User login
S-ICD ‘noninferior’ to transvenous-lead ICD in head-to-head PRAETORIAN trial
by turning in a “noninferior” performance when it was compared with transvenous-lead devices in a first-of-its-kind head-to-head study.
Patients implanted with the subcutaneous-lead S-ICD (Boston Scientific) defibrillator showed a 4-year risk for inappropriate shocks or device-related complications similar to that seen with standard transvenous-lead implantable cardioverter defibrillators (ICD) in a randomized comparison.
At the same time, the S-ICD did its job by showing a highly significant three-fourths reduction in risk for lead-related complications, compared with ICDs with standard leads, in the trial with more than 800 patients, called PRAETORIAN.
The study population represented a mix of patients seen in “real-world” practice who have an ICD indication, of whom about two-thirds had ischemic cardiomyopathy, said Reinoud Knops, MD, PhD, Academic Medical Center, Hilversum, the Netherlands. About 80% received the devices for primary prevention.
Knops, the trial’s principal investigator, presented the results online May 8 as one of the Heart Rhythm Society 2020 Scientific Sessions virtual presentations.
“I think the PRAETORIAN trial has really shown now, in a conventional ICD population – the real-world patients that we treat with ICD therapy, the single-chamber ICD cohort – that the S-ICD is a really good alternative option,” he said to reporters during a media briefing.
“The main conclusion is that the S-ICD should be considered in all patients who need an ICD who do not have a pacing indication,” Knops said.
This latter part is critical, because the S-ICD does not provide pacing therapy, including antitachycardia pacing (ATP) and cardiac resynchronization therapy (CRT), and the trial did not enter patients considered likely to benefit from it. For example, it excluded anyone with bradycardia or treatment-refractory monomorphic ventricular tachycardia (VT) and patients considered appropriate for CRT.
In fact, there are a lot reasons clinicians might prefer a transvenous-lead ICD over the S-ICD, observed Anne B. Curtis, MD, University at Buffalo, State University of New York, who is not associated with PRAETORIAN.
A transvenous-lead system might be preferred in older patients, those with heart failure, and those with a lot of comorbidities. “A lot of these patients already have cardiomyopathies, so they’re more likely to develop atrial fibrillation or a need for CRT,” conditions that might make a transvenous-lead system the better choice, Curtis told theheart.org | Medscape Cardiology.
“For a lot of patients, you’re always thinking that you may have a need for that kind of therapy.”
In contrast, younger patients who perhaps have survived cardiac arrest and probably don’t have heart failure, and so may be less likely to benefit from pacing therapy, Curtis said, “are the kind of patient who you would probably lean very strongly toward for an S-ICD rather than a transvenous ICD.”
Remaining patients, those who might be considered candidates for either kind of device, are actually “a fairly limited subset,” she said.
The trial randomized 849 patients in Europe and the United States, from March 2011 to January 2017, who had a class I or IIa indication for an ICD but no bradycardia or need for CRT or ATP, to be implanted with an S-ICD or a transvenous-lead ICD.
The rates of the primary end point, a composite of device-related complications and inappropriate shocks at a median follow-up of 4 years, were comparable, at 15.1% in the S-ICD group and 15.7% for those with transvenous-lead ICDs.
The incidence of device-related complications numerically favored the S-ICD group, and the incidence of inappropriate shocks numerically favored the transvenous-lead group, but neither difference reached significance.
Knops said the PRAETORIAN researchers are seeking addition funding to extend the follow-up to 8 years. “We will get more insight into the durability of the S-ICD when we follow these patients longer,” he told theheart.org | Medscape Cardiology.
The investigator-initiated trial received support from Boston Scientific. Knops discloses receiving consultancy fees and research grants from Abbott, Boston Scientific, Medtronic, and Cairdac, and holding stock options from AtaCor Medical.
This article first appeared on Medscape.com.
by turning in a “noninferior” performance when it was compared with transvenous-lead devices in a first-of-its-kind head-to-head study.
Patients implanted with the subcutaneous-lead S-ICD (Boston Scientific) defibrillator showed a 4-year risk for inappropriate shocks or device-related complications similar to that seen with standard transvenous-lead implantable cardioverter defibrillators (ICD) in a randomized comparison.
At the same time, the S-ICD did its job by showing a highly significant three-fourths reduction in risk for lead-related complications, compared with ICDs with standard leads, in the trial with more than 800 patients, called PRAETORIAN.
The study population represented a mix of patients seen in “real-world” practice who have an ICD indication, of whom about two-thirds had ischemic cardiomyopathy, said Reinoud Knops, MD, PhD, Academic Medical Center, Hilversum, the Netherlands. About 80% received the devices for primary prevention.
Knops, the trial’s principal investigator, presented the results online May 8 as one of the Heart Rhythm Society 2020 Scientific Sessions virtual presentations.
“I think the PRAETORIAN trial has really shown now, in a conventional ICD population – the real-world patients that we treat with ICD therapy, the single-chamber ICD cohort – that the S-ICD is a really good alternative option,” he said to reporters during a media briefing.
“The main conclusion is that the S-ICD should be considered in all patients who need an ICD who do not have a pacing indication,” Knops said.
This latter part is critical, because the S-ICD does not provide pacing therapy, including antitachycardia pacing (ATP) and cardiac resynchronization therapy (CRT), and the trial did not enter patients considered likely to benefit from it. For example, it excluded anyone with bradycardia or treatment-refractory monomorphic ventricular tachycardia (VT) and patients considered appropriate for CRT.
In fact, there are a lot reasons clinicians might prefer a transvenous-lead ICD over the S-ICD, observed Anne B. Curtis, MD, University at Buffalo, State University of New York, who is not associated with PRAETORIAN.
A transvenous-lead system might be preferred in older patients, those with heart failure, and those with a lot of comorbidities. “A lot of these patients already have cardiomyopathies, so they’re more likely to develop atrial fibrillation or a need for CRT,” conditions that might make a transvenous-lead system the better choice, Curtis told theheart.org | Medscape Cardiology.
“For a lot of patients, you’re always thinking that you may have a need for that kind of therapy.”
In contrast, younger patients who perhaps have survived cardiac arrest and probably don’t have heart failure, and so may be less likely to benefit from pacing therapy, Curtis said, “are the kind of patient who you would probably lean very strongly toward for an S-ICD rather than a transvenous ICD.”
Remaining patients, those who might be considered candidates for either kind of device, are actually “a fairly limited subset,” she said.
The trial randomized 849 patients in Europe and the United States, from March 2011 to January 2017, who had a class I or IIa indication for an ICD but no bradycardia or need for CRT or ATP, to be implanted with an S-ICD or a transvenous-lead ICD.
The rates of the primary end point, a composite of device-related complications and inappropriate shocks at a median follow-up of 4 years, were comparable, at 15.1% in the S-ICD group and 15.7% for those with transvenous-lead ICDs.
The incidence of device-related complications numerically favored the S-ICD group, and the incidence of inappropriate shocks numerically favored the transvenous-lead group, but neither difference reached significance.
Knops said the PRAETORIAN researchers are seeking addition funding to extend the follow-up to 8 years. “We will get more insight into the durability of the S-ICD when we follow these patients longer,” he told theheart.org | Medscape Cardiology.
The investigator-initiated trial received support from Boston Scientific. Knops discloses receiving consultancy fees and research grants from Abbott, Boston Scientific, Medtronic, and Cairdac, and holding stock options from AtaCor Medical.
This article first appeared on Medscape.com.
by turning in a “noninferior” performance when it was compared with transvenous-lead devices in a first-of-its-kind head-to-head study.
Patients implanted with the subcutaneous-lead S-ICD (Boston Scientific) defibrillator showed a 4-year risk for inappropriate shocks or device-related complications similar to that seen with standard transvenous-lead implantable cardioverter defibrillators (ICD) in a randomized comparison.
At the same time, the S-ICD did its job by showing a highly significant three-fourths reduction in risk for lead-related complications, compared with ICDs with standard leads, in the trial with more than 800 patients, called PRAETORIAN.
The study population represented a mix of patients seen in “real-world” practice who have an ICD indication, of whom about two-thirds had ischemic cardiomyopathy, said Reinoud Knops, MD, PhD, Academic Medical Center, Hilversum, the Netherlands. About 80% received the devices for primary prevention.
Knops, the trial’s principal investigator, presented the results online May 8 as one of the Heart Rhythm Society 2020 Scientific Sessions virtual presentations.
“I think the PRAETORIAN trial has really shown now, in a conventional ICD population – the real-world patients that we treat with ICD therapy, the single-chamber ICD cohort – that the S-ICD is a really good alternative option,” he said to reporters during a media briefing.
“The main conclusion is that the S-ICD should be considered in all patients who need an ICD who do not have a pacing indication,” Knops said.
This latter part is critical, because the S-ICD does not provide pacing therapy, including antitachycardia pacing (ATP) and cardiac resynchronization therapy (CRT), and the trial did not enter patients considered likely to benefit from it. For example, it excluded anyone with bradycardia or treatment-refractory monomorphic ventricular tachycardia (VT) and patients considered appropriate for CRT.
In fact, there are a lot reasons clinicians might prefer a transvenous-lead ICD over the S-ICD, observed Anne B. Curtis, MD, University at Buffalo, State University of New York, who is not associated with PRAETORIAN.
A transvenous-lead system might be preferred in older patients, those with heart failure, and those with a lot of comorbidities. “A lot of these patients already have cardiomyopathies, so they’re more likely to develop atrial fibrillation or a need for CRT,” conditions that might make a transvenous-lead system the better choice, Curtis told theheart.org | Medscape Cardiology.
“For a lot of patients, you’re always thinking that you may have a need for that kind of therapy.”
In contrast, younger patients who perhaps have survived cardiac arrest and probably don’t have heart failure, and so may be less likely to benefit from pacing therapy, Curtis said, “are the kind of patient who you would probably lean very strongly toward for an S-ICD rather than a transvenous ICD.”
Remaining patients, those who might be considered candidates for either kind of device, are actually “a fairly limited subset,” she said.
