User login
Grand Rounds: Woman, 22, With Dizziness and Headache
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
A 22-year-old student was brought in to a college student health center in a wheelchair by campus safety personnel. She appeared drowsy and was crying softly. She complained of a severe headache and said she was “tired of going through this all the time.” The woman said she had seen spots and become dizzy, then had gotten “the worst headache of my life” while sitting in class. She rated the headache pain at 8 on a 10-point scale and also complained of nausea and photophobia.
The history revealed dizziness that made her “feel as if I’m tipping over” and similar headaches during the previous year. The patient said she had seen “a few doctors” for her symptoms, but that they “could never find anything.” The headaches usually occurred on the left side of her head, lasted hours to days, and were only partially relieved with acetaminophen. The patient could not remember whether she had eaten breakfast and was unsure of what day it was. She described herself as frustrated and began to weep again.
She was currently under the care of a psychologist but seemed uncertain why; she said that she was sexually active and used condoms. She had undergone an appendectomy at age 12. She denied taking any medications besides acetaminophen. She denied smoking or drug use, history of migraine headaches, vision or hearing changes, facial weakness, depression, or anxiety. Her family history included a grandfather with diabetes and hypertension and an uncle with heart disease. The family history was negative for migraine or psychiatric illness.
Because of the patient’s weakness, she was assisted onto the examination table by a nurse. Physical exam revealed a pale, slightly sweaty, overweight, tearful young woman who was slow to respond. Her blood pressure was measured at 134/104 mm Hg; pulse, 100 beats/min; respirations, 14 breaths/min; and temperature, 97.0ºF. Point-of-care testing of blood glucose was 91 mg/dL, and hemoglobin was measured at 12.3 g/dL. The ophthalmologic exam was positive for photophobia and revealed slightly disconjugate gaze with horizontal nystagmus during testing of cranial nerves (CN) III, IV, and VI. The otoscopic exam revealed a slightly injected right tympanic membrane, and there were no apparent hearing deficits.
The neurologic exam showed patellar and brachial deep tendon reflexes equal, grips weak and equal, and the pupillary response intact. The patient was able to stand without assistance, although her gait was slightly unsteady. Because the patient was of college age, the clinician ruled out meningitis by negative Kernig’s and Brudzinski’s signs and absence of fever. Subarachnoid hemorrhage was also a concern when the patient mentioned the “worst headache of my life,” indicating the need for emergent imaging.
The patient’s presentation, it was felt, warranted a 911 call. The emergency medical team arrived, and its members began to question the patient. Discrepancies in the patient’s history during the paramedics’ reexamination led them to question whether an emergency department (ED) visit was necessary, but at the clinician’s insistence, they agreed to transport the student to the ED.
The following day, the student health center clinician was contacted by a member of the hospital ED staff with an update on the patient’s status. Shortly after her arrival at the hospital, she underwent MRI and was diagnosed with a vestibular schwannoma. She had surgery that same evening, during which the surgeon removed most of the tumor. Although the ED staff was not at liberty to provide more complete information, they did inform the clinician that the patient would require radiation for the remainder of the tumor.
DISCUSSION
Vestibular schwannoma is also known as acoustic schwannoma, acoustic neuroma, acoustic neurinoma, or vestibular neurilemmoma. These tumors arise from perineural elements of Schwann cells, which commonly form and lead to myelination in the vestibular area of CN VIII1 (see figure). They occur with equal frequency on the superior and inferior branches of the vestibular nerve and originate only rarely at the cochlear portion of the eighth cranial nerve. Vestibular schwannomas represent approximately 8% to 10% of brain tumors and 80% to 90% of tumors in the cerebellopontine angle in adults.2 Tumors are distributed evenly across genders, but the majority of diagnosed patients are white.3
Most likely because of improvements in diagnostic technology, the incidence of vestibular schwannoma has increased over the past 30 years. One British research team predicts that one in 1,000 persons will receive a diagnosis of vestibular schwannoma in their lifetime.4 These tumors are most commonly diagnosed in people ages 30 to 60, with a median age of 55.5
A relationship has been demonstrated between neurofibromatosis type 2 (NF2), an autosomal-dominant disease, and the development of vestibular schwannomas.6,7 NF2 has a birth prevalence of one in about 25,000 persons,4,8 and those who inherit the responsible gene inevitably develop vestibular schwannomas.9 Patients with a confirmed diagnosis of vestibular schwannoma should be screened by a geneticist for the NF2 gene; although the tumors are benign, they can cause compression of the vestibular nerve, leading to deafness and balance disorders.10 Schwannomas of the spinal nerves can also occur in persons with NF2.11 Compression of the spinal nerves in these patients can lead to significant morbidity and a shortened average life span.10
NF2 is diagnosed using the following criteria:
1) Bilateral vestibular schwannomas
2) Diagnosis of a family member with either NF2 or unilateral vestibular schwannoma, and
3) Juvenile posterior subscapular lens opacities.9,12,13
Because schwannomas grow slowly, the vestibular system can adapt to the slow destruction of CN VIII. For this reason, patients typically present with unilateral deafness or hearing impairment rather than dizziness.11 Many patients also present with tinnitus and/or vertigo.14,15
Some vestibular tumors remain stable or even regress; others progress, in some cases causing life-threatening complications.16 An extremely rare complication of a vestibular schwannoma was reported in one patient: an intratumoral hemorrhage that led to acute neurologic deterioration and death.17
Since the case patient underwent immediate surgical intervention, it appears she was experiencing significant involvement and it was likely anticipated that without surgical intervention, clinical progression would occur. Her young age could be considered a risk factor for a faster-growing neuroma.18
Clinical Presentation and Diagnosis
Primary care clinicians commonly see patients with complaints of dizziness, lightheadedness, faintness, or a sensation of spinning or tilting. Vestibular schwannoma should be considered in the differential diagnosis of the patient who presents with these complaints, as well as tinnitus or hearing loss.9 The patient with vestibular schwannoma may also have a history of headache, unsteady gait, facial pain, and numbness.19 A partial differential diagnosis is listed in the table20,21). The astute clinician will systematically rule out many of these conditions, since certain other features that may be present (eg, rapid onset, vomiting, fever) do not typically occur in the patient with vestibular schwannoma.
Because the symptoms typically associated with vestibular schwannoma are likely to occur bilaterally in patients with other conditions, unilateral symptoms should alert the clinician to investigate further. The patterns and growth rates of vestibular schwannomas are highly variable and currently unpredictable18 (according to Fortnum et al,14 at least 50% of tumors do not grow within several years after diagnosis); thus, no clear predictors of tumor growth have been identified to assist in the evaluation of an affected patient,16 although faster tumor growth rates have been reported in young patients, and Baser et al18 have called for additional research involving younger persons with vestibular schwannomas.
Standard testing is audiometry followed by MRI, which is considered the most effective means to confirm a diagnosis of vestibular schwannoma.5,14,22
Treatment for Vestibular Schwannoma
Treatment, whether with surgery or radiation, is associated with significant morbidity and possibly decreased quality of life.16 Therefore, distinguishing patients whose tumors will grow and pose a threat to them from those whose tumors are likely to remain stable is central to appropriate management.23
Treatment modalities are considered based on tumor size, growth, presence or absence of tinnitus, and the patient’s preferences and life expectancy.23 In most cases, decision making is complex and should be customized to meet the patient’s individual circumstances. Patients with similar clinical scenarios have been reported to opt for different treatment choices.24
Four treatment options are currently available for patients with vestibular schwannoma:
Serial observation with periodic MRI studies. Since vestibular schwannomas are benign and slow-growing, conservative management can be a reasonable option, particularly if the patient is elderly, the tumor is small, and/or little hearing loss has taken place. However, use of observation is associated with a risk for progressive and permanent hearing loss.2 Between 15% and 50% of patients who opt for serial observation will undergo subsequent surgical intervention, particularly in cases involving worsening tinnitus, balance problems, or hearing loss.23-25
Chemotherapy. Agents including bevacizumab (a humanized monoclonal antibody against vascular endothelial growth factor)8,26,27 and erlotinib (an epidermal growth factor receptor inhibitor) may delay progression or even facilitate regression of vestibular schwannomas.28 Hearing improvement has also been reported in patients with NF2 who were treated with bevacizumab8; research is ongoing.26
Fractionated radiotherapy. Hearing may be preserved in 60% to 95% of patients, depending on levels of dosing to the cochlea, but 3% to 7% of patients will need further treatment.29-31 Radiation treatment is a likely choice in patients with tumors measuring 2.0 cm or less. Larger tumors are considered a surgical disease, and directed radiotherapy may be administered postoperatively (as in the case patient) for residual portions of the tumor.16
Microsurgery. Compared with other treatment modalities, the emphasis of microsurgery is on removing tumors (particularly larger tumors) rather than controlling their growth.29 The three common approaches are retrosigmoid, middle fossa, or translabyrinthine.32-34 Preservation of hearing is reportedly better following retrosigmoid or middle fossa microsurgery, compared with a translabyrinthine procedure (because in the latter, the tumor cannot be exposed without damage to the inner ear).32,35
With any such surgery, risks include cranial nerve damage, leakage of cerebrospinal fluid, and infection.29,32 Postsurgically, about half of patients report frequent headaches, which are persistent in about half of these cases.36-38 Another concern is preservation of the facial nerves, with a risk for temporary facial weakness or dysfunction.3,24,39 Less than 2% of patients who undergo microsurgery require additional treatment.29
Stereotactic radiosurgery. These procedures, which are performed using the Gamma Knife,® the CyberKnife, or the linear accelerator,29,40,41 are considered appropriate for patients with smaller tumors and those who are not candidates for conventional surgery.1 Trigeminal neuropathy, injury to the facial nerves, and hydrocephaly are reported complications of Gamma Knife radiosurgery, but improvements in these technologies are ongoing.1,2,40
Patient Outcomes
The outcome in a patient with vestibular schwannoma depends on the treatment administered, but prolonged follow-up is typically necessary. For patients being managed through observation, annual brain scans are recommended for 10 years, with subsequent scans every three to five years if no tumor growth is seen. For patients who have had surgery, annual brain scans are advised for the successive eight to 10 years, with decreasing frequency if no tumor remains. In patients who undergo radiation, annual scans are recommended for 10 years, then every two years if no tumor growth is detected.36
Psychosocial experiences vary widely among patients who have undergone treatment for vestibular schwannomas. Some are unable to perform necessary or recreational activities, and others must retire early from work.42 Others, however, have minimal disruption in their lives and enjoy a good quality of life. The most difficult consequence of vestibular schwannoma and its treatment, according to patients, is the associated hearing loss.8,19
THE CASE PATIENT
The 22-year-old patient in this case had an atypical presentation of vestibular schwannoma. Although she did present with vertigo, she also complained of headache, nausea, and photophobia—which are rarely reported in investigations of these tumors. She was also younger than the typical patient and did not report tinnitus.
The case patient reportedly underwent surgery and subsequent radiation to treat the remaining portion of her tumor. She suspended her attendance at the college and, as of this writing, has not re-enrolled. She was lost to follow-up.
CONCLUSION
For the primary care provider, diagnostic challenges require great clinical acumen. Vertigo, headache, hearing loss, and tinnitus are all symptoms seen in the primary care setting; when they occur together, the clinician should be alerted to investigate further. A high level of suspicion is appropriate when a patient complains of longstanding auditory symptoms, with or without headache. Unilateral hearing loss is a common symptom in patients with vestibular schwannomas, although some may present with facial weakness or pain, imbalance, and/or vertigo.
In addition to the history and physical exam, experts recommend that audiometry and MRI be considered, particularly if hearing loss is unilateral. Genetic screening for NF2 should be performed if vestibular schwannoma is found on MRI. Referral to a neurologist, a neurosurgeon, or an otolaryngologist is appropriate.
REFERENCES
1. Arthurs BJ, Lamoreaux WT, Giddings NA, et al. Gamma Knife radiosurgery for vestibular schwannoma: case report and review of the literature. World J Surg Oncol. 2009 Dec 18;7:100.
2. Mohammed TA, Ahuja MS, Ju SS, Thomas J. Normal pressure hydrocephalus after Gamma Knife radiosurgery for vestibular schwannoma. J Postgrad Med. 2010;56(3):213-215.
3. Gal TJ, Shinn J, Huang B. Current epidemiology and management trends in acoustic neuroma. Otolaryngol Head Neck Surg. 2010;142(5):677-681.
4. Evans DG, Moran A, King A, et al. Incidence of vestibular schwannoma and neurofibromatosis 2 in the North West of England over a 10-year period: higher incidence than previously thought. Otol Neurotol. 2005;26(1):93-97.
5. Haynes D. Acoustic neuroma diagnosis and treatment options. Hearing Health. 2009;25(3):32. www.drf.org/magazine/36/Summer+2009+Issue/article/272. Accessed May 16, 2011.
6. Sobel RA. Vestibular (acoustic) schwannomas: histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol. 1993;52(2):106-113.
7. Evans DG, Huson SM, Donnai D, et al. A clinical study of type 2 neurofibromatosis. Q J Med. 1992;84(304):603-618.
8. Plotkin SR, Stemmer-Rachamimov AO, Barker FG 2nd, et al. Hearing improvement after bevacizumab in patients with neurofibromatosis type 2. N Engl J Med. 2009;361(4):358-367.
9. Evans DGR, Sainio M, Baser E. Neurofibromatosis type 2. J Med Genet. 2000:37(11):897-904.
10. Gusella JF, Ramesh V, MacCollin M, Jacoby LB. Neurofibromatosis 2: loss of Merlin’s protective spell. Curr Opin Genet Dev. 1996;6(1):87-92.
11. Sagar SM, Israel MA. Ch 374. Primary and metastatic tumors of the nervous system. In: Kasper DL, Braunwald E, Fauci AS, et al. Harrison’s Principles of Internal Medicine. 17th ed. New York, NY: McGraw-Hill Companies, Inc; 2008:2601-2610.
12. Evans DGR. Neurofibromatosis 2 [bilateral acoustic neurofibromatosis, central neurofibromatosis, NF2, neurofibromatosis type II]. Genet Med. 2009;11(9):599-610.
13. Arya R, Sahu JK, Kabra M. Neurofibromatosis type II (Wishart type). J Pediatr Neurol. 2009;7(3): 333-335.
14. Fortnum H, O’Neill C, Taylor R, et al. The role of magnetic resonance imaging in the identification of suspected acoustic neuroma: a systematic review of clinical and cost effectiveness and natural history. Health Technol Assess. 2009;13(18):iii-iv, ix-xi, 1-154.
15. Forton GE, Cremers CW, Offeciers EE. Acoustic neuroma ingrowth in the cochlear nerve: does it influence the clinical presentation? Ann Otol Rhinol Laryngol. 2004;113(7):582-586.
16. Nikolopoulos TP, Fortnum H, O’Donoghue G, Baguley D. Acoustic neuroma growth: a systematic review of the evidence. Otol Neurotol. 2010;31(3):478-485.
17. Yates CW, Weinberg M, Packer MJ, Jacob A. Fatal case of tumor-associated hemorrhage in a large vestibular schwannoma. Ann Otol Rhinol Laryngol. 2010;119(6):402-405.
18. Baser ME, Mautner VF, Parry DM, Evans DGR. Methodological issues in longitudinal studies; vestibular schwannoma growth rates in neurofibromatosis 2. J Med Genet. 2005;42(12):903-906.
19. Brooker J, Burney S, Fletcher J, Dally M. A qualitative exploration of quality of life among individuals diagnosed with an acoustic neuroma. Br J Health Psychol. 2009;14(pt 3):563-578.
