User login
Painful Papules on the Arms
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
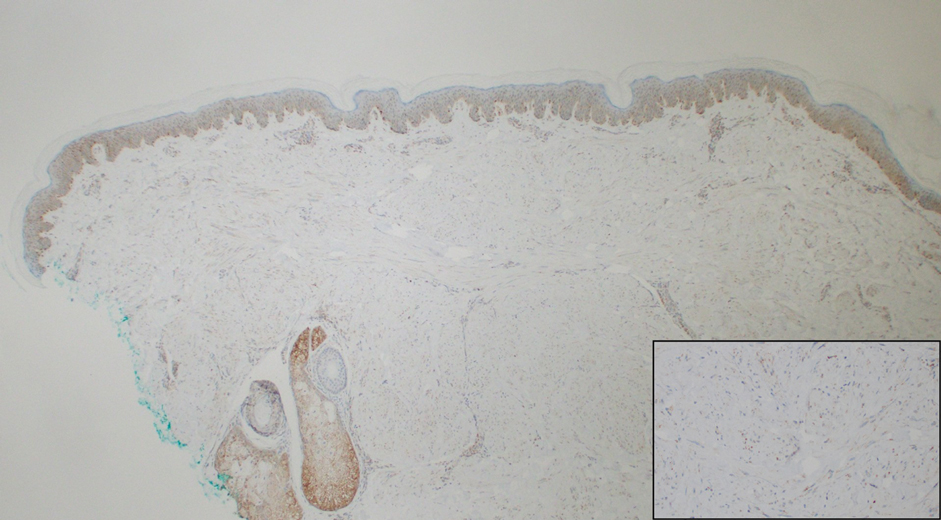
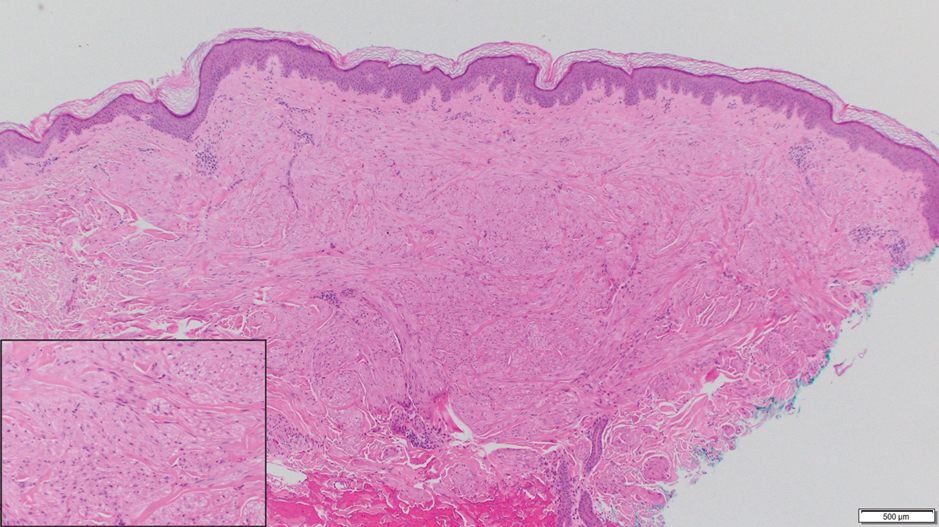
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
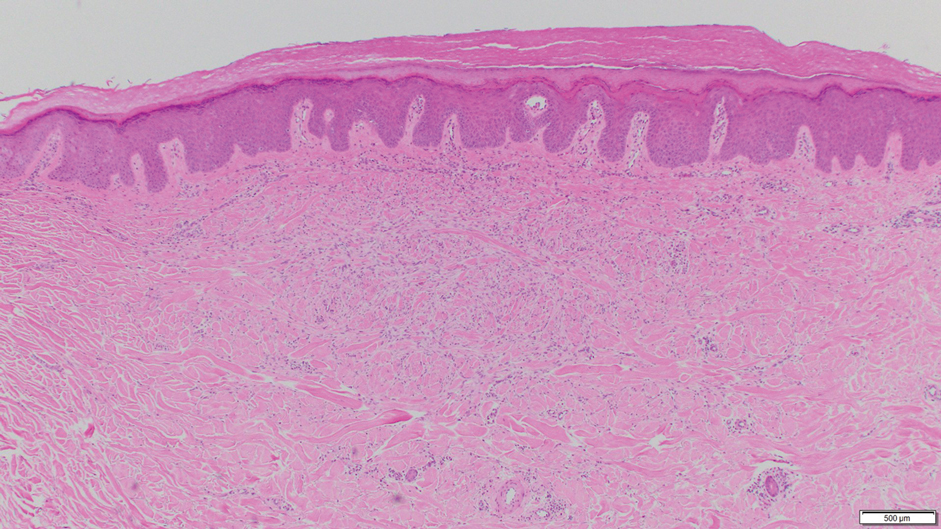
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
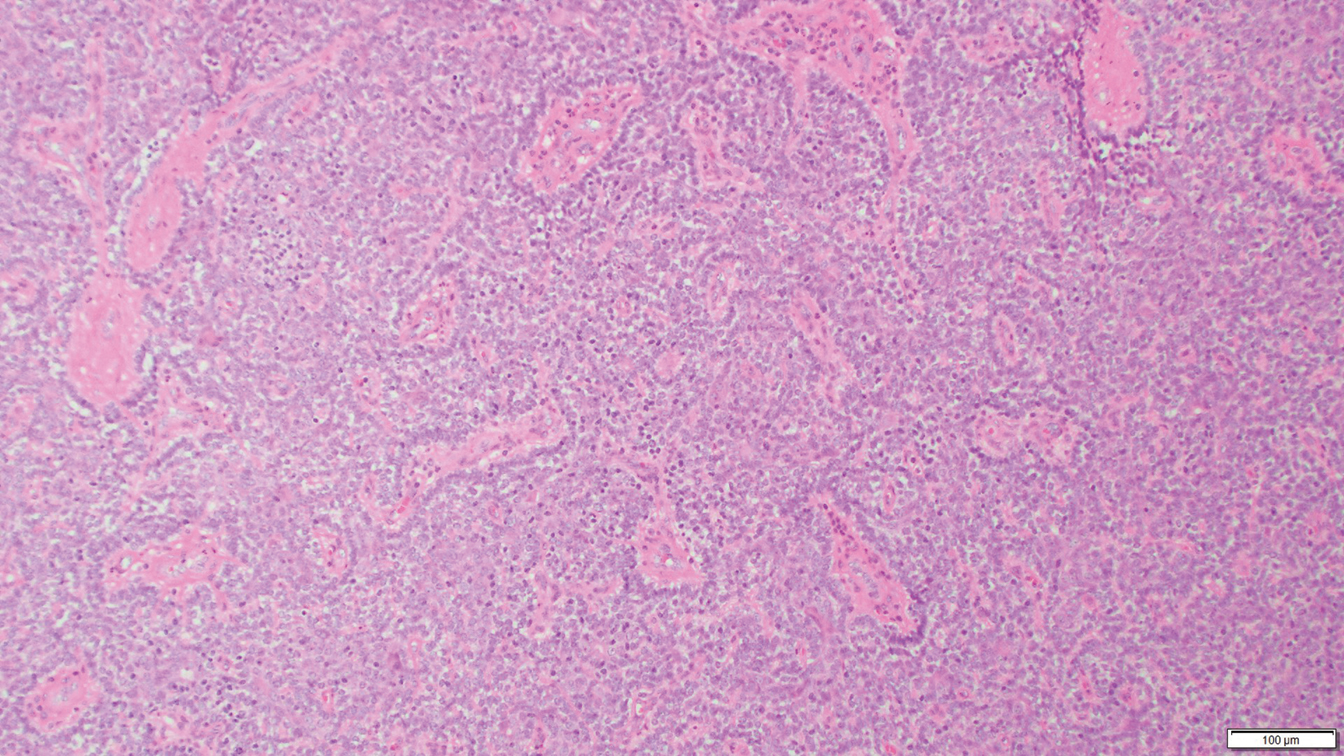
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.

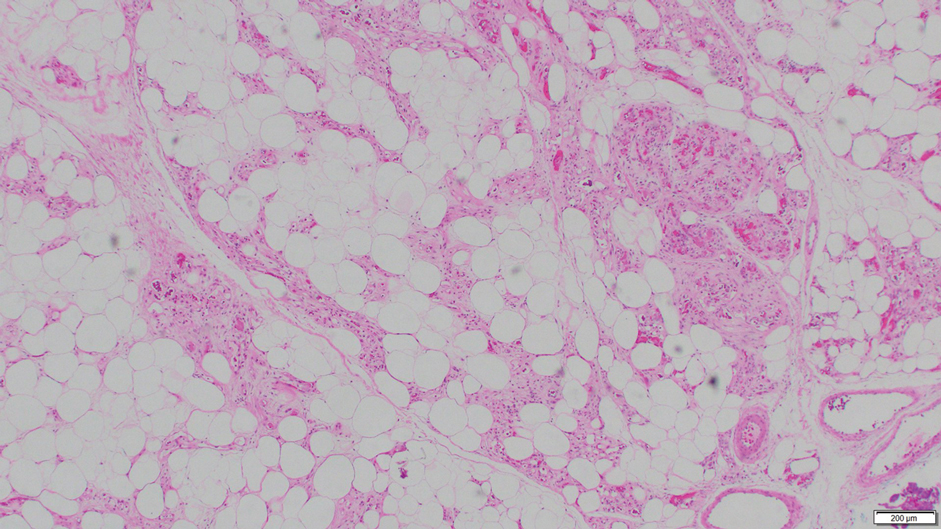
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
The Diagnosis: Piloleiomyoma
Leiomyoma cutis, also known as cutaneous leiomyoma, is a benign smooth muscle tumor first described in 1854.1 Cutaneous leiomyoma is comprised of 3 distinct types that depend on the origin of smooth muscle tumor: piloleiomyoma (arrector pili muscle), angioleiomyoma (tunica media of arteries/veins), and genital leiomyoma (dartos muscle of the scrotum and labia majora, erectile muscle of nipple).2 It affects both sexes equally, though some reports have noted an increased prevalence in females. Piloleiomyomas commonly present on the extensor surfaces of the extremities (solitary) and trunk (multiple).1 Tumors most often present as firm flesh-colored or pink-brown papulonodules. They can be linear, dermatomal, segmental, or diffuse, and often are painful. Clinical differential diagnosis for painful skin tumors is aided by the acronym "BLEND AN EGG": blue rubber bleb nevus, leiomyoma, eccrine spiradenoma, neuroma, dermatofibroma, angiolipoma, neurilemmoma, endometrioma, glomangioma, and granular cell tumor.3 For isolated lesions, surgical excision is the treatment of choice. For numerous lesions in which excision would not be feasible, intralesional corticosteroids, medications (eg, calcium channel blockers, alpha blockers, nitroglycerin), and botulinum toxin have been used for pain relief.4
Notably, multiple cutaneous leiomyomas can be seen in association with uterine leiomyomas in Reed syndrome due to an autosomal-dominant or de novo mutation in the fumarate hydratase gene, FH. Reed syndrome is associated with a lifetime risk for renal cell carcinoma (hereditary leiomyomatosis and renal cell cancer) in 15% of cases with FH mutations.5 In our patient, both immunohistochemical staining and blood testing for FH were performed. Immunohistochemistry revealed notably diminished staining with only weak patchy granular cytoplasmic staining present (Figure 1). Genetic testing revealed heterozygosity for a pathogenic variant of the FH gene, consistent with a diagnosis of Reed syndrome.
Histologically, the differential diagnosis includes other spindle cell tumors, such as dermatofibroma, neurofibroma, and dermatomyofibroma. The histologic appearance varies depending on the type, with piloleiomyoma typically located within the reticular dermis with possible subcutaneous extension. Fascicles of eosinophilic smooth muscle cells in an interlacing arrangement often ramify between neighboring dermal collagen; these smooth muscle cells contain cigar-shaped, blunt-ended nuclei with a perinuclear clear vacuole. Marked epidermal hyperplasia is possible.6 A close association with a nearby hair follicle frequently is noted. Although differentiated smooth muscle cells usually are evident on hematoxylin and eosin, positive staining for smooth muscle actin (SMA) and desmin can aid in diagnosis.7 Immunohistochemical staining for FH has proven to be highly specific (97.6%) with moderate sensitivity (70.0%).8 Angioleiomyomas appear as well-demarcated dermal to subcutaneous tumors composed of smooth muscle cells surrounding thick-walled vaculature.9 Scrotal and vulvar leiomyomas are composed of eosinophilic spindle cells, though vulvar leiomyomas have shown epithelioid differentiation.10 Nipple leiomyomas appear similar to piloleiomyomas on histology with interlacing smooth muscle fiber bundles.