The trial randomized 849 patients in Europe and the United States, from March 2011 to January 2017, who had a class I or IIa indication for an ICD but no bradycardia or need for CRT or ATP, to be implanted with an S-ICD or a transvenous-lead ICD.
The rates of the primary end point, a composite of device-related complications and inappropriate shocks at a median follow-up of 4 years, were comparable, at 15.1% in the S-ICD group and 15.7% for those with transvenous-lead ICDs.
The incidence of device-related complications numerically favored the S-ICD group, and the incidence of inappropriate shocks numerically favored the transvenous-lead group, but neither difference reached significance.
Knops said the PRAETORIAN researchers are seeking addition funding to extend the follow-up to 8 years. “We will get more insight into the durability of the S-ICD when we follow these patients longer,” he told theheart.org | Medscape Cardiology.
The investigator-initiated trial received support from Boston Scientific. Knops discloses receiving consultancy fees and research grants from Abbott, Boston Scientific, Medtronic, and Cairdac, and holding stock options from AtaCor Medical.
This article first appeared on Medscape.com.
Signature STEMI sign may be less diagnostic in the COVID-19 age
The signature electrocardiographic sign indicating ST-segment-elevation MI may be a less-consistent indicator of actual STEMI at a time when patients with COVID-19 have come to overwhelm many hospital ICUs.
Many of the 18 such patients identified at six New York City hospitals who showed ST-segment elevation on their 12-lead ECG in the city’s first month of fighting the pandemic turned out to be free of either obstructive coronary artery disease by angiography or of regional wall-motion abnormalities (RWMA) by ECG, according to a letter published in the New England Journal of Medicine.
Those 10 patients in the 18-case series were said to have noncoronary myocardial injury, perhaps from myocarditis – a prevalent feature of severe COVID-19 – and the remaining 8 patients with obstructive coronary artery disease, RWMA, or both were diagnosed with STEMI. Of the latter patients, six went to the cath lab and five of those underwent percutaneous coronary intervention, Sripal Bangalore, MD, MHA, of New York University, and colleagues reported.
In an interview, Dr. Bangalore framed the case-series report as a caution against substituting fibrinolytic therapy for primary percutaneous coronary intervention in patients with STE while hospitals are unusually burdened by the COVID-19 pandemic and invasive procedures intensify the threat of SARS-CoV-2 exposure to clinicians.
The strategy was recently advanced as an option for highly selected patients in a statement from the American College of Cardiology and Society for Cardiovascular Angiography and Interventions (SCAI).
“During the COVID-19 pandemic, one of the main reasons fibrinolytic therapy has been pushed is to reduce the exposure to the cath-lab staff,” Dr. Bangalore observed. “But if you pursue that route, it’s problematic because more than half may not have obstructive disease and fibrinolytic therapy may not help. And if you give them fibrinolytics, you’re potentially increasing their risk of bleeding complications.
“The take-home from these 18 patients is that it’s very difficult to guess who is going to have obstructive disease and who is going to have nonobstructive disease,” Dr. Bangalore said. “Maybe we should assess these patients with not just an ECG but with a quick echo, then make a decision. Our practice so far has been to take these patients to the cath lab.”
The ACC/SCAI statement proposed that “fibrinolysis can be considered an option for the relatively stable STEMI patient with active COVID-19” after careful consideration of possible patient benefit versus the risks of cath-lab personnel exposure to the virus.
Only six patients in the current series, including five in the STEMI group, are reported to have had chest pain at about the time of STE, observed Michael J. Blaha, MD, MPH, of Johns Hopkins Hospital, Baltimore.
So, he said in an interview, “one of their points is that you have to take ST elevations with a grain of salt in this [COVID-19] era, because there are a lot of people presenting with ST elevations in the absence of chest pain.”
That, and the high prevalence of nonobstructive disease in the series, indeed argues against the use of fibrinolytic therapy in such patients, Dr. Blaha said.
Normally, when there is STE, “the pretest probability of STEMI is so high, and if you can’t make it to the cath lab for some reason, sure, it makes sense to give lytics.” However, he said, “COVID-19 is changing the clinical landscape. Now, with a variety of virus-mediated myocardial injury presentations, including myocarditis, the pretest probability of MI is lower.”
The current report “confirms that, in the COVID era, ST elevations are not diagnostic for MI and must be considered within the totality of clinical evidence, and a conservative approach to going to the cath lab is probably warranted,” Dr. Blaha said in an interview.
However, with the reduced pretest probability of STE for STEMI, he agreed, “I almost don’t see any scenario where I’d be comfortable, based on ECG changes alone, giving lytics at this time.”
Dr. Bangalore pointed out that all of the 18 patients in the series had elevated levels of the fibrin degradation product D-dimer, a biomarker that reflects ongoing hemostatic activation. Levels were higher in the 8 patients who ultimately received a STEMI diagnosis than in the remaining 10 patients.
But COVID-19 patients in general may have elevated D-dimer and “a lot of microthrombi,” he said. “So the question is, are those microthrombi also causal for any of the ECG changes we are also seeing?”
Aside from microthrombi, global hypoxia and myocarditis could be other potential causes of STE in COVID-19 patients in the absence of STEMI, Dr. Bangalore proposed. “At this point we just generally don’t know.”
Dr. Bangalore reported no conflicts; disclosures for the other authors are available at nejm.org. Dr. Blaha disclosed receiving grants from Amgen and serving on advisory boards for Amgen and other pharmaceutical companies.
A version of this article originally appeared on Medscape.com.
The signature electrocardiographic sign indicating ST-segment-elevation MI may be a less-consistent indicator of actual STEMI at a time when patients with COVID-19 have come to overwhelm many hospital ICUs.
Many of the 18 such patients identified at six New York City hospitals who showed ST-segment elevation on their 12-lead ECG in the city’s first month of fighting the pandemic turned out to be free of either obstructive coronary artery disease by angiography or of regional wall-motion abnormalities (RWMA) by ECG, according to a letter published in the New England Journal of Medicine.
Those 10 patients in the 18-case series were said to have noncoronary myocardial injury, perhaps from myocarditis – a prevalent feature of severe COVID-19 – and the remaining 8 patients with obstructive coronary artery disease, RWMA, or both were diagnosed with STEMI. Of the latter patients, six went to the cath lab and five of those underwent percutaneous coronary intervention, Sripal Bangalore, MD, MHA, of New York University, and colleagues reported.
In an interview, Dr. Bangalore framed the case-series report as a caution against substituting fibrinolytic therapy for primary percutaneous coronary intervention in patients with STE while hospitals are unusually burdened by the COVID-19 pandemic and invasive procedures intensify the threat of SARS-CoV-2 exposure to clinicians.
The strategy was recently advanced as an option for highly selected patients in a statement from the American College of Cardiology and Society for Cardiovascular Angiography and Interventions (SCAI).
“During the COVID-19 pandemic, one of the main reasons fibrinolytic therapy has been pushed is to reduce the exposure to the cath-lab staff,” Dr. Bangalore observed. “But if you pursue that route, it’s problematic because more than half may not have obstructive disease and fibrinolytic therapy may not help. And if you give them fibrinolytics, you’re potentially increasing their risk of bleeding complications.
“The take-home from these 18 patients is that it’s very difficult to guess who is going to have obstructive disease and who is going to have nonobstructive disease,” Dr. Bangalore said. “Maybe we should assess these patients with not just an ECG but with a quick echo, then make a decision. Our practice so far has been to take these patients to the cath lab.”
The ACC/SCAI statement proposed that “fibrinolysis can be considered an option for the relatively stable STEMI patient with active COVID-19” after careful consideration of possible patient benefit versus the risks of cath-lab personnel exposure to the virus.
Only six patients in the current series, including five in the STEMI group, are reported to have had chest pain at about the time of STE, observed Michael J. Blaha, MD, MPH, of Johns Hopkins Hospital, Baltimore.
So, he said in an interview, “one of their points is that you have to take ST elevations with a grain of salt in this [COVID-19] era, because there are a lot of people presenting with ST elevations in the absence of chest pain.”
That, and the high prevalence of nonobstructive disease in the series, indeed argues against the use of fibrinolytic therapy in such patients, Dr. Blaha said.
Normally, when there is STE, “the pretest probability of STEMI is so high, and if you can’t make it to the cath lab for some reason, sure, it makes sense to give lytics.” However, he said, “COVID-19 is changing the clinical landscape. Now, with a variety of virus-mediated myocardial injury presentations, including myocarditis, the pretest probability of MI is lower.”
The current report “confirms that, in the COVID era, ST elevations are not diagnostic for MI and must be considered within the totality of clinical evidence, and a conservative approach to going to the cath lab is probably warranted,” Dr. Blaha said in an interview.
However, with the reduced pretest probability of STE for STEMI, he agreed, “I almost don’t see any scenario where I’d be comfortable, based on ECG changes alone, giving lytics at this time.”
Dr. Bangalore pointed out that all of the 18 patients in the series had elevated levels of the fibrin degradation product D-dimer, a biomarker that reflects ongoing hemostatic activation. Levels were higher in the 8 patients who ultimately received a STEMI diagnosis than in the remaining 10 patients.
But COVID-19 patients in general may have elevated D-dimer and “a lot of microthrombi,” he said. “So the question is, are those microthrombi also causal for any of the ECG changes we are also seeing?”
Aside from microthrombi, global hypoxia and myocarditis could be other potential causes of STE in COVID-19 patients in the absence of STEMI, Dr. Bangalore proposed. “At this point we just generally don’t know.”
Dr. Bangalore reported no conflicts; disclosures for the other authors are available at nejm.org. Dr. Blaha disclosed receiving grants from Amgen and serving on advisory boards for Amgen and other pharmaceutical companies.
A version of this article originally appeared on Medscape.com.
The signature electrocardiographic sign indicating ST-segment-elevation MI may be a less-consistent indicator of actual STEMI at a time when patients with COVID-19 have come to overwhelm many hospital ICUs.
