User login
Lucas Franki is an associate editor for MDedge News, and has been with the company since 2014. He has a BA in English from Penn State University and is an Eagle Scout.
Over one-third of psoriasis patients have PsA
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
Over one-third of psoriasis patients have PsA
About two-thirds of patients with psoriasis in a national registry also had psoriatic arthritis (PsA) and/or psoriasis in at least one challenging-to-treat (CTT) area, and one-quarter had both, according to Kristina Callis Duffin, MD, of the University of Utah, Salt Lake City, and her associates.
Their analysis included 2,042 psoriasis patients who were enrolled in the Corrona Psoriasis Registry between April 2015 and May 2018 and initiated biologic treatment during that time. The mean age was 49.6 years, 80% of the patients were white, and 51% were obese. Mean disease duration was 19.9 years and 89.2% of the patients had moderate to severe disease. CTT areas include the scalp, nails, and palmoplantar areas.
A total of 784 people in the cohort (38.4%) had PsA, 778 (38.1%) had scalp psoriasis, 326 (16.0%) had nail psoriasis, 223 (10.9%) had palmoplantar psoriasis, and 535 (26.2%) had both PsA and psoriasis in at least two CTT areas. The most common combinations were PsA plus scalp psoriasis and PsA plus nail and scalp psoriasis.
“These results indicate a need to further characterize patients with psoriasis who have PsA and CTT areas and evaluate the impact of these factors to better understand their treatment needs,” the investigators noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and the study authors reported numerous financial relationships with industry; two authors are Novartis employees.
Secukinumab effective for slowing radiographic progression in active PsA
Treatment with secukinumab significantly reduced radiographic progression in patients with active PsA, according to Désirée van der Heijde, MD, PhD, professor of rheumatology at Leiden University Medical Center, and her associates.
The results come from an analysis of the FUTURE 5 trial, a study of 996 patients with active PsA despite previous NSAID treatment, disease-modifying antirheumatic drug treatment, or anti–tumor necrosis factor (TNF) therapy. Patients were randomized to receive 300 mg subcutaneous secukinumab with loading dose, 150 mg secukinumab with loading dose, 150 mg secukinumab without loading dose, or placebo, at baseline; weeks 1, 2, 3, and 4; then every 4 weeks.
After 24 weeks, the mean change in van der Heijde–modified Total Sharp Score for PsA was 0.08 for the 300-mg secukinumab group (P less than .01), 0.17 for the 150-mg secukinumab with loading dose group (P less than .05), a reduction of 0.09 for the 150-mg secukinumab without loading dose group (P less than .01), and 0.50 for the placebo group. Lower radiographic progression was seen regardless of prior anti-TNF or concomitant methotrexate treatment.
The study was funded by Novartis. The study authors reported financial disclosures with numerous companies; five authors are Novartis employees.
Tildrakizumab sustains efficacy in plaque psoriasis treatment after 1 year
Nearly all patients receiving the interleukin-23 inhibitor tildrakizumab for the treatment of moderate to severe plaque psoriasis maintained or improved their Psoriasis Area and Severity Index (PASI) response rate after 52 weeks of treatment, compared with their response after 28 weeks.
The analysis, conducted by Boni E. Elewski, MD, of the University of Alabama at Birmingham, and her associates, included 352 patients who received 100 mg tildrakizumab and 313 who received 200 mg tildrakizumab. Treatment was received at baseline, at 4 weeks, and then every 12 weeks afterward.
At week 28, the proportions of patients achieving PASI 100, PASI 90-99, PASI 75-89, and PASI 50-74 at week 28 were 25.9%, 38.4%, 25.3%, and 10.5%, respectively, among those treated with the 100-mg dose. The proportions were 24.6%, 24.3%, 19.5%, and 31.6%, respectively, among those treated with the 200-mg dose.
In patients who achieved at least PASI 90 on either dose at week 28, 88.9%-89.4% maintained that response at week 52. For patients with PASI 75-89, 39.3%-40.4% maintained that response and 33.7%-41.0% achieved a PASI 90 response. At week 52, in patients with PASI 50-74, 20.2%-29.7% achieved at least a PASI 90, 52.5%-64.9% achieved PASI 75, and only 2.6% of patients on either dose had fallen below PASI 50.
Four study authors reported being clinical investigators on studies sponsored by Merck and Sun Pharmaceuticals; five authors are employees of Sun Pharmaceuticals.
Halobetasol/tazarotene combination most effective for plaque psoriasis treatment
A fixed combination of halobetasol propionate 0.01% and tazarotene 0.045% lotion provided a synergistic effect over either component on its own for the treatment of plaque psoriasis, according to Leon H. Kircik, MD, of Indiana University, Indianapolis, and his associates.
The investigators performed a post hoc analysis of 212 patients with moderate to severe plaque psoriasis randomized to receive either the halobetasol/tazarotene combination, halobetasol only, tazarotene only, or vehicle only for 8 weeks, with follow-up at 12 weeks. Treatment success was based on the proportion of patients who achieved at least a 2-grade improvement in the Investigator Global Assessment (IGA) score, IGA scores of “clear” or “almost clear,” and percent change from baseline in IGA multiplied by Body Surface Area (BSA) composite score (IGAxBSA). “Synergy was calculated by summing up the contribution of the individual active ingredients (HP and TAZ) to overall efficacy and comparing to the efficacy achieved with HP/TAZ lotion relative to vehicle,” the authors explained.
