User login
Buyer beware
The invitation came to my house, addressed to “residential customer.” It was for my wife and me to attend a free dinner at a swanky local restaurant to learn about “revolutionary” treatments for memory loss. It was presented by a “wellness expert.”
Of course, I just had to check out the website.
The dinner was hosted by an internist pushing an unproven (except for the usual small noncontrolled studies) treatment. Although not stated, I’m sure when you call you’ll find out this is not covered by insurance; not Food and Drug Administration approved to treat, cure, diagnose, prevent any disease; your mileage may vary, etc.

The website was full of testimonials as to how well the treatments worked, primarily from people in their 20s-40s who are, realistically, unlikely to have a pathologically serious cause for memory problems. The site also mentions that you can use it to treat traumatic brain injury, ADHD, learning disorders, obsessive-compulsive disorder, PTSD, Parkinson’s disease, autism, dementia, and stroke, though it does clearly state that such use is not FDA approved.
Prices (I assume all cash pay) for the treatment weren’t listed. I guess you have to come to the “free” dinner for those, or submit an online form to the office.
I’m not going to say the advertised treatment doesn’t work. It might, for at least some of those things. A PubMed search tells me it’s under investigation for several of them.
But that doesn’t mean it works. It might, but a lot of things that look promising in early trials end up failing in the long run. So, at least to me, this is no different than people selling various over-the-counter supplements online with all kinds of extravagant claims and testimonials.
I also have to question a treatment targeting young people for memory loss. In neurology we see a lot of that, and know that true pathology is rare. Most of these patients have root issues with depression, or anxiety, or stress that are affecting their memory. That doesn’t make their memory issues any less real, but they shouldn’t be lumped in with neurodegenerative diseases. They need to be correctly diagnosed and treated for what they are.
Maybe it’s just me, but I often see this sort of thing as kind of sketchy – generating business for unproven treatments by selling fear – you need to do something NOW to keep from getting worse. And, of course there’s always the mysterious “they.” The treatments “they” offer don’t work. Why aren’t “they” telling you what really does?
Looking at the restaurant’s online menu, dinners are around $75 per person and the invitation says “seats are limited.” Doing some mental math gives you an idea how many diners need to come, what percentage of them need to sign up for the treatment, and how much it costs to recoup the investment.
Let the buyer beware.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
The invitation came to my house, addressed to “residential customer.” It was for my wife and me to attend a free dinner at a swanky local restaurant to learn about “revolutionary” treatments for memory loss. It was presented by a “wellness expert.”
Of course, I just had to check out the website.
The dinner was hosted by an internist pushing an unproven (except for the usual small noncontrolled studies) treatment. Although not stated, I’m sure when you call you’ll find out this is not covered by insurance; not Food and Drug Administration approved to treat, cure, diagnose, prevent any disease; your mileage may vary, etc.
The website was full of testimonials as to how well the treatments worked, primarily from people in their 20s-40s who are, realistically, unlikely to have a pathologically serious cause for memory problems. The site also mentions that you can use it to treat traumatic brain injury, ADHD, learning disorders, obsessive-compulsive disorder, PTSD, Parkinson’s disease, autism, dementia, and stroke, though it does clearly state that such use is not FDA approved.
Prices (I assume all cash pay) for the treatment weren’t listed. I guess you have to come to the “free” dinner for those, or submit an online form to the office.
I’m not going to say the advertised treatment doesn’t work. It might, for at least some of those things. A PubMed search tells me it’s under investigation for several of them.
But that doesn’t mean it works. It might, but a lot of things that look promising in early trials end up failing in the long run. So, at least to me, this is no different than people selling various over-the-counter supplements online with all kinds of extravagant claims and testimonials.
I also have to question a treatment targeting young people for memory loss. In neurology we see a lot of that, and know that true pathology is rare. Most of these patients have root issues with depression, or anxiety, or stress that are affecting their memory. That doesn’t make their memory issues any less real, but they shouldn’t be lumped in with neurodegenerative diseases. They need to be correctly diagnosed and treated for what they are.
Maybe it’s just me, but I often see this sort of thing as kind of sketchy – generating business for unproven treatments by selling fear – you need to do something NOW to keep from getting worse. And, of course there’s always the mysterious “they.” The treatments “they” offer don’t work. Why aren’t “they” telling you what really does?
Looking at the restaurant’s online menu, dinners are around $75 per person and the invitation says “seats are limited.” Doing some mental math gives you an idea how many diners need to come, what percentage of them need to sign up for the treatment, and how much it costs to recoup the investment.
Let the buyer beware.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
The invitation came to my house, addressed to “residential customer.” It was for my wife and me to attend a free dinner at a swanky local restaurant to learn about “revolutionary” treatments for memory loss. It was presented by a “wellness expert.”
Of course, I just had to check out the website.
The dinner was hosted by an internist pushing an unproven (except for the usual small noncontrolled studies) treatment. Although not stated, I’m sure when you call you’ll find out this is not covered by insurance; not Food and Drug Administration approved to treat, cure, diagnose, prevent any disease; your mileage may vary, etc.
The website was full of testimonials as to how well the treatments worked, primarily from people in their 20s-40s who are, realistically, unlikely to have a pathologically serious cause for memory problems. The site also mentions that you can use it to treat traumatic brain injury, ADHD, learning disorders, obsessive-compulsive disorder, PTSD, Parkinson’s disease, autism, dementia, and stroke, though it does clearly state that such use is not FDA approved.
Prices (I assume all cash pay) for the treatment weren’t listed. I guess you have to come to the “free” dinner for those, or submit an online form to the office.
I’m not going to say the advertised treatment doesn’t work. It might, for at least some of those things. A PubMed search tells me it’s under investigation for several of them.
But that doesn’t mean it works. It might, but a lot of things that look promising in early trials end up failing in the long run. So, at least to me, this is no different than people selling various over-the-counter supplements online with all kinds of extravagant claims and testimonials.
I also have to question a treatment targeting young people for memory loss. In neurology we see a lot of that, and know that true pathology is rare. Most of these patients have root issues with depression, or anxiety, or stress that are affecting their memory. That doesn’t make their memory issues any less real, but they shouldn’t be lumped in with neurodegenerative diseases. They need to be correctly diagnosed and treated for what they are.
Maybe it’s just me, but I often see this sort of thing as kind of sketchy – generating business for unproven treatments by selling fear – you need to do something NOW to keep from getting worse. And, of course there’s always the mysterious “they.” The treatments “they” offer don’t work. Why aren’t “they” telling you what really does?
Looking at the restaurant’s online menu, dinners are around $75 per person and the invitation says “seats are limited.” Doing some mental math gives you an idea how many diners need to come, what percentage of them need to sign up for the treatment, and how much it costs to recoup the investment.
Let the buyer beware.
Dr. Block has a solo neurology practice in Scottsdale, Ariz.
Treating poikiloderma
and is one of the most frustrating dermatologic problems to treat.
Poikiloderma is an area of mottled pigmentation (hyper and hypo) with telangiectasias and atrophy often present on the V of the chest, lateral neck, and lateral face. It is always present in sun-exposed areas but shaded areas of the neck, such as the area under the chin, are spared. Cumulative UV radiation is the predominant underlying cause; however, postmenopausal hormonal changes and contact sensitization with perfumes and cosmetics can exacerbate the condition.
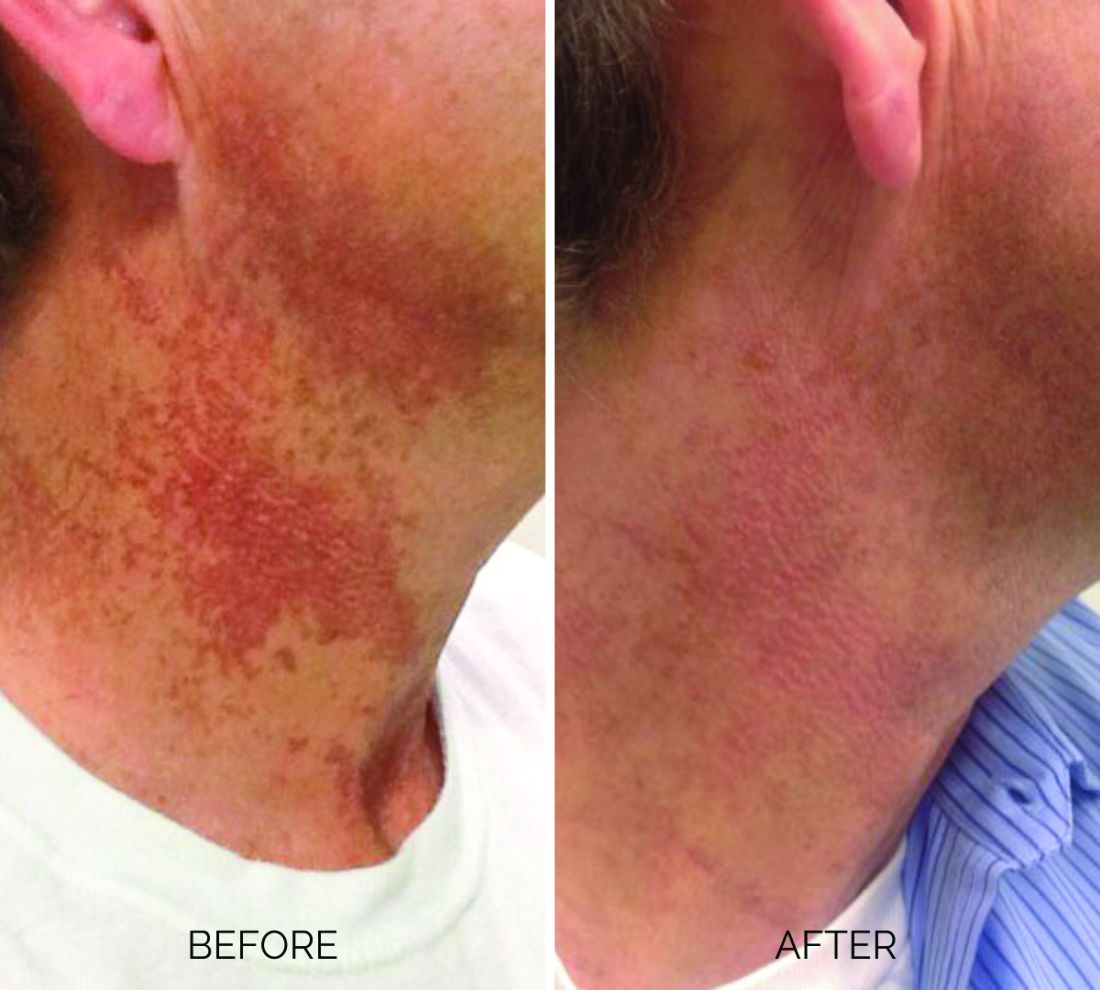
Breaking down the subtypes will help direct the treatment options. There are two main types of poikiloderma – telangiectatic and hyperpigmented – and of course, an overlap between the two. Choosing which subtype is dominant is based primarily on clinical presentation and dermoscopic findings. Atrophy is ubiquitous, thus collagen remodeling is a necessary treatment for both.
In my clinical practice, the pigmentation component of poikiloderma in all skin types is pretreated and posttreated with topical hydroquinone and/or oral tranexamic acid to avoid recurrence after any laser treatment. In the majority of my patients with poikiloderma, I first treat the pigmentation with hydroquinone and tranexamic acid (if the patient is a candidate) to minimize the pigment as much as possible and then treat the telangiectasias with lasers. I try to avoid laser treatment of the hyperpigmentation if at all possible.

Telangiectatic poikiloderma is characterized by a linear and reticular dilated network of vessels. Laser treatment options include IPL, V-beam, and KTP lasers. Multiple treatments are usually necessary and if the patient has concomitant flushing and burning symptoms associated with poikiloderma, topical rosacea treatments such as topical oxymetazoline, as well as avoidance of fragrance, and strict use of a broad spectrum mineral sunscreen, should be initiated prior to laser treatments.
Hyperpigmented poikiloderma is characterized by mottled hyperpigmentation caused by the increased melanin irregularly distributed in the basal layer of the epidermis and melanophages within the dermis. The best treatment for this is with 1,927-nm fractionated resurfacing modalities. Although IPL has been used in this area and is often recommended in the literature for the lentigines, in my experience, the results are transient and it is much harder to blend the color of the skin with the surrounding area of the neck, lateral chest, shoulders, and arms. The 1,927-nm fractionated laser allows for a smoother transition and blending of the skin and also helps with some collagen remodeling of the dermis.
Atrophy is visualized under dermoscopy as a white polka dot–like print with flattened, atrophic epidermis and an elastotic papillary dermis in between the hyperemic telangiectatic network. With every case of poikiloderma, there is some atrophy present; therefore, I combine platelet rich plasma (PRP), PRP with microneedling, or very light treatments with the Fraxel dual (1927/1550) laser to help improve architectural changes of the dermis.
As with any condition of the chest and neck, there is a very fine line between treatment efficacy and complications. All treatments, particularly lasers, should be used with considerable caution and test spots and with the expectation that the treatment will mitigate, not resolve the condition. Sun avoidance, use of daily mineral SPF, and avoidance of fragrance should be emphasized. If expectations are set properly, patients are often satisfied with small improvements as this condition can be very troubling and difficult to treat.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Geronemus R. Arch Dermatol. 1990 Apr;126(4):547-8.