Many of the 18 such patients identified at six New York City hospitals who showed ST-segment elevation on their 12-lead ECG in the city’s first month of fighting the pandemic turned out to be free of either obstructive coronary artery disease by angiography or of regional wall-motion abnormalities (RWMA) by ECG, according to a letter published in the New England Journal of Medicine.
Those 10 patients in the 18-case series were said to have noncoronary myocardial injury, perhaps from myocarditis – a prevalent feature of severe COVID-19 – and the remaining 8 patients with obstructive coronary artery disease, RWMA, or both were diagnosed with STEMI. Of the latter patients, six went to the cath lab and five of those underwent percutaneous coronary intervention, Sripal Bangalore, MD, MHA, of New York University, and colleagues reported.
In an interview, Dr. Bangalore framed the case-series report as a caution against substituting fibrinolytic therapy for primary percutaneous coronary intervention in patients with STE while hospitals are unusually burdened by the COVID-19 pandemic and invasive procedures intensify the threat of SARS-CoV-2 exposure to clinicians.
The strategy was recently advanced as an option for highly selected patients in a statement from the American College of Cardiology and Society for Cardiovascular Angiography and Interventions (SCAI).
“During the COVID-19 pandemic, one of the main reasons fibrinolytic therapy has been pushed is to reduce the exposure to the cath-lab staff,” Dr. Bangalore observed. “But if you pursue that route, it’s problematic because more than half may not have obstructive disease and fibrinolytic therapy may not help. And if you give them fibrinolytics, you’re potentially increasing their risk of bleeding complications.
“The take-home from these 18 patients is that it’s very difficult to guess who is going to have obstructive disease and who is going to have nonobstructive disease,” Dr. Bangalore said. “Maybe we should assess these patients with not just an ECG but with a quick echo, then make a decision. Our practice so far has been to take these patients to the cath lab.”
The ACC/SCAI statement proposed that “fibrinolysis can be considered an option for the relatively stable STEMI patient with active COVID-19” after careful consideration of possible patient benefit versus the risks of cath-lab personnel exposure to the virus.
Only six patients in the current series, including five in the STEMI group, are reported to have had chest pain at about the time of STE, observed Michael J. Blaha, MD, MPH, of Johns Hopkins Hospital, Baltimore.
So, he said in an interview, “one of their points is that you have to take ST elevations with a grain of salt in this [COVID-19] era, because there are a lot of people presenting with ST elevations in the absence of chest pain.”
That, and the high prevalence of nonobstructive disease in the series, indeed argues against the use of fibrinolytic therapy in such patients, Dr. Blaha said.
Normally, when there is STE, “the pretest probability of STEMI is so high, and if you can’t make it to the cath lab for some reason, sure, it makes sense to give lytics.” However, he said, “COVID-19 is changing the clinical landscape. Now, with a variety of virus-mediated myocardial injury presentations, including myocarditis, the pretest probability of MI is lower.”
The current report “confirms that, in the COVID era, ST elevations are not diagnostic for MI and must be considered within the totality of clinical evidence, and a conservative approach to going to the cath lab is probably warranted,” Dr. Blaha said in an interview.
However, with the reduced pretest probability of STE for STEMI, he agreed, “I almost don’t see any scenario where I’d be comfortable, based on ECG changes alone, giving lytics at this time.”
Dr. Bangalore pointed out that all of the 18 patients in the series had elevated levels of the fibrin degradation product D-dimer, a biomarker that reflects ongoing hemostatic activation. Levels were higher in the 8 patients who ultimately received a STEMI diagnosis than in the remaining 10 patients.
But COVID-19 patients in general may have elevated D-dimer and “a lot of microthrombi,” he said. “So the question is, are those microthrombi also causal for any of the ECG changes we are also seeing?”
Aside from microthrombi, global hypoxia and myocarditis could be other potential causes of STE in COVID-19 patients in the absence of STEMI, Dr. Bangalore proposed. “At this point we just generally don’t know.”
Dr. Bangalore reported no conflicts; disclosures for the other authors are available at nejm.org. Dr. Blaha disclosed receiving grants from Amgen and serving on advisory boards for Amgen and other pharmaceutical companies.
A version of this article originally appeared on Medscape.com.
VICTORIA: Vericiguat seen as novel success in tough-to-treat, high-risk heart failure
Not too many years ago, clinicians who treat patients with heart failure, especially those at high risk for decompensation, lamented what seemed a dearth of new drug therapy options.
Now, with the toolbox brimming with new guideline-supported alternatives, .
Importantly, it entered an especially high-risk population with heart failure and reduced ejection fraction (HFrEF); everyone in the trial had experienced a prior, usually quite recent, heart failure exacerbation.
In such patients, the addition of vericiguat (Merck/Bayer) to standard drug and device therapies was followed by a moderately but significantly reduced relative risk for the trial’s primary clinical endpoint over about 11 months.
Recipients benefited with a 10% drop in adjusted risk (P = .019) for cardiovascular (CV) death or first heart failure hospitalization compared to a placebo control group.
But researchers leading the 5050-patient Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction (VICTORIA), as well as unaffiliated experts who have studied the trial, say that in this case, risk reduction in absolute numbers is a more telling outcome.
“Remember who we’re talking about here in terms of the patients who have this degree of morbidity and mortality,” VICTORIA study chair Paul W. Armstrong, MD, University of Alberta, Edmonton, Canada, told theheart.org | Medscape Cardiology, pointing to the “incredible placebo-group event rate and relatively modest follow-up of 10.8 months.”
The control group’s primary-endpoint event rate was 37.8 per 100 patient-years, 4.2 points higher than the rate for patients who received vericiguat. “And from there you get a number needed to treat of 24 to prevent one event, which is low,” Armstrong said.
“Think about the hundreds of thousands of people with this disease and what that means at the public health level.” About one in four patients with heart failure experience such exacerbations each year, he said.
Armstrong is lead author on the 42-country trial’s publication today in the New England Journal of Medicine, timed to coincide with his online presentation for the American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). The annual session was conducted virtually this year following the traditional live meeting’s cancelation due to the COVID-19 pandemic.
The VICTORIA presentation and publication flesh out the cursory top-line primary results that Merck unveiled in November 2019, which had not included the magnitude of the vericiguat relative benefit for the primary endpoint.
The trial represents “another win” for the treatment of heart failure, Clyde W. Yancy, MD, Northwestern University, Chicago, Illinois, said as an invited discussant following Armstrong’s presentation.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
Symptomatic hypotension occurred in less than 10% and syncope in 4% or less of both groups; neither difference between the two groups was significant. Anemia developed more often in patients receiving vericiguat (7.6%) than in the control group (5.7%).
“We think that on balance, vericiguat is a useful alternative option for patients. But certainly the only thing we can say at this point is it works in the high-risk population that we studied,” Armstrong said. “Whether it works in lower-risk populations and how it compares is speculation, of course.”
The drug’s cost, whatever it might be if approved, is another factor affecting how it would be used, noted several observers.
“We don’t know what the cost-effectiveness will be. It should be reasonable because the benefit was on hospitalization. That’s a costly outcome,” Yancy said.
McMurray was also hopeful. “If the treatment is well tolerated and reasonably priced, it may still be a valuable asset for at least a subset of patients.”
VICTORIA was supported by Merck Sharp & Dohme Corp and Bayer AG. Armstrong discloses receiving research grants from Merck, Bayer AG, Sanofi-Aventis, Boehringer Ingelheim, and CSL Ltd and consulting fees from Merck, Bayer AG, AstraZeneca, and Novartis. Y ancy has previously disclosed no relevant financial relationships. Stevenson has previously disclosed receiving research grants from Novartis, consulting or serving on an advisory board for Abbott and travel expenses or meals from Novartis and St Jude Medical. McMurray has previously disclosed nonfinancial support or other support from AstraZeneca, Bayer, Cardiorentis, Amgen, Oxford University/Bayer, Theracos, AbbVie, DalCor, Pfizer, Merck, Novartis, GlaxoSmithKline, Bristol-Myers Squibb, and Vifor-Fresenius.
American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). Presented March 28, 2020. Session 402-08.
N Engl J Med. Published online March 28, 2020. Full text; Circulation. Published online March 28, 2020. Full text.
This article first appeared on Medscape.com.
Not too many years ago, clinicians who treat patients with heart failure, especially those at high risk for decompensation, lamented what seemed a dearth of new drug therapy options.
Now, with the toolbox brimming with new guideline-supported alternatives, .
Importantly, it entered an especially high-risk population with heart failure and reduced ejection fraction (HFrEF); everyone in the trial had experienced a prior, usually quite recent, heart failure exacerbation.
In such patients, the addition of vericiguat (Merck/Bayer) to standard drug and device therapies was followed by a moderately but significantly reduced relative risk for the trial’s primary clinical endpoint over about 11 months.
Recipients benefited with a 10% drop in adjusted risk (P = .019) for cardiovascular (CV) death or first heart failure hospitalization compared to a placebo control group.
But researchers leading the 5050-patient Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction (VICTORIA), as well as unaffiliated experts who have studied the trial, say that in this case, risk reduction in absolute numbers is a more telling outcome.
“Remember who we’re talking about here in terms of the patients who have this degree of morbidity and mortality,” VICTORIA study chair Paul W. Armstrong, MD, University of Alberta, Edmonton, Canada, told theheart.org | Medscape Cardiology, pointing to the “incredible placebo-group event rate and relatively modest follow-up of 10.8 months.”
The control group’s primary-endpoint event rate was 37.8 per 100 patient-years, 4.2 points higher than the rate for patients who received vericiguat. “And from there you get a number needed to treat of 24 to prevent one event, which is low,” Armstrong said.
“Think about the hundreds of thousands of people with this disease and what that means at the public health level.” About one in four patients with heart failure experience such exacerbations each year, he said.
Armstrong is lead author on the 42-country trial’s publication today in the New England Journal of Medicine, timed to coincide with his online presentation for the American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). The annual session was conducted virtually this year following the traditional live meeting’s cancelation due to the COVID-19 pandemic.
