User login
Genotype, need for transfusion predict death in VEXAS syndrome
Among patients with the recently defined severe autoinflammatory syndrome VEXAS, those who are transfusion dependent or have a specific amino acid substitution are at highest risk for death, whereas those with ear chondritis are at significantly lower risk, a multinational team of investigators has found.
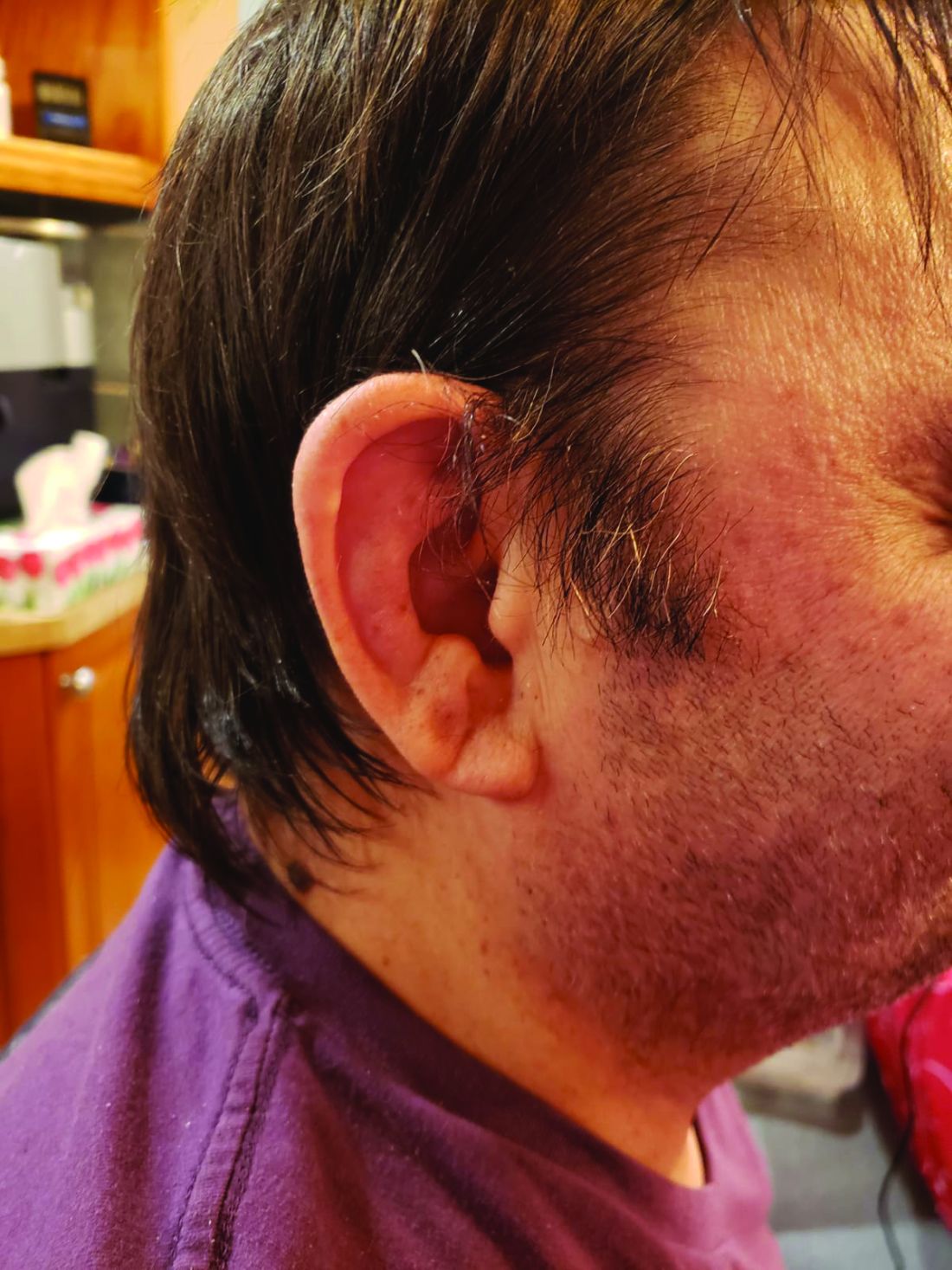
Their study of mortality and predictors of survival among patients with genetically confirmed VEXAS showed that patients with a VEXAS variant resulting in an amino acid substitution of a methionine for a valine had a 3.5-fold higher risk for death, compared with patients with either a methionine-to-threonine substitution or a methionine-to-leucine swap.
Transfusion dependence was an independent predictor of mortality. Patients who became dependent on transfusions after symptom onset had a nearly threefold higher risk for death, reported Marcela A. Ferrada, MD, a clinical fellow at the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
“These findings should inform risk assessment and clinical management in patients with VEXAS syndrome,” she said in an oral abstract presentation during the virtual annual meeting of the American College of Rheumatology.
“These genetic findings have proven right now to be not only diagnostic, but we have shown that they’re also prognostic, and we hope that this is going to help us identify patients who could have more aggressive treatment,” Dr. Ferrada said.
She also discussed her findings in a media briefing held 2 days prior to her plenary presentation. At that briefing, this news organization asked participating clinicians whether they had patients who they suspected may have had undiagnosed VEXAS.
“My answer to that is interesting,” replied moderator Vaneet Sandhu, MD, from Loma Linda (Calif.) University and Riverside University Health System.
“In the last couple of days, I’ve been reading about VEXAS, and actually texted one of my colleagues yesterday and said, ‘Hey, you know these patients we’ve been seeing who have these strange rashes and chondritis and have maybe a diagnosis of leukocytoclastic vasculitis or something else – are we not diagnosing these patients?’ ” she said.
“I think we are looking at every patient with chondritis and reexamining their phenotype. We had dismissed certain symptoms because they didn’t fit the archetype for relapsing polychondritis, for example, but it could be VEXAS,” said Alfred Kim, MD, PhD, of Washington University in St. Louis, who also presented data during the briefing.
Three variants
VEXAS is caused by somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation.
The syndrome’s name is an acronym descriptive of the major features:
- Vacuoles in bone marrow cells.
- E-1 activating enzyme that UBA1 encodes for.
- X-linked.
- Autoinflammatory.
- Somatic mutation featuring hematologic mosaicism.
VEXAS results in rheumatologic, dermatologic, and hematologic symptoms that are often misdiagnosed as being caused by treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, giant cell arteritis, or myelodysplastic syndrome (MDS).
VEXAS was identified as a distinct syndrome within the past year by Dr. Ferrada and other investigators at NIAMS, the National Human Genome Research Institute, and other institutions.
In the study reported at ACR 2021, Dr. Ferrada and colleagues assessed 83 men who had been referred for genetic testing for VEXAS at the National Institutes of Health, in Bethesda, Md., and at Leeds (England) Teaching Hospitals NHS Trust.
All patients were confirmed to have VEXAS-defining genetic mutations in UBA1 by Sanger sequencing of peripheral blood samples. Only those patients with mutations at codon p.Met41 were included in the investigators’ analysis. Mutations at that site account for nearly all cases of VEXAS that have been identified to date.
The most common clinical manifestation of VEXAS was skin involvement, which occurred in all but one of the 83 patients. Other common manifestations included arthritis (58 patients), pulmonary infiltrates (57 patients), and ear chondritis (54 patients).
Fifteen patients were found to have the leucine variant, 18 had the valine variant, and 50 had the threonine variant. The median age at disease onset was 66 years in the leucine and threonine variant groups and 65 in the valine variant group.
The clinical diagnosis differed according to genotype: 4 of 18 patients (22%) with the valine variant were diagnosed with relapsing polychondritis, compared with 8 of 15 (53%) with the leucine variant and 31 of 50 (62%) with the threonine variant (P = .01).
In contrast, 55% of patients with valine genotype were diagnosed with undifferentiated fever, compared with 6% of those with the leucine and 16% with the threonine genotypes (P = .001). More patients with the leucine variant (60%) were diagnosed with Sweet syndrome, compared with 11% and 14% of patients with the valine and threonine variants, respectively (P = .001).
There was no significant difference among the three genotypes in the percentage of patients diagnosed with MDS.
The follow-up period ranged from 1 to 18 years (median, 4.7 years). The median survival time from disease onset for all patients was 10 years.
Among patients with the valine variant, median survival was 9 years, which was significantly less than among patients with the other two variants (P = .01).
In univariable analysis, independent predictors of mortality were ear chondritis (hazard ratio, 0.26; P = .005), transfusion dependence, a time-dependent variable (HR, 2.59; P = .03), and the valine variant (HR, 3.5; P = .008).
The association between VEXAS genotype and phenotype could be explained by the finding that, among patients with the valine variant, there was significantly less translation of the catalytically proficient UBA1b isoform than in patients with the other two variants, Dr. Ferrada said.
Therapeutic options
Dr. Ferrada noted that to date no drugs have been shown to provide consistent therapeutic benefits for patients with VEXAS, but evidence as to the etiology of the syndrome points to possible treatment approaches.
“All of these findings I think are extremely important to help us guide management of these patients, as we know that the mutation is located in the stem cells in the bone marrow. So we suspect that doing a bone marrow transplant in these patients is going to be curative,” Dr. Ferrada said during the briefing.
Investigators are planning a phase 2 trial of allogeneic hematopoietic stem cell transplant for patients with VEXAS.
The study was supported by the National Institutes of Health. Dr. Ferrada, Dr. Sandhu, and Dr. Kim have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among patients with the recently defined severe autoinflammatory syndrome VEXAS, those who are transfusion dependent or have a specific amino acid substitution are at highest risk for death, whereas those with ear chondritis are at significantly lower risk, a multinational team of investigators has found.
Their study of mortality and predictors of survival among patients with genetically confirmed VEXAS showed that patients with a VEXAS variant resulting in an amino acid substitution of a methionine for a valine had a 3.5-fold higher risk for death, compared with patients with either a methionine-to-threonine substitution or a methionine-to-leucine swap.
Transfusion dependence was an independent predictor of mortality. Patients who became dependent on transfusions after symptom onset had a nearly threefold higher risk for death, reported Marcela A. Ferrada, MD, a clinical fellow at the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
“These findings should inform risk assessment and clinical management in patients with VEXAS syndrome,” she said in an oral abstract presentation during the virtual annual meeting of the American College of Rheumatology.
“These genetic findings have proven right now to be not only diagnostic, but we have shown that they’re also prognostic, and we hope that this is going to help us identify patients who could have more aggressive treatment,” Dr. Ferrada said.
She also discussed her findings in a media briefing held 2 days prior to her plenary presentation. At that briefing, this news organization asked participating clinicians whether they had patients who they suspected may have had undiagnosed VEXAS.
“My answer to that is interesting,” replied moderator Vaneet Sandhu, MD, from Loma Linda (Calif.) University and Riverside University Health System.
“In the last couple of days, I’ve been reading about VEXAS, and actually texted one of my colleagues yesterday and said, ‘Hey, you know these patients we’ve been seeing who have these strange rashes and chondritis and have maybe a diagnosis of leukocytoclastic vasculitis or something else – are we not diagnosing these patients?’ ” she said.
“I think we are looking at every patient with chondritis and reexamining their phenotype. We had dismissed certain symptoms because they didn’t fit the archetype for relapsing polychondritis, for example, but it could be VEXAS,” said Alfred Kim, MD, PhD, of Washington University in St. Louis, who also presented data during the briefing.
Three variants
VEXAS is caused by somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation.
The syndrome’s name is an acronym descriptive of the major features:
- Vacuoles in bone marrow cells.
- E-1 activating enzyme that UBA1 encodes for.
- X-linked.
- Autoinflammatory.
- Somatic mutation featuring hematologic mosaicism.
VEXAS results in rheumatologic, dermatologic, and hematologic symptoms that are often misdiagnosed as being caused by treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, giant cell arteritis, or myelodysplastic syndrome (MDS).
VEXAS was identified as a distinct syndrome within the past year by Dr. Ferrada and other investigators at NIAMS, the National Human Genome Research Institute, and other institutions.
In the study reported at ACR 2021, Dr. Ferrada and colleagues assessed 83 men who had been referred for genetic testing for VEXAS at the National Institutes of Health, in Bethesda, Md., and at Leeds (England) Teaching Hospitals NHS Trust.
All patients were confirmed to have VEXAS-defining genetic mutations in UBA1 by Sanger sequencing of peripheral blood samples. Only those patients with mutations at codon p.Met41 were included in the investigators’ analysis. Mutations at that site account for nearly all cases of VEXAS that have been identified to date.
The most common clinical manifestation of VEXAS was skin involvement, which occurred in all but one of the 83 patients. Other common manifestations included arthritis (58 patients), pulmonary infiltrates (57 patients), and ear chondritis (54 patients).
Fifteen patients were found to have the leucine variant, 18 had the valine variant, and 50 had the threonine variant. The median age at disease onset was 66 years in the leucine and threonine variant groups and 65 in the valine variant group.
The clinical diagnosis differed according to genotype: 4 of 18 patients (22%) with the valine variant were diagnosed with relapsing polychondritis, compared with 8 of 15 (53%) with the leucine variant and 31 of 50 (62%) with the threonine variant (P = .01).
In contrast, 55% of patients with valine genotype were diagnosed with undifferentiated fever, compared with 6% of those with the leucine and 16% with the threonine genotypes (P = .001). More patients with the leucine variant (60%) were diagnosed with Sweet syndrome, compared with 11% and 14% of patients with the valine and threonine variants, respectively (P = .001).
There was no significant difference among the three genotypes in the percentage of patients diagnosed with MDS.
The follow-up period ranged from 1 to 18 years (median, 4.7 years). The median survival time from disease onset for all patients was 10 years.
Among patients with the valine variant, median survival was 9 years, which was significantly less than among patients with the other two variants (P = .01).
In univariable analysis, independent predictors of mortality were ear chondritis (hazard ratio, 0.26; P = .005), transfusion dependence, a time-dependent variable (HR, 2.59; P = .03), and the valine variant (HR, 3.5; P = .008).
The association between VEXAS genotype and phenotype could be explained by the finding that, among patients with the valine variant, there was significantly less translation of the catalytically proficient UBA1b isoform than in patients with the other two variants, Dr. Ferrada said.
Therapeutic options
Dr. Ferrada noted that to date no drugs have been shown to provide consistent therapeutic benefits for patients with VEXAS, but evidence as to the etiology of the syndrome points to possible treatment approaches.
“All of these findings I think are extremely important to help us guide management of these patients, as we know that the mutation is located in the stem cells in the bone marrow. So we suspect that doing a bone marrow transplant in these patients is going to be curative,” Dr. Ferrada said during the briefing.
Investigators are planning a phase 2 trial of allogeneic hematopoietic stem cell transplant for patients with VEXAS.
The study was supported by the National Institutes of Health. Dr. Ferrada, Dr. Sandhu, and Dr. Kim have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
Among patients with the recently defined severe autoinflammatory syndrome VEXAS, those who are transfusion dependent or have a specific amino acid substitution are at highest risk for death, whereas those with ear chondritis are at significantly lower risk, a multinational team of investigators has found.
Their study of mortality and predictors of survival among patients with genetically confirmed VEXAS showed that patients with a VEXAS variant resulting in an amino acid substitution of a methionine for a valine had a 3.5-fold higher risk for death, compared with patients with either a methionine-to-threonine substitution or a methionine-to-leucine swap.
Transfusion dependence was an independent predictor of mortality. Patients who became dependent on transfusions after symptom onset had a nearly threefold higher risk for death, reported Marcela A. Ferrada, MD, a clinical fellow at the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
“These findings should inform risk assessment and clinical management in patients with VEXAS syndrome,” she said in an oral abstract presentation during the virtual annual meeting of the American College of Rheumatology.
“These genetic findings have proven right now to be not only diagnostic, but we have shown that they’re also prognostic, and we hope that this is going to help us identify patients who could have more aggressive treatment,” Dr. Ferrada said.
She also discussed her findings in a media briefing held 2 days prior to her plenary presentation. At that briefing, this news organization asked participating clinicians whether they had patients who they suspected may have had undiagnosed VEXAS.
“My answer to that is interesting,” replied moderator Vaneet Sandhu, MD, from Loma Linda (Calif.) University and Riverside University Health System.
