User login
Lucas Franki is an associate editor for MDedge News, and has been with the company since 2014. He has a BA in English from Penn State University and is an Eagle Scout.
FDA approves Seysara for treatment of moderate to severe acne
vulgaris for people aged 9 years and older, according to a press release from Paratek, the drug’s manufacturer.
FDA approval is based on results from two phase 3 clinical trials (NCT02320149 and NCT02322866), in which patients received either sarecycline at 1.5 mg/kg per day or placebo for 12 weeks. Patients who received sarecycline were significantly more likely to reach the primary endpoint of improved acne severity based on absolute change in facial lesion counts and percentage of participants with Investigator Global Assessment success.
Common adverse events reported in the sarecycline group were nausea (3.2%), nasopharyngitis (2.8%), and headache (2.8%). The discontinuation rate due to adverse events among sarecycline-treated patients in both studies combined was 1.4%.
Sarecycline is an oral, narrow spectrum tetracycline-derived antibiotic with anti-inflammatory properties, and is approved for once-daily use, according to Paratek. Seysara will be marketed in the United States by Almirall SA.
vulgaris for people aged 9 years and older, according to a press release from Paratek, the drug’s manufacturer.
FDA approval is based on results from two phase 3 clinical trials (NCT02320149 and NCT02322866), in which patients received either sarecycline at 1.5 mg/kg per day or placebo for 12 weeks. Patients who received sarecycline were significantly more likely to reach the primary endpoint of improved acne severity based on absolute change in facial lesion counts and percentage of participants with Investigator Global Assessment success.
Common adverse events reported in the sarecycline group were nausea (3.2%), nasopharyngitis (2.8%), and headache (2.8%). The discontinuation rate due to adverse events among sarecycline-treated patients in both studies combined was 1.4%.
Sarecycline is an oral, narrow spectrum tetracycline-derived antibiotic with anti-inflammatory properties, and is approved for once-daily use, according to Paratek. Seysara will be marketed in the United States by Almirall SA.
vulgaris for people aged 9 years and older, according to a press release from Paratek, the drug’s manufacturer.
FDA approval is based on results from two phase 3 clinical trials (NCT02320149 and NCT02322866), in which patients received either sarecycline at 1.5 mg/kg per day or placebo for 12 weeks. Patients who received sarecycline were significantly more likely to reach the primary endpoint of improved acne severity based on absolute change in facial lesion counts and percentage of participants with Investigator Global Assessment success.
Common adverse events reported in the sarecycline group were nausea (3.2%), nasopharyngitis (2.8%), and headache (2.8%). The discontinuation rate due to adverse events among sarecycline-treated patients in both studies combined was 1.4%.
Sarecycline is an oral, narrow spectrum tetracycline-derived antibiotic with anti-inflammatory properties, and is approved for once-daily use, according to Paratek. Seysara will be marketed in the United States by Almirall SA.
Emgality approved for migraine prevention in adults
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.

FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.
FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
The Food and Drug Administration has approved galcanezumab-gnlm (Emgality) for the preventive treatment of migraine in adults, according to an announcement from Eli Lilly, the drug’s manufacturer.
FDA approval is based on results of three different phase 3 clinical trials, EVOLVE-1, EVOLVE-2, and REGAIN. EVOLVE-1 and EVOLVE-2 were 6-month, double-blind, placebo-controlled studies that included adults with episodic migraine (4-14 headache days per month). REGAIN was a 3-month, double-blind, placebo-controlled study of adults with chronic migraine (at least 15 headache days per month). The primary endpoint of all trials was mean change from baseline in the number of monthly headache days.
In all trials, patients received either placebo, 120 mg galcanezumab-gnlm after an initial loading dose of 240 mg, or 240 mg galcanezumab-gnlm. In EVOLVE-1 and EVOLVE-2, people who received galcanezumab-gnlm had significantly fewer headache days per month than with placebo, and those who received galcanezumab-gnlm were also more likely to achieve a 50%, 75%, and 100% reduction in headache days.
In REGAIN, patients who received galcanezumab-gnlm experienced fewer headache days than those who received placebo, and were also more likely to achieve a 50% reduction in headache days. There was no difference between groups in likelihood of achieving a 75% or 100% reduction.
The recommended dosage, according to the label, is a monthly, 120-mg subcutaneous injection, with an initial loading dose of 240 mg. The most common adverse event associated with galcanezumab-gnlm was injection site reaction.
