User login
Under new administration, best time to lobby for health care may be now
The ambitious infrastructure bill now being debated in the US Congress might be one of the best immediate opportunities to lobby for legislative or policy changes in delivery of health care during the current Biden administration, according to an analysis delivered at the annual health policy and advocacy conference sponsored by the American College of Chest Physicians.
The infrastructure bill is likely to be pushed forward in the filibuster-proof reconciliation process, which means “that some things might get passed that otherwise would not,” explained Keith S. Studdard, Vice President, Jeffrey J. Kimbell & Associates, Washington, DC.

With few exceptions, the key players in the health care team of President Joe Biden’s new administration are in place, according to Mr. Studdard, who is a lobbyist and health care expert. By moving quickly to fill key positions, the new administration “got off to a good start” for a health care agenda that Mr. Studdard believes will be a focus of the Biden presidency. There is some degree of urgency.
“The amount of time [the Biden administration has] to get their agenda through is fairly limited,” Mr. Studdard reported. The problems include a slim majority of fellow Democrats in the House of Representatives (222 vs 213), no majority of Democrats over Republicans in the Senate (50 vs 50), and mid-term elections that are already looming.
“Midterms historically favor the opposition party,” Mr. Studdard said. He expects party lines to harden as the midterms approach, dissipating the already limited appetite for bipartisan cooperation.
The midterms provide the basis for trying to affect change in advance of legislative gridlock, but the recently announced $2 trillion infrastructure bill is an even more compelling impetus. Infrastructure in this case is not limited to the construction of bridges and roads. Rather, this bill “is a massive package that will almost certainly touch on health care policy,” according to Mr. Studdard.
As the infrastructure bill winds its way through the legislative process, Mr. Studdard expects there will be efforts to include language that favors expansion of services and funding for health care. This includes those related to the Affordable Care Act (ACA) and the temporary modifications permitted under the CARES Act, which was passed during the early months of the COVID-19 pandemic.
For those who think that waivers and exceptions introduced in the CARES Act, such as the expansion of telehealth, should be made permanent, “this will be your main shot on goal,” Mr. Studdard said.
The debates around the ambitious infrastructure bill are “all that we will be hearing about from the legislative standpoint for the next few months,” Mr. Studdard said. He expects major lobbying efforts in regard to this legislation from a vast array of interest groups, not just those with a stake in health care.
If the bill passes, it will likely to be greatly helped by a vote under the reconciliation process. Created in 1974 to allow expedited consideration of spending legislation, the reconciliation process allows bills to be enacted with a simple majority, which is 51 votes in the Senate and 218 votes in the House. Filibustering is not permitted.
This means that the infrastructure bill, like the CARES Act, which was also passed through the reconciliation process, can be made into law even if all 50 Republican senators vote against it. As she has already done three times since taking office—most recently for COVID19 relief bill in early March—Vice President Kamala Harris can break a 50-50 tie with her vote for the administration’s agenda.
Legislation is one of two paths for altering funding and rules regarding health care in the United States. Policy is the other. For reaching decision makers with influence on policy, Mr. Studdard provided a long list of agencies, political appointees, and elected representatives that could be targeted. Many, such as the director of the Centers for Medicare & Medicaid Services (CMS), are well known, but others might be overlooked without a detailed list of the players.
As one example, he pointed to the Center for Medicare and Medicaid Innovation (CMMI), which is a relatively new organization within CMS. Led by Liz Fowler, a former Senate aide involved in writing the ACA, the CMMI has broad authority over several aspects of health policy, such as value-based care.
“The CCMI is something you should put on your radar. It moves with more flexibility than the HHS [Department of Health and Human Services],” Mr. Studdard said.
Mr. Studdard’s detailed overview of the intricacies of how to affect change in health policy and the likely trajectory under the Biden Administration included frequent comments about the traits, background, and goals of the specific decision makers he identified. The implication is that personal relations matter. Mr. Studdard indicated that knowing who to contact is just the first step.
For the Health Policy and Advocacy Committee, this information is critical. In his outline of the numerous paths for influencing health care policy, Mr. Studdard’s comments lead directly to strategies to lobbying goals for CHEST.

“CHEST and its Health Policy and Advocacy Committee are keeping a focus on health care policy to improve access and to improve care for our patients and reduce the burden on our providers,” according to the Chair of the Committee, Neil Freedman, MD, FCCP. Dr, Freedman is the Division Head Pulmonary, Critical Care, Allergy, and Immunology, Northshore University HeatlhSystem, Evanston, Illinois.
“We would hope that, in addition to the proposed infrastructure bill subsidizing some additional costs for the ACA and COBRA [Consolidated Omnibus Budget Reconciliation Act] and enhancing Medicaid eligibility, the bill would also provide some additional funding for the provider relief fund,” he said.
Mr. Studdard or his lobbying firm represent 62 clients with interests in health care policy.
The ambitious infrastructure bill now being debated in the US Congress might be one of the best immediate opportunities to lobby for legislative or policy changes in delivery of health care during the current Biden administration, according to an analysis delivered at the annual health policy and advocacy conference sponsored by the American College of Chest Physicians.
The infrastructure bill is likely to be pushed forward in the filibuster-proof reconciliation process, which means “that some things might get passed that otherwise would not,” explained Keith S. Studdard, Vice President, Jeffrey J. Kimbell & Associates, Washington, DC.
With few exceptions, the key players in the health care team of President Joe Biden’s new administration are in place, according to Mr. Studdard, who is a lobbyist and health care expert. By moving quickly to fill key positions, the new administration “got off to a good start” for a health care agenda that Mr. Studdard believes will be a focus of the Biden presidency. There is some degree of urgency.
“The amount of time [the Biden administration has] to get their agenda through is fairly limited,” Mr. Studdard reported. The problems include a slim majority of fellow Democrats in the House of Representatives (222 vs 213), no majority of Democrats over Republicans in the Senate (50 vs 50), and mid-term elections that are already looming.
“Midterms historically favor the opposition party,” Mr. Studdard said. He expects party lines to harden as the midterms approach, dissipating the already limited appetite for bipartisan cooperation.
The midterms provide the basis for trying to affect change in advance of legislative gridlock, but the recently announced $2 trillion infrastructure bill is an even more compelling impetus. Infrastructure in this case is not limited to the construction of bridges and roads. Rather, this bill “is a massive package that will almost certainly touch on health care policy,” according to Mr. Studdard.
As the infrastructure bill winds its way through the legislative process, Mr. Studdard expects there will be efforts to include language that favors expansion of services and funding for health care. This includes those related to the Affordable Care Act (ACA) and the temporary modifications permitted under the CARES Act, which was passed during the early months of the COVID-19 pandemic.
For those who think that waivers and exceptions introduced in the CARES Act, such as the expansion of telehealth, should be made permanent, “this will be your main shot on goal,” Mr. Studdard said.
The debates around the ambitious infrastructure bill are “all that we will be hearing about from the legislative standpoint for the next few months,” Mr. Studdard said. He expects major lobbying efforts in regard to this legislation from a vast array of interest groups, not just those with a stake in health care.
If the bill passes, it will likely to be greatly helped by a vote under the reconciliation process. Created in 1974 to allow expedited consideration of spending legislation, the reconciliation process allows bills to be enacted with a simple majority, which is 51 votes in the Senate and 218 votes in the House. Filibustering is not permitted.
This means that the infrastructure bill, like the CARES Act, which was also passed through the reconciliation process, can be made into law even if all 50 Republican senators vote against it. As she has already done three times since taking office—most recently for COVID19 relief bill in early March—Vice President Kamala Harris can break a 50-50 tie with her vote for the administration’s agenda.
Legislation is one of two paths for altering funding and rules regarding health care in the United States. Policy is the other. For reaching decision makers with influence on policy, Mr. Studdard provided a long list of agencies, political appointees, and elected representatives that could be targeted. Many, such as the director of the Centers for Medicare & Medicaid Services (CMS), are well known, but others might be overlooked without a detailed list of the players.
As one example, he pointed to the Center for Medicare and Medicaid Innovation (CMMI), which is a relatively new organization within CMS. Led by Liz Fowler, a former Senate aide involved in writing the ACA, the CMMI has broad authority over several aspects of health policy, such as value-based care.
“The CCMI is something you should put on your radar. It moves with more flexibility than the HHS [Department of Health and Human Services],” Mr. Studdard said.
Mr. Studdard’s detailed overview of the intricacies of how to affect change in health policy and the likely trajectory under the Biden Administration included frequent comments about the traits, background, and goals of the specific decision makers he identified. The implication is that personal relations matter. Mr. Studdard indicated that knowing who to contact is just the first step.
For the Health Policy and Advocacy Committee, this information is critical. In his outline of the numerous paths for influencing health care policy, Mr. Studdard’s comments lead directly to strategies to lobbying goals for CHEST.
“CHEST and its Health Policy and Advocacy Committee are keeping a focus on health care policy to improve access and to improve care for our patients and reduce the burden on our providers,” according to the Chair of the Committee, Neil Freedman, MD, FCCP. Dr, Freedman is the Division Head Pulmonary, Critical Care, Allergy, and Immunology, Northshore University HeatlhSystem, Evanston, Illinois.
“We would hope that, in addition to the proposed infrastructure bill subsidizing some additional costs for the ACA and COBRA [Consolidated Omnibus Budget Reconciliation Act] and enhancing Medicaid eligibility, the bill would also provide some additional funding for the provider relief fund,” he said.
Mr. Studdard or his lobbying firm represent 62 clients with interests in health care policy.
The ambitious infrastructure bill now being debated in the US Congress might be one of the best immediate opportunities to lobby for legislative or policy changes in delivery of health care during the current Biden administration, according to an analysis delivered at the annual health policy and advocacy conference sponsored by the American College of Chest Physicians.
The infrastructure bill is likely to be pushed forward in the filibuster-proof reconciliation process, which means “that some things might get passed that otherwise would not,” explained Keith S. Studdard, Vice President, Jeffrey J. Kimbell & Associates, Washington, DC.
With few exceptions, the key players in the health care team of President Joe Biden’s new administration are in place, according to Mr. Studdard, who is a lobbyist and health care expert. By moving quickly to fill key positions, the new administration “got off to a good start” for a health care agenda that Mr. Studdard believes will be a focus of the Biden presidency. There is some degree of urgency.
“The amount of time [the Biden administration has] to get their agenda through is fairly limited,” Mr. Studdard reported. The problems include a slim majority of fellow Democrats in the House of Representatives (222 vs 213), no majority of Democrats over Republicans in the Senate (50 vs 50), and mid-term elections that are already looming.
“Midterms historically favor the opposition party,” Mr. Studdard said. He expects party lines to harden as the midterms approach, dissipating the already limited appetite for bipartisan cooperation.
The midterms provide the basis for trying to affect change in advance of legislative gridlock, but the recently announced $2 trillion infrastructure bill is an even more compelling impetus. Infrastructure in this case is not limited to the construction of bridges and roads. Rather, this bill “is a massive package that will almost certainly touch on health care policy,” according to Mr. Studdard.
As the infrastructure bill winds its way through the legislative process, Mr. Studdard expects there will be efforts to include language that favors expansion of services and funding for health care. This includes those related to the Affordable Care Act (ACA) and the temporary modifications permitted under the CARES Act, which was passed during the early months of the COVID-19 pandemic.
For those who think that waivers and exceptions introduced in the CARES Act, such as the expansion of telehealth, should be made permanent, “this will be your main shot on goal,” Mr. Studdard said.
The debates around the ambitious infrastructure bill are “all that we will be hearing about from the legislative standpoint for the next few months,” Mr. Studdard said. He expects major lobbying efforts in regard to this legislation from a vast array of interest groups, not just those with a stake in health care.
If the bill passes, it will likely to be greatly helped by a vote under the reconciliation process. Created in 1974 to allow expedited consideration of spending legislation, the reconciliation process allows bills to be enacted with a simple majority, which is 51 votes in the Senate and 218 votes in the House. Filibustering is not permitted.
This means that the infrastructure bill, like the CARES Act, which was also passed through the reconciliation process, can be made into law even if all 50 Republican senators vote against it. As she has already done three times since taking office—most recently for COVID19 relief bill in early March—Vice President Kamala Harris can break a 50-50 tie with her vote for the administration’s agenda.
Legislation is one of two paths for altering funding and rules regarding health care in the United States. Policy is the other. For reaching decision makers with influence on policy, Mr. Studdard provided a long list of agencies, political appointees, and elected representatives that could be targeted. Many, such as the director of the Centers for Medicare & Medicaid Services (CMS), are well known, but others might be overlooked without a detailed list of the players.
As one example, he pointed to the Center for Medicare and Medicaid Innovation (CMMI), which is a relatively new organization within CMS. Led by Liz Fowler, a former Senate aide involved in writing the ACA, the CMMI has broad authority over several aspects of health policy, such as value-based care.
“The CCMI is something you should put on your radar. It moves with more flexibility than the HHS [Department of Health and Human Services],” Mr. Studdard said.
Mr. Studdard’s detailed overview of the intricacies of how to affect change in health policy and the likely trajectory under the Biden Administration included frequent comments about the traits, background, and goals of the specific decision makers he identified. The implication is that personal relations matter. Mr. Studdard indicated that knowing who to contact is just the first step.
For the Health Policy and Advocacy Committee, this information is critical. In his outline of the numerous paths for influencing health care policy, Mr. Studdard’s comments lead directly to strategies to lobbying goals for CHEST.
“CHEST and its Health Policy and Advocacy Committee are keeping a focus on health care policy to improve access and to improve care for our patients and reduce the burden on our providers,” according to the Chair of the Committee, Neil Freedman, MD, FCCP. Dr, Freedman is the Division Head Pulmonary, Critical Care, Allergy, and Immunology, Northshore University HeatlhSystem, Evanston, Illinois.
“We would hope that, in addition to the proposed infrastructure bill subsidizing some additional costs for the ACA and COBRA [Consolidated Omnibus Budget Reconciliation Act] and enhancing Medicaid eligibility, the bill would also provide some additional funding for the provider relief fund,” he said.
Mr. Studdard or his lobbying firm represent 62 clients with interests in health care policy.
REPORTING FROM THE CHEST HEALTH POLICY AND ADVOCACY CONFERENCE
Novel study links air pollution to increased risk of rheumatoid arthritis flares
Pollution appears to trigger inflammation
In patients with rheumatoid arthritis, exposure to air pollution is associated with both elevated levels of C-reactive protein (CRP) and increased risk of arthritis flares, according to a novel longitudinal study presented at the annual European Congress of Rheumatology.

The data revealed “a striking association between air pollution and increased CRP levels and risk of an arthritis flare,” reported first author Giovanni Adami, MD, DSc, of the rheumatology unit at the University of Verona (Italy).
The excess risk of elevated CRP and flares began “at very low levels of exposure, even those below commonly used thresholds for risk to human health,” he added.
Study details
Researchers collected data on 888 patients with RA from numerous patient visits in the context of more than 13,000 air pollution records. The CRP levels and RA flares were evaluated in the context of air pollution monitoring that is performed on a daily basis at several sites in the city of Verona where the study was conducted. Verona is an industrial city in northern Italy that has high but variable levels of air pollution based on factory activity and weather conditions.
Patients with RA who provided clinical data for this study were matched by their proximity to specific air pollution monitoring sites. By linking CRP levels and disease activity to air pollution levels over multiple follow-up visits, the design allowed the RA study participants “to serve as their own controls,” Dr. Adami explained.
At each patient visit during the study, CRP levels were measured and disease activity assessed. Patients were considered to have elevated CRP when levels were 5 mg/L or higher. The presence of an RA flare was defined by a 1.2-point increase or more in 28-joint Disease Activity Score using CRP (DAS28-CRP).
Both the CRP level and the presence or absence of a flare were evaluated in relationship to the patient’s specific local air pollution levels in the prior 60 days.
