User login
Updates in the medical management of Parkinson disease
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING

Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
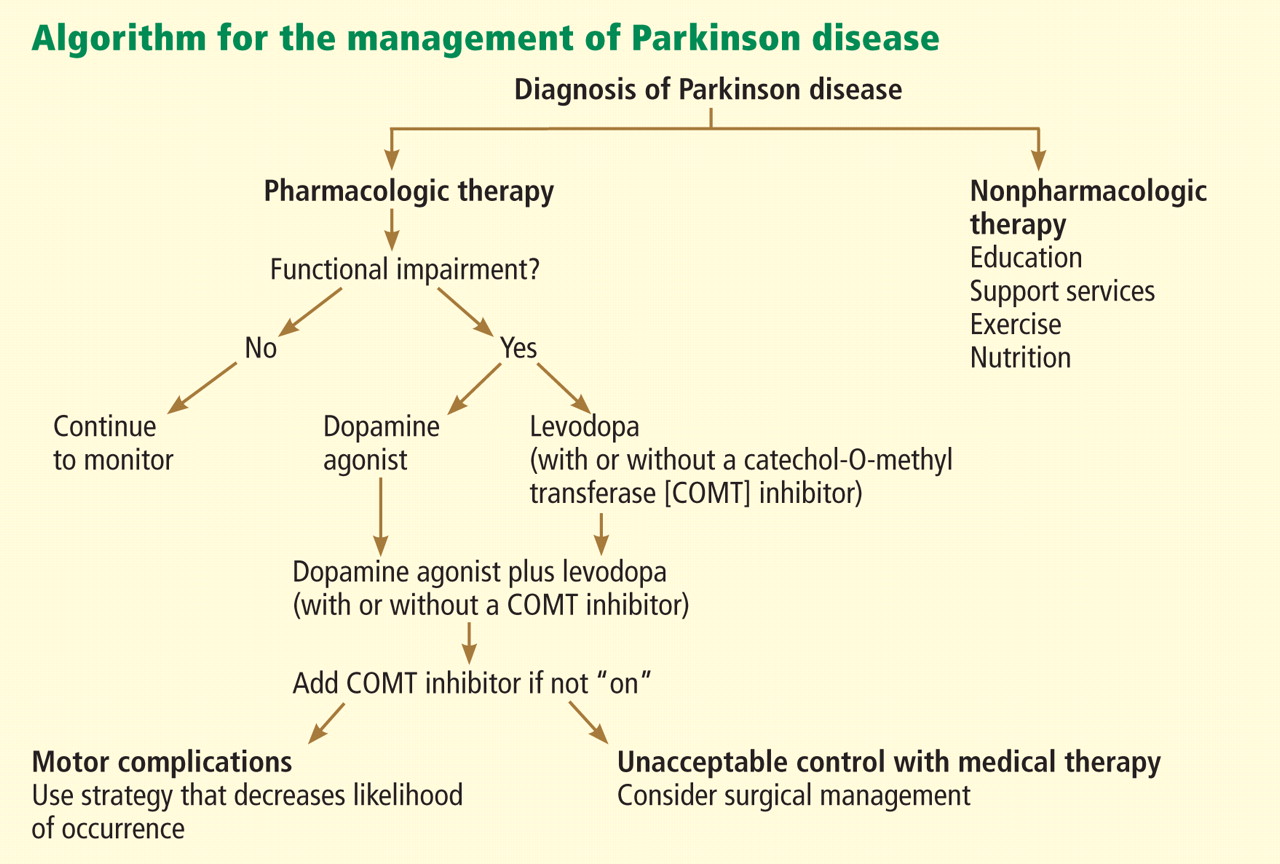
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
More than a dozen drugs have been approved by the US Food and Drug Administration (FDA) for treating Parkinson disease, and more are expected in the near future. Many are currently in clinical trials, with the goals of finding ways to better control the disease with fewer adverse effects and, ultimately, to provide neuroprotection.
This article will review the features of Parkinson disease, the treatment options, and the complications in moderate to advanced disease.
PARKINSON DISEASE IS MULTIFACTORIAL
Although the cure for Parkinson disease is still elusive, much has been learned over the nearly 200 years since it was first described by James Parkinson in 1817. It is now understood to be a progressive neurodegenerative disease of multifactorial etiology: although a small proportion of patients have a direct inherited mutation that causes it, multiple genetic predisposition factors and environmental factors are more commonly involved.
The central pathology is dopaminergic loss in the basal ganglia, but other neurotransmitters are also involved and the disease extends to other areas of the brain.
CARDINAL MOTOR SYMPTOMS
In general, Parkinson disease is easy to identify. The classic patient has1:
- Tremor at rest, which can be subtle—such as only involving a thumb or a few fingers—and is absent in 20% of patients at presentation.
- Rigidity, which is felt by the examiner rather than seen by an observer.
- Bradykinesia (slow movements), which is characteristic of all Parkinson patients.
- Gait and balance problems, which usually arise after a few years, although occasionally patients present with them. Patients typically walk with small steps with occasional freezing, as if their foot were stuck. Balance problems are the most difficult to treat among the motor problems.
Asymmetry of motor problems is apparent in 75% of patients at presentation, although problems become bilateral later in the course of the disease.
NONMOTOR FEATURES CAN BE MORE DISABLING
Pain is common, but years ago it was not recognized as a specific feature of Parkinson disease. The pain from other conditions may also worsen.
Fatigue is very common and, if present, is usually one of the most disabling features.
Neuropsychiatric disturbances are among the most difficult problems, and they become increasingly common as motor symptoms are better controlled with treatment and patients live longer.
INCREASINGLY PREVALENT AS THE POPULATION AGES
Parkinson disease can present from the teenage years up to age 90, but it is most often diagnosed in patients from 60 to 70 years old (mean onset, 62.5 years). A different nomenclature is used depending on the age of onset:
- 10 to 20 years: juvenile-onset
- 21 to 40 years: young-onset.
Parkinson disease is now an epidemic, with an estimated 1 million people having it in the United States, representing 0.3% of the population and 1% of those older than 60 years.2 More people can be expected to develop it as our population ages in the next decades. It is estimated that in 2040 more people will die from Parkinson disease, Alzheimer disease, and amyotrophic lateral sclerosis (all of which are neurodegenerative diseases) than from kidney cancer, malignant melanoma, colon cancer, and lung cancer combined.
DIAGNOSIS IS STILL MAINLY CLINICAL
The diagnosis of Parkinson disease remains clinical. In addition to the motor features, the best test is a clear response to dopaminergic treatment with levodopa. If all these features are present, the diagnosis of Parkinson disease is usually correct.3
Imaging useful in select patients
The FDA recently approved a radiopharmaceutical contrast agent, DaTscan, to use with single-photon emission computed tomography (SPECT) to help diagnose Parkinson disease. DaTscan is a dopamine transporter ligand that tags presynaptic dopaminergic neurons in the basal ganglia; a patient with Parkinson disease has less signal.
The test can be used to distinguish parkinsonian syndromes from disorders that can mimic them, such as essential tremor or a psychogenic disorder. However, it cannot differentiate various Parkinson-plus syndromes (see below) such as multiple system atrophy or progressive nuclear palsy. It also cannot be used to detect drug-induced or vascular parkinsonism.
Check for Wilson disease or brain tumors in young or atypical cases
For most patients, no imaging or blood tests are needed to make the diagnosis. However, in patients younger than 50, Wilson disease, a rare inherited disorder characterized by excess copper accumulation, must be considered. Testing for Wilson disease includes serum ceruloplasmin, 24-hour urinary copper excretion, and an ophthalmologic slit-lamp examination for Kaiser-Fleischer rings.
For patients who do not quite fit the picture of Parkinson disease, such as those who have spasticity with little tremor, or who have a minimal response to levodopa, magnetic resonance imaging should be done to see if a structural lesion is present.
Consider secondary parkinsonism
Although idiopathic Parkinson disease is by far the most common form of parkinsonism in the United States and in most developing countries, secondary causes must also be considered in a patient presenting with symptoms of parkinsonism. They include:
- Dopamine-receptor blocking agents: metoclopramide (Reglan), prochlorperazine (Compazine), haloperidol (Haldol), thioridazine (Mellaril), risperidone (Risperdal), olanzapine (Zyprexa)
- Strokes in the basal ganglia
- Normal pressure hydrocephalus.
Parkinson-plus syndromes
Parkinson-plus syndromes have other features in addition to the classic features of idiopathic Parkinson disease. They occur commonly and can be difficult to distinguish from Parkinson disease and from each other.
Parkinson-plus syndromes include:
- Progressive supranuclear palsy
- Multiple system atrophy
- Corticobasal degeneration
- Lewy body dementia.
Clinical features that suggest a diagnosis other than Parkinson disease include poor response to adequate dosages of levodopa, early onset of postural instability, axial more than appendicular rigidity, early dementia, and inability to look up or down without needing to move the head (supranuclear palsy).4
MANAGING PARKINSON DISEASE
Nonpharmacologic therapy is very important. Because patients tend to live longer because of better treatment, education is particularly important. The benefits of exercise go beyond general conditioning and cardiovascular health. People who exercise vigorously at least three times a week for 30 to 45 minutes are less likely to develop Parkinson disease and, if they develop it, they tend to have slower progression.
Prevention with neuroprotective drugs is not yet an option but hopefully will be in the near future.
Drug treatment generally starts when the patient is functionally impaired. If so, either levodopa or a dopamine agonist is started, depending on the patient’s age and the severity of symptoms. With increasing severity, other drugs can be added, and when those fail to control symptoms, surgery should be considered.
Deep brain stimulation surgery can make a tremendous difference in a patient’s quality of life. Other than levodopa, it is probably the best therapy available; however, it is very expensive and is not without risks.
Levodopa: The most effective drug, until it wears off
All current drugs for Parkinson disease activate dopamine neurotransmission in the brain. The most effective—and the cheapest—is still carbidopa/levodopa (Sinemet, Parcopa, Atamet). Levodopa converts to dopamine both peripherally and after it crosses the blood-brain barrier. Carbidopa prevents the peripheral conversion of levodopa to dopamine, reducing the peripheral adverse effects of levodopa, such as nausea and vomiting. The combination drug is usually given three times a day, with different doses available (10 mg carbidopa/100 mg levodopa, 25/100, 50/200, and 25/250) and as immediate-release and controlled-release formulations as well as an orally dissolving form (Parcopa) for patients with difficulty swallowing.
The major problem with levodopa is that after 4 to 6 years of treatment, about 40% of patients develop motor fluctuations and dyskinesias.5 If treatment is started too soon or at too high a dose, these problems tend to develop even earlier, especially among younger patients.
Motor fluctuations can take many forms: slow wearing-off, abrupt loss of effectiveness, and random on-and-off effectiveness (“yo-yoing”).
Dyskinesias typically involve constant chorea (dance-like) movements and occur at peak dose. Although chorea is easily treated by lowering the dosage, patients generally prefer having these movements rather than the Parkinson symptoms that recur from underdosing.
Dopamine agonists may be best for younger patients in early stages
The next most effective class of drugs are the dopamine agonists: pramipexole (Mirapex), ropinirole (Requip), and bromocriptine (Parlodel). A fourth drug, pergolide, is no longer available because of associated valvular heart complications. Each can be used as monotherapy in mild, early Parkinson disease or as an additional drug for moderate to severe disease. They are longer-acting than levodopa and can be taken once daily. Although they are less likely than levodopa to cause wearing-off or dyskinesias, they are associated with more nonmotor side effects: nausea and vomiting, hallucinations, confusion, somnolence or sleep attacks, low blood pressure, edema, and impulse control disorders.
Multiple clinical trials have been conducted to test the efficacy of dopamine agonists vs levodopa for treating Parkinson disease.6–9 Almost always, levodopa is more effective but involves more wearing-off and dyskinesias. For this reason, for patients with milder parkinsonism who may not need the strongest drug available, trying one of the dopamine agonists first may be worthwhile.
In addition, patients younger than age 60 are more prone to develop motor fluctuations and dyskinesias, so a dopamine agonist should be tried first in patients in that age group. For patients over age 65 for whom cost may be of concern, levodopa is the preferred starting drug.
Anticholinergic drugs for tremor
Before 1969, only anticholinergic drugs were available to treat Parkinson disease. Examples include trihexyphenidyl (Artane, Trihexane) and benztropine (Cogentin). These drugs are effective for treating tremor and drooling but are much less useful against rigidity, bradykinesia, and balance problems. Side effects include confusion, dry mouth, constipation, blurred vision, urinary retention, and cognitive impairment.
Anticholinergics should only be considered for young patients in whom tremor is a large problem and who have not responded well to the traditional Parkinson drugs. Because tremor is mostly a cosmetic problem, anticholinergics can also be useful for treating actors, musicians, and other patients with a public role.
Monoamine oxidase B inhibitors are well tolerated but less effective
In the brain, dopamine is broken down by monoamine oxidase B (MAO-B); therefore, inhibiting this enzyme increases dopamine’s availability. The MAO-B inhibitors selegiline (Eldepryl, Zelapar) and rasagiline (Azilect) are effective for monotherapy for Parkinson disease but are not as effective as levodopa. Most physicians feel MAO-B inhibitors are also less effective than dopamine agonists, although double-blind, randomized clinical trials have not proven this.6,10,11
MAO-B inhibitors have a long half-life, allowing once-daily dosing, and they are very well tolerated, with a side-effect profile similar to that of placebo. As with all MAO inhibitors, caution is needed regarding drug and food interactions.
EFFECTIVE NEUROPROTECTIVE AGENTS REMAIN ELUSIVE
Although numerous drugs are now available to treat the symptoms of Parkinson disease, the ability to slow the progression of the disease remains elusive. The only factor consistently shown by epidemiologic evidence to be protective is cigarette smoking, but we don’t recommend it.
A number of agents have been tested for neuroprotective efficacy:
Coenzyme Q10 has been tested at low and high dosages but was not found to be effective.
Pramipexole, a dopamine agonist, has also been studied without success.
Creatine is currently being studied and shows promise, possibly because of its effects on complex-I, part of the electron transport chain in mitochondria, which may be disrupted in Parkinson disease.
Inosine, which elevates uric acid, is also promising. The link between high uric acid and Parkinson disease was serendipitously discovered: when evaluating numerous blood panels taken from patients with Parkinson disease who were in clinical trials (using what turned out to be ineffective agents), it was noted that patients with the slowest progression of disease tended to have the highest uric acid levels. This has led to trials evaluating the effect of elevating uric acid to a pre-gout threshold.
Calcium channel blockers may be protective, according to epidemiologic evidence. Experiments involving injecting isradipine (DynaCirc) in rat models of Parkinson disease have indicated that the drug is promising.
Rasagiline: Protective effects still unknown
A large study of the neuroprotective effects of the MAO-B inhibitor rasagiline has just been completed, but the results are uncertain.12 A unique “delayed-start” clinical trial design was used to try to evaluate whether this agent that is known to reduce symptoms may also be neuroprotective. More than 1,000 people with untreated Parkinson disease from 14 countries were randomly assigned to receive rasagiline (the early-start group) or placebo (the delayed-start group) for 36 weeks. Afterward, both groups were given rasagiline for another 36 weeks. Rasagiline was given in a daily dose of either 1 mg or 2 mg.
The investigators anticipated that if the benefits of rasagiline were purely symptomatic, the early- and delayed-start groups would have equivalent disease severity at the end of the study. If rasagiline were protective, the early-start group would be better off at the end of the study. Unfortunately, the results were ambiguous: the early- and delayed-start groups were equivalent at the end of the study if they received the 2-mg daily dose, apparently indicating no protective effect. But at the 1-mg daily dose, the delayed-start group developed more severe disease at 36 weeks and did not catch up to the early-start group after treatment with rasagiline, apparently indicating a protective benefit. As a result, no definitive conclusion can be drawn.
EXTENDING TREATMENT EFFECTS IN ADVANCED PARKINSON DISEASE
For most patients, the first 5 years after being diagnosed with Parkinson disease is the “honeymoon phase,” when almost any treatment is effective. During this time, patients tend to have enough surviving dopaminergic neurons to store levodopa, despite its very short half-life of only 60 minutes.
As the disease progresses, fewer dopaminergic neurons survive, the therapeutic window narrows, and dosing becomes a balancing act: too much dopamine causes dyskinesias, hallucinations, delusions, and impulsive behavior, and too little dopamine causes worsening of Parkinson symptoms, freezing, and wearing-off, with ensuing falls and fractures. At this stage, some patients are prescribed levodopa every 1.5 or 2 hours.
Drugs are now available that extend the half-life of levodopa by slowing the breakdown of dopamine.
Catechol-O-methyltransferase (COMT) inhibitors—including tolcapone (Tasmar) and entacapone (Comtan) (also available as combined cardidopa, entacapone, and levodopa [Stalevo])—reduce off periods by about 1 hour per day.13 Given that the price is about $2,500 per year, the cost and benefits to the patient must be considered.14–17
Rasagiline, an MAO-B inhibitor, can also be added to levodopa to extend the “on” time for about 1 hour a day and to reduce freezing of gait. Clinical trials have shown it to be well tolerated, although common side effects include worsening dyskinesias and nausea.18,19
Apomorphine (Apokyn) is a dopamine agonist given by subcutaneous injection, allowing it to avoid first-pass metabolism by the liver. The benefits start just 10 minutes after injection, but only last for about 1 hour. It is a good option for rescue therapy for patients who cannot swallow or who have severe, unpredictable, or painful off-periods. It is also useful for situations in which it is especially inconvenient to have an off-period, such as being away from home.
Many agents have been tested for improving the off-period, but most work for about 1 to 2 hours, which is not nearly as effective as deep brain stimulation.
Managing dyskinesias
Dyskinesias can be managed by giving lower doses of levodopa more often. If wearing-off is a problem, a dopamine agonist or MAO-B inhibitor can be added. For patients at this stage, a specialist should be consulted.
Amantadine (Symmetrel), an N-methyl-d-aspartate (NMDA) receptor antagonist and dopamine-releasing agent used to treat influenza, is also effective against dyskinesias. Adverse effects include anxiety, insomnia, nightmares, anticholinergic effects, and livedo reticularis.20,21
Deep brain stimulation is the best treatment for dyskinesias in a patient for whom the procedure is appropriate and who has medical insurance that covers it.
NONMOTOR FEATURES OF PARKINSON DISEASE
Dementia: One of the most limiting nonmotor features
Often the most limiting nonmotor feature of Parkinson disease is dementia, which develops at about four to six times the rate for age-matched controls. At a given time, about 40% of patients with Parkinson disease have dementia, and the risk is 80% over 15 years of the disease.
If dementia is present, many of the drugs effective against Parkinson disease cannot be used because of exacerbating side effects. Treatment is mainly restricted to levodopa.
The only FDA-approved drug to treat dementia in Parkinson disease is the same drug for Alzheimer disease, rivastigmine (Exelon). Its effects are only modest, and its cholinergic side effects may transiently worsen parkinsonian features.22
Psychosis: Also very common
About half of patients with Parkinson disease have an episode of hallucinations or delusions in their lifetime, and about 20% are actively psychotic at any time. Delusions typically have the theme of spousal infidelity. Psychosis is associated with a higher rate of death compared with patients with Parkinson disease who do not develop it. Rebound psychosis may occur on withdrawal of antipsychotic medication.23–27
Patients who develop psychosis should have a physical examination and laboratory evaluation to determine if an infection or electrolyte imbalance is the cause. Medications should be discontinued in the following order: anticholinergic drug, amantadine, MAO-B inhibitor, dopamine agonist, and COMT inhibitor. Levodopa and carbidopa should be reduced to the minimum tolerable yet effective dosages.
For a patient who still has psychosis despite a minimum Parkinson drug regimen, an atypical antipsychotic drug should be used. Although clozapine (Clozaril, FazaClo) is very effective without worsening parkinsonism, it requires weekly monitoring with a complete blood count because of the small (< 1%) risk of agranulocytosis. For that reason, the first-line drug is quetiapine (Seroquel). Most double-blind studies have not found it to be effective, yet it is the drug most often used. No other antipsychotic drugs are safe to treat Parkinson psychosis.
Many patients with Parkinson disease who are hospitalized become agitated and confused soon after they are admitted to the hospital. The best treatment is quetiapine if an oral drug can be prescribed. A benzodiazepine—eg, clonazepam (Klonopin), lorazepam (Ativan), diazepam (Valium)—at a low dose may also be effective. Haloperidol, risperidone, and olanzapine should not be given, as they block dopamine receptors and worsen rigidity.
Mood disturbances
Depression occurs in about half of patients with Parkinson disease and is a significant cause of functional impairment. About 25% of patients have anxiety, and 20% are apathetic.
Depression appears to be secondary to underlying neuroanatomic degeneration rather than a reaction to disability.28 Fortunately, most antidepressants are effective in patients with Parkinson disease.29,30 Bupropion (Wellbutrin) is a dopamine reuptake inhibitor and so increases the availability of dopamine, and it should also have antiparkinsonian effects, but unfortunately it does not. Conversely, selective serotonin reuptake inhibitors (SSRIs) theoretically can worsen or cause parkinsonism, but evidence shows that they are safe to use in patients with Parkinson disease. Some evidence indicates that tricyclic antidepressants may be superior to SSRIs for treating depression in patients with Parkinson disease, so they might be the better choice in patients who can tolerate them.
Compulsive behaviors such as punding (prolonged performance of repetitive, mechanical tasks, such as disassembling and reassembling household objects) may occur from levodopa.
In addition, impulse control disorders involving pathologic gambling, hypersexuality, compulsive shopping, or binge eating occur in about 8% of patients with Parkinson disease taking dopamine agonists. These behaviors are more likely to arise in young, single patients, who are also more likely to have a family history of impulsive control disorder.31
THE FUTURE OF DRUG THERAPY
Clinical trials are now testing new therapies that work the traditional way through dopaminergic mechanisms, as well as those that work in novel ways.
A large international trial is studying patients with newly diagnosed Parkinson disease to try to discover a biomarker. Parkinson disease is unlike many other diseases in that physicians can only use clinical features to measure improvement, which is very crude. Identifying a biomarker will make evaluating and monitoring treatment a more exact science, and will lead to faster development of effective treatments.
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
- Adler CH, Ahlskog JE. Parkinson’s Disease and Movement Disorders: Diagnosis and Treatment Guidelines for The Practicing Physician. Totowa, NJ: Humana Press; 2000.
- Nutt JG, Wooten GF. Clinical practice. Diagnosis and initial management of Parkinson’s disease. N Engl J Med 2005; 353:1021–1027.
- Litvan I, Bhatia KP, Burn DJ, et al; Movement Disorders Society Scientific Issues Committee. Movement Disorders Society Scientific Issues Committee report: SIC Task Force appraisal of clinical diagnostic criteria for Parkinsonian disorders. Mov Disord 2003; 18:467–486.
- Wenning GK, Ben-Shlomo Y, Hughes A, Daniel SE, Lees A, Quinn NP. What clinical features are most useful to distinguish definite multiple system atrophy from Parkinson’s disease? J Neurol Neurosurg Psychiatry 2000; 68:434–440.
- Ahlskog JE, Muenter MD. Frequency of levodopa-related dyskinesias and motor fluctuations as estimated from the cumulative literature. Mov Disord 2001; 16:448–458.
- Parkinson Study Group. Pramipexole vs levodopa as initial treatment for Parkinson disease: a randomized controlled trial. Parkinson Study Group. JAMA 2000; 284:1931–1938.
- Rascol O, Brooks DJ, Korczyn AD, De Deyn PP, Clarke CE, Lang AE. A five-year study of the incidence of dyskinesia in patients with early Parkinson’s disease who were treated with ropinirole or levodopa. 056 Study Group. N Engl J Med 2000; 342:1484–1491.
- Oertel WH, Wolters E, Sampaio C, et al. Pergolide versus levodopa monotherapy in early Parkinson’s disease patients: The PELMOPET study. Mov Disord 2006; 21:343–353.
- Lees AJ, Katzenschlager R, Head J, Ben-Shlomo Y. Ten-year follow-up of three different initial treatments in de-novo PD: a randomized trial. Neurology 2001; 57:1687–1694.
- Fowler JS, Volkow ND, Logan J, et al. Slow recovery of human brain MAO B after L-deprenyl (selegeline) withdrawal. Synapse 1994; 18:86–93.
