User login
EMPEROR-Preserved spouts torrent of reports on empagliflozin treatment of HFpEF
The featured report from the 6,000-patient EMPEROR-Preserved trial at the virtual annual congress of the European Society of Cardiology drew lots of attention for its headline finding: the first unequivocal demonstration that a medication, empagliflozin, can significantly reduce the rate of cardiovascular death and hospitalization for heart failure in patients with heart failure with preserved ejection fraction (HFpEF, a left ventricular ejection fraction of more than 40%), with the details simultaneously published online.
But at the same time, the EMPEROR-Preserved investigators released four additional reports with a lot more outcome analyses that also deserve some attention.
The puzzling neutral effect on renal events
Perhaps the most surprising and complicated set of findings among the main EMPEROR-Preserved outcomes involved renal outcomes.
The trial’s primary outcome was the combined rate of cardiovascular death or hospitalization for heart failure (HHF), and the results showed that treatment with empagliflozin (Jardiance) for a median of 26 months on top of standard treatment for patients with HFpEF led to a significant 21% relative risk reduction, compared with placebo-treated patients.
The trial had two prespecified secondary outcomes. One was the total number of HHF, which dropped by a significant 27%, compared with placebo. The second was the mean change in slope of estimated glomerular filtration rate (eGFR) on an annualized basis, and the empagliflozin regimen reduced the cumulative annual deficit, compared with placebo by an average of 1.36 mL/min per 1.73 m2, a significant difference.
This preservation of renal function was consistent with results from many prior studies of empagliflozin and all of the other U.S.-approved agents from the sodium-glucose cotransporter 2 inhibitor class. Preservation of renal function and a reduction in renal events has become a hallmark property of all agents in the SGLT2 inhibitor class both in patients with type 2 diabetes, as well as in those without diabetes but with heart failure with reduced ejection fraction (HFrEF) or with chronic kidney disease.
EMPEROR-Preserved threw a wrench into what had been an unbroken history of renal protection by SGLT2 inhibitors. That happened when a prespecified endpoint of the study – a composite renal outcome defined as time to first occurrence of chronic dialysis, renal transplantation, a sustained reduction of at least 40% in eGFR, or a sustained drop in eGFR of more than 10 or 15 mL/min per 1.73 m2 from baseline – yielded an unexpected neutral finding.
For this composite renal outcome, EMPEROR-Preserved showed a nonsignificant 5% reduction, compared with placebo, a result that both differed from what had been seen in essentially all the other SGLT2 inhibitor trials that had looked at this, but which also seemed at odds with the observed significant preservation of renal function that seemed substantial enough to produce a clinically meaningful benefit.
Renal effects blunted in HFpEF
The immediate upshot was a letter published by several EMPEROR-Preserved investigators that spelled out this discrepancy and came to the jolting conclusion that “eGFR slope analysis has limitations as a surrogate for predicting the effect of drugs on renal outcomes in patients with heart failure.”
The same authors, along with some additional associates, also published a second letter that noted a further unexpected twist with the renal outcome: “In prior large-scale clinical trials, the effect of SGLT2 inhibitors on heart failure and renal outcomes had consistently tracked together,” they noted, but in this case it didn’t, a discordance they said was “extraordinarily puzzling”.

This led the study’s leaders to reanalyze the renal outcomes using a different definition, one that Milton Packer, MD, who helped design the trial and oversaw several of its analyses, called “a more conventional definition of renal events,” during his presentation of these findings at the congress. The researchers swapped out a 40% drop from baseline eGFR as an event and replaced it with a 50% decline, a change designed to screen out less severe, and often transient, reductions in kidney function that have less lasting impact on health. They also added an additional component to the composite endpoint, renal death. A revised analysis using this new renal composite outcome appeared in the European Journal of Heart Failure letter.
This change cut the total number of renal events tallied in the trial nearly in half, down to 112, and showed a more robust decline in renal events with empagliflozin treatment compared with the initial analysis, although the drop remained nonsignificant. The revised analysis also showed that the overall, nonsignificant 22% relative reduction in renal events in patients on empagliflozin, compared with placebo, dwindled down to completely nonexistent in the tertile of patients with a left ventricular ejection fraction of 60% or greater. In this tertile the hazard ratio actually showed a nonsignificant point estimate of a 24% increased rate of renal events on empagliflozin, with the caveat that this subgroup now included a total of just 40 total events between the two treatment arms. (Each of the two other tertiles also had roughly the same number of total events.)
The biggest effect on renal-event reduction was in the tertile of patients with an ejection fraction of 41%-49%, in which empagliflozin treatment was linked with a significant 59% cut in renal events, compared with placebo. The analysis also showed significant heterogeneity in thus outcome between this subgroup and the other two tertiles that had higher ejection fractions and showed reduced rates of protection by empagliflozin against renal events.
This apparent blunting of a renal effect despite preservation of renal function seemed to mimic the blunting of the primary cardiovascular outcome effect that also appeared in patients with ejection fractions in the 60%-65% range or above.
“If we knew what blunted the effect of empagliflozin on heart failure outcomes at higher ejection fraction levels, we think the same explanation may also apply to the blunting of effect on renal outcomes, but right now we do not know the answer to either question,” Dr. Packer said in an interview. He’s suggested that one possibility is that many of the enrolled patients identified as having HFpEF, but with these high ejection fractions may have not actually had HFpEF, and their signs and symptoms may have instead resulted from atrial fibrillation.
“Many patients with an ejection fraction of 60%-65% and above had atrial fibrillation,” he noted, with a prevalence at enrollment in this subgroup of about 50%. Atrial fibrillation can cause dyspnea, a hallmark symptom leading to diagnosis of heart failure, and it also increases levels of N-terminal of the prohormone brain natriuretic peptide, a metric that served as a gatekeeper for entry into the trial. “Essentially, we are saying that many of the criteria that we specified to ensure that patients had heart failure probably did not work very well in patients with an ejection fraction of 65% or greater,” said Dr. Packer, a cardiologist at Baylor University Medical Center in Dallas. “We need to figure out who these patients are.”
Some experts not involved with the study voiced skepticism that the renal findings reflected a real issue.
“I’m quite optimistic that in the long-term the effect on eGFR will translate into renal protection,” said Rudolf A. de Boer, MD, PhD, a professor of translational cardiology at University Medical Center Groningen (the Netherlands), and designated discussant at the congress for the presentation by Dr. Packer.

John J.V. McMurray, MD, a professor of cardiology and a heart failure specialist at Glasgow University, speculated that the unexpected renal outcomes data may relate to the initial decline in renal function produced by treatment with SGLT2 inhibitors despite their longer-term enhancement of renal protection.
“If you use a treatment that protects the kidneys in the long-term but causes an initial dip in eGFR, more patients receiving that treatment will have an early ‘event,’ ” he noted in an interview. He also cautioned about the dangers of subgroup analyses that dice the study population into small cohorts.
“Trials are powered to look at the effect of treatment in the overall population. Everything else is exploratory, underpowered, and subject to the play of chance,” Dr. McMurray stressed.
Counting additional cardiovascular disease events allows more analyses
A third auxiliary report from the EMPEROR-Preserved investigators performed several prespecified analyses that depended on adding additional cardiovascular disease endpoints to the core tallies of cardiovascular death or HHF – such as emergent, urgent, and outpatient events that reflected worsening heart failure – and also included information on diuretic and vasopressor use because of worsening heart failure. The increased event numbers allowed the researchers to perform 30 additional analyses included in this report, according to the count kept by Dr. Packer who was the lead author.
He highlighted several of the additional results in this paper that documented benefits from empagliflozin treatment, compared with placebo:
- A significant 29% reduction in the need for admission to a cardiac care unit or intensive care unit during an HHF.
- A nonsignificant 33% reduction in the need for intravenous vasopressors or positive inotropic drugs during HHF.
- A significantly increased rate of patients achieving a higher New York Heart Association functional class. For example, after the first year of treatment patients who received empagliflozin had a 37% higher rate of functional class improvement, compared with patients who received placebo.
Dr. McMurray had his own list of key takeaways from this paper, including:
- Among patients who needed hospitalization, “those treated with empagliflozin were less sick than those in the placebo group.”
- In addition to reducing HHF empagliflozin treatment also reduced episodes of outpatient worsening as reflected by their receipt of intensified diuretic treatment, which occurred a significant 27% less often, compared with patients on placebo.
- Treatment with empagliflozin also linked with a significant 39% relative reduction in emergency or urgent-care visits that required intravenous therapy.
Empagliflozin’s performance relative to sacubitril/valsartan
The fourth additional report focused on a post hoc, cross-trial comparison of the results from EMPEROR-Preserved and from another recent trial that, like EMPEROR-Preserved, assessed in patients with HFpEF a drug previously proven to work quite well in patients with HFrEF. The comparator drug was sacubitril/valsartan (Entresto), which underwent testing in patients with HFpEF in the PARAGON-HF trial.
The primary outcome of PARAGON-HF, which randomized 4,822 patients, was reduction in cardiovascular death and in total HHF. This dropped by a relative 13%, compared with placebo, during a median of 35 months, a between-group difference that came close to but did not achieve significance (P = .06). Despite this limitation, the Food and Drug Administration in February 2021 loosened the indication for using sacubitril/valsartan in patients with heart failure and a “below normal” ejection fraction, a category that can include many patients considered to have HFpEF.
Although the researchers who ran this analysis, including Dr. Packer, who was the first author, admitted that “comparison of effect sizes across trials is fraught with difficulties,” they nonetheless concluded from their analysis that “for all outcomes that included HHF the effect size was larger for empagliflozin than for sacubitril/valsartan.”
Dr. McMurray, a lead instigator for PARAGON-HF, said there was little to take away from this analysis.
“The patient populations were different, and sacubitril/valsartan was compared against an active therapy, valsartan,” while in EMPEROR-Preserved empagliflozin compared against placebo. “Most of us believe that sacubitril/valsartan and SGLT2 inhibitors work in different but complementary ways, and their benefits are additive. You would want patients with HFpEF or HFrEF to take both,” he said in an interview.
Dr. Packer agreed with that approach and added that he would probably also prescribe a third agent, spironolactone, to many patients with HFpEF.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and Eli Lilly, which jointly market empagliflozin (Jardiance). PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. de Boer has research contracts with Boehringer Ingelheim as well as from Abbott, AstraZeneca, Cardior, Ionis, Novo Nordisk, and Roche, and he has been a consultant to Novartis as well as to Abbott, AstraZeneca, Gayer, and Roche. Dr. McMurray led trials of sacubitril/valsartan sponsored by Novartis, and his institution has received compensation for his participation in studies sponsored by Abbvie, AstraZeneca, Cardurion, DalCor, GlaxoSmithKline, Pfizer, and Theracos.
The featured report from the 6,000-patient EMPEROR-Preserved trial at the virtual annual congress of the European Society of Cardiology drew lots of attention for its headline finding: the first unequivocal demonstration that a medication, empagliflozin, can significantly reduce the rate of cardiovascular death and hospitalization for heart failure in patients with heart failure with preserved ejection fraction (HFpEF, a left ventricular ejection fraction of more than 40%), with the details simultaneously published online.
But at the same time, the EMPEROR-Preserved investigators released four additional reports with a lot more outcome analyses that also deserve some attention.
The puzzling neutral effect on renal events
Perhaps the most surprising and complicated set of findings among the main EMPEROR-Preserved outcomes involved renal outcomes.
The trial’s primary outcome was the combined rate of cardiovascular death or hospitalization for heart failure (HHF), and the results showed that treatment with empagliflozin (Jardiance) for a median of 26 months on top of standard treatment for patients with HFpEF led to a significant 21% relative risk reduction, compared with placebo-treated patients.
The trial had two prespecified secondary outcomes. One was the total number of HHF, which dropped by a significant 27%, compared with placebo. The second was the mean change in slope of estimated glomerular filtration rate (eGFR) on an annualized basis, and the empagliflozin regimen reduced the cumulative annual deficit, compared with placebo by an average of 1.36 mL/min per 1.73 m2, a significant difference.
This preservation of renal function was consistent with results from many prior studies of empagliflozin and all of the other U.S.-approved agents from the sodium-glucose cotransporter 2 inhibitor class. Preservation of renal function and a reduction in renal events has become a hallmark property of all agents in the SGLT2 inhibitor class both in patients with type 2 diabetes, as well as in those without diabetes but with heart failure with reduced ejection fraction (HFrEF) or with chronic kidney disease.
EMPEROR-Preserved threw a wrench into what had been an unbroken history of renal protection by SGLT2 inhibitors. That happened when a prespecified endpoint of the study – a composite renal outcome defined as time to first occurrence of chronic dialysis, renal transplantation, a sustained reduction of at least 40% in eGFR, or a sustained drop in eGFR of more than 10 or 15 mL/min per 1.73 m2 from baseline – yielded an unexpected neutral finding.
For this composite renal outcome, EMPEROR-Preserved showed a nonsignificant 5% reduction, compared with placebo, a result that both differed from what had been seen in essentially all the other SGLT2 inhibitor trials that had looked at this, but which also seemed at odds with the observed significant preservation of renal function that seemed substantial enough to produce a clinically meaningful benefit.
Renal effects blunted in HFpEF
The immediate upshot was a letter published by several EMPEROR-Preserved investigators that spelled out this discrepancy and came to the jolting conclusion that “eGFR slope analysis has limitations as a surrogate for predicting the effect of drugs on renal outcomes in patients with heart failure.”
The same authors, along with some additional associates, also published a second letter that noted a further unexpected twist with the renal outcome: “In prior large-scale clinical trials, the effect of SGLT2 inhibitors on heart failure and renal outcomes had consistently tracked together,” they noted, but in this case it didn’t, a discordance they said was “extraordinarily puzzling”.
This led the study’s leaders to reanalyze the renal outcomes using a different definition, one that Milton Packer, MD, who helped design the trial and oversaw several of its analyses, called “a more conventional definition of renal events,” during his presentation of these findings at the congress. The researchers swapped out a 40% drop from baseline eGFR as an event and replaced it with a 50% decline, a change designed to screen out less severe, and often transient, reductions in kidney function that have less lasting impact on health. They also added an additional component to the composite endpoint, renal death. A revised analysis using this new renal composite outcome appeared in the European Journal of Heart Failure letter.
This change cut the total number of renal events tallied in the trial nearly in half, down to 112, and showed a more robust decline in renal events with empagliflozin treatment compared with the initial analysis, although the drop remained nonsignificant. The revised analysis also showed that the overall, nonsignificant 22% relative reduction in renal events in patients on empagliflozin, compared with placebo, dwindled down to completely nonexistent in the tertile of patients with a left ventricular ejection fraction of 60% or greater. In this tertile the hazard ratio actually showed a nonsignificant point estimate of a 24% increased rate of renal events on empagliflozin, with the caveat that this subgroup now included a total of just 40 total events between the two treatment arms. (Each of the two other tertiles also had roughly the same number of total events.)
The biggest effect on renal-event reduction was in the tertile of patients with an ejection fraction of 41%-49%, in which empagliflozin treatment was linked with a significant 59% cut in renal events, compared with placebo. The analysis also showed significant heterogeneity in thus outcome between this subgroup and the other two tertiles that had higher ejection fractions and showed reduced rates of protection by empagliflozin against renal events.
This apparent blunting of a renal effect despite preservation of renal function seemed to mimic the blunting of the primary cardiovascular outcome effect that also appeared in patients with ejection fractions in the 60%-65% range or above.
“If we knew what blunted the effect of empagliflozin on heart failure outcomes at higher ejection fraction levels, we think the same explanation may also apply to the blunting of effect on renal outcomes, but right now we do not know the answer to either question,” Dr. Packer said in an interview. He’s suggested that one possibility is that many of the enrolled patients identified as having HFpEF, but with these high ejection fractions may have not actually had HFpEF, and their signs and symptoms may have instead resulted from atrial fibrillation.
“Many patients with an ejection fraction of 60%-65% and above had atrial fibrillation,” he noted, with a prevalence at enrollment in this subgroup of about 50%. Atrial fibrillation can cause dyspnea, a hallmark symptom leading to diagnosis of heart failure, and it also increases levels of N-terminal of the prohormone brain natriuretic peptide, a metric that served as a gatekeeper for entry into the trial. “Essentially, we are saying that many of the criteria that we specified to ensure that patients had heart failure probably did not work very well in patients with an ejection fraction of 65% or greater,” said Dr. Packer, a cardiologist at Baylor University Medical Center in Dallas. “We need to figure out who these patients are.”
Some experts not involved with the study voiced skepticism that the renal findings reflected a real issue.
“I’m quite optimistic that in the long-term the effect on eGFR will translate into renal protection,” said Rudolf A. de Boer, MD, PhD, a professor of translational cardiology at University Medical Center Groningen (the Netherlands), and designated discussant at the congress for the presentation by Dr. Packer.
John J.V. McMurray, MD, a professor of cardiology and a heart failure specialist at Glasgow University, speculated that the unexpected renal outcomes data may relate to the initial decline in renal function produced by treatment with SGLT2 inhibitors despite their longer-term enhancement of renal protection.
“If you use a treatment that protects the kidneys in the long-term but causes an initial dip in eGFR, more patients receiving that treatment will have an early ‘event,’ ” he noted in an interview. He also cautioned about the dangers of subgroup analyses that dice the study population into small cohorts.
“Trials are powered to look at the effect of treatment in the overall population. Everything else is exploratory, underpowered, and subject to the play of chance,” Dr. McMurray stressed.
Counting additional cardiovascular disease events allows more analyses
A third auxiliary report from the EMPEROR-Preserved investigators performed several prespecified analyses that depended on adding additional cardiovascular disease endpoints to the core tallies of cardiovascular death or HHF – such as emergent, urgent, and outpatient events that reflected worsening heart failure – and also included information on diuretic and vasopressor use because of worsening heart failure. The increased event numbers allowed the researchers to perform 30 additional analyses included in this report, according to the count kept by Dr. Packer who was the lead author.
He highlighted several of the additional results in this paper that documented benefits from empagliflozin treatment, compared with placebo:
- A significant 29% reduction in the need for admission to a cardiac care unit or intensive care unit during an HHF.
- A nonsignificant 33% reduction in the need for intravenous vasopressors or positive inotropic drugs during HHF.
- A significantly increased rate of patients achieving a higher New York Heart Association functional class. For example, after the first year of treatment patients who received empagliflozin had a 37% higher rate of functional class improvement, compared with patients who received placebo.
Dr. McMurray had his own list of key takeaways from this paper, including:
- Among patients who needed hospitalization, “those treated with empagliflozin were less sick than those in the placebo group.”
- In addition to reducing HHF empagliflozin treatment also reduced episodes of outpatient worsening as reflected by their receipt of intensified diuretic treatment, which occurred a significant 27% less often, compared with patients on placebo.
- Treatment with empagliflozin also linked with a significant 39% relative reduction in emergency or urgent-care visits that required intravenous therapy.
Empagliflozin’s performance relative to sacubitril/valsartan
The fourth additional report focused on a post hoc, cross-trial comparison of the results from EMPEROR-Preserved and from another recent trial that, like EMPEROR-Preserved, assessed in patients with HFpEF a drug previously proven to work quite well in patients with HFrEF. The comparator drug was sacubitril/valsartan (Entresto), which underwent testing in patients with HFpEF in the PARAGON-HF trial.
The primary outcome of PARAGON-HF, which randomized 4,822 patients, was reduction in cardiovascular death and in total HHF. This dropped by a relative 13%, compared with placebo, during a median of 35 months, a between-group difference that came close to but did not achieve significance (P = .06). Despite this limitation, the Food and Drug Administration in February 2021 loosened the indication for using sacubitril/valsartan in patients with heart failure and a “below normal” ejection fraction, a category that can include many patients considered to have HFpEF.
Although the researchers who ran this analysis, including Dr. Packer, who was the first author, admitted that “comparison of effect sizes across trials is fraught with difficulties,” they nonetheless concluded from their analysis that “for all outcomes that included HHF the effect size was larger for empagliflozin than for sacubitril/valsartan.”
Dr. McMurray, a lead instigator for PARAGON-HF, said there was little to take away from this analysis.
“The patient populations were different, and sacubitril/valsartan was compared against an active therapy, valsartan,” while in EMPEROR-Preserved empagliflozin compared against placebo. “Most of us believe that sacubitril/valsartan and SGLT2 inhibitors work in different but complementary ways, and their benefits are additive. You would want patients with HFpEF or HFrEF to take both,” he said in an interview.
Dr. Packer agreed with that approach and added that he would probably also prescribe a third agent, spironolactone, to many patients with HFpEF.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and Eli Lilly, which jointly market empagliflozin (Jardiance). PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. de Boer has research contracts with Boehringer Ingelheim as well as from Abbott, AstraZeneca, Cardior, Ionis, Novo Nordisk, and Roche, and he has been a consultant to Novartis as well as to Abbott, AstraZeneca, Gayer, and Roche. Dr. McMurray led trials of sacubitril/valsartan sponsored by Novartis, and his institution has received compensation for his participation in studies sponsored by Abbvie, AstraZeneca, Cardurion, DalCor, GlaxoSmithKline, Pfizer, and Theracos.
The featured report from the 6,000-patient EMPEROR-Preserved trial at the virtual annual congress of the European Society of Cardiology drew lots of attention for its headline finding: the first unequivocal demonstration that a medication, empagliflozin, can significantly reduce the rate of cardiovascular death and hospitalization for heart failure in patients with heart failure with preserved ejection fraction (HFpEF, a left ventricular ejection fraction of more than 40%), with the details simultaneously published online.
But at the same time, the EMPEROR-Preserved investigators released four additional reports with a lot more outcome analyses that also deserve some attention.
The puzzling neutral effect on renal events
Perhaps the most surprising and complicated set of findings among the main EMPEROR-Preserved outcomes involved renal outcomes.
The trial’s primary outcome was the combined rate of cardiovascular death or hospitalization for heart failure (HHF), and the results showed that treatment with empagliflozin (Jardiance) for a median of 26 months on top of standard treatment for patients with HFpEF led to a significant 21% relative risk reduction, compared with placebo-treated patients.
The trial had two prespecified secondary outcomes. One was the total number of HHF, which dropped by a significant 27%, compared with placebo. The second was the mean change in slope of estimated glomerular filtration rate (eGFR) on an annualized basis, and the empagliflozin regimen reduced the cumulative annual deficit, compared with placebo by an average of 1.36 mL/min per 1.73 m2, a significant difference.
This preservation of renal function was consistent with results from many prior studies of empagliflozin and all of the other U.S.-approved agents from the sodium-glucose cotransporter 2 inhibitor class. Preservation of renal function and a reduction in renal events has become a hallmark property of all agents in the SGLT2 inhibitor class both in patients with type 2 diabetes, as well as in those without diabetes but with heart failure with reduced ejection fraction (HFrEF) or with chronic kidney disease.
EMPEROR-Preserved threw a wrench into what had been an unbroken history of renal protection by SGLT2 inhibitors. That happened when a prespecified endpoint of the study – a composite renal outcome defined as time to first occurrence of chronic dialysis, renal transplantation, a sustained reduction of at least 40% in eGFR, or a sustained drop in eGFR of more than 10 or 15 mL/min per 1.73 m2 from baseline – yielded an unexpected neutral finding.
For this composite renal outcome, EMPEROR-Preserved showed a nonsignificant 5% reduction, compared with placebo, a result that both differed from what had been seen in essentially all the other SGLT2 inhibitor trials that had looked at this, but which also seemed at odds with the observed significant preservation of renal function that seemed substantial enough to produce a clinically meaningful benefit.
Renal effects blunted in HFpEF
The immediate upshot was a letter published by several EMPEROR-Preserved investigators that spelled out this discrepancy and came to the jolting conclusion that “eGFR slope analysis has limitations as a surrogate for predicting the effect of drugs on renal outcomes in patients with heart failure.”
The same authors, along with some additional associates, also published a second letter that noted a further unexpected twist with the renal outcome: “In prior large-scale clinical trials, the effect of SGLT2 inhibitors on heart failure and renal outcomes had consistently tracked together,” they noted, but in this case it didn’t, a discordance they said was “extraordinarily puzzling”.
This led the study’s leaders to reanalyze the renal outcomes using a different definition, one that Milton Packer, MD, who helped design the trial and oversaw several of its analyses, called “a more conventional definition of renal events,” during his presentation of these findings at the congress. The researchers swapped out a 40% drop from baseline eGFR as an event and replaced it with a 50% decline, a change designed to screen out less severe, and often transient, reductions in kidney function that have less lasting impact on health. They also added an additional component to the composite endpoint, renal death. A revised analysis using this new renal composite outcome appeared in the European Journal of Heart Failure letter.
This change cut the total number of renal events tallied in the trial nearly in half, down to 112, and showed a more robust decline in renal events with empagliflozin treatment compared with the initial analysis, although the drop remained nonsignificant. The revised analysis also showed that the overall, nonsignificant 22% relative reduction in renal events in patients on empagliflozin, compared with placebo, dwindled down to completely nonexistent in the tertile of patients with a left ventricular ejection fraction of 60% or greater. In this tertile the hazard ratio actually showed a nonsignificant point estimate of a 24% increased rate of renal events on empagliflozin, with the caveat that this subgroup now included a total of just 40 total events between the two treatment arms. (Each of the two other tertiles also had roughly the same number of total events.)
The biggest effect on renal-event reduction was in the tertile of patients with an ejection fraction of 41%-49%, in which empagliflozin treatment was linked with a significant 59% cut in renal events, compared with placebo. The analysis also showed significant heterogeneity in thus outcome between this subgroup and the other two tertiles that had higher ejection fractions and showed reduced rates of protection by empagliflozin against renal events.
This apparent blunting of a renal effect despite preservation of renal function seemed to mimic the blunting of the primary cardiovascular outcome effect that also appeared in patients with ejection fractions in the 60%-65% range or above.
“If we knew what blunted the effect of empagliflozin on heart failure outcomes at higher ejection fraction levels, we think the same explanation may also apply to the blunting of effect on renal outcomes, but right now we do not know the answer to either question,” Dr. Packer said in an interview. He’s suggested that one possibility is that many of the enrolled patients identified as having HFpEF, but with these high ejection fractions may have not actually had HFpEF, and their signs and symptoms may have instead resulted from atrial fibrillation.
“Many patients with an ejection fraction of 60%-65% and above had atrial fibrillation,” he noted, with a prevalence at enrollment in this subgroup of about 50%. Atrial fibrillation can cause dyspnea, a hallmark symptom leading to diagnosis of heart failure, and it also increases levels of N-terminal of the prohormone brain natriuretic peptide, a metric that served as a gatekeeper for entry into the trial. “Essentially, we are saying that many of the criteria that we specified to ensure that patients had heart failure probably did not work very well in patients with an ejection fraction of 65% or greater,” said Dr. Packer, a cardiologist at Baylor University Medical Center in Dallas. “We need to figure out who these patients are.”
Some experts not involved with the study voiced skepticism that the renal findings reflected a real issue.
“I’m quite optimistic that in the long-term the effect on eGFR will translate into renal protection,” said Rudolf A. de Boer, MD, PhD, a professor of translational cardiology at University Medical Center Groningen (the Netherlands), and designated discussant at the congress for the presentation by Dr. Packer.
John J.V. McMurray, MD, a professor of cardiology and a heart failure specialist at Glasgow University, speculated that the unexpected renal outcomes data may relate to the initial decline in renal function produced by treatment with SGLT2 inhibitors despite their longer-term enhancement of renal protection.
“If you use a treatment that protects the kidneys in the long-term but causes an initial dip in eGFR, more patients receiving that treatment will have an early ‘event,’ ” he noted in an interview. He also cautioned about the dangers of subgroup analyses that dice the study population into small cohorts.