Goldman MP and Weiss RA. Plast Reconstr Surg. 2001 May;107(6):1376-81.
Katoulis AC and Stavrianeas NG. Poikiloderma of Civatte. In: Rigopoulos D, Katoulis AC, editors. Hyperpigmentation (Boca Raton, Fla.: CRC Press, 2017). Chapter 12.
and is one of the most frustrating dermatologic problems to treat.
Poikiloderma is an area of mottled pigmentation (hyper and hypo) with telangiectasias and atrophy often present on the V of the chest, lateral neck, and lateral face. It is always present in sun-exposed areas but shaded areas of the neck, such as the area under the chin, are spared. Cumulative UV radiation is the predominant underlying cause; however, postmenopausal hormonal changes and contact sensitization with perfumes and cosmetics can exacerbate the condition.
Breaking down the subtypes will help direct the treatment options. There are two main types of poikiloderma – telangiectatic and hyperpigmented – and of course, an overlap between the two. Choosing which subtype is dominant is based primarily on clinical presentation and dermoscopic findings. Atrophy is ubiquitous, thus collagen remodeling is a necessary treatment for both.
In my clinical practice, the pigmentation component of poikiloderma in all skin types is pretreated and posttreated with topical hydroquinone and/or oral tranexamic acid to avoid recurrence after any laser treatment. In the majority of my patients with poikiloderma, I first treat the pigmentation with hydroquinone and tranexamic acid (if the patient is a candidate) to minimize the pigment as much as possible and then treat the telangiectasias with lasers. I try to avoid laser treatment of the hyperpigmentation if at all possible.
Telangiectatic poikiloderma is characterized by a linear and reticular dilated network of vessels. Laser treatment options include IPL, V-beam, and KTP lasers. Multiple treatments are usually necessary and if the patient has concomitant flushing and burning symptoms associated with poikiloderma, topical rosacea treatments such as topical oxymetazoline, as well as avoidance of fragrance, and strict use of a broad spectrum mineral sunscreen, should be initiated prior to laser treatments.
Hyperpigmented poikiloderma is characterized by mottled hyperpigmentation caused by the increased melanin irregularly distributed in the basal layer of the epidermis and melanophages within the dermis. The best treatment for this is with 1,927-nm fractionated resurfacing modalities. Although IPL has been used in this area and is often recommended in the literature for the lentigines, in my experience, the results are transient and it is much harder to blend the color of the skin with the surrounding area of the neck, lateral chest, shoulders, and arms. The 1,927-nm fractionated laser allows for a smoother transition and blending of the skin and also helps with some collagen remodeling of the dermis.
Atrophy is visualized under dermoscopy as a white polka dot–like print with flattened, atrophic epidermis and an elastotic papillary dermis in between the hyperemic telangiectatic network. With every case of poikiloderma, there is some atrophy present; therefore, I combine platelet rich plasma (PRP), PRP with microneedling, or very light treatments with the Fraxel dual (1927/1550) laser to help improve architectural changes of the dermis.
As with any condition of the chest and neck, there is a very fine line between treatment efficacy and complications. All treatments, particularly lasers, should be used with considerable caution and test spots and with the expectation that the treatment will mitigate, not resolve the condition. Sun avoidance, use of daily mineral SPF, and avoidance of fragrance should be emphasized. If expectations are set properly, patients are often satisfied with small improvements as this condition can be very troubling and difficult to treat.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Geronemus R. Arch Dermatol. 1990 Apr;126(4):547-8.
Goldman MP and Weiss RA. Plast Reconstr Surg. 2001 May;107(6):1376-81.
Katoulis AC and Stavrianeas NG. Poikiloderma of Civatte. In: Rigopoulos D, Katoulis AC, editors. Hyperpigmentation (Boca Raton, Fla.: CRC Press, 2017). Chapter 12.
and is one of the most frustrating dermatologic problems to treat.
Poikiloderma is an area of mottled pigmentation (hyper and hypo) with telangiectasias and atrophy often present on the V of the chest, lateral neck, and lateral face. It is always present in sun-exposed areas but shaded areas of the neck, such as the area under the chin, are spared. Cumulative UV radiation is the predominant underlying cause; however, postmenopausal hormonal changes and contact sensitization with perfumes and cosmetics can exacerbate the condition.
Breaking down the subtypes will help direct the treatment options. There are two main types of poikiloderma – telangiectatic and hyperpigmented – and of course, an overlap between the two. Choosing which subtype is dominant is based primarily on clinical presentation and dermoscopic findings. Atrophy is ubiquitous, thus collagen remodeling is a necessary treatment for both.
In my clinical practice, the pigmentation component of poikiloderma in all skin types is pretreated and posttreated with topical hydroquinone and/or oral tranexamic acid to avoid recurrence after any laser treatment. In the majority of my patients with poikiloderma, I first treat the pigmentation with hydroquinone and tranexamic acid (if the patient is a candidate) to minimize the pigment as much as possible and then treat the telangiectasias with lasers. I try to avoid laser treatment of the hyperpigmentation if at all possible.
Telangiectatic poikiloderma is characterized by a linear and reticular dilated network of vessels. Laser treatment options include IPL, V-beam, and KTP lasers. Multiple treatments are usually necessary and if the patient has concomitant flushing and burning symptoms associated with poikiloderma, topical rosacea treatments such as topical oxymetazoline, as well as avoidance of fragrance, and strict use of a broad spectrum mineral sunscreen, should be initiated prior to laser treatments.
Hyperpigmented poikiloderma is characterized by mottled hyperpigmentation caused by the increased melanin irregularly distributed in the basal layer of the epidermis and melanophages within the dermis. The best treatment for this is with 1,927-nm fractionated resurfacing modalities. Although IPL has been used in this area and is often recommended in the literature for the lentigines, in my experience, the results are transient and it is much harder to blend the color of the skin with the surrounding area of the neck, lateral chest, shoulders, and arms. The 1,927-nm fractionated laser allows for a smoother transition and blending of the skin and also helps with some collagen remodeling of the dermis.
Atrophy is visualized under dermoscopy as a white polka dot–like print with flattened, atrophic epidermis and an elastotic papillary dermis in between the hyperemic telangiectatic network. With every case of poikiloderma, there is some atrophy present; therefore, I combine platelet rich plasma (PRP), PRP with microneedling, or very light treatments with the Fraxel dual (1927/1550) laser to help improve architectural changes of the dermis.
As with any condition of the chest and neck, there is a very fine line between treatment efficacy and complications. All treatments, particularly lasers, should be used with considerable caution and test spots and with the expectation that the treatment will mitigate, not resolve the condition. Sun avoidance, use of daily mineral SPF, and avoidance of fragrance should be emphasized. If expectations are set properly, patients are often satisfied with small improvements as this condition can be very troubling and difficult to treat.
Dr. Talakoub is in private practice in McLean, Va. Write to her at [email protected]. She had no relevant disclosures.
References
Geronemus R. Arch Dermatol. 1990 Apr;126(4):547-8.
Goldman MP and Weiss RA. Plast Reconstr Surg. 2001 May;107(6):1376-81.
Katoulis AC and Stavrianeas NG. Poikiloderma of Civatte. In: Rigopoulos D, Katoulis AC, editors. Hyperpigmentation (Boca Raton, Fla.: CRC Press, 2017). Chapter 12.
The three pillars of perinatal care: Babies, parents, dyadic relationships
Perinatal depression (PND) is the most common obstetric complication in the United States. Even when screening results are positive, mothers often do not receive further evaluation, and even when PND is diagnosed, mothers do not receive evidence-based treatments.

Meta-analytic estimates show that pregnant women suffer from PND at rates from 6.5% to 12.9% across pregnancy to 3-months post partum.1 Women from low-income families and adolescent mothers are at highest risk, where rates are double and triple respectively.
Fathers also suffer from PND, with a prevalence rate from 2% to 25%, increasing to 50% when the mother experiences PND.
The American Academy of Pediatrics issued a Policy Statement (January 2019) about the need to recognize and manage PND. They recommended that pediatric medical homes establish a system to implement the screening of mothers at the 1-, 2-, 4-, and 6-month well-child visits, to use community resources for the treatment and referral of the mother with depression, and to provide support for the maternal-child relationship.2
The American Academy of Pediatrics also recommends advocacy for workforce development for mental health professionals who care for young children and mother-infant dyads, and for promotion of evidence-based interventions focused on healthy attachment and parent-child relationships.
Family research
There is a bidirectional association between family relational stress and PND. Lack of family support is both a predictor and a consequence of perinatal depression. Frequent arguments, conflict because one or both partners did not want the pregnancy, division of labor, poor support following stressful life events, lack of partner availability, and low intimacy are associated with increased perinatal depressive symptoms.
Gender role stress is also included as a risk factor. For example, men may fear performance failure related to work and sex, and women may fear disruption in the couple relationship due to the introduction of a child.
When depressed and nondepressed women at 2 months post delivery were compared, the women with depressive symptoms perceived that their partners did not share similar interests, provided little companionship, expressed disinterest in infant care, did not provide a feeling of connection, did not encourage them to get assistance to cope with difficulties, and expressed disagreement in infant care.3
A high-quality intimate relationship is protective for many illnesses and PND is no exception.4
Assessment
Despite the availability of effective treatments, perinatal mental health utilization rates are strikingly low. There are limited providers and a general lack of awareness of the need for this care. The stigma for assessing and treating PND is high because the perception is that pregnancy is supposed to be a joyous time and with time, PND will pass.
The first step is a timely and accurate assessment of the mother, which should, if possible, include the father and other family support people. The preferred standard for women is the Edinburgh Postnatal Depression Scale (EPDS), a checklist of 10 items (listed below) with a maximum score of 30, and any score over 10 warrants further assessment.5 This scale is used worldwide in obstetric clinics and has been used to identify PND in fathers.
- I have been able to laugh and see the funny side of things.
- I have looked forward with enjoyment to things.
- I have blamed myself unnecessarily when things went wrong.
- I have been anxious or worried for no good reason.
- I have felt scared or panicky for no good reason.
- Things have been getting to me.
- I have been so unhappy that I have had difficulty sleeping.
- I have felt sad or miserable.
- I have been so unhappy that I have been crying.
- The thought of harming myself has occurred to me.
A new ultrabrief tool with only four questions is the Brief Multidimensional Assessment Scale (BMAS), which measures the ability to get things done, emotional support in important relationships, quality of life, and sense of purpose in life. It demonstrates concurrent validity with other measures and discriminates between nonclinical participants and participants from most clinical contexts.6
For those interested in assessing family health, an easy-to-use assessment tool is the 12-item Family Assessment Device (FAD).7
Family therapy interventions
A systematic review and meta-analysis of the current evidence on the usefulness of family therapy interventions in the prevention and treatment of PND identified seven studies.
In these studies, there were statistically significant reductions in depressive symptoms at postintervention in intervention group mothers. Intervention intensity and level of family involvement moderated the impacts of intervention on maternal depression, and there was a trend in improved family functioning in intervention group couples.8
Evidence-based interventions are usually psychoeducational or cognitive-behavioral family therapy models where focused interventions target the following three areas:
- Communication skills related to expectations (including those that pertain to gender roles and the transition to parenthood) and emotional support.
- Conflict management.
- Problem-solving skills related to shared responsibility in infant care and household activities.
Intensive day program for mothers and babies
There is a growing awareness of the effectiveness of specialized mother-baby day hospital programs for women with psychiatric distress during the peripartum period.9
The Women & Infants’ Hospital (WIH) in Providence, R.I., established a mother-baby postpartum depression day program in 2000, adjacent to the obstetrical hospital, the ninth largest obstetrical service in the United States. The day program is integrated with the hospital’s obstetric medicine team and referrals are also accepted from the perinatal practices in the surrounding community. The treatment day includes group, individual, and milieu treatment, as well as consultation with psychiatrists, nutritionists, social workers, lactation specialists and others.
The primary theoretical model utilized by the program is interpersonal psychotherapy (IPT), with essential elements of the program incorporating cognitive behavioral therapy (CBT), and experiential strategies (for instance, mindfulness, breathing, progressive muscle relaxation) to improve self-care and relaxation skills. Patient satisfaction surveys collected from 800 women, (54% identified as White) treated at the program between 2007 and 2012 found that women were highly satisfied with the treatment received, noting that the inclusion of the baby in their treatment is a highly valued aspect of care.
A similar program in Minnesota reported that 328 women who consented to participation in research had significant improvements (P < .001) in self-report scales assessing depression, anxiety, and maternal functioning, improving mental health and parenting functioning.10
Lastly, a recent study out of Brussels, on the benefit of a mother-baby day program analyzed patient data from 2015 and 2020. This clinical population of 92 patients (43% identifying as North African) was comparable to the population of the inpatient mother-baby units in terms of psychosocial fragility except that the parents entering the day program had less severe illnesses, more anxiety disorder, and less postpartum psychosis. In the day program, all the babies improved in terms of symptoms and relationships, except for those with significant developmental difficulties.
