User login
Sickle cell infusion gains FDA breakthrough designation
The in patients with sickle cell disease of all genotypes.

The designation allows the treatment to be reviewed on an expedited schedule.
Crizanlizumab, marketed by Novartis, is a humanized anti–P-selectin monoclonal antibody that has been shown to inhibit interactions between endothelial cells, platelets, red blood cells, sickled red blood cells, and leukocytes.
In the phase 2 SUSTAIN trial, crizanlizumab reduced the median annual rate of vasoocclusive crises that resulted in health care visits by about 45%, compared with placebo (1.63 vs. 2.98; P = .010). The drug also increased the percentage of patients who did not experience any vasoocclusive crises, compared with placebo (35.8% vs. 16.9%; P = .010).
The rates of treatment-emergent and serious adverse events was similar in the drug and placebo arms of the trial.
The in patients with sickle cell disease of all genotypes.
The designation allows the treatment to be reviewed on an expedited schedule.
Crizanlizumab, marketed by Novartis, is a humanized anti–P-selectin monoclonal antibody that has been shown to inhibit interactions between endothelial cells, platelets, red blood cells, sickled red blood cells, and leukocytes.
In the phase 2 SUSTAIN trial, crizanlizumab reduced the median annual rate of vasoocclusive crises that resulted in health care visits by about 45%, compared with placebo (1.63 vs. 2.98; P = .010). The drug also increased the percentage of patients who did not experience any vasoocclusive crises, compared with placebo (35.8% vs. 16.9%; P = .010).
The rates of treatment-emergent and serious adverse events was similar in the drug and placebo arms of the trial.
The in patients with sickle cell disease of all genotypes.
The designation allows the treatment to be reviewed on an expedited schedule.
Crizanlizumab, marketed by Novartis, is a humanized anti–P-selectin monoclonal antibody that has been shown to inhibit interactions between endothelial cells, platelets, red blood cells, sickled red blood cells, and leukocytes.
In the phase 2 SUSTAIN trial, crizanlizumab reduced the median annual rate of vasoocclusive crises that resulted in health care visits by about 45%, compared with placebo (1.63 vs. 2.98; P = .010). The drug also increased the percentage of patients who did not experience any vasoocclusive crises, compared with placebo (35.8% vs. 16.9%; P = .010).
The rates of treatment-emergent and serious adverse events was similar in the drug and placebo arms of the trial.
Poor-prognosis cancers linked to highest suicide risk in first year
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
Suicide risk significantly increases within the first year of a cancer diagnosis, with risk varying by type of cancer, according to investigators who conducted a retrospective analysis representing nearly 4.7 million patients.
Risk of suicide in that first year after diagnosis was especially high in pancreatic and lung cancers, while by contrast, breast and prostate cancer did not increase suicide risk, reported the researchers, led by Hesham Hamoda, MD, MPH, of Boston Children’s Hospital/Harvard Medical School, and Ahmad Alfaar, MBBCh, MSc, of Charité–Universitätsmedizin Berlin.
That variation in suicide risk by cancer type suggests that prognosis and 5-year relative survival play a role in increasing suicide rates, according to Dr. Hamoda, Dr. Alfaar, and their coauthors.
“After the diagnosis, it is important that health care providers be vigilant in screening for suicide and ensuring that patients have access to social and emotional support,” they wrote in a report published in Cancer. Their analysis was based on 4,671,989 patients with a diagnosis of cancer in the Surveillance, Epidemiology, and End Results (SEER) database between 2000 and 2014. Out of 1,005,825 of those patients who died within the first year of diagnosis, the cause of death was suicide for 1,585, or 0.16%.
Overall, the risk of suicide increased significantly among cancer patients versus the general population, with an observed-to-expected (O/E) ratio of 2.51 per 10,000 person-years, the investigators found. The risk was highest in the first 6 months, with an O/E mortality of 3.13 versus 1.8 in the latter 6 months.
The highest ratios were seen for pancreatic cancer, with an O/E ratio of 8.01, and lung cancer, with a ratio of 6.05, the researchers found in further analysis.
Significant increases in suicide risk were also seen for colorectal cancer (2.08) and melanoma (1.45), though rates were not significantly different versus the general population for breast (1.23) and prostate (0.99), according to the reported data.
Suicide risk was relatively high for any cancer with distant metastases (5.63), though still significantly higher at 1.65 in persons with localized/regional disease, the data show.
The increased suicide risk persisted more than 1 year after the cancer diagnosis, though not to the degree observed within that first year, they added.
Most patients with suicide as a cause of death were white (90.2%) and male (87%). Nearly 60% were between the ages of 65 and 84 at the time of suicide.
Social support plays an integral role in suicide prevention among cancer patients, the researchers noted.
Previous studies suggest that support programs may decrease suicide risk by making patients better aware of their prognosis, receptive to decreased social stigma, or less likely to have stress related to cost of care, they said.
“Discussing the quality of life after diagnosis, the effectiveness of therapy, and the prognosis of the disease and maintaining a trusting relationship with health care professionals all decrease the likelihood of suicide immediately after a diagnosis of cancer,” they said.
Dr. Hamoda, Dr. Alfaar, and their coauthors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service (Dr. Alfaar).
SOURCE: Saad AM, et al. Cancer 2019 Jan 7. doi: 10.1002/cncr.31876.
FROM CANCER
Key clinical point: A cancer diagnosis significantly increases risk of suicide in comparison to the general population, particularly for poorer-prognosis cancers.
Major finding: The observed-to-expected mortality ratio was substantially higher for pancreatic cancer (8.01), and lung cancer (6.05), but not significantly increased for breast (1.23) and prostate (0.99).
Study details: A retrospective population-based study of 4,671,989 cancer patients.
Disclosures: The authors reported no conflicts of interest. Funding for the study came in part from the German Academic Exchange Service.
Source: Saad AM et al. Cancer. 2019 Jan 7. doi: 10.1002/cncr.31876.
Tests can identify leukemia risk in newborns with Down syndrome
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.

“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.
“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
SAN DIEGO – Research into hundreds of babies with Down syndrome is providing valuable insight into the genetic roots of leukemia and offering a route to identify newborns at high risk.
“We can now identify children at high risk of developing myeloid leukemia within 4 years” through blood or genetic tests, Irene Roberts, MD, a pediatric hematologist at the University of Oxford’s (England) MRC Weatherall Institute of Molecular Medicine, said at the annual meeting of the American Society of Hematology.
About 2%-3% of children with Down syndrome will develop acute lymphocytic leukemia (ALL) or acute myeloid leukemia (AML), according to the National Cancer Institute, rates that are much higher than in the general population.
Research suggests that among children aged 0-4 years with Down syndrome, the standardized incidence ratio (SIR) of AML is 114, compared with other children, Dr. Roberts said. The SIR of ALL is 27 in children aged 1-4 years, she said.
For people with Down syndrome aged 0-60 years, the SIRs are 12 and 13 in AML and ALL, respectively, she said.
In her presentation, Dr. Roberts focused on AML that appears before age 4 years and is preceded by a neonatal preleukemia – transient abnormal myelopoiesis (TAM) – that only occurs in Down syndrome. In most cases, TAM, which occurs with GATA1 mutations, resolves on its own after birth, she said. But in others, the GATA1 mutations continue and cause AML to develop.
Dr. Roberts highlighted her institution’s Oxford Down Syndrome Cohort Study and offered an update to a 2013 report (Blood. 2013 Dec 5;122[24]:3908–17). The study recruited 471 neonates with Down syndrome and followed them for up to 4 years: 341 with no GATA1 mutation and 130 (28%) with the mutation. Dr. Roberts called the latter number a “very high frequency.”
Of those with the mutation, 7 patients (5%) developed AML at a median age of 16 months. None of those without the mutation developed AML.
Also, among the 130 neonates with the mutation, 42% were considered to have “clinical” TAM (more than 10% blasts) and 58% were considered to have “silent” TAM (fewer than 10% blasts).
“We predicted that these babies with clinical TAM would have more severe clinical disease ... and that in fact turned out to be the case,” Dr. Roberts said.
Why is the GATA1 mutation so significant? Research suggests that platelet production is abnormal in neonates with Down syndrome, compared with neonates without it, regardless of whether they have the mutation, Dr. Roberts said.
The mutation doesn’t reduce further platelet count, but does disrupt megakaryopoiesis – the process of the production of platelets. As a result, giant platelets and megakaryocyte fragments are more common, she explained.
Moving forward, research data can be used to identify which children are most at risk, Dr. Roberts said. Newborns with Down syndrome are more likely to survive without leukemia if they have silent TAM, compared with those who have clinical TAM, and if they have an estimated variant allele frequency above 15%, according to findings from the Oxford study.
Children at high risk of AML before age 4 years can be identified by analyzing the percentage of blasts on a smear and/or by analyzing mutation of GATA1, according to Dr. Roberts. However, this cannot be accomplished by the use of a complete blood count (CBC) test, she said, which is used to check for leukemia.
Dr. Roberts called for the development of more guidelines for screening newborns with Down syndrome for leukemia risk. The British Society for Haematology issued testing guidelines, coauthored by Dr. Roberts, in 2018 (Br J Haematol. 2018 Jul;182[2]:200-11).
Dr. Roberts reported having no financial disclosures.
EXPERT ANALYSIS FROM ASH 2018
ALL chemotherapy looks effective in mixed phenotype leukemia
SAN DIEGO – The majority of pediatric patients with mixed phenotype acute leukemia (MPAL) who were treated with acute lymphoblastic leukemia (ALL)–directed chemotherapy achieved a minimum residual disease (MRD)–negative complete response by the end of consolidation, according to findings from a multicenter retrospective cohort study.