- Elmer LW, Bertoni JM. The increasing role of monoamine oxidase type B inhibitors in Parkinson’s disease therapy. Expert Opin Pharmacother 2008; 9:2759–2772.
- Olanow CW, Rascol O, Hauser R, et al; ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N Engl J Med 2009; 361:1268–1278. Erratum in: N Engl J Med 2011; 364:1882.
- Stocchi F, Barbato L, Nordera G, Bolner A, Caraceni T. Entacapone improves the pharmacokinetic and therapeutic response of controlled release levodopa/carbidopa in Parkinson’s patients. J Neural Transm 2004; 111:173–180.
- Brooks DJ, Sagar HUK-Irish Entacapone Study Group. Entacapone is beneficial in both fluctuating and non-fluctuating patients with Parkinson’s disease: a randomised, placebo controlled, double blind six month study. J Neurol Neurosurg Psychiatry 2003; 74:1071–1079.
- Poewe WH, Deuschl G, Gordin A, Kultalahti ER, Leinonen M; Celomen Study Group. Efficacy and safety of entacapone in Parkinson’s disease patients with soboptimal levodopa response: a 6-month randomized placebo-controlled double-blind study in Germany and Austria (Celomen study). Acta Neurol Scand 2002; 105:245–255.
- Rinne UK, Larsen JP, Siden A, Worm-Petersen J. Entacapone enhances the response to levodopa in parkinsonian patients with motor fluctuations. Nomecomt Study Group. Neurology 1998; 51:1309–1314.
- Entacapone improves motor fluctuations in levodopa-treated Parkinson’s disease patients. Parkinson Study Group. Ann Neurol 1997; 42:747–755.
- Parkinson Study Group. A randomized placebo-controlled trial of rasagiline in levodopa-treated patients with Parkinson disease and motor fluctuations: the PRESTO study. Arch Neurol 2005; 62:241–248.
- Rascol O, Brooks DJ, Melamed E, et al; LARGO study group. Rasagiline as an adjunct to levodopa in patients with Parkinson’s disease and motor fluctuations (LARGO, Lasting effect in Adjunct therapy with Rasagiline Given Once daily, study): a randomised, double-blind, parallel-group trial. Lancet 2005; 365:947–954.
- Metman LV, Del Dotto P, LePoole K, Konitsiotis S, Fang J, Chase TN. Amantadine for levodopa-induced dyskinesias: a 1-year follow-up study. Arch Neurol 1999; 56:1383–1386.
- Snow BJ, Macdonald L, Mcauley D, Wallis W. The effect of amantadine on levodopa-induced dyskinesias in Parkinson’s disease: a double-blind, placebo-controlled study. Clin Neuropharmacol 2000; 23:82–85.
- Almaraz AC, Driver-Dunckley ED, Woodruff BK, et al. Efficacy of rivastigmine for cognitive symptoms in Parkinson disease with dementia. Neurologist 2009; 15:234–237.
- Fénelon G, Mahieux F, Huon R, Ziégler M. Hallucinations in Parkinson’s disease: prevalence, phenomenology and risk factors. Brain 2000; 123:733–745.
- Fernandez HH, Donnelly EM, Friedman JH. Long-term outcome of clozapine use for psychosis in parkinsonian patients. Mov Disord 2004; 19:831–833.
- Goetz CG, Wuu J, Curgian LM, Leurgans S. Hallucinations and sleep disorders in PD: six-year prospective longitudinal study. Neurology 2005; 64:81–86.
- Tollefson GD, Dellva MA, Mattler CA, Kane JM, Wirshing DA, Kinon BJ. Controlled, double-blind investigation of the clozapine discontinuation symptoms with conversion to either olanzapine or placebo. The Collaborative Crossover Study Group. J Clin Psychopharmacol 1999; 19:435–443.
- Fernandez HH, Trieschmann ME, Okun MS. Rebound psychosis: effect of discontinuation of antipsychotics in Parkinson’s disease. Mov Disord 2005; 20:104–105.
- McDonald WM, Richard IH, DeLong MR. Prevalence, etiology, and treatment of depression in Parkinson’s disease. Biol Psychiatry 2003; 54:363–375.
- Devos D, Dujardin K, Poirot I, et al. Comparison of desipramine and citalopram treatments for depression in Parkinson’s disease: a double-blind, randomized, placebo-controlled study. Mov Disord 2008; 23:850–857.
- Menza M, Dobkin RD, Marin H, et al. A controlled trial of antidepressants in patients with Parkinson disease and depression. Neurology 2009; 72:886–892.
- Voon V, Sohr M, Lang AE, et al. Impulse control disorders in Parkinson disease: a multicenter case-control study. Ann Neurol 2011; 69:986–996. .
KEY POINTS
- Parkinson disease can usually be diagnosed on the basis of clinical features: slow movement, resting tremor, rigidity, and asymmetrical presentation, as well as alleviation of symptoms with dopaminergic therapy.
- Early disease can be treated with levodopa, dopamine agonists, anticholinergics, and monoamine oxidase-B inhibitors.
- Advanced Parkinson disease may require a catechol-O-methyltransferase (COMT) inhibitor, apomorphine, and amantadine (Symmetrel). Side effects include motor fluctuations, dyskinesias, and cognitive problems.
Cytomegalovirus colitis
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
A 21-year-old woman with Crohn disease presented to the hospital after 5 days of diffuse abdominal pain, nausea, vomiting, and watery diarrhea despite taking azathioprine (Imuran) 100 mg daily as maintenance therapy. She had been hospitalized 2 weeks previously at another hospital for a Crohn disease flare, which was treated with intravenous methylprednisolone (Solu-Medrol).
On admission to our hospital, her temperature was 38.4°C (101.1°F), heart rate 78 per minute, respiratory rate 18 per minute, blood pressure 110/50 mm Hg, and oxygen saturation 98% while breathing room air. She had diffuse abdominal tenderness without rebound tenderness.
Because Clostridium difficile has a high prevalence in our hospital, treament for C difficile diarrhea was started empirically directly upon hospital admission; it was stopped 48 hours later when stool cultures came back negative for C difficile.
COLITIS AND CYTOMEGALOVIRUS INFECTION
CMV colitis is common in patients with inflammatory bowel disease (ie, Crohn disease or ulcerative colitis) who are on long-term immunosuppressive therapy. Heightened suspicion for it is needed when treating patients with inflammatory bowel disease, as they tend to present with atypical symptoms and signs.
It is also important to keep a wide differential diagnosis in mind, as acute fever and diarrhea in patients with inflammatory bowel disease are not always related to the underlying disease. In these patients, a variety of diagnostic tests may be necessary to exclude an opportunistic infection and an unrelated intercurrent illness.
Human CMV is a member of the family of herpes viruses, which persist for life after a primary infection. In exacerbations of inflammatory bowel disease, it is not clear whether CMV is a nonpathogenic bystander or a true pathogen.1 Most CMV infections in patients with inflammatory bowel disease are due to reactivation of the virus, as levels of inflammatory cytokines such as tumor necrosis factor are increased in the intestinal mucosa in active inflammatory bowel disease, and these cytokines are known to trigger reactivation.2
In patients with chronic inflammatory bowel disease, CMV colitis usually presents with abdominal pain, diarrhea, intestinal bleeding, and fever. The gold standard for diagnosis is immunohistochemical testing of colon biopsy samples using monoclonal antibodies against CMV. Owl’s eye inclusion bodies on histopathologic sections are highly specific for CMV infection. Other diagnostic studies include endoscopy and serologic testing.
The gastrointestinal tract is thought to contain latent CMV after a primary infection, and long-term treatment with immunomodulatory drugs such as azathioprine and corticosteroids can cause local reactivation of the latent virus.3
CMV infection in patients with inflammatory bowel disease is associated with poor outcomes, such as the need for colectomy.4 The prevalence of CMV infection in patients with inflammatory bowel disease has been reported as 5% to 36%, and higher in patients with disease refractory to steroid therapy.1,5
When a patient with inflammatory bowel disease is diagnosed with CMV infection, the immunomodulatory drugs should be stopped and the corticosteroids should be tapered to the lowest possible dose. Treatment of the infection is intravenous ganciclovir at 5 mg per kilogram of body weight twice daily for 14 days, followed by oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks.
After receiving intravenous ganciclovir for 14 days, our patient received oral valacyclovir (Valtrex) 450 mg twice daily for 4 weeks. Her azathioprine was stopped while she was taking the antivirals, and it was resumed the day after she completed the course of valacyclovir.
The response to treatment is monitored with a cytomegalovirus pp 65 antigenemia assay. Immunomodulatory therapy can be reintroduced slowly if needed.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
- Kandiel A, Lashner B. Cytomegalovirus colitis complicating inflammatory bowel disease. Am J Gastroenterol 2006; 101:2857–2865.
- Söderberg-Nauclér C, Fish KN, Nelson JA. Interferon-gamma and tumor necrosis factor-alpha specifically induce formation of cytomegalovirus-permissive monocyte-derived macrophages that are refractory to the antiviral activity of these cytokines. J Clin Invest 1997; 100:3154–3163.
- Goodgame RW. Gastrointestinal cytomegalovirus disease. Ann Intern Med 1993; 119:924–935.
- Cottone M, Pietrosi G, Martorana G, et al. Prevalence of cytomegalovirus infection in severe refractory ulcerative and Crohn’s colitis. Am J Gastroenterol 2001; 96:773–775.
- Kishore J, Ghoshal U, Ghoshal UC, et al. Infection with cytomegalovirus in patients with inflammatory bowel disease: prevalence, clinical significance, and outcome. J Med Microbiol 2004; 53:1155–1160.
Bugs, pundits, evolution, and the New Year

The article on soft-tissue infections in this issue of the Journal by Dr. Sabitha Rajan made me reflect on the relentless march of biology. Pathogens continue to evolve, influenced by human behavior but untouched by self-promoting and partisan dialogue and undaunted by doubting politicians. Several years ago, we could assume that most skin pathogens would readily be controlled by normal body defenses, a few requiring cephalosporin therapy and even fewer needing surgical intervention. But now, environmental pressures, including the zealous use of antibiotics, have altered the microbiology of skin infections. This requires new choices for empiric antibiotic therapy of these infections. With more than just altered susceptibility profiles, these bugs exhibit biologic behaviors distinct from their historic predecessors. The “spider bite” lesion of MRSA and the scarily rapid advance of certain streptococcal infections across tissue planes mandate prompt recognition by astute clinicians—the physical examination still matters.
The brisk evolutionary pace of this new range of infections stokes the urgent need to rapidly develop novel antibiotics, a process caught smack in the middle of our pundits’ political debates. Will the development of drugs for uncommon but serious infections be underwritten by the government, or will companies be required to bear the full expense of developing drugs under the scrutiny of the FDA? Will they then be pressed to price them “affordably” or price them to recoup estimated development costs, only to have payors list them as “third-tier” on the formulary, thus making them unaffordable to many patients? Our ability to medically confront this evolution will be directly affected by the outcome of the current political debate. Will all patients be able to easily access medical care so that early significant infections are recognized for what they are, and will the new antibiotics required for appropriate treatment be affordable? This year is going to be an interesting one.
So, as empiric therapy with cephalexin changes to clindamycin and 2011 rolls into 2012, I and our editorial staff offer our sincere wishes for a healthy, happy, and especially a peaceful New Year.
The article on soft-tissue infections in this issue of the Journal by Dr. Sabitha Rajan made me reflect on the relentless march of biology. Pathogens continue to evolve, influenced by human behavior but untouched by self-promoting and partisan dialogue and undaunted by doubting politicians. Several years ago, we could assume that most skin pathogens would readily be controlled by normal body defenses, a few requiring cephalosporin therapy and even fewer needing surgical intervention. But now, environmental pressures, including the zealous use of antibiotics, have altered the microbiology of skin infections. This requires new choices for empiric antibiotic therapy of these infections. With more than just altered susceptibility profiles, these bugs exhibit biologic behaviors distinct from their historic predecessors. The “spider bite” lesion of MRSA and the scarily rapid advance of certain streptococcal infections across tissue planes mandate prompt recognition by astute clinicians—the physical examination still matters.
The brisk evolutionary pace of this new range of infections stokes the urgent need to rapidly develop novel antibiotics, a process caught smack in the middle of our pundits’ political debates. Will the development of drugs for uncommon but serious infections be underwritten by the government, or will companies be required to bear the full expense of developing drugs under the scrutiny of the FDA? Will they then be pressed to price them “affordably” or price them to recoup estimated development costs, only to have payors list them as “third-tier” on the formulary, thus making them unaffordable to many patients? Our ability to medically confront this evolution will be directly affected by the outcome of the current political debate. Will all patients be able to easily access medical care so that early significant infections are recognized for what they are, and will the new antibiotics required for appropriate treatment be affordable? This year is going to be an interesting one.
So, as empiric therapy with cephalexin changes to clindamycin and 2011 rolls into 2012, I and our editorial staff offer our sincere wishes for a healthy, happy, and especially a peaceful New Year.
The article on soft-tissue infections in this issue of the Journal by Dr. Sabitha Rajan made me reflect on the relentless march of biology. Pathogens continue to evolve, influenced by human behavior but untouched by self-promoting and partisan dialogue and undaunted by doubting politicians. Several years ago, we could assume that most skin pathogens would readily be controlled by normal body defenses, a few requiring cephalosporin therapy and even fewer needing surgical intervention. But now, environmental pressures, including the zealous use of antibiotics, have altered the microbiology of skin infections. This requires new choices for empiric antibiotic therapy of these infections. With more than just altered susceptibility profiles, these bugs exhibit biologic behaviors distinct from their historic predecessors. The “spider bite” lesion of MRSA and the scarily rapid advance of certain streptococcal infections across tissue planes mandate prompt recognition by astute clinicians—the physical examination still matters.
The brisk evolutionary pace of this new range of infections stokes the urgent need to rapidly develop novel antibiotics, a process caught smack in the middle of our pundits’ political debates. Will the development of drugs for uncommon but serious infections be underwritten by the government, or will companies be required to bear the full expense of developing drugs under the scrutiny of the FDA? Will they then be pressed to price them “affordably” or price them to recoup estimated development costs, only to have payors list them as “third-tier” on the formulary, thus making them unaffordable to many patients? Our ability to medically confront this evolution will be directly affected by the outcome of the current political debate. Will all patients be able to easily access medical care so that early significant infections are recognized for what they are, and will the new antibiotics required for appropriate treatment be affordable? This year is going to be an interesting one.
So, as empiric therapy with cephalexin changes to clindamycin and 2011 rolls into 2012, I and our editorial staff offer our sincere wishes for a healthy, happy, and especially a peaceful New Year.
Skin and soft-tissue infections: Classifying and treating a spectrum
Skin and soft-tissue infections (SSTIs) are a common reason for presentation to outpatient practices, emergency rooms, and hospitals.1–5 They account for more than 14 million outpatient visits in the United States each year,1 and visits to the emergency room and admissions to the hospital for them are increasing.2,3 Hospital admissions for SSTIs increased by 29% from 2000 to 2004.3
MORE MRSA NOW, BUT STREPTOCOCCI ARE STILL COMMON
The increase in hospital admissions for SSTIs has been attributed to a rising number of infections with methicillin-resistant Staphylococcus aureus (MRSA).3–5
In addition, strains once seen mostly in the community and other strains that were associated with health care are now being seen more often in both settings. Clinical characteristics do not differ between community-acquired and health-care-associated MRSA, and therefore the distinction between the two is becoming less useful in guiding empiric therapy.6,7
After steadily increasing for several years, the incidence of MRSA has recently stabilized. The US Centers for Disease Control and Prevention maintains a surveillance program and a Web site on MRSA.8

COMPLICATED OR UNCOMPLICATED

The intent of the 1998 guideline was to provide not a clinical framework but rather a guide for industry in designing trials that would include similar groups of infections and therefore be relevant when compared with each other. In 2008, the Anti-Infective Drugs Advisory Committee was convened,11 and subsequently, in August 2010, the FDA released a revision of the guide.12
The revised guidelines specifically exclude many diagnoses, such as bite wounds, bone and joint infections, necrotizing fasciitis, diabetic foot infections, decubitus ulcers, catheter site infections, myonecrosis, and ecthyma gangrenosum. Notably, the word “bacterial” in the title excludes mycobacterial and fungal infections from consideration. The diagnoses that are included include cellulitis, erysipelas, major cutaneous abscess, and burn infections. These are further specified to include 75 cm2 of redness, edema, or induration to standardize the extent of the infection—ie, the infection has to be at least this large or else it is not “complicated.”
The terms “complicated” and “uncomplicated” skin and skin structure infections persist and can be useful adjuncts in describing SSTIs.13–16 However, more specific descriptions of SSTIs based on pathogenesis are more useful to the clinician and are usually the basis for guidelines, such as for preventing surgical site infections or for reducing amputations in diabetic foot infections.
This review will focus on the general categories of SSTI and will not address surgical site infections, pressure ulcers, diabetic foot infections, perirectal wounds, or adjuvant therapies in severe SSTIs, such as negative pressure wound care (vacuum-assisted closure devices) and hyperbaric chambers.
OTHER DISEASES CAN MIMIC SSTIs
SSTIs vary broadly in their location and severity.
Although the classic presentation of erythema, warmth, edema, and tenderness often signals infection, other diseases can mimic SSTIs. Common ones that should be included in the differential diagnosis include gout, thrombophlebitis, deep vein thrombosis, contact dermatitis, carcinoma erysipeloides, drug eruption, and a foreign body reaction.17,18
CLUES FROM THE HISTORY

Wounds. Skin infections are usually precipitated by a break in the skin from a cut, laceration, excoriation, fungal infection, insect or animal bite, or puncture wound.
Impaired response. Patients with diabetes, renal failure, cirrhosis, chronic glucocorticoid use, history of organ transplantation, chronic immunosuppressive therapy, HIV infection, or malnourishment have impaired host responses to infection and are at risk for both more severe infections and recurrent infections. Immunocompromised hosts may also have atypical infections with opportunistic organisms such as Pseudomonas, Proteus, Serratia, Enterobacter, Citrobacter, and anaerobes. Close follow-up of these patients is warranted to ascertain appropriate response to therapy.19
Surgery that includes lymph node dissection or saphenous vein resection for coronary artery bypass can lead to impaired lymphatic drainage and edema, and therefore predisposes patients to SSTIs.
PHYSICAL EXAMINATION
The physical examination should include descriptions of the extent and location of erythema, edema, warmth, and tenderness so that progression or resolution with treatment can be followed in detail.
Crepitus can be felt in gas-forming infections and raises the concern for necrotizing fasciitis and infection with anaerobic organisms such as Clostridium perfringens.
Necrosis can occur in brown recluse spider bites, venous snake bites, or group A streptococcal infections.
Fluctuance indicates fluid and a likely abscess that may need incision and drainage.
Purpura may be present in patients on anticoagulation therapy, but if it is accompanying an SSTI, it also raises the concern for the possibility of sepsis and disseminated intravascular coagulation, especially from streptococcal infections.
Bullae can be seen in impetigo caused by staphylococci or in infection with Vibrio vulnificus or Streptococcus pneumoniae.19
Systemic signs, in addition to fever, can include hypotension and tachycardia, which would prompt closer monitoring and possible hospitalization.
Lymphangitic spread also indicates severe infection.
LABORATORY STUDIES
Simple, localized SSTIs usually do not require laboratory evaluation. Jenkins et al21 recently demonstrated that by using an algorithm for the management of hospitalized patients with cellulitis or cutaneous abscess, they could decrease resource utilization, including laboratory testing, without adversely affecting clinical outcome.
If patients have underlying disease or more extensive infection, then baseline chemistry values, a complete blood cell count, and the C-reactive protein level should be acquired.19 Laboratory findings that suggest more severe disease include low sodium, low bicarbonate (or an anion gap), and high creatinine levels; new anemia; a high or very low white blood cell count; and a high C-reactive protein level. A high C-reactive protein level has been associated with longer hospitalization.22
A score to estimate the risk of necrotizing fasciitis
This tool was developed retrospectively but has been validated prospectively. It has a high sensitivity and a positive predictive value of 92% in patients with a score of six points or more. Its specificity is also high, with a negative predictive value of 96%.20,24
Necrotizing fasciitis has a mortality rate of 23.5%, but this may be reduced to 10% with early detection and prompt surgical intervention.15 Since necrotizing fasciitis is very difficult to diagnose, clinicians must maintain a high level of suspicion and use the LRINEC score to trigger early surgical evaluation. Surgical exploration is the only way to definitively diagnose necrotizing fasciitis.
Blood cultures in some cases
Blood cultures have a low yield and are usually not cost-effective, but they should be obtained in patients who have lymphedema, immune deficiency, fever, pain out of proportion to the findings on examination, tachycardia, or hypotension, as blood cultures are more likely to be positive in more serious infections and can help guide antimicrobial therapy. Blood cultures are also recommended in patients with infections involving specific anatomic sites, such as the mouth and eyes.19
Aspiration, swabs, incision and drainage
Fluid aspirated from abscesses and swabs of debrided ulcerated wounds should be sent for Gram stain and culture. Gram stain and culture have widely varying yields, from less than 5% to 40%, depending on the source and technique.19 Cultures were not routinely obtained before MRSA emerged, but knowing antimicrobial susceptibility is now important to guide antibiotic therapy. Unfortunately, in cellulitis, swabs and aspirates of the leading edge have a low yield of around 10%.25 One prospective study of 25 hospitalized patients did report a higher yield of positive cultures in patients with fever or underlying disease,26 so aspirates may be used in selected cases. In small studies, the yield of punch biopsies was slightly better than that of needle aspirates and was as high as 20% to 30%.27
IMAGING STUDIES
Imaging can be helpful in determining the depth of involvement. Plain radiography can reveal gas or periosteal inflammation and is especially helpful in diabetic foot infections. Ultrasonography can detect abscesses.
Both magnetic resonance imaging (MRI) and computed tomography (CT) are useful to image fascial planes, although MRI is more sensitive. However, in cases of suspected necrotizing fasciitis, imaging should not delay surgical evaluation and debridement or be used as the definitive study. Therefore, the practicality of CT and MRI can be limited.15,16
ANTIMICROBIAL TREATMENT FOR SSTIs IN OUTPATIENTS
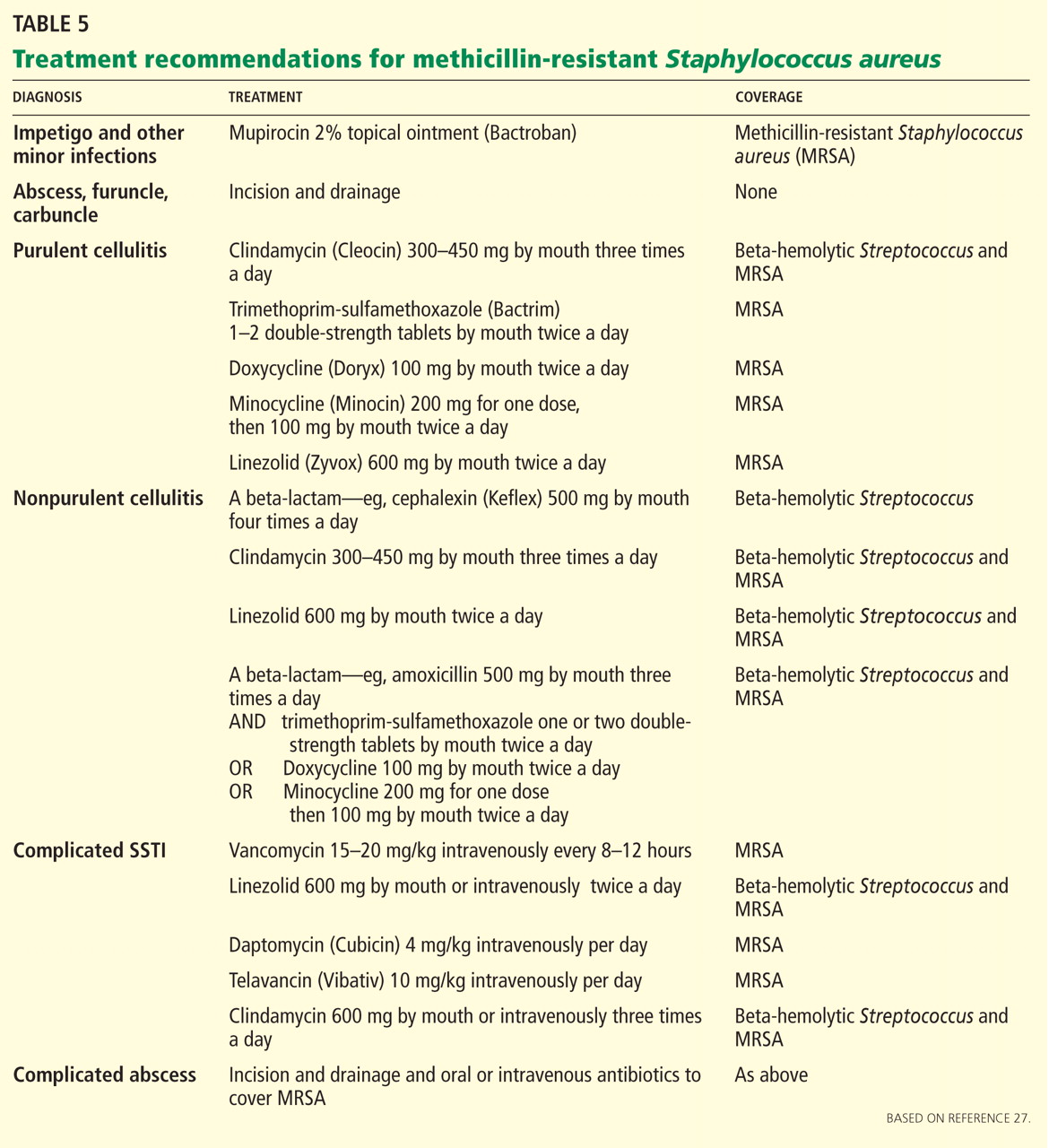
For minor skin infections such as impetigo and secondarily infected skin lesions such as eczema, ulcers, or lacerations, mupirocin 2% topical ointment (Bactroban) can be effective.27
For a simple abscess or boil, incision and drainage is the primary treatment, and antibiotics are not needed.
