User login
Antibody hierarchy may drive development of SLE vs. antiphospholipid syndrome
according to study findings presented at the European Congress of Rheumatology.

Spanish researchers found that the number of antiphospholipid (aPL) antibodies present was important for the development of antiphospholipid syndrome (APS) and that lupus anticoagulant (LA) was the major aPL antibody linked to systemic lupus erythematosus (SLE)–related organ involvement.
“aPL [antibodies] has been extensively associated with an increased risk of thrombosis and poor pregnancy outcomes, mainly in patients with primary APS,” study investigator Leyre Riancho-Zarrabeitia, MD, PhD, explained in an interview ahead of the congress.
“Moreover, aPL [antibody] positivity in SLE has been proposed to be associated with higher damage accrual and with certain manifestations such as valvular heart disease, pulmonary hypertension, and neuropsychiatric manifestations,” she added.
Anticardiolipin antibodies – notably IgG rather than IgM isotypes – also seemed to play an important role in APS and SLE manifestations, Dr. Riancho-Zarrabeitia, of Hospital Sierrallana, Instituto De Investigación Marqués De Valdecilla, and the University of Cantabria (Spain), noted during her oral presentation.
She reported data on 3,651 patients included in the RELESSER registry between October 2011 and August 2012. This large, multicenter, hospital-based registry retrospectively collects immunologic, clinical and demographic data from unselected adult patients with SLE who are attending 45 Spanish rheumatology services within the country’s national health system.
Over one-third (37.5%) of patients, who had a mean age of 47 years and were mostly (90%) women, were positive for aPL. The most frequent aPL detected was IgG anticardiolipin (aCL) antibodies, seen in 25% of patients, followed by LA in 24%, and IgM aCL in 20%.
Of the aPL-positive patients, 20.6% were positive for only one antibody, 12.1% were positive for two antibodies, and 4.8% were positive for three antibodies.
“All types of aPL were associated with classic APS manifestations,” Dr. Riancho-Zarrabeitia said. The associations were strongest for thrombotic events, such as arterial and venous small-vessel thrombosis and recurrent early pregnancy losses.
aCL antibodies conferred the highest risk for arterial thrombosis, she noted (odds ratio, 5.7), whereas LA conferred the highest risk for venous thrombosis (OR, 4.7). Both IgG and IgM isotypes were associated with thrombotic events, fetal death and recurrent pregnancy loss, but the association was stronger with the IgG isotypes.
Having more than one aPL was particularly associated with a higher risk of these APS manifestations. For example, when one antibody was present the OR for arterial thrombosis was 4.45, but when two or more aPL were detected, the ORs rose to 9.23 and 15.6, respectively.
aCL and LA also were associated with thrombocytopenia and hemolytic anemia, with ORs of around 1-2 and 2-3 respectively. There also were antibody associations with cognitive impairments.
Similar results were seen in patients with SLE. “aPL [antibody] positivity in SLE patients influenced the risk for thrombotic and obstetric manifestations,” Dr. Riancho-Zarrabeitia said. LA and aCL were associated with an increased risk of neuropsychiatric manifestations, and LA was linked to an increased risk for renal disease.
The risk for specific SLE manifestations was again higher with IgG isotypes of aCL, notably an increased risk for cardiac and respiratory events.
While increased antibody numbers generally led to a higher risk of complications, the risk for cutaneous manifestations decreased.
“The load of aPL [antibodies] confers a higher risk for APS,” Dr. Riancho-Zarrabeitia said during her conclusion. “Regarding systemic lupus erythematosus, the number of positive antibodies is directly associated with neurological and ophthalmological manifestations, and inversely associated with cutaneous manifestations.”
What these findings show, said Dr. Riancho-Zarrabeitia in the precongress interview, is that individuals who test positive for aPL antibodies need careful monitoring to prevent and treat severe manifestations. “The next step would be to confirm our findings with a prospective study.”
Dr. Riancho-Zarrabeitia has received travel grants from AbbVie, Pfizer, UCB, Merck, GlaxoSmithKline, Amgen, and Roche.
SOURCE: Riancho-Zarrabeitia L et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):136-7. Abstract OP0124. doi: 10.1136/annrheumdis-2019-eular.2485.
according to study findings presented at the European Congress of Rheumatology.
Spanish researchers found that the number of antiphospholipid (aPL) antibodies present was important for the development of antiphospholipid syndrome (APS) and that lupus anticoagulant (LA) was the major aPL antibody linked to systemic lupus erythematosus (SLE)–related organ involvement.
“aPL [antibodies] has been extensively associated with an increased risk of thrombosis and poor pregnancy outcomes, mainly in patients with primary APS,” study investigator Leyre Riancho-Zarrabeitia, MD, PhD, explained in an interview ahead of the congress.
“Moreover, aPL [antibody] positivity in SLE has been proposed to be associated with higher damage accrual and with certain manifestations such as valvular heart disease, pulmonary hypertension, and neuropsychiatric manifestations,” she added.
Anticardiolipin antibodies – notably IgG rather than IgM isotypes – also seemed to play an important role in APS and SLE manifestations, Dr. Riancho-Zarrabeitia, of Hospital Sierrallana, Instituto De Investigación Marqués De Valdecilla, and the University of Cantabria (Spain), noted during her oral presentation.
She reported data on 3,651 patients included in the RELESSER registry between October 2011 and August 2012. This large, multicenter, hospital-based registry retrospectively collects immunologic, clinical and demographic data from unselected adult patients with SLE who are attending 45 Spanish rheumatology services within the country’s national health system.
Over one-third (37.5%) of patients, who had a mean age of 47 years and were mostly (90%) women, were positive for aPL. The most frequent aPL detected was IgG anticardiolipin (aCL) antibodies, seen in 25% of patients, followed by LA in 24%, and IgM aCL in 20%.
Of the aPL-positive patients, 20.6% were positive for only one antibody, 12.1% were positive for two antibodies, and 4.8% were positive for three antibodies.
“All types of aPL were associated with classic APS manifestations,” Dr. Riancho-Zarrabeitia said. The associations were strongest for thrombotic events, such as arterial and venous small-vessel thrombosis and recurrent early pregnancy losses.
aCL antibodies conferred the highest risk for arterial thrombosis, she noted (odds ratio, 5.7), whereas LA conferred the highest risk for venous thrombosis (OR, 4.7). Both IgG and IgM isotypes were associated with thrombotic events, fetal death and recurrent pregnancy loss, but the association was stronger with the IgG isotypes.
Having more than one aPL was particularly associated with a higher risk of these APS manifestations. For example, when one antibody was present the OR for arterial thrombosis was 4.45, but when two or more aPL were detected, the ORs rose to 9.23 and 15.6, respectively.
aCL and LA also were associated with thrombocytopenia and hemolytic anemia, with ORs of around 1-2 and 2-3 respectively. There also were antibody associations with cognitive impairments.
Similar results were seen in patients with SLE. “aPL [antibody] positivity in SLE patients influenced the risk for thrombotic and obstetric manifestations,” Dr. Riancho-Zarrabeitia said. LA and aCL were associated with an increased risk of neuropsychiatric manifestations, and LA was linked to an increased risk for renal disease.
The risk for specific SLE manifestations was again higher with IgG isotypes of aCL, notably an increased risk for cardiac and respiratory events.
While increased antibody numbers generally led to a higher risk of complications, the risk for cutaneous manifestations decreased.
“The load of aPL [antibodies] confers a higher risk for APS,” Dr. Riancho-Zarrabeitia said during her conclusion. “Regarding systemic lupus erythematosus, the number of positive antibodies is directly associated with neurological and ophthalmological manifestations, and inversely associated with cutaneous manifestations.”
What these findings show, said Dr. Riancho-Zarrabeitia in the precongress interview, is that individuals who test positive for aPL antibodies need careful monitoring to prevent and treat severe manifestations. “The next step would be to confirm our findings with a prospective study.”
Dr. Riancho-Zarrabeitia has received travel grants from AbbVie, Pfizer, UCB, Merck, GlaxoSmithKline, Amgen, and Roche.
SOURCE: Riancho-Zarrabeitia L et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):136-7. Abstract OP0124. doi: 10.1136/annrheumdis-2019-eular.2485.
according to study findings presented at the European Congress of Rheumatology.
Spanish researchers found that the number of antiphospholipid (aPL) antibodies present was important for the development of antiphospholipid syndrome (APS) and that lupus anticoagulant (LA) was the major aPL antibody linked to systemic lupus erythematosus (SLE)–related organ involvement.
“aPL [antibodies] has been extensively associated with an increased risk of thrombosis and poor pregnancy outcomes, mainly in patients with primary APS,” study investigator Leyre Riancho-Zarrabeitia, MD, PhD, explained in an interview ahead of the congress.
“Moreover, aPL [antibody] positivity in SLE has been proposed to be associated with higher damage accrual and with certain manifestations such as valvular heart disease, pulmonary hypertension, and neuropsychiatric manifestations,” she added.
Anticardiolipin antibodies – notably IgG rather than IgM isotypes – also seemed to play an important role in APS and SLE manifestations, Dr. Riancho-Zarrabeitia, of Hospital Sierrallana, Instituto De Investigación Marqués De Valdecilla, and the University of Cantabria (Spain), noted during her oral presentation.
She reported data on 3,651 patients included in the RELESSER registry between October 2011 and August 2012. This large, multicenter, hospital-based registry retrospectively collects immunologic, clinical and demographic data from unselected adult patients with SLE who are attending 45 Spanish rheumatology services within the country’s national health system.
