User login
Dipstick Proteinuria Predicts Acute Kidney Injury in Septic Patients
NATIONAL HARBOR, MD. – De novo dipstick proteinuria accurately predicted acute kidney injury among 328 critically ill septic patients, a retrospective chart study has shown.
With sepsis, inflammation results in increased capillary permeability to plasma proteins, manifesting in an increased excretion of albumin into the urine. Because the production of creatinine from the muscle is reduced in septic patients, relying on changes in serum creatinine could delay the diagnosis of this acute kidney injury (AKI), according to Dr. Javier Neyra.
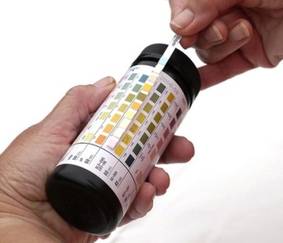
"It is highly important to identify biomarkers that are sensitive, specific, and provide timely and early diagnosis of acute kidney injury before substantial damage has already been done. ... De novo dipstick proteinuria represents a simple, inexpensive biomarker in sepsis with predictive power for AKI," said Dr. Neyra of the Henry Ford Hospital, Detroit.
Charts from a total of 2,252 patients admitted to the intensive care unit for severe sepsis between January 2004 and July 2011 were analyzed retrospectively. Patients with a baseline serum creatinine level greater than 1.5 mg/dL, the presence of dipstick proteinuria within 3 months of the admission date, or common causes of false-positive dipstick tests such as urinary tract infection or gross hematuria were excluded. Of the remaining 470 patients, 328 had undergone dipstick testing on admission. Of those, 46% (152) had dipstick proteinuria.
Serum creatinine increased by at least 0.3 mg/dL in 210 (64%) patients within the first 72 hours of admission, signifying the first stage of acute kidney injury. In this group, new-onset dipstick proteinuria was found in 114 (54%) patients, for a positive predictive value of 75%. Dipstick proteinuria was found in 91 (55%) of 166 patients who met the Acute Kidney Injury Network criteria for AKI, for a positive predictive value of 60%.
After adjustment for age, sex, race, comorbidities, hemodynamic status, and other variables, de novo dipstick proteinuria at the time of admission independently predicted AKI with an odds ratio of 2.3 (95% confidence interval, 1.4-3.8), Dr. Neyra reported in a poster at a meeting sponsored by the National Kidney Foundation.
In an interview, Dr. Neyra explained that such information identifies septic patients who would benefit from more careful monitoring of kidney function and hemodynamic stability. In addition, in those patients it would be important to avoid nephrotoxic agents such as aminoglycoside antibiotics and nonsteroidal anti-inflammatory agents, as well as exposure to contrast material unless it was absolutely necessary.
"The dipstick is a test that you already have in your hospital that you can utilize. It’s simple, inexpensive, and it’s already there," he said.
Dr. Neyra stated that he had no relevant financial disclosures.
NATIONAL HARBOR, MD. – De novo dipstick proteinuria accurately predicted acute kidney injury among 328 critically ill septic patients, a retrospective chart study has shown.
With sepsis, inflammation results in increased capillary permeability to plasma proteins, manifesting in an increased excretion of albumin into the urine. Because the production of creatinine from the muscle is reduced in septic patients, relying on changes in serum creatinine could delay the diagnosis of this acute kidney injury (AKI), according to Dr. Javier Neyra.
"It is highly important to identify biomarkers that are sensitive, specific, and provide timely and early diagnosis of acute kidney injury before substantial damage has already been done. ... De novo dipstick proteinuria represents a simple, inexpensive biomarker in sepsis with predictive power for AKI," said Dr. Neyra of the Henry Ford Hospital, Detroit.
Charts from a total of 2,252 patients admitted to the intensive care unit for severe sepsis between January 2004 and July 2011 were analyzed retrospectively. Patients with a baseline serum creatinine level greater than 1.5 mg/dL, the presence of dipstick proteinuria within 3 months of the admission date, or common causes of false-positive dipstick tests such as urinary tract infection or gross hematuria were excluded. Of the remaining 470 patients, 328 had undergone dipstick testing on admission. Of those, 46% (152) had dipstick proteinuria.
Serum creatinine increased by at least 0.3 mg/dL in 210 (64%) patients within the first 72 hours of admission, signifying the first stage of acute kidney injury. In this group, new-onset dipstick proteinuria was found in 114 (54%) patients, for a positive predictive value of 75%. Dipstick proteinuria was found in 91 (55%) of 166 patients who met the Acute Kidney Injury Network criteria for AKI, for a positive predictive value of 60%.
After adjustment for age, sex, race, comorbidities, hemodynamic status, and other variables, de novo dipstick proteinuria at the time of admission independently predicted AKI with an odds ratio of 2.3 (95% confidence interval, 1.4-3.8), Dr. Neyra reported in a poster at a meeting sponsored by the National Kidney Foundation.
In an interview, Dr. Neyra explained that such information identifies septic patients who would benefit from more careful monitoring of kidney function and hemodynamic stability. In addition, in those patients it would be important to avoid nephrotoxic agents such as aminoglycoside antibiotics and nonsteroidal anti-inflammatory agents, as well as exposure to contrast material unless it was absolutely necessary.
"The dipstick is a test that you already have in your hospital that you can utilize. It’s simple, inexpensive, and it’s already there," he said.
Dr. Neyra stated that he had no relevant financial disclosures.
NATIONAL HARBOR, MD. – De novo dipstick proteinuria accurately predicted acute kidney injury among 328 critically ill septic patients, a retrospective chart study has shown.
With sepsis, inflammation results in increased capillary permeability to plasma proteins, manifesting in an increased excretion of albumin into the urine. Because the production of creatinine from the muscle is reduced in septic patients, relying on changes in serum creatinine could delay the diagnosis of this acute kidney injury (AKI), according to Dr. Javier Neyra.
"It is highly important to identify biomarkers that are sensitive, specific, and provide timely and early diagnosis of acute kidney injury before substantial damage has already been done. ... De novo dipstick proteinuria represents a simple, inexpensive biomarker in sepsis with predictive power for AKI," said Dr. Neyra of the Henry Ford Hospital, Detroit.
Charts from a total of 2,252 patients admitted to the intensive care unit for severe sepsis between January 2004 and July 2011 were analyzed retrospectively. Patients with a baseline serum creatinine level greater than 1.5 mg/dL, the presence of dipstick proteinuria within 3 months of the admission date, or common causes of false-positive dipstick tests such as urinary tract infection or gross hematuria were excluded. Of the remaining 470 patients, 328 had undergone dipstick testing on admission. Of those, 46% (152) had dipstick proteinuria.
Serum creatinine increased by at least 0.3 mg/dL in 210 (64%) patients within the first 72 hours of admission, signifying the first stage of acute kidney injury. In this group, new-onset dipstick proteinuria was found in 114 (54%) patients, for a positive predictive value of 75%. Dipstick proteinuria was found in 91 (55%) of 166 patients who met the Acute Kidney Injury Network criteria for AKI, for a positive predictive value of 60%.
After adjustment for age, sex, race, comorbidities, hemodynamic status, and other variables, de novo dipstick proteinuria at the time of admission independently predicted AKI with an odds ratio of 2.3 (95% confidence interval, 1.4-3.8), Dr. Neyra reported in a poster at a meeting sponsored by the National Kidney Foundation.
In an interview, Dr. Neyra explained that such information identifies septic patients who would benefit from more careful monitoring of kidney function and hemodynamic stability. In addition, in those patients it would be important to avoid nephrotoxic agents such as aminoglycoside antibiotics and nonsteroidal anti-inflammatory agents, as well as exposure to contrast material unless it was absolutely necessary.
"The dipstick is a test that you already have in your hospital that you can utilize. It’s simple, inexpensive, and it’s already there," he said.
Dr. Neyra stated that he had no relevant financial disclosures.
FROM A MEETING SPONSORED BY THE NATIONAL KIDNEY FOUNDATION
Major Finding: Dipstick proteinuria was found in 55% of patients who met the AKIN criteria for acute kidney injury, for a positive predictive value of 60%.
Data Source: The findings come from a retrospective chart study of 328 ICU patients with sepsis who had dipstick testing done on admission.
Disclosures: Dr. Neyra reported having no relevant financial disclosures.
At 2 Years, Resolute Stent Performs Well in Diabetes Patients
PHILADELPHIA – At 2 years post surgery, safety and efficacy outcomes for the Resolute zotarolimus-eluting coronary stent were similar between non–insulin-requiring patients with diabetes and those without diabetes.
Moreover, even the outcomes for the insulin-requiring patients were superior to results seen with other drug-eluting stents (DES) that have been published in the literature, said Dr. Scott W. Lee, of the department of medicine at Loma Linda (Calif.) University and medical director of global clinical research for Medtronic Diabetes, Northridge, Calif.

Patients with diabetes and coronary artery disease have significantly higher event rates because of endothelial dysfunction, impaired platelet function, altered coagulation/fibrinolysis, and increased smooth muscle proliferation. "From a surgery point of view, many patients with diabetes and heart disease were not even candidates for stent placement 20 years ago. They immediately went on to bypass surgery because of the complexity of their coronary artery disease," Dr. Lee commented.
Since then, current revascularization guidelines support the use of drug-eluting stents for the treatment of obstructive coronary artery disease in patients with diabetes. In March of this year, the Food and Drug Administration approved the Resolute stent for patients with symptomatic ischemic heart disease, including those with diabetes.
In a previous pooled analysis of data on 5,130 patients from five of Medtronic’s clinical trials on the Resolute DES, 878 of the total 1,535 patients with diabetes were compared with 1,302 diabetes patients with DES from the literature in order to define a performance standard. The rate of target vessel failure for the 878 Resolute patients at 12 months was found to be 7.8%, significantly less than the 14.5% seen in the literature among diabetes patients who received other drug-eluting stents. At 24 months, target vessel failure was still lower, at 12.1%, Dr. Lee noted.
Now, new 2-year data come from a post hoc descriptive analysis of the 878 from the Medtronic trials who were matched with 1,903 Resolute stent recipients without diabetes. Even with the same extent of vessel disease, the patients with diabetes had higher metabolic risk. They were slightly older (65.2 vs. 63.6 years) and were more likely to have hypertension (88% vs. 73%) and hyperlipidemia (86% vs. 76%). Males made up a smaller proportion of the diabetic group, 66% vs. 74%.
Target lesion failure – defined as cardiac death, target vessel myocardial infarction, or target lesion revascularization – was higher among those with diabetes at 2 years, 9.6% vs. 7.1%. However, that 9.6% is still "far lower" than the 14.5% published rate in the literature for DES in patients with diabetes, Dr. Lee noted.
When the diabetes patients were further divided into the 250 who were insulin requiring (including both type 1 and type 2 patients) and the 628 who were not, there was a continuum of risk: Target lesion revascularization rates by 2 years were similar between the non–insulin-requiring patients, 4.3%, and the nondiabetics, 3.4%. Among the insulin-requiring diabetes patients, 6.5% had target lesion revascularization.
Cardiac death/myocardial infarction rates were also similar between the non–insulin-requiring diabetic group and the nondiabetics, at 3.9% and 4.1% respectively, and were higher among the insulin-requiring diabetes patients, at 8.6%. "Again, this event rate is still lower than what is in the published literature," Dr. Lee noted.
Stent thrombosis rates were low – less than 1% – across all three groups, he said.
"I think it’s reassuring that there are other options available with regard to acute disease management that are specifically outcome driven," Dr. Lee concluded.
There are plans to study the Resolute DES in diabetes patients in a randomized, prospective study, he said in response to an audience member’s question.
In an interview, session moderator Dr. Alan Garber commented, "This study is very interesting. ... Restenosis has really plagued the patients with diabetes. Thus far, drug-eluting stents have not produced the relief [that] we would like to see in patients with diabetes. Things are better, to be sure, but they haven’t normalized restenosis rates in diabetes patients, compared to nondiabetic patients. This looks to be a substantial advancement in improving rates of restenosis that, although not normal, are closer to normal," said Dr. Garber, professor of medicine, biochemistry and molecular biology and molecular and cellular biology at Baylor College, Houston.
But, he added, "It’s not a head-to-head study. It’s a retrospective post-hoc analysis, but it looks to be better and therefore would justify doing a head-to-head prospective trial. I think cardiologists should consider [using Resolute in patients with diabetes], but a definitive randomized prospective head-to-head trial would be required to be sure."
The post hoc analysis was funded by Medtronic, and Dr. Lee is an employee of the company. Dr. Garber is a consultant/advisory board member and/or on the speakers bureaus for Novo Nordisk, Daiichi Sankyo, Merck, Takeda, LipoScience, Boehringer Ingelheim, Sekris, and Lexicon.
PHILADELPHIA – At 2 years post surgery, safety and efficacy outcomes for the Resolute zotarolimus-eluting coronary stent were similar between non–insulin-requiring patients with diabetes and those without diabetes.
Moreover, even the outcomes for the insulin-requiring patients were superior to results seen with other drug-eluting stents (DES) that have been published in the literature, said Dr. Scott W. Lee, of the department of medicine at Loma Linda (Calif.) University and medical director of global clinical research for Medtronic Diabetes, Northridge, Calif.
Patients with diabetes and coronary artery disease have significantly higher event rates because of endothelial dysfunction, impaired platelet function, altered coagulation/fibrinolysis, and increased smooth muscle proliferation. "From a surgery point of view, many patients with diabetes and heart disease were not even candidates for stent placement 20 years ago. They immediately went on to bypass surgery because of the complexity of their coronary artery disease," Dr. Lee commented.
Since then, current revascularization guidelines support the use of drug-eluting stents for the treatment of obstructive coronary artery disease in patients with diabetes. In March of this year, the Food and Drug Administration approved the Resolute stent for patients with symptomatic ischemic heart disease, including those with diabetes.
In a previous pooled analysis of data on 5,130 patients from five of Medtronic’s clinical trials on the Resolute DES, 878 of the total 1,535 patients with diabetes were compared with 1,302 diabetes patients with DES from the literature in order to define a performance standard. The rate of target vessel failure for the 878 Resolute patients at 12 months was found to be 7.8%, significantly less than the 14.5% seen in the literature among diabetes patients who received other drug-eluting stents. At 24 months, target vessel failure was still lower, at 12.1%, Dr. Lee noted.
Now, new 2-year data come from a post hoc descriptive analysis of the 878 from the Medtronic trials who were matched with 1,903 Resolute stent recipients without diabetes. Even with the same extent of vessel disease, the patients with diabetes had higher metabolic risk. They were slightly older (65.2 vs. 63.6 years) and were more likely to have hypertension (88% vs. 73%) and hyperlipidemia (86% vs. 76%). Males made up a smaller proportion of the diabetic group, 66% vs. 74%.
Target lesion failure – defined as cardiac death, target vessel myocardial infarction, or target lesion revascularization – was higher among those with diabetes at 2 years, 9.6% vs. 7.1%. However, that 9.6% is still "far lower" than the 14.5% published rate in the literature for DES in patients with diabetes, Dr. Lee noted.