Eccrine spiradenoma is a relatively uncommon adnexal tumor derived from eccrine sweat glands. It most often presents as a small, painful or tender, intradermal nodule (or rarely as nodules) on the head or ventral trunk.11 There is no sexual predilection. It affects adults at any age but most often from 15 to 35 years. Although rare, malignant transformation is possible. Histologically, eccrine spiradenomas appear as a well-demarcated dermal tumor composed of bland basaloid cells with minimal cytoplasm, often with numerous admixed lymphocytes and variably prominent vasculature (Figure 2). Eosinophilic basement membrane material can be seen within or surrounding the nodules of tumor cells. Multiple spiradenomas can occur in the setting of Brooke-Spiegler syndrome, which is an autosomal-dominant disorder due to an inherited mutation in the CYLD gene. Spiradenomas are benign neoplasms, and surgical excision with clear margins is the treatment of choice.12
Dermatofibroma, also known as cutaneous benign fibrous histiocytoma, is a firm, flesh-colored papule or nodule that most often presents on the lower extremities. It typically is seen in women aged 20 to 40 years.13 The etiology is uncertain, and dermatofibromas often spontaneously develop, though there are inconsistent reports of development with local trauma including insect bites and puncture wounds. The dimple sign refers to skin dimpling with lateral pressure.13 Most commonly, dermatofibromas consist of a dermal proliferation of bland fibroblastic cells with entrapment of dermal collagen bundles at the periphery of the tumors (Figure 3). The fibroblastic cells often are paler and less eosinophilic than smooth muscle cells seen in cutaneous leiomyomas, with tapered nuclei that lack a perinuclear vacuole. Admixed histocytes and other inflammatory cells often are present. Overlying epidermal hyperplasia and/or hyperpigmentation also may be present. Numerous histologic variants have been described, including cellular, epithelioid, aneurysmal, atypical, and hemosiderotic types.14 Immunohistochemical stains may show patchy positive staining for SMA, but h-caldesmon and desmin typically are negative.
Neurofibroma is a tumor derived from neuromesenchymal tissue with nerve axons. They form through neuromesenchyme (eg, Schwann cells, mast cells, perineural cells, endoneural fibroblast) proliferation. Solitary neurofibromas occur most commonly in adults and have no gender predilection. The most common presentation is an asymptomatic, solitary, soft, flesh-colored papulonodule.15 Clinical variants include pigmented, diffuse, and plexiform, with plexiform neurofibromas almost always being consistent with a diagnosis of neurofibromatosis type 1. Histologically, neurofibromas present as dermal or subcutaneous nodules composed of randomly arranged spindle cells with wavy tapered nuclei within a loose collagenous stroma (Figure 4).16 The spindle cells in neurofibromas will stain positively for S-100 protein and SOX-10 and negatively for SMA and desmin.
Angiolipoma is a benign tumor composed of adipocytes that also contains vasculature.17 The majority of cases are of unknown etiology, though familial cases have been described. They typically present as multiple painful or tender (differentiating from lipomas) subcutaneous swellings over the forearms in individuals aged 20 to 30 years.18 On histopathology, angiolipomas appear as well-circumscribed subcutaneous tumors containing mature adipocytes intermixed with small capillary vessels, some of which contain luminal fibrin thrombi (Figure 5).
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
- Malik K, Patel P, Chen J, et al. Leiomyoma cutis: a focused review on presentation, management, and association with malignancy. Am J Clin Dermatol. 2015;16:35-46.
- Malhotra P, Walia H, Singh A, et al. Leiomyoma cutis: a clinicopathological series of 37 cases. Indian J Dermatol. 2010;55:337-341.
- Delfino S, Toto V, Brunetti B, et al. Recurrent atypical eccrine spiradenoma of the forehead. In Vivo. 2008;22:821-823.
- Onder M, Adis¸en E. A new indication of botulinum toxin: leiomyoma-related pain. J Am Acad Dermatol. 2009;60:325-328.
- Menko FH, Maher ER, Schmidt LS, et al. Hereditary leiomyomatosis and renal cell cancer (HLRCC): renal cancer risk, surveillance and treatment. Fam Cancer. 2014;13:637-644.
- Raj S, Calonje E, Kraus M, et al. Cutaneous pilar leiomyoma: clinicopathologic analysis of 53 lesions in 45 patients. Am J Dermatopathol. 1997;19:2-9.
- Choi JH, Ro JY. Cutaneous spindle cell neoplasms: pattern-based diagnostic approach. Arch Pathol Lab Med. 2018;142:958-972.
- Carter CS, Skala SL, Chinnaiyan AM, et al. Immunohistochemical characterization of fumarate hydratase (FH) and succinate dehydrogenase (SDH) in cutaneous leiomyomas for detection of familial cancer syndromes. Am J Surg Pathol. 2017;41:801-809.
- Kanitakis J. Angioleiomyoma of the auricle: an unusual tumor on a rare location. Case Rep Otolaryngol. 2017;2017:1-3.
- Tavassoli FA, Norris HJ. Smooth muscle tumors of the vulva. Obstet Gynecol. 1979;53:213-217.
- Phukan J, Sinha A, Pal S. Fine needle aspiration cytology of eccrine spiradenoma of back: report of a rare case. J Lab Physicians. 2014;6:130.
- Zheng Y, Tian Q, Wang J, et al. Differential diagnosis of eccrine spiradenoma: a case report. Exp Ther Med. 2014;8:1097-1101.
- Bandyopadhyay MR, Besra M, Dutta S, et al. Dermatofibroma: atypical presentations. Indian J Dermatol. 2016;61:121.
- Commons JD, Parish L, Yazdanian S, et al. Dermatofibroma: a curious tumor. Skinmed. 2012;10:268-270.
- Lee YB, Lee JI, Park HJ, et al. Solitary neurofibromas: does an uncommon site exist? Ann Dermatol. 2012;24:101-102.
- Ortonne N, Wolkenstein P, Blakeley JO, et al. Cutaneous neurofibromas: current clinical and pathologic issues. Neurology. 2018;91:S5-S13.
- Howard WR. Angiolipoma. Arch Dermatol. 1960;82:924.
- Ghosh S, Haldar BA. Multiple angiolipomas. Indian J Dermatol Venereol Leprol. 1990;56:143-144.
A 36-year-old woman presented with multiple new-onset, firm, tender, subcutaneous papules and nodules involving the upper arms and shoulders.
Lichen Sclerosus of the Eyelid
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
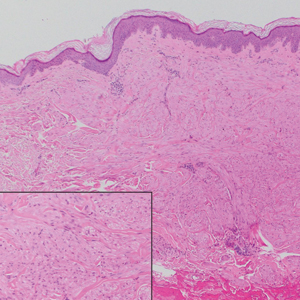
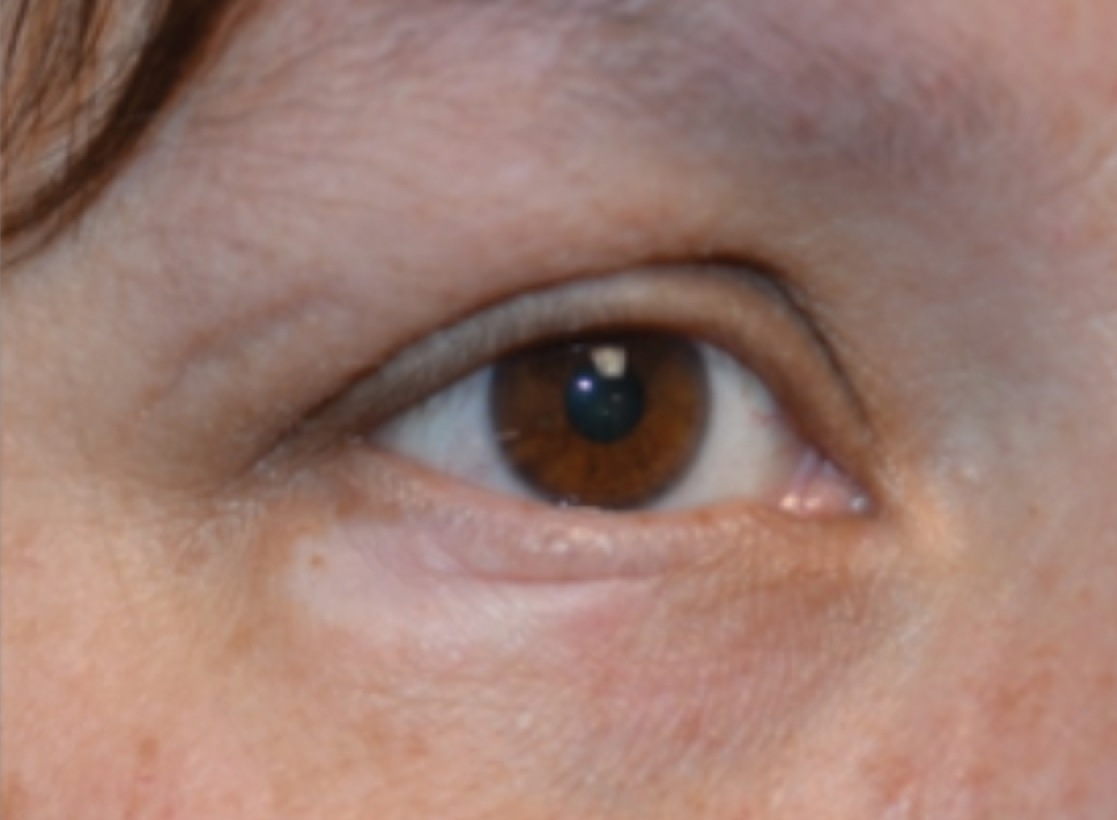
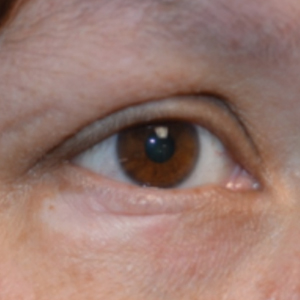
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
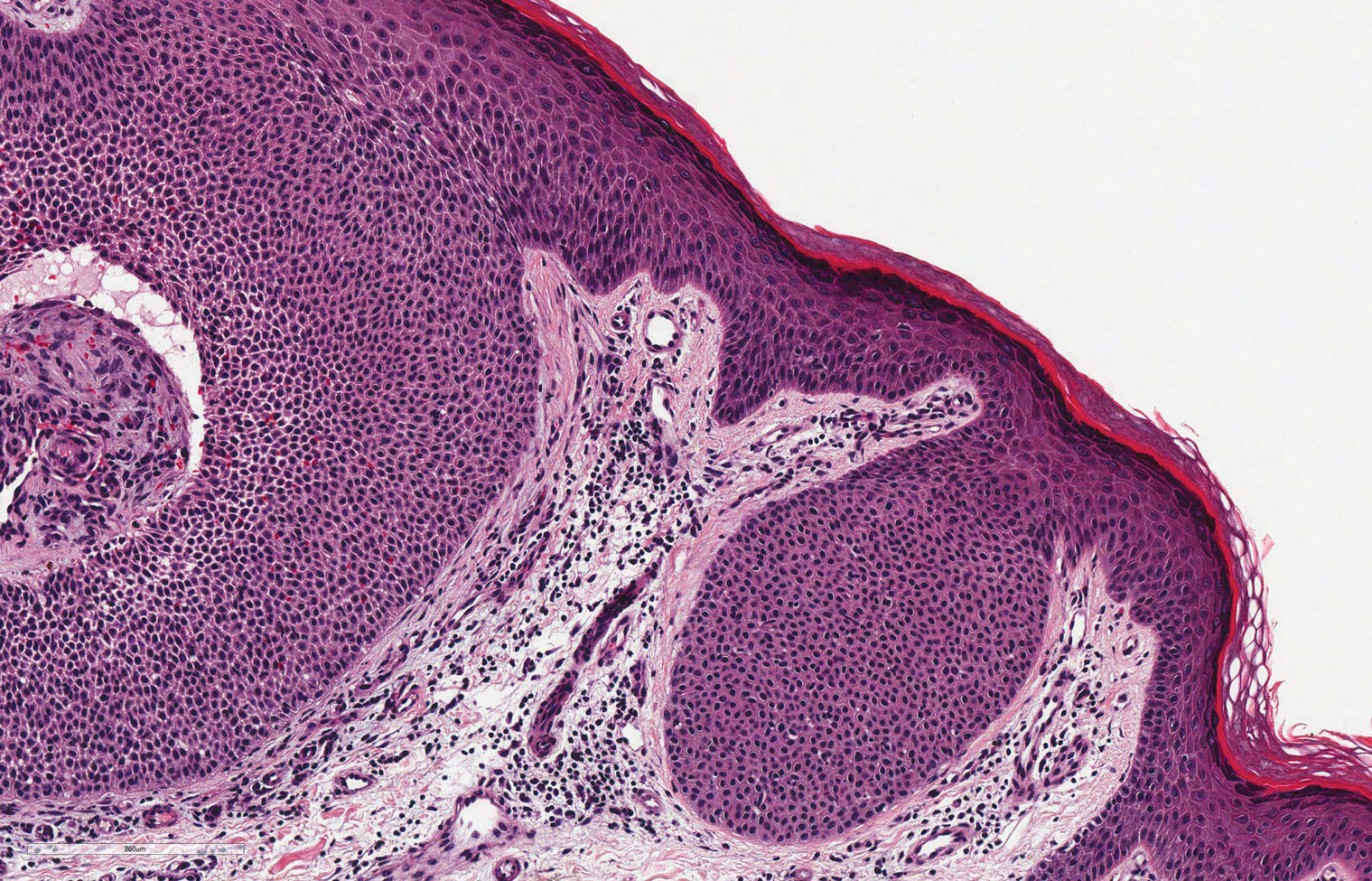
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
To the Editor:
Lichen sclerosus is a chronic inflammatory skin disease of unknown cause that predominantly affects the anogenital region, but isolated extragenital lesions occur in 6% to 15% of patients. The buttocks, thighs, neck, shoulder, upper torso, and wrists most commonly are involved; the face rarely is affected.1,2 Although the etiology of lichen sclerosus remains undetermined, there is growing evidence that autoimmunity may play a role.1 Lichen sclerosus more commonly is seen in women, and the disease can present at any age, with a bimodal onset in prepubertal children and in postmenopausal women and men in the fourth decade of life.1-3 A PubMed search of articles indexed for MEDLINE using the terms lichen and eyelid and manually screened revealed 6 cases of lichen sclerosus involving the eyelid.2-4 We describe a case of lichen sclerosus involving the eyelid and its histopathology.
