User login
Medical causes of back pain
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
To the Editor: In their otherwise excellent review, “Masquerade: Medical causes of back pain” (Cleve Clin J Med 2007; 74:905–913), Dr. Klineberg et al seem to confuse two distinct pathologic processes—aortic dissection and rupture of an aortic aneurysm. Parts of their description seem to fit the pathology of abdominal aortic aneurysm, with a pulsatile abdominal mass, sentinel bleeding, and rupture risk with a size over 6 cm, whereas other parts seem to correspond to aortic dissection, with severe, ripping pain and an association with Marfan syndrome. They also use the terminology “dissecting aortic aneurysm,” which again implies a single entity, when in fact the two conditions rarely occur together. The authors are not alone in their use of this misnomer: a review of the Web sites of renowned universities reveals use of the same terminology. The readers would have been better served if the authors had discussed “acute aortic dissection” and “ruptured aortic aneurysm” as two separate causes of back pain, with a note that in rare cases an aortic aneurysm can develop a dissection.
In reply: Medical causes of back pain
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.
In Reply: We appreciate Dr. Hirsch’s comments and are pleased to expand the discussion of this important point.
He is correct in his assertion that dissection and aneurysm are distinct processes. But the goal of this review was to remind practitioners to consider the aorta as a possible source of pain when it occurs acutely or in an atypical manner.
A number of aortic processes can cause back pain, and aneurysm and dissection are two of them, aneurysm being more common than aortic dissection. But the pain can also be from aortic ulceration, aortitis, contained rupture of an aneurysm, and other more esoteric problems.
Aortic dissection often presents as a tearing, severe, thoracic back pain. Pain from a progressive abdominal aneurysm is more commonly referred to the lower back or flank and can be severe and unrelenting. It is rarely described as a tearing pain like that of dissection.
It is difficult on initial physical examination to distinguish aneurysm from dissection. The key to diagnosing aneurysm is to detect the pulsatile abdominal mass. A pulsatile, tender abdominal mass with hypotension and back pain is classically associated with rupture of an abdominal aortic aneurysm. The combination of back pain, a deficit in peripheral pulses, and hypertension is more often associated with dissection.
Without imaging and appropriate consultation, it is difficult for even an experienced provider to definitively diagnose these disorders. Without a bit of suspicion, even with a careful physical examination either disorder might be overlooked entirely, with disastrous effect. The purpose of our review was to remind the reader that these conditions, while uncommon or even rare, do occur and should be sought out in patients presenting with acute, atypical lumbar and thoracic back pain. As with each of the conditions discussed in this review, the decision to linger a bit over the patient’s history and then perform a basic, focused physical examination can be life-saving.
Should patients on long-term warfarin take aspirin for heart disease?
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
The literature on this topic is limited, but it suggests that the decision to prescribe aspirin to patients already taking warfarin (Coumadin) should be individualized. On one hand, the cardiovascular benefit of starting or continuing aspirin in patients already on warfarin outweighs the increased risk of bleeding in patients presenting with an acute coronary syndrome or those with mechanical heart valves or coronary stents. However, for patients with stable coronary artery disease or at risk of coronary disease, the benefit of adding aspirin is not substantial, and continuing warfarin alone may be the preferred strategy.
In patients with coronary artery disease, aspirin has been shown to reduce the rate of death due to all causes by about 18% and the rate of vascular events by about 25% to 30%.1,2 Warfarin is at least as effective as aspirin in reducing the rate of future cardiovascular events (especially if the target international normalized ratio [INR] is greater than 2.5), albeit with a higher bleeding risk.3–6
The decision to prescribe or continue aspirin in patients with coronary artery disease who also need long-term anticoagulation with warfarin for an unrelated medical problem, such as pulmonary emboli, requires careful assessment of the individual patient’s bleeding risk and cardiovascular benefit.
ESTIMATING THE BLEEDING RISK FOR PATIENTS ON WARFARIN
In patients taking warfarin, the risk of major bleeding (defined in most studies as hospitalization because of bleeding and requiring transfusion of at least two units of packed red cells, or an intracranial, intraperitoneal, or fatal bleeding episode) is reported to be about 2.0% to 3.8% per person-year.7–11 The risk of major bleeding with aspirin alone is estimated to be 0.13% per person-year,12 but when aspirin is combined with warfarin, the risk increases significantly.13 In a meta-analysis of randomized controlled trials,14 the risk of major bleeding was calculated to be about 1.5 times higher with combination therapy with aspirin and warfarin than with warfarin alone.
The individual’s bleeding risk depends on specific risk factors and the intensity of anticoagulation.15 The outpatient Bleeding Risk Index (BRI) can be used to estimate the bleeding risk for patients on warfarin.16 The BRI includes four risk factors for major bleeding, each scored as 1 point:
- Age 65 or older
- History of gastrointestinal bleeding
- History of stroke
- One or more comorbid conditions—recent myocardial infarction, anemia (hematocrit < 30%), renal impairment (serum creatinine level > 1.5 mg/dL), or diabetes mellitus.
The risk is low if the score is 0, moderate if the score is 1 or 2, and high if the score is 3 or more. In a validation study of the BRI, the rate of major bleeding was found to be 0.8%, 2.5%, and 10.6% per person-year on warfarin in the low, intermediate, and high-risk groups, respectively.17 In addition, compared with patients with a target INR of 2.5, those with a target INR higher than 3.0 have a higher frequency of bleeding episodes.10,15
CONDITIONS IN WHICH ADDING ASPIRIN TO WARFARIN IS FAVORABLE
Acute coronary syndromes
Drugs that inhibit platelet function are the mainstay of medical treatment for acute coronary syndromes. The American College of Cardiology/American Heart Association (ACC/AHA) guidelines recommend that aspirin be started in patients who have an acute myocardial infarction even if they have been receiving warfarin long-term and their INR is in the therapeutic range, especially if a percutaneous coronary intervention is anticipated.4
After percutaneous coronary intervention
In patients who have undergone percutaneous coronary intervention with stent implantation, dual antiplatelet therapy with aspirin and a thienopyridine—ie, clopidogrel (Plavix) or ticlopidine (Ticlid)—is superior to aspirin or warfarin alone in reducing the risk of stent thrombosis and major adverse cardiovascular events such as myocardial infarction or urgent revascularization.18,19 If patients have an indication for long-term anticoagulation, triple therapy with aspirin, warfarin, and clopidogrel or ticlopidine may be considered in order to reduce the likelihood of stent thrombosis.4,20,21 In such patients the INR should be maintained between 2.0 and 3.0 to reduce the risk of bleeding.
The duration of triple therapy is guided by the type of stent used. For bare metal stents, aspirin, clopidogrel or ticlopidine, and warfarin should be given for at least 1 month, after which clopidogrel or ticlopidine may be discontinued. If drug-eluting stents are used, the duration of clopidogrel or ticlopidine therapy should be extended to 1 year or more.4,22
Mechanical heart valves
In patients with mechanical heart valves, the combination of aspirin and warfarin has been shown to decrease the frequency of thromboembolism.23 Guidelines recommend adding aspirin (75 to 100 mg per day) to warfarin in all patients with mechanical valves, especially in patients who have had an embolus while on warfarin therapy or who have a history of cerebrovascular or peripheral vascular disease, a hypercoagulable state, or coronary artery disease.24
CONDITIONS IN WHICH WARFARIN ALONE MAY BE SUFFICIENT
At risk of coronary artery disease
Aspirin therapy is generally recommended as primary prevention for patients whose estimated risk of coronary events is 1.5% per year or higher.25 However, warfarin has also been shown to be effective in the primary prevention of coronary artery disease in men,26 and for patients already taking warfarin, the possible benefit of adding aspirin for primary prevention is outweighed by the increased risk of major bleeding.14 The Medical Research Council directly compared low-intensity warfarin therapy (mean INR 1.47), aspirin, and placebo in a two-by-two factorial study of primary prevention of ischemic heart disease in men.26 Warfarin was more effective than aspirin, and men who received warfarin plus aspirin or warfarin plus placebo had a rate of ischemic heart disease that was 21% lower than those who received aspirin plus placebo or double placebo, and their rate of all-cause mortality was 17% lower. Combining aspirin and warfarin for patients at risk of coronary disease led to a higher rate of major bleeding but no difference in cardiovascular events or all-cause mortality (odds ratio 0.98; 95% confidence interval 0.77–1.25).14
Stable coronary artery disease without mechanical heart valves or stents
Large randomized trials have found warfarin to be effective in secondary prevention of coronary artery disease.4–6 For most patients with stable coronary artery disease (ie, who have had no ischemic events or coronary interventions in the last 6 months) who need anticoagulation because of atrial fibrillation or venous thromboembolism, warfarin alone (target INR 2.0–3.0) should provide satisfactory antithrombotic prophylaxis against both cerebral and myocardial ischemic events.27 The addition of an antiplatelet agent is not required unless a patient has a coronary stent, a mechanical valve, or an excessive thrombotic risk.4,24,27
TAKE-HOME POINTS
For patients receiving warfarin therapy, whether to add or continue aspirin to their treatment is a common clinical question. The risk of bleeding is greater with combination therapy than with warfarin alone. The cardiovascular benefit varies depending on the clinical situation:
- In patients who have had an acute coronary syndrome or who have a coronary stent or mechanical valve, combination therapy is usually recommended because the benefits outweigh the risks.
- In patients with stable coronary artery disease or those without coronary artery disease who are at risk of coronary events, the risks outweigh the benefits. Combination therapy is usually not indicated in these patients.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
- Weisman SM, Graham DY. Evaluation of the benefits and risks of low-dose aspirin in the secondary prevention of cardiovascular and cerebrovascular events. Arch Intern Med 2002; 162:2197–2202.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Hurlen M, Abdelnoor M, Smith P, Erikssen J, Arnesen H. Warfarin, aspirin, or both after myocardial infarction. N Engl J Med 2002; 347:969–974.
- Antman EM, Anbe DT, Armstrong PW, et al. ACC/AHA guidelines for the management of patients with ST-elevation myocardial infarction; a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines (Committee to Revise the 1999 Guidelines for the Management of patients with acute myocardial infarction). J Am Coll Cardiol 2004; 44:E1–E211.
- Van Es RF, Jonker JJ, Verheugt FW, et al. Antithrombotics in the Secondary Prevention of Events in Coronary Thrombosis-2 (ASPECT-2) Research Group. Aspirin and coumadin after acute coronary syndromes (the ASPECT-2 study): a randomised controlled trial. Lancet 2002; 360:109–113.
- Anand SS, Yusuf S. Oral anticoagulant therapy in patients with coronary artery disease: a meta-analysis. JAMA 1999; 282:2058–2067.
- Schulman S, Granqvist S, Holmstrom M, et al. The duration of oral anticoagulant therapy after a second episode of venous thromboembolism. The Duration of Anticoagulation Trial Study Group. N Engl J Med 1997; 336:393–398.
- Kearon C, Gent M, Hirsh J, et al. A comparison of three months of anticoagulation with extended anticoagulation for a first episode of idiopathic venous thromboembolism. N Engl J Med 1999; 340:901–907.
- Agnelli G, Prandoni P, Santamaria MG, et al. Three months versus one year of oral anticoagulant therapy for idiopathic deep venous thrombosis. Warfarin Optimal Duration Italian Trial Investigators. N Engl J Med 2001; 345:165–169.
- Levine MN, Raskob G, Beyth RJ, Kearon C, Schulman S. Hemorrhagic complications of anticoagulant treatment: the Seventh ACCP Conference on Antithrombotic and Thrombolytic Therapy. Chest 2004; 126 suppl:287S–310S.
- Linkins LA, Choi PT, Douketis JD. Clinical impact of bleeding in patients taking oral anticoagulant therapy for venous thromboembolism: a meta-analysis. Ann Intern Med 2003; 139:893–900.
- McQuaid KR, Laine L. Systematic review and meta-analysis of adverse events of low-dose aspirin and clopidogrel in randomized controlled trials. Am J Med 2006; 119:624–638.
- Rothberg MB, Celestin C, Fiore LD, Lawler E, Cook JR. Warfarin plus aspirin after myocardial infarction or the acute coronary syndrome: meta-analysis with estimates of risk and benefit. Ann Intern Med 2005; 143:241–250.
- Dentali F, Douketis JD, Lim W, Crowther M. Combined aspirin-oral anticoagulant therapy compared with oral anticoagulant therapy alone among patients at risk for cardiovascular disease: a meta-analysis of randomized trials. Arch Intern Med 2007; 167:117–124.
- Hirsh J, Fuster V, Ansell J, Halperin JL. American Heart Association; American College of Cardiology Foundation. American Heart Association/American College of Cardiology Foundation guide to warfarin therapy. Circulation 2003; 107:1692–1711.
- Beyth RJ, Quinn LM, Landefeld CS. Prospective evaluation of an index for predicting the risk of major bleeding in outpatients treated with warfarin. Am J Med 1998; 105:91–99.
- Aspinall SL, DeSanzo BE, Trilli LE, Good CB. Bleeding Risk Index in an anticoagulation clinic. Assessment by indication and implications for care. J Gen Intern Med 2005; 20:1008–1013.
- Mehta SR, Yusuf S, Peters RJ, et al. Clopidogrel in Unstable angina to prevent Recurrent Events trial (CURE) Investigators. Effects of pretreatment with clopidogrel and aspirin followed by long-term therapy in patients undergoing percutaneous coronary intervention: the PCICURE study. Lancet 2001; 358:527–533.
- Bertrand ME, Legrand V, Boland J, et al. Randomized multicenter comparison of conventional anticoagulation versus antiplatelet therapy in unplanned and elective coronary stenting. The Full Anticoagulation versus Aspirin and Ticlopidine (FANTASTIC) study. Circulation 1998; 98:1597–1603.
- Kushner FG, Antman EM. Oral anticoagulation for atrial fibrillation after ST-elevation myocardial infarction: new evidence to guide clinical practice. Circulation 2005; 112:3212–3214.
- Porter A, Konstantino Y, Iakobishvili Z, Shachar L, Battler A, Hasdai D. Short-term triple therapy with aspirin, warfarin, and a thienopyridine among patients undergoing percutaneous coronary intervention. Catheter Cardiovasc Interv 2006; 68:56–61.
- Anderson JL, Adams CD, Antman EM, et al. ACC/AHA 2007 Guidelines for the management of patients with unstable angina/non-ST-elevation myocardial infarction—executive summary. A report of the ACC-AHA Task Force on Practice Guidelines (Writing Committee to Revise the 2002 Guidelines for the Management of Patients With Unstable Angina/Non-ST-Elevation Myocardial Infarction). J Am Coll Cardiol 2007; 50:652–726.
- Turpie AG, Gent M, Laupacis A, et al. A comparison of aspirin with placebo in patients treated with warfarin after heart-valve replacement. N Engl J Med 1993; 329:524–529.
- Bonow RO, Carabello BA, Kanu C, et al. ACC/AHA 2006 guidelines for the management of patients with valvular heart disease: a report of the ACC/AHA Task Force on Practice Guidelines (writing committee to revise the 1998 Guidelines for the Management of Patients With Valvular Heart Disease). Circulation 2006; 114:e84–e231.
- Lauer MS. Clinical practice. Aspirin for primary prevention of coronary events. N Engl J Med 2002; 346:1468–1474.
- The Medical Research Council’s General Practice Research Framework. Thrombosis prevention trial: randomised trial of low-intensity oral anticoagulation with warfarin and low-dose aspirin in the primary prevention of ischaemic heart disease in men at increased risk. Lancet 1998; 351:233–241.
- Fuster V, Ryden LE, Cannom DS, et al. ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation. Circulation 2006; 114:260–335.
Preventing and managing diabetic complications in elderly patients
In elderly patients, as in all patients, diabetes is much more than the blood glucose level. However, in elderly patients the disease accelerates other common conditions of that population and markedly complicates their management.
Hypertension, coronary artery disease, and cerebrovascular attacks are more common in patients with diabetes.1 Longitudinal studies of elderly and middle-aged people with diabetes show increased rates of cognitive decline and dementia.2–4 Depression, urinary incontinence, and falls are also more common in elderly patients with diabetes. Physical disability is also increased: women with diabetes are half as likely to be able to manage ordinary physical tasks such as walking, climbing stairs, and doing housework as women without diabetes.5
In an earlier paper in this journal,6 we reviewed the management of diabetes per se in elderly patients. In the pages that follow, we review the management of its associated conditions.
HEART RISK TRUMPS BLOOD SUGAR
Coronary artery disease is by far the leading cause of death in elderly people with diabetes: 40% to 50% of patients with type 2 diabetes die of cardiac disease.7–9 The conventional risk factors—hypertension, hyperlipidemia, smoking, and diabetes—remain risk factors throughout old age. Risk reduction should focus on treating hypertension and dyslipidemia, smoking cessation, aspirin therapy, and exercise. While glycemic control reduces the risk of microvascular complications (eg, diabetic retinopathy and nephropathy) after about 8 years of treatment, benefits from control of elevated blood pressure and cholesterol occur after only 2 to 3 years.
Tight control of hypertension confers significant benefit
The United Kingdom Prospective Diabetes Study (UKPDS)10 found that patients who had tight control of blood pressure (mean treated blood pressure 144/82 mm Hg) had 24% fewer diabetes-related end points, 32% fewer diabetes-related deaths, 44% fewer strokes, a 34% reduced risk of deterioration of retinopathy, and a 47% reduced risk of visual deterioration than patients who had usual control (mean treated blood pressure 157/87 mm Hg). The benefit of treating hypertension outweighed the benefits of tight glycemic control.
A strong focus on blood pressure control should be a major focus of any treatment program. The American Geriatrics Society goal for blood pressure is less than 140/80 mm Hg if tolerated. Others have proposed more stringent targets.
Lipid control
Lipid control is integral to managing elderly patients with diabetes. In the Cholesterol and Recurrent Events trial11 and the Heart Protection Study,12 the cardiovascular benefits of reducing serum low-density lipoprotein cholesterol (LDL-C) levels were similar in elderly and younger patients with diabetes. In a meta-analysis of secondary prevention trials, absolute risk reduction was greatest in subjects older than 65 years with either diabetes or diastolic hypertension.
The American Diabetes Association,13 the American Geriatrics Society,14 and the Department of Veterans Affairs15,16 have all set a goal for serum LDL-C of less than 100 mg/dL. In addition, the American Diabetes Association has set goal levels for triglycerides (< 150 mg/dL) and high-density lipoprotein cholesterol (> 40 mg/dL).
Glycemic control
The importance of tight glycemic control in preventing coronary heart disease in the elderly is somewhat controversial. Treatment guidelines for elderly patients with diabetes are mainly extrapolated from the UKPDS, in which patients were a mean of 54 years old at the start of the study. After 10 years, the mean hemoglobin A1c levels were 7.9% in patients receiving conventional control and 7.0% in patients with intensive therapy. Every 1% reduction in hemoglobin A1c was associated with a 37% decline in microvascular complications of diabetes, a 14% decline in myocardial infarctions, and a 21% decline in any diabetes-related outcome.17
In the original trial,18 the rate of myocardial infarction was 17.4% in the conventional treatment group vs 14.7% in the intensive group (P = .052), and the risk of stroke did not differ. No thresholds for realizing benefits from reducing fasting glucose or hemoglobin A1c levels were detected.
A recent cohort study involving about 10,000 participants aged 45 to 79 years found that the risk of cardiovascular disease and death from any cause increased continuously with increasing hemoglobin A1c levels in people with or without diabetes.19 However, the impact of treatment remains to be clarified. The Action to Control Cardiovascular Risk in Diabetes trial will address this question (and others), but results will not be available for several years.
RETINOPATHY IS A MAJOR CAUSE OF BLINDNESS
Diabetic retinopathy, a leading cause of blindness in the United States, is perhaps the most threatening of the chronic microvascular complications of diabetes for elderly patients. The strongest predictor of retinopathy is the duration of diabetes.20–22 Retinopathy is classified as being nonproliferative, preproliferative, or proliferative.