Over one-third (37.5%) of patients, who had a mean age of 47 years and were mostly (90%) women, were positive for aPL. The most frequent aPL detected was IgG anticardiolipin (aCL) antibodies, seen in 25% of patients, followed by LA in 24%, and IgM aCL in 20%.
Of the aPL-positive patients, 20.6% were positive for only one antibody, 12.1% were positive for two antibodies, and 4.8% were positive for three antibodies.
“All types of aPL were associated with classic APS manifestations,” Dr. Riancho-Zarrabeitia said. The associations were strongest for thrombotic events, such as arterial and venous small-vessel thrombosis and recurrent early pregnancy losses.
aCL antibodies conferred the highest risk for arterial thrombosis, she noted (odds ratio, 5.7), whereas LA conferred the highest risk for venous thrombosis (OR, 4.7). Both IgG and IgM isotypes were associated with thrombotic events, fetal death and recurrent pregnancy loss, but the association was stronger with the IgG isotypes.
Having more than one aPL was particularly associated with a higher risk of these APS manifestations. For example, when one antibody was present the OR for arterial thrombosis was 4.45, but when two or more aPL were detected, the ORs rose to 9.23 and 15.6, respectively.
aCL and LA also were associated with thrombocytopenia and hemolytic anemia, with ORs of around 1-2 and 2-3 respectively. There also were antibody associations with cognitive impairments.
Similar results were seen in patients with SLE. “aPL [antibody] positivity in SLE patients influenced the risk for thrombotic and obstetric manifestations,” Dr. Riancho-Zarrabeitia said. LA and aCL were associated with an increased risk of neuropsychiatric manifestations, and LA was linked to an increased risk for renal disease.
The risk for specific SLE manifestations was again higher with IgG isotypes of aCL, notably an increased risk for cardiac and respiratory events.
While increased antibody numbers generally led to a higher risk of complications, the risk for cutaneous manifestations decreased.
“The load of aPL [antibodies] confers a higher risk for APS,” Dr. Riancho-Zarrabeitia said during her conclusion. “Regarding systemic lupus erythematosus, the number of positive antibodies is directly associated with neurological and ophthalmological manifestations, and inversely associated with cutaneous manifestations.”
What these findings show, said Dr. Riancho-Zarrabeitia in the precongress interview, is that individuals who test positive for aPL antibodies need careful monitoring to prevent and treat severe manifestations. “The next step would be to confirm our findings with a prospective study.”
Dr. Riancho-Zarrabeitia has received travel grants from AbbVie, Pfizer, UCB, Merck, GlaxoSmithKline, Amgen, and Roche.
SOURCE: Riancho-Zarrabeitia L et al. Ann Rheum Dis. Jun 2019;78(Suppl 2):136-7. Abstract OP0124. doi: 10.1136/annrheumdis-2019-eular.2485.
REPORTING FROM EULAR 2019 CONGRESS
Cumulative smoking affects skin manifestations of SLE
SAN FRANCISCO – Nnenna Ezeh reported at an international congress on systemic lupus erythematosus.

“We saw in our study a suggestion of a dose-response relationship. If we tell patients, ‘The more you smoke, the more likely you are to have chronic skin disease or skin damage that’s permanent,’ it could be a way to trigger more smoking cessation strategies in their mind,” said Ms. Ezeh, of the University of Wisconsin, Madison. “We know that skin manifestations of lupus have a major negative impact on a patient’s quality of life, so this could be a way to decrease smoking by saying, ‘Not only does smoking impact your heart and put you at risk for cardiovascular disease, it also affects your skin.’ It’s a way to bridge the priorities that physicians have with the priorities that patients have.”
She presented a retrospective study of the medical records of 632 consecutive SLE patients seen at the university medical center’s ambulatory rheumatology clinic. Slightly more than 60% of them were never smokers; 8.7% had a history of low smoking exposure, defined as less than 5 pack-years; 5.8% had a medium-smoking history of 5-10 pack-years; 15% had a high-smoking history, with more than 10 pack-years; and the smoking history of 10% of the patients was unrecorded.
In a multivariate analysis adjusted for age, sex, and race, the low-smoking group was ninefold more likely than never smokers to develop any mucocutaneous manifestations of SLE, including a malar or discoid rash, mucosal ulcers, photosensitivity, alopecia, or scarring. They were also 3.7 times more likely to meet any Systemic Lupus International Collaborating Clinics (SLICC) cutaneous criteria and twofold more likely than never smokers to meet any of the American College of Rheumatology cutaneous criteria. Patients with an intermediate smoking exposure history of 5-10 pack-years were 2.3-fold more likely to meet any SLICC cutaneous criteria.
The risks of meeting SLICC chronic cutaneous criteria and SLICC Damage Index skin damage criteria rose in a linear fashion with the number of pack-years of smoking. Those SLE patients with more than a 10 pack-year smoking history were 4.2-fold more likely than never smokers to fulfill any SLICC Damage Index skin damage criteria, which consist of scarring alopecia, extensive scarring, or skin ulcers. The heaviest smokers were also at 2.1-fold increased risk of discoid lupus and 2.2-fold more likely to meet SLICC chronic cutaneous criteria, according to Ms. Ezeh.
Patients of color, who comprised 18% of the study population, were significantly more likely to smoke than white patients. Independent of their smoking history, however, they had significantly increased risks of chronic cutaneous manifestations of lupus and of irreversible skin damage.
Ms. Ezeh reported having no financial conflicts regarding her study, supported by a grant from the Rheumatology Research Foundation.
SAN FRANCISCO – Nnenna Ezeh reported at an international congress on systemic lupus erythematosus.
“We saw in our study a suggestion of a dose-response relationship. If we tell patients, ‘The more you smoke, the more likely you are to have chronic skin disease or skin damage that’s permanent,’ it could be a way to trigger more smoking cessation strategies in their mind,” said Ms. Ezeh, of the University of Wisconsin, Madison. “We know that skin manifestations of lupus have a major negative impact on a patient’s quality of life, so this could be a way to decrease smoking by saying, ‘Not only does smoking impact your heart and put you at risk for cardiovascular disease, it also affects your skin.’ It’s a way to bridge the priorities that physicians have with the priorities that patients have.”
She presented a retrospective study of the medical records of 632 consecutive SLE patients seen at the university medical center’s ambulatory rheumatology clinic. Slightly more than 60% of them were never smokers; 8.7% had a history of low smoking exposure, defined as less than 5 pack-years; 5.8% had a medium-smoking history of 5-10 pack-years; 15% had a high-smoking history, with more than 10 pack-years; and the smoking history of 10% of the patients was unrecorded.
In a multivariate analysis adjusted for age, sex, and race, the low-smoking group was ninefold more likely than never smokers to develop any mucocutaneous manifestations of SLE, including a malar or discoid rash, mucosal ulcers, photosensitivity, alopecia, or scarring. They were also 3.7 times more likely to meet any Systemic Lupus International Collaborating Clinics (SLICC) cutaneous criteria and twofold more likely than never smokers to meet any of the American College of Rheumatology cutaneous criteria. Patients with an intermediate smoking exposure history of 5-10 pack-years were 2.3-fold more likely to meet any SLICC cutaneous criteria.
The risks of meeting SLICC chronic cutaneous criteria and SLICC Damage Index skin damage criteria rose in a linear fashion with the number of pack-years of smoking. Those SLE patients with more than a 10 pack-year smoking history were 4.2-fold more likely than never smokers to fulfill any SLICC Damage Index skin damage criteria, which consist of scarring alopecia, extensive scarring, or skin ulcers. The heaviest smokers were also at 2.1-fold increased risk of discoid lupus and 2.2-fold more likely to meet SLICC chronic cutaneous criteria, according to Ms. Ezeh.
Patients of color, who comprised 18% of the study population, were significantly more likely to smoke than white patients. Independent of their smoking history, however, they had significantly increased risks of chronic cutaneous manifestations of lupus and of irreversible skin damage.
Ms. Ezeh reported having no financial conflicts regarding her study, supported by a grant from the Rheumatology Research Foundation.
SAN FRANCISCO – Nnenna Ezeh reported at an international congress on systemic lupus erythematosus.
“We saw in our study a suggestion of a dose-response relationship. If we tell patients, ‘The more you smoke, the more likely you are to have chronic skin disease or skin damage that’s permanent,’ it could be a way to trigger more smoking cessation strategies in their mind,” said Ms. Ezeh, of the University of Wisconsin, Madison. “We know that skin manifestations of lupus have a major negative impact on a patient’s quality of life, so this could be a way to decrease smoking by saying, ‘Not only does smoking impact your heart and put you at risk for cardiovascular disease, it also affects your skin.’ It’s a way to bridge the priorities that physicians have with the priorities that patients have.”
She presented a retrospective study of the medical records of 632 consecutive SLE patients seen at the university medical center’s ambulatory rheumatology clinic. Slightly more than 60% of them were never smokers; 8.7% had a history of low smoking exposure, defined as less than 5 pack-years; 5.8% had a medium-smoking history of 5-10 pack-years; 15% had a high-smoking history, with more than 10 pack-years; and the smoking history of 10% of the patients was unrecorded.