Relative to vehicle, treatment success for halobetasol/tazarotene after 8 weeks was 42.8%, 23.6% for halobetasol alone, and 9.0% for tazarotene alone. After 12 weeks, the difference was 31.3%, 14.1%, and 5.9%, respectively. The percent change in IGAxBSA scores from baseline after 8 weeks, relative to vehicle, were 51.6%, 37.3%, and 3.3%, respectively. After 12 weeks, the change was 47.3%, 25.7%, and 8.6%, respectively.
After 8 weeks, the synergy ratio for treatment success and IGAxBSA scores for the halobetasol/tazarotene combination was 1.3. After 12 weeks, the synergy ratio for treatment success was 1.6 and the ratio for IGAxBSA scores was 1.4.
“By combining two agents into one once-daily formulation, this novel formulation reduces the number of product applications and may help patient adherence,” the study authors noted.
Dr. Kircik reported serving as a consultant and investigator for Valeant Pharmaceuticals. One study author is an employee of Bausch Health and Ortho Dermatologics, and another is an employee of Dow Pharmaceutical Sciences (a division of Valeant).
Brodalumab demonstrates low immunogenicity in moderate to severe psoriasis
The immunogenicity of brodalumab in patients with moderate to severe plaque psoriasis was low and did not compromise the efficacy or safety profile of the drug, according to Kristian Reich, MD, of Dermatologikum Berlin and SCIderm Research Institute in Hamburg, Germany, and his associates.
Data from a 12-week, phase 2 trial with a 352-week, open-label extension and three 52-week phase 3 trials were included in the analysis. Antidrug antibodies (ADAs) were tested, and positive samples were further analyzed for neutralizing ADAs by a cell-based assay.
Out of the 4,461 patients who received brodalumab, 122 (2.7%) were positive for ADAs after starting brodalumab. The incidence rate ranged from 1.9% to 3.4% between all dosing groups (140 mg, 210 mg, variable dosing, and 210 mg of brodalumab after ustekinumab). In 58 (1.4%) of patients, ADAs were transient. No patients had neutralizing ADAs, and no evidence of altered pharmacokinetics, loss of efficacy, or changes in the safety profile of brodalumab in subjects positive for ADAs was seen.
No significant difference was seen in the incidence rate of hypersensitivity or injection site reactions in brodalumab, compared with placebo or ustekinumab. The most common injection site reactions were injection site pain, erythema, and bruising.
The study was supported by Amgen. The study authors reported numerous disclosures. Two authors are employees of Leo Pharma, one author is a former employee of the company.
Secukinumab improves patient-reported outcomes in CTT psoriasis
Treatment with secukinumab significantly improved patient-reported outcomes such as fatigue, itch, pain, and quality of life measures in patients with CTT psoriasis after 6 months, according to Jerry Bagel, MD, of the Psoriasis Treatment Center of Central New Jersey, East Windsor, and his associates.
A total of 68 patients with psoriasis localized to at least one CTT area who were enrolled in the Corrona Psoriasis Registry from April 15, 2015, through May 10, 2018, and were receiving secukinumab for the entirety of the 6-month study period were included in the analysis. Patient-reported outcomes included in the analysis were fatigue, itch, pain, Dermatology Quality of Life Index (DLQI) score, and Work Productivity and Activity Impairment (WPAI) scale.
The mean age at enrollment was 51.2 years and almost 80% of patients were white. Mean psoriasis duration was 21.8 years and nearly half had PsA.
Visual analog scale scores improved over baseline for fatigue (mean, 23.2 vs. 33.2; P = .01), itch (20.9 vs. 49.6; P less than .0001), and pain (12.1 vs. 33.8; P less than .0001). DLQI scores also improved (2.9 vs. 8.1; P less than .0001), and the proportion of patients who reported that psoriasis had at least a moderate effect on their life was reduced after 6 months (22.1% vs. 59.7%; P less than .0001).
Based on WPAI results, patients experienced significant improvements in the percentage of daily activities impaired (mean, 9.5% vs 17.5%; P = .0075); of the 42 patients who were employed, both impairment percentage (3.7% vs. 11.2%; P = .0148) and percentage of work hours affected (4.9% vs. 11.9%; P = .0486) were reduced from baseline.
“These results are consistent with previous reports from secukinumab clinical trials; however, additional real-world studies are needed to evaluate the long-term effectiveness of secukinumab for improving [patient-reported outcomes] in patients with psoriasis in CTT areas,” the authors noted.
The Corrona registry has been supported by numerous pharmaceutical companies, and several study authors reported various disclosures with industry. Two authors are Novartis employees. The study was supported by Novartis; the company participated in the interpretation of data and review and approval of the abstract.
These posters were presented at Skin Disease Education Foundation’s annual Las Vegas Dermatology Seminar. SDEF and this news organization are owned by the same parent company.
FROM SDEF LAS VEGAS DERMATOLOGY SEMINAR
FDA approves adalimumab biosimilar Hyrimoz
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.

FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.
FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.
FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
Canagliflozin approved for cardiovascular event risk reduction
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.

The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.
The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
The Food and Drug Administration has approved canagliflozin (Invokana) as a way to reduce the risk of major adverse cardiovascular events in patients with type 2 diabetes and cardiovascular disease, according to Janssen Pharmaceuticals.
The sodium–glucose cotransporter 2 inhibitor was first approved in 2013 to improve glycemic control in adults with type 2 diabetes.