“Trials are powered to look at the effect of treatment in the overall population. Everything else is exploratory, underpowered, and subject to the play of chance,” Dr. McMurray stressed.
Counting additional cardiovascular disease events allows more analyses
A third auxiliary report from the EMPEROR-Preserved investigators performed several prespecified analyses that depended on adding additional cardiovascular disease endpoints to the core tallies of cardiovascular death or HHF – such as emergent, urgent, and outpatient events that reflected worsening heart failure – and also included information on diuretic and vasopressor use because of worsening heart failure. The increased event numbers allowed the researchers to perform 30 additional analyses included in this report, according to the count kept by Dr. Packer who was the lead author.
He highlighted several of the additional results in this paper that documented benefits from empagliflozin treatment, compared with placebo:
- A significant 29% reduction in the need for admission to a cardiac care unit or intensive care unit during an HHF.
- A nonsignificant 33% reduction in the need for intravenous vasopressors or positive inotropic drugs during HHF.
- A significantly increased rate of patients achieving a higher New York Heart Association functional class. For example, after the first year of treatment patients who received empagliflozin had a 37% higher rate of functional class improvement, compared with patients who received placebo.
Dr. McMurray had his own list of key takeaways from this paper, including:
- Among patients who needed hospitalization, “those treated with empagliflozin were less sick than those in the placebo group.”
- In addition to reducing HHF empagliflozin treatment also reduced episodes of outpatient worsening as reflected by their receipt of intensified diuretic treatment, which occurred a significant 27% less often, compared with patients on placebo.
- Treatment with empagliflozin also linked with a significant 39% relative reduction in emergency or urgent-care visits that required intravenous therapy.
Empagliflozin’s performance relative to sacubitril/valsartan
The fourth additional report focused on a post hoc, cross-trial comparison of the results from EMPEROR-Preserved and from another recent trial that, like EMPEROR-Preserved, assessed in patients with HFpEF a drug previously proven to work quite well in patients with HFrEF. The comparator drug was sacubitril/valsartan (Entresto), which underwent testing in patients with HFpEF in the PARAGON-HF trial.
The primary outcome of PARAGON-HF, which randomized 4,822 patients, was reduction in cardiovascular death and in total HHF. This dropped by a relative 13%, compared with placebo, during a median of 35 months, a between-group difference that came close to but did not achieve significance (P = .06). Despite this limitation, the Food and Drug Administration in February 2021 loosened the indication for using sacubitril/valsartan in patients with heart failure and a “below normal” ejection fraction, a category that can include many patients considered to have HFpEF.
Although the researchers who ran this analysis, including Dr. Packer, who was the first author, admitted that “comparison of effect sizes across trials is fraught with difficulties,” they nonetheless concluded from their analysis that “for all outcomes that included HHF the effect size was larger for empagliflozin than for sacubitril/valsartan.”
Dr. McMurray, a lead instigator for PARAGON-HF, said there was little to take away from this analysis.
“The patient populations were different, and sacubitril/valsartan was compared against an active therapy, valsartan,” while in EMPEROR-Preserved empagliflozin compared against placebo. “Most of us believe that sacubitril/valsartan and SGLT2 inhibitors work in different but complementary ways, and their benefits are additive. You would want patients with HFpEF or HFrEF to take both,” he said in an interview.
Dr. Packer agreed with that approach and added that he would probably also prescribe a third agent, spironolactone, to many patients with HFpEF.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and Eli Lilly, which jointly market empagliflozin (Jardiance). PARAGON-HF was sponsored by Novartis, which markets sacubitril/valsartan (Entresto). Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. de Boer has research contracts with Boehringer Ingelheim as well as from Abbott, AstraZeneca, Cardior, Ionis, Novo Nordisk, and Roche, and he has been a consultant to Novartis as well as to Abbott, AstraZeneca, Gayer, and Roche. Dr. McMurray led trials of sacubitril/valsartan sponsored by Novartis, and his institution has received compensation for his participation in studies sponsored by Abbvie, AstraZeneca, Cardurion, DalCor, GlaxoSmithKline, Pfizer, and Theracos.
FROM ESC 2021
Although inconclusive, CV safety study of cancer therapy attracts attention
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.

“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).

In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.
“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).
In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
The first global trial to compare the cardiovascular (CV) safety of two therapies for prostate cancer proved inconclusive because of inadequate enrollment and events, but the study is a harbinger of growth in the emerging specialty of cardio-oncology, according to experts.
“Many new cancer agents have extended patient survival, yet some of these agents have significant potential cardiovascular toxicity,” said Renato D. Lopes, MD, in presenting a study at the annual congress of the European Society of Cardiology.
In the context of improving survival in patients with or at risk for both cancer and cardiovascular disease, he suggested that the prostate cancer study he led could be “a model for interdisciplinary collaboration” needed to address the relative and sometimes competing risks of these disease states.
This point was seconded by several pioneers in cardio-oncology who participated in the discussion of the results of the trial, called PRONOUNCE.
“We know many drugs in oncology increase cardiovascular risk, so these are the types of trials we need,” according Thomas M. Suter, MD, who leads the cardio-oncology service at the University Hospital, Berne, Switzerland. He was the ESC-invited discussant for PRONOUNCE.
More than 100 centers in 12 countries involved
In PRONOUNCE, 545 patients with prostate cancer and established atherosclerotic cardiovascular disease were randomized to degarelix, a gonadotropin-releasing hormone antagonist, or leuprolide, a GnRH agonist. The patients were enrolled at 113 participating centers in 12 countries. All of the patients had an indication for an androgen-deprivation therapy (ADT).
In numerous previous studies, “ADT has been associated with higher CV morbidity and mortality, particularly in men with preexisting CV disease,” explained Dr. Lopes, but the relative cardiovascular safety of GnRH agonists relative to GnRH antagonists has been “controversial.”
The PRONOUNCE study was designed to resolve this issue, but the study was terminated early because of slow enrollment (not related to the COVID-19 pandemic). The planned enrollment was 900 patients.
In addition, the rate of major adverse cardiovascular events (MACE), defined as myocardial infarction, stroke, or death, was lower over the course of follow-up than anticipated in the study design.
No significant difference on primary endpoint
At the end of 12 months, MACE occurred in 11 (4.1%) of patients randomized to leuprolide and 15 (5.5%) of those randomized to degarelix. The greater hazard ratio for MACE in the degarelix group did not approach statistical significance (hazard ratio, 1.28; P = .53).
As a result, the question of the relative CV safety of these drugs “remains unresolved,” according to Dr. Lopes, professor of medicine at Duke University Medical Center, Durham, N.C.
This does not diminish the need to answer this question. In the addition to the fact that cancer is a malignancy primarily of advancing age when CV disease is prevalent – the mean age in this study was 73 years and 44% were over age 75 – it is often an indolent disease with long periods of survival, according to Dr. Lopes. About half of prostate cancer patients have concomitant CV disease, and about half will receive ADT at some point in their treatment.
In patients receiving ADT, leuprolide is far more commonly used than GnRH antagonists, which are offered in only about 4% of patients, according to data cited by Dr. Lopes. The underlying hypothesis of this study was that leuprolide is associated with greater CV risk, which might have been relevant to a risk-benefit calculation, if the hypothesis had been confirmed.
Cancer drugs can increase CV risk
Based on experimental data, “there is concern the leuprolide is involved in plaque destabilization,” said Dr. Lopes, but he noted that ADTs in general are associated with adverse metabolic changes, including increases in LDL cholesterol, insulin resistance, and body fat, all of which could be relevant to CV risk.
It is the improving rates of survival for prostate cancer as well for other types of cancer that have increased attention to the potential for cancer drugs to increase CV risk, another major cause of early mortality. For these competing risks, objective data are needed to evaluate a relative risk-to-benefit ratio for treatment choices.
This dilemma led the ESC to recently establish its Council on Cardio-Oncology, and many centers around the world are also creating interdisciplinary groups to guide treatment choices for patients with both diseases.
“You will certainly get a lot of referrals,” said Rudolf de Boer, MD, professor of translational cardiology, University Medical Center, Groningen, Netherlands. Basing his remark on his own experience starting a cardio-oncology clinic at his institution, he called this work challenging and agreed that the need for objective data is urgent.
“We need data to provide common ground on which to judge relative risks,” Dr. de Boer said. He also praised the PRONOUNCE investigators for their efforts even if the data failed to answer the question posed.
The PRONOUNCE results were published online in Circulation at the time of Dr. Lopes’s presentation.
The study received funding from Ferring Pharmaceuticals. Dr. Lopes reports financial relationships with Bristol-Myers Squibb, GlaxoSmithKline, Medtronic, Pfizer, and Sanofi. Dr. Suter reports financial relationships with Boehringer Ingelheim, GlaxoSmithKline, and Roche. Dr. de Boer reports financial relationships with AstraZeneca, Abbott, Bristol-Myers Squibb, Novartis, Novo Nordisk, and Roche.
FROM ESC 2021
‘High normal’ sodium, poor hydration linked to heart failure
– a heart failure (HF) precursor – and for HF itself, in older age, a new study suggests.

Compared with middle-aged adults in the Atherosclerosis Risk in Communities (ARIC) study with normal serum sodium, those with levels of 142-146 mmol/L were more likely to have left ventricular hypertrophy or HF when they were in their 70s and 80s, independent of other risk factors.
Natalia Dmitrieva, PhD, a research scientist at the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Md., presented the study findings in an e-poster on Aug. 27 at the European Society of Cardiology (ESC) Congress 2021.
“Our study suggests that maintaining good hydration can prevent or at least slow down the changes within the heart that lead to heart failure,” she said in a statement from the ESC.
It “suggests that all adults should aim for eight to ten glasses of liquid [daily] and keep salt intake low,” Dr. Dmitrieva said in an interview.
However, people should not rely completely on thirst, she cautioned, especially in middle age, when thirst sensation starts to deteriorate. And too much fluid intake can be harmful and even dangerous.
Normal serum sodium is usually defined as 135-146 mmol/L, Dr. Dmitrieva explained, and this study involved only patients in ARIC with sodium levels in this range, to try to exclude patients with genetic or water-salt balance diseases.
The findings suggest that a serum sodium level of 142-146 mmol/L, which would not be flagged as abnormal by a test lab, “can be used by clinicians as a warning sign” for a patient’s increased risk for HF, she noted.
Clinicians should explain this risk to patients and advise them to drink at least 2 L per day. However, people should not try to reduce their sodium levels by drinking more than 2 to 3 L per day, she cautioned, which can be harmful and even deadly, and they should consult their doctors.
Watch hydration
“An important finding of this study is that sodium values considered ‘normal’ may also be deleterious,” Jacob Joseph, MD, director, heart failure program, VA Boston Healthcare System, who was not involved with this study, said in an interview.
“These results are similar to studies we have conducted in heart failure with preserved ejection fraction,” noted Dr. Joseph, associate professor of medicine at Harvard Medical School, Boston.
Their studies showed a U-shaped relationship between serum sodium values and adverse outcomes, “indicating an ‘optimal’ range of serum sodium value that was narrower than the accepted normal laboratory value range,” he noted.
The study by Dmitrieva et al. was observational and the findings would need to be verified in a randomized controlled trial, Dr. Joseph pointed out; however, the research “supports the idea that even a high normal sodium level may indicate risk of future heart failure.
“Hence, patients should pay attention to hydration,” he continued, and “clinicians should not assume that a sodium level of 142 mmol/L is appropriate and should ensure that patients are paying attention to hydration.
“In today’s busy and stress-filled lifestyle, it is easy to forget about adequate fluid intake,” Dr. Joseph added.
More than 15,000 adults followed for 25 Years
To investigate the relationship between serum sodium, hydration, and future heart failure, Dr. Dmitrieva and colleagues analyzed data from 15,792 adults in ARIC who were 44-66 years of age at study entry, with serum sodium levels from 135 to 146 mmol/L.
The participants were evaluated over five visits until they reached 70-90 years.
They were divided into four groups based on their average serum sodium concentrations at study visits one and two (conducted in the first 3 years): 135 -139.5 mmol/L, 140-141.5 mmol/L, 142-143.5 mmol/L, and 144-146 mmol/L.
The researchers determined the percentage of people in each group who developed HF and left ventricular hypertrophy at visit five (25 years after study enrollment).
Patients with higher serum sodium levels had a significantly higher risk for HF and left ventricular hypertrophy, after adjustment for other risk factors, including age, blood pressure, kidney function, blood cholesterol, blood glucose, body mass index, sex, and smoking status.
Every 1 mmol/L increase in serum sodium concentration in midlife was associated with 1.20 and 1.11 increased odds of developing left ventricular hypertrophy and HF, respectively, 25 years later.
“More studies are needed to find out what proportion of people with serum sodium 142 mmol/L and higher have this [serum sodium] level because they do not drink enough and will be able to reduce it by making sure they consistently drink 2 to 2.5 L per day,” said Dr. Dmitrieva.
“It is likely that for some people, other factors that are related to genetics or diseases affecting water-salt balance could be causing their increased serum sodium levels,” she speculated.
The study was funded by the Intramural Program of the National Heart, Lung, and Blood Institute. The authors and Dr. Joseph have no relevant financial disclosures.
A version of this article first appeared on Medscape.com.
– a heart failure (HF) precursor – and for HF itself, in older age, a new study suggests.
Compared with middle-aged adults in the Atherosclerosis Risk in Communities (ARIC) study with normal serum sodium, those with levels of 142-146 mmol/L were more likely to have left ventricular hypertrophy or HF when they were in their 70s and 80s, independent of other risk factors.
Natalia Dmitrieva, PhD, a research scientist at the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Md., presented the study findings in an e-poster on Aug. 27 at the European Society of Cardiology (ESC) Congress 2021.
“Our study suggests that maintaining good hydration can prevent or at least slow down the changes within the heart that lead to heart failure,” she said in a statement from the ESC.
It “suggests that all adults should aim for eight to ten glasses of liquid [daily] and keep salt intake low,” Dr. Dmitrieva said in an interview.
However, people should not rely completely on thirst, she cautioned, especially in middle age, when thirst sensation starts to deteriorate. And too much fluid intake can be harmful and even dangerous.
Normal serum sodium is usually defined as 135-146 mmol/L, Dr. Dmitrieva explained, and this study involved only patients in ARIC with sodium levels in this range, to try to exclude patients with genetic or water-salt balance diseases.
The findings suggest that a serum sodium level of 142-146 mmol/L, which would not be flagged as abnormal by a test lab, “can be used by clinicians as a warning sign” for a patient’s increased risk for HF, she noted.
Clinicians should explain this risk to patients and advise them to drink at least 2 L per day. However, people should not try to reduce their sodium levels by drinking more than 2 to 3 L per day, she cautioned, which can be harmful and even deadly, and they should consult their doctors.
Watch hydration
“An important finding of this study is that sodium values considered ‘normal’ may also be deleterious,” Jacob Joseph, MD, director, heart failure program, VA Boston Healthcare System, who was not involved with this study, said in an interview.
“These results are similar to studies we have conducted in heart failure with preserved ejection fraction,” noted Dr. Joseph, associate professor of medicine at Harvard Medical School, Boston.
Their studies showed a U-shaped relationship between serum sodium values and adverse outcomes, “indicating an ‘optimal’ range of serum sodium value that was narrower than the accepted normal laboratory value range,” he noted.
The study by Dmitrieva et al. was observational and the findings would need to be verified in a randomized controlled trial, Dr. Joseph pointed out; however, the research “supports the idea that even a high normal sodium level may indicate risk of future heart failure.
“Hence, patients should pay attention to hydration,” he continued, and “clinicians should not assume that a sodium level of 142 mmol/L is appropriate and should ensure that patients are paying attention to hydration.
“In today’s busy and stress-filled lifestyle, it is easy to forget about adequate fluid intake,” Dr. Joseph added.
More than 15,000 adults followed for 25 Years
To investigate the relationship between serum sodium, hydration, and future heart failure, Dr. Dmitrieva and colleagues analyzed data from 15,792 adults in ARIC who were 44-66 years of age at study entry, with serum sodium levels from 135 to 146 mmol/L.
The participants were evaluated over five visits until they reached 70-90 years.
They were divided into four groups based on their average serum sodium concentrations at study visits one and two (conducted in the first 3 years): 135 -139.5 mmol/L, 140-141.5 mmol/L, 142-143.5 mmol/L, and 144-146 mmol/L.
The researchers determined the percentage of people in each group who developed HF and left ventricular hypertrophy at visit five (25 years after study enrollment).
Patients with higher serum sodium levels had a significantly higher risk for HF and left ventricular hypertrophy, after adjustment for other risk factors, including age, blood pressure, kidney function, blood cholesterol, blood glucose, body mass index, sex, and smoking status.
Every 1 mmol/L increase in serum sodium concentration in midlife was associated with 1.20 and 1.11 increased odds of developing left ventricular hypertrophy and HF, respectively, 25 years later.
“More studies are needed to find out what proportion of people with serum sodium 142 mmol/L and higher have this [serum sodium] level because they do not drink enough and will be able to reduce it by making sure they consistently drink 2 to 2.5 L per day,” said Dr. Dmitrieva.
“It is likely that for some people, other factors that are related to genetics or diseases affecting water-salt balance could be causing their increased serum sodium levels,” she speculated.
The study was funded by the Intramural Program of the National Heart, Lung, and Blood Institute. The authors and Dr. Joseph have no relevant financial disclosures.
A version of this article first appeared on Medscape.com.
– a heart failure (HF) precursor – and for HF itself, in older age, a new study suggests.
Compared with middle-aged adults in the Atherosclerosis Risk in Communities (ARIC) study with normal serum sodium, those with levels of 142-146 mmol/L were more likely to have left ventricular hypertrophy or HF when they were in their 70s and 80s, independent of other risk factors.
Natalia Dmitrieva, PhD, a research scientist at the National Heart, Lung, and Blood Institute, National Institutes of Health, Bethesda, Md., presented the study findings in an e-poster on Aug. 27 at the European Society of Cardiology (ESC) Congress 2021.
“Our study suggests that maintaining good hydration can prevent or at least slow down the changes within the heart that lead to heart failure,” she said in a statement from the ESC.
It “suggests that all adults should aim for eight to ten glasses of liquid [daily] and keep salt intake low,” Dr. Dmitrieva said in an interview.
However, people should not rely completely on thirst, she cautioned, especially in middle age, when thirst sensation starts to deteriorate. And too much fluid intake can be harmful and even dangerous.
Normal serum sodium is usually defined as 135-146 mmol/L, Dr. Dmitrieva explained, and this study involved only patients in ARIC with sodium levels in this range, to try to exclude patients with genetic or water-salt balance diseases.
The findings suggest that a serum sodium level of 142-146 mmol/L, which would not be flagged as abnormal by a test lab, “can be used by clinicians as a warning sign” for a patient’s increased risk for HF, she noted.
Clinicians should explain this risk to patients and advise them to drink at least 2 L per day. However, people should not try to reduce their sodium levels by drinking more than 2 to 3 L per day, she cautioned, which can be harmful and even deadly, and they should consult their doctors.
Watch hydration
“An important finding of this study is that sodium values considered ‘normal’ may also be deleterious,” Jacob Joseph, MD, director, heart failure program, VA Boston Healthcare System, who was not involved with this study, said in an interview.
“These results are similar to studies we have conducted in heart failure with preserved ejection fraction,” noted Dr. Joseph, associate professor of medicine at Harvard Medical School, Boston.
Their studies showed a U-shaped relationship between serum sodium values and adverse outcomes, “indicating an ‘optimal’ range of serum sodium value that was narrower than the accepted normal laboratory value range,” he noted.
The study by Dmitrieva et al. was observational and the findings would need to be verified in a randomized controlled trial, Dr. Joseph pointed out; however, the research “supports the idea that even a high normal sodium level may indicate risk of future heart failure.
“Hence, patients should pay attention to hydration,” he continued, and “clinicians should not assume that a sodium level of 142 mmol/L is appropriate and should ensure that patients are paying attention to hydration.
“In today’s busy and stress-filled lifestyle, it is easy to forget about adequate fluid intake,” Dr. Joseph added.
More than 15,000 adults followed for 25 Years
To investigate the relationship between serum sodium, hydration, and future heart failure, Dr. Dmitrieva and colleagues analyzed data from 15,792 adults in ARIC who were 44-66 years of age at study entry, with serum sodium levels from 135 to 146 mmol/L.
The participants were evaluated over five visits until they reached 70-90 years.
They were divided into four groups based on their average serum sodium concentrations at study visits one and two (conducted in the first 3 years): 135 -139.5 mmol/L, 140-141.5 mmol/L, 142-143.5 mmol/L, and 144-146 mmol/L.
The researchers determined the percentage of people in each group who developed HF and left ventricular hypertrophy at visit five (25 years after study enrollment).
Patients with higher serum sodium levels had a significantly higher risk for HF and left ventricular hypertrophy, after adjustment for other risk factors, including age, blood pressure, kidney function, blood cholesterol, blood glucose, body mass index, sex, and smoking status.
Every 1 mmol/L increase in serum sodium concentration in midlife was associated with 1.20 and 1.11 increased odds of developing left ventricular hypertrophy and HF, respectively, 25 years later.
“More studies are needed to find out what proportion of people with serum sodium 142 mmol/L and higher have this [serum sodium] level because they do not drink enough and will be able to reduce it by making sure they consistently drink 2 to 2.5 L per day,” said Dr. Dmitrieva.
“It is likely that for some people, other factors that are related to genetics or diseases affecting water-salt balance could be causing their increased serum sodium levels,” she speculated.
The study was funded by the Intramural Program of the National Heart, Lung, and Blood Institute. The authors and Dr. Joseph have no relevant financial disclosures.
A version of this article first appeared on Medscape.com.
APAF-CRT: ‘Ablate and pace’ cuts mortality in narrow-QRS HF, permanent AFib
When a patient has permanent atrial fibrillation (AFib) and advanced heart failure (HF), rate control therapy is an option but an “ablate-and-pace” strategy may be better at improving symptoms. The ablate-and-pace approach, compared to pharmacologic rate control, may even prolong survival in a subset of such patients when the accompanying pacemaker provides cardiac resynchronization therapy (CRT), suggests a new randomized trial.
In the APAF-CRT trial, mortality fell more than 70% over 4 years for such patients with HF and narrow QRS intervals who were assigned to ablate-and-pace – that is, CRT after creation of heart block by atrioventricular (AV) junction ablation – compared to those managed medically.
The benefit was seen regardless of left ventricular ejection fraction (LVEF) at the start of the trial and probably stemmed from “the combination of strict rate control and rate regulation achieved by AV-junction ablation together with biventricular pacing,” said Michele Brignole, MD, Istituto Auxologico Italiano, Ospedale San Luca, Milan. The CRT substitution for a standard pacemaker, he explained, is thought to “counteract” the adverse remodeling effects of apical right ventricular (RV) pacing.
Dr. Brignole delivered the remarks at a media presentation before his presentation of the APAF-CRT during the virtual annual congress of the European Society of Cardiology.
The results “support ablation-CRT as a first-line therapy in patients with permanent AFib and narrow QRS who were hospitalized for heart failure,” regardless of ejection fraction, said Dr. Brignole, lead author on the study’s same-day publication in the European Heart Journal.
“The results are not surprising. They are in line with prior studies with shorter follow-up, and they justify a relatively common practice today, to implant CRT in these patients. It has previously been shown to improve heart failure and quality of life, and is now proven to improve survival because of the longer follow-up,” Michael Glikson, MD, Shaare Zedek Medical Center, Jerusalem, said at the media briefing.
“The APAF-CRT mortality trial makes an important contribution to establishment of AV-nodal ablation with CRT as first-line therapy of resistant atrial fibrillation with heart failure, mostly in patients with reduced ejection fraction,” said Dr. Glikson, who was not part of the trial.
However, he added, “the advantage of CRT over RV pacing is still somewhat unclear in patients with normal or preserved ejection fraction,” who were relatively few in APAF-CRT and in whom RV apical pacing after AV nodal ablation has not been shown to make a big difference to ventricular function.
The new analysis covered the trial’s second phase, which featured a mortality primary endpoint, in contrast to the previously reported initial stage that followed the first 102 patients over 2 years for death, worsening HF, or HF hospitalization.
The first phase had halted enrollment before reaching its planned target of 280 patients when an interim analysis showed a significant benefit for ablate and pace. The mortality trial continued to recruit at 11 centers in Europe, reaching 133 patients, who were followed for up to 4 years, the report notes. But its enrollment had also been suspended after an interim analysis saw superiority in the ablate-and-pace arm.
APAF-CRT entered patients with severely symptomatic permanent AFib for longer than 6 months, with a QRS interval no greater than 110 ms, who had at least one HF hospitalization in the last year and were considered poor candidates for AFib ablation. Their mean age was 73 years, and almost half, 47%, were women.
They were randomly assigned to ablate-and-pace with CRT or pharmacologic rate control therapy, 63 and 70 patients, respectively. Patients in either group could be given an implantable defibrillator at physician discretion.
Patients had been followed a median of 29 months when the trial was stopped for efficacy. The hazard ratio (HR) for death from any cause, ablate-and-pace vs. rate control, was 0.26 (95% confidence interval, 0.10-0.65; P = .004), with a number needed to treat to prevent an event of 3.7. The HR was 0.40 (95% CI, 0.22-0.73; P = .002) for the secondary endpoint of death or HF hospitalization.
The new ESC guidelines on cardiac pacing and cardiac resynchronization therapy recommend “that if the ejection fraction is subnormal, they should receive a CRT as the first choice,” Dr. Glikson said. “However, for patients who are undergoing AV nodal ablation and have normal ejection fractions, we thought that RV apical pacing should be okay,” so that was the main recommendation, he said.
“I think that the APAF-CRT study does not really change this approach” because the study was small and there were few data on such patients.
APAF-CRT was an investigator-initiated independent clinical trial, sponsored by a nonprofit organization, Centro Prevenzione Malattie Cardiorespiratorie ‘Nuccia e Vittore Corbella’, Rapallo, Italy, which received an unrestricted research grant from the Boston Scientific Investigator Sponsored Research (ISR) Committee. Dr. Brignole declared no conflicts. Disclosures for the other authors are in the report. Dr. Glikson had no disclosures.
A version of this article first appeared on Medscape.com.
When a patient has permanent atrial fibrillation (AFib) and advanced heart failure (HF), rate control therapy is an option but an “ablate-and-pace” strategy may be better at improving symptoms. The ablate-and-pace approach, compared to pharmacologic rate control, may even prolong survival in a subset of such patients when the accompanying pacemaker provides cardiac resynchronization therapy (CRT), suggests a new randomized trial.
In the APAF-CRT trial, mortality fell more than 70% over 4 years for such patients with HF and narrow QRS intervals who were assigned to ablate-and-pace – that is, CRT after creation of heart block by atrioventricular (AV) junction ablation – compared to those managed medically.
The benefit was seen regardless of left ventricular ejection fraction (LVEF) at the start of the trial and probably stemmed from “the combination of strict rate control and rate regulation achieved by AV-junction ablation together with biventricular pacing,” said Michele Brignole, MD, Istituto Auxologico Italiano, Ospedale San Luca, Milan. The CRT substitution for a standard pacemaker, he explained, is thought to “counteract” the adverse remodeling effects of apical right ventricular (RV) pacing.
Dr. Brignole delivered the remarks at a media presentation before his presentation of the APAF-CRT during the virtual annual congress of the European Society of Cardiology.
The results “support ablation-CRT as a first-line therapy in patients with permanent AFib and narrow QRS who were hospitalized for heart failure,” regardless of ejection fraction, said Dr. Brignole, lead author on the study’s same-day publication in the European Heart Journal.