“In the last couple of days, I’ve been reading about VEXAS, and actually texted one of my colleagues yesterday and said, ‘Hey, you know these patients we’ve been seeing who have these strange rashes and chondritis and have maybe a diagnosis of leukocytoclastic vasculitis or something else – are we not diagnosing these patients?’ ” she said.
“I think we are looking at every patient with chondritis and reexamining their phenotype. We had dismissed certain symptoms because they didn’t fit the archetype for relapsing polychondritis, for example, but it could be VEXAS,” said Alfred Kim, MD, PhD, of Washington University in St. Louis, who also presented data during the briefing.
Three variants
VEXAS is caused by somatic mutations in UBA1, a gene that initiates cytoplasmic ubiquitylation, a process by which misfolded proteins are tagged for degradation.
The syndrome’s name is an acronym descriptive of the major features:
- Vacuoles in bone marrow cells.
- E-1 activating enzyme that UBA1 encodes for.
- X-linked.
- Autoinflammatory.
- Somatic mutation featuring hematologic mosaicism.
VEXAS results in rheumatologic, dermatologic, and hematologic symptoms that are often misdiagnosed as being caused by treatment-refractory relapsing polychondritis, polyarteritis nodosa, Sweet syndrome, giant cell arteritis, or myelodysplastic syndrome (MDS).
VEXAS was identified as a distinct syndrome within the past year by Dr. Ferrada and other investigators at NIAMS, the National Human Genome Research Institute, and other institutions.
In the study reported at ACR 2021, Dr. Ferrada and colleagues assessed 83 men who had been referred for genetic testing for VEXAS at the National Institutes of Health, in Bethesda, Md., and at Leeds (England) Teaching Hospitals NHS Trust.
All patients were confirmed to have VEXAS-defining genetic mutations in UBA1 by Sanger sequencing of peripheral blood samples. Only those patients with mutations at codon p.Met41 were included in the investigators’ analysis. Mutations at that site account for nearly all cases of VEXAS that have been identified to date.
The most common clinical manifestation of VEXAS was skin involvement, which occurred in all but one of the 83 patients. Other common manifestations included arthritis (58 patients), pulmonary infiltrates (57 patients), and ear chondritis (54 patients).
Fifteen patients were found to have the leucine variant, 18 had the valine variant, and 50 had the threonine variant. The median age at disease onset was 66 years in the leucine and threonine variant groups and 65 in the valine variant group.
The clinical diagnosis differed according to genotype: 4 of 18 patients (22%) with the valine variant were diagnosed with relapsing polychondritis, compared with 8 of 15 (53%) with the leucine variant and 31 of 50 (62%) with the threonine variant (P = .01).
In contrast, 55% of patients with valine genotype were diagnosed with undifferentiated fever, compared with 6% of those with the leucine and 16% with the threonine genotypes (P = .001). More patients with the leucine variant (60%) were diagnosed with Sweet syndrome, compared with 11% and 14% of patients with the valine and threonine variants, respectively (P = .001).
There was no significant difference among the three genotypes in the percentage of patients diagnosed with MDS.
The follow-up period ranged from 1 to 18 years (median, 4.7 years). The median survival time from disease onset for all patients was 10 years.
Among patients with the valine variant, median survival was 9 years, which was significantly less than among patients with the other two variants (P = .01).
In univariable analysis, independent predictors of mortality were ear chondritis (hazard ratio, 0.26; P = .005), transfusion dependence, a time-dependent variable (HR, 2.59; P = .03), and the valine variant (HR, 3.5; P = .008).
The association between VEXAS genotype and phenotype could be explained by the finding that, among patients with the valine variant, there was significantly less translation of the catalytically proficient UBA1b isoform than in patients with the other two variants, Dr. Ferrada said.
Therapeutic options
Dr. Ferrada noted that to date no drugs have been shown to provide consistent therapeutic benefits for patients with VEXAS, but evidence as to the etiology of the syndrome points to possible treatment approaches.
“All of these findings I think are extremely important to help us guide management of these patients, as we know that the mutation is located in the stem cells in the bone marrow. So we suspect that doing a bone marrow transplant in these patients is going to be curative,” Dr. Ferrada said during the briefing.
Investigators are planning a phase 2 trial of allogeneic hematopoietic stem cell transplant for patients with VEXAS.
The study was supported by the National Institutes of Health. Dr. Ferrada, Dr. Sandhu, and Dr. Kim have disclosed no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM ACR 2021
Risk-based antenatal type-and-screen blood testing safe and economical
Implementing a selective type-and-screen blood testing policy in the labor and delivery unit was associated with projected annual savings of close to $200,000, a large single-center study found. Furthermore, there was no evidence of increased maternal morbidity in the university-based facility performing more than 4,400 deliveries per year, according to Ashley E. Benson, MD, MA, of the department of obstetrics and gynecology at the University of Utah, Salt Lake City, and colleagues.
The study, published in Obstetrics & Gynecology, evaluated patient safety, resource utilization, and transfusion-related costs after a policy change from universal type and screen to selective, risk-based type and screen on admission to labor and delivery.
“There had been some national interest in moving toward decreased resource utilization, and findings that universal screening was not cost effective,” Dr. Benson, who has since relocated to Oregon Health & Science University, Portland, said in an interview. An earlier cost-effective modeling study at her center had suggested that universal test and screen was not cost effective and likely not safer either. “So based on that data we felt an implementation study was warranted.”
The switch to a selective policy was made in 2018, after which her group compared outcomes from October 2017 to September 2019, looking those both 1 year preimplementation and 1 year post implementation.
One year post implementation, the following outcomes emerged, compared with preimplementation:
- Overall projected saving of $181,000 a year in the maternity unit
- Lower mean monthly type- and screen-related costs, such as those for ABO typing, antibody screen, and antibody workup. cross-matches, hold clots, and transfused products: $9,753 vs. $20,676 in the preimplementation year (P < .001)
- A lower mean monthly cost of total transfusion preparedness: $25,090 vs. $39,211 (P < .001)
- No differences in emergency-release transfusion events (four vs. three, P = .99),the study’s primary safety outcome
- Fewer emergency-release red blood cell units transfused (9 vs. 24, P = .002) and O-negative RBC units transfused (8 vs. 18, P = .016)
- No differences in hysterectomies (0.05% vs. 0.1%, P = .44) and ICU admissions (0.45% vs. 0.51%, P = .43)
“In a year of selective type and screen, we saw a 51% reduction in costs related to type and screen, and a 38% reduction in overall transfusion-related costs,” the authors wrote. “This study supports other literature suggesting that more judicious use of type and screen may be safe and cost effective.”
Dr. Benson said the results were positively received when presented a meeting 2 years ago but the published version has yet to prompt feedback.
The study
Antepartum patients underwent transfusion preparedness tests according to the center’s standard antenatal admission order sets and were risk stratified in alignment with California Maternal Quality Care Collaborative recommendations. The mean maternal age of patients in both time periods was similar at just over 29 years and the mean gestational age at delivery was just under 38 weeks.
Under the new policy, a “hold clot” is obtained for women stratified as low or medium risk on admission. In this instance, a tube of patient blood is held in the blood bank but processed only if needed, as in the event of active hemorrhage or an order for transfusion. A blood cross-match is obtained on all women stratified as high risk or having a prior positive antibody screen.
Relevant costs were the direct costs of transfusion-related testing in the labor and delivery unit from a health system perspective.
Obstetric hemorrhage is the leading cause of maternal death worldwide, the authors pointed out. While transfusion in obstetric patients occurs in only 1% or 2% of all deliveries it is nevertheless difficult to predict which patients will need transfusion, with only 2%-8% of those stratified as high risk ultimately requiring transfusion. Although obstetric hemorrhage safety bundles recommend risk stratification on admission to labor and delivery with selective type and screen for higher-risk individuals, for safety and simplicity’s sake, many labor and delivery units perform universal type and screen.
The authors cautioned that these results occurred in an academic tertiary care center with systems fine-tuned to deal with active hemorrhage and deliver timely transfusable blood. “At the moment we don’t have enough data to say whether the selective approach would be safe in hospitals with more limited blood bank capacity and access and fewer transfusion specialists in a setting optimized to respond to emergent needs, Dr. Benson said.
Katayoun F. M. Fomani, MD, a transfusion medicine specialist and medical director of blood bank and transfusion services at Long Island Jewish Medical Center, New York, agreed. “This approach only works in a controlled environment such as in this study where eligible women were assessed antenatally at the same center, but it would not work at every institution,” she said in an interview. “In addition, all patients were assessed according to the California Collaborative guideline, which itself increases the safety level but is not followed everywhere.”
The obstetric division at her hospital in New York adheres to the universal type and screen. “We have patients coming in from outside whose antenatal testing was not done at our hospital,” she said. “For this selective approach to work you need a controlled population and the electronic resources and personnel to follow each patient carefully.”
The authors indicated no specific funding for this study and disclosed no potential conflicts of interest. Dr. Fomani had no potential competing interests to declare.
Implementing a selective type-and-screen blood testing policy in the labor and delivery unit was associated with projected annual savings of close to $200,000, a large single-center study found. Furthermore, there was no evidence of increased maternal morbidity in the university-based facility performing more than 4,400 deliveries per year, according to Ashley E. Benson, MD, MA, of the department of obstetrics and gynecology at the University of Utah, Salt Lake City, and colleagues.
The study, published in Obstetrics & Gynecology, evaluated patient safety, resource utilization, and transfusion-related costs after a policy change from universal type and screen to selective, risk-based type and screen on admission to labor and delivery.
“There had been some national interest in moving toward decreased resource utilization, and findings that universal screening was not cost effective,” Dr. Benson, who has since relocated to Oregon Health & Science University, Portland, said in an interview. An earlier cost-effective modeling study at her center had suggested that universal test and screen was not cost effective and likely not safer either. “So based on that data we felt an implementation study was warranted.”
The switch to a selective policy was made in 2018, after which her group compared outcomes from October 2017 to September 2019, looking those both 1 year preimplementation and 1 year post implementation.
One year post implementation, the following outcomes emerged, compared with preimplementation:
- Overall projected saving of $181,000 a year in the maternity unit
- Lower mean monthly type- and screen-related costs, such as those for ABO typing, antibody screen, and antibody workup. cross-matches, hold clots, and transfused products: $9,753 vs. $20,676 in the preimplementation year (P < .001)
- A lower mean monthly cost of total transfusion preparedness: $25,090 vs. $39,211 (P < .001)
- No differences in emergency-release transfusion events (four vs. three, P = .99),the study’s primary safety outcome
- Fewer emergency-release red blood cell units transfused (9 vs. 24, P = .002) and O-negative RBC units transfused (8 vs. 18, P = .016)
- No differences in hysterectomies (0.05% vs. 0.1%, P = .44) and ICU admissions (0.45% vs. 0.51%, P = .43)
“In a year of selective type and screen, we saw a 51% reduction in costs related to type and screen, and a 38% reduction in overall transfusion-related costs,” the authors wrote. “This study supports other literature suggesting that more judicious use of type and screen may be safe and cost effective.”
Dr. Benson said the results were positively received when presented a meeting 2 years ago but the published version has yet to prompt feedback.
The study
Antepartum patients underwent transfusion preparedness tests according to the center’s standard antenatal admission order sets and were risk stratified in alignment with California Maternal Quality Care Collaborative recommendations. The mean maternal age of patients in both time periods was similar at just over 29 years and the mean gestational age at delivery was just under 38 weeks.
Under the new policy, a “hold clot” is obtained for women stratified as low or medium risk on admission. In this instance, a tube of patient blood is held in the blood bank but processed only if needed, as in the event of active hemorrhage or an order for transfusion. A blood cross-match is obtained on all women stratified as high risk or having a prior positive antibody screen.
Relevant costs were the direct costs of transfusion-related testing in the labor and delivery unit from a health system perspective.
Obstetric hemorrhage is the leading cause of maternal death worldwide, the authors pointed out. While transfusion in obstetric patients occurs in only 1% or 2% of all deliveries it is nevertheless difficult to predict which patients will need transfusion, with only 2%-8% of those stratified as high risk ultimately requiring transfusion. Although obstetric hemorrhage safety bundles recommend risk stratification on admission to labor and delivery with selective type and screen for higher-risk individuals, for safety and simplicity’s sake, many labor and delivery units perform universal type and screen.
The authors cautioned that these results occurred in an academic tertiary care center with systems fine-tuned to deal with active hemorrhage and deliver timely transfusable blood. “At the moment we don’t have enough data to say whether the selective approach would be safe in hospitals with more limited blood bank capacity and access and fewer transfusion specialists in a setting optimized to respond to emergent needs, Dr. Benson said.
Katayoun F. M. Fomani, MD, a transfusion medicine specialist and medical director of blood bank and transfusion services at Long Island Jewish Medical Center, New York, agreed. “This approach only works in a controlled environment such as in this study where eligible women were assessed antenatally at the same center, but it would not work at every institution,” she said in an interview. “In addition, all patients were assessed according to the California Collaborative guideline, which itself increases the safety level but is not followed everywhere.”
The obstetric division at her hospital in New York adheres to the universal type and screen. “We have patients coming in from outside whose antenatal testing was not done at our hospital,” she said. “For this selective approach to work you need a controlled population and the electronic resources and personnel to follow each patient carefully.”
The authors indicated no specific funding for this study and disclosed no potential conflicts of interest. Dr. Fomani had no potential competing interests to declare.
Implementing a selective type-and-screen blood testing policy in the labor and delivery unit was associated with projected annual savings of close to $200,000, a large single-center study found. Furthermore, there was no evidence of increased maternal morbidity in the university-based facility performing more than 4,400 deliveries per year, according to Ashley E. Benson, MD, MA, of the department of obstetrics and gynecology at the University of Utah, Salt Lake City, and colleagues.
The study, published in Obstetrics & Gynecology, evaluated patient safety, resource utilization, and transfusion-related costs after a policy change from universal type and screen to selective, risk-based type and screen on admission to labor and delivery.
“There had been some national interest in moving toward decreased resource utilization, and findings that universal screening was not cost effective,” Dr. Benson, who has since relocated to Oregon Health & Science University, Portland, said in an interview. An earlier cost-effective modeling study at her center had suggested that universal test and screen was not cost effective and likely not safer either. “So based on that data we felt an implementation study was warranted.”
The switch to a selective policy was made in 2018, after which her group compared outcomes from October 2017 to September 2019, looking those both 1 year preimplementation and 1 year post implementation.
One year post implementation, the following outcomes emerged, compared with preimplementation:
- Overall projected saving of $181,000 a year in the maternity unit
- Lower mean monthly type- and screen-related costs, such as those for ABO typing, antibody screen, and antibody workup. cross-matches, hold clots, and transfused products: $9,753 vs. $20,676 in the preimplementation year (P < .001)
- A lower mean monthly cost of total transfusion preparedness: $25,090 vs. $39,211 (P < .001)
- No differences in emergency-release transfusion events (four vs. three, P = .99),the study’s primary safety outcome
- Fewer emergency-release red blood cell units transfused (9 vs. 24, P = .002) and O-negative RBC units transfused (8 vs. 18, P = .016)
- No differences in hysterectomies (0.05% vs. 0.1%, P = .44) and ICU admissions (0.45% vs. 0.51%, P = .43)
“In a year of selective type and screen, we saw a 51% reduction in costs related to type and screen, and a 38% reduction in overall transfusion-related costs,” the authors wrote. “This study supports other literature suggesting that more judicious use of type and screen may be safe and cost effective.”
Dr. Benson said the results were positively received when presented a meeting 2 years ago but the published version has yet to prompt feedback.
The study
Antepartum patients underwent transfusion preparedness tests according to the center’s standard antenatal admission order sets and were risk stratified in alignment with California Maternal Quality Care Collaborative recommendations. The mean maternal age of patients in both time periods was similar at just over 29 years and the mean gestational age at delivery was just under 38 weeks.
Under the new policy, a “hold clot” is obtained for women stratified as low or medium risk on admission. In this instance, a tube of patient blood is held in the blood bank but processed only if needed, as in the event of active hemorrhage or an order for transfusion. A blood cross-match is obtained on all women stratified as high risk or having a prior positive antibody screen.
Relevant costs were the direct costs of transfusion-related testing in the labor and delivery unit from a health system perspective.