FDA approval was based on results from the CANVAS (Canagliflozin Cardiovascular Assessment Study) trial, which included more than 10,000 adults with type 2 diabetes who either had cardiovascular disease or were at risk for cardiovascular disease. Overall, patients who received canagliflozin had a 14% lower risk of experiencing a major cardiovascular event over the control group, and patients with established cardiovascular disease had an 18% lower risk.
The most common adverse events associated with canagliflozin include female genital mycotic infections, urinary tract infection, and increased urination. Notably, canagliflozin also increases the risk of lower-extremity amputation, especially in those with a history of amputation.
“Americans living with type 2 diabetes are two to three times more likely to die from heart disease than adults without diabetes. With this approval, Invokana now plays an even more important role in the overall treatment mix with its demonstrated ability to reduce the risk of potentially devastating cardiovascular events,” Ralph A. DeFronzo, MD, professor and division chief of medicine and diabetes at the University of Texas, San Antonio, said in the press release.
The new indication applies to all formulations of canagliflozin.
Find the full press release on the Janssen website.
FDA clears Abbott’s Influenza A & B 2, Strep A 2 assays
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
The Food and Drug Administration has cleared Abbott Laboratories’ next-generation Influenza A & B 2 and Strep A 2 molecular assays for point-of-care testing.
The Influenza A & B 2 assay can detect and differentiate influenza A and B in 13 minutes, with a call-out of positive results at 5 minutes. It can be stored at room temperature, simplifying storage and ordering. The Strep A 2 assay detects group A streptococcus bacterial nucleic acid in 6 minutes, with a call-out of positive results at 2 minutes. Both will be the fastest tests currently on the market in their respective fields, according to a corporate press release.
The assays will be available in a variety of inpatient and outpatient settings, particularly in locations where patients commonly access health care services, such as EDs, physician offices, walk-in clinics, and urgent care centers. This will allow health care providers to make a fast, informed diagnosis and provide appropriate treatment within the span of a single patient visit.
“The ability to obtain early call outs for positive test results with molecular accuracy in as little as 5 minutes for influenza and 2 minutes for strep A is a game-changing development that allows prompt treatment decisions at the point of care. Rapid testing may also help reduce improper antibiotic usage, which can occur when treatment is based exclusively on a patient’s symptoms, and contributes to antibiotic resistance,” Gregory J. Berry, PhD, director of molecular diagnostics at Northwell Health Laboratories in Lake Success, N.Y., said in the press release.
Find the full press release on the Abbott Laboratories website.
FDA approves Xofluza for treatment of influenza
The Food and Drug Administration has approved Xofluza (baloxavir marboxil) for the treatment of acute uncomplicated influenza in people aged 12 years or older who have been symptomatic for 48 hours or less.
The FDA approval is based on results from two randomized, clinical trials. In both trials, patients who received Xofluza experienced a shorter duration until alleviation of symptoms, compared with patients who received a placebo. In the second trial, patients who received Xofluza and patients who received another approved antiviral influenza medication experienced similar durations until symptom alleviation.
“When treatment is started within 48 hours of becoming sick with flu symptoms, antiviral drugs can lessen symptoms and shorten the time patients feel sick. Having more treatment options that work in different ways to attack the virus is important because flu viruses can become resistant to antiviral drugs,” Debra Birnkrant, MD, director of the Division of Antiviral Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
The most common adverse events associated with Xofluza were diarrhea and bronchitis.
“This is the first new antiviral flu treatment with a novel mechanism of action approved by the FDA in nearly 20 years,” FDA Commissioner Scott Gottlieb, MD, added. “With thousands of people getting the flu every year, and many people becoming seriously ill, having safe and effective treatment alternatives is critical. This novel drug provides an important, additional treatment option.”
Find the full press release on the FDA website.
The Food and Drug Administration has approved Xofluza (baloxavir marboxil) for the treatment of acute uncomplicated influenza in people aged 12 years or older who have been symptomatic for 48 hours or less.
The FDA approval is based on results from two randomized, clinical trials. In both trials, patients who received Xofluza experienced a shorter duration until alleviation of symptoms, compared with patients who received a placebo. In the second trial, patients who received Xofluza and patients who received another approved antiviral influenza medication experienced similar durations until symptom alleviation.
“When treatment is started within 48 hours of becoming sick with flu symptoms, antiviral drugs can lessen symptoms and shorten the time patients feel sick. Having more treatment options that work in different ways to attack the virus is important because flu viruses can become resistant to antiviral drugs,” Debra Birnkrant, MD, director of the Division of Antiviral Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
The most common adverse events associated with Xofluza were diarrhea and bronchitis.
“This is the first new antiviral flu treatment with a novel mechanism of action approved by the FDA in nearly 20 years,” FDA Commissioner Scott Gottlieb, MD, added. “With thousands of people getting the flu every year, and many people becoming seriously ill, having safe and effective treatment alternatives is critical. This novel drug provides an important, additional treatment option.”
Find the full press release on the FDA website.
The Food and Drug Administration has approved Xofluza (baloxavir marboxil) for the treatment of acute uncomplicated influenza in people aged 12 years or older who have been symptomatic for 48 hours or less.
The FDA approval is based on results from two randomized, clinical trials. In both trials, patients who received Xofluza experienced a shorter duration until alleviation of symptoms, compared with patients who received a placebo. In the second trial, patients who received Xofluza and patients who received another approved antiviral influenza medication experienced similar durations until symptom alleviation.
