User login
Obesity Overview
Principles and Process for Reducing the Need for Insulin in Patients With Type 2 Diabetes

For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
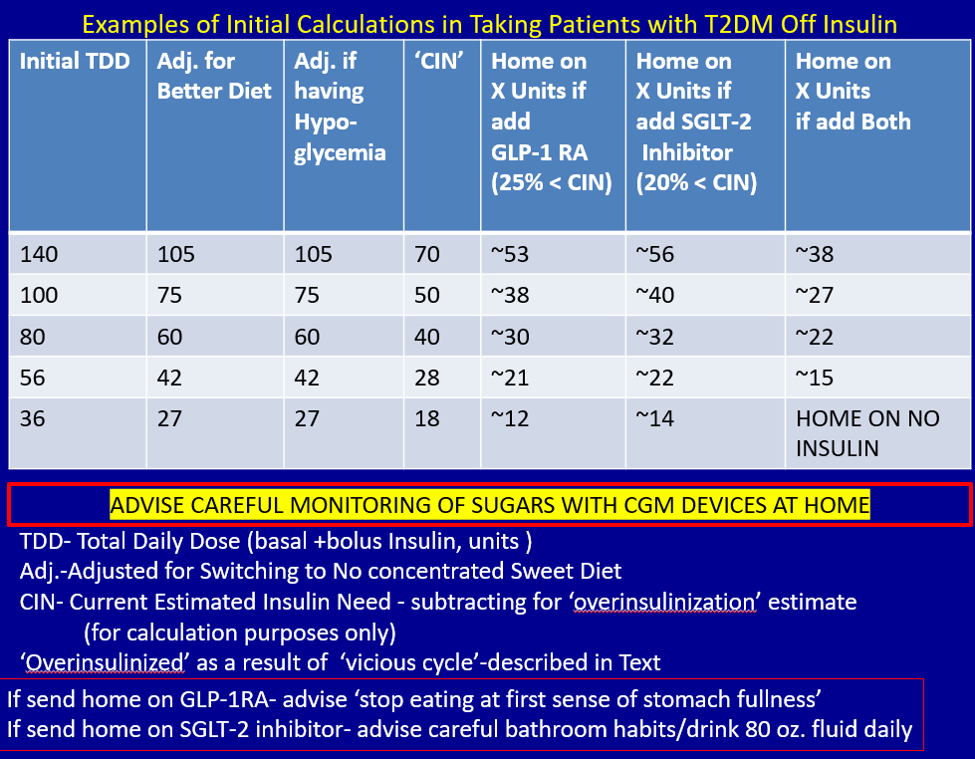
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
Feel free to contact Dr. Schwartz at [email protected] with any questions about his protocol or diet.
For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
Feel free to contact Dr. Schwartz at [email protected] with any questions about his protocol or diet.
For people living with type 2 diabetes mellitus (T2D), exogenous insulin, whether given early or later in T2D diagnosis, can provide many pharmacologically desirable effects. But it has always been clear, and is now more widely recognized, that insulin treatments are not completely risk-free for the patient. There are now newer, non-insulin therapy options that could be used, along with certain patient lifestyle changes in diet and activity levels, that have been shown to achieve desired glucose control—without the associated risks that insulin can bring.
But is it possible to markedly reduce the need for insulin in some 90% of T2D patients and to reduce the doses in the others? Yes—if patients have sufficient beta-cell function and are willing to change their lifestyle. This mode of treatment has been slowly gaining momentum as of late in the medical community because of the benefits it ultimately provides for the patient. In my practice, I personally have done this by using an evidence-based approach that includes thinking inside a larger box. It is a 2-way street, and each should drive the other: the right drugs (in the right doses), and in the right patients.
Why avoid early insulin therapy?
Is the requirement of early insulin therapy in many or most patients a myth?
Yes. It resulted from “old logic,” which was to use insulin early to reduce glucotoxicity and lipotoxicity. The American Diabetes Association guidelines recommend that glycated hemoglobin (HbA1c) should not exceed 8.0% and consider a fasting blood glucose level >250 mg/dL as high, with a need to start insulin treatment right away; other guidelines recommend initiating insulin immediately in patients with HbA1c >9% and postprandial glucose 300 mg/dL. But this was at a time when oral agents were not as effective and took time to titrate or engender good control. We now have agents that are more effective and start working right away.
However, the main problem in early insulin treatment is the significant risk of over-insulinization—a vicious cycle of insulin-caused increased appetite, hypoglycemia-resultant increased weight gain, insulin resistance (poorer control), increased circulating insulin, etc. Moreover, weight gain and individual hypoglycemic events can cause an increase in the risk of cardiovascular (CV) events.
I believe clinicians must start as early as possible in the natural history of T2D to prevent progressive beta-cell failure. Do not believe in “first-, second-, or third-line”; in other words, do not prioritize, so there is no competition between classes. The goal I have for my patients is to provide therapies that aim for the lowest HbA1c possible without hypoglycemia, provide the greatest CV benefit, and assist in weight reduction.
My protocol, “the egregious eleven,” involves using the least number of agents in combinations that treat the greatest number of mechanisms of hyperglycemia—without the use of sulfonylureas (which cause beta-cell apoptosis, hypoglycemia, and weight gain). Fortunately, newer agents, such as glucagon-like peptide 1 receptor agonist (GLP-1 RA) and sodium-glucose cotransporter 1 (SGLT-2) inhibitors, work right away, cause weight reduction, and have side benefits of CV risk reduction—as well as preserve beta-cell function. Metformin remains a valuable agent and has its own potential side benefits, and bromocriptine-QR and pioglitazone have CV side benefits. So, there is really no need for early insulin in true T2D patients (ie, those that are non-ketosis prone and have sufficient beta-cell reserve).
Why reduce insulin in patients who are already on insulin?
Prior protocols resulted in 40%-50% of T2D patients being placed on insulin unnecessarily. As discussed, the side effects of insulin are many; they include weight gain, insulin resistance, hypoglycemia, and CV complications—all of which have been associated with a decline in quality of life.
What is your approach to reduce or eliminate insulin in those already on it (unnecessarily)?
First, I pick the right patient. Physicians should use sound clinical judgment to identify patients with likely residual beta-cell function. It is not just the “insulin-resistant patient," as 30%-50% of type 1 diabetes mellitus patients also have insulin resistance.
It needs to be a definite T2D patient: not ketosis prone, a family history T2D, no islet cell antibodies (if one has any concerns, check for them). They were often started on insulin in the emergency department with no ketosis and never received non-insulin therapy.
Patients need to be willing to commit to my strict, no-concentrated-sweets diet, to perform careful glucose monitoring, and to check their ketones. Patients should be willing to contact me if their sugar level is >250 mg/dL for 2 measurements in a row, while testing 4 times a day or using a continuous glucose-monitoring (CGM) device.
First, estimate a patient’s “current insulin need” (CIN), or the dose they might be on if they had not been subject to over-insulinization (ie, if they had not been subject to the “vicious cycle” discussed above). I do this by taking their total basal and bolus insulin dose, then reducing it by ~25% as the patient changes to a no-concentrated-sweets diet with an additional up-to-25% dose reduction if the patient has been experiencing symptomatic or asymptomatic hypoglycemia.
Next, I reduce this CIN number by ~25% upon starting a rapid-acting subcutaneous GLP-1 RA (liraglutide or oral semaglutide) and reduce the CIN another 20% as they start the SGLT-2 inhibitor. If patients come into my office on <40 U/d, I stop insulin as I start a GLP-1 RA and an SGLT-2 inhibitor and have them monitor home glucose levels to assure reasonable results as they go off the insulin and on their new therapy.
If patients come into my office on >40 U/d, they go home on a GLP-1 RA and an SGLT-2 inhibitor and ~30% of their presenting dose, apportioned between basal/bolus dosing based on when they are currently getting hypoglycemic.
The rapid initial reduction in their insulin doses, with initial adjustments in estimated insulin doses as needed based on home glucose monitoring, and rapid stabilization of glycemic levels by the effectiveness of these 2 agents give patients great motivation to keep up with the diet/program.
Then, as patients lose weight, they are told to report any glucose measurements <80 mg/dL, so that further reduction in insulin doses can be made. When patients achieve a new steady state of glycemia, weight, and GLP-1 RA and SGLT-2 inhibitor doses, you can add bromocriptine-QR, pioglitazone, and/or metformin as needed to allow for a further reduction of insulin. And, as you see the delayed effects of subsequently adding these new agents (eg, glucose <80 mg/dL), you can ultimately stop insulin when they get to <10-12 U/d. The process works very well, even in those starting on up to 300 units of insulin daily. Patients love the outcome and will greatly appreciate your care.
Feel free to contact Dr. Schwartz at [email protected] with any questions about his protocol or diet.
A reason for hope in the face of long COVID
In this issue, Mayo and colleagues1 summarize what we know about patients with long COVID. The report made me pause and realize that it has been 3 years since we heard the very first reports of patients infected with SARS-CoV-2, which would eventually cause the COVID-19 pandemic. I suspect that I am not alone in having been fascinated by the rapid communication of information (of variable quality and veracity) via peer-reviewed papers, pre-print servers, the media, and social media.
The early studies were largely descriptive, focusing on symptom constellations and outbreak data. Much of what we had by way of treatment was supportive and “let’s try anything”—whether reasonable or, in some cases, not. In relatively short order, though, we developed effective vaccines to help protect people from getting seriously ill, being hospitalized, and dying; we also identified targeted therapies for those who became ill.2 But variants continued—or rather, continue—to emerge, and we remain committed to meeting the demands of the day.
The Centers for Disease Control and Prevention reports that more than 98 million Americans have contracted COVID, and more than 1 million have died.3 Besides the astonishingly high total mortality, the ravages of COVID-19 include new-onset respiratory, cardiovascular, neurologic, and psychiatric illnesses.4,5 As many as half of adults hospitalized for COVID report having persistent symptoms.6
As described in this issue, what we know about long COVID appears to be following the early course of its parent illness. As was true then, we are learning about the symptoms, etiology, and best ways to manage our patients. As in the early days of the pandemic, treatment is supportive, and we await definitive therapies.
I am optimistic, though. Why? Because shortly after the first reports of COVID-19, the virus’ DNA sequence was shared online. Based on that information, diagnostic assays were developed. Within 2 years of the outbreak, we had effective vaccines and specific therapies.
Another call to action. If 5% of patients contracting COVID (a very low estimate) develop long COVID, that would translate to 4.9 million people with long COVID in the United States. That is an astounding burden of suffering that I have no doubt will motivate innovation.
Innovation is a strength of the US health care system. I believe we will rise to the next challenge that COVID-19 has put before us. We have reason to be hopeful.
1. Mayo NL, Ellenbogen RL, Mendoza MD, et al. The family physician’s role in long COVID management. J Fam Pract. 2022;71:426-431. doi: 10.12788/jfp.0517
2. Kulshreshtha A, Sizemore S, Barry HC. COVID-19 therapy: What works? What doesn’t? And what’s on the horizon? J Fam Pract. 2022;71:E3-E16. doi: 10.12788/jfp.0474
3. CDC. COVID data tracker. Accessed December 5, 2022. https://covid.cdc.gov/covid-data-tracker/#datatracker-home
4. Taquet M, Geddes JR, Husain M, et al. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8:416-427. doi: 10.1016/s2215-0366(21) 00084-5
5. Ayoubkhani D, Khunti K, Nafilyan V, et al. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. 2021;372:n693. doi: 10.1136/bmj.n693
6. Writing Committee for the Comebac Study Group, Morin L, Savale L, Pham T, et al. Four-month clinical status of a cohort of patients after hospitalization for COVID-19. JAMA. 2021;325:1525-1534. doi: 10.1001/jama.2021.3331
In this issue, Mayo and colleagues1 summarize what we know about patients with long COVID. The report made me pause and realize that it has been 3 years since we heard the very first reports of patients infected with SARS-CoV-2, which would eventually cause the COVID-19 pandemic. I suspect that I am not alone in having been fascinated by the rapid communication of information (of variable quality and veracity) via peer-reviewed papers, pre-print servers, the media, and social media.
The early studies were largely descriptive, focusing on symptom constellations and outbreak data. Much of what we had by way of treatment was supportive and “let’s try anything”—whether reasonable or, in some cases, not. In relatively short order, though, we developed effective vaccines to help protect people from getting seriously ill, being hospitalized, and dying; we also identified targeted therapies for those who became ill.2 But variants continued—or rather, continue—to emerge, and we remain committed to meeting the demands of the day.
The Centers for Disease Control and Prevention reports that more than 98 million Americans have contracted COVID, and more than 1 million have died.3 Besides the astonishingly high total mortality, the ravages of COVID-19 include new-onset respiratory, cardiovascular, neurologic, and psychiatric illnesses.4,5 As many as half of adults hospitalized for COVID report having persistent symptoms.6
As described in this issue, what we know about long COVID appears to be following the early course of its parent illness. As was true then, we are learning about the symptoms, etiology, and best ways to manage our patients. As in the early days of the pandemic, treatment is supportive, and we await definitive therapies.
