization rate among children with COVID held at 0.8%, where it has been for the last 4 weeks. Hospitalization rates as high as 3.8% were recorded early in the pandemic, the AAP and CHA noted.
Customized chemotherapy did not improve survival in early NSCLC
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
The patients were randomized to receive investigator’s choice of platinum-based chemotherapy or treatment tailored according to messenger RNA (mRNA) expression of two molecular markers – excision repair cross complementation 1 (ERCC1) and thymidylate synthase (TS).
There was no significant difference in overall survival or recurrence-free survival between the treatment approaches. However, toxicity was less common among patients who received customized treatment.
These results, from the phase 3 ITACA trial, were presented at the 2020 World Conference on Lung Cancer (Abstract 1820), which was rescheduled to January 2021.
“There is a clear need to define patients most likely to derive survival benefit from adjuvant therapy and spare patients who do not need adjuvant chemotherapy due to the toxicity of such therapy,” said presenter Silvia Novello, MD, PhD, of the University of Turin in Italy. “mRNA expression of different genes has been correlated with the sensitivity or resistance to specific anticancer agents.”
With this in mind, Dr. Novello and colleagues conducted the ITACA trial. The researchers’ primary goal was to determine whether an adjuvant pharmacogenomic-driven approach was able to improve overall survival in completely resected NSCLC.
Patients and treatment
The researchers randomized 773 NSCLC patients within 5-8 weeks after radical surgery. Genomic analyses were performed soon after surgery, and patients were randomly assigned to investigator’s choice of platinum-based chemotherapy or to tailored treatments defined by mRNA levels of ERCC1 and TS.
Patients with high ERCC1 mRNA expression who were randomized to tailored treatment received single-agent docetaxel if their TS level was high or pemetrexed monotherapy if their TS level was low.
Patients with low ERCC1 mRNA expression who were randomized to tailored treatment received cisplatin-gemcitabine if their TS level was high or cisplatin-pemetrexed if their TS was low.
The most frequent doublets used in control patients were cisplatin-gemcitabine and cisplatin-vinorelbine.
The demographic characteristics of the 384 patients randomized to tailored therapy and the 389 control subjects were well-balanced, Dr. Novello said. Two-thirds of patients had stage II disease, 11% were never smokers, and the vast majority had a lobectomy as the resection method.
Results
At a median follow-up of 28.2 months, the median overall survival was 96.4 months in the tailored therapy arm and 83.5 months in the control arm. The median recurrence-free survival was 64.4 months and 41.5 months, respectively.
“Adjuvant chemotherapy customization based on the primary tumor tissue mRNA expression of ERCC1 and TS did not significantly improve overall survival or recurrence-free survival,” Dr. Novello said. “There was a non–statistically significant trend for overall survival favoring the customized arm.”
Dr. Novello noted that, when the final analysis was performed, the study was underpowered, as only 46% of expected events were collected. Assuming the same hazard ratio point estimate and that the expected 336 events were collected, the hazard ratio estimate would be 0.76 (P = .012).
Grade 3/4 toxicities occurred in 32.6% of patients in the tailored therapy arm and 45.9% of those in the control arm (P < .001).
“It is important to underline that the treatment customization significantly improved the toxicity profile without compromising the efficacy,” Dr. Novello said.
She added that “more comprehensive and high-throughput diagnostic techniques will be needed in order to tailor adjuvant chemotherapy, with or without immunotherapy, in completely resected NSCLC.”
“The ITACA study is the largest adjuvant study tailored to ERCC1/TS status, and the results have been long-awaited,” said Tetsuya Mitsudomi, MD, a professor at Kindai University in Japan and president of the International Association for the Study of Lung Cancer.
“This trial should be praised for the mandated genomic analysis that was accomplished within a reasonably short time frame before random assignment. In addition, this trial confirmed that there is no biomarker strong enough to predict the efficacy of cytotoxic chemotherapy. However, the concept of customizing adjuvant therapy according to the genomic status of patients’ tumors is valid, leading to the recent demonstration in the ADAURA study of the superiority of osimertinib in delaying the postoperative recurrence of disease in patients with EGFR-mutated NSCLC.”
The ITACA study was funded by University of Turin and Eli Lilly. Dr. Novello disclosed relationships with Eli Lilly, Amgen, AstraZeneca, Bohringer Ingelheim, Beigene, Pfizer, Roche, Merck, Bristol-Myers Squibb, Takeda, and Sanofi. Dr. Mitsudomi disclosed relationships with Eli Lilly, AstraZeneca, Boehringer-Ingelheim, Chugai, Pfizer, Merck, Ono Pharmaceutical, Bristol-Myers Squibb, Novartis, ThermoFisher, Guardant, Eisai, Amgen, and Johnson & Johnson.