Ischemia is believed to be the major cause of diabetic retinopathy, and glucose control has been shown to be of major benefit. A study of young adults with type 1 diabetes found that intensive therapy reduced the risk of developing retinopathy by 76% and slowed the progression of retinopathy by 54%. Comparable data for patients with type 2 diabetes are lacking.
Of some concern is a study in which retinopathy progressed more rapidly during the first year of aggressive insulin therapy in elderly patients with diabetes and baseline retinopathy.23 Further research is needed to identify which subgroups would benefit most from aggressive glycemic control.
In addition to specific ophthalmologic treatment, managing cardiovascular risk factors may reduce the progression of retinopathy: each cardiovascular risk factor has been found to also be a risk factor for retinopathy. Hypertension is an independent risk factor for any retinopathy, and its tight control reduces progression.20,24 Aspirin therapy has not been found to confer either risk or benefit.25,26
Although guidelines typically call for yearly ophthalmic examinations to screen for retinopathy, whether this is cost-effective has been questioned.27,28 But people older than 65 years with diabetes also have twice the risk of developing cataracts and three times the risk of developing glaucoma than those without diabetes. Considering the effects of visual loss on quality of life as well as the subsequent higher risk of accidents, eye examinations by an ophthalmologist at the time of diagnosis and annually thereafter are recommended. Tight glycemic and blood pressure control remains the cornerstone in the primary prevention of diabetic retinopathy. Panretinal and focal retinal laser photocoagulation reduces the risk of visual loss in patients with severe retinopathy and macular edema, respectively.29
NEUROPATHY PRESENTS IN MANY FORMS
Neuropathy is a particularly distressing complication and can lead to loss of sleep, limitation of activity, and depression.26,30,31 Diabetic neuropathies include focal neuropathies (entrapment syndromes and mono-neuropathies), polyneuropathy, and autonomic neuropathy.
Distal symmetric polyneuropathy (“glove and stocking” sensory symptoms) is the most common neuropathy of elderly people with diabetes. Pain, which can interrupt sleep and limit activity, can be treated with the anticonvulsants gabapentin (Gabarone, Neurontin), phenytoin (Dilantin, Phenytek) and carbamazepine (Carbatrol, Epitol, Equetro, Tegretol), and with tricyclic antidepressants. However, the anticholinergic effects of tricyclic antidepressants limit their use in older patients. Newer agents, such as duloxetine (Cymbalta) and pregabalin (Lyrica) show promise.30,31 Dysesthesia of a burning quality is sometimes treated with topical capsaicin or with oral mexiletine (Mexitil), although their role in treating older patients is not well established.
Patients with distal sensory polyneuropathy are predisposed to develop Charcot joints, which may mimic gout or degenerative joint disease. Plain radiography of the foot can help differentiate these diseases. Distal sensory polyneuropathy also predisposes patients to neuropathic foot ulcer, the leading cause of foot amputation in the United States.32
Feet should be inspected at each office visit. Testing sensation with a monofilament detects sensory neuropathy. Patients should be encouraged to examine their feet daily. Therapeutic shoes, prescribed by a podiatrist and individually designed to prevent blisters, calluses, and ulcers, are covered by Medicare for peripheral neuropathy if any of the following are also present: callus formation, poor circulation, foot deformity, or a history of foot callus, ulcer, or amputation (partial or complete). Medicare will pay for one pair of shoes plus three pairs of inserts per year.
Proximal motor neuropathy (diabetic amyotrophy) primarily affects elderly patients. It begins with unilateral thigh pain, which becomes bilateral and progresses to proximal muscle weakness and wasting. Distal symmetric polyneuropathy may also be present. Treatment includes glycemic control (usually with insulin) and physical therapy. Some forms of amyotrophy respond to immunotherapy.
Autonomic neuropathy, although not painful, can be the most life-threatening form of diabetic neuropathy.33 Tachycardia increases the risk of sudden death, while postural hypotension increases the risk of syncope, falling, and injury. Other forms of autonomic neuropathy include neurogenic bladder, sexual dysfunction, gastropathy (which is particularly sensitive to glycemic control), enteropathy, and gustatory sweating. Patients with autonomic neuropathy are more likely to have hypoglycemic unawareness.
NEPHROPATHY CAN PROGRESS RAPIDLY
Elderly patients with diabetes are especially at risk of developing nephropathy, which progresses from microalbuminuria to overt proteinuria to renal insufficiency and end-stage renal disease. Nephropathy may develop over a shorter time than the typical 10 to 20 years in younger patients. Independent risk factors for proteinuria and renal insufficiency include poor glycemic control over many years, hypertension, longer duration of diabetes, male sex, high serum total cholesterol levels, and smoking. Elderly patients are also at risk of renal insults such as receiving intravenous iodinated contrast agents in the course of radiologic procedures, nephrotoxic drugs, and comorbid illness such as congestive heart failure.
The diagnosis of diabetic nephropathy is usually made clinically and not by renal biopsy. Diabetic nephropathy can be diagnosed with almost 100% specificity in type 1 diabetes and more than 85% specificity in type 2 diabetes by a urinary albumin excretion of more than 300 mg per day and an appropriate time course in the absence of other obvious causes of renal disease. The urinary albumin-to-creatinine ratio can be used to screen for microalbuminuria (the precursor of frank proteinuria and renal insufficiency). A value of more than 30 mg of albumin per gram of creatinine suggests that albumin excretion exceeds 30 mg and that microalbuminuria is present.
Prevention is a cornerstone of management. Good glycemic control reduces the risk of microalbuminuria, the progression of albuminuria, and the development of renal insufficiency. Lowering blood pressure reduces the decline in glomerular filtration rate and albuminuria. Angiotensin-converting enzyme (ACE) inhibitors reduce the rate of progression of proteinuria and reduce the rate of end-stage renal disease, although the data are stronger in patients with type 1 diabetes.34 When side effects such as cough limit the use of ACE inhibitors, angiotensin receptor blockers can be used as an alternative. Blood pressure should be controlled to reduce stroke and cardiovascular complications, regardless of whether microalbuminuria is present.35
End-stage renal disease in elderly patients with diabetes is becoming increasingly frequent. Nephropathy in older patients is different from that in younger patients. In elderly patients, the pathologic findings may suggest ischemia and hypertension, and the classic Kimmelstiel-Wilson lesions may be absent. Patients may present with end-stage renal disease following an episode of acute renal failure that does not resolve, which may occur after a radiologic procedure involving an iodinated contrast agent.
NONKETOTIC HYPEROSMOLAR COMA
Nonketotic hyperosmolar coma occurs predominantly in elderly patients with type 2 diabetes. Predisposing factors include dementia, infection, stroke, and myocardial infarction. Coma results from osmotic diuresis due to hyperglycemia and consequent dehydration. A drop in the glomerular filtration rate promotes further hyperglycemia and dehydration in a vicious circle. Glucose levels commonly reach 600 mg/dL or more, and serum osmolality often exceeds 320 mOsm/L. A fluid deficit of 5 to 10 L is typical.
Fluid replacement is the mainstay of treatment. Because free water is typically lost in an osmotic diuresis, 0.9% (normal) saline is usually given if hemodynamic instability is present or 0.45% (half-normal) saline otherwise. Insulin is also required, as is specific treatment of the precipitating cause, eg, infection. Ketoacidosis may also occur in the elderly.
Recovery from coma or improvement in mental status may lag behind correction of the serum osmolality and may take several days. Mortality rates can be high: severe hyperosmolarity, advanced age, and nursing home residence are the major risk factors for death.
INFECTIONS: SEVERE AND UNUSUAL
Elderly patients with diabetes are at increased risk of developing severe and unusual infections, particularly malignant external otitis. Necrotizing Pseudomonas aeruginosa infection initially involves the external ear canal and progresses to the mastoid air cells, the skull base, or temporal bone. The clinical presentation consists of fever, otalgia, otorrhea, and less commonly, cranial nerve palsy. Treatment involves surgical debridement and antibiotics.
Other infections associated with diabetes include rhinocerebral mucormycosis, necrotizing fasciitis, emphysematous cholecystitis, and emphysematous pyelonephritis. An elderly patient with diabetes is also at increased risk of renal papillary necrosis, which presents as insidious renal failure.
COGNITIVE IMPAIRMENT
Elderly people with diabetes are at increased risk of cognitive impairment, which poses a barrier to taking medications appropriately and performing other tasks of self-management.
Because dementia may go undetected, particularly in the early stages, cognitive function should be assessed in elderly patients when they fail to take therapy correctly or have frequent episodes of hypoglycemia, or if glycemic control deteriorates without an obvious explanation. Caregivers play a critical role in detecting and reporting early cognitive impairment.
DEPRESSION IS OFTEN UNDETECTED
Elderly patients with diabetes have a higher rate of depression than do age-matched controls, but it is commonly underdetected and undertreated.5,36 Depression has been associated with poor glycemic control, and treatment of depression is associated with improved control. Routine screening for depression should be performed; a variety of diagnostic instruments are available. Particular attention should be given to medications that are associated with depression.
POLYPHARMACY
Many elderly patients take multiple medications. Polypharmacy increases the risk of drug side effects, interactions, and nonadherence to taking medications.37–39 This problem is increased in diabetes, in which several medications are necessary to manage hyper-glycemia, hyperlipidemia, hypertension, and other associated conditions.
Patients should keep accurate medication lists, including over-the-counter medications, herbs, and nutritional supplements. Physicians should carefully review each medication to check if it is appropriate and used correctly.
FALLS
Elderly patients with diabetes mellitus are at increased risk of injurious falls, which are associated with high rates of complications, death, and functional decline.40,41 Risk factors include frailty and functional disability, visual impairment, peripheral or autonomic neuropathy, hypoglycemia, and polypharmacy.
Elderly patients should be screened for their risk of falls, and appropriate measures should be instituted. The American Geriatrics Society has guidelines for preventing falls in the elderly.41
URINARY INCONTINENCE
Elderly women with diabetes are at increased risk of developing urinary incontinence. Risk factors include autonomic neuropathy (causing either neurogenic bladder or fecal impaction), polyuria due to hyperglycemia, and urinary tract and vaginal infections. Although evidence is lacking that urinary incontinence affects glycemic control, assessing and treating the condition improves quality of life.
SUMMARY
Diabetes is a common problem in the elderly, accounting for considerable morbidity and mortality. In a large longitudinal analysis (> 50,000 patients), elderly persons newly diagnosed as having diabetes experienced high rates of complications during 10-year follow-up, far in excess of elderly persons without diabetes.42 Diabetes is underdiagnosed in the elderly and is frequently undertreated. Management of the elderly with diabetes presents unique challenges because of associated comorbidities, but with attention to detail and individualized approaches, quality and duration of life can be optimized. The greatest attention should be given to reduction of overall cardiovascular risk. Glycemic goals and the treatment regimens to achieve those goals should be individualized and chosen to control hyperglycemic symptoms and achieve the maximal glycemic control possible while minimizing the risk of hypoglycemia. Diabetes will continue to be a challenge to the patient, the physician, the care team, and the health care system.
- Gregg EW, Engelgau MM, Narayan V. Complications of diabetes in elderly people. BMJ 2002; 325:916–917.
- Knopman D, Boland LL, Mosley T, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001; 56:42–48.
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53:1937–1942.
- Fontbonne A, Berr C, Ducimetiere P, Alperovitch A. Changes in cognitive abilities over a 4-year period are unfavorably affected in elderly diabetic subjects: results of the Epidemiology of Vascular Aging Study. Diabetes Care 2001; 24:366–370.
- Gregg EW, Mangione CM, Cauley JA, et al. Diabetes and incidence of functional disability in older women. Diabetes Care 2002; 25:61–67.
- Hornick T, Aron DC. Managing diabetes in the elderly: go easy, individualize. Cleve Clin J Med 2008; 75:70–78.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bertoni AG, Krop JS, Anderson GF, Brancati FL. Diabetes-related morbidity and mortality in a national sample of U.S. elders. Diabetes Care 2002; 25:471–475.
- Bertoni AG, Kirk JK, Goff DC, Wagenknecht LE. Excess mortality related to diabetes mellitus in elderly Medicare beneficiaries. Ann Epidemiol 2004; 14:362–367.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the Cholesterol and Recurrent Events (CARE) trial. The CARE Investigators. Circulation 1998; 98:2513–2519.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes. Lancet 2003; 361:2005–2016.
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care 2005; 28:S4–S36.
- Brown AF, Mangione CM, Saliba D, Sarkisian CA California Healthcare Foundation/American Geriatrics Society Panel on Improving Care for Elders with Diabetes. Guidelines for improving the care of the older person with diabetes mellitus. J Am Geriatr Soc 2003; 51:S265–S280.
- VA/DoD Clinical Practice Guideline for the Management of Diabetes Mellitus in the Primary Care Setting 2003. Accessed January 4, 2008. www.oqp.med.va.gov/cpg/dm/DM3_cpg/content/introduction.htm.
- Pogach LM, Brietzke SA, Cowan CL, Conlin P, Walder DJ, Sawin CT VA/DoD Diabetes Guideline Development Group. Development of evidence-based clinical practice guidelines for diabetes: the Department of Veterans Affairs/Department of Defense guidelines initiative. Diabetes Care 2004; 27:B82–B89.
- Stratton IM, Asler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321:405–412.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 2004; 141:413–420.
- Matthews DR, Stratton IM, Aldington SJ, Holman RR, Kohner EM UK Prospective Diabetes Study Group. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol 2004; 122:1631–1640.
- Cahill M, Halley A, Codd M, et al. Prevalence of diabetic retinopathy in patients with diabetic mellitus diagnosed after the age of 70 years. Br J Opthalmol 1997; 81:218–222.
- Hirvela H, Laatikainen L. Diabetic retinopathy in people aged 70 years or older. The Oulu Eye Study. Br J Ophthalmol 1997; 81:214–217.
- Tovi J, Ingemansson SO, Engfeldt P. Insulin treatment of elderly type 2 diabetic patients: effects on retinopathy. Diabetes Metab 1998; 24:442–447.
- Schrier RW, Estacio RO, Esler A, Mehler P. Effects of aggressive blood pressure control in normotensive type 2 diabetic patients on albuminuria, retinopathy and strokes. Kidney Int 2002; 61:1086–1097.
- Kohner EM. Aspirin for diabetic retinopathy. BMJ 2003; 327:1060–1061.
- Greene DA, Stevens MJ, Feldman EL. Diabetic neuropathy: scope of the syndrome. Am J Med 1999; 107:2S–8S.
- Hutchinson A, McIntosh A, Peters J, et al. Effectiveness of screening and monitoring tests for diabetic retinopathy—a systematic review. Diabet Med 2000; 17:495–506.
- Vijan S, Hofer TP, Hayward RA. Cost-utility analysis of screening intervals for diabetic retinopathy in patients with type 2 diabetes mellitus. JAMA 2000; 283:889–896.
- Mohamed Q, Gillies MC, Wong TY. Management of diabetic retinopathy: a systematic review. JAMA 2007; 298:902–916.
- Argoff CE, Cole BE, Fishbain DA, Irving GA. Diabetic peripheral neuropathic pain: clinical and quality-of-life issues. Mayo Clin Proc 2006; 81:S3–S11.
- Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ 2007; 335:87: epubl June 11, 2007.
- Bild DE, Selby JV, Sinnock P, Browner WS, Braveman P, Showstack JA. Lower-extremity amputation in people with diabetes. Epidemiology and prevention. Diabetes Care 1989; 12:24–31.
- Wheeler SG, Ahroni JH, Boyko EJ. Prospective study of autonomic neuropathy as a predictor of mortality in patients with diabetes. Diabetes Res Clin Pract 2002; 58:131–138.
- Brenner BM, Cooper ME, de Zeeuw D RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Sinclair AJ, Girling AJ, Bayer AJ. Cognitive dysfunction in older subjects with diabetes mellitus: impact on diabetes self-management and use of care services. All Wales Research into Elderly (AWARE) Study. Diabetes Res Clin Pract 2000; 50:203–212.
- Moisan J, Gaudet M, Gregoire JP, Bouchard R. Non-compliance with drug treatment and reading difficulties with regard to prescription labelling among seniors. Gerontology 2002; 48:44–51.
- Boyd CM, Darer J, Boult C, Fried LP, Boult L, Wu AW. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005; 294:716–724.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Schwartz AV, Hillier TA, Sellmeyer DE, et al. Older women with diabetes have a higher risk of falls: a prospective study. Diabetes Care 2002; 25:1749–1754.
- American Geriatrics Society, British Geriatrics Society, and American Academy of Orthopaedic Surgeons Panel on Falls Prevention. Guideline for the prevention of falls in older persons. J Am Geriatr Soc 2001; 49:664–672.
- Bethel MA, Sloan FA, Belsky D, Feinglos MN. Longitudinal incidence and prevalence of adverse outcomes of diabetes mellitus in elderly patients. Arch Intern Med 2007; 167:921–927.
In elderly patients, as in all patients, diabetes is much more than the blood glucose level. However, in elderly patients the disease accelerates other common conditions of that population and markedly complicates their management.
Hypertension, coronary artery disease, and cerebrovascular attacks are more common in patients with diabetes.1 Longitudinal studies of elderly and middle-aged people with diabetes show increased rates of cognitive decline and dementia.2–4 Depression, urinary incontinence, and falls are also more common in elderly patients with diabetes. Physical disability is also increased: women with diabetes are half as likely to be able to manage ordinary physical tasks such as walking, climbing stairs, and doing housework as women without diabetes.5
In an earlier paper in this journal,6 we reviewed the management of diabetes per se in elderly patients. In the pages that follow, we review the management of its associated conditions.
HEART RISK TRUMPS BLOOD SUGAR
Coronary artery disease is by far the leading cause of death in elderly people with diabetes: 40% to 50% of patients with type 2 diabetes die of cardiac disease.7–9 The conventional risk factors—hypertension, hyperlipidemia, smoking, and diabetes—remain risk factors throughout old age. Risk reduction should focus on treating hypertension and dyslipidemia, smoking cessation, aspirin therapy, and exercise. While glycemic control reduces the risk of microvascular complications (eg, diabetic retinopathy and nephropathy) after about 8 years of treatment, benefits from control of elevated blood pressure and cholesterol occur after only 2 to 3 years.
Tight control of hypertension confers significant benefit
The United Kingdom Prospective Diabetes Study (UKPDS)10 found that patients who had tight control of blood pressure (mean treated blood pressure 144/82 mm Hg) had 24% fewer diabetes-related end points, 32% fewer diabetes-related deaths, 44% fewer strokes, a 34% reduced risk of deterioration of retinopathy, and a 47% reduced risk of visual deterioration than patients who had usual control (mean treated blood pressure 157/87 mm Hg). The benefit of treating hypertension outweighed the benefits of tight glycemic control.
A strong focus on blood pressure control should be a major focus of any treatment program. The American Geriatrics Society goal for blood pressure is less than 140/80 mm Hg if tolerated. Others have proposed more stringent targets.
Lipid control
Lipid control is integral to managing elderly patients with diabetes. In the Cholesterol and Recurrent Events trial11 and the Heart Protection Study,12 the cardiovascular benefits of reducing serum low-density lipoprotein cholesterol (LDL-C) levels were similar in elderly and younger patients with diabetes. In a meta-analysis of secondary prevention trials, absolute risk reduction was greatest in subjects older than 65 years with either diabetes or diastolic hypertension.
The American Diabetes Association,13 the American Geriatrics Society,14 and the Department of Veterans Affairs15,16 have all set a goal for serum LDL-C of less than 100 mg/dL. In addition, the American Diabetes Association has set goal levels for triglycerides (< 150 mg/dL) and high-density lipoprotein cholesterol (> 40 mg/dL).