I am optimistic, though. Why? Because shortly after the first reports of COVID-19, the virus’ DNA sequence was shared online. Based on that information, diagnostic assays were developed. Within 2 years of the outbreak, we had effective vaccines and specific therapies.
Another call to action. If 5% of patients contracting COVID (a very low estimate) develop long COVID, that would translate to 4.9 million people with long COVID in the United States. That is an astounding burden of suffering that I have no doubt will motivate innovation.
Innovation is a strength of the US health care system. I believe we will rise to the next challenge that COVID-19 has put before us. We have reason to be hopeful.
In this issue, Mayo and colleagues1 summarize what we know about patients with long COVID. The report made me pause and realize that it has been 3 years since we heard the very first reports of patients infected with SARS-CoV-2, which would eventually cause the COVID-19 pandemic. I suspect that I am not alone in having been fascinated by the rapid communication of information (of variable quality and veracity) via peer-reviewed papers, pre-print servers, the media, and social media.
The early studies were largely descriptive, focusing on symptom constellations and outbreak data. Much of what we had by way of treatment was supportive and “let’s try anything”—whether reasonable or, in some cases, not. In relatively short order, though, we developed effective vaccines to help protect people from getting seriously ill, being hospitalized, and dying; we also identified targeted therapies for those who became ill.2 But variants continued—or rather, continue—to emerge, and we remain committed to meeting the demands of the day.
The Centers for Disease Control and Prevention reports that more than 98 million Americans have contracted COVID, and more than 1 million have died.3 Besides the astonishingly high total mortality, the ravages of COVID-19 include new-onset respiratory, cardiovascular, neurologic, and psychiatric illnesses.4,5 As many as half of adults hospitalized for COVID report having persistent symptoms.6
As described in this issue, what we know about long COVID appears to be following the early course of its parent illness. As was true then, we are learning about the symptoms, etiology, and best ways to manage our patients. As in the early days of the pandemic, treatment is supportive, and we await definitive therapies.
I am optimistic, though. Why? Because shortly after the first reports of COVID-19, the virus’ DNA sequence was shared online. Based on that information, diagnostic assays were developed. Within 2 years of the outbreak, we had effective vaccines and specific therapies.
Another call to action. If 5% of patients contracting COVID (a very low estimate) develop long COVID, that would translate to 4.9 million people with long COVID in the United States. That is an astounding burden of suffering that I have no doubt will motivate innovation.
Innovation is a strength of the US health care system. I believe we will rise to the next challenge that COVID-19 has put before us. We have reason to be hopeful.
1. Mayo NL, Ellenbogen RL, Mendoza MD, et al. The family physician’s role in long COVID management. J Fam Pract. 2022;71:426-431. doi: 10.12788/jfp.0517
2. Kulshreshtha A, Sizemore S, Barry HC. COVID-19 therapy: What works? What doesn’t? And what’s on the horizon? J Fam Pract. 2022;71:E3-E16. doi: 10.12788/jfp.0474
3. CDC. COVID data tracker. Accessed December 5, 2022. https://covid.cdc.gov/covid-data-tracker/#datatracker-home
4. Taquet M, Geddes JR, Husain M, et al. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8:416-427. doi: 10.1016/s2215-0366(21) 00084-5
5. Ayoubkhani D, Khunti K, Nafilyan V, et al. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. 2021;372:n693. doi: 10.1136/bmj.n693
6. Writing Committee for the Comebac Study Group, Morin L, Savale L, Pham T, et al. Four-month clinical status of a cohort of patients after hospitalization for COVID-19. JAMA. 2021;325:1525-1534. doi: 10.1001/jama.2021.3331
1. Mayo NL, Ellenbogen RL, Mendoza MD, et al. The family physician’s role in long COVID management. J Fam Pract. 2022;71:426-431. doi: 10.12788/jfp.0517
2. Kulshreshtha A, Sizemore S, Barry HC. COVID-19 therapy: What works? What doesn’t? And what’s on the horizon? J Fam Pract. 2022;71:E3-E16. doi: 10.12788/jfp.0474
3. CDC. COVID data tracker. Accessed December 5, 2022. https://covid.cdc.gov/covid-data-tracker/#datatracker-home
4. Taquet M, Geddes JR, Husain M, et al. 6-month neurological and psychiatric outcomes in 236 379 survivors of COVID-19: a retrospective cohort study using electronic health records. Lancet Psychiatry. 2021;8:416-427. doi: 10.1016/s2215-0366(21) 00084-5
5. Ayoubkhani D, Khunti K, Nafilyan V, et al. Post-covid syndrome in individuals admitted to hospital with covid-19: retrospective cohort study. BMJ. 2021;372:n693. doi: 10.1136/bmj.n693
6. Writing Committee for the Comebac Study Group, Morin L, Savale L, Pham T, et al. Four-month clinical status of a cohort of patients after hospitalization for COVID-19. JAMA. 2021;325:1525-1534. doi: 10.1001/jama.2021.3331
Blue-black hyperpigmentation on the extremities
A 68-year-old man with type 2 diabetes presented with progressive hyperpigmentation of the lower extremities and face over the past 3 years. Clinical examination revealed confluent, blue-black hyperpigmentation of the lower extremities (Figure), upper extremities, neck, and face. Laboratory tests and arterial studies were within normal ranges. The patient’s medication list included lisinopril 10 mg/d, metformin 1000 mg twice daily, minocycline 100 mg twice daily, and omeprazole 20 mg/d.

WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
Hyperpigmentation is a rare but not uncommon adverse effect of long-term minocycline use. In this case, our patient had been taking minocycline for more than 5 years. When seen in our clinic, he said he could not remember why he was taking minocycline and incorrectly assumed it was for his diabetes. Chart review of outside records revealed that it had been prescribed, and refilled annually, by his primary physician for rosacea.
Minocycline hyperpigmentation is subdivided into 3 types:
- Type I manifests with blue-black discoloration in previously inflamed areas of skin.
- Type II manifests with blue-gray pigmentation in previously normal skin areas.
- Type III manifests diffusely with muddy-brown hyperpigmentation on photoexposed skin.
Furthermore, noncutaneous manifestations may occur on the sclera, nails, ear cartilage, bone, oral mucosa, teeth, and thyroid gland.1
Diagnosis focuses on identifying the source
Minocycline is one of many drugs that can induce hyperpigmentation of the skin. In addition to history, examination, and review of the patient’s medication list, there are some clues on exam that may suggest a certain type of medication at play.
Continue to: Antimalarials
Antimalarials. Chloroquine, hydroxychloroquine, and quinacrine can cause blue-black skin hyperpigmentation in as many as 25% of patients. Common locations include the shins, face, oral mucosa, and subungual skin. This hyperpigmentation rarely fully resolves.2
Amiodarone. Hyperpigmentation secondary to amiodarone use typically is slate-gray in color and involves photoexposed skin. Patients should be counseled that pigmentation may—but does not always—fade with time after discontinuation of the drug.2
Heavy metals. Argyria results from exposure to silver, either ingested orally or applied externally. A common cause of argyria is ingestion of excessive amounts of silver-containing supplements.3 Affected patients present with diffuse slate-gray discoloration of the skin.
Other metals implicated in skin hyperpigmentation include arsenic, gold, mercury, and iron. Review of all supplements and herbal remedies in patients presenting with skin hyperpigmentation is crucial.
Bleomycin is a chemotherapeutic agent with a rare but unique adverse effect of inducing flagellate hyperpigmentation that favors the chest, abdomen, or back. This may be induced by trauma or scratching and is often transient. Hyperpigmentation can occur secondary to either intravenous or intralesional injection of the medication.2
Continue to: In addition to medication...
In addition to medication- or supplement-induced hyperpigmentation, there is a physiologic source that should be considered when a patient presents with lower-extremity hyperpigmentation:
Stasis hyperpigmentation. Patients with chronic venous insufficiency may present with hyperpigmentation of the lower extremities. Commonly due to dysfunctional venous valves or obstruction, stasis hyperpigmentation manifests with red-brown discoloration from dermal hemosiderin deposition.4
Unlike our patient, those with stasis hyperpigmentation may present symptomatically, with associated dry skin, pruritus, induration, and inflammation. Treatment involves management of the underlying venous insufficiency.4
When there’s no obvious cause, be prepared to dig deeper
At the time of initial assessment, a thorough review of systems and detailed medication history, including over-the-counter supplements, should be obtained. Physical examination revealing diffuse, generalized hyperpigmentation with no reliable culprit medication in the patient’s history warrants further laboratory evaluation. This includes ordering renal and liver studies and tests for thyroid-stimulating hormone and ferritin and cortisol levels to rule out metabolic or endocrine hyperpigmentation disorders.
Stopping the offending medication is the first step
Discontinuation of the offending medication may result in mild improvement in skin hyperpigmentation over time. Some patients may not experience any improvement. If improvement occurs, it is important to educate patients that it can take several months to years. Dermatology guidelines favor discontinuation of antibiotics for acne or rosacea after 3 to 6 months to avoid bacterial resistance.5 Worsening hyperpigmentation despite medication discontinuation warrants further work-up.
Patients who are distressed by persistent hyperpigmentation can be treated using picosecond or Q-switched lasers.6
Our patient was advised to discontinue the minocycline. Three test spots on his face were treated with pulsed-dye laser, carbon dioxide laser, and dermabrasion. The patient noted that the spots responded better to the carbon dioxide laser and dermabrasion compared to the pulsed-dye laser. He did not follow up for further treatment.
1. Wetter DA. Minocycline hyperpigmentation. Mayo Clin Proc. 2012;87:e33. doi: 10.1016/j.mayocp.2012.02.013
2. Chang MW. Chapter 67: Disorders of hyperpigmentation. In: Bolognia J, Schaffer J, Cerroni L, et al (eds). Dermatology. 4th ed. Elsevier; 2018:1122-1124.
3. Bowden LP, Royer MC, Hallman JR, et al. Rapid onset of argyria induced by a silver-containing dietary supplement. J Cutan Pathol. 2011;38:832-835. doi: 10.1111/j.1600-0560.2011.01755.x
4. Patterson J. Stasis dermatitis. In: Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier;2010: 121-153.
5. Zaenglein AL, Pathy AL, Schlosser BJ, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2016;74:945-73.e33. doi: 10.1016/j.jaad.2015.12.037
6. Barrett T, de Zwaan S. Picosecond alexandrite laser is superior to Q-switched Nd:YAG laser in treatment of minocycline-induced hyperpigmentation: a case study and review of the literature. J Cosmet Laser Ther. 2018;20:387-390. doi: 10.1080/14764172.2017.1418514
A 68-year-old man with type 2 diabetes presented with progressive hyperpigmentation of the lower extremities and face over the past 3 years. Clinical examination revealed confluent, blue-black hyperpigmentation of the lower extremities (Figure), upper extremities, neck, and face. Laboratory tests and arterial studies were within normal ranges. The patient’s medication list included lisinopril 10 mg/d, metformin 1000 mg twice daily, minocycline 100 mg twice daily, and omeprazole 20 mg/d.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
Hyperpigmentation is a rare but not uncommon adverse effect of long-term minocycline use. In this case, our patient had been taking minocycline for more than 5 years. When seen in our clinic, he said he could not remember why he was taking minocycline and incorrectly assumed it was for his diabetes. Chart review of outside records revealed that it had been prescribed, and refilled annually, by his primary physician for rosacea.
Minocycline hyperpigmentation is subdivided into 3 types:
- Type I manifests with blue-black discoloration in previously inflamed areas of skin.
- Type II manifests with blue-gray pigmentation in previously normal skin areas.
- Type III manifests diffusely with muddy-brown hyperpigmentation on photoexposed skin.
Furthermore, noncutaneous manifestations may occur on the sclera, nails, ear cartilage, bone, oral mucosa, teeth, and thyroid gland.1
Diagnosis focuses on identifying the source
Minocycline is one of many drugs that can induce hyperpigmentation of the skin. In addition to history, examination, and review of the patient’s medication list, there are some clues on exam that may suggest a certain type of medication at play.
Continue to: Antimalarials
Antimalarials. Chloroquine, hydroxychloroquine, and quinacrine can cause blue-black skin hyperpigmentation in as many as 25% of patients. Common locations include the shins, face, oral mucosa, and subungual skin. This hyperpigmentation rarely fully resolves.2
Amiodarone. Hyperpigmentation secondary to amiodarone use typically is slate-gray in color and involves photoexposed skin. Patients should be counseled that pigmentation may—but does not always—fade with time after discontinuation of the drug.2
Heavy metals. Argyria results from exposure to silver, either ingested orally or applied externally. A common cause of argyria is ingestion of excessive amounts of silver-containing supplements.3 Affected patients present with diffuse slate-gray discoloration of the skin.