When the diabetes patients were further divided into the 250 who were insulin requiring (including both type 1 and type 2 patients) and the 628 who were not, there was a continuum of risk: Target lesion revascularization rates by 2 years were similar between the non–insulin-requiring patients, 4.3%, and the nondiabetics, 3.4%. Among the insulin-requiring diabetes patients, 6.5% had target lesion revascularization.
Cardiac death/myocardial infarction rates were also similar between the non–insulin-requiring diabetic group and the nondiabetics, at 3.9% and 4.1% respectively, and were higher among the insulin-requiring diabetes patients, at 8.6%. "Again, this event rate is still lower than what is in the published literature," Dr. Lee noted.
Stent thrombosis rates were low – less than 1% – across all three groups, he said.
"I think it’s reassuring that there are other options available with regard to acute disease management that are specifically outcome driven," Dr. Lee concluded.
There are plans to study the Resolute DES in diabetes patients in a randomized, prospective study, he said in response to an audience member’s question.
In an interview, session moderator Dr. Alan Garber commented, "This study is very interesting. ... Restenosis has really plagued the patients with diabetes. Thus far, drug-eluting stents have not produced the relief [that] we would like to see in patients with diabetes. Things are better, to be sure, but they haven’t normalized restenosis rates in diabetes patients, compared to nondiabetic patients. This looks to be a substantial advancement in improving rates of restenosis that, although not normal, are closer to normal," said Dr. Garber, professor of medicine, biochemistry and molecular biology and molecular and cellular biology at Baylor College, Houston.
But, he added, "It’s not a head-to-head study. It’s a retrospective post-hoc analysis, but it looks to be better and therefore would justify doing a head-to-head prospective trial. I think cardiologists should consider [using Resolute in patients with diabetes], but a definitive randomized prospective head-to-head trial would be required to be sure."
The post hoc analysis was funded by Medtronic, and Dr. Lee is an employee of the company. Dr. Garber is a consultant/advisory board member and/or on the speakers bureaus for Novo Nordisk, Daiichi Sankyo, Merck, Takeda, LipoScience, Boehringer Ingelheim, Sekris, and Lexicon.
PHILADELPHIA – At 2 years post surgery, safety and efficacy outcomes for the Resolute zotarolimus-eluting coronary stent were similar between non–insulin-requiring patients with diabetes and those without diabetes.
Moreover, even the outcomes for the insulin-requiring patients were superior to results seen with other drug-eluting stents (DES) that have been published in the literature, said Dr. Scott W. Lee, of the department of medicine at Loma Linda (Calif.) University and medical director of global clinical research for Medtronic Diabetes, Northridge, Calif.
Patients with diabetes and coronary artery disease have significantly higher event rates because of endothelial dysfunction, impaired platelet function, altered coagulation/fibrinolysis, and increased smooth muscle proliferation. "From a surgery point of view, many patients with diabetes and heart disease were not even candidates for stent placement 20 years ago. They immediately went on to bypass surgery because of the complexity of their coronary artery disease," Dr. Lee commented.
Since then, current revascularization guidelines support the use of drug-eluting stents for the treatment of obstructive coronary artery disease in patients with diabetes. In March of this year, the Food and Drug Administration approved the Resolute stent for patients with symptomatic ischemic heart disease, including those with diabetes.
In a previous pooled analysis of data on 5,130 patients from five of Medtronic’s clinical trials on the Resolute DES, 878 of the total 1,535 patients with diabetes were compared with 1,302 diabetes patients with DES from the literature in order to define a performance standard. The rate of target vessel failure for the 878 Resolute patients at 12 months was found to be 7.8%, significantly less than the 14.5% seen in the literature among diabetes patients who received other drug-eluting stents. At 24 months, target vessel failure was still lower, at 12.1%, Dr. Lee noted.
Now, new 2-year data come from a post hoc descriptive analysis of the 878 from the Medtronic trials who were matched with 1,903 Resolute stent recipients without diabetes. Even with the same extent of vessel disease, the patients with diabetes had higher metabolic risk. They were slightly older (65.2 vs. 63.6 years) and were more likely to have hypertension (88% vs. 73%) and hyperlipidemia (86% vs. 76%). Males made up a smaller proportion of the diabetic group, 66% vs. 74%.
Target lesion failure – defined as cardiac death, target vessel myocardial infarction, or target lesion revascularization – was higher among those with diabetes at 2 years, 9.6% vs. 7.1%. However, that 9.6% is still "far lower" than the 14.5% published rate in the literature for DES in patients with diabetes, Dr. Lee noted.
When the diabetes patients were further divided into the 250 who were insulin requiring (including both type 1 and type 2 patients) and the 628 who were not, there was a continuum of risk: Target lesion revascularization rates by 2 years were similar between the non–insulin-requiring patients, 4.3%, and the nondiabetics, 3.4%. Among the insulin-requiring diabetes patients, 6.5% had target lesion revascularization.
Cardiac death/myocardial infarction rates were also similar between the non–insulin-requiring diabetic group and the nondiabetics, at 3.9% and 4.1% respectively, and were higher among the insulin-requiring diabetes patients, at 8.6%. "Again, this event rate is still lower than what is in the published literature," Dr. Lee noted.
Stent thrombosis rates were low – less than 1% – across all three groups, he said.
"I think it’s reassuring that there are other options available with regard to acute disease management that are specifically outcome driven," Dr. Lee concluded.
There are plans to study the Resolute DES in diabetes patients in a randomized, prospective study, he said in response to an audience member’s question.
In an interview, session moderator Dr. Alan Garber commented, "This study is very interesting. ... Restenosis has really plagued the patients with diabetes. Thus far, drug-eluting stents have not produced the relief [that] we would like to see in patients with diabetes. Things are better, to be sure, but they haven’t normalized restenosis rates in diabetes patients, compared to nondiabetic patients. This looks to be a substantial advancement in improving rates of restenosis that, although not normal, are closer to normal," said Dr. Garber, professor of medicine, biochemistry and molecular biology and molecular and cellular biology at Baylor College, Houston.
But, he added, "It’s not a head-to-head study. It’s a retrospective post-hoc analysis, but it looks to be better and therefore would justify doing a head-to-head prospective trial. I think cardiologists should consider [using Resolute in patients with diabetes], but a definitive randomized prospective head-to-head trial would be required to be sure."
The post hoc analysis was funded by Medtronic, and Dr. Lee is an employee of the company. Dr. Garber is a consultant/advisory board member and/or on the speakers bureaus for Novo Nordisk, Daiichi Sankyo, Merck, Takeda, LipoScience, Boehringer Ingelheim, Sekris, and Lexicon.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION OF CLINICAL ENDOCRINOLOGISTS
Major Finding: Target lesion failure was higher among those with diabetes at 2 years, 9.6% vs. 7.1%. However, that 9.6% is still lower than the 14.5% published rate in the literature for DES in patients with diabetes.
Data Source: Data were taken from a pooled analysis of five clinical trials comprising a total 5,130 recipients of the Resolute drug-eluting stent.
Disclosures: The study was sponsored by Medtronic, and Dr. Lee is an employee of the company
Linagliptin Found Effective, Safe in African Americans With Type 2 Diabetes
PHILADELPHIA – Linagliptin was associated with significant improvements in hyperglycemia in a multicenter, randomized, placebo-controlled, double-blind trial of 226 African-American patients with type 2 diabetes.
"African Americans have a 77% greater likelihood of developing diabetes yet tend to be underrepresented in clinical trials of antidiabetic drugs," said Dr. James R. Thrasher, director of medical investigation at Arkansas Diabetes and Endocrinology Center, Little Rock. "We wanted to look at this group with higher rates of diabetes and higher risk for complications of diabetes."
Linagliptin (Tradjenta) is a once-daily dipeptidyl peptidase-4 inhibitor, licensed for the treatment of type 2 diabetes. Unlike other DPP-4 inhibitors, it does not require dose adjustment for patients with renal or hepatic impairment, Dr. Thrasher noted in an interview at the annual meeting of the American Association of Clinical Endocrinologists.
Patients were randomized to 24 weeks of 5 mg/day linagliptin or placebo. Overall, 54% of the patients were men. The mean age was 54 years and mean body mass index was 32.7 kg/m2. Nearly two-thirds (72%) had hypertension. In the efficacy analysis set of 100 linagliptin and 111 placebo patients, mean baseline hemoglobin A1c was 8.6% for the linagliptin group and 8.7% for the placebo group. Most patients were already taking metformin or a sulfonylurea, which did not change. Twelve percent were treatment naive.
By 24 weeks, mean HbA1c had dropped by 0.88 percentage points with linagliptin and 0.24 with placebo, a highly significant difference (P = 0.0002). Roughly three times more linagliptin patients achieved an HbA1c of less than 7.0% (28.0% vs. 8.7%) and almost twice as many had an HbA1c reduction of 0.5% or more (55.3% vs. 28.3%), Dr. Thrasher reported.
In the safety analysis set comprising 106 linagliptin and 120 placebo patients, most adverse events were mild or moderated and were considered unrelated to the study drug. The most common of these were hyperglycemia (2.8% in the linagliptin group and 9.2% in the placebo group) and nasopharyngitis (3.8% and 5.0%, respectively). Serious adverse events were reported in one linagliptin patient and two placebo patients. Hypoglycemia occurred in three linagliptin patients and one placebo patient. No event required external assistance, he said.
The responses among the African Americans in this study were comparable to those seen in the pivotal trials for linagliptin. "We know from studies of blood pressure medications that African Americans respond differently to different medications. In the area of diabetes, this is kind of landmark. It’s something new for us to look at ethnic groups specifically with a clinical trial for a drug. ... I hope this opens up the door to more [such] research with other drugs," Dr. Thrasher said.
Dr. Thrasher disclosed that he has received research grants and is a speaker for Boehringer Ingelheim, which manufactures linagliptin, and Lilly.
PHILADELPHIA – Linagliptin was associated with significant improvements in hyperglycemia in a multicenter, randomized, placebo-controlled, double-blind trial of 226 African-American patients with type 2 diabetes.
"African Americans have a 77% greater likelihood of developing diabetes yet tend to be underrepresented in clinical trials of antidiabetic drugs," said Dr. James R. Thrasher, director of medical investigation at Arkansas Diabetes and Endocrinology Center, Little Rock. "We wanted to look at this group with higher rates of diabetes and higher risk for complications of diabetes."
Linagliptin (Tradjenta) is a once-daily dipeptidyl peptidase-4 inhibitor, licensed for the treatment of type 2 diabetes. Unlike other DPP-4 inhibitors, it does not require dose adjustment for patients with renal or hepatic impairment, Dr. Thrasher noted in an interview at the annual meeting of the American Association of Clinical Endocrinologists.
Patients were randomized to 24 weeks of 5 mg/day linagliptin or placebo. Overall, 54% of the patients were men. The mean age was 54 years and mean body mass index was 32.7 kg/m2. Nearly two-thirds (72%) had hypertension. In the efficacy analysis set of 100 linagliptin and 111 placebo patients, mean baseline hemoglobin A1c was 8.6% for the linagliptin group and 8.7% for the placebo group. Most patients were already taking metformin or a sulfonylurea, which did not change. Twelve percent were treatment naive.
By 24 weeks, mean HbA1c had dropped by 0.88 percentage points with linagliptin and 0.24 with placebo, a highly significant difference (P = 0.0002). Roughly three times more linagliptin patients achieved an HbA1c of less than 7.0% (28.0% vs. 8.7%) and almost twice as many had an HbA1c reduction of 0.5% or more (55.3% vs. 28.3%), Dr. Thrasher reported.
In the safety analysis set comprising 106 linagliptin and 120 placebo patients, most adverse events were mild or moderated and were considered unrelated to the study drug. The most common of these were hyperglycemia (2.8% in the linagliptin group and 9.2% in the placebo group) and nasopharyngitis (3.8% and 5.0%, respectively). Serious adverse events were reported in one linagliptin patient and two placebo patients. Hypoglycemia occurred in three linagliptin patients and one placebo patient. No event required external assistance, he said.
The responses among the African Americans in this study were comparable to those seen in the pivotal trials for linagliptin. "We know from studies of blood pressure medications that African Americans respond differently to different medications. In the area of diabetes, this is kind of landmark. It’s something new for us to look at ethnic groups specifically with a clinical trial for a drug. ... I hope this opens up the door to more [such] research with other drugs," Dr. Thrasher said.
Dr. Thrasher disclosed that he has received research grants and is a speaker for Boehringer Ingelheim, which manufactures linagliptin, and Lilly.
PHILADELPHIA – Linagliptin was associated with significant improvements in hyperglycemia in a multicenter, randomized, placebo-controlled, double-blind trial of 226 African-American patients with type 2 diabetes.
"African Americans have a 77% greater likelihood of developing diabetes yet tend to be underrepresented in clinical trials of antidiabetic drugs," said Dr. James R. Thrasher, director of medical investigation at Arkansas Diabetes and Endocrinology Center, Little Rock. "We wanted to look at this group with higher rates of diabetes and higher risk for complications of diabetes."
Linagliptin (Tradjenta) is a once-daily dipeptidyl peptidase-4 inhibitor, licensed for the treatment of type 2 diabetes. Unlike other DPP-4 inhibitors, it does not require dose adjustment for patients with renal or hepatic impairment, Dr. Thrasher noted in an interview at the annual meeting of the American Association of Clinical Endocrinologists.
Patients were randomized to 24 weeks of 5 mg/day linagliptin or placebo. Overall, 54% of the patients were men. The mean age was 54 years and mean body mass index was 32.7 kg/m2. Nearly two-thirds (72%) had hypertension. In the efficacy analysis set of 100 linagliptin and 111 placebo patients, mean baseline hemoglobin A1c was 8.6% for the linagliptin group and 8.7% for the placebo group. Most patients were already taking metformin or a sulfonylurea, which did not change. Twelve percent were treatment naive.
By 24 weeks, mean HbA1c had dropped by 0.88 percentage points with linagliptin and 0.24 with placebo, a highly significant difference (P = 0.0002). Roughly three times more linagliptin patients achieved an HbA1c of less than 7.0% (28.0% vs. 8.7%) and almost twice as many had an HbA1c reduction of 0.5% or more (55.3% vs. 28.3%), Dr. Thrasher reported.
In the safety analysis set comprising 106 linagliptin and 120 placebo patients, most adverse events were mild or moderated and were considered unrelated to the study drug. The most common of these were hyperglycemia (2.8% in the linagliptin group and 9.2% in the placebo group) and nasopharyngitis (3.8% and 5.0%, respectively). Serious adverse events were reported in one linagliptin patient and two placebo patients. Hypoglycemia occurred in three linagliptin patients and one placebo patient. No event required external assistance, he said.
The responses among the African Americans in this study were comparable to those seen in the pivotal trials for linagliptin. "We know from studies of blood pressure medications that African Americans respond differently to different medications. In the area of diabetes, this is kind of landmark. It’s something new for us to look at ethnic groups specifically with a clinical trial for a drug. ... I hope this opens up the door to more [such] research with other drugs," Dr. Thrasher said.
