User login
Lucas Franki is an associate editor for MDedge News, and has been with the company since 2014. He has a BA in English from Penn State University and is an Eagle Scout.
FDA approves rifamycin for treatment of traveler’s diarrhea
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.

FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.
FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has approved rifamycin (Aemcolo) for the treatment of traveler’s diarrhea caused by noninvasive strains of Escherichia coli.
FDA approval was based on results of three clinical trials. The efficacy of rifamycin was shown in a trial of 264 adults with traveler’s diarrhea in Guatemala and Mexico. Compared with placebo, rifamycin significantly reduced symptoms of the condition. The safety of rifamycin was illustrated in a pair of studies including 619 adults with traveler’s diarrhea who took rifamycin orally for 3-4 days. The most common adverse events were headache and constipation.
Traveler’s diarrhea is the most common travel-related illness, affecting 10%-40% of travelers. It can be caused by a multitude of pathogens, but bacteria from food or water is the most common source. High-risk areas include much of Asia, the Middle East, Mexico, Central and South America, and Africa.
Rifamycin was not effective in patients with diarrhea complicated by fever and/or bloody stool or in diarrhea caused by a pathogen other than E. coli.
“Travelers’ diarrhea affects millions of people each year, and having treatment options for this condition can help reduce symptoms of the condition,” Edward Cox, MD, MPH, director of the Office of Antimicrobial Products in the FDA’s Center for Drug Evaluation and Research, said in the press release.
Find the full press release on the FDA website.
ICYMI: Prednisone lowers IRIS risk in patients with HIV
In patients with HIV, treatment with prednisone for 4 weeks after antiretroviral therapy initiation significantly reduced the risk of tuberculosis-associated immune reconstitution inflammatory syndrome (IRIS). The results from the randomized, double-blind, placebo-controlled trial were published in the New England Journal of Medicine (2018 Nov 14. doi: 10.1056/NEJMoa1800762).
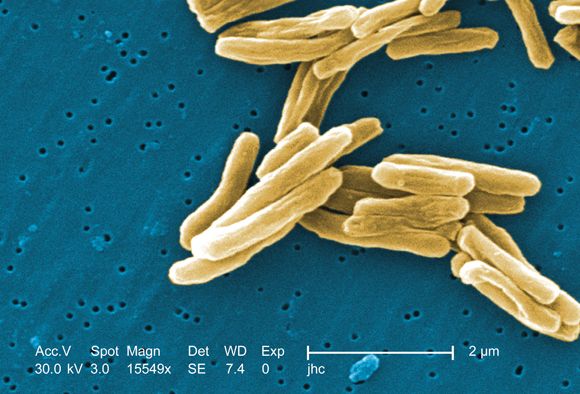
A total of 240 patients were enrolled in the study, with a median age of 36 years; 60% were men, and 73% had microbiologically confirmed tuberculosis. The median CD4 count of the patients was 49 cells/mcL and the median HIV type 1 RNA viral load was 5.5 log10 copies/mL. A total of 120 patients were assigned to each group, with 18 patients lost to follow-up or withdrawn. Tuberculosis-associated IRIS was diagnosed in 39 patients (32.5%) in the prednisone group and in 56 (46.7%) in the placebo group, yielding a relative IRIS risk of 0.70 (95% confidence interval, 0.51-0.96; P = .03), according to the researchers for the PredART (Preventing TB-IRIS in High-Risk Patients: a Randomized Placebo-Controlled Trial of Prednisone) study team.
We covered this story before it was published in the journal. Find our coverage from the Conference on Retroviruses & Opportunistic Infections at the link below.
In patients with HIV, treatment with prednisone for 4 weeks after antiretroviral therapy initiation significantly reduced the risk of tuberculosis-associated immune reconstitution inflammatory syndrome (IRIS). The results from the randomized, double-blind, placebo-controlled trial were published in the New England Journal of Medicine (2018 Nov 14. doi: 10.1056/NEJMoa1800762).