“When treatment is started within 48 hours of becoming sick with flu symptoms, antiviral drugs can lessen symptoms and shorten the time patients feel sick. Having more treatment options that work in different ways to attack the virus is important because flu viruses can become resistant to antiviral drugs,” Debra Birnkrant, MD, director of the Division of Antiviral Products in the FDA’s Center for Drug Evaluation and Research, said in a press release.
The most common adverse events associated with Xofluza were diarrhea and bronchitis.
“This is the first new antiviral flu treatment with a novel mechanism of action approved by the FDA in nearly 20 years,” FDA Commissioner Scott Gottlieb, MD, added. “With thousands of people getting the flu every year, and many people becoming seriously ill, having safe and effective treatment alternatives is critical. This novel drug provides an important, additional treatment option.”
Find the full press release on the FDA website.
ICYMI: Hereditary cancer screening feasible in community gynecologic practices
After a group of community obstetrics and gynecology practices underwent training in hereditary cancer screening, genetic testing rates increased by four times over the immediate preintervention rate and by eight times over the year prior to intervention, according to results of a prospective process intervention study published in Obstetrics & Gynecology (2018 Oct 5. doi: 10.1097/AOG.0000000000002916).
We covered this story before it was published in the journal. Find our conference coverage at the link below.
After a group of community obstetrics and gynecology practices underwent training in hereditary cancer screening, genetic testing rates increased by four times over the immediate preintervention rate and by eight times over the year prior to intervention, according to results of a prospective process intervention study published in Obstetrics & Gynecology (2018 Oct 5. doi: 10.1097/AOG.0000000000002916).
We covered this story before it was published in the journal. Find our conference coverage at the link below.
After a group of community obstetrics and gynecology practices underwent training in hereditary cancer screening, genetic testing rates increased by four times over the immediate preintervention rate and by eight times over the year prior to intervention, according to results of a prospective process intervention study published in Obstetrics & Gynecology (2018 Oct 5. doi: 10.1097/AOG.0000000000002916).
We covered this story before it was published in the journal. Find our conference coverage at the link below.
FROM OBSTETRICS & GYNECOLOGY
HF unlikely in pregnant cancer survivors without history of cardiotoxicity
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
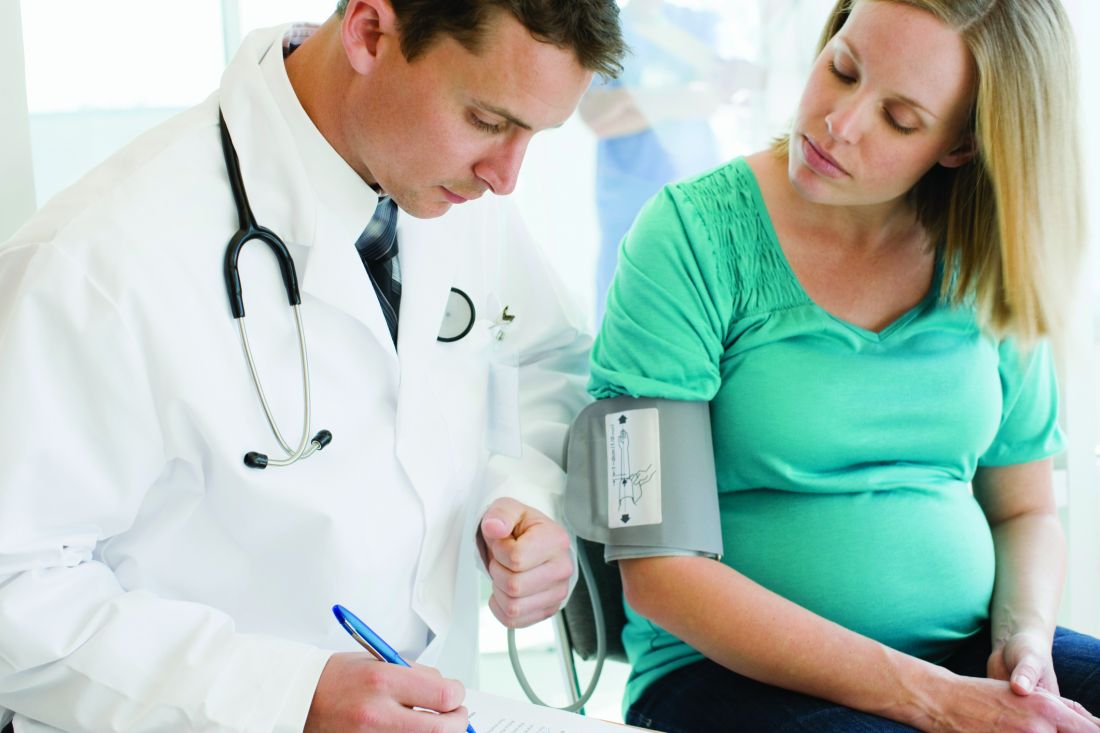
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
The risk of adverse cardiac events in female cancer survivors during pregnancy is low unless there is a history of cardiotoxicity, according to Shiying Liu, MD, of the University of Toronto, and her associates.
In a research letter published in the Journal of the American College of Cardiology, Dr. Liu and her associates reported on a retrospective chart review of 78 women with 94 pregnancies who had previously received cancer therapy who were seen at Mount Sinai Hospital between 2005 and 2015. Of these, 15 pregnancies occurred in 13 women with a prior history of cardiotoxicity. The primary outcome was a composite of cardiac events including cardiac death, heart failure (HF), acute coronary syndrome, and sustained arrhythmia.