20. Strupp M, Brandt T. Diagnosis and treatment of vertigo and dizziness. Dtsch Arzetbl Int. 2008;105(10):173-180.
21. Kerber KA. Dizziness and vertigo. In: Andreoli TE, Griggs RC, Benjamin I , Wing EJ, eds. Andreoli and Carpenter’s Cecil Essentials of Medicine. 8th ed. Philadelphia, PA: Elsevier Inc; 2010:1104-1105.
22. Gimsing S. Vestibular schwannoma: when to look for it? J Laryngol Otol. 2010;124(3):258-264.
23. Agrawal Y, Clark JH, Limb CJ, et al. Predictors of vestibular schwannoma growth and clinical implications. Otol Neurotol. 2010;31(5):807-812.
24. Cheung SW, Aranda D, Driscoll CLW, Parsa AT. Mapping clinical outcomes expectations to treatment decisions: an application to vestibular schwannoma management. Otol Neurotol. 2010;31(2):284-293.
25. Myrseth E, Pedersen PH, Møller P, Lund-Johansen M. Treatment of vestibular schwannomas: why, when and how? Acta Neurochir (Wien). 2007;149(7):647-660.
26. Sidney Kimmel Comprehensive Cancer Center, Massachusetts General Hospital, National Cancer Institute. Bevacizumab for symptomatic vestibular schwannoma in neurofibromatosis type 2 (NF2). http://clinicaltrials.gov/ct2/show/NCT01207687. Accessed May 16, 2011.
27. Mautner VF, Nguyen R, Kutta H, et al. Bevacizumab induces regression of vestibular schwannomas in patients with neurofibromatosis type 2. Neuro Oncol. 2010;12(1):14-18.
28. Plotkin SR, Halpin C, McKenna MJ, et al. Erlotinib for progressive vestibular schwannoma in neurofibromatosis 2 patients. Otol Neurotol. 2010;31(7):1135-1143.
29. Arthurs BJ, Fairbanks RK, Demakas JJ, et al. A review of treatment modalities for vestibular schwannoma. Neurosurg Rev. 2011 Feb 9; [Epub ahead of print].
30. Andrews DW, Werner-Wasik M, Den RB, et al. Toward dose optimization for fractionated stereotactic radiotherapy for acoustic neuromas: comparison of two dose cohorts. Int J Radiat Oncol Biol Phys. 2009;74(2):419-426.
31. Thomas C, Di Maio S, Ma R, et al. Hearing preservation following fractionated stereotactic radiotherapy for vestibular schwannomas: prognostic implications of cochlear dose. J Neurosurg. 2007;107(5):917-926.
32. Samii M, Gerganov V, Samii A. Improved preservation of hearing and facial nerve function in vestibular schwannoma surgery via the retrosigmoid approach in a series of 200 patients. J Neurosurg. 2006;105(4):527-535.
33. Shiobara R, Ohira T, Inoue Y, et al. Extended middle cranial fossa approach for vestibular schwannoma: technical note and surgical results of 896 operations. Prog Neurol Surg. 2008;21:65-72.
34. Schmerber S, Palombi O, Boubagra K, et al. Long-term control of vestibular schwannoma after a translabyrinthine complete removal. Neurosurgery. 2005;57(4):693-698.
35. Phillips DJ, Kobylarz EJ, De Peralta ET, et al. Predictive factors of hearing preservation after surgical resection of small vestibular schwannomas. Otol Neurotol. 2010;31(9):1463-1468.
36. Park JK, Black MP, Vernick DM, Ramakrishna N. Vestibular schwannoma (acoustic neuroma) (2010). www.uptodate.com/contents/vestibular-schwannoma-acoustic-neuroma. Accessed May 16, 2011.
37. Schankin CJ, Gall C, Straube A. Headache syndromes after acoustic neuroma surgery and their implications for quality of life. Cephalalgia. 2009;29(7):760-761.
38. Ryzenman JM, Pensak ML, Tew JM Jr. Headache: a quality of life analysis in a cohort of 1,657 patients undergoing acoustic neuroma surgery: results from the Acoustic Neuroma Association. Laryngoscope. 2005;115(4):703-711.
39. Sriskandan N, Connor SE. The role of radiology in the diagnosis and management of vestibular schwannoma. Clin Radiol. 2010;66(4):357-365.
40. Yang I, Sughrue ME, Han SJ, et al. Facial nerve preservation after vestibular schwannoma Gamma Knife surgery. J Neurooncol. 2009;93(1): 41-48.
41. Unger F, Dominikus K, Haselsberger K. Stereotactic radiosurgery and fractionated stereotactic radiotherapy of acoustic neuromas [in German]. HNO. 2011;59(1):31-37.
42. Tos T, Caye-Thomasen P, Stangerup SE, et al. Long-term socio-economic impact of vestibular schwannoma for patients under observation and after surgery. J Laryngol Otol. 2003;117(12):955-964.
Car Accident and a Language Barrier
ANSWER
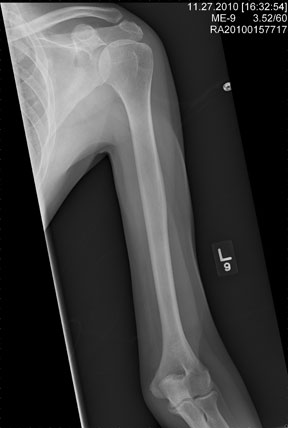
The radiograph shows an obvious deformity in the distal humerus consistent with an old fracture with chronic malunion. There is no evidence of a superimposed acute fracture.
Once family and interpreters became available, it was elicited that the patient, who is originally from Nepal, did sustain a childhood injury and broke his right arm. No acute intervention was required.
ANSWER
The radiograph shows an obvious deformity in the distal humerus consistent with an old fracture with chronic malunion. There is no evidence of a superimposed acute fracture.
Once family and interpreters became available, it was elicited that the patient, who is originally from Nepal, did sustain a childhood injury and broke his right arm. No acute intervention was required.
ANSWER
The radiograph shows an obvious deformity in the distal humerus consistent with an old fracture with chronic malunion. There is no evidence of a superimposed acute fracture.
Once family and interpreters became available, it was elicited that the patient, who is originally from Nepal, did sustain a childhood injury and broke his right arm. No acute intervention was required.
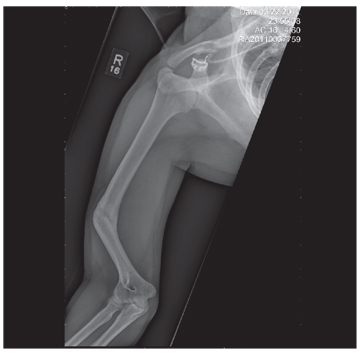
You are asked to see a 41-year-old man complaining of right upper arm pain. He was brought in by EMS from a reported single-vehicle crash, in which he was one of approximately 15 people traveling in a van. The patient speaks little to no English, and details of the accident are sketchy. Best as can be ascertained, the vehicle either went out of control or was hit and ran off the road. There were known fatalities at the scene. Due to language barriers, history is limited. Physical exam shows a middle-aged Asian man who appears quite uncomfortable. He indicates he is hurting in his chest, back, and right arm. His vital signs are normal, and primary survey appears stable, with the patient having multiple abrasions on his face and whole body. Examination of his right arm shows multiple abrasions with some bruising and swelling, as well as a deformity just above the elbow. The patient is able to slowly move his wrist and fingers. Distal pulses and sensation appear intact. Radiograph of the right humerus is shown. What is your impression?
Grand Rounds: Woman, 49, With Dyspnea and Chest Tightness
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
A 49-year-old woman presented to urgent care with complaints of worsening dyspnea for the previous two days. She reported that her symptoms had begun gradually; at the time of her presentation, however, she was also experiencing chest tightness, occasional wheezing, and a nonproductive cough. She had experienced similar symptoms in the past and obtained good results by using her albuterol inhaler. During the current episode, however, she had not had the usual response to inhaler treatment.
The patient’s medical history was positive for environmental allergies, asthma, and GERD. Two weeks earlier, she had undergone dilatation and curettage (D&C) for dysfunctional bleeding, with no associated complications.
In the social history, the patient reported drinking four to six caffeine beverages daily and consuming alcohol moderately (two to four glasses of wine per week). She was following no formal dietary regimen. The patient denied current or past history of tobacco use and had not traveled recently. She had no family history of coronary vascular disease.
Her medications included albuterol and desloratadine as needed, pantoprazole 40 mg/d, and drospirenone/ethinyl estradiol. The patient said she used her albuterol inhaler four to six times per month but more often in the summer and fall. Nighttime awakenings due to asthma symptoms occurred no more than twice per month. She denied prior history of acute asthma exacerbations requiring oral systemic corticosteroids. The patient stated that since her D&C, she had been using ibuprofen almost daily for mild abdominal cramping.
A review of systems was positive for mild fatigue since her D&C. The patient denied fever, chills, headache, sore throat, or cough. She did complain of daily nasal congestion but with no unusual drainage. The patient denied orthopnea, chest pain, palpitations, or peripheral edema, as well as nausea, vomiting, diarrhea, constipation, hematochezia, or melena. She admitted to daily heartburn for the previous two weeks that was relieved somewhat with pantoprazole. She had not experienced urinary frequency or urgency, dysuria, or hematuria. She also denied rash, pruritus, weakness, paresthesias, joint pain, or swelling.
Physical examination revealed an alert, oriented female who appeared slightly anxious but was in no acute distress. Specific findings were pulse, 110 beats/min; blood pressure, 138/88 mm Hg; respirations, 24 breaths/min; temperature, 97.7°F; O2 saturation, 92% on room air. Her height measured 5’2” and weight, 150 lb (BMI, 27.43).
Her conjunctiva were slightly injected, and the tympanic membranes were intact bilaterally with a light reflex; the septum was midline. The mucosa was pale, boggy, and moist with clear drainage and no inflammation. The nasopharynx had no erythema, and the tonsils appeared normal, although a cobblestone appearance was noted in the posterior pharynx. The neck was supple with no adenopathy.
The patient’s heart rate, 110 beats/min, was regular with no murmurs, rubs, or gallops. In the lungs, a prolonged expiratory phase was noted, with diffuse wheezing on chest auscultation bilaterally. Neither retractions nor use of accessory muscles with breathing was observed. The abdomen was soft, rounded, and nontender with no organomegaly. Bowel sounds were evident in all four quadrants. The patient’s skin was free of suspicious lesions or rashes. Her extremities were without edema, and no calf tenderness was noted; Homans’ sign was negative. Superficial varicosities were noted bilaterally.
The top differential diagnosis included:
• Acute asthma (risk factors: history of uncontrolled asthma, as evidenced by frequent use of albuterol)
• Acute anemia (risk factors: history of dysfunctional uterine bleeding, recent D&C)
• Pulmonary embolism (risk factors: recent surgery, recent start of oral contraceptive use).
Additional diagnoses to be considered less likely included:
• Acute coronary syndrome/MI (possible causes of chest tightness, dyspnea, dyspepsia; but no chest pain, diaphoresis, or nausea)
• Acute respiratory distress (history of tachycardia, possible dyspnea; but no diaphoresis, cyanosis, retractions, accessory muscle use, or lung crackles)
• Pneumonia (risk factors: recent surgery, possible cause of nonproductive cough; but no evidence of fever, chills, rales, or pleuritic chest pain).
Diagnostic testing included a 12-lead ECG to evaluate the patient for cardiac arrhythmia or injury; on it, tachycardia was noted, with a regular rate of 106 beats/min. The patient’s chest x-ray yielded normal results.
Laboratory testing included a complete blood count to screen for anemia and infection. Results included a white blood cell count of 8,200/mL (normal range, 4,500 to 11,000/mL); hematocrit, 38.2% (normal range for women, 36.1% to 44.3%); hemoglobin, 13.1 g/dL (normal for women, 12.1 to 15.1 g/dL). A comprehensive metabolic panel was performed to assess electrolyte levels and kidney and liver function; findings were normal. Results of a D-dimer assay, which was obtained to exclude pulmonary embolism,1 were normal at 0.5 mg/L (range, 0.4 to 1.4 mg/L).
In the case of heightened suspicion for MI, the patient would have been transferred to the emergency department (ED) for evaluation, including serial cardiac troponin levels; elevated troponin levels are deemed the standard criterion to define and diagnose MI in a consensus document from the European Society of Cardiology and the American College of Cardiology.2 (Troponin-T and troponin-I are more tissue-specific than the MB fraction of creatine kinase [CK-MB] in detecting MI; positive troponin levels are considered virtually diagnostic of MI.2 Typically, cardiac troponin levels are measured two to three times over a 12- to 16-hour period.)
Peak expiratory flow (PEF), which was measured to evaluate the patient’s respiratory status, was 150 L/min (compared with personal best for a patient of her height and age, approximately 460 L/min). She was given 2.5 mg/3 mL of inhaled albuterol over 15 minutes. Her PEF increased to 350 L/min. O2 saturation improved to 96% on room air, pulse to 104 beats/min, and respirations 20 breaths/min; her blood pressure reading was now 140/90 mm Hg. A prolonged expiratory phase persisted in the lungs, but diffuse wheezing decreased by 40% on chest auscultation.
A second albuterol treatment was administered 20 minutes later, and the patient’s PEF increased to 380 L/min and O2 saturation to 99%. The lungs presently cleared with no further wheezing noted.
In addition, the patient was given a GI cocktail (ie, liquid antacid combined with an anticholinergic agent and viscous lidocaine). Within 10 minutes, her chest tightness was relieved 100%. Her blood pressure was then measured at 135/84 mm Hg; respirations, 18 breaths/min; and pulse rate, 96 beats/min.
According to the National Asthma Education and Prevention Program (NAEPP) 2007 Guidelines for the Diagnosis and Management of Asthma, Expert Panel Report 3 (EPR-3),3 the patient was classified as having intermittent, not-well-controlled asthma with an acute exacerbation. In addition, she was given a diagnosis of uncontrolled GERD.
DISCUSSION
Asthma Incidence and Risk Factors
Asthma affects approximately 300 million people worldwide and remains a global respiratory concern.4 In the United States, this chronic health condition has a prevalence of 8% to 10%. It is estimated that 5% to 10% of asthmatic patients have severe disease that does not respond typically to therapeutic interventions.5
Asthma involves bronchial hyperresponsiveness, airflow obstruction, and underlying inflammation. Acute episodes of asthma, arising from bronchospasm, usually manifest with progressively worsening cough, shortness of breath, chest tightness and wheezing (asthma’s hallmark symptoms), or a combination of symptoms.3
Symptoms of asthma or exacerbations of reactive airway disease vary from patient to patient. In addition to the hallmark symptoms noted, subacute or acute episodes of asthma exacerbation are characterized by decreases in expiratory airflow that can be documented by objective measurements of lung function, such as PEF or spirometry; these measures of airflow indicate the severity of an exacerbation more reliably than does perceived symptom severity.3 The EPR-3 panelists recommend determining asthma severity using a combination of objective criteria and clinical symptoms,3 although few clinicians use the objective criteria.6
Estimates of the prevalence of GERD among patients with asthma have varied from 34% to 89%.7-9 Patients with GERD are 1.97 times more likely than patients without GERD to have asthma10; silent gastroesophageal reflux has been identified in 24% to 62% of patients with asthma, and early studies suggest that treatment for GERD may improve asthma control in patients with severe or difficult-to-control asthma.8,11,12
The exact link between the two conditions is unclear. However, possible explanations why GERD and asthma coincide are that acid flow causes injury to the lining of the throat, airways, and lungs, making inhalation difficult and often causing a persistent cough; or that when acid enters the esophagus, a nerve reflex is triggered that causes the airways to narrow in order to prevent the acid from entering; this can explain dyspnea.8,9
Economic Burden
Asthma is costly to treat, and because there is no cure, the expense is ongoing. According to a 2011 report,13 the average annual direct cost of care (eg, medications, hospital admissions, nonemergency office visits) for one asthma patient between 2002 and 2007 was $3,259. In 2007, the most current data available, the total cost of asthma in the US was $56 billion, with productivity losses due to mortality accounting for $2.1 billion and morbidity-related losses estimated at $3.8 billion.13 The economic consequences of asthma are substantial and can place a considerable burden on affected individuals, their families, the health care system, and society as a whole.3
Current Standard of Care
Based on the scientific literature and the opinions expressed by the NAEPP in the EPR-3,3 clinicians are advised to consider the following general principles and goals for managing asthma: early treatment, special attention to patients at high risk for asthma-related death, and special attention to infants.3 The guidelines emphasize the importance of a clinician/patient partnership to facilitate the asthma patient’s self-management.