A total of 240 patients were enrolled in the study, with a median age of 36 years; 60% were men, and 73% had microbiologically confirmed tuberculosis. The median CD4 count of the patients was 49 cells/mcL and the median HIV type 1 RNA viral load was 5.5 log10 copies/mL. A total of 120 patients were assigned to each group, with 18 patients lost to follow-up or withdrawn. Tuberculosis-associated IRIS was diagnosed in 39 patients (32.5%) in the prednisone group and in 56 (46.7%) in the placebo group, yielding a relative IRIS risk of 0.70 (95% confidence interval, 0.51-0.96; P = .03), according to the researchers for the PredART (Preventing TB-IRIS in High-Risk Patients: a Randomized Placebo-Controlled Trial of Prednisone) study team.
We covered this story before it was published in the journal. Find our coverage from the Conference on Retroviruses & Opportunistic Infections at the link below.
In patients with HIV, treatment with prednisone for 4 weeks after antiretroviral therapy initiation significantly reduced the risk of tuberculosis-associated immune reconstitution inflammatory syndrome (IRIS). The results from the randomized, double-blind, placebo-controlled trial were published in the New England Journal of Medicine (2018 Nov 14. doi: 10.1056/NEJMoa1800762).
A total of 240 patients were enrolled in the study, with a median age of 36 years; 60% were men, and 73% had microbiologically confirmed tuberculosis. The median CD4 count of the patients was 49 cells/mcL and the median HIV type 1 RNA viral load was 5.5 log10 copies/mL. A total of 120 patients were assigned to each group, with 18 patients lost to follow-up or withdrawn. Tuberculosis-associated IRIS was diagnosed in 39 patients (32.5%) in the prednisone group and in 56 (46.7%) in the placebo group, yielding a relative IRIS risk of 0.70 (95% confidence interval, 0.51-0.96; P = .03), according to the researchers for the PredART (Preventing TB-IRIS in High-Risk Patients: a Randomized Placebo-Controlled Trial of Prednisone) study team.
We covered this story before it was published in the journal. Find our coverage from the Conference on Retroviruses & Opportunistic Infections at the link below.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
FDA approves pembrolizumab for sorafenib-intolerant HCC patients
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with hepatocellular carcinoma who were previously treated with sorafenib.

Approval was based on results of KEYNOTE-224, a single-arm, open-label, multicenter trial evaluating pembrolizumab in a group of 104 patients with hepatocellular carcinoma who were either intolerant to or had disease progression with sorafenib, according to a company press release.
The objective response rate was 17%, with a complete response rate of 1% and a partial response rate of 16%. In responding patients, 89% had a response duration of at least 6 months, and 56% had a response duration of at least 12 months.
Adverse events were generally similar to those seen in trials of patients with melanoma or non–small cell lung cancer, and included pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and allogeneic hematopoietic stem cell transplantation complications.
“Hepatocellular carcinoma is the most common type of liver cancer in adults, and while we have seen recent therapeutic advancements, there are still limited treatment options for advanced recurrent disease. Today’s approval of Keytruda is important, as it provides a new treatment option for patients with hepatocellular carcinoma who have been previously treated with sorafenib,” Andrew X. Zhu, MD, lead investigator and director of liver cancer research at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, said in the press release.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with hepatocellular carcinoma who were previously treated with sorafenib.
Approval was based on results of KEYNOTE-224, a single-arm, open-label, multicenter trial evaluating pembrolizumab in a group of 104 patients with hepatocellular carcinoma who were either intolerant to or had disease progression with sorafenib, according to a company press release.
The objective response rate was 17%, with a complete response rate of 1% and a partial response rate of 16%. In responding patients, 89% had a response duration of at least 6 months, and 56% had a response duration of at least 12 months.
Adverse events were generally similar to those seen in trials of patients with melanoma or non–small cell lung cancer, and included pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and allogeneic hematopoietic stem cell transplantation complications.
“Hepatocellular carcinoma is the most common type of liver cancer in adults, and while we have seen recent therapeutic advancements, there are still limited treatment options for advanced recurrent disease. Today’s approval of Keytruda is important, as it provides a new treatment option for patients with hepatocellular carcinoma who have been previously treated with sorafenib,” Andrew X. Zhu, MD, lead investigator and director of liver cancer research at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, said in the press release.
The Food and Drug Administration has approved pembrolizumab (Keytruda) for the treatment of patients with hepatocellular carcinoma who were previously treated with sorafenib.
Approval was based on results of KEYNOTE-224, a single-arm, open-label, multicenter trial evaluating pembrolizumab in a group of 104 patients with hepatocellular carcinoma who were either intolerant to or had disease progression with sorafenib, according to a company press release.