Glycemic control
The importance of tight glycemic control in preventing coronary heart disease in the elderly is somewhat controversial. Treatment guidelines for elderly patients with diabetes are mainly extrapolated from the UKPDS, in which patients were a mean of 54 years old at the start of the study. After 10 years, the mean hemoglobin A1c levels were 7.9% in patients receiving conventional control and 7.0% in patients with intensive therapy. Every 1% reduction in hemoglobin A1c was associated with a 37% decline in microvascular complications of diabetes, a 14% decline in myocardial infarctions, and a 21% decline in any diabetes-related outcome.17
In the original trial,18 the rate of myocardial infarction was 17.4% in the conventional treatment group vs 14.7% in the intensive group (P = .052), and the risk of stroke did not differ. No thresholds for realizing benefits from reducing fasting glucose or hemoglobin A1c levels were detected.
A recent cohort study involving about 10,000 participants aged 45 to 79 years found that the risk of cardiovascular disease and death from any cause increased continuously with increasing hemoglobin A1c levels in people with or without diabetes.19 However, the impact of treatment remains to be clarified. The Action to Control Cardiovascular Risk in Diabetes trial will address this question (and others), but results will not be available for several years.
RETINOPATHY IS A MAJOR CAUSE OF BLINDNESS
Diabetic retinopathy, a leading cause of blindness in the United States, is perhaps the most threatening of the chronic microvascular complications of diabetes for elderly patients. The strongest predictor of retinopathy is the duration of diabetes.20–22 Retinopathy is classified as being nonproliferative, preproliferative, or proliferative.
Ischemia is believed to be the major cause of diabetic retinopathy, and glucose control has been shown to be of major benefit. A study of young adults with type 1 diabetes found that intensive therapy reduced the risk of developing retinopathy by 76% and slowed the progression of retinopathy by 54%. Comparable data for patients with type 2 diabetes are lacking.
Of some concern is a study in which retinopathy progressed more rapidly during the first year of aggressive insulin therapy in elderly patients with diabetes and baseline retinopathy.23 Further research is needed to identify which subgroups would benefit most from aggressive glycemic control.
In addition to specific ophthalmologic treatment, managing cardiovascular risk factors may reduce the progression of retinopathy: each cardiovascular risk factor has been found to also be a risk factor for retinopathy. Hypertension is an independent risk factor for any retinopathy, and its tight control reduces progression.20,24 Aspirin therapy has not been found to confer either risk or benefit.25,26
Although guidelines typically call for yearly ophthalmic examinations to screen for retinopathy, whether this is cost-effective has been questioned.27,28 But people older than 65 years with diabetes also have twice the risk of developing cataracts and three times the risk of developing glaucoma than those without diabetes. Considering the effects of visual loss on quality of life as well as the subsequent higher risk of accidents, eye examinations by an ophthalmologist at the time of diagnosis and annually thereafter are recommended. Tight glycemic and blood pressure control remains the cornerstone in the primary prevention of diabetic retinopathy. Panretinal and focal retinal laser photocoagulation reduces the risk of visual loss in patients with severe retinopathy and macular edema, respectively.29
NEUROPATHY PRESENTS IN MANY FORMS
Neuropathy is a particularly distressing complication and can lead to loss of sleep, limitation of activity, and depression.26,30,31 Diabetic neuropathies include focal neuropathies (entrapment syndromes and mono-neuropathies), polyneuropathy, and autonomic neuropathy.
Distal symmetric polyneuropathy (“glove and stocking” sensory symptoms) is the most common neuropathy of elderly people with diabetes. Pain, which can interrupt sleep and limit activity, can be treated with the anticonvulsants gabapentin (Gabarone, Neurontin), phenytoin (Dilantin, Phenytek) and carbamazepine (Carbatrol, Epitol, Equetro, Tegretol), and with tricyclic antidepressants. However, the anticholinergic effects of tricyclic antidepressants limit their use in older patients. Newer agents, such as duloxetine (Cymbalta) and pregabalin (Lyrica) show promise.30,31 Dysesthesia of a burning quality is sometimes treated with topical capsaicin or with oral mexiletine (Mexitil), although their role in treating older patients is not well established.
Patients with distal sensory polyneuropathy are predisposed to develop Charcot joints, which may mimic gout or degenerative joint disease. Plain radiography of the foot can help differentiate these diseases. Distal sensory polyneuropathy also predisposes patients to neuropathic foot ulcer, the leading cause of foot amputation in the United States.32
Feet should be inspected at each office visit. Testing sensation with a monofilament detects sensory neuropathy. Patients should be encouraged to examine their feet daily. Therapeutic shoes, prescribed by a podiatrist and individually designed to prevent blisters, calluses, and ulcers, are covered by Medicare for peripheral neuropathy if any of the following are also present: callus formation, poor circulation, foot deformity, or a history of foot callus, ulcer, or amputation (partial or complete). Medicare will pay for one pair of shoes plus three pairs of inserts per year.
Proximal motor neuropathy (diabetic amyotrophy) primarily affects elderly patients. It begins with unilateral thigh pain, which becomes bilateral and progresses to proximal muscle weakness and wasting. Distal symmetric polyneuropathy may also be present. Treatment includes glycemic control (usually with insulin) and physical therapy. Some forms of amyotrophy respond to immunotherapy.
Autonomic neuropathy, although not painful, can be the most life-threatening form of diabetic neuropathy.33 Tachycardia increases the risk of sudden death, while postural hypotension increases the risk of syncope, falling, and injury. Other forms of autonomic neuropathy include neurogenic bladder, sexual dysfunction, gastropathy (which is particularly sensitive to glycemic control), enteropathy, and gustatory sweating. Patients with autonomic neuropathy are more likely to have hypoglycemic unawareness.
NEPHROPATHY CAN PROGRESS RAPIDLY
Elderly patients with diabetes are especially at risk of developing nephropathy, which progresses from microalbuminuria to overt proteinuria to renal insufficiency and end-stage renal disease. Nephropathy may develop over a shorter time than the typical 10 to 20 years in younger patients. Independent risk factors for proteinuria and renal insufficiency include poor glycemic control over many years, hypertension, longer duration of diabetes, male sex, high serum total cholesterol levels, and smoking. Elderly patients are also at risk of renal insults such as receiving intravenous iodinated contrast agents in the course of radiologic procedures, nephrotoxic drugs, and comorbid illness such as congestive heart failure.
The diagnosis of diabetic nephropathy is usually made clinically and not by renal biopsy. Diabetic nephropathy can be diagnosed with almost 100% specificity in type 1 diabetes and more than 85% specificity in type 2 diabetes by a urinary albumin excretion of more than 300 mg per day and an appropriate time course in the absence of other obvious causes of renal disease. The urinary albumin-to-creatinine ratio can be used to screen for microalbuminuria (the precursor of frank proteinuria and renal insufficiency). A value of more than 30 mg of albumin per gram of creatinine suggests that albumin excretion exceeds 30 mg and that microalbuminuria is present.
Prevention is a cornerstone of management. Good glycemic control reduces the risk of microalbuminuria, the progression of albuminuria, and the development of renal insufficiency. Lowering blood pressure reduces the decline in glomerular filtration rate and albuminuria. Angiotensin-converting enzyme (ACE) inhibitors reduce the rate of progression of proteinuria and reduce the rate of end-stage renal disease, although the data are stronger in patients with type 1 diabetes.34 When side effects such as cough limit the use of ACE inhibitors, angiotensin receptor blockers can be used as an alternative. Blood pressure should be controlled to reduce stroke and cardiovascular complications, regardless of whether microalbuminuria is present.35
End-stage renal disease in elderly patients with diabetes is becoming increasingly frequent. Nephropathy in older patients is different from that in younger patients. In elderly patients, the pathologic findings may suggest ischemia and hypertension, and the classic Kimmelstiel-Wilson lesions may be absent. Patients may present with end-stage renal disease following an episode of acute renal failure that does not resolve, which may occur after a radiologic procedure involving an iodinated contrast agent.
NONKETOTIC HYPEROSMOLAR COMA
Nonketotic hyperosmolar coma occurs predominantly in elderly patients with type 2 diabetes. Predisposing factors include dementia, infection, stroke, and myocardial infarction. Coma results from osmotic diuresis due to hyperglycemia and consequent dehydration. A drop in the glomerular filtration rate promotes further hyperglycemia and dehydration in a vicious circle. Glucose levels commonly reach 600 mg/dL or more, and serum osmolality often exceeds 320 mOsm/L. A fluid deficit of 5 to 10 L is typical.
Fluid replacement is the mainstay of treatment. Because free water is typically lost in an osmotic diuresis, 0.9% (normal) saline is usually given if hemodynamic instability is present or 0.45% (half-normal) saline otherwise. Insulin is also required, as is specific treatment of the precipitating cause, eg, infection. Ketoacidosis may also occur in the elderly.
Recovery from coma or improvement in mental status may lag behind correction of the serum osmolality and may take several days. Mortality rates can be high: severe hyperosmolarity, advanced age, and nursing home residence are the major risk factors for death.
INFECTIONS: SEVERE AND UNUSUAL
Elderly patients with diabetes are at increased risk of developing severe and unusual infections, particularly malignant external otitis. Necrotizing Pseudomonas aeruginosa infection initially involves the external ear canal and progresses to the mastoid air cells, the skull base, or temporal bone. The clinical presentation consists of fever, otalgia, otorrhea, and less commonly, cranial nerve palsy. Treatment involves surgical debridement and antibiotics.
Other infections associated with diabetes include rhinocerebral mucormycosis, necrotizing fasciitis, emphysematous cholecystitis, and emphysematous pyelonephritis. An elderly patient with diabetes is also at increased risk of renal papillary necrosis, which presents as insidious renal failure.
COGNITIVE IMPAIRMENT
Elderly people with diabetes are at increased risk of cognitive impairment, which poses a barrier to taking medications appropriately and performing other tasks of self-management.
Because dementia may go undetected, particularly in the early stages, cognitive function should be assessed in elderly patients when they fail to take therapy correctly or have frequent episodes of hypoglycemia, or if glycemic control deteriorates without an obvious explanation. Caregivers play a critical role in detecting and reporting early cognitive impairment.
DEPRESSION IS OFTEN UNDETECTED
Elderly patients with diabetes have a higher rate of depression than do age-matched controls, but it is commonly underdetected and undertreated.5,36 Depression has been associated with poor glycemic control, and treatment of depression is associated with improved control. Routine screening for depression should be performed; a variety of diagnostic instruments are available. Particular attention should be given to medications that are associated with depression.
POLYPHARMACY
Many elderly patients take multiple medications. Polypharmacy increases the risk of drug side effects, interactions, and nonadherence to taking medications.37–39 This problem is increased in diabetes, in which several medications are necessary to manage hyper-glycemia, hyperlipidemia, hypertension, and other associated conditions.
Patients should keep accurate medication lists, including over-the-counter medications, herbs, and nutritional supplements. Physicians should carefully review each medication to check if it is appropriate and used correctly.
FALLS
Elderly patients with diabetes mellitus are at increased risk of injurious falls, which are associated with high rates of complications, death, and functional decline.40,41 Risk factors include frailty and functional disability, visual impairment, peripheral or autonomic neuropathy, hypoglycemia, and polypharmacy.
Elderly patients should be screened for their risk of falls, and appropriate measures should be instituted. The American Geriatrics Society has guidelines for preventing falls in the elderly.41
URINARY INCONTINENCE
Elderly women with diabetes are at increased risk of developing urinary incontinence. Risk factors include autonomic neuropathy (causing either neurogenic bladder or fecal impaction), polyuria due to hyperglycemia, and urinary tract and vaginal infections. Although evidence is lacking that urinary incontinence affects glycemic control, assessing and treating the condition improves quality of life.
SUMMARY
Diabetes is a common problem in the elderly, accounting for considerable morbidity and mortality. In a large longitudinal analysis (> 50,000 patients), elderly persons newly diagnosed as having diabetes experienced high rates of complications during 10-year follow-up, far in excess of elderly persons without diabetes.42 Diabetes is underdiagnosed in the elderly and is frequently undertreated. Management of the elderly with diabetes presents unique challenges because of associated comorbidities, but with attention to detail and individualized approaches, quality and duration of life can be optimized. The greatest attention should be given to reduction of overall cardiovascular risk. Glycemic goals and the treatment regimens to achieve those goals should be individualized and chosen to control hyperglycemic symptoms and achieve the maximal glycemic control possible while minimizing the risk of hypoglycemia. Diabetes will continue to be a challenge to the patient, the physician, the care team, and the health care system.
In elderly patients, as in all patients, diabetes is much more than the blood glucose level. However, in elderly patients the disease accelerates other common conditions of that population and markedly complicates their management.
Hypertension, coronary artery disease, and cerebrovascular attacks are more common in patients with diabetes.1 Longitudinal studies of elderly and middle-aged people with diabetes show increased rates of cognitive decline and dementia.2–4 Depression, urinary incontinence, and falls are also more common in elderly patients with diabetes. Physical disability is also increased: women with diabetes are half as likely to be able to manage ordinary physical tasks such as walking, climbing stairs, and doing housework as women without diabetes.5
In an earlier paper in this journal,6 we reviewed the management of diabetes per se in elderly patients. In the pages that follow, we review the management of its associated conditions.
HEART RISK TRUMPS BLOOD SUGAR
Coronary artery disease is by far the leading cause of death in elderly people with diabetes: 40% to 50% of patients with type 2 diabetes die of cardiac disease.7–9 The conventional risk factors—hypertension, hyperlipidemia, smoking, and diabetes—remain risk factors throughout old age. Risk reduction should focus on treating hypertension and dyslipidemia, smoking cessation, aspirin therapy, and exercise. While glycemic control reduces the risk of microvascular complications (eg, diabetic retinopathy and nephropathy) after about 8 years of treatment, benefits from control of elevated blood pressure and cholesterol occur after only 2 to 3 years.
Tight control of hypertension confers significant benefit
The United Kingdom Prospective Diabetes Study (UKPDS)10 found that patients who had tight control of blood pressure (mean treated blood pressure 144/82 mm Hg) had 24% fewer diabetes-related end points, 32% fewer diabetes-related deaths, 44% fewer strokes, a 34% reduced risk of deterioration of retinopathy, and a 47% reduced risk of visual deterioration than patients who had usual control (mean treated blood pressure 157/87 mm Hg). The benefit of treating hypertension outweighed the benefits of tight glycemic control.
A strong focus on blood pressure control should be a major focus of any treatment program. The American Geriatrics Society goal for blood pressure is less than 140/80 mm Hg if tolerated. Others have proposed more stringent targets.
Lipid control
Lipid control is integral to managing elderly patients with diabetes. In the Cholesterol and Recurrent Events trial11 and the Heart Protection Study,12 the cardiovascular benefits of reducing serum low-density lipoprotein cholesterol (LDL-C) levels were similar in elderly and younger patients with diabetes. In a meta-analysis of secondary prevention trials, absolute risk reduction was greatest in subjects older than 65 years with either diabetes or diastolic hypertension.
The American Diabetes Association,13 the American Geriatrics Society,14 and the Department of Veterans Affairs15,16 have all set a goal for serum LDL-C of less than 100 mg/dL. In addition, the American Diabetes Association has set goal levels for triglycerides (< 150 mg/dL) and high-density lipoprotein cholesterol (> 40 mg/dL).
Glycemic control
The importance of tight glycemic control in preventing coronary heart disease in the elderly is somewhat controversial. Treatment guidelines for elderly patients with diabetes are mainly extrapolated from the UKPDS, in which patients were a mean of 54 years old at the start of the study. After 10 years, the mean hemoglobin A1c levels were 7.9% in patients receiving conventional control and 7.0% in patients with intensive therapy. Every 1% reduction in hemoglobin A1c was associated with a 37% decline in microvascular complications of diabetes, a 14% decline in myocardial infarctions, and a 21% decline in any diabetes-related outcome.17
In the original trial,18 the rate of myocardial infarction was 17.4% in the conventional treatment group vs 14.7% in the intensive group (P = .052), and the risk of stroke did not differ. No thresholds for realizing benefits from reducing fasting glucose or hemoglobin A1c levels were detected.
A recent cohort study involving about 10,000 participants aged 45 to 79 years found that the risk of cardiovascular disease and death from any cause increased continuously with increasing hemoglobin A1c levels in people with or without diabetes.19 However, the impact of treatment remains to be clarified. The Action to Control Cardiovascular Risk in Diabetes trial will address this question (and others), but results will not be available for several years.
RETINOPATHY IS A MAJOR CAUSE OF BLINDNESS
Diabetic retinopathy, a leading cause of blindness in the United States, is perhaps the most threatening of the chronic microvascular complications of diabetes for elderly patients. The strongest predictor of retinopathy is the duration of diabetes.20–22 Retinopathy is classified as being nonproliferative, preproliferative, or proliferative.
Ischemia is believed to be the major cause of diabetic retinopathy, and glucose control has been shown to be of major benefit. A study of young adults with type 1 diabetes found that intensive therapy reduced the risk of developing retinopathy by 76% and slowed the progression of retinopathy by 54%. Comparable data for patients with type 2 diabetes are lacking.
Of some concern is a study in which retinopathy progressed more rapidly during the first year of aggressive insulin therapy in elderly patients with diabetes and baseline retinopathy.23 Further research is needed to identify which subgroups would benefit most from aggressive glycemic control.
In addition to specific ophthalmologic treatment, managing cardiovascular risk factors may reduce the progression of retinopathy: each cardiovascular risk factor has been found to also be a risk factor for retinopathy. Hypertension is an independent risk factor for any retinopathy, and its tight control reduces progression.20,24 Aspirin therapy has not been found to confer either risk or benefit.25,26
Although guidelines typically call for yearly ophthalmic examinations to screen for retinopathy, whether this is cost-effective has been questioned.27,28 But people older than 65 years with diabetes also have twice the risk of developing cataracts and three times the risk of developing glaucoma than those without diabetes. Considering the effects of visual loss on quality of life as well as the subsequent higher risk of accidents, eye examinations by an ophthalmologist at the time of diagnosis and annually thereafter are recommended. Tight glycemic and blood pressure control remains the cornerstone in the primary prevention of diabetic retinopathy. Panretinal and focal retinal laser photocoagulation reduces the risk of visual loss in patients with severe retinopathy and macular edema, respectively.29
NEUROPATHY PRESENTS IN MANY FORMS
Neuropathy is a particularly distressing complication and can lead to loss of sleep, limitation of activity, and depression.26,30,31 Diabetic neuropathies include focal neuropathies (entrapment syndromes and mono-neuropathies), polyneuropathy, and autonomic neuropathy.
Distal symmetric polyneuropathy (“glove and stocking” sensory symptoms) is the most common neuropathy of elderly people with diabetes. Pain, which can interrupt sleep and limit activity, can be treated with the anticonvulsants gabapentin (Gabarone, Neurontin), phenytoin (Dilantin, Phenytek) and carbamazepine (Carbatrol, Epitol, Equetro, Tegretol), and with tricyclic antidepressants. However, the anticholinergic effects of tricyclic antidepressants limit their use in older patients. Newer agents, such as duloxetine (Cymbalta) and pregabalin (Lyrica) show promise.30,31 Dysesthesia of a burning quality is sometimes treated with topical capsaicin or with oral mexiletine (Mexitil), although their role in treating older patients is not well established.
Patients with distal sensory polyneuropathy are predisposed to develop Charcot joints, which may mimic gout or degenerative joint disease. Plain radiography of the foot can help differentiate these diseases. Distal sensory polyneuropathy also predisposes patients to neuropathic foot ulcer, the leading cause of foot amputation in the United States.32
Feet should be inspected at each office visit. Testing sensation with a monofilament detects sensory neuropathy. Patients should be encouraged to examine their feet daily. Therapeutic shoes, prescribed by a podiatrist and individually designed to prevent blisters, calluses, and ulcers, are covered by Medicare for peripheral neuropathy if any of the following are also present: callus formation, poor circulation, foot deformity, or a history of foot callus, ulcer, or amputation (partial or complete). Medicare will pay for one pair of shoes plus three pairs of inserts per year.