For a complicated abscess or boil. Patients should be given oral or intravenous antibiotic therapy to cover MRSA and, depending on the severity, should be considered for hospitalization if the abscess is associated with severe disease, rapid progression in the presence of associated cellulitis, septic phlebitis, constitutional symptoms, comorbidity (including immunosuppression), or an abscess or boil in an area difficult to drain, such as the face, hands, or genitalia.27
For purulent cellulitis in outpatients, empiric therapy for community-acquired MRSA is recommended, pending culture results. Empiric therapy for streptococcal infection is likely unnecessary. For empiric coverage of community-acquired MRSA in purulent cellulitis, oral antibiotic options include clindamycin (Cleocin), trimethoprim-sulfamethoxazole (Bactrim), doxycycline (Doryx), minocycline (Minocin), and linezolid (Zyvox).
For nonpurulent cellulitis in outpatients, empiric coverage for beta-hemolytic streptococci is warranted. Coverage for community-acquired MRSA should subsequently be added for patients who do not respond to beta-lactam therapy within 48 to 72 hours or who have chills, fever, a new abscess, increasing erythema, or uncontrolled pain.
Options for coverage of both beta-hemolytic streptococci and community-acquired MRSA for outpatient therapy include clindamycin on its own, trimethoprim-sulfamethoxazole or a tetracycline in combination with a beta-lactam, or linezolid on its own.
Increasing rates of resistance to clindamycin, tetracycline, and trimethoprim-sulfamethoxazole in community-acquired MRSA may limit empiric treatment. In areas where resistance is prevalent, culture with antimicrobial susceptibility testing may be required before starting one of these antibiotics.
The use of rifampin (Rifadin) as a single agent is not recommended because resistance is likely to develop. Also, rifampin is not useful as adjunctive therapy, as evidence does not support its efficacy.19,27,29
ANTIMICROBIAL TREATMENT FOR SSTIs IN HOSPITALIZED PATIENTS
For hospitalized patients with a complicated or severe SSTI, empiric therapy for MRSA should be started pending culture results. FDA-approved options are vancomycin, linezolid, daptomycin (Cubicin), tigecycline (Tygacil), and telavancin (Vibativ). Data on clindamycin are very limited in this population. A beta-lactam antibiotic such as cefazolin (Ancef) may be considered in hospitalized patients with nonpurulent cellulitis, and the regimen can be modified to MRSA-active therapy if there is no clinical response. Linezolid, daptomycin, vancomycin, and telavancin have adequate streptococcal coverage in addition to MRSA coverage.
Clindamycin is approved by the FDA for treating serious infections due to S aureus. It has excellent tissue penetration, particularly in bone and abscesses.
Clindamycin resistance in staphylococci can be either constitutive or inducible, and clinicians must be watchful for signs of resistance.
Diarrhea is the most common adverse effect and occurs in up to 20% of patients. Clostridium difficile colitis may occur more frequently with clindamycin than with other oral agents, but it has also has been reported with fluoroquinolones and can be associated with any antibiotic therapy.30
Trimethoprim-sulfamethoxazole is not FDA-approved for treating any staphylococcal infection. However, because 95% to 100% of community-acquired MRSA strains are susceptible to it in vitro, it has become an important option in the outpatient treatment of SSTIs. Caution is advised when using it in elderly patients, particularly those with chronic renal insufficiency, because of an increased risk of hyperkalemia.
Tetracyclines. Doxycycline is FDA-approved for treating SSTIs due to S aureus, although not specifically for MRSA. Minocycline may be an option even when strains are resistant to doxycycline, since it does not induce its own resistance as doxycycline does.
Tigecycline is a glycylcycline (a tetracycline derivative) and is FDA-approved in adults for complicated SSTIs and intra-abdominal infections. It has a large volume of distribution and achieves high concentrations in tissues and low concentrations in serum.
The FDA recently issued a warning to consider alternative agents in patients with serious infections because of higher rates of all-cause mortality noted in phase III and phase IV clinical trials. Due to this warning and the availability of multiple alternatives active against MRSA, tigecycline was not included in the Infectious Diseases Society of America guidelines.31
Linezolid is a synthetic oxazolidinone and is FDA-approved for treating SSTIs and nosocomial pneumonia caused by MRSA. It has 100% oral bioavailability, so parenteral therapy should only be given if there are problems with gastrointestinal absorption or if the patient is unable to take oral medications.
Long-term use of linezolid (> 2 weeks) is limited by hematologic toxicity, especially thrombocytopenia, which occurs more frequently than anemia and neutropenia. Lactic acidosis and peripheral and optic neuropathy are also limiting toxicities. Although myelosuppression is generally reversible, peripheral and optic neuropathy may not be.
Linezolid should not used in patients taking selective serotonin reuptake inhibitors if they cannot stop taking these antidepressant drugs during therapy, as the combination can lead to the serotonin syndrome.
Vancomycin is still the mainstay of parenteral therapy for MRSA infections. However, its efficacy has come into question, with concerns over its slow bactericidal activity and the emergence of resistant strains. The rate of treatment failure is high in those with infection caused by MRSA having minimum inhibitory concentrations of 1 μg/mL or greater. Vancomycin kills staphylococci more slowly than do beta-lactams in vitro and is clearly inferior to beta-lactams for methicillin-sensitive S aureus bacteremia.
Daptomycin is a lipopeptide antibiotic that is FDA-approved for adults with MRSA bacteremia, right-sided infective endocarditis, and complicated SSTI. Elevations in creatinine phosphokinase, which are rarely treatment-limiting, have occurred in patients receiving 6 mg/kg/day but not in those receiving 4 mg/kg/day. Patients should be observed for development of muscle pain or weakness and should have their creatine phosphokinase levels checked weekly, with more frequent monitoring in those with renal insufficiency or who are receiving concomitant statin therapy.
Telavancin is a parenteral lipoglycopeptide that is bactericidal against MRSA. It is FDA-approved for complicated SSTIs in adults. Creatinine levels should be monitored, and the dosage should be adjusted on the basis of creatinine clearance, because nephrotoxicity was more commonly reported among individuals treated with telavancin than among those treated with vancomycin.
Ceftaroline (Teflaro), a fifth-generation cephalosporin, was approved for SSTIs by the FDA in October 2010. It is active against MRSA and gram-negative pathogens.
Cost is a consideration
Cost is a consideration, as it may limit the availability of and access to treatment. In 2008, the expense for 10 days of treatment with generic vancomycin was $183, compared with $1,661 for daptomycin, $1,362 for tigecycline, and $1,560 for linezolid. For outpatient therapy, the contrast was even starker, as generic trimethoprim-sulfamethoxazole cost $9.40 and generic clindamycin cost $95.10.32
INDICATIONS FOR HOSPITALIZATION
Patients who have evidence of tissue necrosis, fever, hypotension, severe pain, altered mental status, an immunocompromised state, or organ failure (respiratory, renal, or hepatic) must be hospitalized.
Although therapy for MRSA is the mainstay of empiric therapy, polymicrobial infections are not uncommon, and gram-negative and anaerobic coverage should be added as appropriate. One study revealed a longer length of stay for hospitalized patients who had inadequate initial empiric coverage.33
Vigilance should be maintained for overlying cellulitis which can mask necrotizing fasciitis, septic joints, or osteomyelitis.
Perianal abscesses and infections, infected decubitus ulcers, and moderate to severe diabetic foot infections are often polymicrobial and warrant coverage for streptococci, MRSA, aerobic gram-negative bacilli, and anaerobes until culture results can guide therapy.
INDICATIONS FOR SURGICAL REFERRAL
Extensive perianal or multiple abscesses may require surgical drainage and debridement.
Surgical site infections should be referred for consideration of opening the incision for drainage.
Necrotizing infections warrant prompt aggressive surgical debridement. Strongly suggestive clinical signs include bullae, crepitus, gas on radiography, hypotension with systolic blood pressure less than 90 mm Hg, or skin necrosis. However, these are late findings, and fewer than 50% of these patients have one of these. Most cases of necrotizing fasciitis originally have an admitting diagnosis of cellulitis and cases of fasciitis are relatively rare, so the diagnosis is easy to miss.15,16 Patients with an LRINEC score of six or more should have prompt surgical evaluation.20,24,34,35
- Hersh AL, Chambers HF, Maselli JH, Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med 2008; 168:1585–1591.
- Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med 2008; 51:291–298.
- Edelsberg J, Taneja C, Zervos M, et al. Trends in US hospital admissions for skin and soft tissue infections. Emerg Infect Dis 2009; 15:1516–1518.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med 2007; 357:380–390.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Chua K, Laurent F, Coombs G, Grayson ML, Howden BP. Antimicrobial resistance: not community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA)! A clinician’s guide to community MRSA—its evolving antimicrobial resistance and implications for therapy. Clin Infect Dis 2011; 52:99–114.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Centers for Disease Control and Prevention. MRSA Infections. http://www.cdc.gov/mrsa/statistics/MRSA-Surveillance-Summary.html. Accessed December 14, 2011.
- Moet GJ, Jones RN, Biedenbach DJ, Stilwell MG, Fritsche TR. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis 2007; 57:7–13.
- US Department of Health and Human Services. Guidance for Industry: Uncomplicated and Complicated Skin and Skin Structure Infections—Developing Antimicrobial Drugs for Treatment (draft guidance). July 1998. http://www.fda.gov/ohrms/dockets/98fr/2566dft.pdf. Accessed September 7, 2011.
- US Food and Drug Administration. CDER 2008 Meeting Documents. Anti-Infective Drugs Advisory Committee. http://www.fda.gov/ohrms/dockets/ac/cder08.html#AntiInfective. Accessed September 7, 2011.
- US Department of Health and Human Services. Guidance for Industry: Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment (draft guidance). August 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071185.pdf. Accessed December 14, 2011.
- Cornia PB, Davidson HL, Lipsky BA. The evaluation and treatment of complicated skin and skin structure infections. Expert Opin Pharmacother 2008; 9:717–730.
- Ki V, Rotstein C. Bacterial skin and soft tissue infections in adults: a review of their epidemiology, pathogenesis, diagnosis, treatment and site of care. Can J Infect Dis Med Microbiol 2008; 19:173–184.
- May AK, Stafford RE, Bulger EM, et al; Surgical Infection Society. Treatment of complicated skin and soft tissue infections. Surg Infect (Larchmt) 2009; 10:467–499.
- Napolitano LM. Severe soft tissue infections. Infect Dis Clin North Am 2009; 23:571–591.
- Papadavid E, Dalamaga M, Stavrianeas N, Papiris SA. Subcutaneous sarcoidosis masquerading as cellulitis. Dermatology 2008; 217:212–214.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med 2004; 32:1535–1541.
- Jenkins TC, Knepper BC, Sabel AL, et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med 2011; 171:1072–1079.
- Lazzarini L, Conti E, Tositti G, de Lalla F. Erysipelas and cellulitis: clinical and microbiological spectrum in an Italian tertiary care hospital. J Infect 2005; 51:383–389.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis 2007; 44:705–710.
- Hasham S, Matteucci P, Stanley PR, Hart NB. Necrotising fasciitis. BMJ 2005; 330:830–833.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol 1988; 26:401–404.
- Sachs MK. The optimum use of needle aspiration in the bacteriologic diagnosis of cellulitis in adults. Arch Intern Med 1990; 150:1907–1912.
- Liu C, Bayer A, Cosgrove SE, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:e18–e55.
- Hammond SP, Baden LR. Clinical decisions. Management of skin and soft-tissue infection—polling results. N Engl J Med 2008; 359:e20.
- Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med 2008; 168:805–819.
- Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect 1998; 40:1–15.
- US Food and Drug Administration. FDA Drug Safety Communication: increased risk of death with Tygacil (tigecycline) compared to other antibiotics used to treat similar infections. September 2010. http://www.fda.gov/Drugs/DrugSafety/ucm224370.htm. Accessed September 7, 2011.
- Moellering RC. A 39-year-old man with a skin infection. JAMA 2008; 299:79–87.
- Zilberberg MD, Shorr AF, Micek ST, et al. Hospitalizations with healthcare-associated complicated skin and skin structure infections: impact of inappropriate empiric therapy on outcomes. J Hosp Med 2010; 5:535–540.
- Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85:1454–1460.
- Hsiao CT, Weng HH, Yuan YD, Chen CT, Chen IC. Predictors of mortality in patients with necrotizing fasciitis. Am J Emerg Med 2008; 26:170–175.
Skin and soft-tissue infections (SSTIs) are a common reason for presentation to outpatient practices, emergency rooms, and hospitals.1–5 They account for more than 14 million outpatient visits in the United States each year,1 and visits to the emergency room and admissions to the hospital for them are increasing.2,3 Hospital admissions for SSTIs increased by 29% from 2000 to 2004.3
MORE MRSA NOW, BUT STREPTOCOCCI ARE STILL COMMON
The increase in hospital admissions for SSTIs has been attributed to a rising number of infections with methicillin-resistant Staphylococcus aureus (MRSA).3–5
In addition, strains once seen mostly in the community and other strains that were associated with health care are now being seen more often in both settings. Clinical characteristics do not differ between community-acquired and health-care-associated MRSA, and therefore the distinction between the two is becoming less useful in guiding empiric therapy.6,7
After steadily increasing for several years, the incidence of MRSA has recently stabilized. The US Centers for Disease Control and Prevention maintains a surveillance program and a Web site on MRSA.8
COMPLICATED OR UNCOMPLICATED
The intent of the 1998 guideline was to provide not a clinical framework but rather a guide for industry in designing trials that would include similar groups of infections and therefore be relevant when compared with each other. In 2008, the Anti-Infective Drugs Advisory Committee was convened,11 and subsequently, in August 2010, the FDA released a revision of the guide.12
The revised guidelines specifically exclude many diagnoses, such as bite wounds, bone and joint infections, necrotizing fasciitis, diabetic foot infections, decubitus ulcers, catheter site infections, myonecrosis, and ecthyma gangrenosum. Notably, the word “bacterial” in the title excludes mycobacterial and fungal infections from consideration. The diagnoses that are included include cellulitis, erysipelas, major cutaneous abscess, and burn infections. These are further specified to include 75 cm2 of redness, edema, or induration to standardize the extent of the infection—ie, the infection has to be at least this large or else it is not “complicated.”
The terms “complicated” and “uncomplicated” skin and skin structure infections persist and can be useful adjuncts in describing SSTIs.13–16 However, more specific descriptions of SSTIs based on pathogenesis are more useful to the clinician and are usually the basis for guidelines, such as for preventing surgical site infections or for reducing amputations in diabetic foot infections.
This review will focus on the general categories of SSTI and will not address surgical site infections, pressure ulcers, diabetic foot infections, perirectal wounds, or adjuvant therapies in severe SSTIs, such as negative pressure wound care (vacuum-assisted closure devices) and hyperbaric chambers.
OTHER DISEASES CAN MIMIC SSTIs
SSTIs vary broadly in their location and severity.
Although the classic presentation of erythema, warmth, edema, and tenderness often signals infection, other diseases can mimic SSTIs. Common ones that should be included in the differential diagnosis include gout, thrombophlebitis, deep vein thrombosis, contact dermatitis, carcinoma erysipeloides, drug eruption, and a foreign body reaction.17,18
CLUES FROM THE HISTORY
Wounds. Skin infections are usually precipitated by a break in the skin from a cut, laceration, excoriation, fungal infection, insect or animal bite, or puncture wound.
Impaired response. Patients with diabetes, renal failure, cirrhosis, chronic glucocorticoid use, history of organ transplantation, chronic immunosuppressive therapy, HIV infection, or malnourishment have impaired host responses to infection and are at risk for both more severe infections and recurrent infections. Immunocompromised hosts may also have atypical infections with opportunistic organisms such as Pseudomonas, Proteus, Serratia, Enterobacter, Citrobacter, and anaerobes. Close follow-up of these patients is warranted to ascertain appropriate response to therapy.19
Surgery that includes lymph node dissection or saphenous vein resection for coronary artery bypass can lead to impaired lymphatic drainage and edema, and therefore predisposes patients to SSTIs.
PHYSICAL EXAMINATION
The physical examination should include descriptions of the extent and location of erythema, edema, warmth, and tenderness so that progression or resolution with treatment can be followed in detail.
Crepitus can be felt in gas-forming infections and raises the concern for necrotizing fasciitis and infection with anaerobic organisms such as Clostridium perfringens.
Necrosis can occur in brown recluse spider bites, venous snake bites, or group A streptococcal infections.
Fluctuance indicates fluid and a likely abscess that may need incision and drainage.
Purpura may be present in patients on anticoagulation therapy, but if it is accompanying an SSTI, it also raises the concern for the possibility of sepsis and disseminated intravascular coagulation, especially from streptococcal infections.
Bullae can be seen in impetigo caused by staphylococci or in infection with Vibrio vulnificus or Streptococcus pneumoniae.19
Systemic signs, in addition to fever, can include hypotension and tachycardia, which would prompt closer monitoring and possible hospitalization.
Lymphangitic spread also indicates severe infection.
LABORATORY STUDIES
Simple, localized SSTIs usually do not require laboratory evaluation. Jenkins et al21 recently demonstrated that by using an algorithm for the management of hospitalized patients with cellulitis or cutaneous abscess, they could decrease resource utilization, including laboratory testing, without adversely affecting clinical outcome.
If patients have underlying disease or more extensive infection, then baseline chemistry values, a complete blood cell count, and the C-reactive protein level should be acquired.19 Laboratory findings that suggest more severe disease include low sodium, low bicarbonate (or an anion gap), and high creatinine levels; new anemia; a high or very low white blood cell count; and a high C-reactive protein level. A high C-reactive protein level has been associated with longer hospitalization.22
A score to estimate the risk of necrotizing fasciitis
This tool was developed retrospectively but has been validated prospectively. It has a high sensitivity and a positive predictive value of 92% in patients with a score of six points or more. Its specificity is also high, with a negative predictive value of 96%.20,24
Necrotizing fasciitis has a mortality rate of 23.5%, but this may be reduced to 10% with early detection and prompt surgical intervention.15 Since necrotizing fasciitis is very difficult to diagnose, clinicians must maintain a high level of suspicion and use the LRINEC score to trigger early surgical evaluation. Surgical exploration is the only way to definitively diagnose necrotizing fasciitis.
Blood cultures in some cases
Blood cultures have a low yield and are usually not cost-effective, but they should be obtained in patients who have lymphedema, immune deficiency, fever, pain out of proportion to the findings on examination, tachycardia, or hypotension, as blood cultures are more likely to be positive in more serious infections and can help guide antimicrobial therapy. Blood cultures are also recommended in patients with infections involving specific anatomic sites, such as the mouth and eyes.19
Aspiration, swabs, incision and drainage
Fluid aspirated from abscesses and swabs of debrided ulcerated wounds should be sent for Gram stain and culture. Gram stain and culture have widely varying yields, from less than 5% to 40%, depending on the source and technique.19 Cultures were not routinely obtained before MRSA emerged, but knowing antimicrobial susceptibility is now important to guide antibiotic therapy. Unfortunately, in cellulitis, swabs and aspirates of the leading edge have a low yield of around 10%.25 One prospective study of 25 hospitalized patients did report a higher yield of positive cultures in patients with fever or underlying disease,26 so aspirates may be used in selected cases. In small studies, the yield of punch biopsies was slightly better than that of needle aspirates and was as high as 20% to 30%.27
IMAGING STUDIES
Imaging can be helpful in determining the depth of involvement. Plain radiography can reveal gas or periosteal inflammation and is especially helpful in diabetic foot infections. Ultrasonography can detect abscesses.
Both magnetic resonance imaging (MRI) and computed tomography (CT) are useful to image fascial planes, although MRI is more sensitive. However, in cases of suspected necrotizing fasciitis, imaging should not delay surgical evaluation and debridement or be used as the definitive study. Therefore, the practicality of CT and MRI can be limited.15,16
ANTIMICROBIAL TREATMENT FOR SSTIs IN OUTPATIENTS
For minor skin infections such as impetigo and secondarily infected skin lesions such as eczema, ulcers, or lacerations, mupirocin 2% topical ointment (Bactroban) can be effective.27
For a simple abscess or boil, incision and drainage is the primary treatment, and antibiotics are not needed.
For a complicated abscess or boil. Patients should be given oral or intravenous antibiotic therapy to cover MRSA and, depending on the severity, should be considered for hospitalization if the abscess is associated with severe disease, rapid progression in the presence of associated cellulitis, septic phlebitis, constitutional symptoms, comorbidity (including immunosuppression), or an abscess or boil in an area difficult to drain, such as the face, hands, or genitalia.27
For purulent cellulitis in outpatients, empiric therapy for community-acquired MRSA is recommended, pending culture results. Empiric therapy for streptococcal infection is likely unnecessary. For empiric coverage of community-acquired MRSA in purulent cellulitis, oral antibiotic options include clindamycin (Cleocin), trimethoprim-sulfamethoxazole (Bactrim), doxycycline (Doryx), minocycline (Minocin), and linezolid (Zyvox).
For nonpurulent cellulitis in outpatients, empiric coverage for beta-hemolytic streptococci is warranted. Coverage for community-acquired MRSA should subsequently be added for patients who do not respond to beta-lactam therapy within 48 to 72 hours or who have chills, fever, a new abscess, increasing erythema, or uncontrolled pain.
Options for coverage of both beta-hemolytic streptococci and community-acquired MRSA for outpatient therapy include clindamycin on its own, trimethoprim-sulfamethoxazole or a tetracycline in combination with a beta-lactam, or linezolid on its own.
Increasing rates of resistance to clindamycin, tetracycline, and trimethoprim-sulfamethoxazole in community-acquired MRSA may limit empiric treatment. In areas where resistance is prevalent, culture with antimicrobial susceptibility testing may be required before starting one of these antibiotics.
The use of rifampin (Rifadin) as a single agent is not recommended because resistance is likely to develop. Also, rifampin is not useful as adjunctive therapy, as evidence does not support its efficacy.19,27,29
ANTIMICROBIAL TREATMENT FOR SSTIs IN HOSPITALIZED PATIENTS
For hospitalized patients with a complicated or severe SSTI, empiric therapy for MRSA should be started pending culture results. FDA-approved options are vancomycin, linezolid, daptomycin (Cubicin), tigecycline (Tygacil), and telavancin (Vibativ). Data on clindamycin are very limited in this population. A beta-lactam antibiotic such as cefazolin (Ancef) may be considered in hospitalized patients with nonpurulent cellulitis, and the regimen can be modified to MRSA-active therapy if there is no clinical response. Linezolid, daptomycin, vancomycin, and telavancin have adequate streptococcal coverage in addition to MRSA coverage.