The objective response rate was 17%, with a complete response rate of 1% and a partial response rate of 16%. In responding patients, 89% had a response duration of at least 6 months, and 56% had a response duration of at least 12 months.
Adverse events were generally similar to those seen in trials of patients with melanoma or non–small cell lung cancer, and included pneumonitis, colitis, hepatitis, endocrinopathies, nephritis, severe skin reactions, solid organ transplant rejection, and allogeneic hematopoietic stem cell transplantation complications.
“Hepatocellular carcinoma is the most common type of liver cancer in adults, and while we have seen recent therapeutic advancements, there are still limited treatment options for advanced recurrent disease. Today’s approval of Keytruda is important, as it provides a new treatment option for patients with hepatocellular carcinoma who have been previously treated with sorafenib,” Andrew X. Zhu, MD, lead investigator and director of liver cancer research at Massachusetts General Hospital and professor of medicine at Harvard Medical School, both in Boston, said in the press release.
FDA approves Yupelri for COPD maintenance therapy
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).
Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).
Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
The Food and Drug Administration has approved Yupelri (revefenacin) for maintenance therapy of patients with chronic obstructive pulmonary disease (COPD).
Revefenacin is a long-acting muscarinic antagonist aimed at improving the lung function of patients with COPD. Yupelri is an inhalation solution administered once daily through a standard jet nebulizer.
The most common adverse events associated with Yupelri are cough, nasopharyngitis, upper respiratory tract infection, headache, and back pain. Patients receiving other anticholinergic-containing drugs or OATP1B1 and OATP1B3 inhibitors should not receive Yupelri.
“Patients should also be alert for signs and symptoms of acute narrow-angle glaucoma [e.g., eye pain or discomfort, blurred vision, visual changes]. Patients should consult a healthcare professional immediately if any of these signs or symptoms develop,” the FDA said in the press release.
The expanded label for Yupelri can be found on the FDA website.
FDA authorizes emergency use of rapid fingerstick test for Ebola
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
The Food and Drug Administration has issued an emergency use authorization (EUA) for the DPP Ebola Antigen System, a rapid, single-use test for the detection of Ebola virus.
The DPP Ebola Antigen System can provide rapid results in locations where health care providers lack access to authorized Ebola virus nucleic acid tests, which are highly sensitive but require an adequately equipped laboratory setting. The new system is authorized to use blood specimens from capillary whole blood, ethylenediaminetetraacetic acid (EDTA) venous whole blood, and EDTA plasma. It is to be used in individuals with signs and symptoms of Ebola virus disease, in addition to other risk factors, such as living in an area with high Ebola virus prevalence or having had contact with people showing signs or symptoms of the disease.
The system is the second Ebola rapid antigen fingerstick test made available through the EUA, but it is the first to use a portable, battery-operated reader, allowing for easier use in remote areas where patients are likely to be treated.
The FDA noted that a negative result from the DPP Ebola Antigen system does not necessarily indicate a negative diagnosis and should not be taken authoritatively, especially in individuals displaying signs and systems of Ebola virus disease.
“This EUA is part of the agency’s ongoing efforts to help mitigate potential, future threats by making medical products that have the potential to prevent, diagnosis, or treat available as quickly as possible. We’re committed to helping the people of the DRC [Democratic Republic of the Congo] effectively confront and end the current Ebola outbreak. By authorizing the first fingerstick test with a portable reader, we hope to better arm health care providers in the field to more quickly detect the virus in patients and improve patient outcomes,” FDA Commissioner Scott Gottlieb, MD, said in the press release.
Find the full press release on the FDA website.
ICYMI: Elotuzumab reduces progression risk in lenalidomide-refractory multiple myeloma
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
Patients with multiple myeloma who did not respond to treatment with lenalidomide and a proteasome inhibitor had a significantly lower risk of progression or death when receiving elotuzumab plus pomalidomide and dexamethasone, compared with pomalidomide and dexamethasone alone (hazard ratio, 0.54; 95% confidence interval, 0.34-0.86; P = .008), according to results of a multicenter, randomized, open-label, phase 2 trial published in the New England Journal of Medicine 2018 Nov 7. doi: 10.1056/NEJMoa1805762.
Study results of ELOQUENT-3 were presented earlier this year at the Annual Congress of the European Hematology Association.