Increased levels of CRP, a surrogate for inflammatory activity, and increased disease activity, were both associated with elevated exposure to air pollutants prior to an office visit. These associations remained statistically significant when evaluated by specific air pollutants such as carbon monoxide (CO), nitrogen oxides (NO2, NO), small particulate matter (PM10; particles ≤ 10 mcm), and ozone (O3).
The relationship between increased exposure to air pollution contaminants and elevated CRP was supported by a dose effect. In the case of PM10, for example, the odds ratio of having elevated CRP was increased by only about 25% (OR, 1.25) when mean levels were 30 mcg/m3 or lower in the period prior to the office visit. This rose incrementally for higher mean levels of PM10, reaching 70% (OR, 1.70) for levels > 50 mcg/m3.
The researchers detected statistically significant differences in mean and area-under-the curve (AUC) values of most air pollutants in the 60 days prior to office visits when patients had a flare versus when disease activity was low. For example, the difference in mean and AUC levels in the period prior to a flare relative to a period with low disease activity was significant for CO (P = .001 for both) and NO and NO2 (P = .003 for both), and O3 (P = .002 and P = .001, respectively). For PM10, P values were .011 and .005, respectively.
“Remarkably, we found that the cumulative exposure to NO2 in the 60 days preceding a flare was approximately 500 mcg/m3 higher than the low disease activity visit, an exposure that equates to approximately 200 passively smoked cigarettes,” Dr. Adami reported.
Trying to confirm causality of association
Dr. Adami’s study is not the first study to link air pollution to risk of RA. Several have suggested that air pollution is a risk factor for developing joint disease, but a recently published study conducted in Kuwait associated greater disease activity with NO2 and another air pollutant, sulfur dioxide (SO2), although not CO, PM10, or O3.
A coauthor of that study, which evaluated pollution in regard to disease activity on DAS score, Adeeba Al-Herz, MD, a rheumatology consultant at Al-Amiri Hospital, Kuwait City, said in an interview, “We proved the correlation between them but not the causality.”
However, she believes that this is an important area of inquiry.
“We are working now on another paper in which we studied a causal relationship between the two, meaning that we are evaluating whether SO2 and NO2 trigger RA activity,” Dr. Al-Herz said. That study is now complete, and the manuscript is being written.
The magnitude of the association in these two studies suggest that there might be a clinical message if causality can be confirmed, according to Dr. Adami. Although there are many reasons to seek to reduce and avoid air pollution, these data suggest risk of a proinflammatory state might be one of them.
Dr. Adami believes that the evidence of an adverse effect on patients with RA is strong.
“In order to reduce the burden of RA, public and environmental health policy makers should aim to diminish gaseous and particulate matter emissions to a larger extent than currently recommended,” he said.
In an interview after his presentation, Dr. Adami suggested that the risk of an inflammatory response and increases in arthritis flares from air pollution is not surprising. Previous studies have linked cigarette smoking to both.
“The mechanisms underlying the development of inflammation are very similar. Indeed, the toxic components contained in cigarette smoking are largely shared with diesel exhaust and fossil fuel combustion,” he said.
Although causality between air pollution and arthritis flares cannot be confirmed in these data, a basis for suspecting a causal relationship is supported by “plenty of in vitro and animal studies,” according to Dr. Adami.
On the basis of these studies, several mechanisms have been postulated.
“As an example, exposure to air pollution can promote the activation of the bronchus-associated lymphoid tissue (BALT), which can trigger the activation of the transcription factor nuclear factor-kappaB,” he said. This, in turn, can “lead to the secretion of proinflammatory cytokines, such as tumor necrosis factor–alpha and interleukin-1.”
Another theory is that posttranslational modification of proteins in the lung, a process called citrullination, “can lead to production of autoantibodies known to have a pathogenic role in RA,” he added.
Proving a causal relationship, however, is difficult.
“We certainly cannot conduct a randomized clinical trial on that and voluntarily expose some patients to pollution. Thus, we need to rely on observational data,” Dr. Adami said.
Of strategies being considered to generate evidence of a causal relationship between pollution and the exacerbation of RA, “we certainly will try to study those patients that move from a highly polluted area to a greener zone and vice versa,” he said. This will allow us “to explore what happens when the exposure to pollution changes dramatically in a short period of time.”
In the meantime, “given what is known to date, I would certainly advise my RA patients to avoid exposure to air pollution,” Dr. Adami said. He acknowledged there is no proof that this will help patients to reduce the risk of flares, but there are already many good reasons to minimize exposure to air pollution.
Dr. Adami and Dr. Al-Herz report no potential conflicts of interest.
Pollution appears to trigger inflammation
Pollution appears to trigger inflammation
In patients with rheumatoid arthritis, exposure to air pollution is associated with both elevated levels of C-reactive protein (CRP) and increased risk of arthritis flares, according to a novel longitudinal study presented at the annual European Congress of Rheumatology.
The data revealed “a striking association between air pollution and increased CRP levels and risk of an arthritis flare,” reported first author Giovanni Adami, MD, DSc, of the rheumatology unit at the University of Verona (Italy).
The excess risk of elevated CRP and flares began “at very low levels of exposure, even those below commonly used thresholds for risk to human health,” he added.
Study details
Researchers collected data on 888 patients with RA from numerous patient visits in the context of more than 13,000 air pollution records. The CRP levels and RA flares were evaluated in the context of air pollution monitoring that is performed on a daily basis at several sites in the city of Verona where the study was conducted. Verona is an industrial city in northern Italy that has high but variable levels of air pollution based on factory activity and weather conditions.
Patients with RA who provided clinical data for this study were matched by their proximity to specific air pollution monitoring sites. By linking CRP levels and disease activity to air pollution levels over multiple follow-up visits, the design allowed the RA study participants “to serve as their own controls,” Dr. Adami explained.
At each patient visit during the study, CRP levels were measured and disease activity assessed. Patients were considered to have elevated CRP when levels were 5 mg/L or higher. The presence of an RA flare was defined by a 1.2-point increase or more in 28-joint Disease Activity Score using CRP (DAS28-CRP).
Both the CRP level and the presence or absence of a flare were evaluated in relationship to the patient’s specific local air pollution levels in the prior 60 days.
Increased levels of CRP, a surrogate for inflammatory activity, and increased disease activity, were both associated with elevated exposure to air pollutants prior to an office visit. These associations remained statistically significant when evaluated by specific air pollutants such as carbon monoxide (CO), nitrogen oxides (NO2, NO), small particulate matter (PM10; particles ≤ 10 mcm), and ozone (O3).
The relationship between increased exposure to air pollution contaminants and elevated CRP was supported by a dose effect. In the case of PM10, for example, the odds ratio of having elevated CRP was increased by only about 25% (OR, 1.25) when mean levels were 30 mcg/m3 or lower in the period prior to the office visit. This rose incrementally for higher mean levels of PM10, reaching 70% (OR, 1.70) for levels > 50 mcg/m3.
The researchers detected statistically significant differences in mean and area-under-the curve (AUC) values of most air pollutants in the 60 days prior to office visits when patients had a flare versus when disease activity was low. For example, the difference in mean and AUC levels in the period prior to a flare relative to a period with low disease activity was significant for CO (P = .001 for both) and NO and NO2 (P = .003 for both), and O3 (P = .002 and P = .001, respectively). For PM10, P values were .011 and .005, respectively.
“Remarkably, we found that the cumulative exposure to NO2 in the 60 days preceding a flare was approximately 500 mcg/m3 higher than the low disease activity visit, an exposure that equates to approximately 200 passively smoked cigarettes,” Dr. Adami reported.
Trying to confirm causality of association
Dr. Adami’s study is not the first study to link air pollution to risk of RA. Several have suggested that air pollution is a risk factor for developing joint disease, but a recently published study conducted in Kuwait associated greater disease activity with NO2 and another air pollutant, sulfur dioxide (SO2), although not CO, PM10, or O3.
A coauthor of that study, which evaluated pollution in regard to disease activity on DAS score, Adeeba Al-Herz, MD, a rheumatology consultant at Al-Amiri Hospital, Kuwait City, said in an interview, “We proved the correlation between them but not the causality.”
However, she believes that this is an important area of inquiry.
“We are working now on another paper in which we studied a causal relationship between the two, meaning that we are evaluating whether SO2 and NO2 trigger RA activity,” Dr. Al-Herz said. That study is now complete, and the manuscript is being written.
The magnitude of the association in these two studies suggest that there might be a clinical message if causality can be confirmed, according to Dr. Adami. Although there are many reasons to seek to reduce and avoid air pollution, these data suggest risk of a proinflammatory state might be one of them.
Dr. Adami believes that the evidence of an adverse effect on patients with RA is strong.
“In order to reduce the burden of RA, public and environmental health policy makers should aim to diminish gaseous and particulate matter emissions to a larger extent than currently recommended,” he said.
In an interview after his presentation, Dr. Adami suggested that the risk of an inflammatory response and increases in arthritis flares from air pollution is not surprising. Previous studies have linked cigarette smoking to both.
“The mechanisms underlying the development of inflammation are very similar. Indeed, the toxic components contained in cigarette smoking are largely shared with diesel exhaust and fossil fuel combustion,” he said.
Although causality between air pollution and arthritis flares cannot be confirmed in these data, a basis for suspecting a causal relationship is supported by “plenty of in vitro and animal studies,” according to Dr. Adami.
On the basis of these studies, several mechanisms have been postulated.
“As an example, exposure to air pollution can promote the activation of the bronchus-associated lymphoid tissue (BALT), which can trigger the activation of the transcription factor nuclear factor-kappaB,” he said. This, in turn, can “lead to the secretion of proinflammatory cytokines, such as tumor necrosis factor–alpha and interleukin-1.”
Another theory is that posttranslational modification of proteins in the lung, a process called citrullination, “can lead to production of autoantibodies known to have a pathogenic role in RA,” he added.
Proving a causal relationship, however, is difficult.
“We certainly cannot conduct a randomized clinical trial on that and voluntarily expose some patients to pollution. Thus, we need to rely on observational data,” Dr. Adami said.
Of strategies being considered to generate evidence of a causal relationship between pollution and the exacerbation of RA, “we certainly will try to study those patients that move from a highly polluted area to a greener zone and vice versa,” he said. This will allow us “to explore what happens when the exposure to pollution changes dramatically in a short period of time.”
In the meantime, “given what is known to date, I would certainly advise my RA patients to avoid exposure to air pollution,” Dr. Adami said. He acknowledged there is no proof that this will help patients to reduce the risk of flares, but there are already many good reasons to minimize exposure to air pollution.
Dr. Adami and Dr. Al-Herz report no potential conflicts of interest.
In patients with rheumatoid arthritis, exposure to air pollution is associated with both elevated levels of C-reactive protein (CRP) and increased risk of arthritis flares, according to a novel longitudinal study presented at the annual European Congress of Rheumatology.
The data revealed “a striking association between air pollution and increased CRP levels and risk of an arthritis flare,” reported first author Giovanni Adami, MD, DSc, of the rheumatology unit at the University of Verona (Italy).
The excess risk of elevated CRP and flares began “at very low levels of exposure, even those below commonly used thresholds for risk to human health,” he added.
Study details
Researchers collected data on 888 patients with RA from numerous patient visits in the context of more than 13,000 air pollution records. The CRP levels and RA flares were evaluated in the context of air pollution monitoring that is performed on a daily basis at several sites in the city of Verona where the study was conducted. Verona is an industrial city in northern Italy that has high but variable levels of air pollution based on factory activity and weather conditions.
Patients with RA who provided clinical data for this study were matched by their proximity to specific air pollution monitoring sites. By linking CRP levels and disease activity to air pollution levels over multiple follow-up visits, the design allowed the RA study participants “to serve as their own controls,” Dr. Adami explained.
At each patient visit during the study, CRP levels were measured and disease activity assessed. Patients were considered to have elevated CRP when levels were 5 mg/L or higher. The presence of an RA flare was defined by a 1.2-point increase or more in 28-joint Disease Activity Score using CRP (DAS28-CRP).
Both the CRP level and the presence or absence of a flare were evaluated in relationship to the patient’s specific local air pollution levels in the prior 60 days.
Increased levels of CRP, a surrogate for inflammatory activity, and increased disease activity, were both associated with elevated exposure to air pollutants prior to an office visit. These associations remained statistically significant when evaluated by specific air pollutants such as carbon monoxide (CO), nitrogen oxides (NO2, NO), small particulate matter (PM10; particles ≤ 10 mcm), and ozone (O3).
The relationship between increased exposure to air pollution contaminants and elevated CRP was supported by a dose effect. In the case of PM10, for example, the odds ratio of having elevated CRP was increased by only about 25% (OR, 1.25) when mean levels were 30 mcg/m3 or lower in the period prior to the office visit. This rose incrementally for higher mean levels of PM10, reaching 70% (OR, 1.70) for levels > 50 mcg/m3.
The researchers detected statistically significant differences in mean and area-under-the curve (AUC) values of most air pollutants in the 60 days prior to office visits when patients had a flare versus when disease activity was low. For example, the difference in mean and AUC levels in the period prior to a flare relative to a period with low disease activity was significant for CO (P = .001 for both) and NO and NO2 (P = .003 for both), and O3 (P = .002 and P = .001, respectively). For PM10, P values were .011 and .005, respectively.
“Remarkably, we found that the cumulative exposure to NO2 in the 60 days preceding a flare was approximately 500 mcg/m3 higher than the low disease activity visit, an exposure that equates to approximately 200 passively smoked cigarettes,” Dr. Adami reported.
Trying to confirm causality of association
Dr. Adami’s study is not the first study to link air pollution to risk of RA. Several have suggested that air pollution is a risk factor for developing joint disease, but a recently published study conducted in Kuwait associated greater disease activity with NO2 and another air pollutant, sulfur dioxide (SO2), although not CO, PM10, or O3.
A coauthor of that study, which evaluated pollution in regard to disease activity on DAS score, Adeeba Al-Herz, MD, a rheumatology consultant at Al-Amiri Hospital, Kuwait City, said in an interview, “We proved the correlation between them but not the causality.”
However, she believes that this is an important area of inquiry.
“We are working now on another paper in which we studied a causal relationship between the two, meaning that we are evaluating whether SO2 and NO2 trigger RA activity,” Dr. Al-Herz said. That study is now complete, and the manuscript is being written.
The magnitude of the association in these two studies suggest that there might be a clinical message if causality can be confirmed, according to Dr. Adami. Although there are many reasons to seek to reduce and avoid air pollution, these data suggest risk of a proinflammatory state might be one of them.
Dr. Adami believes that the evidence of an adverse effect on patients with RA is strong.
“In order to reduce the burden of RA, public and environmental health policy makers should aim to diminish gaseous and particulate matter emissions to a larger extent than currently recommended,” he said.
In an interview after his presentation, Dr. Adami suggested that the risk of an inflammatory response and increases in arthritis flares from air pollution is not surprising. Previous studies have linked cigarette smoking to both.
“The mechanisms underlying the development of inflammation are very similar. Indeed, the toxic components contained in cigarette smoking are largely shared with diesel exhaust and fossil fuel combustion,” he said.
Although causality between air pollution and arthritis flares cannot be confirmed in these data, a basis for suspecting a causal relationship is supported by “plenty of in vitro and animal studies,” according to Dr. Adami.
On the basis of these studies, several mechanisms have been postulated.
“As an example, exposure to air pollution can promote the activation of the bronchus-associated lymphoid tissue (BALT), which can trigger the activation of the transcription factor nuclear factor-kappaB,” he said. This, in turn, can “lead to the secretion of proinflammatory cytokines, such as tumor necrosis factor–alpha and interleukin-1.”
Another theory is that posttranslational modification of proteins in the lung, a process called citrullination, “can lead to production of autoantibodies known to have a pathogenic role in RA,” he added.
Proving a causal relationship, however, is difficult.