Early treatment is a particularly important component for management of asthma exacerbations. Important elements of early treatment include a written asthma action plan, combined with enhanced awareness of the early indicators of an exacerbation (ie, worsening PEF).3,14 It is believed that if patients are able to monitor their respiratory condition and follow a plan of care based on their PEF and/or signs and symptoms of asthma, they are more likely to achieve optimal management of their disease.15
Written Asthma Action Plan. The EPR-33 recommends that health care providers supply all asthmatic patients with a written asthma action plan that will define and support the patient’s efforts at self-management. Written asthma action plans are particularly beneficial for patients with moderate to severe persistent asthma, poorly controlled asthma, or a history of severe exacerbations.3,14
The written asthma action plan should include instructions for daily management of asthma and ways to recognize and treat worsening asthma, including adjustments to medication dosing. Plans may be based on PEF and/or symptoms. Asthma action plans should be discussed and reevaluated at follow-up visits.3 A sample asthma action plan can be found at www.health.state.ny.us/diseases/asthma/pdf/4850.pdf.16
Peak Expiratory Flow (PEF). The EPR-33 recommends PEF monitoring in all asthma patients, regardless of the severity of their exacerbations.17 PEF-based plans are especially useful for the patient who has difficulty perceiving early signs and symptoms of worsening asthma.3,18 A PEF-based plan instructs the patient to use quick-relief medications if symptoms occur or if PEF drops below 80% of the patient’s personal or predicted best. (Measured personal best is the patient’s highest PEF in the previous two weeks of good asthma control,3,19 whereas predicted best is calculated based on findings from a 1983 study by Knudson et al.3,20)
A PEF between 50% and 79% requires the patient to carefully monitor his or her response to the quick-relief medication and, based on that response, consider whether to contact a health care provider. When PEF falls below 50%, a provider’s immediate intervention is usually recommended.3
In the urgent care or ED setting, according to EPR-3 recommendations,3 the PEF or forced expiratory volume in 1 second (FEV1) is used to indicate the following:
• ≥ 70% predicted PEF or FEV1: goal for discharge
• 40% to 69% predicted PEF or FEV1: incomplete response to treatment, frequent need for treatment in the ED
• 3
Treatment and Management
Asthma management interventions that target the treatment of active disease and predisposing triggers are designed to reduce the severity and/or duration of morbidity associated with asthma—principally, to prevent symptoms and exacerbations (see Table 13).
When patients are discharged following an asthma exacerbation, their medications should include an oral corticosteroid burst and a short-acting b2-agonist (SABA); the clinician should also consider prescribing an inhaled corticosteroid (ICS).3
It is no longer recommended that ICS dosing be doubled in place of an oral steroid burst.3,21 The addition of a leukotriene receptor antagonist (LTRA) may also be considered.3,22
Patients should be given an action plan, and follow-up with a primary care provider should be scheduled within a few days—or even the following day, depending on the severity of the patient’s condition. The importance of follow-up with a primary care provider, a pulmonologist, or an asthma/allergy specialist should be emphasized.3,23
For patients who have difficulty recognizing their symptoms, a peak flow meter may be useful. This device is also recommended for patients with moderate to severe asthma or a history of numerous severe exacerbations.3 Additionally, spacers should always be used with metered dose inhalers (MDIs), because they make it easier for medication to reach the lungs and reduce the amount deposited in the mouth and throat, where it can lead to irritation. At each office visit, use of the peak flow meter and inhaler technique should be observed, and the action plan reevaluated and changed if necessary.3,14
Additional components of patient education include instruction in controlling environmental factors: avoiding environmental tobacco smoke, exposure to insect allergens, and molds. It is also important to stress controlling comorbid conditions that influence asthma, such as allergies or GERD. Patients with symptoms of GERD should be advised to take the steps shown in Table 2.8,24
Clinical Implications
Assessment of the severity of an asthma exacerbation is an essential component of ambulatory asthma care. Underclassification of asthma severity has been associated with increased morbidity and mortality,6 and the NAEPP guidelines recommend that clinicians assess and document asthma severity at each clinic visit.3,25 Patients who receive care based on evidence-based practice guidelines have been shown to experience 28% better outcomes.26
PATIENT OUTCOME
The case patient was discharged on an oral corticosteroid burst and a low-dose ICS. She was instructed how and when to use her SABA and given a prescription for a spacer; use of a peak flow meter was initiated with an estimated personal best goal of 460 L/min. The patient was given a written asthma action plan to help her recognize early signs and symptoms of worsening asthma and was advised to use quick-relief medications if she experienced symptoms or if her PEF dropped below 80% of her predicted best.
The patient’s clinician emphasized the importance of controlling any asthma-triggering environmental factors and reviewed nonpharmacologic interventions to control GERD. The patient was advised to resume desloratadine 5 mg/d and pantoprazole 40 mg/d. She was also instructed to schedule an appointment with her primary care provider within 48 hours and to return to urgent care or the ED with any further exacerbation of respiratory symptoms not controlled by her SABA.
CONCLUSION
Asthma morbidity is a nationally recognized, major public health problem. Given the sharp rise in health care costs and limited resources, health care providers must factor in the comparative effectiveness, comparative cost, and cost-effectiveness of both new and existing health care interventions when making treatment decisions.
Many asthmatic patients face the challenges of health care access and quality. By promoting their self-care and awareness, clinicians can help asthmatic patients achieve better symptom management and use the health care system less often.
REFERENCES
1. Stein PD, Hull RD, Patel KC, et al. D-Dimer for the exclusion of acute venous thrombosis and pulmonary embolism. Ann Intern Med. 2004;140(8):589-602.
2. Alpert JS, Thygesen K, Antman E, Bassand JP. Myocardial infarction redefined: a consensus document of The Joint European Society of Cardiology/American College of Cardiology Committee for the redefinition of myocardial infarction. J Am Coll Cardiol. 2000;36(3):959-969.
3. National Asthma Education and Prevention Program, National Heart, Lung, and Blood Institute. Expert Panel Report 3: Guidelines for the Diagnosis and Management of Asthma. Bethesda, MD: National Heart, Lung, and Blood Institute; 2007. US Department of Health and Human Services publication NIH 07-4051.
4. Lougheed DM. Variability in asthma: symptom perception, care, and outcomes. Can J Physiol Pharmacol. 2007;85(1):149-154.
5. Higgins JC. The ‘crashing asthmatic.’ Am Fam Physician. 2003;67(5):997-1004.
6. Cowen MK, Wakefield DB, Cloutier MM. Classifying asthma severity: objective versus subjective measures. J Asthma. 2007;44(9):711-715.
7. Takenaka R, Matsuno O, Kitajima K, et al. The use of frequency scale for the symptoms of GERD in assessment of gastro-oesophageal reflux symptoms in asthma. Allergol Immunopathol (Madr). 2010;38(1):20-24.
8. Harding SM, Barnes PJ, Hollingsworth H. Gastroesophageal reflux and asthma (2010). www.uptodate.com/contents/gastroesophageal-reflux-and-asthma. Accessed April 5, 2011.
9. Havemann BD, Henderson CA, El-Serag HB. The association between gastro-oesophageal reflux disease and asthma: a systematic review. Gut. 2007;56(12):1654-1664.
10. Tsai MC, Lin HL, Lin CC, et al. Increased risk of concurrent asthma among patients with gastroesophageal reflux disease: a nationwide population-based study. Eur J Gastroenterol Hepatol. 2010;22(10):1169-1173.
11. Harding SM, Richter JE, Guzzo MR, et al. Asthma and gastroesophageal reflux: acid suppressive therapy improves asthma outcome. Am J Med. 1996;100(4):395-405.
12. Gibson PG, Henry RL, Coughlan JL. Gastro-oesophageal reflux treatment for asthma in adults and children. Cochrane Database Syst Rev. 2003;(2):CD001496.
13. Barnett SB, Nurmagambetov TA. Costs of asthma in the United States: 2002-2007. J Allergy Clin Immunol. 2011;127(1):145-152.
14. Walders N, Kercsmar C, Schluchter M, et al. An interdisciplinary intervention for undertreated pediatric asthma. Chest. 2006;129(2):292-299.
15. Morrow R, Fletcher J, Mulvihill M, Park H. The asthma dialogues: a model of interactive education for skills. J Contin Educ Health Prof. 2007;27(1): 49-58.
16. State of New York, Department of Health. Asthma action plan. www.health.state.ny.us/diseases/asthma/pdf/4850.pdf. Accessed April 11, 2011.
17. Picken HA, Greenfield S, Teres D, et al. Effects of local standards on the implementation of national guidelines for asthma: primary care agreement with national asthma guidelines. J Gen Intern Med. 1998;13(10):659-663.
18. Hardie GE, Gold WM, Janson S, et al. Understanding how asthmatics perceive symptom distress during a methacholine challenge. J Asthma. 2002;39(7):611-618.
19. Reddel HK, Marks GB, Jenkins CR. When can personal best peak flow be determined for asthma action plans? Thorax. 2004;59(11):922-924.
20. Knudson RJ, Lebowitz MD, Holberg CJ, Burrows B. Changes in the normal maximal expiratory flow-volume curve with growth and aging. Am Rev Respir Dis. 1983;127(6):725-734.
21. Ind PW, Dal Negro R, Colman NC, et al. Addition of salmeterol to fluticasone propionate treatment in moderate-to-severe asthma. Respir Med. 2003;97(5):555-562.
22. Price DB, Hernandez D, Magyar P, et al; Clinical Outcomes with Montelukast as a Partner Agent to Corticosteroid Therapy (COMPACT) International Study Group. Randomised controlled trial of montelukast plus inhaled budesonide versus double dose inhaled budesonide in adult patients with asthma. Thorax. 2003;58(3):211-216.
23. Schatz M, Zeiger RS, Mosen D, et al. Improved asthma outcomes from allergy specialist care: a population-based cross-sectional analysis. J Allergy Clin Immunol. 2005;116(6):1307-1313.
24. Hampel H, Abraham NS, El-Serag HB. Meta-analysis: obesity and the risk for gastroesophageal reflux disease and its complications. Ann Intern Med. 2005;143(3):199-211.
25. Cabana MD, Bruckman D, Meister K, et al. Documentation of asthma severity in pediatric outpatient clinics. Clin Pediatr (Phila). 2003;42(2):121-125.
26. Heater BS, Becker AM, Olson RK. Nursing interventions and patient outcomes: a meta-analysis of studies. Nurs Res. 1988;37(5):303-307.
Disoriented, Intoxicated, and Hurting All Over
Answer
The radiograph demonstrates a mildly displaced fracture of the right iliac wing. In addition, there is moderate diastasis of the pubic symphysis, with the right pubic symphysis being in superior position to the left. Also, a right sacral fracture is present.
The patient was admitted by the trauma service and initially evaluated by the orthopedic trauma team, who planned to take the patient to surgery for subsequent open reduction and internal fixation of these injuries.
Answer
The radiograph demonstrates a mildly displaced fracture of the right iliac wing. In addition, there is moderate diastasis of the pubic symphysis, with the right pubic symphysis being in superior position to the left. Also, a right sacral fracture is present.
The patient was admitted by the trauma service and initially evaluated by the orthopedic trauma team, who planned to take the patient to surgery for subsequent open reduction and internal fixation of these injuries.
Answer
The radiograph demonstrates a mildly displaced fracture of the right iliac wing. In addition, there is moderate diastasis of the pubic symphysis, with the right pubic symphysis being in superior position to the left. Also, a right sacral fracture is present.
The patient was admitted by the trauma service and initially evaluated by the orthopedic trauma team, who planned to take the patient to surgery for subsequent open reduction and internal fixation of these injuries.
A woman, approximately 30 years old, is airlifted to your facility after being “found in a ditch,” presumably as a result of a motor vehicle collision. Details are sketchy. Upon arrival at your facility, she is awake, crying, and complaining of “hurting all over.” She appears to be intoxicated. She is able to give you her name, but not much else in the way of history. Primary survey shows her vital signs to be: blood pressure, 105/75 mm Hg; heart rate, 102 beats/min; and respiratory rate, 20 breaths/min. She is afebrile. She has a superficial scalp laceration but no other obvious injuries. You obtain preliminary portable radiographs of the pelvis. What is your impression?
Nausea, vomiting, and panic attacks in a 50-year-old woman
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
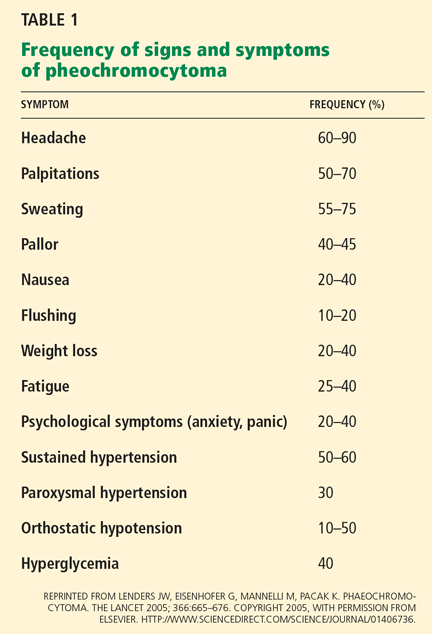
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid

Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
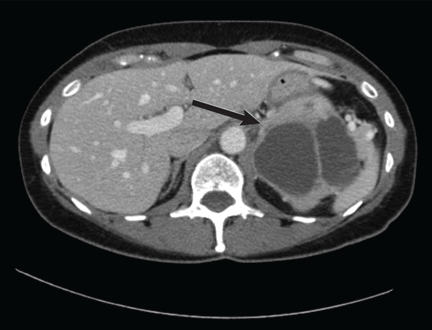
VALUE OF IMAGING STUDIES
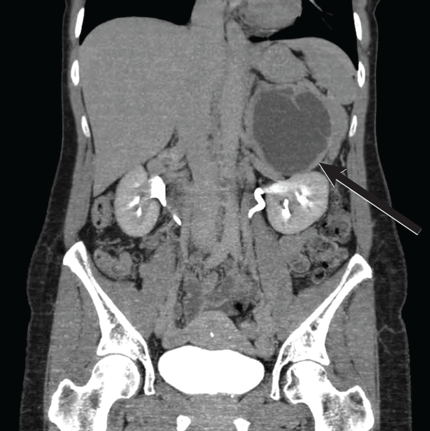
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
A 50-year-old woman presents to the emergency department because of repeated episodes of vomiting over the past 12 hours. She reports eight episodes of non-bloody, nonbilious emesis associated with palpitations and feelings of anxiety, but with no fever or diarrhea. She has not traveled recently and does not have any sick contacts.
She reports that she never had health problems until 6 months ago, when she began having panic attacks that woke her from sleep. The episodes first occurred once or twice per week, usually at night, and involved palpitations and feelings of anxiety that lasted 2 to 4 hours, but no other associated symptoms. After a month, the episodes began to occur more regularly during the day and were accompanied by a pounding headache that began in the back of her neck and extended up and over her head. Her primary care physician prescribed sertraline (Zoloft) and referred her to a neurologist to evaluate the headaches. The neurologic workup included brain magnetic resonance imaging and electroencephalography, both of which were normal.