Galcanezumab-gnlm, a calcitonin gene–related peptide antagonist, is also under final review by the European Commission for approval in Europe.
The U.S. list price of galcanezumab-gnlm is $575 once-monthly, or $6,900 annually, and patients with commercial insurance are candidates to receive it free for up to 12 months as part of Lilly’s patient support program (subject to specific terms and conditions), according to the announcement.
ICYMI: Wearable cardioverter-defibrillator ineffective at reducing arrhythmic death risk
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
Patients who recently experienced an MI and had an ejection fraction of 35% or less who used a wearable cardioverter-defibrillator had no decreased risk of arrhythmic death, compared with people who did not use a wearable cardioverter-defibrillator, according to an analysis of data from the multicenter, randomized, and controlled VEST trial (NCT01446965). The results were published Sept. 26 in the New England Journal of Medicine (doi: 10.1056/NEJMoa1800781).
We covered this story before it was published in the journal. Find our conference coverage at the links below.
FROM NEW ENGLAND JOURNAL OF MEDICINE
FDA approves Ajovy for migraine prevention
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
The .
Fremanezumab-vfrm is an injectable anti–calcitonin gene-related peptide monoclonal antibody, the first ever approved by the FDA. It can be administered at a 225-mg dose every month or at a 675-mg dose every 3 months, according to a press release from Teva, the drug’s manufacturer.
FDA approval was based on results from two, phase 3 trials in patients with disabling migraines, one of which involved fremanezumab-vfrm alone, with the other involving the injection in tandem with an oral treatment.
In both trials, patients experienced a reduction of monthly migraine days over a 12-week period. The most common adverse event in both trials was injection site reactions.
“Migraine is a disabling neurological disease that affects more than 36 million people in the United States. About 40 percent of people living with migraine may be appropriate candidates for preventive treatment, yet the majority of them are untreated. I am pleased to have another treatment option that may allow my patients to experience fewer monthly migraine days,” Stephen Silberstein, MD, director of the Jefferson Headache Center at Thomas Jefferson University Hospital in Philadelphia, said in the Teva press release.
CDC opens Emergency Operations Center in advance of Hurricane Florence
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
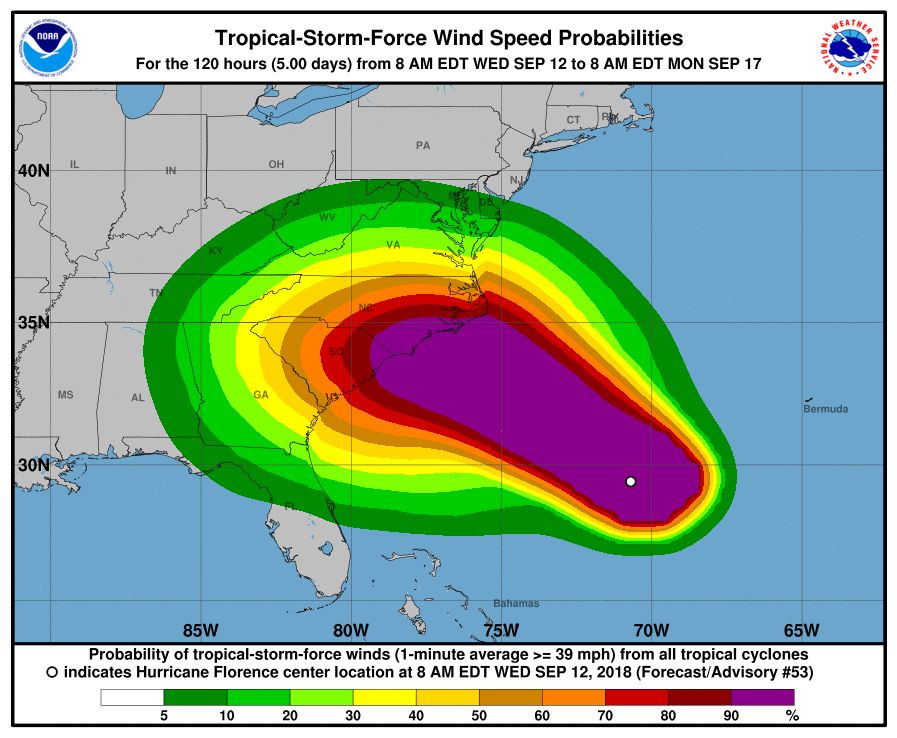
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
The Centers for Disease Control and Prevention is activating its Emergency Operations Center (EOC) in advance of the landfall of Hurricane Florence, according to a CDC media statement.