Other metals implicated in skin hyperpigmentation include arsenic, gold, mercury, and iron. Review of all supplements and herbal remedies in patients presenting with skin hyperpigmentation is crucial.
Bleomycin is a chemotherapeutic agent with a rare but unique adverse effect of inducing flagellate hyperpigmentation that favors the chest, abdomen, or back. This may be induced by trauma or scratching and is often transient. Hyperpigmentation can occur secondary to either intravenous or intralesional injection of the medication.2
Continue to: In addition to medication...
In addition to medication- or supplement-induced hyperpigmentation, there is a physiologic source that should be considered when a patient presents with lower-extremity hyperpigmentation:
Stasis hyperpigmentation. Patients with chronic venous insufficiency may present with hyperpigmentation of the lower extremities. Commonly due to dysfunctional venous valves or obstruction, stasis hyperpigmentation manifests with red-brown discoloration from dermal hemosiderin deposition.4
Unlike our patient, those with stasis hyperpigmentation may present symptomatically, with associated dry skin, pruritus, induration, and inflammation. Treatment involves management of the underlying venous insufficiency.4
When there’s no obvious cause, be prepared to dig deeper
At the time of initial assessment, a thorough review of systems and detailed medication history, including over-the-counter supplements, should be obtained. Physical examination revealing diffuse, generalized hyperpigmentation with no reliable culprit medication in the patient’s history warrants further laboratory evaluation. This includes ordering renal and liver studies and tests for thyroid-stimulating hormone and ferritin and cortisol levels to rule out metabolic or endocrine hyperpigmentation disorders.
Stopping the offending medication is the first step
Discontinuation of the offending medication may result in mild improvement in skin hyperpigmentation over time. Some patients may not experience any improvement. If improvement occurs, it is important to educate patients that it can take several months to years. Dermatology guidelines favor discontinuation of antibiotics for acne or rosacea after 3 to 6 months to avoid bacterial resistance.5 Worsening hyperpigmentation despite medication discontinuation warrants further work-up.
Patients who are distressed by persistent hyperpigmentation can be treated using picosecond or Q-switched lasers.6
Our patient was advised to discontinue the minocycline. Three test spots on his face were treated with pulsed-dye laser, carbon dioxide laser, and dermabrasion. The patient noted that the spots responded better to the carbon dioxide laser and dermabrasion compared to the pulsed-dye laser. He did not follow up for further treatment.
A 68-year-old man with type 2 diabetes presented with progressive hyperpigmentation of the lower extremities and face over the past 3 years. Clinical examination revealed confluent, blue-black hyperpigmentation of the lower extremities (Figure), upper extremities, neck, and face. Laboratory tests and arterial studies were within normal ranges. The patient’s medication list included lisinopril 10 mg/d, metformin 1000 mg twice daily, minocycline 100 mg twice daily, and omeprazole 20 mg/d.
WHAT IS YOUR DIAGNOSIS?
HOW WOULD YOU TREAT THIS PATIENT?
Diagnosis: Minocycline-induced hyperpigmentation
Hyperpigmentation is a rare but not uncommon adverse effect of long-term minocycline use. In this case, our patient had been taking minocycline for more than 5 years. When seen in our clinic, he said he could not remember why he was taking minocycline and incorrectly assumed it was for his diabetes. Chart review of outside records revealed that it had been prescribed, and refilled annually, by his primary physician for rosacea.
Minocycline hyperpigmentation is subdivided into 3 types:
- Type I manifests with blue-black discoloration in previously inflamed areas of skin.
- Type II manifests with blue-gray pigmentation in previously normal skin areas.
- Type III manifests diffusely with muddy-brown hyperpigmentation on photoexposed skin.
Furthermore, noncutaneous manifestations may occur on the sclera, nails, ear cartilage, bone, oral mucosa, teeth, and thyroid gland.1
Diagnosis focuses on identifying the source
Minocycline is one of many drugs that can induce hyperpigmentation of the skin. In addition to history, examination, and review of the patient’s medication list, there are some clues on exam that may suggest a certain type of medication at play.
Continue to: Antimalarials
Antimalarials. Chloroquine, hydroxychloroquine, and quinacrine can cause blue-black skin hyperpigmentation in as many as 25% of patients. Common locations include the shins, face, oral mucosa, and subungual skin. This hyperpigmentation rarely fully resolves.2
Amiodarone. Hyperpigmentation secondary to amiodarone use typically is slate-gray in color and involves photoexposed skin. Patients should be counseled that pigmentation may—but does not always—fade with time after discontinuation of the drug.2
Heavy metals. Argyria results from exposure to silver, either ingested orally or applied externally. A common cause of argyria is ingestion of excessive amounts of silver-containing supplements.3 Affected patients present with diffuse slate-gray discoloration of the skin.
Other metals implicated in skin hyperpigmentation include arsenic, gold, mercury, and iron. Review of all supplements and herbal remedies in patients presenting with skin hyperpigmentation is crucial.
Bleomycin is a chemotherapeutic agent with a rare but unique adverse effect of inducing flagellate hyperpigmentation that favors the chest, abdomen, or back. This may be induced by trauma or scratching and is often transient. Hyperpigmentation can occur secondary to either intravenous or intralesional injection of the medication.2
Continue to: In addition to medication...
In addition to medication- or supplement-induced hyperpigmentation, there is a physiologic source that should be considered when a patient presents with lower-extremity hyperpigmentation:
Stasis hyperpigmentation. Patients with chronic venous insufficiency may present with hyperpigmentation of the lower extremities. Commonly due to dysfunctional venous valves or obstruction, stasis hyperpigmentation manifests with red-brown discoloration from dermal hemosiderin deposition.4
Unlike our patient, those with stasis hyperpigmentation may present symptomatically, with associated dry skin, pruritus, induration, and inflammation. Treatment involves management of the underlying venous insufficiency.4
When there’s no obvious cause, be prepared to dig deeper
At the time of initial assessment, a thorough review of systems and detailed medication history, including over-the-counter supplements, should be obtained. Physical examination revealing diffuse, generalized hyperpigmentation with no reliable culprit medication in the patient’s history warrants further laboratory evaluation. This includes ordering renal and liver studies and tests for thyroid-stimulating hormone and ferritin and cortisol levels to rule out metabolic or endocrine hyperpigmentation disorders.
Stopping the offending medication is the first step
Discontinuation of the offending medication may result in mild improvement in skin hyperpigmentation over time. Some patients may not experience any improvement. If improvement occurs, it is important to educate patients that it can take several months to years. Dermatology guidelines favor discontinuation of antibiotics for acne or rosacea after 3 to 6 months to avoid bacterial resistance.5 Worsening hyperpigmentation despite medication discontinuation warrants further work-up.
Patients who are distressed by persistent hyperpigmentation can be treated using picosecond or Q-switched lasers.6
Our patient was advised to discontinue the minocycline. Three test spots on his face were treated with pulsed-dye laser, carbon dioxide laser, and dermabrasion. The patient noted that the spots responded better to the carbon dioxide laser and dermabrasion compared to the pulsed-dye laser. He did not follow up for further treatment.
1. Wetter DA. Minocycline hyperpigmentation. Mayo Clin Proc. 2012;87:e33. doi: 10.1016/j.mayocp.2012.02.013
2. Chang MW. Chapter 67: Disorders of hyperpigmentation. In: Bolognia J, Schaffer J, Cerroni L, et al (eds). Dermatology. 4th ed. Elsevier; 2018:1122-1124.
3. Bowden LP, Royer MC, Hallman JR, et al. Rapid onset of argyria induced by a silver-containing dietary supplement. J Cutan Pathol. 2011;38:832-835. doi: 10.1111/j.1600-0560.2011.01755.x
4. Patterson J. Stasis dermatitis. In: Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier;2010: 121-153.
5. Zaenglein AL, Pathy AL, Schlosser BJ, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2016;74:945-73.e33. doi: 10.1016/j.jaad.2015.12.037
6. Barrett T, de Zwaan S. Picosecond alexandrite laser is superior to Q-switched Nd:YAG laser in treatment of minocycline-induced hyperpigmentation: a case study and review of the literature. J Cosmet Laser Ther. 2018;20:387-390. doi: 10.1080/14764172.2017.1418514
1. Wetter DA. Minocycline hyperpigmentation. Mayo Clin Proc. 2012;87:e33. doi: 10.1016/j.mayocp.2012.02.013
2. Chang MW. Chapter 67: Disorders of hyperpigmentation. In: Bolognia J, Schaffer J, Cerroni L, et al (eds). Dermatology. 4th ed. Elsevier; 2018:1122-1124.
3. Bowden LP, Royer MC, Hallman JR, et al. Rapid onset of argyria induced by a silver-containing dietary supplement. J Cutan Pathol. 2011;38:832-835. doi: 10.1111/j.1600-0560.2011.01755.x
4. Patterson J. Stasis dermatitis. In: Weedon’s Skin Pathology. 3rd ed. Churchill Livingstone Elsevier;2010: 121-153.
5. Zaenglein AL, Pathy AL, Schlosser BJ, et al. Guidelines of care for the management of acne vulgaris. J Am Acad Dermatol. 2016;74:945-73.e33. doi: 10.1016/j.jaad.2015.12.037
6. Barrett T, de Zwaan S. Picosecond alexandrite laser is superior to Q-switched Nd:YAG laser in treatment of minocycline-induced hyperpigmentation: a case study and review of the literature. J Cosmet Laser Ther. 2018;20:387-390. doi: 10.1080/14764172.2017.1418514

Consider this SGLT2 inhibitor for patients with HF with preserved ejection fraction
ILLUSTRATIVE CASE
A 72-year-old man with a history of hypertension, permanent atrial fibrillation, and heart failure (HF) comes into your clinic for follow-up. He was hospitalized a few months ago for HF requiring diuresis. His echocardiogram at that time showed an EF of 50% and no significant valvular disease. He does not have a history of diabetes or tobacco use. His medication regimen includes metoprolol,
HFpEF was first defined as HF in patients with a left ventricular ejection fraction (LVEF) > 40%. However, HF with an LVEF between 41% and 49% has been reclassified as its own category:
In comparison with HF with reduced ejection fraction (HFrEF), there are limited proven treatment options with cardiovascular (CV) benefit in HFpEF.
STUDY SUMMARY
Confirmation of benefit of empagliflozin for patients with HFpEF
The EMPEROR-Preserved study was a double-blind, placebo-controlled trial that randomized adult patients with HFpEF (defined by an LVEF > 40%) to either placebo or empagliflozin 10 mg/d, in addition to usual therapy. Patients were randomized in a 1:1 ratio stratified by geographic region, diabetes status, renal function (estimated glomerular filtration rate [eGFR] either < 60 or ≥ 60 mL/min/1.73 m2), and LVEF > 40% to < 50% or LVEF ≥ 50%.
Included patients were 18 years or older and had an NT-proBNP level > 300 pg/mL (or > 900 pg/mL if the patient had atrial fibrillation at baseline
The primary outcome was a composite of CV death or first hospitalization for HF. The secondary outcomes were all hospitalizations for HF and the rate of decline in eGFR.
Of the 5988 patients in the trial, 2997 were randomized to receive empagliflozin and 2991 were randomized to placebo. The average age was 72 years in each group, 45% of patients were women, about 76% were White, and 12% were from North America. About 81% of patients were classified as NYHA class II, nearly half had diabetes, and half had an eGFR < 60 mL/min/1.73 m2. The median body mass index (BMI) was 30, and the median LVEF was 54%. At baseline, the groups were similar in BMI, history of HF hospitalization in the past 12 months, history of common risk factors for HFpEF (atrial fibrillation, diabetes, and hypertension), and prescribed CV medications (ACE inhibitor or ARB with or without a neprilysin inhibitor, spironolactone, beta-blocker, digitalis glycosides, aspirin, and statins). Patients were followed for a median of 26.2 months.
Continue to: The primary composite...
The primary composite outcome of death from CV causes or HF-related hospitalization occurred in 415 patients (13.8%) in the empagliflozin group and in 511 patients (17.1%) in the placebo group (hazard ratio [HR] = 0.79; 95% CI, 0.69-0.90; P < .001). The number needed to treat to prevent 1 primary outcome event was 31 (95% CI, 20-69). Hospitalization for HF occurred in 259 patients (8.6%) with empagliflozin vs 352 patients (11.8%) with placebo (HR = 0.71; 95% CI, 0.60-0.83), and CV death occurred in 219 patients (7.3%) with empagliflozin vs 244 patients (8.2%) with placebo (HR = 0.91; 95% CI, 0.76-1.09). The effect was consistent in patients with or without diabetes at baseline; however, the largest reduction in the primary composite outcome was seen in those with an LVEF < 50%, age ≥ 70 years old, BMI < 30, and NYHA class II status.