In a multivariate analysis adjusted for age, sex, and race, the low-smoking group was ninefold more likely than never smokers to develop any mucocutaneous manifestations of SLE, including a malar or discoid rash, mucosal ulcers, photosensitivity, alopecia, or scarring. They were also 3.7 times more likely to meet any Systemic Lupus International Collaborating Clinics (SLICC) cutaneous criteria and twofold more likely than never smokers to meet any of the American College of Rheumatology cutaneous criteria. Patients with an intermediate smoking exposure history of 5-10 pack-years were 2.3-fold more likely to meet any SLICC cutaneous criteria.
The risks of meeting SLICC chronic cutaneous criteria and SLICC Damage Index skin damage criteria rose in a linear fashion with the number of pack-years of smoking. Those SLE patients with more than a 10 pack-year smoking history were 4.2-fold more likely than never smokers to fulfill any SLICC Damage Index skin damage criteria, which consist of scarring alopecia, extensive scarring, or skin ulcers. The heaviest smokers were also at 2.1-fold increased risk of discoid lupus and 2.2-fold more likely to meet SLICC chronic cutaneous criteria, according to Ms. Ezeh.
Patients of color, who comprised 18% of the study population, were significantly more likely to smoke than white patients. Independent of their smoking history, however, they had significantly increased risks of chronic cutaneous manifestations of lupus and of irreversible skin damage.
Ms. Ezeh reported having no financial conflicts regarding her study, supported by a grant from the Rheumatology Research Foundation.
REPORTING FROM LUPUS 2019
AAD issues position statement addressing sexual, gender minority health
The individuals.
Some of the unique dermatologic issues faced by SGM individuals include a disproportionate risk for skin cancer in men who have sex with men; higher rates of sexually transmitted infections such as HIV, anogenital dysplasia, and anal cancer; and management of complications such as acne or scarring stemming from medical and/or surgical gender-affirming treatments for transgender individuals. In addition, racial and ethnic minority persons who identify as SGM or LGBTQ face additional stigma and health care disparities.
“While the precise role of dermatology remains controversial regarding anal cancer screening, treatment, and surveillance in these populations, comprehensive skin examinations as well as appropriate counseling and referrals may be linked to earlier detection and improved outcomes,” the statement notes.
The AAD has already taken some steps in advancing the care of SGM individuals, including dedicated educational sessions and workshops at AAD meetings, formation of the AAD LGBTQ/SGM Health Expert Resource Group, incorporation of LGBTQ/SGM content into online AAD basic dermatology curriculum modules, revision of the AAD position statement on isotretinoin, and forthcoming book chapters and CME articles for the Journal of the American Academy of Dermatology.
In order to further commit to the care of diverse populations, the AAD recognized a series of 11 positions in accordance with the association’s “core values of patient-first medicine and visionary leadership,” such as recognizing and affirming the identity and dignity of LGBTQ/SGM individuals, opposing all bias and discrimination, endorsing policies and initiatives that ensure nondiscrimination, and supporting training in cultural humility and structural competency.
“Adequate training of medical professionals regarding the unique health care needs of LGBTQ/SGM people and ongoing research into best care practices are necessary to provide care that facilitates trust and resilience while ensuring the ability of LGBTQ/SGM individuals to thrive,” the statement says.
Find the full position statement on the AAD website.
The individuals.
Some of the unique dermatologic issues faced by SGM individuals include a disproportionate risk for skin cancer in men who have sex with men; higher rates of sexually transmitted infections such as HIV, anogenital dysplasia, and anal cancer; and management of complications such as acne or scarring stemming from medical and/or surgical gender-affirming treatments for transgender individuals. In addition, racial and ethnic minority persons who identify as SGM or LGBTQ face additional stigma and health care disparities.
“While the precise role of dermatology remains controversial regarding anal cancer screening, treatment, and surveillance in these populations, comprehensive skin examinations as well as appropriate counseling and referrals may be linked to earlier detection and improved outcomes,” the statement notes.
The AAD has already taken some steps in advancing the care of SGM individuals, including dedicated educational sessions and workshops at AAD meetings, formation of the AAD LGBTQ/SGM Health Expert Resource Group, incorporation of LGBTQ/SGM content into online AAD basic dermatology curriculum modules, revision of the AAD position statement on isotretinoin, and forthcoming book chapters and CME articles for the Journal of the American Academy of Dermatology.
In order to further commit to the care of diverse populations, the AAD recognized a series of 11 positions in accordance with the association’s “core values of patient-first medicine and visionary leadership,” such as recognizing and affirming the identity and dignity of LGBTQ/SGM individuals, opposing all bias and discrimination, endorsing policies and initiatives that ensure nondiscrimination, and supporting training in cultural humility and structural competency.
“Adequate training of medical professionals regarding the unique health care needs of LGBTQ/SGM people and ongoing research into best care practices are necessary to provide care that facilitates trust and resilience while ensuring the ability of LGBTQ/SGM individuals to thrive,” the statement says.
Find the full position statement on the AAD website.
The individuals.
Some of the unique dermatologic issues faced by SGM individuals include a disproportionate risk for skin cancer in men who have sex with men; higher rates of sexually transmitted infections such as HIV, anogenital dysplasia, and anal cancer; and management of complications such as acne or scarring stemming from medical and/or surgical gender-affirming treatments for transgender individuals. In addition, racial and ethnic minority persons who identify as SGM or LGBTQ face additional stigma and health care disparities.
“While the precise role of dermatology remains controversial regarding anal cancer screening, treatment, and surveillance in these populations, comprehensive skin examinations as well as appropriate counseling and referrals may be linked to earlier detection and improved outcomes,” the statement notes.
The AAD has already taken some steps in advancing the care of SGM individuals, including dedicated educational sessions and workshops at AAD meetings, formation of the AAD LGBTQ/SGM Health Expert Resource Group, incorporation of LGBTQ/SGM content into online AAD basic dermatology curriculum modules, revision of the AAD position statement on isotretinoin, and forthcoming book chapters and CME articles for the Journal of the American Academy of Dermatology.
In order to further commit to the care of diverse populations, the AAD recognized a series of 11 positions in accordance with the association’s “core values of patient-first medicine and visionary leadership,” such as recognizing and affirming the identity and dignity of LGBTQ/SGM individuals, opposing all bias and discrimination, endorsing policies and initiatives that ensure nondiscrimination, and supporting training in cultural humility and structural competency.
“Adequate training of medical professionals regarding the unique health care needs of LGBTQ/SGM people and ongoing research into best care practices are necessary to provide care that facilitates trust and resilience while ensuring the ability of LGBTQ/SGM individuals to thrive,” the statement says.
Find the full position statement on the AAD website.
Review clarifies depression, anxiety risk among hidradenitis suppurativa patients
and meta-analysis of more than 40,000 adults.
Previous studies of psychiatric comorbidities in HS patients have suggested an increased rate of depression, anxiety, and suicide risk, but have varied in methodology. “Therefore, the exact magnitude of the prevalence and odds of depression and anxiety in patients with HS is unclear,” wrote Myrela O. Machado, MD, of the division of dermatology, Women’s College Hospital, Toronto, and colleagues.
In a review of PubMed/MEDLINE, Embase, and PsycINFO electronic databases through July 2018, the researchers identified 10 studies comprising 40,307 adults with HS. The overall prevalence of depression was 16.9%, and in the studies that included a comparison group, the odds ratio for depression was 1.84 for HS patients, compared with controls who did not have HS. The overall prevalence of anxiety was 4.9%, but data were insufficient to determine an odds ratio for anxiety. The study was published in JAMA Dermatology.
All 10 studies assessed depression; 4 assessed anxiety. In subgroup analyses, the prevalence of depression was 11.9% in studies with a diagnosis based on clinical criteria and 26.8% in studies that used a screening instrument. The prevalence was 25.9% for outpatients with HS.
“Although our findings indicate that depression and anxiety may be common among people with HS, whether there is a causal relationship in those associations remains to be proved,” the researchers wrote.
However, they noted, “the consequences of depression and anxiety on HS-related outcomes have recently received more attention.”
The study findings were limited by several factors including the lack of data from structured diagnostic interviews, variation in methodological quality, and variation in comparison groups across studies, as well as a high level of heterogeneity across studies, the researchers noted. However, the results support the need to recognize and treat psychiatric conditions in HS patients, and to develop new management strategies, they said.
Dr. Machado had no financial conflicts to disclose. Several coauthors disclosed relationships with Galderma, LEO Pharma, Janssen, Novartis, AbbVie, Celgene, Naos, Lilly, Sanofi, Valeant, and La Roche-Posay.
SOURCE: Machado M et al. JAMA Dermatol. 2019 June 5. doi: 10.1001/jamadermatol.2019.0759.
and meta-analysis of more than 40,000 adults.
Previous studies of psychiatric comorbidities in HS patients have suggested an increased rate of depression, anxiety, and suicide risk, but have varied in methodology. “Therefore, the exact magnitude of the prevalence and odds of depression and anxiety in patients with HS is unclear,” wrote Myrela O. Machado, MD, of the division of dermatology, Women’s College Hospital, Toronto, and colleagues.
In a review of PubMed/MEDLINE, Embase, and PsycINFO electronic databases through July 2018, the researchers identified 10 studies comprising 40,307 adults with HS. The overall prevalence of depression was 16.9%, and in the studies that included a comparison group, the odds ratio for depression was 1.84 for HS patients, compared with controls who did not have HS. The overall prevalence of anxiety was 4.9%, but data were insufficient to determine an odds ratio for anxiety. The study was published in JAMA Dermatology.
All 10 studies assessed depression; 4 assessed anxiety. In subgroup analyses, the prevalence of depression was 11.9% in studies with a diagnosis based on clinical criteria and 26.8% in studies that used a screening instrument. The prevalence was 25.9% for outpatients with HS.