A 45-year-old woman was referred to dermatology for evaluation of a right lower eyelid lesion of 3 months’ duration. She first noted a small white patch under the eyelid that had doubled in size and felt firm without bleeding or ulceration. Her medical history was unremarkable, and there was no history of ophthalmic conditions, autoimmune disease, trauma, or cancer. An ophthalmic examination was normal, except for a 20×8-mm, flat, depigmented, firm papule with scalloped borders involving the right lower eyelid margin and extending inferiorly without evidence of madarosis or ulceration (Figure 1). She underwent an incisional biopsy that revealed the diagnosis of lichen sclerosus et atrophicus (Figure 2). A full dermatologic evaluation included a genital examination and did not reveal any additional lesions. Tacrolimus ointment was started to avoid the need for long-term use of periocular steroids and their complications.
Extragenital lichen sclerosus typically is asymptomatic and only rarely presents with pruritus, in contrast to genital lichen sclerosus, which characteristically involves pruritus and dyspareunia. Although eyelid involvement is rare, ophthalmic manifestations of lichen sclerosus have included lid notching, ectropion, acquired Brown syndrome, and associated keratoconjunctivitis sicca.3-5 It characteristically appears as a well-demarcated hypopigmented papule. The differential diagnosis for a hypopigmented papule also includes amelanotic melanoma, basal cell carcinoma, vitiligo, tinea versicolor, lichen simplex chronicus, lichen planus, morphea (localized scleroderma), and systemic scleroderma with eyelid involvement.1,5
Differentiating lichen sclerosus from these conditions is of importance, as some of them can have notable morbidity and/or mortality. Of all the autoimmune connective tissue disorders, systemic sclerosus has the highest disease-specific mortality.6 Morphea, on the other hand, can have considerable morbidity. Morphea involving the head and neck notably increases the risk for neurologic complications such as seizures or central nervous system vasculitis as well as ocular complications such as anterior uveitis.6 Of note, genital lichen sclerosus carries an increased risk for squamous cell carcinoma and verrucous carcinoma; however, there have been no reported cases of malignant transformation with extragenital lesions.2
Histopathology is useful to distinguish among these entities. Although there are no specific features separating lichen sclerosus from a morphea overlap and both entities often are classified by clinical presentation, lichen sclerosus demonstrates epidermal atrophy, follicular plugging, homogenized collagen in the upper dermis with dermal edema, and lichenoid lymphocytic infiltrate (Figure 2).1 Extragenital lesions in particular also have been noted to have more epidermal atrophy and decreased rete ridges.2
First-line treatment of lichen sclerosus includes topical corticosteroids with emollients for supportive therapy. A topical calcineurin inhibitor such as tacrolimus should be considered for patients who do not respond to corticosteroid therapy or in cases in which corticosteroid therapy is contraindicated to avoid steroid-induced glaucoma or undesirable skin atrophy and hypopigmentation.2 A collaborative approach including dermatology and internal medicine can help identify a systemic or multisystem process.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
- Fistarol SK, Itin PH. Diagnosis and treatment of lichen sclerosus: an update. Am J Clin Dermatol. 2013;14:27-47.
- Rosenthal IM, Taube JM, Nelson DL, et al. A case of infraorbital lichen sclerosus. Dermatol Online J. 2013;19:20021.
- Rabinowitz R, Rosenthal G, Yerushalmy J, et al. Keratoconjunctivitis sicca associated with lichen sclerosus et atrophicus. Eye. 2000;14:103-104.
- Olver J, Laidler P. Acquired Brown’s syndrome in a patient with combined lichen sclerosus et atrophicus and morphea. Br J Ophthalmol. 1988;72:552-557.
- El-Baba F, Frangieh GT, Iliff WJ, et al. Morphea of the eyelids. Ophthalmology. 1982;89:125-128.
- Fett N. Scleroderma: nomenclatures, etiology, pathogenesis, prognosis, and treatment: facts and controversies. Clin Dermatol. 2013;31:432-437.
Practice Points
- Lichen sclerosus is not confined to only the anogenital area and can affect the face in rare cases.
Irritated Pigmented Plaque on the Scalp
The Diagnosis: Clonal Melanoacanthoma
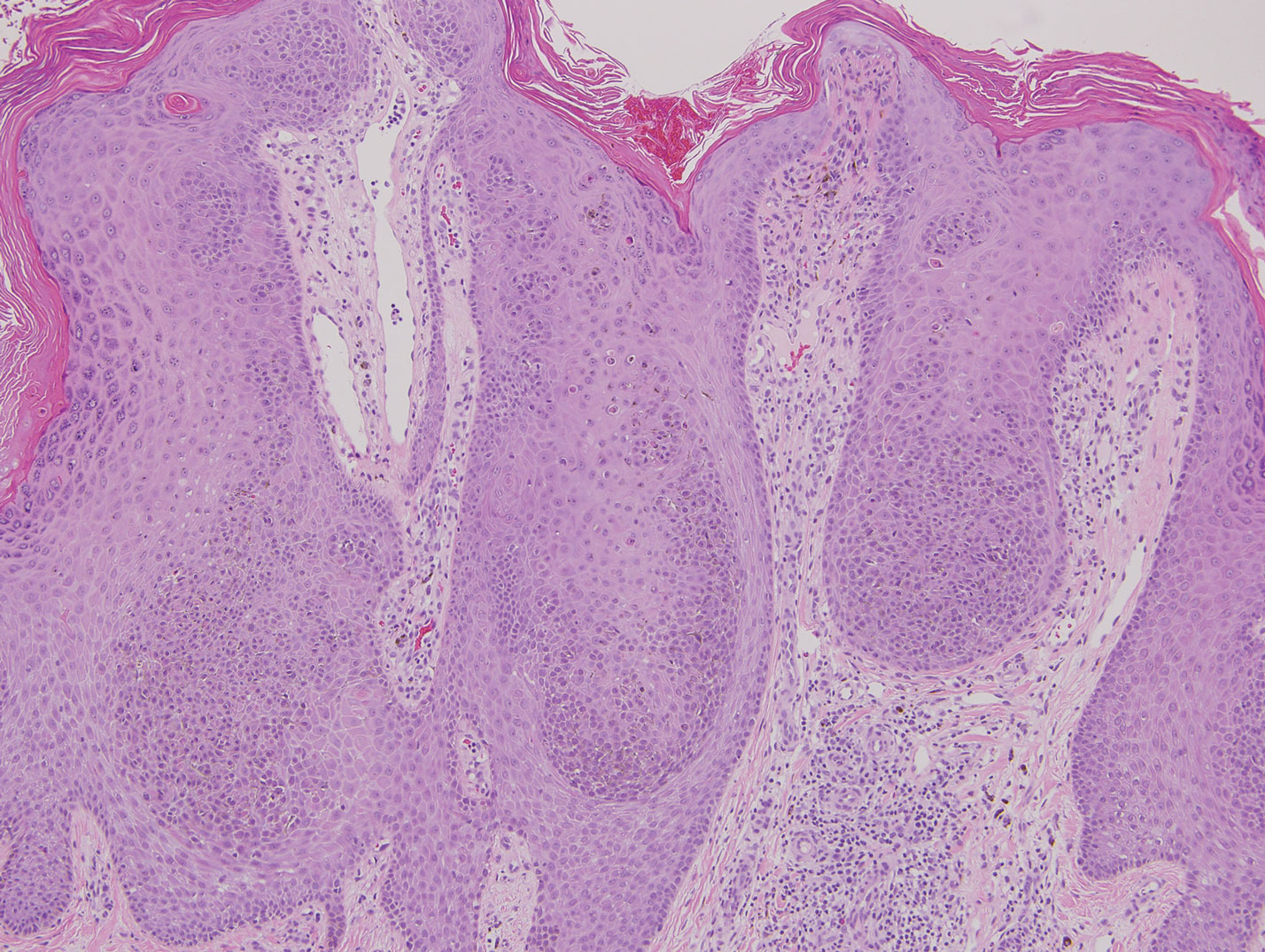
Melanoacanthoma (MA) is an extremely rare, benign, epidermal tumor histologically characterized by keratinocytes and large, pigmented, dendritic melanocytes. These lesions are loosely related to seborrheic keratoses, and the term was first coined by Mishima and Pinkus1 in 1960. It is estimated that the lesion occurs in only 5 of 500,000 individuals and tends to occur in older, light-skinned individuals.2 The majority are slow growing and are present on the head, neck, or upper extremities; however, similar lesions also have been reported on the oral mucosa.3 Melanoacanthomas range in size from 2×2 to 15×15 cm; are clinically pigmented; and present as either a papule, plaque, nodule, or horn.2
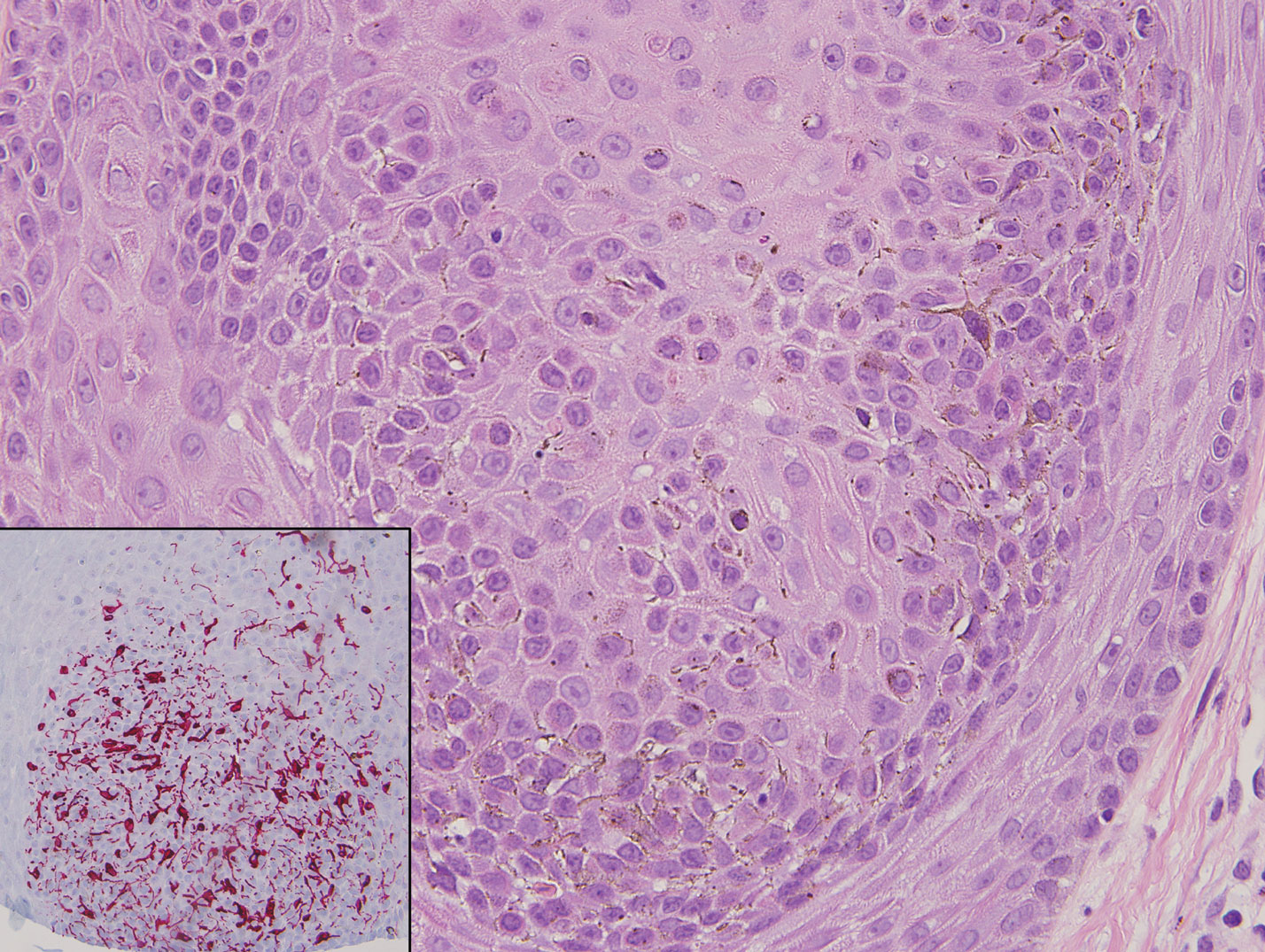
Classic histologic findings of MA include papillomatosis, acanthosis, and hyperkeratosis with heavily pigmented dendritic melanocytes diffusely dispersed throughout all layers of the seborrheic keratosis-like epidermis.3 Other features include keratin-filled pseudocysts, Langerhans cells, reactive spindling of keratinocytes, and an inflammatory infiltrate. In our case, the classic histologic findings also were architecturally arranged in oval to round clones within the epidermis (quiz images 1 and 2). A MART-1 (melanoma antigen recognized by T cells) immunostain was obtained that highlighted the numerous but benign-appearing, dendritic melanocytes (quiz image 2 [inset]). A dual MART-1/Ki67 immunostain later was obtained and demonstrated a negligible proliferation index within the dendritic melanocytes. Therefore, the diagnosis of clonal MA was rendered. This formation of epidermal clones also is called the Borst-Jadassohn phenomenon, which rarely occurs in MAs. This subtype is important to recognize because the clonal pattern can more closely mimic malignant neoplasms such as melanoma.