After 8 weeks on sertraline, the episodes continued to increase in frequency and severity, and her physician switched her to paroxetine (Paxil) and added lorazepam (Ativan), which did not improve her symptoms. Over the past 2 months, during which time she has not been taking any medications, the episodes began to involve nausea and, more recently, vomiting, with episodes occurring as often as once or twice daily, and with intermittent symptom-free days. None of the prior episodes was accompanied by symptoms as severe as those she is currently experiencing.
She is otherwise healthy with no chronic diseases. Her surgical history includes resection of an angiolipoma from her right arm and dilation and curettage for endometrial polyps. She has no personal or family history of psychiatric illness.
PHYSICAL EXAMINATION
The patient is slender and tremulous but does not appear diaphoretic. Her blood pressure is 176/92 mm Hg, pulse 98, temperature 36.5°C (97.7°F), and respiratory rate 20 per minute. Oxygen saturation by pulse oximetry is 98% on room air. She has dry mucus membranes and orthostatic hypotension, but her physical examination is otherwise normal. Electrocardiography (ECG) shows a normal sinus rhythm with a prolonged QTc of 571 ms and peaked P and T waves.
LABORATORY VALUES
- Hemoglobin 15.6 g/dL (reference range 11.5–15.5)
- Hematocrit 47.2% (36.0–46.0)
- Platelet count 448 × 109/L (150–400)
- White cell count 18.65 × 109/L (3.70–11.00)
- Potassium 2.5 mmol/L (3.5–4.0)
- Chloride 97 mmol/L (98–110)
- Bicarbonate 21 mmol/L (23–32)
- Anion gap 20 mmol/L (0–15)
- Glucose 233 mg/dL (65–100).
Sodium, blood urea nitrogen, and creatinine levels are all within normal limits. Urinalysis suggests a urinary tract infection.
IS THIS A PANIC ATTACK?
1. Which of the following is not characteristic of a panic attack?
- Nausea and vomiting
- Onset during sleep
- Palpitations
- Chest pain or discomfort
- Headache
- Trembling or shaking
According to the Diagnostic and Statistical Manual of Mental Disorders (Fourth Edition) (DSM-IV), the diagnosis of panic attack requires the presence of intense fear or discomfort and four or more other symptoms that may come from any of six domains:
- Cardiovascular: palpitations, pounding heart, tachycardia, and chest pain or discomfort
- Autonomic: sweating, chills or hot flushes, and trembling or shaking
- Pulmonary: shortness of breath or a smothering sensation
- Neurologic: dizziness or light-headedness and paresthesias
- Gastrointestinal: choking and nausea or abdominal distress
- Psychological: compass derealization, depersonalization, and the fear of losing control or “going crazy.”1
Two aspects of the patient’s history may be misinterpreted by those unfamiliar with the symptomatology of panic attack. First, although panic disorder carries an increased risk of many comorbidities, including migraine, headache is not typically associated with the panic attacks themselves.2 Second, while not a part of the diagnostic criteria, sleep disturbances are common in patients with panic disorder, and 30% to 45% of patients with the disorder experience recurrent nocturnal panic attacks.3 Therefore, the correct answer is headache.
THE DIFFERENTIAL DIAGNOSIS
When considering a diagnosis of panic attack or panic disorder, the DSM-IV mandates that medical causes of the symptoms must be excluded. Common conditions causing a similar spectrum of symptoms include hyperthyroidism, caffeine and stimulant use or abuse, asthma, cardiac arrhythmias, alcohol withdrawal, and, more rarely, complex partial seizures and pheochromocytoma.2,4 Many of these conditions can be ruled out by the history alone in a reliable patient.
Our patient’s electrocardiogram showed no evidence of ischemia or arrhythmias. Also, her recent negative neurologic workup makes seizure activity less likely.
Many of this patient’s laboratory abnormalities are easily explained by her repeated bouts of vomiting. Specifically, her elevated hemoglobin level and hematocrit are likely secondary to volume contraction, while hypochloremia is seen following losses of HCl with emesis. Typically, however, patients with vomiting have a hypochloremic metabolic alkalosis, and her low serum bicarbonate level is inconsistent with the history.
Three factors might be contributing to this patient’s hypokalemia. First, in a volume-depleted state, the cortical collecting tubules secrete potassium in exchange for increased sodium reabsorption in an attempt to correct volume status. Second, the alkalotic state caused by losses of acid with vomiting results in a transcellular shift of potassium ions into cells in exchange for hydrogen ions. Third, increased levels of epinephrine also cause a shift of potassium ions into cells.5 Potassium is not lost directly through nausea and vomiting.
A state of catecholamine excess, such as during a severe panic attack or in the presence of a catecholamine-secreting tumor, could explain many of her abnormalities. In addition to causing hypokalemia, epinephrine has a gluconeogenic effect, whereas norepinephrine inhibits insulin release, providing a potential explanation for hyperglycemia in a patient with no risk factors for diabetes. Finally, catecholamine excess contributes to lactic acidosis, which could help to explain the low serum bicarbonate level and the elevated anion gap, but unless we take arterial blood gas measurements, the patient’s acid-base status cannot be determined.
While panic attacks do stimulate the sympathetic nervous system, certain elements of her history raise the clinical suspicion for another process. First, the severity of the electrolyte abnormalities is suspicious. Second, a typical panic attack peaks at 10 minutes and begins to subside, whereas this woman’s symptoms have persisted for 12 hours. Finally, the clinical history, in particular the prominence of headaches associated with the symptoms, is inconsistent with classic panic attack. Consequently, an alternative diagnosis, such as pheochromocytoma, deserves more careful evaluation.
Whenever laboratory results do not fit with the clinical scenario or patient, however, one final possibility should always be considered—laboratory error. Errors can be preanalytical (eg, patient misidentification), analytical, or postanalytical. In aggregate, the frequency of errors in laboratory results is 1 in 214 to 8,316.6 Given that even the more conservative estimates show an incidence higher than that of many of the rare diseases for which clinicians may be testing, laboratory error always deserves consideration.
COULD THIS BE PHEOCHROMOCYTOMA?
Pheochromocytoma is a neuroendocrine tumor most commonly arising from the chromaffin cells of the adrenal medulla. However, extra-adrenal pheochromocytoma, generally paraganglioma, accounts for 15% to 20% of these tumors. Although the condition is generally considered very rare, autopsy studies have demonstrated a prevalence of 0.05%, suggesting that many tumors are either missed or are not clinically significant.
The diagnosis is most often sought in hypertensive patients, a population in which pheochromocytoma has a prevalence of 0.1% to 0.6%.7
2. What is the most common presenting symptom of pheochromocytoma?
- Paroxysmal hypertension
- Sustained hypertension
- Nausea
- Cardiomyopathy
- Headache
- Hemorrhagic shock
- Psychological symptoms such as anxiety or panic
Although 50% to 60% of patients with pheochromocytoma have sustained hypertension, it may be absent in patients with primarily epinephrine-secreting tumors or large tumors that degrade catecholamines, leading to normal or low blood pressure.
Cardiomyopathy is a rare consequence of untreated pheochromocytoma, caused by the effects of excess circulating catecholamines over a long period of time.8 As seen in this patient, a prolonged QTc on ECG associated with elevated levels of norepinephrine and normetanephrine may be the only red flag.9
Pheochromocytoma is typically an extremely well-vascularized tumor, and rupture or hemorrhage is a rare but often fatal complication.
IMPORTANT FAMILY HISTORY
The classic “rule of 10s” suggests that 10% of pheochromocytomas are hereditary, but in fact the number may be higher. In a large cohort of patients with apparently sporadic pheochromocytoma, 25% were found to have germ-line mutations.10 This finding highlights the importance not only of obtaining a thorough family history, but also of genetic testing and counseling once the diagnosis has been made.
3. Which hereditary syndrome is not associated with pheochromocytoma?
- Von Hippel-Lindau syndrome
- Neurofibromatosis type 1
- Neurofibromatosis type 2
- Multiple endocrine neoplasia type 2
- Paraganglioma syndromes
Germ-line mutations in five genes related to three hereditary syndromes (von Hippel-Lindau, neurofibromatosis type 1, and multiple endocrine neoplasia type 2) and in two genes related to paraganglioma syndromes are known to be associated with pheochromocytoma.7
Von Hippel-Lindau syndrome
Von Hippel-Lindau syndrome affects 1 in 36,000 live births. It is caused by a mutation of the von Hippel-Lindau gene on chromosome 3, and 10% to 20% of patients with the syndrome have pheochromocytoma. Other associated problems include renal clear-cell carcinomas and cysts, central nervous system and retinal hemangioblastomas, pancreatic tumors and cysts, endolymphatic tumors, and epididymal cysts.
Neurofibromatosis type 1
Neurofibromatosis type 1 affects 1 in 2,500 to 3,000 individuals and is caused by a mutation of the neurofibromatosis type 1 gene on chromosome 17. The disease is diagnosed by the presence of café-au-lait macules, axillary or inguinal freckling (or both), dermal or plexiform neurofibromas, Lisch nodules, or osseous lesions, but the condition is associated with many other pathologic findings, including optic pathway gliomas, cardiovascular abnormalities, and, in up to 5.7% of patients, pheochromocytoma.11
Neurofibromatosis type 2
Neurofibromatosis type 2 affects 1 in 25,000 live births and is caused by a mutation of the neurofibromatosis type 2 gene on chromosome 22. Patients often develop nervous system tumors, ophthalmologic pathology, and cutaneous lesions, but the condition is not associated with pheochromocytoma.12
Multiple endocrine neoplasia type 2
Multiple endocrine neoplasia type 2 affects 1 in 35,000 individuals and is caused by an activating mutation of the RET proto-oncogene on chromosome 21. The syndrome is most worrisome because of the 95% lifetime risk of medullary thyroid carcinoma in affected patients, but it is also associated with a 50% risk of pheochromocytoma and a 20% to 30% risk of primary hyperparathyroidism. Pheochromocytoma is the presenting clinical problem in 10% to 30% of patients.13
Paraganglioma syndromes
Paraganglioma syndromes are caused by mutations in the three genes encoding subunits of the succinate dehydrogenase enzyme. These mutations affect 1 in 30,000 to 100,000 individuals and incur a 70% lifetime risk of developing pheochromocytoma or paraganglioma.14
TESTING FOR AND MANAGING PHEOCHROMOCYTOMA
The consequences of untreated pheochromocytoma are potentially devastating and include progression to metastatic disease, hypertensive crises, cardiomyopathy, and adrenal hemorrhage. Nevertheless, the average patient goes 3 years before receiving the correct diagnosis.7 Consequently, heightened suspicion and tests with both high sensitivity and specificity are needed.
4. Which test for pheochromocytoma has the highest sensitivity?
- Plasma free metanephrines
- Plasma catecholamines
- Urine total metanephrines
- Urine fractionated metanephrines
- Urine catecholamines
- Urine vanillylmandelic acid
Some researchers have also examined plasma total metanephrines and found that any one of these three biochemical markers at a value two times greater than the upper limit of normal provides specificity of around 95%.16
Further laboratory tests in our patient
- Serum dopamine 70 pg/mL (reference range 0–20)
- Norepinephrine 2,018 pg/mL (80–520)
- Epinephrine 2,479 pg/mL (10–200)
- Free normetanephrine 12 pg/mL (< 0.9)
- Free metanephrine 17.8 pg/mL (< 0.5).
VALUE OF IMAGING STUDIES
Computed tomography has a sensitivity of up to 95% for detecting adrenal tumors and is able to detect tumors larger than 0.5 cm, but its specificity may be as low as 50%.17 Studies utilizing modern imaging equipment report a prevalence of adrenal incidentaloma of 4%, of which only 1.5% to 11% are pheochromocytoma.18 Thus, while the simultaneous occurrence of pheochromocytoma-like symptoms and an incidentaloma is not common, the potential for unnecessary surgery precludes diagnosis and treatment based on symptoms and imaging alone.
Magnetic resonance imaging has similar sensitivity and specificity but can better characterize the tumor’s blood supply and relationship to other structures.
Iodine 131 metaiodobenzylguanidine (MIBG) scanning is a physiologic study that uses a radiolabeled amine. Since it can identify pheochromocytoma regardless of location, MIBG scanning is typically used when pheochromocytoma is diagnosed by biochemical testing but CT and MRI fail to locate the lesion, or as a follow-up test in patients in whom recurrence or metastasis is suspected or documented.
The specificity of MIBG scanning is 95% to 100%, but the need to protect the thyroid from ablation and the potential need to repeat scans for up to 72 hours make it a poor choice for the initial evaluation.17
5. What is the next best step in our patient’s management?
- Treat her hypertension with a beta-blocker
- Begin a course of alpha-blockade
- Urgent surgery
- Observation
Because of the high concentration of circulating catecholamines and the instability of the tumor to physical manipulation, appropriate medical management before surgical resection is of paramount importance.
Beta-blockade can lead to malignant hypertension due to the unopposed alpha stimulation and must not be begun until alpha-blockade has been started. The standard of care is to give an alpha-blocker or calcium channel blocker 10 to 14 days before surgery. Typically, oral phenoxybenzamine (Dibenzyline) 10 mg twice daily is started and titrated upward daily by 10 to 20 mg until a target seated blood pressure of 120/80 mm Hg is obtained. Selective alpha-1 blockers such as prazosin (Minipress) and terazosin (Hytrin) have also been used and have the benefit of a preserved alpha-2 catecholamine reuptake mechanism.17
After several days, a beta-blocker may be added, particularly for patients with arrhythmias.7 In patients with refractory hypertension, metyrosine (Demser) can be useful.
During surgery, the patient’s hemodynamic stability and glucose levels can fluctuate rapidly from sudden releases of catecholamines during manipulation of the tumor, as well as from the sudden loss of catecholamines after ligation of draining vessels. Advances in medical care have reduced the perioperative death rate from 50% to less than 3%.7,19
CASE CONCLUSION AND FOLLOW-UP
Two months after her initial presentation, the patient underwent open surgery and had the mass removed without complications. She reports that the “panic attacks” have ceased completely.
The recurrence rate of pheochromocytoma is 13% in patients with sporadic disease and 33% in patients with familial syndromes. The overall recurrence rate with long-term follow-up is 17%, half of recurrences being malignant disease. All patients should therefore be followed in the clinic annually for at least 10 years to identify and treat recurrences early,7 and many experts recommend lifelong follow-up, even for patients without hereditary syndromes.17
Nearly every diagnosis in the DSM-IV includes the caveat that medical causes of disease must be excluded before psychiatric labels can be applied. Although panic disorder and panic attack are far more common than pheochromocytoma, just as essential hypertension is far more common than pheochromocytoma, physicians need to remember that pheochromocytoma can cause symptoms common to both illnesses. Thus, while rare conditions are rare, atypical presentations of common conditions may deserve a second glance.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
- Yates WR. Phenomenology and epidemiology of panic disorder. Ann Clin Psychiatry 2009; 21:95–102.
- Katon WJ. Clinical practice. Panic disorder. N Engl J Med 2006; 354:2360–2367.
- Craske MG, Tsao JC. Assessment and treatment of nocturnal panic attacks. Sleep Med Rev 2005; 9:173–184.
- Roy-Byrne PP, Craske MG, Stein MB. Panic disorder. Lancet 2006; 368:1023–1032.
- Beal AL, Deuser WE, Beilman GJ. A role for epinephrine in post-traumatic hypokalemia. Shock 2007; 27:358–363.
- Kalra J. Medical errors: impact on clinical laboratories and other critical areas. Clin Biochem 2004; 37:1052–1062.