Proximal motor neuropathy (diabetic amyotrophy) primarily affects elderly patients. It begins with unilateral thigh pain, which becomes bilateral and progresses to proximal muscle weakness and wasting. Distal symmetric polyneuropathy may also be present. Treatment includes glycemic control (usually with insulin) and physical therapy. Some forms of amyotrophy respond to immunotherapy.
Autonomic neuropathy, although not painful, can be the most life-threatening form of diabetic neuropathy.33 Tachycardia increases the risk of sudden death, while postural hypotension increases the risk of syncope, falling, and injury. Other forms of autonomic neuropathy include neurogenic bladder, sexual dysfunction, gastropathy (which is particularly sensitive to glycemic control), enteropathy, and gustatory sweating. Patients with autonomic neuropathy are more likely to have hypoglycemic unawareness.
NEPHROPATHY CAN PROGRESS RAPIDLY
Elderly patients with diabetes are especially at risk of developing nephropathy, which progresses from microalbuminuria to overt proteinuria to renal insufficiency and end-stage renal disease. Nephropathy may develop over a shorter time than the typical 10 to 20 years in younger patients. Independent risk factors for proteinuria and renal insufficiency include poor glycemic control over many years, hypertension, longer duration of diabetes, male sex, high serum total cholesterol levels, and smoking. Elderly patients are also at risk of renal insults such as receiving intravenous iodinated contrast agents in the course of radiologic procedures, nephrotoxic drugs, and comorbid illness such as congestive heart failure.
The diagnosis of diabetic nephropathy is usually made clinically and not by renal biopsy. Diabetic nephropathy can be diagnosed with almost 100% specificity in type 1 diabetes and more than 85% specificity in type 2 diabetes by a urinary albumin excretion of more than 300 mg per day and an appropriate time course in the absence of other obvious causes of renal disease. The urinary albumin-to-creatinine ratio can be used to screen for microalbuminuria (the precursor of frank proteinuria and renal insufficiency). A value of more than 30 mg of albumin per gram of creatinine suggests that albumin excretion exceeds 30 mg and that microalbuminuria is present.
Prevention is a cornerstone of management. Good glycemic control reduces the risk of microalbuminuria, the progression of albuminuria, and the development of renal insufficiency. Lowering blood pressure reduces the decline in glomerular filtration rate and albuminuria. Angiotensin-converting enzyme (ACE) inhibitors reduce the rate of progression of proteinuria and reduce the rate of end-stage renal disease, although the data are stronger in patients with type 1 diabetes.34 When side effects such as cough limit the use of ACE inhibitors, angiotensin receptor blockers can be used as an alternative. Blood pressure should be controlled to reduce stroke and cardiovascular complications, regardless of whether microalbuminuria is present.35
End-stage renal disease in elderly patients with diabetes is becoming increasingly frequent. Nephropathy in older patients is different from that in younger patients. In elderly patients, the pathologic findings may suggest ischemia and hypertension, and the classic Kimmelstiel-Wilson lesions may be absent. Patients may present with end-stage renal disease following an episode of acute renal failure that does not resolve, which may occur after a radiologic procedure involving an iodinated contrast agent.
NONKETOTIC HYPEROSMOLAR COMA
Nonketotic hyperosmolar coma occurs predominantly in elderly patients with type 2 diabetes. Predisposing factors include dementia, infection, stroke, and myocardial infarction. Coma results from osmotic diuresis due to hyperglycemia and consequent dehydration. A drop in the glomerular filtration rate promotes further hyperglycemia and dehydration in a vicious circle. Glucose levels commonly reach 600 mg/dL or more, and serum osmolality often exceeds 320 mOsm/L. A fluid deficit of 5 to 10 L is typical.
Fluid replacement is the mainstay of treatment. Because free water is typically lost in an osmotic diuresis, 0.9% (normal) saline is usually given if hemodynamic instability is present or 0.45% (half-normal) saline otherwise. Insulin is also required, as is specific treatment of the precipitating cause, eg, infection. Ketoacidosis may also occur in the elderly.
Recovery from coma or improvement in mental status may lag behind correction of the serum osmolality and may take several days. Mortality rates can be high: severe hyperosmolarity, advanced age, and nursing home residence are the major risk factors for death.
INFECTIONS: SEVERE AND UNUSUAL
Elderly patients with diabetes are at increased risk of developing severe and unusual infections, particularly malignant external otitis. Necrotizing Pseudomonas aeruginosa infection initially involves the external ear canal and progresses to the mastoid air cells, the skull base, or temporal bone. The clinical presentation consists of fever, otalgia, otorrhea, and less commonly, cranial nerve palsy. Treatment involves surgical debridement and antibiotics.
Other infections associated with diabetes include rhinocerebral mucormycosis, necrotizing fasciitis, emphysematous cholecystitis, and emphysematous pyelonephritis. An elderly patient with diabetes is also at increased risk of renal papillary necrosis, which presents as insidious renal failure.
COGNITIVE IMPAIRMENT
Elderly people with diabetes are at increased risk of cognitive impairment, which poses a barrier to taking medications appropriately and performing other tasks of self-management.
Because dementia may go undetected, particularly in the early stages, cognitive function should be assessed in elderly patients when they fail to take therapy correctly or have frequent episodes of hypoglycemia, or if glycemic control deteriorates without an obvious explanation. Caregivers play a critical role in detecting and reporting early cognitive impairment.
DEPRESSION IS OFTEN UNDETECTED
Elderly patients with diabetes have a higher rate of depression than do age-matched controls, but it is commonly underdetected and undertreated.5,36 Depression has been associated with poor glycemic control, and treatment of depression is associated with improved control. Routine screening for depression should be performed; a variety of diagnostic instruments are available. Particular attention should be given to medications that are associated with depression.
POLYPHARMACY
Many elderly patients take multiple medications. Polypharmacy increases the risk of drug side effects, interactions, and nonadherence to taking medications.37–39 This problem is increased in diabetes, in which several medications are necessary to manage hyper-glycemia, hyperlipidemia, hypertension, and other associated conditions.
Patients should keep accurate medication lists, including over-the-counter medications, herbs, and nutritional supplements. Physicians should carefully review each medication to check if it is appropriate and used correctly.
FALLS
Elderly patients with diabetes mellitus are at increased risk of injurious falls, which are associated with high rates of complications, death, and functional decline.40,41 Risk factors include frailty and functional disability, visual impairment, peripheral or autonomic neuropathy, hypoglycemia, and polypharmacy.
Elderly patients should be screened for their risk of falls, and appropriate measures should be instituted. The American Geriatrics Society has guidelines for preventing falls in the elderly.41
URINARY INCONTINENCE
Elderly women with diabetes are at increased risk of developing urinary incontinence. Risk factors include autonomic neuropathy (causing either neurogenic bladder or fecal impaction), polyuria due to hyperglycemia, and urinary tract and vaginal infections. Although evidence is lacking that urinary incontinence affects glycemic control, assessing and treating the condition improves quality of life.
SUMMARY
Diabetes is a common problem in the elderly, accounting for considerable morbidity and mortality. In a large longitudinal analysis (> 50,000 patients), elderly persons newly diagnosed as having diabetes experienced high rates of complications during 10-year follow-up, far in excess of elderly persons without diabetes.42 Diabetes is underdiagnosed in the elderly and is frequently undertreated. Management of the elderly with diabetes presents unique challenges because of associated comorbidities, but with attention to detail and individualized approaches, quality and duration of life can be optimized. The greatest attention should be given to reduction of overall cardiovascular risk. Glycemic goals and the treatment regimens to achieve those goals should be individualized and chosen to control hyperglycemic symptoms and achieve the maximal glycemic control possible while minimizing the risk of hypoglycemia. Diabetes will continue to be a challenge to the patient, the physician, the care team, and the health care system.
- Gregg EW, Engelgau MM, Narayan V. Complications of diabetes in elderly people. BMJ 2002; 325:916–917.
- Knopman D, Boland LL, Mosley T, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001; 56:42–48.
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53:1937–1942.
- Fontbonne A, Berr C, Ducimetiere P, Alperovitch A. Changes in cognitive abilities over a 4-year period are unfavorably affected in elderly diabetic subjects: results of the Epidemiology of Vascular Aging Study. Diabetes Care 2001; 24:366–370.
- Gregg EW, Mangione CM, Cauley JA, et al. Diabetes and incidence of functional disability in older women. Diabetes Care 2002; 25:61–67.
- Hornick T, Aron DC. Managing diabetes in the elderly: go easy, individualize. Cleve Clin J Med 2008; 75:70–78.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bertoni AG, Krop JS, Anderson GF, Brancati FL. Diabetes-related morbidity and mortality in a national sample of U.S. elders. Diabetes Care 2002; 25:471–475.
- Bertoni AG, Kirk JK, Goff DC, Wagenknecht LE. Excess mortality related to diabetes mellitus in elderly Medicare beneficiaries. Ann Epidemiol 2004; 14:362–367.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the Cholesterol and Recurrent Events (CARE) trial. The CARE Investigators. Circulation 1998; 98:2513–2519.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes. Lancet 2003; 361:2005–2016.
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care 2005; 28:S4–S36.
- Brown AF, Mangione CM, Saliba D, Sarkisian CA California Healthcare Foundation/American Geriatrics Society Panel on Improving Care for Elders with Diabetes. Guidelines for improving the care of the older person with diabetes mellitus. J Am Geriatr Soc 2003; 51:S265–S280.
- VA/DoD Clinical Practice Guideline for the Management of Diabetes Mellitus in the Primary Care Setting 2003. Accessed January 4, 2008. www.oqp.med.va.gov/cpg/dm/DM3_cpg/content/introduction.htm.
- Pogach LM, Brietzke SA, Cowan CL, Conlin P, Walder DJ, Sawin CT VA/DoD Diabetes Guideline Development Group. Development of evidence-based clinical practice guidelines for diabetes: the Department of Veterans Affairs/Department of Defense guidelines initiative. Diabetes Care 2004; 27:B82–B89.
- Stratton IM, Asler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321:405–412.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 2004; 141:413–420.
- Matthews DR, Stratton IM, Aldington SJ, Holman RR, Kohner EM UK Prospective Diabetes Study Group. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol 2004; 122:1631–1640.
- Cahill M, Halley A, Codd M, et al. Prevalence of diabetic retinopathy in patients with diabetic mellitus diagnosed after the age of 70 years. Br J Opthalmol 1997; 81:218–222.
- Hirvela H, Laatikainen L. Diabetic retinopathy in people aged 70 years or older. The Oulu Eye Study. Br J Ophthalmol 1997; 81:214–217.
- Tovi J, Ingemansson SO, Engfeldt P. Insulin treatment of elderly type 2 diabetic patients: effects on retinopathy. Diabetes Metab 1998; 24:442–447.
- Schrier RW, Estacio RO, Esler A, Mehler P. Effects of aggressive blood pressure control in normotensive type 2 diabetic patients on albuminuria, retinopathy and strokes. Kidney Int 2002; 61:1086–1097.
- Kohner EM. Aspirin for diabetic retinopathy. BMJ 2003; 327:1060–1061.
- Greene DA, Stevens MJ, Feldman EL. Diabetic neuropathy: scope of the syndrome. Am J Med 1999; 107:2S–8S.
- Hutchinson A, McIntosh A, Peters J, et al. Effectiveness of screening and monitoring tests for diabetic retinopathy—a systematic review. Diabet Med 2000; 17:495–506.
- Vijan S, Hofer TP, Hayward RA. Cost-utility analysis of screening intervals for diabetic retinopathy in patients with type 2 diabetes mellitus. JAMA 2000; 283:889–896.
- Mohamed Q, Gillies MC, Wong TY. Management of diabetic retinopathy: a systematic review. JAMA 2007; 298:902–916.
- Argoff CE, Cole BE, Fishbain DA, Irving GA. Diabetic peripheral neuropathic pain: clinical and quality-of-life issues. Mayo Clin Proc 2006; 81:S3–S11.
- Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ 2007; 335:87: epubl June 11, 2007.
- Bild DE, Selby JV, Sinnock P, Browner WS, Braveman P, Showstack JA. Lower-extremity amputation in people with diabetes. Epidemiology and prevention. Diabetes Care 1989; 12:24–31.
- Wheeler SG, Ahroni JH, Boyko EJ. Prospective study of autonomic neuropathy as a predictor of mortality in patients with diabetes. Diabetes Res Clin Pract 2002; 58:131–138.
- Brenner BM, Cooper ME, de Zeeuw D RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Sinclair AJ, Girling AJ, Bayer AJ. Cognitive dysfunction in older subjects with diabetes mellitus: impact on diabetes self-management and use of care services. All Wales Research into Elderly (AWARE) Study. Diabetes Res Clin Pract 2000; 50:203–212.
- Moisan J, Gaudet M, Gregoire JP, Bouchard R. Non-compliance with drug treatment and reading difficulties with regard to prescription labelling among seniors. Gerontology 2002; 48:44–51.
- Boyd CM, Darer J, Boult C, Fried LP, Boult L, Wu AW. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005; 294:716–724.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Schwartz AV, Hillier TA, Sellmeyer DE, et al. Older women with diabetes have a higher risk of falls: a prospective study. Diabetes Care 2002; 25:1749–1754.
- American Geriatrics Society, British Geriatrics Society, and American Academy of Orthopaedic Surgeons Panel on Falls Prevention. Guideline for the prevention of falls in older persons. J Am Geriatr Soc 2001; 49:664–672.
- Bethel MA, Sloan FA, Belsky D, Feinglos MN. Longitudinal incidence and prevalence of adverse outcomes of diabetes mellitus in elderly patients. Arch Intern Med 2007; 167:921–927.
- Gregg EW, Engelgau MM, Narayan V. Complications of diabetes in elderly people. BMJ 2002; 325:916–917.
- Knopman D, Boland LL, Mosley T, et al. Cardiovascular risk factors and cognitive decline in middle-aged adults. Neurology 2001; 56:42–48.
- Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM. Diabetes mellitus and the risk of dementia: The Rotterdam Study. Neurology 1999; 53:1937–1942.
- Fontbonne A, Berr C, Ducimetiere P, Alperovitch A. Changes in cognitive abilities over a 4-year period are unfavorably affected in elderly diabetic subjects: results of the Epidemiology of Vascular Aging Study. Diabetes Care 2001; 24:366–370.
- Gregg EW, Mangione CM, Cauley JA, et al. Diabetes and incidence of functional disability in older women. Diabetes Care 2002; 25:61–67.
- Hornick T, Aron DC. Managing diabetes in the elderly: go easy, individualize. Cleve Clin J Med 2008; 75:70–78.
- Haffner SM, Lehto S, Ronnemaa T, Pyorala K, Laakso M. Mortality from coronary heart disease in subjects with type 2 diabetes and in nondiabetic subjects with and without prior myocardial infarction. N Engl J Med 1998; 339:229–234.
- Bertoni AG, Krop JS, Anderson GF, Brancati FL. Diabetes-related morbidity and mortality in a national sample of U.S. elders. Diabetes Care 2002; 25:471–475.
- Bertoni AG, Kirk JK, Goff DC, Wagenknecht LE. Excess mortality related to diabetes mellitus in elderly Medicare beneficiaries. Ann Epidemiol 2004; 14:362–367.
- UK Prospective Diabetes Study Group. Tight blood pressure control and risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 38. BMJ 1998; 317:703–713. Erratum in: BMJ 1999; 318:29.
- Goldberg RB, Mellies MJ, Sacks FM, et al. Cardiovascular events and their reduction with pravastatin in diabetic and glucose-intolerant myocardial infarction survivors with average cholesterol levels: subgroup analyses in the Cholesterol and Recurrent Events (CARE) trial. The CARE Investigators. Circulation 1998; 98:2513–2519.
- Collins R, Armitage J, Parish S, Sleigh P, Peto R. MRC/BHF Heart Protection Study of cholesterol-lowering with simvastatin in 5963 people with diabetes. Lancet 2003; 361:2005–2016.
- American Diabetes Association. Standards of medical care in diabetes. Diabetes Care 2005; 28:S4–S36.
- Brown AF, Mangione CM, Saliba D, Sarkisian CA California Healthcare Foundation/American Geriatrics Society Panel on Improving Care for Elders with Diabetes. Guidelines for improving the care of the older person with diabetes mellitus. J Am Geriatr Soc 2003; 51:S265–S280.
- VA/DoD Clinical Practice Guideline for the Management of Diabetes Mellitus in the Primary Care Setting 2003. Accessed January 4, 2008. www.oqp.med.va.gov/cpg/dm/DM3_cpg/content/introduction.htm.
- Pogach LM, Brietzke SA, Cowan CL, Conlin P, Walder DJ, Sawin CT VA/DoD Diabetes Guideline Development Group. Development of evidence-based clinical practice guidelines for diabetes: the Department of Veterans Affairs/Department of Defense guidelines initiative. Diabetes Care 2004; 27:B82–B89.
- Stratton IM, Asler AI, Neil HA, et al. Association of glycaemia with macrovascular and microvascular complications of type 2 diabetes (UKPDS 35): prospective observational study. BMJ 2000; 321:405–412.
- UK Prospective Diabetes Study (UKPDS) Group. Intensive blood-glucose control with sulphonylureas or insulin compared with conventional treatment and risk of complications in patients with type 2 diabetes (UKPDS 33). Lancet 1998; 352:837–853. Erratum in: Lancet 1999; 354:602.
- Khaw KT, Wareham N, Bingham S, Luben R, Welch A, Day N. Association of hemoglobin A1c with cardiovascular disease and mortality in adults: the European prospective investigation into cancer in Norfolk. Ann Intern Med 2004; 141:413–420.
- Matthews DR, Stratton IM, Aldington SJ, Holman RR, Kohner EM UK Prospective Diabetes Study Group. Risks of progression of retinopathy and vision loss related to tight blood pressure control in type 2 diabetes mellitus: UKPDS 69. Arch Ophthalmol 2004; 122:1631–1640.
- Cahill M, Halley A, Codd M, et al. Prevalence of diabetic retinopathy in patients with diabetic mellitus diagnosed after the age of 70 years. Br J Opthalmol 1997; 81:218–222.
- Hirvela H, Laatikainen L. Diabetic retinopathy in people aged 70 years or older. The Oulu Eye Study. Br J Ophthalmol 1997; 81:214–217.
- Tovi J, Ingemansson SO, Engfeldt P. Insulin treatment of elderly type 2 diabetic patients: effects on retinopathy. Diabetes Metab 1998; 24:442–447.
- Schrier RW, Estacio RO, Esler A, Mehler P. Effects of aggressive blood pressure control in normotensive type 2 diabetic patients on albuminuria, retinopathy and strokes. Kidney Int 2002; 61:1086–1097.
- Kohner EM. Aspirin for diabetic retinopathy. BMJ 2003; 327:1060–1061.
- Greene DA, Stevens MJ, Feldman EL. Diabetic neuropathy: scope of the syndrome. Am J Med 1999; 107:2S–8S.
- Hutchinson A, McIntosh A, Peters J, et al. Effectiveness of screening and monitoring tests for diabetic retinopathy—a systematic review. Diabet Med 2000; 17:495–506.
- Vijan S, Hofer TP, Hayward RA. Cost-utility analysis of screening intervals for diabetic retinopathy in patients with type 2 diabetes mellitus. JAMA 2000; 283:889–896.
- Mohamed Q, Gillies MC, Wong TY. Management of diabetic retinopathy: a systematic review. JAMA 2007; 298:902–916.
- Argoff CE, Cole BE, Fishbain DA, Irving GA. Diabetic peripheral neuropathic pain: clinical and quality-of-life issues. Mayo Clin Proc 2006; 81:S3–S11.
- Wong MC, Chung JW, Wong TK. Effects of treatments for symptoms of painful diabetic neuropathy: systematic review. BMJ 2007; 335:87: epubl June 11, 2007.