Clindamycin is approved by the FDA for treating serious infections due to S aureus. It has excellent tissue penetration, particularly in bone and abscesses.
Clindamycin resistance in staphylococci can be either constitutive or inducible, and clinicians must be watchful for signs of resistance.
Diarrhea is the most common adverse effect and occurs in up to 20% of patients. Clostridium difficile colitis may occur more frequently with clindamycin than with other oral agents, but it has also has been reported with fluoroquinolones and can be associated with any antibiotic therapy.30
Trimethoprim-sulfamethoxazole is not FDA-approved for treating any staphylococcal infection. However, because 95% to 100% of community-acquired MRSA strains are susceptible to it in vitro, it has become an important option in the outpatient treatment of SSTIs. Caution is advised when using it in elderly patients, particularly those with chronic renal insufficiency, because of an increased risk of hyperkalemia.
Tetracyclines. Doxycycline is FDA-approved for treating SSTIs due to S aureus, although not specifically for MRSA. Minocycline may be an option even when strains are resistant to doxycycline, since it does not induce its own resistance as doxycycline does.
Tigecycline is a glycylcycline (a tetracycline derivative) and is FDA-approved in adults for complicated SSTIs and intra-abdominal infections. It has a large volume of distribution and achieves high concentrations in tissues and low concentrations in serum.
The FDA recently issued a warning to consider alternative agents in patients with serious infections because of higher rates of all-cause mortality noted in phase III and phase IV clinical trials. Due to this warning and the availability of multiple alternatives active against MRSA, tigecycline was not included in the Infectious Diseases Society of America guidelines.31
Linezolid is a synthetic oxazolidinone and is FDA-approved for treating SSTIs and nosocomial pneumonia caused by MRSA. It has 100% oral bioavailability, so parenteral therapy should only be given if there are problems with gastrointestinal absorption or if the patient is unable to take oral medications.
Long-term use of linezolid (> 2 weeks) is limited by hematologic toxicity, especially thrombocytopenia, which occurs more frequently than anemia and neutropenia. Lactic acidosis and peripheral and optic neuropathy are also limiting toxicities. Although myelosuppression is generally reversible, peripheral and optic neuropathy may not be.
Linezolid should not used in patients taking selective serotonin reuptake inhibitors if they cannot stop taking these antidepressant drugs during therapy, as the combination can lead to the serotonin syndrome.
Vancomycin is still the mainstay of parenteral therapy for MRSA infections. However, its efficacy has come into question, with concerns over its slow bactericidal activity and the emergence of resistant strains. The rate of treatment failure is high in those with infection caused by MRSA having minimum inhibitory concentrations of 1 μg/mL or greater. Vancomycin kills staphylococci more slowly than do beta-lactams in vitro and is clearly inferior to beta-lactams for methicillin-sensitive S aureus bacteremia.
Daptomycin is a lipopeptide antibiotic that is FDA-approved for adults with MRSA bacteremia, right-sided infective endocarditis, and complicated SSTI. Elevations in creatinine phosphokinase, which are rarely treatment-limiting, have occurred in patients receiving 6 mg/kg/day but not in those receiving 4 mg/kg/day. Patients should be observed for development of muscle pain or weakness and should have their creatine phosphokinase levels checked weekly, with more frequent monitoring in those with renal insufficiency or who are receiving concomitant statin therapy.
Telavancin is a parenteral lipoglycopeptide that is bactericidal against MRSA. It is FDA-approved for complicated SSTIs in adults. Creatinine levels should be monitored, and the dosage should be adjusted on the basis of creatinine clearance, because nephrotoxicity was more commonly reported among individuals treated with telavancin than among those treated with vancomycin.
Ceftaroline (Teflaro), a fifth-generation cephalosporin, was approved for SSTIs by the FDA in October 2010. It is active against MRSA and gram-negative pathogens.
Cost is a consideration
Cost is a consideration, as it may limit the availability of and access to treatment. In 2008, the expense for 10 days of treatment with generic vancomycin was $183, compared with $1,661 for daptomycin, $1,362 for tigecycline, and $1,560 for linezolid. For outpatient therapy, the contrast was even starker, as generic trimethoprim-sulfamethoxazole cost $9.40 and generic clindamycin cost $95.10.32
INDICATIONS FOR HOSPITALIZATION
Patients who have evidence of tissue necrosis, fever, hypotension, severe pain, altered mental status, an immunocompromised state, or organ failure (respiratory, renal, or hepatic) must be hospitalized.
Although therapy for MRSA is the mainstay of empiric therapy, polymicrobial infections are not uncommon, and gram-negative and anaerobic coverage should be added as appropriate. One study revealed a longer length of stay for hospitalized patients who had inadequate initial empiric coverage.33
Vigilance should be maintained for overlying cellulitis which can mask necrotizing fasciitis, septic joints, or osteomyelitis.
Perianal abscesses and infections, infected decubitus ulcers, and moderate to severe diabetic foot infections are often polymicrobial and warrant coverage for streptococci, MRSA, aerobic gram-negative bacilli, and anaerobes until culture results can guide therapy.
INDICATIONS FOR SURGICAL REFERRAL
Extensive perianal or multiple abscesses may require surgical drainage and debridement.
Surgical site infections should be referred for consideration of opening the incision for drainage.
Necrotizing infections warrant prompt aggressive surgical debridement. Strongly suggestive clinical signs include bullae, crepitus, gas on radiography, hypotension with systolic blood pressure less than 90 mm Hg, or skin necrosis. However, these are late findings, and fewer than 50% of these patients have one of these. Most cases of necrotizing fasciitis originally have an admitting diagnosis of cellulitis and cases of fasciitis are relatively rare, so the diagnosis is easy to miss.15,16 Patients with an LRINEC score of six or more should have prompt surgical evaluation.20,24,34,35
Skin and soft-tissue infections (SSTIs) are a common reason for presentation to outpatient practices, emergency rooms, and hospitals.1–5 They account for more than 14 million outpatient visits in the United States each year,1 and visits to the emergency room and admissions to the hospital for them are increasing.2,3 Hospital admissions for SSTIs increased by 29% from 2000 to 2004.3
MORE MRSA NOW, BUT STREPTOCOCCI ARE STILL COMMON
The increase in hospital admissions for SSTIs has been attributed to a rising number of infections with methicillin-resistant Staphylococcus aureus (MRSA).3–5
In addition, strains once seen mostly in the community and other strains that were associated with health care are now being seen more often in both settings. Clinical characteristics do not differ between community-acquired and health-care-associated MRSA, and therefore the distinction between the two is becoming less useful in guiding empiric therapy.6,7
After steadily increasing for several years, the incidence of MRSA has recently stabilized. The US Centers for Disease Control and Prevention maintains a surveillance program and a Web site on MRSA.8
COMPLICATED OR UNCOMPLICATED
The intent of the 1998 guideline was to provide not a clinical framework but rather a guide for industry in designing trials that would include similar groups of infections and therefore be relevant when compared with each other. In 2008, the Anti-Infective Drugs Advisory Committee was convened,11 and subsequently, in August 2010, the FDA released a revision of the guide.12
The revised guidelines specifically exclude many diagnoses, such as bite wounds, bone and joint infections, necrotizing fasciitis, diabetic foot infections, decubitus ulcers, catheter site infections, myonecrosis, and ecthyma gangrenosum. Notably, the word “bacterial” in the title excludes mycobacterial and fungal infections from consideration. The diagnoses that are included include cellulitis, erysipelas, major cutaneous abscess, and burn infections. These are further specified to include 75 cm2 of redness, edema, or induration to standardize the extent of the infection—ie, the infection has to be at least this large or else it is not “complicated.”
The terms “complicated” and “uncomplicated” skin and skin structure infections persist and can be useful adjuncts in describing SSTIs.13–16 However, more specific descriptions of SSTIs based on pathogenesis are more useful to the clinician and are usually the basis for guidelines, such as for preventing surgical site infections or for reducing amputations in diabetic foot infections.
This review will focus on the general categories of SSTI and will not address surgical site infections, pressure ulcers, diabetic foot infections, perirectal wounds, or adjuvant therapies in severe SSTIs, such as negative pressure wound care (vacuum-assisted closure devices) and hyperbaric chambers.
OTHER DISEASES CAN MIMIC SSTIs
SSTIs vary broadly in their location and severity.
Although the classic presentation of erythema, warmth, edema, and tenderness often signals infection, other diseases can mimic SSTIs. Common ones that should be included in the differential diagnosis include gout, thrombophlebitis, deep vein thrombosis, contact dermatitis, carcinoma erysipeloides, drug eruption, and a foreign body reaction.17,18
CLUES FROM THE HISTORY
Wounds. Skin infections are usually precipitated by a break in the skin from a cut, laceration, excoriation, fungal infection, insect or animal bite, or puncture wound.
Impaired response. Patients with diabetes, renal failure, cirrhosis, chronic glucocorticoid use, history of organ transplantation, chronic immunosuppressive therapy, HIV infection, or malnourishment have impaired host responses to infection and are at risk for both more severe infections and recurrent infections. Immunocompromised hosts may also have atypical infections with opportunistic organisms such as Pseudomonas, Proteus, Serratia, Enterobacter, Citrobacter, and anaerobes. Close follow-up of these patients is warranted to ascertain appropriate response to therapy.19
Surgery that includes lymph node dissection or saphenous vein resection for coronary artery bypass can lead to impaired lymphatic drainage and edema, and therefore predisposes patients to SSTIs.
PHYSICAL EXAMINATION
The physical examination should include descriptions of the extent and location of erythema, edema, warmth, and tenderness so that progression or resolution with treatment can be followed in detail.
Crepitus can be felt in gas-forming infections and raises the concern for necrotizing fasciitis and infection with anaerobic organisms such as Clostridium perfringens.
Necrosis can occur in brown recluse spider bites, venous snake bites, or group A streptococcal infections.
Fluctuance indicates fluid and a likely abscess that may need incision and drainage.
Purpura may be present in patients on anticoagulation therapy, but if it is accompanying an SSTI, it also raises the concern for the possibility of sepsis and disseminated intravascular coagulation, especially from streptococcal infections.
Bullae can be seen in impetigo caused by staphylococci or in infection with Vibrio vulnificus or Streptococcus pneumoniae.19
Systemic signs, in addition to fever, can include hypotension and tachycardia, which would prompt closer monitoring and possible hospitalization.
Lymphangitic spread also indicates severe infection.
LABORATORY STUDIES
Simple, localized SSTIs usually do not require laboratory evaluation. Jenkins et al21 recently demonstrated that by using an algorithm for the management of hospitalized patients with cellulitis or cutaneous abscess, they could decrease resource utilization, including laboratory testing, without adversely affecting clinical outcome.
If patients have underlying disease or more extensive infection, then baseline chemistry values, a complete blood cell count, and the C-reactive protein level should be acquired.19 Laboratory findings that suggest more severe disease include low sodium, low bicarbonate (or an anion gap), and high creatinine levels; new anemia; a high or very low white blood cell count; and a high C-reactive protein level. A high C-reactive protein level has been associated with longer hospitalization.22
A score to estimate the risk of necrotizing fasciitis
This tool was developed retrospectively but has been validated prospectively. It has a high sensitivity and a positive predictive value of 92% in patients with a score of six points or more. Its specificity is also high, with a negative predictive value of 96%.20,24
Necrotizing fasciitis has a mortality rate of 23.5%, but this may be reduced to 10% with early detection and prompt surgical intervention.15 Since necrotizing fasciitis is very difficult to diagnose, clinicians must maintain a high level of suspicion and use the LRINEC score to trigger early surgical evaluation. Surgical exploration is the only way to definitively diagnose necrotizing fasciitis.
Blood cultures in some cases
Blood cultures have a low yield and are usually not cost-effective, but they should be obtained in patients who have lymphedema, immune deficiency, fever, pain out of proportion to the findings on examination, tachycardia, or hypotension, as blood cultures are more likely to be positive in more serious infections and can help guide antimicrobial therapy. Blood cultures are also recommended in patients with infections involving specific anatomic sites, such as the mouth and eyes.19
Aspiration, swabs, incision and drainage
Fluid aspirated from abscesses and swabs of debrided ulcerated wounds should be sent for Gram stain and culture. Gram stain and culture have widely varying yields, from less than 5% to 40%, depending on the source and technique.19 Cultures were not routinely obtained before MRSA emerged, but knowing antimicrobial susceptibility is now important to guide antibiotic therapy. Unfortunately, in cellulitis, swabs and aspirates of the leading edge have a low yield of around 10%.25 One prospective study of 25 hospitalized patients did report a higher yield of positive cultures in patients with fever or underlying disease,26 so aspirates may be used in selected cases. In small studies, the yield of punch biopsies was slightly better than that of needle aspirates and was as high as 20% to 30%.27
IMAGING STUDIES
Imaging can be helpful in determining the depth of involvement. Plain radiography can reveal gas or periosteal inflammation and is especially helpful in diabetic foot infections. Ultrasonography can detect abscesses.
Both magnetic resonance imaging (MRI) and computed tomography (CT) are useful to image fascial planes, although MRI is more sensitive. However, in cases of suspected necrotizing fasciitis, imaging should not delay surgical evaluation and debridement or be used as the definitive study. Therefore, the practicality of CT and MRI can be limited.15,16
ANTIMICROBIAL TREATMENT FOR SSTIs IN OUTPATIENTS
For minor skin infections such as impetigo and secondarily infected skin lesions such as eczema, ulcers, or lacerations, mupirocin 2% topical ointment (Bactroban) can be effective.27
For a simple abscess or boil, incision and drainage is the primary treatment, and antibiotics are not needed.
For a complicated abscess or boil. Patients should be given oral or intravenous antibiotic therapy to cover MRSA and, depending on the severity, should be considered for hospitalization if the abscess is associated with severe disease, rapid progression in the presence of associated cellulitis, septic phlebitis, constitutional symptoms, comorbidity (including immunosuppression), or an abscess or boil in an area difficult to drain, such as the face, hands, or genitalia.27
For purulent cellulitis in outpatients, empiric therapy for community-acquired MRSA is recommended, pending culture results. Empiric therapy for streptococcal infection is likely unnecessary. For empiric coverage of community-acquired MRSA in purulent cellulitis, oral antibiotic options include clindamycin (Cleocin), trimethoprim-sulfamethoxazole (Bactrim), doxycycline (Doryx), minocycline (Minocin), and linezolid (Zyvox).
For nonpurulent cellulitis in outpatients, empiric coverage for beta-hemolytic streptococci is warranted. Coverage for community-acquired MRSA should subsequently be added for patients who do not respond to beta-lactam therapy within 48 to 72 hours or who have chills, fever, a new abscess, increasing erythema, or uncontrolled pain.
Options for coverage of both beta-hemolytic streptococci and community-acquired MRSA for outpatient therapy include clindamycin on its own, trimethoprim-sulfamethoxazole or a tetracycline in combination with a beta-lactam, or linezolid on its own.
Increasing rates of resistance to clindamycin, tetracycline, and trimethoprim-sulfamethoxazole in community-acquired MRSA may limit empiric treatment. In areas where resistance is prevalent, culture with antimicrobial susceptibility testing may be required before starting one of these antibiotics.
The use of rifampin (Rifadin) as a single agent is not recommended because resistance is likely to develop. Also, rifampin is not useful as adjunctive therapy, as evidence does not support its efficacy.19,27,29
ANTIMICROBIAL TREATMENT FOR SSTIs IN HOSPITALIZED PATIENTS
For hospitalized patients with a complicated or severe SSTI, empiric therapy for MRSA should be started pending culture results. FDA-approved options are vancomycin, linezolid, daptomycin (Cubicin), tigecycline (Tygacil), and telavancin (Vibativ). Data on clindamycin are very limited in this population. A beta-lactam antibiotic such as cefazolin (Ancef) may be considered in hospitalized patients with nonpurulent cellulitis, and the regimen can be modified to MRSA-active therapy if there is no clinical response. Linezolid, daptomycin, vancomycin, and telavancin have adequate streptococcal coverage in addition to MRSA coverage.
Clindamycin is approved by the FDA for treating serious infections due to S aureus. It has excellent tissue penetration, particularly in bone and abscesses.
Clindamycin resistance in staphylococci can be either constitutive or inducible, and clinicians must be watchful for signs of resistance.
Diarrhea is the most common adverse effect and occurs in up to 20% of patients. Clostridium difficile colitis may occur more frequently with clindamycin than with other oral agents, but it has also has been reported with fluoroquinolones and can be associated with any antibiotic therapy.30
Trimethoprim-sulfamethoxazole is not FDA-approved for treating any staphylococcal infection. However, because 95% to 100% of community-acquired MRSA strains are susceptible to it in vitro, it has become an important option in the outpatient treatment of SSTIs. Caution is advised when using it in elderly patients, particularly those with chronic renal insufficiency, because of an increased risk of hyperkalemia.
Tetracyclines. Doxycycline is FDA-approved for treating SSTIs due to S aureus, although not specifically for MRSA. Minocycline may be an option even when strains are resistant to doxycycline, since it does not induce its own resistance as doxycycline does.
Tigecycline is a glycylcycline (a tetracycline derivative) and is FDA-approved in adults for complicated SSTIs and intra-abdominal infections. It has a large volume of distribution and achieves high concentrations in tissues and low concentrations in serum.
The FDA recently issued a warning to consider alternative agents in patients with serious infections because of higher rates of all-cause mortality noted in phase III and phase IV clinical trials. Due to this warning and the availability of multiple alternatives active against MRSA, tigecycline was not included in the Infectious Diseases Society of America guidelines.31
Linezolid is a synthetic oxazolidinone and is FDA-approved for treating SSTIs and nosocomial pneumonia caused by MRSA. It has 100% oral bioavailability, so parenteral therapy should only be given if there are problems with gastrointestinal absorption or if the patient is unable to take oral medications.
Long-term use of linezolid (> 2 weeks) is limited by hematologic toxicity, especially thrombocytopenia, which occurs more frequently than anemia and neutropenia. Lactic acidosis and peripheral and optic neuropathy are also limiting toxicities. Although myelosuppression is generally reversible, peripheral and optic neuropathy may not be.
Linezolid should not used in patients taking selective serotonin reuptake inhibitors if they cannot stop taking these antidepressant drugs during therapy, as the combination can lead to the serotonin syndrome.
Vancomycin is still the mainstay of parenteral therapy for MRSA infections. However, its efficacy has come into question, with concerns over its slow bactericidal activity and the emergence of resistant strains. The rate of treatment failure is high in those with infection caused by MRSA having minimum inhibitory concentrations of 1 μg/mL or greater. Vancomycin kills staphylococci more slowly than do beta-lactams in vitro and is clearly inferior to beta-lactams for methicillin-sensitive S aureus bacteremia.
Daptomycin is a lipopeptide antibiotic that is FDA-approved for adults with MRSA bacteremia, right-sided infective endocarditis, and complicated SSTI. Elevations in creatinine phosphokinase, which are rarely treatment-limiting, have occurred in patients receiving 6 mg/kg/day but not in those receiving 4 mg/kg/day. Patients should be observed for development of muscle pain or weakness and should have their creatine phosphokinase levels checked weekly, with more frequent monitoring in those with renal insufficiency or who are receiving concomitant statin therapy.
Telavancin is a parenteral lipoglycopeptide that is bactericidal against MRSA. It is FDA-approved for complicated SSTIs in adults. Creatinine levels should be monitored, and the dosage should be adjusted on the basis of creatinine clearance, because nephrotoxicity was more commonly reported among individuals treated with telavancin than among those treated with vancomycin.
Ceftaroline (Teflaro), a fifth-generation cephalosporin, was approved for SSTIs by the FDA in October 2010. It is active against MRSA and gram-negative pathogens.
Cost is a consideration
Cost is a consideration, as it may limit the availability of and access to treatment. In 2008, the expense for 10 days of treatment with generic vancomycin was $183, compared with $1,661 for daptomycin, $1,362 for tigecycline, and $1,560 for linezolid. For outpatient therapy, the contrast was even starker, as generic trimethoprim-sulfamethoxazole cost $9.40 and generic clindamycin cost $95.10.32
INDICATIONS FOR HOSPITALIZATION
Patients who have evidence of tissue necrosis, fever, hypotension, severe pain, altered mental status, an immunocompromised state, or organ failure (respiratory, renal, or hepatic) must be hospitalized.
Although therapy for MRSA is the mainstay of empiric therapy, polymicrobial infections are not uncommon, and gram-negative and anaerobic coverage should be added as appropriate. One study revealed a longer length of stay for hospitalized patients who had inadequate initial empiric coverage.33
Vigilance should be maintained for overlying cellulitis which can mask necrotizing fasciitis, septic joints, or osteomyelitis.
Perianal abscesses and infections, infected decubitus ulcers, and moderate to severe diabetic foot infections are often polymicrobial and warrant coverage for streptococci, MRSA, aerobic gram-negative bacilli, and anaerobes until culture results can guide therapy.
INDICATIONS FOR SURGICAL REFERRAL
Extensive perianal or multiple abscesses may require surgical drainage and debridement.
Surgical site infections should be referred for consideration of opening the incision for drainage.
Necrotizing infections warrant prompt aggressive surgical debridement. Strongly suggestive clinical signs include bullae, crepitus, gas on radiography, hypotension with systolic blood pressure less than 90 mm Hg, or skin necrosis. However, these are late findings, and fewer than 50% of these patients have one of these. Most cases of necrotizing fasciitis originally have an admitting diagnosis of cellulitis and cases of fasciitis are relatively rare, so the diagnosis is easy to miss.15,16 Patients with an LRINEC score of six or more should have prompt surgical evaluation.20,24,34,35
- Hersh AL, Chambers HF, Maselli JH, Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med 2008; 168:1585–1591.
- Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med 2008; 51:291–298.
- Edelsberg J, Taneja C, Zervos M, et al. Trends in US hospital admissions for skin and soft tissue infections. Emerg Infect Dis 2009; 15:1516–1518.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med 2007; 357:380–390.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Chua K, Laurent F, Coombs G, Grayson ML, Howden BP. Antimicrobial resistance: not community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA)! A clinician’s guide to community MRSA—its evolving antimicrobial resistance and implications for therapy. Clin Infect Dis 2011; 52:99–114.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Centers for Disease Control and Prevention. MRSA Infections. http://www.cdc.gov/mrsa/statistics/MRSA-Surveillance-Summary.html. Accessed December 14, 2011.
- Moet GJ, Jones RN, Biedenbach DJ, Stilwell MG, Fritsche TR. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis 2007; 57:7–13.
- US Department of Health and Human Services. Guidance for Industry: Uncomplicated and Complicated Skin and Skin Structure Infections—Developing Antimicrobial Drugs for Treatment (draft guidance). July 1998. http://www.fda.gov/ohrms/dockets/98fr/2566dft.pdf. Accessed September 7, 2011.
- US Food and Drug Administration. CDER 2008 Meeting Documents. Anti-Infective Drugs Advisory Committee. http://www.fda.gov/ohrms/dockets/ac/cder08.html#AntiInfective. Accessed September 7, 2011.
- US Department of Health and Human Services. Guidance for Industry: Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment (draft guidance). August 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071185.pdf. Accessed December 14, 2011.
- Cornia PB, Davidson HL, Lipsky BA. The evaluation and treatment of complicated skin and skin structure infections. Expert Opin Pharmacother 2008; 9:717–730.
- Ki V, Rotstein C. Bacterial skin and soft tissue infections in adults: a review of their epidemiology, pathogenesis, diagnosis, treatment and site of care. Can J Infect Dis Med Microbiol 2008; 19:173–184.
- May AK, Stafford RE, Bulger EM, et al; Surgical Infection Society. Treatment of complicated skin and soft tissue infections. Surg Infect (Larchmt) 2009; 10:467–499.
- Napolitano LM. Severe soft tissue infections. Infect Dis Clin North Am 2009; 23:571–591.
- Papadavid E, Dalamaga M, Stavrianeas N, Papiris SA. Subcutaneous sarcoidosis masquerading as cellulitis. Dermatology 2008; 217:212–214.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med 2004; 32:1535–1541.
- Jenkins TC, Knepper BC, Sabel AL, et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med 2011; 171:1072–1079.
- Lazzarini L, Conti E, Tositti G, de Lalla F. Erysipelas and cellulitis: clinical and microbiological spectrum in an Italian tertiary care hospital. J Infect 2005; 51:383–389.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis 2007; 44:705–710.