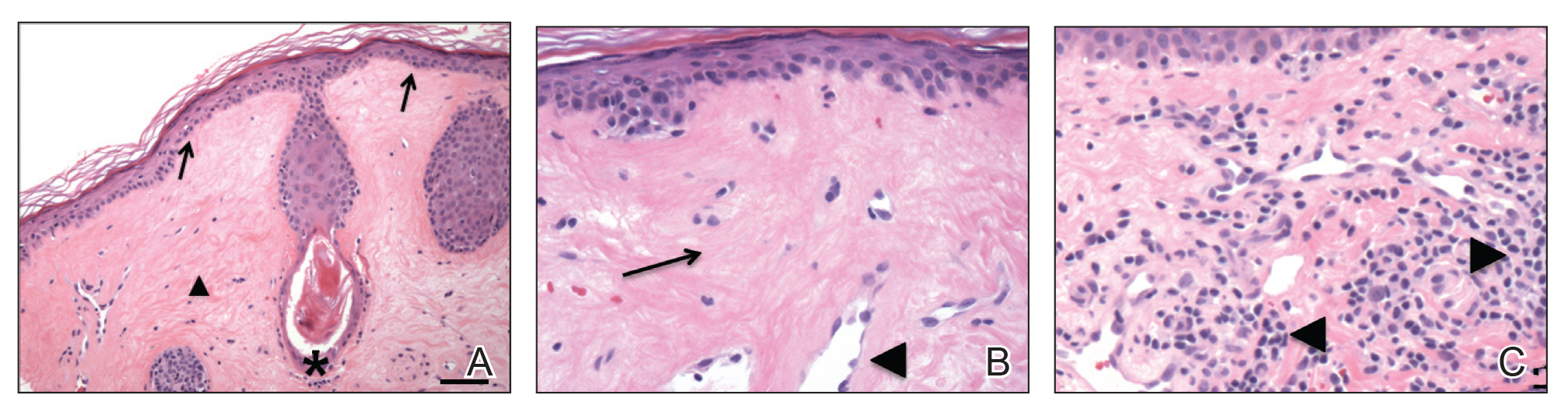
Hidroacanthoma simplex is an intraepidermal variant of eccrine poroma. It is a rare entity that typically occurs in the extremities of women as a hyperkeratotic plaque. These typically clonal epidermal tumors may be heavily pigmented and rarely contain dendritic melanocytes; therefore, they may be confused with MA. However, classic histology will reveal an intraepidermal clonal proliferation of bland, monotonous, cuboidal cells with ample pink cytoplasm, as well as occasional cuticle-lined ducts (Figure 1).4 These ducts will highlight with carcinoembryonic antigen and epithelial membrane antigen immunostaining.
Malignant melanoma typically presents as a growing pigmented lesion and therefore can clinically mimic MA. Histologically, MA could be confused with melanoma due to the increased number of melanocytes plus the appearance of pagetoid spread resulting from the diffuse presence of melanocytes throughout the neoplasm. However, histologic assessment of melanoma should reveal cytologic atypia such as nuclear enlargement, hyperchromasia, molding, pleomorphism, and mitotic activity (Figure 2). Architectural atypia such as poor lateral circumscription of melanocytes, confluence and pagetoid spread of nondendritic atypical junctional melanocytes, production of pigment in deep dermal nests of melanocytes, and lack of maturation and dispersion of dermal melanocytes also should be seen.5 Unlike a melanocytic neoplasm, true melanocytic nests are not seen in MA, and the melanocytes are bland, normal-appearing but heavily pigmented, dendritic melanocytes. Electron microscopy has shown a defect in the transfer of melanin from these highly dendritic melanocytes to the keratinocytes.6

Similar to melanoma, seborrheic keratosis presents as a pigmented growing lesion; therefore, definitive diagnosis often is achieved via skin biopsy. Classic histologic findings include acanthotic or exophytic epidermal growth with a dome-shaped configuration containing multiple cornified hornlike cysts (Figure 3).7 Multiple keratin plugs and variably sized concentric keratin islands are common features. There may be varying degrees of melanin pigment deposition among the proliferating cells, and clonal formation may occur. Melanocyte-specific special stains and immunostains can be used to differentiate MA from seborrheic keratosis by highlighting numerous dendritic melanocytes diffusely spread throughout the epidermis in MA vs a normal distribution of occasional junctional melanocytes in seborrheic keratosis.2,8
Squamous cell carcinoma in situ presents histologically with cytologically atypical keratinocytes encompassing the full thickness of the epidermis and sometimes crushing the basement membrane zone (Figure 4). There is a loss of the granular layer and overlying parakeratosis that often spares the adnexal ostial epithelium.9 Clonal formation can occur as well as increased pigment production. In comparison, bland keratinocytes are seen in MA.
Establishing the diagnosis of MA based on clinical features alone can be difficult. Dermoscopy can prove to be useful and typically will show a sunburst pattern with ridges and fissures.2 However, seborrheic keratoses and melanomas can have similar dermoscopic findings10; therefore, a biopsy often is necessary to establish the diagnosis.
- Mishima Y, Pinkus H. Benign mixed tumor of melanocytes and malpighian cells: melanoacanthoma: its relationship to Bloch's benign non-nevoid melanoepithelioma. Arch Dermatol. 1960;81:539-550.
- Gutierrez N, Erickson C P, Calame A, et al. Melanoacanthoma masquerading as melanoma: case reports and literature review. Cureus. 2019;11:E4998.
- Fornatora ML, Reich RF, Haber S, et al. Oral melanoacanthoma: a report of 10 cases, review of literature, and immunohistochemical analysis for HMB-45 reactivity. Am J Dermatopathol. 2003;25:12-15.
- Rahbari H. Hidroacanthoma simplex--a review of 15 cases. Br J Dermatol. 1983;109:219-225.
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19:S34-S40.
- Mishra DK, Jakati S, Dave TV, et al. A rare pigmented lesion of the eyelid. Int J Trichol. 2019;11:167-169.
- Greco MJ, Mahabadi N, Gossman W. Seborrheic keratosis. StatPearls. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK545285/. Accessed September 18, 2020.
- Kihiczak G, Centurion SA, Schwartz RA, et al. Giant cutaneous melanoacanthoma. Int J Dermatol. 2004;43:936-937.
- Morais P, Schettini A, Junior R. Pigmented squamous cell carcinoma: a case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Chung E, Marqhoob A, Carrera C, et al. Clinical and dermoscopic features of cutaneous melanoacanthoma. JAMA Dermatol. 2015;151:1129-1130.
The Diagnosis: Clonal Melanoacanthoma
Melanoacanthoma (MA) is an extremely rare, benign, epidermal tumor histologically characterized by keratinocytes and large, pigmented, dendritic melanocytes. These lesions are loosely related to seborrheic keratoses, and the term was first coined by Mishima and Pinkus1 in 1960. It is estimated that the lesion occurs in only 5 of 500,000 individuals and tends to occur in older, light-skinned individuals.2 The majority are slow growing and are present on the head, neck, or upper extremities; however, similar lesions also have been reported on the oral mucosa.3 Melanoacanthomas range in size from 2×2 to 15×15 cm; are clinically pigmented; and present as either a papule, plaque, nodule, or horn.2
Classic histologic findings of MA include papillomatosis, acanthosis, and hyperkeratosis with heavily pigmented dendritic melanocytes diffusely dispersed throughout all layers of the seborrheic keratosis-like epidermis.3 Other features include keratin-filled pseudocysts, Langerhans cells, reactive spindling of keratinocytes, and an inflammatory infiltrate. In our case, the classic histologic findings also were architecturally arranged in oval to round clones within the epidermis (quiz images 1 and 2). A MART-1 (melanoma antigen recognized by T cells) immunostain was obtained that highlighted the numerous but benign-appearing, dendritic melanocytes (quiz image 2 [inset]). A dual MART-1/Ki67 immunostain later was obtained and demonstrated a negligible proliferation index within the dendritic melanocytes. Therefore, the diagnosis of clonal MA was rendered. This formation of epidermal clones also is called the Borst-Jadassohn phenomenon, which rarely occurs in MAs. This subtype is important to recognize because the clonal pattern can more closely mimic malignant neoplasms such as melanoma.
Hidroacanthoma simplex is an intraepidermal variant of eccrine poroma. It is a rare entity that typically occurs in the extremities of women as a hyperkeratotic plaque. These typically clonal epidermal tumors may be heavily pigmented and rarely contain dendritic melanocytes; therefore, they may be confused with MA. However, classic histology will reveal an intraepidermal clonal proliferation of bland, monotonous, cuboidal cells with ample pink cytoplasm, as well as occasional cuticle-lined ducts (Figure 1).4 These ducts will highlight with carcinoembryonic antigen and epithelial membrane antigen immunostaining.
Malignant melanoma typically presents as a growing pigmented lesion and therefore can clinically mimic MA. Histologically, MA could be confused with melanoma due to the increased number of melanocytes plus the appearance of pagetoid spread resulting from the diffuse presence of melanocytes throughout the neoplasm. However, histologic assessment of melanoma should reveal cytologic atypia such as nuclear enlargement, hyperchromasia, molding, pleomorphism, and mitotic activity (Figure 2). Architectural atypia such as poor lateral circumscription of melanocytes, confluence and pagetoid spread of nondendritic atypical junctional melanocytes, production of pigment in deep dermal nests of melanocytes, and lack of maturation and dispersion of dermal melanocytes also should be seen.5 Unlike a melanocytic neoplasm, true melanocytic nests are not seen in MA, and the melanocytes are bland, normal-appearing but heavily pigmented, dendritic melanocytes. Electron microscopy has shown a defect in the transfer of melanin from these highly dendritic melanocytes to the keratinocytes.6
Similar to melanoma, seborrheic keratosis presents as a pigmented growing lesion; therefore, definitive diagnosis often is achieved via skin biopsy. Classic histologic findings include acanthotic or exophytic epidermal growth with a dome-shaped configuration containing multiple cornified hornlike cysts (Figure 3).7 Multiple keratin plugs and variably sized concentric keratin islands are common features. There may be varying degrees of melanin pigment deposition among the proliferating cells, and clonal formation may occur. Melanocyte-specific special stains and immunostains can be used to differentiate MA from seborrheic keratosis by highlighting numerous dendritic melanocytes diffusely spread throughout the epidermis in MA vs a normal distribution of occasional junctional melanocytes in seborrheic keratosis.2,8
Squamous cell carcinoma in situ presents histologically with cytologically atypical keratinocytes encompassing the full thickness of the epidermis and sometimes crushing the basement membrane zone (Figure 4). There is a loss of the granular layer and overlying parakeratosis that often spares the adnexal ostial epithelium.9 Clonal formation can occur as well as increased pigment production. In comparison, bland keratinocytes are seen in MA.
Establishing the diagnosis of MA based on clinical features alone can be difficult. Dermoscopy can prove to be useful and typically will show a sunburst pattern with ridges and fissures.2 However, seborrheic keratoses and melanomas can have similar dermoscopic findings10; therefore, a biopsy often is necessary to establish the diagnosis.
The Diagnosis: Clonal Melanoacanthoma
Melanoacanthoma (MA) is an extremely rare, benign, epidermal tumor histologically characterized by keratinocytes and large, pigmented, dendritic melanocytes. These lesions are loosely related to seborrheic keratoses, and the term was first coined by Mishima and Pinkus1 in 1960. It is estimated that the lesion occurs in only 5 of 500,000 individuals and tends to occur in older, light-skinned individuals.2 The majority are slow growing and are present on the head, neck, or upper extremities; however, similar lesions also have been reported on the oral mucosa.3 Melanoacanthomas range in size from 2×2 to 15×15 cm; are clinically pigmented; and present as either a papule, plaque, nodule, or horn.2
Classic histologic findings of MA include papillomatosis, acanthosis, and hyperkeratosis with heavily pigmented dendritic melanocytes diffusely dispersed throughout all layers of the seborrheic keratosis-like epidermis.3 Other features include keratin-filled pseudocysts, Langerhans cells, reactive spindling of keratinocytes, and an inflammatory infiltrate. In our case, the classic histologic findings also were architecturally arranged in oval to round clones within the epidermis (quiz images 1 and 2). A MART-1 (melanoma antigen recognized by T cells) immunostain was obtained that highlighted the numerous but benign-appearing, dendritic melanocytes (quiz image 2 [inset]). A dual MART-1/Ki67 immunostain later was obtained and demonstrated a negligible proliferation index within the dendritic melanocytes. Therefore, the diagnosis of clonal MA was rendered. This formation of epidermal clones also is called the Borst-Jadassohn phenomenon, which rarely occurs in MAs. This subtype is important to recognize because the clonal pattern can more closely mimic malignant neoplasms such as melanoma.