“We certainly cannot conduct a randomized clinical trial on that and voluntarily expose some patients to pollution. Thus, we need to rely on observational data,” Dr. Adami said.
Of strategies being considered to generate evidence of a causal relationship between pollution and the exacerbation of RA, “we certainly will try to study those patients that move from a highly polluted area to a greener zone and vice versa,” he said. This will allow us “to explore what happens when the exposure to pollution changes dramatically in a short period of time.”
In the meantime, “given what is known to date, I would certainly advise my RA patients to avoid exposure to air pollution,” Dr. Adami said. He acknowledged there is no proof that this will help patients to reduce the risk of flares, but there are already many good reasons to minimize exposure to air pollution.
Dr. Adami and Dr. Al-Herz report no potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Gene variant confirmed as strong predictor of lung disease in RA
Carriers have more than twofold greater risk
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
Carriers have more than twofold greater risk
Carriers have more than twofold greater risk
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
Patients with rheumatoid arthritis who carry a specific allele of the gene MUC5B have about double the risk of developing interstitial lung disease when compared with noncarriers, according to a large Finnish biobank study presented at the annual European Congress of Rheumatology.
“The risk difference [or carriers relative to noncarriers] started at about age 65, with a bigger difference [for] men than women,” reported Antti Palomäki, MD, PhD, of the center for rheumatology and clinical immunology at Turku (Finland) University.
The gain-of-function MUC5B variant, which encodes mucin 5B, was first linked to RA-associated interstitial lung disease (ILD) more than 3 years ago. At that time, it was already a known genetic risk factor for idiopathic pulmonary fibrosis in the general population. The new data confirm the association in a longitudinal analysis of a large biobank and suggest the association might have clinical utility.
“This is not ready for clinical practice at the moment. We do not yet know whether we can change therapy to reduce risk,” Dr. Palomäki said, adding “in the future we can look.”
One question that might be asked in clinical studies using MUC5B as a tool to assess and modify risk of ILD in patients with RA is whether one therapy is better than another in avoiding or delaying development of lung fibrosis. Dr. Palomäki noted that biologics, for example, might be a more favorable choice in patients with RA who are at high risk of developing ILD.
The association of the MUC5B variant with increased ILD incidence in patients with RA was drawn from a data set known as FinnGen, a biobank collection of epidemiologic cohorts and hospital samples with genotypes of about 10% of the Finnish population. Follow-up extends to 46 years in some of these individuals.
When 248,4000 individuals in this data set were evaluated, 5,534 had a diagnosis of RA. Of these, 178 (3.2%) developed ILD. About 20% of both those with and without RA were MUC5B variant carriers, meaning the remainder were not.
Sex and age factor into lifetime risk
In patients with RA, the lifetime rate of ILD among MUC5B variant carriers was 16.8% versus only 6.1% among noncarriers. This finding translated into a hazard ratio for ILD of 2.27 (95% confidence interval, 1.75–2.96) for variant carriers versus noncarriers.
The lifetime rate of ILD in patients with RA was greater in men versus women regardless of carrier status (18.5% vs. 8.5%). For women, the lifetime rate was lower for carriers, although the difference relative to female noncarriers was greater (14.5% vs. 4.7%).
ILD, whether in the general population or in patients with RA, is a disease of advancing age. When Dr. Palomäki showed a graph, the rise in ILD incidence did not start in any population, whether those with or without RA and regardless of carrier status, until about age 55. In those without RA and in noncarriers of the variant, ILD incidence remained low and began a discernible climb at around age 70.
In those who did not have RA but were positive for the variant, the rates rose more than twice as fast, particularly after age 70. In people who had RA but not the variant, the rate of ILD was greater than in patients who carried the variant without RA, starting the climb earlier and rising more steeply with age. In those with RA and the variant, the climb in ILD incidence rose rapidly after age 65 years even though the incidence remained fairly similar between all of these groups at age 60.
Putting the findings into context
The need to develop ways to prevent ILD in RA is urgent. ILD is one of the most common extraarticular manifestations of RA, developing in up to 60% of patients with RA in older age groups when evaluated with imaging, according to Dr. Palomäki. Although it develops into a clinically significant complication in only about 10% of these patients, ILD still is a significant cause of illness and death in elderly patients with RA.
In the 2018 study that first linked the MUC5B variant to RA-ILD, the investigators also found that the variant was associated with an increased likelihood of developing the usual interstitial pneumonia type of ILD on imaging. David Schwartz, MD, professor of medicine, pulmonary sciences, and critical care and chair of the department of medicine at the University of Colorado at Denver, Aurora, was a senior author of that study. He said these findings build on the 2018 study.
“While the gain-of-function MUC5B promoter variant is important in predicting who will develop RA-ILD, these findings also suggest that MUC5B may be involved in the etiology of RA-ILD, at least for those with the MUC5B variant,” he said.
“The study also raises the possibility that there are several subtypes of RA-ILD, and the subtype that is driven by MUC5B may respond differently to RA biologics or therapeutic agents to treat ILD,” he added.
In the discussion following the presentation by Dr. Palomäki, others agreed, with that statement including Dr. Palomäki. He expressed interest in clinical studies comparing different classes of RA therapies for their relative impact on the risk of developing ILD.Dr. Palomäki reported financial relationships with AbbVie, Merck, Pfizer, and Sanofi. Dr. Schwartz is the founder of Eleven P15, which is developing methods for early diagnosis and treatment of pulmonary fibrosis.
FROM THE EULAR 2021 CONGRESS
Nintedanib slows interstitial lung disease in RA patients
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.

“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
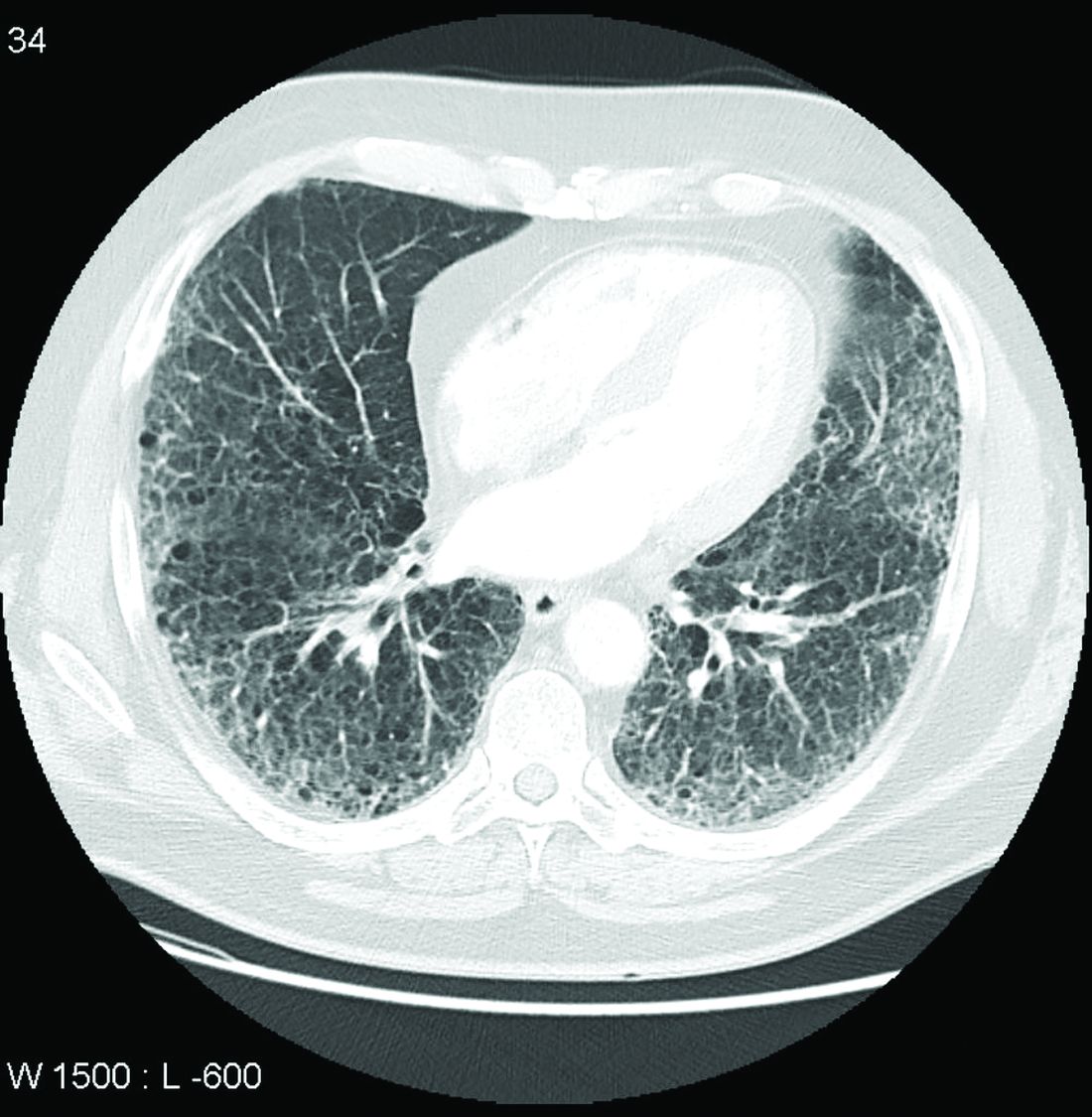
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
Subgroup analysis from INBUILD trial finds results similar to overall study cohort
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.
“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
In a new subgroup analysis of a previously published multinational trial, the preservation of lung function with nintedanib (Ofev) was about the same in patients with interstitial lung disease related to rheumatoid arthritis (RA-ILD) as it was in patients with other etiologies, according to data presented at the annual European Congress of Rheumatology.
“There was no significant heterogeneity across any of several characteristics we evaluated,” reported Clive Kelly, MBBS, of the Institute of Cellular Medicine at Newcastle University (England).
The INBUILD trial, which enrolled more than 600 patients in 15 countries with a range of fibrosing lung diseases, was published almost 2 years ago. On the primary endpoint of rate of decline in forced vital capacity (FVC), the medians were –80.8 mL per year among those randomized to nintedanib and –187.8 mL per year (P < .001) on placebo.
The INBUILD study provided evidence that fibrosing lung diseases have a common pathobiologic mechanism that can be slowed by targeting intracellular kinases. Nintedanib inhibits several growth factor receptors as well as nonreceptor tyrosine kinases, but its exact mechanism for slowing fibrosing lung diseases remains unclear. Initially approved for, nintedanib received approvals from the FDA for systemic sclerosis–associated ILD in 2019 and for chronic fibrosing ILD with progressive phenotypes in 2020 after being initially approved for the treatment of idiopathic pulmonary fibrosis in 2014.
When asked for comment, Paul F. Dellaripa, MD, an associate professor of medicine in the division of rheumatology, immunology, and allergy at Harvard Medical School, Boston, indicated these data are helpful in considering strategies for RA patients with ILD, but he encouraged collaboration between joint and lung specialists.
“Antifibrotic agents for patients with progressive ILD in autoimmune diseases like RA is a welcome addition to our care of this challenging complication,” said Dr. Dellaripa, who has published frequently on the diagnosis and treatment of lung diseases associated with RA. Yet, treatment must be individualized, he added.
“It will be incumbent for rheumatologists to incorporate lung health as a critical part of patient care and work closely with pulmonologists to consider when to institute antifibrotic therapy in patients with ILD,” he said.
Details of subanalysis
In the RA-ILD subpopulation of 89 patients, there was no further decline in FVC from 24 weeks after randomization to the end of 52 weeks for those on nintedanib, but the decline remained steady over the full course of follow-up among those in the placebo group. At 52 weeks, the decline in the placebo group reached –200 mL at the end of 52 weeks. As a result, the between-group relative reduction in FVC at 52 weeks of 116.7 mL favoring nintedanib over placebo (P < .037) slightly exceeded the 107-mL reduction (P < .001) observed in the overall INBUILD study population.
Among other subgroups the investigators evaluated, outcomes with nintedanib did not differ when patients were split into groups with higher or lower baseline levels of high-sensitivity C-reactive protein, regardless of whether the groups were defined by levels above and below 1 mg/L or 3 mg/L. The same was true for those who were taking nonbiologic disease-modifying antirheumatic drugs or glucocorticoids.
However, for these latter analyses, Dr. Kelly conceded that the differences were based on small numbers of patients and so cannot be considered conclusive.
The adverse event most closely associated with nintedanib in the RA-ILD population was diarrhea, just as in the overall study, and it was more than twice as frequent in the RA-ILD patients receiving the active therapy, compared with placebo (54.8% vs. 25.5%). Nausea was also more common (21.4% vs. 10.6%), and so was decreased appetite (11.9% vs. 2.1%) and weight reduction (9.5% vs. 2.1%).
Lung-related adverse events, such as bronchiolitis (21.4% vs. 17.0%) and dyspnea (11.9% vs. 10.6%), were only slightly more frequent in the nintedanib group. Nasopharyngitis (7.1% vs. 12.8%) was less common. Side effects leading to treatment discontinuation were higher on nintedanib (19.0% vs. 12.8%)
The RA-ILD subgroup represented 13.4% of those randomized in INBUILD. The mean time since diagnosis of RA was about 10 years. More than 60% were smokers or former smokers. At baseline, the mean FVC of predicted was 71%. More than 85% had a usual interstitial pneumonia (UIP) radiologic pattern.
Acute exacerbations and death were not evaluated in the RA-ILD subpopulation, but these were secondary endpoints in the published INBUILD study according to the presence or absence of a UIP-like fibrotic pattern. For the combined endpoint of acute exacerbation of ILD or death, the protection associated with nintedanib approached statistical significance for the population overall (odds ratio, 0.68; 95% confidence interval, 0.46-1.01) and reached significance for those with a UIP pattern (OR, 0.61; 95% CI, 0.38-0.98).
Nintedanib led to lower death rates at 52 weeks in the overall population (8.1% vs. 11.5% with placebo) and in the group with a UIP pattern (9.7% vs. 15.0% with placebo).
Dr. Kelly has financial relationships with multiple pharmaceutical companies, including Boehringer Ingelheim, which provided funding for INBUILD and this subpopulation analysis. Dr. Dellaripa reported financial relationships with Bristol-Myers Squibb and Genentech.
FROM THE EULAR 2021 CONGRESS
Intravenous immunoglobulin controls dermatomyositis in phase 3 trial
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
Nearly 50% achieve moderate improvement or better
Nearly 50% achieve moderate improvement or better
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
The first multinational, phase 3, placebo-controlled trial conducted with intravenous immunoglobulin therapy (IVIg) for dermatomyositis has confirmed significant efficacy and acceptable safety, according to data presented at the opening plenary abstract session of the annual European Congress of Rheumatology.
At the week 16 evaluation of the trial, called ProDERM, the response rates were 78.7% and 43.8% (P = .0008) for active therapy and placebo, respectively, reported Rohit Aggarwal, MD, medical director of the Arthritis and Autoimmunity Center at the University of Pittsburgh.
ProDERM is a “much-awaited study,” according to session moderator Hendrik Schulze-Koops, MD, PhD, of the division of rheumatology and clinical immunology at Ludwig Maximilian University of Munich (Germany). He was not involved in the study.
“We all have been doing what we have been doing,” Dr. Schulze-Koops said, referring to the use of IVIg for the control of dermatomyositis, “but we had no evidence for support.”
This statement could apply not only to IVIg, which has long been listed among treatment options by the Myositis Association despite the absence of controlled studies, but also to most immunosuppressive therapies and other options used for this challenging disease.
The proprietary IVIg employed in this study, Octagam 10%, has been approved in the United States for the treatment of chronic immune thrombocytopenic purpura. Its manufacturer, Octagam, plans to file a supplemental new drug application with the Food and Drug Administration for the treatment of dermatomyositis. The agent is already approved for dermatomyositis by the European Medicines Agency, according to Dr. Aggarwal.