The cohort included 94 patients aged 1-21 years who met strict World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions. Most had B/myeloid phenotype (89%), and 87 patients were treated with an ALL regimen, Etan Orgel, MD, reported at the annual meeting of the American Society of Hematology.
Of those 87 patients, 81 (93%) experienced an end-of-induction (EOI) complete response. One patient died during induction and six had induction failures, defined as either disease progression before EOI (two patients) or EOI MRD of 5% or greater (three patients), said Dr. Orgel of the University of Southern California, Los Angeles, and Children’s Hospital Los Angeles.
The MRD-negative rates, defined as MRD less than 0.01%, were 70% at EOI and 86% at EOI or end of consolidation (EOC); 12 of 14 patients who were MRD positive at EOI and continued on ALL therapy achieved an EOC MRD-negative complete response, including 8 of 8 with EOI MRD of 0.01%-0.09% and 4 of 6 with EOI MRD of 1% or greater.
Event-free survival at 5 years in the 78 patients without hematopoietic stem cell transplant at first remission was 75%, and 5-year overall survival was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said. “This is very different from the approach used at many adult centers and many of the adult recommendations.”
Overall 5-year EOI event-free survival was 80% in the 59 patients who were MRD negative at EOI, and 13% in 25 patients who were MRD-positive at EOI. The corresponding overall survival rates were 91% and 84%.
Overall 5-year EOC event-free survival was 77% in 74 patients who were MRD negative at EOC and was unavailable in 3 patients who were MRD positive at EOC, although all three were salvaged. The corresponding EOC overall survival rates were 89% and “not available,” Dr. Orgel reported.
Multivariable analysis confirmed the predictive value of MRD at EOI (hazard ratio for event-free survival and overall survival, 3.77 and 3.54, respectively).
Of note, there was a possible trend toward earlier failure and a trend toward worse overall survival (HR, 4.49, P = .074) for T-lineage–containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” he said.
MRD in pediatric MPAL is rare. Recent studies of MPAL biology show areas of similarity with ALL and AML, and while this could eventually help further subcategorize or classify the disease and lead to biology-driven therapies, it is important to know how to treat the disease today, Dr. Orgel said.
The evolving consensus is that ALL therapy is adequate for most MPAL, but there is no established threshold for MRD to enable a risk-stratified MPAL approach, he added.
The current findings suggest that ALL therapy – without hematopoietic stem cell transplant – may be sufficient to treat most patients with pediatric MPAL, Dr. Orgen reported, noting that clinical trials are necessary to prospectively validate MRD thresholds at EOI and EOC and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” he said.
Dr. Orgel reported having no financial disclosures.
SOURCE: Oberley M et al. ASH 2018, Abstract 558.
SAN DIEGO – The majority of pediatric patients with mixed phenotype acute leukemia (MPAL) who were treated with acute lymphoblastic leukemia (ALL)–directed chemotherapy achieved a minimum residual disease (MRD)–negative complete response by the end of consolidation, according to findings from a multicenter retrospective cohort study.
The cohort included 94 patients aged 1-21 years who met strict World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions. Most had B/myeloid phenotype (89%), and 87 patients were treated with an ALL regimen, Etan Orgel, MD, reported at the annual meeting of the American Society of Hematology.
Of those 87 patients, 81 (93%) experienced an end-of-induction (EOI) complete response. One patient died during induction and six had induction failures, defined as either disease progression before EOI (two patients) or EOI MRD of 5% or greater (three patients), said Dr. Orgel of the University of Southern California, Los Angeles, and Children’s Hospital Los Angeles.
The MRD-negative rates, defined as MRD less than 0.01%, were 70% at EOI and 86% at EOI or end of consolidation (EOC); 12 of 14 patients who were MRD positive at EOI and continued on ALL therapy achieved an EOC MRD-negative complete response, including 8 of 8 with EOI MRD of 0.01%-0.09% and 4 of 6 with EOI MRD of 1% or greater.
Event-free survival at 5 years in the 78 patients without hematopoietic stem cell transplant at first remission was 75%, and 5-year overall survival was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said. “This is very different from the approach used at many adult centers and many of the adult recommendations.”
Overall 5-year EOI event-free survival was 80% in the 59 patients who were MRD negative at EOI, and 13% in 25 patients who were MRD-positive at EOI. The corresponding overall survival rates were 91% and 84%.
Overall 5-year EOC event-free survival was 77% in 74 patients who were MRD negative at EOC and was unavailable in 3 patients who were MRD positive at EOC, although all three were salvaged. The corresponding EOC overall survival rates were 89% and “not available,” Dr. Orgel reported.
Multivariable analysis confirmed the predictive value of MRD at EOI (hazard ratio for event-free survival and overall survival, 3.77 and 3.54, respectively).
Of note, there was a possible trend toward earlier failure and a trend toward worse overall survival (HR, 4.49, P = .074) for T-lineage–containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” he said.
MRD in pediatric MPAL is rare. Recent studies of MPAL biology show areas of similarity with ALL and AML, and while this could eventually help further subcategorize or classify the disease and lead to biology-driven therapies, it is important to know how to treat the disease today, Dr. Orgel said.
The evolving consensus is that ALL therapy is adequate for most MPAL, but there is no established threshold for MRD to enable a risk-stratified MPAL approach, he added.
The current findings suggest that ALL therapy – without hematopoietic stem cell transplant – may be sufficient to treat most patients with pediatric MPAL, Dr. Orgen reported, noting that clinical trials are necessary to prospectively validate MRD thresholds at EOI and EOC and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” he said.
Dr. Orgel reported having no financial disclosures.
SOURCE: Oberley M et al. ASH 2018, Abstract 558.
SAN DIEGO – The majority of pediatric patients with mixed phenotype acute leukemia (MPAL) who were treated with acute lymphoblastic leukemia (ALL)–directed chemotherapy achieved a minimum residual disease (MRD)–negative complete response by the end of consolidation, according to findings from a multicenter retrospective cohort study.
The cohort included 94 patients aged 1-21 years who met strict World Health Organization MPAL criteria and were treated between 2008 and 2016 at one of six U.S. institutions. Most had B/myeloid phenotype (89%), and 87 patients were treated with an ALL regimen, Etan Orgel, MD, reported at the annual meeting of the American Society of Hematology.
Of those 87 patients, 81 (93%) experienced an end-of-induction (EOI) complete response. One patient died during induction and six had induction failures, defined as either disease progression before EOI (two patients) or EOI MRD of 5% or greater (three patients), said Dr. Orgel of the University of Southern California, Los Angeles, and Children’s Hospital Los Angeles.
The MRD-negative rates, defined as MRD less than 0.01%, were 70% at EOI and 86% at EOI or end of consolidation (EOC); 12 of 14 patients who were MRD positive at EOI and continued on ALL therapy achieved an EOC MRD-negative complete response, including 8 of 8 with EOI MRD of 0.01%-0.09% and 4 of 6 with EOI MRD of 1% or greater.
Event-free survival at 5 years in the 78 patients without hematopoietic stem cell transplant at first remission was 75%, and 5-year overall survival was 89%, “thus demonstrating that, for a majority of patients, transplant in first remission may not be necessary,” Dr. Orgel said. “This is very different from the approach used at many adult centers and many of the adult recommendations.”
Overall 5-year EOI event-free survival was 80% in the 59 patients who were MRD negative at EOI, and 13% in 25 patients who were MRD-positive at EOI. The corresponding overall survival rates were 91% and 84%.
Overall 5-year EOC event-free survival was 77% in 74 patients who were MRD negative at EOC and was unavailable in 3 patients who were MRD positive at EOC, although all three were salvaged. The corresponding EOC overall survival rates were 89% and “not available,” Dr. Orgel reported.
Multivariable analysis confirmed the predictive value of MRD at EOI (hazard ratio for event-free survival and overall survival, 3.77 and 3.54, respectively).
Of note, there was a possible trend toward earlier failure and a trend toward worse overall survival (HR, 4.49, P = .074) for T-lineage–containing MPAL.
“That indicates that this might be a group that needs careful scrutiny of which form of ALL therapy they receive,” he said.
MRD in pediatric MPAL is rare. Recent studies of MPAL biology show areas of similarity with ALL and AML, and while this could eventually help further subcategorize or classify the disease and lead to biology-driven therapies, it is important to know how to treat the disease today, Dr. Orgel said.
The evolving consensus is that ALL therapy is adequate for most MPAL, but there is no established threshold for MRD to enable a risk-stratified MPAL approach, he added.
The current findings suggest that ALL therapy – without hematopoietic stem cell transplant – may be sufficient to treat most patients with pediatric MPAL, Dr. Orgen reported, noting that clinical trials are necessary to prospectively validate MRD thresholds at EOI and EOC and to establish the threshold for favorable survival.
“Future research should explore either intensification of therapy or different therapies for patients with persistent MRD,” he said.
Dr. Orgel reported having no financial disclosures.
SOURCE: Oberley M et al. ASH 2018, Abstract 558.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: MRD-negative rates were 70% at end of induction and 86% at end of induction or consolidation.
Study details: A retrospective cohort study of 87 pediatric MPAL patients.
Disclosures: Dr. Orgel reported having no financial disclosures.
Source: Oberley M et al. ASH 2018, Abstract 558.
Immediate acting inhibitors complicate hemophilia A diagnosis
A small but substantial proportion of patients with hemophilia A develop immediate acting factor VIII inhibitors, the diversity and complexity of which create a diagnostic challenge in the laboratory, according to authors of a recent observational study.