The VICTORIA presentation and publication flesh out the cursory top-line primary results that Merck unveiled in November 2019, which had not included the magnitude of the vericiguat relative benefit for the primary endpoint.
The trial represents “another win” for the treatment of heart failure, Clyde W. Yancy, MD, Northwestern University, Chicago, Illinois, said as an invited discussant following Armstrong’s presentation.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
Symptomatic hypotension occurred in less than 10% and syncope in 4% or less of both groups; neither difference between the two groups was significant. Anemia developed more often in patients receiving vericiguat (7.6%) than in the control group (5.7%).
“We think that on balance, vericiguat is a useful alternative option for patients. But certainly the only thing we can say at this point is it works in the high-risk population that we studied,” Armstrong said. “Whether it works in lower-risk populations and how it compares is speculation, of course.”
The drug’s cost, whatever it might be if approved, is another factor affecting how it would be used, noted several observers.
“We don’t know what the cost-effectiveness will be. It should be reasonable because the benefit was on hospitalization. That’s a costly outcome,” Yancy said.
McMurray was also hopeful. “If the treatment is well tolerated and reasonably priced, it may still be a valuable asset for at least a subset of patients.”
VICTORIA was supported by Merck Sharp & Dohme Corp and Bayer AG. Armstrong discloses receiving research grants from Merck, Bayer AG, Sanofi-Aventis, Boehringer Ingelheim, and CSL Ltd and consulting fees from Merck, Bayer AG, AstraZeneca, and Novartis. Y ancy has previously disclosed no relevant financial relationships. Stevenson has previously disclosed receiving research grants from Novartis, consulting or serving on an advisory board for Abbott and travel expenses or meals from Novartis and St Jude Medical. McMurray has previously disclosed nonfinancial support or other support from AstraZeneca, Bayer, Cardiorentis, Amgen, Oxford University/Bayer, Theracos, AbbVie, DalCor, Pfizer, Merck, Novartis, GlaxoSmithKline, Bristol-Myers Squibb, and Vifor-Fresenius.
American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). Presented March 28, 2020. Session 402-08.
N Engl J Med. Published online March 28, 2020. Full text; Circulation. Published online March 28, 2020. Full text.
This article first appeared on Medscape.com.
Not too many years ago, clinicians who treat patients with heart failure, especially those at high risk for decompensation, lamented what seemed a dearth of new drug therapy options.
Now, with the toolbox brimming with new guideline-supported alternatives, .
Importantly, it entered an especially high-risk population with heart failure and reduced ejection fraction (HFrEF); everyone in the trial had experienced a prior, usually quite recent, heart failure exacerbation.
In such patients, the addition of vericiguat (Merck/Bayer) to standard drug and device therapies was followed by a moderately but significantly reduced relative risk for the trial’s primary clinical endpoint over about 11 months.
Recipients benefited with a 10% drop in adjusted risk (P = .019) for cardiovascular (CV) death or first heart failure hospitalization compared to a placebo control group.
But researchers leading the 5050-patient Vericiguat Global Study in Subjects with Heart Failure with Reduced Ejection Fraction (VICTORIA), as well as unaffiliated experts who have studied the trial, say that in this case, risk reduction in absolute numbers is a more telling outcome.
“Remember who we’re talking about here in terms of the patients who have this degree of morbidity and mortality,” VICTORIA study chair Paul W. Armstrong, MD, University of Alberta, Edmonton, Canada, told theheart.org | Medscape Cardiology, pointing to the “incredible placebo-group event rate and relatively modest follow-up of 10.8 months.”
The control group’s primary-endpoint event rate was 37.8 per 100 patient-years, 4.2 points higher than the rate for patients who received vericiguat. “And from there you get a number needed to treat of 24 to prevent one event, which is low,” Armstrong said.
“Think about the hundreds of thousands of people with this disease and what that means at the public health level.” About one in four patients with heart failure experience such exacerbations each year, he said.
Armstrong is lead author on the 42-country trial’s publication today in the New England Journal of Medicine, timed to coincide with his online presentation for the American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). The annual session was conducted virtually this year following the traditional live meeting’s cancelation due to the COVID-19 pandemic.
The VICTORIA presentation and publication flesh out the cursory top-line primary results that Merck unveiled in November 2019, which had not included the magnitude of the vericiguat relative benefit for the primary endpoint.
The trial represents “another win” for the treatment of heart failure, Clyde W. Yancy, MD, Northwestern University, Chicago, Illinois, said as an invited discussant following Armstrong’s presentation.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
“Hospitalization for heart failure generates a major inflection point in the natural history of this condition, with a marked change in the risk for re-hospitalization and death. Up until now, no prior therapies have attenuated this risk, except for more intensive processes and care improvement strategies,” he said.
“Now we have a therapy that may be the first one to change that natural history after a person with heart failure has had a worsening event.”
Interestingly, the primary-endpoint reduction was driven by a significant drop in heart failure hospitalizations, even within a fairly short follow-up time.
“What was fascinating is that the requisite number of events were accrued in less than 12 months — meaning that inexplicably, this is one of the few times we’ve had a trial where the event rate realized was higher than the event rate predicted,” Yancy observed for theheart.org | Medscape Cardiology.
Although the effect size was similar to what was observed for dapagliflozin (Farxiga, AstraZeneca) in DAPA-HF and sacubitril/valsartan (Entresto, Novartis) in PARADIGM-HF, he said, VICTORIA’s population was much sicker and had an “astonishingly high” event rate even while receiving aggressive background heart failure therapy.
It included “triple therapy with renin-angiotensin system inhibitors, β-blockers, and mineralocorticoid receptor antagonists in 60% of patients, and at least double therapy in 90% of patients.” Also, Yancy said, 30% of the population had implantable devices, such as defibrillators and biventricular pacemakers.
Such patients with advanced, late-stage disease are common as the latest therapies for heart failure prolong their survival, notes Lynne W. Stevenson, MD, Vanderbilt University, Nashville, Tennessee, also as an invited discussant after Armstrong’s presentation.
“It’s a unique population with longer disease duration, more severe disease, and narrow options,” one in which personalized approaches are needed. Yet VICTORIA-like patients “have been actively excluded from all the trials that have shown benefit,” she said.
“VICTORIA finally addresses this population of decompensated patients,” she said, and seems to show that vericiguat may help some of them.
At the University of Glasgow, United Kingdom, John J.V. McMurray, MBChB, MD, agreed that the relative risk reduction was “small but significant,” but also that the control group’s event rate was “very high, reflecting the inclusion and exclusion criteria.”
As a result, McMurray told theheart.org | Medscape Cardiology, there was “quite a large absolute risk reduction and small number needed to treat. Also on the positive side: no significant excess of the adverse effects we might have been concerned about,” for example, hypotension.
Vericiguat, if ultimately approved in heart failure, “isn’t going to be first-line or widely used, but it is an additional asset,” he said. “Anything that helps in heart failure is valuable. There are always patients who can’t tolerate treatments, and always people who need more done.”
It’s appealing that the drug works by a long but unfruitfully explored mechanism that has little to do directly with the renin-angiotensin-aldosterone system.
Vericiguat is a soluble guanylate cyclase stimulator that boosts cyclic guanosine monophosphate activity along several pathways, potentiating the salutary pulmonary artery–vasodilating effects of nitric oxide. It improved natriuretic peptide levels in the preceding phase 2 SOCRATES-REDUCED study.
“This is not a me-too drug. It’s a new avenue for heart failure patients,” Armstrong said in an interview. It’s taken once daily, “was relatively easy to titrate up to the target dose, pretty well tolerated, and very safe. And remarkably, you don’t need to measure renal function.”
However, because the drug’s mechanism resides in the same neighborhood of biochemical pathways affected by chronic nitrates and by phosphodiesterase type 5 inhibitors, such as sildenafil and tadalafil, patients taking those drugs were excluded from VICTORIA. Acute nitrates were allowed, however.
Symptomatic hypotension occurred in less than 10% and syncope in 4% or less of both groups; neither difference between the two groups was significant. Anemia developed more often in patients receiving vericiguat (7.6%) than in the control group (5.7%).
“We think that on balance, vericiguat is a useful alternative option for patients. But certainly the only thing we can say at this point is it works in the high-risk population that we studied,” Armstrong said. “Whether it works in lower-risk populations and how it compares is speculation, of course.”
The drug’s cost, whatever it might be if approved, is another factor affecting how it would be used, noted several observers.
“We don’t know what the cost-effectiveness will be. It should be reasonable because the benefit was on hospitalization. That’s a costly outcome,” Yancy said.
McMurray was also hopeful. “If the treatment is well tolerated and reasonably priced, it may still be a valuable asset for at least a subset of patients.”
VICTORIA was supported by Merck Sharp & Dohme Corp and Bayer AG. Armstrong discloses receiving research grants from Merck, Bayer AG, Sanofi-Aventis, Boehringer Ingelheim, and CSL Ltd and consulting fees from Merck, Bayer AG, AstraZeneca, and Novartis. Y ancy has previously disclosed no relevant financial relationships. Stevenson has previously disclosed receiving research grants from Novartis, consulting or serving on an advisory board for Abbott and travel expenses or meals from Novartis and St Jude Medical. McMurray has previously disclosed nonfinancial support or other support from AstraZeneca, Bayer, Cardiorentis, Amgen, Oxford University/Bayer, Theracos, AbbVie, DalCor, Pfizer, Merck, Novartis, GlaxoSmithKline, Bristol-Myers Squibb, and Vifor-Fresenius.
American College of Cardiology 2020 Scientific Session (ACC.20)/World Congress of Cardiology (WCC). Presented March 28, 2020. Session 402-08.
N Engl J Med. Published online March 28, 2020. Full text; Circulation. Published online March 28, 2020. Full text.
This article first appeared on Medscape.com.