The secondary outcome of total number of hospitalizations for HF was 407 with empagliflozin vs 541 with placebo (HR = 0.73; 95% CI, 0.61-0.88; P < .001). The rate of decline in the eGFR per year was –1.25 in the empagliflozin group vs –2.62 in the placebo group (P < .001), indicating that those taking empagliflozin had preserved renal function compared with those taking placebo.
Death from any cause occurred in 422 patients (14.1%) in the empagliflozin group and 427 patients (14.3%) in the placebo group (HR = 1.00; 95% CI, 0.87-1.15). Empagliflozin treatment was associated with higher rates of genital infections (2.2% vs 0.7%; P value not provided), urinary tract infections (9.9% vs 8.1%; P value not provided), and hypotension (10.4% vs 8.6%; P value not provided), compared to placebo.
WHAT’S NEW
Risk of hospitalization significantly reduced for patients with HFpEF
In the EMPEROR-Preserved study, empagliflozin led to a lower incidence of hospitalization for HF in patients with HFpEF but did not significantly reduce the number of deaths from CV disease or other causes. In comparison, in the similarly designed EMPEROR-Reduced trial, treatment with empagliflozin reduced CV and all-cause mortality in individuals with HFrEF.8
CAVEATS
HF criteria, study population may limit generalizability
The reduction in the primary outcome of CV death or first hospitalization was most pronounced in patients with an LVEF > 40% to < 50%, typically defined as HFmrEF, who often have clinical features similar to those with HFrEF. This raises the question of how generalizable these results are for all patients with HFpEF.
Continue to: The study's generalizability...
The study’s generalizability was further limited by its significant exclusion criteria, which included elevated blood pressure, chronic obstructive pulmonary disease on home oxygen, liver disease, renal disease with an eGFR < 20 mL/min/1.73 m2 or requiring dialysis, and BMI ≥ 45.
Finally, only 12% of patients were from North America, and results were not significant for this subgroup (HR = 0.72; 95% CI, 0.52-1.00), which may challenge its external validity. The authors noted that 23% of patients discontinued treatment for reasons other than death, which may have driven the null effect.
CHALLENGES TO IMPLEMENTATION
Empagliflozin is expensive,but coverage may improve
Cost could be a major barrier to implementation. Retail pricing for empagliflozin is estimated to be more than $550 per month, which may be prohibitive for patients with no insurance or with higher-deductible plans.11 However, the US Food and Drug Administration has approved empagliflozin to reduce the risk of CV death and hospitalization for HF in adults,12 which may help to improve insurance coverage.
1. Anker SD, Butler J, Filippatos G, et al; EMPEROR-Preserved Trial Investigators. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451-1461. doi: 10.1056/NEJMoa2107038
2. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi: 10.1161/CIR.0000000000001063
3. Gevaert AB, Kataria R, Zannad F, et al. Heart failure with preserved ejection fraction: recent concepts in diagnosis, mechanisms and management. Heart. 2022;108:1342-1350. doi: 10.1136/heartjnl-2021-319605
4. Vaduganathan M, Claggett BL, Jhund PS, et al. Estimating lifetime benefits of comprehensive disease-modifying pharmacological therapies in patients with heart failure with reduced ejection fraction: a comparative analysis of three randomised controlled trials. Lancet. 2020;396:121-128. doi: 10.1016/S0140-6736(20)30748-0
5. Solomon SD, Claggett B, Lewis EF, et al; TOPCAT Investigators. Influence of ejection fraction on outcomes and efficacy of spironolactone in patients with heart failure with preserved ejection fraction. Eur Heart J. 2016;37:455-462. doi: 10.1093/eurheartj/ehv464
6. Martin N, Manoharan K, Thomas J, et al. Beta-blockers and inhibitors of the renin-angiotensin aldosterone system for chronic heart failure with preserved ejection fraction. Cochrane Database Syst Rev. 2018;6:CD012721. doi: 10.1002/14651858.CD012721.pub2
7. Solomon SD, McMurray JJV, Anand IS, et al; PARAGON-HF Investigators and Committees. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609-1620. doi: 10.1056/NEJMoa1908655
8. Zannad F, Ferreira JP, Pocock SJ, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. 2020;396:819-829. doi: 10.1016/S0140-6736(20)31824-9
9. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536. doi: 10.1161/CIRCULATIONAHA. 119.040130
10. Bhatt DL, Szarek M, Steg PG, et al; SOLOIST-WHF Trial Investigators. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384:117-128. doi: 10.1056/NEJM oa2030183
11. Empagliflozin. GoodRx.com. Accessed June 3, 2022. www.goodrx.com/empagliflozin
12. FDA approves treatment for wider range of patients with heart failure. News release. US Food and Drug Administration; February 24, 2022. Accessed June 3, 2022. www.fda.gov/news-events/press-announcements/fda-approves-treatment-wider-range-patients-heart-failure
ILLUSTRATIVE CASE
A 72-year-old man with a history of hypertension, permanent atrial fibrillation, and heart failure (HF) comes into your clinic for follow-up. He was hospitalized a few months ago for HF requiring diuresis. His echocardiogram at that time showed an EF of 50% and no significant valvular disease. He does not have a history of diabetes or tobacco use. His medication regimen includes metoprolol,
HFpEF was first defined as HF in patients with a left ventricular ejection fraction (LVEF) > 40%. However, HF with an LVEF between 41% and 49% has been reclassified as its own category:
In comparison with HF with reduced ejection fraction (HFrEF), there are limited proven treatment options with cardiovascular (CV) benefit in HFpEF.
STUDY SUMMARY
Confirmation of benefit of empagliflozin for patients with HFpEF
The EMPEROR-Preserved study was a double-blind, placebo-controlled trial that randomized adult patients with HFpEF (defined by an LVEF > 40%) to either placebo or empagliflozin 10 mg/d, in addition to usual therapy. Patients were randomized in a 1:1 ratio stratified by geographic region, diabetes status, renal function (estimated glomerular filtration rate [eGFR] either < 60 or ≥ 60 mL/min/1.73 m2), and LVEF > 40% to < 50% or LVEF ≥ 50%.
Included patients were 18 years or older and had an NT-proBNP level > 300 pg/mL (or > 900 pg/mL if the patient had atrial fibrillation at baseline
The primary outcome was a composite of CV death or first hospitalization for HF. The secondary outcomes were all hospitalizations for HF and the rate of decline in eGFR.
Of the 5988 patients in the trial, 2997 were randomized to receive empagliflozin and 2991 were randomized to placebo. The average age was 72 years in each group, 45% of patients were women, about 76% were White, and 12% were from North America. About 81% of patients were classified as NYHA class II, nearly half had diabetes, and half had an eGFR < 60 mL/min/1.73 m2. The median body mass index (BMI) was 30, and the median LVEF was 54%. At baseline, the groups were similar in BMI, history of HF hospitalization in the past 12 months, history of common risk factors for HFpEF (atrial fibrillation, diabetes, and hypertension), and prescribed CV medications (ACE inhibitor or ARB with or without a neprilysin inhibitor, spironolactone, beta-blocker, digitalis glycosides, aspirin, and statins). Patients were followed for a median of 26.2 months.
Continue to: The primary composite...
The primary composite outcome of death from CV causes or HF-related hospitalization occurred in 415 patients (13.8%) in the empagliflozin group and in 511 patients (17.1%) in the placebo group (hazard ratio [HR] = 0.79; 95% CI, 0.69-0.90; P < .001). The number needed to treat to prevent 1 primary outcome event was 31 (95% CI, 20-69). Hospitalization for HF occurred in 259 patients (8.6%) with empagliflozin vs 352 patients (11.8%) with placebo (HR = 0.71; 95% CI, 0.60-0.83), and CV death occurred in 219 patients (7.3%) with empagliflozin vs 244 patients (8.2%) with placebo (HR = 0.91; 95% CI, 0.76-1.09). The effect was consistent in patients with or without diabetes at baseline; however, the largest reduction in the primary composite outcome was seen in those with an LVEF < 50%, age ≥ 70 years old, BMI < 30, and NYHA class II status.
The secondary outcome of total number of hospitalizations for HF was 407 with empagliflozin vs 541 with placebo (HR = 0.73; 95% CI, 0.61-0.88; P < .001). The rate of decline in the eGFR per year was –1.25 in the empagliflozin group vs –2.62 in the placebo group (P < .001), indicating that those taking empagliflozin had preserved renal function compared with those taking placebo.
Death from any cause occurred in 422 patients (14.1%) in the empagliflozin group and 427 patients (14.3%) in the placebo group (HR = 1.00; 95% CI, 0.87-1.15). Empagliflozin treatment was associated with higher rates of genital infections (2.2% vs 0.7%; P value not provided), urinary tract infections (9.9% vs 8.1%; P value not provided), and hypotension (10.4% vs 8.6%; P value not provided), compared to placebo.
WHAT’S NEW
Risk of hospitalization significantly reduced for patients with HFpEF
In the EMPEROR-Preserved study, empagliflozin led to a lower incidence of hospitalization for HF in patients with HFpEF but did not significantly reduce the number of deaths from CV disease or other causes. In comparison, in the similarly designed EMPEROR-Reduced trial, treatment with empagliflozin reduced CV and all-cause mortality in individuals with HFrEF.8
CAVEATS
HF criteria, study population may limit generalizability
The reduction in the primary outcome of CV death or first hospitalization was most pronounced in patients with an LVEF > 40% to < 50%, typically defined as HFmrEF, who often have clinical features similar to those with HFrEF. This raises the question of how generalizable these results are for all patients with HFpEF.
Continue to: The study's generalizability...
The study’s generalizability was further limited by its significant exclusion criteria, which included elevated blood pressure, chronic obstructive pulmonary disease on home oxygen, liver disease, renal disease with an eGFR < 20 mL/min/1.73 m2 or requiring dialysis, and BMI ≥ 45.
Finally, only 12% of patients were from North America, and results were not significant for this subgroup (HR = 0.72; 95% CI, 0.52-1.00), which may challenge its external validity. The authors noted that 23% of patients discontinued treatment for reasons other than death, which may have driven the null effect.
CHALLENGES TO IMPLEMENTATION
Empagliflozin is expensive,but coverage may improve
Cost could be a major barrier to implementation. Retail pricing for empagliflozin is estimated to be more than $550 per month, which may be prohibitive for patients with no insurance or with higher-deductible plans.11 However, the US Food and Drug Administration has approved empagliflozin to reduce the risk of CV death and hospitalization for HF in adults,12 which may help to improve insurance coverage.
ILLUSTRATIVE CASE
A 72-year-old man with a history of hypertension, permanent atrial fibrillation, and heart failure (HF) comes into your clinic for follow-up. He was hospitalized a few months ago for HF requiring diuresis. His echocardiogram at that time showed an EF of 50% and no significant valvular disease. He does not have a history of diabetes or tobacco use. His medication regimen includes metoprolol,
HFpEF was first defined as HF in patients with a left ventricular ejection fraction (LVEF) > 40%. However, HF with an LVEF between 41% and 49% has been reclassified as its own category:
In comparison with HF with reduced ejection fraction (HFrEF), there are limited proven treatment options with cardiovascular (CV) benefit in HFpEF.
STUDY SUMMARY
Confirmation of benefit of empagliflozin for patients with HFpEF
The EMPEROR-Preserved study was a double-blind, placebo-controlled trial that randomized adult patients with HFpEF (defined by an LVEF > 40%) to either placebo or empagliflozin 10 mg/d, in addition to usual therapy. Patients were randomized in a 1:1 ratio stratified by geographic region, diabetes status, renal function (estimated glomerular filtration rate [eGFR] either < 60 or ≥ 60 mL/min/1.73 m2), and LVEF > 40% to < 50% or LVEF ≥ 50%.
Included patients were 18 years or older and had an NT-proBNP level > 300 pg/mL (or > 900 pg/mL if the patient had atrial fibrillation at baseline
The primary outcome was a composite of CV death or first hospitalization for HF. The secondary outcomes were all hospitalizations for HF and the rate of decline in eGFR.
Of the 5988 patients in the trial, 2997 were randomized to receive empagliflozin and 2991 were randomized to placebo. The average age was 72 years in each group, 45% of patients were women, about 76% were White, and 12% were from North America. About 81% of patients were classified as NYHA class II, nearly half had diabetes, and half had an eGFR < 60 mL/min/1.73 m2. The median body mass index (BMI) was 30, and the median LVEF was 54%. At baseline, the groups were similar in BMI, history of HF hospitalization in the past 12 months, history of common risk factors for HFpEF (atrial fibrillation, diabetes, and hypertension), and prescribed CV medications (ACE inhibitor or ARB with or without a neprilysin inhibitor, spironolactone, beta-blocker, digitalis glycosides, aspirin, and statins). Patients were followed for a median of 26.2 months.
Continue to: The primary composite...