Hidroacanthoma simplex is an intraepidermal variant of eccrine poroma. It is a rare entity that typically occurs in the extremities of women as a hyperkeratotic plaque. These typically clonal epidermal tumors may be heavily pigmented and rarely contain dendritic melanocytes; therefore, they may be confused with MA. However, classic histology will reveal an intraepidermal clonal proliferation of bland, monotonous, cuboidal cells with ample pink cytoplasm, as well as occasional cuticle-lined ducts (Figure 1).4 These ducts will highlight with carcinoembryonic antigen and epithelial membrane antigen immunostaining.
Malignant melanoma typically presents as a growing pigmented lesion and therefore can clinically mimic MA. Histologically, MA could be confused with melanoma due to the increased number of melanocytes plus the appearance of pagetoid spread resulting from the diffuse presence of melanocytes throughout the neoplasm. However, histologic assessment of melanoma should reveal cytologic atypia such as nuclear enlargement, hyperchromasia, molding, pleomorphism, and mitotic activity (Figure 2). Architectural atypia such as poor lateral circumscription of melanocytes, confluence and pagetoid spread of nondendritic atypical junctional melanocytes, production of pigment in deep dermal nests of melanocytes, and lack of maturation and dispersion of dermal melanocytes also should be seen.5 Unlike a melanocytic neoplasm, true melanocytic nests are not seen in MA, and the melanocytes are bland, normal-appearing but heavily pigmented, dendritic melanocytes. Electron microscopy has shown a defect in the transfer of melanin from these highly dendritic melanocytes to the keratinocytes.6
Similar to melanoma, seborrheic keratosis presents as a pigmented growing lesion; therefore, definitive diagnosis often is achieved via skin biopsy. Classic histologic findings include acanthotic or exophytic epidermal growth with a dome-shaped configuration containing multiple cornified hornlike cysts (Figure 3).7 Multiple keratin plugs and variably sized concentric keratin islands are common features. There may be varying degrees of melanin pigment deposition among the proliferating cells, and clonal formation may occur. Melanocyte-specific special stains and immunostains can be used to differentiate MA from seborrheic keratosis by highlighting numerous dendritic melanocytes diffusely spread throughout the epidermis in MA vs a normal distribution of occasional junctional melanocytes in seborrheic keratosis.2,8
Squamous cell carcinoma in situ presents histologically with cytologically atypical keratinocytes encompassing the full thickness of the epidermis and sometimes crushing the basement membrane zone (Figure 4). There is a loss of the granular layer and overlying parakeratosis that often spares the adnexal ostial epithelium.9 Clonal formation can occur as well as increased pigment production. In comparison, bland keratinocytes are seen in MA.
Establishing the diagnosis of MA based on clinical features alone can be difficult. Dermoscopy can prove to be useful and typically will show a sunburst pattern with ridges and fissures.2 However, seborrheic keratoses and melanomas can have similar dermoscopic findings10; therefore, a biopsy often is necessary to establish the diagnosis.
- Mishima Y, Pinkus H. Benign mixed tumor of melanocytes and malpighian cells: melanoacanthoma: its relationship to Bloch's benign non-nevoid melanoepithelioma. Arch Dermatol. 1960;81:539-550.
- Gutierrez N, Erickson C P, Calame A, et al. Melanoacanthoma masquerading as melanoma: case reports and literature review. Cureus. 2019;11:E4998.
- Fornatora ML, Reich RF, Haber S, et al. Oral melanoacanthoma: a report of 10 cases, review of literature, and immunohistochemical analysis for HMB-45 reactivity. Am J Dermatopathol. 2003;25:12-15.
- Rahbari H. Hidroacanthoma simplex--a review of 15 cases. Br J Dermatol. 1983;109:219-225.
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19:S34-S40.
- Mishra DK, Jakati S, Dave TV, et al. A rare pigmented lesion of the eyelid. Int J Trichol. 2019;11:167-169.
- Greco MJ, Mahabadi N, Gossman W. Seborrheic keratosis. StatPearls. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK545285/. Accessed September 18, 2020.
- Kihiczak G, Centurion SA, Schwartz RA, et al. Giant cutaneous melanoacanthoma. Int J Dermatol. 2004;43:936-937.
- Morais P, Schettini A, Junior R. Pigmented squamous cell carcinoma: a case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Chung E, Marqhoob A, Carrera C, et al. Clinical and dermoscopic features of cutaneous melanoacanthoma. JAMA Dermatol. 2015;151:1129-1130.
- Mishima Y, Pinkus H. Benign mixed tumor of melanocytes and malpighian cells: melanoacanthoma: its relationship to Bloch's benign non-nevoid melanoepithelioma. Arch Dermatol. 1960;81:539-550.
- Gutierrez N, Erickson C P, Calame A, et al. Melanoacanthoma masquerading as melanoma: case reports and literature review. Cureus. 2019;11:E4998.
- Fornatora ML, Reich RF, Haber S, et al. Oral melanoacanthoma: a report of 10 cases, review of literature, and immunohistochemical analysis for HMB-45 reactivity. Am J Dermatopathol. 2003;25:12-15.
- Rahbari H. Hidroacanthoma simplex--a review of 15 cases. Br J Dermatol. 1983;109:219-225.
- Smoller BR. Histologic criteria for diagnosing primary cutaneous malignant melanoma. Mod Pathol. 2006;19:S34-S40.
- Mishra DK, Jakati S, Dave TV, et al. A rare pigmented lesion of the eyelid. Int J Trichol. 2019;11:167-169.
- Greco MJ, Mahabadi N, Gossman W. Seborrheic keratosis. StatPearls. Treasure Island, FL: StatPearls Publishing; 2020. https://www.ncbi.nlm.nih.gov/books/NBK545285/. Accessed September 18, 2020.
- Kihiczak G, Centurion SA, Schwartz RA, et al. Giant cutaneous melanoacanthoma. Int J Dermatol. 2004;43:936-937.
- Morais P, Schettini A, Junior R. Pigmented squamous cell carcinoma: a case report and importance of differential diagnosis. An Bras Dermatol. 2018;93:96-98.
- Chung E, Marqhoob A, Carrera C, et al. Clinical and dermoscopic features of cutaneous melanoacanthoma. JAMA Dermatol. 2015;151:1129-1130.
A 49-year-old man with light brown skin and no history of skin cancer presented with a pruritic lesion on the scalp of 3 years’ duration. Physical examination revealed a 7×3-cm, brown, mammillated plaque on the left parietal scalp. A shave biopsy of the scalp lesion was performed.
Rapid Onset of Widespread Nodules and Lymphadenopathy
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
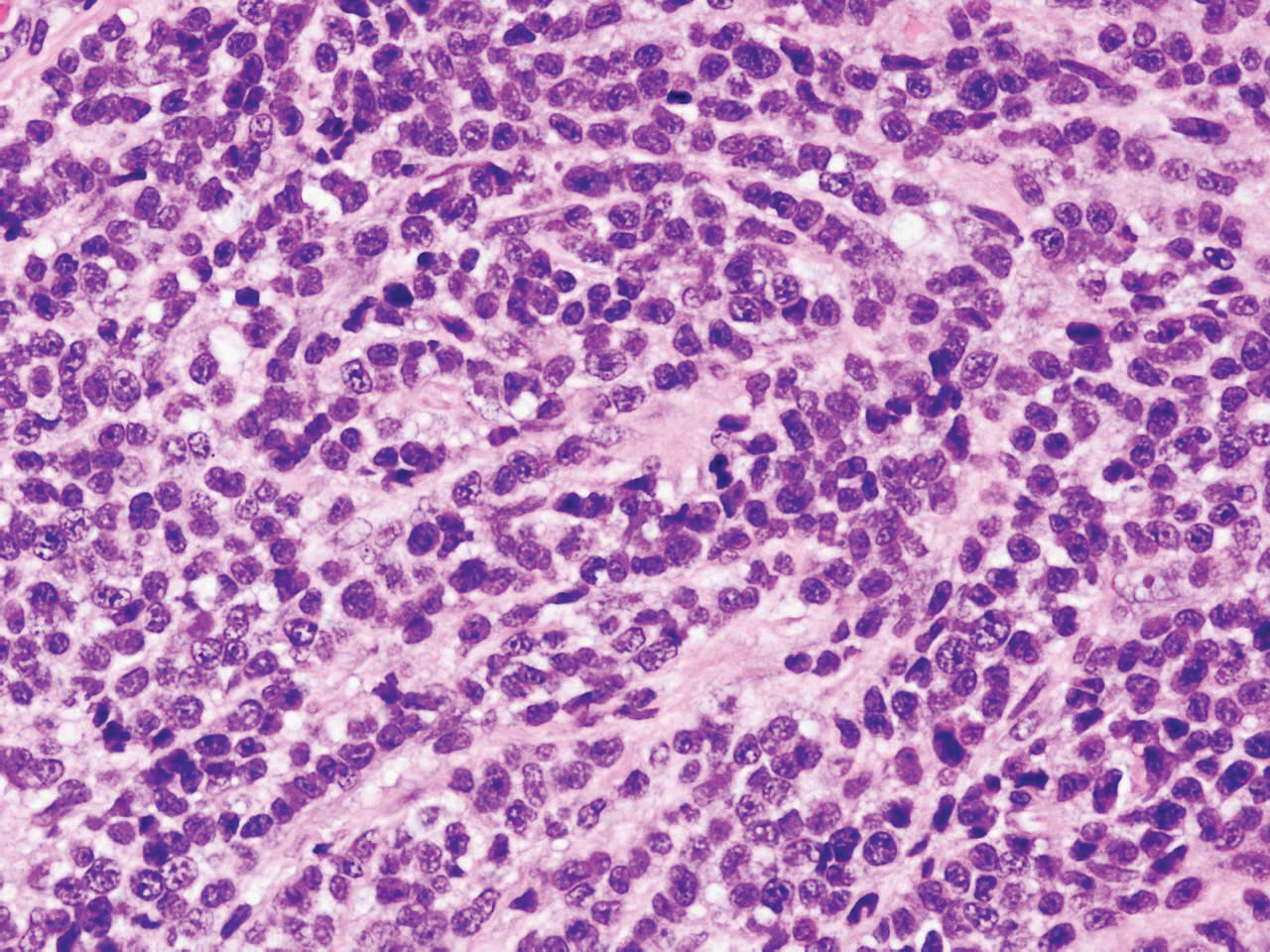
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
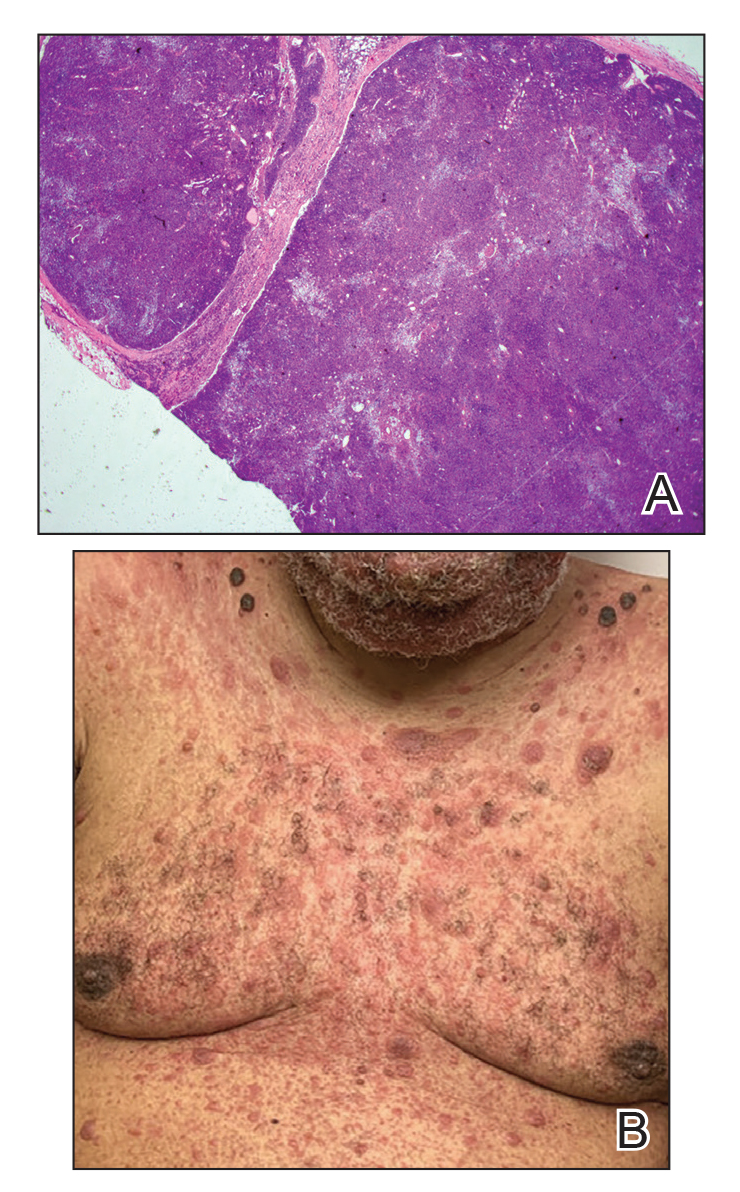
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8

Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
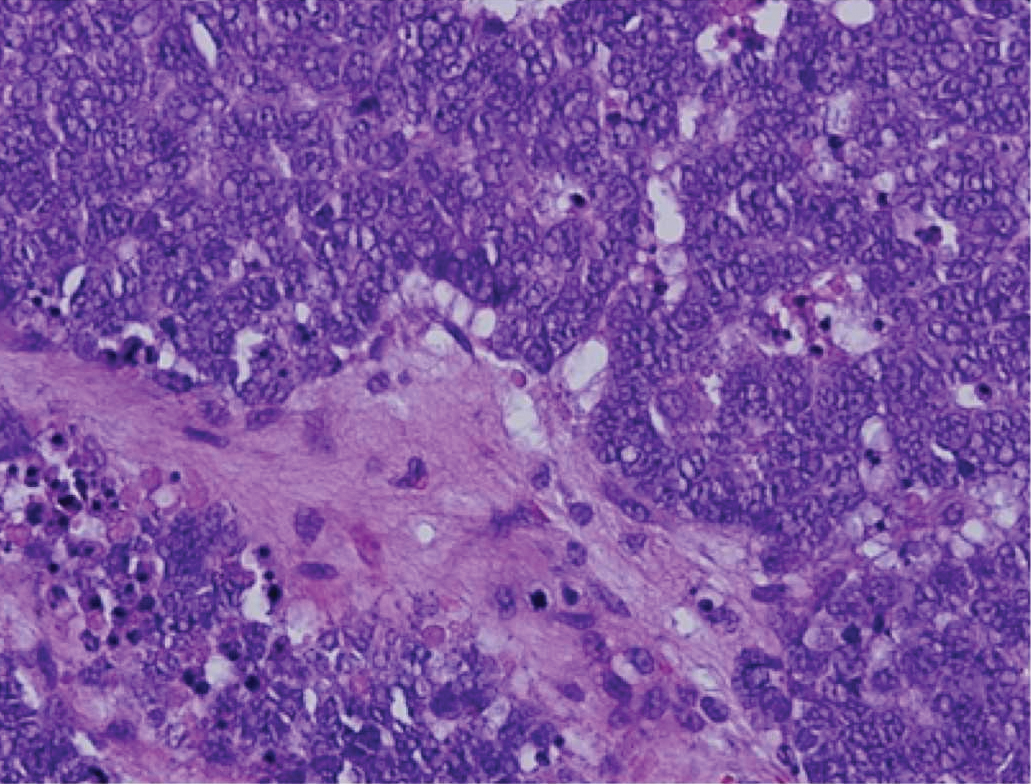
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14

Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8
Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14
Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
The Diagnosis: Primary Cutaneous γδ T-cell Lymphoma
Primary cutaneous γδ T-cell lymphoma (PCGDTL) is a distinct entity that can be confused with other types of cutaneous T-cell lymphomas. Often rapidly fatal, PCGDTL has a broad clinical spectrum that may include indolent variants—subcutaneous, epidermotropic, and dermal.1 Primary cutaneous γδ T-cell lymphoma represents less than 1% of all cutaneous T-cell lymphomas.2 Diagnosis and treatment remain challenging. Patients typically present with nodular lesions that progress to ulceration and necrosis. Early lesions can be confused with erythema nodosum, mycosis fungoides, or infection on clinical examination; biopsy establishes the diagnosis. Typical findings include a cytotoxic phenotype, variable epidermotropism, dermal and subcutaneous involvement, and loss of CD4 and often CD8 expression. Testing for Epstein-Barr virus expression yields negative results. The neoplastic lymphocytes in dermal and subcutaneous PCGDTL typically are T-cell intracellular antigen-1 (TIA-1) and granzyme positive.1
Immunohistochemistry failed to reveal CD8, CD56, granzyme, or T-cell intracellular antigen-1 staining of neoplastic cells in our patient but stained diffusely positive with CD3 and CD4. A CD20 stain decorated only a few dermal cells. The patient’s skin lesions continued to enlarge, and the massive lymphadenopathy made breathing difficult. Computed tomography revealed diffuse systemic involvement. An axillary lymph node biopsy revealed sinusoids with complete diffuse effacement of architecture as well as frequent mitotic figures and karyorrhectic debris (Figure 1A). Negative staining for T-cell receptor beta-F1 of the axillary lymph node biopsy and clonal rearrangement of the T-cell receptor gamma chain supported the diagnosis of PCGDTL. Nuclear staining for Epstein-Barr virus–encoded RNA was negative. Human T-cell leukemia virus type 1 antibodies and polymerase chain reaction also were negative. Flow cytometry demonstrated an atypical population of CD3+, CD4+, and CD7− γδ T lymphocytes, further supporting the diagnosis of lymphoma.