Activation of the center will allow for 24/7 management of all CDC activities before, during, and after the landfall of Hurricane Florence, including deployment of personnel and resources.
All CDC staff should be ready to provide infectious disease outbreak surveillance, public health messages, water/food safety evaluations, mold prevention and treatment, industrial contamination containment, and standing water control. The CDC is also sharing messages with the public on how to protect themselves from threats before, during, and after landfall, such as drowning and floodwater safety, carbon monoxide poisoning, downed power lines, unsafe food and water, and mold.
The EOC is the CDC’s command center for the coordination and monitoring of CDC activity with all other U.S. federal, state, and local agencies. The EOC also works with the Secretary of Health & Human Services Secretary’s Operations Center to ensure awareness and a coordinated public health and medical response, according to the statement.
Additional resources for interested parties can be found on the CDC website.
Female to male transgender teens most likely to attempt suicide
according to a study published in Pediatrics.
Russell B. Toomey, PhD, of the University of Arizona, Tucson, and his associates performed an analysis of data from the Profiles of Student Life: Attitudes and Behaviors survey. Data was collected from June 2012 to May 2015 and included 120,617 adolescents aged 11-19 years. A total of 202 adolescents identified as male to female transgender, 175 identified as female to male transgender, 344 identified as nonbinary transgender, 1,052 identified as questioning, 60,973 identified as female, and 57,871 identified as male.
Male adolescents were least likely to attempt suicide, with 10% reporting at least one attempt, followed by those identifying as female (18%), questioning (28%), male to female transgender (30%), nonbinary transgender (42%), and female to male transgender (51%). All groups were significantly more likely to attempt suicide, compared with male adolescents.
Compared with transgender adolescents who identified as heterosexual only, identifying as non-heterosexual was associated with an increased risk of attempting suicide, except in nonbinary transgender adolescents. There was no association of increased risk of attempting suicide in transgender adolescents based on ethnicity, parental education levels, age, or urbanicity, except parent education level appeared to be a protective factor for questioning adolescents.
“These results should be used to inform suicide prevention and intervention policy and programs that are aimed at reducing ongoing gender identity–related disparities in suicide behavior as well as ongoing research in which authors seek to better understand for whom and why suicide behavior risk exists,” the authors concluded.
The study was supported by the American Foundation for Suicide Prevention and the National Institute of Minority Health and Health Disparities. The authors had no relevant financial disclosures.
SOURCE: Toomey RB et al. Pediatrics. 2018 Sept 11. doi: 10.1542/peds.2017-4218.
according to a study published in Pediatrics.
Russell B. Toomey, PhD, of the University of Arizona, Tucson, and his associates performed an analysis of data from the Profiles of Student Life: Attitudes and Behaviors survey. Data was collected from June 2012 to May 2015 and included 120,617 adolescents aged 11-19 years. A total of 202 adolescents identified as male to female transgender, 175 identified as female to male transgender, 344 identified as nonbinary transgender, 1,052 identified as questioning, 60,973 identified as female, and 57,871 identified as male.
Male adolescents were least likely to attempt suicide, with 10% reporting at least one attempt, followed by those identifying as female (18%), questioning (28%), male to female transgender (30%), nonbinary transgender (42%), and female to male transgender (51%). All groups were significantly more likely to attempt suicide, compared with male adolescents.
Compared with transgender adolescents who identified as heterosexual only, identifying as non-heterosexual was associated with an increased risk of attempting suicide, except in nonbinary transgender adolescents. There was no association of increased risk of attempting suicide in transgender adolescents based on ethnicity, parental education levels, age, or urbanicity, except parent education level appeared to be a protective factor for questioning adolescents.
“These results should be used to inform suicide prevention and intervention policy and programs that are aimed at reducing ongoing gender identity–related disparities in suicide behavior as well as ongoing research in which authors seek to better understand for whom and why suicide behavior risk exists,” the authors concluded.
The study was supported by the American Foundation for Suicide Prevention and the National Institute of Minority Health and Health Disparities. The authors had no relevant financial disclosures.