Dr. Thrasher disclosed that he has received research grants and is a speaker for Boehringer Ingelheim, which manufactures linagliptin, and Lilly.
FROM THE ANNUAL MEETING OF THE AMERICAN ASSOCIATION OF CLINICAL ENDOCRINOLOGISTS
Fidaxomicin Noninferior to Vancomycin as C. diff. Cure, Superior at 4 Weeks
LONDON – Fidaxomicin was noninferior to vancomycin in achieving clinical cure of Clostridium difficile infection after 10 days of treatment but was superior to vancomycin in attaining lower recurrence rates and higher sustained response rates at 4 weeks.
The findings come from a multicenter, randomized, controlled, double-blind study conducted in seven European countries, Canada, and the United States. It was funded by Optimer Pharmaceuticals, manufacturer of fidaxomicin, a novel macrocyclic antibiotic with a narrow spectrum of activity against gram-positive bacteria and minimal activity against normal gut flora. It was approved for treatment of C. difficile infection in the United States in May 2011 and in Europe in December 2011. A previous phase III trial in Canada and the United States showed fidaxomicin to be noninferior to vancomycin for clinical cure and superior for sustained response at 4 weeks after treatment completion (N. Engl. J. Med. 2011;364:422-31).
The current study, identical in design and procedures, compared the efficacy of fidaxomicin in Europe as well as in the United States and Canada, said Dr. Oliver A. Cornely of University Hospital, Cologne, Germany. The findings were published online prior to the ECCMID meeting (Lancet Infect. Dis. 2012;12:281-9).
The 509 patients included in the intent to treat analysis were all aged 16 years or older, had more than three unformed bowel movements per 24 hours, had C. diff infection confirmed by toxin A and/or B in stool, and had a first episode or recurrence within 90 days. They were randomized to either twice-daily 200-mg doses of fidaxomicin with twice-daily intervening placebo or the standard 125-mg doses of vancomycin four times daily.
The 198 patients from Europe were slightly older than those from the United States and Canada (66.9 vs. 61.2 years), and fewer were women (54.5% vs. 65%). More of the European group were inpatients at the time of study enrollment (84.3% vs. 57.9%), but fewer in Europe had had a previous episode of C. diff within the prior 90 days (10.1% vs. 18.0%). As expected, the hypervirulent, fluoroquinolone-resistant C. difficile strain NAP1/BI/027 was more common in the U.S./Canada (45.9% vs. 10.4%).
Use of concomitant antibiotics to treat other infections was more common in Europe than in the United States during both treatment and follow-up (34.8% vs. 26.7% for either time period), Dr. Cornely reported.
For the primary study end point ("resolution of diarrhea and no further need for treatment"), fidaxomicin was noninferior to vancomycin, 87.7% vs. 86.8%, well within the prespecified 10% margin. However, fidaxomicin was superior in recurrence, defined as return of diarrhea within 4 weeks of completing therapy plus a positive toxin test (12.7% vs. 26.9%). Fidaxomicin was also superior for sustained response, defined as clinical cure maintained for 4 weeks (76.6% vs. 63.4%). The median time to resolution of diarrhea did not differ between fidaxomicin and vancomycin (56 vs. 58 hours, respectively). (Time to resolution was defined as the number of hours from the first dose of the study drug to the last unformed bowel movement preceding 2 days of three or fewer unformed stools per day.)
Resolution of diarrhea was significantly delayed among patients who received antibiotics to treat other infections at the same time as they were being treated for CDI (median time to resolution 92 hours vs. 54 hours). Among the patients who were receiving concomitant antibiotics, fidaxomicin was superior to vancomycin in clinical cure rate (90.2% of 51 vs. 73.3% of 45), Dr. Cornely reported.
When Europe and the U.S./Canada were analyzed separately, fidaxomicin remained noninferior for clinical cure and superior for recurrence and sustained response. However, recurrence rates overall were slightly higher and sustained response slightly lower in the U.S./Canada, compared with Europe, although the confidence intervals were broad. Early resolution (in less than 72 hours) was more common in Europe (51 vs. 60 hours), but the overall trend was not significantly different for Europe and North America, he said.
There were no significant differences in cure rates between fidaxomicin and vancomycin for other subgroups, including those stratified by age, prior C. diff. infection, inpatient/outpatient, severity, strain, or location.
No fidaxomicin-resistant clones were isolated at baseline or at the end of treatment, although a single isolate recovered at recurrence in Canada has reduced susceptibility, Dr. Cornely noted.
The study was funded by Optimer Pharmaceuticals. Dr. Cornely disclosed that he has received research grants or lecture honoraria or is an adviser to Optimer and multiple other firms, including Actelion, Astellas, Basilea, Bayer, BioCryst, Celgene, F2G, Genzyme, Gilead, Merck, Miltenyi, Pfizer, Quintiles, and ViroPharma.
Vancomycin has hitherto been the best treatment option for Clostridium difficile infection (CDI). Of every 10 patients with this infection, about 9 usually respond to treatment. However, two or three have recurrent symptoms, primarily in the first month after therapy. Therefore, treatment is inadequate for roughly 4 of every 10 patients with C. difficile infection. Furthermore, elderly people – who often have comorbidities and acute illnesses for which antibiotics are prescribed – are most frequently infected and are most at risk of treatment failure.
Recurrence of CDI might be associated with the amount of perturbation of gut flora, possibly because C. difficile regrowth and toxin expression is not likely when disruption is low. Protective effects of treatment that causes little damage to the gut flora such as fidaxomicin might therefore decrease when baseline flora disruption is high. Patients infected with C. difficile who have several recurrences could be less likely to benefit from the few effects on gut flora associated with fidaxomicin.
Other patients at the greatest risk of recurrent CDI could benefit most from fidaxomicin as a new treatment option. Identification of these individuals is not straightforward but might include age greater than 65 years, severe disease, and receipt of concomitant antibiotics after treatment for CDI.
Generally, the baseline level of antibiotic susceptibility of C. difficile and clinical outcome are not associated. No resistance to either fidaxomicin or vancomycin was detected during treatment in phase III studies. Nevertheless, monitoring of emergence of fidaxomicin resistance will be important. Dedicated surveillance programs will be needed because susceptibility testing of C. difficile isolates is not routine practice.
Improvements are still to be made in treatment of CDI. A prospectively validated, simple but accurate scoring system, or laboratory test, is still needed to predict recurrent infections, to help clinicians to make informed treatment choices, and to justify the high cost of fidaxomicin. Shortened hospital stays or symptom duration, or reduction in excess mortality associated with CDI remain elusive treatment goals. The next new drug for treatment of this infection will probably not be licensed for the next 4-5 years. In the meantime, fidaxomicin could be a much needed step forward.
Dr. Mark H. Wilcox, microbiology department, Old Medical School, Leeds General Infirmary, and University of Leeds, England. He disclosed that in the past 2 years he has been a consultant to or has received research support or lecture honoraria from Optimer, which sponsored the study, as well as Actelion, Astellas, Merck, Novacta, Pfizer, Summit, the Medicines Company, Cubist, Optimer, Pfizer, and Sanofi Pasteur.
Vancomycin has hitherto been the best treatment option for Clostridium difficile infection (CDI). Of every 10 patients with this infection, about 9 usually respond to treatment. However, two or three have recurrent symptoms, primarily in the first month after therapy. Therefore, treatment is inadequate for roughly 4 of every 10 patients with C. difficile infection. Furthermore, elderly people – who often have comorbidities and acute illnesses for which antibiotics are prescribed – are most frequently infected and are most at risk of treatment failure.
Recurrence of CDI might be associated with the amount of perturbation of gut flora, possibly because C. difficile regrowth and toxin expression is not likely when disruption is low. Protective effects of treatment that causes little damage to the gut flora such as fidaxomicin might therefore decrease when baseline flora disruption is high. Patients infected with C. difficile who have several recurrences could be less likely to benefit from the few effects on gut flora associated with fidaxomicin.
Other patients at the greatest risk of recurrent CDI could benefit most from fidaxomicin as a new treatment option. Identification of these individuals is not straightforward but might include age greater than 65 years, severe disease, and receipt of concomitant antibiotics after treatment for CDI.
Generally, the baseline level of antibiotic susceptibility of C. difficile and clinical outcome are not associated. No resistance to either fidaxomicin or vancomycin was detected during treatment in phase III studies. Nevertheless, monitoring of emergence of fidaxomicin resistance will be important. Dedicated surveillance programs will be needed because susceptibility testing of C. difficile isolates is not routine practice.
Improvements are still to be made in treatment of CDI. A prospectively validated, simple but accurate scoring system, or laboratory test, is still needed to predict recurrent infections, to help clinicians to make informed treatment choices, and to justify the high cost of fidaxomicin. Shortened hospital stays or symptom duration, or reduction in excess mortality associated with CDI remain elusive treatment goals. The next new drug for treatment of this infection will probably not be licensed for the next 4-5 years. In the meantime, fidaxomicin could be a much needed step forward.
Dr. Mark H. Wilcox, microbiology department, Old Medical School, Leeds General Infirmary, and University of Leeds, England. He disclosed that in the past 2 years he has been a consultant to or has received research support or lecture honoraria from Optimer, which sponsored the study, as well as Actelion, Astellas, Merck, Novacta, Pfizer, Summit, the Medicines Company, Cubist, Optimer, Pfizer, and Sanofi Pasteur.
Vancomycin has hitherto been the best treatment option for Clostridium difficile infection (CDI). Of every 10 patients with this infection, about 9 usually respond to treatment. However, two or three have recurrent symptoms, primarily in the first month after therapy. Therefore, treatment is inadequate for roughly 4 of every 10 patients with C. difficile infection. Furthermore, elderly people – who often have comorbidities and acute illnesses for which antibiotics are prescribed – are most frequently infected and are most at risk of treatment failure.
Recurrence of CDI might be associated with the amount of perturbation of gut flora, possibly because C. difficile regrowth and toxin expression is not likely when disruption is low. Protective effects of treatment that causes little damage to the gut flora such as fidaxomicin might therefore decrease when baseline flora disruption is high. Patients infected with C. difficile who have several recurrences could be less likely to benefit from the few effects on gut flora associated with fidaxomicin.
Other patients at the greatest risk of recurrent CDI could benefit most from fidaxomicin as a new treatment option. Identification of these individuals is not straightforward but might include age greater than 65 years, severe disease, and receipt of concomitant antibiotics after treatment for CDI.
Generally, the baseline level of antibiotic susceptibility of C. difficile and clinical outcome are not associated. No resistance to either fidaxomicin or vancomycin was detected during treatment in phase III studies. Nevertheless, monitoring of emergence of fidaxomicin resistance will be important. Dedicated surveillance programs will be needed because susceptibility testing of C. difficile isolates is not routine practice.
Improvements are still to be made in treatment of CDI. A prospectively validated, simple but accurate scoring system, or laboratory test, is still needed to predict recurrent infections, to help clinicians to make informed treatment choices, and to justify the high cost of fidaxomicin. Shortened hospital stays or symptom duration, or reduction in excess mortality associated with CDI remain elusive treatment goals. The next new drug for treatment of this infection will probably not be licensed for the next 4-5 years. In the meantime, fidaxomicin could be a much needed step forward.
Dr. Mark H. Wilcox, microbiology department, Old Medical School, Leeds General Infirmary, and University of Leeds, England. He disclosed that in the past 2 years he has been a consultant to or has received research support or lecture honoraria from Optimer, which sponsored the study, as well as Actelion, Astellas, Merck, Novacta, Pfizer, Summit, the Medicines Company, Cubist, Optimer, Pfizer, and Sanofi Pasteur.
LONDON – Fidaxomicin was noninferior to vancomycin in achieving clinical cure of Clostridium difficile infection after 10 days of treatment but was superior to vancomycin in attaining lower recurrence rates and higher sustained response rates at 4 weeks.
The findings come from a multicenter, randomized, controlled, double-blind study conducted in seven European countries, Canada, and the United States. It was funded by Optimer Pharmaceuticals, manufacturer of fidaxomicin, a novel macrocyclic antibiotic with a narrow spectrum of activity against gram-positive bacteria and minimal activity against normal gut flora. It was approved for treatment of C. difficile infection in the United States in May 2011 and in Europe in December 2011. A previous phase III trial in Canada and the United States showed fidaxomicin to be noninferior to vancomycin for clinical cure and superior for sustained response at 4 weeks after treatment completion (N. Engl. J. Med. 2011;364:422-31).
The current study, identical in design and procedures, compared the efficacy of fidaxomicin in Europe as well as in the United States and Canada, said Dr. Oliver A. Cornely of University Hospital, Cologne, Germany. The findings were published online prior to the ECCMID meeting (Lancet Infect. Dis. 2012;12:281-9).
The 509 patients included in the intent to treat analysis were all aged 16 years or older, had more than three unformed bowel movements per 24 hours, had C. diff infection confirmed by toxin A and/or B in stool, and had a first episode or recurrence within 90 days. They were randomized to either twice-daily 200-mg doses of fidaxomicin with twice-daily intervening placebo or the standard 125-mg doses of vancomycin four times daily.
The 198 patients from Europe were slightly older than those from the United States and Canada (66.9 vs. 61.2 years), and fewer were women (54.5% vs. 65%). More of the European group were inpatients at the time of study enrollment (84.3% vs. 57.9%), but fewer in Europe had had a previous episode of C. diff within the prior 90 days (10.1% vs. 18.0%). As expected, the hypervirulent, fluoroquinolone-resistant C. difficile strain NAP1/BI/027 was more common in the U.S./Canada (45.9% vs. 10.4%).
Use of concomitant antibiotics to treat other infections was more common in Europe than in the United States during both treatment and follow-up (34.8% vs. 26.7% for either time period), Dr. Cornely reported.
For the primary study end point ("resolution of diarrhea and no further need for treatment"), fidaxomicin was noninferior to vancomycin, 87.7% vs. 86.8%, well within the prespecified 10% margin. However, fidaxomicin was superior in recurrence, defined as return of diarrhea within 4 weeks of completing therapy plus a positive toxin test (12.7% vs. 26.9%). Fidaxomicin was also superior for sustained response, defined as clinical cure maintained for 4 weeks (76.6% vs. 63.4%). The median time to resolution of diarrhea did not differ between fidaxomicin and vancomycin (56 vs. 58 hours, respectively). (Time to resolution was defined as the number of hours from the first dose of the study drug to the last unformed bowel movement preceding 2 days of three or fewer unformed stools per day.)
Resolution of diarrhea was significantly delayed among patients who received antibiotics to treat other infections at the same time as they were being treated for CDI (median time to resolution 92 hours vs. 54 hours). Among the patients who were receiving concomitant antibiotics, fidaxomicin was superior to vancomycin in clinical cure rate (90.2% of 51 vs. 73.3% of 45), Dr. Cornely reported.