- Lenders JW, Eisenhofer G, Mannelli M, Pacak K. Phaeochromocytoma. Lancet 2005; 366:665–675.
- Leissner KB, Mahmood F, Aragam JR, Amouzgar A, Ortega R. Catecholamine-induced cardiomyopathy and pheochromocytoma. Anesth Analg 2008; 107:410–412.
- Yu R, Furmark L, Wong C. Cardiac abnormalities associated with pheochromocytoma and other adrenal tumors. Endocr Pract 2009; 15:10–16.
- Neumann HP, Bausch B, McWhinney SR, et al; Freiburg-Warsaw-Columbus Pheochromocytoma Study Group. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med 2002; 346:1459–1466.
- Williams VC, Lucas J, Babcock MA, Gutmann DH, Korf B, Maria BL. Neurofibromatosis type 1 revisited. Pediatrics 2009; 123:124–133.
- Asthagiri AR, Parry DM, Butman JA, et al. Neurofibromatosis type 2. Lancet 2009; 373:1974–1986.
- Callender GG, Rich TA, Perrier ND. Multiple endocrine neoplasia syndromes. Surg Clin North Am 2008; 88:863–895.
- Pasini B, Stratakis CA. SDH mutations in tumorigenesis and inherited endocrine tumours: lesson from the phaeochromocytoma-paraganglioma syndromes. J Intern Med 2009; 266:19–42.
- Yu R, Nissen NN, Chopra P, Dhall D, Phillips E, Wei M. Diagnosis and treatment of pheochromocytoma in an academic hospital from 1997 to 2007. Am J Med 2009; 122:85–95.
- Grouzmann E, Drouard-Troalen L, Baudin E, et al. Diagnostic accuracy of free and total metanephrines in plasma and fractionated metanephrines in urine of patients with pheochromocytoma. Eur J Endocrinol 2010; 162:951–960.
- Mittendorf EA, Evans DB, Lee JE, Perrier ND. Pheochromocytoma: advances in genetics, diagnosis, localization, and treatment. Hematol Oncol Clin North Am 2007; 21:509–525.
- Singh PK, Buch HN. Adrenal incidentaloma: evaluation and management. J Clin Pathol 2008; 61:1168–1173.
- Kasturi S, Kutikov A, Guzzo TJ, Smith AL, Wein AJ. Modern management of pheochromocytoma. Nat Clin Pract Urol 2007; 4:630–633.
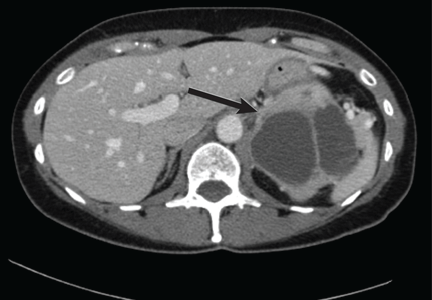
Man, 54, With Delusions and Seizures
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
A 54-year-old African-American man was brought by police officers to the emergency department (ED) after he called 911 several times to report seeing a Rottweiler looking into his second-story window. At the scene, the police were unable to confirm his story, thought the man seemed intoxicated, and brought him to the ED for evaluation.
The patient reported that he had been drinking the previous evening but denied current intoxication or illicit drug use. He denied experiencing symptoms of alcohol withdrawal.
Regarding his medical history, the patient admitted to having had seizures, including two episodes that he said required hospitalization. He described these episodes as right-hand “tingling” (paresthesias), accompanied by right-facial numbness and aphasia. The patient said his physician had instructed him to take “a few phenytoin pills” whenever these episodes occurred. He reported that the medication usually helped resolve his symptoms. He said he had taken phenytoin shortly before his current presentation.
According to friends of the patient who were questioned, he had had noticeable memory problems during the previous six to eight months. They said that he often told the same joke, day after day. His speech had become increasingly slurred, even when he was not drinking.
Once the patient’s medical records were retrieved, it was revealed that he had been hospitalized twice for witnessed grand mal seizures about six months before his current admission; he had been drinking alcohol prior to both episodes. He underwent electroencephalography (EEG) during one of these hospitalizations, with results reported as normal. On both occasions, the patient was discharged with phenytoin and was instructed to follow up with his primary care provider and neurologist.
The patient, who reported working in customer service, had no known allergies. He claimed to drink one or two 40-ounce beers twice per week and admitted to occasional cocaine use. Of significance in his family history was a fatal MI in his mother. Although the patient denied any history of rashes or lesions, his current delirium made it impossible to obtain a reliable sexual history; a friend who was questioned, however, described the patient as promiscuous.
On initial physical examination, the man was afebrile, tachycardic, and somewhat combative with the ED staff. He was fully oriented to self but only partially to place and time.
His right pupil was 3+ and his left pupil was 2+, with neither reactive to light. He spoke with tangential speech and his gait was unsteady, but no other significant abnormalities were noted. A full assessment revealed no rashes or other lesions.
Significant laboratory findings included a low level of phenytoin, a negative blood alcohol level, presence of cocaine on urine drug screening, and normal levels of thyroid-stimulating hormone (TSH), vitamin B12, and folate. The patient’s serum VDRL (venereal disease research laboratory) titer was positive at 1:256.
Electroencephalography showed diffuse slowing, and brain CT performed in the ED showed atrophy that was mild but appropriate for a person of the patient’s age, with no evidence of a cerebrovascular accident (CVA). Aneurysm was ruled out by CT angiography of the brain. MRI revealed persistent increased signal in the subarachnoid space.
The patient was admitted with an initial diagnosis of paranoid delusional psychosis and monitored for alcohol withdrawal. He was given lorazepam as needed for agitation. Consultations were arranged with the psychiatry service regarding his delusions, and with neurology to determine whether to continue phenytoin.
The patient showed little response during the next several days. Based on positive results on serum VDRL with high titer, the presence of Argyll-Robertson pupils on exam, and his history of dementia-like symptoms, a lumbar puncture was performed to rule out neurosyphilis. In the patient’s cerebral spinal fluid (CSF) analysis, the first tube was clear and colorless, with 72 cells (28% neutrophils, 59% lymphocytes); glucose, 64 mg/dL; and total protein, 117 mg/dL. The fourth tube had 34 cells (17% neutrophils, 65% lymphocytes) and a positive VDRL titer at 1:128. Results from a serum syphilis immunoglobulin G (IgG) test were positive, and HIV antibody testing was nonreactive, confirming the diagnosis of neurosyphilis.
The hospital’s infectious disease (ID) team recommended treatment with IV penicillin for 14 days. Once this was completed, the patient was discharged with instructions to follow up at the ID clinic in three months for a repeat CSF VDRL titer to monitor for resolution of the disease. His prescription for phenytoin was discontinued.
At the time of discharge, it was noted that the patient showed no evidence of having regained cognitive function. He was deemed by the psychiatry service to lack decision-making capacity—a likely sequelae of untreated neurosyphilis of unknown duration.
He did return to the ID clinic six months after his discharge. At that visit, a VDRL serum titer was drawn with a result of 1:64, a decrease from 1:128. His syphilis IgG remained positive, however.
Discussion
Definition and Epidemiology
Syphilis is commonly known as a sexually transmitted disease with primary, secondary, and tertiary (early and late latent) stages.1 Neurosyphilis is defined as a manifestation of the inflammatory response to invasion over decades by the Treponema pallidum spirochete in the CSF as a result of untreated primary and/or secondary syphilis.2 About one in 10 patients with untreated syphilis will experience neurologic involvement.3,4 Before 2005, neurosyphilis was required to be reported as a specific stage of syphilis (ie, a manifestation of tertiary syphilis4), but now should be reported as syphilis with neurologic manifestations.5
A reportable infectious disease, syphilis was widespread until the advent of penicillin. According to CDC statistics,6 the number of reported cases of primary and secondary syphilis has declined steadily since 1943. In the late 1970s and early 1980s, the number of tertiary cases also began to plateau, likely as a result of earlier diagnosis and more widespread use of penicillin. Recent case reports suggest greater prevalence of syphilis among men than women and increased incidence among men who have sex with men.7
Pathogenesis
Syphilis is most commonly spread by sexual contact or contact with an infected primary lesion (chancre). Less likely routes of transmission are placental passage or blood transfusion. Infectivity is greatest in the early disease stages.8
Primary syphilis is marked by transmission of the spirochete, ending with development of secondary syphilis (usually two to 12 weeks after transmission). A chancre commonly develops but is often missed by patients because it is painless and can heal spontaneously.7 The chancre is also often confused with two other sources of genital lesions, herpes simplex (genital herpes) and Haemophilus ducreyi (chancroid). In two-thirds of cases of untreated primary syphilis, the infection clears spontaneously, but in the remaining one-third, the disease progresses.8
Secondary syphilis, with or without presence of a chancre, manifests with constitutional symptoms, including lymphadenopathy, fever, headache, and malaise. Patients in this disease phase may also present with a generalized, nonpruritic, macular to maculopapular or pustular rash. The rash can affect the skin of the trunk, the proximal extremities, and the palms and soles. Ocular involvement may occur, especially in patients who are coinfected with HIV.8 In either primary or secondary syphilis, infection can invade the central nervous system.1
During latent syphilis, patients show serologic conversion without overt symptoms. Early latent syphilis is defined as infection within the previous year, as demonstrated by conversion from negative to positive testing, or an increase in titers within the previous year. Any case occurring after one year is defined as late or unknown latent syphilis.8
Tertiary syphilis is marked by complications resulting from untreated syphilis; affected patients commonly experience central nervous system and cardiovascular involvement. Gummatous disease is seen in 15% of patients.1
The early stages of neurosyphilis may be asymptomatic, acute meningeal, and meningovascular.1,4,8,9 Only 5% of patients with early neurosyphilis are symptomatic, with the added potential for cranial neuritis or ocular involvement.1 The late stages of neurosyphilis are detailed in the table.1,4,8
Diagnosis
A diagnosis of syphilis is made by testing blood samples or scrapings from a lesion. In patients with suspected syphilis, rapid plasma reagin (RPR) testing or a VDRL titer is commonly ordered. When results are positive, a serum treponemal test is recommended to confirm a diagnosis of syphilis. Options include the fluorescent treponemal antibody absorption test (FTA-ABS) and the microhemagglutinin assay for antibody to T pallidum (MHA-TP).5
If neurologic symptoms are present, a CSF sample should be obtained, followed by the same testing. A confirmed diagnosis of neurosyphilis is defined by the CDC as syphilis at any stage that meets laboratory criteria for neurosyphilis5; these include increased CSF protein or an elevated CSF leukocyte count with no other known cause, and clinical signs or symptoms without other known causes.7
Treatment
Treatment of syphilis generally consists of penicillin, administered intramuscularly (IM) or IV, depending on the stage. According to 2006 guidelines from the CDC,10,11 treatment for adults with primary and secondary syphilis is a single dose of IM penicillin G, 2.4 million units. If neurosyphilis is suspected, recommended treatment is IV penicillin G, 18 to 24 million units per day divided into six doses (ie, 3 to 4 million units every four hours) or continuous pump infusion for 10 to 14 days.10-12 Follow-up is recommended by monitoring CSF titers to ensure clearance of infection; retreatment may be required if CSF abnormalities persist after two years.11
Patients with a penicillin allergy should undergo desensitization, as penicillin is the preferred agent; the potential exists for cross-reactivity with ceftriaxone, a possible alternative for patients with neurosyphilis.11 All patients diagnosed with syphilis should also be tested for HIV and other sexually transmitted diseases.10-12
The prognosis of patients treated for neurosyphilis is generally good if the condition is diagnosed and treated early. In patients with cerebral atrophy, frontal lesions, dementia, or tabes dorsalis, the potential for recovery decreases.2,13,14
Teaching Points
There are several teaching points to take away from this case:
• Remember to rule out a CVA in any patient who presents with numbness, paresthesias, or slurred speech. In this case, a brain CT and CT angiography of the brain were both obtained in the ED before the patient was admitted. They both yielded negative results; because the patient’s history was consistent with alcohol and drug use and he had a history of seizures, he was monitored closely for signs of withdrawal or further seizure.
• Phenytoin is an antiepileptic agent whose use requires proper patient education and drug level monitoring. Appropriate follow-up must be ensured before phenytoin therapy is begun, as toxicity can result in nystagmus, ataxia, slurred speech, decreased coordination, mental confusion, and possibly death.15,16
• For patients with a suspected acute change in mental status, a workup is required and should be tailored appropriately, based on findings. This should include, but not be limited to, a thorough history and physical exam, CT of the brain (to rule out an acute brain injury17), and, if warranted, MRI of the brain. Also, a urine drug screen and alcohol level, a complete blood count, a TSH level (to evaluate for altered thyroid function that may explain mental status changes), comprehensive panel, RPR testing and/or a VDRL titer should be obtained, depending on the facility’s protocol18,19; at some facilities, a treponemal test, rather than VDRL, is being obtained at the outset.20 Levels of vitamin B12 (as part of the dementia workup), folate, thiamine, and ammonia (in patients with suspected liver disease) can also be obtained in patients with change in mental status.18,19 Urinalysis should not be overlooked to check for a urinary tract infection, especially in elderly patients.21
• If primary syphilis is suspected, treatment must be undertaken.20
Conclusion
Despite the decline seen since the 1940s in cases of primary and secondary syphilis, and the effectiveness of penicillin in treating the infection early, patients with late-stage syphilis, including those with neurosyphilis, may still present to the emergency care, urgent care, or primary care setting. Immediate treatment with penicillin is recommended to achieve an optimal prognosis for the affected patient.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.
1. Golden MR, Marra CM, Holmes KK. Update on syphilis: resurgence of an old problem. JAMA. 2003;290(11):1510-1514.
2. Simon RP. Chapter 20. Neurosyphilis. In: Klausner JD, Hook EW III, eds. Current Diagnosis & Treatment of Sexually Transmitted Diseases. USA: The McGraw-Hill Companies; 2007:130-137.
3. Sanchez FM, Zisselman MH. Treatment of psychiatric symptoms associated with neurosyphilis. Psychosomatics. 2007;48:440-445.
4. Marra CM. Neurosyphilis. Curr Neurol Neurosci Rep. 2004;4(6):435-440.
5. CDC. Sexually transmitted diseases surveillance, 2007: STD surveillance case definitions. www.cdc.gov/std/stats07/app-casedef.htm. Accessed March 23, 2011.
6. CDC. 2008 Sexually Transmitted Diseases Surveillance: Table 1. Cases of sexually transmitted diseases reported by state health departments and rates per 100,000 population: United States, 1941-2008. www.cdc.gov/std/stats08/tables/1.htm. Accessed March 23, 2011.
7. CDC. Sexually transmitted diseases (STDs): Syphilis: CDC fact sheet. www.cdc.gov/std/syphilis/STDfact-syphilis.htm. Accessed March 23, 2011.
8. Tramont EC. Chapter 238. Treponema pallidum (syphilis). In: Mandell GL, Bennett JE, Dolin R, eds. Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases. 7th ed. Philadelphia: Elsevier Churchill Livingstone; 2009.
9. Ghanem KG. Neurosyphilis: a historical perspective and review. CNS Neurosci Ther. 2010; 16(5):e157-e168.
10. Workowski KA, Berman SM; CDC. Sexually transmitted diseases treatment guidelines, 2006. MMWR Recomm Rep. 2006;55(RR-11):1-94.
11. CDC. Sexually transmitted diseases: treatment guidelines 2006. www.cdc.gov/std/treatment/2006/genital-ulcers.htm#genulc6. Accessed March 29, 2011.
12. Drugs for sexually transmitted infections. Treatment Guidelines from the Medical Letter. 2010;95:95a. http://secure.medicalletter.org. Accessed March 23, 2011.