SOURCE: Toomey RB et al. Pediatrics. 2018 Sept 11. doi: 10.1542/peds.2017-4218.
according to a study published in Pediatrics.
Russell B. Toomey, PhD, of the University of Arizona, Tucson, and his associates performed an analysis of data from the Profiles of Student Life: Attitudes and Behaviors survey. Data was collected from June 2012 to May 2015 and included 120,617 adolescents aged 11-19 years. A total of 202 adolescents identified as male to female transgender, 175 identified as female to male transgender, 344 identified as nonbinary transgender, 1,052 identified as questioning, 60,973 identified as female, and 57,871 identified as male.
Male adolescents were least likely to attempt suicide, with 10% reporting at least one attempt, followed by those identifying as female (18%), questioning (28%), male to female transgender (30%), nonbinary transgender (42%), and female to male transgender (51%). All groups were significantly more likely to attempt suicide, compared with male adolescents.
Compared with transgender adolescents who identified as heterosexual only, identifying as non-heterosexual was associated with an increased risk of attempting suicide, except in nonbinary transgender adolescents. There was no association of increased risk of attempting suicide in transgender adolescents based on ethnicity, parental education levels, age, or urbanicity, except parent education level appeared to be a protective factor for questioning adolescents.
“These results should be used to inform suicide prevention and intervention policy and programs that are aimed at reducing ongoing gender identity–related disparities in suicide behavior as well as ongoing research in which authors seek to better understand for whom and why suicide behavior risk exists,” the authors concluded.
The study was supported by the American Foundation for Suicide Prevention and the National Institute of Minority Health and Health Disparities. The authors had no relevant financial disclosures.
SOURCE: Toomey RB et al. Pediatrics. 2018 Sept 11. doi: 10.1542/peds.2017-4218.
FROM PEDIATRICS
Antidepressants not effective in treating depression in patients with dementia
Antidepressants often are used to treat depressive symptoms in patients with dementia, but this practice is not backed by strong evidence, according to a systematic review published in the Cochrane Database of Systematic Reviews.
“On the only measure of efficacy for which we had high-quality evidence (depression rating scale scores), antidepressants showed little or no effect,” reported Robert Dudas, MD, PhD, of the University of Cambridge (England), and his associates. “The evidence on remission rates favored antidepressants but was of moderate quality, so future research may find a different result.”
For the review, a total of 10 studies – which altogether included of 1,592 patients – were examined, of which 8 included enough information to enter into analyses about antidepressant efficacy, Dr. Dudas and his associates reported. The average age in the studies was 75 years, and study participants had mild or moderate dementia.
After 6-13 weeks, little difference in depression symptom rating scale scores was found between antidepressant- and placebo-treated groups (standardized mean difference, –0.10 points; 95% confidence interval, –0.26 to 0.06). Similar results were found after 6-9 months (SMD, 0.59 points; 95% CI, –1.12 to 2.30).
Antidepressants did not significantly improve response rates (odds ratio, 1.71; 95% CI, 0.80-3.67) but were associated with a higher remission rate (OR, 2.57; 95% CI, 1.44-4.59). After 6-13 weeks, no difference was found between antidepressants and placebo on performance of daily activities (SMD, –0.05; 95% CI, –0.36 to 0.25) or in cognition (mean difference of 0.33 in Mini-Mental State Examination scores; 95% CI, –1.31 to 1.96).
In addition to experiencing no significant improvement in depression symptoms, patients who received antidepressants were more likely to drop out of treatment than were those receiving placebo (OR, 1.51; 95% CI, 1.07-2.14) and to experience at least one adverse event (OR, 1.55; 95% CI, 1.21-1.98). Adverse events more likely to occur in patients receiving antidepressants included dry mouth (OR, 1.80; 95% CI, 1.23-2.63) and dizziness (OR, 2.00; 95% CI, 1.34-2.98).
“ There is a need for well-conducted, randomized, controlled trials, using scales validated in older people with depression and dementia, of modern, frequently used drugs and sufficient sample sizes that would allow a study of treatment response and detailed adverse event profile according to dementia etiology and severity and depression severity,” the investigators concluded.
The study was supported by several entities, including the Cambridgeshire and Petersborough NHS Foundation Trust, Collaborations for Leadership in Applied Health Research and Care, and the National Institute for Health Research, all in the United Kingdom. Dr. Dudas and his associates declared no conflicts of interest.