“The results are not surprising. They are in line with prior studies with shorter follow-up, and they justify a relatively common practice today, to implant CRT in these patients. It has previously been shown to improve heart failure and quality of life, and is now proven to improve survival because of the longer follow-up,” Michael Glikson, MD, Shaare Zedek Medical Center, Jerusalem, said at the media briefing.
“The APAF-CRT mortality trial makes an important contribution to establishment of AV-nodal ablation with CRT as first-line therapy of resistant atrial fibrillation with heart failure, mostly in patients with reduced ejection fraction,” said Dr. Glikson, who was not part of the trial.
However, he added, “the advantage of CRT over RV pacing is still somewhat unclear in patients with normal or preserved ejection fraction,” who were relatively few in APAF-CRT and in whom RV apical pacing after AV nodal ablation has not been shown to make a big difference to ventricular function.
The new analysis covered the trial’s second phase, which featured a mortality primary endpoint, in contrast to the previously reported initial stage that followed the first 102 patients over 2 years for death, worsening HF, or HF hospitalization.
The first phase had halted enrollment before reaching its planned target of 280 patients when an interim analysis showed a significant benefit for ablate and pace. The mortality trial continued to recruit at 11 centers in Europe, reaching 133 patients, who were followed for up to 4 years, the report notes. But its enrollment had also been suspended after an interim analysis saw superiority in the ablate-and-pace arm.
APAF-CRT entered patients with severely symptomatic permanent AFib for longer than 6 months, with a QRS interval no greater than 110 ms, who had at least one HF hospitalization in the last year and were considered poor candidates for AFib ablation. Their mean age was 73 years, and almost half, 47%, were women.
They were randomly assigned to ablate-and-pace with CRT or pharmacologic rate control therapy, 63 and 70 patients, respectively. Patients in either group could be given an implantable defibrillator at physician discretion.
Patients had been followed a median of 29 months when the trial was stopped for efficacy. The hazard ratio (HR) for death from any cause, ablate-and-pace vs. rate control, was 0.26 (95% confidence interval, 0.10-0.65; P = .004), with a number needed to treat to prevent an event of 3.7. The HR was 0.40 (95% CI, 0.22-0.73; P = .002) for the secondary endpoint of death or HF hospitalization.
The new ESC guidelines on cardiac pacing and cardiac resynchronization therapy recommend “that if the ejection fraction is subnormal, they should receive a CRT as the first choice,” Dr. Glikson said. “However, for patients who are undergoing AV nodal ablation and have normal ejection fractions, we thought that RV apical pacing should be okay,” so that was the main recommendation, he said.
“I think that the APAF-CRT study does not really change this approach” because the study was small and there were few data on such patients.
APAF-CRT was an investigator-initiated independent clinical trial, sponsored by a nonprofit organization, Centro Prevenzione Malattie Cardiorespiratorie ‘Nuccia e Vittore Corbella’, Rapallo, Italy, which received an unrestricted research grant from the Boston Scientific Investigator Sponsored Research (ISR) Committee. Dr. Brignole declared no conflicts. Disclosures for the other authors are in the report. Dr. Glikson had no disclosures.
A version of this article first appeared on Medscape.com.
When a patient has permanent atrial fibrillation (AFib) and advanced heart failure (HF), rate control therapy is an option but an “ablate-and-pace” strategy may be better at improving symptoms. The ablate-and-pace approach, compared to pharmacologic rate control, may even prolong survival in a subset of such patients when the accompanying pacemaker provides cardiac resynchronization therapy (CRT), suggests a new randomized trial.
In the APAF-CRT trial, mortality fell more than 70% over 4 years for such patients with HF and narrow QRS intervals who were assigned to ablate-and-pace – that is, CRT after creation of heart block by atrioventricular (AV) junction ablation – compared to those managed medically.
The benefit was seen regardless of left ventricular ejection fraction (LVEF) at the start of the trial and probably stemmed from “the combination of strict rate control and rate regulation achieved by AV-junction ablation together with biventricular pacing,” said Michele Brignole, MD, Istituto Auxologico Italiano, Ospedale San Luca, Milan. The CRT substitution for a standard pacemaker, he explained, is thought to “counteract” the adverse remodeling effects of apical right ventricular (RV) pacing.
Dr. Brignole delivered the remarks at a media presentation before his presentation of the APAF-CRT during the virtual annual congress of the European Society of Cardiology.
The results “support ablation-CRT as a first-line therapy in patients with permanent AFib and narrow QRS who were hospitalized for heart failure,” regardless of ejection fraction, said Dr. Brignole, lead author on the study’s same-day publication in the European Heart Journal.
“The results are not surprising. They are in line with prior studies with shorter follow-up, and they justify a relatively common practice today, to implant CRT in these patients. It has previously been shown to improve heart failure and quality of life, and is now proven to improve survival because of the longer follow-up,” Michael Glikson, MD, Shaare Zedek Medical Center, Jerusalem, said at the media briefing.
“The APAF-CRT mortality trial makes an important contribution to establishment of AV-nodal ablation with CRT as first-line therapy of resistant atrial fibrillation with heart failure, mostly in patients with reduced ejection fraction,” said Dr. Glikson, who was not part of the trial.
However, he added, “the advantage of CRT over RV pacing is still somewhat unclear in patients with normal or preserved ejection fraction,” who were relatively few in APAF-CRT and in whom RV apical pacing after AV nodal ablation has not been shown to make a big difference to ventricular function.
The new analysis covered the trial’s second phase, which featured a mortality primary endpoint, in contrast to the previously reported initial stage that followed the first 102 patients over 2 years for death, worsening HF, or HF hospitalization.
The first phase had halted enrollment before reaching its planned target of 280 patients when an interim analysis showed a significant benefit for ablate and pace. The mortality trial continued to recruit at 11 centers in Europe, reaching 133 patients, who were followed for up to 4 years, the report notes. But its enrollment had also been suspended after an interim analysis saw superiority in the ablate-and-pace arm.
APAF-CRT entered patients with severely symptomatic permanent AFib for longer than 6 months, with a QRS interval no greater than 110 ms, who had at least one HF hospitalization in the last year and were considered poor candidates for AFib ablation. Their mean age was 73 years, and almost half, 47%, were women.
They were randomly assigned to ablate-and-pace with CRT or pharmacologic rate control therapy, 63 and 70 patients, respectively. Patients in either group could be given an implantable defibrillator at physician discretion.
Patients had been followed a median of 29 months when the trial was stopped for efficacy. The hazard ratio (HR) for death from any cause, ablate-and-pace vs. rate control, was 0.26 (95% confidence interval, 0.10-0.65; P = .004), with a number needed to treat to prevent an event of 3.7. The HR was 0.40 (95% CI, 0.22-0.73; P = .002) for the secondary endpoint of death or HF hospitalization.
The new ESC guidelines on cardiac pacing and cardiac resynchronization therapy recommend “that if the ejection fraction is subnormal, they should receive a CRT as the first choice,” Dr. Glikson said. “However, for patients who are undergoing AV nodal ablation and have normal ejection fractions, we thought that RV apical pacing should be okay,” so that was the main recommendation, he said.
“I think that the APAF-CRT study does not really change this approach” because the study was small and there were few data on such patients.
APAF-CRT was an investigator-initiated independent clinical trial, sponsored by a nonprofit organization, Centro Prevenzione Malattie Cardiorespiratorie ‘Nuccia e Vittore Corbella’, Rapallo, Italy, which received an unrestricted research grant from the Boston Scientific Investigator Sponsored Research (ISR) Committee. Dr. Brignole declared no conflicts. Disclosures for the other authors are in the report. Dr. Glikson had no disclosures.
A version of this article first appeared on Medscape.com.
FIDELITY: Finerenone benefits patients with T2D across CKD spectrum
New data on using the nonsteroidal mineralocorticoid receptor antagonist (MRA) finerenone to treat patients with type 2 diabetes and chronic kidney disease did more than further confirm this new drug’s efficacy in these patients for slowing progression to end-stage renal disease and reducing hospitalizations for heart failure.

It also strengthened the case for clinicians to be much more proactive in collecting urine specimens from patients with type 2 diabetes (T2D) to find those with albuminuria whose kidney function has not yet dropped below 60 mL/min per 1.73 m2, a population that the data show finerenone can help.
The FIDELITY prespecified meta-analysis combined data from two related pivotal trials of finerenone (Kerendia) in a total of more than 13,000 patients with T2D and chronic kidney disease (CKD). Each of these two trials, FIDELIO-DKD and FIGARO-DKD, identified patients with CKD by either of two methods, or a total of four different criteria.
In sum, the two trials enrolled patients with an estimated glomerular filtration rate (eGFR) of 25-90 mL/min per 1.73 m2 and a urinary albumin-to-creatinine ratio (UACR) of 30-299, or an eGFR of 25-75 mL/min per 1.73 m2 and a UACR of 300-5,000. The result was that 40% of enrolled patients had an eGFR of at least 60, levels that are considered normal, but they also had some level of albuminuria that defined them as having CKD.
The results showed that during a median follow-up of 36 months, patients with a normal eGFR and albuminuria had their combined incidence of cardiovascular disease events (cardiovascular death, MI, stroke, or hospitalization for heart failure) reduced by roughly the same amount as seen in patients with lower levels of eGFR and renal function, a finding that reimagines how clinicians need to routinely screen patients with T2D for CKD, Gerasimos Filippatos, MD, reported at the virtual annual congress of the European Society of Cardiology.
“Measuring UACR in patients with type 2 diabetes is important to identify patients who will benefit from finerenone treatment independent of their eGFR,” said Dr. Filippatos, professor of medicine at the University of Athens and director of the heart failure unit at Attikon University Hospital in Athens.
The combined FIDELITY analysis showed a significant overall cut in the combined cardiovascular disease endpoint of 14% relative to placebo, which reflected a 1.7% absolute reduction in events between the two arms during 3 years of treatment. The primary driver of this benefit was the significant drop in hospitalizations for heart failure on finerenone compared with placebo, which fell by a relative 22% and by an absolute 1.1%, Dr. Filippatos reported.
Routinely screening for albuminuria is ‘practice changing’
“This is really practice changing information for cardiologists,” said Rajiv L. Agarwal, MD, a copresenter of the FIDELITY analysis and a lead investigator of the two finerenone trials.
When cardiologists and possibly other specialists see patients with T2D, they traditionally have focused on measuring left ventricular ejection fraction and checking for other indications of heart failure. The new results from FIDELIO-DKD and FIGARO-DKD showed that finerenone treatment can prevent heart failure onset or worsening in patients with T2D with finerenone, which clinicians can accomplish by “simply measuring UACR,” as well as eGFR, and then treating patients with abnormal levels of either, explained Dr. Agarwal, a nephrologist and professor of medicine at Indiana University in Indianapolis.
“Diabetologists know that when they see patients with diabetes they need to collect a urine sample to check for albuminuria. But when some other clinicians see a patient with type 2 diabetes and a normal eGFR, they often think that the patient is okay and don’t get a urine specimen,” noted Bertram Pitt, MD, another collaborator of the finerenone trials and a heart failure specialist affiliated with the University of Michigan in Ann Arbor.

“We need to pay more attention to UACR and albuminuria; traditionally clinicians have mostly looked at eGFR,” agreed Dipti Itchhaporia, MD, a cardiologist at the Carlton Heart and Vascular Institute of Hoag Hospital in Newport Beach, Calif. UACR “is a marker that should be shared” between endocrinologists, nephrologists, and cardiologists as they together care for patients with T2D, suggested Dr. Itchhaporia, president of the American College of Cardiology.
Two pivotal trials with consistent findings
The FIDELITY analysis combined data from the FIDELIO-DKD trial, reported in 2020, and from the FIGARO-DKD trial that was first reported during the current congress as well as in a simultaneous report published online.
Results from the two trials were very consistent, although the primary endpoint in FIDELIO-DKD was a composite measure of renal disease with the combined cardiovascular disease metric a secondary endpoint, while this got flipped in FIGARO-DKD which had the cardiovascular disease composite as its primary endpoint as the combined renal outcomes as a secondary endpoint.
In addition to showing a consistent, significant reduction in both combined cardiovascular disease events and in the specific endpoint of hospitalization for heart failure, the two trials also showed a consistent benefit for slowing renal disease progression, including significantly fewer patients developing end-stage kidney disease. In the combined FIDELITY analysis, treatment with finerenone cut the incidence of end-stage kidney disease by a significant 20% compared with placebo, and by an absolute reduction of 0.6%.
Another common finding was a relatively low incidence of hyperkalemia compared with what’s usually seen using a steroidal MRA, spironolactone or eplerenone. In the combined analysis treatment with finerenone produced a 14% incidence of any hyperkalemia compared with 7% among placebo-treated patients, and the rate of patients stopping their treatment because of hyperkalemia was 1.7% on finerenone and 0.6% on placebo.
“Finerenone is much better tolerated” than the steroidal MRAs in causing clinically significant hyperkalemia, noted Dr. Pitt. “There are a lot of misconceptions” about the potassium-raising potential of MRAs, and “people get frightened” by the potential. Spreading the message of finerenone’s relative safety “will take a lot of education,” he acknowledged. Routine monitoring of potassium levels is a key step to minimizing the risk for hyperkalemia when using finerenone, he added.
Suggested benefit from combination treatment
Another intriguing observation from FIDELITY derived from the fact that roughly 7% of enrolled patients were also on treatment with a sodium-glucose cotransporter 2 (SGLT2) inhibitor at entry, and about 7% were on treatment with a glucagon-like peptide-1 (GLP-1) receptor agonist, and in both subgroups the incidence of the composite cardiovascular disease endpoint appeared to suggest additive effects of agents from either of these classes when combined with finerenone. Although the numbers of patients on combined treatment were too low to show a definitive result, “our expectation is that we will see an additive effect,” said Dr. Pitt. Ideally, patients with T2D and CKD “should be on both” an SGLT2 inhibitor and finerenone, he predicted.
SGLT2 inhibitors have now been embraced as a key treatment for patients with T2D or with heart failure with reduced ejection fraction, and the preliminary data suggest that combining these agents with finerenone can provide additional benefit, agreed Dr. Itchhaporia. Aside from the need for more evidence to prove this, there are also practical considerations of “How do we pay for all these fantastic therapies?” She expressed optimism that cost-benefit analyses will eventually show that the additive benefits justify the added cost.
Based largely on results from FIDELIO-DKD, finerenone received marketing approval from the Food and Drug Administration in July 2021 for the indication of treating patients with T2D and chronic kidney disease.
FIGARO-DKD, FIDELIO-DKD, and FIDELITY were sponsored by Bayer, the company that markets finerenone. Dr. Filippatos has received lecture fees from Bayer, and has had financial relationships with Amgen, Boehringer Ingelheim, Medtronic, Novartis, Servier, and Vifor. Dr. Agarwal received travel support from and has been a consultant to Bayer and to numerous other companies. Dr. Pitt has been a consultant to Bayer and to numerous other companies. Dr. Itchhaporia had no disclosures.
New data on using the nonsteroidal mineralocorticoid receptor antagonist (MRA) finerenone to treat patients with type 2 diabetes and chronic kidney disease did more than further confirm this new drug’s efficacy in these patients for slowing progression to end-stage renal disease and reducing hospitalizations for heart failure.
It also strengthened the case for clinicians to be much more proactive in collecting urine specimens from patients with type 2 diabetes (T2D) to find those with albuminuria whose kidney function has not yet dropped below 60 mL/min per 1.73 m2, a population that the data show finerenone can help.
The FIDELITY prespecified meta-analysis combined data from two related pivotal trials of finerenone (Kerendia) in a total of more than 13,000 patients with T2D and chronic kidney disease (CKD). Each of these two trials, FIDELIO-DKD and FIGARO-DKD, identified patients with CKD by either of two methods, or a total of four different criteria.
In sum, the two trials enrolled patients with an estimated glomerular filtration rate (eGFR) of 25-90 mL/min per 1.73 m2 and a urinary albumin-to-creatinine ratio (UACR) of 30-299, or an eGFR of 25-75 mL/min per 1.73 m2 and a UACR of 300-5,000. The result was that 40% of enrolled patients had an eGFR of at least 60, levels that are considered normal, but they also had some level of albuminuria that defined them as having CKD.
The results showed that during a median follow-up of 36 months, patients with a normal eGFR and albuminuria had their combined incidence of cardiovascular disease events (cardiovascular death, MI, stroke, or hospitalization for heart failure) reduced by roughly the same amount as seen in patients with lower levels of eGFR and renal function, a finding that reimagines how clinicians need to routinely screen patients with T2D for CKD, Gerasimos Filippatos, MD, reported at the virtual annual congress of the European Society of Cardiology.
“Measuring UACR in patients with type 2 diabetes is important to identify patients who will benefit from finerenone treatment independent of their eGFR,” said Dr. Filippatos, professor of medicine at the University of Athens and director of the heart failure unit at Attikon University Hospital in Athens.
The combined FIDELITY analysis showed a significant overall cut in the combined cardiovascular disease endpoint of 14% relative to placebo, which reflected a 1.7% absolute reduction in events between the two arms during 3 years of treatment. The primary driver of this benefit was the significant drop in hospitalizations for heart failure on finerenone compared with placebo, which fell by a relative 22% and by an absolute 1.1%, Dr. Filippatos reported.
Routinely screening for albuminuria is ‘practice changing’
“This is really practice changing information for cardiologists,” said Rajiv L. Agarwal, MD, a copresenter of the FIDELITY analysis and a lead investigator of the two finerenone trials.
When cardiologists and possibly other specialists see patients with T2D, they traditionally have focused on measuring left ventricular ejection fraction and checking for other indications of heart failure. The new results from FIDELIO-DKD and FIGARO-DKD showed that finerenone treatment can prevent heart failure onset or worsening in patients with T2D with finerenone, which clinicians can accomplish by “simply measuring UACR,” as well as eGFR, and then treating patients with abnormal levels of either, explained Dr. Agarwal, a nephrologist and professor of medicine at Indiana University in Indianapolis.
“Diabetologists know that when they see patients with diabetes they need to collect a urine sample to check for albuminuria. But when some other clinicians see a patient with type 2 diabetes and a normal eGFR, they often think that the patient is okay and don’t get a urine specimen,” noted Bertram Pitt, MD, another collaborator of the finerenone trials and a heart failure specialist affiliated with the University of Michigan in Ann Arbor.
“We need to pay more attention to UACR and albuminuria; traditionally clinicians have mostly looked at eGFR,” agreed Dipti Itchhaporia, MD, a cardiologist at the Carlton Heart and Vascular Institute of Hoag Hospital in Newport Beach, Calif. UACR “is a marker that should be shared” between endocrinologists, nephrologists, and cardiologists as they together care for patients with T2D, suggested Dr. Itchhaporia, president of the American College of Cardiology.
Two pivotal trials with consistent findings
The FIDELITY analysis combined data from the FIDELIO-DKD trial, reported in 2020, and from the FIGARO-DKD trial that was first reported during the current congress as well as in a simultaneous report published online.
Results from the two trials were very consistent, although the primary endpoint in FIDELIO-DKD was a composite measure of renal disease with the combined cardiovascular disease metric a secondary endpoint, while this got flipped in FIGARO-DKD which had the cardiovascular disease composite as its primary endpoint as the combined renal outcomes as a secondary endpoint.
In addition to showing a consistent, significant reduction in both combined cardiovascular disease events and in the specific endpoint of hospitalization for heart failure, the two trials also showed a consistent benefit for slowing renal disease progression, including significantly fewer patients developing end-stage kidney disease. In the combined FIDELITY analysis, treatment with finerenone cut the incidence of end-stage kidney disease by a significant 20% compared with placebo, and by an absolute reduction of 0.6%.
Another common finding was a relatively low incidence of hyperkalemia compared with what’s usually seen using a steroidal MRA, spironolactone or eplerenone. In the combined analysis treatment with finerenone produced a 14% incidence of any hyperkalemia compared with 7% among placebo-treated patients, and the rate of patients stopping their treatment because of hyperkalemia was 1.7% on finerenone and 0.6% on placebo.
“Finerenone is much better tolerated” than the steroidal MRAs in causing clinically significant hyperkalemia, noted Dr. Pitt. “There are a lot of misconceptions” about the potassium-raising potential of MRAs, and “people get frightened” by the potential. Spreading the message of finerenone’s relative safety “will take a lot of education,” he acknowledged. Routine monitoring of potassium levels is a key step to minimizing the risk for hyperkalemia when using finerenone, he added.
Suggested benefit from combination treatment
Another intriguing observation from FIDELITY derived from the fact that roughly 7% of enrolled patients were also on treatment with a sodium-glucose cotransporter 2 (SGLT2) inhibitor at entry, and about 7% were on treatment with a glucagon-like peptide-1 (GLP-1) receptor agonist, and in both subgroups the incidence of the composite cardiovascular disease endpoint appeared to suggest additive effects of agents from either of these classes when combined with finerenone. Although the numbers of patients on combined treatment were too low to show a definitive result, “our expectation is that we will see an additive effect,” said Dr. Pitt. Ideally, patients with T2D and CKD “should be on both” an SGLT2 inhibitor and finerenone, he predicted.
SGLT2 inhibitors have now been embraced as a key treatment for patients with T2D or with heart failure with reduced ejection fraction, and the preliminary data suggest that combining these agents with finerenone can provide additional benefit, agreed Dr. Itchhaporia. Aside from the need for more evidence to prove this, there are also practical considerations of “How do we pay for all these fantastic therapies?” She expressed optimism that cost-benefit analyses will eventually show that the additive benefits justify the added cost.
Based largely on results from FIDELIO-DKD, finerenone received marketing approval from the Food and Drug Administration in July 2021 for the indication of treating patients with T2D and chronic kidney disease.
FIGARO-DKD, FIDELIO-DKD, and FIDELITY were sponsored by Bayer, the company that markets finerenone. Dr. Filippatos has received lecture fees from Bayer, and has had financial relationships with Amgen, Boehringer Ingelheim, Medtronic, Novartis, Servier, and Vifor. Dr. Agarwal received travel support from and has been a consultant to Bayer and to numerous other companies. Dr. Pitt has been a consultant to Bayer and to numerous other companies. Dr. Itchhaporia had no disclosures.
New data on using the nonsteroidal mineralocorticoid receptor antagonist (MRA) finerenone to treat patients with type 2 diabetes and chronic kidney disease did more than further confirm this new drug’s efficacy in these patients for slowing progression to end-stage renal disease and reducing hospitalizations for heart failure.
It also strengthened the case for clinicians to be much more proactive in collecting urine specimens from patients with type 2 diabetes (T2D) to find those with albuminuria whose kidney function has not yet dropped below 60 mL/min per 1.73 m2, a population that the data show finerenone can help.
The FIDELITY prespecified meta-analysis combined data from two related pivotal trials of finerenone (Kerendia) in a total of more than 13,000 patients with T2D and chronic kidney disease (CKD). Each of these two trials, FIDELIO-DKD and FIGARO-DKD, identified patients with CKD by either of two methods, or a total of four different criteria.
In sum, the two trials enrolled patients with an estimated glomerular filtration rate (eGFR) of 25-90 mL/min per 1.73 m2 and a urinary albumin-to-creatinine ratio (UACR) of 30-299, or an eGFR of 25-75 mL/min per 1.73 m2 and a UACR of 300-5,000. The result was that 40% of enrolled patients had an eGFR of at least 60, levels that are considered normal, but they also had some level of albuminuria that defined them as having CKD.
The results showed that during a median follow-up of 36 months, patients with a normal eGFR and albuminuria had their combined incidence of cardiovascular disease events (cardiovascular death, MI, stroke, or hospitalization for heart failure) reduced by roughly the same amount as seen in patients with lower levels of eGFR and renal function, a finding that reimagines how clinicians need to routinely screen patients with T2D for CKD, Gerasimos Filippatos, MD, reported at the virtual annual congress of the European Society of Cardiology.
“Measuring UACR in patients with type 2 diabetes is important to identify patients who will benefit from finerenone treatment independent of their eGFR,” said Dr. Filippatos, professor of medicine at the University of Athens and director of the heart failure unit at Attikon University Hospital in Athens.
The combined FIDELITY analysis showed a significant overall cut in the combined cardiovascular disease endpoint of 14% relative to placebo, which reflected a 1.7% absolute reduction in events between the two arms during 3 years of treatment. The primary driver of this benefit was the significant drop in hospitalizations for heart failure on finerenone compared with placebo, which fell by a relative 22% and by an absolute 1.1%, Dr. Filippatos reported.
Routinely screening for albuminuria is ‘practice changing’
“This is really practice changing information for cardiologists,” said Rajiv L. Agarwal, MD, a copresenter of the FIDELITY analysis and a lead investigator of the two finerenone trials.
When cardiologists and possibly other specialists see patients with T2D, they traditionally have focused on measuring left ventricular ejection fraction and checking for other indications of heart failure. The new results from FIDELIO-DKD and FIGARO-DKD showed that finerenone treatment can prevent heart failure onset or worsening in patients with T2D with finerenone, which clinicians can accomplish by “simply measuring UACR,” as well as eGFR, and then treating patients with abnormal levels of either, explained Dr. Agarwal, a nephrologist and professor of medicine at Indiana University in Indianapolis.
“Diabetologists know that when they see patients with diabetes they need to collect a urine sample to check for albuminuria. But when some other clinicians see a patient with type 2 diabetes and a normal eGFR, they often think that the patient is okay and don’t get a urine specimen,” noted Bertram Pitt, MD, another collaborator of the finerenone trials and a heart failure specialist affiliated with the University of Michigan in Ann Arbor.
“We need to pay more attention to UACR and albuminuria; traditionally clinicians have mostly looked at eGFR,” agreed Dipti Itchhaporia, MD, a cardiologist at the Carlton Heart and Vascular Institute of Hoag Hospital in Newport Beach, Calif. UACR “is a marker that should be shared” between endocrinologists, nephrologists, and cardiologists as they together care for patients with T2D, suggested Dr. Itchhaporia, president of the American College of Cardiology.
Two pivotal trials with consistent findings
The FIDELITY analysis combined data from the FIDELIO-DKD trial, reported in 2020, and from the FIGARO-DKD trial that was first reported during the current congress as well as in a simultaneous report published online.
Results from the two trials were very consistent, although the primary endpoint in FIDELIO-DKD was a composite measure of renal disease with the combined cardiovascular disease metric a secondary endpoint, while this got flipped in FIGARO-DKD which had the cardiovascular disease composite as its primary endpoint as the combined renal outcomes as a secondary endpoint.
In addition to showing a consistent, significant reduction in both combined cardiovascular disease events and in the specific endpoint of hospitalization for heart failure, the two trials also showed a consistent benefit for slowing renal disease progression, including significantly fewer patients developing end-stage kidney disease. In the combined FIDELITY analysis, treatment with finerenone cut the incidence of end-stage kidney disease by a significant 20% compared with placebo, and by an absolute reduction of 0.6%.
Another common finding was a relatively low incidence of hyperkalemia compared with what’s usually seen using a steroidal MRA, spironolactone or eplerenone. In the combined analysis treatment with finerenone produced a 14% incidence of any hyperkalemia compared with 7% among placebo-treated patients, and the rate of patients stopping their treatment because of hyperkalemia was 1.7% on finerenone and 0.6% on placebo.
“Finerenone is much better tolerated” than the steroidal MRAs in causing clinically significant hyperkalemia, noted Dr. Pitt. “There are a lot of misconceptions” about the potassium-raising potential of MRAs, and “people get frightened” by the potential. Spreading the message of finerenone’s relative safety “will take a lot of education,” he acknowledged. Routine monitoring of potassium levels is a key step to minimizing the risk for hyperkalemia when using finerenone, he added.