When Europe and the U.S./Canada were analyzed separately, fidaxomicin remained noninferior for clinical cure and superior for recurrence and sustained response. However, recurrence rates overall were slightly higher and sustained response slightly lower in the U.S./Canada, compared with Europe, although the confidence intervals were broad. Early resolution (in less than 72 hours) was more common in Europe (51 vs. 60 hours), but the overall trend was not significantly different for Europe and North America, he said.
There were no significant differences in cure rates between fidaxomicin and vancomycin for other subgroups, including those stratified by age, prior C. diff. infection, inpatient/outpatient, severity, strain, or location.
No fidaxomicin-resistant clones were isolated at baseline or at the end of treatment, although a single isolate recovered at recurrence in Canada has reduced susceptibility, Dr. Cornely noted.
The study was funded by Optimer Pharmaceuticals. Dr. Cornely disclosed that he has received research grants or lecture honoraria or is an adviser to Optimer and multiple other firms, including Actelion, Astellas, Basilea, Bayer, BioCryst, Celgene, F2G, Genzyme, Gilead, Merck, Miltenyi, Pfizer, Quintiles, and ViroPharma.
LONDON – Fidaxomicin was noninferior to vancomycin in achieving clinical cure of Clostridium difficile infection after 10 days of treatment but was superior to vancomycin in attaining lower recurrence rates and higher sustained response rates at 4 weeks.
The findings come from a multicenter, randomized, controlled, double-blind study conducted in seven European countries, Canada, and the United States. It was funded by Optimer Pharmaceuticals, manufacturer of fidaxomicin, a novel macrocyclic antibiotic with a narrow spectrum of activity against gram-positive bacteria and minimal activity against normal gut flora. It was approved for treatment of C. difficile infection in the United States in May 2011 and in Europe in December 2011. A previous phase III trial in Canada and the United States showed fidaxomicin to be noninferior to vancomycin for clinical cure and superior for sustained response at 4 weeks after treatment completion (N. Engl. J. Med. 2011;364:422-31).
The current study, identical in design and procedures, compared the efficacy of fidaxomicin in Europe as well as in the United States and Canada, said Dr. Oliver A. Cornely of University Hospital, Cologne, Germany. The findings were published online prior to the ECCMID meeting (Lancet Infect. Dis. 2012;12:281-9).
The 509 patients included in the intent to treat analysis were all aged 16 years or older, had more than three unformed bowel movements per 24 hours, had C. diff infection confirmed by toxin A and/or B in stool, and had a first episode or recurrence within 90 days. They were randomized to either twice-daily 200-mg doses of fidaxomicin with twice-daily intervening placebo or the standard 125-mg doses of vancomycin four times daily.
The 198 patients from Europe were slightly older than those from the United States and Canada (66.9 vs. 61.2 years), and fewer were women (54.5% vs. 65%). More of the European group were inpatients at the time of study enrollment (84.3% vs. 57.9%), but fewer in Europe had had a previous episode of C. diff within the prior 90 days (10.1% vs. 18.0%). As expected, the hypervirulent, fluoroquinolone-resistant C. difficile strain NAP1/BI/027 was more common in the U.S./Canada (45.9% vs. 10.4%).
Use of concomitant antibiotics to treat other infections was more common in Europe than in the United States during both treatment and follow-up (34.8% vs. 26.7% for either time period), Dr. Cornely reported.
For the primary study end point ("resolution of diarrhea and no further need for treatment"), fidaxomicin was noninferior to vancomycin, 87.7% vs. 86.8%, well within the prespecified 10% margin. However, fidaxomicin was superior in recurrence, defined as return of diarrhea within 4 weeks of completing therapy plus a positive toxin test (12.7% vs. 26.9%). Fidaxomicin was also superior for sustained response, defined as clinical cure maintained for 4 weeks (76.6% vs. 63.4%). The median time to resolution of diarrhea did not differ between fidaxomicin and vancomycin (56 vs. 58 hours, respectively). (Time to resolution was defined as the number of hours from the first dose of the study drug to the last unformed bowel movement preceding 2 days of three or fewer unformed stools per day.)
Resolution of diarrhea was significantly delayed among patients who received antibiotics to treat other infections at the same time as they were being treated for CDI (median time to resolution 92 hours vs. 54 hours). Among the patients who were receiving concomitant antibiotics, fidaxomicin was superior to vancomycin in clinical cure rate (90.2% of 51 vs. 73.3% of 45), Dr. Cornely reported.
When Europe and the U.S./Canada were analyzed separately, fidaxomicin remained noninferior for clinical cure and superior for recurrence and sustained response. However, recurrence rates overall were slightly higher and sustained response slightly lower in the U.S./Canada, compared with Europe, although the confidence intervals were broad. Early resolution (in less than 72 hours) was more common in Europe (51 vs. 60 hours), but the overall trend was not significantly different for Europe and North America, he said.
There were no significant differences in cure rates between fidaxomicin and vancomycin for other subgroups, including those stratified by age, prior C. diff. infection, inpatient/outpatient, severity, strain, or location.
No fidaxomicin-resistant clones were isolated at baseline or at the end of treatment, although a single isolate recovered at recurrence in Canada has reduced susceptibility, Dr. Cornely noted.
The study was funded by Optimer Pharmaceuticals. Dr. Cornely disclosed that he has received research grants or lecture honoraria or is an adviser to Optimer and multiple other firms, including Actelion, Astellas, Basilea, Bayer, BioCryst, Celgene, F2G, Genzyme, Gilead, Merck, Miltenyi, Pfizer, Quintiles, and ViroPharma.
FROM THE EUROPEAN CONGRESS OF CLINICAL MICROBIOLOGY AND INFECTIOUS DISEASES
Major Finding: Fidaxomicin was noninferior to vancomycin in curing C. difficile, 87.7% vs. 86.8%. However, fidaxomicin was superior in terms of lower recurrence at 4 weeks (12.7% vs. 26.9%) and in sustained response at 4 weeks (76.6% vs. 63.4%).
Data Source: The findings come from a multicenter, randomized, controlled, double-blind study of 509 patients with CDI, conducted in Europe (seven countries), Canada, and the United States.
Disclosures: The study was funded by Optimer Pharmaceuticals. Dr. Cornely disclosed that he has received research grants or lecture honoraria or is an advisor to Optimer, as well as Actelion, Astellas, Basilea, Bayer, Biocryst, Celgene, F2G, Genzyme, Gilead, Merck, Miltenyi, Pfizer, Quintiles, and Viropharma.
Diabetes Death Rates Drop
Excess all-cause mortality and cardiovascular mortality dropped substantially among adults with diabetes between 1997 and 2006, according to a report in Diabetes Care.
Data from the National Health Interview Surveys linked to the National Death Index on a nationally representative sample of 242,383 adults aged 18 years and older with and without diabetes (approximately 30,000 per year) demonstrated a 40% reduction in excess all-cause mortality and a 23% drop in excess cardiovascular disease (CVD) mortality among those with diabetes. These rates of improvement have exceeded those of the nondiabetic population, resulting in more than a 50% reduction in the excess death rates that have been repeatedly attributed to diabetes, Edward W. Gregg, Ph.D., and his associates said (Diabetes Care 2012;35:1252–7 [doi: 10.2337/dc11-1162]).
"These encouraging findings, however, suggest that diabetes prevalence is likely to rise in the future if diabetes incidence is not curtailed," said Dr. Gregg, who is from the Division of Diabetes Translation, Centers for Disease Control and Prevention, and his associates, two of whom are with the National Institute of Diabetes and Digestive and Kidney Diseases.
Among the population with diabetes, there were consistent increases over time in the levels of education, income, and obesity, as well as decreases in the proportion of smokers and those with sedentary behavior and difficulty walking. There were no significant changes in age, race/ethnicity, history of CVD, or diabetes duration. Most of those demographic trends over time were similar for the nondiabetic population.
From 1997-1998 to 2003-2004, 3-year CVD death rates declined among those diagnosed with diabetes by 4 deaths/1,000 person-years, from 9.5 to 5.6. In multivariate analyses adjusting for age, sex, race/ethnicity, and diabetes duration, diabetic adults in the most recent sample (2003-2004) had 40% lower CVD mortality (hazard rate ratio, 0.60) and 23% lower all-cause mortality (0.77) than did those individuals in the earliest sample (1997-1998). There were no significant changes in the rates of cancer mortality in persons either with or without diabetes, Dr. Gregg and his associates noted.
Deaths due to CVD also dropped in the nondiabetic population during that time, but to a lesser degree than in the diabetic population, from 3.7 to 3.3 deaths/1,000. Furthermore, there was not a significant decline in all-cause mortality among those without diabetes. As a result, both excess CVD and all-cause death rates associated with diabetes – as compared to those without diabetes – declined significantly (by 3.5 and 4.7 deaths/1,000 person-years, respectively).
All-cause and CVD death rates in the diabetic population declined among both men and women, although there was a slightly greater magnitude of decline in men than in women. Again, the trends for both men and women paralleled less dramatic reductions in the nondiabetic population.
"Although excess mortality risk remains high – about 2 deaths per 1,000 due to CVD and about 6 all-cause deaths – this excess risk is now considerably lower than previous reports and consistent with improvements in several risk factors, complications, and indicators of medical care and representative of gradual, ongoing improvement in health for people with diagnosed diabetes," Dr. Gregg and his associates wrote.
However, they pointed out, models have indicated that declining mortality among people with diabetes is expected to lead to a substantial increase in the prevalence of people living with the condition. "Thus, the excess mortality associated with diabetes, though declining, will be spread among a considerably greater proportion of the population. Ultimately, this means that the need for vigilant efforts to prevent vascular and neuropathic complication and early mortality associated with diabetes, along with efforts to reduce diabetes incidence, will continue to be major demands into the future."
No potential relevant conflicts of interest were reported.
Excess all-cause mortality and cardiovascular mortality dropped substantially among adults with diabetes between 1997 and 2006, according to a report in Diabetes Care.
Data from the National Health Interview Surveys linked to the National Death Index on a nationally representative sample of 242,383 adults aged 18 years and older with and without diabetes (approximately 30,000 per year) demonstrated a 40% reduction in excess all-cause mortality and a 23% drop in excess cardiovascular disease (CVD) mortality among those with diabetes. These rates of improvement have exceeded those of the nondiabetic population, resulting in more than a 50% reduction in the excess death rates that have been repeatedly attributed to diabetes, Edward W. Gregg, Ph.D., and his associates said (Diabetes Care 2012;35:1252–7 [doi: 10.2337/dc11-1162]).
"These encouraging findings, however, suggest that diabetes prevalence is likely to rise in the future if diabetes incidence is not curtailed," said Dr. Gregg, who is from the Division of Diabetes Translation, Centers for Disease Control and Prevention, and his associates, two of whom are with the National Institute of Diabetes and Digestive and Kidney Diseases.
Among the population with diabetes, there were consistent increases over time in the levels of education, income, and obesity, as well as decreases in the proportion of smokers and those with sedentary behavior and difficulty walking. There were no significant changes in age, race/ethnicity, history of CVD, or diabetes duration. Most of those demographic trends over time were similar for the nondiabetic population.
From 1997-1998 to 2003-2004, 3-year CVD death rates declined among those diagnosed with diabetes by 4 deaths/1,000 person-years, from 9.5 to 5.6. In multivariate analyses adjusting for age, sex, race/ethnicity, and diabetes duration, diabetic adults in the most recent sample (2003-2004) had 40% lower CVD mortality (hazard rate ratio, 0.60) and 23% lower all-cause mortality (0.77) than did those individuals in the earliest sample (1997-1998). There were no significant changes in the rates of cancer mortality in persons either with or without diabetes, Dr. Gregg and his associates noted.
Deaths due to CVD also dropped in the nondiabetic population during that time, but to a lesser degree than in the diabetic population, from 3.7 to 3.3 deaths/1,000. Furthermore, there was not a significant decline in all-cause mortality among those without diabetes. As a result, both excess CVD and all-cause death rates associated with diabetes – as compared to those without diabetes – declined significantly (by 3.5 and 4.7 deaths/1,000 person-years, respectively).
All-cause and CVD death rates in the diabetic population declined among both men and women, although there was a slightly greater magnitude of decline in men than in women. Again, the trends for both men and women paralleled less dramatic reductions in the nondiabetic population.
"Although excess mortality risk remains high – about 2 deaths per 1,000 due to CVD and about 6 all-cause deaths – this excess risk is now considerably lower than previous reports and consistent with improvements in several risk factors, complications, and indicators of medical care and representative of gradual, ongoing improvement in health for people with diagnosed diabetes," Dr. Gregg and his associates wrote.
However, they pointed out, models have indicated that declining mortality among people with diabetes is expected to lead to a substantial increase in the prevalence of people living with the condition. "Thus, the excess mortality associated with diabetes, though declining, will be spread among a considerably greater proportion of the population. Ultimately, this means that the need for vigilant efforts to prevent vascular and neuropathic complication and early mortality associated with diabetes, along with efforts to reduce diabetes incidence, will continue to be major demands into the future."
No potential relevant conflicts of interest were reported.
Excess all-cause mortality and cardiovascular mortality dropped substantially among adults with diabetes between 1997 and 2006, according to a report in Diabetes Care.
Data from the National Health Interview Surveys linked to the National Death Index on a nationally representative sample of 242,383 adults aged 18 years and older with and without diabetes (approximately 30,000 per year) demonstrated a 40% reduction in excess all-cause mortality and a 23% drop in excess cardiovascular disease (CVD) mortality among those with diabetes. These rates of improvement have exceeded those of the nondiabetic population, resulting in more than a 50% reduction in the excess death rates that have been repeatedly attributed to diabetes, Edward W. Gregg, Ph.D., and his associates said (Diabetes Care 2012;35:1252–7 [doi: 10.2337/dc11-1162]).
"These encouraging findings, however, suggest that diabetes prevalence is likely to rise in the future if diabetes incidence is not curtailed," said Dr. Gregg, who is from the Division of Diabetes Translation, Centers for Disease Control and Prevention, and his associates, two of whom are with the National Institute of Diabetes and Digestive and Kidney Diseases.
Among the population with diabetes, there were consistent increases over time in the levels of education, income, and obesity, as well as decreases in the proportion of smokers and those with sedentary behavior and difficulty walking. There were no significant changes in age, race/ethnicity, history of CVD, or diabetes duration. Most of those demographic trends over time were similar for the nondiabetic population.
From 1997-1998 to 2003-2004, 3-year CVD death rates declined among those diagnosed with diabetes by 4 deaths/1,000 person-years, from 9.5 to 5.6. In multivariate analyses adjusting for age, sex, race/ethnicity, and diabetes duration, diabetic adults in the most recent sample (2003-2004) had 40% lower CVD mortality (hazard rate ratio, 0.60) and 23% lower all-cause mortality (0.77) than did those individuals in the earliest sample (1997-1998). There were no significant changes in the rates of cancer mortality in persons either with or without diabetes, Dr. Gregg and his associates noted.
