User login
-
div[contains(@class, 'header__large-screen')]
div[contains(@class, 'read-next-article')]
div[contains(@class, 'main-prefix')]
div[contains(@class, 'nav-primary')]
nav[contains(@class, 'nav-primary')]
section[contains(@class, 'footer-nav-section-wrapper')]
footer[@id='footer']
section[contains(@class, 'nav-hidden')]
div[contains(@class, 'ce-card-content')]
nav[contains(@class, 'nav-ce-stack')]
div[contains(@class, 'view-medstat-quiz-listing-panes')]
div[contains(@class, 'pane-article-sidebar-latest-news')]
FDA blazes path for ‘real-world’ evidence as proof of efficacy
In 2016, results from the LEADER trial of liraglutide in patients with type 2 diabetes helped jump-start awareness of the potential role of this new class of drugs, the glucagonlike peptide–1 receptor agonists, for reducing cardiovascular events. The randomized, placebo-controlled trial enrolled more than 9000 patients at more than 400 sites in over 30 countries, and took nearly 6 years from the start of patient enrollment to publication of the landmark results.
In December 2020, an independent team of researchers published results from a study with a design identical to LEADER, but used data that came not from a massive, global, years-long trial but from already-existing numbers culled from three large U.S. insurance claim databases. The result of this emulation using real-world data was virtually identical to what the actual trial showed, replicating both the direction and statistical significance of the original finding of the randomized, controlled trial (RCT).
What if research proved that this sort of RCT emulation could reliably be done on a regular basis? What might it mean for regulatory decisions on drugs and devices that historically have been based entirely on efficacy evidence from RCTs?
Making the most of a sea of observational data
Medicine in the United States has become increasingly awash in a sea of observational data collected from sources that include electronic health records, insurance claims, and increasingly, personal-health monitoring devices.
The Food and Drug Administration is now in the process of trying to figure out how it can legitimately harness this tsunami of real-world data to make efficacy decisions, essentially creating a new category of evidence to complement traditional data from randomized trials. It’s an opportunity that agency staff and their outside advisors have been keen to seize, especially given the soaring cost of prospective, randomized trials.
Recognition of this untapped resource in part led to a key initiative, among many others, included in the 21st Century Cures Act, passed in December 2016. Among the Act’s mandates was that, by the end of 2021, the FDA would issue guidance on when drug sponsors could use real-world evidence (RWE) to either help support a new indication for an already approved drug or help satisfy postapproval study requirements.
The initiative recognizes that this approach is not appropriate for initial drug approvals, which remain exclusively reliant on evidence from RCTs. Instead, it seems best suited to support expanding indications for already approved drugs.
Although FDA staff have made progress in identifying the challenges and broadening their understanding of how to best handle real-world data that come from observing patients in routine practice, agency leaders stress that this complex issue will likely not be fully resolved by their guidance to be published later this year. The FDA released a draft of the guidance in May 2019.
Can RWE be ‘credible and reliable?’
“Whether observational, nonrandomized data can become credible enough to use is what we’re talking about. These are possibilities that need to be explained and better understood,” said Robert Temple, MD, deputy director for clinical science of the FDA Center for Drug Evaluation and Research.
“Since the 1970s, the FDA has recognized historical controls as legitimate, so it’s possible [for RWE] to be credible. The big test is when is it credible and reliable enough [to assess efficacy]?” wondered Dr. Temple during a 2-day workshop on the topic held mid-February and organized by Duke University’s Margolis Center for Health Policy.
“We’re approaching an inflection point regarding how observational studies are generated and used, but our evidentiary standards will not lower, and it will be a case-by-case decision” by the agency as they review future RWE submissions, said John Concato, MD, the FDA’s associate director for real-world evidence, during the workshop.
“We are working toward guidance development, but also looking down the road to what we need to do to enable this,” said Dr. Concato. “It’s a complicated issue. If it was easy, it would have already been fixed.” He added that the agency will likely release a “portfolio” of guidance for submitting real-world data and RWE. Real-world data are raw information that, when analyzed, become RWE.
In short, the FDA seems headed toward guidance that won’t spell out a pathway that guarantees success using RWE but will at least open the door to consideration of this unprecedented application.
Not like flipping a switch
The guidance will not activate acceptance of RWE all at once. “It’s not like a light switch,” cautioned Adam Kroetsch, MPP, research director for biomedical innovation and regulatory policy at Duke-Margolis in Washington, D.C. “It’s an evolutionary process,” and the upcoming guidance will provide “just a little more clarity” on what sorts of best practices using RWE the FDA will find persuasive. “It’s hard for the FDA to clearly say what it’s looking for until they see some good examples,” Dr. Kroetsch said in an interview.
What will change is that drug sponsors can submit using RWE, and the FDA “will have a more open-minded view,” predicted Sebastian Schneeweiss, MD, ScD, a workshop participant and chief of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital in Boston. “For the first time, a law required [the FDA] to take a serious look” at observational data for efficacy assessment.
“The FDA has had a bias against using RWE for evidence of efficacy but has long used it to understand drug safety. Now the FDA is trying to wrap its arms around how to best use RWE” for efficacy decisions, said Joseph S. Ross, MD, another workshop participant and professor of medicine and public health at Yale University, New Haven, Conn.
The agency’s cautious approach is reassuring, Dr. Ross noted in an interview. “There was worry that the 21st Century Cures Act would open the door to allowing real-world data to be used in ways that weren’t very reliable. Very quickly, the FDA started trying to figure out the best ways to use these data in reasonable ways.”
Duplicating RCTs with RWE
To help better understand the potential use of RWE, the FDA sponsored several demonstration projects. Researchers presented results from three of these projects during the workshop in February. All three examined whether RWE, plugged into the design of an actual RCT, can produce roughly similar results when similar patients are used.
A generally consistent finding from the three demonstration projects was that “when the data are fit for purpose” the emulated or duplicated analyses with RWE “can come to similar conclusions” as the actual RCTs, said Dr. Schneeweiss, who leads one of the demonstration projects, RCT DUPLICATE.
At the workshop he reported results from RWE duplications of 20 different RCTs using insurance claims data from U.S. patients. The findings came from 10 duplications already reported in Circulation in December 2020 (including a duplication of the LEADER trial), and an additional 10 as yet unpublished RCT duplications. In the next few months, the researchers intend to assess a final group of 10 more RCT duplications.
Workshop participants also presented results from two other FDA demonstration projects: the OPERAND program run by the Multi-Regional Clinical Trials Center of Brigham and Women’s Hospital and Harvard; and the CERSI program based at Yale and the Mayo Clinic in Rochester, Minn. Both are smaller in scale than RCT DUPLICATE, incorporate lab data in addition to claims data, and in some cases test how well RWE can emulate RCTs that are not yet completed.
Collectively, results from these demonstration projects suggest that RWE can successfully emulate the results of an RCT, said Dr. Ross, a coinvestigator on the CERSI study. But the CERSI findings also highlighted how an RCT can fall short of clinical relevance.
“One of our most important findings was that RCTs don’t always represent real-world practice,” he said. His group attempted to replicate the 5,000-patient GRADE trial of four different drug options added to metformin in patients with type 2 diabetes. One of the four options included insulin glargine (Lantus), and the attempt to emulate the study with RWE hit the bump that no relevant real-world patients in their US claims database actually received the formulation.
That means the GRADE trial “is almost meaningless. It doesn’t reflect real-world practice,” Dr. Ross noted.
Results from the three demonstration projects “highlight the gaps we still have,” summed up Dr. Kroetsch. “They show where we need better data” from observational sources that function as well as data from RCTs.
Still, the demonstration project results are “an important step forward in establishing the validity of real-world evidence,” commented David Kerr, MBChB, an endocrinologist and director of research and innovation at the Sansum Diabetes Research Institute in Santa Barbara, Calif.
‘Target trials’ tether RWE
The target trial approach to designing an observational study is a key tool for boosting reliability and applicability of the results. The idea is to create a well-designed trial that could be the basis for a conventional RCT, and then use observational data to flesh out the target trial instead of collecting data from prospectively enrolled patients.
Designing observational studies that emulate target trials allows causal inferences, said Miguel A. Hernán, MD, DrPH, a professor of biostatistics and epidemiology at the Harvard School of Public Health, Boston. Plugging real-world data into the framework of an appropriately designed target trial substantially cuts the risk of a biased analysis, he explained during the workshop.
However, the approach has limitations. The target trial must be a pragmatic trial, and the approach does not work for placebo-controlled trials, although it can accommodate a usual-care control arm. It also usually precludes patient blinding, testing treatments not used in routine practice, and close monitoring of patients in ways that are uncommon in usual care.
The target trial approach received broad endorsement during the workshop as the future for observational studies destined for efficacy consideration by the FDA.
“The idea of prespecifying a target trial is a really fantastic place to start,” commented Robert Ball, MD, deputy director of the FDA Office of Surveillance and Epidemiology. “There is still a whole set of questions once the trial is prespecified, but prespecification would be a fantastic step forward,” he said during the workshop.
Participants also endorsed other important steps to boost the value of observational studies for regulatory reviews, including preregistering the study on a site such as clinicaltrials.gov; being fully transparent about the origins of observational data; using data that match the needs of the target trial; not reviewing the data in advance to avoid cherry picking and gaming the analysis; and reporting neutral or negative results when they occur, something often not currently done for observational analyses.
But although there was clear progress and much agreement among thought leaders at the workshop, FDA representatives stressed caution in moving forward.
“No easy answer”
“With more experience, we can learn what works and what doesn’t work in generating valid results from observational studies,” said Dr. Concato. “Although the observational results have upside potential, we need to learn more. There is no easy answer, no checklist for fit-for-use data, no off-the-shelf study design, and no ideal analytic method.”
Dr. Concato acknowledged that the FDA’s goal is clear given the 2016 legislation. “The FDA is embracing our obligations under the 21st Century Cures Act to evaluate use of real-world data and real-world evidence.”
He also suggested that researchers “shy away from a false dichotomy of RCTs or observational studies and instead think about how and when RCTs and observational studies can be designed and conducted to yield trustworthy results.” Dr. Concato’s solution: “a taxonomy of interventional or noninterventional studies.”
“The FDA is under enormous pressure to embrace real-world evidence, both because of the economics of running RCTs and because of the availability of new observational data from electronic health records, wearable devices, claims, etc.,” said Dr. Kerr, who did not participate in the workshop but coauthored an editorial that calls for using real-world data in regulatory decisions for drugs and devices for diabetes. These factors create an “irresistible force” spurring the FDA to consider observational, noninterventional data.
“I think the FDA really wants this to go forward,” Dr. Kerr added in an interview. “The FDA keeps telling us that clinical trials do not have enough women or patients from minority groups. Real-world data is a way to address that. This will not be the death of RCTs, but this work shines a light on the deficiencies of RCTs and how the deficiencies can be dealt with.”
Dr. Kroetsch has reported no relevant financial relationships. Dr. Schneeweiss has reported being a consultant to and holding equity in Aetion and receiving research funding from the FDA. Dr. Ross has reported receiving research funding from the FDA, Johnson & Johnson, and Medtronic. Dr. Hernán has reported being a consultant for Cytel. Dr. Kerr has reported being a consultant for Ascensia, EOFlow, Lifecare, Merck, Novo Nordisk, Roche Diagnostics, and Voluntis. Dr. Temple, Dr. Concato, and Dr. Ball are FDA employees.
A version of this article first appeared on Medscape.com.
In 2016, results from the LEADER trial of liraglutide in patients with type 2 diabetes helped jump-start awareness of the potential role of this new class of drugs, the glucagonlike peptide–1 receptor agonists, for reducing cardiovascular events. The randomized, placebo-controlled trial enrolled more than 9000 patients at more than 400 sites in over 30 countries, and took nearly 6 years from the start of patient enrollment to publication of the landmark results.
In December 2020, an independent team of researchers published results from a study with a design identical to LEADER, but used data that came not from a massive, global, years-long trial but from already-existing numbers culled from three large U.S. insurance claim databases. The result of this emulation using real-world data was virtually identical to what the actual trial showed, replicating both the direction and statistical significance of the original finding of the randomized, controlled trial (RCT).
What if research proved that this sort of RCT emulation could reliably be done on a regular basis? What might it mean for regulatory decisions on drugs and devices that historically have been based entirely on efficacy evidence from RCTs?
Making the most of a sea of observational data
Medicine in the United States has become increasingly awash in a sea of observational data collected from sources that include electronic health records, insurance claims, and increasingly, personal-health monitoring devices.
The Food and Drug Administration is now in the process of trying to figure out how it can legitimately harness this tsunami of real-world data to make efficacy decisions, essentially creating a new category of evidence to complement traditional data from randomized trials. It’s an opportunity that agency staff and their outside advisors have been keen to seize, especially given the soaring cost of prospective, randomized trials.
Recognition of this untapped resource in part led to a key initiative, among many others, included in the 21st Century Cures Act, passed in December 2016. Among the Act’s mandates was that, by the end of 2021, the FDA would issue guidance on when drug sponsors could use real-world evidence (RWE) to either help support a new indication for an already approved drug or help satisfy postapproval study requirements.
The initiative recognizes that this approach is not appropriate for initial drug approvals, which remain exclusively reliant on evidence from RCTs. Instead, it seems best suited to support expanding indications for already approved drugs.
Although FDA staff have made progress in identifying the challenges and broadening their understanding of how to best handle real-world data that come from observing patients in routine practice, agency leaders stress that this complex issue will likely not be fully resolved by their guidance to be published later this year. The FDA released a draft of the guidance in May 2019.
Can RWE be ‘credible and reliable?’
“Whether observational, nonrandomized data can become credible enough to use is what we’re talking about. These are possibilities that need to be explained and better understood,” said Robert Temple, MD, deputy director for clinical science of the FDA Center for Drug Evaluation and Research.
“Since the 1970s, the FDA has recognized historical controls as legitimate, so it’s possible [for RWE] to be credible. The big test is when is it credible and reliable enough [to assess efficacy]?” wondered Dr. Temple during a 2-day workshop on the topic held mid-February and organized by Duke University’s Margolis Center for Health Policy.
“We’re approaching an inflection point regarding how observational studies are generated and used, but our evidentiary standards will not lower, and it will be a case-by-case decision” by the agency as they review future RWE submissions, said John Concato, MD, the FDA’s associate director for real-world evidence, during the workshop.
“We are working toward guidance development, but also looking down the road to what we need to do to enable this,” said Dr. Concato. “It’s a complicated issue. If it was easy, it would have already been fixed.” He added that the agency will likely release a “portfolio” of guidance for submitting real-world data and RWE. Real-world data are raw information that, when analyzed, become RWE.
In short, the FDA seems headed toward guidance that won’t spell out a pathway that guarantees success using RWE but will at least open the door to consideration of this unprecedented application.
Not like flipping a switch
The guidance will not activate acceptance of RWE all at once. “It’s not like a light switch,” cautioned Adam Kroetsch, MPP, research director for biomedical innovation and regulatory policy at Duke-Margolis in Washington, D.C. “It’s an evolutionary process,” and the upcoming guidance will provide “just a little more clarity” on what sorts of best practices using RWE the FDA will find persuasive. “It’s hard for the FDA to clearly say what it’s looking for until they see some good examples,” Dr. Kroetsch said in an interview.
What will change is that drug sponsors can submit using RWE, and the FDA “will have a more open-minded view,” predicted Sebastian Schneeweiss, MD, ScD, a workshop participant and chief of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital in Boston. “For the first time, a law required [the FDA] to take a serious look” at observational data for efficacy assessment.
“The FDA has had a bias against using RWE for evidence of efficacy but has long used it to understand drug safety. Now the FDA is trying to wrap its arms around how to best use RWE” for efficacy decisions, said Joseph S. Ross, MD, another workshop participant and professor of medicine and public health at Yale University, New Haven, Conn.
The agency’s cautious approach is reassuring, Dr. Ross noted in an interview. “There was worry that the 21st Century Cures Act would open the door to allowing real-world data to be used in ways that weren’t very reliable. Very quickly, the FDA started trying to figure out the best ways to use these data in reasonable ways.”
Duplicating RCTs with RWE
To help better understand the potential use of RWE, the FDA sponsored several demonstration projects. Researchers presented results from three of these projects during the workshop in February. All three examined whether RWE, plugged into the design of an actual RCT, can produce roughly similar results when similar patients are used.
A generally consistent finding from the three demonstration projects was that “when the data are fit for purpose” the emulated or duplicated analyses with RWE “can come to similar conclusions” as the actual RCTs, said Dr. Schneeweiss, who leads one of the demonstration projects, RCT DUPLICATE.
At the workshop he reported results from RWE duplications of 20 different RCTs using insurance claims data from U.S. patients. The findings came from 10 duplications already reported in Circulation in December 2020 (including a duplication of the LEADER trial), and an additional 10 as yet unpublished RCT duplications. In the next few months, the researchers intend to assess a final group of 10 more RCT duplications.
Workshop participants also presented results from two other FDA demonstration projects: the OPERAND program run by the Multi-Regional Clinical Trials Center of Brigham and Women’s Hospital and Harvard; and the CERSI program based at Yale and the Mayo Clinic in Rochester, Minn. Both are smaller in scale than RCT DUPLICATE, incorporate lab data in addition to claims data, and in some cases test how well RWE can emulate RCTs that are not yet completed.
Collectively, results from these demonstration projects suggest that RWE can successfully emulate the results of an RCT, said Dr. Ross, a coinvestigator on the CERSI study. But the CERSI findings also highlighted how an RCT can fall short of clinical relevance.
“One of our most important findings was that RCTs don’t always represent real-world practice,” he said. His group attempted to replicate the 5,000-patient GRADE trial of four different drug options added to metformin in patients with type 2 diabetes. One of the four options included insulin glargine (Lantus), and the attempt to emulate the study with RWE hit the bump that no relevant real-world patients in their US claims database actually received the formulation.
That means the GRADE trial “is almost meaningless. It doesn’t reflect real-world practice,” Dr. Ross noted.
Results from the three demonstration projects “highlight the gaps we still have,” summed up Dr. Kroetsch. “They show where we need better data” from observational sources that function as well as data from RCTs.
Still, the demonstration project results are “an important step forward in establishing the validity of real-world evidence,” commented David Kerr, MBChB, an endocrinologist and director of research and innovation at the Sansum Diabetes Research Institute in Santa Barbara, Calif.
‘Target trials’ tether RWE
The target trial approach to designing an observational study is a key tool for boosting reliability and applicability of the results. The idea is to create a well-designed trial that could be the basis for a conventional RCT, and then use observational data to flesh out the target trial instead of collecting data from prospectively enrolled patients.
Designing observational studies that emulate target trials allows causal inferences, said Miguel A. Hernán, MD, DrPH, a professor of biostatistics and epidemiology at the Harvard School of Public Health, Boston. Plugging real-world data into the framework of an appropriately designed target trial substantially cuts the risk of a biased analysis, he explained during the workshop.
However, the approach has limitations. The target trial must be a pragmatic trial, and the approach does not work for placebo-controlled trials, although it can accommodate a usual-care control arm. It also usually precludes patient blinding, testing treatments not used in routine practice, and close monitoring of patients in ways that are uncommon in usual care.
The target trial approach received broad endorsement during the workshop as the future for observational studies destined for efficacy consideration by the FDA.
“The idea of prespecifying a target trial is a really fantastic place to start,” commented Robert Ball, MD, deputy director of the FDA Office of Surveillance and Epidemiology. “There is still a whole set of questions once the trial is prespecified, but prespecification would be a fantastic step forward,” he said during the workshop.
Participants also endorsed other important steps to boost the value of observational studies for regulatory reviews, including preregistering the study on a site such as clinicaltrials.gov; being fully transparent about the origins of observational data; using data that match the needs of the target trial; not reviewing the data in advance to avoid cherry picking and gaming the analysis; and reporting neutral or negative results when they occur, something often not currently done for observational analyses.
But although there was clear progress and much agreement among thought leaders at the workshop, FDA representatives stressed caution in moving forward.
“No easy answer”
“With more experience, we can learn what works and what doesn’t work in generating valid results from observational studies,” said Dr. Concato. “Although the observational results have upside potential, we need to learn more. There is no easy answer, no checklist for fit-for-use data, no off-the-shelf study design, and no ideal analytic method.”
Dr. Concato acknowledged that the FDA’s goal is clear given the 2016 legislation. “The FDA is embracing our obligations under the 21st Century Cures Act to evaluate use of real-world data and real-world evidence.”
He also suggested that researchers “shy away from a false dichotomy of RCTs or observational studies and instead think about how and when RCTs and observational studies can be designed and conducted to yield trustworthy results.” Dr. Concato’s solution: “a taxonomy of interventional or noninterventional studies.”
“The FDA is under enormous pressure to embrace real-world evidence, both because of the economics of running RCTs and because of the availability of new observational data from electronic health records, wearable devices, claims, etc.,” said Dr. Kerr, who did not participate in the workshop but coauthored an editorial that calls for using real-world data in regulatory decisions for drugs and devices for diabetes. These factors create an “irresistible force” spurring the FDA to consider observational, noninterventional data.
“I think the FDA really wants this to go forward,” Dr. Kerr added in an interview. “The FDA keeps telling us that clinical trials do not have enough women or patients from minority groups. Real-world data is a way to address that. This will not be the death of RCTs, but this work shines a light on the deficiencies of RCTs and how the deficiencies can be dealt with.”
Dr. Kroetsch has reported no relevant financial relationships. Dr. Schneeweiss has reported being a consultant to and holding equity in Aetion and receiving research funding from the FDA. Dr. Ross has reported receiving research funding from the FDA, Johnson & Johnson, and Medtronic. Dr. Hernán has reported being a consultant for Cytel. Dr. Kerr has reported being a consultant for Ascensia, EOFlow, Lifecare, Merck, Novo Nordisk, Roche Diagnostics, and Voluntis. Dr. Temple, Dr. Concato, and Dr. Ball are FDA employees.
A version of this article first appeared on Medscape.com.
In 2016, results from the LEADER trial of liraglutide in patients with type 2 diabetes helped jump-start awareness of the potential role of this new class of drugs, the glucagonlike peptide–1 receptor agonists, for reducing cardiovascular events. The randomized, placebo-controlled trial enrolled more than 9000 patients at more than 400 sites in over 30 countries, and took nearly 6 years from the start of patient enrollment to publication of the landmark results.
In December 2020, an independent team of researchers published results from a study with a design identical to LEADER, but used data that came not from a massive, global, years-long trial but from already-existing numbers culled from three large U.S. insurance claim databases. The result of this emulation using real-world data was virtually identical to what the actual trial showed, replicating both the direction and statistical significance of the original finding of the randomized, controlled trial (RCT).
What if research proved that this sort of RCT emulation could reliably be done on a regular basis? What might it mean for regulatory decisions on drugs and devices that historically have been based entirely on efficacy evidence from RCTs?
Making the most of a sea of observational data
Medicine in the United States has become increasingly awash in a sea of observational data collected from sources that include electronic health records, insurance claims, and increasingly, personal-health monitoring devices.
The Food and Drug Administration is now in the process of trying to figure out how it can legitimately harness this tsunami of real-world data to make efficacy decisions, essentially creating a new category of evidence to complement traditional data from randomized trials. It’s an opportunity that agency staff and their outside advisors have been keen to seize, especially given the soaring cost of prospective, randomized trials.
Recognition of this untapped resource in part led to a key initiative, among many others, included in the 21st Century Cures Act, passed in December 2016. Among the Act’s mandates was that, by the end of 2021, the FDA would issue guidance on when drug sponsors could use real-world evidence (RWE) to either help support a new indication for an already approved drug or help satisfy postapproval study requirements.
The initiative recognizes that this approach is not appropriate for initial drug approvals, which remain exclusively reliant on evidence from RCTs. Instead, it seems best suited to support expanding indications for already approved drugs.
Although FDA staff have made progress in identifying the challenges and broadening their understanding of how to best handle real-world data that come from observing patients in routine practice, agency leaders stress that this complex issue will likely not be fully resolved by their guidance to be published later this year. The FDA released a draft of the guidance in May 2019.
Can RWE be ‘credible and reliable?’
“Whether observational, nonrandomized data can become credible enough to use is what we’re talking about. These are possibilities that need to be explained and better understood,” said Robert Temple, MD, deputy director for clinical science of the FDA Center for Drug Evaluation and Research.
“Since the 1970s, the FDA has recognized historical controls as legitimate, so it’s possible [for RWE] to be credible. The big test is when is it credible and reliable enough [to assess efficacy]?” wondered Dr. Temple during a 2-day workshop on the topic held mid-February and organized by Duke University’s Margolis Center for Health Policy.
“We’re approaching an inflection point regarding how observational studies are generated and used, but our evidentiary standards will not lower, and it will be a case-by-case decision” by the agency as they review future RWE submissions, said John Concato, MD, the FDA’s associate director for real-world evidence, during the workshop.
“We are working toward guidance development, but also looking down the road to what we need to do to enable this,” said Dr. Concato. “It’s a complicated issue. If it was easy, it would have already been fixed.” He added that the agency will likely release a “portfolio” of guidance for submitting real-world data and RWE. Real-world data are raw information that, when analyzed, become RWE.
In short, the FDA seems headed toward guidance that won’t spell out a pathway that guarantees success using RWE but will at least open the door to consideration of this unprecedented application.
Not like flipping a switch
The guidance will not activate acceptance of RWE all at once. “It’s not like a light switch,” cautioned Adam Kroetsch, MPP, research director for biomedical innovation and regulatory policy at Duke-Margolis in Washington, D.C. “It’s an evolutionary process,” and the upcoming guidance will provide “just a little more clarity” on what sorts of best practices using RWE the FDA will find persuasive. “It’s hard for the FDA to clearly say what it’s looking for until they see some good examples,” Dr. Kroetsch said in an interview.
What will change is that drug sponsors can submit using RWE, and the FDA “will have a more open-minded view,” predicted Sebastian Schneeweiss, MD, ScD, a workshop participant and chief of pharmacoepidemiology and pharmacoeconomics at Brigham and Women’s Hospital in Boston. “For the first time, a law required [the FDA] to take a serious look” at observational data for efficacy assessment.
“The FDA has had a bias against using RWE for evidence of efficacy but has long used it to understand drug safety. Now the FDA is trying to wrap its arms around how to best use RWE” for efficacy decisions, said Joseph S. Ross, MD, another workshop participant and professor of medicine and public health at Yale University, New Haven, Conn.