HF occurred during five pregnancies in four women; no other adverse cardiac events occurred during the study period. All four of the women who experienced HF had a history of cardiotoxicity. There was no difference in age at cancer diagnosis, age at pregnancy, cancer type, or exposure to anthracyclines between those who did and did not experience HF, but women who developed HF were more likely to have left ventricular systolic dysfunction at the first antenatal visit (75% vs. 8%; P = .004) and to be on cardiac medications (50% vs. 8%; P = .026).
“The risk of developing [HF] during pregnancy is rare in female cancer survivors without a history of cardiotoxicity. These women can be reassured that they are at a very low risk of developing [HF] during pregnancy. Women who have a history of cardiotoxicity have an approximately one in three chance of developing [HF] during pregnancy and should receive close cardiac surveillance during pregnancy at a center with expertise in cardiac disease in pregnancy,” the authors concluded.
Coauthor Paaladinesh Thavendiranathan, MD, reported support from the Canadian Institutes of Health Research New Investigator Award. None of the other authors had any relevant financial disclosures.
SOURCE: Liu S et al. J Am Coll Cardiol. 2018 Oct 15. doi: 10.1016/j.jacc.2018.07.085.
FROM THE JOURNAL OF THE AMERICAN COLLEGE OF CARDIOLOGY
Snake bite detox, kidney stone coaster cure, soporific speakers
At least opioids don’t bite

Remember that song about the old lady who “swallowed” a fly? Well, what if the fly were actually a poisonous snake? And what if she didn’t really swallow it? And what if we stopped asking so many questions? A 33-year-old Indian man who was addicted to alcohol, tobacco, and opioids in the form of raw opium and “puppy husk” (we’re thinking it’s poppy husk, but the case report clearly said “puppy”) heard about something new: letting a poisonous snake bite you on the tongue.
He found some nomadic snake charmers to provide the snake (possibly a cobra, but he’s not sure), and the snake’s bite provides him with an hour of jerky body movements, blurred vision, and unresponsiveness, followed by 3-4 weeks of heightened arousal and a sense of well-being that is “more intense tha[n] the state of high experienced … with any dose of alcohol or opioids,” according to the Indian Journal of Psychological Medicine (2018;40[3]:269-71). Plus, he doesn’t use nearly as much alcohol and opium. (Yes, he has done this more than once. You’re rude to ask.)
The old lady in the song, of course, dies after “swallowing” a horse. Fortunately for this guy, there’s no such thing as a poisonous horse.
Ride out the kidney stones

Disney World, the happiest place on earth – unless you’re a kidney stone. A professor from the College of Osteopathic Medicine at Michigan State University, East Lansing, was inspired when one of his patients told him a recent trip to the Magic Kingdom had successfully dislodged a kidney stone. Armed with a silicone model of the renal system, David Wartinger, DO, spent many grueling hours atop various theme park rides. And, one can only assume, many more grueling hours exploring Cinderella’s Castle.
Dr. Wartinger discovered that the Big Thunder Mountain Railroad was more effective than Space Mountain or the Rock ‘n’ Roller Coaster at rattling around the rider and thus shaking out those kidney stones.
The brave professor’s tireless work has earned him one of 2018’s Ig Nobel prizes. We salute you.
In Russia, bear cures YOU

They do things a bit differently in Russia. Who else but a group of Russians would stare a freshly caught and presumably very angry Siberian brown bear in the mouth and think, “Hmm, yes – the perfect source for new and exciting antibiotics!”
Somehow, they managed to make it work. In an article published in Proceedings of the National Academy of Sciences, Russian scientists demonstrated a new technology that rapidly tests the microbiota within bear saliva for potential antibiotics to use against antibiotic-resistant infections such as Staphylococcus aureus. In one of the thousands of samples taken, all introduced S. aureus bacteria had been eliminated, and further analysis showed the presence of amicoumacin, a previously known antibiotic.
And this is just one bear. Who knows what new and exciting goodies lie in the mouths of other bears? Now, we feel it’s important to note that the saliva of any sufficiently wild animal would have sufficed. But when in Russia, if you can think of an excuse to catch a bear, then by God you had better go catch a bear. Anything less would be an insult to the Motherland!
I need a hug

Don’t we all. A new study from the department of psychology at Carnegie Mellon University in Pittsburgh examined whether hugs actually do anything to improve a bad mood. Unverified rumors purport that the study was commissioned by a cuddly red monster named Elmo.
Researchers concluded that consensual hugs are beneficial after some sort of conflict or negative event during the day and that they positively affect the hug-receiver. Researchers also concluded that hugs given by teddy bears or red pandas are the most beneficial, but human hugs will do if those aren’t available. Despite their bactericidal qualities, hugs from Russian bears aren’t encouraged.
Next slide, pleazzzzzzz

Picture a world in which your smartwatch counted backward. In which the room WiFi is a black hole. In which space-time’s zipper seems hopelessly stuck, with a loose string – or, in fact, PowerPoint slide #14 – wedged into its interlocking teeth. That’s right: You’re in a session at a medical conference.
Now, one meeting attendee has boldly researched what many an attendee has wondered before: Do boring speakers really talk for longer? Taking one for all humanity, that intrepid time traveler endured an inhumane, institutional review board–unapproved study design of 50 12-minute meeting sessions. After 4 minutes, he determined whether the speaker was, scientifically speaking, “boring” or “not boring.” He then clocked the time each speaker took to wrap it up.