SOURCE: Dudas R et al. Cochrane Database Syst Rev. 2018 Aug 31. doi: 10.1002/14651858.CD003944.pub2.
Antidepressants often are used to treat depressive symptoms in patients with dementia, but this practice is not backed by strong evidence, according to a systematic review published in the Cochrane Database of Systematic Reviews.
“On the only measure of efficacy for which we had high-quality evidence (depression rating scale scores), antidepressants showed little or no effect,” reported Robert Dudas, MD, PhD, of the University of Cambridge (England), and his associates. “The evidence on remission rates favored antidepressants but was of moderate quality, so future research may find a different result.”
For the review, a total of 10 studies – which altogether included of 1,592 patients – were examined, of which 8 included enough information to enter into analyses about antidepressant efficacy, Dr. Dudas and his associates reported. The average age in the studies was 75 years, and study participants had mild or moderate dementia.
After 6-13 weeks, little difference in depression symptom rating scale scores was found between antidepressant- and placebo-treated groups (standardized mean difference, –0.10 points; 95% confidence interval, –0.26 to 0.06). Similar results were found after 6-9 months (SMD, 0.59 points; 95% CI, –1.12 to 2.30).
Antidepressants did not significantly improve response rates (odds ratio, 1.71; 95% CI, 0.80-3.67) but were associated with a higher remission rate (OR, 2.57; 95% CI, 1.44-4.59). After 6-13 weeks, no difference was found between antidepressants and placebo on performance of daily activities (SMD, –0.05; 95% CI, –0.36 to 0.25) or in cognition (mean difference of 0.33 in Mini-Mental State Examination scores; 95% CI, –1.31 to 1.96).
In addition to experiencing no significant improvement in depression symptoms, patients who received antidepressants were more likely to drop out of treatment than were those receiving placebo (OR, 1.51; 95% CI, 1.07-2.14) and to experience at least one adverse event (OR, 1.55; 95% CI, 1.21-1.98). Adverse events more likely to occur in patients receiving antidepressants included dry mouth (OR, 1.80; 95% CI, 1.23-2.63) and dizziness (OR, 2.00; 95% CI, 1.34-2.98).
“ There is a need for well-conducted, randomized, controlled trials, using scales validated in older people with depression and dementia, of modern, frequently used drugs and sufficient sample sizes that would allow a study of treatment response and detailed adverse event profile according to dementia etiology and severity and depression severity,” the investigators concluded.
The study was supported by several entities, including the Cambridgeshire and Petersborough NHS Foundation Trust, Collaborations for Leadership in Applied Health Research and Care, and the National Institute for Health Research, all in the United Kingdom. Dr. Dudas and his associates declared no conflicts of interest.
SOURCE: Dudas R et al. Cochrane Database Syst Rev. 2018 Aug 31. doi: 10.1002/14651858.CD003944.pub2.
Antidepressants often are used to treat depressive symptoms in patients with dementia, but this practice is not backed by strong evidence, according to a systematic review published in the Cochrane Database of Systematic Reviews.
“On the only measure of efficacy for which we had high-quality evidence (depression rating scale scores), antidepressants showed little or no effect,” reported Robert Dudas, MD, PhD, of the University of Cambridge (England), and his associates. “The evidence on remission rates favored antidepressants but was of moderate quality, so future research may find a different result.”
For the review, a total of 10 studies – which altogether included of 1,592 patients – were examined, of which 8 included enough information to enter into analyses about antidepressant efficacy, Dr. Dudas and his associates reported. The average age in the studies was 75 years, and study participants had mild or moderate dementia.
After 6-13 weeks, little difference in depression symptom rating scale scores was found between antidepressant- and placebo-treated groups (standardized mean difference, –0.10 points; 95% confidence interval, –0.26 to 0.06). Similar results were found after 6-9 months (SMD, 0.59 points; 95% CI, –1.12 to 2.30).
Antidepressants did not significantly improve response rates (odds ratio, 1.71; 95% CI, 0.80-3.67) but were associated with a higher remission rate (OR, 2.57; 95% CI, 1.44-4.59). After 6-13 weeks, no difference was found between antidepressants and placebo on performance of daily activities (SMD, –0.05; 95% CI, –0.36 to 0.25) or in cognition (mean difference of 0.33 in Mini-Mental State Examination scores; 95% CI, –1.31 to 1.96).