The great majority of the inhibitor-positive patients in the 4,900-patient study had classical FVIII inhibitors, which are typically time- and temperature-dependent and react more slowly in mixing studies, the researchers reported.
By contrast, about 1 in 10 patients demonstrated immediate acting inhibitors, and of those, some had lupus anticoagulants, some had factor VIII inhibitors, and some had both, according to Shrimati Shetty, PhD, of the National Institute of Immunohaematology in Mumbai, India, and her colleagues.
“There is a possibility of misdiagnosis of the patient when they present for the first time,” the researchers wrote. The report is in Thrombosis Research.
In the case of immediate-acting inhibitors, use of ELISA or chromogenic assays alongside lupus anticoagulant testing may help clarify the diagnosis; however, those tests are costly and may not be routinely available.
In the study by Dr. Shetty and her colleagues, patients in India with congenital hemophilia were initially screened for inhibitors. A total of 451 were found to be positive, and of those, 398 were observed to have classical factor VIII inhibitors, while the remaining 53 had immediate-acting inhibitors.
Looking specifically at hemophilia A patients with immediate-acting inhibitors, which comprised 48 of those 53 patients, the majority, or 42 patients, were positive for lupus anticoagulants, and of those, 38 were positive for both lupus anticoagulants and factor VIII inhibitors, while 4 patients were positive for lupus anticoagulants only.
“These are a heterogeneous group of antibodies interfering with all phospholipid dependent reactions,” the researchers wrote.
Properly interpreting factor inhibitor assays is an important step that helps guide later management of inhibitor-positive patients, according to Dr. Shetty and her coauthors.
“Once the patients become positive for inhibitors, they have to opt for alternate modalities of treatment, i.e. bypassing agents like activated prothrombin complex concentrate and activated recombinant factor VII, which are much more expensive,” they wrote.
In light of the diagnostic difficulties they highlighted, Dr. Shetty and her coauthors recommended a “systematic approach” to testing. Both factor VIII and factor IX assays need to be conducted, along with a lupus anticoagulant test. For inhibitor titer, either chromogenic assays or ELISA tests are recommended, they wrote.
Dr. Shetty and her coauthors reported that they had no conflicts of interest.
SOURCE: Patil R et al. Thromb Res. 2018 Dec;172:29-35.
A small but substantial proportion of patients with hemophilia A develop immediate acting factor VIII inhibitors, the diversity and complexity of which create a diagnostic challenge in the laboratory, according to authors of a recent observational study.
The great majority of the inhibitor-positive patients in the 4,900-patient study had classical FVIII inhibitors, which are typically time- and temperature-dependent and react more slowly in mixing studies, the researchers reported.
By contrast, about 1 in 10 patients demonstrated immediate acting inhibitors, and of those, some had lupus anticoagulants, some had factor VIII inhibitors, and some had both, according to Shrimati Shetty, PhD, of the National Institute of Immunohaematology in Mumbai, India, and her colleagues.
“There is a possibility of misdiagnosis of the patient when they present for the first time,” the researchers wrote. The report is in Thrombosis Research.
In the case of immediate-acting inhibitors, use of ELISA or chromogenic assays alongside lupus anticoagulant testing may help clarify the diagnosis; however, those tests are costly and may not be routinely available.
In the study by Dr. Shetty and her colleagues, patients in India with congenital hemophilia were initially screened for inhibitors. A total of 451 were found to be positive, and of those, 398 were observed to have classical factor VIII inhibitors, while the remaining 53 had immediate-acting inhibitors.
Looking specifically at hemophilia A patients with immediate-acting inhibitors, which comprised 48 of those 53 patients, the majority, or 42 patients, were positive for lupus anticoagulants, and of those, 38 were positive for both lupus anticoagulants and factor VIII inhibitors, while 4 patients were positive for lupus anticoagulants only.
“These are a heterogeneous group of antibodies interfering with all phospholipid dependent reactions,” the researchers wrote.
Properly interpreting factor inhibitor assays is an important step that helps guide later management of inhibitor-positive patients, according to Dr. Shetty and her coauthors.
“Once the patients become positive for inhibitors, they have to opt for alternate modalities of treatment, i.e. bypassing agents like activated prothrombin complex concentrate and activated recombinant factor VII, which are much more expensive,” they wrote.
In light of the diagnostic difficulties they highlighted, Dr. Shetty and her coauthors recommended a “systematic approach” to testing. Both factor VIII and factor IX assays need to be conducted, along with a lupus anticoagulant test. For inhibitor titer, either chromogenic assays or ELISA tests are recommended, they wrote.
Dr. Shetty and her coauthors reported that they had no conflicts of interest.
SOURCE: Patil R et al. Thromb Res. 2018 Dec;172:29-35.
A small but substantial proportion of patients with hemophilia A develop immediate acting factor VIII inhibitors, the diversity and complexity of which create a diagnostic challenge in the laboratory, according to authors of a recent observational study.
The great majority of the inhibitor-positive patients in the 4,900-patient study had classical FVIII inhibitors, which are typically time- and temperature-dependent and react more slowly in mixing studies, the researchers reported.
By contrast, about 1 in 10 patients demonstrated immediate acting inhibitors, and of those, some had lupus anticoagulants, some had factor VIII inhibitors, and some had both, according to Shrimati Shetty, PhD, of the National Institute of Immunohaematology in Mumbai, India, and her colleagues.
“There is a possibility of misdiagnosis of the patient when they present for the first time,” the researchers wrote. The report is in Thrombosis Research.
In the case of immediate-acting inhibitors, use of ELISA or chromogenic assays alongside lupus anticoagulant testing may help clarify the diagnosis; however, those tests are costly and may not be routinely available.
In the study by Dr. Shetty and her colleagues, patients in India with congenital hemophilia were initially screened for inhibitors. A total of 451 were found to be positive, and of those, 398 were observed to have classical factor VIII inhibitors, while the remaining 53 had immediate-acting inhibitors.
Looking specifically at hemophilia A patients with immediate-acting inhibitors, which comprised 48 of those 53 patients, the majority, or 42 patients, were positive for lupus anticoagulants, and of those, 38 were positive for both lupus anticoagulants and factor VIII inhibitors, while 4 patients were positive for lupus anticoagulants only.
“These are a heterogeneous group of antibodies interfering with all phospholipid dependent reactions,” the researchers wrote.
Properly interpreting factor inhibitor assays is an important step that helps guide later management of inhibitor-positive patients, according to Dr. Shetty and her coauthors.
“Once the patients become positive for inhibitors, they have to opt for alternate modalities of treatment, i.e. bypassing agents like activated prothrombin complex concentrate and activated recombinant factor VII, which are much more expensive,” they wrote.
In light of the diagnostic difficulties they highlighted, Dr. Shetty and her coauthors recommended a “systematic approach” to testing. Both factor VIII and factor IX assays need to be conducted, along with a lupus anticoagulant test. For inhibitor titer, either chromogenic assays or ELISA tests are recommended, they wrote.
Dr. Shetty and her coauthors reported that they had no conflicts of interest.
SOURCE: Patil R et al. Thromb Res. 2018 Dec;172:29-35.
FROM THROMBOSIS RESEARCH
Key clinical point:
Major finding: Of 48 inhibitor-positive hemophilia A patients with immediate acting inhibitors, 42 were positive for lupus anticoagulants.
Study details: An analysis of 4,900 patients in India with confirmed or suspected congenital hemophilia.
Disclosures: The authors reported that they had no conflicts of interest.
Source: Patil R et al. Thromb Res. 2018 Dec;172:29-35.
Monoclonal gammopathy of undetermined significance: A primary care guide


MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.