The dyadic relationship was measured using “levels of adaptation of the parent–child relationship” scale which has four general levels of adjustment, from well-adjusted to troubled or dangerous relationship. Unlike programs in the United States, this program takes children up to 2.5 years old and the assessment period is up to 8 weeks.11
Prevention of mental illness is best achieved by reducing the known determinants of illness. For PND, the research is clear, so why not start at the earliest possible stage, when we know that change is possible? Pushing health care systems to change is not easy, but as the research accumulates and the positive results grow, our arguments become stronger.
Dr. Heru is a psychiatrist in Aurora, Colo. She is editor of “Working With Families in Medical Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals” (New York: Routledge, 2013). She has no conflicts of interest to disclose. Contact Dr. Heru at [email protected].
References
1. Gavin NI et al. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005 Nov;106(5 Pt 1):1071-83. doi: 10.1097/01.AOG.0000183597.31630.db.
2. Rafferty J et al. Incorporating recognition and management of perinatal depression into pediatric practice. Pediatrics. 2019 Jan;143(1):e20183260. doi: 10.1542/peds.2018-3260.
3. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
4. Kumar SA et al. Promoting resilience to depression among couples during pregnancy: The protective functions of intimate relationship satisfaction and self-compassion. Family Process. 2022 May;62(1):387-405. doi: 10.1111/famp.12788.
5. Cox JL et al. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987 Jun;150:782-6. doi: 10.1192/bjp.150.6.782.
6. Keitner GI et al. The Brief Multidimensional Assessment Scale (BMAS): A broad measure of patient well-being. Am J Psychother. 2023 Feb 1;76(2):75-81. doi: 10.1176/appi.psychotherapy.20220032.
7. Boterhoven de Haan KL et al. Reliability and validity of a short version of the general functioning subscale of the McMaster Family Assessment Device. Fam Process. 2015 Mar;54(1):116-23. doi: 10.1111/famp.12113.
8. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
9. Battle CL, Howard MM. A mother-baby psychiatric day hospital: History, rationale, and why perinatal mental health is important for obstetric medicine. Obstet Med. 2014 Jun;7(2):66-70. doi: 10.1177/1753495X13514402.
10. Kim HG et al. Keeping Parent, Child, and Relationship in Mind: Clinical Effectiveness of a Trauma-informed, Multigenerational, Attachment-Based, Mother-Baby Partial Hospital Program in an Urban Safety Net Hospital. Matern Child Health J. 2021 Nov;25(11):1776-86. doi: 10.1007/s10995-021-03221-4.
11. Moureau A et al. A 5 years’ experience of a parent-baby day unit: impact on baby’s development. Front Psychiatry. 2023 June 15;14. doi: 10.3389/fpsyt.2023.1121894.
Perinatal depression (PND) is the most common obstetric complication in the United States. Even when screening results are positive, mothers often do not receive further evaluation, and even when PND is diagnosed, mothers do not receive evidence-based treatments.
Meta-analytic estimates show that pregnant women suffer from PND at rates from 6.5% to 12.9% across pregnancy to 3-months post partum.1 Women from low-income families and adolescent mothers are at highest risk, where rates are double and triple respectively.
Fathers also suffer from PND, with a prevalence rate from 2% to 25%, increasing to 50% when the mother experiences PND.
The American Academy of Pediatrics issued a Policy Statement (January 2019) about the need to recognize and manage PND. They recommended that pediatric medical homes establish a system to implement the screening of mothers at the 1-, 2-, 4-, and 6-month well-child visits, to use community resources for the treatment and referral of the mother with depression, and to provide support for the maternal-child relationship.2
The American Academy of Pediatrics also recommends advocacy for workforce development for mental health professionals who care for young children and mother-infant dyads, and for promotion of evidence-based interventions focused on healthy attachment and parent-child relationships.
Family research
There is a bidirectional association between family relational stress and PND. Lack of family support is both a predictor and a consequence of perinatal depression. Frequent arguments, conflict because one or both partners did not want the pregnancy, division of labor, poor support following stressful life events, lack of partner availability, and low intimacy are associated with increased perinatal depressive symptoms.
Gender role stress is also included as a risk factor. For example, men may fear performance failure related to work and sex, and women may fear disruption in the couple relationship due to the introduction of a child.
When depressed and nondepressed women at 2 months post delivery were compared, the women with depressive symptoms perceived that their partners did not share similar interests, provided little companionship, expressed disinterest in infant care, did not provide a feeling of connection, did not encourage them to get assistance to cope with difficulties, and expressed disagreement in infant care.3
A high-quality intimate relationship is protective for many illnesses and PND is no exception.4
Assessment
Despite the availability of effective treatments, perinatal mental health utilization rates are strikingly low. There are limited providers and a general lack of awareness of the need for this care. The stigma for assessing and treating PND is high because the perception is that pregnancy is supposed to be a joyous time and with time, PND will pass.
The first step is a timely and accurate assessment of the mother, which should, if possible, include the father and other family support people. The preferred standard for women is the Edinburgh Postnatal Depression Scale (EPDS), a checklist of 10 items (listed below) with a maximum score of 30, and any score over 10 warrants further assessment.5 This scale is used worldwide in obstetric clinics and has been used to identify PND in fathers.
- I have been able to laugh and see the funny side of things.
- I have looked forward with enjoyment to things.
- I have blamed myself unnecessarily when things went wrong.
- I have been anxious or worried for no good reason.
- I have felt scared or panicky for no good reason.
- Things have been getting to me.
- I have been so unhappy that I have had difficulty sleeping.
- I have felt sad or miserable.
- I have been so unhappy that I have been crying.
- The thought of harming myself has occurred to me.
A new ultrabrief tool with only four questions is the Brief Multidimensional Assessment Scale (BMAS), which measures the ability to get things done, emotional support in important relationships, quality of life, and sense of purpose in life. It demonstrates concurrent validity with other measures and discriminates between nonclinical participants and participants from most clinical contexts.6
For those interested in assessing family health, an easy-to-use assessment tool is the 12-item Family Assessment Device (FAD).7
Family therapy interventions
A systematic review and meta-analysis of the current evidence on the usefulness of family therapy interventions in the prevention and treatment of PND identified seven studies.
In these studies, there were statistically significant reductions in depressive symptoms at postintervention in intervention group mothers. Intervention intensity and level of family involvement moderated the impacts of intervention on maternal depression, and there was a trend in improved family functioning in intervention group couples.8
Evidence-based interventions are usually psychoeducational or cognitive-behavioral family therapy models where focused interventions target the following three areas:
- Communication skills related to expectations (including those that pertain to gender roles and the transition to parenthood) and emotional support.
- Conflict management.
- Problem-solving skills related to shared responsibility in infant care and household activities.
Intensive day program for mothers and babies
There is a growing awareness of the effectiveness of specialized mother-baby day hospital programs for women with psychiatric distress during the peripartum period.9
The Women & Infants’ Hospital (WIH) in Providence, R.I., established a mother-baby postpartum depression day program in 2000, adjacent to the obstetrical hospital, the ninth largest obstetrical service in the United States. The day program is integrated with the hospital’s obstetric medicine team and referrals are also accepted from the perinatal practices in the surrounding community. The treatment day includes group, individual, and milieu treatment, as well as consultation with psychiatrists, nutritionists, social workers, lactation specialists and others.
The primary theoretical model utilized by the program is interpersonal psychotherapy (IPT), with essential elements of the program incorporating cognitive behavioral therapy (CBT), and experiential strategies (for instance, mindfulness, breathing, progressive muscle relaxation) to improve self-care and relaxation skills. Patient satisfaction surveys collected from 800 women, (54% identified as White) treated at the program between 2007 and 2012 found that women were highly satisfied with the treatment received, noting that the inclusion of the baby in their treatment is a highly valued aspect of care.
A similar program in Minnesota reported that 328 women who consented to participation in research had significant improvements (P < .001) in self-report scales assessing depression, anxiety, and maternal functioning, improving mental health and parenting functioning.10
Lastly, a recent study out of Brussels, on the benefit of a mother-baby day program analyzed patient data from 2015 and 2020. This clinical population of 92 patients (43% identifying as North African) was comparable to the population of the inpatient mother-baby units in terms of psychosocial fragility except that the parents entering the day program had less severe illnesses, more anxiety disorder, and less postpartum psychosis. In the day program, all the babies improved in terms of symptoms and relationships, except for those with significant developmental difficulties.
The dyadic relationship was measured using “levels of adaptation of the parent–child relationship” scale which has four general levels of adjustment, from well-adjusted to troubled or dangerous relationship. Unlike programs in the United States, this program takes children up to 2.5 years old and the assessment period is up to 8 weeks.11
Prevention of mental illness is best achieved by reducing the known determinants of illness. For PND, the research is clear, so why not start at the earliest possible stage, when we know that change is possible? Pushing health care systems to change is not easy, but as the research accumulates and the positive results grow, our arguments become stronger.
Dr. Heru is a psychiatrist in Aurora, Colo. She is editor of “Working With Families in Medical Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals” (New York: Routledge, 2013). She has no conflicts of interest to disclose. Contact Dr. Heru at [email protected].
References
1. Gavin NI et al. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005 Nov;106(5 Pt 1):1071-83. doi: 10.1097/01.AOG.0000183597.31630.db.
2. Rafferty J et al. Incorporating recognition and management of perinatal depression into pediatric practice. Pediatrics. 2019 Jan;143(1):e20183260. doi: 10.1542/peds.2018-3260.
3. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
4. Kumar SA et al. Promoting resilience to depression among couples during pregnancy: The protective functions of intimate relationship satisfaction and self-compassion. Family Process. 2022 May;62(1):387-405. doi: 10.1111/famp.12788.
5. Cox JL et al. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987 Jun;150:782-6. doi: 10.1192/bjp.150.6.782.
6. Keitner GI et al. The Brief Multidimensional Assessment Scale (BMAS): A broad measure of patient well-being. Am J Psychother. 2023 Feb 1;76(2):75-81. doi: 10.1176/appi.psychotherapy.20220032.
7. Boterhoven de Haan KL et al. Reliability and validity of a short version of the general functioning subscale of the McMaster Family Assessment Device. Fam Process. 2015 Mar;54(1):116-23. doi: 10.1111/famp.12113.
8. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
9. Battle CL, Howard MM. A mother-baby psychiatric day hospital: History, rationale, and why perinatal mental health is important for obstetric medicine. Obstet Med. 2014 Jun;7(2):66-70. doi: 10.1177/1753495X13514402.
10. Kim HG et al. Keeping Parent, Child, and Relationship in Mind: Clinical Effectiveness of a Trauma-informed, Multigenerational, Attachment-Based, Mother-Baby Partial Hospital Program in an Urban Safety Net Hospital. Matern Child Health J. 2021 Nov;25(11):1776-86. doi: 10.1007/s10995-021-03221-4.
11. Moureau A et al. A 5 years’ experience of a parent-baby day unit: impact on baby’s development. Front Psychiatry. 2023 June 15;14. doi: 10.3389/fpsyt.2023.1121894.
Perinatal depression (PND) is the most common obstetric complication in the United States. Even when screening results are positive, mothers often do not receive further evaluation, and even when PND is diagnosed, mothers do not receive evidence-based treatments.
Meta-analytic estimates show that pregnant women suffer from PND at rates from 6.5% to 12.9% across pregnancy to 3-months post partum.1 Women from low-income families and adolescent mothers are at highest risk, where rates are double and triple respectively.
Fathers also suffer from PND, with a prevalence rate from 2% to 25%, increasing to 50% when the mother experiences PND.
The American Academy of Pediatrics issued a Policy Statement (January 2019) about the need to recognize and manage PND. They recommended that pediatric medical homes establish a system to implement the screening of mothers at the 1-, 2-, 4-, and 6-month well-child visits, to use community resources for the treatment and referral of the mother with depression, and to provide support for the maternal-child relationship.2
The American Academy of Pediatrics also recommends advocacy for workforce development for mental health professionals who care for young children and mother-infant dyads, and for promotion of evidence-based interventions focused on healthy attachment and parent-child relationships.
Family research
There is a bidirectional association between family relational stress and PND. Lack of family support is both a predictor and a consequence of perinatal depression. Frequent arguments, conflict because one or both partners did not want the pregnancy, division of labor, poor support following stressful life events, lack of partner availability, and low intimacy are associated with increased perinatal depressive symptoms.
Gender role stress is also included as a risk factor. For example, men may fear performance failure related to work and sex, and women may fear disruption in the couple relationship due to the introduction of a child.
When depressed and nondepressed women at 2 months post delivery were compared, the women with depressive symptoms perceived that their partners did not share similar interests, provided little companionship, expressed disinterest in infant care, did not provide a feeling of connection, did not encourage them to get assistance to cope with difficulties, and expressed disagreement in infant care.3
A high-quality intimate relationship is protective for many illnesses and PND is no exception.4
Assessment
Despite the availability of effective treatments, perinatal mental health utilization rates are strikingly low. There are limited providers and a general lack of awareness of the need for this care. The stigma for assessing and treating PND is high because the perception is that pregnancy is supposed to be a joyous time and with time, PND will pass.
The first step is a timely and accurate assessment of the mother, which should, if possible, include the father and other family support people. The preferred standard for women is the Edinburgh Postnatal Depression Scale (EPDS), a checklist of 10 items (listed below) with a maximum score of 30, and any score over 10 warrants further assessment.5 This scale is used worldwide in obstetric clinics and has been used to identify PND in fathers.