The primary composite outcome of death from CV causes or HF-related hospitalization occurred in 415 patients (13.8%) in the empagliflozin group and in 511 patients (17.1%) in the placebo group (hazard ratio [HR] = 0.79; 95% CI, 0.69-0.90; P < .001). The number needed to treat to prevent 1 primary outcome event was 31 (95% CI, 20-69). Hospitalization for HF occurred in 259 patients (8.6%) with empagliflozin vs 352 patients (11.8%) with placebo (HR = 0.71; 95% CI, 0.60-0.83), and CV death occurred in 219 patients (7.3%) with empagliflozin vs 244 patients (8.2%) with placebo (HR = 0.91; 95% CI, 0.76-1.09). The effect was consistent in patients with or without diabetes at baseline; however, the largest reduction in the primary composite outcome was seen in those with an LVEF < 50%, age ≥ 70 years old, BMI < 30, and NYHA class II status.
The secondary outcome of total number of hospitalizations for HF was 407 with empagliflozin vs 541 with placebo (HR = 0.73; 95% CI, 0.61-0.88; P < .001). The rate of decline in the eGFR per year was –1.25 in the empagliflozin group vs –2.62 in the placebo group (P < .001), indicating that those taking empagliflozin had preserved renal function compared with those taking placebo.
Death from any cause occurred in 422 patients (14.1%) in the empagliflozin group and 427 patients (14.3%) in the placebo group (HR = 1.00; 95% CI, 0.87-1.15). Empagliflozin treatment was associated with higher rates of genital infections (2.2% vs 0.7%; P value not provided), urinary tract infections (9.9% vs 8.1%; P value not provided), and hypotension (10.4% vs 8.6%; P value not provided), compared to placebo.
WHAT’S NEW
Risk of hospitalization significantly reduced for patients with HFpEF
In the EMPEROR-Preserved study, empagliflozin led to a lower incidence of hospitalization for HF in patients with HFpEF but did not significantly reduce the number of deaths from CV disease or other causes. In comparison, in the similarly designed EMPEROR-Reduced trial, treatment with empagliflozin reduced CV and all-cause mortality in individuals with HFrEF.8
CAVEATS
HF criteria, study population may limit generalizability
The reduction in the primary outcome of CV death or first hospitalization was most pronounced in patients with an LVEF > 40% to < 50%, typically defined as HFmrEF, who often have clinical features similar to those with HFrEF. This raises the question of how generalizable these results are for all patients with HFpEF.
Continue to: The study's generalizability...
The study’s generalizability was further limited by its significant exclusion criteria, which included elevated blood pressure, chronic obstructive pulmonary disease on home oxygen, liver disease, renal disease with an eGFR < 20 mL/min/1.73 m2 or requiring dialysis, and BMI ≥ 45.
Finally, only 12% of patients were from North America, and results were not significant for this subgroup (HR = 0.72; 95% CI, 0.52-1.00), which may challenge its external validity. The authors noted that 23% of patients discontinued treatment for reasons other than death, which may have driven the null effect.
CHALLENGES TO IMPLEMENTATION
Empagliflozin is expensive,but coverage may improve
Cost could be a major barrier to implementation. Retail pricing for empagliflozin is estimated to be more than $550 per month, which may be prohibitive for patients with no insurance or with higher-deductible plans.11 However, the US Food and Drug Administration has approved empagliflozin to reduce the risk of CV death and hospitalization for HF in adults,12 which may help to improve insurance coverage.
1. Anker SD, Butler J, Filippatos G, et al; EMPEROR-Preserved Trial Investigators. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451-1461. doi: 10.1056/NEJMoa2107038
2. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi: 10.1161/CIR.0000000000001063
3. Gevaert AB, Kataria R, Zannad F, et al. Heart failure with preserved ejection fraction: recent concepts in diagnosis, mechanisms and management. Heart. 2022;108:1342-1350. doi: 10.1136/heartjnl-2021-319605
4. Vaduganathan M, Claggett BL, Jhund PS, et al. Estimating lifetime benefits of comprehensive disease-modifying pharmacological therapies in patients with heart failure with reduced ejection fraction: a comparative analysis of three randomised controlled trials. Lancet. 2020;396:121-128. doi: 10.1016/S0140-6736(20)30748-0
5. Solomon SD, Claggett B, Lewis EF, et al; TOPCAT Investigators. Influence of ejection fraction on outcomes and efficacy of spironolactone in patients with heart failure with preserved ejection fraction. Eur Heart J. 2016;37:455-462. doi: 10.1093/eurheartj/ehv464
6. Martin N, Manoharan K, Thomas J, et al. Beta-blockers and inhibitors of the renin-angiotensin aldosterone system for chronic heart failure with preserved ejection fraction. Cochrane Database Syst Rev. 2018;6:CD012721. doi: 10.1002/14651858.CD012721.pub2
7. Solomon SD, McMurray JJV, Anand IS, et al; PARAGON-HF Investigators and Committees. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609-1620. doi: 10.1056/NEJMoa1908655
8. Zannad F, Ferreira JP, Pocock SJ, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. 2020;396:819-829. doi: 10.1016/S0140-6736(20)31824-9
9. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536. doi: 10.1161/CIRCULATIONAHA. 119.040130
10. Bhatt DL, Szarek M, Steg PG, et al; SOLOIST-WHF Trial Investigators. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384:117-128. doi: 10.1056/NEJM oa2030183
11. Empagliflozin. GoodRx.com. Accessed June 3, 2022. www.goodrx.com/empagliflozin
12. FDA approves treatment for wider range of patients with heart failure. News release. US Food and Drug Administration; February 24, 2022. Accessed June 3, 2022. www.fda.gov/news-events/press-announcements/fda-approves-treatment-wider-range-patients-heart-failure
1. Anker SD, Butler J, Filippatos G, et al; EMPEROR-Preserved Trial Investigators. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451-1461. doi: 10.1056/NEJMoa2107038
2. Heidenreich PA, Bozkurt B, Aguilar D, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure: a report of the American College of Cardiology/American Heart Association Joint Committee on Clinical Practice Guidelines. Circulation. 2022;145:e895-e1032. doi: 10.1161/CIR.0000000000001063
3. Gevaert AB, Kataria R, Zannad F, et al. Heart failure with preserved ejection fraction: recent concepts in diagnosis, mechanisms and management. Heart. 2022;108:1342-1350. doi: 10.1136/heartjnl-2021-319605
4. Vaduganathan M, Claggett BL, Jhund PS, et al. Estimating lifetime benefits of comprehensive disease-modifying pharmacological therapies in patients with heart failure with reduced ejection fraction: a comparative analysis of three randomised controlled trials. Lancet. 2020;396:121-128. doi: 10.1016/S0140-6736(20)30748-0
5. Solomon SD, Claggett B, Lewis EF, et al; TOPCAT Investigators. Influence of ejection fraction on outcomes and efficacy of spironolactone in patients with heart failure with preserved ejection fraction. Eur Heart J. 2016;37:455-462. doi: 10.1093/eurheartj/ehv464
6. Martin N, Manoharan K, Thomas J, et al. Beta-blockers and inhibitors of the renin-angiotensin aldosterone system for chronic heart failure with preserved ejection fraction. Cochrane Database Syst Rev. 2018;6:CD012721. doi: 10.1002/14651858.CD012721.pub2
7. Solomon SD, McMurray JJV, Anand IS, et al; PARAGON-HF Investigators and Committees. Angiotensin-neprilysin inhibition in heart failure with preserved ejection fraction. N Engl J Med. 2019;381:1609-1620. doi: 10.1056/NEJMoa1908655
8. Zannad F, Ferreira JP, Pocock SJ, et al. SGLT2 inhibitors in patients with heart failure with reduced ejection fraction: a meta-analysis of the EMPEROR-Reduced and DAPA-HF trials. Lancet. 2020;396:819-829. doi: 10.1016/S0140-6736(20)31824-9
9. Kato ET, Silverman MG, Mosenzon O, et al. Effect of dapagliflozin on heart failure and mortality in type 2 diabetes mellitus. Circulation. 2019;139:2528-2536. doi: 10.1161/CIRCULATIONAHA. 119.040130
10. Bhatt DL, Szarek M, Steg PG, et al; SOLOIST-WHF Trial Investigators. Sotagliflozin in patients with diabetes and recent worsening heart failure. N Engl J Med. 2021;384:117-128. doi: 10.1056/NEJM oa2030183
11. Empagliflozin. GoodRx.com. Accessed June 3, 2022. www.goodrx.com/empagliflozin
12. FDA approves treatment for wider range of patients with heart failure. News release. US Food and Drug Administration; February 24, 2022. Accessed June 3, 2022. www.fda.gov/news-events/press-announcements/fda-approves-treatment-wider-range-patients-heart-failure
PRACTICE CHANGER
Consider adding empagliflozin 10 mg to usual therapy to reduce hospitalization of symptomatic patients with
STRENGTH OF RECOMMENDATION
B: Based on a single, good-quality, multicenter, randomized controlled trial.1
Anker SD, Butler J, Filippatos G, et al; EMPEROR-Preserved Trial Investigators. Empagliflozin in heart failure with a preserved ejection fraction. N Engl J Med. 2021;385:1451-1461. doi: 10.1056/NEJM oa2107038

40-year-old woman • fever • rash • arthralgia • Dx?
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8

The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.

At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8
The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.
At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
THE CASE
A 40-year-old woman with no significant medical history sought care at the emergency department for a fever, rash, and arthralgia. On admission, she had worsening bilateral ankle pain and was having difficulty walking. During the previous 3 months, she’d had 3 episodes of tonsillitis, all of which were presumed to be caused by Streptococcus, although no swabs were obtained. Her primary care physician treated her with antibiotics each time: 1 round of amoxicillin 500 mg twice daily for 10 days and 2 rounds of amoxicillin/clavulanate 875 mg twice daily for 7 to 10 days. During the previous month, she’d experienced intermittent fevers ranging from 100.2 °F to 100.8
The patient said that 2 weeks prior to her admission to the hospital, she’d developed a rash on her right arm, which was papular, nondraining, nonpruritic, and not painful (FIGURE 1). Six days later, the rash spread to her left arm, chest, and back, with a few lesions on her legs (FIGURE 2). A few days later, she developed arthralgias in her hips, knees, and ankles. These were associated with the appearance of large, flat, erythematous lesions on her anterior lower extremities (FIGURE 2). About 5 days before she was admitted to our hospital, the patient was seen at another hospital and treated for possible cellulitis with cephalexin (500 mg 4 times daily for 5-7 days), but her symptoms persisted.
At this point, she sought care at our hospital for her worsening lower extremity arthralgia, difficulty walking, and the persistent rash. An initial lab report showed a white blood cell (WBC) count of 12.6 × 103/µL (normal range, 4.0-10.0 × 103/µL) with an absolute neutrophil count of 9.7 × 103/µL (normal, 1.7-7.0 × 103/µL). Her C-reactive protein (CRP) level was elevated (194.7 mg/L; normal, 0.0-5.0 mg/L), as was her erythrocyte sedimentation rate (ESR) (102.0 mm/h; normal, 0.0-20.0 mm/h). A rapid pharyngeal strep test was negative. Her anti-streptolysin O (ASO) titer was elevated (2092.0 IU/mL; normal, < 250.0 IU/mL), and her rheumatic factor was mildly elevated (19.0 IU/mL; normal, 0.0-14.0 IU/mL). An antinuclear antibody panel was positive at 1:80. Further testing was performed, and the patient was found to be negative for Sjögren syndrome A, Sjögren syndrome B, anti-Smith, scleroderma-70, double-stranded DNA, and chromatin AB—making an autoimmune disease unlikely.
THE DIAGNOSIS
The patient met the American Heart Association’s revised Jones criteria for the diagnosis of rheumatic fever: She had a positive ASO titer; polyarthritis and subcutaneous nodules (2 major criteria); and ESR > 60 mm/h and CRP > 3 mg/L (1 minor criterion).1 She started taking naproxen 500 mg twice per day and was given a penicillin G 1.5-million-unit injection. A transthoracic echocardiogram also was performed during her admission to rule out endocarditis; no abnormalities were found.
A few days after starting treatment for rheumatic fever, the patient’s WBC count returned to within normal limits and her joint swelling and pain improved; however, her rash did not go away, leading us to wonder if there was a second disease at work. Dermatology was consulted, and a punch biopsy was obtained. The results showed acute febrile neutrophilic dermatosis, or Sweet syndrome.
DISCUSSION
Sweet syndrome is considered rare, and incidence numbers are elusive.2 It has a worldwide distribution and no racial bias.3 Sweet syndrome usually occurs in women ages 30 to 50 years, although it may also occur in younger adults and children.
Three subtypes have been defined based on etiology: (1) classical (or idiopathic) Sweet syndrome; (2) malignancy-associated Sweet syndrome, which is most often related to acute myelogenous leukemia; and (3) drug-induced Sweet syndrome, which is usually associated with granulocyte colony–stimulating factor treatment.4 Our patient had the most common subtype: classical Sweet syndrome.
Continue to: What you'll see
What you’ll see.