A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?

Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.

Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?

that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.

Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.

The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.
A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?
Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.
Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?
that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.
Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.
The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
MGUS is present in 3% to 4% of the population over age 50 and is more common in older men, African Americans, and Africans.1–6
The overall risk of progression to myeloma and related disorders is less than or equal to 1% per year depending on the subtype of the M protein (higher risk with IgM than non-IgM and light-chain MGUS).7,8 While the risk of malignant transformation is low, multiple myeloma is almost always preceded by the presence of an asymptomatic and often unrecognized monoclonal protein.
WHEN SHOULD WE LOOK FOR AN M PROTEIN?
An M protein is typically an incidental finding when a patient is being assessed for any of a number of presenting symptoms or conditions. A large retrospective study9 found that screening for MGUS was mostly performed by internal medicine physicians. The indications for testing were anemia, bone-related issues, elevated creatinine, elevated erythrocyte sedimentation rate, and neuropathy.
A low anion gap is not a major indicator of an M protein unless in a high concentration, in which case other manifestations would be present, such as renal failure, which would guide the diagnosis. Polyclonal hypergammaglobulinemia as a cause of low anion gap is far more common than MGUS.
HOW SHOULD WE SCREEN FOR AN M PROTEIN?
Serum protein electrophoresis is an initial test used to identify an M protein and has a key role in quantifying it (Figure 2). An M protein appears as a narrow spike on the agarose gel and should be distinguished from the broad band seen in polyclonal gammopathies associated with cirrhosis and chronic infectious and inflammatory conditions, among others.12 A major disadvantage of serum protein electrophoresis is that it cannot detect an M protein in very low concentrations or determine its identity.
Serum immunofixation is more sensitive than serum protein electrophoresis and should always be ordered in conjunction with it, mostly to ensure detecting tiny amounts of M protein and to identify the type of its heavy chain and light-chain components.13
The serum free light-chain assay is also considered an essential part of the screening process to detect light-chain MGUS and light-chain myeloma. As many as 16% of myeloma patients secrete only light chains, which may not be identified on serum immunofixation.3,6,7,10,14,15 In general, a low kappa-lambda ratio (< 0.26) indicates the overproduction of lambda light chains, and a high ratio (> 1.65) indicates the overproduction of kappa light chains.
The serum free light-chain assay helps detect abnormal secretion of monoclonal light chains before they appear in the urine once the kidney tubules become saturated and unable to reabsorb them.
Of note, the free light-chain ratio can be abnormal (< 0.26 or > 1.65) in chronic kidney disease. Thus, it may be challenging to discern whether an abnormal light-chain ratio is related to impaired light-chain clearance by the kidneys or to MGUS. In general, kappa light chains are more elevated than lambda light chains in chronic kidney disease, but the ratio should not be considerably skewed. A kappa-lambda ratio below 0.37 or above 3 is rarely seen in chronic kidney disease and should prompt workup for MGUS.16
Tests in combination. The sensitivity of screening for M proteins ranges from 82% with serum protein electrophoresis alone to 93% with the addition of serum immunofixation and to 98% with the serum free light-chain assay.15 The latter can replace urine protein electrophoresis and immunofixation when screening for M protein, given its higher sensitivity.15,17 An important caveat is that urine dipstick testing does not detect urine light chains.
Table 3 lists the initial laboratory tests required in patients with MGUS.
WHAT IS THE DIFFERENTIAL DIAGNOSIS OF MONOCLONAL GAMMOPATHIES?
that feature an M protein and would otherwise require treatment (Table 4). The differential diagnosis includes smoldering multiple myeloma, symptomatic multiple myeloma, Waldenström macroglobulinemia, light-chain amyloidosis, low-grade B-cell lymphoproliferative disorders, a variety of monoclonal protein-related kidney disorders, and plasmacytomas.10,14
MGUS
Based on the International Myeloma Working Group consensus, a formal diagnosis of MGUS is established when a serum M protein is detected and measured at a concentration less than 3 g/dL on serum protein electrophoresis along with less than 10% clonal plasma cells in the bone marrow.1–6,14,18,19 Nevertheless, bone marrow biopsy can be omitted in certain patients as discussed below. The absence of myeloma-related organ damage—particularly osteolytic bone lesions, anemia, otherwise unexplained renal failure, and hypercalcemia—is fundamental and necessary for a diagnosis of MGUS.
Smoldering multiple myeloma
Compared with patients with MGUS, patients with smoldering multiple myeloma have higher M protein concentrations (≥ 3 g/dL) or 10% or more clonal plasma cells in the marrow or both, and are at higher risk of progression to symptomatic multiple myeloma. Nevertheless, like patients with MGUS, they have no myeloma symptoms or evidence of end-organ damage.
Symptomatic multiple myeloma
By definition, patients with multiple myeloma develop organ damage related to their malignancy and need therapy to halt disease progression. Multiple myeloma causes clinical manifestations through cellular infiltration of the bone and bone marrow (anemia, osteolysis, and hypercalcemia) and light chain-induced toxicity (renal tubular damage and cast nephropathy).
In 2014, the definition of multiple myeloma was updated to include 3 new myeloma-defining events that herald a significantly higher risk of progression from smoldering to symptomatic multiple myeloma, and now constitute an integral part of the diagnosis of symptomatic multiple myeloma. These are:
- Focal lesions (> 1 lesion larger than 5 mm) visible on magnetic resonance imaging
- ≥ 60% clonal plasma cells on bone marrow biopsy
- Ratio of involved to uninvolved serum free light chains ≥ 100 (the involved light chain is the one detected on serum protein electrophoresis and immunofixation).14
Bone pain, symptoms of anemia, and decreased urine output may suggest myeloma, but are not diagnostic. Although the “CRAB” criteria (elevated calcium, renal failure, anemia, and bone lesions) define multiple myeloma, the presence of anemia, hypercalcemia, or renal dysfunction do not by themselves mark transformation from MGUS to multiple myeloma. Thus, other causes need to be considered, since the risk of transformation is so low. Importantly, hyperparathyroidism must be ruled out if hypercalcemia is present in a patient with MGUS.10
Waldenström macroglobulinemia
Waldenström macroglobulinemia, also called lymphoplasmacytic lymphoma, is an indolent non-Hodgkin B-cell lymphoma that can invade the marrow, liver, spleen, and lymph nodes, leading to anemia and organomegaly. It features a monoclonal IgM protein that can be associated with increased blood viscosity, cold agglutinin disease, peripheral neuropathy, and cryoglobulinemia.
Waldenström macroglobulinemia should be suspected in any patient with IgM type M protein and symptoms related to hyperviscosity (headache, blurry vision, lightheadedness, shortness of breath, unexplained epistaxis, gum bleeding); systemic symptoms (fever, weight loss, and night sweats); and abdominal pain (due to organomegaly).23
Monoclonal gammopathy of renal significance
Monoclonal gammopathy of renal significance (MGRS) is a newly recognized entity defined by kidney dysfunction associated with an M protein without evidence of myeloma or other lymphoid disorders.24 Multiple disorders have been included in this category with different underlying mechanisms of kidney injury. This entity is beyond the scope of this discussion.
Light-chain amyloidosis
Misfolded light-chain deposition leading to organ dysfunction is the hallmark of light-chain amyloidosis, which constitutes a subset of MGRS. An abnormal light-chain ratio, especially if skewed toward lambda should trigger an investigation for light-chain amyloidosis.10
Abnormal light chains may infiltrate any organ or tissue, but of greatest concern is infiltration of the myocardium with ensuing heart failure manifestations. N-terminal pro-B-type natriuretic peptide (NT-proBNP) is a sensitive marker for cardiac amyloidosis in the presence of suggestive features on transthoracic echocardiography (eg, left ventricular hypertrophy) but is not specific as it can be elevated in heart failure regardless of the underlying cause.10
Glomerular injury with nephrotic syndrome may also point toward renal involvement by light-chain amyloidosis and establishes a key distinctive factor from myeloma in which tubular injury is the main mechanism of kidney dysfunction.
Clinical clues for light-chain amyloidosis include heart failure symptoms, neuropathy, and macroglossia. If any of these symptoms and signs is present, we recommend electrocardiography (look for low voltage in limb leads), transthoracic echocardiography, measuring the NT-proBNP level, and urinalysis to look for albuminuria. Notably, carpal tunnel syndrome may be a very early clinical manifestation of amyloidosis, but by itself it is nonspecific. Light-chain amyloidosis is a common cause of macroglossia in adults.10,25
Neuropathy associated with M proteins is a clinical entity related to a multitude of disorders that may necessitate treating the underlying cellular clone responsible for the secretion of the toxic M protein. These disorders include light-chain amyloidosis, POEMS (polyneuropathy, organomegaly, endocrinopathy, M protein, and skin changes or sclerotic bone lesions) syndrome, and IgM-related neuropathies with anti-myelin-associated glycoprotein antibodies.3,10,11,14
Notably, weight loss and fatigue in a patient with MGUS may be the first signs of light-chain amyloidosis or Waldenström macroglobulinemia and should prompt further evaluation.25
HOW ARE PATIENTS WITH MGUS RISK-STRATIFIED AND FOLLOWED?
Research has helped to refine the diagnostic workup and recognize subsets of patients with MGUS at different risks of progression to myeloma and related disorders. Factors predicting progression are 1,6,7,26,27:
- The amount of the M protein
- The type of M protein (IgG vs non-IgG)
- An abnormal free light-chain ratio.