- I have been able to laugh and see the funny side of things.
- I have looked forward with enjoyment to things.
- I have blamed myself unnecessarily when things went wrong.
- I have been anxious or worried for no good reason.
- I have felt scared or panicky for no good reason.
- Things have been getting to me.
- I have been so unhappy that I have had difficulty sleeping.
- I have felt sad or miserable.
- I have been so unhappy that I have been crying.
- The thought of harming myself has occurred to me.
A new ultrabrief tool with only four questions is the Brief Multidimensional Assessment Scale (BMAS), which measures the ability to get things done, emotional support in important relationships, quality of life, and sense of purpose in life. It demonstrates concurrent validity with other measures and discriminates between nonclinical participants and participants from most clinical contexts.6
For those interested in assessing family health, an easy-to-use assessment tool is the 12-item Family Assessment Device (FAD).7
Family therapy interventions
A systematic review and meta-analysis of the current evidence on the usefulness of family therapy interventions in the prevention and treatment of PND identified seven studies.
In these studies, there were statistically significant reductions in depressive symptoms at postintervention in intervention group mothers. Intervention intensity and level of family involvement moderated the impacts of intervention on maternal depression, and there was a trend in improved family functioning in intervention group couples.8
Evidence-based interventions are usually psychoeducational or cognitive-behavioral family therapy models where focused interventions target the following three areas:
- Communication skills related to expectations (including those that pertain to gender roles and the transition to parenthood) and emotional support.
- Conflict management.
- Problem-solving skills related to shared responsibility in infant care and household activities.
Intensive day program for mothers and babies
There is a growing awareness of the effectiveness of specialized mother-baby day hospital programs for women with psychiatric distress during the peripartum period.9
The Women & Infants’ Hospital (WIH) in Providence, R.I., established a mother-baby postpartum depression day program in 2000, adjacent to the obstetrical hospital, the ninth largest obstetrical service in the United States. The day program is integrated with the hospital’s obstetric medicine team and referrals are also accepted from the perinatal practices in the surrounding community. The treatment day includes group, individual, and milieu treatment, as well as consultation with psychiatrists, nutritionists, social workers, lactation specialists and others.
The primary theoretical model utilized by the program is interpersonal psychotherapy (IPT), with essential elements of the program incorporating cognitive behavioral therapy (CBT), and experiential strategies (for instance, mindfulness, breathing, progressive muscle relaxation) to improve self-care and relaxation skills. Patient satisfaction surveys collected from 800 women, (54% identified as White) treated at the program between 2007 and 2012 found that women were highly satisfied with the treatment received, noting that the inclusion of the baby in their treatment is a highly valued aspect of care.
A similar program in Minnesota reported that 328 women who consented to participation in research had significant improvements (P < .001) in self-report scales assessing depression, anxiety, and maternal functioning, improving mental health and parenting functioning.10
Lastly, a recent study out of Brussels, on the benefit of a mother-baby day program analyzed patient data from 2015 and 2020. This clinical population of 92 patients (43% identifying as North African) was comparable to the population of the inpatient mother-baby units in terms of psychosocial fragility except that the parents entering the day program had less severe illnesses, more anxiety disorder, and less postpartum psychosis. In the day program, all the babies improved in terms of symptoms and relationships, except for those with significant developmental difficulties.
The dyadic relationship was measured using “levels of adaptation of the parent–child relationship” scale which has four general levels of adjustment, from well-adjusted to troubled or dangerous relationship. Unlike programs in the United States, this program takes children up to 2.5 years old and the assessment period is up to 8 weeks.11
Prevention of mental illness is best achieved by reducing the known determinants of illness. For PND, the research is clear, so why not start at the earliest possible stage, when we know that change is possible? Pushing health care systems to change is not easy, but as the research accumulates and the positive results grow, our arguments become stronger.
Dr. Heru is a psychiatrist in Aurora, Colo. She is editor of “Working With Families in Medical Settings: A Multidisciplinary Guide for Psychiatrists and Other Health Professionals” (New York: Routledge, 2013). She has no conflicts of interest to disclose. Contact Dr. Heru at [email protected].
References
1. Gavin NI et al. Perinatal depression: a systematic review of prevalence and incidence. Obstet Gynecol. 2005 Nov;106(5 Pt 1):1071-83. doi: 10.1097/01.AOG.0000183597.31630.db.
2. Rafferty J et al. Incorporating recognition and management of perinatal depression into pediatric practice. Pediatrics. 2019 Jan;143(1):e20183260. doi: 10.1542/peds.2018-3260.
3. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
4. Kumar SA et al. Promoting resilience to depression among couples during pregnancy: The protective functions of intimate relationship satisfaction and self-compassion. Family Process. 2022 May;62(1):387-405. doi: 10.1111/famp.12788.
5. Cox JL et al. Detection of postnatal depression: Development of the 10-item Edinburgh Postnatal Depression Scale. Br J Psychiatry. 1987 Jun;150:782-6. doi: 10.1192/bjp.150.6.782.
6. Keitner GI et al. The Brief Multidimensional Assessment Scale (BMAS): A broad measure of patient well-being. Am J Psychother. 2023 Feb 1;76(2):75-81. doi: 10.1176/appi.psychotherapy.20220032.
7. Boterhoven de Haan KL et al. Reliability and validity of a short version of the general functioning subscale of the McMaster Family Assessment Device. Fam Process. 2015 Mar;54(1):116-23. doi: 10.1111/famp.12113.
8. Cluxton-Keller F, Bruce ML. Clinical effectiveness of family therapeutic interventions in the prevention and treatment of perinatal depression: A systematic review and meta-analysis. PLoS One. 2018 Jun 14;13(6):e0198730. doi: 10.1371/journal.pone.0198730.
9. Battle CL, Howard MM. A mother-baby psychiatric day hospital: History, rationale, and why perinatal mental health is important for obstetric medicine. Obstet Med. 2014 Jun;7(2):66-70. doi: 10.1177/1753495X13514402.
10. Kim HG et al. Keeping Parent, Child, and Relationship in Mind: Clinical Effectiveness of a Trauma-informed, Multigenerational, Attachment-Based, Mother-Baby Partial Hospital Program in an Urban Safety Net Hospital. Matern Child Health J. 2021 Nov;25(11):1776-86. doi: 10.1007/s10995-021-03221-4.
11. Moureau A et al. A 5 years’ experience of a parent-baby day unit: impact on baby’s development. Front Psychiatry. 2023 June 15;14. doi: 10.3389/fpsyt.2023.1121894.
American Geriatrics Society 2023 updated Beers Criteria highlights
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.

Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.
Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Every 4 years, an interprofessional panel of experts from the American Geriatrics Society provides updated guidelines on safe prescribing of medications in older adults, known as the Beers Criteria. A 2023 update was released in May 2023 after panel review of more 1,500 clinical trials and research studies published since the last update.
Anticoagulants
Notable changes to the 2023 guidelines include updated recommendations for anticoagulation. Warfarin should be avoided as initial therapy for venous thromboembolism or nonvalvular atrial fibrillation unless there are contraindications to direct oral anticoagulants (DOACs) or other substantial barriers to use.
Rivaroxaban should also be avoided, and dabigatran used with caution in favor of apixaban, which is felt to have a better safety profile in older adults. Rivaroxaban may be considered if once daily dosing is deemed to be more clinically appropriate. Financial barriers regarding drug coverage and formulary options were acknowledged as a significant barrier to equitable access to preferred direct oral anticoagulants in older adults.
Diabetes medication
Regarding diabetes management, short-acting sulfonylureas should be avoided in addition to long-acting sulfonylureas, because of the increased risk of hypoglycemia, and cardiovascular and all-cause mortality in older adults. Sodium-glucose cotransporter 2 inhibitors as an entire class are recommended to be used with caution, as older adults are at higher risk of euglycemic ketoacidosis and urogenital infections, particularly in women in the first month of initiating treatment.
Like DOACs, the panel acknowledged that financial considerations may lead to limited options for oral diabetic treatment. In circumstances where a sulfonylurea is used, short-acting forms are preferred over long acting to reduce the risk of prolonged hypoglycemia.
Aspirin for primary prevention
Alongside the U.S. Preventive Services Task Force guideline update in 2022 regarding aspirin for primary prevention of cardiovascular disease and stroke, the Beer’s Criteria recommend against initiation of aspirin for primary prevention in older adults. Ticagrelor and prasugrel should be used with caution because of the increased risk of major bleeding in older adults over the age of 75, compared with clopidogrel. If prasugrel is used, a lower dose of 5 mg is recommended, in line with guidelines by the American College of Cardiology and American Heart Association.
Pain medication
For pain management, the Beer’s Criteria updated recommendations to avoid NSAIDs, particularly when used in combination with steroids or anticoagulants. The panel highlights that even short-term use of NSAIDs is high risk when used in combination with steroids or anticoagulants. If no other alternatives are possible, patients should be placed on a proton pump inhibitor or misoprostol while taking NSAIDs.
Baclofen should be avoided in older adults with renal insufficiency (estimated glomerular filtration rate < 60 mL/min per 1.73 m2) because of the increased risk of encephalopathy, and when used, should be given at the lowest effective dose with close monitoring for mental status changes.
Androgen and estrogen replacement therapy
For androgen replacement therapy, the panel notes that testosterone supplementation should be avoided because of cardiovascular risks unless there is confirmed hypogonadism. The panel revised their recommendation on the basis of emerging data that a history of prostate cancer is not an absolute contraindication for exogenous testosterone. A risk versus benefit discussion about exogenous testosterone should be had with a medical oncologist or urologist in those with a history of prostate cancer.
Regarding estrogen, systemic formulations should not be initiated in women over the age of 60 because of increased risk of cardiovascular events, venous thromboembolism, and dementia. In women with a history of breast cancer, vaginal estrogens are generally felt to be safe to use at low doses, such as less than 25 mcg twice weekly.
Dr. Wang is a geriatrician and general internist at Harborview Medical Center, Seattle.
Morning vs. afternoon exercise debate: A false dichotomy
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
Should we be exercising in the morning or afternoon? Before a meal or after a meal?
Popular media outlets, researchers, and clinicians seem to love these debates. I hate them. For me, it’s a false dichotomy. A false dichotomy is when people argue two sides as if only one option exists. A winner must be crowned, and a loser exists. But
Some but not all research suggests that morning fasted exercise may be the best time of day and condition to work out for weight control and training adaptations. Morning exercise may be a bit better for logistical reasons if you like to get up early. Some of us are indeed early chronotypes who rise early, get as much done as we can, including all our fitness and work-related activities, and then head to bed early (for me that is about 10 PM). Getting an early morning workout seems to fit with our schedules as morning larks.
But if you are a late-day chronotype, early exercise may not be in sync with your low morning energy levels or your preference for leisure-time activities later in the day. And lots of people with diabetes prefer to eat and then exercise. Late chronotypes are less physically active in general, compared with early chronotypes, and those who train in the morning tend to have better training adherence and expend more energy overall throughout the day. According to Dr. Normand Boulé from the University of Alberta, Edmonton, who presented on the topic of exercise time of day at the recent scientific sessions of the American Diabetes Association in San Diego, morning exercise in the fasted state tends to be associated with higher rates of fat oxidation, better weight control, and better skeletal muscle adaptations over time, compared with exercise performed later in the day. Dr Boulé also proposed that fasted exercise might be superior for training adaptations and long-term glycemia if you have type 2 diabetes.
But the argument for morning-only exercise falls short when we look specifically at postmeal glycemia, according to Dr. Jenna Gillen from the University of Toronto, who faced off against Dr. Boulé at a debate at the meeting and also publishes on the topic. She pointed out that mild to moderate intensity exercising done soon after meals typically results in fewer glucose spikes after meals in people with diabetes, and her argument is supported by at least one recent meta-analysis where postmeal walking was best for improving glycemia in those with prediabetes and type 2 diabetes.
The notion that postmeal or afternoon exercise is best for people with type 2 diabetes is also supported by a recent reexamination of the original Look AHEAD Trial of over 2,400 adults with type 2 diabetes, wherein the role of lifestyle intervention on cardiovascular outcomes was the original goal. In this recent secondary analysis of the Look AHEAD Trial, those most active in the afternoon (between 1:43 p.m. and 5:00 p.m.) had the greatest improvements in their overall glucose control after 1 year of the intensive lifestyle intervention, compared with exercise at other times of day. Afternoon exercisers were also more likely to have complete “remission” of their diabetes, as defined by no longer needing any glucose-lowering agents to control their glucose levels. But this was not a study that was designed for determining whether exercise time of day matters for glycemia because the participants were not randomly assigned to a set time of day for their activity, and glycemic control was not the primary endpoint (cardiovascular events were).
But hold on a minute. I said this was a false-dichotomy argument. It is. Just because it may or may not be “better” for your glucose to exercise in the morning vs. afternoon, if you have diabetes, it doesn’t mean you have to choose one or the other. You could choose neither (okay, that’s bad), both, or you could alternate between the two. For me this argument is like saying; “There only one time of day to save money”; “to tell a joke”; “to eat a meal” (okay, that’s another useless debate); or “do my laundry” (my mother once told me it’s technically cheaper after 6 p.m.!).