Corticosteroid therapy is the gold standard for treatment of classical Sweet syndrome. Dosing usually starts with prednisone 1 mg/kg/d, which can be tapered to 10 mg/d within 4 to 6 weeks.5 If steroid treatment is contraindicated in the patient, alternative treatments are colchicine 0.5 mg 3 times daily for 10 to 21 days or enteric-coated potassium iodide 300 mg 3 times daily until the rash subsides.5 Without treatment, symptoms may resolve within weeks to months; with treatment, the rash usually resolves within 2 to 5 days. Some resistant forms may require 2 to 3 months of treatment.
There is a risk of recurrence in approximately one-third of patients after successful treatment of classical Sweet syndrome.5 Recurrence can be caused by another inciting factor (ie, irritable bowel disease, upper respiratory tract infection, malignancy, or a new medication), making a new investigation necessary. However, treatment would entail the same medications.5
The patient was placed on penicillin V 250 mg twice daily for 5 years due to the significant risk of carditis in the setting of rheumatic fever. She started an oral steroid regimen of a prednisone weekly taper, starting with 60 mg/d, for 4 to 6 weeks. Her papular rash improved soon after initiation of steroid therapy.
THE TAKEAWAY
On presentation, this patient’s symptoms met the Jones criteria for rheumatic fever, but she did not respond to treatment. This led us to revisit her case, order additional tests, and identify a second diagnosis—Sweet syndrome—that responded positively to treatment. This case is a reminder that sometimes the signs and symptoms we are looking at are the result of 2 underlying illnesses, with 1 possibly triggering the other. That was likely what occurred in this case.
Farah Leclercq, DO, Department of Family Medicine, University of Florida, 12041 Southwest 1 Lane, Gainesville, FL 32607; [email protected]
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34
1. Gewitz MH, Baltimore SR, Tani LY, et al. Revision of the Jones Criteria for the diagnosis of acute rheumatic fever in the era of doppler echocardiography: a scientific statement from the American Heart Association. Circulation. 2015;131:1806-1818. doi: 10.1161/CIR.0000000000000205
2. Joshi TP, Friske SK, Hsiou DA, Duvic M. New practical aspects of Sweet syndrome. Am J Clin Dermatol. 2022;23:301-318. doi: 10.1007/s40257-022-00673-4
3. Cohen PR, Kurzrock R. Sweets syndrome revisited: a review of disease concepts. Int J Dermatol. 2003;42:761-778. doi: 10.1046/j.1365-4362.2003.01891.x
4. Merola JF. Sweet syndrome (acute febrile neutrophilic dermatosis): pathogenesis, clinical manifestations, and diagnosis. UpToDate. August 9, 2020. Accessed October 27, 2022. www.uptodate.com/contents/sweet-syndrome-acute-febrile-neutrophilic-dermatosis-pathogenesis-clinical-manifestations-and-diagnosis
5. Cohen PR. Sweets syndrome—a comprehensive review of an acute febrile neutrophilic dermatosis. Orphanet J Rare Dis. 2007;2:34. doi: 10.1186/1750-1172-2-34

Whom to screen for anxiety and depression: Updated USPSTF recommendations
In September 2022, the US Preventive Services Task Force (USPSTF) released 2 sets of draft recommendations on screening for 3 mental health conditions in adults: anxiety, depression, and suicide risk.1,2 These draft recommendations are summarized in TABLE 11-4 along with finalized recommendations on the same topics for children and adolescents, published in October 2022.3,4

The recommendations on depression and suicide risk screening in adults are updates of previous recommendations (2016 for depression and 2014 for suicide risk) with no major changes. Screening for anxiety is a topic addressed for the first time this year for adults and for children and adolescents.1,3
The recommendations are fairly consistent between age groups. A “B” recommendation supports screening for major depression in all patients starting at age 12 years, including during pregnancy and the postpartum period. (See TABLE 1 for grade definitions.) For all age groups, evidence was insufficient to recommend screening for suicide risk. A “B” recommendation was also assigned to screening for anxiety in those ages 8 to 64 years. The USPSTF believes the evidence is insufficient to make a recommendation on screening for anxiety among adults ≥ 65 years of age.
The anxiety disorders common to both children and adults included in the USPSTF recommendations are generalized anxiety disorder, social anxiety disorder, panic disorder, separation anxiety disorder, phobias, selective mutism, and anxiety type not specified. For adults, the USPSTF also includes substance/medication-induced anxiety and anxiety due to other medical conditions.
Adults with anxiety often present with generalized complaints such as sleep disturbance, pain, and other somatic disorders that can remain undiagnosed for years. The USPSTF cites a lifetime prevalence of anxiety disorders of 26.4% for men and 40.4% for women, although the data used are 10 years old.5 The cited rate of generalized anxiety in pregnancy is 8.5% to 10.5%, and in the postpartum period, 4.4% to 10.8%.6
The data on direct benefits and harms of screening for anxiety in adults through age 64 are sparse. Nevertheless, the USPSTF deemed that screening tests for anxiety have adequate accuracy and that psychological interventions for anxiety result in moderate reduction of anxiety symptoms. Pharmacologic interventions produce a small benefit, although there is a lack of evidence for pharmacotherapy in pregnant and postpartum women. There is even less evidence of benefit for treatment in adults ≥ 65 years of age.1
How anxiety screening tests compare
Screening tests for anxiety in adults reviewed by the USPSTF included the Generalized Anxiety Disorder (GAD) scale and the Edinburgh Postnatal Depression Scale (EPDS) anxiety subscale.1 The most studied tools are the GAD-2 and GAD-7.
Continue to: The sensitivity and specificity...
The sensitivity and specificity of each test depends on the cutoff used. With the GAD-2, a cutoff of 2 or more resulted in a sensitivity of 94% and a specificity of 68% for detecting generalized anxiety.7 A cutoff of 3 or more resulted in a sensitivity of 81% and a specificity of 86%.7 The GAD-7, using 10 as a cutoff, achieves a sensitivity of 79% and a specificity of 89%.7 Given the similar performance of the 2 options, the GAD-2 (TABLE 28,9) is probably preferable for use in primary care because of its ease of administration.
")
The tests evaluated by the USPSTF for anxiety screening in children and adolescents ≥ 8 years of age included the Screen for Child Anxiety Related Disorders (SCARED) and the Patient Health Questionnaire–Adolescent (PHQ-A).3 These tools ask more questions than the adult screening tools do: 41 for the SCARED and 13 for the PHQ-A. The sensitivity of SCARED for generalized anxiety disorder was 64% and the specificity was 63%.10 The sensitivity of the PHQ-A was 50% and the specificity was 98%.10
Various versions of all of these screening tools can be easily located on the internet. Search for them using the acronyms.
Screening for major depression
The depression screening tests the USPSTF examined were various versions of the Patient Health Questionnaire (PHQ), the Center for Epidemiologic Studies Depression Scale (CES-D), the Geriatric Depression Scale (GDS) in older adults, and the EPDS in postpartum and pregnant persons.7
A 2-question version of the PHQ was found to have a sensitivity of 91% with a specificity of 67%. The 9-question PHQ was found to have a similar sensitivity (88%) but better specificity (85%).7 TABLE 311 lists the 2 questions in the PHQ-2 and explains how to score the results.
")
Continue to: The most commonly...
The most commonly studied screening tool for adolescents is the PHQ-A. Its sensitivity is 73% and specificity is 94%.12
The GAD-2 and PHQ-2 have the same possible answers and scores and can be combined into a 4-question screening tool to assess for anxiety and depression. If an initial screen for anxiety or depression (or both) is positive, further diagnostic testing and follow-up are needed.
Frequency of screening
The USPSTF recognized that limited information on the frequency of screening for both anxiety and depression does not support any recommendation on this matter. It suggested screening everyone once and then basing the need for subsequent screening tests on clinical judgment after considering risk factors and life events, with periodic rescreening of those at high risk. Finally, USPSTF recognized the many challenges to implementing screening tests for mental health conditions in primary care practice, but offered little practical advice on how to do this.
Suicide risk screening
As for the evidence on benefits and harms of screening for suicide risk in all age groups, the USPSTF still regards it as insufficient to make a recommendation. The lack of evidence applies to all aspects of screening, including the accuracy of the various screening tools and the potential benefits and harms of preventive interventions.2,7
Next steps
The recommendations on screening for depression, suicide risk, and anxiety in adults have been published as a draft, and the public comment period will be over by the time of this publication. The USPSTF generally takes 6 to 9 months to consider all the public comments and to publish final recommendations. The final recommendations on these topics for children and adolescents have been published since drafts were made available last April. There were no major changes between the draft and final versions.
1. USPSTF. Screening for anxiety in adults. Draft recommendation statement. Published September 20, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-recommendation/anxiety-adults-screening
2. USPSTF. Screening for depression and suicide risk in adults. Updated September 14, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-update-summary/screening-depression-suicide-risk-adults
3. USPSTF. Anxiety in children and adolescents: screening. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-anxiety-children-adolescents
4. USPSTF. Depression and suicide risk in children and adolescents: screening. Final recommendation statement. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-depression-suicide-risk-children-adolescents
5. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21:169-184. doi: 10.1002/mpr.1359
6. Misri S, Abizadeh J, Sanders S, et al. Perinatal generalized anxiety disorder: assessment and treatment. J Womens Health (Larchmt). 2015;24:762-770. doi: 10.1089/jwh.2014.5150
7. O’Connor E, Henninger M, Perdue LA, et al. Screening for depression, anxiety, and suicide risk in adults: a systematic evidence review for the US Preventive Services Task Force. Accessed November 22, 2022. www.uspreventiveservicestaskforce.org/home/getfilebytoken/dpG5pjV5yCew8fXvctFJNK
8. Sapra A, Bhandari P, Sharma S, et al. Using Generalized Anxiety Disorder-2 (GAD-2) and GAD-7 in a primary care setting. Cureus. 2020;12:e8224. doi: 10.7759/cureus.8224
9. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-1097. doi: 10.1001/archinte.166.10.1092
10. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for anxiety in children and adolescents: evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1445-1455. doi: 10.1001/jama.2022.16303
11. Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire‐2: validity of a two‐item depression screener. Med Care. 2003;41:1284‐1292. doi: 10.1097/01.MLR.0000093487.78664.3C
12. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for depression and suicide risk in children and adolescents: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1543-1556. doi:10.1001/jama.2022.16310
In September 2022, the US Preventive Services Task Force (USPSTF) released 2 sets of draft recommendations on screening for 3 mental health conditions in adults: anxiety, depression, and suicide risk.1,2 These draft recommendations are summarized in TABLE 11-4 along with finalized recommendations on the same topics for children and adolescents, published in October 2022.3,4
The recommendations on depression and suicide risk screening in adults are updates of previous recommendations (2016 for depression and 2014 for suicide risk) with no major changes. Screening for anxiety is a topic addressed for the first time this year for adults and for children and adolescents.1,3
The recommendations are fairly consistent between age groups. A “B” recommendation supports screening for major depression in all patients starting at age 12 years, including during pregnancy and the postpartum period. (See TABLE 1 for grade definitions.) For all age groups, evidence was insufficient to recommend screening for suicide risk. A “B” recommendation was also assigned to screening for anxiety in those ages 8 to 64 years. The USPSTF believes the evidence is insufficient to make a recommendation on screening for anxiety among adults ≥ 65 years of age.
The anxiety disorders common to both children and adults included in the USPSTF recommendations are generalized anxiety disorder, social anxiety disorder, panic disorder, separation anxiety disorder, phobias, selective mutism, and anxiety type not specified. For adults, the USPSTF also includes substance/medication-induced anxiety and anxiety due to other medical conditions.
Adults with anxiety often present with generalized complaints such as sleep disturbance, pain, and other somatic disorders that can remain undiagnosed for years. The USPSTF cites a lifetime prevalence of anxiety disorders of 26.4% for men and 40.4% for women, although the data used are 10 years old.5 The cited rate of generalized anxiety in pregnancy is 8.5% to 10.5%, and in the postpartum period, 4.4% to 10.8%.6
The data on direct benefits and harms of screening for anxiety in adults through age 64 are sparse. Nevertheless, the USPSTF deemed that screening tests for anxiety have adequate accuracy and that psychological interventions for anxiety result in moderate reduction of anxiety symptoms. Pharmacologic interventions produce a small benefit, although there is a lack of evidence for pharmacotherapy in pregnant and postpartum women. There is even less evidence of benefit for treatment in adults ≥ 65 years of age.1
How anxiety screening tests compare
Screening tests for anxiety in adults reviewed by the USPSTF included the Generalized Anxiety Disorder (GAD) scale and the Edinburgh Postnatal Depression Scale (EPDS) anxiety subscale.1 The most studied tools are the GAD-2 and GAD-7.
Continue to: The sensitivity and specificity...