Half of patients with MGUS fall into the low-risk category, which is defined by IgG-type serum M protein in a concentration less than 1.5 g/dL and a normal serum free light-chain ratio (kappa-lambda 0.26–1.65).5,27 The absolute risk of progression at 20 years is only 5% for patients with low-risk MGUS, compared with 58% in patients with high-risk MGUS (positive for all 3 risk factors).5
The presence of less than 10% plasma cells in the bone marrow is required to satisfy the definition of MGUS, but bone marrow biopsy can be omitted for patients with low-risk MGUS, given the slim chance of finding a significant percentage of clonal plasma cells in the marrow and the inherently low risk of progression.5,10 Skeletal surveys are often deferred for low-risk MGUS, but we obtain them in all our patients to ensure the absence of plasmacytomas, which need to be treated (typically with radiotherapy). Importantly, patients with unexplained bone pain (mostly in long bones, ribs, and spine, whereas joints are not typically involved) and a normal skeletal survey should undergo advanced imaging (whole-body magnetic resonance imaging or whole-body positron emission tomography and computed tomography) to detect bone lesions otherwise missed on plain radiography.28,29
Most of the recommendations regarding follow-up are based on expert opinion, given the lack of randomized data. Most experts agree that all patients should be reevaluated 6 months after an M protein is detected, with laboratory surveillance tests (complete blood cell count, serum creatinine, serum calcium level, serum protein electrophoresis, and serum free light chains). Low-risk patients with a stable M protein level can be followed every 2 to 3 years.
Suspect malignant progression if the serum M protein level increases by 50% or more (with an absolute increase of ≥ 0.5 g/dL); the serum M protein level is 3 g/dL or higher; the serum free light-chain ratio is more than 100; or the patient has unexplained anemia, elevated creatinine, bone pain, fracture, or hypercalcemia.
Patients at intermediate or high risk should be followed annually after the initial 6-month visit.5,7,10
A recent study highlighted the importance of risk stratification in reducing the costs associated with an overzealous diagnostic workup of patients with low-risk MGUS.30 These savings are in addition to a reduction in patient anticipation and anxiety that universally occur before invasive procedures.
THE ROLE OF THE PRIMARY CARE PROVIDER AND THE HEMATOLOGIST
Once an M protein is identified, a comprehensive history, physical examination, and laboratory tests (serum protein electrophoresis to quantify the protein, serum immunofixation, serum free light chains, complete blood cell count, calcium, and creatinine) should be done, taking into consideration the differential diagnosis of monoclonal gammopathies discussed above. After MGUS is confirmed, the patient should be risk-stratified to determine the need for bone marrow biopsy and to predict the risk of progression to more serious conditions.
Referral to a hematologist is warranted for patients with intermediate- and high-risk MGUS, patients with abnormal serum free light-chain ratios, and those who show evidence of malignant progression. Patients with intermediate- and high-risk MGUS could be referred for bone marrow biopsy before assessment by a hematologist. The primary care provider may continue to follow patients with low-risk MGUS who do not display clinical or laboratory evidence of myeloma or related disorders.
The importance of educating patients to report any new worrisome symptom (eg, fatigue, neuropathy, weight loss, night sweats, bone pain) cannot be overemphasized, as some patients may progress to myeloma or other disorders between follow-up visits.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
- van de Donk NW, Palumbo A, Johnsen HE, et al; European Myeloma Network. The clinical relevance and management of monoclonal gammopathy of undetermined significance and related disorders: recommendations from the European Myeloma Network. Haematologica 2014; 99(6):984–996. doi:10.3324/haematol.2013.100552
- International Myeloma Working Group. Criteria for the classification of monoclonal gammopathies, multiple myeloma and related disorders: a report of the International Myeloma Working Group. Br J Haematol 2003; 121(5):749–757. pmid:12780789
- Rajan AM, Rajkumar SV. Diagnostic evaluation of monoclonal gammopathy of undetermined significance. Eur J Haematol 2013; 91(6):561–562. doi:10.1111/ejh.12198
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance. Br J Haematol 2006; 134(6):573–589. doi:10.1111/j.1365-2141.2006.06235.x
- Kyle RA, Durie BG, Rajkumar SV, et al; International Myeloma Working Group. Monoclonal gammopathy of undetermined significance (MGUS) and smoldering (asymptomatic) multiple myeloma: IMWG consensus perspectives risk factors for progression and guidelines for monitoring and management. Leukemia 2010; 24(6):1121–1127. doi:10.1038/leu.2010.60
- Bird J, Behrens J, Westin J, et al; Haemato-oncology Task Force of the British Committee for Standards in Haematology, UK Myeloma Forum and Nordic Myeloma Study Group. UK Myeloma Forum (UKMF) and Nordic Myeloma Study Group (NMSG): guidelines for the investigation of newly detected M-proteins and the management of monoclonal gammopathy of undetermined significance (MGUS). Br J Haematol 2009; 147(1):22–42. doi:10.1111/j.1365-2141.2009.07807.x
- Rajkumar SV, Kyle RA, Buadi FK. Advances in the diagnosis, classification, risk stratification, and management of monoclonal gammopathy of undetermined significance: implications for recategorizing disease entities in the presence of evolving scientific evidence. Mayo Clin Proc 2010; 85(10):945–948. doi:10.4065/mcp.2010.0520
- Kyle RA, Therneau TM, Rajkumar SV, et al. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346(8):564–569. doi:10.1056/NEJMoa01133202
- Doyle LM, Gundrum JD, Farnen JP, Wright LJ, Kranig JAI, Go RS. Determining why and which clinicians order serum protein electrophoresis (SPEP), subsequent diagnoses based on indications, and clinical significance of routine follow-up: a study of patients with monoclonal gammopathy of undetermined significance (MGUS). Blood 2009; 114(22):Abstr 4883. www.bloodjournal.org/content/114/22/4883. Accessed December 4, 2018.
- Merlini G, Palladini G. Differential diagnosis of monoclonal gammopathy of undetermined significance. Hematology Am Soc Hematol Educ Program 2012; 2012:595–603. doi:10.1182/asheducation-2012.1.595
- Glavey SV, Leung N. Monoclonal gammopathy: the good, the bad and the ugly. Blood Rev 2016; 30(3):223–231. doi:10.1016/j.blre.2015.12.001
- Dispenzieri A, Gertz MA, Therneau TM, Kyle RA. Retrospective cohort study of 148 patients with polyclonal gammopathy. Mayo Clin Proc 2001; 76(5):476–487. doi:10.4065/76.5.476
- Merlini G, Stone MJ. Dangerous small B-cell clones. Blood 2006; 108(8):2520–2530. doi:10.1182/blood-2006-03-001164
- Rajkumar SV, Dimopoulos MA, Palumbo A, et al. International Myeloma Working Group updated criteria for the diagnosis of multiple myeloma. Lancet Oncol 2014; 15(12):e538–e548. doi:10.1016/S1470-2045(14)70442-5
- Kyle RA, Gertz MA, Witzig TE, et al. Review of 1027 patients with newly diagnosed multiple myeloma. Mayo Clin Proc 2003; 78(1):21–33. doi:10.4065/78.1.21
- Hutchison CA, Harding S, Hewins P, et al. Quantitative assessment of serum and urinary polyclonal free light chains in patients with chronic kidney disease. Clin J Am Soc Nephrol 2008; 3(6):1684–1690. doi:10.2215/CJN.02290508
- Katzmann JA, Dispenzieri A, Kyle RA, et al. Elimination of the need for urine studies in the screening algorithm for monoclonal gammopathies by using serum immunofixation and free light chain assays. Mayo Clin Proc 2006; 81(12):1575–1578. doi:10.4065/81.12.1575
- Berenson JR, Anderson KC, Audell RA, et al. Monoclonal gammopathy of undetermined significance: a consensus statement. Br J Haematol 2010; 150(1):28–38. doi:10.1111/j.1365-2141.2010.08207.x
- Mangiacavalli S, Cocito F, Pochintesta L, et al. Monoclonal gammopathy of undetermined significance: a new proposal of workup. Eur J Haematol 2013; 91(4):356–360. doi:10.1111/ejh.12172
- Bianchi G, Kyle RA, Colby CL, et al. Impact of optimal follow-up of monoclonal gammopathy of undetermined significance on early diagnosis and prevention of myeloma-related complications. Blood 2010;116:2019–2025. doi:10.1182/blood-2010-04-277566
- Rosiñol L, Cibeira MT, Montoto S, et al. Monoclonal gammopathy of undetermined significance: predictors of malignant transformation and recognition of an evolving type characterized by a progressive increase in M protein size. Mayo Clin Proc 2007; 82(4):428–434. doi:10.4065/82.4.428
- Vanderschueren S, Mylle M, Dierickx D, et al. Monoclonal gammopathy of undetermined significance: significant beyond hematology. Mayo Clin Proc 2009; 84(9):842–845. doi:10.4065/84.9.842
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139(5):730–743. doi:10.1111/j.1365-2141.2007.06873.x
- Leung N, Bridoux F, Hutchison CA, et al; International Kidney and Monoclonal Gammopathy Research Group. Monoclonal gammopathy of renal significance: when MGUS is no longer undetermined or insignificant. Blood. 2012; 120(22):4292–4295. doi:10.1182/blood-2012-07-445304
- Merlini G, Wechalekar AD, Palladini G. Systemic light chain amyloidosis: an update for treating physicians. Blood 2013; 121(26):5124–5130. doi:10.1182/blood-2013-01-453001
- Dispenzieri A, Katzmann JA, Kyle RA, et al. Prevalence and risk of progression of light-chain monoclonal gammopathy of undetermined significance: a retrospective population-based cohort study. Lancet 2010; 375(9727):1721–1728. doi:10.1016/S0140-6736(10)60482-5
- Rajkumar SV, Kyle RA, Therneau TM, et al. Serum free light chain ratio is an independent risk factor for progression in monoclonal gammopathy of undetermined significance. Blood 2005; 106(3):812–817. doi:10.1182/blood-2005-03-1038
- Dimopoulos MA, Hillengass J, Usmani S, et al. Role of magnetic resonance imaging in the management of patients with multiple myeloma: a consensus statement. J Clin Oncol 2015; 33(6):657–664. doi:10.1200/JCO.2014.57.9961
- Dimopoulos M, Kyle R, Fermand JP, et al. Consensus recommendations for standard investigative workup: report of the International Myeloma Workshop Consensus Panel 3. Blood 2011; 117(18):4701–4705. doi:10.1182/blood-2010-10-299529
- Pompa T, Maddox M, Woodard A, et al. Cost effectiveness in low risk MGUS patients. Blood 2016; 128:2360. http://www.bloodjournal.org/content/128/22/2360. Accessed December 4, 2018.
KEY POINTS
- MGUS is the most common of the monoclonal gammopathies.
- The overall risk of MGUS progressing to myeloma and other lymphoproliferative disorders is 1% per year.
- Low-risk MGUS is defined by an immunoglobulin G monoclonal protein at a concentration less than 1.5 g/dL and a normal serum free light-chain ratio.
- Low-risk MGUS carries a much lower risk of progression than intermediate- and high-risk MGUS, may not require subspecialty referral, and can be followed by the outpatient provider.
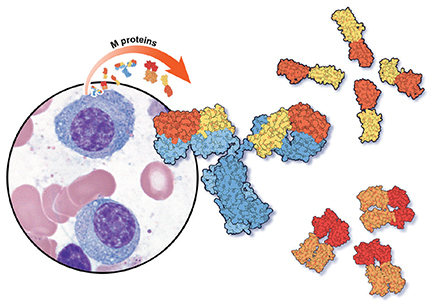
MGUS: It’s about the protein, not just the marrow