Obstetric hemorrhage is the leading cause of maternal death worldwide, the authors pointed out. While transfusion in obstetric patients occurs in only 1% or 2% of all deliveries it is nevertheless difficult to predict which patients will need transfusion, with only 2%-8% of those stratified as high risk ultimately requiring transfusion. Although obstetric hemorrhage safety bundles recommend risk stratification on admission to labor and delivery with selective type and screen for higher-risk individuals, for safety and simplicity’s sake, many labor and delivery units perform universal type and screen.
The authors cautioned that these results occurred in an academic tertiary care center with systems fine-tuned to deal with active hemorrhage and deliver timely transfusable blood. “At the moment we don’t have enough data to say whether the selective approach would be safe in hospitals with more limited blood bank capacity and access and fewer transfusion specialists in a setting optimized to respond to emergent needs, Dr. Benson said.
Katayoun F. M. Fomani, MD, a transfusion medicine specialist and medical director of blood bank and transfusion services at Long Island Jewish Medical Center, New York, agreed. “This approach only works in a controlled environment such as in this study where eligible women were assessed antenatally at the same center, but it would not work at every institution,” she said in an interview. “In addition, all patients were assessed according to the California Collaborative guideline, which itself increases the safety level but is not followed everywhere.”
The obstetric division at her hospital in New York adheres to the universal type and screen. “We have patients coming in from outside whose antenatal testing was not done at our hospital,” she said. “For this selective approach to work you need a controlled population and the electronic resources and personnel to follow each patient carefully.”
The authors indicated no specific funding for this study and disclosed no potential conflicts of interest. Dr. Fomani had no potential competing interests to declare.
FROM OBSTETRICS & GYNECOLOGY
Antithrombotic therapy not warranted in COVID-19 outpatients
Antithrombotic therapy in clinically stable, nonhospitalized COVID-19 patients does not offer protection against adverse cardiovascular or pulmonary events, new randomized clinical trial results suggest.

Antithrombotic therapy has proven useful in acutely ill inpatients with COVID-19, but in this study, treatment with aspirin or apixaban (Eliquis) did not reduce the rate of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary causes in patients ill with COVID-19 but who were not hospitalized.
“Among symptomatic, clinically stable outpatients with COVID-19, treatment with aspirin or apixaban compared with placebo did not reduce the rate of a composite clinical outcome,” the authors conclude. “However, the study was terminated after enrollment of 9% of participants because of a primary event rate lower than anticipated.”
The study, which was led by Jean M. Connors, MD, Brigham and Women’s Hospital, Boston, was published online October 11 in JAMA.
The ACTIV-4B Outpatient Thrombosis Prevention Trial was a randomized, adaptive, double-blind, placebo-controlled trial that sought to compare anticoagulant and antiplatelet therapy among 7,000 symptomatic but clinically stable outpatients with COVID-19.
The trial was conducted at 52 sites in the U.S. between Sept. 2020 and June 2021, with final follow-up this past August 5, and involved minimal face-to-face interactions with study participants.
Patients were randomized in a 1:1:1:1 ratio to aspirin (81 mg orally once daily; n = 164 patients), prophylactic-dose apixaban (2.5 mg orally twice daily; n = 165), therapeutic-dose apixaban (5 mg orally twice daily; n = 164), or placebo (n = 164) for 45 days.
The primary endpoint was a composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause.
The trial was terminated early this past June by the independent data monitoring committee because of lower than anticipated event rates. At the time, just 657 symptomatic outpatients with COVID-19 had been enrolled.
The median age of the study participants was 54 years (Interquartile Range [IQR] 46-59); 59% were women.
The median time from diagnosis to randomization was 7 days, and the median time from randomization to initiation of study medications was 3 days.
The trial’s primary efficacy and safety analyses were restricted to patients who received at least one dose of trial medication, for a final number of 558 patients.
Among these patients, the primary endpoint occurred in 1 patient (0.7%) in the aspirin group, 1 patient (0.7%) in the 2.5 mg apixaban group, 2 patients (1.4%) in the 5-mg apixaban group, and 1 patient (0.7%) in the placebo group.
The researchers found that the absolute risk reductions compared with placebo for the primary outcome were 0.0% (95% confidence interval not calculable) in the aspirin group, 0.7% (95% confidence interval, -2.1% to 4.1%) in the prophylactic-dose apixaban group, and 1.4% (95% CI, -1.5% to 5%) in the therapeutic-dose apixaban group.
No major bleeding events were reported.
The absolute risk differences compared with placebo for clinically relevant nonmajor bleeding events were 2% (95% CI, -2.7% to 6.8%) in the aspirin group, 4.5% (95% CI, -0.7% to 10.2%) in the prophylactic-dose apixaban group, and 6.9% (95% CI, 1.4% to 12.9%) in the therapeutic-dose apixaban group.
Safety and efficacy results were similar in all randomly assigned patients.
The researchers speculated that a combination of two demographic shifts over time may have led to the lower than anticipated rate of events in ACTIV-4B.
“First, the threshold for hospital admission has markedly declined since the beginning of the pandemic, such that hospitalization is no longer limited almost exclusively to those with severe pulmonary distress likely to require mechanical ventilation,” they write. “As a result, the severity of illness among individuals with COVID-19 and destined for outpatient care has declined.”
“Second, at least within the U.S., where the trial was conducted, individuals currently being infected with SARS-CoV-2 tend to be younger and have fewer comorbidities when compared with individuals with incident infection at the onset of the pandemic,” they add.
Further, COVID-19 testing was quite limited early in the pandemic, they note, “and it is possible that the anticipated event rates based on data from registries available at that time were overestimated because the denominator (that is, the number of infected individuals overall) was essentially unknown.”
Robust evidence
“The ACTIV-4B trial is the first randomized trial to generate robust evidence about the effects of antithrombotic therapy in outpatients with COVID-19,” Otavio Berwanger, MD, PhD, director of the Academic Research Organization, Hospital Israelita Albert Einstein, Sao Paulo-SP, Brazil, told this news organization.
“It should be noted that this was a well-designed trial with low risk of bias. On the other hand, the main limitation is the low number of events and, consequently, the limited statistical power,” said Dr. Berwanger, who wrote an accompanying editorial.
The ACTIV-4B trial has immediate implications for clinical practice, he added.
“In this sense, considering the neutral results for major cardiopulmonary outcomes, the use of aspirin or apixaban for the management of outpatients with COVID-19 should not be recommended.”
ACTIV-4B also provides useful information for the steering committees of other ongoing trials of antithrombotic therapy for patients with COVID-19 who are not hospitalized, Dr. Berwanger added.
“In this sense, probably issues like statistical power, outcome choices, recruitment feasibility, and even futility would need to be revisited. And finally, lessons learned from the implementation of an innovative, pragmatic, and decentralized trial design represent an important legacy for future trials in cardiovascular diseases and other common conditions,” he said.
The study was funded by the National Institutes of Health, and the National Heart, Lung, and Blood Institute. Dr. Connors reports financial relationships with Bristol-Myers Squibb, Pfizer, Abbott, Alnylam, Takeda, Roche, and Sanofi. Dr. Berwanger reports financial relationships with AstraZeneca, Amgen, Servier, Bristol-Myers Squibb, Bayer, Novartis, Pfizer, and Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
Antithrombotic therapy in clinically stable, nonhospitalized COVID-19 patients does not offer protection against adverse cardiovascular or pulmonary events, new randomized clinical trial results suggest.
Antithrombotic therapy has proven useful in acutely ill inpatients with COVID-19, but in this study, treatment with aspirin or apixaban (Eliquis) did not reduce the rate of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary causes in patients ill with COVID-19 but who were not hospitalized.
“Among symptomatic, clinically stable outpatients with COVID-19, treatment with aspirin or apixaban compared with placebo did not reduce the rate of a composite clinical outcome,” the authors conclude. “However, the study was terminated after enrollment of 9% of participants because of a primary event rate lower than anticipated.”
The study, which was led by Jean M. Connors, MD, Brigham and Women’s Hospital, Boston, was published online October 11 in JAMA.
The ACTIV-4B Outpatient Thrombosis Prevention Trial was a randomized, adaptive, double-blind, placebo-controlled trial that sought to compare anticoagulant and antiplatelet therapy among 7,000 symptomatic but clinically stable outpatients with COVID-19.
The trial was conducted at 52 sites in the U.S. between Sept. 2020 and June 2021, with final follow-up this past August 5, and involved minimal face-to-face interactions with study participants.
Patients were randomized in a 1:1:1:1 ratio to aspirin (81 mg orally once daily; n = 164 patients), prophylactic-dose apixaban (2.5 mg orally twice daily; n = 165), therapeutic-dose apixaban (5 mg orally twice daily; n = 164), or placebo (n = 164) for 45 days.
The primary endpoint was a composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause.
The trial was terminated early this past June by the independent data monitoring committee because of lower than anticipated event rates. At the time, just 657 symptomatic outpatients with COVID-19 had been enrolled.
The median age of the study participants was 54 years (Interquartile Range [IQR] 46-59); 59% were women.
The median time from diagnosis to randomization was 7 days, and the median time from randomization to initiation of study medications was 3 days.
The trial’s primary efficacy and safety analyses were restricted to patients who received at least one dose of trial medication, for a final number of 558 patients.
Among these patients, the primary endpoint occurred in 1 patient (0.7%) in the aspirin group, 1 patient (0.7%) in the 2.5 mg apixaban group, 2 patients (1.4%) in the 5-mg apixaban group, and 1 patient (0.7%) in the placebo group.
The researchers found that the absolute risk reductions compared with placebo for the primary outcome were 0.0% (95% confidence interval not calculable) in the aspirin group, 0.7% (95% confidence interval, -2.1% to 4.1%) in the prophylactic-dose apixaban group, and 1.4% (95% CI, -1.5% to 5%) in the therapeutic-dose apixaban group.
No major bleeding events were reported.
The absolute risk differences compared with placebo for clinically relevant nonmajor bleeding events were 2% (95% CI, -2.7% to 6.8%) in the aspirin group, 4.5% (95% CI, -0.7% to 10.2%) in the prophylactic-dose apixaban group, and 6.9% (95% CI, 1.4% to 12.9%) in the therapeutic-dose apixaban group.
Safety and efficacy results were similar in all randomly assigned patients.
The researchers speculated that a combination of two demographic shifts over time may have led to the lower than anticipated rate of events in ACTIV-4B.
“First, the threshold for hospital admission has markedly declined since the beginning of the pandemic, such that hospitalization is no longer limited almost exclusively to those with severe pulmonary distress likely to require mechanical ventilation,” they write. “As a result, the severity of illness among individuals with COVID-19 and destined for outpatient care has declined.”
“Second, at least within the U.S., where the trial was conducted, individuals currently being infected with SARS-CoV-2 tend to be younger and have fewer comorbidities when compared with individuals with incident infection at the onset of the pandemic,” they add.
Further, COVID-19 testing was quite limited early in the pandemic, they note, “and it is possible that the anticipated event rates based on data from registries available at that time were overestimated because the denominator (that is, the number of infected individuals overall) was essentially unknown.”
Robust evidence
“The ACTIV-4B trial is the first randomized trial to generate robust evidence about the effects of antithrombotic therapy in outpatients with COVID-19,” Otavio Berwanger, MD, PhD, director of the Academic Research Organization, Hospital Israelita Albert Einstein, Sao Paulo-SP, Brazil, told this news organization.
“It should be noted that this was a well-designed trial with low risk of bias. On the other hand, the main limitation is the low number of events and, consequently, the limited statistical power,” said Dr. Berwanger, who wrote an accompanying editorial.
The ACTIV-4B trial has immediate implications for clinical practice, he added.
“In this sense, considering the neutral results for major cardiopulmonary outcomes, the use of aspirin or apixaban for the management of outpatients with COVID-19 should not be recommended.”
ACTIV-4B also provides useful information for the steering committees of other ongoing trials of antithrombotic therapy for patients with COVID-19 who are not hospitalized, Dr. Berwanger added.
“In this sense, probably issues like statistical power, outcome choices, recruitment feasibility, and even futility would need to be revisited. And finally, lessons learned from the implementation of an innovative, pragmatic, and decentralized trial design represent an important legacy for future trials in cardiovascular diseases and other common conditions,” he said.
The study was funded by the National Institutes of Health, and the National Heart, Lung, and Blood Institute. Dr. Connors reports financial relationships with Bristol-Myers Squibb, Pfizer, Abbott, Alnylam, Takeda, Roche, and Sanofi. Dr. Berwanger reports financial relationships with AstraZeneca, Amgen, Servier, Bristol-Myers Squibb, Bayer, Novartis, Pfizer, and Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
Antithrombotic therapy in clinically stable, nonhospitalized COVID-19 patients does not offer protection against adverse cardiovascular or pulmonary events, new randomized clinical trial results suggest.
Antithrombotic therapy has proven useful in acutely ill inpatients with COVID-19, but in this study, treatment with aspirin or apixaban (Eliquis) did not reduce the rate of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary causes in patients ill with COVID-19 but who were not hospitalized.
“Among symptomatic, clinically stable outpatients with COVID-19, treatment with aspirin or apixaban compared with placebo did not reduce the rate of a composite clinical outcome,” the authors conclude. “However, the study was terminated after enrollment of 9% of participants because of a primary event rate lower than anticipated.”
The study, which was led by Jean M. Connors, MD, Brigham and Women’s Hospital, Boston, was published online October 11 in JAMA.
The ACTIV-4B Outpatient Thrombosis Prevention Trial was a randomized, adaptive, double-blind, placebo-controlled trial that sought to compare anticoagulant and antiplatelet therapy among 7,000 symptomatic but clinically stable outpatients with COVID-19.
The trial was conducted at 52 sites in the U.S. between Sept. 2020 and June 2021, with final follow-up this past August 5, and involved minimal face-to-face interactions with study participants.
Patients were randomized in a 1:1:1:1 ratio to aspirin (81 mg orally once daily; n = 164 patients), prophylactic-dose apixaban (2.5 mg orally twice daily; n = 165), therapeutic-dose apixaban (5 mg orally twice daily; n = 164), or placebo (n = 164) for 45 days.
The primary endpoint was a composite of all-cause mortality, symptomatic venous or arterial thromboembolism, myocardial infarction, stroke, or hospitalization for cardiovascular or pulmonary cause.
The trial was terminated early this past June by the independent data monitoring committee because of lower than anticipated event rates. At the time, just 657 symptomatic outpatients with COVID-19 had been enrolled.
The median age of the study participants was 54 years (Interquartile Range [IQR] 46-59); 59% were women.
The median time from diagnosis to randomization was 7 days, and the median time from randomization to initiation of study medications was 3 days.
The trial’s primary efficacy and safety analyses were restricted to patients who received at least one dose of trial medication, for a final number of 558 patients.
Among these patients, the primary endpoint occurred in 1 patient (0.7%) in the aspirin group, 1 patient (0.7%) in the 2.5 mg apixaban group, 2 patients (1.4%) in the 5-mg apixaban group, and 1 patient (0.7%) in the placebo group.
The researchers found that the absolute risk reductions compared with placebo for the primary outcome were 0.0% (95% confidence interval not calculable) in the aspirin group, 0.7% (95% confidence interval, -2.1% to 4.1%) in the prophylactic-dose apixaban group, and 1.4% (95% CI, -1.5% to 5%) in the therapeutic-dose apixaban group.
No major bleeding events were reported.
The absolute risk differences compared with placebo for clinically relevant nonmajor bleeding events were 2% (95% CI, -2.7% to 6.8%) in the aspirin group, 4.5% (95% CI, -0.7% to 10.2%) in the prophylactic-dose apixaban group, and 6.9% (95% CI, 1.4% to 12.9%) in the therapeutic-dose apixaban group.
Safety and efficacy results were similar in all randomly assigned patients.
The researchers speculated that a combination of two demographic shifts over time may have led to the lower than anticipated rate of events in ACTIV-4B.
“First, the threshold for hospital admission has markedly declined since the beginning of the pandemic, such that hospitalization is no longer limited almost exclusively to those with severe pulmonary distress likely to require mechanical ventilation,” they write. “As a result, the severity of illness among individuals with COVID-19 and destined for outpatient care has declined.”
“Second, at least within the U.S., where the trial was conducted, individuals currently being infected with SARS-CoV-2 tend to be younger and have fewer comorbidities when compared with individuals with incident infection at the onset of the pandemic,” they add.
Further, COVID-19 testing was quite limited early in the pandemic, they note, “and it is possible that the anticipated event rates based on data from registries available at that time were overestimated because the denominator (that is, the number of infected individuals overall) was essentially unknown.”