“Although our findings indicate that depression and anxiety may be common among people with HS, whether there is a causal relationship in those associations remains to be proved,” the researchers wrote.
However, they noted, “the consequences of depression and anxiety on HS-related outcomes have recently received more attention.”
The study findings were limited by several factors including the lack of data from structured diagnostic interviews, variation in methodological quality, and variation in comparison groups across studies, as well as a high level of heterogeneity across studies, the researchers noted. However, the results support the need to recognize and treat psychiatric conditions in HS patients, and to develop new management strategies, they said.
Dr. Machado had no financial conflicts to disclose. Several coauthors disclosed relationships with Galderma, LEO Pharma, Janssen, Novartis, AbbVie, Celgene, Naos, Lilly, Sanofi, Valeant, and La Roche-Posay.
SOURCE: Machado M et al. JAMA Dermatol. 2019 June 5. doi: 10.1001/jamadermatol.2019.0759.
and meta-analysis of more than 40,000 adults.
Previous studies of psychiatric comorbidities in HS patients have suggested an increased rate of depression, anxiety, and suicide risk, but have varied in methodology. “Therefore, the exact magnitude of the prevalence and odds of depression and anxiety in patients with HS is unclear,” wrote Myrela O. Machado, MD, of the division of dermatology, Women’s College Hospital, Toronto, and colleagues.
In a review of PubMed/MEDLINE, Embase, and PsycINFO electronic databases through July 2018, the researchers identified 10 studies comprising 40,307 adults with HS. The overall prevalence of depression was 16.9%, and in the studies that included a comparison group, the odds ratio for depression was 1.84 for HS patients, compared with controls who did not have HS. The overall prevalence of anxiety was 4.9%, but data were insufficient to determine an odds ratio for anxiety. The study was published in JAMA Dermatology.
All 10 studies assessed depression; 4 assessed anxiety. In subgroup analyses, the prevalence of depression was 11.9% in studies with a diagnosis based on clinical criteria and 26.8% in studies that used a screening instrument. The prevalence was 25.9% for outpatients with HS.
“Although our findings indicate that depression and anxiety may be common among people with HS, whether there is a causal relationship in those associations remains to be proved,” the researchers wrote.
However, they noted, “the consequences of depression and anxiety on HS-related outcomes have recently received more attention.”
The study findings were limited by several factors including the lack of data from structured diagnostic interviews, variation in methodological quality, and variation in comparison groups across studies, as well as a high level of heterogeneity across studies, the researchers noted. However, the results support the need to recognize and treat psychiatric conditions in HS patients, and to develop new management strategies, they said.
Dr. Machado had no financial conflicts to disclose. Several coauthors disclosed relationships with Galderma, LEO Pharma, Janssen, Novartis, AbbVie, Celgene, Naos, Lilly, Sanofi, Valeant, and La Roche-Posay.
SOURCE: Machado M et al. JAMA Dermatol. 2019 June 5. doi: 10.1001/jamadermatol.2019.0759.
FROM JAMA DERMATOLOGY
Major increase in hidradenitis suppurativa cases anticipated
CHICAGO – Once thought to be a rare disease and largely neglected as a focus of research, and increase the number of patients seeking care, according to an expert interview conducted at the annual meeting of the Society for Investigational Dermatology.
“For decades ... we’ve really not understood how prevalent it is,” said Haley B. Naik, MD, of the department of dermatology at University of California, San Francisco, in a video interview. “Now, thanks to great population-based studies and new data, we know that HS is common and hugely impacts the lives of the people who suffer with this condition.”
Recapping some of the highlights of an update on this chronic inflammatory skin condition that she presented at the meeting, Dr. Naik said that HS has been mischaracterized as rare. Many patients, embarrassed by the symptoms or failing to receive adequate relief from previous health care encounters, are not currently seeking care.
This will change as treatments improve, according to Dr. Naik, who asserted that HS has become a hot topic. Progress in understanding the underlying pathophysiology has been driving new management strategies. She counted more than 15 clinical trials being conducted with new agents for this disease in a clinicaltrials.gov survey.
In the interview, Dr. Naik calls on dermatologists to increase their awareness of the signs and symptoms of HS so that they can diagnose and intervene earlier, a step that will be made easier as new therapies become available.
CHICAGO – Once thought to be a rare disease and largely neglected as a focus of research, and increase the number of patients seeking care, according to an expert interview conducted at the annual meeting of the Society for Investigational Dermatology.
“For decades ... we’ve really not understood how prevalent it is,” said Haley B. Naik, MD, of the department of dermatology at University of California, San Francisco, in a video interview. “Now, thanks to great population-based studies and new data, we know that HS is common and hugely impacts the lives of the people who suffer with this condition.”
Recapping some of the highlights of an update on this chronic inflammatory skin condition that she presented at the meeting, Dr. Naik said that HS has been mischaracterized as rare. Many patients, embarrassed by the symptoms or failing to receive adequate relief from previous health care encounters, are not currently seeking care.
This will change as treatments improve, according to Dr. Naik, who asserted that HS has become a hot topic. Progress in understanding the underlying pathophysiology has been driving new management strategies. She counted more than 15 clinical trials being conducted with new agents for this disease in a clinicaltrials.gov survey.
In the interview, Dr. Naik calls on dermatologists to increase their awareness of the signs and symptoms of HS so that they can diagnose and intervene earlier, a step that will be made easier as new therapies become available.
CHICAGO – Once thought to be a rare disease and largely neglected as a focus of research, and increase the number of patients seeking care, according to an expert interview conducted at the annual meeting of the Society for Investigational Dermatology.
“For decades ... we’ve really not understood how prevalent it is,” said Haley B. Naik, MD, of the department of dermatology at University of California, San Francisco, in a video interview. “Now, thanks to great population-based studies and new data, we know that HS is common and hugely impacts the lives of the people who suffer with this condition.”
Recapping some of the highlights of an update on this chronic inflammatory skin condition that she presented at the meeting, Dr. Naik said that HS has been mischaracterized as rare. Many patients, embarrassed by the symptoms or failing to receive adequate relief from previous health care encounters, are not currently seeking care.
This will change as treatments improve, according to Dr. Naik, who asserted that HS has become a hot topic. Progress in understanding the underlying pathophysiology has been driving new management strategies. She counted more than 15 clinical trials being conducted with new agents for this disease in a clinicaltrials.gov survey.
In the interview, Dr. Naik calls on dermatologists to increase their awareness of the signs and symptoms of HS so that they can diagnose and intervene earlier, a step that will be made easier as new therapies become available.
REPORTING FROM SID 2019

Molecular profiling shows promise for treating unusual skin rashes
CHICAGO – Although at a relatively early stage of research, according to research that was described at the annual meeting of the Society for Investigative Dermatology.

“We now have several cases that suggest single-cell molecular profiling can provide treatment guidance for atypical rashes, providing an opportunity for a rational treatment choice rather than just improvising in a difficult population,” reported Raymond Cho, MD, PhD, of the department of dermatology at the University of California, San Francisco.
Based on a growing cohort of patients with atypical rashes, one goal is to develop “a library of molecular fingerprints” for classifying rashes that are atypical when defined by morphology, histopathology, or therapeutic response, according to Dr. Cho.
“The big focus now is on expanding this patient cohort. We want to move from anecdotal cases to a larger patient population with which we can statistically prove that we can nominate the best first-line therapy through this approach,” he explained.
In describing work he is performing in collaboration with Jeffrey Cheng, MD, also with the department of dermatology at UCSF, Dr. Cho said the profiles are based on RNA sequencing from single immune cells and epitope measurements. Work already performed in rashes of known etiology supports the approach. For example, the profile for atopic dermatitis includes elevated expression of interleukin (IL)-4 and IL-13, whereas that of psoriasis includes elevated expression of IL-17, which fit with the expected molecular signatures of these diseases.
To be considered for inclusion in the cohort of atypical rashes, patients are required to have an idiopathic skin lesion of at least 6 months’ duration with at least two atypical features defined by such characteristics as morphology or location. Many of these patients have already consulted with multiple providers, have undergone multiple biopsies without a diagnosis, and have failed common treatments, such as steroids.
Examples selected from this cohort have already supported the premise that molecular profiles are relevant to treatment choice. Dr. Cho described one patient with unremitting generalized pruritus and another with nodular lesions on the legs. Both had symptoms of long duration that had failed multiple treatments.
In both cases, immune cell profiling identified lesions high in IL-13 expression. Both achieved complete or near complete resolution of their rash and symptoms when treated with dupilumab, a biologic that targets the IL-13 pathway. In one patient with a large symptom burden, Dr. Cho described the response as “remarkable.”
There are more than 40 patients in the expanding cohort, according to Dr. Cho, who emphasized that this work is timely because of “the armamentarium of immunomodulatory drugs that are coming on line.” He said this type of drug development in dermatology is the basis for a potential paradigm shift.
“Personalized therapy has been used in clinical oncology for almost 10 years now, but this is an approach that needs to find a home in our specialty as well,” Dr. Cho said. He cited data suggesting that nearly 15% of rashes are atypical and represent a major source of frustration to both patients and clinicians when conventional treatments fail.
Asked about cost, he acknowledged that the molecular profiling that he and Dr. Cheng are performing is expensive at the current time, but “we are hopeful that we can find cheaper markers and technologies” to bring this cost down. However, he noted that undiagnosed rashes consume a great deal of time of effort from clinicians while generating significant morbidity for patients, which is justifying novel strategies to find effective therapies.