- Hasham S, Matteucci P, Stanley PR, Hart NB. Necrotising fasciitis. BMJ 2005; 330:830–833.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol 1988; 26:401–404.
- Sachs MK. The optimum use of needle aspiration in the bacteriologic diagnosis of cellulitis in adults. Arch Intern Med 1990; 150:1907–1912.
- Liu C, Bayer A, Cosgrove SE, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:e18–e55.
- Hammond SP, Baden LR. Clinical decisions. Management of skin and soft-tissue infection—polling results. N Engl J Med 2008; 359:e20.
- Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med 2008; 168:805–819.
- Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect 1998; 40:1–15.
- US Food and Drug Administration. FDA Drug Safety Communication: increased risk of death with Tygacil (tigecycline) compared to other antibiotics used to treat similar infections. September 2010. http://www.fda.gov/Drugs/DrugSafety/ucm224370.htm. Accessed September 7, 2011.
- Moellering RC. A 39-year-old man with a skin infection. JAMA 2008; 299:79–87.
- Zilberberg MD, Shorr AF, Micek ST, et al. Hospitalizations with healthcare-associated complicated skin and skin structure infections: impact of inappropriate empiric therapy on outcomes. J Hosp Med 2010; 5:535–540.
- Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85:1454–1460.
- Hsiao CT, Weng HH, Yuan YD, Chen CT, Chen IC. Predictors of mortality in patients with necrotizing fasciitis. Am J Emerg Med 2008; 26:170–175.
- Hersh AL, Chambers HF, Maselli JH, Gonzales R. National trends in ambulatory visits and antibiotic prescribing for skin and soft-tissue infections. Arch Intern Med 2008; 168:1585–1591.
- Pallin DJ, Egan DJ, Pelletier AJ, Espinola JA, Hooper DC, Camargo CA. Increased US emergency department visits for skin and soft tissue infections, and changes in antibiotic choices, during the emergence of community-associated methicillin-resistant Staphylococcus aureus. Ann Emerg Med 2008; 51:291–298.
- Edelsberg J, Taneja C, Zervos M, et al. Trends in US hospital admissions for skin and soft tissue infections. Emerg Infect Dis 2009; 15:1516–1518.
- Daum RS. Clinical practice. Skin and soft-tissue infections caused by methicillin-resistant Staphylococcus aureus. N Engl J Med 2007; 357:380–390.
- Klevens RM, Morrison MA, Nadle J, et al; Active Bacterial Core surveillance (ABCs) MRSA Investigators. Invasive methicillin-resistant Staphylococcus aureus infections in the United States. JAMA 2007; 298:1763–1771.
- Chua K, Laurent F, Coombs G, Grayson ML, Howden BP. Antimicrobial resistance: not community-associated methicillin-resistant Staphylococcus aureus (CA-MRSA)! A clinician’s guide to community MRSA—its evolving antimicrobial resistance and implications for therapy. Clin Infect Dis 2011; 52:99–114.
- Miller LG, Perdreau-Remington F, Bayer AS, et al. Clinical and epidemiologic characteristics cannot distinguish community-associated methicillin-resistant Staphylococcus aureus infection from methicillin-susceptible S. aureus infection: a prospective investigation. Clin Infect Dis 2007; 44:471–482.
- Centers for Disease Control and Prevention. MRSA Infections. http://www.cdc.gov/mrsa/statistics/MRSA-Surveillance-Summary.html. Accessed December 14, 2011.
- Moet GJ, Jones RN, Biedenbach DJ, Stilwell MG, Fritsche TR. Contemporary causes of skin and soft tissue infections in North America, Latin America, and Europe: report from the SENTRY Antimicrobial Surveillance Program (1998–2004). Diagn Microbiol Infect Dis 2007; 57:7–13.
- US Department of Health and Human Services. Guidance for Industry: Uncomplicated and Complicated Skin and Skin Structure Infections—Developing Antimicrobial Drugs for Treatment (draft guidance). July 1998. http://www.fda.gov/ohrms/dockets/98fr/2566dft.pdf. Accessed September 7, 2011.
- US Food and Drug Administration. CDER 2008 Meeting Documents. Anti-Infective Drugs Advisory Committee. http://www.fda.gov/ohrms/dockets/ac/cder08.html#AntiInfective. Accessed September 7, 2011.
- US Department of Health and Human Services. Guidance for Industry: Acute Bacterial Skin and Skin Structure Infections: Developing Drugs for Treatment (draft guidance). August 2010. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm071185.pdf. Accessed December 14, 2011.
- Cornia PB, Davidson HL, Lipsky BA. The evaluation and treatment of complicated skin and skin structure infections. Expert Opin Pharmacother 2008; 9:717–730.
- Ki V, Rotstein C. Bacterial skin and soft tissue infections in adults: a review of their epidemiology, pathogenesis, diagnosis, treatment and site of care. Can J Infect Dis Med Microbiol 2008; 19:173–184.
- May AK, Stafford RE, Bulger EM, et al; Surgical Infection Society. Treatment of complicated skin and soft tissue infections. Surg Infect (Larchmt) 2009; 10:467–499.
- Napolitano LM. Severe soft tissue infections. Infect Dis Clin North Am 2009; 23:571–591.
- Papadavid E, Dalamaga M, Stavrianeas N, Papiris SA. Subcutaneous sarcoidosis masquerading as cellulitis. Dermatology 2008; 217:212–214.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Wong CH, Khin LW, Heng KS, Tan KC, Low CO. The LRINEC (Laboratory Risk Indicator for Necrotizing Fasciitis) score: a tool for distinguishing necrotizing fasciitis from other soft tissue infections. Crit Care Med 2004; 32:1535–1541.
- Jenkins TC, Knepper BC, Sabel AL, et al. Decreased antibiotic utilization after implementation of a guideline for inpatient cellulitis and cutaneous abscess. Arch Intern Med 2011; 171:1072–1079.
- Lazzarini L, Conti E, Tositti G, de Lalla F. Erysipelas and cellulitis: clinical and microbiological spectrum in an Italian tertiary care hospital. J Infect 2005; 51:383–389.
- Anaya DA, Dellinger EP. Necrotizing soft-tissue infection: diagnosis and management. Clin Infect Dis 2007; 44:705–710.
- Hasham S, Matteucci P, Stanley PR, Hart NB. Necrotising fasciitis. BMJ 2005; 330:830–833.
- Newell PM, Norden CW. Value of needle aspiration in bacteriologic diagnosis of cellulitis in adults. J Clin Microbiol 1988; 26:401–404.
- Sachs MK. The optimum use of needle aspiration in the bacteriologic diagnosis of cellulitis in adults. Arch Intern Med 1990; 150:1907–1912.
- Liu C, Bayer A, Cosgrove SE, et al; Infectious Diseases Society of America. Clinical practice guidelines by the Infectious Diseases society of America for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin Infect Dis 2011; 52:e18–e55.
- Hammond SP, Baden LR. Clinical decisions. Management of skin and soft-tissue infection—polling results. N Engl J Med 2008; 359:e20.
- Perlroth J, Kuo M, Tan J, Bayer AS, Miller LG. Adjunctive use of rifampin for the treatment of Staphylococcus aureus infections: a systematic review of the literature. Arch Intern Med 2008; 168:805–819.
- Bignardi GE. Risk factors for Clostridium difficile infection. J Hosp Infect 1998; 40:1–15.
- US Food and Drug Administration. FDA Drug Safety Communication: increased risk of death with Tygacil (tigecycline) compared to other antibiotics used to treat similar infections. September 2010. http://www.fda.gov/Drugs/DrugSafety/ucm224370.htm. Accessed September 7, 2011.
- Moellering RC. A 39-year-old man with a skin infection. JAMA 2008; 299:79–87.
- Zilberberg MD, Shorr AF, Micek ST, et al. Hospitalizations with healthcare-associated complicated skin and skin structure infections: impact of inappropriate empiric therapy on outcomes. J Hosp Med 2010; 5:535–540.
- Wong CH, Chang HC, Pasupathy S, Khin LW, Tan JL, Low CO. Necrotizing fasciitis: clinical presentation, microbiology, and determinants of mortality. J Bone Joint Surg Am 2003; 85:1454–1460.
- Hsiao CT, Weng HH, Yuan YD, Chen CT, Chen IC. Predictors of mortality in patients with necrotizing fasciitis. Am J Emerg Med 2008; 26:170–175.
KEY POINTS
- Categories and definitions of specific subtypes of infections are evolving and have implications for treatment.
- Methicillin-resistant Staphylococcus aureus (MRSA) and streptococci continue to be the predominant organisms in SSTIs.
- A careful history and examination along with clinical attention are needed to elucidate atypical and severe infections.
- Laboratory data can help characterize the severity of disease and determine the probability of necrotizing fasciitis.
- Although cultures are unfortunately not reliably positive, their yield is higher in severe disease and they should be obtained, given the importance of antimicrobial susceptibility.
- The Infectious Diseases Society of America has recently released guidelines on MRSA, and additional guidelines addressing the spectrum of SSTIs are expected within a year.
Dabigatran
To the Editor: The article “Dabigatran: Will it change clinical practice”1 has a dangerous error. In its Key Points, it says “dabigatran is a potent, reversible direct thrombin inhibitor.” In fact, it is not reversible.2
Shamefully poor editing.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- Antithrombotic drugs. Treat Guidel Met Lett 2011; 9:61–66.
To the Editor: The article “Dabigatran: Will it change clinical practice”1 has a dangerous error. In its Key Points, it says “dabigatran is a potent, reversible direct thrombin inhibitor.” In fact, it is not reversible.2
Shamefully poor editing.
To the Editor: The article “Dabigatran: Will it change clinical practice”1 has a dangerous error. In its Key Points, it says “dabigatran is a potent, reversible direct thrombin inhibitor.” In fact, it is not reversible.2
Shamefully poor editing.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- Antithrombotic drugs. Treat Guidel Met Lett 2011; 9:61–66.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- Antithrombotic drugs. Treat Guidel Met Lett 2011; 9:61–66.
In reply: Dabigatran
In Reply: This is not an error. When we1 and others2 said that dabigatran is a reversible direct thrombin inhibitor, we were referring to its effect at the molecular level, the appropriate description of its mechanism of action. However, we suspect that Dr. Smith means that there is no antidote to give in cases of bleeding or overdose. We share his concern and we discussed this in our article.
Unlike heparin, direct thrombin inhibitors act independently of antithrombin and inhibit thrombin bound to fibrin or fibrin degradation products. There are two types of direct thrombin inhibitors: bivalent (eg, hirudin) and univalent (eg, argatroban, ximelagatran, and dabigatran). The bivalent ones block thrombin at its active site and at an exosite and form an irreversible complex with it. The univalent ones interact with only the active site and reversibly inhibit thrombin, eventually dissociating from it and leaving a small amount of free, enzymatically active thrombin available for hemostatic interactions. Therefore, in contrast to the hirudins, they produce relatively transient thrombin inhibition.2–4
As we pointed out in our article, the lack of an antidote for dabigatran and the lack of experience in treating bleeding complications are major concerns. Fortunately, the drug has a short half-life (12–14 hours) so that the treatment is to withhold the next dose while maintaining adequate diuresis and giving transfusions as indicated. Activated charcoal, given orally to reduce absorption, is under evaluation but must be given within 1 or 2 hours after the dabigatran dose.1 Dabigatran can be removed by dialysis (in part because it is a reversible inhibitor), a measure that may be necessary in life-threatening cases. Recombinant activated factor VII or prothrombin complex concentrates may be additional treatment options.1,4 With time will come experience and, we hope, evidence-based guidelines.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Di Nisio M, Middeldorp S, Büller HR. Direct thrombin inhibitors. N Engl J Med 2005; 353:1028–1040.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
In Reply: This is not an error. When we1 and others2 said that dabigatran is a reversible direct thrombin inhibitor, we were referring to its effect at the molecular level, the appropriate description of its mechanism of action. However, we suspect that Dr. Smith means that there is no antidote to give in cases of bleeding or overdose. We share his concern and we discussed this in our article.
Unlike heparin, direct thrombin inhibitors act independently of antithrombin and inhibit thrombin bound to fibrin or fibrin degradation products. There are two types of direct thrombin inhibitors: bivalent (eg, hirudin) and univalent (eg, argatroban, ximelagatran, and dabigatran). The bivalent ones block thrombin at its active site and at an exosite and form an irreversible complex with it. The univalent ones interact with only the active site and reversibly inhibit thrombin, eventually dissociating from it and leaving a small amount of free, enzymatically active thrombin available for hemostatic interactions. Therefore, in contrast to the hirudins, they produce relatively transient thrombin inhibition.2–4
As we pointed out in our article, the lack of an antidote for dabigatran and the lack of experience in treating bleeding complications are major concerns. Fortunately, the drug has a short half-life (12–14 hours) so that the treatment is to withhold the next dose while maintaining adequate diuresis and giving transfusions as indicated. Activated charcoal, given orally to reduce absorption, is under evaluation but must be given within 1 or 2 hours after the dabigatran dose.1 Dabigatran can be removed by dialysis (in part because it is a reversible inhibitor), a measure that may be necessary in life-threatening cases. Recombinant activated factor VII or prothrombin complex concentrates may be additional treatment options.1,4 With time will come experience and, we hope, evidence-based guidelines.
In Reply: This is not an error. When we1 and others2 said that dabigatran is a reversible direct thrombin inhibitor, we were referring to its effect at the molecular level, the appropriate description of its mechanism of action. However, we suspect that Dr. Smith means that there is no antidote to give in cases of bleeding or overdose. We share his concern and we discussed this in our article.
Unlike heparin, direct thrombin inhibitors act independently of antithrombin and inhibit thrombin bound to fibrin or fibrin degradation products. There are two types of direct thrombin inhibitors: bivalent (eg, hirudin) and univalent (eg, argatroban, ximelagatran, and dabigatran). The bivalent ones block thrombin at its active site and at an exosite and form an irreversible complex with it. The univalent ones interact with only the active site and reversibly inhibit thrombin, eventually dissociating from it and leaving a small amount of free, enzymatically active thrombin available for hemostatic interactions. Therefore, in contrast to the hirudins, they produce relatively transient thrombin inhibition.2–4
As we pointed out in our article, the lack of an antidote for dabigatran and the lack of experience in treating bleeding complications are major concerns. Fortunately, the drug has a short half-life (12–14 hours) so that the treatment is to withhold the next dose while maintaining adequate diuresis and giving transfusions as indicated. Activated charcoal, given orally to reduce absorption, is under evaluation but must be given within 1 or 2 hours after the dabigatran dose.1 Dabigatran can be removed by dialysis (in part because it is a reversible inhibitor), a measure that may be necessary in life-threatening cases. Recombinant activated factor VII or prothrombin complex concentrates may be additional treatment options.1,4 With time will come experience and, we hope, evidence-based guidelines.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Di Nisio M, Middeldorp S, Büller HR. Direct thrombin inhibitors. N Engl J Med 2005; 353:1028–1040.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Wartak SA, Bartholomew JR. Dabigatran: Will it change clinical practice? Cleve Clin J Med 2011; 78:657–664.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Di Nisio M, Middeldorp S, Büller HR. Direct thrombin inhibitors. N Engl J Med 2005; 353:1028–1040.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
Bioidentical hormone therapy: Clarifying the misconceptions
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.

WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
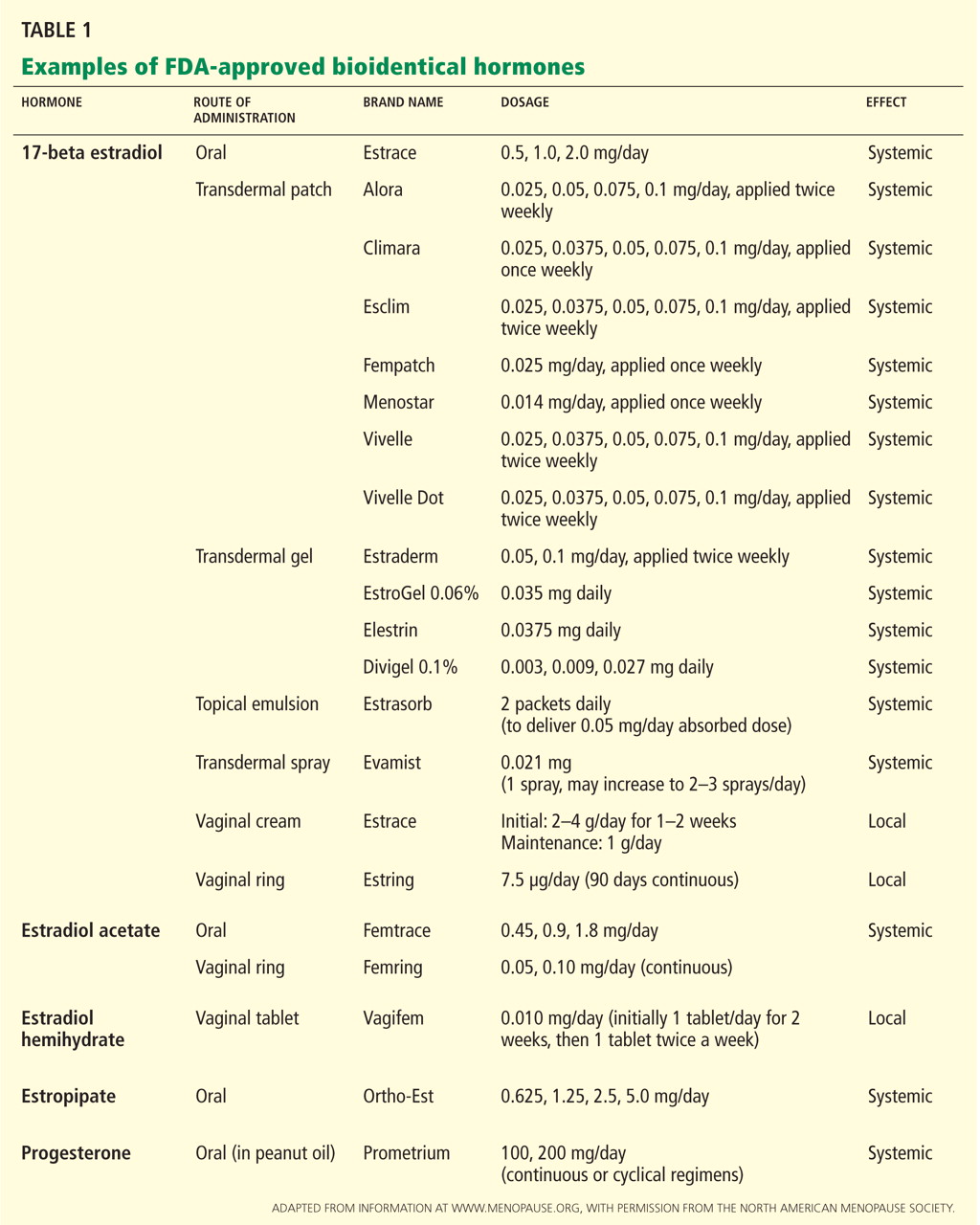
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2

Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
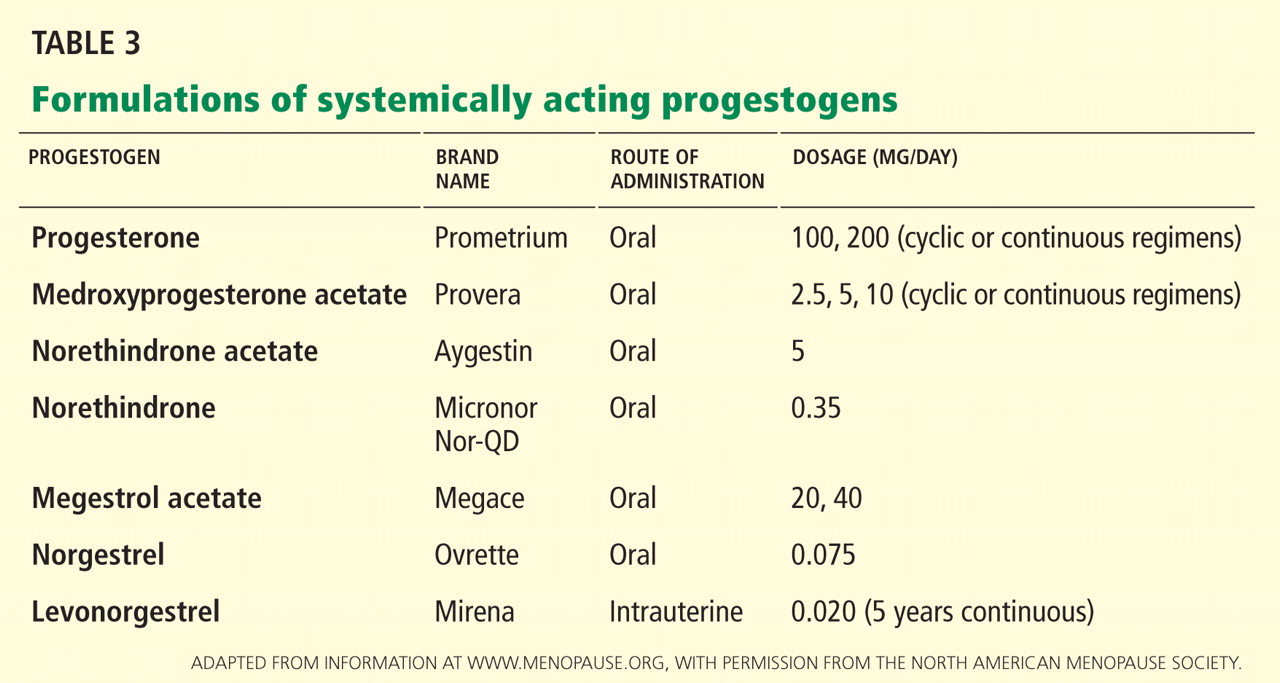
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.
WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2
Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
Recent product endorsements from celebrities on television have brought a new term into the vocabulary of many American women: bioidentical hormone therapy—treatment with hormone products that are identical in molecular structure to those in the human body.
Since 2002, when results of the Women’s Health Initiative1 raised questions about the safety of hormone replacement therapy, women have been inundated by commercials, talk shows, and self-help books that promote bioidentical hormone therapy as a safe and natural way to treat menopausal symptoms—and more.
Although this publicity has helped promote discussion about menopause, it has also perpetuated confusion and misinformation among the lay public and the general medical community concerning menopausal hormone therapy.
Many postmenopausal women suffering from vasomotor symptoms, vaginal dryness, and vaginal atrophy are apprehensive about seeking therapy, owing to concerns resulting from misinterpreted information derived from the Women’s Health Initiative trial.1 (See “What are the known risks of FDA-approved hormone therapy.”2–8) Many others are told to suffer through their symptoms, which may eventually pass. It is not surprising, then, that women turn to unconventional treatments that are claimed to be safer. This unfortunate situation has driven the business of many compounding pharmacies into the multibillion dollar level.
In this paper, we hope to clarify some of the misconceptions surrounding this issue. But first we need to define some terms in what has become a confusing area.
WHAT ARE BIOIDENTICAL HORMONES?
“Bioidentical” means identical in molecular structure to endogenous hormones. However, as we will see, a better distinction should be made between products that are approved and regulated by the US Food and Drug Administration (FDA) and those that are not.
Endogenous reproductive hormones
Women produce various reproductive hormones, including three estrogens—estradiol, estrone, and estriol—as well as progesterone and testosterone.9
17-beta estradiol (E2) is the most bioactive endogenous estrogen. It is primarily produced by the dominant ovarian follicle and the corpus luteum and is synthesized intracellularly through aromatase activity.10,11 The rest of the circulating estradiol is derived from peripheral conversion of estrone to estradiol, and this is the primary source in postmenopausal women not on hormone therapy.11
In postmenopausal women, serum estradiol levels are often below 15 pg/mL. Many physiologic effects of the cellular compartmentalized estradiol contribute to an over-riding force in certain tissues even after menopause.10 With the loss of estradiol, many tissues in postmenopausal women can be affected, particularly resulting in genitourinary atrophy and bone loss.
Estrone (E1), the second dominant human estrogen, is primarily derived from the metabolism of estradiol and from the aromatization of androstenedione in adipose tissue, with a small quantity being secreted directly by the ovary and the adrenal glands.9 In postmenopausal women, mean estrone levels are about 30 pg/mL.11
Estriol (E3), the least active of the endogenous estrogens, is very short-acting.