Robust evidence
“The ACTIV-4B trial is the first randomized trial to generate robust evidence about the effects of antithrombotic therapy in outpatients with COVID-19,” Otavio Berwanger, MD, PhD, director of the Academic Research Organization, Hospital Israelita Albert Einstein, Sao Paulo-SP, Brazil, told this news organization.
“It should be noted that this was a well-designed trial with low risk of bias. On the other hand, the main limitation is the low number of events and, consequently, the limited statistical power,” said Dr. Berwanger, who wrote an accompanying editorial.
The ACTIV-4B trial has immediate implications for clinical practice, he added.
“In this sense, considering the neutral results for major cardiopulmonary outcomes, the use of aspirin or apixaban for the management of outpatients with COVID-19 should not be recommended.”
ACTIV-4B also provides useful information for the steering committees of other ongoing trials of antithrombotic therapy for patients with COVID-19 who are not hospitalized, Dr. Berwanger added.
“In this sense, probably issues like statistical power, outcome choices, recruitment feasibility, and even futility would need to be revisited. And finally, lessons learned from the implementation of an innovative, pragmatic, and decentralized trial design represent an important legacy for future trials in cardiovascular diseases and other common conditions,” he said.
The study was funded by the National Institutes of Health, and the National Heart, Lung, and Blood Institute. Dr. Connors reports financial relationships with Bristol-Myers Squibb, Pfizer, Abbott, Alnylam, Takeda, Roche, and Sanofi. Dr. Berwanger reports financial relationships with AstraZeneca, Amgen, Servier, Bristol-Myers Squibb, Bayer, Novartis, Pfizer, and Boehringer Ingelheim.
A version of this article first appeared on Medscape.com.
Autopsy findings reveal venous thromboembolism in patients with COVID-19
Background: Despite the increased mortality rate of the novel coronavirus compared with influenza, little is understood about its pathogenicity. Prior studies have identified D-dimer levels, high Sequential Organ Failure Assessment score, and older age as markers for more severe disease and mortality. The specific cause of death of COVID-19 remains largely unknown.

Study design: Prospective cohort study.
Setting: Single academic center in Germany.
Synopsis: A complete autopsy was performed on 12 consecutive COVID-19 patient deaths at a single center. Seven had evidence of venous thromboembolism (VTE): three with bilateral lower extremity deep venous thrombosis (DVT) and four with massive pulmonary embolism/associated lower-extremity DVT. Prior to death, VTE was suspected clinically in only a single patient.
This small case series piques interest in the potential underrecognized thromboembolic pathology of COVID-19. While not practice changing, this study highlights the importance of hospitalists staying attuned to further studies regarding VTE prophylaxis in COVID-19.
Bottom line: Autopsies of COVID-19 patients revealed a high incidence of thromboembolic events; COVID-19–induced coagulopathy may play an underrecognized role in pathogenesis.
Citation: Wichmann D et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020;173(4):268-77.
Dr. Fletcher is a hospitalist at the Lexington (Ky.) VA Health Care System.
Background: Despite the increased mortality rate of the novel coronavirus compared with influenza, little is understood about its pathogenicity. Prior studies have identified D-dimer levels, high Sequential Organ Failure Assessment score, and older age as markers for more severe disease and mortality. The specific cause of death of COVID-19 remains largely unknown.
Study design: Prospective cohort study.
Setting: Single academic center in Germany.
Synopsis: A complete autopsy was performed on 12 consecutive COVID-19 patient deaths at a single center. Seven had evidence of venous thromboembolism (VTE): three with bilateral lower extremity deep venous thrombosis (DVT) and four with massive pulmonary embolism/associated lower-extremity DVT. Prior to death, VTE was suspected clinically in only a single patient.
This small case series piques interest in the potential underrecognized thromboembolic pathology of COVID-19. While not practice changing, this study highlights the importance of hospitalists staying attuned to further studies regarding VTE prophylaxis in COVID-19.
Bottom line: Autopsies of COVID-19 patients revealed a high incidence of thromboembolic events; COVID-19–induced coagulopathy may play an underrecognized role in pathogenesis.
Citation: Wichmann D et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020;173(4):268-77.
Dr. Fletcher is a hospitalist at the Lexington (Ky.) VA Health Care System.
Background: Despite the increased mortality rate of the novel coronavirus compared with influenza, little is understood about its pathogenicity. Prior studies have identified D-dimer levels, high Sequential Organ Failure Assessment score, and older age as markers for more severe disease and mortality. The specific cause of death of COVID-19 remains largely unknown.
Study design: Prospective cohort study.
Setting: Single academic center in Germany.
Synopsis: A complete autopsy was performed on 12 consecutive COVID-19 patient deaths at a single center. Seven had evidence of venous thromboembolism (VTE): three with bilateral lower extremity deep venous thrombosis (DVT) and four with massive pulmonary embolism/associated lower-extremity DVT. Prior to death, VTE was suspected clinically in only a single patient.
This small case series piques interest in the potential underrecognized thromboembolic pathology of COVID-19. While not practice changing, this study highlights the importance of hospitalists staying attuned to further studies regarding VTE prophylaxis in COVID-19.
Bottom line: Autopsies of COVID-19 patients revealed a high incidence of thromboembolic events; COVID-19–induced coagulopathy may play an underrecognized role in pathogenesis.
Citation: Wichmann D et al. Autopsy findings and venous thromboembolism in patients with COVID-19. Ann Intern Med. 2020;173(4):268-77.
Dr. Fletcher is a hospitalist at the Lexington (Ky.) VA Health Care System.
Rivaroxaban’s single daily dose may lead to higher bleeding risk than other DOACs
The results, which were published in the Annals of Internal Medicine, could help guide DOAC selection for high-risk groups with a prior history of peptic ulcer disease or major GI bleeding, said lead study authors Arnar Bragi Ingason, MD and Einar S. Björnsson, MD, PhD, in an email.
DOACs treat conditions such as atrial fibrillation, venous thromboembolism, and ischemic stroke and are known to cause GI bleeding. Previous studies have suggested that rivaroxaban poses a higher GI-bleeding risk than other DOACs.
These studies, which used large administrative databases, “had an inherent risk of selection bias due to insurance status, age, and comorbidities due to their origin from insurance/administrative databases. In addition, they lacked phenotypic details on GI bleeding events,” said Dr. Björnsson and Dr. Ingason, who are both of Landspitali University Hospital, Reykjavik, Iceland,
Daily dosage may exacerbate risk
Rivaroxaban is administered as a single daily dose, compared with apixaban’s and dabigatran’s twice-daily regimens. “We hypothesized that this may lead to a greater variance in drug plasma concentration, making these patients more susceptible to GI bleeding,” the lead authors said.
Using data from the Icelandic Medicine Registry, a national database of outpatient prescription information, they compared rates of GI bleeding among new users of apixaban, dabigatran, and rivaroxaban from 2014 to 2019. Overall, 5,868 patients receiving one of the DOACs took part in the study. Among these participants, 3,217 received rivaroxaban, 2,157 received apixaban, and 494 received dabigatran. The researchers used inverse probability weighting, Kaplan–Meier survival estimates, and Cox regression to compare GI bleeding.
Compared with dabigatran, rivaroxaban was associated with a 63%-104% higher overall risk for GI bleeding and 39%-95% higher risk for major GI bleeding. Rivaroxaban also had a 40%-42% higher overall risk for GI bleeding and 49%-50% higher risk for major GI bleeding, compared with apixaban.
The investigators were surprised by the low rate of upper GI bleeding for dabigatran, compared with the other two drugs. “However, these results must be interpreted in the context that the dabigatran group was relatively small,” said Dr. Björnsson and Dr. Ingason via email.
Overall, the study cohort was small, compared with previous registry studies.
Investigators also did not account for account for socioeconomic status or lifestyle factors, such as alcohol consumption or smoking. “However, because the cost of all DOACs is similar in Iceland, selection bias due to socioeconomic status is unlikely,” the investigators reported in their paper. “We are currently working on comparing the rates of thromboembolisms and overall major bleeding events between the drugs,” the lead authors said.
Clinicians should consider location of bleeding
Though retrospective, the study by Ingason et. al. “is likely as close as is feasible to a randomized trial as is possible,” said Don C. Rockey, MD, a professor of medicine at the Medical University of South Carolina, Charleston, in an interview.
“From the clinician’s perspective, it is important to take away that there may be differences among the DOACs in terms of where in the GI tract the bleeding occurs,” said Dr. Rockey. In the study, the greatest differences appeared to be in the upper GI tract, with rivaroxaban outpacing apixaban and dabigatran. In patients who are at risk for upper GI bleeding, it may be reasonable to consider use of dabigatran or apixaban, he suggested.
“A limitation of the study is that it is likely underpowered overall,” said Dr. Rockey. It also wasn’t clear how many deaths occurred either directly from GI bleeding or as a complication of GI bleeding, he said.The study also didn’t differentiate major bleeding among DOACs specifically in the upper or lower GI tract, Dr. Rockey added.
Other studies yield similar results
Dr. Ingason and Dr. Björnsson said their work complements previous studies, and Neena S. Abraham, MD, MSc , who has conducted a similar investigation to the new study, agreed with that statement.
Data from the last 4 years overwhelmingly show that rivaroxaban is most likely to cause GI bleeding, said Dr. Abraham, professor of medicine and a consultant with Mayo Clinic’s division of gastroenterology and hepatology, in an interview.
A comparative safety study Dr. Abraham coauthored in 2017 of rivaroxaban, apixaban, and dabigatran in a much larger U.S. cohort of 372,380 patients revealed that rivaroxaban had the worst GI bleeding profile. Apixaban was 66% safer than rivaroxaban and 64% safer than dabigatran to prevent gastrointestinal bleeding.
“I believe our group was the first to conduct this study and show clinically significant differences in GI safety of the available direct oral anticoagulants,” she said. Other investigators have since published similar results, and the topic of the new study needs no further investigation, according to Dr. Abraham.
“It is time for physicians to choose a better choice when prescribing a direct oral anticoagulant to their atrial fibrillation patients, and that choice is not rivaroxaban,” she said.
The Icelandic Centre for Research and the Landspítali University Hospital Research Fund provided funds for this study. Dr. Ingason, Dr. Björnsson, Dr. Rockey, and Dr. Abraham reported no disclosures.
The results, which were published in the Annals of Internal Medicine, could help guide DOAC selection for high-risk groups with a prior history of peptic ulcer disease or major GI bleeding, said lead study authors Arnar Bragi Ingason, MD and Einar S. Björnsson, MD, PhD, in an email.
DOACs treat conditions such as atrial fibrillation, venous thromboembolism, and ischemic stroke and are known to cause GI bleeding. Previous studies have suggested that rivaroxaban poses a higher GI-bleeding risk than other DOACs.
These studies, which used large administrative databases, “had an inherent risk of selection bias due to insurance status, age, and comorbidities due to their origin from insurance/administrative databases. In addition, they lacked phenotypic details on GI bleeding events,” said Dr. Björnsson and Dr. Ingason, who are both of Landspitali University Hospital, Reykjavik, Iceland,
Daily dosage may exacerbate risk
Rivaroxaban is administered as a single daily dose, compared with apixaban’s and dabigatran’s twice-daily regimens. “We hypothesized that this may lead to a greater variance in drug plasma concentration, making these patients more susceptible to GI bleeding,” the lead authors said.
Using data from the Icelandic Medicine Registry, a national database of outpatient prescription information, they compared rates of GI bleeding among new users of apixaban, dabigatran, and rivaroxaban from 2014 to 2019. Overall, 5,868 patients receiving one of the DOACs took part in the study. Among these participants, 3,217 received rivaroxaban, 2,157 received apixaban, and 494 received dabigatran. The researchers used inverse probability weighting, Kaplan–Meier survival estimates, and Cox regression to compare GI bleeding.
Compared with dabigatran, rivaroxaban was associated with a 63%-104% higher overall risk for GI bleeding and 39%-95% higher risk for major GI bleeding. Rivaroxaban also had a 40%-42% higher overall risk for GI bleeding and 49%-50% higher risk for major GI bleeding, compared with apixaban.
The investigators were surprised by the low rate of upper GI bleeding for dabigatran, compared with the other two drugs. “However, these results must be interpreted in the context that the dabigatran group was relatively small,” said Dr. Björnsson and Dr. Ingason via email.
Overall, the study cohort was small, compared with previous registry studies.
Investigators also did not account for account for socioeconomic status or lifestyle factors, such as alcohol consumption or smoking. “However, because the cost of all DOACs is similar in Iceland, selection bias due to socioeconomic status is unlikely,” the investigators reported in their paper. “We are currently working on comparing the rates of thromboembolisms and overall major bleeding events between the drugs,” the lead authors said.
Clinicians should consider location of bleeding
Though retrospective, the study by Ingason et. al. “is likely as close as is feasible to a randomized trial as is possible,” said Don C. Rockey, MD, a professor of medicine at the Medical University of South Carolina, Charleston, in an interview.
“From the clinician’s perspective, it is important to take away that there may be differences among the DOACs in terms of where in the GI tract the bleeding occurs,” said Dr. Rockey. In the study, the greatest differences appeared to be in the upper GI tract, with rivaroxaban outpacing apixaban and dabigatran. In patients who are at risk for upper GI bleeding, it may be reasonable to consider use of dabigatran or apixaban, he suggested.
“A limitation of the study is that it is likely underpowered overall,” said Dr. Rockey. It also wasn’t clear how many deaths occurred either directly from GI bleeding or as a complication of GI bleeding, he said.The study also didn’t differentiate major bleeding among DOACs specifically in the upper or lower GI tract, Dr. Rockey added.
Other studies yield similar results
Dr. Ingason and Dr. Björnsson said their work complements previous studies, and Neena S. Abraham, MD, MSc , who has conducted a similar investigation to the new study, agreed with that statement.
Data from the last 4 years overwhelmingly show that rivaroxaban is most likely to cause GI bleeding, said Dr. Abraham, professor of medicine and a consultant with Mayo Clinic’s division of gastroenterology and hepatology, in an interview.
A comparative safety study Dr. Abraham coauthored in 2017 of rivaroxaban, apixaban, and dabigatran in a much larger U.S. cohort of 372,380 patients revealed that rivaroxaban had the worst GI bleeding profile. Apixaban was 66% safer than rivaroxaban and 64% safer than dabigatran to prevent gastrointestinal bleeding.
“I believe our group was the first to conduct this study and show clinically significant differences in GI safety of the available direct oral anticoagulants,” she said. Other investigators have since published similar results, and the topic of the new study needs no further investigation, according to Dr. Abraham.
“It is time for physicians to choose a better choice when prescribing a direct oral anticoagulant to their atrial fibrillation patients, and that choice is not rivaroxaban,” she said.
The Icelandic Centre for Research and the Landspítali University Hospital Research Fund provided funds for this study. Dr. Ingason, Dr. Björnsson, Dr. Rockey, and Dr. Abraham reported no disclosures.
The results, which were published in the Annals of Internal Medicine, could help guide DOAC selection for high-risk groups with a prior history of peptic ulcer disease or major GI bleeding, said lead study authors Arnar Bragi Ingason, MD and Einar S. Björnsson, MD, PhD, in an email.
DOACs treat conditions such as atrial fibrillation, venous thromboembolism, and ischemic stroke and are known to cause GI bleeding. Previous studies have suggested that rivaroxaban poses a higher GI-bleeding risk than other DOACs.
These studies, which used large administrative databases, “had an inherent risk of selection bias due to insurance status, age, and comorbidities due to their origin from insurance/administrative databases. In addition, they lacked phenotypic details on GI bleeding events,” said Dr. Björnsson and Dr. Ingason, who are both of Landspitali University Hospital, Reykjavik, Iceland,
Daily dosage may exacerbate risk
Rivaroxaban is administered as a single daily dose, compared with apixaban’s and dabigatran’s twice-daily regimens. “We hypothesized that this may lead to a greater variance in drug plasma concentration, making these patients more susceptible to GI bleeding,” the lead authors said.
Using data from the Icelandic Medicine Registry, a national database of outpatient prescription information, they compared rates of GI bleeding among new users of apixaban, dabigatran, and rivaroxaban from 2014 to 2019. Overall, 5,868 patients receiving one of the DOACs took part in the study. Among these participants, 3,217 received rivaroxaban, 2,157 received apixaban, and 494 received dabigatran. The researchers used inverse probability weighting, Kaplan–Meier survival estimates, and Cox regression to compare GI bleeding.