Spoiler alert: “Not boring” also meant “done sooner” – in a mean time of 11 minutes and 42 seconds versus 13 minutes and 12 seconds for those oscillating in the dullness duality’s boring state. So, if the 4-minute marker heralds a soporific session? There’s more schwag and better WiFi in the exhibit hall.
At least opioids don’t bite
Remember that song about the old lady who “swallowed” a fly? Well, what if the fly were actually a poisonous snake? And what if she didn’t really swallow it? And what if we stopped asking so many questions? A 33-year-old Indian man who was addicted to alcohol, tobacco, and opioids in the form of raw opium and “puppy husk” (we’re thinking it’s poppy husk, but the case report clearly said “puppy”) heard about something new: letting a poisonous snake bite you on the tongue.
He found some nomadic snake charmers to provide the snake (possibly a cobra, but he’s not sure), and the snake’s bite provides him with an hour of jerky body movements, blurred vision, and unresponsiveness, followed by 3-4 weeks of heightened arousal and a sense of well-being that is “more intense tha[n] the state of high experienced … with any dose of alcohol or opioids,” according to the Indian Journal of Psychological Medicine (2018;40[3]:269-71). Plus, he doesn’t use nearly as much alcohol and opium. (Yes, he has done this more than once. You’re rude to ask.)
The old lady in the song, of course, dies after “swallowing” a horse. Fortunately for this guy, there’s no such thing as a poisonous horse.
Ride out the kidney stones
Disney World, the happiest place on earth – unless you’re a kidney stone. A professor from the College of Osteopathic Medicine at Michigan State University, East Lansing, was inspired when one of his patients told him a recent trip to the Magic Kingdom had successfully dislodged a kidney stone. Armed with a silicone model of the renal system, David Wartinger, DO, spent many grueling hours atop various theme park rides. And, one can only assume, many more grueling hours exploring Cinderella’s Castle.
Dr. Wartinger discovered that the Big Thunder Mountain Railroad was more effective than Space Mountain or the Rock ‘n’ Roller Coaster at rattling around the rider and thus shaking out those kidney stones.
The brave professor’s tireless work has earned him one of 2018’s Ig Nobel prizes. We salute you.
In Russia, bear cures YOU
They do things a bit differently in Russia. Who else but a group of Russians would stare a freshly caught and presumably very angry Siberian brown bear in the mouth and think, “Hmm, yes – the perfect source for new and exciting antibiotics!”
Somehow, they managed to make it work. In an article published in Proceedings of the National Academy of Sciences, Russian scientists demonstrated a new technology that rapidly tests the microbiota within bear saliva for potential antibiotics to use against antibiotic-resistant infections such as Staphylococcus aureus. In one of the thousands of samples taken, all introduced S. aureus bacteria had been eliminated, and further analysis showed the presence of amicoumacin, a previously known antibiotic.
And this is just one bear. Who knows what new and exciting goodies lie in the mouths of other bears? Now, we feel it’s important to note that the saliva of any sufficiently wild animal would have sufficed. But when in Russia, if you can think of an excuse to catch a bear, then by God you had better go catch a bear. Anything less would be an insult to the Motherland!
I need a hug
Don’t we all. A new study from the department of psychology at Carnegie Mellon University in Pittsburgh examined whether hugs actually do anything to improve a bad mood. Unverified rumors purport that the study was commissioned by a cuddly red monster named Elmo.
Researchers concluded that consensual hugs are beneficial after some sort of conflict or negative event during the day and that they positively affect the hug-receiver. Researchers also concluded that hugs given by teddy bears or red pandas are the most beneficial, but human hugs will do if those aren’t available. Despite their bactericidal qualities, hugs from Russian bears aren’t encouraged.
Next slide, pleazzzzzzz
Picture a world in which your smartwatch counted backward. In which the room WiFi is a black hole. In which space-time’s zipper seems hopelessly stuck, with a loose string – or, in fact, PowerPoint slide #14 – wedged into its interlocking teeth. That’s right: You’re in a session at a medical conference.
Now, one meeting attendee has boldly researched what many an attendee has wondered before: Do boring speakers really talk for longer? Taking one for all humanity, that intrepid time traveler endured an inhumane, institutional review board–unapproved study design of 50 12-minute meeting sessions. After 4 minutes, he determined whether the speaker was, scientifically speaking, “boring” or “not boring.” He then clocked the time each speaker took to wrap it up.
Spoiler alert: “Not boring” also meant “done sooner” – in a mean time of 11 minutes and 42 seconds versus 13 minutes and 12 seconds for those oscillating in the dullness duality’s boring state. So, if the 4-minute marker heralds a soporific session? There’s more schwag and better WiFi in the exhibit hall.
At least opioids don’t bite
Remember that song about the old lady who “swallowed” a fly? Well, what if the fly were actually a poisonous snake? And what if she didn’t really swallow it? And what if we stopped asking so many questions? A 33-year-old Indian man who was addicted to alcohol, tobacco, and opioids in the form of raw opium and “puppy husk” (we’re thinking it’s poppy husk, but the case report clearly said “puppy”) heard about something new: letting a poisonous snake bite you on the tongue.