Progesterone is a 21-carbon steroid secreted by the human ovary.9 It is formed during the transformation of cholesterol to estrogens and androgens and is no longer produced after menopause.9
Testosterone. In premenopausal women, the androgen testosterone is synthesized by the ovary, the adrenal cortex, and the peripheral conversion of circulating androstenedione and dehydroepiandrosterone (DHEA).9 Over a woman’s life span, her androgen levels decline progressively.10 The rate of decline has not been shown to be appreciably affected by the onset of menopause.10
All these hormone therapy products are synthesized
Many nonmedical women’s health books erroneously classify the forms of estrogen used in hormone therapy as either bioidentical or synthetic. In fact, they are all man-made.
Bioidentical hormones are synthesized by chemically extracting diosgenin from plants such as yams and soy.12 Diosgenin is chemically modified to yield the precursor progesterone, which is then used to synthesize bioidentical estrogens and androgens.10
Nonbioidentical estrogen products include conjugated equine estrogens (CEE), which is extracted from the urine of pregnant mares. The two predominant estrogens found in CEE are equilin sulfate (native to horses) and estrone sulfate.10
Other nonbioidentical products include ethinyl estradiol, which is used in most combined oral contraceptives. It is formed after a minor chemical modification of estradiol that makes it one of the most potent estrogens. The ethinyl group at carbon 17 of ring D of the steroid nucleus greatly slows the hepatic and enzymatic degradation of the molecule and, thereby, makes oral ethinyl estradiol 15 to 20 times more active than oral estradiol.
Mestranol is an inactive prodrug that is converted in the body to ethinyl estradiol.
While many women may find the idea of natural bioidentical hormones derived from sweet potatoes or soybeans more acceptable than taking one made from horse’s urine, all the products undergo extensive chemical processing and modification.
Misconception: FDA-regulated products are not bioidentical
WHAT IS CUSTOMIZED COMPOUNDED HORMONAL THERAPY?
There is often confusion between the terms “bioidentical hormones” and “customized compounded therapy,” which are often used interchangeably. Compounded therapy combines ratios of bioidentical hormones into a particular recipe or mixture. Customized compounding can be done by local compounding pharmacies.2
Compounded bioidentical estrogen products
There are several commonly marketed compounded products.
Tri-estrogen (tri-est) is a compounded hormone preparation made up of a mixture of 80% estriol, 10% estrone, and 10% estradiol.12
Bi-estrogen (bi-est) contains estriol and estradiol in a ratio of 8:1 or 9:1.
Although both tri-est and bi-est are largely composed of estriol, given the low potency of estriol, the effects of these products may be solely mediated by their major bioactive component, estradiol.10,12 No large prospective, well-controlled clinical trial has investigated the compounded ratios of these mixtures of estrogens.10
Tri-est and bi-est are frequently promoted as posing less risk of breast or endometrial cancer than FDA-approved agents, although there is no research to back up this claim.12 In fact, estriol may have a stimulatory effect on the breast and endometrium.9
In addition to these “standard” compounded preparations, women can receive more customized compounds.
Valid uses for customized compounded formulations
Some clinical providers use customized compounded formulations when prescribing hormone therapy to women who have allergies to certain ingredients, such as peanut oil (found in the FDA-regulated oral product Prometrium). Customized compounded formulations have also been used when prescribing hormones currently not FDA-approved for women, such as testosterone and DHEA.12 Before oral micronized progesterone was marketed in the United States as Prometrium, it was frequently prescribed as a compounded hormone.
HORMONE THERAPY COMES IN VARIOUS FORMS
Both FDA-regulated hormone therapy and unregulated compounded hormone therapy come in various doses and dosage forms administered by different routes, allowing for individualization for each woman’s specific characteristics.
Estrogens: Oral, transdermal, others
Estrogen therapy can be given orally, transvaginally (as creams, tablets, and rings), transdermally (as patches, gels, and creams), subcutaneously in pellets, intranasally (in Europe), and by injection.11
Most oral contraceptives contain the synthetic estrogen ethinyl estradiol. Ethinyl estradiol is more potent than human estrogens,11 specifically in increasing the production of hepatic proteins (sex-hormone-binding globulin, renin substrate, corticosteroid-binding globulin, and thyroid-binding globulin).11
Bioidentical estradiol, taken orally in tablet form, is first processed through the liver and converted into estrone.12 This stimulates proteins such as C-reactive protein, activated protein C, and clotting factors, which may increase the risk of clotting.12 Estradiol given transdermally by patch or gel or vaginally bypasses the liver and enters the bloodstream as 17-beta estradiol, therefore avoiding stimulation of these proteins.12 Case-control data have shown an associated lower risk of deep venous thromboembolism with transdermal therapy.3
Subcutaneous pellet therapy is a less common, non-FDA-approved method of hormone therapy to relieve postmenopausal symptoms.10 In an outpatient procedure, the pellet is inserted into the subcutaneous fat of the abdomen.10 The crystalline pellet is biodegradable and contains a mixture of testosterone and 17-beta estradiol.10 It is important to remember that endometrial stimulation may be prolonged with this form of therapy and levels may be supraphysiologic.
Progestogens can also be given by different routes
Oral progesterone has poor gastrointestinal absorption and a short half-life.10 Therefore, it is micronized with oil for better absorption. Reported side effects include sedative and anesthetic effects; therefore, it is recommended that oral progesterone be taken at bedtime.9 Medroxyprogesterone acetate may interfere more with estrogen’s positive effects on cholesterol than micronized progesterone does.13
Topical progesterone preparations vary widely in dosage and formulation. Over-the-counter progesterone creams vary in concentration from no active ingredient to 450 mg or more of progesterone per ounce. Application sites for progesterone cream include the inner arm, chest, and inner thigh. No transdermal hormone should be applied to areas of the body that may allow possible contact and transference to others.
Progestogen products
Progestogen products include “natural” progesterone and synthetic progestins. They should be given concurrently with estrogen therapy in women who have an intact uterus to prevent endometrial hyperplasia.9
Bioidentical progesterone is micronized in the laboratory for better absorption in the gut.2
Misconception: Transdermal progesterone protects the endometrium
In general, transdermal progesterone should be avoided, as it does not protect against endometrial cancer.
Many forms of progesterone are available by prescription at compounding pharmacies as lotions, gels, creams, capsules, trochees, and suppositories.9 Transdermal progesterone creams are also available over the counter at health stores. Some of these creams contain only diosgenin, a progesterone precursor derived from wild yams.10 Diosgenin cannot be converted into progesterone within the body and thus does not provide an adequate amount of absorbable progesterone.9 Therefore, progesterone cream that contains only diosgenin is not effective in preventing endometrial hyperplasia and cancer.
To achieve a physiologic response, progesterone levels must be at least in the nanogram range.10 Transdermal progesterone cream has not been shown to reach this level and may not significantly improve vasomotor symptoms.12 Some practitioners prescribe cream that contains more than 400 mg progesterone per ounce. This may achieve physiologic levels of progesterone, but no improvement has been proven for bone mineral density or endometrial protection. In general, no transdermal progesterone cream can be assumed to protect the endometrium against the stimulatory effects of estrogen.
CUSTOM COMPOUNDING AND SALIVA TESTING TO INDIVIDUALIZE THERAPY
Some clinicians who prescribe compounded hormones order saliva tests. They argue the tests help them to establish which hormones are deficient and therefore to customize therapy.12 The basis for this is that saliva is similar to an ultrafiltrate of blood and, theoretically, hormone levels in saliva should represent the bioavailable hormone in serum.10
Unfortunately, this testing is often unreliable due to poor stability of samples in storage and large interassay variability.10 Many factors may alter hormone levels in saliva and make test results unreproducible, including the time of day the sample is collected and dietary habits.10 The FDA states that there is no scientific basis for using salivary testing to adjust hormone levels.2
Levels of drugs with clearance that varies depending on hepatic enzyme activity and plasma binding (capacity-limited metabolism) such as estradiol and testosterone can be monitored with total blood serum concentrations.10 However, many physiologic effects of estrogens are determined intracellularly at the level of tissues.10 Therefore, although levels during therapy with bioidentical estrogens can be monitored more precisely, the FDA states that hormone therapy should be guided by symptom response and findings on physical examination and not by hormone levels alone.2,12 It may be reasonable to order serum levels of estradiol in women being treated with therapeutic doses of bioidentical estrogen but still not achieving symptom relief. If women are being treated with conjugated equine estrogens, serum levels cannot be monitored. Total estrogen can be monitored as a send-out laboratory test.
MISCONCEPTION: HORMONE THERAPY IS A FOUNTAIN OF YOUTH
Customized compounded hormonal therapy is marketed as being able to help with rejuvenation, improve memory, sexual function, and reverse the aging process, essentially promising to be an elixir or fountain of youth.
These claims are not substantiated. However, the actual benefits of hormone therapy in women who have menopausal symptoms include alleviation of moderate to severe vasomotor symptoms and vaginal atrophy that can result in dyspareunia. By alleviating their symptoms, hormone therapy improves women’s quality of life. It also reduces the incidence of postmenopausal osteoporotic fractures.
A research finding that is often overlooked is that postmenopausal women younger than 60 years who started estrogen or estrogenprogestin therapy soon after menopause had a 30% lower rate of death from all causes.2,14 This difference was statistically significant when the estrogen and estrogen-progestin therapy groups were combined. No reduction in the mortality rate was seen if therapy was started after age 60.
MISCONCEPTION: COMPOUNDED THERAPY IS SAFER
Compounded hormone therapy is often marketed as a safer or more effective alternative to government-regulated and approved therapy. Unfortunately, these claims are often false and misleading, and safety information is not consistently provided to patients as is required with FDA-regulated hormone therapy.2
Since these compounds have not been approved by the FDA, there is no guarantee that the ingredients have been tested for purity, potency, and efficacy. There is no batch standardization. These unregulated therapies may use unapproved ingredients, routes of administration, and mixtures with contaminants such as dyes and preservatives.2
Also, custom-compounded prescriptions are considered experimental. Therefore, they are often not covered by insurance, and many women must pay for them out of pocket.11
The North American Menopause Society does not recommend custom-mixed products over well-tested, government-approved commercial products for most women.2 All bioidentical hormone prescriptions should include a patient package insert,11 identical to that required of FDA-approved products.2
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
- Rossouw JE, Anderson GL, Prentice RL, et al. Risks and benefits of estrogen plus progestin in healthy postmenopausal women: principal results from the Women’s Health Initiative randomized controlled trial. JAMA 2002; 288:321–333.
- North American Menopause Society. Estrogen and progestogen use in postmenopausal women: 2010 position statement of the North American Menopause Society. Menopause 2010; 17:242–255.
- Canonico M, Oger E, Plu-Bureau G; Estrogen and Thromboembolism Risk (ESTHER) Study Group. Hormone therapy and venous thromboembolism among postmenopausal women: impact of the route of estrogen administration and progestogens: the ESTHER study. Circulation 2007; 115:840–845.
- Risks of postmenopausal hormone replacement (letters). JAMA 2002; 288:2819–2825.
- Rossouw JE, Prentice RL, Manson JE, et al. Postmenopausal hormone therapy and risk of cardiovascular disease by age and years since menopause. JAMA 2007; 297:1465–1477.
- Grodstein F, Manson JE, Colditz GA, Willett WC, Speizer FE, Stampfer MJ. A prospective, observational study of postmenopausal hormone therapy and primary prevention of cardiovascular disease. Ann Intern Med 2000; 133:933–941.
- Shumaker SA, Legault C, Rapp SR, et al; WHIMS Investigators. Estrogen plus progestin and the incidence of dementia and mild cognitive impairment in postmenopausal women: the Women’s Health Initiative Memory Study: a randomized controlled trial. JAMA 2003; 289:2651–2662.
- Chlebowski RT, Anderson GL, Gass M, et al; WHI Investigators. Estrogen plus progestin and breast cancer incidence and mortality in postmenopausal women. JAMA 2010; 304:1684–1692.
- Lobo RA. Treatment of the Postmenopausal Woman: Basic and Clinical Aspects. 3rd ed. Burlington, MA: Academic Press; 2007.
- Cirigliano M. Bioidentical hormone therapy: a review of the evidence. J Womens Health (Larchmt) 2007; 16:600–631.
- Menopause Practice: A Clinician’s Guide. 4th ed. Cleveland, OH: The North American Menopause Society; 2010.
- What are bioidentical hormones? Natural. Bioidentical. Compounded. Confusion about these terms is only adding to the confusion over hormone therapy. Harv Womens Health Watch 2006; 13:1–3.
- The Writing Group for the PEPI Trial. Effects of estrogen or estrogen/progestin regimens on heart disease risk factors in postmenopausal women. The Postmenopausal Estrogen/Progestin Interventions (PEPI) Trial. JAMA 1995; 273:199–208.
- Hodis HN, Mack WJ. Postmenopausal hormone therapy in clinical perspective. Menopause 2007; 14:944–957.
KEY POINTS
- Hormone therapy is indicated for relief of menopausal symptoms; claims of reversal of the aging process are unsubstantiated.
- Products that are custom-compounded are not regulated by the US Food and Drug Administration and therefore carry no assurance of purity, safety, or efficacy.
- Transdermal progesterone creams do not achieve high enough serum levels to protect the endometrium.
- Hormone therapy is titrated on the basis of symptom response. Measuring hormone levels in saliva is not called for and is probably not reliable.
The bittersweet of steroid therapy

For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
For many of those long-term effects, such as osteoporosis, cushingoid features, skin fragility, and cataracts, all we can do is hope that they don’t occur, since there is little we can do to screen for or prevent them. We have previously discussed steroid-associated osteoporosis in the Journal,1 and strategies for preventing it have been proposed by specialty societies.2 For other complications such as hypertension, weight gain, and glucose intolerance, we can offer common-sense protective suggestions, monitor for them, and intervene if they occur.
In this issue, Dr. M. Cecilia Lansang and Ms. Leighanne Kramer Hustak3 discuss the management of steroid-induced adrenal suppression and diabetes. They offer practical management suggestions but also point out that the evidence base for our treatment decisions is surprisingly limited.
Nearly all patients chronically receiving high-dose glucocorticoid therapy develop glucose intolerance, but knowing when that is happening is not always easy. In patients destined to develop type 2 diabetes, the laboratory or clinical signs of hyperglycemia appear only when the pancreas can no longer maintain the insulin production necessary to overcome peripheral insulin resistance. Steroid-induced diabetes is characterized by increased gluconeogenesis, insulin resistance, and excessive postprandial surges, so fasting glucose levels are not sensitive for this clinical syndrome.
The degree and duration of the chronic hyperinsulinemia and hyperglycemia dictates the risk of microvascular complications and thus will be linked to duration of steroid therapy (unless the steroid is unmasking preexisting mild diabetes). Although issues surrounding tight control of blood glucose levels in the acute setting remain unresolved, I believe that even short-term significant steroid-induced hyperglycemia should be prevented when reasonably possible, at the least keeping in mind the additive ill effects of hyperglycemia and steroid therapy on the risk of nuisance infections such as oral and vaginal candidiasis and urinary tract infections that, in the setting of high-dose steroid therapy, can rapidly turn nasty.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- American College of Rheumatology Ad Hoc Committee on Glucocorticoid-Induced Osteoporosis. Recommendations for the prevention and treatment of glucocorticoid-induced osteoporosis: 2001 update. Arthritis Rheum 2001; 44:1496–1503.
- Lansang MC, Hustak LK. Glucocorticoid-induced diabetes and adrenal suppression: how to detect and manage them. Cleve Clin J Med 2011; 78:748–756.
Glucocorticoid-induced diabetes and adrenal suppression: How to detect and manage them
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.

If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.

Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:

However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.
If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.
Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:
However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
Glucocorticoids are commonly prescribed by primary care physicians and specialists alike for multiple medical problems, acute as well as chronic.
However, these useful drugs have adverse effects on multiple endocrine systems, effects that include diabetes (or worsening of hyperglycemia in those with known diabetes), Cushing syndrome, adrenal suppression, osteoporosis (reviewed in the Cleveland Clinic Journal of Medicine in August 2010),1 and dyslipidemia. In addition, suppression of gonadotropins, growth hormone, and, acutely, thyrotropin can ensue.
The focus of this review is on the diabetogenic and adrenal suppressive effects of glucocorticoids and their management. We describe the rationale for choosing specific drugs to counter hyperglycemia, tests for determining adrenal suppression and systemic glucocorticoid absorption, and how and why to taper these drugs.
WIDELY USED DRUGS
Although glucocorticoids (often simply called steroids or corticosteroids, although not all steroids are corticosteroids, and not all corticosteroids are glucocorticoids) are the core treatment for adrenal insufficiency, in most cases they are prescribed for their anti-inflammatory effects. They act through multiple pathways at the cellular and molecular levels, suppressing the cascades that would otherwise result in inflammation and promoting pathways that produce anti-inflammatory proteins.2
In addition to formulations that are intended to have systemic effects, other, “local” formulations are made for specific conditions, such as intra-articular injections for arthritis, epidural injections for lumbar disk pain, eye drops for uveitis, nasal sprays for allergic rhinitis, inhalers for asthma, and topical ointments and creams for eczema. However, as we will discuss, even these preparations can have systemic effects.
GLUCOCORTICOID-INDUCED DIABETES IS COMMON
Glucocorticoids are the most common cause of drug-induced diabetes. Though the exact prevalence is not known, a few observations suggest that glucocorticoid-induced diabetes or hyperglycemia is common:
- In patients with rheumatoid arthritis, mean age 62 years, nearly 9% developed diabetes in the 2 years after starting glucocorticoid treatment, which was a higher rate than expected.3
- In nondiabetic patients with primary renal disease treated with prednisolone 0.75 mg/kg/day, 42% were found to have 2-hour post-lunch plasma glucose concentrations higher than 200 mg/dL but normal fasting glucose levels.4
- In a case-control study, the odds ratio of starting an oral hypoglycemic agent or insulin was 1.77 for patients receiving a hydrocortisone-equivalent dose of 1 to 39 mg/day, 3.02 for 40 to 79 mg/day, 5.82 for 80 to 119 mg/day, and 10.34 for 120 mg/day or more.5 (For a full discussion of glucocorticoid equivalents, see the section below on Cushing syndrome and adrenal suppression.)
- In patients with type 1 diabetes, prednisone 60 mg/day raised the blood glucose levels starting 6 hours after the prednisone dose.6
- Diabetic ketoacidosis and hyperosmolar nonketotic syndrome have been reported as a result of glucocorticoid treatment.7–9
GLUCOCORTICOIDS CAUSE DIABETES MAINLY VIA INSULIN RESISTANCE
The mechanism by which glucocorticoids cause diabetes predominantly involves insulin resistance rather than decreased insulin production. In fact, in a study in healthy volunteers, 10 hydrocortisone infusion resulted in higher insulin production than saline infusion did. (In high doses, however, glucocorticoids have been shown to decrease insulin secretion.11)
Normally, in response to insulin, the liver decreases its output of glucose. Glucocorticoids decrease the liver’s sensitivity to insulin, thereby increasing hepatic glucose output.12 They also inhibit glucose uptake in muscle and fat, reducing insulin sensitivity as much as 60% in healthy volunteers. This seems primarily due to a postreceptor effect, ie, inhibition of glucose transport.13–15
THE PEAK EFFECT OCCURS 4 TO 6 HOURS AFTER DOSING
To understand the optimal time for checking plasma glucose and to apply appropriate treatment, we should consider the pharmacokinetic profile of glucocorticoids.
Studied using the whole-blood lymphocyte proliferation technique, prednisone shows a peak effect at about 4 to 6 hours and a duration of action of 13 to 16 hours.16 This closely resembles what we see in terms of glucose excursion with this drug.17 Two studies of intravenous dexamethasone 10 mg showed that glucose levels rose within 4 hours of injection, but did not pursue this beyond that time frame.18,19
PATIENTS WITHOUT A PREVIOUS DIAGNOSIS OF DIABETES
Be alert for new-onset diabetes
For most diseases treated with glucocorticoids, clinicians can estimate in advance how long the patient will need to take the drug. We can arbitrarily classify the projected exposure as either short-term (3 to 4 weeks or less, such as a 6-day course of methylprednisolone for allergic conditions) or long-term (such as in transplant recipients to prevent rejection or to treat graft-vs-host disease). Hyperglycemia is a potential concern with both short-term and long-term treatment. However, guidelines on checking blood sugar levels, as opposed to relying on symptoms alone, are available only for long-term glucocorticoid treatment.
Patients beginning treatment should be warned of typical diabetes symptoms such as thirst and increased urination and, should these occur, to seek medical attention to have their blood glucose level checked. It is also reasonable to have them return in a week for a random postprandial plasma glucose test in the mid-afternoon.
Why this timing? In most once-daily regimens, glucocorticoids are given in the morning to prevent adrenal suppression (discussed below). In our experience, glucose levels start to rise mid-morning and continue to increase until bedtime. Measuring glucose levels 1 to 2 hours after lunch allows for both the glucocorticoid action and the carbohydrate absorption from lunch to reach their peaks. If hyperglycemia is going to happen, it should be detectable by then. A glucose level of 200 mg/dL or higher should prompt the practitioner to pursue this further.
If glucocorticoid treatment is to continue beyond 3 to 4 weeks, the only population for which there are published guidelines on managing glucocorticoid-related diabetes is transplant recipients. International consensus guidelines, published in 2003, suggest checking the fasting plasma glucose level once a week for the first 4 weeks after transplantation, then at 3 months, at 6 months, and then once a year.20
Though practical, this suggestion does not reflect the fact that glucocorticoids often do not affect fasting plasma glucose, especially if given once daily in the morning at doses of 30 mg or less of prednisone or its equivalent. These guidelines thus may not be applicable to other populations with glucocorticoid-induced diabetes.
The transplant guidelines do mention that an oral glucose tolerance test may be more sensitive, but this is often cumbersome to perform. We believe that checking random postprandial plasma glucose levels is helpful in this regard.
If the patient was at risk of developing diabetes even before receiving a glucocorticoid (for example, if he or she is overweight, has a family history of diabetes, or had a previous hemoglobin A1c of 5.7% or higher), then a fasting plasma glucose level of 126 mg/dL or higher or a hemoglobin A1c of 6.5% or higher might suffice to diagnose diabetes. Results should be confirmed on a separate day in the absence of unequivocal hyperglycemia. Fasting hyperglycemia can also be seen in patients receiving higher once-daily glucocorticoid doses—in our experience, an equivalent of prednisone 40 mg once a day in the morning— or twice-daily dosing.
A hemoglobin A1c checked less than 2 to 3 months after starting glucocorticoid treatment will not be sensitive in picking up glucocorticoid-induced diabetes if the patient did not have underlying diabetes.
Diet and exercise may not be practical
Though diet and exercise are important in managing diabetes, the condition for which the patient is receiving a glucocorticoid may prevent him or her from exercising, at least in the acute phase of the illness.
In addition, though the exact mechanism is not known, glucocorticoids increase hunger, and so decreasing food intake is not easy either. Nonetheless, patients should be familiarized with what carbohydrates are and should be advised to reduce their intake of them.
For suspected type 1 diabetes, start insulin
If type 1 diabetes is suspected, for example, in patients who are lean, younger than 30 years, or who had presented with diabetic ketoacidosis, then insulin should be started. In equivocal cases, insulin therapy can commence while testing is done for C-peptide, glutamic acid decarboxylase antibodies, islet cell antibodies, and insulinoma-associated protein antibodies.
Starting oral antidiabetic drugs
Some patients may have contraindications to specific drugs. For example, metformin (Glucophage) is contraindicated if the serum creatinine level is elevated, an abnormality that renal transplant patients may continue to have.
If the patient has no such contraindications, we have found the following medications suitable in view of their efficacy, low risk of hypoglycemia, or lack of distressing side effects. They will often lower glucose levels enough to achieve capillary blood glucose or fingerstick goals (discussed below). None of them has been specifically approved by the US Food and Drug Administration for glucocorticoid-induced diabetes, but they are approved for type 2 diabetes.
Guidelines from the American Association of Clinical Endocrinologists for type 2 diabetes call for starting monotherapy if the hemoglobin A1c is 6.5% to 7.5%, dual therapy if it is 7.6% to 9%, triple therapy if it is higher than 9% and the patient has no symptoms, and insulin if it is higher than 9% and the patient does have symptoms.22
In terms of estimated average glucose levels, these categories correspond to 140 to 169 mg/dL for monotherapy, 171 to 212 mg/dL for dual therapy, and higher than 212 mg/dL for triple therapy or insulin. Since estimated average levels also include fasting glucose levels (which are lower in glucocorticoid-induced diabetes compared with nonfasting levels), and because we use the American Diabetes Association general hemoglobin A1c goal of less than 7%, we believe that our suggestions below are reasonable.