13. Russouw HG, Roberts MC, Emsley RA, et al. Psychiatric manifestations and magnetic resonance imaging in HIV-negative neurosyphilis. Biol Psychiatry. 1997;41(4):467-473.
14. Hooshmand H, Escobar MR, Kopf SW. Neurosyphylis: a study of 241 patients. JAMA. 1972;219 (6):726-729.
15. Miller CA, Joyce DM. Toxicity, phenytoin. http://emedicine.medscape.com/article/816447-overview. Accessed March 23, 2011.
16. Earnest MP, Marx JA, Drury LR. Complications of intravenous phenytoin for acute treatment of seizures: recommendations for usage. JAMA. 1983; 246(6):762-765.
17. Geschwind MD, Shu H, Haman A, et al. Rapidly progressive dementia. Ann Neurol. 2008;64(1): 97-108.
18. Mechem CC. Chapter 143. Altered mental status and coma. In: Ma J, Cline DM, Tintinalli JE, et al, eds. Emergency Medicine Manual, 6e. www.access emergencymedicine.com/content.aspx?aID=2020. Accessed March 23, 2011.
19. Knopman DS, DeKosky ST, Cummings JL, et al; Quality Standards Subcommittee of the American Academy of Neurology. Practice parameter: diagnosis of dementia (an evidence-based review). Neurology. 2001;56(9):1143-1153.
20. CDC. Syphilis testing algorithms using treponemal tests for initial screening—four laboratories, New York City, 2005-2006. MMWR Morb Mortal Wkly Rep. 2008;57(32):872-875.
21. Anderson CA, Filley CM. Chapter 33. Behavioral presentations of medical and neurologic disorders. In: Jacobson JL, Jacobson AM, eds. Psychiatric Secrets. 2nd ed. St. Louis, MO: Hanley & Belfus; 2001.
Unrestrained Passenger Injured in Car Accident
ANSWER
There is a cortical irregularity at the medial margin of the left iliac bone at the level of the acetabulum, strongly suggestive of a fracture. In addition, there may be a nondisplaced fracture within the superior/inferior rami on the left.
CT was recommended to further define these areas (and was already pending to evaluate the patient’s abdomen). Fortunately, there were no fractures within the hip joint, just the nondisplaced rami fracture.
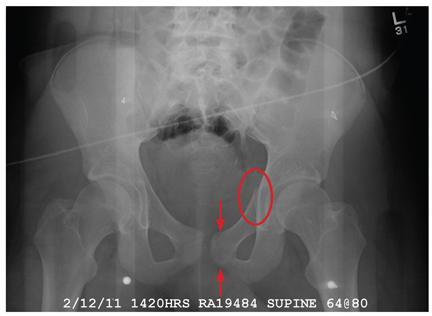
ANSWER
There is a cortical irregularity at the medial margin of the left iliac bone at the level of the acetabulum, strongly suggestive of a fracture. In addition, there may be a nondisplaced fracture within the superior/inferior rami on the left.
CT was recommended to further define these areas (and was already pending to evaluate the patient’s abdomen). Fortunately, there were no fractures within the hip joint, just the nondisplaced rami fracture.
ANSWER
There is a cortical irregularity at the medial margin of the left iliac bone at the level of the acetabulum, strongly suggestive of a fracture. In addition, there may be a nondisplaced fracture within the superior/inferior rami on the left.
CT was recommended to further define these areas (and was already pending to evaluate the patient’s abdomen). Fortunately, there were no fractures within the hip joint, just the nondisplaced rami fracture.
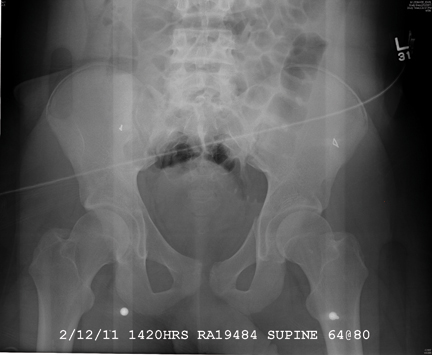
A 19-year-old man is transferred to your facility for injuries he sustained in a motor vehicle collision. He was an unrestrained passenger in a vehicle that went out of control and left the road. At the outside facility, he was found to have a chest injury and a pneumothorax, resulting in his transfer for tertiary level care. On arrival, he is complaining of some chest wall pain, but also states that his hips—especially the left one—are causing quite a bit of discomfort. His medical history is unremarkable except for sickle cell trait. Primary survey reveals stable vital signs and no obvious injury. On closer examination, with stress on his pelvis, he does complain of localized pain on the left side. Radiograph of the pelvis is obtained. What is your impression?
Grand Rounds: Woman, 26, with Kidney Stones
A 26-year-old woman presented to a nephrology office in Virginia for a reevaluation and second opinion regarding her history of kidney stones. This condition had led to uremia and acute kidney failure, requiring hemodialysis.
Her history was significant for recurrent kidney stones and infections, beginning at age 12. Over the next six years, she passed at least five stones and underwent three lithotripsy procedures; according to the patient, however, neither she nor her parents were ever informed of any decrease in her kidney function. The patient said she had been told that her stones were composed of calcium oxalate, and she was placed on potassium citrate therapy but did not take the medication on a regular basis.
After high school, she left the area for college and for several years she frequently and spontaneously passed gravel and stones. She was a runner in high school and college and had two children without experiencing any hypertension, proteinuria, or stone problems during her pregnancies. She had been treated for numerous recurrent urinary tract infections in outpatient clinics and private offices during the 10 years leading up to her current presentation. She had a distant history of a cholecystectomy.
In May 2009, the patient was hospitalized for a kidney infection and underwent cystoscopy with a finding of left ureteral obstruction caused by a stone. A stent was placed, followed by lithotripsy. Her serum creatinine level was measured at 2.2 mg/dL at that time (normal range, 0.6 to 1.5 mg/dL). In August 2009, she was treated again for a kidney infection; a right-sided stone obstruction was noted at that time, and again a stent was placed and lithotripsy was performed. Her serum creatinine level was then 3.3 mg/dL. During these episodes, the patient’s calcium level ranged from 8.2 to 10.1 mg/dL (normal, 4.5 to 5.2 mg/dL). Her phosphorus level was noted to range from 2.6 to 9.5 mg/dL (normal, 2.5 to 4.5 mg/dL). Her intact parathyroid level was 354 pg/mL (normal, 10 to 60 pg/mL). Thus, she had documented secondary hyperparathyroidism, which was treated with paricalcitol and a phosphate binder.
In February 2010, the patient was “feeling poorly” and was taken to a local hospital in South Carolina. She was admitted in acute renal failure and started on dialysis. She did well on hemodialysis with little to no fluid gain and good urine volume. She returned to Virginia temporarily for treatment, to be closer to her family and to prepare for kidney transplantation. She had family members who were willing to donate an organ.
The patient’s family history was negative for gout, kidney disease, or kidney stones. No family member was known to have hypertension, diabetes, or enuresis.
Physical examination showed a thin white woman with a runner’s lean look. She denied laxative use. Her blood pressure was measured at 120/84 mm Hg, and her pulse, 96 beats/min. Findings in the skin/head/eyes/ears/nose/throat exam were within normal limits except for the presence of contact lenses and a subclavicular dialysis indwelling catheter. Neither thyroid enlargement nor supraclavicular adenopathy was noted. Her heart rate was regular without murmurs. The abdomen was soft and nontender without rebound. The extremities showed no edema. Neurologic and vascular findings were intact.
The most recent 24-hour urine study showed a urine creatinine clearance of 4 mL/min (normal, 85 to 125 mL/min), despite a very large urine volume. Renal ultrasonography revealed two small kidneys that were highly echogenic, with evidence of medullary nephrocalcinosis without obstruction bilaterally.
The presentation of a woman with a kidney stone load high enough to cause full kidney failure by age 26 led the nephrologist to suspect the presence of hyperoxaluria type 1 (primary) or type 2 (secondary). The patient’s urine oxalate level was 158 mcmol/L (normal, < 57 mcmol/L), and her plasma oxalate level was 73 mcmol/L (normal, < 10 mcmol/L).
In response to the patient’s high blood and urine oxalate levels and her interest in kidney transplantation, genetic testing was performed to determine whether she had type 1 or type 2 hyperoxaluria. If she was found to have type 1 hyperoxaluria, she would need a liver transplant before her body showered a newly transplanted kidney with stones, causing recurrent kidney failure.
Discussion
Primary hyperoxaluria (PHO) type 1 is a very rare recessive hereditary disease with a prevalence of one to three cases per one million persons.1 Patients typically present with kidney stones at an early age (as did the case patient) or in full kidney failure. It is calculated that PHO is responsible for 1% of all end-stage renal disease among pediatric patients.2,3
Stones are caused by a deficiency of the liver enzyme alanine-glyoxylate aminotransferase (AGXT), which ordinarily converts glyoxylate to glycine.2,4 When AGXT is absent, glyoxylate is converted instead to oxalate, which forms insoluble salts that accumulate in the kidney as oxalate kidney stones. Most patients (ie, 80% to 90%) present in late childhood or early adolescence with systems of recurrent stones and urinary tract infections resulting from blockage.5,6 The natural history of the disease is progression to kidney failure and death from end-stage renal disease unless dialysis is initiated.
While testing of oxalate-to-creatinine molar ratio in a random urine sample may be helpful, this measurement does not stabilize until age 14 to 18—often after kidney damage has already occurred.7 Liver biopsy can confirm whether the enzyme AGXT is absent. Differentiation between PHO and type 2 hyperoxaluria can only be confirmed by genetic testing in which the AGXT gene is identified.8
There is an increased incidence of PHO in Tunisia and Kuwait9-11 and in the Arab and Druze families of Israel12 as a result of intermarriages in this population. Since AGXT is a recessive gene, the child of parents who are both carriers has a 25% chance of having the disease. If either parent carries the genetic variant, there is a 50% chance that the recessive gene will be passed on.
Diagnosis
Early diagnosis of PHO is critical. However, because the disease is so rare, more than 40% of affected patients do not receive a diagnosis until three years after symptoms develop, and 30% are diagnosed only upon presentation with end-stage renal disease.2,13
If PHO is detected early, the key management goal is to minimize renal and skeletal oxalate deposition. Components of medical management are shown in the table.2,14-17 It is important to note that these strategies are effective only if initiated early, that is, before the patient’s glomerular filtration rate drops below 25 mL/min.18
Treatment
Organ transplantation remains the only definitive treatment for PHO14,19—to prevent severe systemic oxalosis or to manage the patient who has progressed to end-stage renal disease. Researchers from the Mayo Clinic in Rochester, Minnesota (where, it should be noted, a National Oxalosis and Hyperoxaluria Registry is maintained under the direction of Dawn S. Milliner, MD), recently published an observational study of outcomes in transplant graft survival among 203 PHO patients. Bergstralh et al20 reported high rates of recurrent oxalosis in patients undergoing kidney transplantation alone, and significantly improved outcomes in patients who underwent both liver and kidney transplantation.
Before 1990, according to a report by the Rare Kidney Stone Consortium,18 the prognosis for PHO transplant patients in the United States was so poor that a donor kidney was considered wasted on these patients. Since the year 2000, however, survival after transplantation has improved greatly, with rates similar to those of all kidney transplant patients nationwide. The explanation for increased survival rates among PHO patients undergoing transplantation was twofold:
• Increased preoperative stone control
• Use of combined liver-kidney transplants.21,22
Since the liver is responsible for the cascade of calcium oxalate stones, the native liver must be fully removed prior to transplantation of a new liver and kidney. Postoperatively, stones will also emerge from where they have lodged in the skeletal tissue to shower the new kidney. Thus, medical management of this cascade of new stones is vital if the transplanted grafts are to survive.23 Calcium oxalate blood levels can remain high for one to two years posttransplantation,2,24 so long-term medical management of oxalate is essential.
The Case Patient
Clinicians engaged in a discussion with the patient and her family regarding a possible diagnosis of PHO. Blood was drawn and sent to the Mayo Clinic for genetic analysis. It was found that the patient had an abnormality in the AGXT gene; with the diagnosis of type 1 hyperoxaluria confirmed, she was flown to Rochester for a full workup.
The patient was the only member of her family with the defective AGXT gene, and her genetic counselors considered this a single mutation. She was accepted for the liver/kidney transplantation list.
Due to the increase in reported survival among patients if they undergo transplantation early in the natural history of stone deposition, the average wait time for PHO patients is only three to four months. The case patient returned to the dialysis unit in Virginia, where she was placed on a dialysis regimen of five-hour treatments, five times per week (nighttime and day); this was determined to be the peak treatment duration for most efficient stone removal, as determined by calcium oxalate measurement during her workup at the Mayo Clinic.
This regimen was continued for three months, at which time the patient was nearing the top of the transplant waiting list. She returned to the Mayo Clinic in September 2010 and underwent transplantation in October; since then, she has regained excellent kidney function and experienced an immediate drop in her calcium oxalate levels. She remained in Rochester until late November, then returned to her home in South Carolina, where she continues to undergo follow-up at a local transplantation center.
The case patient was fortunate that an attending nephrologist at the nephrology office in Virginia developed a high clinical suspicion for her actual condition and started the workup that led to a diagnosis of PHO. She could well have been among the 19% of patients with PHO in whom the correct diagnosis is not reached until after a newly transplanted kidney has been showered with stones again,18,25 necessitating a second kidney transplant following the essential liver transplantation.
Before her current presentation, the patient had been under the care of another nephrologist and had spent six months on a transplant waiting list. If she had proceeded with her original plan, the scheduled kidney transplant (unaccompanied by the essential liver transplant) would have been ineffective, and her donor would have undergone major surgery to no good result.
Conclusion
Type 1 hyperoxaluria is a rare diagnosis that is frequently missed. According to data from the Rare Kidney Stone Consortium,18 nearly one-fifth of patients with PHO do not receive a correct diagnosis until after an unsuccessful kidney transplantation, as liver transplantation is initially required.
The author wishes to extend special thanks to Stephen G. Goldberger, MD, “for being such a good detective.”
References
1. Ajzensztejn MJ, Sebire NJ, Trompeter RS, Marks SD. Primary hyperoxaluria type 1. Arch Dis Child. 2007; 92(3):197.
2. Niaudet P. Primary hyperoxaluria (2010). www.uptodate.com/contents/primary-hyperoxaluria?source=search_result& selectedTitle=1%7E39. Accessed February 17, 2011.
3. Latta K, Brodehl J. Primary hyperoxaluria type I. Eur J Pediatr. 1990;149(8):518-522.
4. Danpure CJ. Advances in the enzymology and molecular genetics of primary hyperoxaluria type 1: prospects for gene therapy. Nephrol Dial Transplant. 1995;10 suppl 8:24-29.
5. Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290-296.
6. Genetics Home Reference. Primary hyperoxaluria. www.ghr.nlm.nih.gov/condition/primary-hyperoxaluria. Accessed February 17, 2011.
7. Remer T, Neubert A, Maser-Gluth C. Anthropometry-based reference values for 24-h urinary creatinine excretion during growth and their use in endocrine and nutritional research. Am J Clin Nutr. 2002;75(3):561-569.
8. Danpure CJ. Molecular and clinical heterogeneity in primary hyperoxaluria type 1. Am J Kidney Dis. 1991;17(4):366-369.
9. Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10(4):479-482.
10. Al-Eisa AA, Samhan M, Naseef M. End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc. 2004;36(6):1788-1791.
11. Cochat P, Liutkus A, Fargue S, et al. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol. 2006;21(8):1075-1081.
12. Rinat C, Wanders RJ, Drukker A, et al. Primary hyperoxaluria type I: a model for multiple mutations in a monogenic disease within a distinct ethnic group. J Am Soc Nephrol. 1999;10(11):2352-2358.
13. Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18(10):986-991.
14. Watts RW. Primary hyperoxaluria type I. QJM. 1994;87(10):593-600.
15. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria: the German experience. Am J Nephrol. 2005;25(3):276-281.
16. Milliner DS, Eickholt JT, Bergstralh EJ, et al. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331(23):1553-1558.
17. Danpure CJ. Primary hyperoxaluria: from gene defects to designer drugs? Nephrol Dial Transplant. 2005;20(8):1525-1529.
18. Rare Kidney Stone Consortium. Primary hyperoxaluria. www.rarekidneystones.org/hyperoxaluria. Accessed February 9, 2011.
19. Brinkert F, Ganschow R, Helmke, K, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87(9):1415:1421.
20. Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10(11):2493-2501.
21. Millan MT, Berquist WE, So SK, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation. 2003;76(10):1458-1463.
22. Watts RWE, Danpure CJ, De Pauw L, Toussaint C; European Study Group on Transplantation in Hyperoxaluria Type 1. Combined liver-kidney and isolated liver transplantations for primary hyperoxaluria type 1: the European experience. Nephrol Dial Transplant. 1991;6(7):502-511.
23. Broyer M, Jouvet P, Niaudet P, et al. Management of oxalosis. Kidney Int Suppl. 1996;53:S93-S98.
24. de Pauw L, Gelin M, Danpure CJ, et al. Combined liver-kidney transplantation in primary hyperoxaluria type 1. Transplantation. 1990;50(5):886-887.
25. Broyer M, Brunner FP, Brynger H, et al. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant. 1990;5(5):332-336.
A 26-year-old woman presented to a nephrology office in Virginia for a reevaluation and second opinion regarding her history of kidney stones. This condition had led to uremia and acute kidney failure, requiring hemodialysis.
Her history was significant for recurrent kidney stones and infections, beginning at age 12. Over the next six years, she passed at least five stones and underwent three lithotripsy procedures; according to the patient, however, neither she nor her parents were ever informed of any decrease in her kidney function. The patient said she had been told that her stones were composed of calcium oxalate, and she was placed on potassium citrate therapy but did not take the medication on a regular basis.
After high school, she left the area for college and for several years she frequently and spontaneously passed gravel and stones. She was a runner in high school and college and had two children without experiencing any hypertension, proteinuria, or stone problems during her pregnancies. She had been treated for numerous recurrent urinary tract infections in outpatient clinics and private offices during the 10 years leading up to her current presentation. She had a distant history of a cholecystectomy.
In May 2009, the patient was hospitalized for a kidney infection and underwent cystoscopy with a finding of left ureteral obstruction caused by a stone. A stent was placed, followed by lithotripsy. Her serum creatinine level was measured at 2.2 mg/dL at that time (normal range, 0.6 to 1.5 mg/dL). In August 2009, she was treated again for a kidney infection; a right-sided stone obstruction was noted at that time, and again a stent was placed and lithotripsy was performed. Her serum creatinine level was then 3.3 mg/dL. During these episodes, the patient’s calcium level ranged from 8.2 to 10.1 mg/dL (normal, 4.5 to 5.2 mg/dL). Her phosphorus level was noted to range from 2.6 to 9.5 mg/dL (normal, 2.5 to 4.5 mg/dL). Her intact parathyroid level was 354 pg/mL (normal, 10 to 60 pg/mL). Thus, she had documented secondary hyperparathyroidism, which was treated with paricalcitol and a phosphate binder.
In February 2010, the patient was “feeling poorly” and was taken to a local hospital in South Carolina. She was admitted in acute renal failure and started on dialysis. She did well on hemodialysis with little to no fluid gain and good urine volume. She returned to Virginia temporarily for treatment, to be closer to her family and to prepare for kidney transplantation. She had family members who were willing to donate an organ.
The patient’s family history was negative for gout, kidney disease, or kidney stones. No family member was known to have hypertension, diabetes, or enuresis.
Physical examination showed a thin white woman with a runner’s lean look. She denied laxative use. Her blood pressure was measured at 120/84 mm Hg, and her pulse, 96 beats/min. Findings in the skin/head/eyes/ears/nose/throat exam were within normal limits except for the presence of contact lenses and a subclavicular dialysis indwelling catheter. Neither thyroid enlargement nor supraclavicular adenopathy was noted. Her heart rate was regular without murmurs. The abdomen was soft and nontender without rebound. The extremities showed no edema. Neurologic and vascular findings were intact.
The most recent 24-hour urine study showed a urine creatinine clearance of 4 mL/min (normal, 85 to 125 mL/min), despite a very large urine volume. Renal ultrasonography revealed two small kidneys that were highly echogenic, with evidence of medullary nephrocalcinosis without obstruction bilaterally.
The presentation of a woman with a kidney stone load high enough to cause full kidney failure by age 26 led the nephrologist to suspect the presence of hyperoxaluria type 1 (primary) or type 2 (secondary). The patient’s urine oxalate level was 158 mcmol/L (normal, < 57 mcmol/L), and her plasma oxalate level was 73 mcmol/L (normal, < 10 mcmol/L).
In response to the patient’s high blood and urine oxalate levels and her interest in kidney transplantation, genetic testing was performed to determine whether she had type 1 or type 2 hyperoxaluria. If she was found to have type 1 hyperoxaluria, she would need a liver transplant before her body showered a newly transplanted kidney with stones, causing recurrent kidney failure.
Discussion
Primary hyperoxaluria (PHO) type 1 is a very rare recessive hereditary disease with a prevalence of one to three cases per one million persons.1 Patients typically present with kidney stones at an early age (as did the case patient) or in full kidney failure. It is calculated that PHO is responsible for 1% of all end-stage renal disease among pediatric patients.2,3
Stones are caused by a deficiency of the liver enzyme alanine-glyoxylate aminotransferase (AGXT), which ordinarily converts glyoxylate to glycine.2,4 When AGXT is absent, glyoxylate is converted instead to oxalate, which forms insoluble salts that accumulate in the kidney as oxalate kidney stones. Most patients (ie, 80% to 90%) present in late childhood or early adolescence with systems of recurrent stones and urinary tract infections resulting from blockage.5,6 The natural history of the disease is progression to kidney failure and death from end-stage renal disease unless dialysis is initiated.
While testing of oxalate-to-creatinine molar ratio in a random urine sample may be helpful, this measurement does not stabilize until age 14 to 18—often after kidney damage has already occurred.7 Liver biopsy can confirm whether the enzyme AGXT is absent. Differentiation between PHO and type 2 hyperoxaluria can only be confirmed by genetic testing in which the AGXT gene is identified.8
There is an increased incidence of PHO in Tunisia and Kuwait9-11 and in the Arab and Druze families of Israel12 as a result of intermarriages in this population. Since AGXT is a recessive gene, the child of parents who are both carriers has a 25% chance of having the disease. If either parent carries the genetic variant, there is a 50% chance that the recessive gene will be passed on.
Diagnosis
Early diagnosis of PHO is critical. However, because the disease is so rare, more than 40% of affected patients do not receive a diagnosis until three years after symptoms develop, and 30% are diagnosed only upon presentation with end-stage renal disease.2,13
If PHO is detected early, the key management goal is to minimize renal and skeletal oxalate deposition. Components of medical management are shown in the table.2,14-17 It is important to note that these strategies are effective only if initiated early, that is, before the patient’s glomerular filtration rate drops below 25 mL/min.18
Treatment
Organ transplantation remains the only definitive treatment for PHO14,19—to prevent severe systemic oxalosis or to manage the patient who has progressed to end-stage renal disease. Researchers from the Mayo Clinic in Rochester, Minnesota (where, it should be noted, a National Oxalosis and Hyperoxaluria Registry is maintained under the direction of Dawn S. Milliner, MD), recently published an observational study of outcomes in transplant graft survival among 203 PHO patients. Bergstralh et al20 reported high rates of recurrent oxalosis in patients undergoing kidney transplantation alone, and significantly improved outcomes in patients who underwent both liver and kidney transplantation.
Before 1990, according to a report by the Rare Kidney Stone Consortium,18 the prognosis for PHO transplant patients in the United States was so poor that a donor kidney was considered wasted on these patients. Since the year 2000, however, survival after transplantation has improved greatly, with rates similar to those of all kidney transplant patients nationwide. The explanation for increased survival rates among PHO patients undergoing transplantation was twofold:
• Increased preoperative stone control
• Use of combined liver-kidney transplants.21,22
Since the liver is responsible for the cascade of calcium oxalate stones, the native liver must be fully removed prior to transplantation of a new liver and kidney. Postoperatively, stones will also emerge from where they have lodged in the skeletal tissue to shower the new kidney. Thus, medical management of this cascade of new stones is vital if the transplanted grafts are to survive.23 Calcium oxalate blood levels can remain high for one to two years posttransplantation,2,24 so long-term medical management of oxalate is essential.
The Case Patient
Clinicians engaged in a discussion with the patient and her family regarding a possible diagnosis of PHO. Blood was drawn and sent to the Mayo Clinic for genetic analysis. It was found that the patient had an abnormality in the AGXT gene; with the diagnosis of type 1 hyperoxaluria confirmed, she was flown to Rochester for a full workup.
The patient was the only member of her family with the defective AGXT gene, and her genetic counselors considered this a single mutation. She was accepted for the liver/kidney transplantation list.
Due to the increase in reported survival among patients if they undergo transplantation early in the natural history of stone deposition, the average wait time for PHO patients is only three to four months. The case patient returned to the dialysis unit in Virginia, where she was placed on a dialysis regimen of five-hour treatments, five times per week (nighttime and day); this was determined to be the peak treatment duration for most efficient stone removal, as determined by calcium oxalate measurement during her workup at the Mayo Clinic.
This regimen was continued for three months, at which time the patient was nearing the top of the transplant waiting list. She returned to the Mayo Clinic in September 2010 and underwent transplantation in October; since then, she has regained excellent kidney function and experienced an immediate drop in her calcium oxalate levels. She remained in Rochester until late November, then returned to her home in South Carolina, where she continues to undergo follow-up at a local transplantation center.
The case patient was fortunate that an attending nephrologist at the nephrology office in Virginia developed a high clinical suspicion for her actual condition and started the workup that led to a diagnosis of PHO. She could well have been among the 19% of patients with PHO in whom the correct diagnosis is not reached until after a newly transplanted kidney has been showered with stones again,18,25 necessitating a second kidney transplant following the essential liver transplantation.
Before her current presentation, the patient had been under the care of another nephrologist and had spent six months on a transplant waiting list. If she had proceeded with her original plan, the scheduled kidney transplant (unaccompanied by the essential liver transplant) would have been ineffective, and her donor would have undergone major surgery to no good result.
Conclusion
Type 1 hyperoxaluria is a rare diagnosis that is frequently missed. According to data from the Rare Kidney Stone Consortium,18 nearly one-fifth of patients with PHO do not receive a correct diagnosis until after an unsuccessful kidney transplantation, as liver transplantation is initially required.
The author wishes to extend special thanks to Stephen G. Goldberger, MD, “for being such a good detective.”
References
1. Ajzensztejn MJ, Sebire NJ, Trompeter RS, Marks SD. Primary hyperoxaluria type 1. Arch Dis Child. 2007; 92(3):197.
2. Niaudet P. Primary hyperoxaluria (2010). www.uptodate.com/contents/primary-hyperoxaluria?source=search_result& selectedTitle=1%7E39. Accessed February 17, 2011.
3. Latta K, Brodehl J. Primary hyperoxaluria type I. Eur J Pediatr. 1990;149(8):518-522.
4. Danpure CJ. Advances in the enzymology and molecular genetics of primary hyperoxaluria type 1: prospects for gene therapy. Nephrol Dial Transplant. 1995;10 suppl 8:24-29.
5. Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290-296.
6. Genetics Home Reference. Primary hyperoxaluria. www.ghr.nlm.nih.gov/condition/primary-hyperoxaluria. Accessed February 17, 2011.
7. Remer T, Neubert A, Maser-Gluth C. Anthropometry-based reference values for 24-h urinary creatinine excretion during growth and their use in endocrine and nutritional research. Am J Clin Nutr. 2002;75(3):561-569.
8. Danpure CJ. Molecular and clinical heterogeneity in primary hyperoxaluria type 1. Am J Kidney Dis. 1991;17(4):366-369.
9. Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10(4):479-482.
10. Al-Eisa AA, Samhan M, Naseef M. End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc. 2004;36(6):1788-1791.
11. Cochat P, Liutkus A, Fargue S, et al. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol. 2006;21(8):1075-1081.
12. Rinat C, Wanders RJ, Drukker A, et al. Primary hyperoxaluria type I: a model for multiple mutations in a monogenic disease within a distinct ethnic group. J Am Soc Nephrol. 1999;10(11):2352-2358.
13. Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18(10):986-991.
14. Watts RW. Primary hyperoxaluria type I. QJM. 1994;87(10):593-600.
15. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria: the German experience. Am J Nephrol. 2005;25(3):276-281.
16. Milliner DS, Eickholt JT, Bergstralh EJ, et al. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331(23):1553-1558.
17. Danpure CJ. Primary hyperoxaluria: from gene defects to designer drugs? Nephrol Dial Transplant. 2005;20(8):1525-1529.
18. Rare Kidney Stone Consortium. Primary hyperoxaluria. www.rarekidneystones.org/hyperoxaluria. Accessed February 9, 2011.
19. Brinkert F, Ganschow R, Helmke, K, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87(9):1415:1421.
20. Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10(11):2493-2501.
21. Millan MT, Berquist WE, So SK, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation. 2003;76(10):1458-1463.
22. Watts RWE, Danpure CJ, De Pauw L, Toussaint C; European Study Group on Transplantation in Hyperoxaluria Type 1. Combined liver-kidney and isolated liver transplantations for primary hyperoxaluria type 1: the European experience. Nephrol Dial Transplant. 1991;6(7):502-511.
23. Broyer M, Jouvet P, Niaudet P, et al. Management of oxalosis. Kidney Int Suppl. 1996;53:S93-S98.
24. de Pauw L, Gelin M, Danpure CJ, et al. Combined liver-kidney transplantation in primary hyperoxaluria type 1. Transplantation. 1990;50(5):886-887.
25. Broyer M, Brunner FP, Brynger H, et al. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant. 1990;5(5):332-336.
A 26-year-old woman presented to a nephrology office in Virginia for a reevaluation and second opinion regarding her history of kidney stones. This condition had led to uremia and acute kidney failure, requiring hemodialysis.
Her history was significant for recurrent kidney stones and infections, beginning at age 12. Over the next six years, she passed at least five stones and underwent three lithotripsy procedures; according to the patient, however, neither she nor her parents were ever informed of any decrease in her kidney function. The patient said she had been told that her stones were composed of calcium oxalate, and she was placed on potassium citrate therapy but did not take the medication on a regular basis.
After high school, she left the area for college and for several years she frequently and spontaneously passed gravel and stones. She was a runner in high school and college and had two children without experiencing any hypertension, proteinuria, or stone problems during her pregnancies. She had been treated for numerous recurrent urinary tract infections in outpatient clinics and private offices during the 10 years leading up to her current presentation. She had a distant history of a cholecystectomy.
In May 2009, the patient was hospitalized for a kidney infection and underwent cystoscopy with a finding of left ureteral obstruction caused by a stone. A stent was placed, followed by lithotripsy. Her serum creatinine level was measured at 2.2 mg/dL at that time (normal range, 0.6 to 1.5 mg/dL). In August 2009, she was treated again for a kidney infection; a right-sided stone obstruction was noted at that time, and again a stent was placed and lithotripsy was performed. Her serum creatinine level was then 3.3 mg/dL. During these episodes, the patient’s calcium level ranged from 8.2 to 10.1 mg/dL (normal, 4.5 to 5.2 mg/dL). Her phosphorus level was noted to range from 2.6 to 9.5 mg/dL (normal, 2.5 to 4.5 mg/dL). Her intact parathyroid level was 354 pg/mL (normal, 10 to 60 pg/mL). Thus, she had documented secondary hyperparathyroidism, which was treated with paricalcitol and a phosphate binder.