He found some nomadic snake charmers to provide the snake (possibly a cobra, but he’s not sure), and the snake’s bite provides him with an hour of jerky body movements, blurred vision, and unresponsiveness, followed by 3-4 weeks of heightened arousal and a sense of well-being that is “more intense tha[n] the state of high experienced … with any dose of alcohol or opioids,” according to the Indian Journal of Psychological Medicine (2018;40[3]:269-71). Plus, he doesn’t use nearly as much alcohol and opium. (Yes, he has done this more than once. You’re rude to ask.)
The old lady in the song, of course, dies after “swallowing” a horse. Fortunately for this guy, there’s no such thing as a poisonous horse.
Ride out the kidney stones
Disney World, the happiest place on earth – unless you’re a kidney stone. A professor from the College of Osteopathic Medicine at Michigan State University, East Lansing, was inspired when one of his patients told him a recent trip to the Magic Kingdom had successfully dislodged a kidney stone. Armed with a silicone model of the renal system, David Wartinger, DO, spent many grueling hours atop various theme park rides. And, one can only assume, many more grueling hours exploring Cinderella’s Castle.
Dr. Wartinger discovered that the Big Thunder Mountain Railroad was more effective than Space Mountain or the Rock ‘n’ Roller Coaster at rattling around the rider and thus shaking out those kidney stones.
The brave professor’s tireless work has earned him one of 2018’s Ig Nobel prizes. We salute you.
In Russia, bear cures YOU
They do things a bit differently in Russia. Who else but a group of Russians would stare a freshly caught and presumably very angry Siberian brown bear in the mouth and think, “Hmm, yes – the perfect source for new and exciting antibiotics!”
Somehow, they managed to make it work. In an article published in Proceedings of the National Academy of Sciences, Russian scientists demonstrated a new technology that rapidly tests the microbiota within bear saliva for potential antibiotics to use against antibiotic-resistant infections such as Staphylococcus aureus. In one of the thousands of samples taken, all introduced S. aureus bacteria had been eliminated, and further analysis showed the presence of amicoumacin, a previously known antibiotic.
And this is just one bear. Who knows what new and exciting goodies lie in the mouths of other bears? Now, we feel it’s important to note that the saliva of any sufficiently wild animal would have sufficed. But when in Russia, if you can think of an excuse to catch a bear, then by God you had better go catch a bear. Anything less would be an insult to the Motherland!
I need a hug
Don’t we all. A new study from the department of psychology at Carnegie Mellon University in Pittsburgh examined whether hugs actually do anything to improve a bad mood. Unverified rumors purport that the study was commissioned by a cuddly red monster named Elmo.
Researchers concluded that consensual hugs are beneficial after some sort of conflict or negative event during the day and that they positively affect the hug-receiver. Researchers also concluded that hugs given by teddy bears or red pandas are the most beneficial, but human hugs will do if those aren’t available. Despite their bactericidal qualities, hugs from Russian bears aren’t encouraged.
Next slide, pleazzzzzzz
Picture a world in which your smartwatch counted backward. In which the room WiFi is a black hole. In which space-time’s zipper seems hopelessly stuck, with a loose string – or, in fact, PowerPoint slide #14 – wedged into its interlocking teeth. That’s right: You’re in a session at a medical conference.
Now, one meeting attendee has boldly researched what many an attendee has wondered before: Do boring speakers really talk for longer? Taking one for all humanity, that intrepid time traveler endured an inhumane, institutional review board–unapproved study design of 50 12-minute meeting sessions. After 4 minutes, he determined whether the speaker was, scientifically speaking, “boring” or “not boring.” He then clocked the time each speaker took to wrap it up.
Spoiler alert: “Not boring” also meant “done sooner” – in a mean time of 11 minutes and 42 seconds versus 13 minutes and 12 seconds for those oscillating in the dullness duality’s boring state. So, if the 4-minute marker heralds a soporific session? There’s more schwag and better WiFi in the exhibit hall.

Acne more common in adults with hidradenitis suppurativa
, according to Sara Wertenteil and her colleagues in the department of dermatology at Hofstra University in Hempstead, N.Y.
Using data collected by IBM Watson Health, the study authors examined a total of 48,050 adults with HS and 16.9 million adults in the general U.S. population. In this study population, 15.2% of adults with HS had acne, compared with only 2.9% of adults in the general population (P less than .0001), the investigators wrote in the Journal of the American Academy of Dermatology.
After adjusting for age, sex, obesity, smoking status, and polycystic ovarian syndrome (PCOS) status, the odds ratio of adults with HS having acne was 4.51 over those without HS (95% confidence interval, 4.40-4.63). In all subgroups measured (male; female; adults aged 18-44 years, 45-64 years, and 65 years and older; white; nonwhite; obese; nonobese; smoker; nonsmoker; positive for PCOS; non-PCOS) adults with HS were significantly more likely to have acne. The strongest association was in patients who were aged 65 years and older (odds ratio, 10.14; 95% CI, 8.97-11.46).
“Patients with HS have an increased prevalence of [acne vulgaris]. Clinicians treating HS patients should be aware of this burden and its potential implications including a further impact on quality of life. Management strategies should include consideration of both conditions, either with treatments that have overlapping efficacy, or with concomitant therapies,” the authors concluded.
The study was sponsored in part by AbbVie. One coauthor reported having served as an advisor for AbbVie, Pfizer, Janssen, and Asana Biosciences.
SOURCE: Wertentiel S et al. J Am Acad Dermatol. 2018 Oct 1. doi: 10.1016/j.jaad.2018.09.040.
, according to Sara Wertenteil and her colleagues in the department of dermatology at Hofstra University in Hempstead, N.Y.