In addition to experiencing no significant improvement in depression symptoms, patients who received antidepressants were more likely to drop out of treatment than were those receiving placebo (OR, 1.51; 95% CI, 1.07-2.14) and to experience at least one adverse event (OR, 1.55; 95% CI, 1.21-1.98). Adverse events more likely to occur in patients receiving antidepressants included dry mouth (OR, 1.80; 95% CI, 1.23-2.63) and dizziness (OR, 2.00; 95% CI, 1.34-2.98).
“ There is a need for well-conducted, randomized, controlled trials, using scales validated in older people with depression and dementia, of modern, frequently used drugs and sufficient sample sizes that would allow a study of treatment response and detailed adverse event profile according to dementia etiology and severity and depression severity,” the investigators concluded.
The study was supported by several entities, including the Cambridgeshire and Petersborough NHS Foundation Trust, Collaborations for Leadership in Applied Health Research and Care, and the National Institute for Health Research, all in the United Kingdom. Dr. Dudas and his associates declared no conflicts of interest.
SOURCE: Dudas R et al. Cochrane Database Syst Rev. 2018 Aug 31. doi: 10.1002/14651858.CD003944.pub2.
FROM THE COCHRANE DATABASE OF SYSTEMATIC REVIEWS
FDA approves first mobile app for contraceptive use
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
The Food and Drug Administration has approved marketing of the first medical mobile application that can be used as a contraceptive, the agency announced in a written statement.
The app, called Natural Cycles, contains an algorithm that calculates the days of the month women are likely to be fertile, based on daily body temperature readings and menstrual cycle information, and is intended for premenopausal women aged 18 years and older. App users are instructed to take their daily body temperatures with a basal body thermometer and enter the information into the app. The more-sensitive basal body thermometer can detect temperature elevations during ovulation, and the app will display a warning on days when users are most fertile. During these days, women should either abstain from sex or use protection.
In clinical studies comprising more than 15,000 women, Natural Cycles had a “perfect use” failure rate of 1.8% and a “normal use” failure rate of 6.5%. This compares favorably with other forms of contraception and birth control: Male condoms have a perfect use failure rate of 2.0% and a normal use failure rate of 18.0%, and combined oral contraceptives have a perfect use failure rate of 0.3% and a normal use failure rate of 9.0%, according to the Association of Reproductive Care Professionals.
“Consumers are increasingly using digital health technologies to inform their everyday health decisions, and this new app can provide an effective method of contraception if it’s used carefully and correctly,” Terri Cornelison, MD, PhD, assistant director for the health of women in the FDA’s Center for Devices and Radiological Health, said in the statement.
The app should not be used by women with a condition in which a pregnancy would present risk to the mother or fetus or by women who are using a birth control or hormonal treatment that inhibits ovulation. Natural Cycles does not protect against sexually transmitted infections.
Parents’ religiosity tied to reduced suicide risk in girls
Girls with parents who considered religion important in their lives were less likely to consider or attempt suicide, according to Connie Svob, PhD, of Columbia University, New York, and her associates.
Data were collected from an ongoing, 30-year, three-generational, observational study at the New York State Psychiatric Institute and Columbia University. In the study, the second and third generations were determined to be at high or low risk for major depressive disorder because of either the presence or absence of MDD in the first generation.
The study included 214 children aged 6-18 years from 112 unique families, who had undergone at least one assessment of religiosity, and a parent from each included family. Among the children, religious importance was associated with reduced suicide risk in girls (OR, 0.48; 95% CI, 0.33-0.70) but not in boys (OR, 1.15; 95% CI, 0.74-1.80).
A similar association was found for religious attendance, where girls saw a reduced risk (OR, 0.64; 95% CI, 0.49-0.84) and boys did not (OR, 0.94; 95% CI, 0.69-1.27), Dr. Svob and her associates reported in JAMA Psychiatry.
“These findings suggest that a parent’s belief in the importance of religion may be a more robust factor than a parent’s attendance at religious services and make one wonder whether religious importance might be more strongly associated with teaching and beliefs about suicide within the home than is service attendance, or whether some other mechanism might be responsible,” the authors noted.
Study limitations included a homogeneous population who were all white and predominantly Roman Catholic or Protestant, as well as a small sample size of children with suicidal behavior or ideation.