The agency’s cautious approach is reassuring, Dr. Ross noted in an interview. “There was worry that the 21st Century Cures Act would open the door to allowing real-world data to be used in ways that weren’t very reliable. Very quickly, the FDA started trying to figure out the best ways to use these data in reasonable ways.”
Duplicating RCTs with RWE
To help better understand the potential use of RWE, the FDA sponsored several demonstration projects. Researchers presented results from three of these projects during the workshop in February. All three examined whether RWE, plugged into the design of an actual RCT, can produce roughly similar results when similar patients are used.
A generally consistent finding from the three demonstration projects was that “when the data are fit for purpose” the emulated or duplicated analyses with RWE “can come to similar conclusions” as the actual RCTs, said Dr. Schneeweiss, who leads one of the demonstration projects, RCT DUPLICATE.
At the workshop he reported results from RWE duplications of 20 different RCTs using insurance claims data from U.S. patients. The findings came from 10 duplications already reported in Circulation in December 2020 (including a duplication of the LEADER trial), and an additional 10 as yet unpublished RCT duplications. In the next few months, the researchers intend to assess a final group of 10 more RCT duplications.
Workshop participants also presented results from two other FDA demonstration projects: the OPERAND program run by the Multi-Regional Clinical Trials Center of Brigham and Women’s Hospital and Harvard; and the CERSI program based at Yale and the Mayo Clinic in Rochester, Minn. Both are smaller in scale than RCT DUPLICATE, incorporate lab data in addition to claims data, and in some cases test how well RWE can emulate RCTs that are not yet completed.
Collectively, results from these demonstration projects suggest that RWE can successfully emulate the results of an RCT, said Dr. Ross, a coinvestigator on the CERSI study. But the CERSI findings also highlighted how an RCT can fall short of clinical relevance.
“One of our most important findings was that RCTs don’t always represent real-world practice,” he said. His group attempted to replicate the 5,000-patient GRADE trial of four different drug options added to metformin in patients with type 2 diabetes. One of the four options included insulin glargine (Lantus), and the attempt to emulate the study with RWE hit the bump that no relevant real-world patients in their US claims database actually received the formulation.
That means the GRADE trial “is almost meaningless. It doesn’t reflect real-world practice,” Dr. Ross noted.
Results from the three demonstration projects “highlight the gaps we still have,” summed up Dr. Kroetsch. “They show where we need better data” from observational sources that function as well as data from RCTs.
Still, the demonstration project results are “an important step forward in establishing the validity of real-world evidence,” commented David Kerr, MBChB, an endocrinologist and director of research and innovation at the Sansum Diabetes Research Institute in Santa Barbara, Calif.
‘Target trials’ tether RWE
The target trial approach to designing an observational study is a key tool for boosting reliability and applicability of the results. The idea is to create a well-designed trial that could be the basis for a conventional RCT, and then use observational data to flesh out the target trial instead of collecting data from prospectively enrolled patients.
Designing observational studies that emulate target trials allows causal inferences, said Miguel A. Hernán, MD, DrPH, a professor of biostatistics and epidemiology at the Harvard School of Public Health, Boston. Plugging real-world data into the framework of an appropriately designed target trial substantially cuts the risk of a biased analysis, he explained during the workshop.
However, the approach has limitations. The target trial must be a pragmatic trial, and the approach does not work for placebo-controlled trials, although it can accommodate a usual-care control arm. It also usually precludes patient blinding, testing treatments not used in routine practice, and close monitoring of patients in ways that are uncommon in usual care.
The target trial approach received broad endorsement during the workshop as the future for observational studies destined for efficacy consideration by the FDA.
“The idea of prespecifying a target trial is a really fantastic place to start,” commented Robert Ball, MD, deputy director of the FDA Office of Surveillance and Epidemiology. “There is still a whole set of questions once the trial is prespecified, but prespecification would be a fantastic step forward,” he said during the workshop.
Participants also endorsed other important steps to boost the value of observational studies for regulatory reviews, including preregistering the study on a site such as clinicaltrials.gov; being fully transparent about the origins of observational data; using data that match the needs of the target trial; not reviewing the data in advance to avoid cherry picking and gaming the analysis; and reporting neutral or negative results when they occur, something often not currently done for observational analyses.
But although there was clear progress and much agreement among thought leaders at the workshop, FDA representatives stressed caution in moving forward.
“No easy answer”
“With more experience, we can learn what works and what doesn’t work in generating valid results from observational studies,” said Dr. Concato. “Although the observational results have upside potential, we need to learn more. There is no easy answer, no checklist for fit-for-use data, no off-the-shelf study design, and no ideal analytic method.”
Dr. Concato acknowledged that the FDA’s goal is clear given the 2016 legislation. “The FDA is embracing our obligations under the 21st Century Cures Act to evaluate use of real-world data and real-world evidence.”
He also suggested that researchers “shy away from a false dichotomy of RCTs or observational studies and instead think about how and when RCTs and observational studies can be designed and conducted to yield trustworthy results.” Dr. Concato’s solution: “a taxonomy of interventional or noninterventional studies.”
“The FDA is under enormous pressure to embrace real-world evidence, both because of the economics of running RCTs and because of the availability of new observational data from electronic health records, wearable devices, claims, etc.,” said Dr. Kerr, who did not participate in the workshop but coauthored an editorial that calls for using real-world data in regulatory decisions for drugs and devices for diabetes. These factors create an “irresistible force” spurring the FDA to consider observational, noninterventional data.
“I think the FDA really wants this to go forward,” Dr. Kerr added in an interview. “The FDA keeps telling us that clinical trials do not have enough women or patients from minority groups. Real-world data is a way to address that. This will not be the death of RCTs, but this work shines a light on the deficiencies of RCTs and how the deficiencies can be dealt with.”
Dr. Kroetsch has reported no relevant financial relationships. Dr. Schneeweiss has reported being a consultant to and holding equity in Aetion and receiving research funding from the FDA. Dr. Ross has reported receiving research funding from the FDA, Johnson & Johnson, and Medtronic. Dr. Hernán has reported being a consultant for Cytel. Dr. Kerr has reported being a consultant for Ascensia, EOFlow, Lifecare, Merck, Novo Nordisk, Roche Diagnostics, and Voluntis. Dr. Temple, Dr. Concato, and Dr. Ball are FDA employees.
A version of this article first appeared on Medscape.com.
Fresh look at ISCHEMIA bolsters conservative message in stable CAD
The more complicated a primary endpoint, the greater a puzzle it can be for clinicians to interpret the results. It’s likely even tougher for patients, who don’t help choose the events studied in clinical trials yet are increasingly sharing in the management decisions they influence.
That creates an opening for a more patient-centered take on one of cardiology’s most influential recent studies, ISCHEMIA, which bolsters the case for conservative, med-oriented management over a more invasive initial strategy for patients with stable coronary artery disease (CAD) and positive stress tests, researchers said.
The new, prespecified analysis replaced the trial’s conventional primary endpoint of major adverse cardiac events (MACE) with one based on “days alive out of hospital” (DAOH) and found an early advantage for the conservative approach, with caveats.
Those assigned to the conservative arm benefited with more out-of-hospital days throughout the next 2 years than those in the invasive-management group, owing to the latter’s protocol-mandated early cath-lab work-up with possible revascularization. The difference averaged more than 6 days for much of that time.
But DAOH evened out for the two groups by the fourth year in the analysis of more than 5,000 patients.
Protocol-determined cath procedures accounted for 61% of hospitalizations in the invasively managed group. A secondary DAOH analysis that excluded such required hospital days, also prespecified, showed no meaningful difference between the two strategies over the 4 years, noted the report published online May 3 in JAMA Cardiology.
DOAH is ‘very, very important’
The DAOH metric has been a far less common consideration in clinical trials, compared with clinical events, yet in some ways it is as “hard” a metric as mortality, encompasses a broader range of outcomes, and may matter more to patients, it’s been argued.
“The thing patients most value is time at home. So they don’t want to be in the hospital, they don’t want to be away from friends, they want to do recreation, or they may want to work,” lead author Harvey D. White, DSc, Green Lane Cardiovascular Services, Auckland (New Zealand) City Hospital, University of Auckland, told this news organization.
“When we need to talk to patients – and we do need to talk to patients – to have a days-out-of-hospital metric is very, very important,” he said. It is not only patient focused, it’s “meaningful in terms of the seriousness of events,” in that length of hospitalization tracks with clinical severity, observed Dr. White, who is slated to present the analysis May 17 during the virtual American College of Cardiology 2021 scientific sessions.
As previously reported, ISCHEMIA showed no significant effect on the primary endpoint of cardiovascular mortality, MI, or hospitalization for unstable angina, heart failure, or resuscitated cardiac arrest by assignment group over a median 3.2 years. Angina and quality of life measures were improved for patients in the invasive arm.
With an invasive initial strategy, “What we know now is that you get nothing of an advantage in terms of the composite endpoint, and you’re going to spend 6 days more in the hospital in the first 2 years, for largely no benefit,” Dr. White said.
That outlook may apply out to 4 years, the analysis suggests, but could conceivably change if DAOH is reassessed later as the ISCHEMIA follow-up continues for what is now a planned total of 10 years.
Meanwhile, the current findings could enhance doctor-patient discussions about the trade-offs between the two strategies for individuals whose considerations will vary.
“This is a very helpful measure to understand the burden of an approach to the patient,” observed E. Magnus Ohman, MD, an interventional cardiologist at Duke University, Durham, N.C., who was not involved in the trial.
With DAOH as an endpoint, “you as a clinician get another aspect of understanding of a treatment’s impact on a multitude of endpoints.” Days out of hospital, he noted, encompasses the effects of clinical events that often go into composite clinical endpoints – not death, but including nonfatal MI, stroke, need for revascularization, and cardiovascular hospitalization.
To patients with stable CAD who ask whether the invasive approach has merits in their case, the DAOH finding “helps you to say, well, at the end of the day, you will probably be spending an equal amount of time in the hospital. Your price up front is a little bit higher, but over time, the group who gets conservative treatment will catch up.”
The DAOH outcome also avoids the limitations of an endpoint based on time to first event, “not the least of which,” said Dr. White, is that it counts only the first of what might be multiple events of varying clinical impact. Misleadingly, “you can have an event that’s a small troponin rise, but that becomes more important in a person than dying the next day.”
The DAOH analysis was based on 5,179 patients from 37 countries who averaged 64 years of age and of whom 23% were women. The endpoint considered only overnight stays in hospitals, skilled nursing facilities, rehabilitation centers, and nursing homes.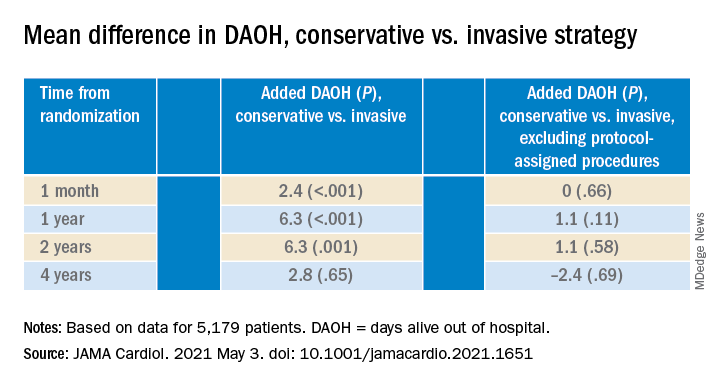
There were many more hospital or extended care facility stays overall in the invasive-management group, 4,002 versus 1,897 for those following the conservative strategy (P < .001), but the numbers flipped after excluding protocol-assigned procedures: 1,568 stays in the invasive group, compared with 1,897 (P = .001)
There were no associations between DAOH and Seattle Angina Questionnaire 7–Angina Frequency scores or DAOH interactions by age, sex, geographic region, or whether the patient had diabetes, prior MI, or heart failure, the report notes.
The primary ISCHEMIA analysis hinted at a possible long-term advantage for the invasive initial strategy in that event curves for the two arms crossed after 2-3 years, Dr. Ohman observed.
Based on that, for younger patients with stable CAD and ischemia at stress testing, “an investment of more hospital days early on might be worth it in the long run.” But ISCHEMIA, he said, “only suggests it, it doesn’t confirm it.”
The study was supported in part by grants from Arbor Pharmaceuticals and AstraZeneca. Devices or medications were provided by Abbott Vascular, Amgen, Arbor, AstraZeneca, Esperion, Medtronic, Merck Sharp & Dohme, Phillips, Omron Healthcare, and Sunovion. Dr. White disclosed receiving grants paid to his institution and fees for serving on a steering committee from Sanofi-Aventis, Regeneron, Eli Lilly, Omthera, American Regent, Eisai, DalCor, CSL Behring, Sanofi-Aventis Australia, and Esperion Therapeutics, and personal fees from Genentech and AstraZeneca. Dr. Ohman reported receiving grants from Abiomed and Cheisi USA, and consulting for Abiomed, Cara Therapeutics, Chiesi USA, Cytokinetics, Imbria Pharmaceuticals, Otsuka Pharmaceuticals, Milestone Pharmaceuticals, and XyloCor Therapeutics.
A version of this article first appeared on Medscape.com.
The more complicated a primary endpoint, the greater a puzzle it can be for clinicians to interpret the results. It’s likely even tougher for patients, who don’t help choose the events studied in clinical trials yet are increasingly sharing in the management decisions they influence.
That creates an opening for a more patient-centered take on one of cardiology’s most influential recent studies, ISCHEMIA, which bolsters the case for conservative, med-oriented management over a more invasive initial strategy for patients with stable coronary artery disease (CAD) and positive stress tests, researchers said.
The new, prespecified analysis replaced the trial’s conventional primary endpoint of major adverse cardiac events (MACE) with one based on “days alive out of hospital” (DAOH) and found an early advantage for the conservative approach, with caveats.
Those assigned to the conservative arm benefited with more out-of-hospital days throughout the next 2 years than those in the invasive-management group, owing to the latter’s protocol-mandated early cath-lab work-up with possible revascularization. The difference averaged more than 6 days for much of that time.
But DAOH evened out for the two groups by the fourth year in the analysis of more than 5,000 patients.
Protocol-determined cath procedures accounted for 61% of hospitalizations in the invasively managed group. A secondary DAOH analysis that excluded such required hospital days, also prespecified, showed no meaningful difference between the two strategies over the 4 years, noted the report published online May 3 in JAMA Cardiology.
DOAH is ‘very, very important’
The DAOH metric has been a far less common consideration in clinical trials, compared with clinical events, yet in some ways it is as “hard” a metric as mortality, encompasses a broader range of outcomes, and may matter more to patients, it’s been argued.
“The thing patients most value is time at home. So they don’t want to be in the hospital, they don’t want to be away from friends, they want to do recreation, or they may want to work,” lead author Harvey D. White, DSc, Green Lane Cardiovascular Services, Auckland (New Zealand) City Hospital, University of Auckland, told this news organization.
“When we need to talk to patients – and we do need to talk to patients – to have a days-out-of-hospital metric is very, very important,” he said. It is not only patient focused, it’s “meaningful in terms of the seriousness of events,” in that length of hospitalization tracks with clinical severity, observed Dr. White, who is slated to present the analysis May 17 during the virtual American College of Cardiology 2021 scientific sessions.
As previously reported, ISCHEMIA showed no significant effect on the primary endpoint of cardiovascular mortality, MI, or hospitalization for unstable angina, heart failure, or resuscitated cardiac arrest by assignment group over a median 3.2 years. Angina and quality of life measures were improved for patients in the invasive arm.
With an invasive initial strategy, “What we know now is that you get nothing of an advantage in terms of the composite endpoint, and you’re going to spend 6 days more in the hospital in the first 2 years, for largely no benefit,” Dr. White said.
That outlook may apply out to 4 years, the analysis suggests, but could conceivably change if DAOH is reassessed later as the ISCHEMIA follow-up continues for what is now a planned total of 10 years.
Meanwhile, the current findings could enhance doctor-patient discussions about the trade-offs between the two strategies for individuals whose considerations will vary.
“This is a very helpful measure to understand the burden of an approach to the patient,” observed E. Magnus Ohman, MD, an interventional cardiologist at Duke University, Durham, N.C., who was not involved in the trial.
With DAOH as an endpoint, “you as a clinician get another aspect of understanding of a treatment’s impact on a multitude of endpoints.” Days out of hospital, he noted, encompasses the effects of clinical events that often go into composite clinical endpoints – not death, but including nonfatal MI, stroke, need for revascularization, and cardiovascular hospitalization.
To patients with stable CAD who ask whether the invasive approach has merits in their case, the DAOH finding “helps you to say, well, at the end of the day, you will probably be spending an equal amount of time in the hospital. Your price up front is a little bit higher, but over time, the group who gets conservative treatment will catch up.”
The DAOH outcome also avoids the limitations of an endpoint based on time to first event, “not the least of which,” said Dr. White, is that it counts only the first of what might be multiple events of varying clinical impact. Misleadingly, “you can have an event that’s a small troponin rise, but that becomes more important in a person than dying the next day.”
The DAOH analysis was based on 5,179 patients from 37 countries who averaged 64 years of age and of whom 23% were women. The endpoint considered only overnight stays in hospitals, skilled nursing facilities, rehabilitation centers, and nursing homes.
There were many more hospital or extended care facility stays overall in the invasive-management group, 4,002 versus 1,897 for those following the conservative strategy (P < .001), but the numbers flipped after excluding protocol-assigned procedures: 1,568 stays in the invasive group, compared with 1,897 (P = .001)
There were no associations between DAOH and Seattle Angina Questionnaire 7–Angina Frequency scores or DAOH interactions by age, sex, geographic region, or whether the patient had diabetes, prior MI, or heart failure, the report notes.
The primary ISCHEMIA analysis hinted at a possible long-term advantage for the invasive initial strategy in that event curves for the two arms crossed after 2-3 years, Dr. Ohman observed.
Based on that, for younger patients with stable CAD and ischemia at stress testing, “an investment of more hospital days early on might be worth it in the long run.” But ISCHEMIA, he said, “only suggests it, it doesn’t confirm it.”
The study was supported in part by grants from Arbor Pharmaceuticals and AstraZeneca. Devices or medications were provided by Abbott Vascular, Amgen, Arbor, AstraZeneca, Esperion, Medtronic, Merck Sharp & Dohme, Phillips, Omron Healthcare, and Sunovion. Dr. White disclosed receiving grants paid to his institution and fees for serving on a steering committee from Sanofi-Aventis, Regeneron, Eli Lilly, Omthera, American Regent, Eisai, DalCor, CSL Behring, Sanofi-Aventis Australia, and Esperion Therapeutics, and personal fees from Genentech and AstraZeneca. Dr. Ohman reported receiving grants from Abiomed and Cheisi USA, and consulting for Abiomed, Cara Therapeutics, Chiesi USA, Cytokinetics, Imbria Pharmaceuticals, Otsuka Pharmaceuticals, Milestone Pharmaceuticals, and XyloCor Therapeutics.
A version of this article first appeared on Medscape.com.
The more complicated a primary endpoint, the greater a puzzle it can be for clinicians to interpret the results. It’s likely even tougher for patients, who don’t help choose the events studied in clinical trials yet are increasingly sharing in the management decisions they influence.
That creates an opening for a more patient-centered take on one of cardiology’s most influential recent studies, ISCHEMIA, which bolsters the case for conservative, med-oriented management over a more invasive initial strategy for patients with stable coronary artery disease (CAD) and positive stress tests, researchers said.
The new, prespecified analysis replaced the trial’s conventional primary endpoint of major adverse cardiac events (MACE) with one based on “days alive out of hospital” (DAOH) and found an early advantage for the conservative approach, with caveats.
Those assigned to the conservative arm benefited with more out-of-hospital days throughout the next 2 years than those in the invasive-management group, owing to the latter’s protocol-mandated early cath-lab work-up with possible revascularization. The difference averaged more than 6 days for much of that time.
But DAOH evened out for the two groups by the fourth year in the analysis of more than 5,000 patients.
Protocol-determined cath procedures accounted for 61% of hospitalizations in the invasively managed group. A secondary DAOH analysis that excluded such required hospital days, also prespecified, showed no meaningful difference between the two strategies over the 4 years, noted the report published online May 3 in JAMA Cardiology.
DOAH is ‘very, very important’
The DAOH metric has been a far less common consideration in clinical trials, compared with clinical events, yet in some ways it is as “hard” a metric as mortality, encompasses a broader range of outcomes, and may matter more to patients, it’s been argued.
“The thing patients most value is time at home. So they don’t want to be in the hospital, they don’t want to be away from friends, they want to do recreation, or they may want to work,” lead author Harvey D. White, DSc, Green Lane Cardiovascular Services, Auckland (New Zealand) City Hospital, University of Auckland, told this news organization.
“When we need to talk to patients – and we do need to talk to patients – to have a days-out-of-hospital metric is very, very important,” he said. It is not only patient focused, it’s “meaningful in terms of the seriousness of events,” in that length of hospitalization tracks with clinical severity, observed Dr. White, who is slated to present the analysis May 17 during the virtual American College of Cardiology 2021 scientific sessions.
As previously reported, ISCHEMIA showed no significant effect on the primary endpoint of cardiovascular mortality, MI, or hospitalization for unstable angina, heart failure, or resuscitated cardiac arrest by assignment group over a median 3.2 years. Angina and quality of life measures were improved for patients in the invasive arm.
With an invasive initial strategy, “What we know now is that you get nothing of an advantage in terms of the composite endpoint, and you’re going to spend 6 days more in the hospital in the first 2 years, for largely no benefit,” Dr. White said.
That outlook may apply out to 4 years, the analysis suggests, but could conceivably change if DAOH is reassessed later as the ISCHEMIA follow-up continues for what is now a planned total of 10 years.
Meanwhile, the current findings could enhance doctor-patient discussions about the trade-offs between the two strategies for individuals whose considerations will vary.
“This is a very helpful measure to understand the burden of an approach to the patient,” observed E. Magnus Ohman, MD, an interventional cardiologist at Duke University, Durham, N.C., who was not involved in the trial.
With DAOH as an endpoint, “you as a clinician get another aspect of understanding of a treatment’s impact on a multitude of endpoints.” Days out of hospital, he noted, encompasses the effects of clinical events that often go into composite clinical endpoints – not death, but including nonfatal MI, stroke, need for revascularization, and cardiovascular hospitalization.
To patients with stable CAD who ask whether the invasive approach has merits in their case, the DAOH finding “helps you to say, well, at the end of the day, you will probably be spending an equal amount of time in the hospital. Your price up front is a little bit higher, but over time, the group who gets conservative treatment will catch up.”
The DAOH outcome also avoids the limitations of an endpoint based on time to first event, “not the least of which,” said Dr. White, is that it counts only the first of what might be multiple events of varying clinical impact. Misleadingly, “you can have an event that’s a small troponin rise, but that becomes more important in a person than dying the next day.”
The DAOH analysis was based on 5,179 patients from 37 countries who averaged 64 years of age and of whom 23% were women. The endpoint considered only overnight stays in hospitals, skilled nursing facilities, rehabilitation centers, and nursing homes.
There were many more hospital or extended care facility stays overall in the invasive-management group, 4,002 versus 1,897 for those following the conservative strategy (P < .001), but the numbers flipped after excluding protocol-assigned procedures: 1,568 stays in the invasive group, compared with 1,897 (P = .001)
There were no associations between DAOH and Seattle Angina Questionnaire 7–Angina Frequency scores or DAOH interactions by age, sex, geographic region, or whether the patient had diabetes, prior MI, or heart failure, the report notes.
The primary ISCHEMIA analysis hinted at a possible long-term advantage for the invasive initial strategy in that event curves for the two arms crossed after 2-3 years, Dr. Ohman observed.
Based on that, for younger patients with stable CAD and ischemia at stress testing, “an investment of more hospital days early on might be worth it in the long run.” But ISCHEMIA, he said, “only suggests it, it doesn’t confirm it.”
The study was supported in part by grants from Arbor Pharmaceuticals and AstraZeneca. Devices or medications were provided by Abbott Vascular, Amgen, Arbor, AstraZeneca, Esperion, Medtronic, Merck Sharp & Dohme, Phillips, Omron Healthcare, and Sunovion. Dr. White disclosed receiving grants paid to his institution and fees for serving on a steering committee from Sanofi-Aventis, Regeneron, Eli Lilly, Omthera, American Regent, Eisai, DalCor, CSL Behring, Sanofi-Aventis Australia, and Esperion Therapeutics, and personal fees from Genentech and AstraZeneca. Dr. Ohman reported receiving grants from Abiomed and Cheisi USA, and consulting for Abiomed, Cara Therapeutics, Chiesi USA, Cytokinetics, Imbria Pharmaceuticals, Otsuka Pharmaceuticals, Milestone Pharmaceuticals, and XyloCor Therapeutics.
A version of this article first appeared on Medscape.com.
Small increase seen in new COVID-19 cases among children
After 2 consecutive weeks of declines, the number of new COVID-19 cases in children rose slightly, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
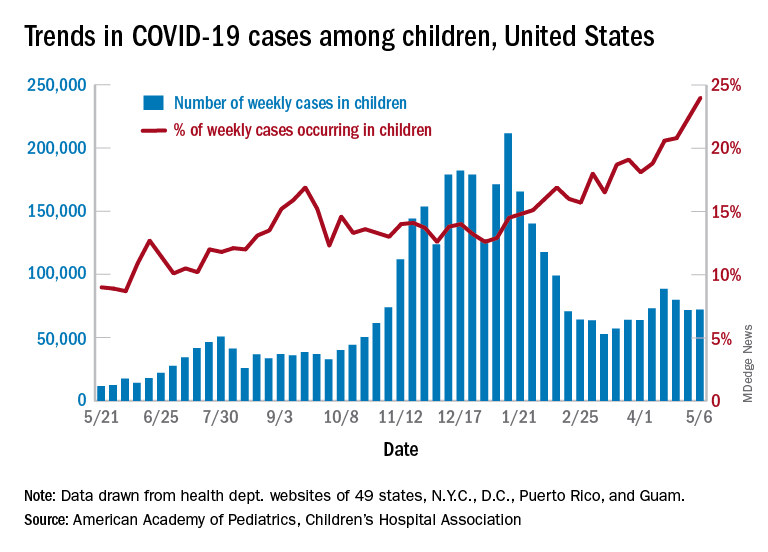
higher than at any other time during the pandemic, the AAP and CHA data show.
It is worth noting, however, that Rhode Island experienced a 30% increase in the last week, adding about 4,900 cases because of data revision and a lag in reporting, the AAP and CHA said in their weekly COVID-19 report.