FROM THE NEW ENGLAND JOURNAL OF MEDICINE
Dulaglutide meets primary endpoint in REWIND
Eli Lilly has announced top line results from REWIND, a multicenter, randomized, double-blind, placebo-controlled trial designed to study dulaglutide (Trulicity). The cardiovascular outcomes trial, which had a median follow-up period of more than 5 years, evaluated cardiovascular outcomes in nearly 10,000 patients with type 2 diabetes.
The primary endpoint of REWIND was time until the first occurrence of a major adverse cardiovascular event. Patients who received dulaglutide had a significantly reduced incidence of these events, compared with placebo, meeting the primary trial endpoint. No specific results were announced.
REWIND was distinct in that it had limited participation from people with established cardiovascular disease, which allowed dulaglutide’s effect to be measured in a broad population of people with type 2 diabetes, Eli Lilly said in their press release.
Dulaglutide is a GLP-1 receptor agonist, and trial participants received a 1.5-mg dose once a week.
Full results of the REWIND trial will be presented at the annual scientific sessions of the American Diabetes Association in June 2019.
Eli Lilly has announced top line results from REWIND, a multicenter, randomized, double-blind, placebo-controlled trial designed to study dulaglutide (Trulicity). The cardiovascular outcomes trial, which had a median follow-up period of more than 5 years, evaluated cardiovascular outcomes in nearly 10,000 patients with type 2 diabetes.
The primary endpoint of REWIND was time until the first occurrence of a major adverse cardiovascular event. Patients who received dulaglutide had a significantly reduced incidence of these events, compared with placebo, meeting the primary trial endpoint. No specific results were announced.
REWIND was distinct in that it had limited participation from people with established cardiovascular disease, which allowed dulaglutide’s effect to be measured in a broad population of people with type 2 diabetes, Eli Lilly said in their press release.
Dulaglutide is a GLP-1 receptor agonist, and trial participants received a 1.5-mg dose once a week.
Full results of the REWIND trial will be presented at the annual scientific sessions of the American Diabetes Association in June 2019.
Eli Lilly has announced top line results from REWIND, a multicenter, randomized, double-blind, placebo-controlled trial designed to study dulaglutide (Trulicity). The cardiovascular outcomes trial, which had a median follow-up period of more than 5 years, evaluated cardiovascular outcomes in nearly 10,000 patients with type 2 diabetes.
The primary endpoint of REWIND was time until the first occurrence of a major adverse cardiovascular event. Patients who received dulaglutide had a significantly reduced incidence of these events, compared with placebo, meeting the primary trial endpoint. No specific results were announced.
REWIND was distinct in that it had limited participation from people with established cardiovascular disease, which allowed dulaglutide’s effect to be measured in a broad population of people with type 2 diabetes, Eli Lilly said in their press release.
Dulaglutide is a GLP-1 receptor agonist, and trial participants received a 1.5-mg dose once a week.
Full results of the REWIND trial will be presented at the annual scientific sessions of the American Diabetes Association in June 2019.
FDA approves adalimumab biosimilar Hyrimoz
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.
FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.
FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
The Food and Drug Administration has approved the adalimumab biosimilar Hyrimoz (adalimumab-adaz) for a variety of conditions, according to Sandoz, the drug’s manufacturer and a division of Novartis.
FDA approval for Hyrimoz is based on a randomized, double-blind, three-arm, parallel biosimilarity study that demonstrated equivalence for all primary pharmacokinetic parameters, according to the press release. A second study confirmed these results in patients with moderate to severe plaque psoriasis, with Hyrimoz having a safety profile similar to that of adalimumab. Hyrimoz was approved in Europe in July 2018.
Hyrimoz has been approved to treat rheumatoid arthritis, juvenile idiopathic arthritis in patients aged 4 years and older, psoriatic arthritis, ankylosing spondylitis, adult Crohn’s disease, ulcerative colitis, and plaque psoriasis. The most common adverse events associated with the drug, according to the label, are infections, injection site reactions, headache, and rash.
Hyrimoz is the third adalimumab biosimilar approved by the FDA.