Deaths due to CVD also dropped in the nondiabetic population during that time, but to a lesser degree than in the diabetic population, from 3.7 to 3.3 deaths/1,000. Furthermore, there was not a significant decline in all-cause mortality among those without diabetes. As a result, both excess CVD and all-cause death rates associated with diabetes – as compared to those without diabetes – declined significantly (by 3.5 and 4.7 deaths/1,000 person-years, respectively).
All-cause and CVD death rates in the diabetic population declined among both men and women, although there was a slightly greater magnitude of decline in men than in women. Again, the trends for both men and women paralleled less dramatic reductions in the nondiabetic population.
"Although excess mortality risk remains high – about 2 deaths per 1,000 due to CVD and about 6 all-cause deaths – this excess risk is now considerably lower than previous reports and consistent with improvements in several risk factors, complications, and indicators of medical care and representative of gradual, ongoing improvement in health for people with diagnosed diabetes," Dr. Gregg and his associates wrote.
However, they pointed out, models have indicated that declining mortality among people with diabetes is expected to lead to a substantial increase in the prevalence of people living with the condition. "Thus, the excess mortality associated with diabetes, though declining, will be spread among a considerably greater proportion of the population. Ultimately, this means that the need for vigilant efforts to prevent vascular and neuropathic complication and early mortality associated with diabetes, along with efforts to reduce diabetes incidence, will continue to be major demands into the future."
No potential relevant conflicts of interest were reported.
FROM DIABETES CARE
Hospital-Acquired Infections Common and Costly
LONDON – Hospital-acquired infections occurred in more than one in four intensive care unit stays and increased the cost per stay by nearly $16,000, a retrospective analysis of data from a large U.S. hospital database revealed.
"The prevalence and burden of hospital-acquired infections in the ICU are high and clearly associated with a major economic impact," said Florence Joly, Pharm.D., of Sanofi R&D, France.

The study was conducted by Sanofi to inform the development of a new drug to combat hospital-associated infections (HAIs) including bloodstream infections, hospital-acquired pneumonia (HAP), and surgical site infections (SSIs). The data came from Premier Perspective, a large U.S. hospital database containing information from more than 700 hospitals and representing about 20% of all hospital discharges in the United States.
The analysis included 463,491 adults aged 18 years and older who were admitted in 2007 for a total of 511,815 ICU stays of more than 48 hours. The patients were 53% male and had a mean age of 64 years. A third (33%) had diabetes, 26% had a central venous catheter/central line, and 22% were on mechanical ventilation. Most of the patients (91.5%) had just one ICU stay, but 7% had two stays and 1.5% had three or more stays.
At least one HAI occurred in 27% of all ICU stays, including pneumonia in 17%, bloodstream infections in 14.5%, and SSIs in 1.5%. In addition, severe sepsis occurred in 40%, she reported at the European Congress of Clinical Microbiology and Infectious Diseases.
Length of stay (LOS) was 23 days for SSIs, at a cost of $59,905 per stay. For bloodstream infections, the stay was 18 days at $43,071 per stay, and for HAP, 15 days at $36,467/stay. Mortality was 25% for bloodstream infections, 17% for HAP, and 11% for SSIs. In total, the length of stay for all that involved HAI was 16 days at a cost of $37,539 per stay, compared with just 8 days at $21,541 per stay for those without HAI. Overall mortality rates were 19% with HAI vs. 4.5% without. All of the HAI/no HAI differences were highly statistically significant, she noted.
Similar results were found using data from 2008 and 2009.
"The increase in mortality rates and longer LOS findings as the drivers of higher ICU costs indicate the need for specific measures to reduce the prevalence of these major types of hospital-acquired infections," Dr. Joly concluded.
In a separate poster presentation by Dr. Caroline Amand-Bourdon, also of Sanofi R&D, the same database of 463,491 patients was analyzed for risk factors predicting HAI for just the first hospitalization per patient. In all, 26% of the patients (119,616) experienced a HAI, including 17% with pneumonia, 14% with a bloodstream infection, and 1% with an SSI. Compared with the 343,875 patients who did not have a HAI, patients who did were older (mean age 66 vs. 63 years) and had more device procedures, including central catheter placements (49% vs. 18%) and mechanical ventilation hookups (42% vs. 15%). The HAI patients were also more often admitted via the emergency department (71% vs. 56%).
Central catheter and mechanical ventilation were identified as the two main risk factors for HAI, with adjusted odds ratios of 3.4 and 2.8, respectively, Dr. Amand-Bourdon and her associates reported.
"These findings illustrate the changing nature of hospital inpatient populations over the years that are more vulnerable to HAI," according to the investigators.
Both Dr. Joly and Dr. Amand-Bourdon are employees of Sanofi, which funded and conducted the study.
LONDON – Hospital-acquired infections occurred in more than one in four intensive care unit stays and increased the cost per stay by nearly $16,000, a retrospective analysis of data from a large U.S. hospital database revealed.
"The prevalence and burden of hospital-acquired infections in the ICU are high and clearly associated with a major economic impact," said Florence Joly, Pharm.D., of Sanofi R&D, France.
The study was conducted by Sanofi to inform the development of a new drug to combat hospital-associated infections (HAIs) including bloodstream infections, hospital-acquired pneumonia (HAP), and surgical site infections (SSIs). The data came from Premier Perspective, a large U.S. hospital database containing information from more than 700 hospitals and representing about 20% of all hospital discharges in the United States.
The analysis included 463,491 adults aged 18 years and older who were admitted in 2007 for a total of 511,815 ICU stays of more than 48 hours. The patients were 53% male and had a mean age of 64 years. A third (33%) had diabetes, 26% had a central venous catheter/central line, and 22% were on mechanical ventilation. Most of the patients (91.5%) had just one ICU stay, but 7% had two stays and 1.5% had three or more stays.
At least one HAI occurred in 27% of all ICU stays, including pneumonia in 17%, bloodstream infections in 14.5%, and SSIs in 1.5%. In addition, severe sepsis occurred in 40%, she reported at the European Congress of Clinical Microbiology and Infectious Diseases.
Length of stay (LOS) was 23 days for SSIs, at a cost of $59,905 per stay. For bloodstream infections, the stay was 18 days at $43,071 per stay, and for HAP, 15 days at $36,467/stay. Mortality was 25% for bloodstream infections, 17% for HAP, and 11% for SSIs. In total, the length of stay for all that involved HAI was 16 days at a cost of $37,539 per stay, compared with just 8 days at $21,541 per stay for those without HAI. Overall mortality rates were 19% with HAI vs. 4.5% without. All of the HAI/no HAI differences were highly statistically significant, she noted.
Similar results were found using data from 2008 and 2009.
"The increase in mortality rates and longer LOS findings as the drivers of higher ICU costs indicate the need for specific measures to reduce the prevalence of these major types of hospital-acquired infections," Dr. Joly concluded.
In a separate poster presentation by Dr. Caroline Amand-Bourdon, also of Sanofi R&D, the same database of 463,491 patients was analyzed for risk factors predicting HAI for just the first hospitalization per patient. In all, 26% of the patients (119,616) experienced a HAI, including 17% with pneumonia, 14% with a bloodstream infection, and 1% with an SSI. Compared with the 343,875 patients who did not have a HAI, patients who did were older (mean age 66 vs. 63 years) and had more device procedures, including central catheter placements (49% vs. 18%) and mechanical ventilation hookups (42% vs. 15%). The HAI patients were also more often admitted via the emergency department (71% vs. 56%).
Central catheter and mechanical ventilation were identified as the two main risk factors for HAI, with adjusted odds ratios of 3.4 and 2.8, respectively, Dr. Amand-Bourdon and her associates reported.
"These findings illustrate the changing nature of hospital inpatient populations over the years that are more vulnerable to HAI," according to the investigators.
Both Dr. Joly and Dr. Amand-Bourdon are employees of Sanofi, which funded and conducted the study.
LONDON – Hospital-acquired infections occurred in more than one in four intensive care unit stays and increased the cost per stay by nearly $16,000, a retrospective analysis of data from a large U.S. hospital database revealed.
"The prevalence and burden of hospital-acquired infections in the ICU are high and clearly associated with a major economic impact," said Florence Joly, Pharm.D., of Sanofi R&D, France.
The study was conducted by Sanofi to inform the development of a new drug to combat hospital-associated infections (HAIs) including bloodstream infections, hospital-acquired pneumonia (HAP), and surgical site infections (SSIs). The data came from Premier Perspective, a large U.S. hospital database containing information from more than 700 hospitals and representing about 20% of all hospital discharges in the United States.
The analysis included 463,491 adults aged 18 years and older who were admitted in 2007 for a total of 511,815 ICU stays of more than 48 hours. The patients were 53% male and had a mean age of 64 years. A third (33%) had diabetes, 26% had a central venous catheter/central line, and 22% were on mechanical ventilation. Most of the patients (91.5%) had just one ICU stay, but 7% had two stays and 1.5% had three or more stays.
At least one HAI occurred in 27% of all ICU stays, including pneumonia in 17%, bloodstream infections in 14.5%, and SSIs in 1.5%. In addition, severe sepsis occurred in 40%, she reported at the European Congress of Clinical Microbiology and Infectious Diseases.
Length of stay (LOS) was 23 days for SSIs, at a cost of $59,905 per stay. For bloodstream infections, the stay was 18 days at $43,071 per stay, and for HAP, 15 days at $36,467/stay. Mortality was 25% for bloodstream infections, 17% for HAP, and 11% for SSIs. In total, the length of stay for all that involved HAI was 16 days at a cost of $37,539 per stay, compared with just 8 days at $21,541 per stay for those without HAI. Overall mortality rates were 19% with HAI vs. 4.5% without. All of the HAI/no HAI differences were highly statistically significant, she noted.
Similar results were found using data from 2008 and 2009.
"The increase in mortality rates and longer LOS findings as the drivers of higher ICU costs indicate the need for specific measures to reduce the prevalence of these major types of hospital-acquired infections," Dr. Joly concluded.
In a separate poster presentation by Dr. Caroline Amand-Bourdon, also of Sanofi R&D, the same database of 463,491 patients was analyzed for risk factors predicting HAI for just the first hospitalization per patient. In all, 26% of the patients (119,616) experienced a HAI, including 17% with pneumonia, 14% with a bloodstream infection, and 1% with an SSI. Compared with the 343,875 patients who did not have a HAI, patients who did were older (mean age 66 vs. 63 years) and had more device procedures, including central catheter placements (49% vs. 18%) and mechanical ventilation hookups (42% vs. 15%). The HAI patients were also more often admitted via the emergency department (71% vs. 56%).
Central catheter and mechanical ventilation were identified as the two main risk factors for HAI, with adjusted odds ratios of 3.4 and 2.8, respectively, Dr. Amand-Bourdon and her associates reported.
"These findings illustrate the changing nature of hospital inpatient populations over the years that are more vulnerable to HAI," according to the investigators.
Both Dr. Joly and Dr. Amand-Bourdon are employees of Sanofi, which funded and conducted the study.
FROM THE EUROPEAN CONGRESS OF CLINICAL MICROBIOLOGY AND INFECTIOUS DISEASES
Breast Brachytherapy Judged Superior in Tumor Bed Control
Accelerated partial breast radiation with MammoSite balloon brachytherapy appears to control the tumor bed more effectively than whole breast irradiation, investigators reported at the annual meeting of the American Society of Breast Surgeons.
Dr. Peter Beitsch and his colleagues compared data from the society’s MammoSite Registry with prior findings from studies of whole-breast irradiation (WBI). They said the proportion of recurrences occurring in the tumor bed was much smaller with accelerated partial breast radiation (APBI) – 28% vs. about 69% in the earlier WBI data.

"While it made common sense to a lot of us that APBI should offer better rates of local control, compared to WBI, since the radiation therapy with APBI is delivered directly to the tumor site, this is the first study to have actually proven this hypothesis," Dr. Beitsch, director of the Dallas Breast Center, said in an interview.
"WBI has been held as the ‘gold standard’ for post-lumpectomy radiation therapy, and our data may change that line of thought," he added.
Randomized trials demonstrate that lumpectomy with WBI yields survival rates equivalent to those seen with mastectomy; they also show that WBI has no impact on the ipsilateral occurrence of new "elsewhere" cancers in quadrants away from the primary tumor quadrant. Dr. Beitsch explained during a press briefing.
In all such trials, thus far, tumor bed recurrence rates have been higher than were the rates of ipsilateral "elsewhere" cancers, he said.
Now, however, 5-year actuarial data from 1,449 cases in 1,440 patients in the MammoSite registry show the opposite. The patients were treated at 97 institutions between May 2002 and July 2004. Most patients, 87%, were diagnosed with invasive breast cancer and the rest, 13%, with ductal carcinoma in situ (DCIS). Median follow up was 60 months.
Dr. Beitsch reported there have been 50 (3.5%) ipsilateral breast tumor recurrences: 14 (1.0%) at the initial tumor site and 36 (2.5%) elsewhere in the breast. The total actuarial rate of ipsilateral breast tumor recurrence was 3.61% (3.65 % for invasive disease and 3.36 % for DCIS). Tumor bed recurrences accounted for 28% of all recurrences, whereas recurrences elsewhere added up to 72%.
In contrast, historical data on whole-breast irradiation (WBI) from six studies demonstrate that tumor bed recurrences are about twice as common as recurrences elsewhere, approximately 69% vs. about 31% elsewhere, said Dr. Beitsch, co-principal investigator for the registry and lead author on the study.
The new findings contrast with a controversial retrospective study of breast brachytherapy in nearly 93,000 older women with invasive breast cancer. In that study, the mastectomy rate 5 years later was about twice as high in women treated with brachytherapy – cumulative incidence 3.95% vs. 2.18% with WBI. The difference persisted after a multivariate adjustment, with a hazard ratio of 2.19, according to a report published May 1, 2012, in JAMA.
Moreover, short-term and long-term complications, including breast pain, were significantly more common in women who had radiation delivered by brachytherapy. Overall survival was not significantly different, however, at about 87% in both groups studied, Dr. Dr. Grace L. Smith of the University of Texas M.D. Anderson Cancer Center in Houston and her coauthors reported (JAMA 2012;307:1827-37).
In a press release asserting that the new study contradicts the JAMA report, Dr. Beitsch called attention to limitations of the M.D. Anderson study: He noted that it is based on Medicare claims data, which often do not provide an accurate clinical picture. In addition, many of the end points are "soft," poorly defined and difficult to quantify, he said, adding that the reported complication rates after breast surgery and radiotherapy vary widely, and are subject to under- or over-reporting.
Finally, "the authors’ inferences of harm to patients from breast brachytherapy are speculative," he said.

The American Society of Breast Surgeons (ASBrS) is one of three groups that previously issued rebuttals to the retrospective study. In the same press release, ASBrS executive committee member Dr. Hiram S. Cody III said that ASBrS continues to support its Consensus Statement on APBI and guidelines for patient selection, which was revised Aug. 15, 2011.
"APBI appears to be safe and effective treatment for properly selected breast conservation patients," said Dr. Cody, an attending surgeon at Memorial Sloan-Kettering Cancer Center and professor of clinical surgery at Cornell University, both in New York.