In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316
In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
In the past decade, it has been increasingly recognized that these clonally produced proteins—entire immunoglobulins or free light chains—may be directly pathogenic, independent of any pathologic effect of cellular clonal expansion and infiltration. Brouet class 1 cryoglobulinemia (in which a monoclonal paraprotein precipitates in cooler temperatures and acts as a source of complement, activating the immune complex) and light chain (usually lambda)-related amyloidosis have been recognized for much longer. But a newer concept, monoclonal gammopathy of renal significance (MGRS), has attracted significant attention and to some extent has modified our approach to patients with either known MGUS or unexplained chronic kidney disease.
Finding MGUS still warrants a parsimonious evaluation for possible progression to myeloma or other proliferative disorder, as discussed by Khouri et al in this issue of the Journal. But it should also prompt a thoughtful assessment of renal function, including estimating the glomerular filtration rate and looking for proteinuria, hematuria, and unexplained glucosuria or inappropriate urine pH. While typical light chain-induced renal tubular injury is usually associated with high levels of proteins such as those seen with myeloma, other patterns of glomerular, vascular, and mixed renal disease are associated with deposition of proteins that, once considered in the differential diagnosis, warrant renal biopsy to diagnose and direct appropriate therapy. That MGUS and MGRS occur more frequently in older patients, who are already at greater risk of multiple common causes of kidney disease, complicates clinical decision-making.1 Some of these disorders are associated with other initially subtle or seemingly disconnected clinical symptoms such as polyneuropathy, rash, and carpal tunnel syndrome, but many are at least initially limited to the kidneys.
As we enter a new calendar year, we at the Journal send our best wishes to all of our readers, authors, and peer reviewers, and we thank you for sharing in our medical education ventures. I personally hope that we have added some joy, enthusiasm—and some knowledge—to your professional activities, and I hope that we all can participate in some way to refashion a more civil and peaceful world in 2019.
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316
- Rosner MH, Edeani A, Yanagita M, et al. Paraprotein-related kidney disease: diagnosing and treating monoclonal gammopathy of renal significance. Clin J Am Soc Neph 2016; 11(12):2280–2287. doi:10.2215/CJN.02920316

FDA approves ravulizumab for treatment of paroxysmal nocturnal hemoglobinuria
The Food and Drug Administration has approved ravulizumab (Ultomiris) injection for the treatment of adult patients with paroxysmal nocturnal hemoglobinuria (PNH).