All the new cases bring the total national count to just over 3.54 million in children, which represents 14.0% of all cases in 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. The cumulative case rate as of May 6 was 5,122 per 100,000 children, the two organizations said.
All the new cases that were added to Rhode Island’s total give it the highest cumulative rate in the country: 9,614 cases per 100,000 children. North Dakota is right behind with 9,526 per 100,000, followed by Tennessee (8,898), Connecticut (8,281), and South Carolina (8,274). Vermont has the highest proportion of cases in children at 22.4%, with Alaska next at 20.3% and South Carolina third at 18.7%, according to the AAP and CHA.
Hawaii just reported its first COVID-19–related death in a child, which drops the number of states with zero deaths in children from 10 to 9. Two other new deaths in children from April 30 to May 6 bring the total number to 306 in the 43 states, along with New York City, Puerto Rico, and Guam, that are reporting the age distribution of deaths.
In a separate statement, AAP president Lee Savio Beers acknowledged the Food and Drug Administration’s authorization of the Pfizer-BioNTech vaccine for children aged 12-15 years as “a critically important step in bringing lifesaving vaccines to children and adolescents. ... We look forward to the discussion by the Advisory Committee on Immunization Practices of the CDC, which will make recommendations about the use of this vaccine in adolescents.”
After 2 consecutive weeks of declines, the number of new COVID-19 cases in children rose slightly, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
higher than at any other time during the pandemic, the AAP and CHA data show.
It is worth noting, however, that Rhode Island experienced a 30% increase in the last week, adding about 4,900 cases because of data revision and a lag in reporting, the AAP and CHA said in their weekly COVID-19 report.
All the new cases bring the total national count to just over 3.54 million in children, which represents 14.0% of all cases in 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. The cumulative case rate as of May 6 was 5,122 per 100,000 children, the two organizations said.
All the new cases that were added to Rhode Island’s total give it the highest cumulative rate in the country: 9,614 cases per 100,000 children. North Dakota is right behind with 9,526 per 100,000, followed by Tennessee (8,898), Connecticut (8,281), and South Carolina (8,274). Vermont has the highest proportion of cases in children at 22.4%, with Alaska next at 20.3% and South Carolina third at 18.7%, according to the AAP and CHA.
Hawaii just reported its first COVID-19–related death in a child, which drops the number of states with zero deaths in children from 10 to 9. Two other new deaths in children from April 30 to May 6 bring the total number to 306 in the 43 states, along with New York City, Puerto Rico, and Guam, that are reporting the age distribution of deaths.
In a separate statement, AAP president Lee Savio Beers acknowledged the Food and Drug Administration’s authorization of the Pfizer-BioNTech vaccine for children aged 12-15 years as “a critically important step in bringing lifesaving vaccines to children and adolescents. ... We look forward to the discussion by the Advisory Committee on Immunization Practices of the CDC, which will make recommendations about the use of this vaccine in adolescents.”
After 2 consecutive weeks of declines, the number of new COVID-19 cases in children rose slightly, according to a report from the American Academy of Pediatrics and the Children’s Hospital Association.
higher than at any other time during the pandemic, the AAP and CHA data show.
It is worth noting, however, that Rhode Island experienced a 30% increase in the last week, adding about 4,900 cases because of data revision and a lag in reporting, the AAP and CHA said in their weekly COVID-19 report.
All the new cases bring the total national count to just over 3.54 million in children, which represents 14.0% of all cases in 49 states (excluding New York), the District of Columbia, New York City, Puerto Rico, and Guam. The cumulative case rate as of May 6 was 5,122 per 100,000 children, the two organizations said.
All the new cases that were added to Rhode Island’s total give it the highest cumulative rate in the country: 9,614 cases per 100,000 children. North Dakota is right behind with 9,526 per 100,000, followed by Tennessee (8,898), Connecticut (8,281), and South Carolina (8,274). Vermont has the highest proportion of cases in children at 22.4%, with Alaska next at 20.3% and South Carolina third at 18.7%, according to the AAP and CHA.
Hawaii just reported its first COVID-19–related death in a child, which drops the number of states with zero deaths in children from 10 to 9. Two other new deaths in children from April 30 to May 6 bring the total number to 306 in the 43 states, along with New York City, Puerto Rico, and Guam, that are reporting the age distribution of deaths.
In a separate statement, AAP president Lee Savio Beers acknowledged the Food and Drug Administration’s authorization of the Pfizer-BioNTech vaccine for children aged 12-15 years as “a critically important step in bringing lifesaving vaccines to children and adolescents. ... We look forward to the discussion by the Advisory Committee on Immunization Practices of the CDC, which will make recommendations about the use of this vaccine in adolescents.”
Dr. Fauci: Feds may ease indoor mask mandates soon
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
Federal guidance on indoor mask use may change soon, Anthony S. Fauci, MD, director of the National Institute of Allergy and Infectious Diseases, said on May 9.
He was asked whether it’s time to start relaxing indoor mask requirements.
“I think so, and I think you’re going to probably be seeing that as we go along and as more people get vaccinated,” Dr. Fauci said on ABC News’s This Week.Nearly 150 million adults in the United States – or about 58% of the adult population – have received at least one COVID-19 vaccine dose, according to the latest CDC tally. About 113 million adults, or 44%, are considered fully vaccinated.
“The CDC will be, you know, almost in real time … updating their recommendations and their guidelines,” Dr. Fauci said.
In April, the CDC relaxed its guidance for those who have been vaccinated against COVID-19. Those who have gotten a shot don’t need to wear a mask outdoors or in small indoor gatherings with other vaccinated people, but both vaccinated and unvaccinated people are still advised to wear masks in indoor public spaces.
“We do need to start being more liberal as we get more people vaccinated,” Dr. Fauci said. “As you get more people vaccinated, the number of cases per day will absolutely go down.”
The United States is averaging about 43,000 cases per day, he said, adding that the cases need to be “much, much lower.” When the case numbers drop and vaccination numbers increase, the risk of infection will fall dramatically indoors and outdoors, he said.
Even after the pandemic, though, wearing masks could become a seasonal habit, Dr. Fauci said May 9 on NBC News’s Meet the Press.“I think people have gotten used to the fact that wearing masks, clearly if you look at the data, it diminishes respiratory diseases. We’ve had practically a nonexistent flu season this year,” he said.
“So it is conceivable that as we go on, a year or 2 or more from now, that during certain seasonal periods when you have respiratory-borne viruses like the flu, people might actually elect to wear masks to diminish the likelihood that you’ll spread these respiratory-borne diseases,” he said.
Dr. Fauci was asked about indoor mask guidelines on May 9 after former FDA Commissioner Scott Gottlieb, MD, said face mask requirements should be relaxed.
“Certainly outdoors, we shouldn’t be putting limits on gatherings anymore,” Dr. Gottlieb said on CBS News’s Face the Nation.“The states where prevalence is low, vaccination rates are high, we have good testing in place, and we’re identifying infections, I think we could start lifting these restrictions indoors as well, on a broad basis,” he said.
Lifting pandemic-related restrictions in areas where they’re no longer necessary could also encourage people to implement them again if cases increase during future surges, such as this fall or winter, Dr. Gottlieb said.
At the same time, Americans should continue to follow CDC guidance and wait for new guidelines before changing their indoor mask use, Jeffrey Zients, the White House COVID-19 response coordinator, said on CNN’s State of the Union on May 9.
“We all want to get back to a normal lifestyle,” he said. “I think we’re on the path to do that, but stay disciplined, and let’s take advantage of the new privilege of being vaccinated and not wearing masks outdoors, for example, unless you’re in a crowded place.”
Mr. Zients pointed to President Joe Biden’s goal for 70% of adults to receive at least one vaccine dose by July 4.
“As we all move toward that 70% goal, there will be more and more advantages to being vaccinated,” he said. “And if you’re not vaccinated, you’re not protected.”
A version of this article first appeared on WebMD.com.
FDA authorizes Pfizer COVID vaccine for teens 12-15
The Food and Drug Administration on May 10 granted emergency use authorization (EUA) for the Pfizer coronavirus vaccine to be given to children 12-15 years old.
The much-expected decision increases the likelihood that schools in the United States will fully reopen in the fall – a goal of both the Biden and Trump administrations.
Acting FDA Commissioner Janet Woodcock, MD, called the decision “a significant step” in “returning to a sense of normalcy.”
“Today’s action allows for a younger population to be protected from COVID-19, bringing us closer to returning to a sense of normalcy and to ending the pandemic,” she said in a statement. “Parents and guardians can rest assured that the agency undertook a rigorous and thorough review of all available data, as we have with all of our COVID-19 vaccine emergency use authorizations.”
The Pfizer adolescent vaccine is not yet a done deal, though.
Next, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices will decide on May 12 whether to recommend use of the vaccine in this age group. After that, CDC Director Rochelle Walensky, MD, will decide whether to give the green light for the vaccine to be administered to that age group.
The FDA action on May 10 amends the Dec. 11, 2020, emergency use authorization that allowed the Pfizer vaccine to be given to people 16 and older. Pfizer was the first company to receive an EUA for its adult vaccine and is the first to receive authorization for its adolescent vaccine. Pfizer is conducting clinical trials on much younger children, too.
The Moderna and Johnson & Johnson vaccines are authorized for people 18 and up. Moderna also has launched clinical trials in children.
Most health experts have said the United States needs to vaccinate children before the COVID-19 pandemic can truly be brought under control. The 12- to 15-year-old group represents 17 million people, about 5% of the population. Thus far, 58% of U.S. adults have had at least one dose of a vaccine and 34.8% of all Americans are fully vaccinated.
American Academy of Pediatrics President Lee Savio Beers, MD, praised the agency’s decision, calling it a “critically important step in bringing life-saving vaccines to children and adolescents. Our youngest generations have shouldered heavy burdens over the past year, and the vaccine is a hopeful sign that they will be able to begin to experience all the activities that are so important for their health and development.”
President Joe Biden recently announced a new strategy for expanding vaccinations in which vaccinating 12- to 15-year-olds was a key component. He said the administration was ready to ship the adolescent vaccine directly to pharmacies and pediatricians to speed up the vaccination rate.
In March, Anthony S. Fauci, MD, told a Senate committee, “We don’t really know what that magical point of herd immunity is, but we do know that if we get the overwhelming population vaccinated, we’re going to be in good shape. … We ultimately would like to get and have to get children into that mix.”
Pfizer submitted data to the FDA in late March showing its mRNA vaccine was 100% effective at preventing COVID-19 infection in children ages 12-15 in clinical trials.
Though most children have milder symptoms when infected with the coronavirus, about 1.5 million cases in children aged 11-17 were reported to the CDC between March 1, 2020, and April 30 of this year, the FDA news release said.
Albert Bourla, CEO of Pfizer, tweeted that “today brings very encouraging news for families and adolescents across the United States.
“While this is a meaningful step forward, we are still in a critical period of combating #COVID19 around the world. In the coming weeks, we hope to continue to receive authorizations from global regulators to support worldwide vaccination efforts,” he said.
“It’s essential for children to be vaccinated against COVID-19. According to data compiled by the AAP and Children’s Hospital Association, more than 3.8 million children have tested positive for COVID-19 in the United States since the start of the pandemic,” said Dr. Savio Beers. “While fewer children than adults have suffered the most severe disease, this is not a benign disease in children. Thousands of children have been hospitalized, and hundreds have died. We will soon have a very safe, highly effective vaccine that can prevent so much suffering. I encourage parents to talk with their pediatricians about how to get the vaccine for their adolescents as soon as they are eligible.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on May 10 granted emergency use authorization (EUA) for the Pfizer coronavirus vaccine to be given to children 12-15 years old.
The much-expected decision increases the likelihood that schools in the United States will fully reopen in the fall – a goal of both the Biden and Trump administrations.
Acting FDA Commissioner Janet Woodcock, MD, called the decision “a significant step” in “returning to a sense of normalcy.”
“Today’s action allows for a younger population to be protected from COVID-19, bringing us closer to returning to a sense of normalcy and to ending the pandemic,” she said in a statement. “Parents and guardians can rest assured that the agency undertook a rigorous and thorough review of all available data, as we have with all of our COVID-19 vaccine emergency use authorizations.”
The Pfizer adolescent vaccine is not yet a done deal, though.
Next, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices will decide on May 12 whether to recommend use of the vaccine in this age group. After that, CDC Director Rochelle Walensky, MD, will decide whether to give the green light for the vaccine to be administered to that age group.
The FDA action on May 10 amends the Dec. 11, 2020, emergency use authorization that allowed the Pfizer vaccine to be given to people 16 and older. Pfizer was the first company to receive an EUA for its adult vaccine and is the first to receive authorization for its adolescent vaccine. Pfizer is conducting clinical trials on much younger children, too.
The Moderna and Johnson & Johnson vaccines are authorized for people 18 and up. Moderna also has launched clinical trials in children.
Most health experts have said the United States needs to vaccinate children before the COVID-19 pandemic can truly be brought under control. The 12- to 15-year-old group represents 17 million people, about 5% of the population. Thus far, 58% of U.S. adults have had at least one dose of a vaccine and 34.8% of all Americans are fully vaccinated.
American Academy of Pediatrics President Lee Savio Beers, MD, praised the agency’s decision, calling it a “critically important step in bringing life-saving vaccines to children and adolescents. Our youngest generations have shouldered heavy burdens over the past year, and the vaccine is a hopeful sign that they will be able to begin to experience all the activities that are so important for their health and development.”
President Joe Biden recently announced a new strategy for expanding vaccinations in which vaccinating 12- to 15-year-olds was a key component. He said the administration was ready to ship the adolescent vaccine directly to pharmacies and pediatricians to speed up the vaccination rate.
In March, Anthony S. Fauci, MD, told a Senate committee, “We don’t really know what that magical point of herd immunity is, but we do know that if we get the overwhelming population vaccinated, we’re going to be in good shape. … We ultimately would like to get and have to get children into that mix.”
Pfizer submitted data to the FDA in late March showing its mRNA vaccine was 100% effective at preventing COVID-19 infection in children ages 12-15 in clinical trials.
Though most children have milder symptoms when infected with the coronavirus, about 1.5 million cases in children aged 11-17 were reported to the CDC between March 1, 2020, and April 30 of this year, the FDA news release said.
Albert Bourla, CEO of Pfizer, tweeted that “today brings very encouraging news for families and adolescents across the United States.
“While this is a meaningful step forward, we are still in a critical period of combating #COVID19 around the world. In the coming weeks, we hope to continue to receive authorizations from global regulators to support worldwide vaccination efforts,” he said.
“It’s essential for children to be vaccinated against COVID-19. According to data compiled by the AAP and Children’s Hospital Association, more than 3.8 million children have tested positive for COVID-19 in the United States since the start of the pandemic,” said Dr. Savio Beers. “While fewer children than adults have suffered the most severe disease, this is not a benign disease in children. Thousands of children have been hospitalized, and hundreds have died. We will soon have a very safe, highly effective vaccine that can prevent so much suffering. I encourage parents to talk with their pediatricians about how to get the vaccine for their adolescents as soon as they are eligible.”
A version of this article first appeared on Medscape.com.
The Food and Drug Administration on May 10 granted emergency use authorization (EUA) for the Pfizer coronavirus vaccine to be given to children 12-15 years old.
The much-expected decision increases the likelihood that schools in the United States will fully reopen in the fall – a goal of both the Biden and Trump administrations.
Acting FDA Commissioner Janet Woodcock, MD, called the decision “a significant step” in “returning to a sense of normalcy.”
“Today’s action allows for a younger population to be protected from COVID-19, bringing us closer to returning to a sense of normalcy and to ending the pandemic,” she said in a statement. “Parents and guardians can rest assured that the agency undertook a rigorous and thorough review of all available data, as we have with all of our COVID-19 vaccine emergency use authorizations.”
The Pfizer adolescent vaccine is not yet a done deal, though.
Next, the Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices will decide on May 12 whether to recommend use of the vaccine in this age group. After that, CDC Director Rochelle Walensky, MD, will decide whether to give the green light for the vaccine to be administered to that age group.
The FDA action on May 10 amends the Dec. 11, 2020, emergency use authorization that allowed the Pfizer vaccine to be given to people 16 and older. Pfizer was the first company to receive an EUA for its adult vaccine and is the first to receive authorization for its adolescent vaccine. Pfizer is conducting clinical trials on much younger children, too.
The Moderna and Johnson & Johnson vaccines are authorized for people 18 and up. Moderna also has launched clinical trials in children.
Most health experts have said the United States needs to vaccinate children before the COVID-19 pandemic can truly be brought under control. The 12- to 15-year-old group represents 17 million people, about 5% of the population. Thus far, 58% of U.S. adults have had at least one dose of a vaccine and 34.8% of all Americans are fully vaccinated.
American Academy of Pediatrics President Lee Savio Beers, MD, praised the agency’s decision, calling it a “critically important step in bringing life-saving vaccines to children and adolescents. Our youngest generations have shouldered heavy burdens over the past year, and the vaccine is a hopeful sign that they will be able to begin to experience all the activities that are so important for their health and development.”
President Joe Biden recently announced a new strategy for expanding vaccinations in which vaccinating 12- to 15-year-olds was a key component. He said the administration was ready to ship the adolescent vaccine directly to pharmacies and pediatricians to speed up the vaccination rate.
In March, Anthony S. Fauci, MD, told a Senate committee, “We don’t really know what that magical point of herd immunity is, but we do know that if we get the overwhelming population vaccinated, we’re going to be in good shape. … We ultimately would like to get and have to get children into that mix.”
Pfizer submitted data to the FDA in late March showing its mRNA vaccine was 100% effective at preventing COVID-19 infection in children ages 12-15 in clinical trials.
Though most children have milder symptoms when infected with the coronavirus, about 1.5 million cases in children aged 11-17 were reported to the CDC between March 1, 2020, and April 30 of this year, the FDA news release said.
Albert Bourla, CEO of Pfizer, tweeted that “today brings very encouraging news for families and adolescents across the United States.
“While this is a meaningful step forward, we are still in a critical period of combating #COVID19 around the world. In the coming weeks, we hope to continue to receive authorizations from global regulators to support worldwide vaccination efforts,” he said.
“It’s essential for children to be vaccinated against COVID-19. According to data compiled by the AAP and Children’s Hospital Association, more than 3.8 million children have tested positive for COVID-19 in the United States since the start of the pandemic,” said Dr. Savio Beers. “While fewer children than adults have suffered the most severe disease, this is not a benign disease in children. Thousands of children have been hospitalized, and hundreds have died. We will soon have a very safe, highly effective vaccine that can prevent so much suffering. I encourage parents to talk with their pediatricians about how to get the vaccine for their adolescents as soon as they are eligible.”
A version of this article first appeared on Medscape.com.
NSAIDs don’t make COVID-19 worse in hospitalized patients
NSAIDs don’t boost the risk of more severe disease or death in hospitalized patients with COVID-19, a new study finds.

“To our knowledge, our prospective study includes the largest number of patients admitted to hospital with COVID-19 to date, and adds to the literature on the safety of NSAIDs and in-hospital outcomes. NSAIDs do not appear to increase the risk of worse in-hospital outcomes ...” the study authors wrote. “NSAIDs are an important analgesic modality and have a vital opioid-sparing role in pain management. Patients and clinicians should be reassured by these findings that NSAIDs are safe in the context of the pandemic.”
The report was published online May 7 in The Lancet Rheumatology and led by clinical research fellow Thomas M. Drake, MBChB, of the University of Edinburgh’s Usher Institute.
For more than a year, researchers worldwide have debated about whether NSAIDs spell trouble for people at risk of COVID-19. In March 2020, French health officials announced that use of the painkillers such as NSAIDs may increase the severity of the disease, and they recommended that patients take acetaminophen instead. The National Health Service in the United Kingdom made a similar recommendation. But other agencies didn’t believe there was enough evidence to support ditching NSAIDs, and recent research studies published in Annals of the Rheumatic Diseases and PLoS Medicine suggested they may be right.
For the new study, researchers identified 72,179 patients who were treated for COVID-19 in British hospitals during January-August 2020. About 56% were men, 74% were White, and 6% took NSAIDs on a regular basis before they entered the hospital. The average age was 70.
The researchers examined whether the patients in either group were more or less likely to die in the hospital, be admitted into a critical care unit, need oxygen treatment, need a ventilator, or suffer kidney injury.
In terms of outcomes, there weren’t any major gaps between the groups overall. The differences in most comparisons were statistically insignificant. For example, 31% of those who didn’t take NSAIDs died vs. 30% of those who did (P = .227). In both groups, 14% required critical care admission (P = .476).
The researchers then focused on two matched groups of 4,205 patients: One group used NSAIDs regularly, and the other group didn’t. The difference in risk of death in those who took NSAIDs vs. those who didn’t was statistically insignificant (odds ratio, 0.95; 95% confidence interval, 0.84-1.07; P = .35). Other comparisons were also statistically insignificant.
The findings offer insight into whether the use of NSAIDs might actually be helpful for patients who develop COVID-19. Scientists believe that COVID-19 is linked to inflammation in the body, and NSAIDs, of course, reduce inflammation. But the researchers didn’t turn up any sign of a benefit.
The new study has some weaknesses: It doesn’t say anything about whether NSAIDs have an impact on whether people get COVID-19 in the first place. Researchers don’t know if high use of NSAIDs may affect the severity of the disease. And it doesn’t examine the potential effect of acetaminophen, although other research suggests the drug also may not cause harm in patients with COVID-19.
Still, the researchers say the study is the largest of its kind to look at the use of NSAIDs by patients who are admitted to the hospital with COVID-19. “Considering all the evidence, if there was an extreme effect of NSAIDs on COVID-19 outcomes or severity, this would have been observed in one or more of the studies that have been done, including the present study,” they wrote.
In a commentary that accompanied the study, three physicians from hospitals in Denmark, led by Kristian Kragholm, MD, of Aalborg University Hospital, praised the research and wrote that it adds to “a growing body of evidence” that NSAIDs don’t make things worse for patients with COVID-19.
The study was funded by the U.K. National Institute for Health Research and the U.K. Medical Research Council. The study and commentary authors reported no relevant disclosures.
NSAIDs don’t boost the risk of more severe disease or death in hospitalized patients with COVID-19, a new study finds.
“To our knowledge, our prospective study includes the largest number of patients admitted to hospital with COVID-19 to date, and adds to the literature on the safety of NSAIDs and in-hospital outcomes. NSAIDs do not appear to increase the risk of worse in-hospital outcomes ...” the study authors wrote. “NSAIDs are an important analgesic modality and have a vital opioid-sparing role in pain management. Patients and clinicians should be reassured by these findings that NSAIDs are safe in the context of the pandemic.”
The report was published online May 7 in The Lancet Rheumatology and led by clinical research fellow Thomas M. Drake, MBChB, of the University of Edinburgh’s Usher Institute.
For more than a year, researchers worldwide have debated about whether NSAIDs spell trouble for people at risk of COVID-19. In March 2020, French health officials announced that use of the painkillers such as NSAIDs may increase the severity of the disease, and they recommended that patients take acetaminophen instead. The National Health Service in the United Kingdom made a similar recommendation. But other agencies didn’t believe there was enough evidence to support ditching NSAIDs, and recent research studies published in Annals of the Rheumatic Diseases and PLoS Medicine suggested they may be right.
For the new study, researchers identified 72,179 patients who were treated for COVID-19 in British hospitals during January-August 2020. About 56% were men, 74% were White, and 6% took NSAIDs on a regular basis before they entered the hospital. The average age was 70.
The researchers examined whether the patients in either group were more or less likely to die in the hospital, be admitted into a critical care unit, need oxygen treatment, need a ventilator, or suffer kidney injury.
In terms of outcomes, there weren’t any major gaps between the groups overall. The differences in most comparisons were statistically insignificant. For example, 31% of those who didn’t take NSAIDs died vs. 30% of those who did (P = .227). In both groups, 14% required critical care admission (P = .476).
The researchers then focused on two matched groups of 4,205 patients: One group used NSAIDs regularly, and the other group didn’t. The difference in risk of death in those who took NSAIDs vs. those who didn’t was statistically insignificant (odds ratio, 0.95; 95% confidence interval, 0.84-1.07; P = .35). Other comparisons were also statistically insignificant.
The findings offer insight into whether the use of NSAIDs might actually be helpful for patients who develop COVID-19. Scientists believe that COVID-19 is linked to inflammation in the body, and NSAIDs, of course, reduce inflammation. But the researchers didn’t turn up any sign of a benefit.
The new study has some weaknesses: It doesn’t say anything about whether NSAIDs have an impact on whether people get COVID-19 in the first place. Researchers don’t know if high use of NSAIDs may affect the severity of the disease. And it doesn’t examine the potential effect of acetaminophen, although other research suggests the drug also may not cause harm in patients with COVID-19.
Still, the researchers say the study is the largest of its kind to look at the use of NSAIDs by patients who are admitted to the hospital with COVID-19. “Considering all the evidence, if there was an extreme effect of NSAIDs on COVID-19 outcomes or severity, this would have been observed in one or more of the studies that have been done, including the present study,” they wrote.
In a commentary that accompanied the study, three physicians from hospitals in Denmark, led by Kristian Kragholm, MD, of Aalborg University Hospital, praised the research and wrote that it adds to “a growing body of evidence” that NSAIDs don’t make things worse for patients with COVID-19.
The study was funded by the U.K. National Institute for Health Research and the U.K. Medical Research Council. The study and commentary authors reported no relevant disclosures.
NSAIDs don’t boost the risk of more severe disease or death in hospitalized patients with COVID-19, a new study finds.
“To our knowledge, our prospective study includes the largest number of patients admitted to hospital with COVID-19 to date, and adds to the literature on the safety of NSAIDs and in-hospital outcomes. NSAIDs do not appear to increase the risk of worse in-hospital outcomes ...” the study authors wrote. “NSAIDs are an important analgesic modality and have a vital opioid-sparing role in pain management. Patients and clinicians should be reassured by these findings that NSAIDs are safe in the context of the pandemic.”
The report was published online May 7 in The Lancet Rheumatology and led by clinical research fellow Thomas M. Drake, MBChB, of the University of Edinburgh’s Usher Institute.
For more than a year, researchers worldwide have debated about whether NSAIDs spell trouble for people at risk of COVID-19. In March 2020, French health officials announced that use of the painkillers such as NSAIDs may increase the severity of the disease, and they recommended that patients take acetaminophen instead. The National Health Service in the United Kingdom made a similar recommendation. But other agencies didn’t believe there was enough evidence to support ditching NSAIDs, and recent research studies published in Annals of the Rheumatic Diseases and PLoS Medicine suggested they may be right.