Suggested benefit from combination treatment
Another intriguing observation from FIDELITY derived from the fact that roughly 7% of enrolled patients were also on treatment with a sodium-glucose cotransporter 2 (SGLT2) inhibitor at entry, and about 7% were on treatment with a glucagon-like peptide-1 (GLP-1) receptor agonist, and in both subgroups the incidence of the composite cardiovascular disease endpoint appeared to suggest additive effects of agents from either of these classes when combined with finerenone. Although the numbers of patients on combined treatment were too low to show a definitive result, “our expectation is that we will see an additive effect,” said Dr. Pitt. Ideally, patients with T2D and CKD “should be on both” an SGLT2 inhibitor and finerenone, he predicted.
SGLT2 inhibitors have now been embraced as a key treatment for patients with T2D or with heart failure with reduced ejection fraction, and the preliminary data suggest that combining these agents with finerenone can provide additional benefit, agreed Dr. Itchhaporia. Aside from the need for more evidence to prove this, there are also practical considerations of “How do we pay for all these fantastic therapies?” She expressed optimism that cost-benefit analyses will eventually show that the additive benefits justify the added cost.
Based largely on results from FIDELIO-DKD, finerenone received marketing approval from the Food and Drug Administration in July 2021 for the indication of treating patients with T2D and chronic kidney disease.
FIGARO-DKD, FIDELIO-DKD, and FIDELITY were sponsored by Bayer, the company that markets finerenone. Dr. Filippatos has received lecture fees from Bayer, and has had financial relationships with Amgen, Boehringer Ingelheim, Medtronic, Novartis, Servier, and Vifor. Dr. Agarwal received travel support from and has been a consultant to Bayer and to numerous other companies. Dr. Pitt has been a consultant to Bayer and to numerous other companies. Dr. Itchhaporia had no disclosures.
FROM ESC CONGRESS 2021
Dapagliflozin in HFrEF may cut arrhythmias, sudden death: DAPA-HF
Dapagliflozin might reduce the risk for ventricular arrhythmias and sudden death in patients with heart failure and reduced ejection fraction (HFrEF), a post hoc analysis of the DAPA-HF trial suggests.

The addition of dapagliflozin to standard therapy reduced the relative risk for the primary composite endpoint of any serious ventricular arrhythmia, resuscitated cardiac arrest, or sudden death by 21%, compared with placebo (hazard ratio, 0.79; 95% confidence interval, 0.63-0.99). The absolute risk reduction was 1.5% (5.9% vs. 7.4%).
The benefit was consistent in a competing-risks analysis that included all-cause mortality (HR, 0.80; P = .043) and across the individual components of the composite outcome, James Curtain, MD, Cardiovascular Research Centre of Glasgow, said at the annual congress of the European Society of Cardiology.
As previously reported from the main trial, treatment with the sodium-glucose cotransporter 2 (SGLT2) inhibitor cut the primary endpoint of cardiovascular death or worsening heart failure by 26% among 4,744 patients with HFrEF and in New York Heart Association functional class 2-4.
Cochair of the late-breaking science session, Lars Lund, MD, Karolinska Institute, Stockholm, pointed out that dapagliflozin reduced sudden cardiac deaths and related events to an extent similar to that observed for cardiovascular deaths, total mortality, and the main trial’s primary endpoint.
“So does that mean it has any particular effect on arrhythmic events or does it mean, such as a beta-blocker, for example, [it] reduces calcium transience and improves handling of calcium, or does it have an effect simply by improving heart failure?” he asked.
Dr. Curtain replied they are still trying to understand the effects of this new class of drug but that studies have shown dapagliflozin and other SGLT2 inhibitors have favorable effects on adverse cardiac remodeling, which contributes to sudden death and ventricular arrhythmia. They’ve also been shown to reduce cardiac chamber size, left ventricular hypertrophy, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels over time, consistent with a reduction in myocardial wall stress. “So it could indeed be one of several mechanisms by which they may exert a beneficial cardiac effect.”
Speaking with this news organization, Dr. Curtain pointed out that the Kaplan-Meier curves for the composite outcome began to separate early on, but that the clearest separation was after 9 months, suggestive of a positive action on adverse cardiac remodeling over time.
“This would improve the patients’ heart failure situation, but also thick ventricles are a key risk factor for the occurrence of sudden death and ventricular arrhythmias,” he said. “The effects on adverse cardiac remodeling, given its plausibility in terms of our Kaplan-Meier curves, are one [mechanism] that I’d look to in the first instance, but I’m sure there are more than one actions at play.”
According to the new analysis, the primary outcome occurred in 315 (6.6%) patients; there were 203 adjudicated sudden deaths (64%), 104 investigator-reported ventricular arrhythmias (33%), and 8 resuscitated cardiac arrests (3%). Independent predictors of the primary outcome were higher NT-proBNP levels (odds ratio, 1.54), previous ventricular arrhythmia (OR, 1.93), previous myocardial infarction (OR, 1.42), male sex (OR, 1.53), and higher body mass index (OR, 1.03).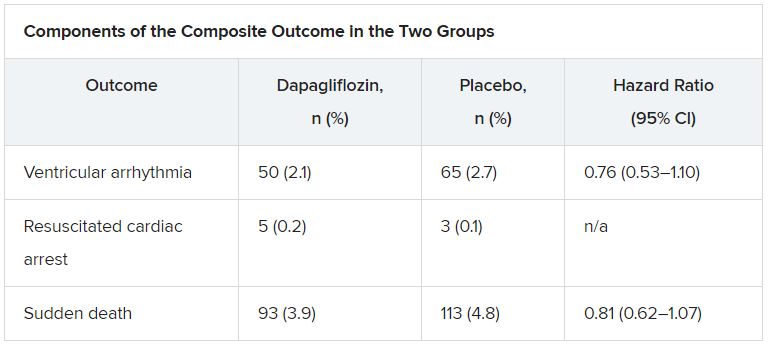
The effect of dapagliflozin on the primary outcome was consistent in several sensitivity analyses and “generally consistent” across key subgroups, Dr. Curtain said.
During a discussion of the results, session cochair Mitja Lainscak, MD, General Hospital Murska Sobota, Slovenia, called out two exceptions. “With regard to patients with implanted ICDs, the effect was neutral, and in the patients without diabetes, the benefit was less than in diabetic patients. Any explanations for that?”
Dr. Curtain responded that “it’s important to note that in the subgroup analyses the point estimates were all on the side favoring dapagliflozin and the interaction test was not significant in that subgroup. The numbers of patients who were in the defibrillator group were modest, and there was a relatively smaller number of events, so it may be harder to show benefit in that group.”
In the dapagliflozin and placebo groups, the event rates per 100 person-years were 3.9 and 5.8, respectively, in patients with diabetes, and 4.1 and 4.7, respectively, in those without diabetes (P for interaction = .273).
Event rates per 100 person-years were 5.8 and 5.9, respectively, in patients with a defibrillator at baseline, and 3.5 and 4.9, respectively, in those without a defibrillator (P for interaction = .174).
Asked to comment on the study, which was simultaneously published in the European Heart Journal, Milton Packer, MD, Baylor University Medical Center, Dallas, said he had “very little confidence” in the findings.
“This was entirely post hoc and the investigators combined events – with markedly different levels of clinical importance – in order to achieve a P value less than 0.05,” he told this news organization. “If one takes asymptomatic ventricular arrhythmias out of the analysis, the effect is no longer statistically significant. Furthermore, half of sudden deaths in patients with heart failure are not related to a ventricular arrhythmia.”
The authors note in their report that the analysis was not prespecified and the findings should be regarded as “hypothesis generating and require confirmation,” but also point out that a recent meta-analysis showed that SGLT2 inhibitor use was associated with a lower risk for ventricular tachycardia. Other limitations to the post hoc analysis are that adverse-event reporting likely underestimated the true prevalence of ventricular arrhythmias, and that these events were not adjudicated.
DAPA-HF was funded by AstraZeneca. Dr. Curtain reports no relevant financial relationships. Disclosures for the coauthors are listed in the paper.
A version of this article first appeared on Medscape.com.
Dapagliflozin might reduce the risk for ventricular arrhythmias and sudden death in patients with heart failure and reduced ejection fraction (HFrEF), a post hoc analysis of the DAPA-HF trial suggests.
The addition of dapagliflozin to standard therapy reduced the relative risk for the primary composite endpoint of any serious ventricular arrhythmia, resuscitated cardiac arrest, or sudden death by 21%, compared with placebo (hazard ratio, 0.79; 95% confidence interval, 0.63-0.99). The absolute risk reduction was 1.5% (5.9% vs. 7.4%).
The benefit was consistent in a competing-risks analysis that included all-cause mortality (HR, 0.80; P = .043) and across the individual components of the composite outcome, James Curtain, MD, Cardiovascular Research Centre of Glasgow, said at the annual congress of the European Society of Cardiology.
As previously reported from the main trial, treatment with the sodium-glucose cotransporter 2 (SGLT2) inhibitor cut the primary endpoint of cardiovascular death or worsening heart failure by 26% among 4,744 patients with HFrEF and in New York Heart Association functional class 2-4.
Cochair of the late-breaking science session, Lars Lund, MD, Karolinska Institute, Stockholm, pointed out that dapagliflozin reduced sudden cardiac deaths and related events to an extent similar to that observed for cardiovascular deaths, total mortality, and the main trial’s primary endpoint.
“So does that mean it has any particular effect on arrhythmic events or does it mean, such as a beta-blocker, for example, [it] reduces calcium transience and improves handling of calcium, or does it have an effect simply by improving heart failure?” he asked.
Dr. Curtain replied they are still trying to understand the effects of this new class of drug but that studies have shown dapagliflozin and other SGLT2 inhibitors have favorable effects on adverse cardiac remodeling, which contributes to sudden death and ventricular arrhythmia. They’ve also been shown to reduce cardiac chamber size, left ventricular hypertrophy, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels over time, consistent with a reduction in myocardial wall stress. “So it could indeed be one of several mechanisms by which they may exert a beneficial cardiac effect.”
Speaking with this news organization, Dr. Curtain pointed out that the Kaplan-Meier curves for the composite outcome began to separate early on, but that the clearest separation was after 9 months, suggestive of a positive action on adverse cardiac remodeling over time.
“This would improve the patients’ heart failure situation, but also thick ventricles are a key risk factor for the occurrence of sudden death and ventricular arrhythmias,” he said. “The effects on adverse cardiac remodeling, given its plausibility in terms of our Kaplan-Meier curves, are one [mechanism] that I’d look to in the first instance, but I’m sure there are more than one actions at play.”
According to the new analysis, the primary outcome occurred in 315 (6.6%) patients; there were 203 adjudicated sudden deaths (64%), 104 investigator-reported ventricular arrhythmias (33%), and 8 resuscitated cardiac arrests (3%). Independent predictors of the primary outcome were higher NT-proBNP levels (odds ratio, 1.54), previous ventricular arrhythmia (OR, 1.93), previous myocardial infarction (OR, 1.42), male sex (OR, 1.53), and higher body mass index (OR, 1.03).
The effect of dapagliflozin on the primary outcome was consistent in several sensitivity analyses and “generally consistent” across key subgroups, Dr. Curtain said.
During a discussion of the results, session cochair Mitja Lainscak, MD, General Hospital Murska Sobota, Slovenia, called out two exceptions. “With regard to patients with implanted ICDs, the effect was neutral, and in the patients without diabetes, the benefit was less than in diabetic patients. Any explanations for that?”
Dr. Curtain responded that “it’s important to note that in the subgroup analyses the point estimates were all on the side favoring dapagliflozin and the interaction test was not significant in that subgroup. The numbers of patients who were in the defibrillator group were modest, and there was a relatively smaller number of events, so it may be harder to show benefit in that group.”
In the dapagliflozin and placebo groups, the event rates per 100 person-years were 3.9 and 5.8, respectively, in patients with diabetes, and 4.1 and 4.7, respectively, in those without diabetes (P for interaction = .273).
Event rates per 100 person-years were 5.8 and 5.9, respectively, in patients with a defibrillator at baseline, and 3.5 and 4.9, respectively, in those without a defibrillator (P for interaction = .174).
Asked to comment on the study, which was simultaneously published in the European Heart Journal, Milton Packer, MD, Baylor University Medical Center, Dallas, said he had “very little confidence” in the findings.
“This was entirely post hoc and the investigators combined events – with markedly different levels of clinical importance – in order to achieve a P value less than 0.05,” he told this news organization. “If one takes asymptomatic ventricular arrhythmias out of the analysis, the effect is no longer statistically significant. Furthermore, half of sudden deaths in patients with heart failure are not related to a ventricular arrhythmia.”
The authors note in their report that the analysis was not prespecified and the findings should be regarded as “hypothesis generating and require confirmation,” but also point out that a recent meta-analysis showed that SGLT2 inhibitor use was associated with a lower risk for ventricular tachycardia. Other limitations to the post hoc analysis are that adverse-event reporting likely underestimated the true prevalence of ventricular arrhythmias, and that these events were not adjudicated.
DAPA-HF was funded by AstraZeneca. Dr. Curtain reports no relevant financial relationships. Disclosures for the coauthors are listed in the paper.
A version of this article first appeared on Medscape.com.
Dapagliflozin might reduce the risk for ventricular arrhythmias and sudden death in patients with heart failure and reduced ejection fraction (HFrEF), a post hoc analysis of the DAPA-HF trial suggests.
The addition of dapagliflozin to standard therapy reduced the relative risk for the primary composite endpoint of any serious ventricular arrhythmia, resuscitated cardiac arrest, or sudden death by 21%, compared with placebo (hazard ratio, 0.79; 95% confidence interval, 0.63-0.99). The absolute risk reduction was 1.5% (5.9% vs. 7.4%).
The benefit was consistent in a competing-risks analysis that included all-cause mortality (HR, 0.80; P = .043) and across the individual components of the composite outcome, James Curtain, MD, Cardiovascular Research Centre of Glasgow, said at the annual congress of the European Society of Cardiology.
As previously reported from the main trial, treatment with the sodium-glucose cotransporter 2 (SGLT2) inhibitor cut the primary endpoint of cardiovascular death or worsening heart failure by 26% among 4,744 patients with HFrEF and in New York Heart Association functional class 2-4.
Cochair of the late-breaking science session, Lars Lund, MD, Karolinska Institute, Stockholm, pointed out that dapagliflozin reduced sudden cardiac deaths and related events to an extent similar to that observed for cardiovascular deaths, total mortality, and the main trial’s primary endpoint.
“So does that mean it has any particular effect on arrhythmic events or does it mean, such as a beta-blocker, for example, [it] reduces calcium transience and improves handling of calcium, or does it have an effect simply by improving heart failure?” he asked.
Dr. Curtain replied they are still trying to understand the effects of this new class of drug but that studies have shown dapagliflozin and other SGLT2 inhibitors have favorable effects on adverse cardiac remodeling, which contributes to sudden death and ventricular arrhythmia. They’ve also been shown to reduce cardiac chamber size, left ventricular hypertrophy, and N-terminal pro-B-type natriuretic peptide (NT-proBNP) levels over time, consistent with a reduction in myocardial wall stress. “So it could indeed be one of several mechanisms by which they may exert a beneficial cardiac effect.”
Speaking with this news organization, Dr. Curtain pointed out that the Kaplan-Meier curves for the composite outcome began to separate early on, but that the clearest separation was after 9 months, suggestive of a positive action on adverse cardiac remodeling over time.
“This would improve the patients’ heart failure situation, but also thick ventricles are a key risk factor for the occurrence of sudden death and ventricular arrhythmias,” he said. “The effects on adverse cardiac remodeling, given its plausibility in terms of our Kaplan-Meier curves, are one [mechanism] that I’d look to in the first instance, but I’m sure there are more than one actions at play.”
According to the new analysis, the primary outcome occurred in 315 (6.6%) patients; there were 203 adjudicated sudden deaths (64%), 104 investigator-reported ventricular arrhythmias (33%), and 8 resuscitated cardiac arrests (3%). Independent predictors of the primary outcome were higher NT-proBNP levels (odds ratio, 1.54), previous ventricular arrhythmia (OR, 1.93), previous myocardial infarction (OR, 1.42), male sex (OR, 1.53), and higher body mass index (OR, 1.03).
The effect of dapagliflozin on the primary outcome was consistent in several sensitivity analyses and “generally consistent” across key subgroups, Dr. Curtain said.
During a discussion of the results, session cochair Mitja Lainscak, MD, General Hospital Murska Sobota, Slovenia, called out two exceptions. “With regard to patients with implanted ICDs, the effect was neutral, and in the patients without diabetes, the benefit was less than in diabetic patients. Any explanations for that?”
Dr. Curtain responded that “it’s important to note that in the subgroup analyses the point estimates were all on the side favoring dapagliflozin and the interaction test was not significant in that subgroup. The numbers of patients who were in the defibrillator group were modest, and there was a relatively smaller number of events, so it may be harder to show benefit in that group.”
In the dapagliflozin and placebo groups, the event rates per 100 person-years were 3.9 and 5.8, respectively, in patients with diabetes, and 4.1 and 4.7, respectively, in those without diabetes (P for interaction = .273).
Event rates per 100 person-years were 5.8 and 5.9, respectively, in patients with a defibrillator at baseline, and 3.5 and 4.9, respectively, in those without a defibrillator (P for interaction = .174).
Asked to comment on the study, which was simultaneously published in the European Heart Journal, Milton Packer, MD, Baylor University Medical Center, Dallas, said he had “very little confidence” in the findings.
“This was entirely post hoc and the investigators combined events – with markedly different levels of clinical importance – in order to achieve a P value less than 0.05,” he told this news organization. “If one takes asymptomatic ventricular arrhythmias out of the analysis, the effect is no longer statistically significant. Furthermore, half of sudden deaths in patients with heart failure are not related to a ventricular arrhythmia.”
The authors note in their report that the analysis was not prespecified and the findings should be regarded as “hypothesis generating and require confirmation,” but also point out that a recent meta-analysis showed that SGLT2 inhibitor use was associated with a lower risk for ventricular tachycardia. Other limitations to the post hoc analysis are that adverse-event reporting likely underestimated the true prevalence of ventricular arrhythmias, and that these events were not adjudicated.
DAPA-HF was funded by AstraZeneca. Dr. Curtain reports no relevant financial relationships. Disclosures for the coauthors are listed in the paper.
A version of this article first appeared on Medscape.com.
FROM ESC CONGRESS 2021
ICMs detect serious arrhythmias in high-risk post-MI patients: SMART-MI
Prevention strategies may be next
After a myocardial infarction, implantable cardiac monitors (ICMs) are sensitive for detecting serious arrhythmias in patients with cardiac autonomic dysfunction but only moderately reduced left ventricular ejection fraction (LVEF), according to results of the randomized SMART-MI trial.

When remote monitoring with the ICM was compared with conventional follow-up in this group of patients, serious arrhythmic events were detected at a nearly sixfold greater rate, reported Axel Bauer, MD, at the annual congress of the European Society of Cardiology.
The study further showed that these events were closely associated with subsequent major adverse cardiac and cerebrovascular events (MACCE).
“SMART-MI is the first study to test an implantable device in high-risk MI patients with a LVEF greater than 35%,” reported Dr. Bauer, a cardiologist and director of the internal medicine clinic, University of Innsbruck (Austria). It showed that the types and frequency of arrhythmias were “comparable to those of post-MI patients with reduced LVEF.”
The ability to assess risk is potentially significant because “the majority of cardiovascular complications, including sudden death, occur in patients with only moderately reduced LVEF,” explained Dr. Bauer.
Despite the greater risk, “there are no preventive strategies so far” currently available for this group, he said.
The SMART-MI study confirms the need for treatments, confirms a method for monitoring risk, and might provide the basis for trials designed to test treatments to modify this risk, he added.
ECG used to define autonomic dysfunction
In the SMART MI protocol, 1,305 survivors of MI with LVEF of 36%-50% at 33 participating centers in Austria and Germany were evaluated with a 20-minute high resolution electrocardiogram. They were enrolled and randomized if they demonstrated cardiac autonomic dysfunction on at least two validated ECG biomarkers.
The 400 participants were randomized to implantation of a ICM, which transmitted daily reports to a ICM core laboratory, or to conventional follow-up.
After a median follow-up of 21 months, serious events were detected in 60 of the 201 patients in the ICM group and 12 of the 199 patients in the control group (29% vs. 6%). Serious adverse events were defined as those that would typically warrant therapy, such as prolonged atrial fibrillation (at least 6 minutes) high-degree atrioventricular block, and sustained ventricular tachycardia.
The difference in the detection rate, which was the primary endpoint, was highly significant (P < .0001), but the study was also able to confirm that these events predicted MACCE, a secondary study endpoint. In those with a serious arrhythmia, the hazard ratio for subsequent MACCE was approximately sevenfold greater relative to those without a serious arrhythmia. This was true of those in the ICM group (HR, 6.8; P < .001) and controls (HR 7.3; P < .001).
Arrhythmias warn of impending complications
“The data show that the prognostic impact of detecting a serious arrhythmia does not depend on the mode of detection,” Dr. Bauer reported. The data also confirm that “subclinical serious arrhythmia events are a warning signal for an impending complication.”
Although more interventions – including pacemakers, catheter ablations, and oral anticoagulants – were offered to patients in the experimental arm, “the study was not powered to show differences in outcomes,” and, in fact, no significant differences were observed, according to Dr. Bauer. However, the evidence that ICM is effective for detecting arrhythmias does provide a structure on which to build clinical trials.
“We now need the trials to see if ICM can change practice and improve outcomes,” said Carlos Aguiar, MD, a staff cardiologist at the Hospital Santa Cruz, Lisbon. He acknowledged that this study proves that ICM can detect serious arrhythmias in patients with moderate left ventricular dysfunction, but “we need to develop and test treatment paths.”
Dr. Aguiar considers SMART-MI an important study that “goes to the heart” of a common clinical dilemma.
“In clinical practice, we see patients with LVEF that is not that suppressed and so do not have a class I indication for ICM, but there are often features that might have you concerned and make you think it would be great if the LVEF was 35% or lower [to justify intervention],” Dr. Aguiar said.
Data provide insight on unaddressed risk group
SMART-MI confirms earlier evidence that post-MI patients with cardiac autonomic dysfunction are at high risk. Currently, this relative increase in risk goes “unaddressed,” according to Dr. Bauer. Although he contended that the risk itself “could be an indication for ICM in a high-risk patient group without classically defined left ventricular dysfunction,” he agreed that the ultimate value of this trial might be that it “opens a window” for a rationale to test preventive strategies.

An invited ESC discussant, Gerhard Hindricks, MD, PhD, praised the study for drawing attention to the risk of events in a subset of post-MI patients with LVEF of 35% or greater. However, he suggested that criteria other than those based on ECG might be more sensitive for selecting patients who might benefit from intervention.
“We do not know whether additional methods of establishing risk, such as imaging, might be valuable,” said Dr. Hindricks, chief of the department of arrhythmology in the Heart Institute of the University of Leipzig (Germany). He believes work in this area is needed to ensure appropriate entry criteria for interventional trials designed to modify risk in post-MI patients who do not meet the traditional definition of reduced ejection fraction.
Dr. Bauer reports financial relationships with Medtronic, which sponsored this study, as well as Bayer, Boehringer Ingelheim, Edwards, and Novartis. Dr. Aguiar reports no relevant financial conflicts.
Prevention strategies may be next
Prevention strategies may be next
After a myocardial infarction, implantable cardiac monitors (ICMs) are sensitive for detecting serious arrhythmias in patients with cardiac autonomic dysfunction but only moderately reduced left ventricular ejection fraction (LVEF), according to results of the randomized SMART-MI trial.
When remote monitoring with the ICM was compared with conventional follow-up in this group of patients, serious arrhythmic events were detected at a nearly sixfold greater rate, reported Axel Bauer, MD, at the annual congress of the European Society of Cardiology.
The study further showed that these events were closely associated with subsequent major adverse cardiac and cerebrovascular events (MACCE).
“SMART-MI is the first study to test an implantable device in high-risk MI patients with a LVEF greater than 35%,” reported Dr. Bauer, a cardiologist and director of the internal medicine clinic, University of Innsbruck (Austria). It showed that the types and frequency of arrhythmias were “comparable to those of post-MI patients with reduced LVEF.”
The ability to assess risk is potentially significant because “the majority of cardiovascular complications, including sudden death, occur in patients with only moderately reduced LVEF,” explained Dr. Bauer.
Despite the greater risk, “there are no preventive strategies so far” currently available for this group, he said.
The SMART-MI study confirms the need for treatments, confirms a method for monitoring risk, and might provide the basis for trials designed to test treatments to modify this risk, he added.
ECG used to define autonomic dysfunction
In the SMART MI protocol, 1,305 survivors of MI with LVEF of 36%-50% at 33 participating centers in Austria and Germany were evaluated with a 20-minute high resolution electrocardiogram. They were enrolled and randomized if they demonstrated cardiac autonomic dysfunction on at least two validated ECG biomarkers.
The 400 participants were randomized to implantation of a ICM, which transmitted daily reports to a ICM core laboratory, or to conventional follow-up.
After a median follow-up of 21 months, serious events were detected in 60 of the 201 patients in the ICM group and 12 of the 199 patients in the control group (29% vs. 6%). Serious adverse events were defined as those that would typically warrant therapy, such as prolonged atrial fibrillation (at least 6 minutes) high-degree atrioventricular block, and sustained ventricular tachycardia.
The difference in the detection rate, which was the primary endpoint, was highly significant (P < .0001), but the study was also able to confirm that these events predicted MACCE, a secondary study endpoint. In those with a serious arrhythmia, the hazard ratio for subsequent MACCE was approximately sevenfold greater relative to those without a serious arrhythmia. This was true of those in the ICM group (HR, 6.8; P < .001) and controls (HR 7.3; P < .001).
Arrhythmias warn of impending complications
“The data show that the prognostic impact of detecting a serious arrhythmia does not depend on the mode of detection,” Dr. Bauer reported. The data also confirm that “subclinical serious arrhythmia events are a warning signal for an impending complication.”
Although more interventions – including pacemakers, catheter ablations, and oral anticoagulants – were offered to patients in the experimental arm, “the study was not powered to show differences in outcomes,” and, in fact, no significant differences were observed, according to Dr. Bauer. However, the evidence that ICM is effective for detecting arrhythmias does provide a structure on which to build clinical trials.
“We now need the trials to see if ICM can change practice and improve outcomes,” said Carlos Aguiar, MD, a staff cardiologist at the Hospital Santa Cruz, Lisbon. He acknowledged that this study proves that ICM can detect serious arrhythmias in patients with moderate left ventricular dysfunction, but “we need to develop and test treatment paths.”
Dr. Aguiar considers SMART-MI an important study that “goes to the heart” of a common clinical dilemma.
“In clinical practice, we see patients with LVEF that is not that suppressed and so do not have a class I indication for ICM, but there are often features that might have you concerned and make you think it would be great if the LVEF was 35% or lower [to justify intervention],” Dr. Aguiar said.
Data provide insight on unaddressed risk group
SMART-MI confirms earlier evidence that post-MI patients with cardiac autonomic dysfunction are at high risk. Currently, this relative increase in risk goes “unaddressed,” according to Dr. Bauer. Although he contended that the risk itself “could be an indication for ICM in a high-risk patient group without classically defined left ventricular dysfunction,” he agreed that the ultimate value of this trial might be that it “opens a window” for a rationale to test preventive strategies.
An invited ESC discussant, Gerhard Hindricks, MD, PhD, praised the study for drawing attention to the risk of events in a subset of post-MI patients with LVEF of 35% or greater. However, he suggested that criteria other than those based on ECG might be more sensitive for selecting patients who might benefit from intervention.