Prescription cascade more likely after CCBs than other hypertension meds
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.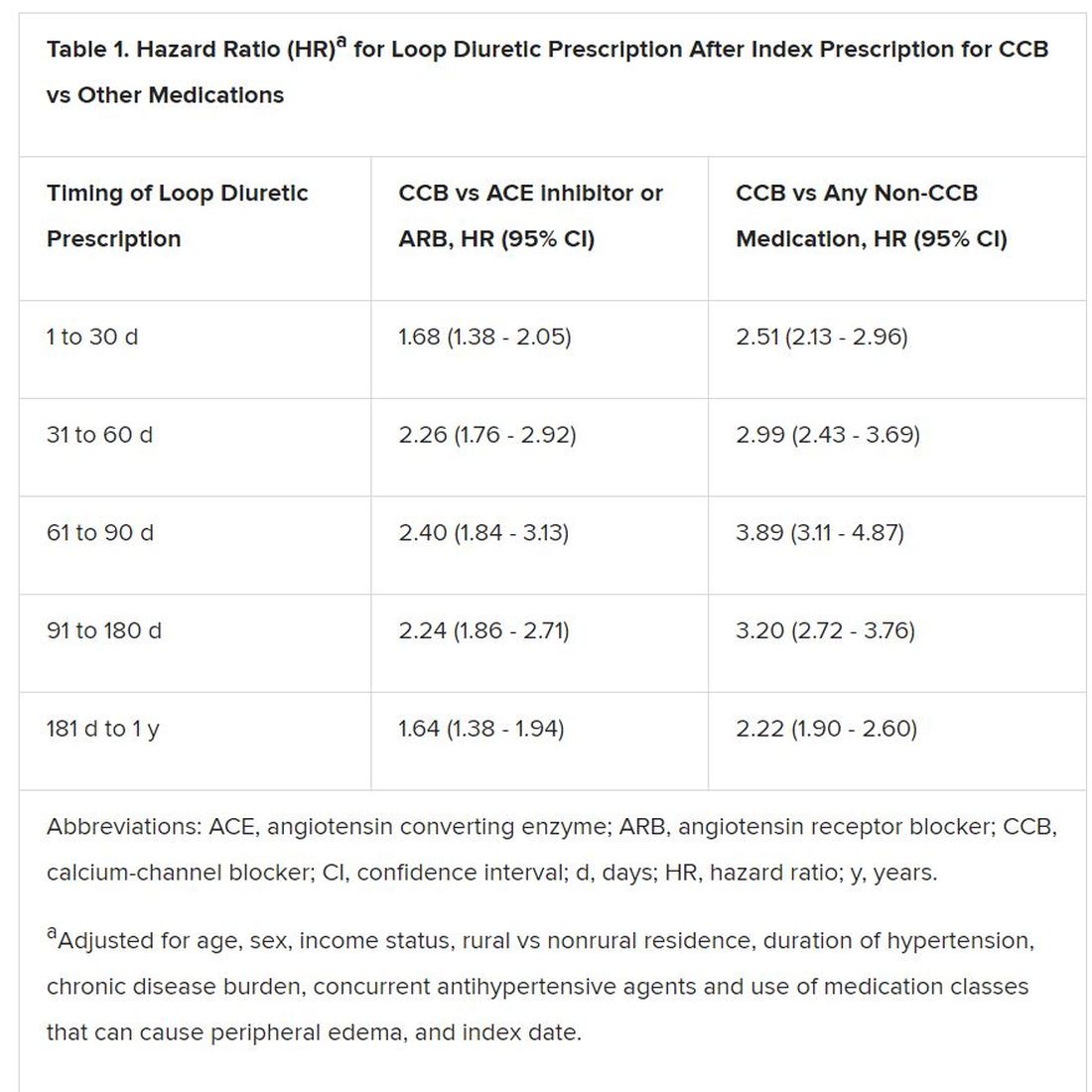
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Elderly adults with hypertension who are newly prescribed a calcium-channel blocker (CCB), compared to other antihypertensive agents, are at least twice as likely to be given a loop diuretic over the following months, a large cohort study suggests.
The likelihood remained elevated for as long as a year after the start of a CCB and was more pronounced when comparing CCBs to any other kind of medication.
“Our findings suggest that many older adults who begin taking a CCB may subsequently experience a prescribing cascade” when loop diuretics are prescribed for peripheral edema, a known CCB adverse effect, that is misinterpreted as a new medical condition, Rachel D. Savage, PhD, Women’s College Hospital, Toronto, Canada, told theheart.org/Medscape Cardiology.
Edema caused by CCBs is caused by fluid redistribution, not overload, and “treating euvolemic individuals with a diuretic places them at increased risk of overdiuresis, leading to falls, urinary incontinence, acute kidney injury, electrolyte imbalances, and a cascade of other downstream consequences to which older adults are especially vulnerable,” explain Savage and coauthors of the analysis published online February 24 in JAMA Internal Medicine.
However, 1.4% of the cohort had been prescribed a loop diuretic, and 4.5% had been given any diuretic within 90 days after the start of CCBs. The corresponding rates were 0.7% and 3.4%, respectively, for patients who had started on ACE inhibitors or angiotensin receptor blocker (ARB) rather than a CCB.
Also, Savage observed, “the likelihood of being prescribed a loop diuretic following initiation of a CCB changed over time and was greatest 61 to 90 days postinitiation.” At that point, it was increased 2.4 times compared with initiation of an ACE inhibitor or an ARB in an adjusted analysis and increased almost 4 times compared with starting on any non-CCB agent.
Importantly, the actual prevalence of peripheral edema among those started on CCBs, ACE inhibitors, ARBs, or any non-CCB medication was not available in the data sets.
However, “the main message for clinicians is to consider medication side effects as a potential cause for new symptoms when patients present. We also encourage patients to ask prescribers about whether new symptoms could be caused by a medication,” senior author Lisa M. McCarthy, PharmD, told theheart.org/Medscape Cardiology.
“If a patient experiences peripheral edema while taking a CCB, we would encourage clinicians to consider whether the calcium-channel blocker is still necessary, whether it could be discontinued or the dose reduced, or whether the patient can be switched to another therapy,” she said.
Based on the current analysis, if the rate of CCB-induced peripheral edema is assumed to be 10%, which is consistent with the literature, then “potentially 7% to 14% of people who develop edema while taking a calcium channel blocker may then receive a loop diuretic,” an accompanying editorial notes.
“Patients with polypharmacy are at heightened risk of being exposed to [a] series of prescribing cascades if their current use of medications is not carefully discussed before the decision to add a new antihypertensive,” observe Timothy S. Anderson, MD, Beth Israel Deaconess Medical Center, Boston, Massachusetts, and Michael A. Steinman, MD, San Francisco Veterans Affairs Medical Center and University of California, San Francisco.
“The initial prescribing cascade can set off many other negative consequences, including adverse drug events, potentially avoidable diagnostic testing, and hospitalizations,” the editorialists caution.
“Identifying prescribing cascades and their consequences is an important step to stem the tide of polypharmacy and inform deprescribing efforts.”
The analysis was based on administrative data from almost 340,000 adults in the community aged 66 years or older with hypertension and new drug prescriptions over 5 years ending in September 2016, the report notes. Their mean age was 74.5 years and 56.5% were women.
The data set included 41,086 patients who were newly prescribed a CCB; 66,494 who were newly prescribed an ACE inhibitor or ARB; and 231,439 newly prescribed any medication other than a CCB. The prescribed CCB was amlodipine in 79.6% of patients.
Although loop diuretics could possibly have been prescribed sometimes as a second-tier antihypertensive in the absence of peripheral edema, “we made efforts, through the design of our study, to limit this where possible,” Savage said in an interview.
For example, the focus was on loop diuretics, which aren’t generally recommended for blood-pressure lowering. Also, patients with heart failure and those with a recent history of diuretic or other antihypertensive medication use had been excluded, she said.
“As such, our cohort comprised individuals with new-onset or milder hypertension for whom diuretics would unlikely to be prescribed as part of guideline-based hypertension management.”
Although amlodipine was the most commonly prescribed CCB, the potential for a prescribing cascade seemed to be a class effect and to apply at a range of dosages.
That was unexpected, McCarthy observed, because “peripheral edema occurs more commonly in people taking dihydropyridine CCBs, like amlodipine, compared to non–dihydropyridine CCBs, such as verapamil and diltiazem.”
Savage, McCarthy, their coauthors, and the editorialists have disclosed no relevant financial relationships.
This article first appeared on Medscape.com.
Societies dig in to EXCEL trial controversy
Thoracic surgery societies on both sides of the Atlantic have released new statements on a continuing controversy dogging the EXCEL trial, one that has fueled a highly public war of words over how the study was conducted, interpreted, and reported by its investigators.

In a statement dated Dec. 19, 2019, the European Association for Cardio-Thoracic Surgery (EACTS) offered new details on why it withdrew its endorsement of the 2018 EACTS-European Society of Cardiology (ESC) clinical guidelines section covering left-main coronary artery disease.
That part of the guideline had relied in part on 3-year outcomes from EXCEL, which were published in the New England Journal of Medicine in 2016 (2016 Dec 8;375[23]:2223-35) and are central to the ongoing dispute. The trial, in essence, was a comparison of percutaneous coronary intervention (PCI) and coronary bypass surgery (CABG) in left-main disease. In that report, PCI was noninferior to CABG with respect to the composite endpoint of death, stroke, or myocardial infarction at 3 years in patients with left-main disease and low or intermediate anatomical complexity.
The new statement, signed by the society’s secretary general Domenico Pagano, MD, also calls for a new EACTS-ESC evidence review and development of updated recommendations for left-main disease “as a matter of urgency.”
For its part, the ESC had earlier declared its continuing support for the full guideline but hinted that might change pending further details on EXCEL yet to be made public.
The EACTS statement follows the society’s earlier announcement that it would pull support of the guideline section on left-main disease in response to a Dec. 9, 2019, news report from BBC Newsnight that was critical of the EXCEL trial’s methodology and reporting.
The news story made a number of allegations regarding the interpretation and reporting of EXCEL based largely on unpublished data it had obtained through unofficial channels.