"We wish to emphasize that, although the 6-year results of APBI are encouraging, they do not conclusively establish equivalence with WBI, for which the supporting data include multiple randomized trials with follow-up exceeding 20 years, and meta-analyses that conclusively link local control and survival," Dr. Cody stated.
"APBI must ultimately be held to the same standard, and a randomized trial, NSABP [National Surgical Adjuvant Breast and Bowel Project] B-39, directly compares partial breast irradiation (by interstitial catheters, balloon devices, strut-based devices, or external beam) with WBI and promises to better define the ultimate role of APBI."
Dr. Beitsch and Dr. Cody stated that they have no disclosures.
Accelerated partial breast radiation with MammoSite balloon brachytherapy appears to control the tumor bed more effectively than whole breast irradiation, investigators reported at the annual meeting of the American Society of Breast Surgeons.
Dr. Peter Beitsch and his colleagues compared data from the society’s MammoSite Registry with prior findings from studies of whole-breast irradiation (WBI). They said the proportion of recurrences occurring in the tumor bed was much smaller with accelerated partial breast radiation (APBI) – 28% vs. about 69% in the earlier WBI data.
"While it made common sense to a lot of us that APBI should offer better rates of local control, compared to WBI, since the radiation therapy with APBI is delivered directly to the tumor site, this is the first study to have actually proven this hypothesis," Dr. Beitsch, director of the Dallas Breast Center, said in an interview.
"WBI has been held as the ‘gold standard’ for post-lumpectomy radiation therapy, and our data may change that line of thought," he added.
Randomized trials demonstrate that lumpectomy with WBI yields survival rates equivalent to those seen with mastectomy; they also show that WBI has no impact on the ipsilateral occurrence of new "elsewhere" cancers in quadrants away from the primary tumor quadrant. Dr. Beitsch explained during a press briefing.
In all such trials, thus far, tumor bed recurrence rates have been higher than were the rates of ipsilateral "elsewhere" cancers, he said.
Now, however, 5-year actuarial data from 1,449 cases in 1,440 patients in the MammoSite registry show the opposite. The patients were treated at 97 institutions between May 2002 and July 2004. Most patients, 87%, were diagnosed with invasive breast cancer and the rest, 13%, with ductal carcinoma in situ (DCIS). Median follow up was 60 months.
Dr. Beitsch reported there have been 50 (3.5%) ipsilateral breast tumor recurrences: 14 (1.0%) at the initial tumor site and 36 (2.5%) elsewhere in the breast. The total actuarial rate of ipsilateral breast tumor recurrence was 3.61% (3.65 % for invasive disease and 3.36 % for DCIS). Tumor bed recurrences accounted for 28% of all recurrences, whereas recurrences elsewhere added up to 72%.
In contrast, historical data on whole-breast irradiation (WBI) from six studies demonstrate that tumor bed recurrences are about twice as common as recurrences elsewhere, approximately 69% vs. about 31% elsewhere, said Dr. Beitsch, co-principal investigator for the registry and lead author on the study.
The new findings contrast with a controversial retrospective study of breast brachytherapy in nearly 93,000 older women with invasive breast cancer. In that study, the mastectomy rate 5 years later was about twice as high in women treated with brachytherapy – cumulative incidence 3.95% vs. 2.18% with WBI. The difference persisted after a multivariate adjustment, with a hazard ratio of 2.19, according to a report published May 1, 2012, in JAMA.
Moreover, short-term and long-term complications, including breast pain, were significantly more common in women who had radiation delivered by brachytherapy. Overall survival was not significantly different, however, at about 87% in both groups studied, Dr. Dr. Grace L. Smith of the University of Texas M.D. Anderson Cancer Center in Houston and her coauthors reported (JAMA 2012;307:1827-37).
In a press release asserting that the new study contradicts the JAMA report, Dr. Beitsch called attention to limitations of the M.D. Anderson study: He noted that it is based on Medicare claims data, which often do not provide an accurate clinical picture. In addition, many of the end points are "soft," poorly defined and difficult to quantify, he said, adding that the reported complication rates after breast surgery and radiotherapy vary widely, and are subject to under- or over-reporting.
Finally, "the authors’ inferences of harm to patients from breast brachytherapy are speculative," he said.
The American Society of Breast Surgeons (ASBrS) is one of three groups that previously issued rebuttals to the retrospective study. In the same press release, ASBrS executive committee member Dr. Hiram S. Cody III said that ASBrS continues to support its Consensus Statement on APBI and guidelines for patient selection, which was revised Aug. 15, 2011.
"APBI appears to be safe and effective treatment for properly selected breast conservation patients," said Dr. Cody, an attending surgeon at Memorial Sloan-Kettering Cancer Center and professor of clinical surgery at Cornell University, both in New York.
"We wish to emphasize that, although the 6-year results of APBI are encouraging, they do not conclusively establish equivalence with WBI, for which the supporting data include multiple randomized trials with follow-up exceeding 20 years, and meta-analyses that conclusively link local control and survival," Dr. Cody stated.
"APBI must ultimately be held to the same standard, and a randomized trial, NSABP [National Surgical Adjuvant Breast and Bowel Project] B-39, directly compares partial breast irradiation (by interstitial catheters, balloon devices, strut-based devices, or external beam) with WBI and promises to better define the ultimate role of APBI."
Dr. Beitsch and Dr. Cody stated that they have no disclosures.
Accelerated partial breast radiation with MammoSite balloon brachytherapy appears to control the tumor bed more effectively than whole breast irradiation, investigators reported at the annual meeting of the American Society of Breast Surgeons.
Dr. Peter Beitsch and his colleagues compared data from the society’s MammoSite Registry with prior findings from studies of whole-breast irradiation (WBI). They said the proportion of recurrences occurring in the tumor bed was much smaller with accelerated partial breast radiation (APBI) – 28% vs. about 69% in the earlier WBI data.
"While it made common sense to a lot of us that APBI should offer better rates of local control, compared to WBI, since the radiation therapy with APBI is delivered directly to the tumor site, this is the first study to have actually proven this hypothesis," Dr. Beitsch, director of the Dallas Breast Center, said in an interview.
"WBI has been held as the ‘gold standard’ for post-lumpectomy radiation therapy, and our data may change that line of thought," he added.
Randomized trials demonstrate that lumpectomy with WBI yields survival rates equivalent to those seen with mastectomy; they also show that WBI has no impact on the ipsilateral occurrence of new "elsewhere" cancers in quadrants away from the primary tumor quadrant. Dr. Beitsch explained during a press briefing.
In all such trials, thus far, tumor bed recurrence rates have been higher than were the rates of ipsilateral "elsewhere" cancers, he said.
Now, however, 5-year actuarial data from 1,449 cases in 1,440 patients in the MammoSite registry show the opposite. The patients were treated at 97 institutions between May 2002 and July 2004. Most patients, 87%, were diagnosed with invasive breast cancer and the rest, 13%, with ductal carcinoma in situ (DCIS). Median follow up was 60 months.
Dr. Beitsch reported there have been 50 (3.5%) ipsilateral breast tumor recurrences: 14 (1.0%) at the initial tumor site and 36 (2.5%) elsewhere in the breast. The total actuarial rate of ipsilateral breast tumor recurrence was 3.61% (3.65 % for invasive disease and 3.36 % for DCIS). Tumor bed recurrences accounted for 28% of all recurrences, whereas recurrences elsewhere added up to 72%.
In contrast, historical data on whole-breast irradiation (WBI) from six studies demonstrate that tumor bed recurrences are about twice as common as recurrences elsewhere, approximately 69% vs. about 31% elsewhere, said Dr. Beitsch, co-principal investigator for the registry and lead author on the study.
The new findings contrast with a controversial retrospective study of breast brachytherapy in nearly 93,000 older women with invasive breast cancer. In that study, the mastectomy rate 5 years later was about twice as high in women treated with brachytherapy – cumulative incidence 3.95% vs. 2.18% with WBI. The difference persisted after a multivariate adjustment, with a hazard ratio of 2.19, according to a report published May 1, 2012, in JAMA.
Moreover, short-term and long-term complications, including breast pain, were significantly more common in women who had radiation delivered by brachytherapy. Overall survival was not significantly different, however, at about 87% in both groups studied, Dr. Dr. Grace L. Smith of the University of Texas M.D. Anderson Cancer Center in Houston and her coauthors reported (JAMA 2012;307:1827-37).
In a press release asserting that the new study contradicts the JAMA report, Dr. Beitsch called attention to limitations of the M.D. Anderson study: He noted that it is based on Medicare claims data, which often do not provide an accurate clinical picture. In addition, many of the end points are "soft," poorly defined and difficult to quantify, he said, adding that the reported complication rates after breast surgery and radiotherapy vary widely, and are subject to under- or over-reporting.
Finally, "the authors’ inferences of harm to patients from breast brachytherapy are speculative," he said.
The American Society of Breast Surgeons (ASBrS) is one of three groups that previously issued rebuttals to the retrospective study. In the same press release, ASBrS executive committee member Dr. Hiram S. Cody III said that ASBrS continues to support its Consensus Statement on APBI and guidelines for patient selection, which was revised Aug. 15, 2011.
"APBI appears to be safe and effective treatment for properly selected breast conservation patients," said Dr. Cody, an attending surgeon at Memorial Sloan-Kettering Cancer Center and professor of clinical surgery at Cornell University, both in New York.
"We wish to emphasize that, although the 6-year results of APBI are encouraging, they do not conclusively establish equivalence with WBI, for which the supporting data include multiple randomized trials with follow-up exceeding 20 years, and meta-analyses that conclusively link local control and survival," Dr. Cody stated.
"APBI must ultimately be held to the same standard, and a randomized trial, NSABP [National Surgical Adjuvant Breast and Bowel Project] B-39, directly compares partial breast irradiation (by interstitial catheters, balloon devices, strut-based devices, or external beam) with WBI and promises to better define the ultimate role of APBI."
Dr. Beitsch and Dr. Cody stated that they have no disclosures.
FROM THE ANNUAL MEETING OF THE AMERICAN SOCIETY OF BREAST SURGEONS
Major Finding: Tumor bed recurrence accounted for 28% of all recurrences after accelerated partial breast radiation vs. about 69% in previous studies of whole breast radiation.
Data Source: The findings come from a comparison of 5-year actuarial data from 1,440 patients in the American Society of Breast Surgeons’ MammoSite Registry with historical data.
Disclosures: Dr. Beitsch and Dr. Cody stated that they have no disclosures. The MammoSite Registry is maintained by the American Society of Breast Surgeons.
Obesity Increases Surgical Site Infection Risk
LONDON – Being obese increased the risk of surgical site infection nearly fourfold among patients who underwent operations in the United Kingdom from 2006 through 2010.
The findings pose questions such as whether preoperative dosing of antibiotics might be adjusted upward or whether preoperative weight loss should be advocated, said Dr. Simon Thelwall of the Health Protection Agency, London.

The analysis was done using nationwide data from the UK’s Health Protection Agency (HPA) Surgical Site Infection Surveillance Service, comprising data submitted from all 212 of the National Health Service hospitals in England on a cumulative total of 326,880 adult patients who underwent one of five operations: abdominal hysterectomy, coronary artery bypass graft (CABG), hip replacement, knee replacement, and large bowel surgery.
Of those, surgical site infections (SSIs) were detected in inpatients and at readmission for 4,453, and body mass index (BMI) data were available for 43%. Of these 112,048 (79.3%) were overweight or obese, said Dr. Thelwall.
The rates of SSIs didn’t differ among those with and without available body mass index data except among CABG patients, for whom the rate of SSIs was double among those with and without BMI data (5.17% vs. 2.71%). The CABG patients with BMI data were also significantly more likely to have received implants (85% vs. 61%), to have undergone emergency operations (1.29% vs. 0.76%), and to have had significantly longer operations (205 vs. 220 minutes).
Thus, "CABG patients with BMI data are more likely to have risk factors predisposing them to SSI," Dr. Thelwall noted.
Obesity significantly increased the risk for SSI in all surgical groups except for abdominal hysterectomy, with risk ratios ranging from 1.62 for knee replacement to 1.87 for large bowel surgery. Obesity still increased the SSI risk among abdominal hysterectomy patients with a risk ratio of 1.81, but that did not reach statistical significance, he said.
Overall, the SSI risk increased with increasing BMI. In multivariate analysis adjusting for a variety of potential confounders including age, trauma, category of surgery, implant, and emergency surgery, the odds ratios for SSI increased from 1.44 among the overweight patients to 1.89 for those with BMIs of 30-34.99 kg/m2, to 2.94 for BMIs of 35-39.99 kg/m2, and to 3.73 for BMIs greater than 40 kg/m2. In the highest BMI category, even the SSI rate for abdominal hysterectomy became significantly greater, compared with that of patients of normal weight (risk ratio, 3.90), Dr. Thelwall said.
Among the morbidly obese (BMI greater than 40 kg/m2), between 65% and 78% of the risk for SSI was attributable to being very obese. And in that group of morbidly obese patients, the risk of SSI among abdominal hysterectomy patients became significantly elevated three- to fivefold, compared with that of women of normal weight, he noted.
Dr. Thelwall declared that he had no disclosures.
LONDON – Being obese increased the risk of surgical site infection nearly fourfold among patients who underwent operations in the United Kingdom from 2006 through 2010.
The findings pose questions such as whether preoperative dosing of antibiotics might be adjusted upward or whether preoperative weight loss should be advocated, said Dr. Simon Thelwall of the Health Protection Agency, London.
The analysis was done using nationwide data from the UK’s Health Protection Agency (HPA) Surgical Site Infection Surveillance Service, comprising data submitted from all 212 of the National Health Service hospitals in England on a cumulative total of 326,880 adult patients who underwent one of five operations: abdominal hysterectomy, coronary artery bypass graft (CABG), hip replacement, knee replacement, and large bowel surgery.
Of those, surgical site infections (SSIs) were detected in inpatients and at readmission for 4,453, and body mass index (BMI) data were available for 43%. Of these 112,048 (79.3%) were overweight or obese, said Dr. Thelwall.
The rates of SSIs didn’t differ among those with and without available body mass index data except among CABG patients, for whom the rate of SSIs was double among those with and without BMI data (5.17% vs. 2.71%). The CABG patients with BMI data were also significantly more likely to have received implants (85% vs. 61%), to have undergone emergency operations (1.29% vs. 0.76%), and to have had significantly longer operations (205 vs. 220 minutes).
Thus, "CABG patients with BMI data are more likely to have risk factors predisposing them to SSI," Dr. Thelwall noted.
Obesity significantly increased the risk for SSI in all surgical groups except for abdominal hysterectomy, with risk ratios ranging from 1.62 for knee replacement to 1.87 for large bowel surgery. Obesity still increased the SSI risk among abdominal hysterectomy patients with a risk ratio of 1.81, but that did not reach statistical significance, he said.