For the new study, researchers identified 72,179 patients who were treated for COVID-19 in British hospitals during January-August 2020. About 56% were men, 74% were White, and 6% took NSAIDs on a regular basis before they entered the hospital. The average age was 70.
The researchers examined whether the patients in either group were more or less likely to die in the hospital, be admitted into a critical care unit, need oxygen treatment, need a ventilator, or suffer kidney injury.
In terms of outcomes, there weren’t any major gaps between the groups overall. The differences in most comparisons were statistically insignificant. For example, 31% of those who didn’t take NSAIDs died vs. 30% of those who did (P = .227). In both groups, 14% required critical care admission (P = .476).
The researchers then focused on two matched groups of 4,205 patients: One group used NSAIDs regularly, and the other group didn’t. The difference in risk of death in those who took NSAIDs vs. those who didn’t was statistically insignificant (odds ratio, 0.95; 95% confidence interval, 0.84-1.07; P = .35). Other comparisons were also statistically insignificant.
The findings offer insight into whether the use of NSAIDs might actually be helpful for patients who develop COVID-19. Scientists believe that COVID-19 is linked to inflammation in the body, and NSAIDs, of course, reduce inflammation. But the researchers didn’t turn up any sign of a benefit.
The new study has some weaknesses: It doesn’t say anything about whether NSAIDs have an impact on whether people get COVID-19 in the first place. Researchers don’t know if high use of NSAIDs may affect the severity of the disease. And it doesn’t examine the potential effect of acetaminophen, although other research suggests the drug also may not cause harm in patients with COVID-19.
Still, the researchers say the study is the largest of its kind to look at the use of NSAIDs by patients who are admitted to the hospital with COVID-19. “Considering all the evidence, if there was an extreme effect of NSAIDs on COVID-19 outcomes or severity, this would have been observed in one or more of the studies that have been done, including the present study,” they wrote.
In a commentary that accompanied the study, three physicians from hospitals in Denmark, led by Kristian Kragholm, MD, of Aalborg University Hospital, praised the research and wrote that it adds to “a growing body of evidence” that NSAIDs don’t make things worse for patients with COVID-19.
The study was funded by the U.K. National Institute for Health Research and the U.K. Medical Research Council. The study and commentary authors reported no relevant disclosures.
FROM THE LANCET RHEUMATOLOGY
A ‘mess’ of a diagnosis: Is it type 2 MI or a nonischemic imposter?
Survival gains in the management of acute myocardial infarction in recent decades don’t apply to one increasingly common category of MI.
Type 2 MI, triggered by a surge in myocardial oxygen demand or a drop in its supply, is on the rise and might be more prognostically serious than the “classic” atherothrombotic type 1 form, for which there have been such impressive strides in therapy.
Strategies for assessing and treating type 2 MI and another condition it can resemble clinically – nonischemic myocardial injury – have been less rigorously explored and are far less settled.
That could be partly because recent iterations of the consensus-based universal definition of MI define type 1 MI primarily by the atherothrombotic process, whereas “demand” type 2 MI is characterized as secondary to other disorders. The list of potential primary conditions, cardiac and noncardiac, is long.
As a result, patients with type 1 MI are clinically well defined, but those with type 2 MI have so far defied efforts to be clinically characterized in a consistent way. However, recent efforts might change that, given growing appreciation that all-cause and cardiovascular (CV) mortality outcomes are actually worse for patients with type 2 MI.
“That’s because we have lots of treatments for type 1 MI. Type 2 and myocardial injury? We don’t know how to treat them,” David E. Newby, MD, PhD, University of Edinburgh, said in an interview.
Dr. Newby pointed to a widely cited 2018 publication, of which he is a coauthor, documenting 5-year outcomes of 2,122 patients with type 1 MI, type 2 MI, or nonischemic myocardial injury per the newly minted fourth universal definition.
Risk-factor profiles for patients with the latter two conditions contrasted with those of patients with type 1 MI, he observed. They were “a lot older,” were less likely to be smokers, had more hypertension and previous stroke, and a less prominent CV family history.
“So they’re a different beast,” Dr. Newby said. And their prognosis tended to be worse: all-cause mortality was about 62% for patients with type 2 MI and 72% with nonischemic myocardial injury, but only 37% for patients with type 1 MI. The difference between the two types of infarction was driven by an excess of noncardiovascular death after type 2 MI.
Mortality in patients with type 2 MI is “quite high, but it may well be a marker of the fact that you’ve got other serious diseases on board that are associated with poorer outcome,” he said.
Risk varies
The degree of risk in type 2 MI seems to vary with the underlying condition, a recent cohort study suggests. In about 3,800 patients with cardiac troponin (cTn) elevations qualifying as MI – a younger group; most were in their 30s and 40s – mortality at 10 years was 12% for those with type 1 MI, but 34% for those with type 2 MI and 46% for the remainder with nonischemic myocardial injury.
Underlying precipitating conditions varied widely among the patients with type 2 MI or nonischemic myocardial injury, and there was broad variation in mortality by etiology among those with type 2 MI. Sepsis and anemia entailed some of the highest risk, and hypertension and arrhythmias some of the lowest.
A prospective, community-based study of 5,460 patients with type 1 MI or type 2 MI reached a similar conclusion, but with a twist. Five-year all-cause mortality contrasted significantly between types of MI at 31% and 52%, respectively, but CV mortality rates were similar in this study.
Mortality in type 2 MI again varied by the precipitating etiology, suggesting that patients can be risk stratified according to pathophysiological mechanism behind their demand infarction, the authors concluded, “underscoring that type 2 MI is not a single entity, rather a group of phenotypic clusters.”
The usually high comorbidity burden and CV risk in patients with type 2 MI, one of those authors said in an interview, suggest there are “opportunities to see whether we can reduce that risk.”
Formal recommendations consistently say that, in patients with type 2 MI, “your first and foremost target should be to treat the underlying trigger and cause,” said Yader Sandoval, MD, Mayo Clinic, Rochester, Minn. That means such opportunities for further CV risk reduction tend to be “underappreciated.”

“In principle, treating the inciting cause of type 2 MI or the injury is important,” said James L. Januzzi, MD, Massachusetts General Hospital, Boston, in an interview, “but I feel quite strongly that there must be more that we can do for these folks.”
Dr. Januzzi is senior author on a recent analysis based on more than 200,000 admissions across the United States that saw a 43% lower risk for in-hospital death and 54% lower risk for 30-day MI readmission for patients with type 2 MI than those with type 1, adjusted for risk factors and comorbidities.
But, “it is important to emphasize that type 2 MI patients had a substantial risk for adverse outcome, nonetheless, and lack a clear management approach,” Dr. Januzzi and colleagues stated in their publication, as reported by this news organization.
“Due to the high rates of long-term cardiovascular events experienced by the frequently encountered type 2 MI patients,” they wrote, “identifying evidence-based therapies represents a major unmet need.”
That such patients tend to be sick with multiple comorbidities and have not yet been clinically well characterized, Dr. Januzzi said, “has stymied our ability to develop a treatment strategy.”
Role of the universal definitions
That challenge might in some ways be complicated by the universal definition, especially version 4, in which the definitions for type 1 MI, type 2 MI, and nonischemic myocardial injury are unified biochemically.
This version, published in 2018 in the European Heart Journal and Circulation, introduced a formal definition of myocardial injury, which was hailed as an innovation: cTn elevation to the 99th percentile of the upper limit of normal in a reference population.
It differentiates type 1 MI from type 2 MI by the separate pathophysiology of the ischemia – plaque rupture with intracoronary thrombosis and myocardial oxygen supply–demand mismatch, respectively. In both cases, however, there must be symptoms or objective evidence of ischemia. Absent signs of ischemia, the determination would be nonischemic myocardial injury.
Yet clinically and prognostically, type 2 MI and nonischemic myocardial injury in some ways are more similar to each other than either is to type 1 MI. Both occur secondary to other conditions across diverse clinical settings and can be a challenge to tell apart.
The universal definition’s perspective of the three events – so heavily dependent on cTn levels and myocardial ischemia – fails to account for the myriad complexities of individual patients in practice, some say, and so can muddle the process of risk assessment and therapy.
“Abnormal troponin identifies injury, but it doesn’t identify mechanism. Type 2 MI is highly prevalent, but there are other things that cause abnormal troponins,” Dr. Januzzi said. That’s why it’s important to explore and map out the clinical variables associated with the two conditions, to “understand who has a type 2 MI and who has cardiac injury. And believe it or not, it’s actually harder than it sounds to sort that out.”
“Practically speaking, the differentiation between these events is clinical,” Dr. Sandoval agreed. “There’s not always perfect agreement on what we’re calling what.”
Consequently, the universal definitions might categorize some events in ways that seem inconsistent from a management perspective. For example, they make a sharp distinction between coronary atherothrombotic and coronary nonatherothrombotic MI etiologies. Some clinicians would group MI caused by coronary spasm, coronary embolism, or spontaneous coronary artery dissection along with MI from coronary plaque rupture and thrombosis. But, Dr. Sandoval said, “even though these are coronary issues, they would fall into the type 2 bin.”
Also, about half of cases identified as type 2 MI are caused by tachyarrhythmias, which can elevate troponin and cause ECG changes and possibly symptoms resembling angina, Dr. Newby observed. “But that is completely different from other types of myocardial infarction, which are much more serious.”
So, “it’s a real mess of a diagnosis – acute myocardial injury, type 2 and type 1 MI – and it can be quite difficult to disentangle,” he said. “I think that the definition certainly has let us down.”
The diversity of type 2 MI clinical settings might also be a challenge. Myocardial injury according to cTn, with or without ischemia, occurs widely during critical illnesses and acute conditions, including respiratory distress, sepsis, internal bleeding, stroke, and pulmonary embolism.
Early in the COVID-19 pandemic, much was made of elevated troponin levels and myocarditis as an apparently frequent complication among hospitalized patients. “I raised my hand and said, we’ve been seeing abnormal troponins in people with influenza for 20 years,” Dr. Januzzi said. “Critical illness, infection, toxicity from drugs, from chemotherapy, from alcohol – there are all sorts of potential triggers of myocardial injury.”
Troponin ‘overdependence’
With many clinical settings in common and the presence or absence of myocardial ischemia to primarily distinguish them, type 2 MI and nonischemic myocardial injury both can be mistaken for the other. That can send management decisions in inappropriate directions.
A 2019 study looked at 633 cases that had been coded as type 2 MI at a major center and readjudicated them according to the fourth universal definition. Only 57% met all the type 2 criteria, 42% were reclassified as nonischemic myocardial injury, and a few were determined to have unstable angina.
“There’s overdependence on the easiest tool in the universal definition,” said Dr. Januzzi, a coauthor on that study. “Frequently people get seduced by the rise in a troponin value and immediately call it a myocardial infarction, lacking the other components of the universal definition that require evidence for coronary ischemia. That happens every day, where someone with an abnormal troponin is incorrectly branded as having an MI.”
It may not help that the current ICD-10-CM system features a diagnostic code for type 2 MI but not for myocardial injury.
“Instead, the new ICD-10-CM coding includes a proxy called ‘non-MI troponin elevation due to an underlying cause,’ ” wrote Kristian Thygesen, MD, DSc, and Allan S. Jaffe, MD, in a recent editorial. They caution against “using this code for myocardial injury because it is not specific for an elevated cTn value and could represent any abnormal laboratory measurements.” The code could be “misleading,” thereby worsening the potential for miscoding and “misattribution of MI diagnoses.”
That potential suggests there could be a growing population of patients who have been told they had an MI, which then becomes part of their medical record, when, actually, they experienced nonischemic myocardial injury.
“Having seen this occur,” Dr. Januzzi explained, “it affects people emotionally to think they’ve had an MI. Precision in diagnosis is important, which is why the universal definition is so valuable. If people would adhere to it more assiduously, we could reduce the frequency of people getting a misdiagnosis of MI when in fact they had injury.”
Still, he added, “if someone has an illness severe enough to cause myocardial injury, they’re at risk for a bad outcome regardless of whether they did or didn’t have an MI.”
The uncertain role of angiography
Angiography isn’t ordered nearly as often for patients ultimately diagnosed with type 2 MI or myocardial injury as for those with type 1 MI. Type 2 MI can hit some patients who have remained symptom free despite possibly unrecognized obstructive coronary artery disease (CAD) when myocardial demand is pushed past supply by a critical illness, tachyarrhythmia, or other acute conditions.
In such cases, “it’s reasonable to hypothesize that revascularization, something that really is not done in the vast majority of patients with type 2 MI, might actually be of benefit,” Dr. Januzzi said.
Whether these patients should routinely have angiography remains an open question. Without intervention, any newly identified obstructive CAD would continue to lurk in the background as a potential threat.
In efforts to differentiate type 2 MI from nonischemic injury, it can be “incredibly hard to know whether or not there’s actual ischemia in the mix. And that’s the only thing that defines the difference before taking an angiogram,” Derek P. Chew, MBBS, MPH, Flinders Medical Centre, Bedford Park, Australia, said in an interview.
Dr. Chew is principal investigator for the ongoing ACT-2 trial that is enrolling hospitalized, hemodynamically stable patients with cTn elevations but no suspicion of type 1 MI and “an unequivocal acute intercurrent diagnosis.” Qualifying diagnoses are prespecified on a list that includes sepsis, pneumonia, septicemia, a systemic inflammatory response, anemia, atrial tachycardia, acute kidney injury, and recent noncardiac surgery.
The patients are randomly assigned to a strategy of routine, usually invasive coronary angiography with discretionary revascularization, or to conservative care with noninvasive functional testing as appropriate. The sicker the patient, the greater the competing risk from other conditions and the less revascularization is likely to improve outcomes, Dr. Chew observed. Importantly, therefore, outcomes in the trial will be stratified by patient risk from comorbidities, measured with baseline GRACE and APACHE III scores.
Dr. Chew said the study aims to determine whether routine angiography is of benefit in patients at some identifiable level of risk, if not the whole range. One possible result, he said, is that there could be a risk-profile “sweet spot” associated with better outcomes in those assigned to angiography.
Enrollment in the trial started about 3 years ago, but “the process has been slow,” he said, because many potentially referring clinicians have a “bias on one side or another,” with about half of them preferring the angiography approach and the other half conservative management.
The unsettled role of drug therapy
With their often-complicated clinical profile, patients with type 2 MI or nonischemic myocardial injury tend to be medically undertreated, yet there is observational evidence they can benefit from familiar drug therapies.
In the previously noted cohort study of 3,800 younger patients with one of the three forms of myocardial injury, less than half of patients with type 2 MI received any form of CAD secondary prevention therapy at discharge, the researchers, with first author Avinainder Singh, MD, from Yale University, New Haven, Conn, wrote.
The finding, consistent with Dr. Newby’s study from 2018, suggests that “categorizing the type of MI in young subjects might inform long-term cardiovascular prognosis,” and “emphasizes the need to identify and implement secondary prevention strategies to mitigate the high rate of cardiovascular death in patients with type 2 MI,” they concluded.
Further, outcomes varied with the number of discharge CV meds in an older cohort of patients with myocardial injury. Those with type 2 MI or acute or chronic nonischemic myocardial injury were far less likely than patients with type 1 MI to be prescribed guideline-based drugs. Survival was greater for those on two or three classes of CV medications, compared with one or none, in patients with acute or chronic nonischemic injury.
The investigators urged that patients with nonischemic myocardial injury or type 2 MI “be treated with cardiovascular medication to a larger degree than what is done today.”
When there is documented CAD in patients with type 2 MI, “it would be reasonable to suggest that preventative secondary prevention approaches, such as such lipid-reduction therapy or aspirin, would be beneficial,” Dr. Sandoval said. “But the reality is, there are no randomized trials, there are no prospective studies. ACT-2 is one of the few and early studies that’s really trying to address this.”
“The great majority of these people are not going to the cath lab, but when they do, there seems to be a signal of potential benefit,” Dr. Januzzi said. “For someone with a type 2 MI, it’s quite possible revascularization might help. Then more long-term treatment with medications that are proven in randomized trials to reduce risk would be a very plausible intervention.”
“We’ve actually proposed a number of potential therapeutic interventions to explore, both in people with type 2 MI and in people with injury without MI,” he said. “They might include sodium glucose cotransporter 2 inhibitors. They might include antithrombotic therapy or more aggressive lipid lowering, possibly for the pleiotropic effects rather than the effects on atherosclerosis.”
Any such therapies that prove successful in well-designed trials could well earn both type 2 MI and nonischemic myocardial injury, neglected as disorders in their own right, the kind of respect in clinical care pathways that they likely deserve.
Dr. Newby has disclosed receiving consulting fees or honoraria from Eli Lilly, Roche, Toshiba, Jansen, Reckitt Benckiser Pharmaceuticals, Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, CellProthera, and Oncoarendi; and conducting research or receiving grants from Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Merck, Boehringer Ingelheim, and Inositec. Sandoval reports serving on an advisory board and as a speaker for Abbott Diagnostics and on an advisory board for Roche Diagnostics. Dr. Januzzi has disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on endpoint committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda. Dr. Chew has reported receiving grants from AstraZeneca and Edwards Life Sciences. ACT-2 is sponsored by the National Medical and Health Research Council of Australia.
A version of this article first appeared on Medscape.com.
Survival gains in the management of acute myocardial infarction in recent decades don’t apply to one increasingly common category of MI.
Type 2 MI, triggered by a surge in myocardial oxygen demand or a drop in its supply, is on the rise and might be more prognostically serious than the “classic” atherothrombotic type 1 form, for which there have been such impressive strides in therapy.
Strategies for assessing and treating type 2 MI and another condition it can resemble clinically – nonischemic myocardial injury – have been less rigorously explored and are far less settled.
That could be partly because recent iterations of the consensus-based universal definition of MI define type 1 MI primarily by the atherothrombotic process, whereas “demand” type 2 MI is characterized as secondary to other disorders. The list of potential primary conditions, cardiac and noncardiac, is long.
As a result, patients with type 1 MI are clinically well defined, but those with type 2 MI have so far defied efforts to be clinically characterized in a consistent way. However, recent efforts might change that, given growing appreciation that all-cause and cardiovascular (CV) mortality outcomes are actually worse for patients with type 2 MI.
“That’s because we have lots of treatments for type 1 MI. Type 2 and myocardial injury? We don’t know how to treat them,” David E. Newby, MD, PhD, University of Edinburgh, said in an interview.
Dr. Newby pointed to a widely cited 2018 publication, of which he is a coauthor, documenting 5-year outcomes of 2,122 patients with type 1 MI, type 2 MI, or nonischemic myocardial injury per the newly minted fourth universal definition.
Risk-factor profiles for patients with the latter two conditions contrasted with those of patients with type 1 MI, he observed. They were “a lot older,” were less likely to be smokers, had more hypertension and previous stroke, and a less prominent CV family history.
“So they’re a different beast,” Dr. Newby said. And their prognosis tended to be worse: all-cause mortality was about 62% for patients with type 2 MI and 72% with nonischemic myocardial injury, but only 37% for patients with type 1 MI. The difference between the two types of infarction was driven by an excess of noncardiovascular death after type 2 MI.
Mortality in patients with type 2 MI is “quite high, but it may well be a marker of the fact that you’ve got other serious diseases on board that are associated with poorer outcome,” he said.
Risk varies
The degree of risk in type 2 MI seems to vary with the underlying condition, a recent cohort study suggests. In about 3,800 patients with cardiac troponin (cTn) elevations qualifying as MI – a younger group; most were in their 30s and 40s – mortality at 10 years was 12% for those with type 1 MI, but 34% for those with type 2 MI and 46% for the remainder with nonischemic myocardial injury.
Underlying precipitating conditions varied widely among the patients with type 2 MI or nonischemic myocardial injury, and there was broad variation in mortality by etiology among those with type 2 MI. Sepsis and anemia entailed some of the highest risk, and hypertension and arrhythmias some of the lowest.
A prospective, community-based study of 5,460 patients with type 1 MI or type 2 MI reached a similar conclusion, but with a twist. Five-year all-cause mortality contrasted significantly between types of MI at 31% and 52%, respectively, but CV mortality rates were similar in this study.
Mortality in type 2 MI again varied by the precipitating etiology, suggesting that patients can be risk stratified according to pathophysiological mechanism behind their demand infarction, the authors concluded, “underscoring that type 2 MI is not a single entity, rather a group of phenotypic clusters.”
The usually high comorbidity burden and CV risk in patients with type 2 MI, one of those authors said in an interview, suggest there are “opportunities to see whether we can reduce that risk.”
Formal recommendations consistently say that, in patients with type 2 MI, “your first and foremost target should be to treat the underlying trigger and cause,” said Yader Sandoval, MD, Mayo Clinic, Rochester, Minn. That means such opportunities for further CV risk reduction tend to be “underappreciated.”
“In principle, treating the inciting cause of type 2 MI or the injury is important,” said James L. Januzzi, MD, Massachusetts General Hospital, Boston, in an interview, “but I feel quite strongly that there must be more that we can do for these folks.”
Dr. Januzzi is senior author on a recent analysis based on more than 200,000 admissions across the United States that saw a 43% lower risk for in-hospital death and 54% lower risk for 30-day MI readmission for patients with type 2 MI than those with type 1, adjusted for risk factors and comorbidities.
But, “it is important to emphasize that type 2 MI patients had a substantial risk for adverse outcome, nonetheless, and lack a clear management approach,” Dr. Januzzi and colleagues stated in their publication, as reported by this news organization.
“Due to the high rates of long-term cardiovascular events experienced by the frequently encountered type 2 MI patients,” they wrote, “identifying evidence-based therapies represents a major unmet need.”
That such patients tend to be sick with multiple comorbidities and have not yet been clinically well characterized, Dr. Januzzi said, “has stymied our ability to develop a treatment strategy.”
Role of the universal definitions
That challenge might in some ways be complicated by the universal definition, especially version 4, in which the definitions for type 1 MI, type 2 MI, and nonischemic myocardial injury are unified biochemically.
This version, published in 2018 in the European Heart Journal and Circulation, introduced a formal definition of myocardial injury, which was hailed as an innovation: cTn elevation to the 99th percentile of the upper limit of normal in a reference population.
It differentiates type 1 MI from type 2 MI by the separate pathophysiology of the ischemia – plaque rupture with intracoronary thrombosis and myocardial oxygen supply–demand mismatch, respectively. In both cases, however, there must be symptoms or objective evidence of ischemia. Absent signs of ischemia, the determination would be nonischemic myocardial injury.
Yet clinically and prognostically, type 2 MI and nonischemic myocardial injury in some ways are more similar to each other than either is to type 1 MI. Both occur secondary to other conditions across diverse clinical settings and can be a challenge to tell apart.
The universal definition’s perspective of the three events – so heavily dependent on cTn levels and myocardial ischemia – fails to account for the myriad complexities of individual patients in practice, some say, and so can muddle the process of risk assessment and therapy.
“Abnormal troponin identifies injury, but it doesn’t identify mechanism. Type 2 MI is highly prevalent, but there are other things that cause abnormal troponins,” Dr. Januzzi said. That’s why it’s important to explore and map out the clinical variables associated with the two conditions, to “understand who has a type 2 MI and who has cardiac injury. And believe it or not, it’s actually harder than it sounds to sort that out.”
“Practically speaking, the differentiation between these events is clinical,” Dr. Sandoval agreed. “There’s not always perfect agreement on what we’re calling what.”
Consequently, the universal definitions might categorize some events in ways that seem inconsistent from a management perspective. For example, they make a sharp distinction between coronary atherothrombotic and coronary nonatherothrombotic MI etiologies. Some clinicians would group MI caused by coronary spasm, coronary embolism, or spontaneous coronary artery dissection along with MI from coronary plaque rupture and thrombosis. But, Dr. Sandoval said, “even though these are coronary issues, they would fall into the type 2 bin.”
Also, about half of cases identified as type 2 MI are caused by tachyarrhythmias, which can elevate troponin and cause ECG changes and possibly symptoms resembling angina, Dr. Newby observed. “But that is completely different from other types of myocardial infarction, which are much more serious.”
So, “it’s a real mess of a diagnosis – acute myocardial injury, type 2 and type 1 MI – and it can be quite difficult to disentangle,” he said. “I think that the definition certainly has let us down.”
The diversity of type 2 MI clinical settings might also be a challenge. Myocardial injury according to cTn, with or without ischemia, occurs widely during critical illnesses and acute conditions, including respiratory distress, sepsis, internal bleeding, stroke, and pulmonary embolism.
Early in the COVID-19 pandemic, much was made of elevated troponin levels and myocarditis as an apparently frequent complication among hospitalized patients. “I raised my hand and said, we’ve been seeing abnormal troponins in people with influenza for 20 years,” Dr. Januzzi said. “Critical illness, infection, toxicity from drugs, from chemotherapy, from alcohol – there are all sorts of potential triggers of myocardial injury.”
Troponin ‘overdependence’
With many clinical settings in common and the presence or absence of myocardial ischemia to primarily distinguish them, type 2 MI and nonischemic myocardial injury both can be mistaken for the other. That can send management decisions in inappropriate directions.
A 2019 study looked at 633 cases that had been coded as type 2 MI at a major center and readjudicated them according to the fourth universal definition. Only 57% met all the type 2 criteria, 42% were reclassified as nonischemic myocardial injury, and a few were determined to have unstable angina.
“There’s overdependence on the easiest tool in the universal definition,” said Dr. Januzzi, a coauthor on that study. “Frequently people get seduced by the rise in a troponin value and immediately call it a myocardial infarction, lacking the other components of the universal definition that require evidence for coronary ischemia. That happens every day, where someone with an abnormal troponin is incorrectly branded as having an MI.”
It may not help that the current ICD-10-CM system features a diagnostic code for type 2 MI but not for myocardial injury.
“Instead, the new ICD-10-CM coding includes a proxy called ‘non-MI troponin elevation due to an underlying cause,’ ” wrote Kristian Thygesen, MD, DSc, and Allan S. Jaffe, MD, in a recent editorial. They caution against “using this code for myocardial injury because it is not specific for an elevated cTn value and could represent any abnormal laboratory measurements.” The code could be “misleading,” thereby worsening the potential for miscoding and “misattribution of MI diagnoses.”
That potential suggests there could be a growing population of patients who have been told they had an MI, which then becomes part of their medical record, when, actually, they experienced nonischemic myocardial injury.
“Having seen this occur,” Dr. Januzzi explained, “it affects people emotionally to think they’ve had an MI. Precision in diagnosis is important, which is why the universal definition is so valuable. If people would adhere to it more assiduously, we could reduce the frequency of people getting a misdiagnosis of MI when in fact they had injury.”