“We do not know whether additional methods of establishing risk, such as imaging, might be valuable,” said Dr. Hindricks, chief of the department of arrhythmology in the Heart Institute of the University of Leipzig (Germany). He believes work in this area is needed to ensure appropriate entry criteria for interventional trials designed to modify risk in post-MI patients who do not meet the traditional definition of reduced ejection fraction.
Dr. Bauer reports financial relationships with Medtronic, which sponsored this study, as well as Bayer, Boehringer Ingelheim, Edwards, and Novartis. Dr. Aguiar reports no relevant financial conflicts.
After a myocardial infarction, implantable cardiac monitors (ICMs) are sensitive for detecting serious arrhythmias in patients with cardiac autonomic dysfunction but only moderately reduced left ventricular ejection fraction (LVEF), according to results of the randomized SMART-MI trial.
When remote monitoring with the ICM was compared with conventional follow-up in this group of patients, serious arrhythmic events were detected at a nearly sixfold greater rate, reported Axel Bauer, MD, at the annual congress of the European Society of Cardiology.
The study further showed that these events were closely associated with subsequent major adverse cardiac and cerebrovascular events (MACCE).
“SMART-MI is the first study to test an implantable device in high-risk MI patients with a LVEF greater than 35%,” reported Dr. Bauer, a cardiologist and director of the internal medicine clinic, University of Innsbruck (Austria). It showed that the types and frequency of arrhythmias were “comparable to those of post-MI patients with reduced LVEF.”
The ability to assess risk is potentially significant because “the majority of cardiovascular complications, including sudden death, occur in patients with only moderately reduced LVEF,” explained Dr. Bauer.
Despite the greater risk, “there are no preventive strategies so far” currently available for this group, he said.
The SMART-MI study confirms the need for treatments, confirms a method for monitoring risk, and might provide the basis for trials designed to test treatments to modify this risk, he added.
ECG used to define autonomic dysfunction
In the SMART MI protocol, 1,305 survivors of MI with LVEF of 36%-50% at 33 participating centers in Austria and Germany were evaluated with a 20-minute high resolution electrocardiogram. They were enrolled and randomized if they demonstrated cardiac autonomic dysfunction on at least two validated ECG biomarkers.
The 400 participants were randomized to implantation of a ICM, which transmitted daily reports to a ICM core laboratory, or to conventional follow-up.
After a median follow-up of 21 months, serious events were detected in 60 of the 201 patients in the ICM group and 12 of the 199 patients in the control group (29% vs. 6%). Serious adverse events were defined as those that would typically warrant therapy, such as prolonged atrial fibrillation (at least 6 minutes) high-degree atrioventricular block, and sustained ventricular tachycardia.
The difference in the detection rate, which was the primary endpoint, was highly significant (P < .0001), but the study was also able to confirm that these events predicted MACCE, a secondary study endpoint. In those with a serious arrhythmia, the hazard ratio for subsequent MACCE was approximately sevenfold greater relative to those without a serious arrhythmia. This was true of those in the ICM group (HR, 6.8; P < .001) and controls (HR 7.3; P < .001).
Arrhythmias warn of impending complications
“The data show that the prognostic impact of detecting a serious arrhythmia does not depend on the mode of detection,” Dr. Bauer reported. The data also confirm that “subclinical serious arrhythmia events are a warning signal for an impending complication.”
Although more interventions – including pacemakers, catheter ablations, and oral anticoagulants – were offered to patients in the experimental arm, “the study was not powered to show differences in outcomes,” and, in fact, no significant differences were observed, according to Dr. Bauer. However, the evidence that ICM is effective for detecting arrhythmias does provide a structure on which to build clinical trials.
“We now need the trials to see if ICM can change practice and improve outcomes,” said Carlos Aguiar, MD, a staff cardiologist at the Hospital Santa Cruz, Lisbon. He acknowledged that this study proves that ICM can detect serious arrhythmias in patients with moderate left ventricular dysfunction, but “we need to develop and test treatment paths.”
Dr. Aguiar considers SMART-MI an important study that “goes to the heart” of a common clinical dilemma.
“In clinical practice, we see patients with LVEF that is not that suppressed and so do not have a class I indication for ICM, but there are often features that might have you concerned and make you think it would be great if the LVEF was 35% or lower [to justify intervention],” Dr. Aguiar said.
Data provide insight on unaddressed risk group
SMART-MI confirms earlier evidence that post-MI patients with cardiac autonomic dysfunction are at high risk. Currently, this relative increase in risk goes “unaddressed,” according to Dr. Bauer. Although he contended that the risk itself “could be an indication for ICM in a high-risk patient group without classically defined left ventricular dysfunction,” he agreed that the ultimate value of this trial might be that it “opens a window” for a rationale to test preventive strategies.
An invited ESC discussant, Gerhard Hindricks, MD, PhD, praised the study for drawing attention to the risk of events in a subset of post-MI patients with LVEF of 35% or greater. However, he suggested that criteria other than those based on ECG might be more sensitive for selecting patients who might benefit from intervention.
“We do not know whether additional methods of establishing risk, such as imaging, might be valuable,” said Dr. Hindricks, chief of the department of arrhythmology in the Heart Institute of the University of Leipzig (Germany). He believes work in this area is needed to ensure appropriate entry criteria for interventional trials designed to modify risk in post-MI patients who do not meet the traditional definition of reduced ejection fraction.
Dr. Bauer reports financial relationships with Medtronic, which sponsored this study, as well as Bayer, Boehringer Ingelheim, Edwards, and Novartis. Dr. Aguiar reports no relevant financial conflicts.
FROM ESC CONGRESS 2021
GUIDE-HF: CardioMEMS-guided meds fall short in mild to moderate heart failure
Medical therapy for heart failure guided by an implanted pulmonary artery pressure (PAP) sensor didn’t improve survival or risk for HF events like hospitalization over a year in a major randomized trial that entered a broad range of patients with mild to moderate disease.
But medical therapy adjustments based on PAP readings from the miniature CardioMEMS (Abbott) implant might well have surpassed conventional HF management for outcomes had the world not been turned upside down by SARS-CoV-2 and the pandemic lockdowns, assert researchers from the GUIDE-HF trial.
Something about the crisis, they concluded – although not without some pushback – led to better outcomes in the standard-care control group, apparently muddling any potential differences from those on PAP-guided management.
Working with regulators, the team conducted a “pre–COVID-19 impact analysis” that compared outcomes before the March 2020 national COVID-19 emergency declaration that forced much of the United States with shelter in place.
By that time, all of the trial’s patients had been followed for at least 3 months, and about three-fourths of its endpoints had already been counted, JoAnn Lindenfeld, MD, Vanderbilt University Medical Center, Nashville, Tenn., said at a media briefing prior to unveiling GUIDE-HF at the all-virtual European Society of Cardiology Congress 2021.
The pre–COVID-19 analysis, approved several months before the end of the trial – while the data were still blinded – had been “suggested by both regulatory agencies and professional societies in Europe and in the United States,” Dr. Lindenfeld said.
It pointed to a possible benefit for the CardioMEMS-guided strategy, a barely significant 19% drop in risk (P = .049) for the primary endpoint of death, HF hospitalization, or urgent HF hospital visit. The effect was driven by a 24% decline in HF events (P = .014), with no significant contribution from mortality.
“The benefits of hemodynamic monitoring and management in reducing heart failure hospitalizations extended to patients with less severe heart failure”; that is, those in New York Heart Association class 2 and any in NYHA class 3 with “elevated natriuretic peptides but no previous hospitalization,” said Dr. Lindenfeld, who is also lead author on the GUIDE-HF report published in the Lancet.
Such benefits would suggest that CardioMEMS-guided management can improve outcomes in an HF population much broader than the device’s current indication.
But as it happens, the trial’s prospectively defined 12-month primary outcomes were less impressive. A 12% decline in risk for the composite endpoint among patients managed by CardioMEMS failed to reach significance compared with standard management (P = .16).
“Several factors could explain the considerable loss of benefit of hemodynamic-guided management during the COVID-19 pandemic,” the Lancet report explained. They include “improved patient compliance with medical and dietary regimens, reduced respiratory infections, altered health-care provider behavior, changes in disease progression due to COVID-19, or other as yet unknown effects of a major pandemic.”
Expanded population
Importantly, GUIDE-HF had entered 1,000 patients in NYHA class 2-4 and either an HF hospitalization in the previous year or elevated natriuretic peptide levels. About 44% of the entrants in NYHA class 3 did not have a 1-year history of HF hospitalization.
That’s a more heterogeneous and potentially lower-risk cohort than patients in the randomized CHAMPION study of 11 years ago, which led to the implant’s approval on both sides of the Atlantic.
In that trial, CardioMEMS-guided management was followed by 30% drop in risk for HF hospitalization over 6 months (P < .001). But CHAMPION was limited to patients in NYHA class 3 with a history of HF hospitalization, the device’s current indication in both the United States and Europe.
The GUIDE-HF findings “reinforce that patients with class 3 heart failure and prior heart failure hospitalization are those in whom there is the clearest benefit, based on the prior CHAMPION trial. These are the patients where this monitoring strategy may be best targeted,” Gregg C. Fonarow, MD, University of California Los Angeles Medical Center, said in an interview.
Although GUIDE-HF didn’t show a significant benefit for NYHA class 2 patients with elevated biomarkers, who aren’t covered by the device’s current labeling, that group showed “some suggestions of potential benefit,” noted Dr. Fonarow, who isn’t a coauthor on the Lancet report. So, “there may be select patients with class 2 heart failure where monitoring could be considered on a case-by-case basis.”
In an interview, Larry A. Allen, MD, MHS, said that, “while the technology is pretty amazing, the real question is whether it tells us something that we didn’t already know that leads to improved care. Unfortunately, as tested here, it doesn’t, or at least not enough to make a big difference.”
The pre–COVID-19 impact analysis “should be interpreted with caution, and not as the primary finding,” Dr. Allen, from the University of Colorado at Denver, Aurora, who is not a GUIDE-HF coauthor, said in an interview.
One might hypothesize, he said, “that, in the setting of limited in-person visits with loss of physical examination, perhaps CardioMEMS would be more – not less – helpful during the pandemic. And yet the opposite was seen.”
The pandemic has “markedly altered all kinds of aspects of patient care and trial conduct, but that doesn’t make the data derived during that period uninformative,” Dr. Allen said. “And as we are increasingly reminded, the future will be a new normal, not a prepandemic normal.”
A third group
The GUIDE-HF trial includes, in addition to the 1,000 randomized patients, a single-group observational cohort of 2,600 patients, whose outcomes will be reported at another time, noted the published report.
But in the randomized comparison, conducted at 118 centers in North America, all patients were implanted with the CardioMEMS device and blinded as to their assigned strategy. Enrollment took place between March 2018 and Dec. 20, 2019.
Of the 1,000 successfully implanted patients, 497 were assigned to the pressure-guided strategy, in which “titration of diuretics was recommended if pulmonary artery pressure provided evidence of excess intravascular volume, and titration of vasodilators was recommended if elevated vascular resistance was evident,” the report stated.
The remaining 503 patients assigned to standard care served as control subjects, for whom “investigators were aware of treatment assignment but did not have access to PAP data.”
The hazard ratio for the primary endpoint in the pressure-guided group, compared with the control group, was 0.88 (95% confidence interval, 0.74-1.05; P = .16) over a median follow-up of 11.7 months.
But in the sensitivity analysis comparing outcomes before and after the COVID-19 lockdowns, using established methodology, the report stated, the primary-endpoint HR was 0.81 (95% CI, 0.66-1.00; P = .049).
The difference is owed to improved outcomes in the control group under pandemic conditions, the researchers concluded. Patients assigned to conventional management –whatever that meant during shelter-in-place – experienced 21% fewer primary-endpoint events than their own rate before the pandemic. After the COVID-19 emergency was declared, there was no significant difference in event rates between the two randomization groups.
In the primary 12-month analysis, the HR for HF events in the guided-therapy was not significant reduced, at 0.85 (95% CI, 0.70-1.03; P = .096). But in the pre-COVID-19 analysis, that risk fell significantly with CardioMEMS-guided management, for an HR of 0.76 (95% CI, 0.61-0.95; P = .014).
An editorial accompanying the GUIDE-HF publication (Lancet. 2021 Aug 27. doi: 10.1016/S0140-6736[21]01914-0) asserts that the trial “did not enroll an ideal group of patients for showing the efficacy of pulmonary artery pressure monitoring, since many had baseline pressures in the target range with little possibility of short-term gain.”
Also, wrote John G. F. Cleland, MD, PhD, University of Glasgow, and Pierpaolo Pellicori, MD, Imperial College London, “follow-up was too short, and interventions did not substantially change pulmonary artery pressure.”
They continue: “Monitoring alone cannot improve outcome, but consequent actions might. The GUIDE-HF results are encouraging but inconclusive, and should inform further research, possibly a large, simple, open-label trial to investigate a system of care rather than a single technology.”
GUIDE-HF was funded by Abbott. Dr. Lindenfeld disclosed receiving research grants from AstraZeneca, Sensible Medical, and Volumetrix; and consulting for Abbott, Alleviant Medical, AstraZeneca, Boehringer Ingelheim, Boston Scientific, CVRx, Edwards, Impulse Dynamics, and VWave. Dr. Fonarow reported consulting for Abbott and that his institution has participated in the GUIDE-HF trial; he has elsewhere disclosed consulting for Amgen, AstraZeneca, CHF Solutions Lifesciences, Janssen, Medtronic, and Novartis. Dr. Allen had elsewhere reported consulting for Abbott, Amgen, Boston Scientific, and Novartis. Dr. Cleland disclosed receiving personal fees from Abbott for serving on an advisory board for the MitraClip device, unrelated to the CardioMEMS device. Dr. Pellicori reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
Medical therapy for heart failure guided by an implanted pulmonary artery pressure (PAP) sensor didn’t improve survival or risk for HF events like hospitalization over a year in a major randomized trial that entered a broad range of patients with mild to moderate disease.
But medical therapy adjustments based on PAP readings from the miniature CardioMEMS (Abbott) implant might well have surpassed conventional HF management for outcomes had the world not been turned upside down by SARS-CoV-2 and the pandemic lockdowns, assert researchers from the GUIDE-HF trial.
Something about the crisis, they concluded – although not without some pushback – led to better outcomes in the standard-care control group, apparently muddling any potential differences from those on PAP-guided management.
Working with regulators, the team conducted a “pre–COVID-19 impact analysis” that compared outcomes before the March 2020 national COVID-19 emergency declaration that forced much of the United States with shelter in place.
By that time, all of the trial’s patients had been followed for at least 3 months, and about three-fourths of its endpoints had already been counted, JoAnn Lindenfeld, MD, Vanderbilt University Medical Center, Nashville, Tenn., said at a media briefing prior to unveiling GUIDE-HF at the all-virtual European Society of Cardiology Congress 2021.
The pre–COVID-19 analysis, approved several months before the end of the trial – while the data were still blinded – had been “suggested by both regulatory agencies and professional societies in Europe and in the United States,” Dr. Lindenfeld said.
It pointed to a possible benefit for the CardioMEMS-guided strategy, a barely significant 19% drop in risk (P = .049) for the primary endpoint of death, HF hospitalization, or urgent HF hospital visit. The effect was driven by a 24% decline in HF events (P = .014), with no significant contribution from mortality.
“The benefits of hemodynamic monitoring and management in reducing heart failure hospitalizations extended to patients with less severe heart failure”; that is, those in New York Heart Association class 2 and any in NYHA class 3 with “elevated natriuretic peptides but no previous hospitalization,” said Dr. Lindenfeld, who is also lead author on the GUIDE-HF report published in the Lancet.
Such benefits would suggest that CardioMEMS-guided management can improve outcomes in an HF population much broader than the device’s current indication.
But as it happens, the trial’s prospectively defined 12-month primary outcomes were less impressive. A 12% decline in risk for the composite endpoint among patients managed by CardioMEMS failed to reach significance compared with standard management (P = .16).
“Several factors could explain the considerable loss of benefit of hemodynamic-guided management during the COVID-19 pandemic,” the Lancet report explained. They include “improved patient compliance with medical and dietary regimens, reduced respiratory infections, altered health-care provider behavior, changes in disease progression due to COVID-19, or other as yet unknown effects of a major pandemic.”
Expanded population
Importantly, GUIDE-HF had entered 1,000 patients in NYHA class 2-4 and either an HF hospitalization in the previous year or elevated natriuretic peptide levels. About 44% of the entrants in NYHA class 3 did not have a 1-year history of HF hospitalization.
That’s a more heterogeneous and potentially lower-risk cohort than patients in the randomized CHAMPION study of 11 years ago, which led to the implant’s approval on both sides of the Atlantic.
In that trial, CardioMEMS-guided management was followed by 30% drop in risk for HF hospitalization over 6 months (P < .001). But CHAMPION was limited to patients in NYHA class 3 with a history of HF hospitalization, the device’s current indication in both the United States and Europe.
The GUIDE-HF findings “reinforce that patients with class 3 heart failure and prior heart failure hospitalization are those in whom there is the clearest benefit, based on the prior CHAMPION trial. These are the patients where this monitoring strategy may be best targeted,” Gregg C. Fonarow, MD, University of California Los Angeles Medical Center, said in an interview.
Although GUIDE-HF didn’t show a significant benefit for NYHA class 2 patients with elevated biomarkers, who aren’t covered by the device’s current labeling, that group showed “some suggestions of potential benefit,” noted Dr. Fonarow, who isn’t a coauthor on the Lancet report. So, “there may be select patients with class 2 heart failure where monitoring could be considered on a case-by-case basis.”
In an interview, Larry A. Allen, MD, MHS, said that, “while the technology is pretty amazing, the real question is whether it tells us something that we didn’t already know that leads to improved care. Unfortunately, as tested here, it doesn’t, or at least not enough to make a big difference.”
The pre–COVID-19 impact analysis “should be interpreted with caution, and not as the primary finding,” Dr. Allen, from the University of Colorado at Denver, Aurora, who is not a GUIDE-HF coauthor, said in an interview.
One might hypothesize, he said, “that, in the setting of limited in-person visits with loss of physical examination, perhaps CardioMEMS would be more – not less – helpful during the pandemic. And yet the opposite was seen.”
The pandemic has “markedly altered all kinds of aspects of patient care and trial conduct, but that doesn’t make the data derived during that period uninformative,” Dr. Allen said. “And as we are increasingly reminded, the future will be a new normal, not a prepandemic normal.”
A third group
The GUIDE-HF trial includes, in addition to the 1,000 randomized patients, a single-group observational cohort of 2,600 patients, whose outcomes will be reported at another time, noted the published report.
But in the randomized comparison, conducted at 118 centers in North America, all patients were implanted with the CardioMEMS device and blinded as to their assigned strategy. Enrollment took place between March 2018 and Dec. 20, 2019.
Of the 1,000 successfully implanted patients, 497 were assigned to the pressure-guided strategy, in which “titration of diuretics was recommended if pulmonary artery pressure provided evidence of excess intravascular volume, and titration of vasodilators was recommended if elevated vascular resistance was evident,” the report stated.
The remaining 503 patients assigned to standard care served as control subjects, for whom “investigators were aware of treatment assignment but did not have access to PAP data.”
The hazard ratio for the primary endpoint in the pressure-guided group, compared with the control group, was 0.88 (95% confidence interval, 0.74-1.05; P = .16) over a median follow-up of 11.7 months.
But in the sensitivity analysis comparing outcomes before and after the COVID-19 lockdowns, using established methodology, the report stated, the primary-endpoint HR was 0.81 (95% CI, 0.66-1.00; P = .049).
The difference is owed to improved outcomes in the control group under pandemic conditions, the researchers concluded. Patients assigned to conventional management –whatever that meant during shelter-in-place – experienced 21% fewer primary-endpoint events than their own rate before the pandemic. After the COVID-19 emergency was declared, there was no significant difference in event rates between the two randomization groups.
In the primary 12-month analysis, the HR for HF events in the guided-therapy was not significant reduced, at 0.85 (95% CI, 0.70-1.03; P = .096). But in the pre-COVID-19 analysis, that risk fell significantly with CardioMEMS-guided management, for an HR of 0.76 (95% CI, 0.61-0.95; P = .014).
An editorial accompanying the GUIDE-HF publication (Lancet. 2021 Aug 27. doi: 10.1016/S0140-6736[21]01914-0) asserts that the trial “did not enroll an ideal group of patients for showing the efficacy of pulmonary artery pressure monitoring, since many had baseline pressures in the target range with little possibility of short-term gain.”
Also, wrote John G. F. Cleland, MD, PhD, University of Glasgow, and Pierpaolo Pellicori, MD, Imperial College London, “follow-up was too short, and interventions did not substantially change pulmonary artery pressure.”
They continue: “Monitoring alone cannot improve outcome, but consequent actions might. The GUIDE-HF results are encouraging but inconclusive, and should inform further research, possibly a large, simple, open-label trial to investigate a system of care rather than a single technology.”
GUIDE-HF was funded by Abbott. Dr. Lindenfeld disclosed receiving research grants from AstraZeneca, Sensible Medical, and Volumetrix; and consulting for Abbott, Alleviant Medical, AstraZeneca, Boehringer Ingelheim, Boston Scientific, CVRx, Edwards, Impulse Dynamics, and VWave. Dr. Fonarow reported consulting for Abbott and that his institution has participated in the GUIDE-HF trial; he has elsewhere disclosed consulting for Amgen, AstraZeneca, CHF Solutions Lifesciences, Janssen, Medtronic, and Novartis. Dr. Allen had elsewhere reported consulting for Abbott, Amgen, Boston Scientific, and Novartis. Dr. Cleland disclosed receiving personal fees from Abbott for serving on an advisory board for the MitraClip device, unrelated to the CardioMEMS device. Dr. Pellicori reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
Medical therapy for heart failure guided by an implanted pulmonary artery pressure (PAP) sensor didn’t improve survival or risk for HF events like hospitalization over a year in a major randomized trial that entered a broad range of patients with mild to moderate disease.
But medical therapy adjustments based on PAP readings from the miniature CardioMEMS (Abbott) implant might well have surpassed conventional HF management for outcomes had the world not been turned upside down by SARS-CoV-2 and the pandemic lockdowns, assert researchers from the GUIDE-HF trial.
Something about the crisis, they concluded – although not without some pushback – led to better outcomes in the standard-care control group, apparently muddling any potential differences from those on PAP-guided management.
Working with regulators, the team conducted a “pre–COVID-19 impact analysis” that compared outcomes before the March 2020 national COVID-19 emergency declaration that forced much of the United States with shelter in place.
By that time, all of the trial’s patients had been followed for at least 3 months, and about three-fourths of its endpoints had already been counted, JoAnn Lindenfeld, MD, Vanderbilt University Medical Center, Nashville, Tenn., said at a media briefing prior to unveiling GUIDE-HF at the all-virtual European Society of Cardiology Congress 2021.
The pre–COVID-19 analysis, approved several months before the end of the trial – while the data were still blinded – had been “suggested by both regulatory agencies and professional societies in Europe and in the United States,” Dr. Lindenfeld said.
It pointed to a possible benefit for the CardioMEMS-guided strategy, a barely significant 19% drop in risk (P = .049) for the primary endpoint of death, HF hospitalization, or urgent HF hospital visit. The effect was driven by a 24% decline in HF events (P = .014), with no significant contribution from mortality.
“The benefits of hemodynamic monitoring and management in reducing heart failure hospitalizations extended to patients with less severe heart failure”; that is, those in New York Heart Association class 2 and any in NYHA class 3 with “elevated natriuretic peptides but no previous hospitalization,” said Dr. Lindenfeld, who is also lead author on the GUIDE-HF report published in the Lancet.
Such benefits would suggest that CardioMEMS-guided management can improve outcomes in an HF population much broader than the device’s current indication.
But as it happens, the trial’s prospectively defined 12-month primary outcomes were less impressive. A 12% decline in risk for the composite endpoint among patients managed by CardioMEMS failed to reach significance compared with standard management (P = .16).
“Several factors could explain the considerable loss of benefit of hemodynamic-guided management during the COVID-19 pandemic,” the Lancet report explained. They include “improved patient compliance with medical and dietary regimens, reduced respiratory infections, altered health-care provider behavior, changes in disease progression due to COVID-19, or other as yet unknown effects of a major pandemic.”
Expanded population
Importantly, GUIDE-HF had entered 1,000 patients in NYHA class 2-4 and either an HF hospitalization in the previous year or elevated natriuretic peptide levels. About 44% of the entrants in NYHA class 3 did not have a 1-year history of HF hospitalization.
That’s a more heterogeneous and potentially lower-risk cohort than patients in the randomized CHAMPION study of 11 years ago, which led to the implant’s approval on both sides of the Atlantic.
In that trial, CardioMEMS-guided management was followed by 30% drop in risk for HF hospitalization over 6 months (P < .001). But CHAMPION was limited to patients in NYHA class 3 with a history of HF hospitalization, the device’s current indication in both the United States and Europe.
The GUIDE-HF findings “reinforce that patients with class 3 heart failure and prior heart failure hospitalization are those in whom there is the clearest benefit, based on the prior CHAMPION trial. These are the patients where this monitoring strategy may be best targeted,” Gregg C. Fonarow, MD, University of California Los Angeles Medical Center, said in an interview.
Although GUIDE-HF didn’t show a significant benefit for NYHA class 2 patients with elevated biomarkers, who aren’t covered by the device’s current labeling, that group showed “some suggestions of potential benefit,” noted Dr. Fonarow, who isn’t a coauthor on the Lancet report. So, “there may be select patients with class 2 heart failure where monitoring could be considered on a case-by-case basis.”
In an interview, Larry A. Allen, MD, MHS, said that, “while the technology is pretty amazing, the real question is whether it tells us something that we didn’t already know that leads to improved care. Unfortunately, as tested here, it doesn’t, or at least not enough to make a big difference.”
The pre–COVID-19 impact analysis “should be interpreted with caution, and not as the primary finding,” Dr. Allen, from the University of Colorado at Denver, Aurora, who is not a GUIDE-HF coauthor, said in an interview.
One might hypothesize, he said, “that, in the setting of limited in-person visits with loss of physical examination, perhaps CardioMEMS would be more – not less – helpful during the pandemic. And yet the opposite was seen.”
The pandemic has “markedly altered all kinds of aspects of patient care and trial conduct, but that doesn’t make the data derived during that period uninformative,” Dr. Allen said. “And as we are increasingly reminded, the future will be a new normal, not a prepandemic normal.”
A third group
The GUIDE-HF trial includes, in addition to the 1,000 randomized patients, a single-group observational cohort of 2,600 patients, whose outcomes will be reported at another time, noted the published report.
But in the randomized comparison, conducted at 118 centers in North America, all patients were implanted with the CardioMEMS device and blinded as to their assigned strategy. Enrollment took place between March 2018 and Dec. 20, 2019.
Of the 1,000 successfully implanted patients, 497 were assigned to the pressure-guided strategy, in which “titration of diuretics was recommended if pulmonary artery pressure provided evidence of excess intravascular volume, and titration of vasodilators was recommended if elevated vascular resistance was evident,” the report stated.
The remaining 503 patients assigned to standard care served as control subjects, for whom “investigators were aware of treatment assignment but did not have access to PAP data.”
The hazard ratio for the primary endpoint in the pressure-guided group, compared with the control group, was 0.88 (95% confidence interval, 0.74-1.05; P = .16) over a median follow-up of 11.7 months.
But in the sensitivity analysis comparing outcomes before and after the COVID-19 lockdowns, using established methodology, the report stated, the primary-endpoint HR was 0.81 (95% CI, 0.66-1.00; P = .049).