FROM WCLC 2020
U.K. COVID-19 variant doubling every 10 days in the U.S.: Study
The SARS-CoV-2 variant first detected in the United Kingdom is rapidly becoming the dominant strain in several countries and is doubling every 10 days in the United States, according to new data.
The findings by Nicole L. Washington, PhD, associate director of research at the genomics company Helix, and colleagues were posted Feb. 7, 2021, on the preprint server medRxiv. The paper has not been peer-reviewed in a scientific journal.
The researchers also found that the transmission rate in the United States of the variant, labeled B.1.1.7, is 30%-40% higher than that of more common lineages.
While clinical outcomes initially were thought to be similar to those of other SARS-CoV-2 variants, early reports suggest that infection with the B.1.1.7 variant may increase death risk by about 30%.
A coauthor of the current study, Kristian Andersen, PhD, told the New York Times , “Nothing in this paper is surprising, but people need to see it.”
Dr. Andersen, a virologist at the Scripps Research Institute in La Jolla, Calif., added that “we should probably prepare for this being the predominant lineage in most places in the United States by March.”
The study of the B.1.1.7 variant adds support for the Centers for Disease Control and Prevention prediction in January that it would dominate by March.
“Our study shows that the U.S. is on a similar trajectory as other countries where B.1.1.7 rapidly became the dominant SARS-CoV-2 variant, requiring immediate and decisive action to minimize COVID-19 morbidity and mortality,” the researchers wrote.
The authors pointed out that the B.1.1.7 variant became the dominant SARS-CoV-2 strain in the United Kingdom within a couple of months of its detection.
“Since then, the variant has been increasingly observed across many European countries, including Portugal and Ireland, which, like the U.K., observed devastating waves of COVID-19 after B.1.1.7 became dominant,” the authors wrote.
“Category 5” storm
The B.1.1.7 variant has likely been spreading between U.S. states since at least December, they wrote.
This news organization reported on Jan. 15 that, as of Jan. 13, the B.1.1.7 variant was seen in 76 cases across 12 U.S. states, according to an early release of the CDC’s Morbidity and Mortality Weekly Report.
As of Feb. 7, there were 690 cases of the B.1.1.7 variant in the US in 33 states, according to the CDC.
Dr. Washington and colleagues examined more than 500,000 coronavirus test samples from cases across the United States that were tested at San Mateo, Calif.–based Helix facilities since July.
In the study, they found inconsistent prevalence of the variant across states. By the last week in January, the researchers estimated the proportion of B.1.1.7 in the U.S. population to be about 2.1% of all COVID-19 cases, though they found it made up about 2% of all COVID-19 cases in California and about 4.5% of cases in Florida. The authors acknowledged that their data is less robust outside of those two states.
Though that seems a relatively low frequency, “our estimates show that its growth rate is at least 35%-45% increased and doubling every week and a half,” the authors wrote.
“Because laboratories in the U.S. are only sequencing a small subset of SARS-CoV-2 samples, the true sequence diversity of SARS-CoV-2 in this country is still unknown,” they noted.
Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis, said last week that the United States is facing a “Category 5” storm with the spread of the B.1.1.7 variant as well as the variants first identified in South Africa and Brazil.
“We are going to see something like we have not seen yet in this country,” Dr. Osterholm said recently on NBC’s Meet the Press.
Lead author Nicole L. Washington and many of the coauthors are employees of Helix. Other coauthors are employees of Illumina. Three coauthors own stock in ILMN. The work was funded by Illumina, Helix, the Innovative Genomics Institute, and the New Frontiers in Research Fund provided by the Canadian Institutes of Health Research.
A version of this article first appeared on Medscape.com.
The SARS-CoV-2 variant first detected in the United Kingdom is rapidly becoming the dominant strain in several countries and is doubling every 10 days in the United States, according to new data.
The findings by Nicole L. Washington, PhD, associate director of research at the genomics company Helix, and colleagues were posted Feb. 7, 2021, on the preprint server medRxiv. The paper has not been peer-reviewed in a scientific journal.
The researchers also found that the transmission rate in the United States of the variant, labeled B.1.1.7, is 30%-40% higher than that of more common lineages.