Compared with dabigatran, rivaroxaban was associated with a 63%-104% higher overall risk for GI bleeding and 39%-95% higher risk for major GI bleeding. Rivaroxaban also had a 40%-42% higher overall risk for GI bleeding and 49%-50% higher risk for major GI bleeding, compared with apixaban.
The investigators were surprised by the low rate of upper GI bleeding for dabigatran, compared with the other two drugs. “However, these results must be interpreted in the context that the dabigatran group was relatively small,” said Dr. Björnsson and Dr. Ingason via email.
Overall, the study cohort was small, compared with previous registry studies.
Investigators also did not account for account for socioeconomic status or lifestyle factors, such as alcohol consumption or smoking. “However, because the cost of all DOACs is similar in Iceland, selection bias due to socioeconomic status is unlikely,” the investigators reported in their paper. “We are currently working on comparing the rates of thromboembolisms and overall major bleeding events between the drugs,” the lead authors said.
Clinicians should consider location of bleeding
Though retrospective, the study by Ingason et. al. “is likely as close as is feasible to a randomized trial as is possible,” said Don C. Rockey, MD, a professor of medicine at the Medical University of South Carolina, Charleston, in an interview.
“From the clinician’s perspective, it is important to take away that there may be differences among the DOACs in terms of where in the GI tract the bleeding occurs,” said Dr. Rockey. In the study, the greatest differences appeared to be in the upper GI tract, with rivaroxaban outpacing apixaban and dabigatran. In patients who are at risk for upper GI bleeding, it may be reasonable to consider use of dabigatran or apixaban, he suggested.
“A limitation of the study is that it is likely underpowered overall,” said Dr. Rockey. It also wasn’t clear how many deaths occurred either directly from GI bleeding or as a complication of GI bleeding, he said.The study also didn’t differentiate major bleeding among DOACs specifically in the upper or lower GI tract, Dr. Rockey added.
Other studies yield similar results
Dr. Ingason and Dr. Björnsson said their work complements previous studies, and Neena S. Abraham, MD, MSc , who has conducted a similar investigation to the new study, agreed with that statement.
Data from the last 4 years overwhelmingly show that rivaroxaban is most likely to cause GI bleeding, said Dr. Abraham, professor of medicine and a consultant with Mayo Clinic’s division of gastroenterology and hepatology, in an interview.
A comparative safety study Dr. Abraham coauthored in 2017 of rivaroxaban, apixaban, and dabigatran in a much larger U.S. cohort of 372,380 patients revealed that rivaroxaban had the worst GI bleeding profile. Apixaban was 66% safer than rivaroxaban and 64% safer than dabigatran to prevent gastrointestinal bleeding.
“I believe our group was the first to conduct this study and show clinically significant differences in GI safety of the available direct oral anticoagulants,” she said. Other investigators have since published similar results, and the topic of the new study needs no further investigation, according to Dr. Abraham.
“It is time for physicians to choose a better choice when prescribing a direct oral anticoagulant to their atrial fibrillation patients, and that choice is not rivaroxaban,” she said.
The Icelandic Centre for Research and the Landspítali University Hospital Research Fund provided funds for this study. Dr. Ingason, Dr. Björnsson, Dr. Rockey, and Dr. Abraham reported no disclosures.
FROM ANNALS OF INTERNAL MEDICINE
Unexpected thrombocytosis could flag occult cancer
A routine blood test may pack a bigger punch than previously suspected, suggests a recent analysis of over 3 million Canadian patient records.
A finding of thrombocytosis (platelet count >450 x 109/L) was associated with a greatly increased risk for some cancers up to 5 years later.
Overall, a high platelet count increased by 2.7 times the odds of receiving a solid-tumor cancer diagnosis within 2 years (95% confidence interval, 2.6-2.8).
The cancers most likely to be associated with unexpected thrombocytosis were those notorious for late-stage diagnosis due to a lack of early symptoms.
The risk was highest (23.3 times) for ovarian cancer. The risk was 3.8 times higher for pancreatic cancer and 3.5 times higher for cervical cancer.
Lung cancer was 4.4 times more likely within 2 years among patients with thrombocytosis compared to patients with normal platelet counts.
Conversely, breast, prostate, and thyroid cancers were not linked to the finding of thrombocytosis.
The study results were published online in JAMA Network Open on Aug. 12).
One of the authors of the article, Stephen A. Narod, MD, director of the Familial Breast Cancer Research Unit at the Women’s College Research Institute, Toronto, said the results were not unexpected but “very striking.”
“I had a hunch we were going to see this because I’ve seen this in other databases,” said Dr. Narod. “I think what struck me about it was how ubiquitous it was.”
Dr. Narod urged physicians, especially those in primary care, to take note: “If the platelets are high, I would certainly have a concern about lung cancer, colon cancer, and ovarian cancer.”
Dr. Narod and coauthor Vasily Giannakeas, a PhD candidate, pointed out that in their analysis that they were unable to single out cases in which a blood test was performed because the patient complained of symptoms that are associated with cancer. In those cases, thrombocytosis may have been diagnostic, rather than a lifesaving serendipitous finding.
Similar findings were reported recently from the United Kingdom.
A study by Sarah Bailey, PhD, MPH, and colleagues that was published last year in the British Journal of General Practice also found a connection between cancer incidence and platelet count. Dr. Bailey is a senior research fellow at the University of Exeter, England.
However, unlike in the Canadian study, the team led by Dr. Bailey was able to distinguish those patients for whom there were alarm symptoms for cancer. Dr. Bailey and colleagues found that two-thirds of men older than 65 had “no recorded alarm features of cancer in the 21 days before their index platelet count.”
Although this suggests that a routine finding of thrombocytosis could uncover unsuspected cancers, Dr. Bailey is cautious about hailing platelet counts as a new cancer-screening tool.
In emailed comments, Dr. Bailey said, “The crucial part of our study is that it was conducted with patients who were ill enough to see their GP [general practitioner]. Opportunistic measurement in patients who are asymptomatic would be quite a different thing. We would have to study the platelet count and subsequent cancers in asymptomatic patients to know if that was worth doing.”
Perhaps most helpfully, the U.K. study showed that cancer risk was increased even among some patients with normal platelet counts. For example, for men aged 60 and older, lung cancer was 4.7 times more likely among those with high-normal counts (≥326 x 109/L).
Because of this somewhat alarming finding, Dr. Bailey suggested moving away from a focus on absolute values. Rising platelet counts might be more clinically useful, she said.
“Physicians should be on the lookout for any unexplained increase in an individual’s platelet count, irrespective of whether the increased value is over or under the local threshold that is applied to define thrombocytosis,” concluded Dr. Bailey.
Dr. Narod has disclosed no relevant financial relationships. Dr. Bailey is a research fellow of the CanTest Collaborative.
A version of this article first appeared on Medscape.com.
A routine blood test may pack a bigger punch than previously suspected, suggests a recent analysis of over 3 million Canadian patient records.
A finding of thrombocytosis (platelet count >450 x 109/L) was associated with a greatly increased risk for some cancers up to 5 years later.
Overall, a high platelet count increased by 2.7 times the odds of receiving a solid-tumor cancer diagnosis within 2 years (95% confidence interval, 2.6-2.8).
The cancers most likely to be associated with unexpected thrombocytosis were those notorious for late-stage diagnosis due to a lack of early symptoms.
The risk was highest (23.3 times) for ovarian cancer. The risk was 3.8 times higher for pancreatic cancer and 3.5 times higher for cervical cancer.
Lung cancer was 4.4 times more likely within 2 years among patients with thrombocytosis compared to patients with normal platelet counts.
Conversely, breast, prostate, and thyroid cancers were not linked to the finding of thrombocytosis.
The study results were published online in JAMA Network Open on Aug. 12).
One of the authors of the article, Stephen A. Narod, MD, director of the Familial Breast Cancer Research Unit at the Women’s College Research Institute, Toronto, said the results were not unexpected but “very striking.”
“I had a hunch we were going to see this because I’ve seen this in other databases,” said Dr. Narod. “I think what struck me about it was how ubiquitous it was.”
Dr. Narod urged physicians, especially those in primary care, to take note: “If the platelets are high, I would certainly have a concern about lung cancer, colon cancer, and ovarian cancer.”
Dr. Narod and coauthor Vasily Giannakeas, a PhD candidate, pointed out that in their analysis that they were unable to single out cases in which a blood test was performed because the patient complained of symptoms that are associated with cancer. In those cases, thrombocytosis may have been diagnostic, rather than a lifesaving serendipitous finding.
Similar findings were reported recently from the United Kingdom.
A study by Sarah Bailey, PhD, MPH, and colleagues that was published last year in the British Journal of General Practice also found a connection between cancer incidence and platelet count. Dr. Bailey is a senior research fellow at the University of Exeter, England.
However, unlike in the Canadian study, the team led by Dr. Bailey was able to distinguish those patients for whom there were alarm symptoms for cancer. Dr. Bailey and colleagues found that two-thirds of men older than 65 had “no recorded alarm features of cancer in the 21 days before their index platelet count.”
Although this suggests that a routine finding of thrombocytosis could uncover unsuspected cancers, Dr. Bailey is cautious about hailing platelet counts as a new cancer-screening tool.
In emailed comments, Dr. Bailey said, “The crucial part of our study is that it was conducted with patients who were ill enough to see their GP [general practitioner]. Opportunistic measurement in patients who are asymptomatic would be quite a different thing. We would have to study the platelet count and subsequent cancers in asymptomatic patients to know if that was worth doing.”
Perhaps most helpfully, the U.K. study showed that cancer risk was increased even among some patients with normal platelet counts. For example, for men aged 60 and older, lung cancer was 4.7 times more likely among those with high-normal counts (≥326 x 109/L).
Because of this somewhat alarming finding, Dr. Bailey suggested moving away from a focus on absolute values. Rising platelet counts might be more clinically useful, she said.
“Physicians should be on the lookout for any unexplained increase in an individual’s platelet count, irrespective of whether the increased value is over or under the local threshold that is applied to define thrombocytosis,” concluded Dr. Bailey.
Dr. Narod has disclosed no relevant financial relationships. Dr. Bailey is a research fellow of the CanTest Collaborative.
A version of this article first appeared on Medscape.com.
A routine blood test may pack a bigger punch than previously suspected, suggests a recent analysis of over 3 million Canadian patient records.
A finding of thrombocytosis (platelet count >450 x 109/L) was associated with a greatly increased risk for some cancers up to 5 years later.
Overall, a high platelet count increased by 2.7 times the odds of receiving a solid-tumor cancer diagnosis within 2 years (95% confidence interval, 2.6-2.8).
The cancers most likely to be associated with unexpected thrombocytosis were those notorious for late-stage diagnosis due to a lack of early symptoms.
The risk was highest (23.3 times) for ovarian cancer. The risk was 3.8 times higher for pancreatic cancer and 3.5 times higher for cervical cancer.
Lung cancer was 4.4 times more likely within 2 years among patients with thrombocytosis compared to patients with normal platelet counts.
Conversely, breast, prostate, and thyroid cancers were not linked to the finding of thrombocytosis.
The study results were published online in JAMA Network Open on Aug. 12).
One of the authors of the article, Stephen A. Narod, MD, director of the Familial Breast Cancer Research Unit at the Women’s College Research Institute, Toronto, said the results were not unexpected but “very striking.”
“I had a hunch we were going to see this because I’ve seen this in other databases,” said Dr. Narod. “I think what struck me about it was how ubiquitous it was.”
Dr. Narod urged physicians, especially those in primary care, to take note: “If the platelets are high, I would certainly have a concern about lung cancer, colon cancer, and ovarian cancer.”
Dr. Narod and coauthor Vasily Giannakeas, a PhD candidate, pointed out that in their analysis that they were unable to single out cases in which a blood test was performed because the patient complained of symptoms that are associated with cancer. In those cases, thrombocytosis may have been diagnostic, rather than a lifesaving serendipitous finding.
Similar findings were reported recently from the United Kingdom.
A study by Sarah Bailey, PhD, MPH, and colleagues that was published last year in the British Journal of General Practice also found a connection between cancer incidence and platelet count. Dr. Bailey is a senior research fellow at the University of Exeter, England.
However, unlike in the Canadian study, the team led by Dr. Bailey was able to distinguish those patients for whom there were alarm symptoms for cancer. Dr. Bailey and colleagues found that two-thirds of men older than 65 had “no recorded alarm features of cancer in the 21 days before their index platelet count.”
Although this suggests that a routine finding of thrombocytosis could uncover unsuspected cancers, Dr. Bailey is cautious about hailing platelet counts as a new cancer-screening tool.
In emailed comments, Dr. Bailey said, “The crucial part of our study is that it was conducted with patients who were ill enough to see their GP [general practitioner]. Opportunistic measurement in patients who are asymptomatic would be quite a different thing. We would have to study the platelet count and subsequent cancers in asymptomatic patients to know if that was worth doing.”
Perhaps most helpfully, the U.K. study showed that cancer risk was increased even among some patients with normal platelet counts. For example, for men aged 60 and older, lung cancer was 4.7 times more likely among those with high-normal counts (≥326 x 109/L).
Because of this somewhat alarming finding, Dr. Bailey suggested moving away from a focus on absolute values. Rising platelet counts might be more clinically useful, she said.
“Physicians should be on the lookout for any unexplained increase in an individual’s platelet count, irrespective of whether the increased value is over or under the local threshold that is applied to define thrombocytosis,” concluded Dr. Bailey.
Dr. Narod has disclosed no relevant financial relationships. Dr. Bailey is a research fellow of the CanTest Collaborative.
A version of this article first appeared on Medscape.com.
VA Turns to Telehealth to Address Delays in Genetic Counseling
The U.S. Department of Veteran Affairs (VA) has been unable to provide genetic counseling to veterans at the same level as the civilian community, and other gaps exist, a genetic counselor told oncologist and hematologist colleagues. The good news is that telemedicine is turning out to be a valuable and proven way to reach veterans who need this kind of care, she said, although certain patients are being left behind.
“To me, telehealth is no doubt the way to go. But it is really important that we continue to look into these disparities, what's causing them, and how we can find a path forward,” said
Deborah Hartzfeld, MS, CGC, of the Genomic Medicine Service based in Salt Lake City, Utah. She spoke in a presentation at the 2021 annual meeting of the Association of VA Hematology/Oncology (AVAHO) that was held virtually and in person in Denver, Colorado, from September 24 to September 26, 2021.
As Harzfeld explained, the genetic counselor workforce is expanding along with the number of indications for genetic testing, especially in cancer, “where the need for germline genetic testing for inherited cancer genes becomes broader every year.”
Genetic counselors are a homogenous group, she said, as revealed by a 2021 survey of most of the nation’s 5,629 certified generic counselors. The North American survey, by the National Society of Genetic Counselors, found that 94% of respondents identified themselves as female, and 90% were white/non-Hispanic.
The survey report also noted that “the genetic counseling profession has grown by over 100% in the last 10 years and is expected to grow another 100% over the next 10 years. By 2025 there should be nearly 7,500 certified genetic counselors, and by 2030 there are likely to be over 10,000.”
Genetic counseling within the VA has also grown rapidly. In 2010, Harzfeld said, about 737 veterans were referred for the service. In 2020, the number was about 10,000, with about half referred for personal or family history of cancer.
The VA has 18 genetic counselors, not all of whom are actively seeing patients or working full time, she said. “Per the National Society of Genetic Counselors, there's one clinical genetic counselor per 100,000 people in the general population,” she said. “It's one for about 474,000 in the VA.”
Wait times for genetic counseling within the VA exceed Mission Act standards outside of urgent referrals in matters such as surgical or medical management, she said. “We usually see those patients within a week, but other folks have to wait or are referred into the community. It remains unclear how many of our patients could access care easily in the community or what the wait times at any individual VA will be.”
Fortunately, she said, telemedicine has increased access to genetic counseling within the general population and the VA, Harzfeld said. “A recent systematic evidence review found providing genetic counseling via video or telephone is comparable to in-person care, it increases access and it's likely feasible and acceptable to major stakeholders. It's worth noting that the data in this evidence review was collected prior to COVID-19 when fewer programs were using telehealth.”