Still, he added, “if someone has an illness severe enough to cause myocardial injury, they’re at risk for a bad outcome regardless of whether they did or didn’t have an MI.”
The uncertain role of angiography
Angiography isn’t ordered nearly as often for patients ultimately diagnosed with type 2 MI or myocardial injury as for those with type 1 MI. Type 2 MI can hit some patients who have remained symptom free despite possibly unrecognized obstructive coronary artery disease (CAD) when myocardial demand is pushed past supply by a critical illness, tachyarrhythmia, or other acute conditions.
In such cases, “it’s reasonable to hypothesize that revascularization, something that really is not done in the vast majority of patients with type 2 MI, might actually be of benefit,” Dr. Januzzi said.
Whether these patients should routinely have angiography remains an open question. Without intervention, any newly identified obstructive CAD would continue to lurk in the background as a potential threat.
In efforts to differentiate type 2 MI from nonischemic injury, it can be “incredibly hard to know whether or not there’s actual ischemia in the mix. And that’s the only thing that defines the difference before taking an angiogram,” Derek P. Chew, MBBS, MPH, Flinders Medical Centre, Bedford Park, Australia, said in an interview.
Dr. Chew is principal investigator for the ongoing ACT-2 trial that is enrolling hospitalized, hemodynamically stable patients with cTn elevations but no suspicion of type 1 MI and “an unequivocal acute intercurrent diagnosis.” Qualifying diagnoses are prespecified on a list that includes sepsis, pneumonia, septicemia, a systemic inflammatory response, anemia, atrial tachycardia, acute kidney injury, and recent noncardiac surgery.
The patients are randomly assigned to a strategy of routine, usually invasive coronary angiography with discretionary revascularization, or to conservative care with noninvasive functional testing as appropriate. The sicker the patient, the greater the competing risk from other conditions and the less revascularization is likely to improve outcomes, Dr. Chew observed. Importantly, therefore, outcomes in the trial will be stratified by patient risk from comorbidities, measured with baseline GRACE and APACHE III scores.
Dr. Chew said the study aims to determine whether routine angiography is of benefit in patients at some identifiable level of risk, if not the whole range. One possible result, he said, is that there could be a risk-profile “sweet spot” associated with better outcomes in those assigned to angiography.
Enrollment in the trial started about 3 years ago, but “the process has been slow,” he said, because many potentially referring clinicians have a “bias on one side or another,” with about half of them preferring the angiography approach and the other half conservative management.
The unsettled role of drug therapy
With their often-complicated clinical profile, patients with type 2 MI or nonischemic myocardial injury tend to be medically undertreated, yet there is observational evidence they can benefit from familiar drug therapies.
In the previously noted cohort study of 3,800 younger patients with one of the three forms of myocardial injury, less than half of patients with type 2 MI received any form of CAD secondary prevention therapy at discharge, the researchers, with first author Avinainder Singh, MD, from Yale University, New Haven, Conn, wrote.
The finding, consistent with Dr. Newby’s study from 2018, suggests that “categorizing the type of MI in young subjects might inform long-term cardiovascular prognosis,” and “emphasizes the need to identify and implement secondary prevention strategies to mitigate the high rate of cardiovascular death in patients with type 2 MI,” they concluded.
Further, outcomes varied with the number of discharge CV meds in an older cohort of patients with myocardial injury. Those with type 2 MI or acute or chronic nonischemic myocardial injury were far less likely than patients with type 1 MI to be prescribed guideline-based drugs. Survival was greater for those on two or three classes of CV medications, compared with one or none, in patients with acute or chronic nonischemic injury.
The investigators urged that patients with nonischemic myocardial injury or type 2 MI “be treated with cardiovascular medication to a larger degree than what is done today.”
When there is documented CAD in patients with type 2 MI, “it would be reasonable to suggest that preventative secondary prevention approaches, such as such lipid-reduction therapy or aspirin, would be beneficial,” Dr. Sandoval said. “But the reality is, there are no randomized trials, there are no prospective studies. ACT-2 is one of the few and early studies that’s really trying to address this.”
“The great majority of these people are not going to the cath lab, but when they do, there seems to be a signal of potential benefit,” Dr. Januzzi said. “For someone with a type 2 MI, it’s quite possible revascularization might help. Then more long-term treatment with medications that are proven in randomized trials to reduce risk would be a very plausible intervention.”
“We’ve actually proposed a number of potential therapeutic interventions to explore, both in people with type 2 MI and in people with injury without MI,” he said. “They might include sodium glucose cotransporter 2 inhibitors. They might include antithrombotic therapy or more aggressive lipid lowering, possibly for the pleiotropic effects rather than the effects on atherosclerosis.”
Any such therapies that prove successful in well-designed trials could well earn both type 2 MI and nonischemic myocardial injury, neglected as disorders in their own right, the kind of respect in clinical care pathways that they likely deserve.
Dr. Newby has disclosed receiving consulting fees or honoraria from Eli Lilly, Roche, Toshiba, Jansen, Reckitt Benckiser Pharmaceuticals, Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, CellProthera, and Oncoarendi; and conducting research or receiving grants from Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Merck, Boehringer Ingelheim, and Inositec. Sandoval reports serving on an advisory board and as a speaker for Abbott Diagnostics and on an advisory board for Roche Diagnostics. Dr. Januzzi has disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on endpoint committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda. Dr. Chew has reported receiving grants from AstraZeneca and Edwards Life Sciences. ACT-2 is sponsored by the National Medical and Health Research Council of Australia.
A version of this article first appeared on Medscape.com.
Survival gains in the management of acute myocardial infarction in recent decades don’t apply to one increasingly common category of MI.
Type 2 MI, triggered by a surge in myocardial oxygen demand or a drop in its supply, is on the rise and might be more prognostically serious than the “classic” atherothrombotic type 1 form, for which there have been such impressive strides in therapy.
Strategies for assessing and treating type 2 MI and another condition it can resemble clinically – nonischemic myocardial injury – have been less rigorously explored and are far less settled.
That could be partly because recent iterations of the consensus-based universal definition of MI define type 1 MI primarily by the atherothrombotic process, whereas “demand” type 2 MI is characterized as secondary to other disorders. The list of potential primary conditions, cardiac and noncardiac, is long.
As a result, patients with type 1 MI are clinically well defined, but those with type 2 MI have so far defied efforts to be clinically characterized in a consistent way. However, recent efforts might change that, given growing appreciation that all-cause and cardiovascular (CV) mortality outcomes are actually worse for patients with type 2 MI.
“That’s because we have lots of treatments for type 1 MI. Type 2 and myocardial injury? We don’t know how to treat them,” David E. Newby, MD, PhD, University of Edinburgh, said in an interview.
Dr. Newby pointed to a widely cited 2018 publication, of which he is a coauthor, documenting 5-year outcomes of 2,122 patients with type 1 MI, type 2 MI, or nonischemic myocardial injury per the newly minted fourth universal definition.
Risk-factor profiles for patients with the latter two conditions contrasted with those of patients with type 1 MI, he observed. They were “a lot older,” were less likely to be smokers, had more hypertension and previous stroke, and a less prominent CV family history.
“So they’re a different beast,” Dr. Newby said. And their prognosis tended to be worse: all-cause mortality was about 62% for patients with type 2 MI and 72% with nonischemic myocardial injury, but only 37% for patients with type 1 MI. The difference between the two types of infarction was driven by an excess of noncardiovascular death after type 2 MI.
Mortality in patients with type 2 MI is “quite high, but it may well be a marker of the fact that you’ve got other serious diseases on board that are associated with poorer outcome,” he said.
Risk varies
The degree of risk in type 2 MI seems to vary with the underlying condition, a recent cohort study suggests. In about 3,800 patients with cardiac troponin (cTn) elevations qualifying as MI – a younger group; most were in their 30s and 40s – mortality at 10 years was 12% for those with type 1 MI, but 34% for those with type 2 MI and 46% for the remainder with nonischemic myocardial injury.
Underlying precipitating conditions varied widely among the patients with type 2 MI or nonischemic myocardial injury, and there was broad variation in mortality by etiology among those with type 2 MI. Sepsis and anemia entailed some of the highest risk, and hypertension and arrhythmias some of the lowest.
A prospective, community-based study of 5,460 patients with type 1 MI or type 2 MI reached a similar conclusion, but with a twist. Five-year all-cause mortality contrasted significantly between types of MI at 31% and 52%, respectively, but CV mortality rates were similar in this study.
Mortality in type 2 MI again varied by the precipitating etiology, suggesting that patients can be risk stratified according to pathophysiological mechanism behind their demand infarction, the authors concluded, “underscoring that type 2 MI is not a single entity, rather a group of phenotypic clusters.”
The usually high comorbidity burden and CV risk in patients with type 2 MI, one of those authors said in an interview, suggest there are “opportunities to see whether we can reduce that risk.”
Formal recommendations consistently say that, in patients with type 2 MI, “your first and foremost target should be to treat the underlying trigger and cause,” said Yader Sandoval, MD, Mayo Clinic, Rochester, Minn. That means such opportunities for further CV risk reduction tend to be “underappreciated.”
“In principle, treating the inciting cause of type 2 MI or the injury is important,” said James L. Januzzi, MD, Massachusetts General Hospital, Boston, in an interview, “but I feel quite strongly that there must be more that we can do for these folks.”
Dr. Januzzi is senior author on a recent analysis based on more than 200,000 admissions across the United States that saw a 43% lower risk for in-hospital death and 54% lower risk for 30-day MI readmission for patients with type 2 MI than those with type 1, adjusted for risk factors and comorbidities.
But, “it is important to emphasize that type 2 MI patients had a substantial risk for adverse outcome, nonetheless, and lack a clear management approach,” Dr. Januzzi and colleagues stated in their publication, as reported by this news organization.
“Due to the high rates of long-term cardiovascular events experienced by the frequently encountered type 2 MI patients,” they wrote, “identifying evidence-based therapies represents a major unmet need.”
That such patients tend to be sick with multiple comorbidities and have not yet been clinically well characterized, Dr. Januzzi said, “has stymied our ability to develop a treatment strategy.”
Role of the universal definitions
That challenge might in some ways be complicated by the universal definition, especially version 4, in which the definitions for type 1 MI, type 2 MI, and nonischemic myocardial injury are unified biochemically.
This version, published in 2018 in the European Heart Journal and Circulation, introduced a formal definition of myocardial injury, which was hailed as an innovation: cTn elevation to the 99th percentile of the upper limit of normal in a reference population.
It differentiates type 1 MI from type 2 MI by the separate pathophysiology of the ischemia – plaque rupture with intracoronary thrombosis and myocardial oxygen supply–demand mismatch, respectively. In both cases, however, there must be symptoms or objective evidence of ischemia. Absent signs of ischemia, the determination would be nonischemic myocardial injury.
Yet clinically and prognostically, type 2 MI and nonischemic myocardial injury in some ways are more similar to each other than either is to type 1 MI. Both occur secondary to other conditions across diverse clinical settings and can be a challenge to tell apart.
The universal definition’s perspective of the three events – so heavily dependent on cTn levels and myocardial ischemia – fails to account for the myriad complexities of individual patients in practice, some say, and so can muddle the process of risk assessment and therapy.
“Abnormal troponin identifies injury, but it doesn’t identify mechanism. Type 2 MI is highly prevalent, but there are other things that cause abnormal troponins,” Dr. Januzzi said. That’s why it’s important to explore and map out the clinical variables associated with the two conditions, to “understand who has a type 2 MI and who has cardiac injury. And believe it or not, it’s actually harder than it sounds to sort that out.”
“Practically speaking, the differentiation between these events is clinical,” Dr. Sandoval agreed. “There’s not always perfect agreement on what we’re calling what.”
Consequently, the universal definitions might categorize some events in ways that seem inconsistent from a management perspective. For example, they make a sharp distinction between coronary atherothrombotic and coronary nonatherothrombotic MI etiologies. Some clinicians would group MI caused by coronary spasm, coronary embolism, or spontaneous coronary artery dissection along with MI from coronary plaque rupture and thrombosis. But, Dr. Sandoval said, “even though these are coronary issues, they would fall into the type 2 bin.”
Also, about half of cases identified as type 2 MI are caused by tachyarrhythmias, which can elevate troponin and cause ECG changes and possibly symptoms resembling angina, Dr. Newby observed. “But that is completely different from other types of myocardial infarction, which are much more serious.”
So, “it’s a real mess of a diagnosis – acute myocardial injury, type 2 and type 1 MI – and it can be quite difficult to disentangle,” he said. “I think that the definition certainly has let us down.”
The diversity of type 2 MI clinical settings might also be a challenge. Myocardial injury according to cTn, with or without ischemia, occurs widely during critical illnesses and acute conditions, including respiratory distress, sepsis, internal bleeding, stroke, and pulmonary embolism.
Early in the COVID-19 pandemic, much was made of elevated troponin levels and myocarditis as an apparently frequent complication among hospitalized patients. “I raised my hand and said, we’ve been seeing abnormal troponins in people with influenza for 20 years,” Dr. Januzzi said. “Critical illness, infection, toxicity from drugs, from chemotherapy, from alcohol – there are all sorts of potential triggers of myocardial injury.”
Troponin ‘overdependence’
With many clinical settings in common and the presence or absence of myocardial ischemia to primarily distinguish them, type 2 MI and nonischemic myocardial injury both can be mistaken for the other. That can send management decisions in inappropriate directions.
A 2019 study looked at 633 cases that had been coded as type 2 MI at a major center and readjudicated them according to the fourth universal definition. Only 57% met all the type 2 criteria, 42% were reclassified as nonischemic myocardial injury, and a few were determined to have unstable angina.
“There’s overdependence on the easiest tool in the universal definition,” said Dr. Januzzi, a coauthor on that study. “Frequently people get seduced by the rise in a troponin value and immediately call it a myocardial infarction, lacking the other components of the universal definition that require evidence for coronary ischemia. That happens every day, where someone with an abnormal troponin is incorrectly branded as having an MI.”
It may not help that the current ICD-10-CM system features a diagnostic code for type 2 MI but not for myocardial injury.
“Instead, the new ICD-10-CM coding includes a proxy called ‘non-MI troponin elevation due to an underlying cause,’ ” wrote Kristian Thygesen, MD, DSc, and Allan S. Jaffe, MD, in a recent editorial. They caution against “using this code for myocardial injury because it is not specific for an elevated cTn value and could represent any abnormal laboratory measurements.” The code could be “misleading,” thereby worsening the potential for miscoding and “misattribution of MI diagnoses.”
That potential suggests there could be a growing population of patients who have been told they had an MI, which then becomes part of their medical record, when, actually, they experienced nonischemic myocardial injury.
“Having seen this occur,” Dr. Januzzi explained, “it affects people emotionally to think they’ve had an MI. Precision in diagnosis is important, which is why the universal definition is so valuable. If people would adhere to it more assiduously, we could reduce the frequency of people getting a misdiagnosis of MI when in fact they had injury.”
Still, he added, “if someone has an illness severe enough to cause myocardial injury, they’re at risk for a bad outcome regardless of whether they did or didn’t have an MI.”
The uncertain role of angiography
Angiography isn’t ordered nearly as often for patients ultimately diagnosed with type 2 MI or myocardial injury as for those with type 1 MI. Type 2 MI can hit some patients who have remained symptom free despite possibly unrecognized obstructive coronary artery disease (CAD) when myocardial demand is pushed past supply by a critical illness, tachyarrhythmia, or other acute conditions.
In such cases, “it’s reasonable to hypothesize that revascularization, something that really is not done in the vast majority of patients with type 2 MI, might actually be of benefit,” Dr. Januzzi said.
Whether these patients should routinely have angiography remains an open question. Without intervention, any newly identified obstructive CAD would continue to lurk in the background as a potential threat.
In efforts to differentiate type 2 MI from nonischemic injury, it can be “incredibly hard to know whether or not there’s actual ischemia in the mix. And that’s the only thing that defines the difference before taking an angiogram,” Derek P. Chew, MBBS, MPH, Flinders Medical Centre, Bedford Park, Australia, said in an interview.
Dr. Chew is principal investigator for the ongoing ACT-2 trial that is enrolling hospitalized, hemodynamically stable patients with cTn elevations but no suspicion of type 1 MI and “an unequivocal acute intercurrent diagnosis.” Qualifying diagnoses are prespecified on a list that includes sepsis, pneumonia, septicemia, a systemic inflammatory response, anemia, atrial tachycardia, acute kidney injury, and recent noncardiac surgery.
The patients are randomly assigned to a strategy of routine, usually invasive coronary angiography with discretionary revascularization, or to conservative care with noninvasive functional testing as appropriate. The sicker the patient, the greater the competing risk from other conditions and the less revascularization is likely to improve outcomes, Dr. Chew observed. Importantly, therefore, outcomes in the trial will be stratified by patient risk from comorbidities, measured with baseline GRACE and APACHE III scores.
Dr. Chew said the study aims to determine whether routine angiography is of benefit in patients at some identifiable level of risk, if not the whole range. One possible result, he said, is that there could be a risk-profile “sweet spot” associated with better outcomes in those assigned to angiography.
Enrollment in the trial started about 3 years ago, but “the process has been slow,” he said, because many potentially referring clinicians have a “bias on one side or another,” with about half of them preferring the angiography approach and the other half conservative management.
The unsettled role of drug therapy
With their often-complicated clinical profile, patients with type 2 MI or nonischemic myocardial injury tend to be medically undertreated, yet there is observational evidence they can benefit from familiar drug therapies.
In the previously noted cohort study of 3,800 younger patients with one of the three forms of myocardial injury, less than half of patients with type 2 MI received any form of CAD secondary prevention therapy at discharge, the researchers, with first author Avinainder Singh, MD, from Yale University, New Haven, Conn, wrote.
The finding, consistent with Dr. Newby’s study from 2018, suggests that “categorizing the type of MI in young subjects might inform long-term cardiovascular prognosis,” and “emphasizes the need to identify and implement secondary prevention strategies to mitigate the high rate of cardiovascular death in patients with type 2 MI,” they concluded.
Further, outcomes varied with the number of discharge CV meds in an older cohort of patients with myocardial injury. Those with type 2 MI or acute or chronic nonischemic myocardial injury were far less likely than patients with type 1 MI to be prescribed guideline-based drugs. Survival was greater for those on two or three classes of CV medications, compared with one or none, in patients with acute or chronic nonischemic injury.
The investigators urged that patients with nonischemic myocardial injury or type 2 MI “be treated with cardiovascular medication to a larger degree than what is done today.”
When there is documented CAD in patients with type 2 MI, “it would be reasonable to suggest that preventative secondary prevention approaches, such as such lipid-reduction therapy or aspirin, would be beneficial,” Dr. Sandoval said. “But the reality is, there are no randomized trials, there are no prospective studies. ACT-2 is one of the few and early studies that’s really trying to address this.”
“The great majority of these people are not going to the cath lab, but when they do, there seems to be a signal of potential benefit,” Dr. Januzzi said. “For someone with a type 2 MI, it’s quite possible revascularization might help. Then more long-term treatment with medications that are proven in randomized trials to reduce risk would be a very plausible intervention.”
“We’ve actually proposed a number of potential therapeutic interventions to explore, both in people with type 2 MI and in people with injury without MI,” he said. “They might include sodium glucose cotransporter 2 inhibitors. They might include antithrombotic therapy or more aggressive lipid lowering, possibly for the pleiotropic effects rather than the effects on atherosclerosis.”
Any such therapies that prove successful in well-designed trials could well earn both type 2 MI and nonischemic myocardial injury, neglected as disorders in their own right, the kind of respect in clinical care pathways that they likely deserve.
Dr. Newby has disclosed receiving consulting fees or honoraria from Eli Lilly, Roche, Toshiba, Jansen, Reckitt Benckiser Pharmaceuticals, Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, CellProthera, and Oncoarendi; and conducting research or receiving grants from Pfizer, AstraZeneca, Bristol-Myers Squibb, GlaxoSmithKline, Merck, Boehringer Ingelheim, and Inositec. Sandoval reports serving on an advisory board and as a speaker for Abbott Diagnostics and on an advisory board for Roche Diagnostics. Dr. Januzzi has disclosed receiving grant support from Novartis, Applied Therapeutics, and Innolife; consulting for Abbott Diagnostics, Janssen, Novartis, Quidel, and Roche Diagnostics; and serving on endpoint committees or data safety monitoring boards for trials supported by Abbott, AbbVie, Amgen, CVRx, Janssen, MyoKardia, and Takeda. Dr. Chew has reported receiving grants from AstraZeneca and Edwards Life Sciences. ACT-2 is sponsored by the National Medical and Health Research Council of Australia.
A version of this article first appeared on Medscape.com.
‘Malicious peer review’ destroyed doc’s career, he says
Cardiothoracic surgeon J. Marvin Smith III, MD, had always thrived on a busy practice schedule, often performing 20-30 surgeries a week. A practicing surgeon for more than 40 years, Dr. Smith said he had no plans to slow down anytime soon.
But Dr. Smith said his career was derailed when leaders at Methodist Healthcare System of San Antonio initiated a sudden peer review proceeding against him. The hospital system alleged certain surgeries performed by Dr. Smith had excessive mortality rates. When he proved the data inaccurate, Dr. Smith said administrators next claimed he was cognitively impaired and wasn’t safe to practice.
Dr. Smith has now been embroiled in a peer review dispute with the hospital system for more than 2 years and says the conflict has essentially forced him out of surgical practice. He believes the peer review was “malicious” and was really launched because of complaints he made about nurse staffing and other issues at the hospital.
“I think it is absolutely in bad faith and is disingenuous what they’ve told me along the way,” said Dr. Smith, 73. “It’s because I pointed out deficiencies in nursing care, and they want to get rid of me. It would be a lot easier for them if I had a contract and they could control me better. But the fact that I was independent, meant they had to resort to a malicious peer review to try and push me out.”
Dr. Smith had a peer review hearing with Methodist in March 2021, and in April, a panel found in Dr. Smith’s favor, according to Dr. Smith. The findings were sent to the hospital’s medical board for review, which issued a decision in early May.
Eric A. Pullen, an attorney for Dr. Smith, said he could not go into detail about the board’s decision for legal reasons, but that “the medical board’s decision did not completely resolve the matter, and Dr. Smith intends to exercise his procedural rights, which could include an appeal.”
Methodist Hospital Texsan and its parent company, Methodist Health System of San Antonio, did not respond to messages seeking comment about the case. Without hearing from the hospital system, its side is unknown and it is unclear if there is more to the story from Methodist’s view.
The problem is not new, but some experts, such as Lawrence Huntoon, MD, PhD, say the practice has become more common in recent years, particularly against independent doctors.
Dr. Huntoon believes there is a nationwide trend at many hospitals to get rid of independent physicians and replace them with employed doctors, he said.
However, because most sham peer reviews go on behind closed doors, there are no data to pinpoint its prevalence or measure its growth.
“Independent physicians are basically being purged from medical staffs across the United States,” said Dr. Huntoon, who is chair of the Association of American Physicians and Surgeons’ Committee to Combat Sham Peer Review. “The hospitals want more control over how physicians practice and who they refer to, and they do that by having employees.”
Anthony P. Weiss, MD, MBA, chief medical officer for Beth Israel Deaconess Medical Center said it has not been his experience that independent physicians are being targeted in such a way. Dr. Weiss responded to an inquiry sent to the American Hospital Association for this story.
“As the authority for peer review rests with the organized medical staff (i.e., physicians), and not formally with the hospital per se, the peer review lever is not typically available as a management tool for hospital administration,” said Dr. Weiss, who is a former member of the AHA’s Committee on Clinical Leadership, but who was speaking on behalf of himself.
A spokesman for the AHA said the organization stands behinds Dr. Weiss’ comments.
Peer review remains a foundational aspect of overseeing the safety and appropriateness of healthcare provided by physicians, Dr. Weiss said. Peer review likely varies from hospital to hospital, he added, although the Healthcare Quality Improvement Act provides some level of guidance as does the American Medical Association Code of Medical Ethics (section 9.4.1).
“In essence, both require that the evaluation be conducted in good faith with the intention to improve care, by physicians with adequate training and knowledge, using a process that is fair and inclusive of the physician under review,” he said. “I believe that most medical staffs abide by these ethical principles, but we have little data to confirm this supposition.”
Did hospital target doc for being vocal?
When members of Methodist’s medical staff first approached Dr. Smith with concerns about his surgery outcomes in November 2018, the physician says he was surprised, but that he was open to an assessment.
“They came to me and said they thought my numbers were bad, and I said: ‘Well my gosh, I certainly don’t want that to be the case. I need to see what numbers you are talking about,’ ” Dr. Smith recalled. “I’ve been president of the Bexar County Medical Society; I’ve been involved with standards and ethics for the Society of Thoracic Surgeons. Quality health care means a whole lot to me.”
The statistical information provided by hospital administrators indicated that Dr. Smith’s mortality rates for coronary artery surgery in 2018 were “excessive” and that his rates for aortic surgery were “unacceptable,” according to a lawsuit Dr. Smith filed against the hospital system. Dr. Smith, who is double boarded with the American Board of Surgery and the American Board of Thoracic Surgery, said his outcomes had never come into question in the past. Dr. Smith said the timing was suspicious to him, however, considering he had recently raised concerns with the hospital through letters about nursing performance, staffing, and compensation.
A peer review investigation was initiated. In the meantime, Dr. Smith agreed to intensivist consults on his postoperative patients and consults with the hospital’s “Heart Team” on all preoperative cardiac, valve, and aortic cases. A vocal critic of the Heart Team, Dr. Smith had long contended the entity provided no meaningful benefit to his patients in most cases and, rather, increased hospital stays and raised medical expenses. Despite his agreement, Dr. Smith was later asked to voluntarily stop performing surgeries at the hospital.
“I agreed, convinced that we’d get this all settled,” he said.
Another report issued by the hospital in 2019 also indicated elevated mortality rates associated with some of Smith’s surgeries, although the document differed from the first report, according to the lawsuit. Dr. Smith says he was ignored when he pointed out problems with the data, including a lack of appropriate risk stratification in the report, departure from Society of Thoracic Surgeons data rules, and improper inclusion of his cases in the denominator of the ratio when a comparison was made of his outcomes with those hospitalwide. A subsequent report from Methodist in March 2019 indicated Dr. Smith’s surgery outcomes were “within the expected parameters of performance,” according to court documents.
The surgery accusations were dropped, but the peer review proceeding against Dr. Smith wasn’t over. The hospital next requested that Dr. Smith undergo a competency evaluation.