“,” Dr. Svob and her associates wrote. “These include conducting a brief spiritual history with parents of offspring being brought in for psychiatric consultations, as well as assessing an offspring’s own beliefs (religious importance) and behaviors (religious service attendance), particularly with girls.”
Dr. Svob reported no disclosures. The study was supported partly by the John Templeton Foundation and the National Institute of Mental Health.
SOURCE: Svob C et al. JAMA Psychiatry. 2018 Aug 8. doi: 10.1001/jamapsychiatry.2018.2060.
Girls with parents who considered religion important in their lives were less likely to consider or attempt suicide, according to Connie Svob, PhD, of Columbia University, New York, and her associates.
Data were collected from an ongoing, 30-year, three-generational, observational study at the New York State Psychiatric Institute and Columbia University. In the study, the second and third generations were determined to be at high or low risk for major depressive disorder because of either the presence or absence of MDD in the first generation.
The study included 214 children aged 6-18 years from 112 unique families, who had undergone at least one assessment of religiosity, and a parent from each included family. Among the children, religious importance was associated with reduced suicide risk in girls (OR, 0.48; 95% CI, 0.33-0.70) but not in boys (OR, 1.15; 95% CI, 0.74-1.80).
A similar association was found for religious attendance, where girls saw a reduced risk (OR, 0.64; 95% CI, 0.49-0.84) and boys did not (OR, 0.94; 95% CI, 0.69-1.27), Dr. Svob and her associates reported in JAMA Psychiatry.
“These findings suggest that a parent’s belief in the importance of religion may be a more robust factor than a parent’s attendance at religious services and make one wonder whether religious importance might be more strongly associated with teaching and beliefs about suicide within the home than is service attendance, or whether some other mechanism might be responsible,” the authors noted.
Study limitations included a homogeneous population who were all white and predominantly Roman Catholic or Protestant, as well as a small sample size of children with suicidal behavior or ideation.
“,” Dr. Svob and her associates wrote. “These include conducting a brief spiritual history with parents of offspring being brought in for psychiatric consultations, as well as assessing an offspring’s own beliefs (religious importance) and behaviors (religious service attendance), particularly with girls.”
Dr. Svob reported no disclosures. The study was supported partly by the John Templeton Foundation and the National Institute of Mental Health.
SOURCE: Svob C et al. JAMA Psychiatry. 2018 Aug 8. doi: 10.1001/jamapsychiatry.2018.2060.
Girls with parents who considered religion important in their lives were less likely to consider or attempt suicide, according to Connie Svob, PhD, of Columbia University, New York, and her associates.
Data were collected from an ongoing, 30-year, three-generational, observational study at the New York State Psychiatric Institute and Columbia University. In the study, the second and third generations were determined to be at high or low risk for major depressive disorder because of either the presence or absence of MDD in the first generation.
The study included 214 children aged 6-18 years from 112 unique families, who had undergone at least one assessment of religiosity, and a parent from each included family. Among the children, religious importance was associated with reduced suicide risk in girls (OR, 0.48; 95% CI, 0.33-0.70) but not in boys (OR, 1.15; 95% CI, 0.74-1.80).
A similar association was found for religious attendance, where girls saw a reduced risk (OR, 0.64; 95% CI, 0.49-0.84) and boys did not (OR, 0.94; 95% CI, 0.69-1.27), Dr. Svob and her associates reported in JAMA Psychiatry.
“These findings suggest that a parent’s belief in the importance of religion may be a more robust factor than a parent’s attendance at religious services and make one wonder whether religious importance might be more strongly associated with teaching and beliefs about suicide within the home than is service attendance, or whether some other mechanism might be responsible,” the authors noted.
Study limitations included a homogeneous population who were all white and predominantly Roman Catholic or Protestant, as well as a small sample size of children with suicidal behavior or ideation.
“,” Dr. Svob and her associates wrote. “These include conducting a brief spiritual history with parents of offspring being brought in for psychiatric consultations, as well as assessing an offspring’s own beliefs (religious importance) and behaviors (religious service attendance), particularly with girls.”
Dr. Svob reported no disclosures. The study was supported partly by the John Templeton Foundation and the National Institute of Mental Health.
SOURCE: Svob C et al. JAMA Psychiatry. 2018 Aug 8. doi: 10.1001/jamapsychiatry.2018.2060.