I live with diabetes, and I take insulin. I like how morning exercise in the form of a run with my dog wakes me up, sets me up for the day with positive thoughts, helps generate lots of creative ideas, and perhaps more importantly for me, it tends not to result in hypoglycemia because my insulin on board is lowest then.
Exercise later in the day is tricky when taking insulin because it tends to result in a higher insulin “potency effect” with prandial insulins. However, I still like midday activity and late-day exercise. For example, taking an activity break after lunch blunts the rise in my glucose and breaks up my prolonged sitting time in the office. After-dinner exercise allows me to spend a little more time with my wife, dog, or friends outdoors as the hot summer day begins to cool off. On Monday nights, I play basketball because that’s the only time we can book the gymnasium and that may not end until 9:45 p.m. (15 minutes before I want to go to bed; if you remember, I am a lark). That can result in two frustrating things related to my diabetes: It can cause an immediate rise in my glucose because of a competitive stress response and then a drop in my glucose overnight when I’m sleeping. But I still do it. I know that the training I’m doing at any point of the day will benefit me in lots of little ways, and I think we all need to take as many opportunities to be physically active as we possibly can. My kids and I coin this our daily “fitness opportunities,” and it does not matter to me if its morning, noon, or night!
It’s time to make the headlines and arguments stop. There is no wrong time of day to exercise. At least not in my opinion.
Dr. Riddle is a full professor in the school of kinesiology and health science at York University and senior scientist at LMC Diabetes & Endocrinology, both in Toronto. He has disclosed financial relationships with Dexcom, Eli Lilly, Indigo Diabetes, Insulet, Novo Nordisk, Sanofi, Supersapiens, and Zucara Therapeutics.
A version of this article first appeared on Medscape.com.
What can you do during a mass shooting? This MD found out
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
Sunday night. Las Vegas. Jason Aldean had just started playing.
My wife and I were at the 2017 Route 91 Harvest Festival with three other couples; two of them were our close friends. We were sitting in the VIP section, a tented area right next to the stage. We started hearing what I was convinced were fireworks.
I’ve been in the Army for 20 some years. I’ve been deployed and shot at multiple times. But these shots were far away. And you don’t expect people to be shooting at you at a concert.
I was on the edge of the VIP area, so I could see around the corner of the tent. I looked up at the Mandalay Bay and saw the muzzle flash in the hotel window. That’s when I knew.
I screamed: “Somebody’s shooting at us! Everybody get down!”
It took a while for people to realize what was going on. When the first couple volleys sprayed into the crowd, nobody understood. But once enough people had been hit and dropped, everyone knew, and it was just mass exodus.
People screamed and ran everywhere. Some of them tried to jump over the front barrier so they could get underneath the stage. Others were trying to pick up loved ones who’d been shot.
The next 15 minutes are a little foggy. I was helping my wife and the people around us to get down. Funny things come back to you afterward. One of my friends was carrying a 16-ounce beer in his hand. Somebody’s shooting at him and he’s walking around with his beer like he’s afraid to put it down. It was so surreal.
We got everybody underneath the tent, and then we just sat there. There would be shooting and then a pause. You’d think it was over. And then there would be more shooting and another pause. It felt like it never was going to stop.
After a short period of time, somebody came in with an official badge, maybe FBI, who knows. They said: “Okay, everybody up. We’ve got to get you out of here.” So, we all got up and headed across the stage. The gate they were taking us to was in full view of the shooter, so it wasn’t very safe.
As I got up, I looked out at the field. Bodies were scattered everywhere. I’m a trauma surgeon by trade. I couldn’t just leave.
I told my two best friends to take my wife with them. My wife lost her mind at that point. She didn’t want me to run out on the field. But I had to. I saw the injured and they needed help. Another buddy and I jumped over the fence and started taking care of people.
The feeling of being out on the field was one of complete frustration. I was in sandals, shorts, and a t-shirt. We had no stretchers, no medical supplies, no nothing. I didn’t have a belt to use as a tourniquet. I didn’t even have a bandage.
Worse: We were seeing high-velocity gunshot wounds that I’ve seen for 20 years in the Army. I know how to take care of them. I know how to fix them. But there wasn’t a single thing I could do.
We had to get people off the field, so we started gathering up as many as we could. We didn’t know if we were going to get shot at again, so we were trying to hide behind things as we ran. Our main objective was just to get people to a place of safety.
A lot of it is a blur. But a few patients stick out in my mind.
A father and son. The father had been shot through the abdomen, exited out through his back. He was in severe pain and couldn’t walk.
A young girl shot in the arm. Her parents carrying her.
A group of people doing CPR on a young lady. She had a gunshot wound to the head or neck. She was obviously dead. But they were still doing chest compressions in the middle of the field. I had to say to them: “She’s dead. You can’t save her. You need to get off the field.” But they wouldn’t stop. We picked her up and took her out while they continued to do CPR.
Later, I realized I knew that woman. She was part of a group of friends that we would see at the festival. I hadn’t recognized her. I also didn’t know that my friend Marco was there. A month or 2 later, we figured out that he was one of the people doing CPR. And I was the guy who came up and said his friend was dead.
Some people were so badly injured we couldn’t lift them. We started tearing apart the fencing used to separate the crowd and slid sections of the barricades under the wounded to carry them. We also carried off a bunch of people who were dead.
We were moving patients to a covered bar area where we thought they would be safer. What we didn’t know was there was an ambulance rally point at the very far end of the field. Unfortunately, we had no idea it was there.
I saw a lot of other first responders out there, people from the fire department, corpsmen from the Navy, medics. I ran into an anesthesia provider and a series of nurses.
When we got everybody off the field, we started moving them into vehicles. People were bringing their trucks up. One guy even stole a truck so he could drive people to the ED. There wasn’t a lot of triage. We were just stacking whoever we could into the backs of these pickups.
I tried to help a nurse taking care of a lady who had been shot in the neck. She was sitting sort of half upright with the patient lying in her arms. When I reached to help her, she said: “You can’t move her.”
“We need to get her to the hospital,” I replied.
“This is the only position that this lady has an airway,” she said. “You’re going to have to move both of us together. If I move at all, she loses her airway.”
So, a group of us managed to slide something underneath and lift them into the back of a truck.
Loading the wounded went on for a while. And then, just like that, everybody was gone.
I walked back out onto this field which not too long ago held 30,000 people. It was as if aliens had just suddenly beamed everyone out.
There was stuff on the ground everywhere – blankets, clothing, single boots, wallets, purses. I walked past a food stand with food still cooking on the grill. There was a beer tap still running. It was the weirdest feeling I’d ever had in my life.
After that, things got a little crazy again. There had been a report of a second shooter, and no one knew if it was real or not. The police started herding a group of us across the street to the Tropicana. We were still trying to take cover as we walked there. We went past a big lion statue in front of one of the casinos. I have a picture from two years earlier of me sitting on the back of that lion. I remember thinking: Now I’m hunkered down behind the same lion hiding from a shooter. Times change.
They brought about 50 of us into a food court, which was closed. They wouldn’t tell us what was going on. And they wouldn’t let us leave. This went on for hours. Meanwhile, I had dropped my cell phone on the field, so my wife couldn’t get hold of me, and later she told me she assumed I’d been shot. I was just hoping that she was safe.
People were huddled together, crying, holding each other. Most were wearing Western concert–going stuff, which for a lot of them wasn’t very much clothing. The hotel eventually brought some blankets.
I was covered in blood. My shirt, shorts, and sandals were soaked. It was running down my legs. I couldn’t find anything to eat or drink. At one point, I sat down at a slot machine, put a hundred dollars in, and started playing slots. I didn’t know what else to do. It didn’t take me very long to lose it all.
Finally, I started looking for a way to get out. I checked all the exits, but there were security and police there. Then I ran into a guy who said he had found a fire exit. When we opened the fire door, there was a big security guard there, and he said: “You can’t leave.”
We said: “Try to stop us. We’re out of here.”
Another thing I’ll always remember – after I broke out of the Tropicana, I was low crawling through the bushes along the Strip toward my hotel. I got a block away and stood up to cross the street. I pushed the crosswalk button and waited. There were no cars, no people. I’ve just broken all the rules, violated police orders, and now I’m standing there waiting for a blinking light to allow me to cross the street!
I made it back to my hotel room around 3:30 or 4:00 in the morning. My wife was hysterical because I hadn’t been answering my cell phone. I came in, and she gave me a big hug, and I got in the shower. Our plane was leaving in a few hours, so we laid down, but didn’t sleep.
As we were getting ready to leave, my wife’s phone rang, and it was my number. A guy at the same hotel had found my phone on the field and called the “in case of emergency” number. So, I got my phone back.
It wasn’t easy to deal with the aftermath. It really affected everybody’s life. To this day, I’m particular about where we sit at concerts. My wife isn’t comfortable if she can’t see an exit. I now have a med bag in my car with tourniquets, pressure dressings, airway masks for CPR.
I’ll never forget that feeling of absolute frustration. That lady without an airway – I could’ve put a trach in her very quickly and made a difference. Were they able to keep her airway? Did she live?
The father and son – did the father make it? I have no idea what happened to any of them. Later, I went through and looked at the pictures of all the people who had died, but I couldn’t recognize anybody.
The hardest part was being there with my wife. I’ve been in places where people are shooting at you, in vehicles that are getting bombed. I’ve always believed that when it’s your time, it’s your time. If I get shot, well, okay, that happens. But if she got shot or my friends ... that would be really tough.
A year later, I gave a talk about it at a conference. I thought I had worked through everything. But all of those feelings, all of that helplessness, that anger, everything came roaring back to the surface again. They asked me how I deal with it, and I said: “Well ... poorly.” I’m the guy who sticks it in a box in the back of his brain, tucks it in and buries it with a bunch of other boxes, and hopes it never comes out again. But every once in a while, it does.
There were all kinds of people out on that field, some with medical training, some without, all determined to help, trying to get those injured people where they needed to be. In retrospect, it does make you feel good. Somebody was shooting at us, but people were still willing to stand up and risk their lives to help others.
We still talk with our friends about what happened that night. Over the years, it’s become less and less. But there’s still a text sent out every year on that day: “Today is the anniversary. Glad we’re all alive. Thanks for being our friends.”
Dr. Sebesta is a bariatric surgeon with MultiCare Health System in Tacoma, Wash.
A version of this article first appeared on Medscape.com.
Two historical events that changed the field of gastroenterology

The first event took place in 1822 at Fort Mackinac, which today is known as Mackinac Island on northern Lake Huron in Michigan. Alexis St. Martin, a French-Canadian fur trapper, was standing outside of the general store when a shotgun blast accidentally struck him in the stomach. Ordinarily, this would have been a fatal wound, but St. Martin miraculously survived--but with a gastric fistula that permanently exposed the interior of his stomach.
William Beaumont, the post surgeon at Fort Mackinac, engaged in a series of experiments – purportedly 238 – to study human digestion. In one experiment, Dr. Beaumont would pull food in and out of the stomach to study digestion. In another, he would withdraw fluid from the stomach to observe digestion outside of the body. The experiments caused St. Martin considerable discomfort. He eventually returned to Canada, but returned later when the U.S. Army agreed to compensate him for some of his expenses. Today, the experiments would be called into question as having crossed ethical boundaries. Dr. Beaumont published the results from his experiments in a book that established the fundamental basics of our current beliefs about digestion. The experiments arguably mark the first example of gastrointestinal research in the United States.
The second historical event – the invention of the fiber-optic endoscope – also occurred in Michigan. At the University of Michigan, Basil Hirschowitz, MD, invented a flexible, fiber-optic instrument that could be used to look into the stomach, and perhaps even the duodenum. He first tried the invention on himself, and in 1957, he demonstrated it at the national meeting of the American Gastroscopic Society by reading a telephone directory through the new device.
The instrument was soon adopted for clinical use by physicians. Whether the fiber-optic machine was superior for visualizing the stomach was hotly debated, but what was very clear was that the fiber-optic tool was more comfortable for patients. By the mid-1960s, the fiber-optic invention had become the instrument of choice for gastrointestinal endoscopy. Many advances have since been made to the original instrument.
Dr. Howell is the Elizabeth Farrand Professor and a professor of internal medicine, history, and health management and policy at the University of Michigan, Ann Arbor. He has no financial disclosures.
The first event took place in 1822 at Fort Mackinac, which today is known as Mackinac Island on northern Lake Huron in Michigan. Alexis St. Martin, a French-Canadian fur trapper, was standing outside of the general store when a shotgun blast accidentally struck him in the stomach. Ordinarily, this would have been a fatal wound, but St. Martin miraculously survived--but with a gastric fistula that permanently exposed the interior of his stomach.
William Beaumont, the post surgeon at Fort Mackinac, engaged in a series of experiments – purportedly 238 – to study human digestion. In one experiment, Dr. Beaumont would pull food in and out of the stomach to study digestion. In another, he would withdraw fluid from the stomach to observe digestion outside of the body. The experiments caused St. Martin considerable discomfort. He eventually returned to Canada, but returned later when the U.S. Army agreed to compensate him for some of his expenses. Today, the experiments would be called into question as having crossed ethical boundaries. Dr. Beaumont published the results from his experiments in a book that established the fundamental basics of our current beliefs about digestion. The experiments arguably mark the first example of gastrointestinal research in the United States.