“These are not happy patients,” Dr. Cho said. Although there are technical challenges for building a molecular library that has practical utility across the substantial heterogeneity of idiopathic rashes, he suggested that a larger patient sample is considered one of the important steps toward overcoming hurdles.
Dr. Cho reports no potential conflicts of interest.
CHICAGO – Although at a relatively early stage of research, according to research that was described at the annual meeting of the Society for Investigative Dermatology.
“We now have several cases that suggest single-cell molecular profiling can provide treatment guidance for atypical rashes, providing an opportunity for a rational treatment choice rather than just improvising in a difficult population,” reported Raymond Cho, MD, PhD, of the department of dermatology at the University of California, San Francisco.
Based on a growing cohort of patients with atypical rashes, one goal is to develop “a library of molecular fingerprints” for classifying rashes that are atypical when defined by morphology, histopathology, or therapeutic response, according to Dr. Cho.
“The big focus now is on expanding this patient cohort. We want to move from anecdotal cases to a larger patient population with which we can statistically prove that we can nominate the best first-line therapy through this approach,” he explained.
In describing work he is performing in collaboration with Jeffrey Cheng, MD, also with the department of dermatology at UCSF, Dr. Cho said the profiles are based on RNA sequencing from single immune cells and epitope measurements. Work already performed in rashes of known etiology supports the approach. For example, the profile for atopic dermatitis includes elevated expression of interleukin (IL)-4 and IL-13, whereas that of psoriasis includes elevated expression of IL-17, which fit with the expected molecular signatures of these diseases.
To be considered for inclusion in the cohort of atypical rashes, patients are required to have an idiopathic skin lesion of at least 6 months’ duration with at least two atypical features defined by such characteristics as morphology or location. Many of these patients have already consulted with multiple providers, have undergone multiple biopsies without a diagnosis, and have failed common treatments, such as steroids.
Examples selected from this cohort have already supported the premise that molecular profiles are relevant to treatment choice. Dr. Cho described one patient with unremitting generalized pruritus and another with nodular lesions on the legs. Both had symptoms of long duration that had failed multiple treatments.
In both cases, immune cell profiling identified lesions high in IL-13 expression. Both achieved complete or near complete resolution of their rash and symptoms when treated with dupilumab, a biologic that targets the IL-13 pathway. In one patient with a large symptom burden, Dr. Cho described the response as “remarkable.”
There are more than 40 patients in the expanding cohort, according to Dr. Cho, who emphasized that this work is timely because of “the armamentarium of immunomodulatory drugs that are coming on line.” He said this type of drug development in dermatology is the basis for a potential paradigm shift.
“Personalized therapy has been used in clinical oncology for almost 10 years now, but this is an approach that needs to find a home in our specialty as well,” Dr. Cho said. He cited data suggesting that nearly 15% of rashes are atypical and represent a major source of frustration to both patients and clinicians when conventional treatments fail.
Asked about cost, he acknowledged that the molecular profiling that he and Dr. Cheng are performing is expensive at the current time, but “we are hopeful that we can find cheaper markers and technologies” to bring this cost down. However, he noted that undiagnosed rashes consume a great deal of time of effort from clinicians while generating significant morbidity for patients, which is justifying novel strategies to find effective therapies.
“These are not happy patients,” Dr. Cho said. Although there are technical challenges for building a molecular library that has practical utility across the substantial heterogeneity of idiopathic rashes, he suggested that a larger patient sample is considered one of the important steps toward overcoming hurdles.
Dr. Cho reports no potential conflicts of interest.
CHICAGO – Although at a relatively early stage of research, according to research that was described at the annual meeting of the Society for Investigative Dermatology.
“We now have several cases that suggest single-cell molecular profiling can provide treatment guidance for atypical rashes, providing an opportunity for a rational treatment choice rather than just improvising in a difficult population,” reported Raymond Cho, MD, PhD, of the department of dermatology at the University of California, San Francisco.
Based on a growing cohort of patients with atypical rashes, one goal is to develop “a library of molecular fingerprints” for classifying rashes that are atypical when defined by morphology, histopathology, or therapeutic response, according to Dr. Cho.
“The big focus now is on expanding this patient cohort. We want to move from anecdotal cases to a larger patient population with which we can statistically prove that we can nominate the best first-line therapy through this approach,” he explained.
In describing work he is performing in collaboration with Jeffrey Cheng, MD, also with the department of dermatology at UCSF, Dr. Cho said the profiles are based on RNA sequencing from single immune cells and epitope measurements. Work already performed in rashes of known etiology supports the approach. For example, the profile for atopic dermatitis includes elevated expression of interleukin (IL)-4 and IL-13, whereas that of psoriasis includes elevated expression of IL-17, which fit with the expected molecular signatures of these diseases.
To be considered for inclusion in the cohort of atypical rashes, patients are required to have an idiopathic skin lesion of at least 6 months’ duration with at least two atypical features defined by such characteristics as morphology or location. Many of these patients have already consulted with multiple providers, have undergone multiple biopsies without a diagnosis, and have failed common treatments, such as steroids.
Examples selected from this cohort have already supported the premise that molecular profiles are relevant to treatment choice. Dr. Cho described one patient with unremitting generalized pruritus and another with nodular lesions on the legs. Both had symptoms of long duration that had failed multiple treatments.
In both cases, immune cell profiling identified lesions high in IL-13 expression. Both achieved complete or near complete resolution of their rash and symptoms when treated with dupilumab, a biologic that targets the IL-13 pathway. In one patient with a large symptom burden, Dr. Cho described the response as “remarkable.”
There are more than 40 patients in the expanding cohort, according to Dr. Cho, who emphasized that this work is timely because of “the armamentarium of immunomodulatory drugs that are coming on line.” He said this type of drug development in dermatology is the basis for a potential paradigm shift.
“Personalized therapy has been used in clinical oncology for almost 10 years now, but this is an approach that needs to find a home in our specialty as well,” Dr. Cho said. He cited data suggesting that nearly 15% of rashes are atypical and represent a major source of frustration to both patients and clinicians when conventional treatments fail.
Asked about cost, he acknowledged that the molecular profiling that he and Dr. Cheng are performing is expensive at the current time, but “we are hopeful that we can find cheaper markers and technologies” to bring this cost down. However, he noted that undiagnosed rashes consume a great deal of time of effort from clinicians while generating significant morbidity for patients, which is justifying novel strategies to find effective therapies.
“These are not happy patients,” Dr. Cho said. Although there are technical challenges for building a molecular library that has practical utility across the substantial heterogeneity of idiopathic rashes, he suggested that a larger patient sample is considered one of the important steps toward overcoming hurdles.
Dr. Cho reports no potential conflicts of interest.
EXPERT ANALYSIS FROM SID 2019
By the numbers: Readmissions for skin conditions
Almost 10% of patients
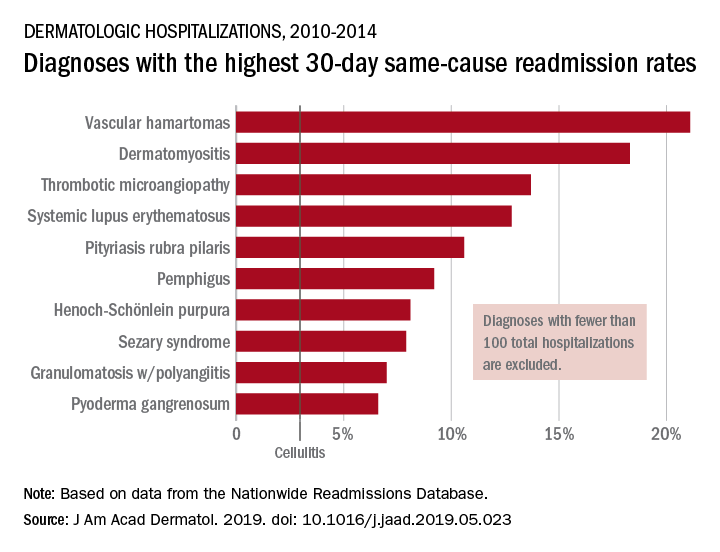
Data from the Nationwide Readmissions Database also showed that the same-cause readmission rate was 3.3% after 30 days and 7.8% within the calendar year (CY) over the 5-year study period of 2010-2014, Myron Zhang, MD, of the department of dermatology at Weill Cornell Medicine, New York, and his associates reported in the Journal of the American Academy of Dermatology.
The total cost of the CY readmissions was $2.54 billion, which works out to $508 million per year or $8,995 per visit. The most common dermatologic diagnosis – cellulitis made up 83.6% of all hospitalizations – was also the most expensive in terms of readmissions, resulting in $1.9 billion in CY costs, Dr. Zhang and associates wrote.
Overall readmission rates for cellulitis were not provided, but annual rates ranged from 9.1% to 9.3% (30-day all cause), from 7.7% to 8.1% (CY same cause), and from 3.1% to 3.3% (30-day same cause), they wrote.
The dermatologic diagnosis with the highest 30-day same-cause readmission rate was vascular hamartomas at 21.1%, followed by dermatomyositis (18.3%) and thrombotic microangiopathy (13.7%). Dermatomyositis had the highest CY same-cause readmission rate (30.8%) and mycosis fungoides had the highest 30-day all-cause rate (32.3%), according to the investigators.