“When they realized the data was bad, they then changed their argument in the peer review proceeding and essentially started to argue that Dr. Smith had some sort of cognitive disability that prevented him from continuing to practice,” said Mr. Pullen. “The way I look at it, when the initial basis for the peer review was proven false, the hospital found something else and some other reason to try to keep Dr. Smith from practicing.”
Thus began a lengthy disagreement about which entity would conduct the evaluation, who would pay, and the type of acceptable assessment. An evaluation by the hospital’s preferred organization resulted in a finding of mild cognitive impairment, Dr. Smith said. He hired his own experts who conducted separate evaluations, finding no impairment and no basis for the former evaluation’s conclusion.
“Literally, the determinant as to whether I was normal or below normal on their test was one point, which was associated with a finding that I didn’t draw a clock correctly,” Dr. Smith claimed. “The reviewer said my minute hand was a little too short and docked me a point. It was purely subjective. To me, the gold standard of whether you are learned in thoracic surgery is the American Board of Thoracic Surgery’s test. The board’s test shows my cognitive ability is entirely in keeping with my practice. That contrasts with the one point off I got for drawing a clock wrong in somebody’s estimation.”
Conflict leads to legal case
In September 2020, Dr. Smith filed a lawsuit against Methodist Healthcare System of San Antonio, alleging business disparagement by Methodist for allegedly publishing false and disparaging information about Dr. Smith and tortious interference with business relations. The latter claim stems from Methodist refusing to provide documents to other hospitals about the status of Dr. Smith’s privileges at Methodist, Mr. Pullen said.
Because Methodist refused to confirm his status, the renewal process for Baptist Health System could not be completed and Dr. Smith lost his privileges at Baptist Health System facilities, according to the lawsuit.
Notably, Dr. Smith’s legal challenge also asks the court to take a stance against alleged amendments by Methodist to its Unified Medical Staff Bylaws. The hospital allegedly proposed changes that would prevent physicians from seeking legal action against the hospital for malicious peer review, according to Dr. Smith’s lawsuit.
The amendments would make the peer review process itself the “sole and exclusive remedy with respect to any action or recommendation taken at the hospital affecting medical staff appointment and/or clinical privileges,” according to an excerpt of the proposed amendments included in Dr. Smith’s lawsuit. In addition, the changes would hold practitioners liable for lost revenues if the doctor initiates “any type of legal action challenging credentialing, privileging, or other medical peer review or professional review activity,” according to the lawsuit.
Dr. Smith’s lawsuit seeks a declaration that the proposed amendments to the bylaws are “void as against public policy,” and a declaration that the proposed amendments to the bylaws cannot take away physicians’ statutory right to bring litigation against Methodist for malicious peer review.
“The proposed amendments have a tendency to and will injure the public good,” Dr. Smith argued in the lawsuit. “The proposed amendments allow Methodist to act with malice and in bad faith in conducting peer review proceedings and face no legal repercussions.”
Regardless of the final outcome of the peer review proceeding, Mr. Pullen said the harm Dr. Smith has already endured cannot be reversed.
“Even if comes out in his favor, the damage is already done,” he said. “It will not remedy the damage Dr. Smith has incurred.”
Fighting sham peer review is difficult
Battling a malicious peer review has long been an uphill battle for physicians, according to Dr. Huntoon. That’s because the Health Care Quality Improvement Act (HCQIA), a federal law passed in 1986, provides near absolute immunity to hospitals and peer reviewers in legal disputes.
The HCQIA was created by Congress to extend immunity to good-faith peer review of doctors and to increase overall participation in peer review by removing fear of litigation. However, the act has also enabled abuse of peer review by shielding bad-faith reviewers from accountability, said Dr. Huntoon.
“The Health Care Quality Improvement Act presumes that what the hospital did was warranted and reasonable and shifts the burden to the physician to prove his innocence by a preponderance of evidence,” he said. “That’s an entirely foreign concept to most people who think a person should be considered innocent until proven guilty. Here, it’s the exact opposite.”
The HCQIA has been challenged numerous times over the years and tested at the appellate level, but continues to survive and remain settled law, added Richard B. Willner, DPM, founder and director of the Center for Peer Review Justice, which assists and counsels physicians about sham peer review.
In 2011, former Rep. Joe Heck, DO, (R-Nev.) introduced a bill that would have amended the HCQIA to prohibit a professional review entity from submitting a report to the National Practitioner Data Bank (NPDB) while the doctor was still under investigation and before the doctor was afforded adequate notice and a hearing. Although the measure had 16 cosponsors and plenty of support from the physician community, it failed.
In addition to a heavy legal burden, physicians who experience malicious peer reviews also face ramifications from being reported to the NPDB. Peer review organizations are required to report certain negative actions or findings to the NPDB.
“A databank entry is a scarlet letter on your forehead,” Dr. Willner said. “The rules at a lot of institutions are not to take anyone who has been databanked, rightfully or wrongfully. And what is the evidence necessary to databank you? None. There’s no evidence needed to databank somebody.”
Despite the bleak landscape, experts say progress has been made on a case-by-case basis by physicians who have succeeded in fighting back against questionable peer reviews in recent years.
In January 2020, Indiana ob.gyn. Rebecca Denman, MD, prevailed in her defamation lawsuit against St Vincent Carmel Hospital and St Vincent Carmel Medical Group, winning $4.75 million in damages. Dr. Denman alleged administrators failed to conduct a proper peer review investigation after a false allegation by a nurse that she was under the influence while on the job.
Indianapolis attorney Kathleen A. DeLaney, who represented Dr. Denman, said hospital leaders misled Dr. Denman into believing a peer review had occurred when no formal peer review hearing or proceeding took place.
“The CMO of the medical group claimed that he performed a peer review ‘screening,’ but he never informed the other members of the peer review executive committee of the matter until after he had placed Dr. Denman on administrative leave,” Ms. DeLaney said. “He also neglected to tell the peer review executive committee that the substance abuse policy had not been followed, or that Dr. Denman had not been tested for alcohol use – due to the 12-hour delay in report.”
Dr. Denman was ultimately required to undergo an alcohol abuse evaluation, enter a treatment program, and sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit. She claimed repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.
She sued the hospital in July 2018, alleging fraud, defamation, tortious interference with an employment relationship, and negligent misrepresentation. After a 4-day trial, jurors found in her favor, awarding Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
A hospital spokesperson said Ascension St Vincent is pursuing an appeal, and that it looks “forward to the opportunity to bring this matter before the Indiana Court of Appeals in June.”
In another case, South Dakota surgeon Linda Miller, MD, was awarded $1.1 million in 2017 after a federal jury found Huron Regional Medical Center breached her contract and violated her due process rights. Dr. Miller became the subject of a peer review at Huron Regional Medical Center when the hospital began analyzing some of her surgery outcomes.
Ken Barker, an attorney for Dr. Miller, said he feels it became evident at trial that the campaign to force Dr. Miller to either resign or lose her privileges was led by the lay board of directors of the hospital and upper-level administration at the hospital.
“They began the process by ordering an unprecedented 90-day review of her medical charts, looking for errors in the medical care she provided patients,” he said. “They could find nothing, so they did a second 90-day review, waiting for a patient’s ‘bad outcome.’ As any general surgeon will say, a ‘bad outcome’ is inevitable. And so it was. Upon that occurrence, they had a medical review committee review the patient’s chart and use it as an excuse to force her to reduce her privileges. Unbeknown to Dr. Miller, an external review had been conducted on another patient’s chart, in which the external review found her care above the standards and, in some measure, ‘exemplary.’ ”
Dr. Miller was eventually pressured to resign, according to her claim. Because of reports made to the NPDB by the medical center, including a patient complication that was allegedly falsified by the hospital, Dr. Miller said she was unable to find work as a general surgeon and went to work as a wound care doctor. At trial, jurors awarded Dr. Miller $586,617 in lost wages, $343,640 for lost future earning capacity, and $250,000 for mental anguish. (The mental anguish award was subsequently struck by a district court.)
Attorneys for Huron Regional Medical Center argued the jury improperly awarded damages and requested a new trial, which was denied by an appeals court.
In the end, the evidence came to light and the jury’s verdict spoke loudly that the hospital had taken unfair advantage of Dr. Miller, Mr. Barker said. But he emphasized that such cases often end differently.
“There are a handful of cases in which physicians like Dr. Miller have challenged the system and won,” he said. “In most cases, however, it is a ‘David vs. Goliath’ scenario where the giant prevails.”
What to do if faced with malicious peer review
An important step when doctors encounter a peer review that they believe is malicious is to consult with an experienced attorney as early as possible, Dr. Huntoon said. “Not all attorneys who set themselves out to be health law attorneys necessarily have knowledge and expertise in sham peer review. And before such a thing happens, I always encourage physicians to read their medical staff bylaws. That’s where everything is set forth, [such as] the corrective action section that tells how peer review is to take place.”
Mr. Barker added that documentation is also key in the event of a potential malicious peer review.
“When a physician senses [the] administration has targeted them, they should start documenting their conversations and actions very carefully, and if possible, recruit another ‘observer’ who can provide a third-party perspective, if necessary,” Mr. Barker said.
Dr. Huntoon recently wrote an article with advice about preparedness and defense of sham peer reviews. The guidance includes that physicians educate themselves about the tactics used by some hospitals to conduct sham peer reviews and the factors that place doctors more at risk. Factors that may raise a doctor’s danger of being targeted include being in solo practice or a small group, being new on staff, or being an older physician approaching retirement as some bad-actor hospitals may view older physicians as being less likely to fight back, said Dr. Huntoon.
Doctors should also keep detailed records and a timeline in the event of a malicious peer review and insist that an independent court reporter record all peer review hearings, even if that means the physician has to pay for the reporter him or herself, according to the guidance. An independent record is invaluable should the physician ultimately issue a future legal challenge against the hospital.
Mr. Willner encourages physicians to call the Center for Peer Review Justice hotline at (504) 621-1670 or visit the website for help with peer review and NPDB issues.
As for Dr. Smith, his days are much quieter and slower today, compared with the active practice he was accustomed to for more than half his life. He misses the fast pace, the patients, and the work that always brought him great joy.
“I hope to get back to doing surgeries eventually,” he said. “I graduated medical school in 1972. Practicing surgery has been my whole life and my career. They have taken my identity and my livelihood away from me based on false numbers and false premises. I want it back.”
A version of this article first appeared on Medscape.com.
Cardiothoracic surgeon J. Marvin Smith III, MD, had always thrived on a busy practice schedule, often performing 20-30 surgeries a week. A practicing surgeon for more than 40 years, Dr. Smith said he had no plans to slow down anytime soon.
But Dr. Smith said his career was derailed when leaders at Methodist Healthcare System of San Antonio initiated a sudden peer review proceeding against him. The hospital system alleged certain surgeries performed by Dr. Smith had excessive mortality rates. When he proved the data inaccurate, Dr. Smith said administrators next claimed he was cognitively impaired and wasn’t safe to practice.
Dr. Smith has now been embroiled in a peer review dispute with the hospital system for more than 2 years and says the conflict has essentially forced him out of surgical practice. He believes the peer review was “malicious” and was really launched because of complaints he made about nurse staffing and other issues at the hospital.
“I think it is absolutely in bad faith and is disingenuous what they’ve told me along the way,” said Dr. Smith, 73. “It’s because I pointed out deficiencies in nursing care, and they want to get rid of me. It would be a lot easier for them if I had a contract and they could control me better. But the fact that I was independent, meant they had to resort to a malicious peer review to try and push me out.”
Dr. Smith had a peer review hearing with Methodist in March 2021, and in April, a panel found in Dr. Smith’s favor, according to Dr. Smith. The findings were sent to the hospital’s medical board for review, which issued a decision in early May.
Eric A. Pullen, an attorney for Dr. Smith, said he could not go into detail about the board’s decision for legal reasons, but that “the medical board’s decision did not completely resolve the matter, and Dr. Smith intends to exercise his procedural rights, which could include an appeal.”
Methodist Hospital Texsan and its parent company, Methodist Health System of San Antonio, did not respond to messages seeking comment about the case. Without hearing from the hospital system, its side is unknown and it is unclear if there is more to the story from Methodist’s view.
The problem is not new, but some experts, such as Lawrence Huntoon, MD, PhD, say the practice has become more common in recent years, particularly against independent doctors.
Dr. Huntoon believes there is a nationwide trend at many hospitals to get rid of independent physicians and replace them with employed doctors, he said.
However, because most sham peer reviews go on behind closed doors, there are no data to pinpoint its prevalence or measure its growth.
“Independent physicians are basically being purged from medical staffs across the United States,” said Dr. Huntoon, who is chair of the Association of American Physicians and Surgeons’ Committee to Combat Sham Peer Review. “The hospitals want more control over how physicians practice and who they refer to, and they do that by having employees.”
Anthony P. Weiss, MD, MBA, chief medical officer for Beth Israel Deaconess Medical Center said it has not been his experience that independent physicians are being targeted in such a way. Dr. Weiss responded to an inquiry sent to the American Hospital Association for this story.
“As the authority for peer review rests with the organized medical staff (i.e., physicians), and not formally with the hospital per se, the peer review lever is not typically available as a management tool for hospital administration,” said Dr. Weiss, who is a former member of the AHA’s Committee on Clinical Leadership, but who was speaking on behalf of himself.
A spokesman for the AHA said the organization stands behinds Dr. Weiss’ comments.
Peer review remains a foundational aspect of overseeing the safety and appropriateness of healthcare provided by physicians, Dr. Weiss said. Peer review likely varies from hospital to hospital, he added, although the Healthcare Quality Improvement Act provides some level of guidance as does the American Medical Association Code of Medical Ethics (section 9.4.1).
“In essence, both require that the evaluation be conducted in good faith with the intention to improve care, by physicians with adequate training and knowledge, using a process that is fair and inclusive of the physician under review,” he said. “I believe that most medical staffs abide by these ethical principles, but we have little data to confirm this supposition.”
Did hospital target doc for being vocal?
When members of Methodist’s medical staff first approached Dr. Smith with concerns about his surgery outcomes in November 2018, the physician says he was surprised, but that he was open to an assessment.
“They came to me and said they thought my numbers were bad, and I said: ‘Well my gosh, I certainly don’t want that to be the case. I need to see what numbers you are talking about,’ ” Dr. Smith recalled. “I’ve been president of the Bexar County Medical Society; I’ve been involved with standards and ethics for the Society of Thoracic Surgeons. Quality health care means a whole lot to me.”
The statistical information provided by hospital administrators indicated that Dr. Smith’s mortality rates for coronary artery surgery in 2018 were “excessive” and that his rates for aortic surgery were “unacceptable,” according to a lawsuit Dr. Smith filed against the hospital system. Dr. Smith, who is double boarded with the American Board of Surgery and the American Board of Thoracic Surgery, said his outcomes had never come into question in the past. Dr. Smith said the timing was suspicious to him, however, considering he had recently raised concerns with the hospital through letters about nursing performance, staffing, and compensation.
A peer review investigation was initiated. In the meantime, Dr. Smith agreed to intensivist consults on his postoperative patients and consults with the hospital’s “Heart Team” on all preoperative cardiac, valve, and aortic cases. A vocal critic of the Heart Team, Dr. Smith had long contended the entity provided no meaningful benefit to his patients in most cases and, rather, increased hospital stays and raised medical expenses. Despite his agreement, Dr. Smith was later asked to voluntarily stop performing surgeries at the hospital.
“I agreed, convinced that we’d get this all settled,” he said.
Another report issued by the hospital in 2019 also indicated elevated mortality rates associated with some of Smith’s surgeries, although the document differed from the first report, according to the lawsuit. Dr. Smith says he was ignored when he pointed out problems with the data, including a lack of appropriate risk stratification in the report, departure from Society of Thoracic Surgeons data rules, and improper inclusion of his cases in the denominator of the ratio when a comparison was made of his outcomes with those hospitalwide. A subsequent report from Methodist in March 2019 indicated Dr. Smith’s surgery outcomes were “within the expected parameters of performance,” according to court documents.
The surgery accusations were dropped, but the peer review proceeding against Dr. Smith wasn’t over. The hospital next requested that Dr. Smith undergo a competency evaluation.
“When they realized the data was bad, they then changed their argument in the peer review proceeding and essentially started to argue that Dr. Smith had some sort of cognitive disability that prevented him from continuing to practice,” said Mr. Pullen. “The way I look at it, when the initial basis for the peer review was proven false, the hospital found something else and some other reason to try to keep Dr. Smith from practicing.”
Thus began a lengthy disagreement about which entity would conduct the evaluation, who would pay, and the type of acceptable assessment. An evaluation by the hospital’s preferred organization resulted in a finding of mild cognitive impairment, Dr. Smith said. He hired his own experts who conducted separate evaluations, finding no impairment and no basis for the former evaluation’s conclusion.
“Literally, the determinant as to whether I was normal or below normal on their test was one point, which was associated with a finding that I didn’t draw a clock correctly,” Dr. Smith claimed. “The reviewer said my minute hand was a little too short and docked me a point. It was purely subjective. To me, the gold standard of whether you are learned in thoracic surgery is the American Board of Thoracic Surgery’s test. The board’s test shows my cognitive ability is entirely in keeping with my practice. That contrasts with the one point off I got for drawing a clock wrong in somebody’s estimation.”
Conflict leads to legal case
In September 2020, Dr. Smith filed a lawsuit against Methodist Healthcare System of San Antonio, alleging business disparagement by Methodist for allegedly publishing false and disparaging information about Dr. Smith and tortious interference with business relations. The latter claim stems from Methodist refusing to provide documents to other hospitals about the status of Dr. Smith’s privileges at Methodist, Mr. Pullen said.
Because Methodist refused to confirm his status, the renewal process for Baptist Health System could not be completed and Dr. Smith lost his privileges at Baptist Health System facilities, according to the lawsuit.
Notably, Dr. Smith’s legal challenge also asks the court to take a stance against alleged amendments by Methodist to its Unified Medical Staff Bylaws. The hospital allegedly proposed changes that would prevent physicians from seeking legal action against the hospital for malicious peer review, according to Dr. Smith’s lawsuit.
The amendments would make the peer review process itself the “sole and exclusive remedy with respect to any action or recommendation taken at the hospital affecting medical staff appointment and/or clinical privileges,” according to an excerpt of the proposed amendments included in Dr. Smith’s lawsuit. In addition, the changes would hold practitioners liable for lost revenues if the doctor initiates “any type of legal action challenging credentialing, privileging, or other medical peer review or professional review activity,” according to the lawsuit.
Dr. Smith’s lawsuit seeks a declaration that the proposed amendments to the bylaws are “void as against public policy,” and a declaration that the proposed amendments to the bylaws cannot take away physicians’ statutory right to bring litigation against Methodist for malicious peer review.
“The proposed amendments have a tendency to and will injure the public good,” Dr. Smith argued in the lawsuit. “The proposed amendments allow Methodist to act with malice and in bad faith in conducting peer review proceedings and face no legal repercussions.”
Regardless of the final outcome of the peer review proceeding, Mr. Pullen said the harm Dr. Smith has already endured cannot be reversed.
“Even if comes out in his favor, the damage is already done,” he said. “It will not remedy the damage Dr. Smith has incurred.”
Fighting sham peer review is difficult
Battling a malicious peer review has long been an uphill battle for physicians, according to Dr. Huntoon. That’s because the Health Care Quality Improvement Act (HCQIA), a federal law passed in 1986, provides near absolute immunity to hospitals and peer reviewers in legal disputes.
The HCQIA was created by Congress to extend immunity to good-faith peer review of doctors and to increase overall participation in peer review by removing fear of litigation. However, the act has also enabled abuse of peer review by shielding bad-faith reviewers from accountability, said Dr. Huntoon.
“The Health Care Quality Improvement Act presumes that what the hospital did was warranted and reasonable and shifts the burden to the physician to prove his innocence by a preponderance of evidence,” he said. “That’s an entirely foreign concept to most people who think a person should be considered innocent until proven guilty. Here, it’s the exact opposite.”
The HCQIA has been challenged numerous times over the years and tested at the appellate level, but continues to survive and remain settled law, added Richard B. Willner, DPM, founder and director of the Center for Peer Review Justice, which assists and counsels physicians about sham peer review.
In 2011, former Rep. Joe Heck, DO, (R-Nev.) introduced a bill that would have amended the HCQIA to prohibit a professional review entity from submitting a report to the National Practitioner Data Bank (NPDB) while the doctor was still under investigation and before the doctor was afforded adequate notice and a hearing. Although the measure had 16 cosponsors and plenty of support from the physician community, it failed.
In addition to a heavy legal burden, physicians who experience malicious peer reviews also face ramifications from being reported to the NPDB. Peer review organizations are required to report certain negative actions or findings to the NPDB.
“A databank entry is a scarlet letter on your forehead,” Dr. Willner said. “The rules at a lot of institutions are not to take anyone who has been databanked, rightfully or wrongfully. And what is the evidence necessary to databank you? None. There’s no evidence needed to databank somebody.”
Despite the bleak landscape, experts say progress has been made on a case-by-case basis by physicians who have succeeded in fighting back against questionable peer reviews in recent years.
In January 2020, Indiana ob.gyn. Rebecca Denman, MD, prevailed in her defamation lawsuit against St Vincent Carmel Hospital and St Vincent Carmel Medical Group, winning $4.75 million in damages. Dr. Denman alleged administrators failed to conduct a proper peer review investigation after a false allegation by a nurse that she was under the influence while on the job.
Indianapolis attorney Kathleen A. DeLaney, who represented Dr. Denman, said hospital leaders misled Dr. Denman into believing a peer review had occurred when no formal peer review hearing or proceeding took place.
“The CMO of the medical group claimed that he performed a peer review ‘screening,’ but he never informed the other members of the peer review executive committee of the matter until after he had placed Dr. Denman on administrative leave,” Ms. DeLaney said. “He also neglected to tell the peer review executive committee that the substance abuse policy had not been followed, or that Dr. Denman had not been tested for alcohol use – due to the 12-hour delay in report.”
Dr. Denman was ultimately required to undergo an alcohol abuse evaluation, enter a treatment program, and sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit. She claimed repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.
She sued the hospital in July 2018, alleging fraud, defamation, tortious interference with an employment relationship, and negligent misrepresentation. After a 4-day trial, jurors found in her favor, awarding Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
A hospital spokesperson said Ascension St Vincent is pursuing an appeal, and that it looks “forward to the opportunity to bring this matter before the Indiana Court of Appeals in June.”
In another case, South Dakota surgeon Linda Miller, MD, was awarded $1.1 million in 2017 after a federal jury found Huron Regional Medical Center breached her contract and violated her due process rights. Dr. Miller became the subject of a peer review at Huron Regional Medical Center when the hospital began analyzing some of her surgery outcomes.
Ken Barker, an attorney for Dr. Miller, said he feels it became evident at trial that the campaign to force Dr. Miller to either resign or lose her privileges was led by the lay board of directors of the hospital and upper-level administration at the hospital.
“They began the process by ordering an unprecedented 90-day review of her medical charts, looking for errors in the medical care she provided patients,” he said. “They could find nothing, so they did a second 90-day review, waiting for a patient’s ‘bad outcome.’ As any general surgeon will say, a ‘bad outcome’ is inevitable. And so it was. Upon that occurrence, they had a medical review committee review the patient’s chart and use it as an excuse to force her to reduce her privileges. Unbeknown to Dr. Miller, an external review had been conducted on another patient’s chart, in which the external review found her care above the standards and, in some measure, ‘exemplary.’ ”
Dr. Miller was eventually pressured to resign, according to her claim. Because of reports made to the NPDB by the medical center, including a patient complication that was allegedly falsified by the hospital, Dr. Miller said she was unable to find work as a general surgeon and went to work as a wound care doctor. At trial, jurors awarded Dr. Miller $586,617 in lost wages, $343,640 for lost future earning capacity, and $250,000 for mental anguish. (The mental anguish award was subsequently struck by a district court.)
Attorneys for Huron Regional Medical Center argued the jury improperly awarded damages and requested a new trial, which was denied by an appeals court.
In the end, the evidence came to light and the jury’s verdict spoke loudly that the hospital had taken unfair advantage of Dr. Miller, Mr. Barker said. But he emphasized that such cases often end differently.
“There are a handful of cases in which physicians like Dr. Miller have challenged the system and won,” he said. “In most cases, however, it is a ‘David vs. Goliath’ scenario where the giant prevails.”
What to do if faced with malicious peer review
An important step when doctors encounter a peer review that they believe is malicious is to consult with an experienced attorney as early as possible, Dr. Huntoon said. “Not all attorneys who set themselves out to be health law attorneys necessarily have knowledge and expertise in sham peer review. And before such a thing happens, I always encourage physicians to read their medical staff bylaws. That’s where everything is set forth, [such as] the corrective action section that tells how peer review is to take place.”
Mr. Barker added that documentation is also key in the event of a potential malicious peer review.
“When a physician senses [the] administration has targeted them, they should start documenting their conversations and actions very carefully, and if possible, recruit another ‘observer’ who can provide a third-party perspective, if necessary,” Mr. Barker said.
Dr. Huntoon recently wrote an article with advice about preparedness and defense of sham peer reviews. The guidance includes that physicians educate themselves about the tactics used by some hospitals to conduct sham peer reviews and the factors that place doctors more at risk. Factors that may raise a doctor’s danger of being targeted include being in solo practice or a small group, being new on staff, or being an older physician approaching retirement as some bad-actor hospitals may view older physicians as being less likely to fight back, said Dr. Huntoon.
Doctors should also keep detailed records and a timeline in the event of a malicious peer review and insist that an independent court reporter record all peer review hearings, even if that means the physician has to pay for the reporter him or herself, according to the guidance. An independent record is invaluable should the physician ultimately issue a future legal challenge against the hospital.
Mr. Willner encourages physicians to call the Center for Peer Review Justice hotline at (504) 621-1670 or visit the website for help with peer review and NPDB issues.
As for Dr. Smith, his days are much quieter and slower today, compared with the active practice he was accustomed to for more than half his life. He misses the fast pace, the patients, and the work that always brought him great joy.
“I hope to get back to doing surgeries eventually,” he said. “I graduated medical school in 1972. Practicing surgery has been my whole life and my career. They have taken my identity and my livelihood away from me based on false numbers and false premises. I want it back.”
A version of this article first appeared on Medscape.com.
Cardiothoracic surgeon J. Marvin Smith III, MD, had always thrived on a busy practice schedule, often performing 20-30 surgeries a week. A practicing surgeon for more than 40 years, Dr. Smith said he had no plans to slow down anytime soon.