The difference is owed to improved outcomes in the control group under pandemic conditions, the researchers concluded. Patients assigned to conventional management –whatever that meant during shelter-in-place – experienced 21% fewer primary-endpoint events than their own rate before the pandemic. After the COVID-19 emergency was declared, there was no significant difference in event rates between the two randomization groups.
In the primary 12-month analysis, the HR for HF events in the guided-therapy was not significant reduced, at 0.85 (95% CI, 0.70-1.03; P = .096). But in the pre-COVID-19 analysis, that risk fell significantly with CardioMEMS-guided management, for an HR of 0.76 (95% CI, 0.61-0.95; P = .014).
An editorial accompanying the GUIDE-HF publication (Lancet. 2021 Aug 27. doi: 10.1016/S0140-6736[21]01914-0) asserts that the trial “did not enroll an ideal group of patients for showing the efficacy of pulmonary artery pressure monitoring, since many had baseline pressures in the target range with little possibility of short-term gain.”
Also, wrote John G. F. Cleland, MD, PhD, University of Glasgow, and Pierpaolo Pellicori, MD, Imperial College London, “follow-up was too short, and interventions did not substantially change pulmonary artery pressure.”
They continue: “Monitoring alone cannot improve outcome, but consequent actions might. The GUIDE-HF results are encouraging but inconclusive, and should inform further research, possibly a large, simple, open-label trial to investigate a system of care rather than a single technology.”
GUIDE-HF was funded by Abbott. Dr. Lindenfeld disclosed receiving research grants from AstraZeneca, Sensible Medical, and Volumetrix; and consulting for Abbott, Alleviant Medical, AstraZeneca, Boehringer Ingelheim, Boston Scientific, CVRx, Edwards, Impulse Dynamics, and VWave. Dr. Fonarow reported consulting for Abbott and that his institution has participated in the GUIDE-HF trial; he has elsewhere disclosed consulting for Amgen, AstraZeneca, CHF Solutions Lifesciences, Janssen, Medtronic, and Novartis. Dr. Allen had elsewhere reported consulting for Abbott, Amgen, Boston Scientific, and Novartis. Dr. Cleland disclosed receiving personal fees from Abbott for serving on an advisory board for the MitraClip device, unrelated to the CardioMEMS device. Dr. Pellicori reported no relevant conflicts.
A version of this article first appeared on Medscape.com.
EMPEROR-Preserved: Empagliflozin scores HFpEF breakthrough
Updated August 30, 2021
The SGLT2 inhibitor empagliflozin achieved in EMPEROR-Preserved what no other agent could previously do: unequivocally cut the incidence of cardiovascular death or hospitalization in patients with heart failure and preserved ejection fraction (HFpEF).

Treatment with empagliflozin (Jardiance) led to a significant 21% relative reduction in the rate of cardiovascular death or hospitalization for heart failure (HHF), compared with placebo, among 5,988 randomized patients with HFpEF during a median 26 months of follow-up, proving that patients with HFpEF finally have a treatment that gives them clinically meaningful benefit, and paving the way to an abrupt change in management of these patients, experts said.
“This is the first trial to show unequivocal benefits of any drug on major heart failure outcomes in patients with HFpEF,” Stefan D. Anker, MD, PhD, declared at the virtual annual congress of the European Society of Cardiology.
The 21% relative reduction, which reflected a cut in the absolute rate of the trial’s primary composite endpoint of 3.3% compared with placebo, was driven mainly by a significant 27% relative reduction in the incidence of HHF (P < .001). Empagliflozin treatment, on top of standard therapy for patients with HFpEF, also resulted in a nonsignificant 9% relative risk reduction in the incidence of cardiovascular death, but it had no discernible impact on the rate of death from any cause, said Dr. Anker, professor of cardiology at Charité Medical University in Berlin.
Concurrently with his talk at the meeting, the results were published online in the New England Journal of Medicine.
Practice will change ‘quickly’
“This will definitely change our practice, and quite quickly,” said Carlos Aguiar, MD, chair of the Advanced Heart Failure and Heart Transplantation Unit at Hospital Santa Cruz in Carnaxide, Portugal, who was not involved in the study.
Transition to routine use of empagliflozin in patients with HFpEF should be swift because it has already become a mainstay of treatment for patients with heart failure with reduced ejection fraction (HFrEF) based on evidence for empagliflozin in EMPEROR-Reduced. A second sodium-glucose cotransporter 2 (SGLT2 ) inhibitor, dapagliflozin (Farxiga), is also an option for treating HFrEF based on results in the DAPA-HF trial, and the DELIVER trial, still in progress, is testing dapagliflozin as a HFpEF treatment in about 6,000 patients, with results expected in 2022.
About half of the patients in EMPEROR-Preserved had diabetes, and the treatment effects on HFpEF were similar regardless of patients’ diabetes status. Empagliflozin, like other members of the SGLT2 inhibitor class, boosts urinary excretion of glucose and received initial regulatory approval as an agent for glycemic control in patients with type 2 diabetes. Empagliflozin also has U.S.-approved marketing indications for treating patients with HFrEF whether or not they also have diabetes, and for reducing cardiovascular death in patients with type 2 diabetes and cardiovascular disease.
“We already use this drug class in cardiovascular medicine and to treat patients with type 2 diabetes, and we have been eager to find a treatment for patients with HFpEF. This is something that will be really significant,” said Dr. Aguiar.
Heart failure clinicians have “become familiar prescribing” SGLT2 inhibitors following approval of HFrEF indications for some of these agents, noted Mary Norine Walsh, MD, a heart failure specialist with Ascension Medical Group in Indianapolis. The new results “are good news because there have been so few options” for patients with HFpEF, she said in an interview.
EMPEROR-Preserved “is the first phase 3 clinical trial that exclusively enrolled patients with heart failure and an ejection fraction of more than 40% to meet its primary outcome,” and the results “represent a major win against a medical condition that had previously proven formidable,” Mark H. Drazner, MD, said in an editorial that accompanied the published results.
The trial’s findings “should contribute to a change in clinical practice given the paucity of therapeutic options available for patients with HFpEF,” wrote Dr. Drazner, a heart failure specialist who is professor and clinical chief of cardiology at UT Southwestern Medical Center in Dallas.
Theresa A, McDonagh, MD, MBChB, who chaired the panel that just released revised guidelines from the European Society of Cardiology for managing patients with heart failure, predicted that empagliflozin treatment for patients with HFpEF will soon show up in guidelines. It will likely receive a “should be considered” ranking despite being a single study because of the impressive size of the treatment effect and lack of well-supported alternative treatments, she commented as a discussant of the trial during its presentation at the congress. If the DELIVER trial with dapagliflozin shows a similar effect, the recommendation would likely become even stronger, added Dr. McDonagh, a heart failure specialist and professor of cardiology at King’s College, London.
More women enrolled than ever before
EMPEROR-Preserved enrolled adults with chronic HFpEF in New York Heart Association functional class II-IV and a left ventricular ejection fraction greater than 40% starting in 2017 at more than 600 sites in more than 20 countries worldwide including the United States. As background therapy, more than 80% of patients received treatment with either an ACE inhibitor or angiotensin receptor blocker (in some instances in the form of sacubitril/valsartan), more than 80% were on a beta-blocker, and about a third were taking a mineralocorticoid receptor antagonist, making them “very well treated HFpEF patients,” Dr. Anker said.
One of the most notable features of enrollment was that 45% of participants were women, giving this trial the highest inclusion of women compared with all prior studies in patients with HFpEF or with HFrEF, said Dr. Walsh. “HFpEF is very prevalent in woman,” she noted, and having this high participation rate of women in the study increases its relevance to these patients. “It’s important to be able to tell women that patients like you were in the study so we can more easily apply the lessons from the trial to you. That can’t be stressed enough,” she said.
The primary outcome occurred in 415 (13.8%) of the 2,997 patients in the empagliflozin group and in 511 (17.1%) of 2,991 patients who received placebo (hazard ratio, 0.79; 95% confidence interval, 0.69-0.90; P < .001).
The study showed a safety profile consistent with prior experience with empagliflozin, Dr. Anker added.
Pooling EMPEROR-Preserved with EMPEROR-Reduced
The investigators who ran EMPEROR-Preserved designed the trial to closely parallel the EMPEROR-Reduced trial in patients with HFrEF, and they included a prespecified analysis (EMPEROR-Pooled) that combined the more than 9,700 patients in the two studies. This showed a consistent and robust benefit from empagliflozin for reducing HHF across a wide spectrum of patients with heart failure, ranging from patients with left ventricular ejection fractions of less than 25% to patients with ejection fractions as high as 64%. However, the analysis also showed that patients with ejection fractions of 65% or greater received no discernible benefit from empagliflozin, Milton Packer, MD, reported in a separate talk at the congress.

“The findings demonstrate the benefits of empagliflozin across a broad range of patients with heart failure who have ejection fractions of less than 60%-65%,” said Dr. Packer, a researcher at Baylor University Medical Center in Dallas.
This apparent attenuation of an effect at higher ejection fractions “has been observed in other HFpEF trials, most recently in the PARAGON-HF trial” of sacubitril/valsartan (Entresto), he noted. Additional analyses led by Dr. Packer showed that in patients with ejection fractions below 65% the HHF benefit from empagliflozin consistently surpassed the benefit seen with sacubitril/valsartan in PARAGON-HF. But he recommended using both drugs in patients with HFpEF and an ejection fraction up to about 60%.
“If I had a patient with HFpEF I would use both drugs as well as beta-blockers and mineralocorticoid receptor antagonists,” he said during a press briefing.
Another finding from analysis of the EMPEROR-Reduced and EMPEROR-Preserved trials together was that patients with reduced ejection fractions showed a significant 49% relative reduction in the incidence of serious renal outcomes, but this effect was completely blunted in EMPEROR-Preserved.
“Ejection fraction influences the effects of empagliflozin on major renal outcomes,” concluded Dr. Packer in a report on this analysis published simultaneously with the main EMPEROR-Preserved findings (N Engl J Med. 2021 Aug 27. doi: 10.1056/NEJMc2112411). “These data from the EMPEROR trials are unique. We have no comparable data” from any of the other reported studies of SGLT2 inhibitors,” he said.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and by Eli Lilly, the two companies that jointly market empagliflozin (Jardiance). Dr. Anker has received personal fees from Boehringer Ingelheim and from several other companies, and he has received grants and personal fees from Abbott Vascular and Vifor. Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. McDonagh has has recent financial relationships with AstraZeneca, Cprpus, Novartis, Pfizer, and Vifor. Dr. Aguiar and Dr. Walsh had no disclosures.
Updated August 30, 2021
The SGLT2 inhibitor empagliflozin achieved in EMPEROR-Preserved what no other agent could previously do: unequivocally cut the incidence of cardiovascular death or hospitalization in patients with heart failure and preserved ejection fraction (HFpEF).
Treatment with empagliflozin (Jardiance) led to a significant 21% relative reduction in the rate of cardiovascular death or hospitalization for heart failure (HHF), compared with placebo, among 5,988 randomized patients with HFpEF during a median 26 months of follow-up, proving that patients with HFpEF finally have a treatment that gives them clinically meaningful benefit, and paving the way to an abrupt change in management of these patients, experts said.
“This is the first trial to show unequivocal benefits of any drug on major heart failure outcomes in patients with HFpEF,” Stefan D. Anker, MD, PhD, declared at the virtual annual congress of the European Society of Cardiology.
The 21% relative reduction, which reflected a cut in the absolute rate of the trial’s primary composite endpoint of 3.3% compared with placebo, was driven mainly by a significant 27% relative reduction in the incidence of HHF (P < .001). Empagliflozin treatment, on top of standard therapy for patients with HFpEF, also resulted in a nonsignificant 9% relative risk reduction in the incidence of cardiovascular death, but it had no discernible impact on the rate of death from any cause, said Dr. Anker, professor of cardiology at Charité Medical University in Berlin.
Concurrently with his talk at the meeting, the results were published online in the New England Journal of Medicine.
Practice will change ‘quickly’
“This will definitely change our practice, and quite quickly,” said Carlos Aguiar, MD, chair of the Advanced Heart Failure and Heart Transplantation Unit at Hospital Santa Cruz in Carnaxide, Portugal, who was not involved in the study.
Transition to routine use of empagliflozin in patients with HFpEF should be swift because it has already become a mainstay of treatment for patients with heart failure with reduced ejection fraction (HFrEF) based on evidence for empagliflozin in EMPEROR-Reduced. A second sodium-glucose cotransporter 2 (SGLT2 ) inhibitor, dapagliflozin (Farxiga), is also an option for treating HFrEF based on results in the DAPA-HF trial, and the DELIVER trial, still in progress, is testing dapagliflozin as a HFpEF treatment in about 6,000 patients, with results expected in 2022.
About half of the patients in EMPEROR-Preserved had diabetes, and the treatment effects on HFpEF were similar regardless of patients’ diabetes status. Empagliflozin, like other members of the SGLT2 inhibitor class, boosts urinary excretion of glucose and received initial regulatory approval as an agent for glycemic control in patients with type 2 diabetes. Empagliflozin also has U.S.-approved marketing indications for treating patients with HFrEF whether or not they also have diabetes, and for reducing cardiovascular death in patients with type 2 diabetes and cardiovascular disease.
“We already use this drug class in cardiovascular medicine and to treat patients with type 2 diabetes, and we have been eager to find a treatment for patients with HFpEF. This is something that will be really significant,” said Dr. Aguiar.
Heart failure clinicians have “become familiar prescribing” SGLT2 inhibitors following approval of HFrEF indications for some of these agents, noted Mary Norine Walsh, MD, a heart failure specialist with Ascension Medical Group in Indianapolis. The new results “are good news because there have been so few options” for patients with HFpEF, she said in an interview.
EMPEROR-Preserved “is the first phase 3 clinical trial that exclusively enrolled patients with heart failure and an ejection fraction of more than 40% to meet its primary outcome,” and the results “represent a major win against a medical condition that had previously proven formidable,” Mark H. Drazner, MD, said in an editorial that accompanied the published results.
The trial’s findings “should contribute to a change in clinical practice given the paucity of therapeutic options available for patients with HFpEF,” wrote Dr. Drazner, a heart failure specialist who is professor and clinical chief of cardiology at UT Southwestern Medical Center in Dallas.
Theresa A, McDonagh, MD, MBChB, who chaired the panel that just released revised guidelines from the European Society of Cardiology for managing patients with heart failure, predicted that empagliflozin treatment for patients with HFpEF will soon show up in guidelines. It will likely receive a “should be considered” ranking despite being a single study because of the impressive size of the treatment effect and lack of well-supported alternative treatments, she commented as a discussant of the trial during its presentation at the congress. If the DELIVER trial with dapagliflozin shows a similar effect, the recommendation would likely become even stronger, added Dr. McDonagh, a heart failure specialist and professor of cardiology at King’s College, London.
More women enrolled than ever before
EMPEROR-Preserved enrolled adults with chronic HFpEF in New York Heart Association functional class II-IV and a left ventricular ejection fraction greater than 40% starting in 2017 at more than 600 sites in more than 20 countries worldwide including the United States. As background therapy, more than 80% of patients received treatment with either an ACE inhibitor or angiotensin receptor blocker (in some instances in the form of sacubitril/valsartan), more than 80% were on a beta-blocker, and about a third were taking a mineralocorticoid receptor antagonist, making them “very well treated HFpEF patients,” Dr. Anker said.
One of the most notable features of enrollment was that 45% of participants were women, giving this trial the highest inclusion of women compared with all prior studies in patients with HFpEF or with HFrEF, said Dr. Walsh. “HFpEF is very prevalent in woman,” she noted, and having this high participation rate of women in the study increases its relevance to these patients. “It’s important to be able to tell women that patients like you were in the study so we can more easily apply the lessons from the trial to you. That can’t be stressed enough,” she said.
The primary outcome occurred in 415 (13.8%) of the 2,997 patients in the empagliflozin group and in 511 (17.1%) of 2,991 patients who received placebo (hazard ratio, 0.79; 95% confidence interval, 0.69-0.90; P < .001).
The study showed a safety profile consistent with prior experience with empagliflozin, Dr. Anker added.
Pooling EMPEROR-Preserved with EMPEROR-Reduced
The investigators who ran EMPEROR-Preserved designed the trial to closely parallel the EMPEROR-Reduced trial in patients with HFrEF, and they included a prespecified analysis (EMPEROR-Pooled) that combined the more than 9,700 patients in the two studies. This showed a consistent and robust benefit from empagliflozin for reducing HHF across a wide spectrum of patients with heart failure, ranging from patients with left ventricular ejection fractions of less than 25% to patients with ejection fractions as high as 64%. However, the analysis also showed that patients with ejection fractions of 65% or greater received no discernible benefit from empagliflozin, Milton Packer, MD, reported in a separate talk at the congress.
“The findings demonstrate the benefits of empagliflozin across a broad range of patients with heart failure who have ejection fractions of less than 60%-65%,” said Dr. Packer, a researcher at Baylor University Medical Center in Dallas.
This apparent attenuation of an effect at higher ejection fractions “has been observed in other HFpEF trials, most recently in the PARAGON-HF trial” of sacubitril/valsartan (Entresto), he noted. Additional analyses led by Dr. Packer showed that in patients with ejection fractions below 65% the HHF benefit from empagliflozin consistently surpassed the benefit seen with sacubitril/valsartan in PARAGON-HF. But he recommended using both drugs in patients with HFpEF and an ejection fraction up to about 60%.
“If I had a patient with HFpEF I would use both drugs as well as beta-blockers and mineralocorticoid receptor antagonists,” he said during a press briefing.
Another finding from analysis of the EMPEROR-Reduced and EMPEROR-Preserved trials together was that patients with reduced ejection fractions showed a significant 49% relative reduction in the incidence of serious renal outcomes, but this effect was completely blunted in EMPEROR-Preserved.
“Ejection fraction influences the effects of empagliflozin on major renal outcomes,” concluded Dr. Packer in a report on this analysis published simultaneously with the main EMPEROR-Preserved findings (N Engl J Med. 2021 Aug 27. doi: 10.1056/NEJMc2112411). “These data from the EMPEROR trials are unique. We have no comparable data” from any of the other reported studies of SGLT2 inhibitors,” he said.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and by Eli Lilly, the two companies that jointly market empagliflozin (Jardiance). Dr. Anker has received personal fees from Boehringer Ingelheim and from several other companies, and he has received grants and personal fees from Abbott Vascular and Vifor. Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. McDonagh has has recent financial relationships with AstraZeneca, Cprpus, Novartis, Pfizer, and Vifor. Dr. Aguiar and Dr. Walsh had no disclosures.
Updated August 30, 2021
The SGLT2 inhibitor empagliflozin achieved in EMPEROR-Preserved what no other agent could previously do: unequivocally cut the incidence of cardiovascular death or hospitalization in patients with heart failure and preserved ejection fraction (HFpEF).
Treatment with empagliflozin (Jardiance) led to a significant 21% relative reduction in the rate of cardiovascular death or hospitalization for heart failure (HHF), compared with placebo, among 5,988 randomized patients with HFpEF during a median 26 months of follow-up, proving that patients with HFpEF finally have a treatment that gives them clinically meaningful benefit, and paving the way to an abrupt change in management of these patients, experts said.
“This is the first trial to show unequivocal benefits of any drug on major heart failure outcomes in patients with HFpEF,” Stefan D. Anker, MD, PhD, declared at the virtual annual congress of the European Society of Cardiology.
The 21% relative reduction, which reflected a cut in the absolute rate of the trial’s primary composite endpoint of 3.3% compared with placebo, was driven mainly by a significant 27% relative reduction in the incidence of HHF (P < .001). Empagliflozin treatment, on top of standard therapy for patients with HFpEF, also resulted in a nonsignificant 9% relative risk reduction in the incidence of cardiovascular death, but it had no discernible impact on the rate of death from any cause, said Dr. Anker, professor of cardiology at Charité Medical University in Berlin.
Concurrently with his talk at the meeting, the results were published online in the New England Journal of Medicine.
Practice will change ‘quickly’
“This will definitely change our practice, and quite quickly,” said Carlos Aguiar, MD, chair of the Advanced Heart Failure and Heart Transplantation Unit at Hospital Santa Cruz in Carnaxide, Portugal, who was not involved in the study.
Transition to routine use of empagliflozin in patients with HFpEF should be swift because it has already become a mainstay of treatment for patients with heart failure with reduced ejection fraction (HFrEF) based on evidence for empagliflozin in EMPEROR-Reduced. A second sodium-glucose cotransporter 2 (SGLT2 ) inhibitor, dapagliflozin (Farxiga), is also an option for treating HFrEF based on results in the DAPA-HF trial, and the DELIVER trial, still in progress, is testing dapagliflozin as a HFpEF treatment in about 6,000 patients, with results expected in 2022.
About half of the patients in EMPEROR-Preserved had diabetes, and the treatment effects on HFpEF were similar regardless of patients’ diabetes status. Empagliflozin, like other members of the SGLT2 inhibitor class, boosts urinary excretion of glucose and received initial regulatory approval as an agent for glycemic control in patients with type 2 diabetes. Empagliflozin also has U.S.-approved marketing indications for treating patients with HFrEF whether or not they also have diabetes, and for reducing cardiovascular death in patients with type 2 diabetes and cardiovascular disease.
“We already use this drug class in cardiovascular medicine and to treat patients with type 2 diabetes, and we have been eager to find a treatment for patients with HFpEF. This is something that will be really significant,” said Dr. Aguiar.
Heart failure clinicians have “become familiar prescribing” SGLT2 inhibitors following approval of HFrEF indications for some of these agents, noted Mary Norine Walsh, MD, a heart failure specialist with Ascension Medical Group in Indianapolis. The new results “are good news because there have been so few options” for patients with HFpEF, she said in an interview.
EMPEROR-Preserved “is the first phase 3 clinical trial that exclusively enrolled patients with heart failure and an ejection fraction of more than 40% to meet its primary outcome,” and the results “represent a major win against a medical condition that had previously proven formidable,” Mark H. Drazner, MD, said in an editorial that accompanied the published results.
The trial’s findings “should contribute to a change in clinical practice given the paucity of therapeutic options available for patients with HFpEF,” wrote Dr. Drazner, a heart failure specialist who is professor and clinical chief of cardiology at UT Southwestern Medical Center in Dallas.
Theresa A, McDonagh, MD, MBChB, who chaired the panel that just released revised guidelines from the European Society of Cardiology for managing patients with heart failure, predicted that empagliflozin treatment for patients with HFpEF will soon show up in guidelines. It will likely receive a “should be considered” ranking despite being a single study because of the impressive size of the treatment effect and lack of well-supported alternative treatments, she commented as a discussant of the trial during its presentation at the congress. If the DELIVER trial with dapagliflozin shows a similar effect, the recommendation would likely become even stronger, added Dr. McDonagh, a heart failure specialist and professor of cardiology at King’s College, London.
More women enrolled than ever before
EMPEROR-Preserved enrolled adults with chronic HFpEF in New York Heart Association functional class II-IV and a left ventricular ejection fraction greater than 40% starting in 2017 at more than 600 sites in more than 20 countries worldwide including the United States. As background therapy, more than 80% of patients received treatment with either an ACE inhibitor or angiotensin receptor blocker (in some instances in the form of sacubitril/valsartan), more than 80% were on a beta-blocker, and about a third were taking a mineralocorticoid receptor antagonist, making them “very well treated HFpEF patients,” Dr. Anker said.
One of the most notable features of enrollment was that 45% of participants were women, giving this trial the highest inclusion of women compared with all prior studies in patients with HFpEF or with HFrEF, said Dr. Walsh. “HFpEF is very prevalent in woman,” she noted, and having this high participation rate of women in the study increases its relevance to these patients. “It’s important to be able to tell women that patients like you were in the study so we can more easily apply the lessons from the trial to you. That can’t be stressed enough,” she said.
The primary outcome occurred in 415 (13.8%) of the 2,997 patients in the empagliflozin group and in 511 (17.1%) of 2,991 patients who received placebo (hazard ratio, 0.79; 95% confidence interval, 0.69-0.90; P < .001).
The study showed a safety profile consistent with prior experience with empagliflozin, Dr. Anker added.
Pooling EMPEROR-Preserved with EMPEROR-Reduced
The investigators who ran EMPEROR-Preserved designed the trial to closely parallel the EMPEROR-Reduced trial in patients with HFrEF, and they included a prespecified analysis (EMPEROR-Pooled) that combined the more than 9,700 patients in the two studies. This showed a consistent and robust benefit from empagliflozin for reducing HHF across a wide spectrum of patients with heart failure, ranging from patients with left ventricular ejection fractions of less than 25% to patients with ejection fractions as high as 64%. However, the analysis also showed that patients with ejection fractions of 65% or greater received no discernible benefit from empagliflozin, Milton Packer, MD, reported in a separate talk at the congress.
“The findings demonstrate the benefits of empagliflozin across a broad range of patients with heart failure who have ejection fractions of less than 60%-65%,” said Dr. Packer, a researcher at Baylor University Medical Center in Dallas.
This apparent attenuation of an effect at higher ejection fractions “has been observed in other HFpEF trials, most recently in the PARAGON-HF trial” of sacubitril/valsartan (Entresto), he noted. Additional analyses led by Dr. Packer showed that in patients with ejection fractions below 65% the HHF benefit from empagliflozin consistently surpassed the benefit seen with sacubitril/valsartan in PARAGON-HF. But he recommended using both drugs in patients with HFpEF and an ejection fraction up to about 60%.
“If I had a patient with HFpEF I would use both drugs as well as beta-blockers and mineralocorticoid receptor antagonists,” he said during a press briefing.
Another finding from analysis of the EMPEROR-Reduced and EMPEROR-Preserved trials together was that patients with reduced ejection fractions showed a significant 49% relative reduction in the incidence of serious renal outcomes, but this effect was completely blunted in EMPEROR-Preserved.
“Ejection fraction influences the effects of empagliflozin on major renal outcomes,” concluded Dr. Packer in a report on this analysis published simultaneously with the main EMPEROR-Preserved findings (N Engl J Med. 2021 Aug 27. doi: 10.1056/NEJMc2112411). “These data from the EMPEROR trials are unique. We have no comparable data” from any of the other reported studies of SGLT2 inhibitors,” he said.
EMPEROR-Preserved was sponsored by Boehringer Ingelheim and by Eli Lilly, the two companies that jointly market empagliflozin (Jardiance). Dr. Anker has received personal fees from Boehringer Ingelheim and from several other companies, and he has received grants and personal fees from Abbott Vascular and Vifor. Dr. Packer has received consulting fees from Boehringer Ingelheim and from numerous other companies. Dr. McDonagh has has recent financial relationships with AstraZeneca, Cprpus, Novartis, Pfizer, and Vifor. Dr. Aguiar and Dr. Walsh had no disclosures.
FROM ESC CONGRESS 2021
Eyes on ESC ‘21: Hope for EMPEROR-Preserved, guidelines remade
There will be so much more to the annual congress of the European Society of Cardiology, which begins Aug. 27 with an all-virtual format, than detailed primary results of EMPEROR-Preserved, a trial that could mark a turning point for heart failure (HF) medical therapy.
Also among the featured Hot Line and Late-Breaking Science sessions are – along with many other studies – explorations of arrhythmia management (ablation or guided by loop recorder); secondary prevention, including by vaccination; oral anticoagulation, notably after transcatheter valve procedures; and colchicine or thrombosis prophylaxis in hospitalized patients with COVID-19.
There will even be a head-to-head comparison of two long-familiar left atrial appendage (LAA) occluders, and a population-based, randomized trial of sodium restriction through wide-scale use of a potassium-based salt substitute.