Genetic counseling works especially well via telehealth because counselors don’t perform physical examinations, she said. “Prior to COVID, service probably saw maybe 4 VVC [VA Video Connect] appointments per month for genetic counseling. Now, VVC makes up about 70% of our new patient encounters. About 25% are telephone and about 5% are clinical video telehealth where the veteran goes into their clinic to be seated in front of the machine.”
Research has suggested that non-White patients are 40 to 50% less likely to be referred to telehealth for genetic counseling vs. in-person encounters, she said, although women in general (including black women) are more likely to be referred.
Harzfeld highlighted several challenges facing genetic counseling in the VA. She notes that contracted laboratories aren’t “really set up to be experts in germline genetic testing, so they’re not as nimble, and their test catalogs are not most likely going to be as comprehensive enough for what is needed.” Also, she said, “test ordering can be quite burdensome.”
“We need to continue working with various partners to increase access and the ease of ordering genetic testing,” she said.
Hartzfeld reports no disclosures.
The U.S. Department of Veteran Affairs (VA) has been unable to provide genetic counseling to veterans at the same level as the civilian community, and other gaps exist, a genetic counselor told oncologist and hematologist colleagues. The good news is that telemedicine is turning out to be a valuable and proven way to reach veterans who need this kind of care, she said, although certain patients are being left behind.
“To me, telehealth is no doubt the way to go. But it is really important that we continue to look into these disparities, what's causing them, and how we can find a path forward,” said
Deborah Hartzfeld, MS, CGC, of the Genomic Medicine Service based in Salt Lake City, Utah. She spoke in a presentation at the 2021 annual meeting of the Association of VA Hematology/Oncology (AVAHO) that was held virtually and in person in Denver, Colorado, from September 24 to September 26, 2021.
As Harzfeld explained, the genetic counselor workforce is expanding along with the number of indications for genetic testing, especially in cancer, “where the need for germline genetic testing for inherited cancer genes becomes broader every year.”
Genetic counselors are a homogenous group, she said, as revealed by a 2021 survey of most of the nation’s 5,629 certified generic counselors. The North American survey, by the National Society of Genetic Counselors, found that 94% of respondents identified themselves as female, and 90% were white/non-Hispanic.
The survey report also noted that “the genetic counseling profession has grown by over 100% in the last 10 years and is expected to grow another 100% over the next 10 years. By 2025 there should be nearly 7,500 certified genetic counselors, and by 2030 there are likely to be over 10,000.”
Genetic counseling within the VA has also grown rapidly. In 2010, Harzfeld said, about 737 veterans were referred for the service. In 2020, the number was about 10,000, with about half referred for personal or family history of cancer.
The VA has 18 genetic counselors, not all of whom are actively seeing patients or working full time, she said. “Per the National Society of Genetic Counselors, there's one clinical genetic counselor per 100,000 people in the general population,” she said. “It's one for about 474,000 in the VA.”
Wait times for genetic counseling within the VA exceed Mission Act standards outside of urgent referrals in matters such as surgical or medical management, she said. “We usually see those patients within a week, but other folks have to wait or are referred into the community. It remains unclear how many of our patients could access care easily in the community or what the wait times at any individual VA will be.”
Fortunately, she said, telemedicine has increased access to genetic counseling within the general population and the VA, Harzfeld said. “A recent systematic evidence review found providing genetic counseling via video or telephone is comparable to in-person care, it increases access and it's likely feasible and acceptable to major stakeholders. It's worth noting that the data in this evidence review was collected prior to COVID-19 when fewer programs were using telehealth.”
Genetic counseling works especially well via telehealth because counselors don’t perform physical examinations, she said. “Prior to COVID, service probably saw maybe 4 VVC [VA Video Connect] appointments per month for genetic counseling. Now, VVC makes up about 70% of our new patient encounters. About 25% are telephone and about 5% are clinical video telehealth where the veteran goes into their clinic to be seated in front of the machine.”
Research has suggested that non-White patients are 40 to 50% less likely to be referred to telehealth for genetic counseling vs. in-person encounters, she said, although women in general (including black women) are more likely to be referred.
Harzfeld highlighted several challenges facing genetic counseling in the VA. She notes that contracted laboratories aren’t “really set up to be experts in germline genetic testing, so they’re not as nimble, and their test catalogs are not most likely going to be as comprehensive enough for what is needed.” Also, she said, “test ordering can be quite burdensome.”
“We need to continue working with various partners to increase access and the ease of ordering genetic testing,” she said.
Hartzfeld reports no disclosures.
The U.S. Department of Veteran Affairs (VA) has been unable to provide genetic counseling to veterans at the same level as the civilian community, and other gaps exist, a genetic counselor told oncologist and hematologist colleagues. The good news is that telemedicine is turning out to be a valuable and proven way to reach veterans who need this kind of care, she said, although certain patients are being left behind.
“To me, telehealth is no doubt the way to go. But it is really important that we continue to look into these disparities, what's causing them, and how we can find a path forward,” said
Deborah Hartzfeld, MS, CGC, of the Genomic Medicine Service based in Salt Lake City, Utah. She spoke in a presentation at the 2021 annual meeting of the Association of VA Hematology/Oncology (AVAHO) that was held virtually and in person in Denver, Colorado, from September 24 to September 26, 2021.
As Harzfeld explained, the genetic counselor workforce is expanding along with the number of indications for genetic testing, especially in cancer, “where the need for germline genetic testing for inherited cancer genes becomes broader every year.”
Genetic counselors are a homogenous group, she said, as revealed by a 2021 survey of most of the nation’s 5,629 certified generic counselors. The North American survey, by the National Society of Genetic Counselors, found that 94% of respondents identified themselves as female, and 90% were white/non-Hispanic.
The survey report also noted that “the genetic counseling profession has grown by over 100% in the last 10 years and is expected to grow another 100% over the next 10 years. By 2025 there should be nearly 7,500 certified genetic counselors, and by 2030 there are likely to be over 10,000.”
Genetic counseling within the VA has also grown rapidly. In 2010, Harzfeld said, about 737 veterans were referred for the service. In 2020, the number was about 10,000, with about half referred for personal or family history of cancer.
The VA has 18 genetic counselors, not all of whom are actively seeing patients or working full time, she said. “Per the National Society of Genetic Counselors, there's one clinical genetic counselor per 100,000 people in the general population,” she said. “It's one for about 474,000 in the VA.”
Wait times for genetic counseling within the VA exceed Mission Act standards outside of urgent referrals in matters such as surgical or medical management, she said. “We usually see those patients within a week, but other folks have to wait or are referred into the community. It remains unclear how many of our patients could access care easily in the community or what the wait times at any individual VA will be.”
Fortunately, she said, telemedicine has increased access to genetic counseling within the general population and the VA, Harzfeld said. “A recent systematic evidence review found providing genetic counseling via video or telephone is comparable to in-person care, it increases access and it's likely feasible and acceptable to major stakeholders. It's worth noting that the data in this evidence review was collected prior to COVID-19 when fewer programs were using telehealth.”
Genetic counseling works especially well via telehealth because counselors don’t perform physical examinations, she said. “Prior to COVID, service probably saw maybe 4 VVC [VA Video Connect] appointments per month for genetic counseling. Now, VVC makes up about 70% of our new patient encounters. About 25% are telephone and about 5% are clinical video telehealth where the veteran goes into their clinic to be seated in front of the machine.”
Research has suggested that non-White patients are 40 to 50% less likely to be referred to telehealth for genetic counseling vs. in-person encounters, she said, although women in general (including black women) are more likely to be referred.
Harzfeld highlighted several challenges facing genetic counseling in the VA. She notes that contracted laboratories aren’t “really set up to be experts in germline genetic testing, so they’re not as nimble, and their test catalogs are not most likely going to be as comprehensive enough for what is needed.” Also, she said, “test ordering can be quite burdensome.”
“We need to continue working with various partners to increase access and the ease of ordering genetic testing,” she said.
Hartzfeld reports no disclosures.
CVST after COVID-19 vaccine: New data confirm high mortality rate
, confirming the severity of the reaction and the associated high mortality rate.
The new series comes from an international registry of consecutive patients who experienced CVST within 28 days of COVID-19 vaccination between March 29 and June 18, 2021, from 81 hospitals in 19 countries.
The cases are described in an article published online on Sept. 28. in JAMA Neurology.
“This is a reliable description on the clinical condition of these patients with CVST associated with COVID-19 vaccination. It is striking that this a much worse condition than CVST not associated with COVID-19 vaccination, with a much higher rate of intracerebral hemorrhage and coma and a much higher mortality rate,” senior author Jonathan M. Coutinho, MD, Amsterdam University Medical Centers, told this news organization.
These data confirm the observations from an earlier U.K. cohort in which cases of cerebral venous thrombosis linked to COVID-19 vaccination occurred.
“This is the biggest series, and as an international series, it gives a broader perspective from a larger range of countries,” Dr. Coutinho said. “All the data together show that, although this side effect is rare, the consequences are very severe,” he added.
In the current study, the researchers regarded CVST as being linked to the vaccine if it was accompanied by thrombosis with thrombocytopenia syndrome (TTS), as evidenced by thrombosis and new-onset thrombocytopenia.
In the cohort of 116 patients with CVST after COVID-19 vaccination, 78 (67.2%) had thrombosis with TTS and were thus classified as having had a vaccine-related adverse event. These patients were frequently comatose at presentation (24%) and often had intracerebral hemorrhage (68%) and concomitant thromboembolism (36%); 47% died during hospitalization.
These patients were compared with the 38 patients in the same cohort who had CVST but in whom there was no indication of concomitant thrombosis and thrombocytopenia. The case patients were also compared with a control group of 207 patients with CVST who were included in a separate international registry before the COVID-19 pandemic.
Mortality rates were much higher among the patients deemed to have had a vaccine-related CVST. The in-hospital mortality rate was 47%, compared with 5% among the patients in the same cohort who did not have TTS and 3.9% among the prepandemic control group.
The mortality rate was even higher (61%) among patients in the TTS group for whom the diagnosis was made before the condition garnered attention in the scientific community. The mortality rate was 42% among patients diagnosed later.
Of the 78 patients in whom CVST and TTS occurred after COVID-19 vaccination in this cohort, 76 had received the AstraZeneca vaccine (in 75 patients, CVST and TTS occurred after the first vaccination; in one patient, they occurred after the second vaccination). One patient had received the Johnson & Johnson vaccine, and one had received the Pfizer vaccine.
“After more analysis, the case after the Pfizer vaccination is not believed to be caused by the vaccine,” Dr. Coutinho said. “In that case, the patient had a platelet count just below the lower limit and was taking an immunomodulator drug that is known to be associated with thrombocytopenia.”
For two patients who received the AstraZeneca vaccine, there was also an alternative explanation for the thrombocytopenia.
Dr. Coutinho also pointed out that the Johnson & Johnson vaccine has been used mainly in the United States, and these data were largely from other countries.
The median time from vaccination to CVST symptom onset was 9 days in the TTS group. The median platelet count at hospital admission among patients with postvaccination CVST-TTS was 45. Three patients presented with a normal platelet count and developed thrombocytopenia during admission; two patients presented with mild thrombocytopenia, 30 presented with moderate thrombocytopenia, and 43 presented with severe thrombocytopenia.
Antibodies against platelet factor 4 (PF4) were measured in 69 patients with TTS, of whom 63 (91%) tested positive (the one patient in whom TTS occurred after the patient received the Pfizer vaccine did not test positive). However, the researchers note that sensitivity varies among different PF4 ELISA tests. Findings of platelet activation assays were positive in all 36 tested patients.
In the TTS group, 52 patients (67%) received immunomodulation therapy, most often intravenous immunoglobulins (IVIG). Among patients treated with IVIG, the mortality rate was lower (28%).
Different from CVST linked to natural COVID-19 infection
Dr. Coutinho noted that CVST can occur in natural SARS-CoV-2 infection but that vaccine-associated CVST is very different.
“In natural COVID-19 infection, there is an increased risk of thrombosis, and some patients can get CVST as a part of this, but in these cases, this is not accompanied by thrombocytopenia. While the CVST in natural COVID-19 infection is also associated with a bad prognosis, this is more to do with the underlying disease. It is normally the very sick COVID patients who develop CVST, and these patients usually die from the underlying disease rather than the CVST itself,” he explained.
“Clinicians need to be aware of vaccine-related CVST, as it requires very specific and rapid treatment,” Dr. Coutinho stressed.
“Patients presenting with an extremely severe headache (unlike any headache they’ve had before) or with seizures or a focal deficit (weakness in arm or problems with speaking or vision) within 4 weeks of an adenovirus COVID-19 vaccination should ring alarm bells. It is important to do diagnostics quickly, with a platelet count the most important first step, and a rapid CT/MRI scan,” he said.
Other tests that should be conducted are D-dimer for thrombosis and the PF4 antibody test. But results for the PF4 antibody test can take days to come back, and clinicians shouldn’t wait for that, Dr. Coutinho notes.
“Specific treatment needs to be given immediately – with anticoagulation (preferably nonheparin) and immunomodulation with IVIG to stop the immune reaction. Platelets should not be given – that may seem counterintuitive in patients with a low platelet count, but giving platelets makes it worse,” he said.
Is there a geographic difference?
Dr. Coutinho pointed out that fewer cases of this vaccine-related CVST are being reported at the current time.
“We are not sure why this is the case. These adenovirus vaccines are not being used much now in Western countries, but our collaboration covers many less developed countries in South America and Asia, which are relying heavily on these vaccines. We are now shifting focus to these countries, but so far we have only seen a handful of cases from these areas,” he said.
He suggested that this may be because these countries started their vaccination programs later and are vaccinating their elderly (who are not so susceptible to this side effect) first, or it may be because of some environmental or genetic factor that has not yet been discovered.
“This is now an important research question – is the risk of vaccine-induced CVST the same in different countries or ethnicities? This could influence decisions on future vaccine strategies,” Dr. Coutinho said.
“So far, female sex is the strongest risk factor for vaccine-induced CVST. In our cohort, 81% of cases were in women. In addition, 95% were White, but that doesn’t allow us to conclude that this is a risk factor, as the majority of people who have been vaccinated are White. So, we have no clear insight into that yet,” he said.
In a comment for this news organization, the lead author of the previous U.K. report of a series of 70 cases of cerebral venous thrombosis linked to COVID-19 vaccination, Richard Perry, PhD, University College Hospital, London, described this new report as “an excellent study, with many of the same strengths and weaknesses as our study and has very similar results.”
Dr. Perry noted that the two studies used slightly different definitions of vaccine-induced thrombotic thrombocytopenia, but the cases reported appear to be very similar overall. “It is reassuring and gratifying to see that they have made such similar observations,” he said.
“And as they have drawn their cases from a broad range of countries whereas ours were all from the U.K., this provides evidence that the observations from both studies are reasonably generalizable,” he added.
Dr. Perry pointed out that this new report states that TTS occurred in one patient after the patient had received a second dose of the AstraZeneca vaccine. “I would like to know more about this case, because we didn’t see any cases after a second dose in our cohort,” he said.
Dr. Coutinho responded that he didn’t believe this was the first reported case after the second dose.
The study did not receive any specific funding. Dr. Coutinho has received grants paid to his institution from Boehringer Ingelheim and Bayer and payments paid to his institution for data safety monitoring board participation by Bayer.
A version of this article first appeared on Medscape.com.
, confirming the severity of the reaction and the associated high mortality rate.
The new series comes from an international registry of consecutive patients who experienced CVST within 28 days of COVID-19 vaccination between March 29 and June 18, 2021, from 81 hospitals in 19 countries.
The cases are described in an article published online on Sept. 28. in JAMA Neurology.
“This is a reliable description on the clinical condition of these patients with CVST associated with COVID-19 vaccination. It is striking that this a much worse condition than CVST not associated with COVID-19 vaccination, with a much higher rate of intracerebral hemorrhage and coma and a much higher mortality rate,” senior author Jonathan M. Coutinho, MD, Amsterdam University Medical Centers, told this news organization.
These data confirm the observations from an earlier U.K. cohort in which cases of cerebral venous thrombosis linked to COVID-19 vaccination occurred.
“This is the biggest series, and as an international series, it gives a broader perspective from a larger range of countries,” Dr. Coutinho said. “All the data together show that, although this side effect is rare, the consequences are very severe,” he added.
In the current study, the researchers regarded CVST as being linked to the vaccine if it was accompanied by thrombosis with thrombocytopenia syndrome (TTS), as evidenced by thrombosis and new-onset thrombocytopenia.