“The approval of Ultomiris will change the way that patients with PNH are treated. Prior to this approval, the only approved therapy for PNH required treatment every 2 weeks, which can be burdensome for patients and their families. Ultomiris uses a novel formulation so patients only need treatment every 8 weeks, without compromising efficacy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a press release from the agency.
Patients with PNH, a rare disorder, lack a protein which protects red blood cells from being destroyed in the immune system. Episodes can be triggered by stresses on the body such as infection or physical exertion, and symptoms include severe anemia, profound fatigue, shortness of breath, intermittent episodes of dark-colored urine, kidney disease, or recurrent pain.
FDA approval for ravulizumab is based on results from a pair of clinical trials. In the first, 246 treatment-naive PNH patients received either ravulizumab or eculizumab, the current standard of care; ravulizumab was noninferior, with no patients undergoing a transfusion and all patients having similar incidence of hemolysis. In the second trial, 195 patients who had clinically stable PNH after receiving eculizumab for 6 months were randomized to receive ravulizumab or continue eculizumab; again, ravulizumab was noninferior.
The most common adverse events associated with ravulizumab were headache and respiratory tract infection. Caution is recommended when prescribing ravulizumab to patients with any type of infection.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ravulizumab (Ultomiris) injection for the treatment of adult patients with paroxysmal nocturnal hemoglobinuria (PNH).
“The approval of Ultomiris will change the way that patients with PNH are treated. Prior to this approval, the only approved therapy for PNH required treatment every 2 weeks, which can be burdensome for patients and their families. Ultomiris uses a novel formulation so patients only need treatment every 8 weeks, without compromising efficacy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a press release from the agency.
Patients with PNH, a rare disorder, lack a protein which protects red blood cells from being destroyed in the immune system. Episodes can be triggered by stresses on the body such as infection or physical exertion, and symptoms include severe anemia, profound fatigue, shortness of breath, intermittent episodes of dark-colored urine, kidney disease, or recurrent pain.
FDA approval for ravulizumab is based on results from a pair of clinical trials. In the first, 246 treatment-naive PNH patients received either ravulizumab or eculizumab, the current standard of care; ravulizumab was noninferior, with no patients undergoing a transfusion and all patients having similar incidence of hemolysis. In the second trial, 195 patients who had clinically stable PNH after receiving eculizumab for 6 months were randomized to receive ravulizumab or continue eculizumab; again, ravulizumab was noninferior.
The most common adverse events associated with ravulizumab were headache and respiratory tract infection. Caution is recommended when prescribing ravulizumab to patients with any type of infection.
Find the full press release on the FDA website.
The Food and Drug Administration has approved ravulizumab (Ultomiris) injection for the treatment of adult patients with paroxysmal nocturnal hemoglobinuria (PNH).
“The approval of Ultomiris will change the way that patients with PNH are treated. Prior to this approval, the only approved therapy for PNH required treatment every 2 weeks, which can be burdensome for patients and their families. Ultomiris uses a novel formulation so patients only need treatment every 8 weeks, without compromising efficacy,” Richard Pazdur, MD, director of the FDA’s Oncology Center of Excellence, said in a press release from the agency.
Patients with PNH, a rare disorder, lack a protein which protects red blood cells from being destroyed in the immune system. Episodes can be triggered by stresses on the body such as infection or physical exertion, and symptoms include severe anemia, profound fatigue, shortness of breath, intermittent episodes of dark-colored urine, kidney disease, or recurrent pain.
FDA approval for ravulizumab is based on results from a pair of clinical trials. In the first, 246 treatment-naive PNH patients received either ravulizumab or eculizumab, the current standard of care; ravulizumab was noninferior, with no patients undergoing a transfusion and all patients having similar incidence of hemolysis. In the second trial, 195 patients who had clinically stable PNH after receiving eculizumab for 6 months were randomized to receive ravulizumab or continue eculizumab; again, ravulizumab was noninferior.
The most common adverse events associated with ravulizumab were headache and respiratory tract infection. Caution is recommended when prescribing ravulizumab to patients with any type of infection.
Find the full press release on the FDA website.
Survivors of childhood Hodgkin lymphoma face 14-fold risk of second cancers
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
Survivors of childhood Hodgkin lymphoma have a 14-fold greater risk for second cancers, compared with the general population, according to newly published data.
The subsequent malignant neoplasms (SMNs) tend to follow specific patterns depending on the patient’s age at treatment, sex, treatment modality, and body region treated.
And although the risk of SMNs appears to be somewhat lower for patients treated in more recent decades, it is still significantly elevated, compared with that of the general population, according to Anna S. Holmqvist, MD, PhD, from Lund University (Sweden), and her colleagues.
“A major goal of the current study was to develop evidence with which to guide the screening of survivors of HL for the development of [solid] SMNs,” the investigators wrote in Cancer.
They examined at data from the Late Effects Study Group, a multinational cohort of patients aged 16 years or younger who were treated for Hodgkin lymphoma and other cancers from 1955 to 1986.
The current report is the third update from an expanded cohort, including data on 1,136 patients with a median follow-up of 26.6 years. The median patient age at diagnosis was 11 years and the patients were followed for 23,212 person-years following the Hodgkin lymphoma diagnosis.
In all, 162 patients developed a total of 196 solid SMNs, including breast cancer in 54 patients, basal cell carcinoma in 34 patients, thyroid cancer in 30, colorectal cancer in 15, lung cancer in 11, other malignancies in 40, and disease site not available in 12 patients.
The cumulative incidence of any solid SMN 40 years after a diagnosis of Hodgkin lymphoma was 26.4%. The standardized incidence ratio for the entire cohort was 14.0, compared with the general population as derived from the Surveillance, Epidemiology and End Results database.
Predisposing factors for breast cancer in females included a Hodgkin lymphoma diagnosis from the ages of 10-16 years, and treatment with radiotherapy to the chest.
The patients at highest risk for subsequent development of lung cancer were males treated with chest radiotherapy before age 10 years. Those at highest risk for colorectal cancer were males and females who had received abdominal/pelvic radiotherapy and high-dose alkylating agents. Patients at highest risk for thyroid cancers were females who had been treated with radiotherapy to the neck before the age of 10.
The cumulative incidence for breast cancer by age 50 years for those at highest risk was 45.3%. The respective cumulative incidences for lung, colorectal, and thyroid cancers by age 50 were 4.2%, 9.5%, and 17.3%.
The investigators noted that patients treated more recently are likely to have received lower doses and volumes of radiotherapy, compared with patients treated in 1970s and earlier. “However, for the cohort of patients treated between 1955 and 1986, it is clear that continued surveillance for [solid] SMNs is essential because their risk continues to increase as these survivors enter their fourth and subsequent decades of life.”
No specific funding source for the study was reported. The authors made no financial disclosures.
SOURCE: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
FROM CANCER
Key clinical point:
Major finding: The risk for a subsequent malignant neoplasm among survivors of childhood Hodgkin lymphoma was 14-fold higher than that of the general population.
Study details: The third update of data on a cohort of 1,136 childhood Hodgkin lymphoma survivors followed for a median of 26.6 years.
Disclosures: No specific funding source for the study was reported. The authors made no financial disclosures.
Source: Holmqvist AS et al. Cancer. 2018 Dec 17. doi: 10.1002/cncr.31807.
GO-8: Early promise for novel FVIII variant in hemophilia A
SAN DIEGO – A novel human factor VIII variant shows promise for the treatment of severe hemophilia A, according to preliminary findings from the ongoing Gene Therapy for Hemophilia A (GO-8) phase 1/2 dose-escalation study.