Multiple response criteria favor IVIg
In the trial, 95 patients with dermatomyositis were randomized to 2 g/kg of IVIg (Octagam 10%) or placebo administered every 4 weeks. In a subsequent open-label extension study in which patients on placebo were switched to active therapy, the same every-4-week treatment schedule was used. The patients’ mean age was 53; 75% were women, and 92% were White.
The primary endpoint was at least minimal improvement on 2016 ACR/EULAR (American College of Rheumatology/European Alliance of Associations for Rheumatology) myositis response criteria, defined as a 20-point or greater gain in the Total Improvement Score (TIS) and no clinical worsening at two consecutive visits. But IVIg also provided a large relative benefit over placebo using more rigorous definitions of improvement. For moderate improvement, defined as at least a 40-point TIS improvement, there was a 45.2% relative advantage for IVIg over placebo (68.1% vs. 22.9%; P < .0001). For major improvement, defined as at least a 60-point TIS improvement, the relative advantage was 23.6% (31.9% vs. 8.3%; P < .0062).
At 16 weeks, the mean TIS score was more than twice as high in those receiving IVIg than in those randomized to placebo (48.4 vs. 21.6). At that point, an open-label extension was initiated. Those in the IVIg group were permitted to remain on therapy for an additional 24 weeks if they had not worsened in the blinded phase.
The mean TIS score in the IVIg group continued to rise during the extension phase. By 12 weeks in this phase, it reached 54.0. Over the same period, mean TIS scores climbed steeply among the placebo-treated patients who had switched to active therapy, reaching 44.4.
At the end of 24 weeks of the extension trial, when patients initiated on IVIg had been on active therapy for 40 weeks, the mean TIS score advantage of starting on IVIg rather than placebo was relatively modest (55.4 vs. 51.1).
Benefit is significant for skin and muscle
Changes in the two major components of dermatomyositis were tracked individually. For skin symptoms, patients were evaluated with the Cutaneous Dermatomyositis Disease Areas and Severity Index (CDASI). For muscle involvement, symptoms were evaluated with the 8-item Manual Muscle Testing (MMT-8) tool.
“The effects of IVIg on the muscle and the skin were both highly statistically significant,” Dr. Aggarwal reported. He said the CDASI score was reduced by almost half at the end of 16 weeks among those treated with IVIg relative to those treated with placebo. Improvement in MMT-8 scores were also clinically as well as statistically significant.
The IVIg therapy was well tolerated. The most common adverse effects in this study, like those reported with IVIg when used to treat other diseases, were headache, pyrexia, and nausea, but Dr. Aggarwal reported that these were generally mild.
Serious adverse events, particularly thromboembolism, did occur over the course of the study, but the rate of events was only slightly higher in the group receiving active therapy (5.8% vs. 4.2%).
Patients who entered the study were permitted to remain on most immunosuppressive therapies, such as methotrexate, mycophenolate, tacrolimus, and glucocorticoids. Dr. Aggarwal said that the majority of patients were taking a glucocorticoid and at least one nonglucocorticoid immunosuppressant.
Effect on associated conditions is planned
The data from this trial have not yet been analyzed for the impact of IVIg on conditions that occur frequently in association with dermatomyositis, such as interstitial lung disease (ILD) and dysphagia, but Dr. Aggarwal reported that there are plans to do so. Although severe ILD was a trial exclusion, the presence of mild to moderate ILD and dysphagia were evaluated at baseline, so the impact of treatment can be assessed.
There are also plans to evaluate how the presence or absence of myositis-specific antibodies, which were also evaluated at baseline, affected response to IVIg.
Dr. Aggarwal has financial relationships with more than 15 pharmaceutical companies, including Octapharma, which provided financial support for this trial. Dr. Schulze-Koops reported no relevant potential conflicts of interest.
FROM THE EULAR 2021 CONGRESS
Benefit from cooling temps for cardiac arrest does not differ in randomized trial
The first randomized controlled trial to compare specific temperatures for therapeutic hypothermia in comatose survivors of out-of-hospital cardiac arrest showed no differences in major outcomes, according to a single-center, double-blind study.

In the CAPITAL-CHILL trial, cooling temperatures of 31° C and 34° C were compared to explore the hypothesis that a lower temperature would improve major outcomes, explained Michel Le May, MD.
No differences for the primary composite outcome of all-cause mortality or poor neurologic outcome at 180 days were observed, he reported at the annual scientific sessions of the American College of Cardiology.
The study was completed over a period of almost 7 years in patients presumed to have had an out-of-hospital cardiac arrest and who were unconscious when they reached a center affiliated with the Ottawa Heart Institute, where Dr. Le May directs the regional STEMI (ST-elevation myocardial infarction) program. The initial rhythm at the time of the cardiac arrest was not an entry criterion.
Of 389 patients enrolled, the intention-to-treat analysis included 184 randomized to a cooling temperature of 31° C group and 183 to a temperature of 34° C. The assigned target temperature, reached with an endovascular device, was known only by the managing nurses.
31° C and 34° C are equivalent
There was a small numerical disadvantage for the lower temperature assignment, but none reached statistical significance. This was true of the primary outcome (48.4% vs. 45.4% for the higher temperature) and its components of mortality (43.5% vs. 41.0%) and poor neurologic outcome (4.9% vs. 4.4%). Poor neurologic outcome was defined as a Disability Rating Scale score of greater than 5.
Deaths were most common in the early part of the 180-day follow-up in both arms. On a Kaplan-Meier survival graph, Dr. Le May showed curves that he characterized as “almost superimposable.”
There were no significant differences for any subgroup stratifications, such as age 75 years or older versus younger, males versus females, presence versus absence or an initial shockable rhythm, percutaneous coronary intervention (PCI) within 24 hours versus later, and STEMI versus non-STEMI. In these analyses, the higher temperature was associated with a potential trend for benefit among females and those with a shockable rhythm.
There was no signal for a difference in neurologic outcomes on the Disability Rating Scale or the Modified Rankin Scale. On the latter, for example, 46% of those in the 31° C group and 44% of these in the 34° C group had a score of four or greater at the end of follow-up.
The baseline characteristics of the two groups were similar. About 80% were male; the average age was roughly 62 years. More than 80% of the cardiac arrests were witnessed with CPR being administered by bystanders in nearly 70%. Nearly 40% had a STEMI.
Interventions were similar. Almost all patients underwent coronary angiography, of which nearly 60% received a percutaneous coronary intervention. More than 50% received a stent. The time from arrest to randomization was slightly longer in the 31° C group (228 vs. 204 minutes). The time to balloon inflation from arrival at the cardiac center was also slightly longer (73 vs. 60 minutes).
There was a trend for an increased rate of seizures in the 31° C group (12.5% vs. 7.1%; P = .08), but other secondary outcomes, including pneumonia (67.8% vs. 63.4%), renal replacement therapy (9.2% vs. 9.3%), and stroke (4.4% vs. 1.6%), were similar in the 31° C and 34° C groups, respectively.
Bleeding, whether measured by transfusion (19.6% vs. 22.4%) or TIMI major bleed (23.4% vs. 19.7%) were similar in the 31° C and 34° C groups, respectively. Thrombosis, whether measured by stent thrombosis (1.2% vs. 2.2%) or deep venous thrombosis (11.4% vs. 10.9%) were similar in these two groups, respectively.
The length of stay in the cardiac intensive care unit was significantly greater in the 31° C group (10 vs. 7 days; P = .004). Some of this increased length of stay can be attributed to the longer rewarming process required for the greater cooling, according to Dr. Le May, but he acknowledged that it is not clear this provides a full explanation.
More trials like CAPITAL-CHILL needed
The validity of these findings is supported by several strengths of the methodology, according to Jeanne E. Poole, MD, director of the arrhythmia service and electrophysiology laboratory, University of Washington, Seattle. This includes the reliance of an endovascular device, which can accelerate the time to the target temperature and assure the precision with which it is reached and maintained.
Dr. Poole did note that many of the primary and secondary measures, including the rates of stroke, seizures, and major bleeds, even though not significantly different, favored the higher temperature. The slightly longer door-to-balloon times might have been a factor. For the higher rate of pneumonia in the 31° C group, she questioned whether the longer period of ventilation linked to a longer period of rewarming might have been a factor.
However, Dr. Poole praised the CAPITAL-CHILL trial for drawing attention to a group of patients for whom survival rates remain “dismally low.” She indicated that these types of high-level trials are needed to look for strategies to improve outcomes.
Dr. Le May and Dr. Poole report no potential conflicts of interest.
The first randomized controlled trial to compare specific temperatures for therapeutic hypothermia in comatose survivors of out-of-hospital cardiac arrest showed no differences in major outcomes, according to a single-center, double-blind study.
In the CAPITAL-CHILL trial, cooling temperatures of 31° C and 34° C were compared to explore the hypothesis that a lower temperature would improve major outcomes, explained Michel Le May, MD.
No differences for the primary composite outcome of all-cause mortality or poor neurologic outcome at 180 days were observed, he reported at the annual scientific sessions of the American College of Cardiology.
The study was completed over a period of almost 7 years in patients presumed to have had an out-of-hospital cardiac arrest and who were unconscious when they reached a center affiliated with the Ottawa Heart Institute, where Dr. Le May directs the regional STEMI (ST-elevation myocardial infarction) program. The initial rhythm at the time of the cardiac arrest was not an entry criterion.
Of 389 patients enrolled, the intention-to-treat analysis included 184 randomized to a cooling temperature of 31° C group and 183 to a temperature of 34° C. The assigned target temperature, reached with an endovascular device, was known only by the managing nurses.
31° C and 34° C are equivalent
There was a small numerical disadvantage for the lower temperature assignment, but none reached statistical significance. This was true of the primary outcome (48.4% vs. 45.4% for the higher temperature) and its components of mortality (43.5% vs. 41.0%) and poor neurologic outcome (4.9% vs. 4.4%). Poor neurologic outcome was defined as a Disability Rating Scale score of greater than 5.
Deaths were most common in the early part of the 180-day follow-up in both arms. On a Kaplan-Meier survival graph, Dr. Le May showed curves that he characterized as “almost superimposable.”
There were no significant differences for any subgroup stratifications, such as age 75 years or older versus younger, males versus females, presence versus absence or an initial shockable rhythm, percutaneous coronary intervention (PCI) within 24 hours versus later, and STEMI versus non-STEMI. In these analyses, the higher temperature was associated with a potential trend for benefit among females and those with a shockable rhythm.
There was no signal for a difference in neurologic outcomes on the Disability Rating Scale or the Modified Rankin Scale. On the latter, for example, 46% of those in the 31° C group and 44% of these in the 34° C group had a score of four or greater at the end of follow-up.
The baseline characteristics of the two groups were similar. About 80% were male; the average age was roughly 62 years. More than 80% of the cardiac arrests were witnessed with CPR being administered by bystanders in nearly 70%. Nearly 40% had a STEMI.
Interventions were similar. Almost all patients underwent coronary angiography, of which nearly 60% received a percutaneous coronary intervention. More than 50% received a stent. The time from arrest to randomization was slightly longer in the 31° C group (228 vs. 204 minutes). The time to balloon inflation from arrival at the cardiac center was also slightly longer (73 vs. 60 minutes).
There was a trend for an increased rate of seizures in the 31° C group (12.5% vs. 7.1%; P = .08), but other secondary outcomes, including pneumonia (67.8% vs. 63.4%), renal replacement therapy (9.2% vs. 9.3%), and stroke (4.4% vs. 1.6%), were similar in the 31° C and 34° C groups, respectively.
Bleeding, whether measured by transfusion (19.6% vs. 22.4%) or TIMI major bleed (23.4% vs. 19.7%) were similar in the 31° C and 34° C groups, respectively. Thrombosis, whether measured by stent thrombosis (1.2% vs. 2.2%) or deep venous thrombosis (11.4% vs. 10.9%) were similar in these two groups, respectively.
The length of stay in the cardiac intensive care unit was significantly greater in the 31° C group (10 vs. 7 days; P = .004). Some of this increased length of stay can be attributed to the longer rewarming process required for the greater cooling, according to Dr. Le May, but he acknowledged that it is not clear this provides a full explanation.
More trials like CAPITAL-CHILL needed
The validity of these findings is supported by several strengths of the methodology, according to Jeanne E. Poole, MD, director of the arrhythmia service and electrophysiology laboratory, University of Washington, Seattle. This includes the reliance of an endovascular device, which can accelerate the time to the target temperature and assure the precision with which it is reached and maintained.
Dr. Poole did note that many of the primary and secondary measures, including the rates of stroke, seizures, and major bleeds, even though not significantly different, favored the higher temperature. The slightly longer door-to-balloon times might have been a factor. For the higher rate of pneumonia in the 31° C group, she questioned whether the longer period of ventilation linked to a longer period of rewarming might have been a factor.
However, Dr. Poole praised the CAPITAL-CHILL trial for drawing attention to a group of patients for whom survival rates remain “dismally low.” She indicated that these types of high-level trials are needed to look for strategies to improve outcomes.
Dr. Le May and Dr. Poole report no potential conflicts of interest.
The first randomized controlled trial to compare specific temperatures for therapeutic hypothermia in comatose survivors of out-of-hospital cardiac arrest showed no differences in major outcomes, according to a single-center, double-blind study.
In the CAPITAL-CHILL trial, cooling temperatures of 31° C and 34° C were compared to explore the hypothesis that a lower temperature would improve major outcomes, explained Michel Le May, MD.
No differences for the primary composite outcome of all-cause mortality or poor neurologic outcome at 180 days were observed, he reported at the annual scientific sessions of the American College of Cardiology.
The study was completed over a period of almost 7 years in patients presumed to have had an out-of-hospital cardiac arrest and who were unconscious when they reached a center affiliated with the Ottawa Heart Institute, where Dr. Le May directs the regional STEMI (ST-elevation myocardial infarction) program. The initial rhythm at the time of the cardiac arrest was not an entry criterion.
Of 389 patients enrolled, the intention-to-treat analysis included 184 randomized to a cooling temperature of 31° C group and 183 to a temperature of 34° C. The assigned target temperature, reached with an endovascular device, was known only by the managing nurses.
31° C and 34° C are equivalent
There was a small numerical disadvantage for the lower temperature assignment, but none reached statistical significance. This was true of the primary outcome (48.4% vs. 45.4% for the higher temperature) and its components of mortality (43.5% vs. 41.0%) and poor neurologic outcome (4.9% vs. 4.4%). Poor neurologic outcome was defined as a Disability Rating Scale score of greater than 5.
Deaths were most common in the early part of the 180-day follow-up in both arms. On a Kaplan-Meier survival graph, Dr. Le May showed curves that he characterized as “almost superimposable.”
There were no significant differences for any subgroup stratifications, such as age 75 years or older versus younger, males versus females, presence versus absence or an initial shockable rhythm, percutaneous coronary intervention (PCI) within 24 hours versus later, and STEMI versus non-STEMI. In these analyses, the higher temperature was associated with a potential trend for benefit among females and those with a shockable rhythm.
There was no signal for a difference in neurologic outcomes on the Disability Rating Scale or the Modified Rankin Scale. On the latter, for example, 46% of those in the 31° C group and 44% of these in the 34° C group had a score of four or greater at the end of follow-up.
The baseline characteristics of the two groups were similar. About 80% were male; the average age was roughly 62 years. More than 80% of the cardiac arrests were witnessed with CPR being administered by bystanders in nearly 70%. Nearly 40% had a STEMI.
Interventions were similar. Almost all patients underwent coronary angiography, of which nearly 60% received a percutaneous coronary intervention. More than 50% received a stent. The time from arrest to randomization was slightly longer in the 31° C group (228 vs. 204 minutes). The time to balloon inflation from arrival at the cardiac center was also slightly longer (73 vs. 60 minutes).