In the cohort of 116 patients with CVST after COVID-19 vaccination, 78 (67.2%) had thrombosis with TTS and were thus classified as having had a vaccine-related adverse event. These patients were frequently comatose at presentation (24%) and often had intracerebral hemorrhage (68%) and concomitant thromboembolism (36%); 47% died during hospitalization.
These patients were compared with the 38 patients in the same cohort who had CVST but in whom there was no indication of concomitant thrombosis and thrombocytopenia. The case patients were also compared with a control group of 207 patients with CVST who were included in a separate international registry before the COVID-19 pandemic.
Mortality rates were much higher among the patients deemed to have had a vaccine-related CVST. The in-hospital mortality rate was 47%, compared with 5% among the patients in the same cohort who did not have TTS and 3.9% among the prepandemic control group.
The mortality rate was even higher (61%) among patients in the TTS group for whom the diagnosis was made before the condition garnered attention in the scientific community. The mortality rate was 42% among patients diagnosed later.
Of the 78 patients in whom CVST and TTS occurred after COVID-19 vaccination in this cohort, 76 had received the AstraZeneca vaccine (in 75 patients, CVST and TTS occurred after the first vaccination; in one patient, they occurred after the second vaccination). One patient had received the Johnson & Johnson vaccine, and one had received the Pfizer vaccine.
“After more analysis, the case after the Pfizer vaccination is not believed to be caused by the vaccine,” Dr. Coutinho said. “In that case, the patient had a platelet count just below the lower limit and was taking an immunomodulator drug that is known to be associated with thrombocytopenia.”
For two patients who received the AstraZeneca vaccine, there was also an alternative explanation for the thrombocytopenia.
Dr. Coutinho also pointed out that the Johnson & Johnson vaccine has been used mainly in the United States, and these data were largely from other countries.
The median time from vaccination to CVST symptom onset was 9 days in the TTS group. The median platelet count at hospital admission among patients with postvaccination CVST-TTS was 45. Three patients presented with a normal platelet count and developed thrombocytopenia during admission; two patients presented with mild thrombocytopenia, 30 presented with moderate thrombocytopenia, and 43 presented with severe thrombocytopenia.
Antibodies against platelet factor 4 (PF4) were measured in 69 patients with TTS, of whom 63 (91%) tested positive (the one patient in whom TTS occurred after the patient received the Pfizer vaccine did not test positive). However, the researchers note that sensitivity varies among different PF4 ELISA tests. Findings of platelet activation assays were positive in all 36 tested patients.
In the TTS group, 52 patients (67%) received immunomodulation therapy, most often intravenous immunoglobulins (IVIG). Among patients treated with IVIG, the mortality rate was lower (28%).
Different from CVST linked to natural COVID-19 infection
Dr. Coutinho noted that CVST can occur in natural SARS-CoV-2 infection but that vaccine-associated CVST is very different.
“In natural COVID-19 infection, there is an increased risk of thrombosis, and some patients can get CVST as a part of this, but in these cases, this is not accompanied by thrombocytopenia. While the CVST in natural COVID-19 infection is also associated with a bad prognosis, this is more to do with the underlying disease. It is normally the very sick COVID patients who develop CVST, and these patients usually die from the underlying disease rather than the CVST itself,” he explained.
“Clinicians need to be aware of vaccine-related CVST, as it requires very specific and rapid treatment,” Dr. Coutinho stressed.
“Patients presenting with an extremely severe headache (unlike any headache they’ve had before) or with seizures or a focal deficit (weakness in arm or problems with speaking or vision) within 4 weeks of an adenovirus COVID-19 vaccination should ring alarm bells. It is important to do diagnostics quickly, with a platelet count the most important first step, and a rapid CT/MRI scan,” he said.
Other tests that should be conducted are D-dimer for thrombosis and the PF4 antibody test. But results for the PF4 antibody test can take days to come back, and clinicians shouldn’t wait for that, Dr. Coutinho notes.
“Specific treatment needs to be given immediately – with anticoagulation (preferably nonheparin) and immunomodulation with IVIG to stop the immune reaction. Platelets should not be given – that may seem counterintuitive in patients with a low platelet count, but giving platelets makes it worse,” he said.
Is there a geographic difference?
Dr. Coutinho pointed out that fewer cases of this vaccine-related CVST are being reported at the current time.
“We are not sure why this is the case. These adenovirus vaccines are not being used much now in Western countries, but our collaboration covers many less developed countries in South America and Asia, which are relying heavily on these vaccines. We are now shifting focus to these countries, but so far we have only seen a handful of cases from these areas,” he said.
He suggested that this may be because these countries started their vaccination programs later and are vaccinating their elderly (who are not so susceptible to this side effect) first, or it may be because of some environmental or genetic factor that has not yet been discovered.
“This is now an important research question – is the risk of vaccine-induced CVST the same in different countries or ethnicities? This could influence decisions on future vaccine strategies,” Dr. Coutinho said.
“So far, female sex is the strongest risk factor for vaccine-induced CVST. In our cohort, 81% of cases were in women. In addition, 95% were White, but that doesn’t allow us to conclude that this is a risk factor, as the majority of people who have been vaccinated are White. So, we have no clear insight into that yet,” he said.
In a comment for this news organization, the lead author of the previous U.K. report of a series of 70 cases of cerebral venous thrombosis linked to COVID-19 vaccination, Richard Perry, PhD, University College Hospital, London, described this new report as “an excellent study, with many of the same strengths and weaknesses as our study and has very similar results.”
Dr. Perry noted that the two studies used slightly different definitions of vaccine-induced thrombotic thrombocytopenia, but the cases reported appear to be very similar overall. “It is reassuring and gratifying to see that they have made such similar observations,” he said.
“And as they have drawn their cases from a broad range of countries whereas ours were all from the U.K., this provides evidence that the observations from both studies are reasonably generalizable,” he added.
Dr. Perry pointed out that this new report states that TTS occurred in one patient after the patient had received a second dose of the AstraZeneca vaccine. “I would like to know more about this case, because we didn’t see any cases after a second dose in our cohort,” he said.
Dr. Coutinho responded that he didn’t believe this was the first reported case after the second dose.
The study did not receive any specific funding. Dr. Coutinho has received grants paid to his institution from Boehringer Ingelheim and Bayer and payments paid to his institution for data safety monitoring board participation by Bayer.
A version of this article first appeared on Medscape.com.
, confirming the severity of the reaction and the associated high mortality rate.
The new series comes from an international registry of consecutive patients who experienced CVST within 28 days of COVID-19 vaccination between March 29 and June 18, 2021, from 81 hospitals in 19 countries.
The cases are described in an article published online on Sept. 28. in JAMA Neurology.
“This is a reliable description on the clinical condition of these patients with CVST associated with COVID-19 vaccination. It is striking that this a much worse condition than CVST not associated with COVID-19 vaccination, with a much higher rate of intracerebral hemorrhage and coma and a much higher mortality rate,” senior author Jonathan M. Coutinho, MD, Amsterdam University Medical Centers, told this news organization.
These data confirm the observations from an earlier U.K. cohort in which cases of cerebral venous thrombosis linked to COVID-19 vaccination occurred.
“This is the biggest series, and as an international series, it gives a broader perspective from a larger range of countries,” Dr. Coutinho said. “All the data together show that, although this side effect is rare, the consequences are very severe,” he added.
In the current study, the researchers regarded CVST as being linked to the vaccine if it was accompanied by thrombosis with thrombocytopenia syndrome (TTS), as evidenced by thrombosis and new-onset thrombocytopenia.
In the cohort of 116 patients with CVST after COVID-19 vaccination, 78 (67.2%) had thrombosis with TTS and were thus classified as having had a vaccine-related adverse event. These patients were frequently comatose at presentation (24%) and often had intracerebral hemorrhage (68%) and concomitant thromboembolism (36%); 47% died during hospitalization.
These patients were compared with the 38 patients in the same cohort who had CVST but in whom there was no indication of concomitant thrombosis and thrombocytopenia. The case patients were also compared with a control group of 207 patients with CVST who were included in a separate international registry before the COVID-19 pandemic.
Mortality rates were much higher among the patients deemed to have had a vaccine-related CVST. The in-hospital mortality rate was 47%, compared with 5% among the patients in the same cohort who did not have TTS and 3.9% among the prepandemic control group.
The mortality rate was even higher (61%) among patients in the TTS group for whom the diagnosis was made before the condition garnered attention in the scientific community. The mortality rate was 42% among patients diagnosed later.
Of the 78 patients in whom CVST and TTS occurred after COVID-19 vaccination in this cohort, 76 had received the AstraZeneca vaccine (in 75 patients, CVST and TTS occurred after the first vaccination; in one patient, they occurred after the second vaccination). One patient had received the Johnson & Johnson vaccine, and one had received the Pfizer vaccine.
“After more analysis, the case after the Pfizer vaccination is not believed to be caused by the vaccine,” Dr. Coutinho said. “In that case, the patient had a platelet count just below the lower limit and was taking an immunomodulator drug that is known to be associated with thrombocytopenia.”
For two patients who received the AstraZeneca vaccine, there was also an alternative explanation for the thrombocytopenia.
Dr. Coutinho also pointed out that the Johnson & Johnson vaccine has been used mainly in the United States, and these data were largely from other countries.
The median time from vaccination to CVST symptom onset was 9 days in the TTS group. The median platelet count at hospital admission among patients with postvaccination CVST-TTS was 45. Three patients presented with a normal platelet count and developed thrombocytopenia during admission; two patients presented with mild thrombocytopenia, 30 presented with moderate thrombocytopenia, and 43 presented with severe thrombocytopenia.
Antibodies against platelet factor 4 (PF4) were measured in 69 patients with TTS, of whom 63 (91%) tested positive (the one patient in whom TTS occurred after the patient received the Pfizer vaccine did not test positive). However, the researchers note that sensitivity varies among different PF4 ELISA tests. Findings of platelet activation assays were positive in all 36 tested patients.
In the TTS group, 52 patients (67%) received immunomodulation therapy, most often intravenous immunoglobulins (IVIG). Among patients treated with IVIG, the mortality rate was lower (28%).
Different from CVST linked to natural COVID-19 infection
Dr. Coutinho noted that CVST can occur in natural SARS-CoV-2 infection but that vaccine-associated CVST is very different.
“In natural COVID-19 infection, there is an increased risk of thrombosis, and some patients can get CVST as a part of this, but in these cases, this is not accompanied by thrombocytopenia. While the CVST in natural COVID-19 infection is also associated with a bad prognosis, this is more to do with the underlying disease. It is normally the very sick COVID patients who develop CVST, and these patients usually die from the underlying disease rather than the CVST itself,” he explained.
“Clinicians need to be aware of vaccine-related CVST, as it requires very specific and rapid treatment,” Dr. Coutinho stressed.
“Patients presenting with an extremely severe headache (unlike any headache they’ve had before) or with seizures or a focal deficit (weakness in arm or problems with speaking or vision) within 4 weeks of an adenovirus COVID-19 vaccination should ring alarm bells. It is important to do diagnostics quickly, with a platelet count the most important first step, and a rapid CT/MRI scan,” he said.
Other tests that should be conducted are D-dimer for thrombosis and the PF4 antibody test. But results for the PF4 antibody test can take days to come back, and clinicians shouldn’t wait for that, Dr. Coutinho notes.
“Specific treatment needs to be given immediately – with anticoagulation (preferably nonheparin) and immunomodulation with IVIG to stop the immune reaction. Platelets should not be given – that may seem counterintuitive in patients with a low platelet count, but giving platelets makes it worse,” he said.
Is there a geographic difference?
Dr. Coutinho pointed out that fewer cases of this vaccine-related CVST are being reported at the current time.
“We are not sure why this is the case. These adenovirus vaccines are not being used much now in Western countries, but our collaboration covers many less developed countries in South America and Asia, which are relying heavily on these vaccines. We are now shifting focus to these countries, but so far we have only seen a handful of cases from these areas,” he said.
He suggested that this may be because these countries started their vaccination programs later and are vaccinating their elderly (who are not so susceptible to this side effect) first, or it may be because of some environmental or genetic factor that has not yet been discovered.
“This is now an important research question – is the risk of vaccine-induced CVST the same in different countries or ethnicities? This could influence decisions on future vaccine strategies,” Dr. Coutinho said.
“So far, female sex is the strongest risk factor for vaccine-induced CVST. In our cohort, 81% of cases were in women. In addition, 95% were White, but that doesn’t allow us to conclude that this is a risk factor, as the majority of people who have been vaccinated are White. So, we have no clear insight into that yet,” he said.
In a comment for this news organization, the lead author of the previous U.K. report of a series of 70 cases of cerebral venous thrombosis linked to COVID-19 vaccination, Richard Perry, PhD, University College Hospital, London, described this new report as “an excellent study, with many of the same strengths and weaknesses as our study and has very similar results.”
Dr. Perry noted that the two studies used slightly different definitions of vaccine-induced thrombotic thrombocytopenia, but the cases reported appear to be very similar overall. “It is reassuring and gratifying to see that they have made such similar observations,” he said.
“And as they have drawn their cases from a broad range of countries whereas ours were all from the U.K., this provides evidence that the observations from both studies are reasonably generalizable,” he added.
Dr. Perry pointed out that this new report states that TTS occurred in one patient after the patient had received a second dose of the AstraZeneca vaccine. “I would like to know more about this case, because we didn’t see any cases after a second dose in our cohort,” he said.
Dr. Coutinho responded that he didn’t believe this was the first reported case after the second dose.
The study did not receive any specific funding. Dr. Coutinho has received grants paid to his institution from Boehringer Ingelheim and Bayer and payments paid to his institution for data safety monitoring board participation by Bayer.
A version of this article first appeared on Medscape.com.
Intracranial hemorrhaging a high risk for patients with hemophilia, especially neonates
The observed rates of intracranial hemorrhaging (ICH) in patients with hemophilia were higher compared to the general populations among all age groups examined, according to a meta-analysis of studies reported online ahead of print in Blood.
As previously reported, the risk seemed higher in the group of infants and toddlers, and neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, in the current study.
The researchers performed a literature review and assessed 45 studies that represented 54,470 patients, 809,151 person-years and 5,326 live births of patients with hemophilia. Pooled ICH incidence and mortality were calculated for three age groups: persons of all ages with hemophilia; children and young adults below 25 years of age with hemophilia; and neonates with hemophilia.
Overall results
Among the persons of all ages, the pooled ICH incidence and mortality rates were 2.3 (95% confidence interval, 1.2-4.8) and 0.8 (95% CI, 0.5-1.2) per 1,000 person-years, respectively, according to the authors. They found that in children and young adults, the pooled ICH incidence and mortality rates were 7.4 (95% CI, 4.9-11.1) and 0.5 (95% CI, 0.3-0.9) per 1,000 person-years, respectively. In neonates, the pooled cumulative ICH incidence was 2.1% (95% CI, 1.5-2.8) per 100 live births and the pooled ICH cumulative mortality was 0.2% (95% CI, 0.0-1.2) per 100 live births.
Overall, the occurrence of ICH was classified as spontaneous in 35%-58% of cases.
Neonates at risk
The observed ICH rates in hemophilia were higher compared to the general populations among the age groups assessed. Neonates showed the highest risk of ICH, which is confirmed by other studies in severe hemophilia demonstrating that neonates were at 11.2 times higher risk for ICH compared with 1- to 12-month-old children, and is also strongly increased compared to neonates in the general populations, the researchers stated.
A previous large study of term infants reported 361 intracranial bleeding episodes per 583,340 live births (0.062% per 100 live births), and comparing this to the current pooled estimate of 2.1% per 100 live births, neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, according to the researchers.
Monitoring and follow-up
“Our findings suggest that adequate follow-up and monitoring of patients is warranted among all ages, especially in the presence of risk factors. Prophylaxis seems to halve ICH risk in children and adults with severe hemophilia, which supports existing recommendations encouraging early initiation of prophylactic treatment,” the authors advised.
Accurate capture of the true frequency of ICH is challenged by considerable clinical heterogeneity, limiting the precision and generalizability of the pooled estimates, leading to the likelihood that ICH and mortality were underdiagnosed in this analysis, according to the authors.
“We found high ICH incidence and mortality rates in patients with hemophilia. Our findings suggest that ICH is still an important problem in hemophilia requiring adequate counseling of patients of all ages,” the researchers concluded.