The second historical event – the invention of the fiber-optic endoscope – also occurred in Michigan. At the University of Michigan, Basil Hirschowitz, MD, invented a flexible, fiber-optic instrument that could be used to look into the stomach, and perhaps even the duodenum. He first tried the invention on himself, and in 1957, he demonstrated it at the national meeting of the American Gastroscopic Society by reading a telephone directory through the new device.
The instrument was soon adopted for clinical use by physicians. Whether the fiber-optic machine was superior for visualizing the stomach was hotly debated, but what was very clear was that the fiber-optic tool was more comfortable for patients. By the mid-1960s, the fiber-optic invention had become the instrument of choice for gastrointestinal endoscopy. Many advances have since been made to the original instrument.
Dr. Howell is the Elizabeth Farrand Professor and a professor of internal medicine, history, and health management and policy at the University of Michigan, Ann Arbor. He has no financial disclosures.
The first event took place in 1822 at Fort Mackinac, which today is known as Mackinac Island on northern Lake Huron in Michigan. Alexis St. Martin, a French-Canadian fur trapper, was standing outside of the general store when a shotgun blast accidentally struck him in the stomach. Ordinarily, this would have been a fatal wound, but St. Martin miraculously survived--but with a gastric fistula that permanently exposed the interior of his stomach.
William Beaumont, the post surgeon at Fort Mackinac, engaged in a series of experiments – purportedly 238 – to study human digestion. In one experiment, Dr. Beaumont would pull food in and out of the stomach to study digestion. In another, he would withdraw fluid from the stomach to observe digestion outside of the body. The experiments caused St. Martin considerable discomfort. He eventually returned to Canada, but returned later when the U.S. Army agreed to compensate him for some of his expenses. Today, the experiments would be called into question as having crossed ethical boundaries. Dr. Beaumont published the results from his experiments in a book that established the fundamental basics of our current beliefs about digestion. The experiments arguably mark the first example of gastrointestinal research in the United States.
The second historical event – the invention of the fiber-optic endoscope – also occurred in Michigan. At the University of Michigan, Basil Hirschowitz, MD, invented a flexible, fiber-optic instrument that could be used to look into the stomach, and perhaps even the duodenum. He first tried the invention on himself, and in 1957, he demonstrated it at the national meeting of the American Gastroscopic Society by reading a telephone directory through the new device.
The instrument was soon adopted for clinical use by physicians. Whether the fiber-optic machine was superior for visualizing the stomach was hotly debated, but what was very clear was that the fiber-optic tool was more comfortable for patients. By the mid-1960s, the fiber-optic invention had become the instrument of choice for gastrointestinal endoscopy. Many advances have since been made to the original instrument.
Dr. Howell is the Elizabeth Farrand Professor and a professor of internal medicine, history, and health management and policy at the University of Michigan, Ann Arbor. He has no financial disclosures.
A 75-year-old White woman presented with diffuse erythema, scale, and pruritus on her scalp
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.

Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The classical presentation includes symmetric proximal muscle weakness and underlying malignancy and is very common in adult patients. The etiology is unknown, however.
Some studies suggest people with certain HLA subtypes are at higher risk, and various infectious and pharmacological triggers are suspected to play a role in the pathogenesis of dermatomyositis. Infectious causes include Coxsackie B, enterovirus, and parvovirus. Drugs such as antineoplastic agents, antibiotics, and NSAIDs have been found to be triggers.
The pathogenesis of dermatomyositis involves immune-mediated damage to muscle capillaries and the endothelium of arterioles. In the typical humoral immune response, complement activation occurs. One mechanism of damage in dermatomyositis occurs when the membrane attack complex formed at the end of the complement process deposits in blood vessels, causing inflammation. B cells, autoantibodies, and interferon overexpression may also play a role in damaging the vasculature and muscle fibers. Hypoxia leads to muscular atrophy, resulting in degeneration and death of the fibers. On muscle biopsy, a perivascular and perimysial inflammatory infiltrate, perifascicular atrophy, and microangiopathy may be present. Skin histology reveals vacuolar changes in the basal layer, a lymphocytic infiltrate, and increased mucin production in the dermis.
On clinical examination, patients will have proximal muscle weakness and a skin rash that may include Gottron’s papules, heliotrope erythema, V-sign, shawl sign, holster sign, scalp erythema, midfacial erythema, and photosensitivity. Scalp erythema in dermatomyositis is highly linked to pruritus, alopecia, and telogen effluvium. Patients may experience small fiber neuropathy in dermatomyositis.
Serologies for this patient, who had previously been diagnosed and treated for dermatomyositis, were significant for a positive ANA 1:2560. Anti-Jo-1 antibody was negative. Her liver function tests, aldolase, creatinine kinase, sedimentation rate, C-reactive protein, and serum protein electrophoresis were normal. Imaging revealed mild chronic interstitial lung disease. A malignancy workup was negative.
Treatment of dermatomyositis involves lifestyle changes and pharmacologic therapy. Because of the intense photosensitivity, patients should be diligent with their sun protection. Methotrexate, azathioprine, and mycophenolate mofetil are considered first-line therapies for dermatomyositis. Therapies such as cyclophosphamide, rituximab, IVIg, and plasmapheresis may also be indicated in severe or refractory cases. Additionally, patients with pulmonary involvement should be given systemic steroids. The side effects of these drugs must be considered in the context of the patient’s demographics, comorbidities and lifestyle.
This case and the photos were submitted by Lucas Shapiro, BS, of Nova Southeastern University College of Osteopathic Medicine, Fort Lauderdale, Fla., and Natalie Y. Nasser, MD, of Kaiser Permanente Riverside Medical Center, Riverside, Calif. The column was edited by Dr. Bilu Martin.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
References
1. Qudsiya Z and Waseem M. Dermatomyositis, in “StatPearls.” Treasure Island, Fla.: StatPearls Publishing, 2023 Jan.
2. Kamperman RG et al. Int J Mol Sci. 2022 Apr 13;23(8):4301.
3. Kassamali B et al. Int J WomensDermatol. 2021 Sep 24;7(5Part A):576-82.
4. Vázquez-Herrera NE et al. Skin Appendage Disord. 2018 Aug;4(3):187-99.
The good, bad, and ugly of direct-to-consumer advertising
Case 1: A 48-year-old female with a 10-year history of left-sided Ulcerative Colitis (UC) has been well controlled on an injectable biologic for 5 years. Her last colonoscopy one year ago was normal without erythema or friability. She presents for an interim visit due to an increase in stool frequency from 2 to 4 bowel movements a day. She denies urgency, nocturnal bowel movements, or blood in her stool. She is concerned about a disease flare and wonders if she is on the right medication. She just saw a TV ad for a new oral UC medication and wants to switch because she prefers an oral medication to an injectable one. Physical exam was normal and in-office flexible sigmoidoscopy demonstrates no change in her colon appearance. You advise her to stay on the biologic because she is still considered well-controlled. She insists on being switched to the new oral medicine. When you probe her more, she says that the TV ad she saw shows people getting the medicine leading normal lives, which has enormous appeal to her.
Case 2: A 52-year-old healthy male is referred for colonoscopy evaluation. He reports no change in bowel habits with rare blood in his stool and thinks his uncle had colon cancer at age 48. He is anxious and not very receptive to having a procedure. He recently saw a TV advertisement promoting non-colonoscopy-based colon cancer screening. You recommend a colonoscopy based on his family history, but he insists on stool-based screening.

Case 3: A 32-year-old female with moderately to well-controlled IBD asks you to review a new “multi-omic” profile of her gut microbiome that she saw advertised on social media. The test report she provides contains a snapshot of her microbiome including abundances of single species and predicted functions for these bacteria from a single stool sample collected and submitted 6 months ago. You counsel her on the role of the gut microbiome in IBD and explain that currently there is not enough knowledge or technology to incorporate these test results into clinical care yet. The patient is frustrated and wants to know why you’re “behind the times.”
These cases may sound familiar to a practicing gastroenterologist. The platform driving all three of these clinical encounters, direct-to-consumer advertising (DTCA), is a legal mechanism by which a commercial entity can communicate directly with the consumer about a medicine or device, bypassing a health care professional.
In the 1960s, Congress granted the Food and Drug Administration regulatory authority over prescription drug labeling and advertising. This included ensuring that ads (1) were not false or misleading, (2) presented a “fair balance” of drug risks and benefits, (3) included facts “material” to a drug’s advertised uses, and (4) included a “brief summary” of all risks described in the drug’s labeling.
Direct-to-consumer advertising increased dramatically in the late 1990s after the FDA eased regulations around risk information by requiring ads to include only “major risks” and provide resources directing consumers to full risk information.
In 2022, the top 10 pharmaceutical ad spenders combined for a total of about $1.7 billion in TV ad spend, with the two top categories being inflammation and diabetes.
The role of the FDA in regulating DTCA of at-home tests is still evolving and largely depends on the intended use of the test results and the health claims used to market the test (that is, whether the test is designed to simply return general information, as in case 3 where DTCA regulations may not apply, or is marketed with specific medical claims or diagnostic interpretations, as in case 2 with clear applications for DTCA regulations.).
It has both potential benefits and potential risks. DTCA can serve to increase disease awareness (e.g., the need for colon cancer screening). It may also prompt patients who might otherwise disregard “red flag” signs and symptoms to seek medical evaluation (e.g., rectal bleeding). DTCA can also alert healthcare providers to new treatment options for diseases within their scope of practice and encourage them to expand their armamentarium.

In bioethics terms, DTCA can be beneficial in facilitating patient autonomy and promoting justice. For example, DTCA can “even the playing field” by ensuring that patients have equitable access to information about available treatments regardless of their socioeconomic status. In doing so, it can empower patients and allow them to have more meaningful discussions with their health care providers and make more informed decisions. In addition, patients may be introduced to alternative testing modalities (i.e., stool-based CRC screening) that, while not necessarily the best screening modality given individual risk (as in Case 2), may offer benefit with greater acceptance compared to inaction (i.e., no screening). Last, the idea of direct-to-consumer “omics” profiling has empowered patients as “citizen scientists” and led to the advent of “biohacking” among the lay population. In doing so, it has challenged the previous bounds of patient autonomy in healthcare by broadening the types of personal health data available to individuals, even when the clinical utility of this data may not yet be clear.
On the flip side, it is undeniable that DTCA of medical products is driven by commercial interests. Branded drugs are primarily, if not exclusively, promoted in DTCA, but these drugs may not always align with standard of care treatment recommendations. A study published in February 2023 in JAMA found that drugs with lower added clinical benefit and higher total sales were associated with higher DTCA spending.
With patients entering medical encounters with preconceived notions of what drugs they want to be prescribed based on media exposure, the ability of health care providers to provide sound medical advice regarding treatment may be circumvented. A patient’s preferred therapy based on exposure to DTCA may sharply contrast with their provider’s recommendation based on their experience and expertise and knowledge of the patient’s unique clinical history.
Unreasonable expectations
An additional potential downside of DTCA is that it can instill unreasonable expectations on the part of patients that the advertised product will benefit them. While DTCA is required to be fair and balanced in reporting benefits and risks, it is difficult to meaningfully address nuanced clinical risks in a brief TV ad and patients may come away with a skewed view of the risk-benefit equation. Furthermore, social media advertising and associated formats may not provide the same level of digestible information as other forms of media and are targeted to individuals likely to identify with the product. Finally, as stated above, only branded products (vs. generics) are advertised. Branded products are generally more costly, and where less expensive and equally effective therapies exist societal costs need to be considered. This can lead to inequities in distributive justice which is the societal allocation of resources. The more the healthcare market is driven towards higher costs in one segment, the less resources are available in another. This may affect regions where healthcare resources are limited.
Shared decision-making
Returning to the 3 cases above, in case 1 the UC patient’s awareness of new treatment options has prompted a shared decision-making discussion. She has a renewed interest in exploring a different route of medication administration because of DTCA. In spite of the patient seemingly well-controlled on her current IBD therapy and minor fluctuations in symptoms that might otherwise be a reason to observe more closely, the patient sees this as a reason to change her treatment based on her impression from the DTCA.
Regarding case 2, disease awareness and CRC screening acceptance is itself a positive outcome. Although commercially driven, the outcome/benefit to society leads to a decrease in disease burden and is a ready alternative with established benefits compared to no screening.
Regarding the proactive “omics-curious” IBD patient in case 3, despite the patient’s disappointment with the DCTA-promoted test’s limited clinical utility at this time, the patient may be communicating a general openness to novel approaches to treating IBD and to advancing her understanding of her disease.
So where does that leave you as a clinician?