While clinical outcomes initially were thought to be similar to those of other SARS-CoV-2 variants, early reports suggest that infection with the B.1.1.7 variant may increase death risk by about 30%.
A coauthor of the current study, Kristian Andersen, PhD, told the New York Times , “Nothing in this paper is surprising, but people need to see it.”
Dr. Andersen, a virologist at the Scripps Research Institute in La Jolla, Calif., added that “we should probably prepare for this being the predominant lineage in most places in the United States by March.”
The study of the B.1.1.7 variant adds support for the Centers for Disease Control and Prevention prediction in January that it would dominate by March.
“Our study shows that the U.S. is on a similar trajectory as other countries where B.1.1.7 rapidly became the dominant SARS-CoV-2 variant, requiring immediate and decisive action to minimize COVID-19 morbidity and mortality,” the researchers wrote.
The authors pointed out that the B.1.1.7 variant became the dominant SARS-CoV-2 strain in the United Kingdom within a couple of months of its detection.
“Since then, the variant has been increasingly observed across many European countries, including Portugal and Ireland, which, like the U.K., observed devastating waves of COVID-19 after B.1.1.7 became dominant,” the authors wrote.
“Category 5” storm
The B.1.1.7 variant has likely been spreading between U.S. states since at least December, they wrote.
This news organization reported on Jan. 15 that, as of Jan. 13, the B.1.1.7 variant was seen in 76 cases across 12 U.S. states, according to an early release of the CDC’s Morbidity and Mortality Weekly Report.
As of Feb. 7, there were 690 cases of the B.1.1.7 variant in the US in 33 states, according to the CDC.
Dr. Washington and colleagues examined more than 500,000 coronavirus test samples from cases across the United States that were tested at San Mateo, Calif.–based Helix facilities since July.
In the study, they found inconsistent prevalence of the variant across states. By the last week in January, the researchers estimated the proportion of B.1.1.7 in the U.S. population to be about 2.1% of all COVID-19 cases, though they found it made up about 2% of all COVID-19 cases in California and about 4.5% of cases in Florida. The authors acknowledged that their data is less robust outside of those two states.
Though that seems a relatively low frequency, “our estimates show that its growth rate is at least 35%-45% increased and doubling every week and a half,” the authors wrote.
“Because laboratories in the U.S. are only sequencing a small subset of SARS-CoV-2 samples, the true sequence diversity of SARS-CoV-2 in this country is still unknown,” they noted.
Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis, said last week that the United States is facing a “Category 5” storm with the spread of the B.1.1.7 variant as well as the variants first identified in South Africa and Brazil.
“We are going to see something like we have not seen yet in this country,” Dr. Osterholm said recently on NBC’s Meet the Press.
Lead author Nicole L. Washington and many of the coauthors are employees of Helix. Other coauthors are employees of Illumina. Three coauthors own stock in ILMN. The work was funded by Illumina, Helix, the Innovative Genomics Institute, and the New Frontiers in Research Fund provided by the Canadian Institutes of Health Research.
A version of this article first appeared on Medscape.com.
The SARS-CoV-2 variant first detected in the United Kingdom is rapidly becoming the dominant strain in several countries and is doubling every 10 days in the United States, according to new data.
The findings by Nicole L. Washington, PhD, associate director of research at the genomics company Helix, and colleagues were posted Feb. 7, 2021, on the preprint server medRxiv. The paper has not been peer-reviewed in a scientific journal.
The researchers also found that the transmission rate in the United States of the variant, labeled B.1.1.7, is 30%-40% higher than that of more common lineages.
While clinical outcomes initially were thought to be similar to those of other SARS-CoV-2 variants, early reports suggest that infection with the B.1.1.7 variant may increase death risk by about 30%.
A coauthor of the current study, Kristian Andersen, PhD, told the New York Times , “Nothing in this paper is surprising, but people need to see it.”
Dr. Andersen, a virologist at the Scripps Research Institute in La Jolla, Calif., added that “we should probably prepare for this being the predominant lineage in most places in the United States by March.”
The study of the B.1.1.7 variant adds support for the Centers for Disease Control and Prevention prediction in January that it would dominate by March.
“Our study shows that the U.S. is on a similar trajectory as other countries where B.1.1.7 rapidly became the dominant SARS-CoV-2 variant, requiring immediate and decisive action to minimize COVID-19 morbidity and mortality,” the researchers wrote.
The authors pointed out that the B.1.1.7 variant became the dominant SARS-CoV-2 strain in the United Kingdom within a couple of months of its detection.