Overall, the SSI risk increased with increasing BMI. In multivariate analysis adjusting for a variety of potential confounders including age, trauma, category of surgery, implant, and emergency surgery, the odds ratios for SSI increased from 1.44 among the overweight patients to 1.89 for those with BMIs of 30-34.99 kg/m2, to 2.94 for BMIs of 35-39.99 kg/m2, and to 3.73 for BMIs greater than 40 kg/m2. In the highest BMI category, even the SSI rate for abdominal hysterectomy became significantly greater, compared with that of patients of normal weight (risk ratio, 3.90), Dr. Thelwall said.
Among the morbidly obese (BMI greater than 40 kg/m2), between 65% and 78% of the risk for SSI was attributable to being very obese. And in that group of morbidly obese patients, the risk of SSI among abdominal hysterectomy patients became significantly elevated three- to fivefold, compared with that of women of normal weight, he noted.
Dr. Thelwall declared that he had no disclosures.
LONDON – Being obese increased the risk of surgical site infection nearly fourfold among patients who underwent operations in the United Kingdom from 2006 through 2010.
The findings pose questions such as whether preoperative dosing of antibiotics might be adjusted upward or whether preoperative weight loss should be advocated, said Dr. Simon Thelwall of the Health Protection Agency, London.
The analysis was done using nationwide data from the UK’s Health Protection Agency (HPA) Surgical Site Infection Surveillance Service, comprising data submitted from all 212 of the National Health Service hospitals in England on a cumulative total of 326,880 adult patients who underwent one of five operations: abdominal hysterectomy, coronary artery bypass graft (CABG), hip replacement, knee replacement, and large bowel surgery.
Of those, surgical site infections (SSIs) were detected in inpatients and at readmission for 4,453, and body mass index (BMI) data were available for 43%. Of these 112,048 (79.3%) were overweight or obese, said Dr. Thelwall.
The rates of SSIs didn’t differ among those with and without available body mass index data except among CABG patients, for whom the rate of SSIs was double among those with and without BMI data (5.17% vs. 2.71%). The CABG patients with BMI data were also significantly more likely to have received implants (85% vs. 61%), to have undergone emergency operations (1.29% vs. 0.76%), and to have had significantly longer operations (205 vs. 220 minutes).
Thus, "CABG patients with BMI data are more likely to have risk factors predisposing them to SSI," Dr. Thelwall noted.
Obesity significantly increased the risk for SSI in all surgical groups except for abdominal hysterectomy, with risk ratios ranging from 1.62 for knee replacement to 1.87 for large bowel surgery. Obesity still increased the SSI risk among abdominal hysterectomy patients with a risk ratio of 1.81, but that did not reach statistical significance, he said.
Overall, the SSI risk increased with increasing BMI. In multivariate analysis adjusting for a variety of potential confounders including age, trauma, category of surgery, implant, and emergency surgery, the odds ratios for SSI increased from 1.44 among the overweight patients to 1.89 for those with BMIs of 30-34.99 kg/m2, to 2.94 for BMIs of 35-39.99 kg/m2, and to 3.73 for BMIs greater than 40 kg/m2. In the highest BMI category, even the SSI rate for abdominal hysterectomy became significantly greater, compared with that of patients of normal weight (risk ratio, 3.90), Dr. Thelwall said.
Among the morbidly obese (BMI greater than 40 kg/m2), between 65% and 78% of the risk for SSI was attributable to being very obese. And in that group of morbidly obese patients, the risk of SSI among abdominal hysterectomy patients became significantly elevated three- to fivefold, compared with that of women of normal weight, he noted.
Dr. Thelwall declared that he had no disclosures.
FROM THE EUROPEAN CONGRESS OF CLINICAL MICROBIOLOGY AND INFECTIOUS DISEASES
Major Finding: Obesity significantly increased the risk for surgical site infection in all surgical groups except for abdominal hysterectomy, with risk ratios ranging from 1.62 for knee replacement to 1.87 for large bowel surgery.
Data Source: This data analysis was based on data from the UK’s Health Protection Agency Surgical Site Infection Surveillance, which included 326,880 adult patients who underwent abdominal hysterectomy, coronary artery bypass graft, hip replacement, knee replacement, or large bowel surgery.
Disclosures: Dr. Thelwall is employed by the UK’s Health Protection Agency and has no financial disclosures.
Panel Recommends Tofacitinib Approval for Refractory RA
SILVER SPRING, MD. – There soon may be a new oral disease-modifying antirheumatic drug in town, the first in more than 10 years to be approved for the treatment of rheumatoid arthritis.
The drug is tofacitinib, and it is the first in a new class of agents for RA patients.
On May 9 the Food and Drug Administration’s Arthritis Advisory Committee voted 8 to 2 in favor of approving tofacitinib for RA patients who have had inadequate response to one or more disease-modifying antirheumatic drugs (DMARDs).
A small-molecule inhibitor of the Janus kinase (JAK) pathways, Pfizer’s tofacitinib blocks inflammatory cytokines that play a role in the pathogenesis of RA. If approved, tofacitinib would be the first JAK inhibitor for RA. Another JAK inhibitor, ruxolitinib (Incyte’s Jakafi) was approved to treat myelofibrosis in November.
Pfizer’s application was based on five randomized controlled phase III efficacy trials involving a total 3,315 patients. The application also included safety data for a total of 4,816 patients from the phase II and III studies, of whom 4,053 were treated for at least 6 months and 567 for more than 3 years, up to 42 months. Among the trials was one that compared tofacitinib with placebo in patients with moderate to severe RA who had incomplete responses to tumor necrosis factor (TNF) inhibitors, and four studies of patients who had inadequate responses to other DMARDs. One study included an arm with adalimumab as an active comparator, and one evaluated radiographic response. In three studies, the participants were taking background methotrexate, and in one study, tofacitinib was used as monotherapy. The studies were conducted in a total of 44 countries.
All five of the phase III studies were designed to show the superiority of 5-mg and 10-mg twice-daily doses of tofacitinib to placebo for the primary end points: a 20% or better increase in the American College of Rheumatology’s definition of improvement in the signs and symptoms of RA (ACR20), improvement in Health Assessment Questionnaire-Disability Index (HAQ-DI) scores, and a Disease Activity Score-28 (DAS28) of less than 2.6. In most of the assessments conducted at 6 and 12 months, both doses exhibited statistical superiority over placebo, with the 10-mg dose showing greater improvement than the 5-mg dose.
However, the fact that only one study evaluated radiographic evidence, which the FDA reviewers deemed inconclusive, was cause for concern for the FDA reviewers and committee members. According to the FDA’s Dr. Yongman Kim, 18% of the 278 patients on 5 mg of tofacitinib, 16% of the 290 with 10 mg of tofacitinib, and 14% of the 140 placebo patients showed an "improved" modified total sharp score (mTSS) of less than 0, suggesting it inhibited joint damage.
The FDA was also concerned about dose- and time-dependent elevations in malignancies, and particularly lymphomas, with a standardized incidence ratio of 2.35 at a median 9 months duration of exposure. Of the seven total patients who developed lymphoma, three were on 5 mg of tofacitinib, three were on 10 mg, and one was in a still-blinded arm of the trial. Overall malignancy rates increased with time, from 0.79 per 100 patient-years at 0-6 months to 1.06 at 12-18 months to 1.43 beyond 24 months.
Several of the committee members who voted in favor of tofacitinib said that they did so because they wanted a new oral option for RA patients and that they don’t anticipate any better data from clinical trials. They strongly urged postmarketing studies of both radiologic efficacy and safety. Pfizer has proposed a Risk Evaluation and Mitigation Strategy that will include, alongside labeling provisions, a Medication Guide, a "Dear Health Care Professional" letter, and routine pharmacovigilance. Additional clinical studies that are ongoing or planned include long-term extension studies to assess cardiovascular safety, serious and opportunistic infections, malignancies, and gastrointestinal perforations; studies to monitor any unidentified risks; a cholesterol kinetic study; and studies to evaluate the effect of tofacitinib on the immune response to pneumococcal and influenza vaccines as well as to further delineate the effects of tofacitinib on lymphocyte subsets.
The panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver but not at this meeting.
Martin Bermin-Gorvine of "The Pink Sheet" contributed to this story. This news service and "The Pink Sheet" are both owned by Elsevier.
SILVER SPRING, MD. – There soon may be a new oral disease-modifying antirheumatic drug in town, the first in more than 10 years to be approved for the treatment of rheumatoid arthritis.
The drug is tofacitinib, and it is the first in a new class of agents for RA patients.
On May 9 the Food and Drug Administration’s Arthritis Advisory Committee voted 8 to 2 in favor of approving tofacitinib for RA patients who have had inadequate response to one or more disease-modifying antirheumatic drugs (DMARDs).
A small-molecule inhibitor of the Janus kinase (JAK) pathways, Pfizer’s tofacitinib blocks inflammatory cytokines that play a role in the pathogenesis of RA. If approved, tofacitinib would be the first JAK inhibitor for RA. Another JAK inhibitor, ruxolitinib (Incyte’s Jakafi) was approved to treat myelofibrosis in November.
Pfizer’s application was based on five randomized controlled phase III efficacy trials involving a total 3,315 patients. The application also included safety data for a total of 4,816 patients from the phase II and III studies, of whom 4,053 were treated for at least 6 months and 567 for more than 3 years, up to 42 months. Among the trials was one that compared tofacitinib with placebo in patients with moderate to severe RA who had incomplete responses to tumor necrosis factor (TNF) inhibitors, and four studies of patients who had inadequate responses to other DMARDs. One study included an arm with adalimumab as an active comparator, and one evaluated radiographic response. In three studies, the participants were taking background methotrexate, and in one study, tofacitinib was used as monotherapy. The studies were conducted in a total of 44 countries.
All five of the phase III studies were designed to show the superiority of 5-mg and 10-mg twice-daily doses of tofacitinib to placebo for the primary end points: a 20% or better increase in the American College of Rheumatology’s definition of improvement in the signs and symptoms of RA (ACR20), improvement in Health Assessment Questionnaire-Disability Index (HAQ-DI) scores, and a Disease Activity Score-28 (DAS28) of less than 2.6. In most of the assessments conducted at 6 and 12 months, both doses exhibited statistical superiority over placebo, with the 10-mg dose showing greater improvement than the 5-mg dose.
However, the fact that only one study evaluated radiographic evidence, which the FDA reviewers deemed inconclusive, was cause for concern for the FDA reviewers and committee members. According to the FDA’s Dr. Yongman Kim, 18% of the 278 patients on 5 mg of tofacitinib, 16% of the 290 with 10 mg of tofacitinib, and 14% of the 140 placebo patients showed an "improved" modified total sharp score (mTSS) of less than 0, suggesting it inhibited joint damage.
The FDA was also concerned about dose- and time-dependent elevations in malignancies, and particularly lymphomas, with a standardized incidence ratio of 2.35 at a median 9 months duration of exposure. Of the seven total patients who developed lymphoma, three were on 5 mg of tofacitinib, three were on 10 mg, and one was in a still-blinded arm of the trial. Overall malignancy rates increased with time, from 0.79 per 100 patient-years at 0-6 months to 1.06 at 12-18 months to 1.43 beyond 24 months.
Several of the committee members who voted in favor of tofacitinib said that they did so because they wanted a new oral option for RA patients and that they don’t anticipate any better data from clinical trials. They strongly urged postmarketing studies of both radiologic efficacy and safety. Pfizer has proposed a Risk Evaluation and Mitigation Strategy that will include, alongside labeling provisions, a Medication Guide, a "Dear Health Care Professional" letter, and routine pharmacovigilance. Additional clinical studies that are ongoing or planned include long-term extension studies to assess cardiovascular safety, serious and opportunistic infections, malignancies, and gastrointestinal perforations; studies to monitor any unidentified risks; a cholesterol kinetic study; and studies to evaluate the effect of tofacitinib on the immune response to pneumococcal and influenza vaccines as well as to further delineate the effects of tofacitinib on lymphocyte subsets.
The panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver but not at this meeting.
Martin Bermin-Gorvine of "The Pink Sheet" contributed to this story. This news service and "The Pink Sheet" are both owned by Elsevier.
SILVER SPRING, MD. – There soon may be a new oral disease-modifying antirheumatic drug in town, the first in more than 10 years to be approved for the treatment of rheumatoid arthritis.
The drug is tofacitinib, and it is the first in a new class of agents for RA patients.
On May 9 the Food and Drug Administration’s Arthritis Advisory Committee voted 8 to 2 in favor of approving tofacitinib for RA patients who have had inadequate response to one or more disease-modifying antirheumatic drugs (DMARDs).
A small-molecule inhibitor of the Janus kinase (JAK) pathways, Pfizer’s tofacitinib blocks inflammatory cytokines that play a role in the pathogenesis of RA. If approved, tofacitinib would be the first JAK inhibitor for RA. Another JAK inhibitor, ruxolitinib (Incyte’s Jakafi) was approved to treat myelofibrosis in November.
Pfizer’s application was based on five randomized controlled phase III efficacy trials involving a total 3,315 patients. The application also included safety data for a total of 4,816 patients from the phase II and III studies, of whom 4,053 were treated for at least 6 months and 567 for more than 3 years, up to 42 months. Among the trials was one that compared tofacitinib with placebo in patients with moderate to severe RA who had incomplete responses to tumor necrosis factor (TNF) inhibitors, and four studies of patients who had inadequate responses to other DMARDs. One study included an arm with adalimumab as an active comparator, and one evaluated radiographic response. In three studies, the participants were taking background methotrexate, and in one study, tofacitinib was used as monotherapy. The studies were conducted in a total of 44 countries.
All five of the phase III studies were designed to show the superiority of 5-mg and 10-mg twice-daily doses of tofacitinib to placebo for the primary end points: a 20% or better increase in the American College of Rheumatology’s definition of improvement in the signs and symptoms of RA (ACR20), improvement in Health Assessment Questionnaire-Disability Index (HAQ-DI) scores, and a Disease Activity Score-28 (DAS28) of less than 2.6. In most of the assessments conducted at 6 and 12 months, both doses exhibited statistical superiority over placebo, with the 10-mg dose showing greater improvement than the 5-mg dose.
However, the fact that only one study evaluated radiographic evidence, which the FDA reviewers deemed inconclusive, was cause for concern for the FDA reviewers and committee members. According to the FDA’s Dr. Yongman Kim, 18% of the 278 patients on 5 mg of tofacitinib, 16% of the 290 with 10 mg of tofacitinib, and 14% of the 140 placebo patients showed an "improved" modified total sharp score (mTSS) of less than 0, suggesting it inhibited joint damage.
The FDA was also concerned about dose- and time-dependent elevations in malignancies, and particularly lymphomas, with a standardized incidence ratio of 2.35 at a median 9 months duration of exposure. Of the seven total patients who developed lymphoma, three were on 5 mg of tofacitinib, three were on 10 mg, and one was in a still-blinded arm of the trial. Overall malignancy rates increased with time, from 0.79 per 100 patient-years at 0-6 months to 1.06 at 12-18 months to 1.43 beyond 24 months.