As of today, if you live in the U.S., DTCA is a reality. As you navigate day-to-day patient interactions, it is important to keep in mind that your first obligation is to your patients and their best medical interests. Having a well-informed and engaged patient is a positive effect of DTCA over the past 30 years despite the challenging discussions you sometimes are forced to have. In many cases, patients are more self-aware of and engaged in their health and are acting on it due to direct acquisition of information from DTCA platforms. As a clinician, you have an ethical obligation to educate your patients and manage expectations, even when those expectations may be formed on the basis of DTCA with inherent conflicts of interest for promoting a product. Moreover, though certain products may be trendy or promoted on a popular science basis, the underlying technology (i.e. stool-based screening or metagenomics) and/or resultant data are likely not well understood by the typical lay patient, such that it may be difficult for a patient to comprehend how the product may not be particularly informative or inappropriate with respect to their personal medical history without additional counseling. Despite the potentially awkward clinician-patient dynamic precipitated by DTCA, these moments do offer an opportunity for you to gain rapport with your patients by taking the time to fill in the gaps of their understanding of their treatment plans, alternatives, individual risk factors and/or disease while gaining a greater appreciation for what they may personally prioritize with respect to their own health. Ultimately, as we transition further toward precision medicine approaches in healthcare, shared interest in individualized health decisions, at least partially informed by DTCA, is a positive outcome.
Dr. Sloan is a chief medical officer at Abivax. He is a member of the AGA Ethics Committee for COI. David A. Drew, PhD is assistant professor of medicine, Harvard Medical School, Boston. He is director of the Massachusetts General Hospital Biobanking, Clinical & Translational Epidemiology Unit, Boston. He is a member of the AGA Ethics Committee.
Case 1: A 48-year-old female with a 10-year history of left-sided Ulcerative Colitis (UC) has been well controlled on an injectable biologic for 5 years. Her last colonoscopy one year ago was normal without erythema or friability. She presents for an interim visit due to an increase in stool frequency from 2 to 4 bowel movements a day. She denies urgency, nocturnal bowel movements, or blood in her stool. She is concerned about a disease flare and wonders if she is on the right medication. She just saw a TV ad for a new oral UC medication and wants to switch because she prefers an oral medication to an injectable one. Physical exam was normal and in-office flexible sigmoidoscopy demonstrates no change in her colon appearance. You advise her to stay on the biologic because she is still considered well-controlled. She insists on being switched to the new oral medicine. When you probe her more, she says that the TV ad she saw shows people getting the medicine leading normal lives, which has enormous appeal to her.
Case 2: A 52-year-old healthy male is referred for colonoscopy evaluation. He reports no change in bowel habits with rare blood in his stool and thinks his uncle had colon cancer at age 48. He is anxious and not very receptive to having a procedure. He recently saw a TV advertisement promoting non-colonoscopy-based colon cancer screening. You recommend a colonoscopy based on his family history, but he insists on stool-based screening.
Case 3: A 32-year-old female with moderately to well-controlled IBD asks you to review a new “multi-omic” profile of her gut microbiome that she saw advertised on social media. The test report she provides contains a snapshot of her microbiome including abundances of single species and predicted functions for these bacteria from a single stool sample collected and submitted 6 months ago. You counsel her on the role of the gut microbiome in IBD and explain that currently there is not enough knowledge or technology to incorporate these test results into clinical care yet. The patient is frustrated and wants to know why you’re “behind the times.”
These cases may sound familiar to a practicing gastroenterologist. The platform driving all three of these clinical encounters, direct-to-consumer advertising (DTCA), is a legal mechanism by which a commercial entity can communicate directly with the consumer about a medicine or device, bypassing a health care professional.
In the 1960s, Congress granted the Food and Drug Administration regulatory authority over prescription drug labeling and advertising. This included ensuring that ads (1) were not false or misleading, (2) presented a “fair balance” of drug risks and benefits, (3) included facts “material” to a drug’s advertised uses, and (4) included a “brief summary” of all risks described in the drug’s labeling.
Direct-to-consumer advertising increased dramatically in the late 1990s after the FDA eased regulations around risk information by requiring ads to include only “major risks” and provide resources directing consumers to full risk information.
In 2022, the top 10 pharmaceutical ad spenders combined for a total of about $1.7 billion in TV ad spend, with the two top categories being inflammation and diabetes.
The role of the FDA in regulating DTCA of at-home tests is still evolving and largely depends on the intended use of the test results and the health claims used to market the test (that is, whether the test is designed to simply return general information, as in case 3 where DTCA regulations may not apply, or is marketed with specific medical claims or diagnostic interpretations, as in case 2 with clear applications for DTCA regulations.).
It has both potential benefits and potential risks. DTCA can serve to increase disease awareness (e.g., the need for colon cancer screening). It may also prompt patients who might otherwise disregard “red flag” signs and symptoms to seek medical evaluation (e.g., rectal bleeding). DTCA can also alert healthcare providers to new treatment options for diseases within their scope of practice and encourage them to expand their armamentarium.
In bioethics terms, DTCA can be beneficial in facilitating patient autonomy and promoting justice. For example, DTCA can “even the playing field” by ensuring that patients have equitable access to information about available treatments regardless of their socioeconomic status. In doing so, it can empower patients and allow them to have more meaningful discussions with their health care providers and make more informed decisions. In addition, patients may be introduced to alternative testing modalities (i.e., stool-based CRC screening) that, while not necessarily the best screening modality given individual risk (as in Case 2), may offer benefit with greater acceptance compared to inaction (i.e., no screening). Last, the idea of direct-to-consumer “omics” profiling has empowered patients as “citizen scientists” and led to the advent of “biohacking” among the lay population. In doing so, it has challenged the previous bounds of patient autonomy in healthcare by broadening the types of personal health data available to individuals, even when the clinical utility of this data may not yet be clear.
On the flip side, it is undeniable that DTCA of medical products is driven by commercial interests. Branded drugs are primarily, if not exclusively, promoted in DTCA, but these drugs may not always align with standard of care treatment recommendations. A study published in February 2023 in JAMA found that drugs with lower added clinical benefit and higher total sales were associated with higher DTCA spending.
With patients entering medical encounters with preconceived notions of what drugs they want to be prescribed based on media exposure, the ability of health care providers to provide sound medical advice regarding treatment may be circumvented. A patient’s preferred therapy based on exposure to DTCA may sharply contrast with their provider’s recommendation based on their experience and expertise and knowledge of the patient’s unique clinical history.
Unreasonable expectations
An additional potential downside of DTCA is that it can instill unreasonable expectations on the part of patients that the advertised product will benefit them. While DTCA is required to be fair and balanced in reporting benefits and risks, it is difficult to meaningfully address nuanced clinical risks in a brief TV ad and patients may come away with a skewed view of the risk-benefit equation. Furthermore, social media advertising and associated formats may not provide the same level of digestible information as other forms of media and are targeted to individuals likely to identify with the product. Finally, as stated above, only branded products (vs. generics) are advertised. Branded products are generally more costly, and where less expensive and equally effective therapies exist societal costs need to be considered. This can lead to inequities in distributive justice which is the societal allocation of resources. The more the healthcare market is driven towards higher costs in one segment, the less resources are available in another. This may affect regions where healthcare resources are limited.
Shared decision-making
Returning to the 3 cases above, in case 1 the UC patient’s awareness of new treatment options has prompted a shared decision-making discussion. She has a renewed interest in exploring a different route of medication administration because of DTCA. In spite of the patient seemingly well-controlled on her current IBD therapy and minor fluctuations in symptoms that might otherwise be a reason to observe more closely, the patient sees this as a reason to change her treatment based on her impression from the DTCA.
Regarding case 2, disease awareness and CRC screening acceptance is itself a positive outcome. Although commercially driven, the outcome/benefit to society leads to a decrease in disease burden and is a ready alternative with established benefits compared to no screening.
Regarding the proactive “omics-curious” IBD patient in case 3, despite the patient’s disappointment with the DCTA-promoted test’s limited clinical utility at this time, the patient may be communicating a general openness to novel approaches to treating IBD and to advancing her understanding of her disease.
So where does that leave you as a clinician?
As of today, if you live in the U.S., DTCA is a reality. As you navigate day-to-day patient interactions, it is important to keep in mind that your first obligation is to your patients and their best medical interests. Having a well-informed and engaged patient is a positive effect of DTCA over the past 30 years despite the challenging discussions you sometimes are forced to have. In many cases, patients are more self-aware of and engaged in their health and are acting on it due to direct acquisition of information from DTCA platforms. As a clinician, you have an ethical obligation to educate your patients and manage expectations, even when those expectations may be formed on the basis of DTCA with inherent conflicts of interest for promoting a product. Moreover, though certain products may be trendy or promoted on a popular science basis, the underlying technology (i.e. stool-based screening or metagenomics) and/or resultant data are likely not well understood by the typical lay patient, such that it may be difficult for a patient to comprehend how the product may not be particularly informative or inappropriate with respect to their personal medical history without additional counseling. Despite the potentially awkward clinician-patient dynamic precipitated by DTCA, these moments do offer an opportunity for you to gain rapport with your patients by taking the time to fill in the gaps of their understanding of their treatment plans, alternatives, individual risk factors and/or disease while gaining a greater appreciation for what they may personally prioritize with respect to their own health. Ultimately, as we transition further toward precision medicine approaches in healthcare, shared interest in individualized health decisions, at least partially informed by DTCA, is a positive outcome.
Dr. Sloan is a chief medical officer at Abivax. He is a member of the AGA Ethics Committee for COI. David A. Drew, PhD is assistant professor of medicine, Harvard Medical School, Boston. He is director of the Massachusetts General Hospital Biobanking, Clinical & Translational Epidemiology Unit, Boston. He is a member of the AGA Ethics Committee.
Case 1: A 48-year-old female with a 10-year history of left-sided Ulcerative Colitis (UC) has been well controlled on an injectable biologic for 5 years. Her last colonoscopy one year ago was normal without erythema or friability. She presents for an interim visit due to an increase in stool frequency from 2 to 4 bowel movements a day. She denies urgency, nocturnal bowel movements, or blood in her stool. She is concerned about a disease flare and wonders if she is on the right medication. She just saw a TV ad for a new oral UC medication and wants to switch because she prefers an oral medication to an injectable one. Physical exam was normal and in-office flexible sigmoidoscopy demonstrates no change in her colon appearance. You advise her to stay on the biologic because she is still considered well-controlled. She insists on being switched to the new oral medicine. When you probe her more, she says that the TV ad she saw shows people getting the medicine leading normal lives, which has enormous appeal to her.
Case 2: A 52-year-old healthy male is referred for colonoscopy evaluation. He reports no change in bowel habits with rare blood in his stool and thinks his uncle had colon cancer at age 48. He is anxious and not very receptive to having a procedure. He recently saw a TV advertisement promoting non-colonoscopy-based colon cancer screening. You recommend a colonoscopy based on his family history, but he insists on stool-based screening.
Case 3: A 32-year-old female with moderately to well-controlled IBD asks you to review a new “multi-omic” profile of her gut microbiome that she saw advertised on social media. The test report she provides contains a snapshot of her microbiome including abundances of single species and predicted functions for these bacteria from a single stool sample collected and submitted 6 months ago. You counsel her on the role of the gut microbiome in IBD and explain that currently there is not enough knowledge or technology to incorporate these test results into clinical care yet. The patient is frustrated and wants to know why you’re “behind the times.”
These cases may sound familiar to a practicing gastroenterologist. The platform driving all three of these clinical encounters, direct-to-consumer advertising (DTCA), is a legal mechanism by which a commercial entity can communicate directly with the consumer about a medicine or device, bypassing a health care professional.
In the 1960s, Congress granted the Food and Drug Administration regulatory authority over prescription drug labeling and advertising. This included ensuring that ads (1) were not false or misleading, (2) presented a “fair balance” of drug risks and benefits, (3) included facts “material” to a drug’s advertised uses, and (4) included a “brief summary” of all risks described in the drug’s labeling.
Direct-to-consumer advertising increased dramatically in the late 1990s after the FDA eased regulations around risk information by requiring ads to include only “major risks” and provide resources directing consumers to full risk information.
In 2022, the top 10 pharmaceutical ad spenders combined for a total of about $1.7 billion in TV ad spend, with the two top categories being inflammation and diabetes.
The role of the FDA in regulating DTCA of at-home tests is still evolving and largely depends on the intended use of the test results and the health claims used to market the test (that is, whether the test is designed to simply return general information, as in case 3 where DTCA regulations may not apply, or is marketed with specific medical claims or diagnostic interpretations, as in case 2 with clear applications for DTCA regulations.).
It has both potential benefits and potential risks. DTCA can serve to increase disease awareness (e.g., the need for colon cancer screening). It may also prompt patients who might otherwise disregard “red flag” signs and symptoms to seek medical evaluation (e.g., rectal bleeding). DTCA can also alert healthcare providers to new treatment options for diseases within their scope of practice and encourage them to expand their armamentarium.
In bioethics terms, DTCA can be beneficial in facilitating patient autonomy and promoting justice. For example, DTCA can “even the playing field” by ensuring that patients have equitable access to information about available treatments regardless of their socioeconomic status. In doing so, it can empower patients and allow them to have more meaningful discussions with their health care providers and make more informed decisions. In addition, patients may be introduced to alternative testing modalities (i.e., stool-based CRC screening) that, while not necessarily the best screening modality given individual risk (as in Case 2), may offer benefit with greater acceptance compared to inaction (i.e., no screening). Last, the idea of direct-to-consumer “omics” profiling has empowered patients as “citizen scientists” and led to the advent of “biohacking” among the lay population. In doing so, it has challenged the previous bounds of patient autonomy in healthcare by broadening the types of personal health data available to individuals, even when the clinical utility of this data may not yet be clear.