There was a trend for an increased rate of seizures in the 31° C group (12.5% vs. 7.1%; P = .08), but other secondary outcomes, including pneumonia (67.8% vs. 63.4%), renal replacement therapy (9.2% vs. 9.3%), and stroke (4.4% vs. 1.6%), were similar in the 31° C and 34° C groups, respectively.
Bleeding, whether measured by transfusion (19.6% vs. 22.4%) or TIMI major bleed (23.4% vs. 19.7%) were similar in the 31° C and 34° C groups, respectively. Thrombosis, whether measured by stent thrombosis (1.2% vs. 2.2%) or deep venous thrombosis (11.4% vs. 10.9%) were similar in these two groups, respectively.
The length of stay in the cardiac intensive care unit was significantly greater in the 31° C group (10 vs. 7 days; P = .004). Some of this increased length of stay can be attributed to the longer rewarming process required for the greater cooling, according to Dr. Le May, but he acknowledged that it is not clear this provides a full explanation.
More trials like CAPITAL-CHILL needed
The validity of these findings is supported by several strengths of the methodology, according to Jeanne E. Poole, MD, director of the arrhythmia service and electrophysiology laboratory, University of Washington, Seattle. This includes the reliance of an endovascular device, which can accelerate the time to the target temperature and assure the precision with which it is reached and maintained.
Dr. Poole did note that many of the primary and secondary measures, including the rates of stroke, seizures, and major bleeds, even though not significantly different, favored the higher temperature. The slightly longer door-to-balloon times might have been a factor. For the higher rate of pneumonia in the 31° C group, she questioned whether the longer period of ventilation linked to a longer period of rewarming might have been a factor.
However, Dr. Poole praised the CAPITAL-CHILL trial for drawing attention to a group of patients for whom survival rates remain “dismally low.” She indicated that these types of high-level trials are needed to look for strategies to improve outcomes.
Dr. Le May and Dr. Poole report no potential conflicts of interest.
FROM ACC 2021
GALACTIC-HF: Novel drug most effective in sickest HFrEF patients
The greatest relative benefit from omecamtiv mecarbil, a member of the novel myotropic drug class that improves cardiac performance, is produced in heart failure patients with the lowest left ventricular ejection fraction (LVEF), a new analysis of the recently published phase 3 GALACTIC-HF trial has found.

The findings reinforce the potential for this drug to be helpful in the management of the most advanced stages of heart failure with reduced ejection fraction (HFrEF), reported John R. Teerlink, MD, director of heart failure at San Francisco Veterans Affairs Medical Center, at the annual scientific sessions of the American College of Cardiology.
The phase 3 multinational GALACTIC-HF trial, published earlier this year, linked omecamtiv mecarbil with an 8% reduction in the risk of a heart failure–related events or cardiovascular death, relative to placebo, which was the primary outcome. For entry, HFrEF patients were required to have a LVEF of 35% or less.
Drilling down on ejection fraction
The new analysis divided participants into quartiles of baseline LVEF and then compared relative outcomes and safety.
In the lowest quartile, defined by a LVEF of 22% or lower, the reduction in risk of events reached 17% (hazard ratio, 0.83; 95% confidence interval, 0.73-0.95) for omecamtiv mecarbil relative to placebo. In the highest, defined by a LVEF of 33% or greater, the benefit fell short of significance (HR 0.99; 95% CI, 0.84-1.16). Across quartiles, LVEF was the “strongest modifier of the treatment effect,” emerging in this analysis as a statistically significant (P = .004) continuous variable.
The comparison by LVEF quartiles also provided an opportunity to show that omecamtiv mecarbil was as safe and well tolerated in those with the most advanced disease as in those less sick. At the lowest levels of LVEF, like the higher levels, omecamtiv mecarbil did not produce any adverse effects on blood pressure, heart rate, potassium homeostasis, or renal function.
In GALACTIC-HF, 8,256 HFrEF patients with LVEF 35% or less were randomized to omecamtiv mecarbil or placebo. The primary composite outcome of hospitalization or urgent visit for heart failure or death from cardiovascular causes was evaluated after a median of 21.8 months on therapy.
When incidence rate per 100 patient years was graphed against the range of LVEF, the relative advantage of omecamtiv mecarbil became visible just below an LVEF of 30%, climbing steadily even to the lowest LVEF, which reached 10%.
Perhaps relevant to the reduction in events, there were also greater relative reductions in NT-proBNP (NT-proB-type natriuretic peptide) for omecamtiv mecarbil at lower relative to higher LVEF. Although omecamtiv mecarbil is not associated with any direct vascular, electrophysiologic, or neurohormonal effects, according to Dr. Teerlink, the indirect effects of selective binding to cardiac myosin has been associated with lower NT-proBNP and other biomarkers of cardiac remodeling in prior clinical studies.
Although Dr. Teerlink acknowledged that relatively few patients in GALACTIC-HF received an angiotensin-receptor neprilysin inhibitor (ARNI) or a sodium glucose cotransporter-2 (SGLT2) inhibitor, he said there is “every reason to believe that omecamtiv mecarbil would be complementary to these therapies.” He said the mechanism of action of omecamtiv mecarbil, which improves systolic function, has no overlap with these drugs.
Importantly, there is a particular need for new treatment options in patients with advanced LVEF, according to Dr. Teerlink, who cited evidence, for example, that “the beneficial effect of [the ARNI] sacubitril valsartan, while still significant, decreases in patients with LVEF less than 35%.”
Overall, based on these results, “we believe that omecamtiv mecarbil represents a novel therapy that holds the promise of improving clinical outcomes in patients with severely reduced ejection fraction, which are the very patients that are most challenging for us to treat,” Dr. Teerlink said.
Omecamtiv mecarbil may ‘buy you some time’
Ileana Piña, MD, clinical professor of medicine, Central Michigan University, Mount Pleasant, Mich., agreed. She said that omecamtiv mecarbil, if approved, will be an option for the type of HFrEF patients who are being considered for heart transplant or mechanical-assist devices.

“We are very loath to use inotropes in this population, because we know that ultimately the inotrope is not going to do well,” said Dr. Piña, calling these therapies a “Band-Aid.” Based on the evidence from GALACTIC-HF, she thinks that omecamtiv mecarbil will be more versatile.
“This drug does not increase myocardial oxygen demand as do the inotropes, and it can be given in the outpatient setting if need be, so I see this as a real advance,” Dr. Piña said. Although Dr. Piña acknowledged that omecamtiv mecarbil did not reduce mortality in the GALACTIC-HF trial, “at least it will buy you some time.”
Dr. Teerlink has financial relationships with multiple pharmaceutical companies, including Amgen, Cytogenetics, and Servier, which provided funding for the GALACTIC-HF trial. Dr. Piña reports no potential conflicts of interest.
The greatest relative benefit from omecamtiv mecarbil, a member of the novel myotropic drug class that improves cardiac performance, is produced in heart failure patients with the lowest left ventricular ejection fraction (LVEF), a new analysis of the recently published phase 3 GALACTIC-HF trial has found.
The findings reinforce the potential for this drug to be helpful in the management of the most advanced stages of heart failure with reduced ejection fraction (HFrEF), reported John R. Teerlink, MD, director of heart failure at San Francisco Veterans Affairs Medical Center, at the annual scientific sessions of the American College of Cardiology.
The phase 3 multinational GALACTIC-HF trial, published earlier this year, linked omecamtiv mecarbil with an 8% reduction in the risk of a heart failure–related events or cardiovascular death, relative to placebo, which was the primary outcome. For entry, HFrEF patients were required to have a LVEF of 35% or less.
Drilling down on ejection fraction
The new analysis divided participants into quartiles of baseline LVEF and then compared relative outcomes and safety.
In the lowest quartile, defined by a LVEF of 22% or lower, the reduction in risk of events reached 17% (hazard ratio, 0.83; 95% confidence interval, 0.73-0.95) for omecamtiv mecarbil relative to placebo. In the highest, defined by a LVEF of 33% or greater, the benefit fell short of significance (HR 0.99; 95% CI, 0.84-1.16). Across quartiles, LVEF was the “strongest modifier of the treatment effect,” emerging in this analysis as a statistically significant (P = .004) continuous variable.
The comparison by LVEF quartiles also provided an opportunity to show that omecamtiv mecarbil was as safe and well tolerated in those with the most advanced disease as in those less sick. At the lowest levels of LVEF, like the higher levels, omecamtiv mecarbil did not produce any adverse effects on blood pressure, heart rate, potassium homeostasis, or renal function.
In GALACTIC-HF, 8,256 HFrEF patients with LVEF 35% or less were randomized to omecamtiv mecarbil or placebo. The primary composite outcome of hospitalization or urgent visit for heart failure or death from cardiovascular causes was evaluated after a median of 21.8 months on therapy.
When incidence rate per 100 patient years was graphed against the range of LVEF, the relative advantage of omecamtiv mecarbil became visible just below an LVEF of 30%, climbing steadily even to the lowest LVEF, which reached 10%.
Perhaps relevant to the reduction in events, there were also greater relative reductions in NT-proBNP (NT-proB-type natriuretic peptide) for omecamtiv mecarbil at lower relative to higher LVEF. Although omecamtiv mecarbil is not associated with any direct vascular, electrophysiologic, or neurohormonal effects, according to Dr. Teerlink, the indirect effects of selective binding to cardiac myosin has been associated with lower NT-proBNP and other biomarkers of cardiac remodeling in prior clinical studies.
Although Dr. Teerlink acknowledged that relatively few patients in GALACTIC-HF received an angiotensin-receptor neprilysin inhibitor (ARNI) or a sodium glucose cotransporter-2 (SGLT2) inhibitor, he said there is “every reason to believe that omecamtiv mecarbil would be complementary to these therapies.” He said the mechanism of action of omecamtiv mecarbil, which improves systolic function, has no overlap with these drugs.
Importantly, there is a particular need for new treatment options in patients with advanced LVEF, according to Dr. Teerlink, who cited evidence, for example, that “the beneficial effect of [the ARNI] sacubitril valsartan, while still significant, decreases in patients with LVEF less than 35%.”
Overall, based on these results, “we believe that omecamtiv mecarbil represents a novel therapy that holds the promise of improving clinical outcomes in patients with severely reduced ejection fraction, which are the very patients that are most challenging for us to treat,” Dr. Teerlink said.
Omecamtiv mecarbil may ‘buy you some time’
Ileana Piña, MD, clinical professor of medicine, Central Michigan University, Mount Pleasant, Mich., agreed. She said that omecamtiv mecarbil, if approved, will be an option for the type of HFrEF patients who are being considered for heart transplant or mechanical-assist devices.
“We are very loath to use inotropes in this population, because we know that ultimately the inotrope is not going to do well,” said Dr. Piña, calling these therapies a “Band-Aid.” Based on the evidence from GALACTIC-HF, she thinks that omecamtiv mecarbil will be more versatile.
“This drug does not increase myocardial oxygen demand as do the inotropes, and it can be given in the outpatient setting if need be, so I see this as a real advance,” Dr. Piña said. Although Dr. Piña acknowledged that omecamtiv mecarbil did not reduce mortality in the GALACTIC-HF trial, “at least it will buy you some time.”
Dr. Teerlink has financial relationships with multiple pharmaceutical companies, including Amgen, Cytogenetics, and Servier, which provided funding for the GALACTIC-HF trial. Dr. Piña reports no potential conflicts of interest.
The greatest relative benefit from omecamtiv mecarbil, a member of the novel myotropic drug class that improves cardiac performance, is produced in heart failure patients with the lowest left ventricular ejection fraction (LVEF), a new analysis of the recently published phase 3 GALACTIC-HF trial has found.
The findings reinforce the potential for this drug to be helpful in the management of the most advanced stages of heart failure with reduced ejection fraction (HFrEF), reported John R. Teerlink, MD, director of heart failure at San Francisco Veterans Affairs Medical Center, at the annual scientific sessions of the American College of Cardiology.
The phase 3 multinational GALACTIC-HF trial, published earlier this year, linked omecamtiv mecarbil with an 8% reduction in the risk of a heart failure–related events or cardiovascular death, relative to placebo, which was the primary outcome. For entry, HFrEF patients were required to have a LVEF of 35% or less.
Drilling down on ejection fraction
The new analysis divided participants into quartiles of baseline LVEF and then compared relative outcomes and safety.
In the lowest quartile, defined by a LVEF of 22% or lower, the reduction in risk of events reached 17% (hazard ratio, 0.83; 95% confidence interval, 0.73-0.95) for omecamtiv mecarbil relative to placebo. In the highest, defined by a LVEF of 33% or greater, the benefit fell short of significance (HR 0.99; 95% CI, 0.84-1.16). Across quartiles, LVEF was the “strongest modifier of the treatment effect,” emerging in this analysis as a statistically significant (P = .004) continuous variable.
The comparison by LVEF quartiles also provided an opportunity to show that omecamtiv mecarbil was as safe and well tolerated in those with the most advanced disease as in those less sick. At the lowest levels of LVEF, like the higher levels, omecamtiv mecarbil did not produce any adverse effects on blood pressure, heart rate, potassium homeostasis, or renal function.
In GALACTIC-HF, 8,256 HFrEF patients with LVEF 35% or less were randomized to omecamtiv mecarbil or placebo. The primary composite outcome of hospitalization or urgent visit for heart failure or death from cardiovascular causes was evaluated after a median of 21.8 months on therapy.
When incidence rate per 100 patient years was graphed against the range of LVEF, the relative advantage of omecamtiv mecarbil became visible just below an LVEF of 30%, climbing steadily even to the lowest LVEF, which reached 10%.
Perhaps relevant to the reduction in events, there were also greater relative reductions in NT-proBNP (NT-proB-type natriuretic peptide) for omecamtiv mecarbil at lower relative to higher LVEF. Although omecamtiv mecarbil is not associated with any direct vascular, electrophysiologic, or neurohormonal effects, according to Dr. Teerlink, the indirect effects of selective binding to cardiac myosin has been associated with lower NT-proBNP and other biomarkers of cardiac remodeling in prior clinical studies.
Although Dr. Teerlink acknowledged that relatively few patients in GALACTIC-HF received an angiotensin-receptor neprilysin inhibitor (ARNI) or a sodium glucose cotransporter-2 (SGLT2) inhibitor, he said there is “every reason to believe that omecamtiv mecarbil would be complementary to these therapies.” He said the mechanism of action of omecamtiv mecarbil, which improves systolic function, has no overlap with these drugs.
Importantly, there is a particular need for new treatment options in patients with advanced LVEF, according to Dr. Teerlink, who cited evidence, for example, that “the beneficial effect of [the ARNI] sacubitril valsartan, while still significant, decreases in patients with LVEF less than 35%.”
Overall, based on these results, “we believe that omecamtiv mecarbil represents a novel therapy that holds the promise of improving clinical outcomes in patients with severely reduced ejection fraction, which are the very patients that are most challenging for us to treat,” Dr. Teerlink said.
Omecamtiv mecarbil may ‘buy you some time’
Ileana Piña, MD, clinical professor of medicine, Central Michigan University, Mount Pleasant, Mich., agreed. She said that omecamtiv mecarbil, if approved, will be an option for the type of HFrEF patients who are being considered for heart transplant or mechanical-assist devices.
“We are very loath to use inotropes in this population, because we know that ultimately the inotrope is not going to do well,” said Dr. Piña, calling these therapies a “Band-Aid.” Based on the evidence from GALACTIC-HF, she thinks that omecamtiv mecarbil will be more versatile.
“This drug does not increase myocardial oxygen demand as do the inotropes, and it can be given in the outpatient setting if need be, so I see this as a real advance,” Dr. Piña said. Although Dr. Piña acknowledged that omecamtiv mecarbil did not reduce mortality in the GALACTIC-HF trial, “at least it will buy you some time.”