- Bild DE, Selby JV, Sinnock P, Browner WS, Braveman P, Showstack JA. Lower-extremity amputation in people with diabetes. Epidemiology and prevention. Diabetes Care 1989; 12:24–31.
- Wheeler SG, Ahroni JH, Boyko EJ. Prospective study of autonomic neuropathy as a predictor of mortality in patients with diabetes. Diabetes Res Clin Pract 2002; 58:131–138.
- Brenner BM, Cooper ME, de Zeeuw D RENAAL Study Investigators. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345:861–869.
- UK Prospective Diabetes Study Group. Efficacy of atenolol and captopril in reducing risk of macrovascular and microvascular complications in type 2 diabetes: UKPDS 39. BMJ 1998; 317:713–720.
- Sinclair AJ, Girling AJ, Bayer AJ. Cognitive dysfunction in older subjects with diabetes mellitus: impact on diabetes self-management and use of care services. All Wales Research into Elderly (AWARE) Study. Diabetes Res Clin Pract 2000; 50:203–212.
- Moisan J, Gaudet M, Gregoire JP, Bouchard R. Non-compliance with drug treatment and reading difficulties with regard to prescription labelling among seniors. Gerontology 2002; 48:44–51.
- Boyd CM, Darer J, Boult C, Fried LP, Boult L, Wu AW. Clinical practice guidelines and quality of care for older patients with multiple comorbid diseases: implications for pay for performance. JAMA 2005; 294:716–724.
- Jackevicius CA, Mamdani M, Tu JV. Adherence with statin therapy in elderly patients with and without acute coronary syndromes. JAMA 2002; 288:462–467.
- Schwartz AV, Hillier TA, Sellmeyer DE, et al. Older women with diabetes have a higher risk of falls: a prospective study. Diabetes Care 2002; 25:1749–1754.
- American Geriatrics Society, British Geriatrics Society, and American Academy of Orthopaedic Surgeons Panel on Falls Prevention. Guideline for the prevention of falls in older persons. J Am Geriatr Soc 2001; 49:664–672.
- Bethel MA, Sloan FA, Belsky D, Feinglos MN. Longitudinal incidence and prevalence of adverse outcomes of diabetes mellitus in elderly patients. Arch Intern Med 2007; 167:921–927.
KEY POINTS
- Compared with strict glycemic control, treating cardiovascular risk factors offers more benefit in a shorter time and should be a higher priority.
- Diabetic retinopathy is a leading cause of blindness. Yearly eye examinations are recommended.
- Elderly patients with diabetes are prone to rapidly progressive nephropathy, especially after receiving iodinated contrast agents. Good glycemic control and control of blood pressure, especially with angiotensin-converting enzyme inhibitors, reduce the risk and the rate of progression.
- Elderly patients with diabetes are at higher risk of cognitive decline, depression, and polypharmacy, all of which impede good diabetes management.
Screen for portopulmonary hypertension, especially in liver transplant candidates
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
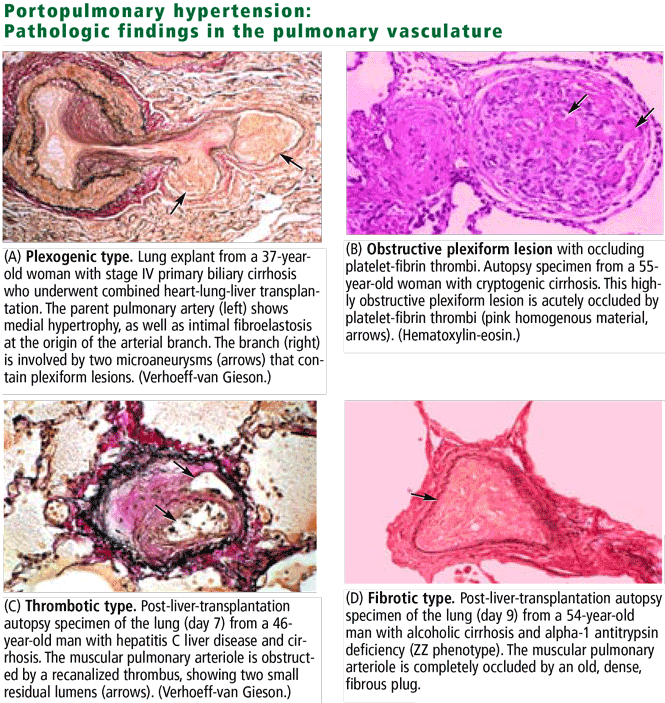
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
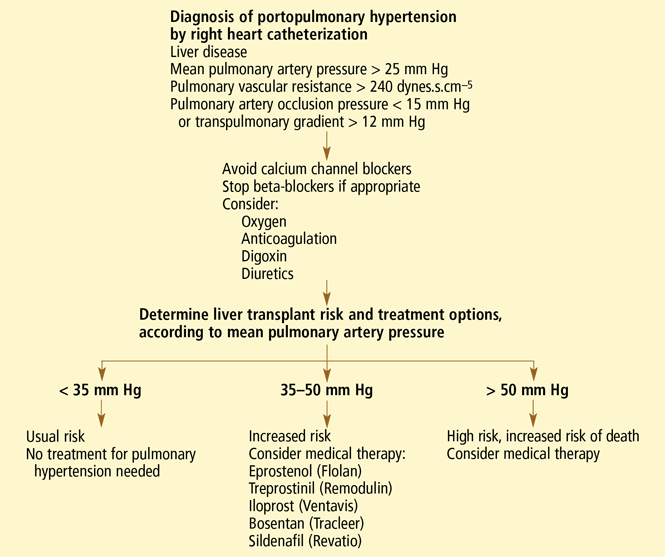
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
Portopulmonary hypertension poses difficulties for patients with liver disease. The elevated pulmonary artery pressure in this disorder makes liver transplantation more dangerous and in fact may rule out the procedure, although in a selected few patients, medical treatment may enable transplantation to proceed. In any event, portopulmonary hypertension should be looked for in patients with liver disease, especially if liver transplantation is being considered.
In this article we discuss the definition, pathophysiology, clinical features, diagnosis, and management of portopulmonary hypertension.
DEFINED BY HEMODYNAMIC CRITERIA
Portopulmonary hypertension—elevated pulmonary artery pressure due to increased resistance to blood flow in patients with portal hypertension—is one of several pulmonary complications of liver disease. A few others to be aware of are pleural effusions (hepatic hydrothorax), dilatation of the pulmonary vasculature with shunting and hypoxemia (hepatopulmonary syndrome), and elevation in pulmonary pressures due to the high cardiac output usually seen in liver disease (flow phenomenon).
The definition of portopulmonary hypertension has evolved as the various hemodynamic profiles that occur in liver disease and their consequences have been described. Currently, it is defined by the following criteria (obtained by right heart catheterization) in a patient with portal hypertension1:
- Elevated mean pulmonary artery pressure (> 25 mm Hg at rest, > 30 mm Hg with exercise);
- Increased pulmonary vascular resistance (> 240 dynes.s.cm−5; pulmonary vascular resistance = [(mean pulmonary artery pressure minus pulmonary artery occlusion pressure) /cardiac output] times 80); and
- Normal pulmonary artery occlusion pressure (< 15 mm Hg) or an elevated transpulmonary gradient (the mean pulmonary artery pressure minus the pulmonary artery occlusion pressure; abnormal is > 12 mm Hg).
The transpulmonary gradient sometimes helps in further assessing the resistance to blood flow in cases that do not meet the other criteria.2 For example, how should we classify a patient whose mean pulmonary artery pressure is 45 mm Hg but whose pulmonary vascular resistance is only 432 dynes.s.cm−5 and whose pulmonary artery occlusion pressure is slightly high at 18 mm Hg? Although this patient does not meet the hemodynamic criteria for portopulmonary hypertension listed above, intuitively, we should not exclude the diagnosis, as the transpulmonary gradient is high at 27 mm Hg.
FLOW PHENOMENON VS TRUE PORTOPULMONARY HYPERTENSION
The cardiopulmonary hemodynamic profile is different in patients with liver disease than in those without liver disease. Understanding the “normal” hemodynamics in liver disease is paramount in understanding the abnormal hemodynamics that occur in portopulmonary hypertension. In general, patients with liver disease have a high cardiac output at baseline (high flow). They may also have an increased blood volume due to fluid shifts (elevated pulmonary artery occlusion pressure).
Right heart catheterization is necessary to make the diagnosis of portopulmonary hypertension, as pulmonary artery pressures may be increased simply from increases in cardiac output and blood volume without an increase in pulmonary vascular resistance.
Consider, for example, a patient whose mean pulmonary artery pressure is 38 mm Hg, pulmonary artery occlusion pressure 14 mm Hg, and cardiac output 8.8 L/minute. In this case, the pulmonary vascular resistance is 218 dynes.s.cm−5. About 30% to 50% of patients with cirrhosis have this type of hyperdynamic pattern, with high cardiac output, low systemic vascular resistance, and low pulmonary vascular resistance.1,3,4 These patients typically have a much better prognosis than those with portopulmonary hypertension and do well with liver transplantation.
Right heart catheterization is also helpful in assessing whether elevated pulmonary pressures are due to increased volume (increased pulmonary artery occlusion pressure), in which case the patient might benefit from more aggressive diuresis.
In true portopulmonary hypertension, the pulmonary vascular resistance is increased due to obstruction of arterial blood flow. Cardiac output may be elevated initially and then decline as pulmonary hypertension becomes more severe. These hemodynamic patterns have different treatment implications and are important when liver transplantation is being considered.5
HOW COMMON IS PORTOPULMONARY HYPERTENSION?
The incidence and prevalence of portopulmonary hypertension is difficult to assess, as many of the estimates are in patients with severe liver disease undergoing evaluation for liver transplantation. Its prevalence in patients with cirrhosis and refractory ascites has been documented at 16.1%,6 while its prevalence in patients with cirrhosis without refractory ascites has been in the range of 0.25% to 4%.7–9
Overall, about 8% of candidates for liver transplantation have portopulmonary hypertension and are at risk of its complications.10 In view of this figure, screening for it should be performed before proceeding with liver transplantation.
VASOCONSTRICTION, REMODELING, THROMBOSIS
The pathogenesis of portopulmonary hypertension is not completely understood but likely involves a complex interaction of several mechanisms, including an imbalance of vascular mediators favoring vasoconstriction,11–13 endothelial damage with vascular remodeling due to excessive pulmonary blood flow,14,15 smooth muscle proliferation, and microvascular thrombosis.16,17
The pulmonary endothelium is a complex, dynamic organ capable of influencing a variety of vascular mediators and adapting to changes in pulmonary volume as necessary. Endothelial dysfunction may initiate the vascular changes seen in portopulmonary hypertension.
Endothelin-1 (ET-1) is a potent vasoconstrictor that has been implicated in the pathogenesis of idiopathic pulmonary artery hypertension. ET-1 levels are also increased in cirrhotic patients with refractory ascites.6
Other mediators favoring vasoconstriction include serotonin, angiotensin II, and norepinephrine. Whether these mediators influence the development of portopulmonary hypertension is not clear.
At the same time, production of vasodilatory mediators such as nitric oxide and prostacyclin may be decreased in portopulmonary hypertension, facilitating vascular remodeling and a proliferative vascular response. Prostacyclin is a potent vasodilator normally found in high concentrations in the lungs. Prostacyclin synthase is the precursor enzyme for the production of prostacyclin and is decreased in the lungs of patients with portopulmonary hypertension.18
Another way that portal hypertension may influence lung vascular tone is that endotoxin, cytokines, or both, released from the splanchnic circulation, may bypass the liver and get into the lungs.19 Evidence in support of this is that patients with portosystemic shunting can develop similar pathologic changes in the pulmonary vascular bed that normalize when the shunt is reversed. To date, however, no substance has been definitively identified.
Yet another proposed mechanism is shear stress on the pulmonary endothelium from the hyperdynamic cardiac output, with resultant vascular remodeling; however, other mechanisms must be involved, as not everyone with liver disease develops portopulmonary hypertension (see below).
These changes are identical to those in idiopathic and familial pulmonary arterial hypertension,21 and indeed, the World Health Organization now classifies portopulmonary hypertension in the same category as these primary forms of pulmonary hypertension rather than in the secondary forms.3
Why doesn’t everyone with liver disease develop portopulmonary hypertension?
The severity of liver disease or degree of portal hypertension does not appear to correlate with the severity of pulmonary hypertension,4 and portopulmonary hypertension does not develop in all patients with portal hypertension. Therefore, it is likely that some patients have a genetic or environmental susceptibility or suffer a “second hit” that triggers dysregulated pulmonary vascular proliferation and contributes to the development of pulmonary hypertension.
Whether genetic mutations play a role in portopulmonary hypertension remains unknown. Such a mutation could be similar to the one identified in the bone morphogenetic protein receptor type 2 gene (BMPR2) in familial pulmonary artery hypertension or the mutation in the activin-like kinase gene (ALK1) seen in pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia.22
Current studies are investigating the role that bone-marrow-derived progenitor cells might play in the pathogenesis of portopulmonary hypertension.
CLINICAL FEATURES MAY NOT BE OBVIOUS AT FIRST
In the early stages of portopulmonary hypertension, patients may have no symptoms or only symptoms of liver disease, so it is important to have a high index of suspicion and screen for pulmonary hypertension. As its severity increases, symptoms may include fatigue, dyspnea, abdominal bloating, palpitations, chest pain or pressure, and syncope. The most common presenting symptom is dyspnea on exertion.
Similarly, the findings on physical examination also depend on the severity of pulmonary hypertension. Patients with mild portopulmonary hypertension may have only signs suggesting liver disease, such as spider telangiectases, jaundice, mild lower extremity edema, and ascites. As the severity of portopulmonary hypertension increases, however, findings of right heart pressure-and-volume overload become more obvious. These include peripheral edema, elevation of the jugular venous pressure, a right ventricular lift, a loud pulmonic valve closure, increased split of the second heart sound, a pulsatile liver, or a right-sided third or fourth heart sound.
SCREEN LIVER TRANSPLANT CANDIDATES
Screening for portopulmonary hypertension should be mandatory in patients undergoing evaluation for liver transplantation. This condition increases the risk of perioperative death, so it is not acceptable to make the diagnosis in the operating room!5
Electrocardiographic abnormalities that may raise the suspicion of portopulmonary hypertension include right atrial or ventricular enlargement and a right bundle branch pattern.
Chest radiographic signs are enlarged central pulmonary arteries and cardiomegaly. These electrocardiographic and radiographic signs tend to reflect advanced pulmonary hypertension.
Pulmonary function testing is not generally helpful, but the diffusing capacity may be decreased.
B-type natriuretic peptide (BNP) measurement may be helpful. BNP is released from the ventricles when the ventricles become dilated (due to pressure or volume overload), as in left or right heart failure. BNP testing is clinically useful in monitoring the severity of disease and the efficacy of treatment in patients with pulmonary hypertension; its role in portopulmonary hypertension requires prospective study.23
Transthoracic Doppler echocardiography is an excellent screening test and should be performed in patients undergoing evaluation for liver transplantation to exclude pulmonary hypertension.1 Findings on echocardiography that suggest portopulmonary hypertension include elevation of right ventricular systolic pressure (RVSP), which is calculated from the peak tricuspid regurgitant velocity (TRV) using the modified Bernoulli equation and an estimate of right atrial pressure (RAP):
RVSP = 4(TRV)2 + RAP.
Right atrial pressure is estimated from the filling characteristics of the inferior vena cava.
Transthoracic Doppler echocardiography has a sensitivity of 97% and a specificity of 77% in diagnosing moderate to severe pulmonary hypertension in patients undergoing evaluation for liver transplantation.24 Using an RVSP cutoff of 40 mm Hg, the sensitivity of Doppler echocardiography is about 80%, specificity 96%, positive predictive value 60%, and negative predictive value 98%.25
At Mayo Clinic, patients with an estimated RVSP greater than 50 mm Hg undergo right heart catheterization (see below). Such patients should also have repeat echocardiography at 1-year intervals to monitor for increasing pulmonary artery pressures5; for those on the waiting list for liver transplantation, the interval should probably be every 6 to 12 months.
RIGHT HEART CATHETERIZATION CONFIRMS THE DIAGNOSIS
The diagnosis of portopulmonary hypertension is confirmed with right heart catheterization to accurately measure pulmonary artery pressures, pulmonary artery occlusion pressure (to exclude volume overload), cardiac output (to exclude high-output pulmonary hypertension), and pulmonary vascular resistance. One study in patients with decompensated cirrhosis and refractory ascites found that a right atrial pressure of 14 mm Hg or greater had a positive predictive value of 83% for pulmonary hypertension.6
Other, potentially treatable causes of pulmonary hypertension must be excluded before diagnosing portopulmonary hypertension. These include thromboembolic disease, interstitial lung disease, connective tissue disease, untreated obstructive sleep apnea, and elevated pulmonary artery pressures due to increased cardiac output.
Vasodilator studies are being done less frequently in patients with portopulmonary hypertension, as they generally cannot tolerate calcium channel blocker therapy. Calcium channel blocker therapy is usually started in patients with idiopathic pulmonary artery hypertension who exhibit a positive vasodilator response. A positive vasodilator response also does not predict survival with or without liver transplantation. Unlike those with idiopathic pulmonary artery hypertension, many patients with portopulmonary hypertension cannot tolerate calcium channel blockers, as some of these drugs can exacerbate edema and portal hypertension.
GENERAL MANAGEMENT
Treatment of mild portopulmonary hypertension (mean pulmonary artery pressure < 35 mm Hg) is debatable. In these cases many patients do not have any symptoms attributable to portopulmonary hypertension, but only symptoms of liver disease, and they have a good functional status. As a group, such patients have not been formally studied to date.
Anticoagulation is often contraindicated in portopulmonary hypertension because of gastroesophageal varices, thrombocytopenia, or other coagulation abnormalities related to liver disease. If contraindications to anticoagulation do not exist, it should be considered.
Diuretics are a mainstay in the treatment of portopulmonary hypertension, both for the pulmonary hypertension and for the liver disease, especially if ascites or peripheral edema is present.
Oxygen should be given to patients with hypoxemia to keep the saturation greater than 90%.
Beta-blockers: A dilemma
Beta-blockers are used in many patients with liver disease as both primary and secondary prophylaxis of variceal bleeding.
However, one study has shown that in patients with moderate to severe portopulmonary hypertension, beta-blockers are associated with significant worsening of exercise capacity and pulmonary hemodynamic measurements.26 After beta-blockers were withdrawn, the 6-minute walking distance increased in 9 of 10 patients, and cardiac output increased with no change in mean pulmonary artery pressure, resulting in a 19% decrease in pulmonary vascular resistance. The increases in cardiac output were related to a 25% increase in heart rate. Long-term follow-up was not reported, and it remains unclear whether rates of gastrointestinal bleeding may increase when beta-blockers are withdrawn.
Beta-blocker therapy in portopulmonary hypertension needs to be carefully considered and if at all possible should be avoided.
VASODILATOR THERAPY
Several vasodilating or vasomodulating drugs are available. However, much of the information about them comes from studies in patients with idiopathic pulmonary artery hypertension or pulmonary hypertension due to connective tissue disease, and no randomized controlled trials in portopulmonary hypertension have been performed.
Prostanoids
Prostanoids have been used successfully to lower pulmonary pressures in portopulmonary hypertension.
Epoprostenol (Flolan) is a pulmonary and systemic vasodilator as well as an inhibitor of platelet aggregation. It is given as a continuous intravenous infusion via an indwelling central venous catheter and a portable infusion pump. It has a very short half-life, requires mixing, and must be kept cold with ice packs, making it somewhat cumbersome to administer.