But Dr. Smith said his career was derailed when leaders at Methodist Healthcare System of San Antonio initiated a sudden peer review proceeding against him. The hospital system alleged certain surgeries performed by Dr. Smith had excessive mortality rates. When he proved the data inaccurate, Dr. Smith said administrators next claimed he was cognitively impaired and wasn’t safe to practice.
Dr. Smith has now been embroiled in a peer review dispute with the hospital system for more than 2 years and says the conflict has essentially forced him out of surgical practice. He believes the peer review was “malicious” and was really launched because of complaints he made about nurse staffing and other issues at the hospital.
“I think it is absolutely in bad faith and is disingenuous what they’ve told me along the way,” said Dr. Smith, 73. “It’s because I pointed out deficiencies in nursing care, and they want to get rid of me. It would be a lot easier for them if I had a contract and they could control me better. But the fact that I was independent, meant they had to resort to a malicious peer review to try and push me out.”
Dr. Smith had a peer review hearing with Methodist in March 2021, and in April, a panel found in Dr. Smith’s favor, according to Dr. Smith. The findings were sent to the hospital’s medical board for review, which issued a decision in early May.
Eric A. Pullen, an attorney for Dr. Smith, said he could not go into detail about the board’s decision for legal reasons, but that “the medical board’s decision did not completely resolve the matter, and Dr. Smith intends to exercise his procedural rights, which could include an appeal.”
Methodist Hospital Texsan and its parent company, Methodist Health System of San Antonio, did not respond to messages seeking comment about the case. Without hearing from the hospital system, its side is unknown and it is unclear if there is more to the story from Methodist’s view.
The problem is not new, but some experts, such as Lawrence Huntoon, MD, PhD, say the practice has become more common in recent years, particularly against independent doctors.
Dr. Huntoon believes there is a nationwide trend at many hospitals to get rid of independent physicians and replace them with employed doctors, he said.
However, because most sham peer reviews go on behind closed doors, there are no data to pinpoint its prevalence or measure its growth.
“Independent physicians are basically being purged from medical staffs across the United States,” said Dr. Huntoon, who is chair of the Association of American Physicians and Surgeons’ Committee to Combat Sham Peer Review. “The hospitals want more control over how physicians practice and who they refer to, and they do that by having employees.”
Anthony P. Weiss, MD, MBA, chief medical officer for Beth Israel Deaconess Medical Center said it has not been his experience that independent physicians are being targeted in such a way. Dr. Weiss responded to an inquiry sent to the American Hospital Association for this story.
“As the authority for peer review rests with the organized medical staff (i.e., physicians), and not formally with the hospital per se, the peer review lever is not typically available as a management tool for hospital administration,” said Dr. Weiss, who is a former member of the AHA’s Committee on Clinical Leadership, but who was speaking on behalf of himself.
A spokesman for the AHA said the organization stands behinds Dr. Weiss’ comments.
Peer review remains a foundational aspect of overseeing the safety and appropriateness of healthcare provided by physicians, Dr. Weiss said. Peer review likely varies from hospital to hospital, he added, although the Healthcare Quality Improvement Act provides some level of guidance as does the American Medical Association Code of Medical Ethics (section 9.4.1).
“In essence, both require that the evaluation be conducted in good faith with the intention to improve care, by physicians with adequate training and knowledge, using a process that is fair and inclusive of the physician under review,” he said. “I believe that most medical staffs abide by these ethical principles, but we have little data to confirm this supposition.”
Did hospital target doc for being vocal?
When members of Methodist’s medical staff first approached Dr. Smith with concerns about his surgery outcomes in November 2018, the physician says he was surprised, but that he was open to an assessment.
“They came to me and said they thought my numbers were bad, and I said: ‘Well my gosh, I certainly don’t want that to be the case. I need to see what numbers you are talking about,’ ” Dr. Smith recalled. “I’ve been president of the Bexar County Medical Society; I’ve been involved with standards and ethics for the Society of Thoracic Surgeons. Quality health care means a whole lot to me.”
The statistical information provided by hospital administrators indicated that Dr. Smith’s mortality rates for coronary artery surgery in 2018 were “excessive” and that his rates for aortic surgery were “unacceptable,” according to a lawsuit Dr. Smith filed against the hospital system. Dr. Smith, who is double boarded with the American Board of Surgery and the American Board of Thoracic Surgery, said his outcomes had never come into question in the past. Dr. Smith said the timing was suspicious to him, however, considering he had recently raised concerns with the hospital through letters about nursing performance, staffing, and compensation.
A peer review investigation was initiated. In the meantime, Dr. Smith agreed to intensivist consults on his postoperative patients and consults with the hospital’s “Heart Team” on all preoperative cardiac, valve, and aortic cases. A vocal critic of the Heart Team, Dr. Smith had long contended the entity provided no meaningful benefit to his patients in most cases and, rather, increased hospital stays and raised medical expenses. Despite his agreement, Dr. Smith was later asked to voluntarily stop performing surgeries at the hospital.
“I agreed, convinced that we’d get this all settled,” he said.
Another report issued by the hospital in 2019 also indicated elevated mortality rates associated with some of Smith’s surgeries, although the document differed from the first report, according to the lawsuit. Dr. Smith says he was ignored when he pointed out problems with the data, including a lack of appropriate risk stratification in the report, departure from Society of Thoracic Surgeons data rules, and improper inclusion of his cases in the denominator of the ratio when a comparison was made of his outcomes with those hospitalwide. A subsequent report from Methodist in March 2019 indicated Dr. Smith’s surgery outcomes were “within the expected parameters of performance,” according to court documents.
The surgery accusations were dropped, but the peer review proceeding against Dr. Smith wasn’t over. The hospital next requested that Dr. Smith undergo a competency evaluation.
“When they realized the data was bad, they then changed their argument in the peer review proceeding and essentially started to argue that Dr. Smith had some sort of cognitive disability that prevented him from continuing to practice,” said Mr. Pullen. “The way I look at it, when the initial basis for the peer review was proven false, the hospital found something else and some other reason to try to keep Dr. Smith from practicing.”
Thus began a lengthy disagreement about which entity would conduct the evaluation, who would pay, and the type of acceptable assessment. An evaluation by the hospital’s preferred organization resulted in a finding of mild cognitive impairment, Dr. Smith said. He hired his own experts who conducted separate evaluations, finding no impairment and no basis for the former evaluation’s conclusion.
“Literally, the determinant as to whether I was normal or below normal on their test was one point, which was associated with a finding that I didn’t draw a clock correctly,” Dr. Smith claimed. “The reviewer said my minute hand was a little too short and docked me a point. It was purely subjective. To me, the gold standard of whether you are learned in thoracic surgery is the American Board of Thoracic Surgery’s test. The board’s test shows my cognitive ability is entirely in keeping with my practice. That contrasts with the one point off I got for drawing a clock wrong in somebody’s estimation.”
Conflict leads to legal case
In September 2020, Dr. Smith filed a lawsuit against Methodist Healthcare System of San Antonio, alleging business disparagement by Methodist for allegedly publishing false and disparaging information about Dr. Smith and tortious interference with business relations. The latter claim stems from Methodist refusing to provide documents to other hospitals about the status of Dr. Smith’s privileges at Methodist, Mr. Pullen said.
Because Methodist refused to confirm his status, the renewal process for Baptist Health System could not be completed and Dr. Smith lost his privileges at Baptist Health System facilities, according to the lawsuit.
Notably, Dr. Smith’s legal challenge also asks the court to take a stance against alleged amendments by Methodist to its Unified Medical Staff Bylaws. The hospital allegedly proposed changes that would prevent physicians from seeking legal action against the hospital for malicious peer review, according to Dr. Smith’s lawsuit.
The amendments would make the peer review process itself the “sole and exclusive remedy with respect to any action or recommendation taken at the hospital affecting medical staff appointment and/or clinical privileges,” according to an excerpt of the proposed amendments included in Dr. Smith’s lawsuit. In addition, the changes would hold practitioners liable for lost revenues if the doctor initiates “any type of legal action challenging credentialing, privileging, or other medical peer review or professional review activity,” according to the lawsuit.
Dr. Smith’s lawsuit seeks a declaration that the proposed amendments to the bylaws are “void as against public policy,” and a declaration that the proposed amendments to the bylaws cannot take away physicians’ statutory right to bring litigation against Methodist for malicious peer review.
“The proposed amendments have a tendency to and will injure the public good,” Dr. Smith argued in the lawsuit. “The proposed amendments allow Methodist to act with malice and in bad faith in conducting peer review proceedings and face no legal repercussions.”
Regardless of the final outcome of the peer review proceeding, Mr. Pullen said the harm Dr. Smith has already endured cannot be reversed.
“Even if comes out in his favor, the damage is already done,” he said. “It will not remedy the damage Dr. Smith has incurred.”
Fighting sham peer review is difficult
Battling a malicious peer review has long been an uphill battle for physicians, according to Dr. Huntoon. That’s because the Health Care Quality Improvement Act (HCQIA), a federal law passed in 1986, provides near absolute immunity to hospitals and peer reviewers in legal disputes.
The HCQIA was created by Congress to extend immunity to good-faith peer review of doctors and to increase overall participation in peer review by removing fear of litigation. However, the act has also enabled abuse of peer review by shielding bad-faith reviewers from accountability, said Dr. Huntoon.
“The Health Care Quality Improvement Act presumes that what the hospital did was warranted and reasonable and shifts the burden to the physician to prove his innocence by a preponderance of evidence,” he said. “That’s an entirely foreign concept to most people who think a person should be considered innocent until proven guilty. Here, it’s the exact opposite.”
The HCQIA has been challenged numerous times over the years and tested at the appellate level, but continues to survive and remain settled law, added Richard B. Willner, DPM, founder and director of the Center for Peer Review Justice, which assists and counsels physicians about sham peer review.
In 2011, former Rep. Joe Heck, DO, (R-Nev.) introduced a bill that would have amended the HCQIA to prohibit a professional review entity from submitting a report to the National Practitioner Data Bank (NPDB) while the doctor was still under investigation and before the doctor was afforded adequate notice and a hearing. Although the measure had 16 cosponsors and plenty of support from the physician community, it failed.
In addition to a heavy legal burden, physicians who experience malicious peer reviews also face ramifications from being reported to the NPDB. Peer review organizations are required to report certain negative actions or findings to the NPDB.
“A databank entry is a scarlet letter on your forehead,” Dr. Willner said. “The rules at a lot of institutions are not to take anyone who has been databanked, rightfully or wrongfully. And what is the evidence necessary to databank you? None. There’s no evidence needed to databank somebody.”
Despite the bleak landscape, experts say progress has been made on a case-by-case basis by physicians who have succeeded in fighting back against questionable peer reviews in recent years.
In January 2020, Indiana ob.gyn. Rebecca Denman, MD, prevailed in her defamation lawsuit against St Vincent Carmel Hospital and St Vincent Carmel Medical Group, winning $4.75 million in damages. Dr. Denman alleged administrators failed to conduct a proper peer review investigation after a false allegation by a nurse that she was under the influence while on the job.
Indianapolis attorney Kathleen A. DeLaney, who represented Dr. Denman, said hospital leaders misled Dr. Denman into believing a peer review had occurred when no formal peer review hearing or proceeding took place.
“The CMO of the medical group claimed that he performed a peer review ‘screening,’ but he never informed the other members of the peer review executive committee of the matter until after he had placed Dr. Denman on administrative leave,” Ms. DeLaney said. “He also neglected to tell the peer review executive committee that the substance abuse policy had not been followed, or that Dr. Denman had not been tested for alcohol use – due to the 12-hour delay in report.”
Dr. Denman was ultimately required to undergo an alcohol abuse evaluation, enter a treatment program, and sign a 5-year monitoring contract with the Indiana State Medical Association as a condition of her employment, according to the lawsuit. She claimed repercussions from the false allegation resulted in lost compensation, out-of-pocket expenses, emotional distress, and damage to her professional reputation.
She sued the hospital in July 2018, alleging fraud, defamation, tortious interference with an employment relationship, and negligent misrepresentation. After a 4-day trial, jurors found in her favor, awarding Dr. Denman $2 million for her defamation claims, $2 million for her claims of fraud and constructive fraud, $500,000 for her claim of tortious interference with an employment relationship, and $250,000 for her claim of negligent misrepresentation.
A hospital spokesperson said Ascension St Vincent is pursuing an appeal, and that it looks “forward to the opportunity to bring this matter before the Indiana Court of Appeals in June.”
In another case, South Dakota surgeon Linda Miller, MD, was awarded $1.1 million in 2017 after a federal jury found Huron Regional Medical Center breached her contract and violated her due process rights. Dr. Miller became the subject of a peer review at Huron Regional Medical Center when the hospital began analyzing some of her surgery outcomes.
Ken Barker, an attorney for Dr. Miller, said he feels it became evident at trial that the campaign to force Dr. Miller to either resign or lose her privileges was led by the lay board of directors of the hospital and upper-level administration at the hospital.
“They began the process by ordering an unprecedented 90-day review of her medical charts, looking for errors in the medical care she provided patients,” he said. “They could find nothing, so they did a second 90-day review, waiting for a patient’s ‘bad outcome.’ As any general surgeon will say, a ‘bad outcome’ is inevitable. And so it was. Upon that occurrence, they had a medical review committee review the patient’s chart and use it as an excuse to force her to reduce her privileges. Unbeknown to Dr. Miller, an external review had been conducted on another patient’s chart, in which the external review found her care above the standards and, in some measure, ‘exemplary.’ ”
Dr. Miller was eventually pressured to resign, according to her claim. Because of reports made to the NPDB by the medical center, including a patient complication that was allegedly falsified by the hospital, Dr. Miller said she was unable to find work as a general surgeon and went to work as a wound care doctor. At trial, jurors awarded Dr. Miller $586,617 in lost wages, $343,640 for lost future earning capacity, and $250,000 for mental anguish. (The mental anguish award was subsequently struck by a district court.)
Attorneys for Huron Regional Medical Center argued the jury improperly awarded damages and requested a new trial, which was denied by an appeals court.
In the end, the evidence came to light and the jury’s verdict spoke loudly that the hospital had taken unfair advantage of Dr. Miller, Mr. Barker said. But he emphasized that such cases often end differently.
“There are a handful of cases in which physicians like Dr. Miller have challenged the system and won,” he said. “In most cases, however, it is a ‘David vs. Goliath’ scenario where the giant prevails.”
What to do if faced with malicious peer review
An important step when doctors encounter a peer review that they believe is malicious is to consult with an experienced attorney as early as possible, Dr. Huntoon said. “Not all attorneys who set themselves out to be health law attorneys necessarily have knowledge and expertise in sham peer review. And before such a thing happens, I always encourage physicians to read their medical staff bylaws. That’s where everything is set forth, [such as] the corrective action section that tells how peer review is to take place.”
Mr. Barker added that documentation is also key in the event of a potential malicious peer review.
“When a physician senses [the] administration has targeted them, they should start documenting their conversations and actions very carefully, and if possible, recruit another ‘observer’ who can provide a third-party perspective, if necessary,” Mr. Barker said.
Dr. Huntoon recently wrote an article with advice about preparedness and defense of sham peer reviews. The guidance includes that physicians educate themselves about the tactics used by some hospitals to conduct sham peer reviews and the factors that place doctors more at risk. Factors that may raise a doctor’s danger of being targeted include being in solo practice or a small group, being new on staff, or being an older physician approaching retirement as some bad-actor hospitals may view older physicians as being less likely to fight back, said Dr. Huntoon.
Doctors should also keep detailed records and a timeline in the event of a malicious peer review and insist that an independent court reporter record all peer review hearings, even if that means the physician has to pay for the reporter him or herself, according to the guidance. An independent record is invaluable should the physician ultimately issue a future legal challenge against the hospital.
Mr. Willner encourages physicians to call the Center for Peer Review Justice hotline at (504) 621-1670 or visit the website for help with peer review and NPDB issues.
As for Dr. Smith, his days are much quieter and slower today, compared with the active practice he was accustomed to for more than half his life. He misses the fast pace, the patients, and the work that always brought him great joy.
“I hope to get back to doing surgeries eventually,” he said. “I graduated medical school in 1972. Practicing surgery has been my whole life and my career. They have taken my identity and my livelihood away from me based on false numbers and false premises. I want it back.”
A version of this article first appeared on Medscape.com.
Carbon monoxide diffusion with COPD declines more in women
Single breath diffusion capacity for carbon monoxide shows greater decline over time in COPD patients compared with controls, but declines significantly more in women compared with men, according to data from 602 adults with a history of smoking.
In previous studies, diffusion capacity for carbon monoxide (DLco) has been associated with decreased exercise capacity and poor health status in patients with COPD, but its association as a measure of disease progression has not been well studied, wrote Ciro Casanova, MD, of Hospital Universitario La Candelaria, Spain, and colleagues.
In a study published in the journal CHEST®, the researchers identified 506 adult smokers with COPD and 96 adult smoker controls without COPD. Lung function based on single breath DLco was measured each year for 5 years. The study population was part of the COPH History Assessment in SpaiN (CHAIN), an ongoing observational study of adults with COPD. COPD was defined as a history of at least 10 pack-years of smoking and a post-bronchodilator FEV1/FVC greater than 0.7 after 400 micrograms of albuterol, the researchers said.
During the 5-year period, the average overall annual decline in DLco was 1.34% in COPD patients, compared with .04% in non-COPD controls (P = .004). Among COPD patients, age, body mass index, FEV1%, and active smoking were not associated with longitudinal change in DLco values, the researchers said.
Notably, women with COPD at baseline had lower baseline DLco values compared with men (11.37%) and a significantly steeper decline in DLco (.89%) compared with men (P = .039). “Being a woman was the only factor that related to the annual rate of change in DLco,” the researchers said.
In a subgroup analysis, the researchers identified 305 COPD patients and 69 non-COPD controls who had at least 3 DLco measurements over the 5-year study period. In this group, 16.4% patients with COPD and 4.3% smokers without COPD showed significant yearly declines in DLco of –4.139% and –4.440%, respectively. Among COPD patients, significantly more women than men showed significant DLco declines (26% vs. 14%, P = .005). No significant differences were observed in mortality or hospitalizations per patient-year for COPD patients with and without DLco decline, the researchers said.
The study findings were limited by several factors including the lack of annual measurements of DLco among some patients, potential variability in the instruments used to measure DLco, and the absence of computerized tomography data for the chest, the researchers noted. However, the results support the value of the test for COPD progression when conducted at 3- to 4-year intervals, given the slow pace of the decline, they said. More research is needed, but “women seem to have a different susceptibility to cigarette smoke in the alveolar or pulmonary vascular domains,” they added.
DLco remains a valuable marker
The study is important because the usual longitudinal decline of diffusion capacity, an important physiological parameter in patients with COPD, was unknown, Juan P. de Torres, MD, of Queen’s University, Kingston, Ont., said in an interview.
“The finding of a different longitudinal decline of DLco in women was a surprise,” said Dr. de Torres, who was a coauthor on the study. “We knew from previous works from our group that COPD has a different clinical and prognostic behavior in women with COPD, but this specific finding is novel and important,” he said.
“These results provide information about the testing frequency (3-4 years) needed to use DLco as a marker of COPD progression in clinical practice,” Dr. de Torres added.
“What is the driving cause of this sex difference is unknown. We speculate that different causes of low DLco in COPD such as degree of emphysema, interstitial lung abnormalities, and pulmonary hypertension, may have a different prevalence and progression in women with COPD,” he said.
Looking ahead, “Large studies including an adequate sample of women with COPD is urgently needed because they will be the main face of COPD in the near future,” said Dr. de Torres. “Sex difference in their physiological characteristics, the reason to explain those differences and how they behave longitudinally is also urgently needed,” he added.
The study was supported in part by AstraZeneca and by the COPD research program of the Spanish Respiratory Society. The researchers and Dr. de Torres had no financial conflicts to disclose.
Single breath diffusion capacity for carbon monoxide shows greater decline over time in COPD patients compared with controls, but declines significantly more in women compared with men, according to data from 602 adults with a history of smoking.
In previous studies, diffusion capacity for carbon monoxide (DLco) has been associated with decreased exercise capacity and poor health status in patients with COPD, but its association as a measure of disease progression has not been well studied, wrote Ciro Casanova, MD, of Hospital Universitario La Candelaria, Spain, and colleagues.
In a study published in the journal CHEST®, the researchers identified 506 adult smokers with COPD and 96 adult smoker controls without COPD. Lung function based on single breath DLco was measured each year for 5 years. The study population was part of the COPH History Assessment in SpaiN (CHAIN), an ongoing observational study of adults with COPD. COPD was defined as a history of at least 10 pack-years of smoking and a post-bronchodilator FEV1/FVC greater than 0.7 after 400 micrograms of albuterol, the researchers said.
During the 5-year period, the average overall annual decline in DLco was 1.34% in COPD patients, compared with .04% in non-COPD controls (P = .004). Among COPD patients, age, body mass index, FEV1%, and active smoking were not associated with longitudinal change in DLco values, the researchers said.
Notably, women with COPD at baseline had lower baseline DLco values compared with men (11.37%) and a significantly steeper decline in DLco (.89%) compared with men (P = .039). “Being a woman was the only factor that related to the annual rate of change in DLco,” the researchers said.
In a subgroup analysis, the researchers identified 305 COPD patients and 69 non-COPD controls who had at least 3 DLco measurements over the 5-year study period. In this group, 16.4% patients with COPD and 4.3% smokers without COPD showed significant yearly declines in DLco of –4.139% and –4.440%, respectively. Among COPD patients, significantly more women than men showed significant DLco declines (26% vs. 14%, P = .005). No significant differences were observed in mortality or hospitalizations per patient-year for COPD patients with and without DLco decline, the researchers said.
The study findings were limited by several factors including the lack of annual measurements of DLco among some patients, potential variability in the instruments used to measure DLco, and the absence of computerized tomography data for the chest, the researchers noted. However, the results support the value of the test for COPD progression when conducted at 3- to 4-year intervals, given the slow pace of the decline, they said. More research is needed, but “women seem to have a different susceptibility to cigarette smoke in the alveolar or pulmonary vascular domains,” they added.
DLco remains a valuable marker
The study is important because the usual longitudinal decline of diffusion capacity, an important physiological parameter in patients with COPD, was unknown, Juan P. de Torres, MD, of Queen’s University, Kingston, Ont., said in an interview.
“The finding of a different longitudinal decline of DLco in women was a surprise,” said Dr. de Torres, who was a coauthor on the study. “We knew from previous works from our group that COPD has a different clinical and prognostic behavior in women with COPD, but this specific finding is novel and important,” he said.
“These results provide information about the testing frequency (3-4 years) needed to use DLco as a marker of COPD progression in clinical practice,” Dr. de Torres added.
“What is the driving cause of this sex difference is unknown. We speculate that different causes of low DLco in COPD such as degree of emphysema, interstitial lung abnormalities, and pulmonary hypertension, may have a different prevalence and progression in women with COPD,” he said.
Looking ahead, “Large studies including an adequate sample of women with COPD is urgently needed because they will be the main face of COPD in the near future,” said Dr. de Torres. “Sex difference in their physiological characteristics, the reason to explain those differences and how they behave longitudinally is also urgently needed,” he added.
The study was supported in part by AstraZeneca and by the COPD research program of the Spanish Respiratory Society. The researchers and Dr. de Torres had no financial conflicts to disclose.
Single breath diffusion capacity for carbon monoxide shows greater decline over time in COPD patients compared with controls, but declines significantly more in women compared with men, according to data from 602 adults with a history of smoking.
In previous studies, diffusion capacity for carbon monoxide (DLco) has been associated with decreased exercise capacity and poor health status in patients with COPD, but its association as a measure of disease progression has not been well studied, wrote Ciro Casanova, MD, of Hospital Universitario La Candelaria, Spain, and colleagues.
In a study published in the journal CHEST®, the researchers identified 506 adult smokers with COPD and 96 adult smoker controls without COPD. Lung function based on single breath DLco was measured each year for 5 years. The study population was part of the COPH History Assessment in SpaiN (CHAIN), an ongoing observational study of adults with COPD. COPD was defined as a history of at least 10 pack-years of smoking and a post-bronchodilator FEV1/FVC greater than 0.7 after 400 micrograms of albuterol, the researchers said.
During the 5-year period, the average overall annual decline in DLco was 1.34% in COPD patients, compared with .04% in non-COPD controls (P = .004). Among COPD patients, age, body mass index, FEV1%, and active smoking were not associated with longitudinal change in DLco values, the researchers said.
Notably, women with COPD at baseline had lower baseline DLco values compared with men (11.37%) and a significantly steeper decline in DLco (.89%) compared with men (P = .039). “Being a woman was the only factor that related to the annual rate of change in DLco,” the researchers said.
In a subgroup analysis, the researchers identified 305 COPD patients and 69 non-COPD controls who had at least 3 DLco measurements over the 5-year study period. In this group, 16.4% patients with COPD and 4.3% smokers without COPD showed significant yearly declines in DLco of –4.139% and –4.440%, respectively. Among COPD patients, significantly more women than men showed significant DLco declines (26% vs. 14%, P = .005). No significant differences were observed in mortality or hospitalizations per patient-year for COPD patients with and without DLco decline, the researchers said.
The study findings were limited by several factors including the lack of annual measurements of DLco among some patients, potential variability in the instruments used to measure DLco, and the absence of computerized tomography data for the chest, the researchers noted. However, the results support the value of the test for COPD progression when conducted at 3- to 4-year intervals, given the slow pace of the decline, they said. More research is needed, but “women seem to have a different susceptibility to cigarette smoke in the alveolar or pulmonary vascular domains,” they added.
DLco remains a valuable marker
The study is important because the usual longitudinal decline of diffusion capacity, an important physiological parameter in patients with COPD, was unknown, Juan P. de Torres, MD, of Queen’s University, Kingston, Ont., said in an interview.
“The finding of a different longitudinal decline of DLco in women was a surprise,” said Dr. de Torres, who was a coauthor on the study. “We knew from previous works from our group that COPD has a different clinical and prognostic behavior in women with COPD, but this specific finding is novel and important,” he said.
“These results provide information about the testing frequency (3-4 years) needed to use DLco as a marker of COPD progression in clinical practice,” Dr. de Torres added.
“What is the driving cause of this sex difference is unknown. We speculate that different causes of low DLco in COPD such as degree of emphysema, interstitial lung abnormalities, and pulmonary hypertension, may have a different prevalence and progression in women with COPD,” he said.