Dr. Teerlink has financial relationships with multiple pharmaceutical companies, including Amgen, Cytogenetics, and Servier, which provided funding for the GALACTIC-HF trial. Dr. Piña reports no potential conflicts of interest.
FROM ACC 2021
SAFE-PAD: Endovascular paclitaxel-coated devices exonerated in real-world analysis
A cohort analysis using advanced strategies to minimize the impact of confounders has concluded that the current Food and Drug Administration warning about paclitaxel-coated devices used for femoropopliteal endovascular treatment should be lifted, according to investigators of a study called SAFE-PAD.

In early 2019, an FDA letter to clinicians warned that endovascular stents and balloons coated with paclitaxel might increase mortality, recounted the principal investigator of SAFE-PAD, Eric A. Secemsky, MD, director of vascular intervention, Beth Israel Deaconess Hospital, Boston.
An FDA advisory committee that was subsequently convened in 2019 did not elect to remove these devices from the market, but it did call for restrictions and for the collection of more safety data. In the absence of a clear mechanism of risk, and in the context of perceived problems with data suggesting harm, Dr. Secemsky said that there was interest in a conclusive answer.
The problem was that a randomized controlled trial, even if funding were available, was considered impractical, he noted in presenting SAFE-PAD at the annual scientific sessions of the American College of Cardiology.
In the initial meta-analysis that suggested an increased mortality risk, no risk was seen in the first year after exposure, and it climbed to only 3.5% after 2 years. As a result, the definitive 2-year study with sufficient power to produce conclusive results was an estimated 40,000 patients. Even if extended to 5 years, 20,000 patients would be needed, according to Dr. Secemsky.
SAFE-PAD born of collaboration
An alternative solution was required, which is why “we became engaged with the FDA to design a real-world study for use in making a regulatory decision,” Dr. Secemsky said.
SAFE-PAD, designed with feedback from the FDA, employed sophisticated methodologies to account for known and unknown confounding in the Medicare cohort data used for this study.
Of 168,553 Medicare fee-for-service patients undergoing femoropopliteal artery revascularization with a stent, a balloon, or both at 2,978 institutions, 70,584 (42%) were treated with a paclitaxel drug-coated device (DCD) and the remainder were managed with a non–drug-coated device (NDCD).
The groups were compared with a primary outcome of all-cause mortality in a design to evaluate DCD for noninferiority. Several secondary outcomes, such as repeated lower extremity revascularization, were also evaluated.
To create balanced groups, inverse probability of treatment weighting (IPTW) blinded to outcome was the primary analytic strategy. In addition, several sensitivity analyses were applied, including a technique that tests for the impact of a hypothetical variable that allows adjustment for an unknown confounder.
After a median follow-up of 2.7 years (longest more than 5 years), the cumulative mortality after weighting was 53.8% in the DCD group and 55.1% in the NDCD group. The 5% advantage for the DCD group (hazard ratio, 0.95; 95% confidence interval, 0.94-0.97) ensured noninferiority (P < .001).
On unweighted analysis, the mortality difference favoring DCD was even greater (HR, 0.85; 95% CI, 0.82–0.85).
None of the sensitivity analyses – including a multivariable Cox regression analysis, an instrumental variable analysis, and a falsification endpoints analysis that employed myocardial infarction, pneumonia, and heart failure – altered the conclusion. The hypothetical variable analysis produced the same result.
“A missing confounder would need to be more prevalent and more strongly associated to outcome than any measured variable in this analysis,” reported Dr. Secemsky, indicating that this ruled out essentially any probability of this occurring.
A subgroup analysis told the same story. By hazard ratio for the outcome of mortality, DCD was consistently favored over NDCD for groups characterized by low risk (HR, 0.98), stent implantation (HR, 0.97), receipt of balloon angioplasty alone (HR, 0.94), having critical limb ischemia (HR, 0.95) or no critical limb ischemia (HR, 0.97), and being managed inpatient (HR, 0.97) or outpatient (HR, 0.95).
The results of SAFE-PAD were simultaneously published with Dr. Secemsky’s ACC presentation.
Value of revascularization questioned
In an accompanying editorial, the coauthors Rita F. Redberg, MD, of the University of California, San Francisco, and Mary M. McDermott, MD, of Northwestern University, Chicago, reiterated the findings and the conclusions, but used the forum to draw attention to the low survival rates.

“Thus, while this well-done observational study provides new information,” they wrote, “a major conclusion should be that mortality is high among Medicare beneficiaries undergoing revascularization [for peripheral artery disease] with any devices.”
‘Very impressive’ methods
Marc P. Bonaca, MD, director of vascular research, University of Colorado at Denver, Aurora, called the methods to ensure the validity of the conclusions of this study “very impressive.” In situations where prospective randomized trials are impractical, he suggested that this type of approach might answer an unmet need.

“We have always desired the ability to look at these large datasets with a lot of power to answer important questions,” he said. While “the issue has always been residual confounding,” he expressed interest in further verifications that this type of methodology can serve as a template for data analysis to guide other regulatory decisions.
Dr. Secemsky reports financial relationships with Abbott, Bayer, Boston Scientific, Cook, CSI, Inari, Janssen, Medtronic, and Phillips. Dr. Redford reports no potential conflicts of interest. Dr. McDermott reports a financial relationship with Regeneron. Dr. Bonaca reports financial relationships with Amgen, AstraZeneca, Bayer, Janssen Merck, Novo Nordisk, Pfizer, and Sanofi.
A cohort analysis using advanced strategies to minimize the impact of confounders has concluded that the current Food and Drug Administration warning about paclitaxel-coated devices used for femoropopliteal endovascular treatment should be lifted, according to investigators of a study called SAFE-PAD.
In early 2019, an FDA letter to clinicians warned that endovascular stents and balloons coated with paclitaxel might increase mortality, recounted the principal investigator of SAFE-PAD, Eric A. Secemsky, MD, director of vascular intervention, Beth Israel Deaconess Hospital, Boston.
An FDA advisory committee that was subsequently convened in 2019 did not elect to remove these devices from the market, but it did call for restrictions and for the collection of more safety data. In the absence of a clear mechanism of risk, and in the context of perceived problems with data suggesting harm, Dr. Secemsky said that there was interest in a conclusive answer.
The problem was that a randomized controlled trial, even if funding were available, was considered impractical, he noted in presenting SAFE-PAD at the annual scientific sessions of the American College of Cardiology.
In the initial meta-analysis that suggested an increased mortality risk, no risk was seen in the first year after exposure, and it climbed to only 3.5% after 2 years. As a result, the definitive 2-year study with sufficient power to produce conclusive results was an estimated 40,000 patients. Even if extended to 5 years, 20,000 patients would be needed, according to Dr. Secemsky.
SAFE-PAD born of collaboration
An alternative solution was required, which is why “we became engaged with the FDA to design a real-world study for use in making a regulatory decision,” Dr. Secemsky said.
SAFE-PAD, designed with feedback from the FDA, employed sophisticated methodologies to account for known and unknown confounding in the Medicare cohort data used for this study.
Of 168,553 Medicare fee-for-service patients undergoing femoropopliteal artery revascularization with a stent, a balloon, or both at 2,978 institutions, 70,584 (42%) were treated with a paclitaxel drug-coated device (DCD) and the remainder were managed with a non–drug-coated device (NDCD).
The groups were compared with a primary outcome of all-cause mortality in a design to evaluate DCD for noninferiority. Several secondary outcomes, such as repeated lower extremity revascularization, were also evaluated.
To create balanced groups, inverse probability of treatment weighting (IPTW) blinded to outcome was the primary analytic strategy. In addition, several sensitivity analyses were applied, including a technique that tests for the impact of a hypothetical variable that allows adjustment for an unknown confounder.
After a median follow-up of 2.7 years (longest more than 5 years), the cumulative mortality after weighting was 53.8% in the DCD group and 55.1% in the NDCD group. The 5% advantage for the DCD group (hazard ratio, 0.95; 95% confidence interval, 0.94-0.97) ensured noninferiority (P < .001).
On unweighted analysis, the mortality difference favoring DCD was even greater (HR, 0.85; 95% CI, 0.82–0.85).
None of the sensitivity analyses – including a multivariable Cox regression analysis, an instrumental variable analysis, and a falsification endpoints analysis that employed myocardial infarction, pneumonia, and heart failure – altered the conclusion. The hypothetical variable analysis produced the same result.
“A missing confounder would need to be more prevalent and more strongly associated to outcome than any measured variable in this analysis,” reported Dr. Secemsky, indicating that this ruled out essentially any probability of this occurring.
A subgroup analysis told the same story. By hazard ratio for the outcome of mortality, DCD was consistently favored over NDCD for groups characterized by low risk (HR, 0.98), stent implantation (HR, 0.97), receipt of balloon angioplasty alone (HR, 0.94), having critical limb ischemia (HR, 0.95) or no critical limb ischemia (HR, 0.97), and being managed inpatient (HR, 0.97) or outpatient (HR, 0.95).
The results of SAFE-PAD were simultaneously published with Dr. Secemsky’s ACC presentation.
Value of revascularization questioned
In an accompanying editorial, the coauthors Rita F. Redberg, MD, of the University of California, San Francisco, and Mary M. McDermott, MD, of Northwestern University, Chicago, reiterated the findings and the conclusions, but used the forum to draw attention to the low survival rates.
“Thus, while this well-done observational study provides new information,” they wrote, “a major conclusion should be that mortality is high among Medicare beneficiaries undergoing revascularization [for peripheral artery disease] with any devices.”
‘Very impressive’ methods
Marc P. Bonaca, MD, director of vascular research, University of Colorado at Denver, Aurora, called the methods to ensure the validity of the conclusions of this study “very impressive.” In situations where prospective randomized trials are impractical, he suggested that this type of approach might answer an unmet need.
“We have always desired the ability to look at these large datasets with a lot of power to answer important questions,” he said. While “the issue has always been residual confounding,” he expressed interest in further verifications that this type of methodology can serve as a template for data analysis to guide other regulatory decisions.
Dr. Secemsky reports financial relationships with Abbott, Bayer, Boston Scientific, Cook, CSI, Inari, Janssen, Medtronic, and Phillips. Dr. Redford reports no potential conflicts of interest. Dr. McDermott reports a financial relationship with Regeneron. Dr. Bonaca reports financial relationships with Amgen, AstraZeneca, Bayer, Janssen Merck, Novo Nordisk, Pfizer, and Sanofi.
A cohort analysis using advanced strategies to minimize the impact of confounders has concluded that the current Food and Drug Administration warning about paclitaxel-coated devices used for femoropopliteal endovascular treatment should be lifted, according to investigators of a study called SAFE-PAD.
In early 2019, an FDA letter to clinicians warned that endovascular stents and balloons coated with paclitaxel might increase mortality, recounted the principal investigator of SAFE-PAD, Eric A. Secemsky, MD, director of vascular intervention, Beth Israel Deaconess Hospital, Boston.
An FDA advisory committee that was subsequently convened in 2019 did not elect to remove these devices from the market, but it did call for restrictions and for the collection of more safety data. In the absence of a clear mechanism of risk, and in the context of perceived problems with data suggesting harm, Dr. Secemsky said that there was interest in a conclusive answer.
The problem was that a randomized controlled trial, even if funding were available, was considered impractical, he noted in presenting SAFE-PAD at the annual scientific sessions of the American College of Cardiology.
In the initial meta-analysis that suggested an increased mortality risk, no risk was seen in the first year after exposure, and it climbed to only 3.5% after 2 years. As a result, the definitive 2-year study with sufficient power to produce conclusive results was an estimated 40,000 patients. Even if extended to 5 years, 20,000 patients would be needed, according to Dr. Secemsky.
SAFE-PAD born of collaboration
An alternative solution was required, which is why “we became engaged with the FDA to design a real-world study for use in making a regulatory decision,” Dr. Secemsky said.
SAFE-PAD, designed with feedback from the FDA, employed sophisticated methodologies to account for known and unknown confounding in the Medicare cohort data used for this study.
Of 168,553 Medicare fee-for-service patients undergoing femoropopliteal artery revascularization with a stent, a balloon, or both at 2,978 institutions, 70,584 (42%) were treated with a paclitaxel drug-coated device (DCD) and the remainder were managed with a non–drug-coated device (NDCD).
The groups were compared with a primary outcome of all-cause mortality in a design to evaluate DCD for noninferiority. Several secondary outcomes, such as repeated lower extremity revascularization, were also evaluated.
To create balanced groups, inverse probability of treatment weighting (IPTW) blinded to outcome was the primary analytic strategy. In addition, several sensitivity analyses were applied, including a technique that tests for the impact of a hypothetical variable that allows adjustment for an unknown confounder.
After a median follow-up of 2.7 years (longest more than 5 years), the cumulative mortality after weighting was 53.8% in the DCD group and 55.1% in the NDCD group. The 5% advantage for the DCD group (hazard ratio, 0.95; 95% confidence interval, 0.94-0.97) ensured noninferiority (P < .001).
On unweighted analysis, the mortality difference favoring DCD was even greater (HR, 0.85; 95% CI, 0.82–0.85).
None of the sensitivity analyses – including a multivariable Cox regression analysis, an instrumental variable analysis, and a falsification endpoints analysis that employed myocardial infarction, pneumonia, and heart failure – altered the conclusion. The hypothetical variable analysis produced the same result.
“A missing confounder would need to be more prevalent and more strongly associated to outcome than any measured variable in this analysis,” reported Dr. Secemsky, indicating that this ruled out essentially any probability of this occurring.
A subgroup analysis told the same story. By hazard ratio for the outcome of mortality, DCD was consistently favored over NDCD for groups characterized by low risk (HR, 0.98), stent implantation (HR, 0.97), receipt of balloon angioplasty alone (HR, 0.94), having critical limb ischemia (HR, 0.95) or no critical limb ischemia (HR, 0.97), and being managed inpatient (HR, 0.97) or outpatient (HR, 0.95).
The results of SAFE-PAD were simultaneously published with Dr. Secemsky’s ACC presentation.
Value of revascularization questioned
In an accompanying editorial, the coauthors Rita F. Redberg, MD, of the University of California, San Francisco, and Mary M. McDermott, MD, of Northwestern University, Chicago, reiterated the findings and the conclusions, but used the forum to draw attention to the low survival rates.
“Thus, while this well-done observational study provides new information,” they wrote, “a major conclusion should be that mortality is high among Medicare beneficiaries undergoing revascularization [for peripheral artery disease] with any devices.”
‘Very impressive’ methods
Marc P. Bonaca, MD, director of vascular research, University of Colorado at Denver, Aurora, called the methods to ensure the validity of the conclusions of this study “very impressive.” In situations where prospective randomized trials are impractical, he suggested that this type of approach might answer an unmet need.
“We have always desired the ability to look at these large datasets with a lot of power to answer important questions,” he said. While “the issue has always been residual confounding,” he expressed interest in further verifications that this type of methodology can serve as a template for data analysis to guide other regulatory decisions.
Dr. Secemsky reports financial relationships with Abbott, Bayer, Boston Scientific, Cook, CSI, Inari, Janssen, Medtronic, and Phillips. Dr. Redford reports no potential conflicts of interest. Dr. McDermott reports a financial relationship with Regeneron. Dr. Bonaca reports financial relationships with Amgen, AstraZeneca, Bayer, Janssen Merck, Novo Nordisk, Pfizer, and Sanofi.
FROM ACC 2021
Dapagliflozin misses as treatment for COVID-19 but leaves intriguing signal for benefit
In patients hospitalized with COVID-19 infection, the sodium-glucose transporter 2 inhibitor dapagliflozin showed a trend for benefit relative to placebo on multiple outcomes, including the primary outcome of time to organ failure or death, according to results from the randomized DARE-19 trial.

Because of the failure to reach statistical significance, these results have no immediate relevance, but the trends support interest in further testing SGLT2 inhibitors in acute diseases posing a high risk for organ failure, according to Mikhail Kosiborod, MD.