The median life expectancy for patients with dermal or subcutaneous PCGDTL is 10 to 15 months after diagnosis.3 The 5-year life expectancy for PCGDTL is approximately 11%.2 Limited treatment options contribute to the poor outcome. Chemotherapy regimens such as CHOP (cyclophosphamide, doxorubicin, vincristine, prednisolone) and EPOCH (etoposide phosphate, prednisone, vincristine sulfate, cyclophosphamide, doxorubicin hydrochloride) have yielded inconsistent results. Stem cell transplant has been tried in progressive disease and also has yielded mixed results.2 Brentuximab is indicated for individuals whose tumors express CD30.4 Associated hemophagic lymphohistiocytosis portends a poor prognosis.5
Despite treatment with etoposide, vincristine, doxorubicin, and high-dose oral steroids, our patient developed progressive difficulty breathing, stridor, kidney injury, and anemia. Our patient died less than 1 month after diagnosis—after only 1 round of chemotherapy—secondary to progressive disease and an uncontrollable gastrointestinal tract bleed. The leonine facies (Figure 1B) encountered in our patient can raise a differential diagnosis that includes infectious as well as neoplastic etiologies; however, most infectious etiologies associated with leonine facies manifest in a chronic fashion rather than with a sudden eruption, as noted in our patient.
Leprosy is caused by Mycobacterium leprae, a grampositive bacillus. The condition manifests across a spectrum, with the poles being tuberculoid and lepromatous, and borderline variants in between.6-8 Lepromatous leprosy arises in individuals who are unable to mount cellular immunity against M leprae secondary to anergy.6 Lepromatous leprosy often presents with numerous papules and nodules. Aside from cutaneous manifestations, lepromatous leprosy has a predilection for peripheral nerves and specifically Schwann cells. Histologically, biopsy reveals a flat epidermis and a cell-free subepidermal grenz zone. Within the dermis, there is a diffuse histiocytic infiltrate that typically is not centered around nerves (Figure 2).6,7 Mycobacterium leprae can appear scattered throughout or clustered in globi. Mycobacterium leprae stains red with Ziehl-Neelsen or Wade-Fite stains.6,7 Immunohistochemistry reveals a CD4+ helper T cell (TH2) predominance, supported by the increased expression of type 2 reaction cytokines such as IL-4, IL-5, IL-10, and IL-13.8
Diffuse large B-cell lymphoma (DLBCL) embodies 10% to 20% of all primary cutaneous lymphomas; it is more prevalent in older adults (age range, 70–82 years) and women. Clinically, DLBCL presents as either single or multiple rapidly progressing nodules or plaques, usually violaceous or blue-red in color.9,10 The most common area of presentation is on the legs, though it also can surface at other sites.9 On histology, DLBCL has clearly malignant features including frequent mitotic figures, large immunoblasts, and involvement throughout the dermis as well as perivascularly (Figure 3). Spindle-shaped cells and anaplastic features can be present. Immunohistochemically, DLBCL stains strongly positive for CD20 and B-cell lymphoma 2 (Bcl-2) along with other pan–B-cell markers.9-11 The aggressive leg type of DLBCL stains positively for multiple myeloma oncogene 1 (MUM-1).9,11
Cutaneous metastatic adenocarcinoma from internal malignancies occurs in approximately 5% of cancer patients with metastatic spread.12 Most of these cutaneous lesions develop in close proximity to the primary tumor such as on the trunk, head, or neck. All cutaneous metastases carry a poor prognosis. Clinical presentation can vary greatly, ranging from painless, firm, or elastic nodules to lesions that mimic inflammatory skin conditions such as erysipelas or scleroderma. The majority of these metastases develop as painless firm nodules that are flesh colored, pink, red-brown, or purple.12,13 The histopathology of metastatic adenocarcinoma demonstrates an infiltrative nodular appearance, though there rarely are well-circumscribed nodules found.13 The lesion originates in the dermis or subcutaneous tissue. It is a glandulartype lesion that may reflect the tissue of the primary tumor (Figure 4).12,14 Immunohistochemical stains likely will remain consistent with those of the primary tumor, which is not always the case.14
Merkel cell carcinoma (MCC) is an aggressive cutaneous malignancy of epithelial and neuroendocrine origin, first described as trabecular carcinoma due to the arrangement of tumor resembling cancellous bone.15,16 Merkel cells are mechanoreceptors found near nerve terminals.17 Approximately 80% of MCCs are associated with Merkel cell polyomavirus, which is a small, double-stranded DNA virus with an icosahedral capsid.17,18 Merkel cell polyomavirus–positive cases of MCC tend to have a better prognosis. In Merkel cell polyomavirus–negative MCC, there is an association with UV damage and increased chromosomal aberrations.18 Merkel cell carcinoma is known for its high rate of recurrence as well as local and distant metastasis. Nodal involvement is the most important prognostic indicator.15 Clinically, MCC is associated with the AEIOU mnemonic (asymptomatic, expanding rapidly, immunosuppression, older than 50 years, UV exposed/fair skin).15-17 Lesions appear as red-blue papules on sun-exposed skin and usually are smaller than 2 cm by their greatest dimension. On histopathology, MCC demonstrates small, round, blue cells arranged in sheets or nests originating in the dermis and occasionally can infiltrate the subcutis and lymphovascular surroundings (Figure 5).16-19 Cells have scant eosinophilic cytoplasm and may have fine granular chromatin. Numerous mitotic figures and apoptotic cells also are present. On immunohistochemistry, these cells will stain positive for cytokeratin AE1/AE3, anticytokeratin (CAM 5.2), CK20, and CD56. Due to their neuroendocrine derivation, they also are commonly synaptophysin, neuron-specific enolase, and chromogranin A positive. Notably, MCC will stain negative for leukocyte common antigen, CD20, CD3, CD34, and thyroid transcription factor 1 (TTF-1).16,17
Primary cutaneous γδ T-cell lymphoma can be difficult to diagnose and requires urgent treatment. Clinicians and dermatopathologists need to work together to establish the diagnosis. There is a high mortality rate associated with PCGDTL, making prompt recognition and timely treatment critical. Acknowledgments—Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
Acknowledgments
Thank you to our colleagues with the Penn State Health Hematology/Oncology Department (Hershey, Pennsylvania) for comanagement of this patient.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
- Merrill ED, Agbay R, Miranda RN, et al. Primary cutaneous T-cell lymphomas showing gamma-delta (γδ) phenotype and predominantly epidermotropic pattern are clinicopathologically distinct from classic primary cutaneous γδ T-cell lymphomas. Am J Surg Pathol. 2017;41:204-215.
- Foppoli M, Ferreri AJ. Gamma‐delta T‐cell lymphomas. Eur J Haematol. 2015;94:206-218.
- Toro JR, Liewehr DJ, Pabby N, et al. Gamma-delta T-cell phenotype is associated with significantly decreased survival in cutaneous T-cell lymphoma. Blood. 2003;101:3407-3412.
- Rubio-Gonzalez B, Zain J, Garcia L, et al. Cutaneous gamma-delta T-cell lymphoma successfully treated with brentuximab vedotin. JAMA Dermatol. 2016;152:1388-1390.
- Tong H, Ren Y, Liu H, et al. Clinical characteristics of T-cell lymphoma associated with hemophagocytic syndrome: comparison of T-cell lymphoma with and without hemophagocytic syndrome. Leuk Lymphoma. 2008;49:81-87.
- Brehmer-Andersson E. Leprosy. Dermatopathology. New York, NY: Springer; 2006:110-113.
- Massone C, Belachew WA, Schettini A. Histopathology of the lepromatous skin biopsy. Clin Dermatol. 2015;33:38-45.
- Naafs B, Noto S. Reactions in leprosy. In: Nunzi E, Massone C, eds. Leprosy: A Practical Guide. Milan, Italy: Springer; 2012:219-239.
- Hope CB, Pincus LB. Primary cutaneous B-cell lymphomas. Clin Lab Med. 2017;37:547-574.
- Billero VL, LaSenna CE, Romanelli M, et al. Primary cutaneous diffuse large B-cell lymphoma presenting as chronic non-healing ulcer. Int Wound J. 2017;14:830-832.