“Since then, the variant has been increasingly observed across many European countries, including Portugal and Ireland, which, like the U.K., observed devastating waves of COVID-19 after B.1.1.7 became dominant,” the authors wrote.
“Category 5” storm
The B.1.1.7 variant has likely been spreading between U.S. states since at least December, they wrote.
This news organization reported on Jan. 15 that, as of Jan. 13, the B.1.1.7 variant was seen in 76 cases across 12 U.S. states, according to an early release of the CDC’s Morbidity and Mortality Weekly Report.
As of Feb. 7, there were 690 cases of the B.1.1.7 variant in the US in 33 states, according to the CDC.
Dr. Washington and colleagues examined more than 500,000 coronavirus test samples from cases across the United States that were tested at San Mateo, Calif.–based Helix facilities since July.
In the study, they found inconsistent prevalence of the variant across states. By the last week in January, the researchers estimated the proportion of B.1.1.7 in the U.S. population to be about 2.1% of all COVID-19 cases, though they found it made up about 2% of all COVID-19 cases in California and about 4.5% of cases in Florida. The authors acknowledged that their data is less robust outside of those two states.
Though that seems a relatively low frequency, “our estimates show that its growth rate is at least 35%-45% increased and doubling every week and a half,” the authors wrote.
“Because laboratories in the U.S. are only sequencing a small subset of SARS-CoV-2 samples, the true sequence diversity of SARS-CoV-2 in this country is still unknown,” they noted.
Michael Osterholm, PhD, MPH, director of the Center for Infectious Disease Research and Policy at the University of Minnesota, Minneapolis, said last week that the United States is facing a “Category 5” storm with the spread of the B.1.1.7 variant as well as the variants first identified in South Africa and Brazil.
“We are going to see something like we have not seen yet in this country,” Dr. Osterholm said recently on NBC’s Meet the Press.
Lead author Nicole L. Washington and many of the coauthors are employees of Helix. Other coauthors are employees of Illumina. Three coauthors own stock in ILMN. The work was funded by Illumina, Helix, the Innovative Genomics Institute, and the New Frontiers in Research Fund provided by the Canadian Institutes of Health Research.
A version of this article first appeared on Medscape.com.
Neoadjuvant atezolizumab safe for patients with resectable lung cancer
Small pilot studies previously suggested that preoperative immune checkpoint inhibitor (ICI) therapy may benefit patients with resectable non–small cell lung cancer (NSCLC).
The LCMC3 study is “unique” because it is the largest monotherapy trial of checkpoint inhibition in resectable NSCLC, and it’s “a landmark study” because it validated results from smaller trials and can serve as a benchmark for future ones, said Jay M. Lee, MD, of the University of California, Los Angeles.
Dr. Lee presented results from LCMC3 at the 2020 World Congress on Lung Cancer (Abstract PS01.05), which was rescheduled for January 2021.
The study included 181 patients, median age 65 years, with stage IB-IIIB NSCLC. The vast majority (90%) of patients were current/former smokers, and two-thirds had a nonsquamous histology. Patients were categorized in the following stages: 17 patients were staged at IB, 20 were IIA, 55 were IIB, 72 were IIIA, and 17 were IIIB.
Patients received 1,200 mg of neoadjuvant atezolizumab intravenously every 3 weeks for two cycles followed by resection between 30 and 50 days from the first cycle. Patients who benefited from the therapy continued adjuvant atezolizumab for 12 months.
The primary endpoint was major pathological response, defined as no more than 10% viable tumor cells at surgery, in patients without epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
Results
Following atezolizumab treatment, 43% of patients were down-staged, and 19% were up-staged. Some degree of pathological regression was observed in all but 3 of the 159 patients who underwent resection.
Among the 144 patients included in the efficacy analysis, the major pathological response rate was 21%, with 7% of patients achieving a complete pathological response.
“We demonstrated that more than half of patients resected with a minimally invasive operation. Remarkably, only 15% required thoracotomy. The 92% complete resection rate is comparable, if not superior to, preoperative chemotherapy trials,” Dr. Lee said.
The majority (88%) of patients underwent surgical resection within a 20-day protocol window. The median time from end of neoadjuvant therapy to surgery was 22 days.
“Historically, the neoadjuvant chemotherapy window is much later for surgery, 3 weeks from neoadjuvant therapy, and that can be stretched to up to 56 days,” Dr. Lee said.