In a trial that did not meet its primary endpoint, Dr. Kosiborod acknowledged that positive interpretations are speculative, but he does believe that there is one immediate take-home message.
“Our results do not support discontinuation of SGLT2 inhibitors in the setting of COVID-19 as long as patients are monitored,” said Dr. Kosiborod, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.
At many institutions, it has been common to discontinue SGLT2 inhibitors in patients admitted with COVID-19. One reason was the concern that drugs in this class could exacerbate organ damage, particularly if they were to induced ketoacidosis. However, only 2 (0.003%) of 613 patients treated with dapagliflozin developed ketoacidosis, and the signal for organ protection overall, although not significant, was consistent.
“Numerically, fewer patients treated with dapagliflozin experienced organ failure and death, and this was consistent across systems, including the kidney,” Dr. Kosiborod said in presenting the study at the annual scientific sessions of the American College of Cardiology.
Overall, the study suggests that, in the context of COVID-19, dapagliflozin did not show harm and might have potential benefit, he added.
DARE-19 was rapidly conceived, designed, and implemented during the early stages of the COVID-19 pandemic. Based on prior evidence that SGLT2 inhibitors “favorably affect a number of pathophysiologic pathways disrupted during acute illness” and that drugs in this class have provided organ protection in the context of heart failure, chronic kidney disease, and other cardiometabolic conditions, the study was designed to test the hypothesis that this mechanism might improve outcomes in patients hospitalized with COVID-19, Dr. Kosiborod said.
The entry criteria included confirmed or suspected COVID-19 with an onset of 4 days of fewer and one additional risk factor, such as atherosclerotic cardiovascular disease, hypertension, or type 2 diabetes. Patients with significant renal impairment or a history of diabetic ketoacidosis were excluded.
On top of standard treatments for COVID-19, patients were randomized to 10 mg dapagliflozin or placebo once daily. There were two primary endpoints. That of prevention was time to criteria for respiratory, cardiovascular, or renal organ failure or death. The second primary outcome, for recovery, was a hierarchical composite for four endpoints: death, organ failure, status at 30 days if hospitalized, and time to discharge if this occurred before day 30.
Of the 1,250 patients randomized at 95 sites in seven countries, 617 in the dapagliflozin group and 620 patients in the placebo group completed the study. Baseline characteristics, which included a mean of age of 62 years; types of comorbidities; and types of treatments were similar.
Results for two primary endpoints
The curves for the primary outcome of prevention had already separated by day 3 and continued to widen over the 30 days in which outcomes were compared. At the end of 30 days, 11.2% of the dapagliflozin group and 13.8% of the placebo group had an event. By hazard ratio, dapagliflozin was linked to 20% nonsignificant relative protection from events (hazard ratio, 0.80; 95% confidence interval, 0.58-1.10).
The trend (P = .168) for the primary endpoint for prevention was reflected in the individual components. For dapagliflozin related to placebo, there were generally similar or greater reductions in new or worsening organ failure (HR, 0.80), cardiac decompensation (HR, 0.81), respiratory decompensation (HR, 0.85), and kidney decompensation (HR, 0.65). None were statistically significant, but the confidence intervals were tight with the upper end never exceeding 1.20.
Moreover, the relative risk reduction for all-cause mortality moved in the same direction (HR, 0.77; 95% CI, 0.52-1.16).
In the hierarchical composite endpoint of recovery, there was no significant difference in the time to discharge, but again many recovery metrics numerically favored dapagliflozin with an overall difference producing a statistical trend (P = .14) similar to organ failure events and death.
In safety analyses, dapagliflozin consistently outperformed placebo across a broad array of safety measure, including any severe adverse event (65% vs. 82%), any adverse event with an outcome of death (32% vs. 48%), discontinuation caused by an adverse event (44% vs. 55%), and acute kidney injury (21% vs. 34%).
Data could fuel related studies
According to Ana Barac, MD, PhD, director of the cardio-oncology program in the Medstar Heart and Vascular Institute, Washington, these data are “thought provoking.” Although this was a negative trial, she said that it generates an “exciting hypothesis” about the potential of SGLT2 inhibitors to provide organ protection. She called for studies to pursue this path of research.

More immediately, Dr. Barac agreed that these data argue against stopping SGLT2 inhibitors in patients admitted to a hospital for COVID-19 infection.
“These data show that these drugs are not going to lead to harm, but they might lead to benefit,” she said.
For James Januzzi, MD, a cardiologist at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, DARE-19 was perhaps most impressive because of its rigorous design and execution in the midst of a pandemic.
Over the past year, “the medical literature was flooded with grossly underpowered, poorly designed, single-center studies” yielding results that have been hard to interpret, Dr. Januzzi said. Despite the fact that this study failed to confirm its hypothesis, he said the investigators deserve praise for the quality of the work.

Dr. Januzzi also believes the study is not without clinically relevant findings, particularly the fact that dapagliflozin was associated with a lower rate of adverse events than placebo. This, at least, provides reassurance about the safety of this drug in the setting of COVID-19 infection.
Dr. Kosiborod reported financial relationships with more than 10 pharmaceutical companies, including AstraZeneca, which provided funding for DARE-19. Dr. Barac reported financial relationships with Bristol-Myers Squibb and CTI BioPharma. Dr. Januzzi reported financial relationships with Boehringer Ingelheim, GE Healthcare, Johnson & Johnson, Merck, Novartis, Pfizer, and Roche.
In patients hospitalized with COVID-19 infection, the sodium-glucose transporter 2 inhibitor dapagliflozin showed a trend for benefit relative to placebo on multiple outcomes, including the primary outcome of time to organ failure or death, according to results from the randomized DARE-19 trial.
Because of the failure to reach statistical significance, these results have no immediate relevance, but the trends support interest in further testing SGLT2 inhibitors in acute diseases posing a high risk for organ failure, according to Mikhail Kosiborod, MD.
In a trial that did not meet its primary endpoint, Dr. Kosiborod acknowledged that positive interpretations are speculative, but he does believe that there is one immediate take-home message.
“Our results do not support discontinuation of SGLT2 inhibitors in the setting of COVID-19 as long as patients are monitored,” said Dr. Kosiborod, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.
At many institutions, it has been common to discontinue SGLT2 inhibitors in patients admitted with COVID-19. One reason was the concern that drugs in this class could exacerbate organ damage, particularly if they were to induced ketoacidosis. However, only 2 (0.003%) of 613 patients treated with dapagliflozin developed ketoacidosis, and the signal for organ protection overall, although not significant, was consistent.
“Numerically, fewer patients treated with dapagliflozin experienced organ failure and death, and this was consistent across systems, including the kidney,” Dr. Kosiborod said in presenting the study at the annual scientific sessions of the American College of Cardiology.
Overall, the study suggests that, in the context of COVID-19, dapagliflozin did not show harm and might have potential benefit, he added.
DARE-19 was rapidly conceived, designed, and implemented during the early stages of the COVID-19 pandemic. Based on prior evidence that SGLT2 inhibitors “favorably affect a number of pathophysiologic pathways disrupted during acute illness” and that drugs in this class have provided organ protection in the context of heart failure, chronic kidney disease, and other cardiometabolic conditions, the study was designed to test the hypothesis that this mechanism might improve outcomes in patients hospitalized with COVID-19, Dr. Kosiborod said.
The entry criteria included confirmed or suspected COVID-19 with an onset of 4 days of fewer and one additional risk factor, such as atherosclerotic cardiovascular disease, hypertension, or type 2 diabetes. Patients with significant renal impairment or a history of diabetic ketoacidosis were excluded.
On top of standard treatments for COVID-19, patients were randomized to 10 mg dapagliflozin or placebo once daily. There were two primary endpoints. That of prevention was time to criteria for respiratory, cardiovascular, or renal organ failure or death. The second primary outcome, for recovery, was a hierarchical composite for four endpoints: death, organ failure, status at 30 days if hospitalized, and time to discharge if this occurred before day 30.
Of the 1,250 patients randomized at 95 sites in seven countries, 617 in the dapagliflozin group and 620 patients in the placebo group completed the study. Baseline characteristics, which included a mean of age of 62 years; types of comorbidities; and types of treatments were similar.
Results for two primary endpoints
The curves for the primary outcome of prevention had already separated by day 3 and continued to widen over the 30 days in which outcomes were compared. At the end of 30 days, 11.2% of the dapagliflozin group and 13.8% of the placebo group had an event. By hazard ratio, dapagliflozin was linked to 20% nonsignificant relative protection from events (hazard ratio, 0.80; 95% confidence interval, 0.58-1.10).
The trend (P = .168) for the primary endpoint for prevention was reflected in the individual components. For dapagliflozin related to placebo, there were generally similar or greater reductions in new or worsening organ failure (HR, 0.80), cardiac decompensation (HR, 0.81), respiratory decompensation (HR, 0.85), and kidney decompensation (HR, 0.65). None were statistically significant, but the confidence intervals were tight with the upper end never exceeding 1.20.
Moreover, the relative risk reduction for all-cause mortality moved in the same direction (HR, 0.77; 95% CI, 0.52-1.16).
In the hierarchical composite endpoint of recovery, there was no significant difference in the time to discharge, but again many recovery metrics numerically favored dapagliflozin with an overall difference producing a statistical trend (P = .14) similar to organ failure events and death.
In safety analyses, dapagliflozin consistently outperformed placebo across a broad array of safety measure, including any severe adverse event (65% vs. 82%), any adverse event with an outcome of death (32% vs. 48%), discontinuation caused by an adverse event (44% vs. 55%), and acute kidney injury (21% vs. 34%).
Data could fuel related studies
According to Ana Barac, MD, PhD, director of the cardio-oncology program in the Medstar Heart and Vascular Institute, Washington, these data are “thought provoking.” Although this was a negative trial, she said that it generates an “exciting hypothesis” about the potential of SGLT2 inhibitors to provide organ protection. She called for studies to pursue this path of research.
More immediately, Dr. Barac agreed that these data argue against stopping SGLT2 inhibitors in patients admitted to a hospital for COVID-19 infection.
“These data show that these drugs are not going to lead to harm, but they might lead to benefit,” she said.
For James Januzzi, MD, a cardiologist at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, DARE-19 was perhaps most impressive because of its rigorous design and execution in the midst of a pandemic.
Over the past year, “the medical literature was flooded with grossly underpowered, poorly designed, single-center studies” yielding results that have been hard to interpret, Dr. Januzzi said. Despite the fact that this study failed to confirm its hypothesis, he said the investigators deserve praise for the quality of the work.
Dr. Januzzi also believes the study is not without clinically relevant findings, particularly the fact that dapagliflozin was associated with a lower rate of adverse events than placebo. This, at least, provides reassurance about the safety of this drug in the setting of COVID-19 infection.
Dr. Kosiborod reported financial relationships with more than 10 pharmaceutical companies, including AstraZeneca, which provided funding for DARE-19. Dr. Barac reported financial relationships with Bristol-Myers Squibb and CTI BioPharma. Dr. Januzzi reported financial relationships with Boehringer Ingelheim, GE Healthcare, Johnson & Johnson, Merck, Novartis, Pfizer, and Roche.
In patients hospitalized with COVID-19 infection, the sodium-glucose transporter 2 inhibitor dapagliflozin showed a trend for benefit relative to placebo on multiple outcomes, including the primary outcome of time to organ failure or death, according to results from the randomized DARE-19 trial.
Because of the failure to reach statistical significance, these results have no immediate relevance, but the trends support interest in further testing SGLT2 inhibitors in acute diseases posing a high risk for organ failure, according to Mikhail Kosiborod, MD.
In a trial that did not meet its primary endpoint, Dr. Kosiborod acknowledged that positive interpretations are speculative, but he does believe that there is one immediate take-home message.
“Our results do not support discontinuation of SGLT2 inhibitors in the setting of COVID-19 as long as patients are monitored,” said Dr. Kosiborod, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo.
At many institutions, it has been common to discontinue SGLT2 inhibitors in patients admitted with COVID-19. One reason was the concern that drugs in this class could exacerbate organ damage, particularly if they were to induced ketoacidosis. However, only 2 (0.003%) of 613 patients treated with dapagliflozin developed ketoacidosis, and the signal for organ protection overall, although not significant, was consistent.
“Numerically, fewer patients treated with dapagliflozin experienced organ failure and death, and this was consistent across systems, including the kidney,” Dr. Kosiborod said in presenting the study at the annual scientific sessions of the American College of Cardiology.
Overall, the study suggests that, in the context of COVID-19, dapagliflozin did not show harm and might have potential benefit, he added.
DARE-19 was rapidly conceived, designed, and implemented during the early stages of the COVID-19 pandemic. Based on prior evidence that SGLT2 inhibitors “favorably affect a number of pathophysiologic pathways disrupted during acute illness” and that drugs in this class have provided organ protection in the context of heart failure, chronic kidney disease, and other cardiometabolic conditions, the study was designed to test the hypothesis that this mechanism might improve outcomes in patients hospitalized with COVID-19, Dr. Kosiborod said.
The entry criteria included confirmed or suspected COVID-19 with an onset of 4 days of fewer and one additional risk factor, such as atherosclerotic cardiovascular disease, hypertension, or type 2 diabetes. Patients with significant renal impairment or a history of diabetic ketoacidosis were excluded.
On top of standard treatments for COVID-19, patients were randomized to 10 mg dapagliflozin or placebo once daily. There were two primary endpoints. That of prevention was time to criteria for respiratory, cardiovascular, or renal organ failure or death. The second primary outcome, for recovery, was a hierarchical composite for four endpoints: death, organ failure, status at 30 days if hospitalized, and time to discharge if this occurred before day 30.
Of the 1,250 patients randomized at 95 sites in seven countries, 617 in the dapagliflozin group and 620 patients in the placebo group completed the study. Baseline characteristics, which included a mean of age of 62 years; types of comorbidities; and types of treatments were similar.
Results for two primary endpoints
The curves for the primary outcome of prevention had already separated by day 3 and continued to widen over the 30 days in which outcomes were compared. At the end of 30 days, 11.2% of the dapagliflozin group and 13.8% of the placebo group had an event. By hazard ratio, dapagliflozin was linked to 20% nonsignificant relative protection from events (hazard ratio, 0.80; 95% confidence interval, 0.58-1.10).
The trend (P = .168) for the primary endpoint for prevention was reflected in the individual components. For dapagliflozin related to placebo, there were generally similar or greater reductions in new or worsening organ failure (HR, 0.80), cardiac decompensation (HR, 0.81), respiratory decompensation (HR, 0.85), and kidney decompensation (HR, 0.65). None were statistically significant, but the confidence intervals were tight with the upper end never exceeding 1.20.
Moreover, the relative risk reduction for all-cause mortality moved in the same direction (HR, 0.77; 95% CI, 0.52-1.16).
In the hierarchical composite endpoint of recovery, there was no significant difference in the time to discharge, but again many recovery metrics numerically favored dapagliflozin with an overall difference producing a statistical trend (P = .14) similar to organ failure events and death.
In safety analyses, dapagliflozin consistently outperformed placebo across a broad array of safety measure, including any severe adverse event (65% vs. 82%), any adverse event with an outcome of death (32% vs. 48%), discontinuation caused by an adverse event (44% vs. 55%), and acute kidney injury (21% vs. 34%).
Data could fuel related studies
According to Ana Barac, MD, PhD, director of the cardio-oncology program in the Medstar Heart and Vascular Institute, Washington, these data are “thought provoking.” Although this was a negative trial, she said that it generates an “exciting hypothesis” about the potential of SGLT2 inhibitors to provide organ protection. She called for studies to pursue this path of research.
More immediately, Dr. Barac agreed that these data argue against stopping SGLT2 inhibitors in patients admitted to a hospital for COVID-19 infection.
“These data show that these drugs are not going to lead to harm, but they might lead to benefit,” she said.