Key among them was that reanalysis of myocardial infarction outcomes using the Third Universal Definition of MI, rather than the primarily enzymatic definition on which the reported outcomes were based, substantially raised the MI count in the PCI group, compared with those who had CABG.
The data for that alternative analysis, which had not been publicly reported, seemed to recast the published EXCEL primary outcome from one of parity for PCI and CABG in left-main disease to one that significantly favored CABG, noted the BBC Newsnight story.
Also, the news story claimed that EXCEL investigators had promised to publicly release the trial’s data based on the Third Universal Definition of MI, but had not done so, and had not adequately heeded concerns raised by its Data Safety Monitoring Board (DSMB) over signs of an apparently increased mortality risk from PCI.
Another society weighs in
The unreported data and other issues have led the American Association for Thoracic Surgery (AATS) to issue a statement acknowledging the possibility of misguided treatment recommendations, and therefore patient care, stemming from incomplete reporting of EXCEL.
If there are serious concerns about the “presentation or interpretation” of clinical trials, “then the best way forward is the public release of all trial data for an independent analysis to confirm that the original trial conclusions are valid,” says the statement, signed by AATS president Vaughn A. Starnes, MD, and secretary David R. Jones, MD.
“The AATS agrees with others that all of the data should be made publicly available for analysis and interpretation, as a way to resolve the current controversy around the EXCEL trial, in order to provide patients with the best possible counsel and informed consent,” it states.
The BBC Newsnight story “raised legitimate questions regarding what data was/was not presented to the other EXCEL investigators and to [the] ESC/EACTS guideline committee, and what, when, and to whom were safety warnings raised by the DSMB,” David Taggart, MD, PhD, wrote in an email interview.
“Until these issues are resolved, both EACTS and AATS have expressed concerns about what has happened and, most importantly, the potential implications for patient safety. This stance underpins their sincerity that patient safety, genuine informed consent, and scientific integrity are amongst their highest priorities. Consequently they have my complete support,” said Dr. Taggart, of the University of Oxford (England) a former EXCEL trialist who has been among the most vocal critics of how the EXCEL leadership has interpreted and reported the trial’s outcomes.
“Personally, I do not feel that the current controversy over the EXCEL trial will be resolved until there is full and independent reanalysis of its data. I feel that this would be absolutely crucial in reassuring our patients, the wider medical community, and the general public of the validity of current recommendations.”
An EXCEL principal investigator and prominent public voice for the trial, Gregg W. Stone, MD, of Icahn School of Medicine at Mount Sinai, New York, has not responded to requests for comment on the new society statements.
Point-counterpoint
As previously reported, allegations about EXCEL in news reports and the sometimes fiery public debate led the trialists to release a long and wide-ranging public communique that forcefully disputes the charges. Among them, that they were either remiss or willfully deceptive in not reporting an analysis based on the Third Universal Definition of MI.
In response, Dr. Taggart provided a toughly worded statement that disputes the EXCEL leadership’s missive nearly point by point. It variously describes the assertions as “simplistic,” seemingly “illogical,” “disingenuous,” and “factually completely incorrect,” among other terms.
The document provides Dr. Taggart’s perspective on how MI was defined and interpreted while he was an active member of the EXCEL trial’s leadership, and alleged shortfalls in how outcomes were interpreted and reported.
In it, Taggart also wonders whether or not EXCEL leadership had possibly been aware of a tilt favoring CABG in the analysis based on Third Universal Definition of MI but “decided to suppress it,” and also whether the trial’s sponsor, Abbott Vascular, had influenced the trial’s conduct.
Despite the EXCEL leadership’s communique, “my profound concerns remain the same and, in my opinion, the very long rebuttal response by the EXCEL investigators does not adequately respond to the core issues,” Dr. Taggart writes.
He withdrew his name as an author on the trial’s 5-year outcomes publication, Dr. Taggart says, because “I believed, and still do, that the final interpretation of the actual data in the [New England Journal of Medicine] manuscript did not appropriately reflect its clinical reality, and especially with regards to mortality, and would therefore have potential to do real harm to patients.”
Dr. Taggart has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.
Thoracic surgery societies on both sides of the Atlantic have released new statements on a continuing controversy dogging the EXCEL trial, one that has fueled a highly public war of words over how the study was conducted, interpreted, and reported by its investigators.
In a statement dated Dec. 19, 2019, the European Association for Cardio-Thoracic Surgery (EACTS) offered new details on why it withdrew its endorsement of the 2018 EACTS-European Society of Cardiology (ESC) clinical guidelines section covering left-main coronary artery disease.
That part of the guideline had relied in part on 3-year outcomes from EXCEL, which were published in the New England Journal of Medicine in 2016 (2016 Dec 8;375[23]:2223-35) and are central to the ongoing dispute. The trial, in essence, was a comparison of percutaneous coronary intervention (PCI) and coronary bypass surgery (CABG) in left-main disease. In that report, PCI was noninferior to CABG with respect to the composite endpoint of death, stroke, or myocardial infarction at 3 years in patients with left-main disease and low or intermediate anatomical complexity.
The new statement, signed by the society’s secretary general Domenico Pagano, MD, also calls for a new EACTS-ESC evidence review and development of updated recommendations for left-main disease “as a matter of urgency.”
For its part, the ESC had earlier declared its continuing support for the full guideline but hinted that might change pending further details on EXCEL yet to be made public.
The EACTS statement follows the society’s earlier announcement that it would pull support of the guideline section on left-main disease in response to a Dec. 9, 2019, news report from BBC Newsnight that was critical of the EXCEL trial’s methodology and reporting.
The news story made a number of allegations regarding the interpretation and reporting of EXCEL based largely on unpublished data it had obtained through unofficial channels.
Key among them was that reanalysis of myocardial infarction outcomes using the Third Universal Definition of MI, rather than the primarily enzymatic definition on which the reported outcomes were based, substantially raised the MI count in the PCI group, compared with those who had CABG.
The data for that alternative analysis, which had not been publicly reported, seemed to recast the published EXCEL primary outcome from one of parity for PCI and CABG in left-main disease to one that significantly favored CABG, noted the BBC Newsnight story.
Also, the news story claimed that EXCEL investigators had promised to publicly release the trial’s data based on the Third Universal Definition of MI, but had not done so, and had not adequately heeded concerns raised by its Data Safety Monitoring Board (DSMB) over signs of an apparently increased mortality risk from PCI.
Another society weighs in
The unreported data and other issues have led the American Association for Thoracic Surgery (AATS) to issue a statement acknowledging the possibility of misguided treatment recommendations, and therefore patient care, stemming from incomplete reporting of EXCEL.
If there are serious concerns about the “presentation or interpretation” of clinical trials, “then the best way forward is the public release of all trial data for an independent analysis to confirm that the original trial conclusions are valid,” says the statement, signed by AATS president Vaughn A. Starnes, MD, and secretary David R. Jones, MD.
“The AATS agrees with others that all of the data should be made publicly available for analysis and interpretation, as a way to resolve the current controversy around the EXCEL trial, in order to provide patients with the best possible counsel and informed consent,” it states.
The BBC Newsnight story “raised legitimate questions regarding what data was/was not presented to the other EXCEL investigators and to [the] ESC/EACTS guideline committee, and what, when, and to whom were safety warnings raised by the DSMB,” David Taggart, MD, PhD, wrote in an email interview.
“Until these issues are resolved, both EACTS and AATS have expressed concerns about what has happened and, most importantly, the potential implications for patient safety. This stance underpins their sincerity that patient safety, genuine informed consent, and scientific integrity are amongst their highest priorities. Consequently they have my complete support,” said Dr. Taggart, of the University of Oxford (England) a former EXCEL trialist who has been among the most vocal critics of how the EXCEL leadership has interpreted and reported the trial’s outcomes.
“Personally, I do not feel that the current controversy over the EXCEL trial will be resolved until there is full and independent reanalysis of its data. I feel that this would be absolutely crucial in reassuring our patients, the wider medical community, and the general public of the validity of current recommendations.”
An EXCEL principal investigator and prominent public voice for the trial, Gregg W. Stone, MD, of Icahn School of Medicine at Mount Sinai, New York, has not responded to requests for comment on the new society statements.
Point-counterpoint
As previously reported, allegations about EXCEL in news reports and the sometimes fiery public debate led the trialists to release a long and wide-ranging public communique that forcefully disputes the charges. Among them, that they were either remiss or willfully deceptive in not reporting an analysis based on the Third Universal Definition of MI.
In response, Dr. Taggart provided a toughly worded statement that disputes the EXCEL leadership’s missive nearly point by point. It variously describes the assertions as “simplistic,” seemingly “illogical,” “disingenuous,” and “factually completely incorrect,” among other terms.
The document provides Dr. Taggart’s perspective on how MI was defined and interpreted while he was an active member of the EXCEL trial’s leadership, and alleged shortfalls in how outcomes were interpreted and reported.
In it, Taggart also wonders whether or not EXCEL leadership had possibly been aware of a tilt favoring CABG in the analysis based on Third Universal Definition of MI but “decided to suppress it,” and also whether the trial’s sponsor, Abbott Vascular, had influenced the trial’s conduct.
Despite the EXCEL leadership’s communique, “my profound concerns remain the same and, in my opinion, the very long rebuttal response by the EXCEL investigators does not adequately respond to the core issues,” Dr. Taggart writes.
He withdrew his name as an author on the trial’s 5-year outcomes publication, Dr. Taggart says, because “I believed, and still do, that the final interpretation of the actual data in the [New England Journal of Medicine] manuscript did not appropriately reflect its clinical reality, and especially with regards to mortality, and would therefore have potential to do real harm to patients.”
Dr. Taggart has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.
Thoracic surgery societies on both sides of the Atlantic have released new statements on a continuing controversy dogging the EXCEL trial, one that has fueled a highly public war of words over how the study was conducted, interpreted, and reported by its investigators.
In a statement dated Dec. 19, 2019, the European Association for Cardio-Thoracic Surgery (EACTS) offered new details on why it withdrew its endorsement of the 2018 EACTS-European Society of Cardiology (ESC) clinical guidelines section covering left-main coronary artery disease.