In February 2010, the patient was “feeling poorly” and was taken to a local hospital in South Carolina. She was admitted in acute renal failure and started on dialysis. She did well on hemodialysis with little to no fluid gain and good urine volume. She returned to Virginia temporarily for treatment, to be closer to her family and to prepare for kidney transplantation. She had family members who were willing to donate an organ.
The patient’s family history was negative for gout, kidney disease, or kidney stones. No family member was known to have hypertension, diabetes, or enuresis.
Physical examination showed a thin white woman with a runner’s lean look. She denied laxative use. Her blood pressure was measured at 120/84 mm Hg, and her pulse, 96 beats/min. Findings in the skin/head/eyes/ears/nose/throat exam were within normal limits except for the presence of contact lenses and a subclavicular dialysis indwelling catheter. Neither thyroid enlargement nor supraclavicular adenopathy was noted. Her heart rate was regular without murmurs. The abdomen was soft and nontender without rebound. The extremities showed no edema. Neurologic and vascular findings were intact.
The most recent 24-hour urine study showed a urine creatinine clearance of 4 mL/min (normal, 85 to 125 mL/min), despite a very large urine volume. Renal ultrasonography revealed two small kidneys that were highly echogenic, with evidence of medullary nephrocalcinosis without obstruction bilaterally.
The presentation of a woman with a kidney stone load high enough to cause full kidney failure by age 26 led the nephrologist to suspect the presence of hyperoxaluria type 1 (primary) or type 2 (secondary). The patient’s urine oxalate level was 158 mcmol/L (normal, < 57 mcmol/L), and her plasma oxalate level was 73 mcmol/L (normal, < 10 mcmol/L).
In response to the patient’s high blood and urine oxalate levels and her interest in kidney transplantation, genetic testing was performed to determine whether she had type 1 or type 2 hyperoxaluria. If she was found to have type 1 hyperoxaluria, she would need a liver transplant before her body showered a newly transplanted kidney with stones, causing recurrent kidney failure.
Discussion
Primary hyperoxaluria (PHO) type 1 is a very rare recessive hereditary disease with a prevalence of one to three cases per one million persons.1 Patients typically present with kidney stones at an early age (as did the case patient) or in full kidney failure. It is calculated that PHO is responsible for 1% of all end-stage renal disease among pediatric patients.2,3
Stones are caused by a deficiency of the liver enzyme alanine-glyoxylate aminotransferase (AGXT), which ordinarily converts glyoxylate to glycine.2,4 When AGXT is absent, glyoxylate is converted instead to oxalate, which forms insoluble salts that accumulate in the kidney as oxalate kidney stones. Most patients (ie, 80% to 90%) present in late childhood or early adolescence with systems of recurrent stones and urinary tract infections resulting from blockage.5,6 The natural history of the disease is progression to kidney failure and death from end-stage renal disease unless dialysis is initiated.
While testing of oxalate-to-creatinine molar ratio in a random urine sample may be helpful, this measurement does not stabilize until age 14 to 18—often after kidney damage has already occurred.7 Liver biopsy can confirm whether the enzyme AGXT is absent. Differentiation between PHO and type 2 hyperoxaluria can only be confirmed by genetic testing in which the AGXT gene is identified.8
There is an increased incidence of PHO in Tunisia and Kuwait9-11 and in the Arab and Druze families of Israel12 as a result of intermarriages in this population. Since AGXT is a recessive gene, the child of parents who are both carriers has a 25% chance of having the disease. If either parent carries the genetic variant, there is a 50% chance that the recessive gene will be passed on.
Diagnosis
Early diagnosis of PHO is critical. However, because the disease is so rare, more than 40% of affected patients do not receive a diagnosis until three years after symptoms develop, and 30% are diagnosed only upon presentation with end-stage renal disease.2,13
If PHO is detected early, the key management goal is to minimize renal and skeletal oxalate deposition. Components of medical management are shown in the table.2,14-17 It is important to note that these strategies are effective only if initiated early, that is, before the patient’s glomerular filtration rate drops below 25 mL/min.18
Treatment
Organ transplantation remains the only definitive treatment for PHO14,19—to prevent severe systemic oxalosis or to manage the patient who has progressed to end-stage renal disease. Researchers from the Mayo Clinic in Rochester, Minnesota (where, it should be noted, a National Oxalosis and Hyperoxaluria Registry is maintained under the direction of Dawn S. Milliner, MD), recently published an observational study of outcomes in transplant graft survival among 203 PHO patients. Bergstralh et al20 reported high rates of recurrent oxalosis in patients undergoing kidney transplantation alone, and significantly improved outcomes in patients who underwent both liver and kidney transplantation.
Before 1990, according to a report by the Rare Kidney Stone Consortium,18 the prognosis for PHO transplant patients in the United States was so poor that a donor kidney was considered wasted on these patients. Since the year 2000, however, survival after transplantation has improved greatly, with rates similar to those of all kidney transplant patients nationwide. The explanation for increased survival rates among PHO patients undergoing transplantation was twofold:
• Increased preoperative stone control
• Use of combined liver-kidney transplants.21,22
Since the liver is responsible for the cascade of calcium oxalate stones, the native liver must be fully removed prior to transplantation of a new liver and kidney. Postoperatively, stones will also emerge from where they have lodged in the skeletal tissue to shower the new kidney. Thus, medical management of this cascade of new stones is vital if the transplanted grafts are to survive.23 Calcium oxalate blood levels can remain high for one to two years posttransplantation,2,24 so long-term medical management of oxalate is essential.
The Case Patient
Clinicians engaged in a discussion with the patient and her family regarding a possible diagnosis of PHO. Blood was drawn and sent to the Mayo Clinic for genetic analysis. It was found that the patient had an abnormality in the AGXT gene; with the diagnosis of type 1 hyperoxaluria confirmed, she was flown to Rochester for a full workup.
The patient was the only member of her family with the defective AGXT gene, and her genetic counselors considered this a single mutation. She was accepted for the liver/kidney transplantation list.
Due to the increase in reported survival among patients if they undergo transplantation early in the natural history of stone deposition, the average wait time for PHO patients is only three to four months. The case patient returned to the dialysis unit in Virginia, where she was placed on a dialysis regimen of five-hour treatments, five times per week (nighttime and day); this was determined to be the peak treatment duration for most efficient stone removal, as determined by calcium oxalate measurement during her workup at the Mayo Clinic.
This regimen was continued for three months, at which time the patient was nearing the top of the transplant waiting list. She returned to the Mayo Clinic in September 2010 and underwent transplantation in October; since then, she has regained excellent kidney function and experienced an immediate drop in her calcium oxalate levels. She remained in Rochester until late November, then returned to her home in South Carolina, where she continues to undergo follow-up at a local transplantation center.
The case patient was fortunate that an attending nephrologist at the nephrology office in Virginia developed a high clinical suspicion for her actual condition and started the workup that led to a diagnosis of PHO. She could well have been among the 19% of patients with PHO in whom the correct diagnosis is not reached until after a newly transplanted kidney has been showered with stones again,18,25 necessitating a second kidney transplant following the essential liver transplantation.
Before her current presentation, the patient had been under the care of another nephrologist and had spent six months on a transplant waiting list. If she had proceeded with her original plan, the scheduled kidney transplant (unaccompanied by the essential liver transplant) would have been ineffective, and her donor would have undergone major surgery to no good result.
Conclusion
Type 1 hyperoxaluria is a rare diagnosis that is frequently missed. According to data from the Rare Kidney Stone Consortium,18 nearly one-fifth of patients with PHO do not receive a correct diagnosis until after an unsuccessful kidney transplantation, as liver transplantation is initially required.
The author wishes to extend special thanks to Stephen G. Goldberger, MD, “for being such a good detective.”
References
1. Ajzensztejn MJ, Sebire NJ, Trompeter RS, Marks SD. Primary hyperoxaluria type 1. Arch Dis Child. 2007; 92(3):197.
2. Niaudet P. Primary hyperoxaluria (2010). www.uptodate.com/contents/primary-hyperoxaluria?source=search_result& selectedTitle=1%7E39. Accessed February 17, 2011.
3. Latta K, Brodehl J. Primary hyperoxaluria type I. Eur J Pediatr. 1990;149(8):518-522.
4. Danpure CJ. Advances in the enzymology and molecular genetics of primary hyperoxaluria type 1: prospects for gene therapy. Nephrol Dial Transplant. 1995;10 suppl 8:24-29.
5. Lieske JC, Monico CG, Holmes WS, et al. International registry for primary hyperoxaluria. Am J Nephrol. 2005;25(3):290-296.
6. Genetics Home Reference. Primary hyperoxaluria. www.ghr.nlm.nih.gov/condition/primary-hyperoxaluria. Accessed February 17, 2011.
7. Remer T, Neubert A, Maser-Gluth C. Anthropometry-based reference values for 24-h urinary creatinine excretion during growth and their use in endocrine and nutritional research. Am J Clin Nutr. 2002;75(3):561-569.
8. Danpure CJ. Molecular and clinical heterogeneity in primary hyperoxaluria type 1. Am J Kidney Dis. 1991;17(4):366-369.
9. Kamoun A, Lakhoua R. End-stage renal disease of the Tunisian child: epidemiology, etiologies, and outcome. Pediatr Nephrol. 1996;10(4):479-482.
10. Al-Eisa AA, Samhan M, Naseef M. End-stage renal disease in Kuwaiti children: an 8-year experience. Transplant Proc. 2004;36(6):1788-1791.
11. Cochat P, Liutkus A, Fargue S, et al. Primary hyperoxaluria type 1: still challenging! Pediatr Nephrol. 2006;21(8):1075-1081.
12. Rinat C, Wanders RJ, Drukker A, et al. Primary hyperoxaluria type I: a model for multiple mutations in a monogenic disease within a distinct ethnic group. J Am Soc Nephrol. 1999;10(11):2352-2358.
13. Hoppe B, Langman CB. A United States survey on diagnosis, treatment, and outcome of primary hyperoxaluria. Pediatr Nephrol. 2003;18(10):986-991.
14. Watts RW. Primary hyperoxaluria type I. QJM. 1994;87(10):593-600.
15. Hoppe B, Latta K, von Schnakenburg C, Kemper MJ. Primary hyperoxaluria: the German experience. Am J Nephrol. 2005;25(3):276-281.
16. Milliner DS, Eickholt JT, Bergstralh EJ, et al. Results of long-term treatment with orthophosphate and pyridoxine in patients with primary hyperoxaluria. N Engl J Med. 1994;331(23):1553-1558.
17. Danpure CJ. Primary hyperoxaluria: from gene defects to designer drugs? Nephrol Dial Transplant. 2005;20(8):1525-1529.
18. Rare Kidney Stone Consortium. Primary hyperoxaluria. www.rarekidneystones.org/hyperoxaluria. Accessed February 9, 2011.
19. Brinkert F, Ganschow R, Helmke, K, et al. Transplantation procedures in children with primary hyperoxaluria type 1: outcome and longitudinal growth. Transplantation. 2009;87(9):1415:1421.
20. Bergstralh EJ, Monico CG, Lieske JC, et al; IPHR Investigators. Transplantation outcomes in primary hyperoxaluria. Am J Transplant. 2010;10(11):2493-2501.
21. Millan MT, Berquist WE, So SK, et al. One hundred percent patient and kidney allograft survival with simultaneous liver and kidney transplantation in infants with primary hyperoxaluria: a single-center experience. Transplantation. 2003;76(10):1458-1463.
22. Watts RWE, Danpure CJ, De Pauw L, Toussaint C; European Study Group on Transplantation in Hyperoxaluria Type 1. Combined liver-kidney and isolated liver transplantations for primary hyperoxaluria type 1: the European experience. Nephrol Dial Transplant. 1991;6(7):502-511.
23. Broyer M, Jouvet P, Niaudet P, et al. Management of oxalosis. Kidney Int Suppl. 1996;53:S93-S98.
24. de Pauw L, Gelin M, Danpure CJ, et al. Combined liver-kidney transplantation in primary hyperoxaluria type 1. Transplantation. 1990;50(5):886-887.
25. Broyer M, Brunner FP, Brynger H, et al. Kidney transplantation in primary oxalosis: data from the EDTA Registry. Nephrol Dial Transplant. 1990;5(5):332-336.
Woman with Severe Ankle Pain After Fall
ANSWER
There is an obvious oblique fracture of the distal fibula. In addition, there is widening of the mortise medially—usually suggestive of a ligament injury. Also, there may be a small avulsion at the inferior aspect of the medial malleolus.
The patient was admitted with a plan for her to undergo an open reduction and internal fixation procedure.
ANSWER
There is an obvious oblique fracture of the distal fibula. In addition, there is widening of the mortise medially—usually suggestive of a ligament injury. Also, there may be a small avulsion at the inferior aspect of the medial malleolus.
The patient was admitted with a plan for her to undergo an open reduction and internal fixation procedure.
ANSWER
There is an obvious oblique fracture of the distal fibula. In addition, there is widening of the mortise medially—usually suggestive of a ligament injury. Also, there may be a small avulsion at the inferior aspect of the medial malleolus.
The patient was admitted with a plan for her to undergo an open reduction and internal fixation procedure.
A 39-year-old woman presents to your emergency department complaining of right ankle pain following an injury last night. She states that she got out of bed during the night, somehow misstepped, and fell to the floor. She is unable to bear weight on the ankle and is experiencing severe pain. Her medical history is significant for being one month status post Chiari decompression. She also has a history of bipolar disorder. Examination of the patient’s right ankle shows it to be moderately swollen with some bruising laterally. The patient has limited flexion and extension, and her ankle is extremely tender to palpation. Her distal pulses and her sensation are intact. Radiographs of the right ankle are shown. What is your impression?
Shoulder Pain in Man Hospitalized for Brain Mass
ANSWER
There is no evidence of an acute fracture or dislocation. However, there is a focal lytic lesion within the scapula. Given a presumed history of renal cell carcinoma, this finding is strongly suspicious for metastasis and must be worked up.
ANSWER
There is no evidence of an acute fracture or dislocation. However, there is a focal lytic lesion within the scapula. Given a presumed history of renal cell carcinoma, this finding is strongly suspicious for metastasis and must be worked up.
ANSWER
There is no evidence of an acute fracture or dislocation. However, there is a focal lytic lesion within the scapula. Given a presumed history of renal cell carcinoma, this finding is strongly suspicious for metastasis and must be worked up.
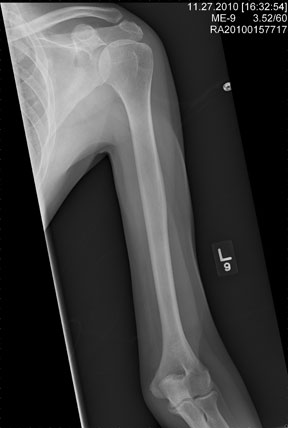
A 53-year-old man is admitted with a possible brain mass. He gives a two-week history of headaches and intermittent left-sided weakness. Outpatient CT of the head showed a possible right frontal mass. His medical history is significant for mild hypertension and for the removal of one kidney seven years ago, due to cancer. He states he received no adjuvant therapy. When you check on the patient today, he mentions that his left shoulder has been bothering him for some time. He denies any injury to it. He states he has decreased range of motion and pain with range of motion. You order a radiograph of the shoulder (shown). What is your impression?