For James Januzzi, MD, a cardiologist at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, DARE-19 was perhaps most impressive because of its rigorous design and execution in the midst of a pandemic.
Over the past year, “the medical literature was flooded with grossly underpowered, poorly designed, single-center studies” yielding results that have been hard to interpret, Dr. Januzzi said. Despite the fact that this study failed to confirm its hypothesis, he said the investigators deserve praise for the quality of the work.
Dr. Januzzi also believes the study is not without clinically relevant findings, particularly the fact that dapagliflozin was associated with a lower rate of adverse events than placebo. This, at least, provides reassurance about the safety of this drug in the setting of COVID-19 infection.
Dr. Kosiborod reported financial relationships with more than 10 pharmaceutical companies, including AstraZeneca, which provided funding for DARE-19. Dr. Barac reported financial relationships with Bristol-Myers Squibb and CTI BioPharma. Dr. Januzzi reported financial relationships with Boehringer Ingelheim, GE Healthcare, Johnson & Johnson, Merck, Novartis, Pfizer, and Roche.
FROM ACC 2021
FLOWER-MI: FFR-guided complete revascularization shows no advantage in STEMI
For patients with ST-elevated myocardial infarction (STEMI) undergoing complete revascularization, percutaneous coronary interventions (PCI) guided by fractional flow reserve (FFR) relative to angiography-guided PCI do not result in significantly lower risk of death or events, according to data from the randomized FLOWER-MI trial.

Rather, the events at 1 year were numerically lower among those randomized to the angiography-guided approach, according to the principal investigator of the trial, Etienne Puymirat, MD, PhD.
Prior studies showing an advantage for FFR-guided PCI in patients with coronary syndromes provided the hypothesis that FFR-guided PCI would also be superior for guiding PCI in STEMI patients. In the multicenter FAME trial, for example, FFR-guided PCI for patients with multivessel disease was associated with fewer stent placements (P < .001) and a nearly 30% lower rate of events at 1 year (P = .02).
While the advantage of complete revascularization, meaning PCI treatment of nonculprit as well as culprit lesions, has already been shown to be a better strategy than treatment of culprit lesions alone, FLOWER-MI is the first large study to compare FFR to angiography for guiding this approach to STEMI patients with multivessel disease, said Dr. Puymirat of Hôpital Européen George Pompidou, Paris, at the annual scientific sessions of the American College of Cardiology.
In this trial, involving multiple centers in France, STEMI patients were eligible for randomization if they had successful PCI of a culprit lesion and 50% or greater stenosis in at least one additional nonculprit lesion. The complete revascularization, whether patients were randomized to PCI guided by angiography or FFR, was performed during the index hospital admission. Patient management and follow-up was otherwise the same.
After a small number of exclusions, the intention-to-treat populations were 577 patients in the angiography-guided group and 586 in the FFR-guided group. The characteristics of the groups were well matched with an average age of about 62 years and similar rates of risk factors, such as hypertension and diabetes.
Angiography guidance just as good
The primary outcome was a composite of all-cause mortality, nonfatal MI, and unplanned revascularization. By hazard ratio, the risk of having one of these events within 1 year of PCI was numerically greater, at 32 in the FFR-guided group and 24 in the angiography-guided group, but the difference was not statistically significant (1.32; P = .31).
However, the total rate of events was low (5.5% vs. 4.2% for the angiography-guided and FFR-guided groups, respectively) and the confidence intervals were wide (95% CI, 0.78-2.23). This was also true of the components of the primary outcome.
No signal for a difference between strategies could be derived from these components, which included a higher rate of MI in the FFR-guided group (3.1% vs. 1.7%) but a lower rate of death (1.5% vs. 1.7%).
Unplanned hospitalizations leading to revascularization rates were also low (1.9% and 2.6% for angiography-guided and FFR-guided PCI, respectively), although it was reported that the rate of revascularization for nonculprit lesions was about twice as high in the FFR group (53.3% vs. 27.3%).
At 1 year, there were also low rates and no significant differences in a list of secondary outcomes that included hospitalization for recurrent ischemia or heart failure, stent thrombosis, and revascularization. As within the primary composite outcome, no pattern could be seen in the secondary events, some of which were numerically more common in the FFR-guided group and some numerically lower.
In a cost-efficacy analysis, the median per-patient cost of the FFR-guided strategy was about 500 Euros ($607) greater (8,832 vs. 8,322; P < .01), leading Dr. Puymirat to conclude that “the use of FFR for nonculprit lesions appears to be less effective but more expensive,” at least by costs derived in France.
Lack of statistical power limits interpretation
The conclusion of FLOWER-MI is that FFR-guided PCI in complete revascularization of nonculprit lesions in STEMI patients is not superior to an angiography-guided approach, but Dr. Puymirat cautioned that the low number of events precludes a definitive message.
William Fearon, MD, professor of cardiovascular medicine at Stanford (Calif.) University Medical Center, agreed. Based on his calculations, the trial was substantially underpowered. Evaluating the details of treatment in the FFR group, Dr. Fearon pointed out that a nonculprit lesion with a FFR of 0.80 or less was identified in about 55% of patients. Ultimately, 66% in the FFR group received PCI, eliminating the key distinction between strategies for the majority of patients enrolled.
“Only about one-third of the FFR-guided patients, or about 200 patients, did not receive nonculprit PCI, and therefore only in this small group could we expect a difference in outcomes from the angio-guided group,” Dr. Fearon said.
Fewer stents were placed in the FFR-guided than angiography-guided group (1.01 vs. 1.5), but Dr. Fearon suggested that it would be very difficult to show a difference in risk of events in a study of this size when event rates at 1 year reached only about 5%.
In response, Dr. Puymirat acknowledged that the rate of events for this trial, which was designed in 2015, were lower than expected. In recalculating the power needed based on the rate of events observed in FLOWER-MI, he estimated that about 8,000 patients would have been needed to show a meaningful difference in these PCI strategies.
Dr. Puymirat reports financial relationships with more than a dozen pharmaceutical companies, including Abbott, which provided some of the funding for this trial. Dr. Fearon reports financial relationships with Abbott, CathWorks, HeartFlow, and Medtronic.
For patients with ST-elevated myocardial infarction (STEMI) undergoing complete revascularization, percutaneous coronary interventions (PCI) guided by fractional flow reserve (FFR) relative to angiography-guided PCI do not result in significantly lower risk of death or events, according to data from the randomized FLOWER-MI trial.
Rather, the events at 1 year were numerically lower among those randomized to the angiography-guided approach, according to the principal investigator of the trial, Etienne Puymirat, MD, PhD.
Prior studies showing an advantage for FFR-guided PCI in patients with coronary syndromes provided the hypothesis that FFR-guided PCI would also be superior for guiding PCI in STEMI patients. In the multicenter FAME trial, for example, FFR-guided PCI for patients with multivessel disease was associated with fewer stent placements (P < .001) and a nearly 30% lower rate of events at 1 year (P = .02).
While the advantage of complete revascularization, meaning PCI treatment of nonculprit as well as culprit lesions, has already been shown to be a better strategy than treatment of culprit lesions alone, FLOWER-MI is the first large study to compare FFR to angiography for guiding this approach to STEMI patients with multivessel disease, said Dr. Puymirat of Hôpital Européen George Pompidou, Paris, at the annual scientific sessions of the American College of Cardiology.
In this trial, involving multiple centers in France, STEMI patients were eligible for randomization if they had successful PCI of a culprit lesion and 50% or greater stenosis in at least one additional nonculprit lesion. The complete revascularization, whether patients were randomized to PCI guided by angiography or FFR, was performed during the index hospital admission. Patient management and follow-up was otherwise the same.
After a small number of exclusions, the intention-to-treat populations were 577 patients in the angiography-guided group and 586 in the FFR-guided group. The characteristics of the groups were well matched with an average age of about 62 years and similar rates of risk factors, such as hypertension and diabetes.
Angiography guidance just as good
The primary outcome was a composite of all-cause mortality, nonfatal MI, and unplanned revascularization. By hazard ratio, the risk of having one of these events within 1 year of PCI was numerically greater, at 32 in the FFR-guided group and 24 in the angiography-guided group, but the difference was not statistically significant (1.32; P = .31).
However, the total rate of events was low (5.5% vs. 4.2% for the angiography-guided and FFR-guided groups, respectively) and the confidence intervals were wide (95% CI, 0.78-2.23). This was also true of the components of the primary outcome.
No signal for a difference between strategies could be derived from these components, which included a higher rate of MI in the FFR-guided group (3.1% vs. 1.7%) but a lower rate of death (1.5% vs. 1.7%).
Unplanned hospitalizations leading to revascularization rates were also low (1.9% and 2.6% for angiography-guided and FFR-guided PCI, respectively), although it was reported that the rate of revascularization for nonculprit lesions was about twice as high in the FFR group (53.3% vs. 27.3%).
At 1 year, there were also low rates and no significant differences in a list of secondary outcomes that included hospitalization for recurrent ischemia or heart failure, stent thrombosis, and revascularization. As within the primary composite outcome, no pattern could be seen in the secondary events, some of which were numerically more common in the FFR-guided group and some numerically lower.
In a cost-efficacy analysis, the median per-patient cost of the FFR-guided strategy was about 500 Euros ($607) greater (8,832 vs. 8,322; P < .01), leading Dr. Puymirat to conclude that “the use of FFR for nonculprit lesions appears to be less effective but more expensive,” at least by costs derived in France.
Lack of statistical power limits interpretation
The conclusion of FLOWER-MI is that FFR-guided PCI in complete revascularization of nonculprit lesions in STEMI patients is not superior to an angiography-guided approach, but Dr. Puymirat cautioned that the low number of events precludes a definitive message.
William Fearon, MD, professor of cardiovascular medicine at Stanford (Calif.) University Medical Center, agreed. Based on his calculations, the trial was substantially underpowered. Evaluating the details of treatment in the FFR group, Dr. Fearon pointed out that a nonculprit lesion with a FFR of 0.80 or less was identified in about 55% of patients. Ultimately, 66% in the FFR group received PCI, eliminating the key distinction between strategies for the majority of patients enrolled.
“Only about one-third of the FFR-guided patients, or about 200 patients, did not receive nonculprit PCI, and therefore only in this small group could we expect a difference in outcomes from the angio-guided group,” Dr. Fearon said.
Fewer stents were placed in the FFR-guided than angiography-guided group (1.01 vs. 1.5), but Dr. Fearon suggested that it would be very difficult to show a difference in risk of events in a study of this size when event rates at 1 year reached only about 5%.
In response, Dr. Puymirat acknowledged that the rate of events for this trial, which was designed in 2015, were lower than expected. In recalculating the power needed based on the rate of events observed in FLOWER-MI, he estimated that about 8,000 patients would have been needed to show a meaningful difference in these PCI strategies.
Dr. Puymirat reports financial relationships with more than a dozen pharmaceutical companies, including Abbott, which provided some of the funding for this trial. Dr. Fearon reports financial relationships with Abbott, CathWorks, HeartFlow, and Medtronic.
For patients with ST-elevated myocardial infarction (STEMI) undergoing complete revascularization, percutaneous coronary interventions (PCI) guided by fractional flow reserve (FFR) relative to angiography-guided PCI do not result in significantly lower risk of death or events, according to data from the randomized FLOWER-MI trial.
Rather, the events at 1 year were numerically lower among those randomized to the angiography-guided approach, according to the principal investigator of the trial, Etienne Puymirat, MD, PhD.
Prior studies showing an advantage for FFR-guided PCI in patients with coronary syndromes provided the hypothesis that FFR-guided PCI would also be superior for guiding PCI in STEMI patients. In the multicenter FAME trial, for example, FFR-guided PCI for patients with multivessel disease was associated with fewer stent placements (P < .001) and a nearly 30% lower rate of events at 1 year (P = .02).
While the advantage of complete revascularization, meaning PCI treatment of nonculprit as well as culprit lesions, has already been shown to be a better strategy than treatment of culprit lesions alone, FLOWER-MI is the first large study to compare FFR to angiography for guiding this approach to STEMI patients with multivessel disease, said Dr. Puymirat of Hôpital Européen George Pompidou, Paris, at the annual scientific sessions of the American College of Cardiology.
In this trial, involving multiple centers in France, STEMI patients were eligible for randomization if they had successful PCI of a culprit lesion and 50% or greater stenosis in at least one additional nonculprit lesion. The complete revascularization, whether patients were randomized to PCI guided by angiography or FFR, was performed during the index hospital admission. Patient management and follow-up was otherwise the same.
After a small number of exclusions, the intention-to-treat populations were 577 patients in the angiography-guided group and 586 in the FFR-guided group. The characteristics of the groups were well matched with an average age of about 62 years and similar rates of risk factors, such as hypertension and diabetes.
Angiography guidance just as good
The primary outcome was a composite of all-cause mortality, nonfatal MI, and unplanned revascularization. By hazard ratio, the risk of having one of these events within 1 year of PCI was numerically greater, at 32 in the FFR-guided group and 24 in the angiography-guided group, but the difference was not statistically significant (1.32; P = .31).
However, the total rate of events was low (5.5% vs. 4.2% for the angiography-guided and FFR-guided groups, respectively) and the confidence intervals were wide (95% CI, 0.78-2.23). This was also true of the components of the primary outcome.
No signal for a difference between strategies could be derived from these components, which included a higher rate of MI in the FFR-guided group (3.1% vs. 1.7%) but a lower rate of death (1.5% vs. 1.7%).
Unplanned hospitalizations leading to revascularization rates were also low (1.9% and 2.6% for angiography-guided and FFR-guided PCI, respectively), although it was reported that the rate of revascularization for nonculprit lesions was about twice as high in the FFR group (53.3% vs. 27.3%).
At 1 year, there were also low rates and no significant differences in a list of secondary outcomes that included hospitalization for recurrent ischemia or heart failure, stent thrombosis, and revascularization. As within the primary composite outcome, no pattern could be seen in the secondary events, some of which were numerically more common in the FFR-guided group and some numerically lower.
In a cost-efficacy analysis, the median per-patient cost of the FFR-guided strategy was about 500 Euros ($607) greater (8,832 vs. 8,322; P < .01), leading Dr. Puymirat to conclude that “the use of FFR for nonculprit lesions appears to be less effective but more expensive,” at least by costs derived in France.
Lack of statistical power limits interpretation
The conclusion of FLOWER-MI is that FFR-guided PCI in complete revascularization of nonculprit lesions in STEMI patients is not superior to an angiography-guided approach, but Dr. Puymirat cautioned that the low number of events precludes a definitive message.
William Fearon, MD, professor of cardiovascular medicine at Stanford (Calif.) University Medical Center, agreed. Based on his calculations, the trial was substantially underpowered. Evaluating the details of treatment in the FFR group, Dr. Fearon pointed out that a nonculprit lesion with a FFR of 0.80 or less was identified in about 55% of patients. Ultimately, 66% in the FFR group received PCI, eliminating the key distinction between strategies for the majority of patients enrolled.
“Only about one-third of the FFR-guided patients, or about 200 patients, did not receive nonculprit PCI, and therefore only in this small group could we expect a difference in outcomes from the angio-guided group,” Dr. Fearon said.
Fewer stents were placed in the FFR-guided than angiography-guided group (1.01 vs. 1.5), but Dr. Fearon suggested that it would be very difficult to show a difference in risk of events in a study of this size when event rates at 1 year reached only about 5%.
In response, Dr. Puymirat acknowledged that the rate of events for this trial, which was designed in 2015, were lower than expected. In recalculating the power needed based on the rate of events observed in FLOWER-MI, he estimated that about 8,000 patients would have been needed to show a meaningful difference in these PCI strategies.
Dr. Puymirat reports financial relationships with more than a dozen pharmaceutical companies, including Abbott, which provided some of the funding for this trial. Dr. Fearon reports financial relationships with Abbott, CathWorks, HeartFlow, and Medtronic.
FROM ACC 2021