- Testo N, Olson L, Subramaniyam S, et al. Primary cutaneous diffuse large B-cell lymphoma with a MYC-IGH rearrangement and gain of BCL2: expanding the spectrum of MYC/BCL2 double hit lymphomas. Am J Dermatopathol. 2016;38:769-774.
- Boyd AS. Pulmonary signet-ring cell adenocarcinoma metastatic to the skin. Am J Dermatopathol. 2017;39:E66-E68.
- Guanziroli E, Coggi A, Venegoni L, et al. Cutaneous metastases of internal malignancies: an experience from a single institution. Eur J Dermatol. 2017;27:609-614.
- Fernandez-Flores A, Cassarino DS. Cutaneous metastasis of adenocarcinoma of the ampulla of Vater. Am J Dermatopathol. 2018;40:758-761.
- Trinidad CM, Torres-Cabala CA, Prieto VG, et. Al. Update on eighth edition American Joint Committee on Cancer classification for Merkel Cell carcinoma and histopathological parameters that determine prognosis. J Clin Pathol. 2017;72:337-340.
- Bandino JP, Purvis CG, Shaffer BR, et al. A comparison of the histopathologic growth patterns between non-Merkel cell small round blue cell tumors and Merkel cell carcinoma. Am J Dermatopathol. 2018;40:815-818.
- Mauzo SH, Rerrarotto R, Bell D, et al. Molecular characteristics and potential therapeutic targets in Merkel cell carcinoma. J Clin Pathol. 2016;69:382-390.
- Lowe G, Brewer J, Bordeaux J. Epidemiology and genetics. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:26-28.
- North J, McCalmont T. Histopathologic diagnosis. In: Alam M, Bordeaux JS, Yu SS, eds. Merkel Cell Carcinoma. New York, NY: Springer; 2013:66-69.
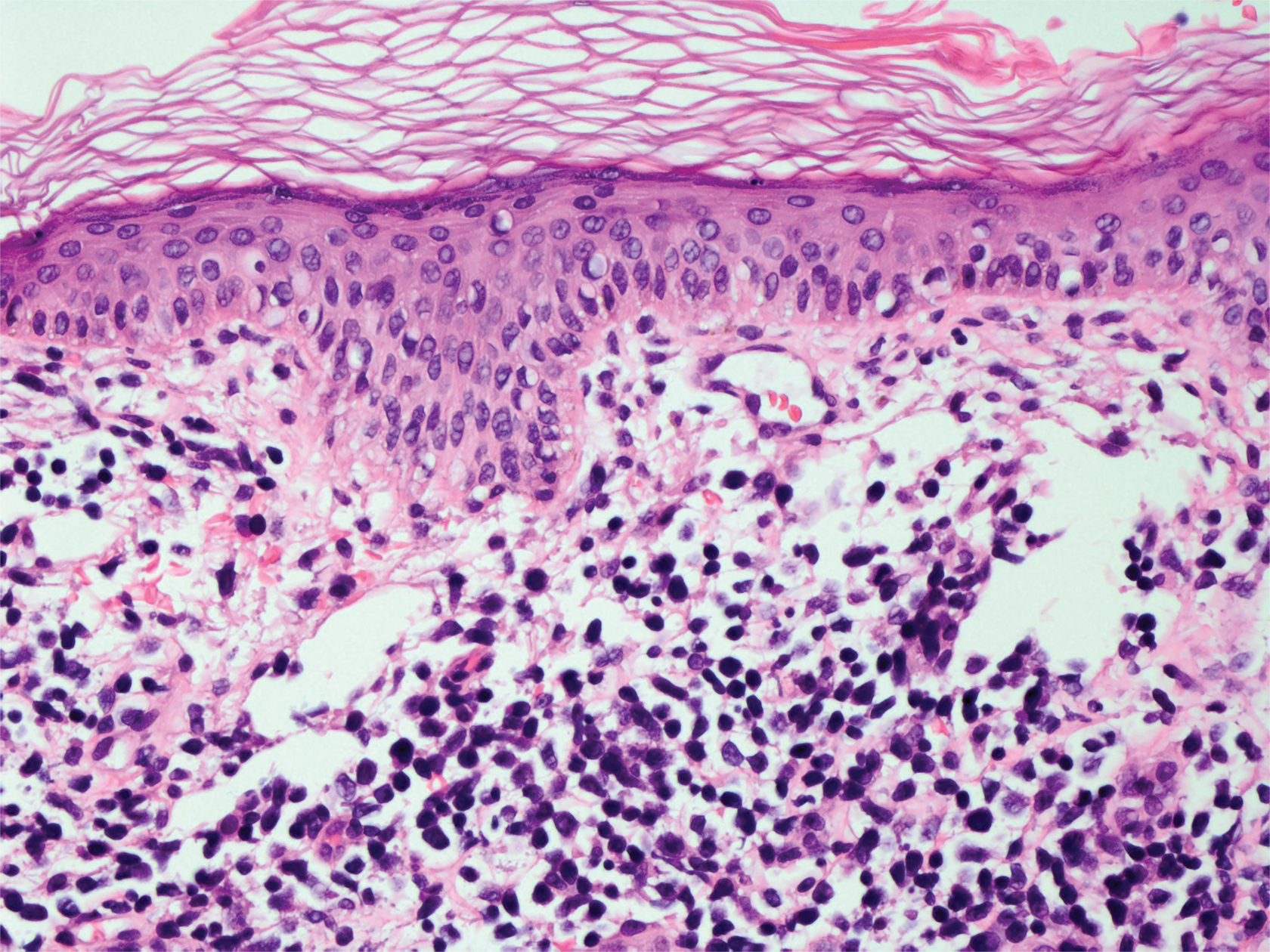
A 71-year-old man presented with an eruption on the face, shoulders, upper back, and arms of 3 weeks’ duration. The lesions were asymptomatic, and he denied fever, chills, or weight loss. He had a history of type 2 diabetes mellitus, hypertension, and hypercholesterolemia. Physical examination revealed coarse facial features with purple-pink nodules on the face and trunk and ulcerated nodules on the upper extremities. Mucous membrane involvement was noted, and there was marked occipital and submandibular lymphadenopathy. A biopsy of an arm nodule revealed a superficial and deep dermal and periadnexal lymphocytic infiltrate of atypical CD3+ cells.
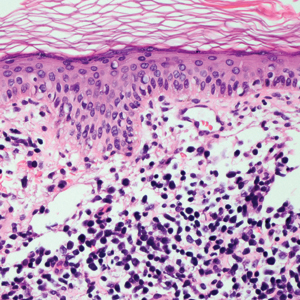
Depressed Shiny Scars and Crusted Erosions
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
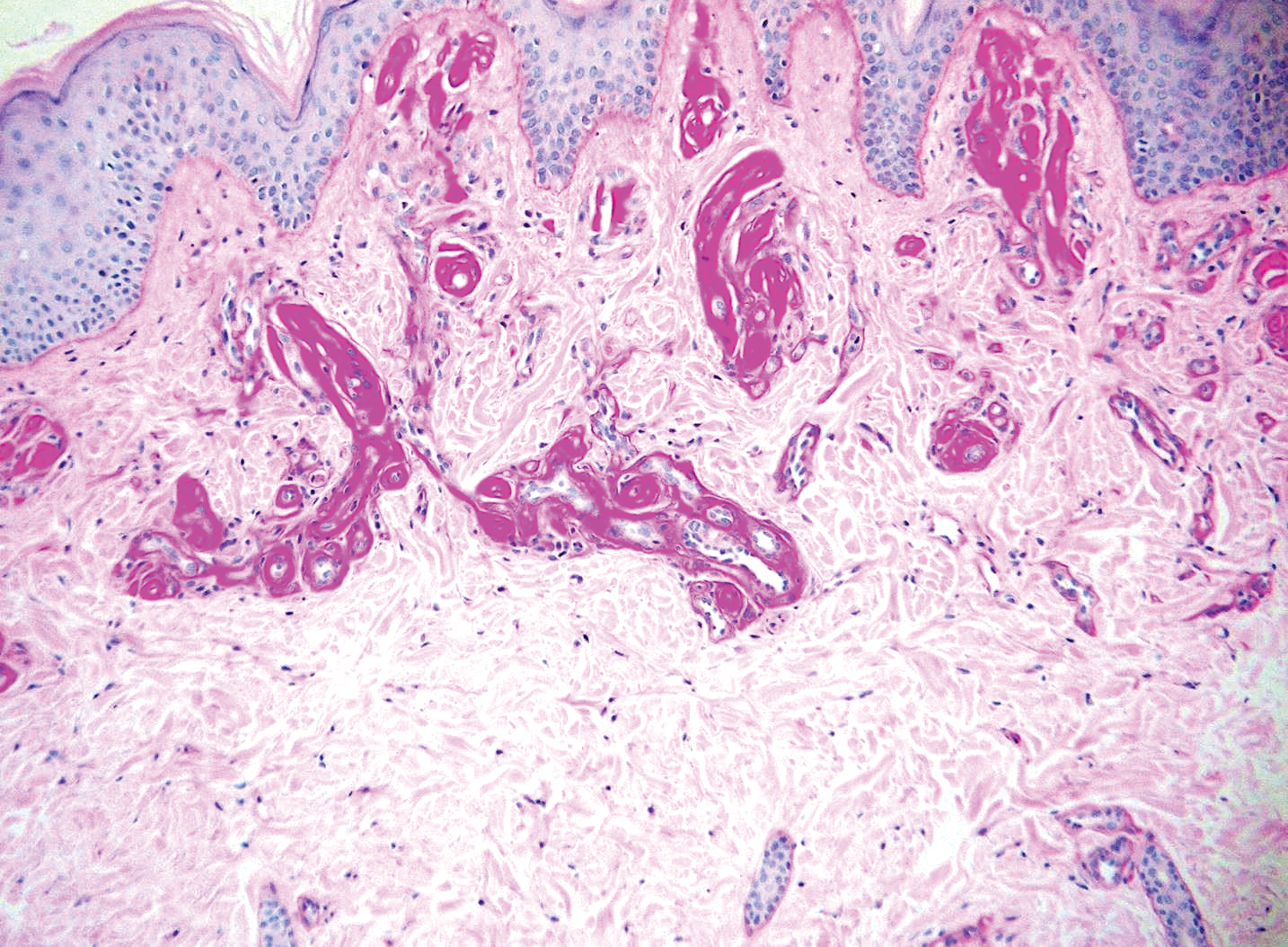
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
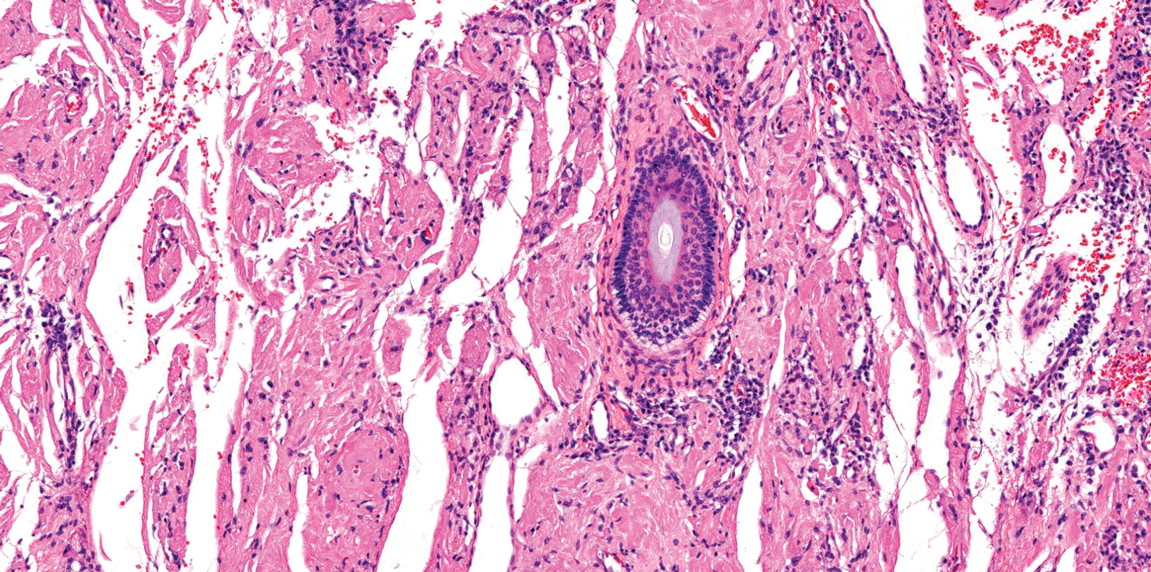
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
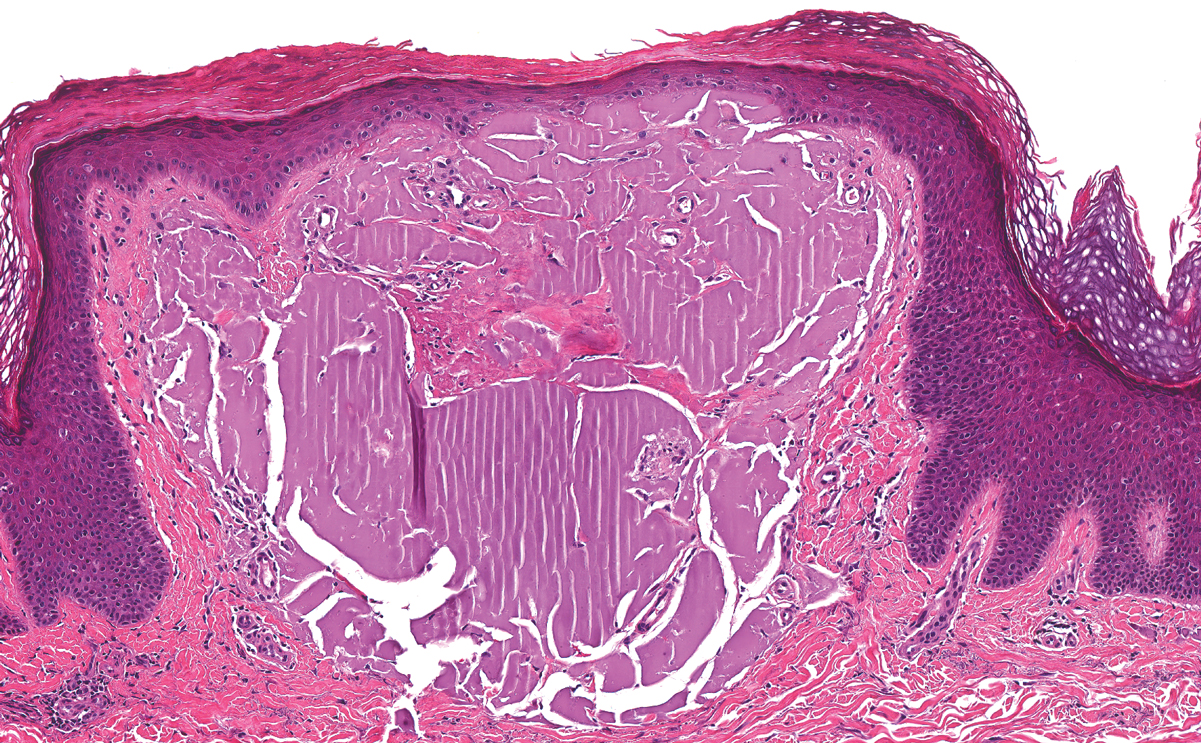
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
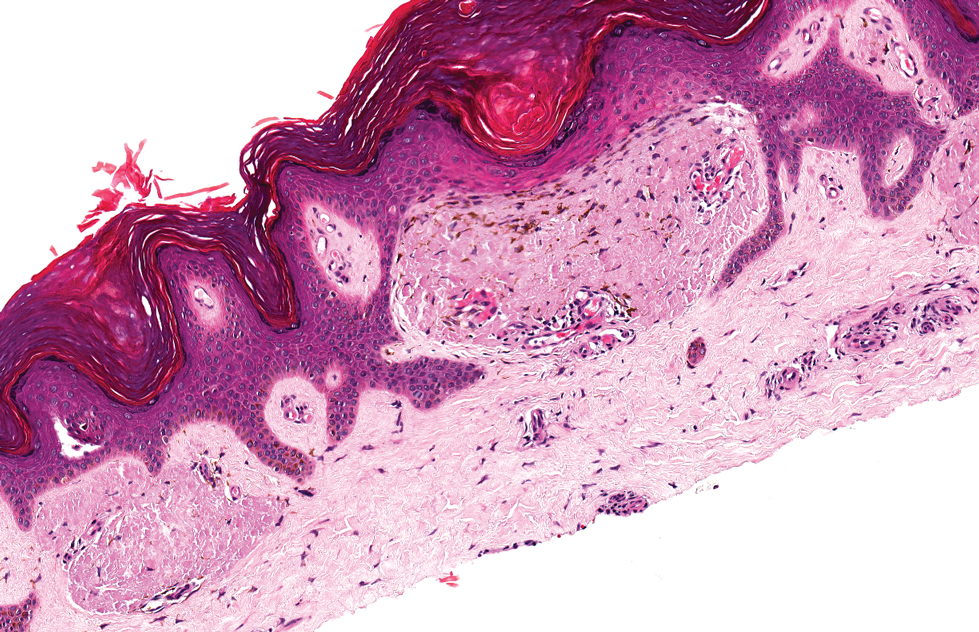
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
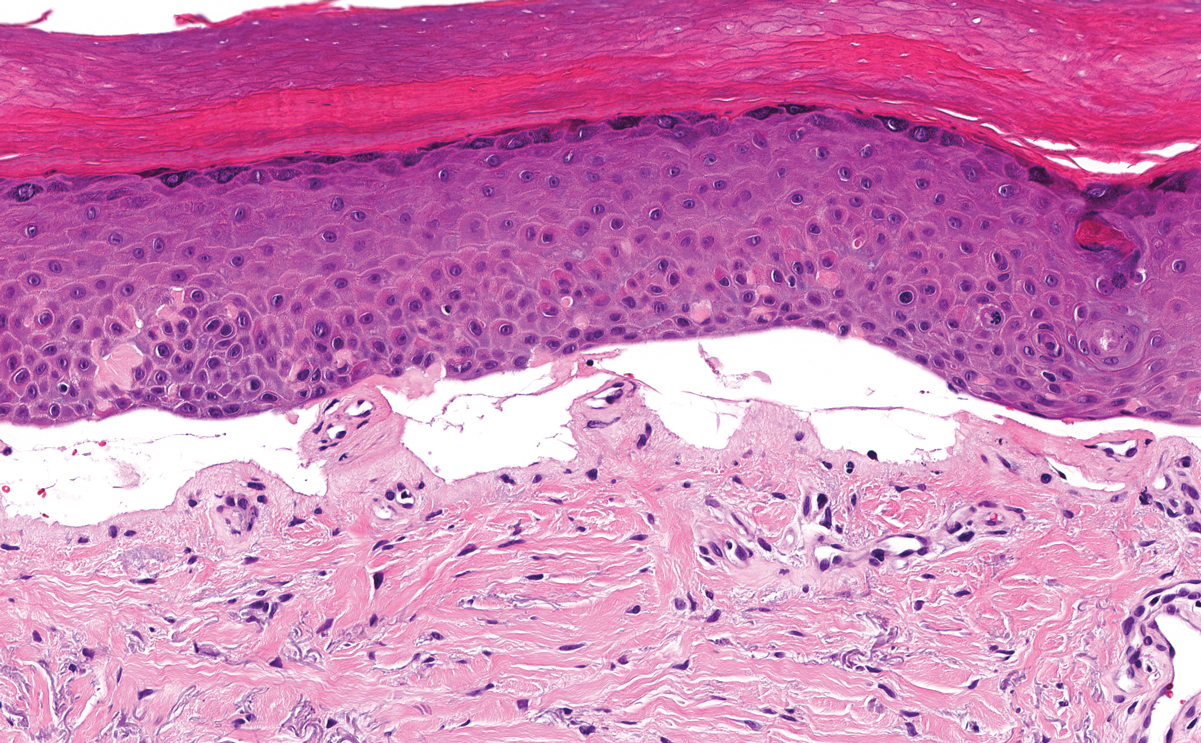
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
The Diagnosis: Erythropoietic Protoporphyria
Erythropoietic protoporphyria (EPP) is an autosomal-recessive photodermatosis that results from loss of activity of ferrochelatase, the last enzyme in the heme biosynthetic pathway.1 Erythropoietic protoporphyria normally involves sun-exposed areas of the body. Skin that is exposed to sunlight develops intense burning and stinging pain followed by erythema, edema, crusting, and petechiae that develops into waxy scarring over time. In contrast to other porphyrias, blistering generally is not seen.2 Accurate diagnosis often can be delayed by a decade or more following symptom onset due to the prominence of subjective pain as the presenting sign.
The histologic appearance of EPP differs depending on the chronicity of lesions. Biopsies of acute lesions show vacuolization of epidermal cells with intercellular edema, vacuolization and cytolysis of endothelial cells in superficial blood vessels, and focal red blood cell extravasation.3,4 A largely neutrophilic inflammatory infiltrate can be present.5 Hyaline cuffing develops over time in and around vessels in the papillary and superficial reticular dermis with notable sparing of adnexal structures. The perivascular deposits are strongly periodic acid-Schiff (PAS) positive and diastase resistant (Figure 1). Direct immunofluorescence shows mainly IgG and some IgM, fibrinogen, and C3 outlining characteristic donut-shaped blood vessels in the papillary dermis.6 The prominent thickness of the perivascular hyaline material depositions and the absence of subepidermal blistering can help differentiate EPP from porphyria cutanea tarda (PCT) and pseudoporphyria.6,7 When the deposition is extensive and involves the surrounding dermis, EPP can mimic colloid milium. Additional histologic differential diagnoses of EPP include other dermal depositional diseases such as lipoid proteinosis and amyloidosis.