The sensitivity and specificity of each test depends on the cutoff used. With the GAD-2, a cutoff of 2 or more resulted in a sensitivity of 94% and a specificity of 68% for detecting generalized anxiety.7 A cutoff of 3 or more resulted in a sensitivity of 81% and a specificity of 86%.7 The GAD-7, using 10 as a cutoff, achieves a sensitivity of 79% and a specificity of 89%.7 Given the similar performance of the 2 options, the GAD-2 (TABLE 28,9) is probably preferable for use in primary care because of its ease of administration.
The tests evaluated by the USPSTF for anxiety screening in children and adolescents ≥ 8 years of age included the Screen for Child Anxiety Related Disorders (SCARED) and the Patient Health Questionnaire–Adolescent (PHQ-A).3 These tools ask more questions than the adult screening tools do: 41 for the SCARED and 13 for the PHQ-A. The sensitivity of SCARED for generalized anxiety disorder was 64% and the specificity was 63%.10 The sensitivity of the PHQ-A was 50% and the specificity was 98%.10
Various versions of all of these screening tools can be easily located on the internet. Search for them using the acronyms.
Screening for major depression
The depression screening tests the USPSTF examined were various versions of the Patient Health Questionnaire (PHQ), the Center for Epidemiologic Studies Depression Scale (CES-D), the Geriatric Depression Scale (GDS) in older adults, and the EPDS in postpartum and pregnant persons.7
A 2-question version of the PHQ was found to have a sensitivity of 91% with a specificity of 67%. The 9-question PHQ was found to have a similar sensitivity (88%) but better specificity (85%).7 TABLE 311 lists the 2 questions in the PHQ-2 and explains how to score the results.
Continue to: The most commonly...
The most commonly studied screening tool for adolescents is the PHQ-A. Its sensitivity is 73% and specificity is 94%.12
The GAD-2 and PHQ-2 have the same possible answers and scores and can be combined into a 4-question screening tool to assess for anxiety and depression. If an initial screen for anxiety or depression (or both) is positive, further diagnostic testing and follow-up are needed.
Frequency of screening
The USPSTF recognized that limited information on the frequency of screening for both anxiety and depression does not support any recommendation on this matter. It suggested screening everyone once and then basing the need for subsequent screening tests on clinical judgment after considering risk factors and life events, with periodic rescreening of those at high risk. Finally, USPSTF recognized the many challenges to implementing screening tests for mental health conditions in primary care practice, but offered little practical advice on how to do this.
Suicide risk screening
As for the evidence on benefits and harms of screening for suicide risk in all age groups, the USPSTF still regards it as insufficient to make a recommendation. The lack of evidence applies to all aspects of screening, including the accuracy of the various screening tools and the potential benefits and harms of preventive interventions.2,7
Next steps
The recommendations on screening for depression, suicide risk, and anxiety in adults have been published as a draft, and the public comment period will be over by the time of this publication. The USPSTF generally takes 6 to 9 months to consider all the public comments and to publish final recommendations. The final recommendations on these topics for children and adolescents have been published since drafts were made available last April. There were no major changes between the draft and final versions.
In September 2022, the US Preventive Services Task Force (USPSTF) released 2 sets of draft recommendations on screening for 3 mental health conditions in adults: anxiety, depression, and suicide risk.1,2 These draft recommendations are summarized in TABLE 11-4 along with finalized recommendations on the same topics for children and adolescents, published in October 2022.3,4
The recommendations on depression and suicide risk screening in adults are updates of previous recommendations (2016 for depression and 2014 for suicide risk) with no major changes. Screening for anxiety is a topic addressed for the first time this year for adults and for children and adolescents.1,3
The recommendations are fairly consistent between age groups. A “B” recommendation supports screening for major depression in all patients starting at age 12 years, including during pregnancy and the postpartum period. (See TABLE 1 for grade definitions.) For all age groups, evidence was insufficient to recommend screening for suicide risk. A “B” recommendation was also assigned to screening for anxiety in those ages 8 to 64 years. The USPSTF believes the evidence is insufficient to make a recommendation on screening for anxiety among adults ≥ 65 years of age.
The anxiety disorders common to both children and adults included in the USPSTF recommendations are generalized anxiety disorder, social anxiety disorder, panic disorder, separation anxiety disorder, phobias, selective mutism, and anxiety type not specified. For adults, the USPSTF also includes substance/medication-induced anxiety and anxiety due to other medical conditions.
Adults with anxiety often present with generalized complaints such as sleep disturbance, pain, and other somatic disorders that can remain undiagnosed for years. The USPSTF cites a lifetime prevalence of anxiety disorders of 26.4% for men and 40.4% for women, although the data used are 10 years old.5 The cited rate of generalized anxiety in pregnancy is 8.5% to 10.5%, and in the postpartum period, 4.4% to 10.8%.6
The data on direct benefits and harms of screening for anxiety in adults through age 64 are sparse. Nevertheless, the USPSTF deemed that screening tests for anxiety have adequate accuracy and that psychological interventions for anxiety result in moderate reduction of anxiety symptoms. Pharmacologic interventions produce a small benefit, although there is a lack of evidence for pharmacotherapy in pregnant and postpartum women. There is even less evidence of benefit for treatment in adults ≥ 65 years of age.1
How anxiety screening tests compare
Screening tests for anxiety in adults reviewed by the USPSTF included the Generalized Anxiety Disorder (GAD) scale and the Edinburgh Postnatal Depression Scale (EPDS) anxiety subscale.1 The most studied tools are the GAD-2 and GAD-7.
Continue to: The sensitivity and specificity...
The sensitivity and specificity of each test depends on the cutoff used. With the GAD-2, a cutoff of 2 or more resulted in a sensitivity of 94% and a specificity of 68% for detecting generalized anxiety.7 A cutoff of 3 or more resulted in a sensitivity of 81% and a specificity of 86%.7 The GAD-7, using 10 as a cutoff, achieves a sensitivity of 79% and a specificity of 89%.7 Given the similar performance of the 2 options, the GAD-2 (TABLE 28,9) is probably preferable for use in primary care because of its ease of administration.
The tests evaluated by the USPSTF for anxiety screening in children and adolescents ≥ 8 years of age included the Screen for Child Anxiety Related Disorders (SCARED) and the Patient Health Questionnaire–Adolescent (PHQ-A).3 These tools ask more questions than the adult screening tools do: 41 for the SCARED and 13 for the PHQ-A. The sensitivity of SCARED for generalized anxiety disorder was 64% and the specificity was 63%.10 The sensitivity of the PHQ-A was 50% and the specificity was 98%.10
Various versions of all of these screening tools can be easily located on the internet. Search for them using the acronyms.
Screening for major depression
The depression screening tests the USPSTF examined were various versions of the Patient Health Questionnaire (PHQ), the Center for Epidemiologic Studies Depression Scale (CES-D), the Geriatric Depression Scale (GDS) in older adults, and the EPDS in postpartum and pregnant persons.7
A 2-question version of the PHQ was found to have a sensitivity of 91% with a specificity of 67%. The 9-question PHQ was found to have a similar sensitivity (88%) but better specificity (85%).7 TABLE 311 lists the 2 questions in the PHQ-2 and explains how to score the results.
Continue to: The most commonly...
The most commonly studied screening tool for adolescents is the PHQ-A. Its sensitivity is 73% and specificity is 94%.12
The GAD-2 and PHQ-2 have the same possible answers and scores and can be combined into a 4-question screening tool to assess for anxiety and depression. If an initial screen for anxiety or depression (or both) is positive, further diagnostic testing and follow-up are needed.
Frequency of screening
The USPSTF recognized that limited information on the frequency of screening for both anxiety and depression does not support any recommendation on this matter. It suggested screening everyone once and then basing the need for subsequent screening tests on clinical judgment after considering risk factors and life events, with periodic rescreening of those at high risk. Finally, USPSTF recognized the many challenges to implementing screening tests for mental health conditions in primary care practice, but offered little practical advice on how to do this.
Suicide risk screening
As for the evidence on benefits and harms of screening for suicide risk in all age groups, the USPSTF still regards it as insufficient to make a recommendation. The lack of evidence applies to all aspects of screening, including the accuracy of the various screening tools and the potential benefits and harms of preventive interventions.2,7
Next steps
The recommendations on screening for depression, suicide risk, and anxiety in adults have been published as a draft, and the public comment period will be over by the time of this publication. The USPSTF generally takes 6 to 9 months to consider all the public comments and to publish final recommendations. The final recommendations on these topics for children and adolescents have been published since drafts were made available last April. There were no major changes between the draft and final versions.
1. USPSTF. Screening for anxiety in adults. Draft recommendation statement. Published September 20, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-recommendation/anxiety-adults-screening
2. USPSTF. Screening for depression and suicide risk in adults. Updated September 14, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-update-summary/screening-depression-suicide-risk-adults
3. USPSTF. Anxiety in children and adolescents: screening. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-anxiety-children-adolescents
4. USPSTF. Depression and suicide risk in children and adolescents: screening. Final recommendation statement. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-depression-suicide-risk-children-adolescents
5. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21:169-184. doi: 10.1002/mpr.1359
6. Misri S, Abizadeh J, Sanders S, et al. Perinatal generalized anxiety disorder: assessment and treatment. J Womens Health (Larchmt). 2015;24:762-770. doi: 10.1089/jwh.2014.5150
7. O’Connor E, Henninger M, Perdue LA, et al. Screening for depression, anxiety, and suicide risk in adults: a systematic evidence review for the US Preventive Services Task Force. Accessed November 22, 2022. www.uspreventiveservicestaskforce.org/home/getfilebytoken/dpG5pjV5yCew8fXvctFJNK
8. Sapra A, Bhandari P, Sharma S, et al. Using Generalized Anxiety Disorder-2 (GAD-2) and GAD-7 in a primary care setting. Cureus. 2020;12:e8224. doi: 10.7759/cureus.8224
9. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-1097. doi: 10.1001/archinte.166.10.1092
10. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for anxiety in children and adolescents: evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1445-1455. doi: 10.1001/jama.2022.16303
11. Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire‐2: validity of a two‐item depression screener. Med Care. 2003;41:1284‐1292. doi: 10.1097/01.MLR.0000093487.78664.3C
12. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for depression and suicide risk in children and adolescents: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1543-1556. doi:10.1001/jama.2022.16310
1. USPSTF. Screening for anxiety in adults. Draft recommendation statement. Published September 20, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-recommendation/anxiety-adults-screening
2. USPSTF. Screening for depression and suicide risk in adults. Updated September 14, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/draft-update-summary/screening-depression-suicide-risk-adults
3. USPSTF. Anxiety in children and adolescents: screening. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-anxiety-children-adolescents
4. USPSTF. Depression and suicide risk in children and adolescents: screening. Final recommendation statement. Published October 11, 2022. Accessed November 22, 2022. https://uspreventiveservicestaskforce.org/uspstf/recommendation/screening-depression-suicide-risk-children-adolescents
5. Kessler RC, Petukhova M, Sampson NA, et al. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. Int J Methods Psychiatr Res. 2012;21:169-184. doi: 10.1002/mpr.1359
6. Misri S, Abizadeh J, Sanders S, et al. Perinatal generalized anxiety disorder: assessment and treatment. J Womens Health (Larchmt). 2015;24:762-770. doi: 10.1089/jwh.2014.5150
7. O’Connor E, Henninger M, Perdue LA, et al. Screening for depression, anxiety, and suicide risk in adults: a systematic evidence review for the US Preventive Services Task Force. Accessed November 22, 2022. www.uspreventiveservicestaskforce.org/home/getfilebytoken/dpG5pjV5yCew8fXvctFJNK
8. Sapra A, Bhandari P, Sharma S, et al. Using Generalized Anxiety Disorder-2 (GAD-2) and GAD-7 in a primary care setting. Cureus. 2020;12:e8224. doi: 10.7759/cureus.8224
9. Spitzer RL, Kroenke K, Williams JBW, et al. A brief measure for assessing generalized anxiety disorder: the GAD-7. Arch Intern Med. 2006;166:1092-1097. doi: 10.1001/archinte.166.10.1092
10. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for anxiety in children and adolescents: evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1445-1455. doi: 10.1001/jama.2022.16303
11. Kroenke K, Spitzer RL, Williams JB. The Patient Health Questionnaire‐2: validity of a two‐item depression screener. Med Care. 2003;41:1284‐1292. doi: 10.1097/01.MLR.0000093487.78664.3C
12. Viswanathan M, Wallace IF, Middleton JC, et al. Screening for depression and suicide risk in children and adolescents: updated evidence report and systematic review for the US Preventive Services Task Force. JAMA. 2022;328:1543-1556. doi:10.1001/jama.2022.16310

Incidence of cardiovascular events in patients with moderate-to-severe atopic dermatitis
Key clinical point: Patients with moderate-to-severe atopic dermatitis (AD) had the highest incidence rate (IR) per 1000 person-years for major adverse cardiovascular events (MACE), followed by venous thrombotic events (VTE), deep vein thrombosis (DVT), and pulmonary embolism (PE).