On the flip side, it is undeniable that DTCA of medical products is driven by commercial interests. Branded drugs are primarily, if not exclusively, promoted in DTCA, but these drugs may not always align with standard of care treatment recommendations. A study published in February 2023 in JAMA found that drugs with lower added clinical benefit and higher total sales were associated with higher DTCA spending.
With patients entering medical encounters with preconceived notions of what drugs they want to be prescribed based on media exposure, the ability of health care providers to provide sound medical advice regarding treatment may be circumvented. A patient’s preferred therapy based on exposure to DTCA may sharply contrast with their provider’s recommendation based on their experience and expertise and knowledge of the patient’s unique clinical history.
Unreasonable expectations
An additional potential downside of DTCA is that it can instill unreasonable expectations on the part of patients that the advertised product will benefit them. While DTCA is required to be fair and balanced in reporting benefits and risks, it is difficult to meaningfully address nuanced clinical risks in a brief TV ad and patients may come away with a skewed view of the risk-benefit equation. Furthermore, social media advertising and associated formats may not provide the same level of digestible information as other forms of media and are targeted to individuals likely to identify with the product. Finally, as stated above, only branded products (vs. generics) are advertised. Branded products are generally more costly, and where less expensive and equally effective therapies exist societal costs need to be considered. This can lead to inequities in distributive justice which is the societal allocation of resources. The more the healthcare market is driven towards higher costs in one segment, the less resources are available in another. This may affect regions where healthcare resources are limited.
Shared decision-making
Returning to the 3 cases above, in case 1 the UC patient’s awareness of new treatment options has prompted a shared decision-making discussion. She has a renewed interest in exploring a different route of medication administration because of DTCA. In spite of the patient seemingly well-controlled on her current IBD therapy and minor fluctuations in symptoms that might otherwise be a reason to observe more closely, the patient sees this as a reason to change her treatment based on her impression from the DTCA.
Regarding case 2, disease awareness and CRC screening acceptance is itself a positive outcome. Although commercially driven, the outcome/benefit to society leads to a decrease in disease burden and is a ready alternative with established benefits compared to no screening.
Regarding the proactive “omics-curious” IBD patient in case 3, despite the patient’s disappointment with the DCTA-promoted test’s limited clinical utility at this time, the patient may be communicating a general openness to novel approaches to treating IBD and to advancing her understanding of her disease.
So where does that leave you as a clinician?
As of today, if you live in the U.S., DTCA is a reality. As you navigate day-to-day patient interactions, it is important to keep in mind that your first obligation is to your patients and their best medical interests. Having a well-informed and engaged patient is a positive effect of DTCA over the past 30 years despite the challenging discussions you sometimes are forced to have. In many cases, patients are more self-aware of and engaged in their health and are acting on it due to direct acquisition of information from DTCA platforms. As a clinician, you have an ethical obligation to educate your patients and manage expectations, even when those expectations may be formed on the basis of DTCA with inherent conflicts of interest for promoting a product. Moreover, though certain products may be trendy or promoted on a popular science basis, the underlying technology (i.e. stool-based screening or metagenomics) and/or resultant data are likely not well understood by the typical lay patient, such that it may be difficult for a patient to comprehend how the product may not be particularly informative or inappropriate with respect to their personal medical history without additional counseling. Despite the potentially awkward clinician-patient dynamic precipitated by DTCA, these moments do offer an opportunity for you to gain rapport with your patients by taking the time to fill in the gaps of their understanding of their treatment plans, alternatives, individual risk factors and/or disease while gaining a greater appreciation for what they may personally prioritize with respect to their own health. Ultimately, as we transition further toward precision medicine approaches in healthcare, shared interest in individualized health decisions, at least partially informed by DTCA, is a positive outcome.
Dr. Sloan is a chief medical officer at Abivax. He is a member of the AGA Ethics Committee for COI. David A. Drew, PhD is assistant professor of medicine, Harvard Medical School, Boston. He is director of the Massachusetts General Hospital Biobanking, Clinical & Translational Epidemiology Unit, Boston. He is a member of the AGA Ethics Committee.
First-line therapy in T2D: Has metformin been ‘dethroned’?
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.
Initially approved by the U.S. Food and Drug Administration (FDA) in 1994, metformin has been the preferred first-line glucose-lowering agent for patients with type 2 diabetes (T2D) owing to its effectiveness, low hypoglycemia risk, weight neutrality, long clinical track record of safety, and affordability. However, the advent of newer glucose-lowering agents with evidence-based cardiovascular (CV) and renal benefits calls into question whether metformin should continue to be the initial pharmacotherapy for all patients with T2D.
Cardiovascular outcome trials transform standard of care
In 2008, the FDA issued guidance to industry to ensure that CV risk is more thoroughly addressed during development of T2D therapies. This guidance document required dedicated trials to establish CV safety of new glucose-lowering therapies. Findings from subsequent cardiovascular outcome trials (CVOTs) and subsequent large renal and heart failure (HF) outcome trials have since prompted frequent and substantial updates to major guidelines. On the basis of recent evidence from CVOT and renal trials, contemporary clinical practice guidelines have transitioned from a traditional glucocentric treatment approach to a holistic management approach that emphasizes organ protection through heart-kidney-metabolic risk reduction.
Per the 2008 FDA guidance, dipeptidyl peptidase-4 (DPP-4) inhibitors, glucagonlike peptide-1 (GLP-1) receptor agonists, and sodium-glucose cotransporter-2 (SGLT2) inhibitors were evaluated in large dedicated CVOTs. Findings from several CVOTs established GLP-1 receptor agonist and SGLT2 inhibitor CV safety, and unexpectedly demonstrated reduced rates of major adverse cardiovascular events (MACE) relative to placebo. The LEADER and EMPA-REG OUTCOME trials were the first CVOTs to report cardioprotective benefits of the GLP-1 receptor agonist liraglutide and the SGLT2 inhibitor empagliflozin, respectively. The LEADER trial reported a 13% significant relative risk reduction for its primary composite MACE outcome, and the EMPA-REG OUTCOME trial similarly reported a 14% relative risk reduction for MACE. After CVOTs on other GLP-1 receptor agonists and SGLT2 inhibitors reported CV benefit, clinical practice guidelines began to recommend use of these agents in at-risk patients to mitigate CV risk.
During the period when most CVOTs were designed and conducted, a majority of trial participants were receiving metformin at baseline. Inclusion of a small subset of metformin-naive participants in these trials allowed for several post hoc and meta-analyses investigating the impact of background metformin use on the overall CV benefits reported. Depending on the trial, baseline metformin use in large GLP-1 receptor agonist CVOTs ranged from 66% to 81%. For instance, 76% of participants in the LEADER trial were receiving metformin at baseline, but a post hoc analysis found no heterogeneity for the observed CV benefit based on background metformin use. Similarly, a subgroup analysis of pooled data from the SUSTAIN-6 and PIONEER 6 trials of injectable and oral formulations of semaglutide, respectively, reported similar CV outcomes for participants, regardless of concomitant metformin use. When looking at the GLP-1 receptor agonist class overall, a meta-analysis of seven CVOTs, which included participants with established atherosclerotic cardiovascular disease (ASCVD) and those with multiple ASCVD risk factors, concluded that GLP-1 receptor agonist therapy reduced the overall incidence of MACE in participants not receiving concomitant metformin at baseline.
Similar analyses have examined the impact of background metformin use on CV outcomes with SGLT2 inhibitors. An analysis of EMPA-REG OUTCOME found that empagliflozin improved CV outcomes and reduced mortality irrespective of background metformin, sulfonylurea, or insulin use. Of note, this analysis suggested a greater risk reduction for incident or worsening nephropathy in patients not on concomitant metformin (hazard ratio, 0.47; 95% confidence interval, 0.37-0.59; P = .01), when compared with those taking metformin at baseline (HR, 0.68; 95% CI, 0.58-0.79; P = .01). In addition, a meta-analysis of six large outcome trials found consistent benefits of SGLT2 inhibition on CV, kidney, and mortality outcomes regardless of background metformin treatment. Therefore, although CVOTs on GLP-1 receptor agonists and SGLT2 inhibitors were not designed to assess the impact of background metformin use on CV outcomes, available evidence supports the CV benefits of these agents independent of metformin use.
Individualizing care to attain cardiorenal-metabolic goals
Three dedicated SGLT2 inhibitor renal outcome trials have been published to date: CREDENCE, DAPA-CKD, and EMPA-KIDNEY. All three studies confirmed the positive secondary renal outcomes observed in SGLT2 inhibitor CVOTs: reduced progression of kidney disease, HF-associated hospital admissions, and CV-related death. The observed renal and CV benefits from the CREDENCE trial were consistent across different levels of kidney function. Similarly, a meta-analysis of five SGLT2 inhibitor trials of patients with HF demonstrated a decreased risk for CV-related death and admission for HF, irrespective of baseline heart function. The ongoing FLOW is the first dedicated kidney-outcome trial to evaluate the effectiveness of a GLP-1 receptor agonist (semaglutide) in slowing the progression and worsening of chronic kidney disease (CKD) in patients with T2D.
As previously noted, findings from the LEADER and EMPA-REG OUTCOME trials demonstrated the beneficial effects of GLP-1 receptor agonists and SGLT2 inhibitors not only on MACE but also on secondary HF and kidney disease outcomes. These findings have supported a series of dedicated HF and kidney outcome trials further informing the standard of care for patients with these key comorbidities. Indeed, the American Diabetes Association’s 2023 Standards of Care in Diabetes updated its recommendations and algorithm for the use of glucose-lowering medications in the management of T2D. The current ADA recommendations stress cardiorenal risk reduction while concurrently achieving and maintaining glycemic and weight management goals. On the basis of evolving outcome trial data, GLP-1 receptor agonists and SGLT2 inhibitors with evidence of benefit are recommended for patients with established or at high risk for ASCVD. Further, the Standards preferentially recommend SGLT2 inhibitors for patients with HF and/or CKD. Because evidence suggests no heterogeneity of benefit based on hemoglobin A1c for MACE outcomes with GLP-1 receptor agonists and no heterogeneity of benefit for HF or CKD benefits with SGLT2 inhibitors, these agents are recommended for cardiorenal risk reduction regardless of the need to lower glucose.
The 2023 update to the American Association of Clinical Endocrinology Consensus Statement: Type 2 Diabetes Management Algorithm similarly recommends the use of GLP-1 receptor agonists and SGLT2 inhibitors to improve cardiorenal outcomes. To further emphasize the importance of prescribing agents with proven organ-protective benefits, the AACE consensus statement provides a complications-centric algorithm to guide therapeutic decisions for risk reduction in patients with key comorbidities (for instance, ASCVD, HF, CKD) and a separate glucocentric algorithm to guide selection and intensification of glucose-lowering agents in patients without key comorbidities to meet individualized glycemic targets. Within the complications-centric algorithm, AACE recommends GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment for cardiorenal risk reduction regardless of background metformin use or A1c level.
In addition to the emphasis on the use of GLP-1 receptor agonists and SGLT2 inhibitors for organ protection, guidelines now recommend SGLT2 inhibitors as the standard-of-care therapy in patients with T2D and CKD with an estimated glomerular filtration rate ≥ 20 mL/min per 1.73 m2, and irrespective of ejection fraction or a diagnosis of diabetes in the setting of HF. Overall, a common thread within current guidelines is the importance of individualized therapy based on patient- and medication-specific factors.
Optimizing guideline-directed medical therapy
Results from the DISCOVER trial found that GLP-1 receptor agonist and SGLT2 inhibitor use was less likely in the key patient subgroups most likely to benefit from therapy, including patients with peripheral artery disease and CKD. Factors contributing to underutilization of newer cardiorenal protective glucose-lowering therapies range from cost and access barriers to clinician-level barriers (for example, lack of knowledge on CKD, lack of familiarity with CKD practice guidelines). Addressing these issues and helping patients work through financial and other access barriers is essential to optimize the utilization of these therapies and improve cardiorenal and metabolic outcomes.
So, has metformin been “dethroned” as a first-line therapy for T2D? As is often the case in medicine, the answer depends on the individual patient and clinical situation. Metformin remains an important first-line treatment in combination with lifestyle interventions to help patients with T2D without key cardiorenal comorbidities achieve individualized glycemic targets. However, based on evidence demonstrating cardiorenal protective benefits and improved glycemia and weight loss, GLP-1 agonists and SGLT2 inhibitors may be considered as first-line treatment for patients with T2D with or at high risk for ASCVD, HF, or CKD, regardless of the need for additional glucose-lowering agents and independent of background metformin. Ultimately, the choice of first-line therapy for patients with T2D should be informed by individualized treatment goals, preferences, and cost-related access. Continued efforts to increase patient access to GLP-1 receptor agonists and SGLT2 inhibitors as first-line treatment when indicated are essential to ensure optimal treatment and outcomes.
Dr. Neumiller is professor, department of pharmacotherapy, Washington State University, Spokane. He disclosed ties with Bayer, Boehringer Ingelheim, and Eli Lilly. Dr. Alicic is clinical professor, department of medicine, University of Washington; and associate director of research, Inland Northwest Washington, Providence St. Joseph Health, Spokane. She disclosed ties with Providence St. Joseph Health, Boehringer Ingelheim/Lilly, and Bayer.
A version of this article appeared on Medscape.com.