The congress will also introduce four guideline documents at sessions throughout the Congress, one on each day. They cover new and modified recommendations for heart failure; pacing, including cardiac resynchronization therapy (CRT); cardiovascular (CV) disease prevention; and, with cosponsorship from the European Association for Cardio-Thoracic Surgery, valvular heart disease.
The virtues of virtual
That next year’s Congress is slated for Aug. 27-30 in Barcelona should be welcome news for anyone whose “what if” curiosity about all-virtual conferences has already been satisfied. But with experience comes wisdom, as the medical societies have learned that online scientific meetings have some winning qualities that may be worth keeping, as least for a while.
“I think there is no doubt that the digital format will continue, for several reasons. One is that this pandemic is not over,” ESC Congress program committee chair Stephan Windecker, MD, Bern (Switzerland) University Hospital, , told this news organization. “As long as it is not over, the digital format is here to stay.”
But it also appears that people who haven’t been able to attend the congress in person are keen to log in and engage online, Dr. Windecker said. The 2020 all-virtual conference drew a much younger pool of registrants, on average, than did the live conferences before the pandemic.
“I think that’s an indication of people that may be in training, in early stages of their career, or they don’t have the support from departments or from their practice, or other financial means.” But they are able to participate via computer, tablet, or smartphone, he said.
“Another advantage is that the recorded content can be replayed at the convenience of whoever wants to consume it at a later point in time,” he added. “Those are just some examples why the digital format is likely to stay,” on its own or in a new age of hybrid meetings.
New and updated guidelines
Leading off the guideline series is the document on diagnosis and treatment of acute and chronic HF, which leveraged the past few busy years of HF clinical trials to arrive at a number of new recommendations and strengthened level-of-evidence ratings. It covers both drug and device therapy of HF with reduced ejection fraction (HFrEF) and acute decompensated HF, and tweaks and further enshrines the concept of HF with mildly reduced ejection fraction (HFmrEF).
Several updated recommendations for both long-used and novel medications, notably the sodium-glucose cotransporter 2 inhibitors, will be included because of the recently appreciated evidence-based impact in HFrEF, Dr. Windecker noted.
“I think it will be particularly interesting to look for the SGLT2 inhibitors as not a completely new class of drugs, but certainly one where there has been a lot of new evidence, to look at how those drugs will be integrated in the overall care pathway.”
A top-line preview of the new HF guideline limited to drug therapy, presented at July’s Heart Failure Association of the European Society of Cardiology (ESC-HFA), provided a simple answer to a common question in the new, bountiful age of HFrEF medications: Which meds, initiated in what order?
As it happens, the new recommendation for first-line HFrEF drug therapy is not a silver bullet, but a shotgun – prompt initiation of at least four meds, one from each of four drug classes: renin-angiotensin system inhibitors, beta-blockers, mineralocorticoid receptor antagonists (MRA), and SGLT2 inhibitors. Each class, as described in the document, is to be started as soon as safely feasible, in a sequence deemed appropriate for each individual patient.
Spotlight on EMPEROR-Preserved
The world already knows that the trial, which tested the SGLT2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) on top of standard therapy, “met” its primary endpoint in almost 6,000 patients with HF with preserved ejection fraction (HFpEF), who included some with HFmrEF by more contemporary definitions.
That means patients in EMPEROR-Preserved assigned to take empagliflozin showed significantly fewer events that made up the study’s primary endpoint, a composite of CV death or HF hospitalization. It appears to be the first clearly significant overall medical therapy benefit for a clinical primary endpoint in a major randomized HFpEF drug trial.
And that, pending fuller presentation of trial results at the Congress on Aug. 27, could be a huge deal for the half of HF patients with left ventricular ejection fractions (LVEF) higher than the HFrEF range.
Those early top-line results weren’t a decisive bombshell for a field now filled with hope for a practice-changing empagliflozin outcome in EMPEROR-Preserved, which isn’t a certainty. They were more like the “boom” of a mortar launching a rocket of fireworks that may explode into a chrysanthemum or green comet or, sometimes, turn out to be no more than a dud. The promise of the early cursory results critically depends on further details.

“Provided there is a compelling benefit, this is what everyone has been waiting for in this condition for decades,” Mikhail N. Kosiborod, MD, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo., said.
“Already knowing that the trial met the primary endpoint is obviously very intriguing and encouraging,” he added. “But there are things we don’t know, such as: What is the magnitude of benefit? And whether that benefit, whatever the magnitude, is driven by reductions in both heart failure hospitalizations and cardiovascular death, or only one of the two.”
For example: “If we see an impressive benefit for reduction of hospitalizations, but not a significant reduction in death, that would still be a huge advance. That’s because, to date, we don’t have any drug for HFpEF that has convincingly demonstrated a compelling reduction in heart failure hospitalization or improvement in symptoms, function, or quality of life,” observed Dr. Kosiborod, who wasn’t part of EMPEROR-Preserved.
There have been “suggestions” from HFrEF trials that empagliflozin and dapagliflozin (Farxiga, AstraZeneca) “have very comparable effects on at least the endpoint of cardiovascular death or hospitalization for heart failure,” he said. “So, my expectation would be that whatever is observed in EMPEROR-Preserved is likely a class effect, as well.”
Following EMPEROR-Preserved on the agenda is EMPEROR-Pooled, a patient-level combined analysis of the EMPEROR series of trials that spans the range of HF, regardless of ejection fraction or diabetes status, primarily exploring the effects of empagliflozin on renal function.
Other offerings, Friday, Aug. 27
Scheduled immediately after EMPEROR-Preserved is a presentation on the SMART-MI trial, which should clarify whether management guided by continuous ambulatory monitoring is effective in patients considered at especially high arrhythmic risk. Entry called for recent myocardial infarction and an LVEF of 36%-50% with evidence of cardiac autonomic dysfunction.
The trial randomly assigned 400 such patients to be or not be implanted with a Reveal LINQ (Medtronic) loop recorder and followed them for up to 18 months, primarily for detection of potentially serious arrhythmic events. Endpoints that involved mortality, hospitalization or other clinical events were secondary.
In a time slot preceding both SMART-MI and EMPEROR-Preserved, the GUIDE-HF trial is following a projected 3,600 patients with HF implanted with a CardioMEMS HF System (Abbott) pulmonary artery (PA) pressure sensor to explore the its value for guiding management.
The trial’s three cohorts, followed for at least 12 months, include randomized sensor-monitored and control groups of patients with New York Heart Association class 2-4 symptoms, as well as a third observational set of patients in NYHA class 3. That’s the indication for which the CardioMEMS monitor gained approval in the United States in 2014 based on the 2011 CHAMPION trial, and which fared just as well in the 2017 CHAMPION Post-Approval Study.
The Friday Hot Lines also include Dal-GenE, which has entered about 6,000 patients with recent MI to test the once-abandoned cholesterol ester transfer protein (CETP) inhibitor dalcetrapib (DalCor) for any secondary-prevention benefits when used selectively. The trial’s hook: All its patients are confirmed to have the AA genotype of the rs1967309 variant in the ADCY9 gene, which has been associated with a pronounced clinical response to CETP inhibition.
Saturday, Aug. 28
The direct oral anticoagulants (DOACs) have largely replaced vitamin K antagonists in patients with nonvalvular atrial fibrillation (AFib). But whether DOACs are similarly preferable in the growing world population of people who have undergone transcatheter aortic valve replacement (TAVR or TAVI), an issue explored with variable results in the ATLANTIS and GALILEO trials, is far from settled.
The ENVISAGE-TAVI AF trial explored the question for the factor X inhibitor edoxaban (Savaysa, Lixiana, Daiichi-Sankyo) in 1,400 patients with AFib and a transfemoral TAVR in the previous 5 days, who were randomly assigned to the DOAC or standard management along with discretionary antiplatelet therapy. They’ve been followed for up to 3 years for a composite endpoint of clinical events – including death, MI, and stroke – and for major bleeding.
The day will also feature MASTER DAPT, a comparison of two dual-antiplatelet therapy (DAPT) regimens in an estimated 4,300 patients considered to be high-risk for bleeding who had received the sirolimus-eluting Ultimaster (Terumo) coronary stent, which has a bioresorbable polymer coating.
Investigators have randomly assigned patients to receive either very-short-duration DAPT, for about a month after stenting, followed by a P2Y12 inhibitor alone for up to a year after the procedure; or a more conventional regimen of a P2Y12 inhibitor for 6-12 months with aspirin maintained for a total of 12 months.
Later that day, investigators from the FIGARO-DKD trial will present their results based on 7,437 patients with type 2 diabetes and chronic kidney disease (CKD), a much fuller version than the top-line findings announced by sponsor Bayer 3 months ago.
Those top-line results suggested that patients assigned to receive the nonsteroidal nonselective mineralocorticoid receptor antagonist (MRA) finerenone (Kerendia) on top of standard care benefited with a drop in risk for the primary endpoint of CV death or nonfatal CV events.
Finerenone was recently approved in the United States for treating patients with both type 2 diabetes and CKD based on the published FIDELIO-DKD trial, which had seen less CKD progression and fewer CV events in such patients who took the novel MRA.
Although similar in design to FIGARO-DKD, FIDELIO-DKD had entered fewer patients with early-stage diabetic kidney disease (DKD). That led researchers to pool the two trials’ populations to create a cohort that spans the spectrum of DKD severity. An analysis of the pooled cohort, dubbed FIDELITY, is on the schedule after FIGARO-DKD.
After FIDELITY is the prospective APAF-CRT trial that is following a projected 1,830 patients with permanent, symptomatic AFib and a recent hospitalization for AFib or HF and who were not good candidates for standard ablation. They were assigned to receive either atrioventricular junctional ablation followed by CRT, with or without a defibrillation, on top of optimal meds – a so-called “ablate-and-pace” strategy – or an implantable cardioverter defibrillator with rate-control drug therapy.
The new analysis represents the trial’s second phase in which mortality was followed for 4 years as the primary endpoint, in contrast to the previously reported initial phase that followed the first 102 patients for 2 years for the composite primary endpoint of death, worsening HF, and HF hospitalization. The first phase had halted enrollment before reaching its planned target of 280 patients after an interim analysis showed a significant benefit for ablate and pace.
Next up: DECAAF 2, a randomized assessment of whether catheter ablation for AFib guided by delayed gadolinium enhancement on MRI, a proxy for scar tissue, can be more effective than standard AFib ablation by pulmonary vein isolation alone. An estimated 900 patients with persistent AFib who had never before undergone ablation for the arrhythmia were randomly assigned to one strategy or the other and followed for AFib recurrence over 18 months.
Sunday, Aug. 29
The TOMAHAWK trial aimed to clarify the optimal timing of invasive coronary angiography for resuscitated patients with non–ST-segment elevation out-of-hospital cardiac arrest, a broad population in a setting for which there is little randomized-trial guidance. Investigators randomly assigned 558 such patients to undergo immediate invasive angiography or to direct intensive care unit admission for initial standard care with discretionary delayed angiography. Patients were followed for all-cause mortality, with other clinical events and neurologic outcomes as secondary endpoints.
Next on the schedule, the RIPCORD-2 trial randomly assigned 1,100 patients with stable known or suspected coronary artery disease (CAD) to undergo conventional angiography alone or with added direct pressure-wire measurement of fractional flow reserve to guide management decisions. Primary outcomes include health care costs and patient-reported quality of life at 1 year.
Slated for later that day, the Asymptomatic Carotid Surgery Trial-2 (ACST-2) has entered an estimated 3600 patients with a substantial carotid artery narrowing not associated with symptoms but for which either carotid endarterectomy (CEA) or carotid artery stenting (CAS) was considered anatomically feasible. There also must have been “substantial uncertainty” regarding the optimal procedure choice.
The trial, conducted in 40 countries primarily in Europe and North America and launched in 2008, randomly assigned the patients to undergo either CEA or CAS, in both cases with appropriate medical therapy, and followed them for periprocedural events and up to 10 years for strokes and stroke-related events.
The LOOP study, which is to directly follow ACST-2, has explored whether screening for AFib using the Medtronic Reveal LINQ monitor in older patients with non-AFib stroke risk factors – with oral anticoagulation prescribed for those who test positive – can lower their risk for stroke or systemic embolism. It randomly assigned 6,000 such patients to care guided by the loop recorder or to standard care.
On a somewhat larger scale, the Salt Substitute and Stroke Study (SSaSS) randomly assigned a total of 20,996 people in about 600 villages across northern China and Tibet to sodium-restriction intervention and control groups by village. All participants had a history of stroke or were aged at least 60 years with uncontrolled hypertension.
As described by the trial’s online portal, participants in villages assigned to the intervention group were given a supply of a low-sodium, potassium-supplementing salt substitute to replace their own salt supplies, along with education on the health benefits of sodium restriction. Participants in control villages continued their normal diets and, at the trial’s beginning, received “advice to reduce their salt intake.” All were required to own a telephone.
Clinical events, including strokes and hospitalizations throughout a 5-year follow-up, were tracked by phone calls made to all participants every 6 months and were documented at follow-up home visits.
Sunday is also to feature a Late-Breaking Trials session with a focus on COVID-19, which leads off with COLCOVID, a test of colchicine in patients hospitalized for suspected SARS-CoV-2 infection and in acute respiratory distress.
The 1,279 participants in Argentina were randomly assigned to receive or not receive the potent anti-inflammatory agent on top of antivirals and other standard management and followed for death or new need for mechanical ventilation. A successful outcome would contrast with the RECOVERY trial, which terminated a colchicine group of patients hospitalized with COVID-19 because of a lack of efficacy earlier this year.
COLCOVID is to be followed by the MICHELLE trial of rivaroxaban (Xarelto, Bayer/Janssen) prophylaxis, compared with no preventive oral anticoagulant, in 320 patients who, when hospitalized with COVID-19, had been on parenteral anticoagulants because of an elevated risk for venous thromboembolism. The trial, conducted in Brazil, called for postdischarge rivaroxaban at a once-daily dosage of 10 mg for about 1 month.
The session also includes a presentation called “Insights into the Effects of the COVID-19 Pandemic: Comprehensive Analysis from the GUIDE-HF Trial,” the primary outcomes of which will be reported on the first day of the Congress.
Following is a presentation on the PREPARE-IT study of icosapent ethyl (Vascepa, Amarin), given at high dosages intended to be anti-inflammatory, compared with placebo, in an estimated 4,000 adults. The trial has two groups: A prevention group of adults living and circulating in the community; and a treatment group of patients aged at least 40 years with confirmed symptomatic SARS-CoV-2 infection for whom the need for hospitalization isn’t clear.
Monday, Aug. 30
The final day of the Congress features a trial called Influenza Vaccination after Myocardial Infarction (IAMI), which has tested the secondary preventive effect of influenza vaccination by randomly assigning 2,571 patients to receive a standard vaccine or a saline placebo injection on one occasion.
Entry to the international trial called for a diagnosis of MI with or without ST-segment elevation, or stable CAD and age at least 75 years with other risk factors. The patients were followed for death, MI, stent thrombosis, and a slew of secondary endpoints over 12 months.
Monday offerings continue later in a time block leading off with the STEP trial, which has randomly assigned an estimated 8,000 patients at 40 centers in China who are 60 to 80 years of age with a systolic blood pressure of 140 to <190 mm Hg to be on standard guideline-based therapy or an intensive drug-management strategy.
The systolic BP goals are 130 to <150 mm Hg for standard care and 110 to <130 mm Hg for the intensive regimen. The composite primary endpoint includes death and clinical events related to acute coronary syndromes, HF, revascularization, and stroke.
Following on heels of STEP, the Amulet IDE trial – the first major randomized comparison of two transcatheter LAA closure devices – entered 1,878 patients with nonvalvular AFib who were considered high-risk for bleeding and stroke or systemic embolism.
They were randomly assigned in the noninferiority trial to receive either the AMPLATZER Amulet (Abbott Medical Devices) or the WATCHMAN (Boston Scientific) closure devices and were followed for safety and efficacy for up to 5 years.
Both LAA closure devices, intended to make patients with AFib less reliant on oral anticoagulation, are now available on both sides of the Atlantic – as well as many other countries – after the Amulet’s United States market approval on Aug. 16, based largely on the Amulet IDE trial.
Rounding out the final Hot Line set is one of the latest efforts to show the efficacy and safety of a very short DAPT period after coronary stenting in patients with acute coronary syndromes, the STOPDAPT-2 ACS trial.
The study assigned 3,008 patients in Japan to receive aspirin and clopidogrel for either 1 month or 1 year after implantation with an everolimus-eluting cobalt-chromium stent and followed them for up to 5 years for a composite of MI, CV death, stent thrombosis, stroke, and bleeding.
The trial follows the published STOPDAPT-2 trial that showed superiority for the 1-month DAPT regimen in a predominantly stable-CAD population treated with the same kind of stent.
Program structure and format
A total of 15 online channels are to be available in the morning, European time, their schedules running in parallel. Presentations often are prerecorded, but also include live sessions at 8:00 a.m. Central time and 12 p.m. CET (2:00 a.m. and 6:00 a.m. Eastern time) to liven up the channel offerings, Dr. Windecker observed, and to make them more immediate and potentially interactive.
Many of the parallel channels are devoted throughout the Congress to particular silos of cardiology; for example, arrhythmias and device therapy is on channel 3; CAD and acute care is on 5; HF is on 6; and preventive cardiology is on 9.
Other channels swing across different topics from day to day, such as channel 1, which covers COVID-19 topics on the first and third day of the meeting, “advances in science” on day 2, and “digital health, public health, health economics” on day 4.
The focus each day, starting at 2:00 p.m. CET (8:00 a.m. ET) and continuing into the evening in Europe, shifts over to the Prime Time live program, which features the Hot Line and guideline presentations and many of the live abstract presentations.
Dr. Kosiborod, not a researcher with the EMPEROR trials, is chair of the Dapagliflozin in Preserved Ejection Fraction Heart Failure ( PRESERVED-HF ) trial, which is scheduled for presentation at the September 2021 Heart Failure Society of American meeting.
A version of this article first appeared on Medscape.com.
There will be so much more to the annual congress of the European Society of Cardiology, which begins Aug. 27 with an all-virtual format, than detailed primary results of EMPEROR-Preserved, a trial that could mark a turning point for heart failure (HF) medical therapy.
Also among the featured Hot Line and Late-Breaking Science sessions are – along with many other studies – explorations of arrhythmia management (ablation or guided by loop recorder); secondary prevention, including by vaccination; oral anticoagulation, notably after transcatheter valve procedures; and colchicine or thrombosis prophylaxis in hospitalized patients with COVID-19.
There will even be a head-to-head comparison of two long-familiar left atrial appendage (LAA) occluders, and a population-based, randomized trial of sodium restriction through wide-scale use of a potassium-based salt substitute.
The congress will also introduce four guideline documents at sessions throughout the Congress, one on each day. They cover new and modified recommendations for heart failure; pacing, including cardiac resynchronization therapy (CRT); cardiovascular (CV) disease prevention; and, with cosponsorship from the European Association for Cardio-Thoracic Surgery, valvular heart disease.
The virtues of virtual
That next year’s Congress is slated for Aug. 27-30 in Barcelona should be welcome news for anyone whose “what if” curiosity about all-virtual conferences has already been satisfied. But with experience comes wisdom, as the medical societies have learned that online scientific meetings have some winning qualities that may be worth keeping, as least for a while.
“I think there is no doubt that the digital format will continue, for several reasons. One is that this pandemic is not over,” ESC Congress program committee chair Stephan Windecker, MD, Bern (Switzerland) University Hospital, , told this news organization. “As long as it is not over, the digital format is here to stay.”
But it also appears that people who haven’t been able to attend the congress in person are keen to log in and engage online, Dr. Windecker said. The 2020 all-virtual conference drew a much younger pool of registrants, on average, than did the live conferences before the pandemic.
“I think that’s an indication of people that may be in training, in early stages of their career, or they don’t have the support from departments or from their practice, or other financial means.” But they are able to participate via computer, tablet, or smartphone, he said.
“Another advantage is that the recorded content can be replayed at the convenience of whoever wants to consume it at a later point in time,” he added. “Those are just some examples why the digital format is likely to stay,” on its own or in a new age of hybrid meetings.
New and updated guidelines
Leading off the guideline series is the document on diagnosis and treatment of acute and chronic HF, which leveraged the past few busy years of HF clinical trials to arrive at a number of new recommendations and strengthened level-of-evidence ratings. It covers both drug and device therapy of HF with reduced ejection fraction (HFrEF) and acute decompensated HF, and tweaks and further enshrines the concept of HF with mildly reduced ejection fraction (HFmrEF).
Several updated recommendations for both long-used and novel medications, notably the sodium-glucose cotransporter 2 inhibitors, will be included because of the recently appreciated evidence-based impact in HFrEF, Dr. Windecker noted.
“I think it will be particularly interesting to look for the SGLT2 inhibitors as not a completely new class of drugs, but certainly one where there has been a lot of new evidence, to look at how those drugs will be integrated in the overall care pathway.”
A top-line preview of the new HF guideline limited to drug therapy, presented at July’s Heart Failure Association of the European Society of Cardiology (ESC-HFA), provided a simple answer to a common question in the new, bountiful age of HFrEF medications: Which meds, initiated in what order?
As it happens, the new recommendation for first-line HFrEF drug therapy is not a silver bullet, but a shotgun – prompt initiation of at least four meds, one from each of four drug classes: renin-angiotensin system inhibitors, beta-blockers, mineralocorticoid receptor antagonists (MRA), and SGLT2 inhibitors. Each class, as described in the document, is to be started as soon as safely feasible, in a sequence deemed appropriate for each individual patient.
Spotlight on EMPEROR-Preserved
The world already knows that the trial, which tested the SGLT2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) on top of standard therapy, “met” its primary endpoint in almost 6,000 patients with HF with preserved ejection fraction (HFpEF), who included some with HFmrEF by more contemporary definitions.
That means patients in EMPEROR-Preserved assigned to take empagliflozin showed significantly fewer events that made up the study’s primary endpoint, a composite of CV death or HF hospitalization. It appears to be the first clearly significant overall medical therapy benefit for a clinical primary endpoint in a major randomized HFpEF drug trial.
And that, pending fuller presentation of trial results at the Congress on Aug. 27, could be a huge deal for the half of HF patients with left ventricular ejection fractions (LVEF) higher than the HFrEF range.
Those early top-line results weren’t a decisive bombshell for a field now filled with hope for a practice-changing empagliflozin outcome in EMPEROR-Preserved, which isn’t a certainty. They were more like the “boom” of a mortar launching a rocket of fireworks that may explode into a chrysanthemum or green comet or, sometimes, turn out to be no more than a dud. The promise of the early cursory results critically depends on further details.
“Provided there is a compelling benefit, this is what everyone has been waiting for in this condition for decades,” Mikhail N. Kosiborod, MD, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo., said.
“Already knowing that the trial met the primary endpoint is obviously very intriguing and encouraging,” he added. “But there are things we don’t know, such as: What is the magnitude of benefit? And whether that benefit, whatever the magnitude, is driven by reductions in both heart failure hospitalizations and cardiovascular death, or only one of the two.”
For example: “If we see an impressive benefit for reduction of hospitalizations, but not a significant reduction in death, that would still be a huge advance. That’s because, to date, we don’t have any drug for HFpEF that has convincingly demonstrated a compelling reduction in heart failure hospitalization or improvement in symptoms, function, or quality of life,” observed Dr. Kosiborod, who wasn’t part of EMPEROR-Preserved.
There have been “suggestions” from HFrEF trials that empagliflozin and dapagliflozin (Farxiga, AstraZeneca) “have very comparable effects on at least the endpoint of cardiovascular death or hospitalization for heart failure,” he said. “So, my expectation would be that whatever is observed in EMPEROR-Preserved is likely a class effect, as well.”
Following EMPEROR-Preserved on the agenda is EMPEROR-Pooled, a patient-level combined analysis of the EMPEROR series of trials that spans the range of HF, regardless of ejection fraction or diabetes status, primarily exploring the effects of empagliflozin on renal function.
Other offerings, Friday, Aug. 27
Scheduled immediately after EMPEROR-Preserved is a presentation on the SMART-MI trial, which should clarify whether management guided by continuous ambulatory monitoring is effective in patients considered at especially high arrhythmic risk. Entry called for recent myocardial infarction and an LVEF of 36%-50% with evidence of cardiac autonomic dysfunction.
The trial randomly assigned 400 such patients to be or not be implanted with a Reveal LINQ (Medtronic) loop recorder and followed them for up to 18 months, primarily for detection of potentially serious arrhythmic events. Endpoints that involved mortality, hospitalization or other clinical events were secondary.
In a time slot preceding both SMART-MI and EMPEROR-Preserved, the GUIDE-HF trial is following a projected 3,600 patients with HF implanted with a CardioMEMS HF System (Abbott) pulmonary artery (PA) pressure sensor to explore the its value for guiding management.
The trial’s three cohorts, followed for at least 12 months, include randomized sensor-monitored and control groups of patients with New York Heart Association class 2-4 symptoms, as well as a third observational set of patients in NYHA class 3. That’s the indication for which the CardioMEMS monitor gained approval in the United States in 2014 based on the 2011 CHAMPION trial, and which fared just as well in the 2017 CHAMPION Post-Approval Study.
The Friday Hot Lines also include Dal-GenE, which has entered about 6,000 patients with recent MI to test the once-abandoned cholesterol ester transfer protein (CETP) inhibitor dalcetrapib (DalCor) for any secondary-prevention benefits when used selectively. The trial’s hook: All its patients are confirmed to have the AA genotype of the rs1967309 variant in the ADCY9 gene, which has been associated with a pronounced clinical response to CETP inhibition.
Saturday, Aug. 28
The direct oral anticoagulants (DOACs) have largely replaced vitamin K antagonists in patients with nonvalvular atrial fibrillation (AFib). But whether DOACs are similarly preferable in the growing world population of people who have undergone transcatheter aortic valve replacement (TAVR or TAVI), an issue explored with variable results in the ATLANTIS and GALILEO trials, is far from settled.
The ENVISAGE-TAVI AF trial explored the question for the factor X inhibitor edoxaban (Savaysa, Lixiana, Daiichi-Sankyo) in 1,400 patients with AFib and a transfemoral TAVR in the previous 5 days, who were randomly assigned to the DOAC or standard management along with discretionary antiplatelet therapy. They’ve been followed for up to 3 years for a composite endpoint of clinical events – including death, MI, and stroke – and for major bleeding.
The day will also feature MASTER DAPT, a comparison of two dual-antiplatelet therapy (DAPT) regimens in an estimated 4,300 patients considered to be high-risk for bleeding who had received the sirolimus-eluting Ultimaster (Terumo) coronary stent, which has a bioresorbable polymer coating.
Investigators have randomly assigned patients to receive either very-short-duration DAPT, for about a month after stenting, followed by a P2Y12 inhibitor alone for up to a year after the procedure; or a more conventional regimen of a P2Y12 inhibitor for 6-12 months with aspirin maintained for a total of 12 months.
Later that day, investigators from the FIGARO-DKD trial will present their results based on 7,437 patients with type 2 diabetes and chronic kidney disease (CKD), a much fuller version than the top-line findings announced by sponsor Bayer 3 months ago.
Those top-line results suggested that patients assigned to receive the nonsteroidal nonselective mineralocorticoid receptor antagonist (MRA) finerenone (Kerendia) on top of standard care benefited with a drop in risk for the primary endpoint of CV death or nonfatal CV events.
Finerenone was recently approved in the United States for treating patients with both type 2 diabetes and CKD based on the published FIDELIO-DKD trial, which had seen less CKD progression and fewer CV events in such patients who took the novel MRA.