This medication has been shown to improve cardiopulmonary hemodynamics and exercise capacity in portopulmonary hypertension, although a survival advantage has not been documented to date.27 In several case series, some patients with portopulmonary hypertension treated with intravenous epoprostenol responded with a reduction in pulmonary pressures and successfully underwent liver transplantation.28–31
Complications of intravenous epoprostenol therapy include central venous catheter thrombosis, infection, and infusion pump failure; a backup pump must be available at all times. Patients with portopulmonary hypertension may also develop progressive splenomegaly and thrombocytopenia that may be due to increased blood flow in the splanchnic circulation.32
Treprostinil (Remodulin) has a longer half-life and does not have to be kept cold. It is given as a 24-hour intravenous or subcutaneous infusion, using an infusion pump that is smaller than that used with epoprostenol.
Although treprostinil is easier for patients to use, larger doses are necessary to achieve the same effect as with epoprostenol. With subcutaneous administration, the biggest drawback is site pain. Prostacyclin-related side effects include flushing, diarrhea, jaw discomfort, and lower extremity pain.
Iloprost (Ventavis) has the advantage of being given by inhalation. It is very short-acting, however, and requires six to nine inhalations per day.
Endothelin receptor blockers
Bosentan (Tracleer) is an oral agent that has been approved by the US Food and Drug Administration (FDA) for the treatment of pulmonary hypertension, including in patients with portopulmonary hypertension who have mild hepatic derangement. This medication is a dual endothelin receptor antagonist, nonselectively blocking the endothelin A and B receptors on the endothelial and vascular smooth muscle cells so that ET-1 cannot bind and cause vasoconstriction.
In approximately 10% of patients, bosentan can cause elevations in aminotransferase, alkaline phosphatase, and bilirubin levels, which therefore must be checked monthly.33 Irreversible hepatic toxicity is uncommon; in most cases, liver function abnormalities return to baseline levels when the medication is stopped. The presumed mechanism is impairment of bile-salt transporters, leading to bile-salt accumulation in the liver.34 Bosentan’s use in patients with liver disease has not been well studied, although several case reports have described its use in patients with portopulmonary hypertension.35–38
Ambrisentan (Letairis) is a selective endothelin receptor-A blocker that has just received FDA approval for the treatment of pulmonary artery hypertension. It has not yet been studied in portopulmonary hypertension. Elevations in liver enzymes and bilirubin may also occur, and monthly monitoring is indicated.
Sildenafil
Another oral agent that might be effective in portopulmonary hypertension is sildenafil (Revatio). A phosphodiesterase-5 inhibitor, it selectively inhibits the cyclic guanosine monophosphatase-specific phosphodiesterase type 5 enzyme that is found in large concentrations in pulmonary artery smooth muscle cells.
In other forms of pulmonary hypertension, sildenafil has been shown to increase cardiac output and decrease pulmonary artery pressures and pulmonary vascular resistance without serious adverse events.39–41
In one reported case, treatment with sildenafil in a patient with portopulmonary hypertension decreased the mean pulmonary artery pressure from 56 mm Hg to 28 to 31 mm Hg, and the patient underwent successful liver transplantation.42 A recent case series of 14 patients with portopulmonary hypertension treated with sildenafil documents some improvement in 6-minute walking distance, suggesting that sildenafil as monotherapy or in combination therapy might be effective in portopulmonary hypertension.43 However, in 3 of these patients, the cardiac index decreased and pulmonary vascular resistance increased.44
We must emphasize that controlled studies in portopulmonary hypertension need to be done to find the optimal therapy.
LIVER TRANSPLANTATION MAY BENEFIT A FEW PATIENTS
Liver transplantation may be beneficial in highly selected patients with portopulmonary hypertension. However, this condition increases the risk of intraoperative and immediate postoperative complications of liver transplantation, so patients should be carefully evaluated5,45 at a liver transplantation center experienced in its management, including medical treatment with well-defined protocols regarding timing of liver transplantation.
Patients with mean pulmonary artery pressures greater than 50 mm Hg should not undergo liver transplantation. Those with mean pulmonary artery pressure between 35 and 50 mm Hg also have an increased mortality rate and may benefit from prolonged treatment for pulmonary hypertension.5,46
One successful case of living-related liver transplantation in a patient with portopulmonary hypertension has been published.47 (Most other successful transplants were from unrelated cadaver donors.)
Some patients who initially cannot undergo liver transplantation owing to severe pulmonary hypertension may eventually be able to do so if they receive medical therapy that improves their pulmonary hemodynamic profile, decreasing their mean pulmonary artery pressure and pulmonary vascular resistance. This would apply to a small subset of patients with portopulmonary hypertension.
When patients without pulmonary hypertension undergo liver transplantation, right ventricular function is preserved throughout all phases of the surgery.48 Patients with portopulmonary hypertension, however, may develop hemodynamic instability during liver transplantation. The most critical times are the induction of anesthesia, during and after graft reperfusion, and the immediate postoperative period.49,50
During the surgery, patients may require vasodilators if they have worsening pulmonary hypertension, or inotropic medications if they have right ventricular dysfunction and heart failure. In one study,51 eight patients with portopulmonary hypertension diagnosed at anesthesia induction for liver transplantation all required intraoperative vasodilator therapy after graft reperfusion because of marked increases in pulmonary artery pressures and pulmonary vascular resistance.
The increase in blood flow following reperfusion or necessary fluid challenges may exacerbate pulmonary hypertension, resulting in worsening right heart function and backup into the transplanted liver. Infusion of 1 liter of crystalloid over 10 minutes has been shown to increase mean pulmonary artery pressure and pulmonary artery occlusion pressure in liver transplantation candidates without pulmonary hypertension52; this response may be exaggerated in portopulmonary hypertension.
PROGNOSIS VARIES WITH SEVERITY OF DISEASE
The natural history of untreated portopulmonary hypertension varies with the degree of liver disease and the severity of pulmonary hypertension. Transplant-free survival was 85% at 1 year and 38% at 3 years in one study.45 The cardiac index appears to be the most significant prognostic variable.20
In a retrospective study of 78 patients with portopulmonary hypertension treated conservatively (before prostanoids were available) the median survival was 6 months (range 0–84 months) from the time of diagnosis.53 Causes of death included right heart failure, sudden death, gastrointestinal bleeding, and small bowel perforation.
Most of the data on outcomes of drug treatment and liver transplantation in patients with portopulmonary hypertension come from case series and retrospective reviews; prospective trials have been lacking.
If right ventricular function is normal and pulmonary hypertension is mild (mean pulmonary artery pressure < 35 mm Hg), patients tend to do well with liver transplantation.9
Outcomes are worse if pulmonary hypertension is more severe. In a database54 from 10 liver transplant centers from 1996 to 2001, 13 (36%) of 36 patients undergoing liver transplantation died in the hospital, emphasizing the importance of accurately assessing the severity of pulmonary hypertension before attempting liver transplantation.46 The rate was even higher—92%—in those with a mean pulmonary artery pressure greater than 35 mm Hg. The cause of death in severe pulmonary hypertension was failure of the right ventricle.
However, some patients with moderate to severe portopulmonary hypertension have been bridged with medications to lower pulmonary artery pressures and pulmonary vascular resistance so that liver transplantation can be safely done, and some have even been able to discontinue medications because their pulmonary hypertension resolved.29,31,41,42,47
Unlike in hepatopulmonary syndrome, liver transplantation is not the treatment of choice for portopulmonary hypertension, and pulmonary hypertension does not always resolve after liver transplantation. Many patients continue therapy for pulmonary hypertension after liver transplantation. Pulmonary hypertension may resolve, persist, or even develop de novo after liver transplantation.1 If pulmonary hypertension resolves, it does so over a prolonged time—months to years—favoring a vascular remodeling hypothesis as opposed to simply reversing vasoconstriction.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
- Rodriguez-Roisin R, Krowka MJ, Hervé P, Fallon MB; ERS Task Force Pulmonary-Hepatic Vascular Disorders (PHD) Scientific Committee. Pulmonary-hepatic vascular disorders (PHD). Eur Respir J 2004; 24:861–880.
- Krowka MJ, Swanson KL, Frantz RP, et al. Portopulmonary hypertension: results from a 10-year screening algorithm. Hepatology 2006; 44:1502–1510.
- Simonneau G, Galie N, Rubin LJ, et al. Clinical classification of pulmonary hypertension. J Am Coll Cardiol 2004; 43:5S–12S.
- Hadengue A, Benhayoun MK, Lebrec D, et al. Pulmonary hypertension complicating portal hypertension: prevalence and relation to splanchnic hemodynamics. Gastroenterology 1991; 100:520–528.
- Krowka MJ, Plevak DJ, Findlay JY, et al. Pulmonary hemodynamics and perioperative cardiopulmonary-related mortality in patients with portopulmonary hypertension undergoing liver transplantation. Liver Transplant 2000; 6:443–450.
- Benjaminov FS, Prentice M, Sniderman KW, et al. Portopulmonary hypertension in decompensated cirrhosis with refractory ascites. Gut 2003; 52:1355–1362.
- McDonnell PJ, Toye PA, Hutchins GM. Primary pulmonary hypertension and cirrhosis: are they related? Am Rev Respir Dis 1983; 127:437–441.
- Cheng EY, Woehlck H. Pulmonary artery hypertension complicating anesthesia for liver transplantation. Anesthesiology 1992; 77:375–378.
- Castro M, Krowka MJ, Schroeder DR, et al. Frequency and clinical implications of increased pulmonary artery pressures in liver transplantation. Mayo Clin Proc 1996; 71:543–551.
- Ramsay MA, Simpson BR, Nguyen AT, et al. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Kiely DG, Cargill RI, Struthers AD, et al. Cardiopulmonary effects of endothelin-1 in man. Cardiovasc Res 1997; 33:378–386.
- Panos RJ, Baker SK. Mediators, cytokines, and growth factors in liver-lung interactions. Clin Chest Med 1996; 17:151–169.
- Higgenbottam T. Pathophysiology of pulmonary hypertension. Chest 1994; 105:7S–12S.
- Krowka MJ. Hepatopulmonary syndrome and portopulmonary hypertension: distinction and dilemmas. Hepatology 1997; 25:1282–1284.
- Hongqun L, Lee SS. Cardiopulmonary dysfunction in cirrhosis. Hepatology 2000; 14:600–608.
- Lebrec D, Brenot F, Simonneau G, et al. Pulmonary arterial hypertension in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Herve P, Lebrec D, Brenot F, et al. Pulmonary vascular disorders in portal hypertension. Eur Respir J 1998; 11:1153–1166.
- Tuder RM, Cool CD, Geraci MW, et al. Prostacyclin synthase expression is decreased in lungs from patients with severe pulmonary hypertension. Am J Respir Crit Care Med 1999; 159:1925–1932.
- Hoeper MM, Krowka MJ, Strassburg CP. Portopulmonary hypertension and hepatopulmonary syndrome. Lancet 2004; 363:1461–1468.
- Edwards B, Weir K, Edwards WD, et al. Coexistent pulmonary and portal hypertension: morphologic and clinical features. J Am Coll Cardiol 1987; 10:1233–1238.
- Ramsay MAE, Simpson BR, Nguyen AT, Ramsay KJ, East C, Klintmalm GB. Severe pulmonary hypertension in liver transplant candidates. Liver Transplant Surg 1997; 3:494–500.
- Trembath RC. Clinical and molecular genetic features of pulmonary hypertension in patients with hereditary hemorrhagic telangiectasia. N Engl J Med 2001; 345:325–334.
- Leuchte HH, Holzapfel M, Baumgartner RA, et al. Clinical significance of brain natriuretic peptide in primary pulmonary hypertension. J Am Coll Cardiol 2004; 43:764–770.
- Kim WR, Krowka MJ, Plevak DJ, et al. Accuracy of Doppler echocardiography in the assessment of pulmonary hypertension in liver transplant candidates. Liver Transplant 2000; 6:453–458.
- Colle IO, Moreau R, Godinho E, et al. Diagnosis of portopulmonary hypertension in candidates for liver transplantation: a prospective study. Hepatology 2003; 37:401–409.
- Provencher S, Herve P, Jais X, et al. Deleterious effects of beta-blockers on exercise capacity and hemodynamics in patients with portopulmonary hypertension. Gastroenterology 2006; 130:120–126.
- Swanson KL, McGoon MD, Krowka MJ. Survival in patients with portopulmonary hypertension [abstract]. Am J Respir Crit Care Med 2003; 167:A693.
- Kuo PC, Johnson LB, Plotkin JS, et al. Continuous intravenous infusion of epoprostenol for the treatment of portopulmonary hypertension. Transplantation 1997; 63:604–616.
- Krowka MJ, Frantz RP, McGoon MD, et al. Improvement in pulmonary hemodynamics during intravenous epoprostenol (prostacyclin): A study of 15 patients with moderate to severe portopulmonary hypertension. Hepatology 1999; 30:641–648.
- Kähler CM, Graziadei I, Wiedermann CJ, Kneussl MP, Vogel W. Successful use of continuous intravenous prostacyclin in a patient with severe portopulmonary hypertension. Wien Klin Wochenschr 2000; 112:637–640.
- Sussman N, Kaza V, Barshes N, et al. Successful liver transplantation following medical management of portopulmonary hypertension: a single-center series. Am J Transplant 2006; 6:2177–2182.
- Findlay JY, Plevak DJ, Krowka MJ, et al. Progressive splenomegaly after epoprostenol therapy in portopulmonary hypertension. Liver Transplant Surg 1999; 5:381–387.
- Rubin LJ, Roux S. Bosentan: a dual endothelin receptor antagonist. Expert Opin Invest Drugs 2002; 11:991–1002.
- Fattinger K, Funk C, Pantze M, et al. The endothelin antagonist bosentan inhibits the canalicular bile salt export pump: a potential mechanism for hepatic adverse reactions. Clin Pharmacol Ther 2001; 69:223–231.
- Hinterhuber L, Graziadei IW, Kahler CM, et al. Endothelin-receptor anatgonist treatment of portopulmonary hypertension. Clin Gastroenterol Hepatol 2004; 2:1039–1042.
- Clift PF, Townend JN, Bramhall S, et al. Successful treatment of severe portopulmonary hypertension after liver transplantation by bosentan. Transplantation 2004; 77:1774–1775.
- Halank M, Miehlke S, Hoeffken G, et al. Use of oral endothelin-receptor antagonist bosentan in the treatment of portopulmonary hypertension. Transplantation 2004; 77:1775–1776.
- Kuntzen C, Gulberg V, Gerbes AL. Use of a mixed endothelin receptor antagonist in portopulmonary hypertension: a safe and effective therapy? Gastroenterology 2005; 128:164–168.
- Watanabe H, Ohashi K, Takeuchi K, et al. Sildenafil for primary and secondary pulmonary hypertension. Clin Pharmacol Ther 2002; 71:398–402.
- Michelakis E, Tymchak W, Lien D, et al. Oral sildenafil is an effective and specific pulmonary vasodilator in patients with pulmonary arterial hypertension: comparison with inhaled nitric oxide. Circulation 2002; 105:2398–2403.
- Ghofrani HA, Wiedemann R, Rose F, et al. Sildenafil for treatment of lung fibrosis and pulmonary hypertension: a randomised controlled trial. Lancet 2002; 360:895–900.
- Makisalo H, Koivusalo A, Vakkuri A, et al. Sildenafil for portopulmonary hypertension in a patient undergoing liver transplantation. Liver Transplant 2004; 10:945–950.
- Reichengerger F, Voswinckel R, Steveling E, et al. Sildenafil treatment for portopulmonary hypertension. Eur Respir J 2006; 28:563–567.
- Krowka MJ, Swanson KL. How should we treat portopulmonary hypertension? Eur Respir J 2006; 28:466–467.
- Kawut SM, Taichman DB, Ahya VN, et al. Hemodynamics and survival of patients with portopulmonary hypertension. Liver Transplant 2005; 11:1107–1111.
- Krowka MJ, Mandell MS, Ramsay MA, et al. Hepatopulmonary syndrome and portopulmonary hypertension: a report of the multicenter liver transplant database. Liver Transplant 2004; 10:174–182.
- Sulica R, Emre S, Poon M. Medical management of portopulmonary hypertension and right heart failure prior to living-related liver transplantation. Congest Heart Fail 2004; 10:192–194.
- De Wolf AM, Begliomini B, Gasior TA, et al. Right ventricular function during orthotopic liver transplantation. Anesthes Analges 1993; 76:562–568.
- Csete M. Intraoperative management of liver transplant patients with pulmonary hypertension. Liver Transplant Surg 1997; 3:454–455.
- Acosta F, Sansano T, Palenciano CG, et al. Portopulmonary hypertension and liver transplantation: hemodynamic consequences at reperfusion. Transplant Proc 2005; 37:3865–3866.
- Taura P, Garcia-Valdecasas JC, Beltran J, et al. Moderate primary pulmonary hypertension in patients undergoing liver transplantation. Anesthes Analges 1996; 83:675–680.
- Kuo PC, Schroeder RA, Vagelos RH, et al. Volume-mediated pulmonary responses in liver transplant candidates. Clin Transplant 1996; 10:521–527.
- Robalino BD, Moodie DS. Association between primary pulmonary hypertension and portal hypertension: analysis of its pathophysiology and clinical, laboratory and hemodynamic manifestations. J Am Coll Cardiol 1991; 17:492–498.
- Mandell MS, Krowka MJ. Formation of a national database on pulmonary hypertension and hepatopulmonary syndrome in chronic liver disease. Anesthesiology 1997; 87:450–451.
KEY POINTS
- In portopulmonary hypertension, the pulmonary artery pressures, pulmonary vascular resistance, and portal venous pressure are all elevated.
- All candidates for liver transplantation should undergo echocardiography to screen for portopulmonary hypertension. If the echocardiogram shows elevated pulmonary pressures, right heart catheterization must be performed to confirm the diagnosis.
- The ideal medical regimen remains to be determined. Although drug treatment may lower pulmonary artery pressures in selected patients so that liver transplantation can be safely done, morbidity and mortality rates remain higher in patients with moderate to severe portopulmonary hypertension.
- Liver transplantation is not the treatment of choice for portopulmonary hypertension.
Should all patients with chronic kidney disease take a statin?
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
We think some patients with chronic kidney disease should take a statin, particularly those in stages 1 through 4 (ie, not yet on dialysis1)* who have low-density lipoprotein cholesterol (LDL-C) levels higher than 100 mg/dL. However, few studies have addressed this question.
*Stages of chronic kidney disease1:
Stage 1—kidney damage with normal or high glomerular filtration rate (GFR ≥ 90 mL/min/1.73 m2)
Stage 2—kidney damage with mildly decreased GFR (60–89 mL/min/1.73 m2)
Stage 3—moderately decreased GFR (30–59 mL/min/1.73 m2)
Stage 4—severely decreased GFR (15–29 mL/min/1.73 m2)
Stage 5—kidney failure (GFR < 15 mL/min/1.73 m2 or dialysis)
The answer is murkier in patients on dialysis. Only one study has been done in this population, and it found no benefit from statin therapy. However, we would prescribe a statin for a dialysis patient who had known coronary artery disease and an LDL-C level higher than 100 mg/dL.
RATIONALE FOR STATIN USE: KIDNEY PATIENTS ARE AT RISK
Cardiovascular disease is common among patients with chronic kidney disease. While the risks of cardiovascular disease and death are highest among those requiring dialysis, earlier stages of chronic kidney disease also are associated with cardiovascular disease.2–4
The prevalence of traditional risk factors, particularly diabetes and hypertension, is high in all stages of kidney disease, and dyslipidemia is extremely common. Patients with chronic kidney disease who are not on dialysis tend to have lower levels of high-density lipoprotein cholesterol and higher levels of triglycerides, lipoprotein remnants, lipoprotein(a), and LDL-C. The lipid profile of dialysis patients is more complex, as malnutrition and inflammation in this population may lead to low cholesterol levels.