A single peripheral vein infusion of the factor VIII (FVIII) variant resulted in FVIII activity levels of about 6% versus levels of no more than 1% of normal at study entry in the first four patients, Pratima Chowdary, MD, reported at the annual meeting of the American Society of Hematology.
The variant, known as scAAV2/8-LP1-hFIXco, is being investigated for safety and efficacy in the GO-8 investigator-led, open-label, nonrandomized trial at a low, mid, and high dose (2 x 1011 vector genomes/kg, 6 x 1011 vector genomes/kg, and 2 x 1012 vector genomes/kg), said Dr. Chowdary, a consultant hematologist at Royal Free Hospital London.
The main study period is 6 months and 15 years of follow-up are offered.
The first patient received the low dose and achieved FVIII of about 6% within 1 week. That level persisted for about 6 weeks when the patient developed a transaminitis, which promptly responded to steroids.
His steady-state FVIII within a few weeks was 7% by one-stage assay and about 3% by chromogenic assay, Dr. Chowdary said.
The remaining patients received the mid dose and also achieved FVIII levels of about 6% within a week. Patient 2 started on prophylactic steroids at week 6, per protocol, and did not experience transaminitis, but also had no increase in FVIII level, compared with the low-dose patient, which may be explained by the potential drug half-life, she noted.
Patient 3 reached a FVIII level of about 30% by week 4. He developed transaminitis at that time, which was about 2 weeks before planned prophylactic drug administration, but the transaminitis was controlled by steroids over a period of about 8-10 weeks.
“His steady-state FVIII level by one stage was 34% and by chromogenic assay was 17%. He has not had any bleeds since his gene transfer and has not required any FVIII concentrate either,” she said.
Patient 4 reached a FVIII level of about 40% by week 4. He was given prophylactic steroids at that time because of the occurrence of transaminitis at week 4 in Patient 3.
The patient developed transaminitis during steroid taper about 4 weeks later, perhaps because of the rapid taper, Dr. Chowdary said, adding that the transaminitis was well controlled with steroids, but follow-up in this patient has only been about 12 weeks.
“The characteristics of FVIII expression in this patient are very similar to the previous patient. ... We suspect he will have a steady-state level of about 30%,” she said. “Again, he’s had no bleeds since his gene transfer and has not required any FVIII concentrate.”
The single infusion of this novel vector was well tolerated in each patient, with no evidence of infusion-related reactions, neutralizing anti-FVIII antibodies, or vector-related adverse events.
“The transgene expression was achieved in all patients and at both vector dosages,” Dr. Chowdary said. “What is very important is that the levels of less than 10% had only a modest impact on the bleed rates and FVIII usage, whereas an expression of more than 10% resulted in zero bleeds and the patient did not require any additional FVIII treatment.”
The data are “encouraging,” she said. “We look forward to escalating the dose in the next patient.”
Dr. Chowdary reported financial relationships with Bayer, CSL Behring, Baxalta, Baxter, Biogen, Freeline, Novo Nordisk, Pfizer, Roche, Shire, and SOBI.
SOURCE: Chowdary P et al. ASH 2018, Abstract 489.
SAN DIEGO – A novel human factor VIII variant shows promise for the treatment of severe hemophilia A, according to preliminary findings from the ongoing Gene Therapy for Hemophilia A (GO-8) phase 1/2 dose-escalation study.
A single peripheral vein infusion of the factor VIII (FVIII) variant resulted in FVIII activity levels of about 6% versus levels of no more than 1% of normal at study entry in the first four patients, Pratima Chowdary, MD, reported at the annual meeting of the American Society of Hematology.
The variant, known as scAAV2/8-LP1-hFIXco, is being investigated for safety and efficacy in the GO-8 investigator-led, open-label, nonrandomized trial at a low, mid, and high dose (2 x 1011 vector genomes/kg, 6 x 1011 vector genomes/kg, and 2 x 1012 vector genomes/kg), said Dr. Chowdary, a consultant hematologist at Royal Free Hospital London.
The main study period is 6 months and 15 years of follow-up are offered.
The first patient received the low dose and achieved FVIII of about 6% within 1 week. That level persisted for about 6 weeks when the patient developed a transaminitis, which promptly responded to steroids.
His steady-state FVIII within a few weeks was 7% by one-stage assay and about 3% by chromogenic assay, Dr. Chowdary said.
The remaining patients received the mid dose and also achieved FVIII levels of about 6% within a week. Patient 2 started on prophylactic steroids at week 6, per protocol, and did not experience transaminitis, but also had no increase in FVIII level, compared with the low-dose patient, which may be explained by the potential drug half-life, she noted.
Patient 3 reached a FVIII level of about 30% by week 4. He developed transaminitis at that time, which was about 2 weeks before planned prophylactic drug administration, but the transaminitis was controlled by steroids over a period of about 8-10 weeks.
“His steady-state FVIII level by one stage was 34% and by chromogenic assay was 17%. He has not had any bleeds since his gene transfer and has not required any FVIII concentrate either,” she said.
Patient 4 reached a FVIII level of about 40% by week 4. He was given prophylactic steroids at that time because of the occurrence of transaminitis at week 4 in Patient 3.
The patient developed transaminitis during steroid taper about 4 weeks later, perhaps because of the rapid taper, Dr. Chowdary said, adding that the transaminitis was well controlled with steroids, but follow-up in this patient has only been about 12 weeks.
“The characteristics of FVIII expression in this patient are very similar to the previous patient. ... We suspect he will have a steady-state level of about 30%,” she said. “Again, he’s had no bleeds since his gene transfer and has not required any FVIII concentrate.”
The single infusion of this novel vector was well tolerated in each patient, with no evidence of infusion-related reactions, neutralizing anti-FVIII antibodies, or vector-related adverse events.
“The transgene expression was achieved in all patients and at both vector dosages,” Dr. Chowdary said. “What is very important is that the levels of less than 10% had only a modest impact on the bleed rates and FVIII usage, whereas an expression of more than 10% resulted in zero bleeds and the patient did not require any additional FVIII treatment.”
The data are “encouraging,” she said. “We look forward to escalating the dose in the next patient.”
Dr. Chowdary reported financial relationships with Bayer, CSL Behring, Baxalta, Baxter, Biogen, Freeline, Novo Nordisk, Pfizer, Roche, Shire, and SOBI.
SOURCE: Chowdary P et al. ASH 2018, Abstract 489.
SAN DIEGO – A novel human factor VIII variant shows promise for the treatment of severe hemophilia A, according to preliminary findings from the ongoing Gene Therapy for Hemophilia A (GO-8) phase 1/2 dose-escalation study.
A single peripheral vein infusion of the factor VIII (FVIII) variant resulted in FVIII activity levels of about 6% versus levels of no more than 1% of normal at study entry in the first four patients, Pratima Chowdary, MD, reported at the annual meeting of the American Society of Hematology.
The variant, known as scAAV2/8-LP1-hFIXco, is being investigated for safety and efficacy in the GO-8 investigator-led, open-label, nonrandomized trial at a low, mid, and high dose (2 x 1011 vector genomes/kg, 6 x 1011 vector genomes/kg, and 2 x 1012 vector genomes/kg), said Dr. Chowdary, a consultant hematologist at Royal Free Hospital London.
The main study period is 6 months and 15 years of follow-up are offered.
The first patient received the low dose and achieved FVIII of about 6% within 1 week. That level persisted for about 6 weeks when the patient developed a transaminitis, which promptly responded to steroids.
His steady-state FVIII within a few weeks was 7% by one-stage assay and about 3% by chromogenic assay, Dr. Chowdary said.
The remaining patients received the mid dose and also achieved FVIII levels of about 6% within a week. Patient 2 started on prophylactic steroids at week 6, per protocol, and did not experience transaminitis, but also had no increase in FVIII level, compared with the low-dose patient, which may be explained by the potential drug half-life, she noted.
Patient 3 reached a FVIII level of about 30% by week 4. He developed transaminitis at that time, which was about 2 weeks before planned prophylactic drug administration, but the transaminitis was controlled by steroids over a period of about 8-10 weeks.
“His steady-state FVIII level by one stage was 34% and by chromogenic assay was 17%. He has not had any bleeds since his gene transfer and has not required any FVIII concentrate either,” she said.
Patient 4 reached a FVIII level of about 40% by week 4. He was given prophylactic steroids at that time because of the occurrence of transaminitis at week 4 in Patient 3.
The patient developed transaminitis during steroid taper about 4 weeks later, perhaps because of the rapid taper, Dr. Chowdary said, adding that the transaminitis was well controlled with steroids, but follow-up in this patient has only been about 12 weeks.
“The characteristics of FVIII expression in this patient are very similar to the previous patient. ... We suspect he will have a steady-state level of about 30%,” she said. “Again, he’s had no bleeds since his gene transfer and has not required any FVIII concentrate.”
The single infusion of this novel vector was well tolerated in each patient, with no evidence of infusion-related reactions, neutralizing anti-FVIII antibodies, or vector-related adverse events.
“The transgene expression was achieved in all patients and at both vector dosages,” Dr. Chowdary said. “What is very important is that the levels of less than 10% had only a modest impact on the bleed rates and FVIII usage, whereas an expression of more than 10% resulted in zero bleeds and the patient did not require any additional FVIII treatment.”
The data are “encouraging,” she said. “We look forward to escalating the dose in the next patient.”
Dr. Chowdary reported financial relationships with Bayer, CSL Behring, Baxalta, Baxter, Biogen, Freeline, Novo Nordisk, Pfizer, Roche, Shire, and SOBI.
SOURCE: Chowdary P et al. ASH 2018, Abstract 489.
REPORTING FROM ASH 2018
Key clinical point:
Major finding: A single infusion of the factor VIII variant resulted in activity levels of about 6%, compared with 1% or less at baseline.
Study details: The findings were from the first four patients in a phase 1/2 dose-escalation study.
Disclosures: Dr. Chowdary reported financial relationships with Bayer, CSL Behring, Baxalta, Baxter, Biogen, Freeline, Novo Nordisk, Pfizer, Roche, Shire, and SOBI.
Source: Chowdary P et al. ASH 2018, Abstract 489.