Although similar in design to FIGARO-DKD, FIDELIO-DKD had entered fewer patients with early-stage diabetic kidney disease (DKD). That led researchers to pool the two trials’ populations to create a cohort that spans the spectrum of DKD severity. An analysis of the pooled cohort, dubbed FIDELITY, is on the schedule after FIGARO-DKD.
After FIDELITY is the prospective APAF-CRT trial that is following a projected 1,830 patients with permanent, symptomatic AFib and a recent hospitalization for AFib or HF and who were not good candidates for standard ablation. They were assigned to receive either atrioventricular junctional ablation followed by CRT, with or without a defibrillation, on top of optimal meds – a so-called “ablate-and-pace” strategy – or an implantable cardioverter defibrillator with rate-control drug therapy.
The new analysis represents the trial’s second phase in which mortality was followed for 4 years as the primary endpoint, in contrast to the previously reported initial phase that followed the first 102 patients for 2 years for the composite primary endpoint of death, worsening HF, and HF hospitalization. The first phase had halted enrollment before reaching its planned target of 280 patients after an interim analysis showed a significant benefit for ablate and pace.
Next up: DECAAF 2, a randomized assessment of whether catheter ablation for AFib guided by delayed gadolinium enhancement on MRI, a proxy for scar tissue, can be more effective than standard AFib ablation by pulmonary vein isolation alone. An estimated 900 patients with persistent AFib who had never before undergone ablation for the arrhythmia were randomly assigned to one strategy or the other and followed for AFib recurrence over 18 months.
Sunday, Aug. 29
The TOMAHAWK trial aimed to clarify the optimal timing of invasive coronary angiography for resuscitated patients with non–ST-segment elevation out-of-hospital cardiac arrest, a broad population in a setting for which there is little randomized-trial guidance. Investigators randomly assigned 558 such patients to undergo immediate invasive angiography or to direct intensive care unit admission for initial standard care with discretionary delayed angiography. Patients were followed for all-cause mortality, with other clinical events and neurologic outcomes as secondary endpoints.
Next on the schedule, the RIPCORD-2 trial randomly assigned 1,100 patients with stable known or suspected coronary artery disease (CAD) to undergo conventional angiography alone or with added direct pressure-wire measurement of fractional flow reserve to guide management decisions. Primary outcomes include health care costs and patient-reported quality of life at 1 year.
Slated for later that day, the Asymptomatic Carotid Surgery Trial-2 (ACST-2) has entered an estimated 3600 patients with a substantial carotid artery narrowing not associated with symptoms but for which either carotid endarterectomy (CEA) or carotid artery stenting (CAS) was considered anatomically feasible. There also must have been “substantial uncertainty” regarding the optimal procedure choice.
The trial, conducted in 40 countries primarily in Europe and North America and launched in 2008, randomly assigned the patients to undergo either CEA or CAS, in both cases with appropriate medical therapy, and followed them for periprocedural events and up to 10 years for strokes and stroke-related events.
The LOOP study, which is to directly follow ACST-2, has explored whether screening for AFib using the Medtronic Reveal LINQ monitor in older patients with non-AFib stroke risk factors – with oral anticoagulation prescribed for those who test positive – can lower their risk for stroke or systemic embolism. It randomly assigned 6,000 such patients to care guided by the loop recorder or to standard care.
On a somewhat larger scale, the Salt Substitute and Stroke Study (SSaSS) randomly assigned a total of 20,996 people in about 600 villages across northern China and Tibet to sodium-restriction intervention and control groups by village. All participants had a history of stroke or were aged at least 60 years with uncontrolled hypertension.
As described by the trial’s online portal, participants in villages assigned to the intervention group were given a supply of a low-sodium, potassium-supplementing salt substitute to replace their own salt supplies, along with education on the health benefits of sodium restriction. Participants in control villages continued their normal diets and, at the trial’s beginning, received “advice to reduce their salt intake.” All were required to own a telephone.
Clinical events, including strokes and hospitalizations throughout a 5-year follow-up, were tracked by phone calls made to all participants every 6 months and were documented at follow-up home visits.
Sunday is also to feature a Late-Breaking Trials session with a focus on COVID-19, which leads off with COLCOVID, a test of colchicine in patients hospitalized for suspected SARS-CoV-2 infection and in acute respiratory distress.
The 1,279 participants in Argentina were randomly assigned to receive or not receive the potent anti-inflammatory agent on top of antivirals and other standard management and followed for death or new need for mechanical ventilation. A successful outcome would contrast with the RECOVERY trial, which terminated a colchicine group of patients hospitalized with COVID-19 because of a lack of efficacy earlier this year.
COLCOVID is to be followed by the MICHELLE trial of rivaroxaban (Xarelto, Bayer/Janssen) prophylaxis, compared with no preventive oral anticoagulant, in 320 patients who, when hospitalized with COVID-19, had been on parenteral anticoagulants because of an elevated risk for venous thromboembolism. The trial, conducted in Brazil, called for postdischarge rivaroxaban at a once-daily dosage of 10 mg for about 1 month.
The session also includes a presentation called “Insights into the Effects of the COVID-19 Pandemic: Comprehensive Analysis from the GUIDE-HF Trial,” the primary outcomes of which will be reported on the first day of the Congress.
Following is a presentation on the PREPARE-IT study of icosapent ethyl (Vascepa, Amarin), given at high dosages intended to be anti-inflammatory, compared with placebo, in an estimated 4,000 adults. The trial has two groups: A prevention group of adults living and circulating in the community; and a treatment group of patients aged at least 40 years with confirmed symptomatic SARS-CoV-2 infection for whom the need for hospitalization isn’t clear.
Monday, Aug. 30
The final day of the Congress features a trial called Influenza Vaccination after Myocardial Infarction (IAMI), which has tested the secondary preventive effect of influenza vaccination by randomly assigning 2,571 patients to receive a standard vaccine or a saline placebo injection on one occasion.
Entry to the international trial called for a diagnosis of MI with or without ST-segment elevation, or stable CAD and age at least 75 years with other risk factors. The patients were followed for death, MI, stent thrombosis, and a slew of secondary endpoints over 12 months.
Monday offerings continue later in a time block leading off with the STEP trial, which has randomly assigned an estimated 8,000 patients at 40 centers in China who are 60 to 80 years of age with a systolic blood pressure of 140 to <190 mm Hg to be on standard guideline-based therapy or an intensive drug-management strategy.
The systolic BP goals are 130 to <150 mm Hg for standard care and 110 to <130 mm Hg for the intensive regimen. The composite primary endpoint includes death and clinical events related to acute coronary syndromes, HF, revascularization, and stroke.
Following on heels of STEP, the Amulet IDE trial – the first major randomized comparison of two transcatheter LAA closure devices – entered 1,878 patients with nonvalvular AFib who were considered high-risk for bleeding and stroke or systemic embolism.
They were randomly assigned in the noninferiority trial to receive either the AMPLATZER Amulet (Abbott Medical Devices) or the WATCHMAN (Boston Scientific) closure devices and were followed for safety and efficacy for up to 5 years.
Both LAA closure devices, intended to make patients with AFib less reliant on oral anticoagulation, are now available on both sides of the Atlantic – as well as many other countries – after the Amulet’s United States market approval on Aug. 16, based largely on the Amulet IDE trial.
Rounding out the final Hot Line set is one of the latest efforts to show the efficacy and safety of a very short DAPT period after coronary stenting in patients with acute coronary syndromes, the STOPDAPT-2 ACS trial.
The study assigned 3,008 patients in Japan to receive aspirin and clopidogrel for either 1 month or 1 year after implantation with an everolimus-eluting cobalt-chromium stent and followed them for up to 5 years for a composite of MI, CV death, stent thrombosis, stroke, and bleeding.
The trial follows the published STOPDAPT-2 trial that showed superiority for the 1-month DAPT regimen in a predominantly stable-CAD population treated with the same kind of stent.
Program structure and format
A total of 15 online channels are to be available in the morning, European time, their schedules running in parallel. Presentations often are prerecorded, but also include live sessions at 8:00 a.m. Central time and 12 p.m. CET (2:00 a.m. and 6:00 a.m. Eastern time) to liven up the channel offerings, Dr. Windecker observed, and to make them more immediate and potentially interactive.
Many of the parallel channels are devoted throughout the Congress to particular silos of cardiology; for example, arrhythmias and device therapy is on channel 3; CAD and acute care is on 5; HF is on 6; and preventive cardiology is on 9.
Other channels swing across different topics from day to day, such as channel 1, which covers COVID-19 topics on the first and third day of the meeting, “advances in science” on day 2, and “digital health, public health, health economics” on day 4.
The focus each day, starting at 2:00 p.m. CET (8:00 a.m. ET) and continuing into the evening in Europe, shifts over to the Prime Time live program, which features the Hot Line and guideline presentations and many of the live abstract presentations.
Dr. Kosiborod, not a researcher with the EMPEROR trials, is chair of the Dapagliflozin in Preserved Ejection Fraction Heart Failure ( PRESERVED-HF ) trial, which is scheduled for presentation at the September 2021 Heart Failure Society of American meeting.
A version of this article first appeared on Medscape.com.
There will be so much more to the annual congress of the European Society of Cardiology, which begins Aug. 27 with an all-virtual format, than detailed primary results of EMPEROR-Preserved, a trial that could mark a turning point for heart failure (HF) medical therapy.
Also among the featured Hot Line and Late-Breaking Science sessions are – along with many other studies – explorations of arrhythmia management (ablation or guided by loop recorder); secondary prevention, including by vaccination; oral anticoagulation, notably after transcatheter valve procedures; and colchicine or thrombosis prophylaxis in hospitalized patients with COVID-19.
There will even be a head-to-head comparison of two long-familiar left atrial appendage (LAA) occluders, and a population-based, randomized trial of sodium restriction through wide-scale use of a potassium-based salt substitute.
The congress will also introduce four guideline documents at sessions throughout the Congress, one on each day. They cover new and modified recommendations for heart failure; pacing, including cardiac resynchronization therapy (CRT); cardiovascular (CV) disease prevention; and, with cosponsorship from the European Association for Cardio-Thoracic Surgery, valvular heart disease.
The virtues of virtual
That next year’s Congress is slated for Aug. 27-30 in Barcelona should be welcome news for anyone whose “what if” curiosity about all-virtual conferences has already been satisfied. But with experience comes wisdom, as the medical societies have learned that online scientific meetings have some winning qualities that may be worth keeping, as least for a while.
“I think there is no doubt that the digital format will continue, for several reasons. One is that this pandemic is not over,” ESC Congress program committee chair Stephan Windecker, MD, Bern (Switzerland) University Hospital, , told this news organization. “As long as it is not over, the digital format is here to stay.”
But it also appears that people who haven’t been able to attend the congress in person are keen to log in and engage online, Dr. Windecker said. The 2020 all-virtual conference drew a much younger pool of registrants, on average, than did the live conferences before the pandemic.
“I think that’s an indication of people that may be in training, in early stages of their career, or they don’t have the support from departments or from their practice, or other financial means.” But they are able to participate via computer, tablet, or smartphone, he said.
“Another advantage is that the recorded content can be replayed at the convenience of whoever wants to consume it at a later point in time,” he added. “Those are just some examples why the digital format is likely to stay,” on its own or in a new age of hybrid meetings.
New and updated guidelines
Leading off the guideline series is the document on diagnosis and treatment of acute and chronic HF, which leveraged the past few busy years of HF clinical trials to arrive at a number of new recommendations and strengthened level-of-evidence ratings. It covers both drug and device therapy of HF with reduced ejection fraction (HFrEF) and acute decompensated HF, and tweaks and further enshrines the concept of HF with mildly reduced ejection fraction (HFmrEF).
Several updated recommendations for both long-used and novel medications, notably the sodium-glucose cotransporter 2 inhibitors, will be included because of the recently appreciated evidence-based impact in HFrEF, Dr. Windecker noted.
“I think it will be particularly interesting to look for the SGLT2 inhibitors as not a completely new class of drugs, but certainly one where there has been a lot of new evidence, to look at how those drugs will be integrated in the overall care pathway.”
A top-line preview of the new HF guideline limited to drug therapy, presented at July’s Heart Failure Association of the European Society of Cardiology (ESC-HFA), provided a simple answer to a common question in the new, bountiful age of HFrEF medications: Which meds, initiated in what order?
As it happens, the new recommendation for first-line HFrEF drug therapy is not a silver bullet, but a shotgun – prompt initiation of at least four meds, one from each of four drug classes: renin-angiotensin system inhibitors, beta-blockers, mineralocorticoid receptor antagonists (MRA), and SGLT2 inhibitors. Each class, as described in the document, is to be started as soon as safely feasible, in a sequence deemed appropriate for each individual patient.
Spotlight on EMPEROR-Preserved
The world already knows that the trial, which tested the SGLT2 inhibitor empagliflozin (Jardiance, Boehringer Ingelheim/Eli Lilly) on top of standard therapy, “met” its primary endpoint in almost 6,000 patients with HF with preserved ejection fraction (HFpEF), who included some with HFmrEF by more contemporary definitions.
That means patients in EMPEROR-Preserved assigned to take empagliflozin showed significantly fewer events that made up the study’s primary endpoint, a composite of CV death or HF hospitalization. It appears to be the first clearly significant overall medical therapy benefit for a clinical primary endpoint in a major randomized HFpEF drug trial.
And that, pending fuller presentation of trial results at the Congress on Aug. 27, could be a huge deal for the half of HF patients with left ventricular ejection fractions (LVEF) higher than the HFrEF range.
Those early top-line results weren’t a decisive bombshell for a field now filled with hope for a practice-changing empagliflozin outcome in EMPEROR-Preserved, which isn’t a certainty. They were more like the “boom” of a mortar launching a rocket of fireworks that may explode into a chrysanthemum or green comet or, sometimes, turn out to be no more than a dud. The promise of the early cursory results critically depends on further details.
“Provided there is a compelling benefit, this is what everyone has been waiting for in this condition for decades,” Mikhail N. Kosiborod, MD, director of cardiometabolic research at Saint Luke’s Mid-America Heart Institute, Kansas City, Mo., said.
“Already knowing that the trial met the primary endpoint is obviously very intriguing and encouraging,” he added. “But there are things we don’t know, such as: What is the magnitude of benefit? And whether that benefit, whatever the magnitude, is driven by reductions in both heart failure hospitalizations and cardiovascular death, or only one of the two.”
For example: “If we see an impressive benefit for reduction of hospitalizations, but not a significant reduction in death, that would still be a huge advance. That’s because, to date, we don’t have any drug for HFpEF that has convincingly demonstrated a compelling reduction in heart failure hospitalization or improvement in symptoms, function, or quality of life,” observed Dr. Kosiborod, who wasn’t part of EMPEROR-Preserved.
There have been “suggestions” from HFrEF trials that empagliflozin and dapagliflozin (Farxiga, AstraZeneca) “have very comparable effects on at least the endpoint of cardiovascular death or hospitalization for heart failure,” he said. “So, my expectation would be that whatever is observed in EMPEROR-Preserved is likely a class effect, as well.”
Following EMPEROR-Preserved on the agenda is EMPEROR-Pooled, a patient-level combined analysis of the EMPEROR series of trials that spans the range of HF, regardless of ejection fraction or diabetes status, primarily exploring the effects of empagliflozin on renal function.
Other offerings, Friday, Aug. 27
Scheduled immediately after EMPEROR-Preserved is a presentation on the SMART-MI trial, which should clarify whether management guided by continuous ambulatory monitoring is effective in patients considered at especially high arrhythmic risk. Entry called for recent myocardial infarction and an LVEF of 36%-50% with evidence of cardiac autonomic dysfunction.
The trial randomly assigned 400 such patients to be or not be implanted with a Reveal LINQ (Medtronic) loop recorder and followed them for up to 18 months, primarily for detection of potentially serious arrhythmic events. Endpoints that involved mortality, hospitalization or other clinical events were secondary.
In a time slot preceding both SMART-MI and EMPEROR-Preserved, the GUIDE-HF trial is following a projected 3,600 patients with HF implanted with a CardioMEMS HF System (Abbott) pulmonary artery (PA) pressure sensor to explore the its value for guiding management.
The trial’s three cohorts, followed for at least 12 months, include randomized sensor-monitored and control groups of patients with New York Heart Association class 2-4 symptoms, as well as a third observational set of patients in NYHA class 3. That’s the indication for which the CardioMEMS monitor gained approval in the United States in 2014 based on the 2011 CHAMPION trial, and which fared just as well in the 2017 CHAMPION Post-Approval Study.
The Friday Hot Lines also include Dal-GenE, which has entered about 6,000 patients with recent MI to test the once-abandoned cholesterol ester transfer protein (CETP) inhibitor dalcetrapib (DalCor) for any secondary-prevention benefits when used selectively. The trial’s hook: All its patients are confirmed to have the AA genotype of the rs1967309 variant in the ADCY9 gene, which has been associated with a pronounced clinical response to CETP inhibition.
Saturday, Aug. 28
The direct oral anticoagulants (DOACs) have largely replaced vitamin K antagonists in patients with nonvalvular atrial fibrillation (AFib). But whether DOACs are similarly preferable in the growing world population of people who have undergone transcatheter aortic valve replacement (TAVR or TAVI), an issue explored with variable results in the ATLANTIS and GALILEO trials, is far from settled.
The ENVISAGE-TAVI AF trial explored the question for the factor X inhibitor edoxaban (Savaysa, Lixiana, Daiichi-Sankyo) in 1,400 patients with AFib and a transfemoral TAVR in the previous 5 days, who were randomly assigned to the DOAC or standard management along with discretionary antiplatelet therapy. They’ve been followed for up to 3 years for a composite endpoint of clinical events – including death, MI, and stroke – and for major bleeding.
The day will also feature MASTER DAPT, a comparison of two dual-antiplatelet therapy (DAPT) regimens in an estimated 4,300 patients considered to be high-risk for bleeding who had received the sirolimus-eluting Ultimaster (Terumo) coronary stent, which has a bioresorbable polymer coating.
Investigators have randomly assigned patients to receive either very-short-duration DAPT, for about a month after stenting, followed by a P2Y12 inhibitor alone for up to a year after the procedure; or a more conventional regimen of a P2Y12 inhibitor for 6-12 months with aspirin maintained for a total of 12 months.
Later that day, investigators from the FIGARO-DKD trial will present their results based on 7,437 patients with type 2 diabetes and chronic kidney disease (CKD), a much fuller version than the top-line findings announced by sponsor Bayer 3 months ago.
Those top-line results suggested that patients assigned to receive the nonsteroidal nonselective mineralocorticoid receptor antagonist (MRA) finerenone (Kerendia) on top of standard care benefited with a drop in risk for the primary endpoint of CV death or nonfatal CV events.
Finerenone was recently approved in the United States for treating patients with both type 2 diabetes and CKD based on the published FIDELIO-DKD trial, which had seen less CKD progression and fewer CV events in such patients who took the novel MRA.
Although similar in design to FIGARO-DKD, FIDELIO-DKD had entered fewer patients with early-stage diabetic kidney disease (DKD). That led researchers to pool the two trials’ populations to create a cohort that spans the spectrum of DKD severity. An analysis of the pooled cohort, dubbed FIDELITY, is on the schedule after FIGARO-DKD.
After FIDELITY is the prospective APAF-CRT trial that is following a projected 1,830 patients with permanent, symptomatic AFib and a recent hospitalization for AFib or HF and who were not good candidates for standard ablation. They were assigned to receive either atrioventricular junctional ablation followed by CRT, with or without a defibrillation, on top of optimal meds – a so-called “ablate-and-pace” strategy – or an implantable cardioverter defibrillator with rate-control drug therapy.
The new analysis represents the trial’s second phase in which mortality was followed for 4 years as the primary endpoint, in contrast to the previously reported initial phase that followed the first 102 patients for 2 years for the composite primary endpoint of death, worsening HF, and HF hospitalization. The first phase had halted enrollment before reaching its planned target of 280 patients after an interim analysis showed a significant benefit for ablate and pace.
Next up: DECAAF 2, a randomized assessment of whether catheter ablation for AFib guided by delayed gadolinium enhancement on MRI, a proxy for scar tissue, can be more effective than standard AFib ablation by pulmonary vein isolation alone. An estimated 900 patients with persistent AFib who had never before undergone ablation for the arrhythmia were randomly assigned to one strategy or the other and followed for AFib recurrence over 18 months.
Sunday, Aug. 29
The TOMAHAWK trial aimed to clarify the optimal timing of invasive coronary angiography for resuscitated patients with non–ST-segment elevation out-of-hospital cardiac arrest, a broad population in a setting for which there is little randomized-trial guidance. Investigators randomly assigned 558 such patients to undergo immediate invasive angiography or to direct intensive care unit admission for initial standard care with discretionary delayed angiography. Patients were followed for all-cause mortality, with other clinical events and neurologic outcomes as secondary endpoints.
Next on the schedule, the RIPCORD-2 trial randomly assigned 1,100 patients with stable known or suspected coronary artery disease (CAD) to undergo conventional angiography alone or with added direct pressure-wire measurement of fractional flow reserve to guide management decisions. Primary outcomes include health care costs and patient-reported quality of life at 1 year.
Slated for later that day, the Asymptomatic Carotid Surgery Trial-2 (ACST-2) has entered an estimated 3600 patients with a substantial carotid artery narrowing not associated with symptoms but for which either carotid endarterectomy (CEA) or carotid artery stenting (CAS) was considered anatomically feasible. There also must have been “substantial uncertainty” regarding the optimal procedure choice.
The trial, conducted in 40 countries primarily in Europe and North America and launched in 2008, randomly assigned the patients to undergo either CEA or CAS, in both cases with appropriate medical therapy, and followed them for periprocedural events and up to 10 years for strokes and stroke-related events.
The LOOP study, which is to directly follow ACST-2, has explored whether screening for AFib using the Medtronic Reveal LINQ monitor in older patients with non-AFib stroke risk factors – with oral anticoagulation prescribed for those who test positive – can lower their risk for stroke or systemic embolism. It randomly assigned 6,000 such patients to care guided by the loop recorder or to standard care.
On a somewhat larger scale, the Salt Substitute and Stroke Study (SSaSS) randomly assigned a total of 20,996 people in about 600 villages across northern China and Tibet to sodium-restriction intervention and control groups by village. All participants had a history of stroke or were aged at least 60 years with uncontrolled hypertension.
As described by the trial’s online portal, participants in villages assigned to the intervention group were given a supply of a low-sodium, potassium-supplementing salt substitute to replace their own salt supplies, along with education on the health benefits of sodium restriction. Participants in control villages continued their normal diets and, at the trial’s beginning, received “advice to reduce their salt intake.” All were required to own a telephone.
Clinical events, including strokes and hospitalizations throughout a 5-year follow-up, were tracked by phone calls made to all participants every 6 months and were documented at follow-up home visits.
Sunday is also to feature a Late-Breaking Trials session with a focus on COVID-19, which leads off with COLCOVID, a test of colchicine in patients hospitalized for suspected SARS-CoV-2 infection and in acute respiratory distress.
The 1,279 participants in Argentina were randomly assigned to receive or not receive the potent anti-inflammatory agent on top of antivirals and other standard management and followed for death or new need for mechanical ventilation. A successful outcome would contrast with the RECOVERY trial, which terminated a colchicine group of patients hospitalized with COVID-19 because of a lack of efficacy earlier this year.
COLCOVID is to be followed by the MICHELLE trial of rivaroxaban (Xarelto, Bayer/Janssen) prophylaxis, compared with no preventive oral anticoagulant, in 320 patients who, when hospitalized with COVID-19, had been on parenteral anticoagulants because of an elevated risk for venous thromboembolism. The trial, conducted in Brazil, called for postdischarge rivaroxaban at a once-daily dosage of 10 mg for about 1 month.
The session also includes a presentation called “Insights into the Effects of the COVID-19 Pandemic: Comprehensive Analysis from the GUIDE-HF Trial,” the primary outcomes of which will be reported on the first day of the Congress.
Following is a presentation on the PREPARE-IT study of icosapent ethyl (Vascepa, Amarin), given at high dosages intended to be anti-inflammatory, compared with placebo, in an estimated 4,000 adults. The trial has two groups: A prevention group of adults living and circulating in the community; and a treatment group of patients aged at least 40 years with confirmed symptomatic SARS-CoV-2 infection for whom the need for hospitalization isn’t clear.
Monday, Aug. 30
The final day of the Congress features a trial called Influenza Vaccination after Myocardial Infarction (IAMI), which has tested the secondary preventive effect of influenza vaccination by randomly assigning 2,571 patients to receive a standard vaccine or a saline placebo injection on one occasion.
Entry to the international trial called for a diagnosis of MI with or without ST-segment elevation, or stable CAD and age at least 75 years with other risk factors. The patients were followed for death, MI, stent thrombosis, and a slew of secondary endpoints over 12 months.
Monday offerings continue later in a time block leading off with the STEP trial, which has randomly assigned an estimated 8,000 patients at 40 centers in China who are 60 to 80 years of age with a systolic blood pressure of 140 to <190 mm Hg to be on standard guideline-based therapy or an intensive drug-management strategy.
The systolic BP goals are 130 to <150 mm Hg for standard care and 110 to <130 mm Hg for the intensive regimen. The composite primary endpoint includes death and clinical events related to acute coronary syndromes, HF, revascularization, and stroke.
Following on heels of STEP, the Amulet IDE trial – the first major randomized comparison of two transcatheter LAA closure devices – entered 1,878 patients with nonvalvular AFib who were considered high-risk for bleeding and stroke or systemic embolism.
They were randomly assigned in the noninferiority trial to receive either the AMPLATZER Amulet (Abbott Medical Devices) or the WATCHMAN (Boston Scientific) closure devices and were followed for safety and efficacy for up to 5 years.
Both LAA closure devices, intended to make patients with AFib less reliant on oral anticoagulation, are now available on both sides of the Atlantic – as well as many other countries – after the Amulet’s United States market approval on Aug. 16, based largely on the Amulet IDE trial.
Rounding out the final Hot Line set is one of the latest efforts to show the efficacy and safety of a very short DAPT period after coronary stenting in patients with acute coronary syndromes, the STOPDAPT-2 ACS trial.
The study assigned 3,008 patients in Japan to receive aspirin and clopidogrel for either 1 month or 1 year after implantation with an everolimus-eluting cobalt-chromium stent and followed them for up to 5 years for a composite of MI, CV death, stent thrombosis, stroke, and bleeding.
The trial follows the published STOPDAPT-2 trial that showed superiority for the 1-month DAPT regimen in a predominantly stable-CAD population treated with the same kind of stent.
Program structure and format
A total of 15 online channels are to be available in the morning, European time, their schedules running in parallel. Presentations often are prerecorded, but also include live sessions at 8:00 a.m. Central time and 12 p.m. CET (2:00 a.m. and 6:00 a.m. Eastern time) to liven up the channel offerings, Dr. Windecker observed, and to make them more immediate and potentially interactive.
Many of the parallel channels are devoted throughout the Congress to particular silos of cardiology; for example, arrhythmias and device therapy is on channel 3; CAD and acute care is on 5; HF is on 6; and preventive cardiology is on 9.
Other channels swing across different topics from day to day, such as channel 1, which covers COVID-19 topics on the first and third day of the meeting, “advances in science” on day 2, and “digital health, public health, health economics” on day 4.
The focus each day, starting at 2:00 p.m. CET (8:00 a.m. ET) and continuing into the evening in Europe, shifts over to the Prime Time live program, which features the Hot Line and guideline presentations and many of the live abstract presentations.
Dr. Kosiborod, not a researcher with the EMPEROR trials, is chair of the Dapagliflozin in Preserved Ejection Fraction Heart Failure ( PRESERVED-HF ) trial, which is scheduled for presentation at the September 2021 Heart Failure Society of American meeting.
A version of this article first appeared on Medscape.com.