Since statins are effective for primary and secondary prevention of cardiovascular events in those in the general population with high LDL-C,5 we could expect that the same holds true for patients with chronic kidney disease. Furthermore, if kidney disease were considered a coronary heart disease equivalent, more than 85% of those with stage 3, 4, or 5 disease would qualify for lipid-lowering therapy by LDL-C criteria.6
However, compared with the large body of evidence in those without kidney disease, we have few data on the effect of statins on cardiovascular outcomes in those with kidney disease. Five of seven major trials of statins excluded patients with chronic kidney disease by using a creatinine cutoff or by excluding patients with known kidney disease.7
Renoprotective effects
Besides their cardiovascular effects, statins may slow the progression of kidney disease.
A subgroup analysis of the Greek Atorvastatin and Coronary Heart Disease Evaluation (GREACE) trial8 showed a 12% increase in creatinine clearance in the group receiving atorvastatin (Lipitor) (P = .0001). In comparison, creatinine clearance decreased by 4% in the placebo group.
A subgroup analysis of the Cholesterol and Recurrent Events (CARE) trial, a secondary prevention trial of pravastatin (Pravachol) vs placebo, showed a similar effect for patients with a glomerular filtration rate (GFR) less than 60 mL/min/1.73 m2 at baseline.9
A meta-analysis of 27 randomized trials (39,704 participants) concluded that, compared with no treatment, statins slowed the loss of GFR by a mean of 1.22 mL/min/year (95% confidence interval 0.44–2.0).10
Statins may confer this benefit independently of lipid-lowering. These drugs seem to decrease proteinuria, possibly by improving endothelial function or decreasing inflammation.11 A meta-analysis (1,384 patients) noted that 13 of 15 published studies found an antiproteinuric effect, with a greater effect in those with greater baseline proteinuria.12
The Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients With Progressive Renal Disease Trial (PLANET) will enroll 345 diabetic patients with protein-uria and hypercholesterolemia and examine the effects of rosuvastatin (Crestor) and atorvastatin on proteinuria and GFR.13
Cardioprotective effects in stages 1–4
Since patients with chronic kidney disease were excluded from most of the major statin trials, the best evidence in those with non-dialysis-dependent disease comes from post hoc analysis of data from the CARE study.14 While this trial excluded patients with more than 2+ proteinuria on dipstick analysis and those with creatinine values greater than 1.5 times the upper limit of normal, 1,711 of the initial 4,159 patients had a creatinine clearance of less than 75 mL/min; the mean creatinine clearance in this subgroup was 61. In this subgroup, pravastatin therapy was associated with a significantly lower risk of cardiovascular death or recurrent nonfatal myocardial infarction (MI) (hazard ratio 0.72, P < 0.05).
Similarly, in the 4,491 patients with chronic kidney disease (mean GFR 55 mL/min/1.73 m2) in the Pravastatin Pooling Project, the hazard of new MI, cardiovascular death, or cardiac intervention was nearly 25% lower in the pravastatin group.15
The ongoing Study of Heart and Renal Protection (SHARP),16 a randomized trial of ezetimibe/simvastatin (Vytorin) that enrolled 6,000 people with stages 3 to 4 kidney disease and 3,000 dialysis patients, will help in determining whether statin therapy prevents new vascular events. The study was launched in 2003 and has now completed enrollment. The primary outcome measure will be the time to first vascular event; secondary analyses will address whether statins decrease proteinuria or slow the progression of kidney disease.
Cardioprotective effects in dialysis patients
The only major randomized trial of statins ever conducted in dialysis patients with diabetes, the German Diabetes and Dialysis Study (4D), did not find atorvastatin 20 mg to have any benefit compared with placebo in reducing a composite end point of death from cardiac causes, stroke, and nonfatal MI over a median of 4 years of follow-up, despite a decrease in LDL-C of over 40% in the treatment group.17 Adverse events were similar in the two groups. The lack of a detectable benefit may be due to differences in the cardiovascular milieu in dialysis patients, who may have more advanced disease, with preexisting cardiac remodeling and congestive heart failure, which may not be modified to the same extent by statin therapy. Alternatively, the dose of atorvastatin may have been too low, or 4 years of treatment may not be sufficient to detect a benefit in these patients.
An ongoing prospective, randomized, placebo-controlled trial in 3,000 hemodialysis patients, called A Study to Evaluate the Use of Rosuvastatin in Subjects on Regular Haemodialysis: an Assessment of Survival and Cardiovascular Events (AURORA),18 will help to clarify the role of statins in this population.
CONCLUSION
The National Kidney Foundation guidelines1,19 note that people with chronic kidney disease are at high risk of cardiovascular disease and therefore should be treated according to guidelines for treating traditional risk factors in high-risk groups. We believe that those with dyslipidemia who are in stages 1 through 4, particularly those with other risk factors for coronary heart disease, should receive a statin, with an LDL-C target of less than 100 mg/dL, even though we have few data from large trials focused on this population and even though LDL-C may not be the only reason to consider statin use. The pleiotropic effects of statins on proteinuria and progression of kidney function loss may be of benefit in this population as well, although we would not recommend starting a statin solely for these effects until more data are available.
Despite the negative results of the 4D trial, given the relative safety of statins and the lack of any trial data suggesting harm in patients with chronic kidney disease, in our practice we treat dialysis patients with known cardiovascular disease with a statin, with a target LDL-C level less than 100 mg/dL. In dialysis patients without known cardiovascular disease, the use of a statin is even more controversial, and decisions should be made on an individual basis.
Results from the SHARP, AURORA, and PLANET trials, each of which is focused on patients with chronic kidney disease, will help determine whether statins benefit patients at this stage of disease.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
- National Kidney Foundation. K/DOQI clinical practice guidelines for chronic kidney disease: evaluation, classification, and stratification. Am J Kidney Dis 2002; 39 suppl 1:S1–S266.
- Shlipak MG, Sarnak MJ, Katz R, et al. Cystatin C and the risk of death and cardiovascular events among elderly persons. N Engl J Med 2005; 352:2049–2060.
- Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis 1998; 32 suppl 3:S112–S119.
- Manjunath G, Tighiouart H, Coresh J, et al. Level of kidney function as a risk factor for cardiovascular outcomes in the elderly. Kidney Int 2003; 63:1121–1129.
- Baigent C, Keech A, Kearney PM, et al Cholesterol Treatment Trialists’ (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. Lancet 2005; 366:1267–1278.
- Hyre AD, Fox CS, Astor BC, Cohen AJ, Muntner P. The impact of reclassifying moderate CKD as a coronary heart disease risk equivalent on the number of US adults recommended lipid-lowering treatment. Am J Kidney Dis 2007; 49:37–45.
- Coca SG, Krumholz HM, Garg AX, Parikh CR. Underrepresentation of renal disease in randomized controlled trials of cardiovascular disease. JAMA 2006; 296:1377–1384.
- Athyros VG, Mikhailidis DP, Papageorgiou AA, et al. The effect of statins versus untreated dyslipidaemia on renal function in patients with coronary heart disease. A subgroup analysis of the Greek atorvastatin and coronary heart disease evaluation (GREACE) study. J Clin Pathol 2004; 57:728–734.
- Tonelli M, Moye L, Sacks FM, Cole T, Curhan GC Cholesterol and Recurrent Events Trial Investigators. Effect of pravastatin on loss of renal function in people with moderate chronic renal insufficiency and cardiovascular disease. J Am Soc Nephrol 2003; 14:1605–1613.
- Sandhu S, Wiebe N, Fried LF, Tonelli M. Statins for improving renal outcomes: a meta-analysis. J Am Soc Nephrol 2006; 17:2006–2016.
- Balk EM, Lau J, Goudas LC, et al. Effects of statins on nonlipid serum markers associated with cardiovascular disease: a systematic review. Ann Intern Med 2003; 139:670–682.
- Douglas K, O’Malley PG, Jackson JL. Meta-analysis: the effect of statins on albuminuria. Ann Intern Med 2006; 145:117–124.
- US National Institutes of Health. Prospective Evaluation of Proteinuria and Renal Function in Diabetic Patients with Progressive Renal Disease (PLANET 1). Accessed December 6, 2007. www.clinicaltrials.gov/ct/show/NCT00296374?order=1.
- Tonelli M, Moye L, Sacks FM, Kiberd B, Curhan G Cholesterol and Recurrent Events (CARE) Trial Investigators. Pravastatin for secondary prevention of cardiovascular events in persons with mild chronic renal insufficiency. Ann Intern Med 2003; 138:98–104.
- Tonelli M, Isles C, Curhan GC, et al. Effect of pravastatin on cardiovascular events in people with chronic kidney disease. Circulation 2004; 110:1557–1563.
- Baigent C, Landry M. Study of Heart and Renal Protection (SHARP). Kidney Int Suppl 2003; 84:S207–S210.
- Wanner C, Krane V, Marz W, et al German Diabetes and Dialysis Study Investigators. Atorvastatin in patients with type 2 diabetes mellitus undergoing hemodialysis. N Engl J Med 2005; 353:238–248.
- Fellstrom BC, Holdaas H, Jardine AG. Why do we need a statin trial in hemodialysis patients? Kidney Int 2003; 84 suppl:S204–S206.
- KDOQI Clinical Practice Guidelines and Clinical Practice Recommendations for Diabetes and Chronic Kidney Disease. Am J Kidney Dis 2007; 49 suppl 2:S12–154.
Acute aortic syndromes: Time to talk of many things
“The time has come,” the Walrus said,
“To talk of many things:
Of shoes—and ships—and sealing-wax—
Of cabbages—and kings—
And why the sea is boiling hot
And whether pigs have wings.”
—Lewis Carroll, The Walrus and the Carpenter (from Through the Looking-Glass and What Alice Found There, 1872).
Lewis Carroll's poem of 1872 is a useful starting point for identifying issues resulting from confusion over the variously described acute aortic syndromes—and, for oysters, the dangers of listening to walruses.
TALK OF MANY THINGS
In cases of aortic dissection (splitting or separation of the layers of the aortic wall), it is important to establish the type (ie, the location and extent) and class (ie, the structure) of the dissection, because these distinctions determine the treatment.1 Similarly, in cases of painful or leaking degenerative aneurysms, we need to know the location of the aneurysm and whether the presenting pain is from compression of surrounding tissue, particularly of the vertebral bodies, or from leakage.
The location and extent of an aortic dissection can be classified in three ways (see Figure 3 in Smith and Schoenhagen’s excellent review of the use of computed tomography [CT] in acute aortic syndromes in this issue of the Cleveland Clinic Journal of Medicine2):
- The DeBakey system (type I, II, or III)
- The Stanford system (type A or B)
- Distal or proximal to the left subclavian artery.
Of note, the DeBakey system does not include tears in the arch that extend distally without ascending involvement. The original Stanford system included arch tears with distal extension in type B; hence, type B excluded all patients without ascending involvement.
The importance of the extent of dissection is that most patients with Stanford type A or DeBakey type I or II dissections should undergo immediate surgery, as most of them would die without it. Surgery is also indicated for arch tears (non-DeBakey, original Stanford type B).
Because these classifications are somewhat confusing, the simplest approach is to note whether the dissection extends proximal or distal to the left subclavian artery, because proximal dissections need surgery and distal ones are first managed medically.
The classes of dissection also have bearing on treatment.1 These are:
- Class I—classic aortic dissection in the media with two lumens separated by a “flap” or septum
- Class II—intramural hematoma in the aortic wall from dissection in which the intimal tear cannot be imaged (these are nearly always found duringsurgery or autopsy)
- Class III—localized confined intimal tears without extensive undermining of the intima or flap formation. These are often seen with Marfan syndrome and can rupture or cause tamponade, as can any type of proximal dissection. The typical appearance is of a bulging bubble in the aortic wall.
- Class IV—penetrating atherosclerotic ulcers with localized dissections or wall hematomas, often with calcium at the base of a mushroom-shaped area of extraluminal contrast. Of note, the plane of dissection is often between the media and adventitia.
- Class V—iatrogenic or posttraumatic dissection.
All class I to class IV tears of the proximal aorta require surgery, whereas distal class IV and V tears may require either open or endovascular surgical intervention. Surgery is also indicated for patients with distal dissections who have severe narrowing of the true lumen, distal ischemia, uncontrolled pain, severe hypertension, or evidence of leaking, particularly with class IV tears.
In distal dissections that are subacute (2–6 week sold), the Investigation of Stent grafts in Patients With Type B Aortic Dissection (INSTEAD) trial found that inserting a stent prophylactically provided no benefit. Further-more, there is no proof that stenting is beneficial if the aortic dissection is chronic, ie, more than 6 weeks old.1,3–5
WHICH SHOE FITS?
There is no ideal procedure to detect dissection, although the trend is towards CT angiography, as Smith and Schoenhagen report.2 Although some investigators have optimistically estimated CT’s sensitivity and specificity as 100%, cardiovascular surgeons are well aware of both false-positive and false-negative CT studies. Thus, for emergency repairs of proximal dissections, transesophageal echocardiography should be done after intubation and before opening a patient’s chest if time allows. Magnetic resonance imaging, CT, and transesophageal echocardiography may all miss class III tears, but these are frequently evidenced by eccentric “bubbles”or “ballooning.”1
SHIPS
Patients with either acute aortic dissection or severe pain associated with degenerative aneurysms need to be “shipped” promptly to a tertiary medical center after diagnosis, since larger volumes of procedures appear to be associated with better outcomes.
SEALING WAX
Using current surgical methods, the aortic valve can be preserved during aortic dissection repair unless the valve is bicuspid or the patient has Marfan syndrome.1,3,4,6–8
Sealing wax in the form of biological glues, rather than for letters, is a new innovation. A caveat remains, however: we have seen patients who have required reoperation for false aneurysms or infection. Hence, glues should be used with caution.
CABBAGES
A dilemma is whether patients should undergo coronary catheterization (or CT angiography—a separate question) and subsequent coronary artery bypass grafting (CABG), if needed, at the time of aortic dissection repair. The problem is that approximately one-third of patients have coronary artery disease that may require CABG, but the delay for catheterization increases the risk of rupture or tamponade before surgery.
Indeed, 40% of patients with proximal dissections die immediately, and 1% to 3% die in the hour before surgery. The short-term (in-hospital and 30-day) mortality rates range from 3.4% (Cleveland Clinic 2006 data) to 25%, and of the survivors only about 50% area live 5 years after surgery.
Though dismal, the prognosis is improving. In 162 patients with aortic dissection and Marfan syndrome or connective tissue disorders who underwent surgery at Cleveland Clinic in the years 1978–2003, the 5-year survival rate in those with aortic dissection was 75% and the 10-year rate was 55%.7 In those without dissection, the 10-year survival rate was approximately 90% (P < .001).
KINGS
Noted personalities who have had aortic dissection include King George II of England (who died in 1760), Lucille Ball, Conway Twitty, Jan Larson, and most recently John Ritter. None of these famous people survived their aortic dissections. Indeed, dissection and diseases of the aorta or its branches cause between 43,000 and 47,000 deaths annually,9 more than from breast cancer, murders, or motor vehicle accidents. The main reason for these dismal statistics is that the disease is often misdiagnosed at the time of presentation.
BOILING SEA
Careful studies from Olmsted County, Minnesota,10 have shown a tripling of the incidence of aortic disease, particularly in women, even though the rate of deaths from coronary artery disease has been decreasing. Furthermore, Olsson et al11 report that the incidence of aortic dissection in men in Sweden increased to approximately16 per 100,000 per year from 1987 to 2002, a 52% increase. The aging of the population must play a large role, but other factors may exist that are not well understood or defined and require further research.
PIGS HAVE WINGS
Will it be possible to overcome this rising problem? The answer is a definite yes. The results of aortic surgery have never been better. Many new innovations are available, such as aortic root preservation and endovascular stenting procedures. It may be possible to slow the growth of or prevent some aneurysms and aortic dissections, particularly with beta-blockers and, potentially, with losartan (Cozaar) for Marfan syndrome patients.
One of the keys to preventing aortic catastrophes and aortic dissection is to repair aortic aneurysms. The threshold for surgery, however, depends on a surgeon’s experience and results, the underlying pathology, and the aortic size.
We observed that 12.5% of dissections in patients with bicuspid valves and 15% of those in patients with Marfan syndrome were in aortas smaller than 5.0 cm in diameter, that aortic dissection occurred at smaller diameters in shorter patients, and that the risk of dissection increased exponentially with the size of the aorta. Subsequently, we found that a better measure of risk is the maximal aortic cross-sectional area in cm2 divided by the patient’s height in meters; if this ratio exceeds 10, then surgery is recommended.12
Results of surgery are good in experienced hands. In patients who undergo surgical repair of bicuspid aortic valves with or without concurrent repair of the ascending aorta (mostly in patients with an aortic cross-section-to-height ratio > 10), the perioperative mortality rate is about 1.0% for both groups, and at 10 years about 98% of patients are free from re-operation on the aorta and more than 90% are free from re-operation on the aortic valve.8 This is important because these are typically young patients who would do better without biological replacement valves (which are not very durable) or mechanical valves (which necessitate lifelong anticoagulation). Results are also good in surgery of the aortic arch and even better in patients with tricuspid aortic valves.4,6,8
Increasingly, in patients at high risk, we are inserting thoracic, abdominal, and thoracoabdominal stent grafts, with excellent early results. An even newer innovation is to replace the aortic valve in high-risk patients via a transcatheter balloon-expandable valve stent inserted through the groin or left ventricular apex.
These treatment innovations have been big strides, but aortic disease continues to increase. Indeed, our volume of thoracic aortic surgery at Cleveland Clinic increased from 190 procedures in 1999 to 717 in 2006. Early detection—before acute emergency surgery is required, with its concomitant high risk of death—is the key to successful surgical outcome and long-term survival.
- Svensson LG, Labib SB, Eisenhauer AC, Butterly JR. Intimal tear without hematoma: an important variant of aortic dissection that can elude current imaging techniques. Circulation 1999; 99:1331–1336.
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75:7–24.
- Svensson LG, Nadolny EM, Kimmel WA. Multimodal protocol influence on stroke and neurocognitive deficit prevention after ascending/arch aortic operations. Ann Thorac Surg 2002; 74:2040–2046.
- Svensson LG, Kim KH, Blackstone EH, et al. Elephant trunk procedure: newer indications and uses. Ann Thorac Surg 2004; 78:109–116.
- Greenberg RK, Haddad F, Svensson L, et al. Hybrid approaches to thoracic aortic aneurysms: the role of endovascular elephant trunk completion. Circulation 2005; 112:2619–2626.
- Svensson LG. Sizing for modified David’s reimplantation procedure. Ann Thorac Surg 2003; 76:1751–1753.
- Svensson LG, Blackstone EH, Feng J, et al. Are Marfan syndrome and marfanoid patients distinguishable on long-term follow-up? Ann Thorac Surg 2007; 83:1067–1074.
- Svensson LG, Blackstone EH, Cosgrove DM 3rd. Surgical options in young adults with aortic valve disease. Curr Probl Cardiol 2003; 28:417–480.
- Svensson LG, Rodriguez ER. Aortic organ disease epidemic, and why do balloons pop? Circulation 2005; 112:1082–1084.
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280:1926–1929.
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114:2611–2618.
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection inpatients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003;126:892–893.
“The time has come,” the Walrus said,
“To talk of many things:
Of shoes—and ships—and sealing-wax—
Of cabbages—and kings—
And why the sea is boiling hot
And whether pigs have wings.”
—Lewis Carroll, The Walrus and the Carpenter (from Through the Looking-Glass and What Alice Found There, 1872).
Lewis Carroll's poem of 1872 is a useful starting point for identifying issues resulting from confusion over the variously described acute aortic syndromes—and, for oysters, the dangers of listening to walruses.
TALK OF MANY THINGS
In cases of aortic dissection (splitting or separation of the layers of the aortic wall), it is important to establish the type (ie, the location and extent) and class (ie, the structure) of the dissection, because these distinctions determine the treatment.1 Similarly, in cases of painful or leaking degenerative aneurysms, we need to know the location of the aneurysm and whether the presenting pain is from compression of surrounding tissue, particularly of the vertebral bodies, or from leakage.