This work was supported by a grant from Sobi. Some of the authors reported research, consulting or lecturing fees from a variety of pharmaceutical companies, including Sobi.
The observed rates of intracranial hemorrhaging (ICH) in patients with hemophilia were higher compared to the general populations among all age groups examined, according to a meta-analysis of studies reported online ahead of print in Blood.
As previously reported, the risk seemed higher in the group of infants and toddlers, and neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, in the current study.
The researchers performed a literature review and assessed 45 studies that represented 54,470 patients, 809,151 person-years and 5,326 live births of patients with hemophilia. Pooled ICH incidence and mortality were calculated for three age groups: persons of all ages with hemophilia; children and young adults below 25 years of age with hemophilia; and neonates with hemophilia.
Overall results
Among the persons of all ages, the pooled ICH incidence and mortality rates were 2.3 (95% confidence interval, 1.2-4.8) and 0.8 (95% CI, 0.5-1.2) per 1,000 person-years, respectively, according to the authors. They found that in children and young adults, the pooled ICH incidence and mortality rates were 7.4 (95% CI, 4.9-11.1) and 0.5 (95% CI, 0.3-0.9) per 1,000 person-years, respectively. In neonates, the pooled cumulative ICH incidence was 2.1% (95% CI, 1.5-2.8) per 100 live births and the pooled ICH cumulative mortality was 0.2% (95% CI, 0.0-1.2) per 100 live births.
Overall, the occurrence of ICH was classified as spontaneous in 35%-58% of cases.
Neonates at risk
The observed ICH rates in hemophilia were higher compared to the general populations among the age groups assessed. Neonates showed the highest risk of ICH, which is confirmed by other studies in severe hemophilia demonstrating that neonates were at 11.2 times higher risk for ICH compared with 1- to 12-month-old children, and is also strongly increased compared to neonates in the general populations, the researchers stated.
A previous large study of term infants reported 361 intracranial bleeding episodes per 583,340 live births (0.062% per 100 live births), and comparing this to the current pooled estimate of 2.1% per 100 live births, neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, according to the researchers.
Monitoring and follow-up
“Our findings suggest that adequate follow-up and monitoring of patients is warranted among all ages, especially in the presence of risk factors. Prophylaxis seems to halve ICH risk in children and adults with severe hemophilia, which supports existing recommendations encouraging early initiation of prophylactic treatment,” the authors advised.
Accurate capture of the true frequency of ICH is challenged by considerable clinical heterogeneity, limiting the precision and generalizability of the pooled estimates, leading to the likelihood that ICH and mortality were underdiagnosed in this analysis, according to the authors.
“We found high ICH incidence and mortality rates in patients with hemophilia. Our findings suggest that ICH is still an important problem in hemophilia requiring adequate counseling of patients of all ages,” the researchers concluded.
This work was supported by a grant from Sobi. Some of the authors reported research, consulting or lecturing fees from a variety of pharmaceutical companies, including Sobi.
The observed rates of intracranial hemorrhaging (ICH) in patients with hemophilia were higher compared to the general populations among all age groups examined, according to a meta-analysis of studies reported online ahead of print in Blood.
As previously reported, the risk seemed higher in the group of infants and toddlers, and neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, in the current study.
The researchers performed a literature review and assessed 45 studies that represented 54,470 patients, 809,151 person-years and 5,326 live births of patients with hemophilia. Pooled ICH incidence and mortality were calculated for three age groups: persons of all ages with hemophilia; children and young adults below 25 years of age with hemophilia; and neonates with hemophilia.
Overall results
Among the persons of all ages, the pooled ICH incidence and mortality rates were 2.3 (95% confidence interval, 1.2-4.8) and 0.8 (95% CI, 0.5-1.2) per 1,000 person-years, respectively, according to the authors. They found that in children and young adults, the pooled ICH incidence and mortality rates were 7.4 (95% CI, 4.9-11.1) and 0.5 (95% CI, 0.3-0.9) per 1,000 person-years, respectively. In neonates, the pooled cumulative ICH incidence was 2.1% (95% CI, 1.5-2.8) per 100 live births and the pooled ICH cumulative mortality was 0.2% (95% CI, 0.0-1.2) per 100 live births.
Overall, the occurrence of ICH was classified as spontaneous in 35%-58% of cases.
Neonates at risk
The observed ICH rates in hemophilia were higher compared to the general populations among the age groups assessed. Neonates showed the highest risk of ICH, which is confirmed by other studies in severe hemophilia demonstrating that neonates were at 11.2 times higher risk for ICH compared with 1- to 12-month-old children, and is also strongly increased compared to neonates in the general populations, the researchers stated.
A previous large study of term infants reported 361 intracranial bleeding episodes per 583,340 live births (0.062% per 100 live births), and comparing this to the current pooled estimate of 2.1% per 100 live births, neonates with hemophilia showed a 33-fold higher risk of ICH than newborns in the general population, according to the researchers.
Monitoring and follow-up
“Our findings suggest that adequate follow-up and monitoring of patients is warranted among all ages, especially in the presence of risk factors. Prophylaxis seems to halve ICH risk in children and adults with severe hemophilia, which supports existing recommendations encouraging early initiation of prophylactic treatment,” the authors advised.
Accurate capture of the true frequency of ICH is challenged by considerable clinical heterogeneity, limiting the precision and generalizability of the pooled estimates, leading to the likelihood that ICH and mortality were underdiagnosed in this analysis, according to the authors.
“We found high ICH incidence and mortality rates in patients with hemophilia. Our findings suggest that ICH is still an important problem in hemophilia requiring adequate counseling of patients of all ages,” the researchers concluded.
This work was supported by a grant from Sobi. Some of the authors reported research, consulting or lecturing fees from a variety of pharmaceutical companies, including Sobi.
FROM BLOOD
Study gives bleeding risk estimates for VTE patients on anticoagulants
The meta-analysis of data from 27 studies with 17,202 patients was published in the Annals of Internal Medicine. According to two of the paper’s coauthors, Faizan Khan, MSc, and Marc A. Rodger, MD, it “provides best available estimates of long-term bleeding risk with different anticoagulants in patients with unprovoked VTE,” including subgroups at increased risk.
Patients at increased risk for major bleeding include those who are older; those using antiplatelet therapy; and patients with kidney disease, a history of bleeding, or anemia, noted the coauthors, who work for the Ottawa Hospital Research Institute.
The researchers focused on randomized controlled trials (RCTs) and prospective cohort studies that reported major bleeding among patients with a first unprovoked or weakly provoked VTE who received oral anticoagulation for at least 6 months beyond an initial anticoagulant treatment course of at least 3 months.
The investigators analyzed data from 14 RCTs and 13 cohort studies. In all, 9,982 patients received a vitamin K antagonist (VKA), and 7,220 received a direct oral anticoagulant (DOAC).
The incidence of major bleeding per 100 person-years was 1.7 events with VKAs, compared with 1.1 events with DOACs. The researchers estimated that the 5-year cumulative incidence of major bleeding with VKAs was 6.3%. The available data for DOACs were insufficient to estimate the incidence of major bleeding beyond 1 year.
“This information can help clinicians counsel patients and inform shared decision-making about extended therapy,” the researchers said.
Risks of serious bleeding ‘not trivial’
Margaret Fang, MD, with the University of California, San Francisco, agreed that the study can help clinicians and patients weigh the risks of extended anticoagulation for common types of VTE.
The study also “highlights that the risks of serious bleeding are not trivial” and points out gaps in the literature regarding the long-term use of DOACs for extended VTE therapy, Dr. Fang said.
Better ways to predict which patients will develop bleeding on anticoagulants are needed, Dr. Fang added. “It will also be important to establish which of the various therapies for preventing recurrent VTE – full dose versus lowered dose, or even aspirin – has the best balance of safety and efficacy,” she said.
‘Standardized approach’ for identifying high-risk patients lacking
Clinical practice guidelines recommend indefinite anticoagulation for an unprovoked VTE, except when patients are at high risk of bleeding, the authors noted. But clinicians lack a “standardized approach to identify patients at high risk of bleeding,” Mr. Khan and Dr. Rodger said. “Evidence from randomized trials on net long-term benefit of extended therapy is limited, and current guideline recommendations are largely based on expert consensus opinion. Major bleeding events are two to three times more likely to be fatal than recurrent VTE events, so extended therapy is not always associated with a net mortality benefit, particularly in patients at low risk of recurrent VTE or high risk of bleeding.”
The analysis indicates that there is “a clinically meaningful difference in long-term risk for anticoagulant-related major bleeding among patients with a first unprovoked VTE stratified according to presence or absence of the following risk factors: age older than 65 years, creatinine clearance less than 50 mL/min, history of bleeding, concomitant use of antiplatelet therapy, and hemoglobin level less than 100 g/L,” the authors said.
For example, the researchers found that the incidence of major bleeding was higher among those older than 65 years, compared with younger patients (incidence rate ratio, 1.84 with VKAs and 2.92 with DOACs), and among those with creatinine clearance less than 50 mL/min (IRR, 2.83 with VKAs and 3.71 with DOACs).
The case-fatality rate of major bleeding was 8.3% with VKAs and 9.7% with DOACs.
The study received funding from the Canadian Institutes of Health Research. Some of the coauthors are employees of or have financial ties to pharmaceutical companies. Mr. Khan, Dr. Rodger, and Dr. Fang had no relevant disclosures.
The meta-analysis of data from 27 studies with 17,202 patients was published in the Annals of Internal Medicine. According to two of the paper’s coauthors, Faizan Khan, MSc, and Marc A. Rodger, MD, it “provides best available estimates of long-term bleeding risk with different anticoagulants in patients with unprovoked VTE,” including subgroups at increased risk.
Patients at increased risk for major bleeding include those who are older; those using antiplatelet therapy; and patients with kidney disease, a history of bleeding, or anemia, noted the coauthors, who work for the Ottawa Hospital Research Institute.
The researchers focused on randomized controlled trials (RCTs) and prospective cohort studies that reported major bleeding among patients with a first unprovoked or weakly provoked VTE who received oral anticoagulation for at least 6 months beyond an initial anticoagulant treatment course of at least 3 months.
The investigators analyzed data from 14 RCTs and 13 cohort studies. In all, 9,982 patients received a vitamin K antagonist (VKA), and 7,220 received a direct oral anticoagulant (DOAC).
The incidence of major bleeding per 100 person-years was 1.7 events with VKAs, compared with 1.1 events with DOACs. The researchers estimated that the 5-year cumulative incidence of major bleeding with VKAs was 6.3%. The available data for DOACs were insufficient to estimate the incidence of major bleeding beyond 1 year.
“This information can help clinicians counsel patients and inform shared decision-making about extended therapy,” the researchers said.
Risks of serious bleeding ‘not trivial’
Margaret Fang, MD, with the University of California, San Francisco, agreed that the study can help clinicians and patients weigh the risks of extended anticoagulation for common types of VTE.
The study also “highlights that the risks of serious bleeding are not trivial” and points out gaps in the literature regarding the long-term use of DOACs for extended VTE therapy, Dr. Fang said.
Better ways to predict which patients will develop bleeding on anticoagulants are needed, Dr. Fang added. “It will also be important to establish which of the various therapies for preventing recurrent VTE – full dose versus lowered dose, or even aspirin – has the best balance of safety and efficacy,” she said.
‘Standardized approach’ for identifying high-risk patients lacking
Clinical practice guidelines recommend indefinite anticoagulation for an unprovoked VTE, except when patients are at high risk of bleeding, the authors noted. But clinicians lack a “standardized approach to identify patients at high risk of bleeding,” Mr. Khan and Dr. Rodger said. “Evidence from randomized trials on net long-term benefit of extended therapy is limited, and current guideline recommendations are largely based on expert consensus opinion. Major bleeding events are two to three times more likely to be fatal than recurrent VTE events, so extended therapy is not always associated with a net mortality benefit, particularly in patients at low risk of recurrent VTE or high risk of bleeding.”
The analysis indicates that there is “a clinically meaningful difference in long-term risk for anticoagulant-related major bleeding among patients with a first unprovoked VTE stratified according to presence or absence of the following risk factors: age older than 65 years, creatinine clearance less than 50 mL/min, history of bleeding, concomitant use of antiplatelet therapy, and hemoglobin level less than 100 g/L,” the authors said.
For example, the researchers found that the incidence of major bleeding was higher among those older than 65 years, compared with younger patients (incidence rate ratio, 1.84 with VKAs and 2.92 with DOACs), and among those with creatinine clearance less than 50 mL/min (IRR, 2.83 with VKAs and 3.71 with DOACs).
The case-fatality rate of major bleeding was 8.3% with VKAs and 9.7% with DOACs.
The study received funding from the Canadian Institutes of Health Research. Some of the coauthors are employees of or have financial ties to pharmaceutical companies. Mr. Khan, Dr. Rodger, and Dr. Fang had no relevant disclosures.
The meta-analysis of data from 27 studies with 17,202 patients was published in the Annals of Internal Medicine. According to two of the paper’s coauthors, Faizan Khan, MSc, and Marc A. Rodger, MD, it “provides best available estimates of long-term bleeding risk with different anticoagulants in patients with unprovoked VTE,” including subgroups at increased risk.
Patients at increased risk for major bleeding include those who are older; those using antiplatelet therapy; and patients with kidney disease, a history of bleeding, or anemia, noted the coauthors, who work for the Ottawa Hospital Research Institute.
The researchers focused on randomized controlled trials (RCTs) and prospective cohort studies that reported major bleeding among patients with a first unprovoked or weakly provoked VTE who received oral anticoagulation for at least 6 months beyond an initial anticoagulant treatment course of at least 3 months.
The investigators analyzed data from 14 RCTs and 13 cohort studies. In all, 9,982 patients received a vitamin K antagonist (VKA), and 7,220 received a direct oral anticoagulant (DOAC).
The incidence of major bleeding per 100 person-years was 1.7 events with VKAs, compared with 1.1 events with DOACs. The researchers estimated that the 5-year cumulative incidence of major bleeding with VKAs was 6.3%. The available data for DOACs were insufficient to estimate the incidence of major bleeding beyond 1 year.
“This information can help clinicians counsel patients and inform shared decision-making about extended therapy,” the researchers said.
Risks of serious bleeding ‘not trivial’
Margaret Fang, MD, with the University of California, San Francisco, agreed that the study can help clinicians and patients weigh the risks of extended anticoagulation for common types of VTE.
The study also “highlights that the risks of serious bleeding are not trivial” and points out gaps in the literature regarding the long-term use of DOACs for extended VTE therapy, Dr. Fang said.
Better ways to predict which patients will develop bleeding on anticoagulants are needed, Dr. Fang added. “It will also be important to establish which of the various therapies for preventing recurrent VTE – full dose versus lowered dose, or even aspirin – has the best balance of safety and efficacy,” she said.
‘Standardized approach’ for identifying high-risk patients lacking
Clinical practice guidelines recommend indefinite anticoagulation for an unprovoked VTE, except when patients are at high risk of bleeding, the authors noted. But clinicians lack a “standardized approach to identify patients at high risk of bleeding,” Mr. Khan and Dr. Rodger said. “Evidence from randomized trials on net long-term benefit of extended therapy is limited, and current guideline recommendations are largely based on expert consensus opinion. Major bleeding events are two to three times more likely to be fatal than recurrent VTE events, so extended therapy is not always associated with a net mortality benefit, particularly in patients at low risk of recurrent VTE or high risk of bleeding.”
The analysis indicates that there is “a clinically meaningful difference in long-term risk for anticoagulant-related major bleeding among patients with a first unprovoked VTE stratified according to presence or absence of the following risk factors: age older than 65 years, creatinine clearance less than 50 mL/min, history of bleeding, concomitant use of antiplatelet therapy, and hemoglobin level less than 100 g/L,” the authors said.
For example, the researchers found that the incidence of major bleeding was higher among those older than 65 years, compared with younger patients (incidence rate ratio, 1.84 with VKAs and 2.92 with DOACs), and among those with creatinine clearance less than 50 mL/min (IRR, 2.83 with VKAs and 3.71 with DOACs).
The case-fatality rate of major bleeding was 8.3% with VKAs and 9.7% with DOACs.
The study received funding from the Canadian Institutes of Health Research. Some of the coauthors are employees of or have financial ties to pharmaceutical companies. Mr. Khan, Dr. Rodger, and Dr. Fang had no relevant disclosures.
FROM ANNALS OF INTERNAL MEDICINE