Major finding: The IR per 1000 person-years for MACE, VTE, DVT, and PE were 2.6 (95% CI 2.1-3.2), 2.0 (95% CI 1.5-2.5), 1.6 (95% CI 1.2-2.1), and 0.7 (95% CI 0.5-1.0), respectively.
Study details: This retrospective cohort study included 8197 patients aged ≥12 years with moderate-to-severe AD.
Disclosures: This study was funded by Pfizer, Inc. Some authors declared receiving grant funding from Pfizer. Two authors declared being current or former employees and shareholders of Pfizer.
Source: Hedderson MM et al. Rates of cardiovascular events among patients with moderate-to-severe atopic dermatitis in an integrated health care system: A retrospective cohort study. PLoS One. 2022;17(11):e0277469 (Nov 17). Doi: 10.1371/journal.pone.0277469
Key clinical point: Patients with moderate-to-severe atopic dermatitis (AD) had the highest incidence rate (IR) per 1000 person-years for major adverse cardiovascular events (MACE), followed by venous thrombotic events (VTE), deep vein thrombosis (DVT), and pulmonary embolism (PE).
Major finding: The IR per 1000 person-years for MACE, VTE, DVT, and PE were 2.6 (95% CI 2.1-3.2), 2.0 (95% CI 1.5-2.5), 1.6 (95% CI 1.2-2.1), and 0.7 (95% CI 0.5-1.0), respectively.
Study details: This retrospective cohort study included 8197 patients aged ≥12 years with moderate-to-severe AD.
Disclosures: This study was funded by Pfizer, Inc. Some authors declared receiving grant funding from Pfizer. Two authors declared being current or former employees and shareholders of Pfizer.
Source: Hedderson MM et al. Rates of cardiovascular events among patients with moderate-to-severe atopic dermatitis in an integrated health care system: A retrospective cohort study. PLoS One. 2022;17(11):e0277469 (Nov 17). Doi: 10.1371/journal.pone.0277469
Key clinical point: Patients with moderate-to-severe atopic dermatitis (AD) had the highest incidence rate (IR) per 1000 person-years for major adverse cardiovascular events (MACE), followed by venous thrombotic events (VTE), deep vein thrombosis (DVT), and pulmonary embolism (PE).
Major finding: The IR per 1000 person-years for MACE, VTE, DVT, and PE were 2.6 (95% CI 2.1-3.2), 2.0 (95% CI 1.5-2.5), 1.6 (95% CI 1.2-2.1), and 0.7 (95% CI 0.5-1.0), respectively.
Study details: This retrospective cohort study included 8197 patients aged ≥12 years with moderate-to-severe AD.
Disclosures: This study was funded by Pfizer, Inc. Some authors declared receiving grant funding from Pfizer. Two authors declared being current or former employees and shareholders of Pfizer.
Source: Hedderson MM et al. Rates of cardiovascular events among patients with moderate-to-severe atopic dermatitis in an integrated health care system: A retrospective cohort study. PLoS One. 2022;17(11):e0277469 (Nov 17). Doi: 10.1371/journal.pone.0277469
Baricitinib a promising treatment option for difficult-to-treat atopic dermatitis in daily practice
Key clinical point: Baricitinib could serve as an effective treatment option for patients with difficult-to-treat moderate-to-severe atopic dermatitis (AD), including those unresponsive to dupilumab treatment; however, a high discontinuation rate indicates its rather heterogenous efficacy.
Major finding: At week 16, the mean Eczema Area and Severity Index score and numerical rating scale-pruritis significantly decreased from 18.3 to 11.1 (P < .0001) and from 6.6 to 5.3 (P < .0001), respectively. The most frequent adverse events (AE) were nausea (11.8%), urinary tract infection (9.8%), and herpes simplex infections (7.8%). Baricitinib treatment was discontinued by 43.2% of patients due to ineffectiveness or AE.
Study details: This multicenter prospective observational study included 51 adult patients with moderate-to-severe AD from the BioDay registry who received baricitinib over 16 weeks.
Disclosures: This study did not report a source of funding. Some authors declared serving as speakers, consultants, advisory board members, or investigators for various organizations.
Source: Boesjes CM et al. Daily practice experience of baricitinib treatment for patients with difficult-to-treat atopic dermatitis: Results from the BioDay registry. Acta Derm Venereol. 2022;102:adv00820 (Nov 24). Doi: 10.2340/actadv.v102.3978
Key clinical point: Baricitinib could serve as an effective treatment option for patients with difficult-to-treat moderate-to-severe atopic dermatitis (AD), including those unresponsive to dupilumab treatment; however, a high discontinuation rate indicates its rather heterogenous efficacy.
Major finding: At week 16, the mean Eczema Area and Severity Index score and numerical rating scale-pruritis significantly decreased from 18.3 to 11.1 (P < .0001) and from 6.6 to 5.3 (P < .0001), respectively. The most frequent adverse events (AE) were nausea (11.8%), urinary tract infection (9.8%), and herpes simplex infections (7.8%). Baricitinib treatment was discontinued by 43.2% of patients due to ineffectiveness or AE.
Study details: This multicenter prospective observational study included 51 adult patients with moderate-to-severe AD from the BioDay registry who received baricitinib over 16 weeks.
Disclosures: This study did not report a source of funding. Some authors declared serving as speakers, consultants, advisory board members, or investigators for various organizations.
Source: Boesjes CM et al. Daily practice experience of baricitinib treatment for patients with difficult-to-treat atopic dermatitis: Results from the BioDay registry. Acta Derm Venereol. 2022;102:adv00820 (Nov 24). Doi: 10.2340/actadv.v102.3978
Key clinical point: Baricitinib could serve as an effective treatment option for patients with difficult-to-treat moderate-to-severe atopic dermatitis (AD), including those unresponsive to dupilumab treatment; however, a high discontinuation rate indicates its rather heterogenous efficacy.
Major finding: At week 16, the mean Eczema Area and Severity Index score and numerical rating scale-pruritis significantly decreased from 18.3 to 11.1 (P < .0001) and from 6.6 to 5.3 (P < .0001), respectively. The most frequent adverse events (AE) were nausea (11.8%), urinary tract infection (9.8%), and herpes simplex infections (7.8%). Baricitinib treatment was discontinued by 43.2% of patients due to ineffectiveness or AE.
Study details: This multicenter prospective observational study included 51 adult patients with moderate-to-severe AD from the BioDay registry who received baricitinib over 16 weeks.
Disclosures: This study did not report a source of funding. Some authors declared serving as speakers, consultants, advisory board members, or investigators for various organizations.
Source: Boesjes CM et al. Daily practice experience of baricitinib treatment for patients with difficult-to-treat atopic dermatitis: Results from the BioDay registry. Acta Derm Venereol. 2022;102:adv00820 (Nov 24). Doi: 10.2340/actadv.v102.3978
Incidental skin finding

This patient was given a diagnosis of cutaneous mastocytosis. The condition was previously known as urticaria pigmentosa, but in 2016 the World Health Organization reclassified the disease to better suit its pathophysiology as a myeloid cell disorder.1
As the name suggests, this condition—which involves lesions in a sporadic, truncal distribution—involves overactivation of mastocytes at the tissue level from various stimuli, resulting in histocyte degranulation and hyperpigmentation. The exact cause is unknown. What is known is that it is often associated with other allergic or immunologic conditions and is thought to be related to mutations in the gene for CD-117’s receptor for tyrosine kinase.1 Incidence is similar to that of asthma, in that it occurs more often in younger patients; a majority of affected individuals will grow out of the disease by adolescence.1
Although most patients do not experience severe symptomatology, it is still important to differentiate cutaneous vs systemic mastocytosis. If a patient presents with inexplicable systemic symptoms of malaise, vague abdominal pain, heartburn, or flushing, the physician should consider systemic mastocytosis, idiopathic anaphylaxis, or hereditary alpha-tryptasemia.2
The test of choice is a serum tryptase test; levels will be elevated with systemic mastocytosis. Consider obtaining a skin biopsy if lesions are ambiguous or nondistinct.
There is no definitive cure for systemic or cutaneous mastocytosis, so treatment is directed at symptoms. Start by advising patients to avoid triggers and to refrain from scratching the affected areas. Topical antihistamines and oral nonsedating antihistamines can be helpful. If symptoms are more severe, refer the patient to an allergist/immunologist or to a hematologist for further medical management.2
The patient in this case had no systemic symptoms, so she was advised to continue taking oral loratadine 10 mg/d, which had been helpful, and to avoid rubbing her skin.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Murtaza Rizvi, MD, and Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405. doi: 10.1182/blood-2016-03-643544.
2. Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2016; 137:35-45. doi: 10.1016/j.jaci.2015.08.034
This patient was given a diagnosis of cutaneous mastocytosis. The condition was previously known as urticaria pigmentosa, but in 2016 the World Health Organization reclassified the disease to better suit its pathophysiology as a myeloid cell disorder.1
As the name suggests, this condition—which involves lesions in a sporadic, truncal distribution—involves overactivation of mastocytes at the tissue level from various stimuli, resulting in histocyte degranulation and hyperpigmentation. The exact cause is unknown. What is known is that it is often associated with other allergic or immunologic conditions and is thought to be related to mutations in the gene for CD-117’s receptor for tyrosine kinase.1 Incidence is similar to that of asthma, in that it occurs more often in younger patients; a majority of affected individuals will grow out of the disease by adolescence.1
Although most patients do not experience severe symptomatology, it is still important to differentiate cutaneous vs systemic mastocytosis. If a patient presents with inexplicable systemic symptoms of malaise, vague abdominal pain, heartburn, or flushing, the physician should consider systemic mastocytosis, idiopathic anaphylaxis, or hereditary alpha-tryptasemia.2
The test of choice is a serum tryptase test; levels will be elevated with systemic mastocytosis. Consider obtaining a skin biopsy if lesions are ambiguous or nondistinct.
There is no definitive cure for systemic or cutaneous mastocytosis, so treatment is directed at symptoms. Start by advising patients to avoid triggers and to refrain from scratching the affected areas. Topical antihistamines and oral nonsedating antihistamines can be helpful. If symptoms are more severe, refer the patient to an allergist/immunologist or to a hematologist for further medical management.2
The patient in this case had no systemic symptoms, so she was advised to continue taking oral loratadine 10 mg/d, which had been helpful, and to avoid rubbing her skin.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Murtaza Rizvi, MD, and Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
This patient was given a diagnosis of cutaneous mastocytosis. The condition was previously known as urticaria pigmentosa, but in 2016 the World Health Organization reclassified the disease to better suit its pathophysiology as a myeloid cell disorder.1
As the name suggests, this condition—which involves lesions in a sporadic, truncal distribution—involves overactivation of mastocytes at the tissue level from various stimuli, resulting in histocyte degranulation and hyperpigmentation. The exact cause is unknown. What is known is that it is often associated with other allergic or immunologic conditions and is thought to be related to mutations in the gene for CD-117’s receptor for tyrosine kinase.1 Incidence is similar to that of asthma, in that it occurs more often in younger patients; a majority of affected individuals will grow out of the disease by adolescence.1
Although most patients do not experience severe symptomatology, it is still important to differentiate cutaneous vs systemic mastocytosis. If a patient presents with inexplicable systemic symptoms of malaise, vague abdominal pain, heartburn, or flushing, the physician should consider systemic mastocytosis, idiopathic anaphylaxis, or hereditary alpha-tryptasemia.2
The test of choice is a serum tryptase test; levels will be elevated with systemic mastocytosis. Consider obtaining a skin biopsy if lesions are ambiguous or nondistinct.
There is no definitive cure for systemic or cutaneous mastocytosis, so treatment is directed at symptoms. Start by advising patients to avoid triggers and to refrain from scratching the affected areas. Topical antihistamines and oral nonsedating antihistamines can be helpful. If symptoms are more severe, refer the patient to an allergist/immunologist or to a hematologist for further medical management.2
The patient in this case had no systemic symptoms, so she was advised to continue taking oral loratadine 10 mg/d, which had been helpful, and to avoid rubbing her skin.
Image courtesy of Daniel Stulberg, MD. Text courtesy of Murtaza Rizvi, MD, and Daniel Stulberg, MD, FAAFP, Professor and Chair, Department of Family and Community Medicine, Western Michigan University Homer Stryker, MD School of Medicine, Kalamazoo.
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405. doi: 10.1182/blood-2016-03-643544.
2. Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2016; 137:35-45. doi: 10.1016/j.jaci.2015.08.034
1. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391-2405. doi: 10.1182/blood-2016-03-643544.
2. Hartmann K, Escribano L, Grattan C, et al. Cutaneous manifestations in patients with mastocytosis: Consensus report of the European Competence Network on Mastocytosis; the American Academy of Allergy, Asthma & Immunology; and the European Academy of Allergology and Clinical Immunology. J Allergy Clin Immunol 2016; 137:35-45. doi: 10.1016/j.jaci.2015.08.034