“Diseases, characteristics, and comorbidities associated with high readmission rates should trigger hospitals to consider dermatology consultation, coordinate outpatient follow-up, and support underinsured outpatient access. These measures have been shown to reduce readmissions or hospital visits in general dermatologic settings, but outcomes in individual diseases are not well studied,” Dr. Zhang and associates wrote. They noted that there have been “very few prior studies of readmissions for skin diseases.”
[email protected]
SOURCE: Zhang M et al. J Am Acad. Dermatol. 2019. doi: 10.1016/j.jaad.2019.05.023. .
Almost 10% of patients
Data from the Nationwide Readmissions Database also showed that the same-cause readmission rate was 3.3% after 30 days and 7.8% within the calendar year (CY) over the 5-year study period of 2010-2014, Myron Zhang, MD, of the department of dermatology at Weill Cornell Medicine, New York, and his associates reported in the Journal of the American Academy of Dermatology.
The total cost of the CY readmissions was $2.54 billion, which works out to $508 million per year or $8,995 per visit. The most common dermatologic diagnosis – cellulitis made up 83.6% of all hospitalizations – was also the most expensive in terms of readmissions, resulting in $1.9 billion in CY costs, Dr. Zhang and associates wrote.
Overall readmission rates for cellulitis were not provided, but annual rates ranged from 9.1% to 9.3% (30-day all cause), from 7.7% to 8.1% (CY same cause), and from 3.1% to 3.3% (30-day same cause), they wrote.
The dermatologic diagnosis with the highest 30-day same-cause readmission rate was vascular hamartomas at 21.1%, followed by dermatomyositis (18.3%) and thrombotic microangiopathy (13.7%). Dermatomyositis had the highest CY same-cause readmission rate (30.8%) and mycosis fungoides had the highest 30-day all-cause rate (32.3%), according to the investigators.
“Diseases, characteristics, and comorbidities associated with high readmission rates should trigger hospitals to consider dermatology consultation, coordinate outpatient follow-up, and support underinsured outpatient access. These measures have been shown to reduce readmissions or hospital visits in general dermatologic settings, but outcomes in individual diseases are not well studied,” Dr. Zhang and associates wrote. They noted that there have been “very few prior studies of readmissions for skin diseases.”
[email protected]
SOURCE: Zhang M et al. J Am Acad. Dermatol. 2019. doi: 10.1016/j.jaad.2019.05.023. .
Almost 10% of patients
Data from the Nationwide Readmissions Database also showed that the same-cause readmission rate was 3.3% after 30 days and 7.8% within the calendar year (CY) over the 5-year study period of 2010-2014, Myron Zhang, MD, of the department of dermatology at Weill Cornell Medicine, New York, and his associates reported in the Journal of the American Academy of Dermatology.
The total cost of the CY readmissions was $2.54 billion, which works out to $508 million per year or $8,995 per visit. The most common dermatologic diagnosis – cellulitis made up 83.6% of all hospitalizations – was also the most expensive in terms of readmissions, resulting in $1.9 billion in CY costs, Dr. Zhang and associates wrote.
Overall readmission rates for cellulitis were not provided, but annual rates ranged from 9.1% to 9.3% (30-day all cause), from 7.7% to 8.1% (CY same cause), and from 3.1% to 3.3% (30-day same cause), they wrote.
The dermatologic diagnosis with the highest 30-day same-cause readmission rate was vascular hamartomas at 21.1%, followed by dermatomyositis (18.3%) and thrombotic microangiopathy (13.7%). Dermatomyositis had the highest CY same-cause readmission rate (30.8%) and mycosis fungoides had the highest 30-day all-cause rate (32.3%), according to the investigators.
“Diseases, characteristics, and comorbidities associated with high readmission rates should trigger hospitals to consider dermatology consultation, coordinate outpatient follow-up, and support underinsured outpatient access. These measures have been shown to reduce readmissions or hospital visits in general dermatologic settings, but outcomes in individual diseases are not well studied,” Dr. Zhang and associates wrote. They noted that there have been “very few prior studies of readmissions for skin diseases.”
[email protected]
SOURCE: Zhang M et al. J Am Acad. Dermatol. 2019. doi: 10.1016/j.jaad.2019.05.023. .
FROM THE JOURNAL OF THE AMERICAN ACADEMY OF DERMATOLOGY
Nintedanib cut lung function decline in interstitial lung disease with systemic sclerosis
DALLAS – Nintedanib, a tyrosine kinase inhibitor, decreased by 44% the annual rate of lung function decline among patients with interstitial lung disease associated with systemic sclerosis, a year-long study has found.

In a placebo-controlled 52-week trial, forced vital capacity (FVC) in patients who took nintedanib (Ofev) declined by a mean of 52 mL – significantly less than the mean 93 mL decline seen among those who were given placebo, Oliver Distler, MD, said at the annual meeting of the American Thoracic Society.
“These are people in their mid-40s and -50s,” said Dr. Distler of the University of Zürich. “They have a long time to go. If there is an annual preservation of lung function by 40%, if you have that every year, it becomes very surely clinically significant. A decline in FVC is also a good surrogate marker of mortality in interstitial lung disease associated with systemic sclerosis. Assuming the effects are ongoing above the 1 year we looked at, then indeed these results are clinically important.”
The study was simultaneously published in the New England Journal of Medicine. Nintedanib is already approved for idiopathic pulmonary fibrosis. But some data suggest that it also exerts antifibrotic and anti-inflammatory effects in animal models of systemic sclerosis and inflammatory lung disease (ILD). SENSCIS (the Safety and Efficacy of Nintedanib in Systemic Sclerosis trial) investigated the molecule’s use in patients with ILD associated with systemic sclerosis.
Conducted in 32 countries, SENSCIS comprised 576 patients with the disorder, whose sclerosis affected at least 10% of their lungs. They were assigned to 52 weeks of either placebo or 150 mg nintedanib twice weekly. However, patients stayed on their blinded treatment until the last patient enrolled had finished the year of treatment; some patients took the drug for 100 weeks, Dr. Distler said. The primary endpoint was annual rate of decline in the forced vital capacity (FEV). Secondary endpoints included changes of the modified Rodnan skin score and in the total score on the St. George’s Respiratory Questionnaire.
Patients were a mean of 54 years old, with a mean disease duration of about 3 years. About half had diffuse cutaneous systemic sclerosis; the sclerosis was limited in the remainder. The mean extent of lung fibrosis was about 36%. Half were taking mycophenolate at baseline, which was allowed as background treatment, along with up to 10 mg/day of prednisone. Any patient who experienced clinically significant lung function deterioration could receive additional therapy at the investigator’s discretion.
The mean baseline FEV for these patients was 72.5% of predicted value. The mean diffusing capacity of the lungs for carbon monoxide was 53% of expected capacity.
Most patients completed the study (80% of the active group and 89% of the placebo group). The mean drug exposure duration was 10 months in the active group and 11 in the placebo group.
Improvement began early in treatment, with the efficacy curves separating by week 12 and continuing to diverge. After 52 weeks of therapy, the annual rate of change was 41 mL less in the active group than in the placebo group (–54.4 mL vs. –93.3 mL). The mean adjusted absolute change from baseline was –54.6 mL in the active group and –101 mL in the placebo at week 52. Significantly fewer patients taking nintedanib also lost more than 10% of FVC by week 52 (16.7% vs. 18%).
The St. George’s Respiratory Questionnaire score improved about one point in the active group and declined about one point in the placebo group.
Nintedanib was equally effective across a number of subgroups, including those divided by sex, age, and race. Antitopoisomerase antibodies and so-called antitopoisomerase I antibody status did not affect nintedanib’s action. Nintedanib also significantly improved scores on the Health Assessment Questionnaire without Disability Index and dyspnea.
More patients in the active group than in on placebo discontinued treatment because of a serious adverse event (16% vs. 8.7%). The most common of these were diarrhea (75.7% vs. 31%), nausea (31.6% vs. 13.5%), and vomiting (24.7% vs.10.4%). Skin ulcers occurred in about 18% of each group. Patients in the active group were significantly more likely to develop elevated alanine and aspartate aminotransferase of up to three times normal levels (4.9% vs. 0.7%).
Treatment did not significantly affect mortality rates, however. Over the treatment period, 10 patients in the nintedanib group and 9 in the placebo group died (3.5% vs. 3.1%).
The study was sponsored by Boehringer Ingelheim. Dr. Distler was the primary investigator on the trial.
SOURCE: Distler O et al. ATS 2019, Abstract A7360.
DALLAS – Nintedanib, a tyrosine kinase inhibitor, decreased by 44% the annual rate of lung function decline among patients with interstitial lung disease associated with systemic sclerosis, a year-long study has found.
In a placebo-controlled 52-week trial, forced vital capacity (FVC) in patients who took nintedanib (Ofev) declined by a mean of 52 mL – significantly less than the mean 93 mL decline seen among those who were given placebo, Oliver Distler, MD, said at the annual meeting of the American Thoracic Society.
“These are people in their mid-40s and -50s,” said Dr. Distler of the University of Zürich. “They have a long time to go. If there is an annual preservation of lung function by 40%, if you have that every year, it becomes very surely clinically significant. A decline in FVC is also a good surrogate marker of mortality in interstitial lung disease associated with systemic sclerosis. Assuming the effects are ongoing above the 1 year we looked at, then indeed these results are clinically important.”