That part of the guideline had relied in part on 3-year outcomes from EXCEL, which were published in the New England Journal of Medicine in 2016 (2016 Dec 8;375[23]:2223-35) and are central to the ongoing dispute. The trial, in essence, was a comparison of percutaneous coronary intervention (PCI) and coronary bypass surgery (CABG) in left-main disease. In that report, PCI was noninferior to CABG with respect to the composite endpoint of death, stroke, or myocardial infarction at 3 years in patients with left-main disease and low or intermediate anatomical complexity.
The new statement, signed by the society’s secretary general Domenico Pagano, MD, also calls for a new EACTS-ESC evidence review and development of updated recommendations for left-main disease “as a matter of urgency.”
For its part, the ESC had earlier declared its continuing support for the full guideline but hinted that might change pending further details on EXCEL yet to be made public.
The EACTS statement follows the society’s earlier announcement that it would pull support of the guideline section on left-main disease in response to a Dec. 9, 2019, news report from BBC Newsnight that was critical of the EXCEL trial’s methodology and reporting.
The news story made a number of allegations regarding the interpretation and reporting of EXCEL based largely on unpublished data it had obtained through unofficial channels.
Key among them was that reanalysis of myocardial infarction outcomes using the Third Universal Definition of MI, rather than the primarily enzymatic definition on which the reported outcomes were based, substantially raised the MI count in the PCI group, compared with those who had CABG.
The data for that alternative analysis, which had not been publicly reported, seemed to recast the published EXCEL primary outcome from one of parity for PCI and CABG in left-main disease to one that significantly favored CABG, noted the BBC Newsnight story.
Also, the news story claimed that EXCEL investigators had promised to publicly release the trial’s data based on the Third Universal Definition of MI, but had not done so, and had not adequately heeded concerns raised by its Data Safety Monitoring Board (DSMB) over signs of an apparently increased mortality risk from PCI.
Another society weighs in
The unreported data and other issues have led the American Association for Thoracic Surgery (AATS) to issue a statement acknowledging the possibility of misguided treatment recommendations, and therefore patient care, stemming from incomplete reporting of EXCEL.
If there are serious concerns about the “presentation or interpretation” of clinical trials, “then the best way forward is the public release of all trial data for an independent analysis to confirm that the original trial conclusions are valid,” says the statement, signed by AATS president Vaughn A. Starnes, MD, and secretary David R. Jones, MD.
“The AATS agrees with others that all of the data should be made publicly available for analysis and interpretation, as a way to resolve the current controversy around the EXCEL trial, in order to provide patients with the best possible counsel and informed consent,” it states.
The BBC Newsnight story “raised legitimate questions regarding what data was/was not presented to the other EXCEL investigators and to [the] ESC/EACTS guideline committee, and what, when, and to whom were safety warnings raised by the DSMB,” David Taggart, MD, PhD, wrote in an email interview.
“Until these issues are resolved, both EACTS and AATS have expressed concerns about what has happened and, most importantly, the potential implications for patient safety. This stance underpins their sincerity that patient safety, genuine informed consent, and scientific integrity are amongst their highest priorities. Consequently they have my complete support,” said Dr. Taggart, of the University of Oxford (England) a former EXCEL trialist who has been among the most vocal critics of how the EXCEL leadership has interpreted and reported the trial’s outcomes.
“Personally, I do not feel that the current controversy over the EXCEL trial will be resolved until there is full and independent reanalysis of its data. I feel that this would be absolutely crucial in reassuring our patients, the wider medical community, and the general public of the validity of current recommendations.”
An EXCEL principal investigator and prominent public voice for the trial, Gregg W. Stone, MD, of Icahn School of Medicine at Mount Sinai, New York, has not responded to requests for comment on the new society statements.
Point-counterpoint
As previously reported, allegations about EXCEL in news reports and the sometimes fiery public debate led the trialists to release a long and wide-ranging public communique that forcefully disputes the charges. Among them, that they were either remiss or willfully deceptive in not reporting an analysis based on the Third Universal Definition of MI.
In response, Dr. Taggart provided a toughly worded statement that disputes the EXCEL leadership’s missive nearly point by point. It variously describes the assertions as “simplistic,” seemingly “illogical,” “disingenuous,” and “factually completely incorrect,” among other terms.
The document provides Dr. Taggart’s perspective on how MI was defined and interpreted while he was an active member of the EXCEL trial’s leadership, and alleged shortfalls in how outcomes were interpreted and reported.
In it, Taggart also wonders whether or not EXCEL leadership had possibly been aware of a tilt favoring CABG in the analysis based on Third Universal Definition of MI but “decided to suppress it,” and also whether the trial’s sponsor, Abbott Vascular, had influenced the trial’s conduct.
Despite the EXCEL leadership’s communique, “my profound concerns remain the same and, in my opinion, the very long rebuttal response by the EXCEL investigators does not adequately respond to the core issues,” Dr. Taggart writes.
He withdrew his name as an author on the trial’s 5-year outcomes publication, Dr. Taggart says, because “I believed, and still do, that the final interpretation of the actual data in the [New England Journal of Medicine] manuscript did not appropriately reflect its clinical reality, and especially with regards to mortality, and would therefore have potential to do real harm to patients.”
Dr. Taggart has disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.
FDA panel rejects vernakalant bid for AFib cardioversion indication
for cardioversion of recent-onset atrial fibrillation (AFib).

It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
for cardioversion of recent-onset atrial fibrillation (AFib).
It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
for cardioversion of recent-onset atrial fibrillation (AFib).
It was the second time before an FDA advisory panel for vernakalant (Brinavess, Correvio International Sàrl), which the agency had declined to approve in 2008 due to safety concerns. That time, however, its advisors had given the agency a decidedly positive recommendation.
Since then, registry data collected for the drug’s resubmission seemed only to raise further safety issues, especially evidence that a single infusion may cause severe hypotension and suppress left ventricular function.
Some members of the Cardiovascular and Renal Drugs Advisory Committee (CRDAC), including a number who voted against approval, expressed hopes for further research aimed at identifying specific AFib patient groups who might safely benefit from vernakalant.
Of note, the drug has long been available for AFib cardioversion in Europe, where there are a number of other pharmacologic options, and was recently approved in Canada.
“We do recognize there’s a significant clinical need here,” observed Paul M. Ridker, MD, MPH, of Harvard Medical School and Brigham and Women’s Hospital Boston, a CRDAC panelist.
The results of the safety study that Correvio presented to the panel were “pretty marginal,” Dr. Ridker said. Given the negative safety signals and the available cardioversion alternatives, he questioned whether vernakalant represented a “substantial advance versus just another option. Right now, I’m not convinced it’s a substantial advance.”
FDA representatives were skeptical about vernakalant when they walked into the meeting room, as noted in briefing documents they had circulated beforehand. The drug’s safety experience under consideration included one case of ventricular arrhythmia and cardiogenic shock in a treated patient without apparent structural heart disease, who subsequently died. That case was much discussed throughout the meeting.
In its resubmission of vernakalant to regulators, Correvio also pointed to a significant unmet need for AFib cardioversion options in the United States, given the few alternatives.
For example, ibutilide is FDA-approved for recent-onset AFib or atrial flutter; but as the company and panelists noted, the drug isn’t often used for that indication. Patients with recent-onset AFib are often put on rate-control meds without cardioversion. Or clinicians may resort to electrical cardioversion, which can be logistically cumbersome and require anesthesia and generally a hospital stay.
Oral or intravenous amiodarone and oral dofetilide, flecainide, and propafenone are guideline-recommended but not actually FDA-approved for recent-onset AFib, the company noted.
Correvio made its “pre-infusion checklist” a core feature of its case. It was designed to guide selection of patients for vernakalant cardioversion based on contraindications such as a systolic blood pressure under 100 mm Hg, severe heart failure, aortic stenosis, severe bradycardia or heart block, or a prolonged QT interval.
In his presentation to the panel, FDA medical officer Preston Dunnmon, MD, said the safety results from the SPECTRUM registry, another main pillar of support for the vernakalant resubmission, “are not reassuring.”
As reasons, Dr. Dunnmon cited likely patient-selection bias and its high proportion of patients who were not prospectively enrolled; 21% were retrospectively entered from records.
Moreover, “the proposed preinfusion checklist will not reliably predict which subjects will experience serious cardiovascular adverse events with vernakalant,” he said.
“Vernakalant has induced harm that cannot be reliably predicted, prevented, or in some cases, treated. In contrast to vernakalant, electrical cardioversion and ibutilide pharmacologic cardioversion can cause adverse events, but these are transient and treatable,” he said.
Many on the panel agreed. “I thought the totality of evidence supported the hypothesis that this drug has a potential for a fatal side effect in a disease that you can live with, potentially, and that there are other treatments for,” said Julia B. Lewis, MD, Vanderbilt Medical Center, Nashville, Tenn., who chaired the CRDAC panel.
“The drug clearly converts atrial fibrillation, although it’s only transient,” observed John H. Alexander, MD, MHSc, Duke University, Durham, N.C., one of the two panelists who voted to recommend approval of vernakalant.
“And, there clearly is a serious safety signal in some populations of patients,” he agreed. “However, I was more reassured by the SPECTRUM data.” There is likely to be a low-risk group of patients for whom vernakalant could represent an important option that “outweighs the relatively low risk of serious complications,” Dr. Alexander said.
“So more work needs to be done to clarify who are the low risk patients where it would be favorable.”
Panelist Matthew Needleman, MD, Walter Reed National Military Medical Center, Bethesda, Md., also voted in favor of approval.
“We’ve all known patients with normal ejection fractions who keep coming in with symptomatic atrial fib, want to get out of it quickly, and get back to their lives. So having an option like this I think would be good for a very select group of patients,” Dr. Needleman said.
But the preinfusion checklist and other potential ways to select low-risk patients for vernakalant could potentially backfire, warned John M. Mandrola, MD, Baptist Medical Associates, Louisville, Ky., from the panel.