Several of the committee members who voted in favor of tofacitinib said that they did so because they wanted a new oral option for RA patients and that they don’t anticipate any better data from clinical trials. They strongly urged postmarketing studies of both radiologic efficacy and safety. Pfizer has proposed a Risk Evaluation and Mitigation Strategy that will include, alongside labeling provisions, a Medication Guide, a "Dear Health Care Professional" letter, and routine pharmacovigilance. Additional clinical studies that are ongoing or planned include long-term extension studies to assess cardiovascular safety, serious and opportunistic infections, malignancies, and gastrointestinal perforations; studies to monitor any unidentified risks; a cholesterol kinetic study; and studies to evaluate the effect of tofacitinib on the immune response to pneumococcal and influenza vaccines as well as to further delineate the effects of tofacitinib on lymphocyte subsets.
The panelists have been cleared of potential conflicts of interest related to the topic of the meeting. Occasionally, a panelist may be given a waiver but not at this meeting.
Martin Bermin-Gorvine of "The Pink Sheet" contributed to this story. This news service and "The Pink Sheet" are both owned by Elsevier.
Meta-Analysis Links Acid Suppression to C. difficile Risk
LONDON – The use of acid-suppression therapy was associated with a significantly increased risk for Clostridium difficile infection in a systematic review and meta-analysis of data from 30 studies.
Previous observational studies have suggested a link between proton pump inhibitor (PPI) use and an increased risk of developing Clostridium difficile infection (CDI). In February, the Food and Drug Administration issued an alert about the use of PPIs because of the increased CDI risk.
This new meta-analysis supports this association, with a significantly increased risk of CDI with acid suppression (odds ratio, 1.58; P less than .05), Dr. Iman Tleyjeh said at the European Congress of Clinical Microbiology and Infectious Diseases.
"We found a robust association between acid-suppression therapy and the appearance of CDI," said Dr. Tleyjeh, director of the research and scientific publication center and a consultant in infectious disease at King Fahad Medical City, Riyadh, Saudi Arabia.
"Our findings have major global implications, particularly that most of the use of acid-suppression therapy [is] unjustified based on available evidence," he added.
The analysis included studies published through January 2012. Inclusion criteria were that each study had to be an analytical study reporting the effect of acid suppression on CDI incidence, and had to be adjusted for confounders such as antibiotic use.
A total of 30 studies from the United States, United Kingdom, Canada, and Israel met the criteria. There were no randomized clinical trials. Twenty-one were cohort studies, and nine were case-control studies, with a total of 52 effect estimates. All of the included studies were of very good quality, said Dr. Tleyjeh, who is also with the Mayo Clinic, Rochester, Minn.
Separate analyses were done for the different types of studies, and the investigators addressed statistical issues including heterogeneity, publication bias, and residual confounding. Twenty-four studies were from single centers, five were multicenter, two were from general practice research databases, and one was from a general practice clinic. Five studies exclusively addressed community-acquired CDI, 24 addressed hospital-acquired CDI, 1 covered both community- and hospital-acquired CDI, and 1 examined CDI rates in a long-term care facility.
A random-effect meta-analysis revealed that use of acid-suppression therapy (PPIs or H2 blockers) was associated with a significantly increased risk for CDI, with an odds ratio of 1.58 (95% confidence interval [CI], 1.38-1.80). The pooled proportion of CDI patients who were exposed to antibiotics was 0.70 (95% CI, 0.64-0.75). Thus, on average, 30% of CDI patients were not exposed to antibiotics, based on pharmacy records, discharge summaries, or other data sources, Dr. Tleyjeh noted.
Each of the separate analyses showed a significant association (P less than 0.05) between acid suppression and CDI, with pooled effect estimates of 1.67 for studies investigating PPIs, 1.44 for those looking at H2 blockers, 1.46 for the cohort studies, and 1.63 for the case-control studies.
Analysis of the heterogeneity between studies revealed that the main driver was an overall lower pooled effect size for the Canadian studies compared with those from the other three countries. Among the Canadian studies, the odds ratio for the effect of acid suppression on CDI was just 1.08, compared with 1.79 for the U.S. studies, 1.91 for the U.K. studies, and 2.52 for the Israeli studies.
The "meta-regression analysis" technique was used to further explore the heterogeneity between studies with regard to study design, study location, journal impact factor, method of ascertainment of antibiotic use, and proportion with antibiotic exposure.
That analysis revealed that the country where the study was performed was a significant independent variable, whereas patient’s sex had a small independent impact. A comparison of different country-specific study characteristics revealed that CDI cases in Canadian studies had a higher exposure to antibiotics than did those in the three other countries, 90% vs. 72%.
Further analysis also revealed that potential publication bias, the tendency for negative studies to remain unpublished, and adjustment for that factor using a novel regression-based method resulted in an adjusted average odds ratio of 1.38 (95% CI, 1.21-1.58). "So, there is resistant association even if we assume there is [publication] bias and we adjust for it," Dr. Tleyjeh said.
Because the included studies were all observational, a final sensitivity analysis was done to assess the possible effect of unknown and unmeasured confounders. This revealed that an unmeasured confounder would have to be severely imbalanced between the acid-suppression users and nonusers, by an odds ratio of 10, or would have to increase the risk of CDI by at least twofold to account for this association. Such a confounder would have to be even stronger than antibiotic use, he commented.
The exact reason for the association between acid suppression and CDI has not been clearly elucidated. Gastric acid does not kill C. difficile spores, but it does kill the vegetative form of C. difficile, the survival of which with acid-suppression therapy could play a role in pathogenesis. Acid-suppression therapy has also been shown to delay gastric emptying, which could improve survival of the vegetative form, he explained.
Dr. Tleyjeh stated that he had no relevant financial disclosures.
LONDON – The use of acid-suppression therapy was associated with a significantly increased risk for Clostridium difficile infection in a systematic review and meta-analysis of data from 30 studies.
Previous observational studies have suggested a link between proton pump inhibitor (PPI) use and an increased risk of developing Clostridium difficile infection (CDI). In February, the Food and Drug Administration issued an alert about the use of PPIs because of the increased CDI risk.
This new meta-analysis supports this association, with a significantly increased risk of CDI with acid suppression (odds ratio, 1.58; P less than .05), Dr. Iman Tleyjeh said at the European Congress of Clinical Microbiology and Infectious Diseases.
"We found a robust association between acid-suppression therapy and the appearance of CDI," said Dr. Tleyjeh, director of the research and scientific publication center and a consultant in infectious disease at King Fahad Medical City, Riyadh, Saudi Arabia.
"Our findings have major global implications, particularly that most of the use of acid-suppression therapy [is] unjustified based on available evidence," he added.
The analysis included studies published through January 2012. Inclusion criteria were that each study had to be an analytical study reporting the effect of acid suppression on CDI incidence, and had to be adjusted for confounders such as antibiotic use.
A total of 30 studies from the United States, United Kingdom, Canada, and Israel met the criteria. There were no randomized clinical trials. Twenty-one were cohort studies, and nine were case-control studies, with a total of 52 effect estimates. All of the included studies were of very good quality, said Dr. Tleyjeh, who is also with the Mayo Clinic, Rochester, Minn.
Separate analyses were done for the different types of studies, and the investigators addressed statistical issues including heterogeneity, publication bias, and residual confounding. Twenty-four studies were from single centers, five were multicenter, two were from general practice research databases, and one was from a general practice clinic. Five studies exclusively addressed community-acquired CDI, 24 addressed hospital-acquired CDI, 1 covered both community- and hospital-acquired CDI, and 1 examined CDI rates in a long-term care facility.
A random-effect meta-analysis revealed that use of acid-suppression therapy (PPIs or H2 blockers) was associated with a significantly increased risk for CDI, with an odds ratio of 1.58 (95% confidence interval [CI], 1.38-1.80). The pooled proportion of CDI patients who were exposed to antibiotics was 0.70 (95% CI, 0.64-0.75). Thus, on average, 30% of CDI patients were not exposed to antibiotics, based on pharmacy records, discharge summaries, or other data sources, Dr. Tleyjeh noted.
Each of the separate analyses showed a significant association (P less than 0.05) between acid suppression and CDI, with pooled effect estimates of 1.67 for studies investigating PPIs, 1.44 for those looking at H2 blockers, 1.46 for the cohort studies, and 1.63 for the case-control studies.
Analysis of the heterogeneity between studies revealed that the main driver was an overall lower pooled effect size for the Canadian studies compared with those from the other three countries. Among the Canadian studies, the odds ratio for the effect of acid suppression on CDI was just 1.08, compared with 1.79 for the U.S. studies, 1.91 for the U.K. studies, and 2.52 for the Israeli studies.
The "meta-regression analysis" technique was used to further explore the heterogeneity between studies with regard to study design, study location, journal impact factor, method of ascertainment of antibiotic use, and proportion with antibiotic exposure.
That analysis revealed that the country where the study was performed was a significant independent variable, whereas patient’s sex had a small independent impact. A comparison of different country-specific study characteristics revealed that CDI cases in Canadian studies had a higher exposure to antibiotics than did those in the three other countries, 90% vs. 72%.
Further analysis also revealed that potential publication bias, the tendency for negative studies to remain unpublished, and adjustment for that factor using a novel regression-based method resulted in an adjusted average odds ratio of 1.38 (95% CI, 1.21-1.58). "So, there is resistant association even if we assume there is [publication] bias and we adjust for it," Dr. Tleyjeh said.
Because the included studies were all observational, a final sensitivity analysis was done to assess the possible effect of unknown and unmeasured confounders. This revealed that an unmeasured confounder would have to be severely imbalanced between the acid-suppression users and nonusers, by an odds ratio of 10, or would have to increase the risk of CDI by at least twofold to account for this association. Such a confounder would have to be even stronger than antibiotic use, he commented.
The exact reason for the association between acid suppression and CDI has not been clearly elucidated. Gastric acid does not kill C. difficile spores, but it does kill the vegetative form of C. difficile, the survival of which with acid-suppression therapy could play a role in pathogenesis. Acid-suppression therapy has also been shown to delay gastric emptying, which could improve survival of the vegetative form, he explained.
Dr. Tleyjeh stated that he had no relevant financial disclosures.
LONDON – The use of acid-suppression therapy was associated with a significantly increased risk for Clostridium difficile infection in a systematic review and meta-analysis of data from 30 studies.
Previous observational studies have suggested a link between proton pump inhibitor (PPI) use and an increased risk of developing Clostridium difficile infection (CDI). In February, the Food and Drug Administration issued an alert about the use of PPIs because of the increased CDI risk.
This new meta-analysis supports this association, with a significantly increased risk of CDI with acid suppression (odds ratio, 1.58; P less than .05), Dr. Iman Tleyjeh said at the European Congress of Clinical Microbiology and Infectious Diseases.
"We found a robust association between acid-suppression therapy and the appearance of CDI," said Dr. Tleyjeh, director of the research and scientific publication center and a consultant in infectious disease at King Fahad Medical City, Riyadh, Saudi Arabia.
"Our findings have major global implications, particularly that most of the use of acid-suppression therapy [is] unjustified based on available evidence," he added.
The analysis included studies published through January 2012. Inclusion criteria were that each study had to be an analytical study reporting the effect of acid suppression on CDI incidence, and had to be adjusted for confounders such as antibiotic use.
A total of 30 studies from the United States, United Kingdom, Canada, and Israel met the criteria. There were no randomized clinical trials. Twenty-one were cohort studies, and nine were case-control studies, with a total of 52 effect estimates. All of the included studies were of very good quality, said Dr. Tleyjeh, who is also with the Mayo Clinic, Rochester, Minn.
Separate analyses were done for the different types of studies, and the investigators addressed statistical issues including heterogeneity, publication bias, and residual confounding. Twenty-four studies were from single centers, five were multicenter, two were from general practice research databases, and one was from a general practice clinic. Five studies exclusively addressed community-acquired CDI, 24 addressed hospital-acquired CDI, 1 covered both community- and hospital-acquired CDI, and 1 examined CDI rates in a long-term care facility.
A random-effect meta-analysis revealed that use of acid-suppression therapy (PPIs or H2 blockers) was associated with a significantly increased risk for CDI, with an odds ratio of 1.58 (95% confidence interval [CI], 1.38-1.80). The pooled proportion of CDI patients who were exposed to antibiotics was 0.70 (95% CI, 0.64-0.75). Thus, on average, 30% of CDI patients were not exposed to antibiotics, based on pharmacy records, discharge summaries, or other data sources, Dr. Tleyjeh noted.
Each of the separate analyses showed a significant association (P less than 0.05) between acid suppression and CDI, with pooled effect estimates of 1.67 for studies investigating PPIs, 1.44 for those looking at H2 blockers, 1.46 for the cohort studies, and 1.63 for the case-control studies.
Analysis of the heterogeneity between studies revealed that the main driver was an overall lower pooled effect size for the Canadian studies compared with those from the other three countries. Among the Canadian studies, the odds ratio for the effect of acid suppression on CDI was just 1.08, compared with 1.79 for the U.S. studies, 1.91 for the U.K. studies, and 2.52 for the Israeli studies.
The "meta-regression analysis" technique was used to further explore the heterogeneity between studies with regard to study design, study location, journal impact factor, method of ascertainment of antibiotic use, and proportion with antibiotic exposure.
That analysis revealed that the country where the study was performed was a significant independent variable, whereas patient’s sex had a small independent impact. A comparison of different country-specific study characteristics revealed that CDI cases in Canadian studies had a higher exposure to antibiotics than did those in the three other countries, 90% vs. 72%.
Further analysis also revealed that potential publication bias, the tendency for negative studies to remain unpublished, and adjustment for that factor using a novel regression-based method resulted in an adjusted average odds ratio of 1.38 (95% CI, 1.21-1.58). "So, there is resistant association even if we assume there is [publication] bias and we adjust for it," Dr. Tleyjeh said.
Because the included studies were all observational, a final sensitivity analysis was done to assess the possible effect of unknown and unmeasured confounders. This revealed that an unmeasured confounder would have to be severely imbalanced between the acid-suppression users and nonusers, by an odds ratio of 10, or would have to increase the risk of CDI by at least twofold to account for this association. Such a confounder would have to be even stronger than antibiotic use, he commented.
The exact reason for the association between acid suppression and CDI has not been clearly elucidated. Gastric acid does not kill C. difficile spores, but it does kill the vegetative form of C. difficile, the survival of which with acid-suppression therapy could play a role in pathogenesis. Acid-suppression therapy has also been shown to delay gastric emptying, which could improve survival of the vegetative form, he explained.
Dr. Tleyjeh stated that he had no relevant financial disclosures.
FROM THE EUROPEAN CONGRESS OF CLINICAL MICROBIOLOGY AND INFECTIOUS DISEASES
Major Finding: A random-effect meta-analysis revealed that acid-suppression therapy, with PPIs and/or H2 blockers, was associated with a significantly increased risk for C. difficile infection (OR, 1.58).
Data Source: This meta-analysis included 30 studies from the United States, United Kingdom, Canada, and Israel (21 cohort studies and 9 case-control studies), with a total of 52 effect estimates.
Disclosures: Dr. Tleyjeh reported having no relevant financial relationships.