The study was simultaneously published in the New England Journal of Medicine. Nintedanib is already approved for idiopathic pulmonary fibrosis. But some data suggest that it also exerts antifibrotic and anti-inflammatory effects in animal models of systemic sclerosis and inflammatory lung disease (ILD). SENSCIS (the Safety and Efficacy of Nintedanib in Systemic Sclerosis trial) investigated the molecule’s use in patients with ILD associated with systemic sclerosis.
Conducted in 32 countries, SENSCIS comprised 576 patients with the disorder, whose sclerosis affected at least 10% of their lungs. They were assigned to 52 weeks of either placebo or 150 mg nintedanib twice weekly. However, patients stayed on their blinded treatment until the last patient enrolled had finished the year of treatment; some patients took the drug for 100 weeks, Dr. Distler said. The primary endpoint was annual rate of decline in the forced vital capacity (FEV). Secondary endpoints included changes of the modified Rodnan skin score and in the total score on the St. George’s Respiratory Questionnaire.
Patients were a mean of 54 years old, with a mean disease duration of about 3 years. About half had diffuse cutaneous systemic sclerosis; the sclerosis was limited in the remainder. The mean extent of lung fibrosis was about 36%. Half were taking mycophenolate at baseline, which was allowed as background treatment, along with up to 10 mg/day of prednisone. Any patient who experienced clinically significant lung function deterioration could receive additional therapy at the investigator’s discretion.
The mean baseline FEV for these patients was 72.5% of predicted value. The mean diffusing capacity of the lungs for carbon monoxide was 53% of expected capacity.
Most patients completed the study (80% of the active group and 89% of the placebo group). The mean drug exposure duration was 10 months in the active group and 11 in the placebo group.
Improvement began early in treatment, with the efficacy curves separating by week 12 and continuing to diverge. After 52 weeks of therapy, the annual rate of change was 41 mL less in the active group than in the placebo group (–54.4 mL vs. –93.3 mL). The mean adjusted absolute change from baseline was –54.6 mL in the active group and –101 mL in the placebo at week 52. Significantly fewer patients taking nintedanib also lost more than 10% of FVC by week 52 (16.7% vs. 18%).
The St. George’s Respiratory Questionnaire score improved about one point in the active group and declined about one point in the placebo group.
Nintedanib was equally effective across a number of subgroups, including those divided by sex, age, and race. Antitopoisomerase antibodies and so-called antitopoisomerase I antibody status did not affect nintedanib’s action. Nintedanib also significantly improved scores on the Health Assessment Questionnaire without Disability Index and dyspnea.
More patients in the active group than in on placebo discontinued treatment because of a serious adverse event (16% vs. 8.7%). The most common of these were diarrhea (75.7% vs. 31%), nausea (31.6% vs. 13.5%), and vomiting (24.7% vs.10.4%). Skin ulcers occurred in about 18% of each group. Patients in the active group were significantly more likely to develop elevated alanine and aspartate aminotransferase of up to three times normal levels (4.9% vs. 0.7%).
Treatment did not significantly affect mortality rates, however. Over the treatment period, 10 patients in the nintedanib group and 9 in the placebo group died (3.5% vs. 3.1%).
The study was sponsored by Boehringer Ingelheim. Dr. Distler was the primary investigator on the trial.
SOURCE: Distler O et al. ATS 2019, Abstract A7360.
DALLAS – Nintedanib, a tyrosine kinase inhibitor, decreased by 44% the annual rate of lung function decline among patients with interstitial lung disease associated with systemic sclerosis, a year-long study has found.
In a placebo-controlled 52-week trial, forced vital capacity (FVC) in patients who took nintedanib (Ofev) declined by a mean of 52 mL – significantly less than the mean 93 mL decline seen among those who were given placebo, Oliver Distler, MD, said at the annual meeting of the American Thoracic Society.
“These are people in their mid-40s and -50s,” said Dr. Distler of the University of Zürich. “They have a long time to go. If there is an annual preservation of lung function by 40%, if you have that every year, it becomes very surely clinically significant. A decline in FVC is also a good surrogate marker of mortality in interstitial lung disease associated with systemic sclerosis. Assuming the effects are ongoing above the 1 year we looked at, then indeed these results are clinically important.”
The study was simultaneously published in the New England Journal of Medicine. Nintedanib is already approved for idiopathic pulmonary fibrosis. But some data suggest that it also exerts antifibrotic and anti-inflammatory effects in animal models of systemic sclerosis and inflammatory lung disease (ILD). SENSCIS (the Safety and Efficacy of Nintedanib in Systemic Sclerosis trial) investigated the molecule’s use in patients with ILD associated with systemic sclerosis.
Conducted in 32 countries, SENSCIS comprised 576 patients with the disorder, whose sclerosis affected at least 10% of their lungs. They were assigned to 52 weeks of either placebo or 150 mg nintedanib twice weekly. However, patients stayed on their blinded treatment until the last patient enrolled had finished the year of treatment; some patients took the drug for 100 weeks, Dr. Distler said. The primary endpoint was annual rate of decline in the forced vital capacity (FEV). Secondary endpoints included changes of the modified Rodnan skin score and in the total score on the St. George’s Respiratory Questionnaire.
Patients were a mean of 54 years old, with a mean disease duration of about 3 years. About half had diffuse cutaneous systemic sclerosis; the sclerosis was limited in the remainder. The mean extent of lung fibrosis was about 36%. Half were taking mycophenolate at baseline, which was allowed as background treatment, along with up to 10 mg/day of prednisone. Any patient who experienced clinically significant lung function deterioration could receive additional therapy at the investigator’s discretion.
The mean baseline FEV for these patients was 72.5% of predicted value. The mean diffusing capacity of the lungs for carbon monoxide was 53% of expected capacity.
Most patients completed the study (80% of the active group and 89% of the placebo group). The mean drug exposure duration was 10 months in the active group and 11 in the placebo group.
Improvement began early in treatment, with the efficacy curves separating by week 12 and continuing to diverge. After 52 weeks of therapy, the annual rate of change was 41 mL less in the active group than in the placebo group (–54.4 mL vs. –93.3 mL). The mean adjusted absolute change from baseline was –54.6 mL in the active group and –101 mL in the placebo at week 52. Significantly fewer patients taking nintedanib also lost more than 10% of FVC by week 52 (16.7% vs. 18%).
The St. George’s Respiratory Questionnaire score improved about one point in the active group and declined about one point in the placebo group.
Nintedanib was equally effective across a number of subgroups, including those divided by sex, age, and race. Antitopoisomerase antibodies and so-called antitopoisomerase I antibody status did not affect nintedanib’s action. Nintedanib also significantly improved scores on the Health Assessment Questionnaire without Disability Index and dyspnea.
More patients in the active group than in on placebo discontinued treatment because of a serious adverse event (16% vs. 8.7%). The most common of these were diarrhea (75.7% vs. 31%), nausea (31.6% vs. 13.5%), and vomiting (24.7% vs.10.4%). Skin ulcers occurred in about 18% of each group. Patients in the active group were significantly more likely to develop elevated alanine and aspartate aminotransferase of up to three times normal levels (4.9% vs. 0.7%).
Treatment did not significantly affect mortality rates, however. Over the treatment period, 10 patients in the nintedanib group and 9 in the placebo group died (3.5% vs. 3.1%).
The study was sponsored by Boehringer Ingelheim. Dr. Distler was the primary investigator on the trial.
SOURCE: Distler O et al. ATS 2019, Abstract A7360.
AT ATS 2019
Key clinical point: The tyrosine kinase inhibitor nintedanib may be a useful treatment for interstitial lung disease associated with systemic sclerosis (SS-ILD).
Major finding: Nintedanib decreased the annual rate of lung function decline by 44% among patients with SS-ILD.
Study details: The randomized, placebo-controlled study comprised 576 patients.
Disclosures: The trial was sponsored by Boehringer Ingelheim. Dr. Distler is the primary investigator.
Source: Distler O et al. ATS 2019, Abstract A7360.
Relatively high starting infliximab doses recommended for hidradenitis suppurativa
CHICAGO – In patients with hidradenitis suppurativa (HS), infliximab started at 7.5 mg/kg was highly effective and well tolerated and may be a more appropriate starting dose than the 5 mg/kg that is recommended in the treatment of plaque psoriasis, according to a chart review presented as a late breaker at the annual meeting of the Society for Investigative Dermatology.
_Ghias_web.jpg)
“Based on these data, we believe that a high starting dose of infliximab should be the new paradigm for this disease,” reported Justine Potemkin, a medical student at Albert Einstein College of Medicine, New York. The analysis was performed under the mentorship of Steven R. Cohen, MD, professor of dermatology and chief of the division of dermatology at Albert Einstein.
Infliximab, a tumor necrosis factor (TNF) blocker, is not approved for the treatment of HS, but it has been widely used following numerous reports of benefit in the literature, including a phase 2 randomized study published several years ago (J Am Acad Dermatol. 2010 Feb;62[2]:205-17), according to Ms. Potemkin.
“Based on dosing recommended for psoriasis, many hidradenitis suppurativa patients are started on 5 mg/kg, but doses of up to 10 mg/kg are within labeling for psoriasis, and our data support more aggressive initial treatment in patients with HS,” said Mondana Ghias, a research assistant in the department of dermatology at Albert Einstein, who was also involved in this study and joined Ms. Potemkin in presenting the data at the meeting.
In the chart review, 58 patients with moderate to severe HS initiated on 7.5 mg/kg of intravenous infliximab were identified. Due to a suboptimal response, 16 of these patients received 10 mg/kg infliximab in subsequent infusions, but the average response scores at 4 weeks compared favorably to previous reports with lower doses of infliximab, without increased toxicity.