We divide our recommendations according to initial random (ideally, 1- to 2-hour postprandial) plasma glucose levels.
If the random or 1- to 2-hour post-meal plasma glucose is lower than 220 mg/dL
In this situation the choices are:
- Metformin
- Dipeptidyl peptidase-4 (DPP-4) inhibitors (“gliptins”)
- Meglitinides (“glinides”). The guidelines on new-onset diabetes after transplantation point out that meglitinides may be the safest agents apart from insulin in the renal transplant population, but does acknowledge that efficacies of different oral agents have not been compared in this group.20
- Glucagon-like protein-1 (GLP-1) agonists
- Sulfonylureas. However, the longer-acting forms such as glimepiride (Amaryl) are not suitable if the fasting plasma glucose is not affected.
We have not used thiazolidinediones (“glitazones”) routinely because they can cause weight gain and edema—problems that are already seen with the use of steroids—and have a slower onset of action.
If the random or 1- to 2-hour post-meal plasma glucose is 220 to 300 mg/dL
Often, a combination of drugs or insulin (see below) is needed. However, you can start with one agent and add a second agent within 2 or 3 months (as is recommended for type 2 diabetes).22,23 The following combinations of the agents listed above are supported by published guidelines for type 2 diabetes:
- Metformin plus a sulfonylurea22,23
- Metformin plus a glinide22
- Metformin plus a GLP-1 agonist23
- Metformin plus a DPP-4 inhibitor.22
If the random or 1- to 2-hour post-meal plasma glucose is higher than 300 mg/dL
In our experience, if their plasma glucose levels are this high, patients are experiencing frank symptoms of hyperglycemia.
Insulin addresses those symptoms and avoids the prolonged wait that often results from unsuccessfully starting one agent and then adding another. Of all the available drugs, insulin is the only one that can be used despite multiple underlying illnesses; it does not cause a lot of drug interactions, and the dose can be adjusted upward and downward in increments to fit the patient’s needs, especially when a larger glucocorticoid load is given up front and then is tapered either slowly or rapidly. However, it can cause hypoglycemia and weight gain.
The initial total daily dose of insulin can be based on the patient’s weight. A starting total daily dose of 0.15 to 0.3 U/kg is reasonable— on the lower end if only the postprandial glucose levels are elevated, and on the higher end if both fasting and postprandial glucose levels are affected.
If fasting glucose levels are not elevated, then Neutral Protamine Hagedorn insulin (which is intermediate-acting) or a premixed combination of an intermediate-acting plus a fast- or short-acting insulin can be given once a day before breakfast, or even before lunch if the glucose levels start to rise only after lunch.
If both the fasting and the postprandial glucose levels are elevated, regimens similar to those for type 1 or insulin-requiring type 2 diabetes can be used, except that the ratios of the doses are tilted more toward covering postprandial than fasting hyperglycemia:
- Long-acting insulin plus prandial insulin, in a ratio of 30:70 to 50:50. As glucocorticoids are tapered, the long-acting insulin may have to be discontinued while the prandial doses are continued, since the fasting glucose level decreases first.
- Premixed insulins, with one-half to two-thirds of the dose given before breakfast and the rest before the evening meal, with the possibility of a third injection before lunch. As glucocorticoids are tapered, the evening dose is tapered first.
- Intermediate-acting insulin plus short- or fast-acting insulin in the morning (these two will make up one-half to two-thirds of the total daily dose), short- or fast-acting insulin before the evening meal, and intermediate-acting insulin at bedtime. As glucocorticoids are tapered, the bedtime insulin is tapered first.
Capillary blood glucose (fingerstick) checks
The timing and frequency of fingerstick checks depend on the treatment.
Though postprandial testing is ideal, it is often not practical or convenient. Before lunch, before dinner, and at bedtime are good alternatives since they reflect the pattern of glucose rise throughout the day. For patients on diet and exercise with or without agents other than insulin, testing once or twice a day is reasonable, rotating times before meals (including fasting if this time is affected) and at bedtime.
For patients on insulin, checking two to four times a day initially would help match insulin doses with glucose excursions. For continued care, the American Diabetes Association recommends fingerstick checks three times daily in patients on multiple insulin injections, but it has no specific recommendations for those on once-a-day insulin.21 We have been recommending that our patients on once-daily insulin check at least twice a day.
Goal fingerstick glucose levels that we use are in accordance with the American Diabetes Association guidelines for diabetes in general21:
- Before meals 70 to 130 mg/dL or
- 1 to 2 hours after meals < 180 mg/dL.
During steroid taper, if the glucocorticoid dose is in the lower range (eg, a prednisone-equivalent dose of approximately 7.5 mg per day or less), the fingerstick glucose levels are at the lower end of the target range, and the patient is on a single antidiabetic agent that does not often cause hypoglycemia (eg, metformin), then it is reasonable to ask the patient to not take the antidiabetic medication for 3 to 7 days while continuing to check fingersticks to see if it needs to be resumed. Patients on agents that can cause hypoglycemia need to check more often during the 1 to 3 days after the glucocorticoid dose reduction, as it may take this much time for the glycemic effect to diminish and to adjust the diabetes medication to the appropriate dose.
STARTING GLUCOCORTICOIDS IN PATIENTS WITH KNOWN DIABETES
Fingerstick checks more often
Most patients will already have a glucose meter. They should be instructed to check as discussed above if they do not have a previous diagnosis of diabetes, or to continue as they are doing if they are already checking more often. Patients who have been checking only fasting levels should be instructed to check later in the day, either before or 1 to 2 hours after meals, as discussed above. Patients on oral medications may need additional oral agents or insulin.
Adjust medications if glucose is not at goal
Patients with type 2 diabetes treated with diet and exercise alone can be started on the medications discussed above if their fingerstick readings are not at goal.
If they are already on insulin, we advise them to increase the short- or fast-acting insulins and the morning intermediate-acting insulin by at least 10% to 20% as soon as an elevation in glucose is detected. Long-acting insulin or nighttime intermediate-acting insulin should be increased if fasting glucose levels are affected.
Insulin requirements can double depending on the glucocorticoid dose. In patients with type 1 diabetes who were given prednisone 60 mg orally for 3 days, mean blood glucose levels increased from a baseline of 110 mg/dL at baseline to 149 mg/dL on the days on prednisone.6 The average blood glucose level remained elevated at 141 mg/dL on the day after the last dose of prednisone. The insulin dose increased by 31% to 102% (mean 69%).
CUSHING SYNDROME AND ADRENAL SUPPRESSION
Unlike glucocorticoid-induced diabetes, in which the dilemma is often when to initiate antidiabetic treatment, the question for patients in whom Cushing syndrome or adrenal suppression has developed is when to discontinue glucocorticoids.
Adrenal suppression for the most part goes hand in hand with exogenous Cushing syndrome. If cushingoid features develop, we can infer that the dose of exogenous glucocorticoid exceeds the physiologic needs. This supraphysiologic dosing also leads to suppression of endogenous cortisol production. The suppression occurs at the level of the hypothalamus and pituitary gland, with subsequent atrophy of the part of the adrenal cortex that produces endogenous glucocorticoids.
To understand further the concept of supraphysiologic dosing, the following interconversion of systemic glucocorticoid effects is helpful24,25:
However, there is not much information on interconversion for the local preparations (intra-articular, epidural, inhaled, topical).
Moreover, the definition of supraphysiologic dosing seems to be evolving. Though a total hydrocortisone-equivalent dose of 30 mg/day is still often touted as physiologic replacement, many patients require less. Several studies in the early 1990s, mostly in children and adolescents, showed the mean daily cortisol production rate to be 4.8 to 6.8 mg/m2/day, or closer to 10 to 15 mg/day.26–28 For purposes of this discussion, a physiologic dose will be defined as up to 30 mg hydrocortisone per day or its equivalent.
Adrenal suppression vs insufficiency
Adrenal suppression is often confused with adrenal insufficiency.
Adrenal suppression occurs when cortisol production is decreased because of the presence of exogenous glucocorticoids or other drugs, such as megestrol acetate (Megace), that act on the glucocorticoid receptor. Another situation beyond the scope of this review is excess endogenous cortisol production by an adrenal adenoma or adrenal carcinoma that causes suppression of the contralateral adrenal gland.29
In contrast, adrenal insufficiency is caused by failure of the adrenal gland to produce cortisol as a result of an innate disorder of the adrenal gland (eg, Addison disease) or pituitary gland (eg, pituitary surgery).
Hence, endogenous cortisol production in a patient taking supraphysiologic doses of exogenous glucocorticoids may be suppressed. Recovery of endogenous cortisol production is expected after stopping the exogenous glucocorticoid, though the time to recovery can vary and the patient can be adrenally insufficient if the glucocorticoid is stopped abruptly.
In addition, during times of intercurrent illness, a patient with adrenal suppression may be relatively adrenally insufficient and may need larger doses (“stress doses”) of glucocorticoids, since the adrenal glands may be unable to mount a stress response.29
Local steroids can suppress the adrenal glands
Glucocorticoids are the most common cause of Cushing syndrome. Oral formulations such as dexamethasone, prednisone, and hydrocortisone taken in supraphysiologic doses and for prolonged durations are easily recognized as obvious causes of Cushing syndrome. However, intra-articular, epidural, inhaled, nasal, ocular, and topical steroids—so-called local preparations—have also been linked to Cushing syndrome, and physicians are less likely to recognize them as causes.30–38
In a study in 16 pediatric patients with asthma and 48 controls, inhaled beclomethasone dipropionate (Qvar) 300 to 500 μg daily resulted in adrenal suppression in 100% of patients after 6 to 42 months, as determined by an insulin tolerance test.30
The topical steroid betamethasone (Diprosone) carries a warning that systemic absorption of topical steroids can cause adrenal suppression.39 Intra-articular, intranasal, epidural, and ocular routes are also reported to cause adrenal suppression.32–38
When is adrenal suppression more likely?
Adrenal suppression is more likely in the following situations:
- Longer duration of treatment. Studies have shown that exposure to supraphysiologic steroid doses for 2 weeks or less might already suppress the adrenal glands, but the clinical significance of this is unclear since some recovery already occurs a few days after the glucocorticoids are discontinued.31,40
- Supraphysiologic doses, stronger formulations, longer-acting formulations.41
When is adrenal suppression less likely?
Adrenal suppression is less likely in the following situations:
- Regimens that mimic the diurnal rhythm of cortisol (higher dose in the morning, lower dose in the afternoon)42
- Alternate-day dosing of steroids.43
Steroid withdrawal vs adrenal insufficiency
Another phenomenon that can be confused with adrenal insufficiency or glucocorticoid insufficiency is steroid withdrawal, in which patients experience lethargy, muscle aches, nausea, vomiting, and postural hypotension as glucocorticoids are tapered and their effects wane.42 Increasing the glucocorticoid dose for presumed adrenal insufficiency may delay recovery of the adrenal function and would have to be weighed against the patient’s symptoms.
The following may help distinguish the two: if the patient is on supraphysiologic glucocorticoid doses, then he or she is not glucocorticoid-deficient and is likely suffering from steroid withdrawal. At this point, patients may just need reassurance, symptomatic treatment, or if necessary, a brief (1-week) increase of the previous lowest dose, followed by reevaluation.
With local glucocorticoid preparations that may be systemically absorbed, however, there is no good way of estimating dose equivalence. In these situations, the decision to simply reassure the patient or give symptomatic treatment—as opposed to giving low-dose oral glucocorticoids such as hydrocortisone 5 to 10 mg daily for a week followed by reevaluation— depends on the severity of symptoms and whether the patient has quick access to medical attention should he or she develop an intercurrent illness.
Identifying patients at risk of adrenal suppression
Patients presenting with weight gain or symptoms suggesting Cushing syndrome should be asked about steroid intake and should be prompted to recall possible nonoral routes. In addition, patients presenting with muscle aches and fatigue—symptoms of steroid withdrawal— may have received unrecognized local glucocorticoids that were systemically absorbed, now with diminishing effects.
The ACTH stimulation test for adrenal recovery
Testing can be done to see if the adrenal glands have recovered and glucocorticoid therapy can be discontinued (see Tapering from glucocorticoids, below).
The test most often used is the corticotropin (ACTH) stimulation test. Since the suppression is at the level of the hypothalamus and the pituitary gland, the ACTH stimulation test is an indirect method of assessing hypothalamic and pituitary function in the context of glucocorticoid-induced adrenal suppression. It has good correlation with the insulin tolerance test, the gold-standard test for an intact hypothalamic-pituitary-adrenal axis.
The synthetic ACTH cosyntropin (Cortrosyn) 250 μg is injected intravenously or intramuscularly, and a cortisol level is drawn at baseline and 30 and 60 minutes later. Other doses such as 1 μg or 10 μg have been reported but are not yet widely accepted. A cortisol level of greater than 18 to 20 μg/dL at any time point shows that the adrenals have regained function and the steroids may be discontinued.42 If adrenal suppression persists, weaning from steroids should continue.
In reality, it may not be possible or practical to do an ACTH stimulation test, as not all physicians’ offices have a supply of cosyntropin or the manpower to perform the test correctly. In these cases, weaning can progress with monitoring of symptoms.
Testing for synthetic glucocorticoids in the urine and serum can demonstrate systemic absorption and may be helpful in patients who do not recall receiving steroids.33
Tapering from glucocorticoids
Several tapering schedules have been suggested (although not necessarily validated). Whether and how to taper depend on how long the glucocorticoid has been taken.
If taken for less than 1 week, glucocorticoids can be stopped without tapering, regardless of the dose.
If taken for 1 to 3 weeks, the decision to taper depends on the clinician’s assessment of the patient’s general health or constitution and the illness for which the glucocorticoid was prescribed. For example, if the underlying disease is less likely to flare with a gradual dose reduction, then tapering would be suitable.44
If taken for more than 3 weeks, the practice has been a more rapid taper at the beginning until a physiologic dose is reached. How quickly to reduce the dose depends on whether the underlying illness is expected to flare up, or if the patient might experience steroid withdrawal symptoms.
One schedule is to lower the glucocorticoid dose by an amount equivalent to prednisolone 2.5 mg every 3 to 4 days when above the physiologic dose, then to taper more slowly by 1 mg every 2 to 4 weeks.44 Once the physiologic dose is reached, one can switch to the equivalent dose of hydrocortisone and decrease the dose by 2.5 mg a week until a daily dose of 10 mg a day is reached and maintained for 2 to 3 months, and then perform a test of adrenal function (see above).44 Passing the test implies that the adrenal glands have recovered and the glucocorticoid can be stopped.
Another option is to switch to alternate-day therapy once a physiologic dose is reached and to test 8:00 am cortisol levels, continuing the glucocorticoid and retesting in 4 to 6 weeks if the value is less than 3 μg/dL; stopping the glucocorticoid if the value is higher than 20 μg/dL; and performing an ACTH stimulation test for values in between.45
A review of other tapering regimens for chronic diseases, mostly pulmonary, did not find enough evidence to recommend one particular schedule over another.46 The tapering schedule may have to be adjusted to prevent disease flare and symptoms of steroid withdrawal.
Locally administered steroids. Since the equivalence of systemically absorbed local glucocorticoids is not known, these patients are likely to present when they have symptoms of steroid withdrawal. In this situation, testing adrenal function will help.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
- Dore RK. How to prevent glucocorticoid-induced osteoporosis. Cleve Clin J Med 2010; 77:529–536.
- Rhen T, Cidlowski JA. Antiinflammatory action of glucocorticoids—new mechanisms for old drugs. N Engl J Med 2005; 353:1711–1723.
- Panthakalam S, Bhatnagar D, Klimiuk P. The prevalence and management of hyperglycaemia in patients with rheumatoid arthritis on corticosteroid therapy. Scott Med J 2004; 49:139–141.
- Uzu T, Harada T, Sakaguchi M, et al. Glucocorticoid-induced diabetes mellitus: prevalence and risk factors in primary renal diseases. Nephron Clin Pract 2007; 105:c54–c57.
- Gurwitz JH, Bohn RL, Glynn RJ, Monane M, Mogun H, Avorn J. Glucocorticoids and the risk for initiation of hypoglycemic therapy. Arch Intern Med 1994; 154:97–101.
- Bevier WC, Zisser HC, Jovanovic L, et al. Use of continuous glucose monitoring to estimate insulin requirements in patients with type 1 diabetes mellitus during a short course of prednisone. J Diabetes Sci Technol 2008; 2:578–583.
- Cagdas DN, Paç FA, Cakal E. Glucocorticoid-induced diabetic ketoacidosis in acute rheumatic fever. J Cardiovasc Pharmacol Ther 2008; 13:298–300.
- Bedalov A, Balasubramanyam A. Glucocorticoid-induced ketoacidosis in gestational diabetes: sequela of the acute treatment of preterm labor. A case report. Diabetes Care 1997; 20:922–924.
- Yang JY, Cui XL, He XJ. Non-ketotic hyperosmolar coma complicating steroid treatment in childhood nephrosis. Pediatr Nephrol 1995; 9:621–622.
- Nielsen MF, Caumo A, Chandramouli V, et al. Impaired basal glucose effectiveness but unaltered fasting glucose release and gluconeogenesis during short-term hypercortisolemia in healthy subjects. Am J Physiol Endocrinol Metab 2004; 286:E102–E110.
- Matsumoto K, Yamasaki H, Akazawa S, et al. High-dose but not low-dose dexamethasone impairs glucose tolerance by inducing compensatory failure of pancreatic beta-cells in normal men. J Clin Endocrinol Metab 1996; 81:2621–2626.
- Rizza RA, Mandarino LJ, Gerich JE. Cortisol-induced insulin resistance in man: impaired suppression of glucose production and stimulation of glucose utilization due to a postreceptor detect of insulin action. J Clin Endocrinol Metab 1982; 54:131–138.
- Meyuhas O, Reshef L, Gunn JM, Hanson RW, Ballard FJ. Regulation of phosphoenolpyruvate carboxykinase (GTP) in adipose tissue in vivo by glucocorticoids and insulin. Biochem J 1976; 158:1–7.
- Tappy L, Randin D, Vollenweider P, et al. Mechanisms of dexamethasone-induced insulin resistance in healthy humans. J Clin Endocrinol Metab 1994; 79:1063–1069.
- Pagano G, Cavallo-Perin P, Cassader M, et al. An in vivo and in vitro study of the mechanism of prednisone-induced insulin resistance in healthy subjects. J Clin Invest 1983; 72:1814–1820.
- Magee MH, Blum RA, Lates CD, Jusko WJ. Pharmacokinetic/pharmaco-dynamic model for prednisolone inhibition of whole blood lymphocyte proliferation. Br J Clin Pharmacol 2002; 53:474–484.
- Burt MG, Roberts GW, Aguilar-Loza NR, Frith P, Stranks SN. Continuous monitoring of circadian glycemic patterns in patients receiving prednisolone for COPD. J Clin Endocrinol Metab 2011; 96:1789–1796.
- Hans P, Vanthuyne A, Dewandre PY, Brichant JF, Bonhomme V. Blood glucose concentration profile after 10 mg dexamethasone in non-diabetic and type 2 diabetic patients undergoing abdominal surgery. Br J Anaesth 2006; 97:164–170.
- Pasternak JJ, McGregor DG, Lanier WL. Effect of single-dose dexamethasone on blood glucose concentration in patients undergoing craniotomy. J Neurosurg Anesthesiol 2004; 16:122–125.
- Davidson J, Wilkinson A, Dantal J, et al; International Expert Panel. New-onset diabetes after transplantation: 2003 international consensus guidelines. Proceedings of an international expert panel meeting. Barcelona, Spain, 19 February 2003. Transplantation 2003; 75(suppl 10):SS3–SS24.
- American Diabetes Association. Standards of medical care in diabetes— 2011. Diabetes Care 2011; 34(suppl 1):S11–S61.
- Rodbard HW, Jellinger PS, Davidson JA, et al. Statement by an American Association of Clinical Endocrinologists/American College of Endocrinology consensus panel on type 2 diabetes mellitus: an algorithm for glycemic control. Endocr Pract 2009; 15:540–559.
- Nathan DM, Buse JB, Davidson MB, et al; American Diabetes Association; European Association for Study of Diabetes. Medical management of hyperglycemia in type 2 diabetes: a consensus algorithm for the initiation and adjustment of therapy: a consensus statement of the American Diabetes Association and the European Association for the Study of Diabetes. Diabetes Care 2009; 32:193–203.
- Axelrod L. Corticosteroid therapy. In:Becker KL, editor. Principles and Practice of Endocrinology and Metabolism. 3rd ed. Philadelphia, PA: Lippincott Williams and Wilkins; 2000:752–763.
- Ferri FF, editor. Practical Guide to the Care of the Medical Patient. 8th ed. Philadelphia, PA: Mosby/Elsevier; 2011.
- Kerrigan JR, Veldhuis JD, Leyo SA, Iranmanesh A, Rogol AD. Estimation of daily cortisol production and clearance rates in normal pubertal males by deconvolution analysis. J Clin Endocrinol Metab 1993; 76:1505–1510.
- Linder BL, Esteban NV, Yergey AL, Winterer JC, Loriaux DL, Cassorla F. Cortisol production rate in childhood and adolescence. J Pediatr 1990; 117:892–896.
- Esteban NV, Loughlin T, Yergey AL, et al. Daily cortisol production rate in man determined by stable isotope dilution/mass spectrometry. J Clin Endocrinol Metab 1991; 72:39–45.
- Lansang MC, Quinn SL. Adrenal suppression. BMJ BestPractice 2010. http://bestpractice.bmj.com/best-practice/monograph/863/diagnosis/stepby-step.html. Accessed August 19, 2011.
- Zöllner EW. Hypothalamic-pituitary-adrenal axis suppression in asthmatic children on inhaled corticosteroids (part 2)—the risk as determined by gold standard adrenal function tests: a systematic review. Pediatr Allergy Immunol 2007; 18:469–474.
- Schuetz P, Christ-Crain M, Schild U, et al. Effect of a 14-day course of systemic corticosteroids on the hypothalamic-pituitary-adrenal-axis in patients with acute exacerbation of chronic obstructive pulmonary disease. BMC Pulm Med 2008; 8:1.
- Kay J, Findling JW, Raff H. Epidural triamcinolone suppresses the pituitary-adrenal axis in human subjects. Anesth Analg 1994; 79:501–505.
- Lansang MC, Farmer T, Kennedy L. Diagnosing the unrecognized systemic absorption of intra-articular and epidural steroid injections. Endocr Pract 2009; 15:225–228.
- Duclos M, Guinot M, Colsy M, et al. High risk of adrenal insufficiency after a single articular steroid injection in athletes. Med Sci Sports Exerc 2007; 39:1036–1043.
- Bong JL, Connell JM, Lever R. Intranasal betamethasone induced acne and adrenal suppression. Br J Dermatol 2000; 142:579–580.
- Atabek ME, Pirgon O, Unal E. Pituitary-adrenal axis suppression due to topical steroid administration in an infant. Pediatr Int 2007; 49:242–244.
- Ozerdem U, Levi L, Cheng L, Song MK, Scher C, Freeman WR. Systemic toxicity of topical and periocular corticosteroid therapy in an 11-year-old male with posterior uveitis. Am J Ophthalmol 2000; 130:240–241.
- Chiang MY, Sarkar M, Koppens JM, Milles J, Shah P. Exogenous Cushing’s syndrome and topical ocular steroids. Eye (Lond) 2006; 20:725–727.
- Diprolene prescribing information. Schering Corp 2005. www.theodora.com/drugs/diprolene_gel_005_schering.html. Accessed September 27, 2011.
- Villabona CV, Koh C, Panergo J, Reddy A, Fogelfeld L. Adrenocorticotropic hormone stimulation test during high-dose glucocorticoid therapy. Endocr Pract 2009; 15:122–127.
- Ortega E, Rodriguez C, Strand LJ, Segre E. Effects of cloprednol and other corticosteroids on hypothalamic-pituitary-adrenal axis function. J Int Med Res 1976; 4:326–337.
- Axelrod L. Glucocorticoid therapy. Medicine (Baltimore) 1976; 55:39–65.