“Biosimilars can help people suffering from chronic, debilitating conditions gain expanded access to important medicines that may change the outcome of their disease. With the FDA approval of Hyrimoz, Sandoz is one step closer to offering U.S. patients with autoimmune diseases the same critical access already available in Europe,” Stefan Hendriks, global head of biopharmaceuticals at Sandoz, said in the press release.
Find the full press release on the Novartis website.
AGA is taking the lead in educating health care providers and patients about biosimilars and how they can be used for IBD patient care. Learn more at www.gastro.org/biosimilars.
FDA approves sufentanil for adults with acute pain
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.

Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
The Food and Drug Administration on Nov. 2 approved sufentanil (Dsuvia) for managing acute pain in adult patients in certified, medically supervised health care settings.
Sufentanil, an opioid analgesic manufactured by AcelRx Pharmaceuticals, was approved as a 30-mcg sublingual tablet. The efficacy of Dsuvia was shown in a randomized, clinical trial where patients who received the drug demonstrated significantly greater pain relief after both 15 minutes and 12 hours, compared with placebo.
“As a single-dose, noninvasive medication with a rapid reduction in pain intensity, Dsuvia represents an important alternative for health care providers to offer patients for acute pain management,” David Leiman, MD, of the department of surgery at the University of Texas, Houston, said in the AcelRx press statement.
FDA Commissioner Scott Gottlieb, MD, commented on the approval amid concerns expressed by some, such as the advocacy group Public Citizen, that the drug is “more than 1,000 times more potent than morphine,” and that approval could lead to diversion and abuse – particularly in light of the U.S. opioid epidemic.
In his statement, Dr. Gottlieb identified one broad, significant issue. “Why do we need an oral formulation of sufentanil – a more potent form of fentanyl that’s been approved for intravenous and epidural use in the U.S. since 1984 – on the market?”
In particular, he focused on the needs of the military. The Department of Defense has taken interest in sufentanil as it fulfills a small but specific battlefield need, namely as a means of pain relief in battlefield situations where soldiers cannot swallow oral medication and access to intravenous medication is limited.
Dr. Gottlieb made clear that sufentanil was meant only to be taken in controlled settings and will have strong limitations on its use. It cannot be prescribed for home use, and treatment should be limited to 72 hours. It can only be delivered by health care professionals using a single-dose applicator and will not be available in pharmacies. It is only to be used in patients who have not tolerated or are expected not to tolerate alternative methods of pain management.
“The FDA has implemented a REMS [Risk Evaluation and Mitigation Strategy] that reflects the potential risks associated with this product and mandates that Dsuvia will only be made available for use in a certified medically supervised heath care setting, including its use on the battlefield,” Dr. Gottlieb said.
However, he recognized that the debate runs deeper than how the FDA should mitigate risk over a new drug, and “as a public health agency, we have an obligation to address this question openly and directly. As a physician and regulator, I won’t bypass legitimate questions and concerns related to our role in addressing the opioid crisis,” he said.
Find Dr. Gottlieb’s full statement on the FDA website.
Yoga feasible, provides modest benefits for women with urinary incontinence
according to Alison J. Huang, MD, of the University of California, San Francisco, and her associates.

In a small study published in the American Journal of Obstetrics and Gynecology, 28 women enrolled in a 3-month yoga intervention program and 28 were enrolled in a control program consisting of nonspecific muscle stretching and strengthening. The mean age was 65 years, baseline urinary incontinence was 3.5 episodes/day, and 37 women had urgency-predominant incontinence.
Of those who completed the study, 89% of 27 patients in the yoga group attended at least 80% of classes, compared with 87% in the control group; over 90% of women in the yoga group completed at least 9 home practice hours.
Overall incontinence frequency was reduced by 76% in the yoga group and by 56% in the control group. Incontinence caused by stress was significantly reduced in the yoga group, compared with the control group (61% vs. 35%; P = .045), but the rate of incontinence caused by urgency did not noticeably differ. A total of 48 nonserious adverse events were reported over the 3-month period (23 in the yoga and 25 in the control group), but none were associated with either yoga or control treatment.
“Yoga may be useful for incontinent women in the community who lack access to incontinence specialists, are unable to use clinical therapies, or wish to enhance conventional care. Since yoga can be practiced in a group setting without continuous supervision by health care specialists, it offers a potentially cost-effective, community-based self-management strategy for incontinence, provided that it can be taught with appropriate attention to safety and to patients’ clinical needs,” the authors noted.