FROM JAMA PSYCHIATRY
Clinical trial: Robotic versus laparoscopic ventral hernia repair
The Robotic Versus Laparoscopic Ventral Hernia Repair study is an interventional trial recruiting patients undergoing elective ventral hernia repair appropriate for minimally invasive surgery.
The trial will compare outcomes of laparoscopic and robotic approaches to ventral hernia repair. compared with the laparoscopic approach, and has been endorsed by the American Hernia Society. However, this evidence is based on database and cohort studies, and more randomized, controlled trials are needed to assess the true effects of the robotic approach.
Patients will be included if they are scheduled for a ventral hernia repair that has been deemed appropriate for minimally invasive surgery. Exclusion criteria include being unlikely to survive for 2 years post surgery, being unlikely to follow up, having advanced chronic obstructive pulmonary disease or congestive heart failure, having a history of open abdomen or extensive lysis of adhesions, having ascites caused by cirrhosis or malignancy, having an active infection, and having a hernia defect size larger than 12 cm.
The primary outcome measure is total number of days spent in the hospital. Secondary outcomes include rates of surgical site infection, rates of surgical site occurrence, rates of hernia reoccurrence, patient-centered outcomes collected using HerQLes (a hernia quality of life measuring instrument), patient-centered outcomes collected using the EQ-5D questionnaire, and cost from a health care perspective.
The primary completion date is April 30, 2020, and the study completion date is April 30, 2023. About 120 people are expected to be recruited.
Find more information on the study page at Clinicaltrials.gov.
The Robotic Versus Laparoscopic Ventral Hernia Repair study is an interventional trial recruiting patients undergoing elective ventral hernia repair appropriate for minimally invasive surgery.
The trial will compare outcomes of laparoscopic and robotic approaches to ventral hernia repair. compared with the laparoscopic approach, and has been endorsed by the American Hernia Society. However, this evidence is based on database and cohort studies, and more randomized, controlled trials are needed to assess the true effects of the robotic approach.
Patients will be included if they are scheduled for a ventral hernia repair that has been deemed appropriate for minimally invasive surgery. Exclusion criteria include being unlikely to survive for 2 years post surgery, being unlikely to follow up, having advanced chronic obstructive pulmonary disease or congestive heart failure, having a history of open abdomen or extensive lysis of adhesions, having ascites caused by cirrhosis or malignancy, having an active infection, and having a hernia defect size larger than 12 cm.
The primary outcome measure is total number of days spent in the hospital. Secondary outcomes include rates of surgical site infection, rates of surgical site occurrence, rates of hernia reoccurrence, patient-centered outcomes collected using HerQLes (a hernia quality of life measuring instrument), patient-centered outcomes collected using the EQ-5D questionnaire, and cost from a health care perspective.
The primary completion date is April 30, 2020, and the study completion date is April 30, 2023. About 120 people are expected to be recruited.
Find more information on the study page at Clinicaltrials.gov.
The Robotic Versus Laparoscopic Ventral Hernia Repair study is an interventional trial recruiting patients undergoing elective ventral hernia repair appropriate for minimally invasive surgery.
The trial will compare outcomes of laparoscopic and robotic approaches to ventral hernia repair. compared with the laparoscopic approach, and has been endorsed by the American Hernia Society. However, this evidence is based on database and cohort studies, and more randomized, controlled trials are needed to assess the true effects of the robotic approach.
Patients will be included if they are scheduled for a ventral hernia repair that has been deemed appropriate for minimally invasive surgery. Exclusion criteria include being unlikely to survive for 2 years post surgery, being unlikely to follow up, having advanced chronic obstructive pulmonary disease or congestive heart failure, having a history of open abdomen or extensive lysis of adhesions, having ascites caused by cirrhosis or malignancy, having an active infection, and having a hernia defect size larger than 12 cm.
The primary outcome measure is total number of days spent in the hospital. Secondary outcomes include rates of surgical site infection, rates of surgical site occurrence, rates of hernia reoccurrence, patient-centered outcomes collected using HerQLes (a hernia quality of life measuring instrument), patient-centered outcomes collected using the EQ-5D questionnaire, and cost from a health care perspective.
The primary completion date is April 30, 2020, and the study completion date is April 30, 2023. About 120 people are expected to be recruited.
Find more information on the study page at Clinicaltrials.gov.