Using data collected by IBM Watson Health, the study authors examined a total of 48,050 adults with HS and 16.9 million adults in the general U.S. population. In this study population, 15.2% of adults with HS had acne, compared with only 2.9% of adults in the general population (P less than .0001), the investigators wrote in the Journal of the American Academy of Dermatology.
After adjusting for age, sex, obesity, smoking status, and polycystic ovarian syndrome (PCOS) status, the odds ratio of adults with HS having acne was 4.51 over those without HS (95% confidence interval, 4.40-4.63). In all subgroups measured (male; female; adults aged 18-44 years, 45-64 years, and 65 years and older; white; nonwhite; obese; nonobese; smoker; nonsmoker; positive for PCOS; non-PCOS) adults with HS were significantly more likely to have acne. The strongest association was in patients who were aged 65 years and older (odds ratio, 10.14; 95% CI, 8.97-11.46).
“Patients with HS have an increased prevalence of [acne vulgaris]. Clinicians treating HS patients should be aware of this burden and its potential implications including a further impact on quality of life. Management strategies should include consideration of both conditions, either with treatments that have overlapping efficacy, or with concomitant therapies,” the authors concluded.
The study was sponsored in part by AbbVie. One coauthor reported having served as an advisor for AbbVie, Pfizer, Janssen, and Asana Biosciences.
SOURCE: Wertentiel S et al. J Am Acad Dermatol. 2018 Oct 1. doi: 10.1016/j.jaad.2018.09.040.
, according to Sara Wertenteil and her colleagues in the department of dermatology at Hofstra University in Hempstead, N.Y.
Using data collected by IBM Watson Health, the study authors examined a total of 48,050 adults with HS and 16.9 million adults in the general U.S. population. In this study population, 15.2% of adults with HS had acne, compared with only 2.9% of adults in the general population (P less than .0001), the investigators wrote in the Journal of the American Academy of Dermatology.
After adjusting for age, sex, obesity, smoking status, and polycystic ovarian syndrome (PCOS) status, the odds ratio of adults with HS having acne was 4.51 over those without HS (95% confidence interval, 4.40-4.63). In all subgroups measured (male; female; adults aged 18-44 years, 45-64 years, and 65 years and older; white; nonwhite; obese; nonobese; smoker; nonsmoker; positive for PCOS; non-PCOS) adults with HS were significantly more likely to have acne. The strongest association was in patients who were aged 65 years and older (odds ratio, 10.14; 95% CI, 8.97-11.46).
“Patients with HS have an increased prevalence of [acne vulgaris]. Clinicians treating HS patients should be aware of this burden and its potential implications including a further impact on quality of life. Management strategies should include consideration of both conditions, either with treatments that have overlapping efficacy, or with concomitant therapies,” the authors concluded.
The study was sponsored in part by AbbVie. One coauthor reported having served as an advisor for AbbVie, Pfizer, Janssen, and Asana Biosciences.
SOURCE: Wertentiel S et al. J Am Acad Dermatol. 2018 Oct 1. doi: 10.1016/j.jaad.2018.09.040.
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
FDA expands CoolSculpting indication to submandibular area
FDA clearance of the expanded indication was based on results of a 22-week study in which patients achieved a mean 33% reduction in fat layer thickness after two treatments. In addition, 85% of patients reported satisfaction with their treatment in that and two other studies. The data were provided in Allergan’s press release announcing the clearance for the cryolipolysis device.
Adverse events associated with the CoolSculpting treatment include temporary redness, swelling, blanching, bruising, firmness, tingling, stinging, tenderness, cramping, aching, itching, and skin sensitivity. People with cryoglobulinemia, cold agglutinin disease, or paroxysmal cold hemoglobinuria should not receive CoolSculpting treatments, according to the company.
The FDA also expanded the CoolSculpting indication to people with a body mass index up to 46.2 kg/m2 when treating the submental and submandibular areas.
FDA clearance of the expanded indication was based on results of a 22-week study in which patients achieved a mean 33% reduction in fat layer thickness after two treatments. In addition, 85% of patients reported satisfaction with their treatment in that and two other studies. The data were provided in Allergan’s press release announcing the clearance for the cryolipolysis device.
Adverse events associated with the CoolSculpting treatment include temporary redness, swelling, blanching, bruising, firmness, tingling, stinging, tenderness, cramping, aching, itching, and skin sensitivity. People with cryoglobulinemia, cold agglutinin disease, or paroxysmal cold hemoglobinuria should not receive CoolSculpting treatments, according to the company.
The FDA also expanded the CoolSculpting indication to people with a body mass index up to 46.2 kg/m2 when treating the submental and submandibular areas.
FDA clearance of the expanded indication was based on results of a 22-week study in which patients achieved a mean 33% reduction in fat layer thickness after two treatments. In addition, 85% of patients reported satisfaction with their treatment in that and two other studies. The data were provided in Allergan’s press release announcing the clearance for the cryolipolysis device.
Adverse events associated with the CoolSculpting treatment include temporary redness, swelling, blanching, bruising, firmness, tingling, stinging, tenderness, cramping, aching, itching, and skin sensitivity. People with cryoglobulinemia, cold agglutinin disease, or paroxysmal cold hemoglobinuria should not receive CoolSculpting treatments, according to the company.
The FDA also expanded the CoolSculpting indication to people with a body mass index up to 46.2 kg/m2 when treating the submental and submandibular areas.