Looking ahead, “Large studies including an adequate sample of women with COPD is urgently needed because they will be the main face of COPD in the near future,” said Dr. de Torres. “Sex difference in their physiological characteristics, the reason to explain those differences and how they behave longitudinally is also urgently needed,” he added.
The study was supported in part by AstraZeneca and by the COPD research program of the Spanish Respiratory Society. The researchers and Dr. de Torres had no financial conflicts to disclose.
FROM CHEST
Dr. Topol talks: COVID-19 variants are innocent until proven guilty
Editor in Chief of this news organization Eric Topol, MD, founder and director of the Scripps Research Translational Institute in La Jolla, Calif., and professor of molecular medicine, has been closely following COVID-19 data since the pandemic began. He spoke with writer Miriam E. Tucker about the latest on SARS-CoV-2 variants and their impact on vaccine efficacy. The conversation serves as a follow-up to his April 13, 2021, New York Times opinion piece, in which he advised readers that “all variants are innocent until proven guilty.”
You have expressed overall confidence in the efficacy of the vaccines thus far despite the emergence of variants, with some caveats. How do you see the current situation?
The Centers for Disease Control and Prevention has designated five “variants of concern,” but only three of them are real concerns – B.1.1.7, first detected in the United Kingdom; P.1, in Brazil and Japan; and B.1.351, in South Africa. Yet, all three are susceptible to our current vaccines.
The U.K. B.1.1.7 is the worst variant of all because it’s hypertransmissible, so I call it a “superspreader strain.” It also causes more severe illness independent of the spread, so it’s a double whammy. It’s clear that it also causes more deaths. The only arguable point is whether it’s 30% or 50% more deaths, but regardless, it’s more lethal and more transmissible.
The B.1.1.7 is going to be the dominant strain worldwide. It could develop new mutations within it that could come back to haunt us. We must keep watch.
But for now, it’s fully responsive to all the vaccines, which is great because if we didn’t have them, we wouldn’t have gotten through this U.S. pandemic like we have, and neither would Israel and the United Kingdom and other countries that have been able to get out of the crisis. We met the enemy and put it in check.
As for the South Africa variant of concern, B.1.351, we just got some encouraging news showing that it›s very responsive to the Pfizer/BioNTech mRNA vaccine in large numbers of people. The study was conducted in Qatar following that country’s mass immunization campaign in which a total of 385,853 people had received at least one vaccine dose and 265,410 had completed the two doses as of March 31, 2021.
At 2 weeks past the second dose, the vaccine was 75% effective at preventing any documented infection with the B.1.351 variant and 89.5% effective against B.1.1.7. The vaccine’s effectiveness against severe, critical, or fatal COVID-19 was greater than 97.4% for all circulating strains in Qatar, where B.1.1.7 and B.1.351 are most prominent.
We also know that B.1.351 is very responsive to the Johnson & Johnson vaccine and the Novavax [vaccine in development] to a lesser degree. It is the most immune-evading variant we’ve seen thus far, with the highest likelihood of providing some vaccine resistance, yet not enough to interfere with vaccination campaigns. So that’s great news.
The caveats here are that you definitely need two doses of the mRNA vaccines to combat the B.1.351 variant. Also, the AstraZeneca vaccine failed to prevent it in South Africa. However, that study was hard to judge because it was underpowered for number of people with mild infections. So, it didn’t look as if it had any efficacy, but maybe it would if tested in a real trial.
The P.1 (Brazil) variant is the second-highest concern after B.1.1.7 because it’s the only one in the United States that’s still headed up. It seems to be competing a bit with B.1.1.7 here. We know it was associated with the crisis in Brazil, in Chile, and some other South American countries. It has some immune escape, but not as bad as B.1.351. It also appears to have somewhat greater transmissibility but not as much as B.1.1.7.
With P.1, we just don’t know enough yet. It was difficult to assess in Brazil because they were in the midst of a catastrophe – like India is now – and you don’t know how much of it is dragged by the catastrophe vs driving it.
We have to respond to P.1 carefully. There are some good data that it does respond to the Chinese vaccine Sinovac and the AstraZeneca vaccine, and it appears to respond to the others as well, based on serum studies. So it doesn’t look like vaccines will be the worry with this variant. Rather, it could be competing with B.1.1.7 and could lead to breakthrough infections in vaccinated people or reinfections in unvaccinated people who had COVID-19. We need several more weeks to sort it out.
Although the B.1.427 and B.1.429 variants initially seen in California remain on the CDC’s concern list, I’m not worried about them.
You mentioned the current COVID-19 crisis in India, where a new variant has been described as a “double mutant,” but on Twitter you called it a “scariant.” Why?
First of all, the B.1.617 variant isn’t a double mutant. It has 15 mutations. It’s a stupid term, focusing on two mutations which largely have been put aside as to concern. One of them is the L452R, which is the same as one of the California variants, and that hasn’t proved to be particularly serious or concerning. The other is the 484Q, and it’s not clear whether that has any function.
The B.1.617 is not the driver of the catastrophe in India. It may be contributing a small amount, but it has been overhyped as the double mutant that’s causing it all. Adding to that are what I call “scariant” headlines here in the United States when a few cases of that variant have been seen.
I coined the term scariant in early February because it was a pretty clear trend. People don’t know what variants are. They know a little bit about mutations but not variants, and they’re scared. A few variants are concerning, but we keep learning more and more things to decrease the concern. That’s why I wrote the New York Times op-ed, to try to provide some reassurance, since there’s such paranoia.
Do you think booster vaccinations will be necessary? If so, will those be of the original vaccines or new ones that incorporate the variants?
As we go forward, there’s still potential for new variants that we haven’t seen yet that combine the worst of all features – transmissibility and immune evasion – especially since we have a world where COVID-19 is unchecked. So, we’re not out of it yet, but at least for the moment, we have vaccines that are capable of protecting against all variants.
In most people, the immune response against SARS-CoV-2 is very durable and strong and may well last for years. With the most closely related SARS-CoV-1, people still had immune responses up to 18 years later. However, some people will have less robust vaccine responses, including the elderly and the immunocompromised. If they don’t have great responses to the vaccine to start with, over time they’re likely to become more vulnerable, especially if they’re exposed to the variants with some degree of immune evasion.
I think we need to study these individuals post vaccination. A lot of people fit into those categories, including seniors, people being treated for cancer or autoimmune conditions, or post organ transplant. We could set up a prospective study to see whether they develop symptomatic COVID-19 and if so, from what – the original strain, B.1.1.7, or the newer variants.
That’s where I think booster shots may be needed. They may not be necessary across the board, but perhaps just in these special subgroups.
All of the current vaccines can be tweaked to include new variants, but the need for that is uncertain as of now. Moderna is working on a so-called bivalent vaccine that includes the original SARS-CoV-2 strain plus the B.1.351 variant, but it isn’t clear that that’s going to be necessary.
Currently, at least 200 COVID-19 vaccines are in development. There will be vaccines you can inhale, room temperature mRNA vaccines, and potentially even oral vaccines.
In the near future, Novavax is close, and there will likely be a two-dose Johnson & Johnson version that has the same potency as the mRNA vaccines. There are a lot of moving parts here.
There may be a step down in efficacy from mRNA to the others, though, and that shouldn’t be discounted. All of the available vaccines so far protect very well against severe disease and death, but some are less effective against mild to moderate infections, which may then lead to long COVID. We don’t yet know whether those who get mild infection post vaccination can still get long COVID.
What do you think it will take to achieve herd immunity?
I prefer the term “containment.” It’s quantitative. If you get to an infection rate of less than 1 in 100,000 people, as they’ve done in Israel, with 0.8 per 100,000, then you have the virus in check, and there will be very little spread when it’s at that controlled rate, with no outbreaks. The United States is currently at about 15 per 100,000. California is at 4. That still has to get lower.
It will be a challenge to get to President Biden’s goal of having 70% of U.S. adults given at least one dose by July 4. We’re now at about 57%. To get that next 13% of adults is going to take an all-out effort: mobile units, going to homes, making it ultraconvenient, education for people with safety concerns, incentivization, and days off.
We also need to get employers, universities, and health systems to get to the mandatory level. We haven’t done that yet. Some universities have mandated it for students, faculty, and staff. We need it in more health care systems. Right now, we only have a couple. We mandate flu shots, and flu is nothing, compared with COVID-19. And the COVID-19 vaccine is far more efficacious – flu shots are 40% efficacious, while these are 95%. COVID-19 is a tenfold more lethal and serious disease, and much more spreadable.
People are using the lack of full licensure by the Food and Drug Administration – as opposed to emergency use authorization – as an excuse not to get vaccinated. A biologics license application takes time to approve. Meanwhile, we have hundreds of millions of doses that have been well tolerated and incredibly effective.
Another aspect to consider regarding containment is that about 110 million Americans have already had COVID-19, even though only about 30 million cases have been confirmed. Most of these people have immune protection, although it’s not as good as if they have one vaccine dose. But they have enough protection to be part of the story here of the wall against COVID-19 and will help us get through this.
That’s a silver lining of having an unchecked epidemic for the entire year of 2020. The good part is that’s helping to get us to achieve an incredible level of containment when we haven’t even been close. Right now, we’re as good as the country has been in the pandemic, but we still have a long gap to get down to that 1 per 100,000. That’s what we should be working toward, and we can get there.
A version of this article first appeared on Medscape.com.
Editor in Chief of this news organization Eric Topol, MD, founder and director of the Scripps Research Translational Institute in La Jolla, Calif., and professor of molecular medicine, has been closely following COVID-19 data since the pandemic began. He spoke with writer Miriam E. Tucker about the latest on SARS-CoV-2 variants and their impact on vaccine efficacy. The conversation serves as a follow-up to his April 13, 2021, New York Times opinion piece, in which he advised readers that “all variants are innocent until proven guilty.”
You have expressed overall confidence in the efficacy of the vaccines thus far despite the emergence of variants, with some caveats. How do you see the current situation?
The Centers for Disease Control and Prevention has designated five “variants of concern,” but only three of them are real concerns – B.1.1.7, first detected in the United Kingdom; P.1, in Brazil and Japan; and B.1.351, in South Africa. Yet, all three are susceptible to our current vaccines.
The U.K. B.1.1.7 is the worst variant of all because it’s hypertransmissible, so I call it a “superspreader strain.” It also causes more severe illness independent of the spread, so it’s a double whammy. It’s clear that it also causes more deaths. The only arguable point is whether it’s 30% or 50% more deaths, but regardless, it’s more lethal and more transmissible.
The B.1.1.7 is going to be the dominant strain worldwide. It could develop new mutations within it that could come back to haunt us. We must keep watch.
But for now, it’s fully responsive to all the vaccines, which is great because if we didn’t have them, we wouldn’t have gotten through this U.S. pandemic like we have, and neither would Israel and the United Kingdom and other countries that have been able to get out of the crisis. We met the enemy and put it in check.
As for the South Africa variant of concern, B.1.351, we just got some encouraging news showing that it›s very responsive to the Pfizer/BioNTech mRNA vaccine in large numbers of people. The study was conducted in Qatar following that country’s mass immunization campaign in which a total of 385,853 people had received at least one vaccine dose and 265,410 had completed the two doses as of March 31, 2021.
At 2 weeks past the second dose, the vaccine was 75% effective at preventing any documented infection with the B.1.351 variant and 89.5% effective against B.1.1.7. The vaccine’s effectiveness against severe, critical, or fatal COVID-19 was greater than 97.4% for all circulating strains in Qatar, where B.1.1.7 and B.1.351 are most prominent.
We also know that B.1.351 is very responsive to the Johnson & Johnson vaccine and the Novavax [vaccine in development] to a lesser degree. It is the most immune-evading variant we’ve seen thus far, with the highest likelihood of providing some vaccine resistance, yet not enough to interfere with vaccination campaigns. So that’s great news.
The caveats here are that you definitely need two doses of the mRNA vaccines to combat the B.1.351 variant. Also, the AstraZeneca vaccine failed to prevent it in South Africa. However, that study was hard to judge because it was underpowered for number of people with mild infections. So, it didn’t look as if it had any efficacy, but maybe it would if tested in a real trial.
The P.1 (Brazil) variant is the second-highest concern after B.1.1.7 because it’s the only one in the United States that’s still headed up. It seems to be competing a bit with B.1.1.7 here. We know it was associated with the crisis in Brazil, in Chile, and some other South American countries. It has some immune escape, but not as bad as B.1.351. It also appears to have somewhat greater transmissibility but not as much as B.1.1.7.
With P.1, we just don’t know enough yet. It was difficult to assess in Brazil because they were in the midst of a catastrophe – like India is now – and you don’t know how much of it is dragged by the catastrophe vs driving it.
We have to respond to P.1 carefully. There are some good data that it does respond to the Chinese vaccine Sinovac and the AstraZeneca vaccine, and it appears to respond to the others as well, based on serum studies. So it doesn’t look like vaccines will be the worry with this variant. Rather, it could be competing with B.1.1.7 and could lead to breakthrough infections in vaccinated people or reinfections in unvaccinated people who had COVID-19. We need several more weeks to sort it out.
Although the B.1.427 and B.1.429 variants initially seen in California remain on the CDC’s concern list, I’m not worried about them.
You mentioned the current COVID-19 crisis in India, where a new variant has been described as a “double mutant,” but on Twitter you called it a “scariant.” Why?
First of all, the B.1.617 variant isn’t a double mutant. It has 15 mutations. It’s a stupid term, focusing on two mutations which largely have been put aside as to concern. One of them is the L452R, which is the same as one of the California variants, and that hasn’t proved to be particularly serious or concerning. The other is the 484Q, and it’s not clear whether that has any function.
The B.1.617 is not the driver of the catastrophe in India. It may be contributing a small amount, but it has been overhyped as the double mutant that’s causing it all. Adding to that are what I call “scariant” headlines here in the United States when a few cases of that variant have been seen.
I coined the term scariant in early February because it was a pretty clear trend. People don’t know what variants are. They know a little bit about mutations but not variants, and they’re scared. A few variants are concerning, but we keep learning more and more things to decrease the concern. That’s why I wrote the New York Times op-ed, to try to provide some reassurance, since there’s such paranoia.
Do you think booster vaccinations will be necessary? If so, will those be of the original vaccines or new ones that incorporate the variants?
As we go forward, there’s still potential for new variants that we haven’t seen yet that combine the worst of all features – transmissibility and immune evasion – especially since we have a world where COVID-19 is unchecked. So, we’re not out of it yet, but at least for the moment, we have vaccines that are capable of protecting against all variants.
In most people, the immune response against SARS-CoV-2 is very durable and strong and may well last for years. With the most closely related SARS-CoV-1, people still had immune responses up to 18 years later. However, some people will have less robust vaccine responses, including the elderly and the immunocompromised. If they don’t have great responses to the vaccine to start with, over time they’re likely to become more vulnerable, especially if they’re exposed to the variants with some degree of immune evasion.
I think we need to study these individuals post vaccination. A lot of people fit into those categories, including seniors, people being treated for cancer or autoimmune conditions, or post organ transplant. We could set up a prospective study to see whether they develop symptomatic COVID-19 and if so, from what – the original strain, B.1.1.7, or the newer variants.
That’s where I think booster shots may be needed. They may not be necessary across the board, but perhaps just in these special subgroups.
All of the current vaccines can be tweaked to include new variants, but the need for that is uncertain as of now. Moderna is working on a so-called bivalent vaccine that includes the original SARS-CoV-2 strain plus the B.1.351 variant, but it isn’t clear that that’s going to be necessary.
Currently, at least 200 COVID-19 vaccines are in development. There will be vaccines you can inhale, room temperature mRNA vaccines, and potentially even oral vaccines.
In the near future, Novavax is close, and there will likely be a two-dose Johnson & Johnson version that has the same potency as the mRNA vaccines. There are a lot of moving parts here.
There may be a step down in efficacy from mRNA to the others, though, and that shouldn’t be discounted. All of the available vaccines so far protect very well against severe disease and death, but some are less effective against mild to moderate infections, which may then lead to long COVID. We don’t yet know whether those who get mild infection post vaccination can still get long COVID.
What do you think it will take to achieve herd immunity?
I prefer the term “containment.” It’s quantitative. If you get to an infection rate of less than 1 in 100,000 people, as they’ve done in Israel, with 0.8 per 100,000, then you have the virus in check, and there will be very little spread when it’s at that controlled rate, with no outbreaks. The United States is currently at about 15 per 100,000. California is at 4. That still has to get lower.
It will be a challenge to get to President Biden’s goal of having 70% of U.S. adults given at least one dose by July 4. We’re now at about 57%. To get that next 13% of adults is going to take an all-out effort: mobile units, going to homes, making it ultraconvenient, education for people with safety concerns, incentivization, and days off.
We also need to get employers, universities, and health systems to get to the mandatory level. We haven’t done that yet. Some universities have mandated it for students, faculty, and staff. We need it in more health care systems. Right now, we only have a couple. We mandate flu shots, and flu is nothing, compared with COVID-19. And the COVID-19 vaccine is far more efficacious – flu shots are 40% efficacious, while these are 95%. COVID-19 is a tenfold more lethal and serious disease, and much more spreadable.
People are using the lack of full licensure by the Food and Drug Administration – as opposed to emergency use authorization – as an excuse not to get vaccinated. A biologics license application takes time to approve. Meanwhile, we have hundreds of millions of doses that have been well tolerated and incredibly effective.
Another aspect to consider regarding containment is that about 110 million Americans have already had COVID-19, even though only about 30 million cases have been confirmed. Most of these people have immune protection, although it’s not as good as if they have one vaccine dose. But they have enough protection to be part of the story here of the wall against COVID-19 and will help us get through this.
That’s a silver lining of having an unchecked epidemic for the entire year of 2020. The good part is that’s helping to get us to achieve an incredible level of containment when we haven’t even been close. Right now, we’re as good as the country has been in the pandemic, but we still have a long gap to get down to that 1 per 100,000. That’s what we should be working toward, and we can get there.
A version of this article first appeared on Medscape.com.
Editor in Chief of this news organization Eric Topol, MD, founder and director of the Scripps Research Translational Institute in La Jolla, Calif., and professor of molecular medicine, has been closely following COVID-19 data since the pandemic began. He spoke with writer Miriam E. Tucker about the latest on SARS-CoV-2 variants and their impact on vaccine efficacy. The conversation serves as a follow-up to his April 13, 2021, New York Times opinion piece, in which he advised readers that “all variants are innocent until proven guilty.”
You have expressed overall confidence in the efficacy of the vaccines thus far despite the emergence of variants, with some caveats. How do you see the current situation?
The Centers for Disease Control and Prevention has designated five “variants of concern,” but only three of them are real concerns – B.1.1.7, first detected in the United Kingdom; P.1, in Brazil and Japan; and B.1.351, in South Africa. Yet, all three are susceptible to our current vaccines.
The U.K. B.1.1.7 is the worst variant of all because it’s hypertransmissible, so I call it a “superspreader strain.” It also causes more severe illness independent of the spread, so it’s a double whammy. It’s clear that it also causes more deaths. The only arguable point is whether it’s 30% or 50% more deaths, but regardless, it’s more lethal and more transmissible.
The B.1.1.7 is going to be the dominant strain worldwide. It could develop new mutations within it that could come back to haunt us. We must keep watch.
But for now, it’s fully responsive to all the vaccines, which is great because if we didn’t have them, we wouldn’t have gotten through this U.S. pandemic like we have, and neither would Israel and the United Kingdom and other countries that have been able to get out of the crisis. We met the enemy and put it in check.
As for the South Africa variant of concern, B.1.351, we just got some encouraging news showing that it›s very responsive to the Pfizer/BioNTech mRNA vaccine in large numbers of people. The study was conducted in Qatar following that country’s mass immunization campaign in which a total of 385,853 people had received at least one vaccine dose and 265,410 had completed the two doses as of March 31, 2021.
At 2 weeks past the second dose, the vaccine was 75% effective at preventing any documented infection with the B.1.351 variant and 89.5% effective against B.1.1.7. The vaccine’s effectiveness against severe, critical, or fatal COVID-19 was greater than 97.4% for all circulating strains in Qatar, where B.1.1.7 and B.1.351 are most prominent.
We also know that B.1.351 is very responsive to the Johnson & Johnson vaccine and the Novavax [vaccine in development] to a lesser degree. It is the most immune-evading variant we’ve seen thus far, with the highest likelihood of providing some vaccine resistance, yet not enough to interfere with vaccination campaigns. So that’s great news.
The caveats here are that you definitely need two doses of the mRNA vaccines to combat the B.1.351 variant. Also, the AstraZeneca vaccine failed to prevent it in South Africa. However, that study was hard to judge because it was underpowered for number of people with mild infections. So, it didn’t look as if it had any efficacy, but maybe it would if tested in a real trial.
The P.1 (Brazil) variant is the second-highest concern after B.1.1.7 because it’s the only one in the United States that’s still headed up. It seems to be competing a bit with B.1.1.7 here. We know it was associated with the crisis in Brazil, in Chile, and some other South American countries. It has some immune escape, but not as bad as B.1.351. It also appears to have somewhat greater transmissibility but not as much as B.1.1.7.
With P.1, we just don’t know enough yet. It was difficult to assess in Brazil because they were in the midst of a catastrophe – like India is now – and you don’t know how much of it is dragged by the catastrophe vs driving it.
We have to respond to P.1 carefully. There are some good data that it does respond to the Chinese vaccine Sinovac and the AstraZeneca vaccine, and it appears to respond to the others as well, based on serum studies. So it doesn’t look like vaccines will be the worry with this variant. Rather, it could be competing with B.1.1.7 and could lead to breakthrough infections in vaccinated people or reinfections in unvaccinated people who had COVID-19. We need several more weeks to sort it out.
Although the B.1.427 and B.1.429 variants initially seen in California remain on the CDC’s concern list, I’m not worried about them.
You mentioned the current COVID-19 crisis in India, where a new variant has been described as a “double mutant,” but on Twitter you called it a “scariant.” Why?
First of all, the B.1.617 variant isn’t a double mutant. It has 15 mutations. It’s a stupid term, focusing on two mutations which largely have been put aside as to concern. One of them is the L452R, which is the same as one of the California variants, and that hasn’t proved to be particularly serious or concerning. The other is the 484Q, and it’s not clear whether that has any function.
The B.1.617 is not the driver of the catastrophe in India. It may be contributing a small amount, but it has been overhyped as the double mutant that’s causing it all. Adding to that are what I call “scariant” headlines here in the United States when a few cases of that variant have been seen.
I coined the term scariant in early February because it was a pretty clear trend. People don’t know what variants are. They know a little bit about mutations but not variants, and they’re scared. A few variants are concerning, but we keep learning more and more things to decrease the concern. That’s why I wrote the New York Times op-ed, to try to provide some reassurance, since there’s such paranoia.
Do you think booster vaccinations will be necessary? If so, will those be of the original vaccines or new ones that incorporate the variants?
As we go forward, there’s still potential for new variants that we haven’t seen yet that combine the worst of all features – transmissibility and immune evasion – especially since we have a world where COVID-19 is unchecked. So, we’re not out of it yet, but at least for the moment, we have vaccines that are capable of protecting against all variants.
In most people, the immune response against SARS-CoV-2 is very durable and strong and may well last for years. With the most closely related SARS-CoV-1, people still had immune responses up to 18 years later. However, some people will have less robust vaccine responses, including the elderly and the immunocompromised. If they don’t have great responses to the vaccine to start with, over time they’re likely to become more vulnerable, especially if they’re exposed to the variants with some degree of immune evasion.
I think we need to study these individuals post vaccination. A lot of people fit into those categories, including seniors, people being treated for cancer or autoimmune conditions, or post organ transplant. We could set up a prospective study to see whether they develop symptomatic COVID-19 and if so, from what – the original strain, B.1.1.7, or the newer variants.
That’s where I think booster shots may be needed. They may not be necessary across the board, but perhaps just in these special subgroups.
All of the current vaccines can be tweaked to include new variants, but the need for that is uncertain as of now. Moderna is working on a so-called bivalent vaccine that includes the original SARS-CoV-2 strain plus the B.1.351 variant, but it isn’t clear that that’s going to be necessary.
Currently, at least 200 COVID-19 vaccines are in development. There will be vaccines you can inhale, room temperature mRNA vaccines, and potentially even oral vaccines.
In the near future, Novavax is close, and there will likely be a two-dose Johnson & Johnson version that has the same potency as the mRNA vaccines. There are a lot of moving parts here.
There may be a step down in efficacy from mRNA to the others, though, and that shouldn’t be discounted. All of the available vaccines so far protect very well against severe disease and death, but some are less effective against mild to moderate infections, which may then lead to long COVID. We don’t yet know whether those who get mild infection post vaccination can still get long COVID.
What do you think it will take to achieve herd immunity?
I prefer the term “containment.” It’s quantitative. If you get to an infection rate of less than 1 in 100,000 people, as they’ve done in Israel, with 0.8 per 100,000, then you have the virus in check, and there will be very little spread when it’s at that controlled rate, with no outbreaks. The United States is currently at about 15 per 100,000. California is at 4. That still has to get lower.
It will be a challenge to get to President Biden’s goal of having 70% of U.S. adults given at least one dose by July 4. We’re now at about 57%. To get that next 13% of adults is going to take an all-out effort: mobile units, going to homes, making it ultraconvenient, education for people with safety concerns, incentivization, and days off.
We also need to get employers, universities, and health systems to get to the mandatory level. We haven’t done that yet. Some universities have mandated it for students, faculty, and staff. We need it in more health care systems. Right now, we only have a couple. We mandate flu shots, and flu is nothing, compared with COVID-19. And the COVID-19 vaccine is far more efficacious – flu shots are 40% efficacious, while these are 95%. COVID-19 is a tenfold more lethal and serious disease, and much more spreadable.
People are using the lack of full licensure by the Food and Drug Administration – as opposed to emergency use authorization – as an excuse not to get vaccinated. A biologics license application takes time to approve. Meanwhile, we have hundreds of millions of doses that have been well tolerated and incredibly effective.
Another aspect to consider regarding containment is that about 110 million Americans have already had COVID-19, even though only about 30 million cases have been confirmed. Most of these people have immune protection, although it’s not as good as if they have one vaccine dose. But they have enough protection to be part of the story here of the wall against COVID-19 and will help us get through this.
That’s a silver lining of having an unchecked epidemic for the entire year of 2020. The good part is that’s helping to get us to achieve an incredible level of containment when we haven’t even been close. Right now, we’re as good as the country has been in the pandemic, but we still have a long gap to get down to that 1 per 100,000. That’s what we should be working toward, and we can get there.
A version of this article first appeared on Medscape.com.