“Starting with the 7.5 mg/kg dose with dose escalation if needed appears to provide a maximal response with minimal safety issues, based on our experience,” Ms. Potemkin reported.
At 4 weeks, a 10-point pain scale fell from a baseline mean of 5.65 to 1.34 (P less than .01). It reached 0.52 at week 12. The HS Physician Global Assessment (HS-PGA) fell from a baseline mean of 4.2 to 2.1 at week 4 (P less than .01) and then to 1.9 at week 12. The control of disease activity correlated with significant reductions in C-reactive protein and erythrocyte sedimentation rate.
The mean age in this patient population was 34.6 years. Slightly more than half (53.3%) were female. The average body mass index was 34.
“Obese patients also achieved a good response, which is consistent with weight-based dosing,” Ms. Potemkin reported.
The TNF blocker adalimumab is the only biologic and the only systemic therapy given regulatory approval for the treatment of HS, but Ms. Potemkin indicated that infliximab remains a widely used treatment option. She suggested that the responses observed in this patient series suggest infliximab is effective when used aggressively.
“No previous reports have examined the efficacy of high-dose infliximab,” Ms. Potemkin noted. “We think these findings are relevant to HS management.”
CHICAGO – In patients with hidradenitis suppurativa (HS), infliximab started at 7.5 mg/kg was highly effective and well tolerated and may be a more appropriate starting dose than the 5 mg/kg that is recommended in the treatment of plaque psoriasis, according to a chart review presented as a late breaker at the annual meeting of the Society for Investigative Dermatology.
“Based on these data, we believe that a high starting dose of infliximab should be the new paradigm for this disease,” reported Justine Potemkin, a medical student at Albert Einstein College of Medicine, New York. The analysis was performed under the mentorship of Steven R. Cohen, MD, professor of dermatology and chief of the division of dermatology at Albert Einstein.
Infliximab, a tumor necrosis factor (TNF) blocker, is not approved for the treatment of HS, but it has been widely used following numerous reports of benefit in the literature, including a phase 2 randomized study published several years ago (J Am Acad Dermatol. 2010 Feb;62[2]:205-17), according to Ms. Potemkin.
“Based on dosing recommended for psoriasis, many hidradenitis suppurativa patients are started on 5 mg/kg, but doses of up to 10 mg/kg are within labeling for psoriasis, and our data support more aggressive initial treatment in patients with HS,” said Mondana Ghias, a research assistant in the department of dermatology at Albert Einstein, who was also involved in this study and joined Ms. Potemkin in presenting the data at the meeting.
In the chart review, 58 patients with moderate to severe HS initiated on 7.5 mg/kg of intravenous infliximab were identified. Due to a suboptimal response, 16 of these patients received 10 mg/kg infliximab in subsequent infusions, but the average response scores at 4 weeks compared favorably to previous reports with lower doses of infliximab, without increased toxicity.
“Starting with the 7.5 mg/kg dose with dose escalation if needed appears to provide a maximal response with minimal safety issues, based on our experience,” Ms. Potemkin reported.
At 4 weeks, a 10-point pain scale fell from a baseline mean of 5.65 to 1.34 (P less than .01). It reached 0.52 at week 12. The HS Physician Global Assessment (HS-PGA) fell from a baseline mean of 4.2 to 2.1 at week 4 (P less than .01) and then to 1.9 at week 12. The control of disease activity correlated with significant reductions in C-reactive protein and erythrocyte sedimentation rate.
The mean age in this patient population was 34.6 years. Slightly more than half (53.3%) were female. The average body mass index was 34.
“Obese patients also achieved a good response, which is consistent with weight-based dosing,” Ms. Potemkin reported.
The TNF blocker adalimumab is the only biologic and the only systemic therapy given regulatory approval for the treatment of HS, but Ms. Potemkin indicated that infliximab remains a widely used treatment option. She suggested that the responses observed in this patient series suggest infliximab is effective when used aggressively.
“No previous reports have examined the efficacy of high-dose infliximab,” Ms. Potemkin noted. “We think these findings are relevant to HS management.”
CHICAGO – In patients with hidradenitis suppurativa (HS), infliximab started at 7.5 mg/kg was highly effective and well tolerated and may be a more appropriate starting dose than the 5 mg/kg that is recommended in the treatment of plaque psoriasis, according to a chart review presented as a late breaker at the annual meeting of the Society for Investigative Dermatology.
“Based on these data, we believe that a high starting dose of infliximab should be the new paradigm for this disease,” reported Justine Potemkin, a medical student at Albert Einstein College of Medicine, New York. The analysis was performed under the mentorship of Steven R. Cohen, MD, professor of dermatology and chief of the division of dermatology at Albert Einstein.
Infliximab, a tumor necrosis factor (TNF) blocker, is not approved for the treatment of HS, but it has been widely used following numerous reports of benefit in the literature, including a phase 2 randomized study published several years ago (J Am Acad Dermatol. 2010 Feb;62[2]:205-17), according to Ms. Potemkin.
“Based on dosing recommended for psoriasis, many hidradenitis suppurativa patients are started on 5 mg/kg, but doses of up to 10 mg/kg are within labeling for psoriasis, and our data support more aggressive initial treatment in patients with HS,” said Mondana Ghias, a research assistant in the department of dermatology at Albert Einstein, who was also involved in this study and joined Ms. Potemkin in presenting the data at the meeting.
In the chart review, 58 patients with moderate to severe HS initiated on 7.5 mg/kg of intravenous infliximab were identified. Due to a suboptimal response, 16 of these patients received 10 mg/kg infliximab in subsequent infusions, but the average response scores at 4 weeks compared favorably to previous reports with lower doses of infliximab, without increased toxicity.
“Starting with the 7.5 mg/kg dose with dose escalation if needed appears to provide a maximal response with minimal safety issues, based on our experience,” Ms. Potemkin reported.
At 4 weeks, a 10-point pain scale fell from a baseline mean of 5.65 to 1.34 (P less than .01). It reached 0.52 at week 12. The HS Physician Global Assessment (HS-PGA) fell from a baseline mean of 4.2 to 2.1 at week 4 (P less than .01) and then to 1.9 at week 12. The control of disease activity correlated with significant reductions in C-reactive protein and erythrocyte sedimentation rate.
The mean age in this patient population was 34.6 years. Slightly more than half (53.3%) were female. The average body mass index was 34.
“Obese patients also achieved a good response, which is consistent with weight-based dosing,” Ms. Potemkin reported.
The TNF blocker adalimumab is the only biologic and the only systemic therapy given regulatory approval for the treatment of HS, but Ms. Potemkin indicated that infliximab remains a widely used treatment option. She suggested that the responses observed in this patient series suggest infliximab is effective when used aggressively.
“No previous reports have examined the efficacy of high-dose infliximab,” Ms. Potemkin noted. “We think these findings are relevant to HS management.”
REPORTING FROM SID 2019

A 72-year-old white male with a history of psoriatic arthritis presented with a 1-year history of multiple, intermittently pruritic papules on his face and trunk
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.

The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.
The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].
Scleromyxedema
area, characteristically involving the glabella and ears. In some patients, the skin may be intensely pruritic, but this is not a universal finding and varies considerably among patients. In addition to affecting the skin, scleromyxedema has variable multisystem effects on the gastrointestinal tract, and musculoskeletal, pulmonary, cardiovascular, renal, and central nervous systems. The most common symptoms are proximal muscle weakness, dysphagia, and dyspnea on exertion. Scleromyxedema can also be associated with a paraproteinemia, mainly immunoglobulin G-lambda type.
Scleromyxedema shares some features with other cutaneous diseases, and the main differential diagnosis includes localized scleromyxedema, also known as lichen myxedematosus. Lichen myxedematosus presents with waxy, firm papules and plaques. Systemic involvement and monoclonal gammopathy are characteristically absent. Scleroderma differs given the increase in fibrosis of cutaneous lesions, a higher percentage of Raynaud’s phenomenon, prominent lung disease, and autoantibodies.
On histopathologic review, scleromyxedema is associated with papular and mucin deposition, and increased fibroblast proliferation. The punch biopsy of the exhibited patient demonstrated a dome-shaped dermal nodule composed of fibroblasts in an edematous stroma. An Alcian blue stain highlighted increased mucin in the dermis and S-100 staining highlighted rare cells in the dermis.
The treatment of choice for scleromyxedema varies but includes intravenous immunoglobulins, systemic glucocorticosteroids, thalidomide, or immunosuppressant medications. In this patient, an IgG paraproteinemia was found on serum protein electrophoresis. The patient was evaluated by hematology-oncology, and no underlying myeloproliferative or dysplastic disease was found. The patient was started on intravenous immunoglobulin infusions with near complete resolution of his eruption and arthritic symptoms.
This case and photo were submitted by Jennifer Maldonado, a medical student at Nova Southeastern University, Ft. Lauderdale, Fla., and Kate Oberlin, MD, and Brian Morrison, MD, of the department of dermatology and cutaneous surgery, University of Miami; and Michelle Demory Beckler, PhD, of the College of Osteopathic Medicine and the department of microbiology, College of Medical Sciences, Nova Southeastern University.
Dr. Bilu Martin is a board-certified dermatologist in private practice at Premier Dermatology, MD, in Aventura, Fla. More diagnostic cases are available at mdedge.com/dermatology. To submit a case for possible publication, send an email to [email protected].