In an exploratory analysis, the 1.5-year overall survival rate was 91% for stage I and II disease and 87% for stage III disease. The survival in both cohorts was superior to that expected historically, Dr. Lee noted.
Intraoperative complications were rare (3%). Postoperative adverse reactions correlated with fewer viable tumor cells in the resected specimen.
One patient died following surgery after the first 30 days, which was deemed unrelated to treatment. Another patient died between 30 and 90 days from treatment-related pneumonitis.
“The LCMC3 study successfully met its primary endpoint of achieving major pathological response,” Dr. Lee concluded. “Neoadjuvant atezolizumab monotherapy was well tolerated, and resection was performed with low perioperative morbidity and mortality, usually within a narrow protocol window and with a short time frame from completion of atezolizumab and with a correspondingly high complete resection rate.”
The study’s results suggest that “neoadjuvant atezolizumab monotherapy is effective, well tolerated, and surgically acceptable,” said study discussant Shinichi Toyooka, MD, of Okayama (Japan) University Hospital.
“I would consider single-agent ICI neoadjuvant therapy for patients with early-stage disease and poor performance status, and an ICI plus chemotherapy for more advanced resectable cases, like locally advanced disease,” Dr. Toyooka said.
The LCMC3 study is sponsored by Genentech. Dr. Lee disclosed relationships with Genentech/Roche, AstraZeneca, Bristol-Myers Squibb, Merck, and Novartis. Dr. Toyooka disclosed relationships with AstraZeneca, Chugai, Taiho Pharmaceutical Group, and Ono Pharmaceutical.
Small pilot studies previously suggested that preoperative immune checkpoint inhibitor (ICI) therapy may benefit patients with resectable non–small cell lung cancer (NSCLC).
The LCMC3 study is “unique” because it is the largest monotherapy trial of checkpoint inhibition in resectable NSCLC, and it’s “a landmark study” because it validated results from smaller trials and can serve as a benchmark for future ones, said Jay M. Lee, MD, of the University of California, Los Angeles.
Dr. Lee presented results from LCMC3 at the 2020 World Congress on Lung Cancer (Abstract PS01.05), which was rescheduled for January 2021.
The study included 181 patients, median age 65 years, with stage IB-IIIB NSCLC. The vast majority (90%) of patients were current/former smokers, and two-thirds had a nonsquamous histology. Patients were categorized in the following stages: 17 patients were staged at IB, 20 were IIA, 55 were IIB, 72 were IIIA, and 17 were IIIB.
Patients received 1,200 mg of neoadjuvant atezolizumab intravenously every 3 weeks for two cycles followed by resection between 30 and 50 days from the first cycle. Patients who benefited from the therapy continued adjuvant atezolizumab for 12 months.
The primary endpoint was major pathological response, defined as no more than 10% viable tumor cells at surgery, in patients without epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
Results
Following atezolizumab treatment, 43% of patients were down-staged, and 19% were up-staged. Some degree of pathological regression was observed in all but 3 of the 159 patients who underwent resection.
Among the 144 patients included in the efficacy analysis, the major pathological response rate was 21%, with 7% of patients achieving a complete pathological response.
“We demonstrated that more than half of patients resected with a minimally invasive operation. Remarkably, only 15% required thoracotomy. The 92% complete resection rate is comparable, if not superior to, preoperative chemotherapy trials,” Dr. Lee said.
The majority (88%) of patients underwent surgical resection within a 20-day protocol window. The median time from end of neoadjuvant therapy to surgery was 22 days.
“Historically, the neoadjuvant chemotherapy window is much later for surgery, 3 weeks from neoadjuvant therapy, and that can be stretched to up to 56 days,” Dr. Lee said.
In an exploratory analysis, the 1.5-year overall survival rate was 91% for stage I and II disease and 87% for stage III disease. The survival in both cohorts was superior to that expected historically, Dr. Lee noted.
Intraoperative complications were rare (3%). Postoperative adverse reactions correlated with fewer viable tumor cells in the resected specimen.
One patient died following surgery after the first 30 days, which was deemed unrelated to treatment. Another patient died between 30 and 90 days from treatment-related pneumonitis.
“The LCMC3 study successfully met its primary endpoint of achieving major pathological response,” Dr. Lee concluded. “Neoadjuvant atezolizumab monotherapy was well tolerated, and resection was performed with low perioperative morbidity and mortality, usually within a narrow protocol window and with a short time frame from completion of atezolizumab and with a correspondingly high complete resection rate.”