The location and extent of an aortic dissection can be classified in three ways (see Figure 3 in Smith and Schoenhagen’s excellent review of the use of computed tomography [CT] in acute aortic syndromes in this issue of the Cleveland Clinic Journal of Medicine2):
- The DeBakey system (type I, II, or III)
- The Stanford system (type A or B)
- Distal or proximal to the left subclavian artery.
Of note, the DeBakey system does not include tears in the arch that extend distally without ascending involvement. The original Stanford system included arch tears with distal extension in type B; hence, type B excluded all patients without ascending involvement.
The importance of the extent of dissection is that most patients with Stanford type A or DeBakey type I or II dissections should undergo immediate surgery, as most of them would die without it. Surgery is also indicated for arch tears (non-DeBakey, original Stanford type B).
Because these classifications are somewhat confusing, the simplest approach is to note whether the dissection extends proximal or distal to the left subclavian artery, because proximal dissections need surgery and distal ones are first managed medically.
The classes of dissection also have bearing on treatment.1 These are:
- Class I—classic aortic dissection in the media with two lumens separated by a “flap” or septum
- Class II—intramural hematoma in the aortic wall from dissection in which the intimal tear cannot be imaged (these are nearly always found duringsurgery or autopsy)
- Class III—localized confined intimal tears without extensive undermining of the intima or flap formation. These are often seen with Marfan syndrome and can rupture or cause tamponade, as can any type of proximal dissection. The typical appearance is of a bulging bubble in the aortic wall.
- Class IV—penetrating atherosclerotic ulcers with localized dissections or wall hematomas, often with calcium at the base of a mushroom-shaped area of extraluminal contrast. Of note, the plane of dissection is often between the media and adventitia.
- Class V—iatrogenic or posttraumatic dissection.
All class I to class IV tears of the proximal aorta require surgery, whereas distal class IV and V tears may require either open or endovascular surgical intervention. Surgery is also indicated for patients with distal dissections who have severe narrowing of the true lumen, distal ischemia, uncontrolled pain, severe hypertension, or evidence of leaking, particularly with class IV tears.
In distal dissections that are subacute (2–6 week sold), the Investigation of Stent grafts in Patients With Type B Aortic Dissection (INSTEAD) trial found that inserting a stent prophylactically provided no benefit. Further-more, there is no proof that stenting is beneficial if the aortic dissection is chronic, ie, more than 6 weeks old.1,3–5
WHICH SHOE FITS?
There is no ideal procedure to detect dissection, although the trend is towards CT angiography, as Smith and Schoenhagen report.2 Although some investigators have optimistically estimated CT’s sensitivity and specificity as 100%, cardiovascular surgeons are well aware of both false-positive and false-negative CT studies. Thus, for emergency repairs of proximal dissections, transesophageal echocardiography should be done after intubation and before opening a patient’s chest if time allows. Magnetic resonance imaging, CT, and transesophageal echocardiography may all miss class III tears, but these are frequently evidenced by eccentric “bubbles”or “ballooning.”1
SHIPS
Patients with either acute aortic dissection or severe pain associated with degenerative aneurysms need to be “shipped” promptly to a tertiary medical center after diagnosis, since larger volumes of procedures appear to be associated with better outcomes.
SEALING WAX
Using current surgical methods, the aortic valve can be preserved during aortic dissection repair unless the valve is bicuspid or the patient has Marfan syndrome.1,3,4,6–8
Sealing wax in the form of biological glues, rather than for letters, is a new innovation. A caveat remains, however: we have seen patients who have required reoperation for false aneurysms or infection. Hence, glues should be used with caution.
CABBAGES
A dilemma is whether patients should undergo coronary catheterization (or CT angiography—a separate question) and subsequent coronary artery bypass grafting (CABG), if needed, at the time of aortic dissection repair. The problem is that approximately one-third of patients have coronary artery disease that may require CABG, but the delay for catheterization increases the risk of rupture or tamponade before surgery.
Indeed, 40% of patients with proximal dissections die immediately, and 1% to 3% die in the hour before surgery. The short-term (in-hospital and 30-day) mortality rates range from 3.4% (Cleveland Clinic 2006 data) to 25%, and of the survivors only about 50% area live 5 years after surgery.
Though dismal, the prognosis is improving. In 162 patients with aortic dissection and Marfan syndrome or connective tissue disorders who underwent surgery at Cleveland Clinic in the years 1978–2003, the 5-year survival rate in those with aortic dissection was 75% and the 10-year rate was 55%.7 In those without dissection, the 10-year survival rate was approximately 90% (P < .001).
KINGS
Noted personalities who have had aortic dissection include King George II of England (who died in 1760), Lucille Ball, Conway Twitty, Jan Larson, and most recently John Ritter. None of these famous people survived their aortic dissections. Indeed, dissection and diseases of the aorta or its branches cause between 43,000 and 47,000 deaths annually,9 more than from breast cancer, murders, or motor vehicle accidents. The main reason for these dismal statistics is that the disease is often misdiagnosed at the time of presentation.
BOILING SEA
Careful studies from Olmsted County, Minnesota,10 have shown a tripling of the incidence of aortic disease, particularly in women, even though the rate of deaths from coronary artery disease has been decreasing. Furthermore, Olsson et al11 report that the incidence of aortic dissection in men in Sweden increased to approximately16 per 100,000 per year from 1987 to 2002, a 52% increase. The aging of the population must play a large role, but other factors may exist that are not well understood or defined and require further research.
PIGS HAVE WINGS
Will it be possible to overcome this rising problem? The answer is a definite yes. The results of aortic surgery have never been better. Many new innovations are available, such as aortic root preservation and endovascular stenting procedures. It may be possible to slow the growth of or prevent some aneurysms and aortic dissections, particularly with beta-blockers and, potentially, with losartan (Cozaar) for Marfan syndrome patients.
One of the keys to preventing aortic catastrophes and aortic dissection is to repair aortic aneurysms. The threshold for surgery, however, depends on a surgeon’s experience and results, the underlying pathology, and the aortic size.
We observed that 12.5% of dissections in patients with bicuspid valves and 15% of those in patients with Marfan syndrome were in aortas smaller than 5.0 cm in diameter, that aortic dissection occurred at smaller diameters in shorter patients, and that the risk of dissection increased exponentially with the size of the aorta. Subsequently, we found that a better measure of risk is the maximal aortic cross-sectional area in cm2 divided by the patient’s height in meters; if this ratio exceeds 10, then surgery is recommended.12
Results of surgery are good in experienced hands. In patients who undergo surgical repair of bicuspid aortic valves with or without concurrent repair of the ascending aorta (mostly in patients with an aortic cross-section-to-height ratio > 10), the perioperative mortality rate is about 1.0% for both groups, and at 10 years about 98% of patients are free from re-operation on the aorta and more than 90% are free from re-operation on the aortic valve.8 This is important because these are typically young patients who would do better without biological replacement valves (which are not very durable) or mechanical valves (which necessitate lifelong anticoagulation). Results are also good in surgery of the aortic arch and even better in patients with tricuspid aortic valves.4,6,8
Increasingly, in patients at high risk, we are inserting thoracic, abdominal, and thoracoabdominal stent grafts, with excellent early results. An even newer innovation is to replace the aortic valve in high-risk patients via a transcatheter balloon-expandable valve stent inserted through the groin or left ventricular apex.
These treatment innovations have been big strides, but aortic disease continues to increase. Indeed, our volume of thoracic aortic surgery at Cleveland Clinic increased from 190 procedures in 1999 to 717 in 2006. Early detection—before acute emergency surgery is required, with its concomitant high risk of death—is the key to successful surgical outcome and long-term survival.
“The time has come,” the Walrus said,
“To talk of many things:
Of shoes—and ships—and sealing-wax—
Of cabbages—and kings—
And why the sea is boiling hot
And whether pigs have wings.”
—Lewis Carroll, The Walrus and the Carpenter (from Through the Looking-Glass and What Alice Found There, 1872).
Lewis Carroll's poem of 1872 is a useful starting point for identifying issues resulting from confusion over the variously described acute aortic syndromes—and, for oysters, the dangers of listening to walruses.
TALK OF MANY THINGS
In cases of aortic dissection (splitting or separation of the layers of the aortic wall), it is important to establish the type (ie, the location and extent) and class (ie, the structure) of the dissection, because these distinctions determine the treatment.1 Similarly, in cases of painful or leaking degenerative aneurysms, we need to know the location of the aneurysm and whether the presenting pain is from compression of surrounding tissue, particularly of the vertebral bodies, or from leakage.
The location and extent of an aortic dissection can be classified in three ways (see Figure 3 in Smith and Schoenhagen’s excellent review of the use of computed tomography [CT] in acute aortic syndromes in this issue of the Cleveland Clinic Journal of Medicine2):
- The DeBakey system (type I, II, or III)
- The Stanford system (type A or B)
- Distal or proximal to the left subclavian artery.
Of note, the DeBakey system does not include tears in the arch that extend distally without ascending involvement. The original Stanford system included arch tears with distal extension in type B; hence, type B excluded all patients without ascending involvement.
The importance of the extent of dissection is that most patients with Stanford type A or DeBakey type I or II dissections should undergo immediate surgery, as most of them would die without it. Surgery is also indicated for arch tears (non-DeBakey, original Stanford type B).
Because these classifications are somewhat confusing, the simplest approach is to note whether the dissection extends proximal or distal to the left subclavian artery, because proximal dissections need surgery and distal ones are first managed medically.
The classes of dissection also have bearing on treatment.1 These are:
- Class I—classic aortic dissection in the media with two lumens separated by a “flap” or septum
- Class II—intramural hematoma in the aortic wall from dissection in which the intimal tear cannot be imaged (these are nearly always found duringsurgery or autopsy)
- Class III—localized confined intimal tears without extensive undermining of the intima or flap formation. These are often seen with Marfan syndrome and can rupture or cause tamponade, as can any type of proximal dissection. The typical appearance is of a bulging bubble in the aortic wall.
- Class IV—penetrating atherosclerotic ulcers with localized dissections or wall hematomas, often with calcium at the base of a mushroom-shaped area of extraluminal contrast. Of note, the plane of dissection is often between the media and adventitia.
- Class V—iatrogenic or posttraumatic dissection.
All class I to class IV tears of the proximal aorta require surgery, whereas distal class IV and V tears may require either open or endovascular surgical intervention. Surgery is also indicated for patients with distal dissections who have severe narrowing of the true lumen, distal ischemia, uncontrolled pain, severe hypertension, or evidence of leaking, particularly with class IV tears.
In distal dissections that are subacute (2–6 week sold), the Investigation of Stent grafts in Patients With Type B Aortic Dissection (INSTEAD) trial found that inserting a stent prophylactically provided no benefit. Further-more, there is no proof that stenting is beneficial if the aortic dissection is chronic, ie, more than 6 weeks old.1,3–5
WHICH SHOE FITS?
There is no ideal procedure to detect dissection, although the trend is towards CT angiography, as Smith and Schoenhagen report.2 Although some investigators have optimistically estimated CT’s sensitivity and specificity as 100%, cardiovascular surgeons are well aware of both false-positive and false-negative CT studies. Thus, for emergency repairs of proximal dissections, transesophageal echocardiography should be done after intubation and before opening a patient’s chest if time allows. Magnetic resonance imaging, CT, and transesophageal echocardiography may all miss class III tears, but these are frequently evidenced by eccentric “bubbles”or “ballooning.”1
SHIPS
Patients with either acute aortic dissection or severe pain associated with degenerative aneurysms need to be “shipped” promptly to a tertiary medical center after diagnosis, since larger volumes of procedures appear to be associated with better outcomes.
SEALING WAX
Using current surgical methods, the aortic valve can be preserved during aortic dissection repair unless the valve is bicuspid or the patient has Marfan syndrome.1,3,4,6–8
Sealing wax in the form of biological glues, rather than for letters, is a new innovation. A caveat remains, however: we have seen patients who have required reoperation for false aneurysms or infection. Hence, glues should be used with caution.
CABBAGES
A dilemma is whether patients should undergo coronary catheterization (or CT angiography—a separate question) and subsequent coronary artery bypass grafting (CABG), if needed, at the time of aortic dissection repair. The problem is that approximately one-third of patients have coronary artery disease that may require CABG, but the delay for catheterization increases the risk of rupture or tamponade before surgery.
Indeed, 40% of patients with proximal dissections die immediately, and 1% to 3% die in the hour before surgery. The short-term (in-hospital and 30-day) mortality rates range from 3.4% (Cleveland Clinic 2006 data) to 25%, and of the survivors only about 50% area live 5 years after surgery.
Though dismal, the prognosis is improving. In 162 patients with aortic dissection and Marfan syndrome or connective tissue disorders who underwent surgery at Cleveland Clinic in the years 1978–2003, the 5-year survival rate in those with aortic dissection was 75% and the 10-year rate was 55%.7 In those without dissection, the 10-year survival rate was approximately 90% (P < .001).
KINGS
Noted personalities who have had aortic dissection include King George II of England (who died in 1760), Lucille Ball, Conway Twitty, Jan Larson, and most recently John Ritter. None of these famous people survived their aortic dissections. Indeed, dissection and diseases of the aorta or its branches cause between 43,000 and 47,000 deaths annually,9 more than from breast cancer, murders, or motor vehicle accidents. The main reason for these dismal statistics is that the disease is often misdiagnosed at the time of presentation.
BOILING SEA
Careful studies from Olmsted County, Minnesota,10 have shown a tripling of the incidence of aortic disease, particularly in women, even though the rate of deaths from coronary artery disease has been decreasing. Furthermore, Olsson et al11 report that the incidence of aortic dissection in men in Sweden increased to approximately16 per 100,000 per year from 1987 to 2002, a 52% increase. The aging of the population must play a large role, but other factors may exist that are not well understood or defined and require further research.
PIGS HAVE WINGS
Will it be possible to overcome this rising problem? The answer is a definite yes. The results of aortic surgery have never been better. Many new innovations are available, such as aortic root preservation and endovascular stenting procedures. It may be possible to slow the growth of or prevent some aneurysms and aortic dissections, particularly with beta-blockers and, potentially, with losartan (Cozaar) for Marfan syndrome patients.
One of the keys to preventing aortic catastrophes and aortic dissection is to repair aortic aneurysms. The threshold for surgery, however, depends on a surgeon’s experience and results, the underlying pathology, and the aortic size.
We observed that 12.5% of dissections in patients with bicuspid valves and 15% of those in patients with Marfan syndrome were in aortas smaller than 5.0 cm in diameter, that aortic dissection occurred at smaller diameters in shorter patients, and that the risk of dissection increased exponentially with the size of the aorta. Subsequently, we found that a better measure of risk is the maximal aortic cross-sectional area in cm2 divided by the patient’s height in meters; if this ratio exceeds 10, then surgery is recommended.12
Results of surgery are good in experienced hands. In patients who undergo surgical repair of bicuspid aortic valves with or without concurrent repair of the ascending aorta (mostly in patients with an aortic cross-section-to-height ratio > 10), the perioperative mortality rate is about 1.0% for both groups, and at 10 years about 98% of patients are free from re-operation on the aorta and more than 90% are free from re-operation on the aortic valve.8 This is important because these are typically young patients who would do better without biological replacement valves (which are not very durable) or mechanical valves (which necessitate lifelong anticoagulation). Results are also good in surgery of the aortic arch and even better in patients with tricuspid aortic valves.4,6,8
Increasingly, in patients at high risk, we are inserting thoracic, abdominal, and thoracoabdominal stent grafts, with excellent early results. An even newer innovation is to replace the aortic valve in high-risk patients via a transcatheter balloon-expandable valve stent inserted through the groin or left ventricular apex.
These treatment innovations have been big strides, but aortic disease continues to increase. Indeed, our volume of thoracic aortic surgery at Cleveland Clinic increased from 190 procedures in 1999 to 717 in 2006. Early detection—before acute emergency surgery is required, with its concomitant high risk of death—is the key to successful surgical outcome and long-term survival.
- Svensson LG, Labib SB, Eisenhauer AC, Butterly JR. Intimal tear without hematoma: an important variant of aortic dissection that can elude current imaging techniques. Circulation 1999; 99:1331–1336.
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75:7–24.
- Svensson LG, Nadolny EM, Kimmel WA. Multimodal protocol influence on stroke and neurocognitive deficit prevention after ascending/arch aortic operations. Ann Thorac Surg 2002; 74:2040–2046.
- Svensson LG, Kim KH, Blackstone EH, et al. Elephant trunk procedure: newer indications and uses. Ann Thorac Surg 2004; 78:109–116.
- Greenberg RK, Haddad F, Svensson L, et al. Hybrid approaches to thoracic aortic aneurysms: the role of endovascular elephant trunk completion. Circulation 2005; 112:2619–2626.
- Svensson LG. Sizing for modified David’s reimplantation procedure. Ann Thorac Surg 2003; 76:1751–1753.
- Svensson LG, Blackstone EH, Feng J, et al. Are Marfan syndrome and marfanoid patients distinguishable on long-term follow-up? Ann Thorac Surg 2007; 83:1067–1074.
- Svensson LG, Blackstone EH, Cosgrove DM 3rd. Surgical options in young adults with aortic valve disease. Curr Probl Cardiol 2003; 28:417–480.
- Svensson LG, Rodriguez ER. Aortic organ disease epidemic, and why do balloons pop? Circulation 2005; 112:1082–1084.
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280:1926–1929.
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114:2611–2618.
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection inpatients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003;126:892–893.
- Svensson LG, Labib SB, Eisenhauer AC, Butterly JR. Intimal tear without hematoma: an important variant of aortic dissection that can elude current imaging techniques. Circulation 1999; 99:1331–1336.
- Smith AD, Schoenhagen P. CT imaging for acute aortic syndrome. Cleve Clin J Med 2008; 75:7–24.
- Svensson LG, Nadolny EM, Kimmel WA. Multimodal protocol influence on stroke and neurocognitive deficit prevention after ascending/arch aortic operations. Ann Thorac Surg 2002; 74:2040–2046.
- Svensson LG, Kim KH, Blackstone EH, et al. Elephant trunk procedure: newer indications and uses. Ann Thorac Surg 2004; 78:109–116.
- Greenberg RK, Haddad F, Svensson L, et al. Hybrid approaches to thoracic aortic aneurysms: the role of endovascular elephant trunk completion. Circulation 2005; 112:2619–2626.
- Svensson LG. Sizing for modified David’s reimplantation procedure. Ann Thorac Surg 2003; 76:1751–1753.
- Svensson LG, Blackstone EH, Feng J, et al. Are Marfan syndrome and marfanoid patients distinguishable on long-term follow-up? Ann Thorac Surg 2007; 83:1067–1074.
- Svensson LG, Blackstone EH, Cosgrove DM 3rd. Surgical options in young adults with aortic valve disease. Curr Probl Cardiol 2003; 28:417–480.
- Svensson LG, Rodriguez ER. Aortic organ disease epidemic, and why do balloons pop? Circulation 2005; 112:1082–1084.
- Clouse WD, Hallett JW Jr, Schaff HV, Gayari MM, Ilstrup DM, Melton LJ 3rd. Improved prognosis of thoracic aortic aneurysms: a population-based study. JAMA 1998; 280:1926–1929.
- Olsson C, Thelin S, Ståhle E, Ekbom A, Granath F. Thoracic aortic aneurysm and dissection: increasing prevalence and improved outcomes reported in a nationwide population-based study of more than 14,000 cases from 1987 to 2002. Circulation 2006; 114:2611–2618.
- Svensson LG, Kim KH, Lytle BW, Cosgrove DM. Relationship of aortic cross-sectional area to height ratio and the risk of aortic dissection inpatients with bicuspid aortic valves. J Thorac Cardiovasc Surg 2003;126:892–893.