- Schürmeyer TH, Tsokos GC, Avgerinos PC, et al. Pituitary-adrenal responsiveness to corticotropin-releasing hormone in patients receiving chronic, alternate day glucocorticoid therapy. J Clin Endocrinol Metab 1985; 61:22–27.
- Stewart PM. The adrenal cortex. In:Kronenberg HM, editor. Williams Textbook of Endocrinology. 11th ed. Philadelphia, PA: Saunders/Elsevier; 2008.
- Hopkins RL, Leinung MC. Exogenous Cushing’s syndrome and glucocorticoid withdrawal. Endocrinol Metab Clin North Am 2005; 34:371–384.
- Richter B, Neises G, Clar C. Glucocorticoid withdrawal schemes in chronic medical disorders. A systematic review. Endocrinol Metab Clin North Am 2002; 31:751–778.
KEY POINTS
- Nonfasting plasma glucose levels are more sensitive than fasting levels for detecting glucocorticoid-induced diabetes, and antidiabetic agents that have greater effects on random postprandial plasma glucose levels are more suitable than those that mostly affect fasting levels.
- Even those glucocorticoid formulations that are not intended to have systemic effects (eg, eye drops, inhaled corticosteroids, creams, intra-articular injections) can cause adrenal suppression and, therefore, if they are discontinued, steroid withdrawal and adrenal insufficiency.
- Needed are studies comparing antidiabetic regimens for glucocorticoid-induced hyperglycemia and studies comparing glucocorticoid tapering schedules for adrenal suppression to determine the best way to manage these adverse effects.
Dabigatran: Will it change clinical practice?
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16

DABIGATRAN, A THROMBIN INHIBITOR

A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN

A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16
DABIGATRAN, A THROMBIN INHIBITOR
A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN
A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
Dabigatran etexilate (Pradaxa) is a new oral anticoagulant that has distinct advantages over warfarin (Coumadin) in terms of its ease of administration, efficacy, and safety.
In the Randomized Evaluation of Long-Term Anticoagulation Therapy (RE-LY trial),1 in patients with nonvalvular atrial fibrillation, dabigatran 110 mg twice a day was found to be as good as warfarin in preventing systemic embolization and stroke (the primary outcome of the study), and at 150 mg twice a day it was superior.1 It has also shown efficacy in treating acute deep vein thrombosis and pulmonary embolism and in preventing these complications in orthopedic surgical patients.2–4
Dabigatran has been approved in 75 countries. It carries the trade name Pradaxa in Europe and the United States and Pradax in Canada. In October 2010, the US Food and Drug Administration (FDA) Cardiovascular and Renal Drugs Advisory Committee endorsed two twice-daily doses (75 mg and 150 mg) of dabigatran for the prevention of systemic embolization and stroke in patients with nonvalvular atrial fibrillation.
However, dabigatran is relatively expensive, and its current high cost might be a barrier to its wider use.
MANY PATIENTS NEED ANTICOAGULATION
Anticoagulation plays a vital role in the primary and secondary prevention of stroke in patients with atrial fibrillation and of pulmonary embolism in patients with venous thromboembolism. It is also used during cardiothoracic and vascular surgery, endovascular procedures, and dialysis and in patients with mechanical heart valves and hypercoagulable conditions.
Atrial fibrillation affects 3.03 million people in the United States (2005 figures), and this number is predicted to be as high as 7.56 million by 2050.5 More than 10% of people over the age of 80 years have it, and the lifetime risk of developing it is approximately 25%.6,7 Its most serious complication is ischemic stroke (the risk of which increases with age) and systemic embolization.5,8
Until the recent introduction of dabigatran, the only oral anticoagulant available in the United States for treating patients with atrial fibrillation was warfarin. Although warfarin has a number of disadvantages (see below), it is actually very effective for preventing ischemic stroke, reducing the incidence by as much as 65%.9,10
Venous thromboembolism is the third most common cardiovascular disorder after myocardial infarction and stroke.11 Although its exact incidence is unknown, nearly 1 million cases of it (incident or recurrent, fatal and nonfatal events) occur in the United States each year.12 Many patients with venous thromboembolism need oral anticoagulation long-term, and currently warfarin remains the only option for them as well.
NEEDED: A BETTER ANTICOAGULANT
Warfarin has been the most commonly prescribed oral anticoagulant in the United States for more than 60 years. As of 2004, more than 30 million outpatient prescriptions for it were filled annually in this country alone.13 However, warfarin has several important limitations.
Warfarin has a narrow therapeutic index. Patients taking it require monitoring of their international normalized ratio (INR) and frequent dose adjustments, and this is time-consuming and inconvenient. The target INR for patients with venous thromboembolism and atrial fibrillation is 2.0 to 3.0, whereas patients with a mechanical heart valve need a higher INR (2.5 to 3.5). If the INR is below these ranges, warfarin is less effective, with a risk of new thrombosis. On the other hand, if the INR is too high, there is a risk of bleeding.14 In fact, the most important side effect of warfarin is the risk of major and minor bleeding.13 However, even in well-designed clinical trials in which patients are closely managed, only 55% to 60% of patients regularly achieve their therapeutic target INR.1,2,14,15
Warfarin also interacts with many drugs and with some foods. Compliance is difficult. It has a slow onset of action. Genetic variations require dose adjustments. When switching from a parenteral anticoagulant, overlapping is required. Skin necrosis is a possible side effect. And warfarin is teratogenic.
Despite these limitations, the American College of Chest Physicians endorses warfarin to prevent or treat venous thromboembolism, and to prevent stroke in patients with atrial fibrillation.16
DABIGATRAN, A THROMBIN INHIBITOR
A prodrug, dabigatran is rapidly absorbed and converted to its active form. Its plasma concentration reaches a peak 1.5 to 3 hours after an oral dose, and it has an elimination half-life of 12 to 14 hours. About 80% of its excretion is by the kidneys and the remaining 20% is through bile.
Dabigatran is not metabolized by cytochrome P450 isoenzymes, and therefore it has few major interactions with other drugs. An exception is rifampin, a P-glycoprotein inducer that blocks dabigatran’s absorption in the gut, so this combination should be avoided. Another is quinidine, a strong P-glycoprotein inhibitor that is contraindicated for use with dabigatran. Also, amiodarone (Cordarone), another P-glycoprotein inhibitor, increases blood levels of dabigatran, and therefore a lower dose of dabigatran is recommended if these drugs are given together.18–20
DOES DABIGATRAN NEED MONITORING? CAN IT EVEN BE MONITORED?
Dabigatran has a predictable pharmacodynamic effect, and current data indicate it does not need regular monitoring.18–20 However, one may need to be able to measure the drug’s activity in certain situations, such as suspected overdose, bleeding, need for emergency surgery, impaired renal function, pregnancy, and obesity, and in children.20
Dabigatran has little effect on the prothrombin time or the INR, even at therapeutic concentrations.19 Further, its effect on the activated partial thromboplastin time (aPTT) is neither linear nor dose-dependent, and the aPTT reaches a plateau and becomes less sensitive at very high concentrations. Therefore, the aPTT does not appear to be an appropriate test to monitor dabigatran’s therapeutic anticoagulant effect, although it does provide a qualitative indication of anticoagulant activity.18,19
The thrombin time is a very sensitive method for determining if dabigatran is present, but the test lacks standardization; the ecarin clotting time provides better evidence of the dose but is not readily available at most institutions.18,19,21
EVALUATED IN CLINICAL TRIALS
DABIGATRAN IS EXPENSIVE BUT MAY BE COST-EFFECTIVE
The estimated price of dabigatran 150 mg twice a day in the United States is about $6.75 to $8.00 per day.26,27
Warfarin, in contrast, costs as little as $50 per year.28 However, this low price does not include the cost of monitoring the INR (office visits and laboratory testing), and these combined expenses are much higher than the price of the warfarin itself.29 In addition, warfarin requires time-consuming management when bridging to a parenteral anticoagulant (for reversal of its anticoagulant action) before routine health maintenance procedures such as dental work and colonoscopy and interventional procedures and surgery. Any bleeding complication will also add to its cost and will be associated with a decrease in the patient’s perceived health and quality of life, but this is true for both drugs.30
In today’s health care environment, controlling costs is a universal priority, but it may be unfair to compare the cost of dabigatran with that of warfarin alone. The expense and morbidity associated with stroke and intracranial bleeding are high, and if patients on dabigatran have fewer strokes (as seen in the RE-LY trial with dabigatran 150 mg twice a day) and no added expense of monitoring, then dabigatran may be cost-effective.
Freeman et al31 analyzed the cost-effectiveness of dabigatran, using an estimated cost of $13.70 per day and data from the RE-LY trial. They concluded that dabigatran may be a cost-effective alternative to warfarin in preventing ischemic stroke in patients considered at higher risk for ischemic stroke or intracranial hemorrhage, ie, those with a CHADS2 score of 1 or higher or equivalent. (The CHADS2 score is calculated as 1 point each for congestive heart failure, hypertension, age 75 or older, and diabetes mellitus; 2 points for prior stroke or transient ischemic attack.)
As more new-generation oral anticoagulants become available (see below), the price of dabigatran will undoubtedly decrease. Until then, warfarin will remain a cost-effective and cost-saving drug that cannot yet be considered obsolete.
WHO SHOULD RECEIVE DABIGATRAN?
The ideal patient for dabigatran treatment is not yet defined. The decision to convert a patient’s treatment from warfarin to dabigatran will likely depend on several factors, including the patient’s response to warfarin and the physician’s comfort with this new drug.
Many patients do extremely well with warfarin, requiring infrequent monitoring to maintain a therapeutic INR and having no bleeding complications. For them, it would be more practical to continue warfarin. Another reason for staying with warfarin would be if twice-a-day dosing would pose a problem.
Dabigatran would be a reasonable choice for a patient whose INR is erratic, who requires more frequent monitoring, for whom cost is not an issue, and for whom there is concern about dietary or drug interactions.
Another consideration is whether the patient has access to a health care facility for warfarin monitoring: this is difficult for those who cannot drive, who depend on others for transportation, and who live in rural areas.
Additionally, dabigatran may be a cost-effective alternative to warfarin for a patient with a high CHADS2 score who is considered at a higher risk for stroke.31
In all cases, the option should be considered only after an open discussion with the patient about the risks and benefits of this new drug.
WHO SHOULD NOT RECEIVE IT?
Dabigatran is a twice-daily drug with a short half-life. No patient with a history of poor compliance will be a good candidate for dabigatran. Since there are no practical laboratory tests for monitoring compliance, one will have to reinforce at every visit the importance of taking this medication according to instructions.
Patients with underlying kidney disease will need close monitoring of their creatinine clearance, with dose adjustment if renal function deteriorates.
Additionally, one should use caution when prescribing dabigatran to obese patients, pregnant women, or children until more is known about its use in these populations.
ADVANTAGES AND DISADVANTAGES OF DABIGATRAN
A reason may be that patients with atrial fibrillation and poor INR control have higher rates of death, stroke, myocardial infarction, and major bleeding.14 In most clinical trials, only 55% to 60% of patients achieve a therapeutic INR on warfarin, leaving them at risk of thrombosis or, conversely, bleeding.1,2,15,32 Dabigatran has predictable pharmacokinetics, and its twice-daily dosing allows for less variability in its anticoagulant effect, making it more consistently therapeutic with less potential for bleeding or thrombosis.1
The Canadian Cardiovascular Society included dabigatran in its 2010 guidelines on atrial fibrillation, recommending it or warfarin.33 The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society now give dabigatran a class I B recommendation (benefit greater than risk, but limited populations studied) in secondary stroke prevention.34
On the other hand, major concerns are the lack of an antidote for dabigatran and a lack of experience in treating bleeding complications. Since dabigatran is not monitored, physicians may be uncertain if we are overdosing or undertreating. As we gain experience, we will learn how to treat bleeding complications. Until then, it will be important to anticipate this problem and to develop an algorithm based on the best available evidence in managing this complication.
Although the overall rates of bleeding in the RE-LY trial were lower with dabigatran than with warfarin, there were more gastrointestinal bleeding events with the 150-mg dose of dabigatran, which was not readily explained.
Further, the rate of dyspepsia was almost twice as high with dabigatran than with warfarin, regardless of the dose of dabigatran. There were also more dropouts in the 2nd year of follow-up in the dabigatran groups, with gastrointestinal intolerance being one of the major reasons. Therefore, dyspepsia may cause intolerance and noncompliance.1
Dabigatran must be taken twice a day and has a relatively short half-life. For a noncompliant patient, missing one or two doses will cause a reversal of its anticoagulation effect, leaving the patient susceptible to thrombosis. In comparison, warfarin has a longer half-life and is taken once a day, so missing a dose is less likely to result in a similar reversal of its anticoagulant effect.
SPECIAL CONDITIONS
Switching from other anticoagulants to dabigatran
When making the transition from a subcutaneously administered anticoagulant, ie, a low-molecular-weight heparin or the anti-Xa inhibitor fondaparinux (Arixtra), dabigatran should be started 0 to 2 hours before the next subcutaneous dose of the parenteral anticoagulant was to be given.21,35
When switching from unfractionated heparin given by continuous intravenous infusion, the first dose of dabigatran should be given at the time the infusion is stopped.
When switching from warfarin, dabigatran should be started once the patient’s INR is less than 2.0.
Switching from dabigatran to a parenteral anticoagulant
When switching from dabigatran back to a parenteral anticoagulant, allow 12 to 24 hours after the last dabigatran dose before starting the parenteral agent.21,35
Elective surgery or invasive procedures
The manufacturer recommends stopping dabigatran 1 to 2 days before elective surgery for patients who have normal renal function and a low risk of bleeding, or 3 to 5 days before surgery for patients who have a creatinine clearance of 50 mL/min or less. Before major surgery or placement of a spinal or epidural catheter, the manufacturer recommends that dabigatran be held even longer.35
If emergency surgery is needed
If emergency surgery is needed, the clinician must use his or her judgment as to the risks of bleeding vs those of postponing the surgery.21,35
Overdose or bleeding
No antidote for dabigatran is currently available. It has a short half-life (12–14 hours), and the treatment for overdose or bleeding is to discontinue it immediately, maintain adequate diuresis, and transfuse fresh-frozen plasma or red blood cells as indicated.
The role of activated charcoal given orally to reduce absorption is under evaluation, but the charcoal must be given within 1 to 2 hours after the overdose is taken.21
Dabigatran does not bind very much to plasma proteins and hence is dialyzable—an approach that may be necessary in cases of persistent or life-threatening bleeding.
Recombinant activated factor VII or prothrombin complex concentrates may be additional options in cases of severe bleeding.18,21
TOPICS OF FUTURE RESEARCH
A limitation of the dabigatran trials was that they did not enroll patients who had renal or liver impairment, cancer, or other comorbidities; pregnant women; or children. Other topics of future research include its use in patients weighing less than 48 kg or more than 110 kg, its efficacy in patients with thrombophilia, in patients with mechanical heart valves, and in long-term follow-up and the use of thrombolytics in patients with acute stroke who are on dabigatran.
WILL DABIGATRAN CHANGE CLINICAL PRACTICE?
Despite some of the challenges listed above, we believe that dabigatran is likely to change medical practice in patients requiring anticoagulation.
Dabigatran’s biggest use will most likely be in patients with atrial fibrillation, mainly because this is the largest group of people receiving anticoagulation. In addition, the incidence of atrial fibrillation rises with age, the US population is living longer, and patients generally require life-long anticoagulation once this condition develops.
Dabigatran may be approved for additional indications in the near future. It has already shown efficacy in primary and secondary prevention of venous thromboembolism. Other important areas to be studied include its use in patients with mechanical heart valves and thrombophilia.
Whether dabigatran will be a worthy substitute for the parenteral anticoagulants (heparin, low-molecular-weight heparins, or factor Xa inhibitors) is not yet known, but it will have an enormous impact on anticoagulation management if proved efficacious.
If dabigatran becomes a major substitute for warfarin, it will affect the anticoagulation clinics, with their well-trained staff, that are currently monitoring millions of patients in the United States. These clinics would no longer be needed, and laboratory and technical costs could be saved. A downside is that patients on dabigatran will not be as closely supervised and reminded to take their medication as patients on warfarin are now at these clinics. Instead, they will likely be supervised by their own physician (or assistants), who will need to become familiar with this anticoagulant. This may affect compliance with dabigatran.
OTHER NEW ORAL ANTICOAGULANTS ARE ON THE WAY
Other oral anticoagulants, including rivaroxaban (Xarelto) and apixaban (Eliquis), have been under study and show promise in preventing both thrombotic stroke and venous thromboembolism. They will likely compete with dabigatran once they are approved.
Rivaroxaban, an oral direct factor Xa inhibitor, is being investigated for stroke prevention in patients with atrial fibrillation. It has also been shown to be not inferior to (and to be less expensive than) enoxaparin in treating and preventing venous thromboembolism in patients undergoing hip or knee arthroplasty.32,36,37 Rivaroxaban has recently been approved by the FDA for this indication.
Apixaban, another direct factor Xa inhibitor, is also being studied for the prevention of stroke and systemic embolism in patients with nonvalvular atrial fibrillation. To date, there are no head-to-head trials comparing dabigatran with either of these new oral anticoagulants.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Schulman S, Kearon C, Kakkar AK, et al. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- RE-MOBILIZE Writing Committee; Ginsberg JS, Davidson BL, Comp PC, et al. Oral thrombin inhibitor dabigatran etexilate vs North American enoxaparin regimen for prevention of venous thromboembolism after knee arthroplasty surgery. J Arthroplasty 2009; 24:1–9.
- Wolowacz SE, Roskell NS, Plumb JM, Caprini JA, Eriksson BI. Efficacy and safety of dabigatran etexilate for the prevention of venous thromboembolism following total hip or knee arthroplasty. A meta-analysis. Thromb Haemost 2009; 101:77–85.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Krahn AD, Manfreda J, Tate RB, Mathewson FA, Cuddy TE. The natural history of atrial fibrillation: incidence, risk factors, and prognosis in the Manitoba follow-up study. Am J Med 1995; 98:476–484.
- Lloyd-Jones DM, Wang TJ, Leip EP, et al. Lifetime risk for development of atrial fibrillation: the Framingham Heart Study. Circulation 2004; 110:1042–1046.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: the Framingham study. Stroke 1991; 22:983–988.
- Go AS, Hylek EM, Chang Y, et al. Anticoagulation therapy for stroke prevention in atrial fibrillation: how well do randomized trials translate into clinical practice? JAMA 2003; 290:2685–2692.
- Singer DE, Chang Y, Fang MC, et al. The net clinical benefit of warfarin anticoagulation in atrial fibrillation. Ann Intern Med 2009; 151:297–305.
- Goldhaber SZ. Pulmonary embolism thrombolysis: a clarion call for international collaboration. J Am Coll Cardiol 1992; 19:246–247.
- Heit JA. The epidemiology of venous thromboembolism in the community. Arterioscler Thromb Vasc Biol 2008; 28:370–372.
- Wysowski DK, Nourjah P, Swartz L. Bleeding complications with warfarin use: a prevalent adverse effect resulting in regulatory action. Arch Intern Med 2007; 167:1414–1419.
- White HD, Gruber M, Feyzi J, et al. Comparison of outcomes among patients randomized to warfarin therapy according to anticoagulant control: results from SPORTIF III and V. Arch Intern Med 2007; 167:239–245.
- ACTIVE Writing Group of the ACTIVE Investigators; Connolly S, Pogue J, Hart R, et al. Clopidogrel plus aspirin versus oral anticoagulation for atrial fibrillation in the Atrial Fibrillation Clopidogrel Trial with Irbesartan for prevention of Vascular Events (ACTIVE W): a randomised controlled trial. Lancet 2006; 367:1903–1912.
- Geerts WH, Bergqvist D, Pineo GF, et al. Prevention of venous thromboembolism: American College of Chest Physicians evidence-based clinical practice guidelines (8th edition). Chest 2008; 133(6 suppl):381S–453S.
- Mungall D. BIBR-1048 Boehringer Ingelheim. Curr Opin Investig Drugs 2002; 3:905–907.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Eisert WG, Hauel N, Stangier J, Wienen W, Clemens A, van Ryn J. Dabigatran: an oral novel potent reversible nonpeptide inhibitor of thrombin. Arterioscler Thromb Vasc Biol 2010; 30:1885–1889.
- Bounameaux H, Reber G. New oral antithrombotics: a need for laboratory monitoring. Against. J Thromb Haemost 2010; 8:627–630.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Eriksson BI, Dahl OE, Buller HR, et al. A new oral direct thrombin inhibitor, dabigatran etexilate, compared with enoxaparin for prevention of thromboembolic events following total hip or knee replacement: the BISTRO II randomized trial. J Thromb Haemost 2005; 3:103–111.
- Eriksson BI, Dahl OE, Rosencher N, et al. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Dahl OE, Rosencher N, et al. Oral dabigatran etexilate vs. subcutaneous enoxaparin for the prevention of venous thromboembolism after total knee replacement: the RE-MODEL randomized trial. J Thromb Haemost 2007; 5:2178–2185.
- Ezekowitz MD, Reilly PA, Nehmiz G, et al. Dabigatran with or without concomitant aspirin compared with warfarin alone in patients with nonvalvular atrial fibrillation (PETRO study). Am J Cardiol 2007; 100:1419–1426.
- Burger L. Bayer rival Boehringer prices blood pill at $6.75. Reuters, October 26, 2010. Available at http://www.reuters.com. Accessed September 12, 2011.
- Drugstore.com. Pradaxa. http://www.drugstore.com/pradaxa/bottle-60-150mg-capsules/qxn00597013554. Accessed September 10, 2011.
- Wal-Mart Stores, Inc. Retail Prescription Program Drug List. http://i.walmartimages.com/i/if/hmp/fusion/customer_list.pdf. Accessed September 10, 2011.
- Teachey DT. Dabigatran versus warfarin for venous thromboembolism (letter). N Engl J Med 2010; 362:1050; author reply1050–1051.
- Lancaster TR, Singer DE, Sheehan MA, et al. The impact of long-term warfarin therapy on quality of life. Evidence from a randomized trial. Boston Area Anticoagulation Trial for Atrial Fibrillation Investigators. Arch Intern Med 1991; 151:1944–1949.
- Freeman JV, Zhu RP, Owens DK, et al. Cost-effectiveness of dabigatran compared with warfarin for stroke prevention in atrial fibrillation. Ann Intern Med 2011; 154:1–11.
- EINSTEIN Investigators; Bauersachs R, Berkowitz SD, Brenner B, et al. Oral rivaroxaban for symptomatic venous thromboembolism. N Engl J Med 2010; 363:2499–2510.
- Cairns JA, Connolly S, McMurtry S, Stephenson M, Talajic M; CCS Atrial Fibrillation Guidelines Committee. Canadian Cardiovascular Society atrial fibrillation guidelines 2010: prevention of stroke and systemic embolization in atrial fibrillation and flutter. Can J Cardiol 2011; 27:74–90.
- Wann LS, Curtis AB, January CT, et al. 2011 ACCF/AHA/HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2011; 123:1144–1150.
- Boehringer Ingelheim. Pradaxa prescribing information. http://www.pradaxa.com. Accessed September 8, 2011.
- Huisman MV, Quinlan DJ, Dahl OE, Schulman S. Enoxaparin versus dabigatran or rivaroxaban for thromboprophylaxis after hip or knee arthroplasty: results of separate pooled analyses of phase III multicenter randomized trials. Circ Cardiovasc Qual Outcomes 2010; 3:652–660.
- McCullagh L, Tilson L, Walsh C, Barry M. A cost-effectiveness model comparing rivaroxaban and dabigatran etexilate with enoxaparin sodium as thromboprophylaxis after total hip and total knee replacement in the Irish healthcare setting. Pharmacoeconomics 2009; 27:829–846.
KEY POINTS
- Dabigatran is a potent, reversible, direct thrombin inhibitor. Available only in oral form, it has a rapid onset of action, a predictable anticoagulant response, and few major interactions.
- Dabigatran does not require dose adjustments (except for renal insufficiency) or monitoring of its effect during treatment.
- In trials in patients with nonvalvular atrial fibrillation, two different doses of dabigatran were compared with warfarin. Less bleeding occurred with the lower dose than with warfarin, while the higher dose was more effective than warfarin in preventing stroke and systemic embolization.
- The American College of Cardiology, the American Heart Association, and the Heart Rhythm Society have given dabigatran a class I B recommendation for secondary stroke prevention in patients with nonvalvular atrial fibrillation.