The study’s results suggest that “neoadjuvant atezolizumab monotherapy is effective, well tolerated, and surgically acceptable,” said study discussant Shinichi Toyooka, MD, of Okayama (Japan) University Hospital.
“I would consider single-agent ICI neoadjuvant therapy for patients with early-stage disease and poor performance status, and an ICI plus chemotherapy for more advanced resectable cases, like locally advanced disease,” Dr. Toyooka said.
The LCMC3 study is sponsored by Genentech. Dr. Lee disclosed relationships with Genentech/Roche, AstraZeneca, Bristol-Myers Squibb, Merck, and Novartis. Dr. Toyooka disclosed relationships with AstraZeneca, Chugai, Taiho Pharmaceutical Group, and Ono Pharmaceutical.
Small pilot studies previously suggested that preoperative immune checkpoint inhibitor (ICI) therapy may benefit patients with resectable non–small cell lung cancer (NSCLC).
The LCMC3 study is “unique” because it is the largest monotherapy trial of checkpoint inhibition in resectable NSCLC, and it’s “a landmark study” because it validated results from smaller trials and can serve as a benchmark for future ones, said Jay M. Lee, MD, of the University of California, Los Angeles.
Dr. Lee presented results from LCMC3 at the 2020 World Congress on Lung Cancer (Abstract PS01.05), which was rescheduled for January 2021.
The study included 181 patients, median age 65 years, with stage IB-IIIB NSCLC. The vast majority (90%) of patients were current/former smokers, and two-thirds had a nonsquamous histology. Patients were categorized in the following stages: 17 patients were staged at IB, 20 were IIA, 55 were IIB, 72 were IIIA, and 17 were IIIB.
Patients received 1,200 mg of neoadjuvant atezolizumab intravenously every 3 weeks for two cycles followed by resection between 30 and 50 days from the first cycle. Patients who benefited from the therapy continued adjuvant atezolizumab for 12 months.
The primary endpoint was major pathological response, defined as no more than 10% viable tumor cells at surgery, in patients without epidermal growth factor receptor or anaplastic lymphoma kinase mutations.
Results
Following atezolizumab treatment, 43% of patients were down-staged, and 19% were up-staged. Some degree of pathological regression was observed in all but 3 of the 159 patients who underwent resection.
Among the 144 patients included in the efficacy analysis, the major pathological response rate was 21%, with 7% of patients achieving a complete pathological response.
“We demonstrated that more than half of patients resected with a minimally invasive operation. Remarkably, only 15% required thoracotomy. The 92% complete resection rate is comparable, if not superior to, preoperative chemotherapy trials,” Dr. Lee said.
The majority (88%) of patients underwent surgical resection within a 20-day protocol window. The median time from end of neoadjuvant therapy to surgery was 22 days.
“Historically, the neoadjuvant chemotherapy window is much later for surgery, 3 weeks from neoadjuvant therapy, and that can be stretched to up to 56 days,” Dr. Lee said.
In an exploratory analysis, the 1.5-year overall survival rate was 91% for stage I and II disease and 87% for stage III disease. The survival in both cohorts was superior to that expected historically, Dr. Lee noted.
Intraoperative complications were rare (3%). Postoperative adverse reactions correlated with fewer viable tumor cells in the resected specimen.
One patient died following surgery after the first 30 days, which was deemed unrelated to treatment. Another patient died between 30 and 90 days from treatment-related pneumonitis.
“The LCMC3 study successfully met its primary endpoint of achieving major pathological response,” Dr. Lee concluded. “Neoadjuvant atezolizumab monotherapy was well tolerated, and resection was performed with low perioperative morbidity and mortality, usually within a narrow protocol window and with a short time frame from completion of atezolizumab and with a correspondingly high complete resection rate.”
The study’s results suggest that “neoadjuvant atezolizumab monotherapy is effective, well tolerated, and surgically acceptable,” said study discussant Shinichi Toyooka, MD, of Okayama (Japan) University Hospital.
“I would consider single-agent ICI neoadjuvant therapy for patients with early-stage disease and poor performance status, and an ICI plus chemotherapy for more advanced resectable cases, like locally advanced disease,” Dr. Toyooka said.
The LCMC3 study is sponsored by Genentech. Dr. Lee disclosed relationships with Genentech/Roche, AstraZeneca, Bristol-Myers Squibb, Merck, and Novartis. Dr. Toyooka disclosed relationships with AstraZeneca, Chugai, Taiho Pharmaceutical Group, and Ono Pharmaceutical.
FROM WCLC 2020