The study was supported with grants from the National Center for Complementary and Integrative Medicine and the UCSF Osher Center for Integrative Medicine’s Bradley fund. One of the authors received a grant from the National Institute of Diabetes and Digestive and Kidney Disorders. Two study authors reported receiving funding from Pfizer and Astellas to conduct research unrelated to the current study.
SOURCE: Huang AJ et al. Am J Obstet Gynecol. 2018 Oct 26. doi: 10.1016/j.ajog.2018.10.031.
according to Alison J. Huang, MD, of the University of California, San Francisco, and her associates.
In a small study published in the American Journal of Obstetrics and Gynecology, 28 women enrolled in a 3-month yoga intervention program and 28 were enrolled in a control program consisting of nonspecific muscle stretching and strengthening. The mean age was 65 years, baseline urinary incontinence was 3.5 episodes/day, and 37 women had urgency-predominant incontinence.
Of those who completed the study, 89% of 27 patients in the yoga group attended at least 80% of classes, compared with 87% in the control group; over 90% of women in the yoga group completed at least 9 home practice hours.
Overall incontinence frequency was reduced by 76% in the yoga group and by 56% in the control group. Incontinence caused by stress was significantly reduced in the yoga group, compared with the control group (61% vs. 35%; P = .045), but the rate of incontinence caused by urgency did not noticeably differ. A total of 48 nonserious adverse events were reported over the 3-month period (23 in the yoga and 25 in the control group), but none were associated with either yoga or control treatment.
“Yoga may be useful for incontinent women in the community who lack access to incontinence specialists, are unable to use clinical therapies, or wish to enhance conventional care. Since yoga can be practiced in a group setting without continuous supervision by health care specialists, it offers a potentially cost-effective, community-based self-management strategy for incontinence, provided that it can be taught with appropriate attention to safety and to patients’ clinical needs,” the authors noted.
The study was supported with grants from the National Center for Complementary and Integrative Medicine and the UCSF Osher Center for Integrative Medicine’s Bradley fund. One of the authors received a grant from the National Institute of Diabetes and Digestive and Kidney Disorders. Two study authors reported receiving funding from Pfizer and Astellas to conduct research unrelated to the current study.
SOURCE: Huang AJ et al. Am J Obstet Gynecol. 2018 Oct 26. doi: 10.1016/j.ajog.2018.10.031.
according to Alison J. Huang, MD, of the University of California, San Francisco, and her associates.
In a small study published in the American Journal of Obstetrics and Gynecology, 28 women enrolled in a 3-month yoga intervention program and 28 were enrolled in a control program consisting of nonspecific muscle stretching and strengthening. The mean age was 65 years, baseline urinary incontinence was 3.5 episodes/day, and 37 women had urgency-predominant incontinence.
Of those who completed the study, 89% of 27 patients in the yoga group attended at least 80% of classes, compared with 87% in the control group; over 90% of women in the yoga group completed at least 9 home practice hours.
Overall incontinence frequency was reduced by 76% in the yoga group and by 56% in the control group. Incontinence caused by stress was significantly reduced in the yoga group, compared with the control group (61% vs. 35%; P = .045), but the rate of incontinence caused by urgency did not noticeably differ. A total of 48 nonserious adverse events were reported over the 3-month period (23 in the yoga and 25 in the control group), but none were associated with either yoga or control treatment.
“Yoga may be useful for incontinent women in the community who lack access to incontinence specialists, are unable to use clinical therapies, or wish to enhance conventional care. Since yoga can be practiced in a group setting without continuous supervision by health care specialists, it offers a potentially cost-effective, community-based self-management strategy for incontinence, provided that it can be taught with appropriate attention to safety and to patients’ clinical needs,” the authors noted.
The study was supported with grants from the National Center for Complementary and Integrative Medicine and the UCSF Osher Center for Integrative Medicine’s Bradley fund. One of the authors received a grant from the National Institute of Diabetes and Digestive and Kidney Disorders. Two study authors reported receiving funding from Pfizer and Astellas to conduct research unrelated to the current study.
SOURCE: Huang AJ et al. Am J Obstet Gynecol. 2018 Oct 26. doi: 10.1016/j.ajog.2018.10.031.
FROM THE AMERICAN JOURNAL OF OBSTETRICS AND GYNECOLOGY