The FDA representatives had presented evidence that the drug can seriously depress ventricular function, and that the lower cardiac output is what leads to hypotension, he elaborated in an interview after the meeting.
If the checklist is used to exclude hemodynamically unstable patients from receiving vernakalant, he said, “Then you’re really giving this drug to relatively healthy patients for convenience, to decrease hospitalization or the hospital stay.”
The signal for substantial harm, Dr. Mandrola said, has to be balanced against that modest benefit.
Moreover, those in whom the drug doesn’t work may be left in a worse situation, he proposed. Only about half of patients are successfully converted on vernakalant, the company and FDA data suggested. The other half of patients who don’t achieve sinus rhythm on the drug still must face the significant hazards of depressed ejection fraction and hypotension, a high price to pay for an unsuccessful treatment.
Dr. Mandrola is Chief Cardiology Correspondent for theheart.org | Medscape Cardiology; his disclosure statement states no relevant financial relationships.
This article first appeared on Medscape.com.
DAPA-HF: Dapagliflozin benefits regardless of age, HF severity
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.

“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.

Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.

After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).

The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).
The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
In DAPA-HF, treatment with dapagliflozin met the three critical goals of heart failure management. When used on top of current guideline-directed medical therapy, the treatment reduced mortality, cut hospitalizations, and improved heart failure–related health status – all to a similar extent regardless of patients’ age or symptom severity at entry. These new, post hoc findings provide important, additional data supporting inhibition of sodium-glucose cotransporter (SGLT) 2 with dapagliflozin as the newest foundational pillar of treatment for heart failure with reduced ejection fraction (HFrEF).
The results of the analysis by baseline symptoms severity as measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ) showed similar treatment effects from dapagliflozin regardless of a patient’s baseline KCCQ score, suggesting that the prior report of a blunted effect of dapagliflozin in patients classified at baseline as being in New York Heart Association functional class III or IV compared with class I and II patients was likely a chance finding.
Both the analyses by age and by KCCQ scores were limited by their post hoc status using data collected in a single study. No evidence addresses whether these are class effects for all drugs in the SGLT2-inhibitor class, whether these findings from DAPA-HF are generalizable to real world practice, or whether treatment with dapagliflozin would have similar effects on outcomes if it had been used more often in combination with sacubitril/valsartan. In DAPA-HF, 11% of patients also received sacubitril/valsartan even though existing management guidelines recommend sacubitril/valsartan as the preferred agent for inhibiting the renin-angiotensin system.
It’s also unclear whether patient-reported outcomes such as those measured by the KCCQ will help in sequencing the introduction of drugs for HFrEF patients, or drug selection by patients, providers, payers, and in guidelines.
Carolyn S.P. Lam, MD, is professor of medicine at Duke-National University of Singapore. She has been a consultant to and has received research funding from AstraZeneca and several other companies. She made these comments as designated discussant for the two reports.
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.
“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.
Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.
After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
PHILADELPHIA – The substantial benefits from adding dapagliflozin to guideline-directed medical therapy for patients with heart failure with reduced ejection fraction enrolled in the DAPA-HF trial applied to patients regardless of their age or baseline health status, a pair of new post hoc analyses suggest.
These findings emerged a day after a report that more fully delineated dapagliflozin’s consistent safety and efficacy in patients with heart failure with reduced ejection fraction (HFrEF) regardless of whether they also had type 2 diabetes. One of the new, post hoc analyses reported at the American Heart Association scientific sessions suggested that even the most elderly enrolled patients, 75 years and older, had a similar cut in mortality and acute heart failure exacerbations, compared with younger patients. A second post hoc analysis indicated that patients with severe heart failure symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with mild baseline symptoms, measured by the Kansas City Cardiomyopathy Questionnaire (KCCQ).
The primary results from the DAPA-HF (Dapagliflozin and Prevention of Adverse Outcomes in Heart Failure) trial, first reported in August 2019, showed that among more than 4,700 patients with HFrEF randomized to receive the sodium-glucose cotransporter 2 (SGLT2) inhibitor dapagliflozin (Farxiga) on top of standard HFrEF medications or placebo, those who received dapagliflozin had a statistically significant, 26% decrease in their incidence of the primary study endpoint over a median 18 months, regardless of diabetes status (N Engl J Med. 2019 Nov 21;381[21]:1995-2008).
“These benefits were entirely consistent across the range of ages studied,” extending from patients younger than 55 years to those older than 75 years, John McMurray, MD, said at the meeting. “In many parts of the world, particularly North America and Western Europe, we have an increasingly elderly population. Many patients with heart failure are much older than in clinical trials,” he said.
“The thing of concern is whether elderly patients get as much benefit and tolerate treatment as well as younger patients,” said Dr. McMurray, professor of medical cardiology at the University of Glasgow.
“Dapagliflozin worked across all ages, including some very elderly patients enrolled in the trial,” said Mary Norine Walsh, MD, medical director of the heart failure and transplant program at St. Vincent Heart Center of Indiana in Indianapolis. “Many trials have not looked at age like this. I hope this is a new way to analyze trials to produce more information that can help patients,” she said in an interview.
Quality-of-life outcomes
The other new, post hoc analysis showed that patients with severe HF symptoms at entry into the trial received about as much benefit from the addition of dapagliflozin as did patients with milder baseline symptoms and less impaired function, measured by the KCCQ. Dapagliflozin treatment “improved cardiovascular death and worsening heart failure to a similar extent across the entire range of KCCQ at baseline,” Mikhail N. Kosiborod, MD, said in a separate talk at the meeting. In addition, dapagliflozin treatment increased the rate of small, moderate, and large clinically meaningful improvements in patients’ KCCQ scores across all key domains of the metric, which scores symptom frequency and severity, physical and social limitations, and quality of life, said Dr. Kosiborod, a cardiologist and professor of medicine at the University of Missouri–Kansas City.
After the first 8 months of treatment in the DAPA-HF trial, 58% of the 2,373 patients who received dapagliflozin had a clinically meaningful improvement in their total KCCQ symptom score of at least 5 points, compared with a 51% rate in the 2,371 patients in the control arm, a statistically significant difference. This meant that the number needed to treat with dapagliflozin was 14 patients to produce one additional patient with at least a 5-point KCCQ improvement compared with controls, a “very small” number needed to treat, Dr. Kosiborod said in an interview.
Addition of the KCCQ to the panel of assessments that patients underwent during DAPA-HF reflected an evolved approach to measuring efficacy outcomes in clinical trials by including patient-reported outcomes. Earlier in 2019, the Food and Drug Administration released draft guidance for heart failure drug development that explicitly called for efficacy endpoints in pivotal studies that measure how patients feel and function, and stating that these endpoints can be the basis for new drug approvals.
“To many patients, how they feel matters as much if not more than how long they live,” Dr. Kosiborod noted. The goals of heart failure treatments are not only to extend survival and reduced hospitalizations, but also to improve symptoms, function, and quality of life, he said.
“There is a lot of interest now in having outcomes in heart failure trials that are more meaningful to patients, like feeling better and being able to do more,” noted Dr. Walsh.
The DAPA-HF results also showed that patients had similar rates of reduction in death, heart failure hospitalization, or urgent clinical visits, regardless of how severely they were affected by their heart failure when they began dapagliflozin treatment. The researchers ran an analysis that divided the entire trial population into tertiles based on their KCCQ score on entering the study. Patients in the most severely-affected tertile had a 30% cut in their rate of death or acute heart failure exacerbation on dapagliflozin compared with placebo, while patients in the tertile with the mildest symptoms at baseline had a 38% reduction in their primary outcome incidence compared with controls who received placebo. Concurrently with Dr. Kosiborod’s report, the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044138).
Outcomes by age
Not surprisingly in DAPA-HF, the older patients were, the sicker, Dr. McMurray observed. Of the study’s 1,149 patients (24% of the study cohort) who were at least 75 years old, 62% had chronic kidney disease, compared with a 14% prevalence among the 636 patients younger than age 55. The 75-and-older group showed a steeper, 32% decline in incidence of the primary endpoint – a composite of cardiovascular (CV) death, HF hospitalization, or urgent HF visit requiring intravenous therapy – than in the other studied age groups: a 24% decline in those 65-74 years old, a 29% cut in those 55-64 years old, and a 13% drop in patients younger than 55 years old.
In addition, patients aged 75 years or greater were just as likely as the overall group to show at least a 5-point improvement in their KCCQ Total Symptom Score on dapagliflozin, as well as about the same reduced rate of deterioration compared with placebo as tracked with the KCCQ.
Patients “got as much benefit in terms of symptoms as well as morbidity and mortality,” Dr. McMurray concluded. Concurrently with the meeting report the results appeared in an article online (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044133).
“These data are of critical importance, as improving patient-reported outcomes in heart failure, especially in highly symptomatic patients, is an important goal in drug development,” G. Michael Felker, MD, wrote in an editorial accompanying the two published analyses (Circulation. 2019 Nov 17. doi: 10.1161/CIRCULATIONAHA.119.044578). These new analyses also highlight another attractive feature of dapagliflozin and, apparently, the entire class of SGLT2 inhibitors: They “ ‘play well with others’ when it comes to overlapping intolerances that often limit (either in reality or in perception) optimization of GDMT [guideline-directed medical therapy]. Although SGLT2 inhibitor therapy may lead to volume depletion and require adjustment of diuretics, the SGLT2 inhibitors generally lack some of the other dose-limiting adverse effects (such as renal dysfunction, hyperkalemia, and hypotension) that can make aggressive up-titration of GDMT problematic, particularly in older patients or those with more advanced disease,“ wrote Dr. Felker, professor of medicine at Duke University in Durham, N.C. “We stand at the beginning of a new era of ‘quadruple therapy’ for HFrEF with beta-blockers, an angiotensin receptor neprilysin inhibitor, mineralocorticoid receptor antagonists, and SGLT2 inhibitors,” he concluded.
A version of this article also appears on Medscape.com
REPORTING FROM THE AHA SCIENTIFIC SESSIONS