Lipoid proteinosis is an autosomal-recessive multisystem genodermatosis caused by mutations in extracellular matrix gene 1, ECM1. The first clinical sign can be a hoarse cry in infancy due to infiltration of vocal cords.3 Development of papulonodular lesions along the eyelids can result in a string-of-beads appearance called moniliform blepharosis, which is pathognomonic for lipoid proteinosis.6 With chronicity, the involved skin can become yellow, waxy, and thickened, particularly in the flexures or areas of trauma. Histologically, the dermis in lipoid proteinosis becomes diffusely thickened due to deposition of PAS-positive eosinophilic hyaline material that stains weakly with Congo red and thioflavin T.6 Early lesions demonstrate pale pink, hyalinelike thickening of the papillary dermal capillaries. Chronic lesions reveal an acanthotic epidermis, occasional papillomatosis with overlying hyperkeratosis, and a thickened dermis where diffuse thick bundles of pink hyaline deposits are oriented perpendicularly to the dermoepidermal junction.1,6 Lipoid proteinosis can be differentiated from EPP by the involvement of adnexal structures such as hair follicles, sebaceous glands, and arrector pili muscles (Figure 2), as opposed to EPP where adnexal structures are spared.1 Additionally, depositions in lipoid proteinosis are centered around both superficial and deep vessels with an onion skin-like pattern, while EPP involves mainly superficial vessels with more mild and focal hyalinization.
Juvenile colloid milium (JCM) is a rare condition that presents before puberty with discrete, yellow-brown, translucent papules predominantly located on the cheeks and nose and around the mouth. A gelatinous material can be expressed after puncturing a lesion.6 Gingival deposits and ligneous conjunctivitis also can be present. On histopathology, JCM shows degeneration of epidermal keratinocytes that form colloid bodies within the superficial dermis following apoptosis.6 Hematoxylin and eosin staining shows amorphous, fissured, pale pink deposits completely filling and expanding the superficial to mid dermis with clefting and no inflammation (Figure 3). Spindle-shaped fibroblasts may be seen within the lines of colloid fissuring and dispersed throughout the deposits.1 Histologically, JCM can be differentiated from EPP because deposits in EPP are distributed around and within superficial blood vessel walls, causing prominent vascular thickening not seen in JCM.6 The adult variant of colloid milium also can be distinguished from EPP by the presence of solar elastosis, which is absent in EPP due to a history of sun avoidance.3,7
Lichen amyloidosis presents with highly pruritic, red-brown, hyperkeratotic papules that commonly are found on the anterior lower legs and extensor forearms.1 The calves, ankles, dorsal aspects of the feet, thighs, and trunk also may be affected. Excoriations, lichenification, and nodular prurigo-like lesions due to chronic scratching can be present.6 Lichen amyloidosis is characterized by large, pink, amorphous deposits in the papillary dermis with epidermal acanthosis, hypergranulosis, and hyperkeratosis (Figure 4).6 Perivascular deposits are not a feature of primary cutaneous localized amyloid lesions.6 The diagnosis can be confirmed with Congo red staining under polarized light, which classically demonstrates apple green birefringence.1 For cases of amyloid that are not detected by Congo red or are not clear-cut, direct immunofluorescence and immunohistochemistry can be used as adjuncts for diagnosis. Amyloid deposits fluoresce positively for immunoglobulins or complements, particularly IgM and C3,8 and immunohistochemistry confirms the presence of keratin epitopes in deposits.9
Porphyria cutanea tarda can appear histologically similar to EPP. Caterpillar bodies, or linearly arranged eosinophilic PAS-positive globules in the epidermis overlying subepidermal bullae, are a diagnostic histopathologic finding in both PCT and EPP but are seen in less than half of both cases.7,10 Compared to EPP, the perivascular deposits in PCT typically are less pronounced and limited to the vessel wall with smaller hyaline cuffs (Figure 5).7 Additionally, solar elastosis can be seen in PCT lesions but not in EPP, as patients with PCT tend to be older and have increased cumulative sun damage.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
- Touart DM, Sau P. Cutaneous deposition diseases. part I. J Am Acad Dermatol. 1998;39(2, pt 1):149-171; quiz 172-144.
- Lim HW. Pathogenesis of photosensitivity in the cutaneous porphyrias. J Invest Dermatol. 2005;124:xvi-xvii.
- In: Alikhan A, Hocker TLH, eds. Review of Dermatology. China: Elsevier; 2017.
- Horner ME, Alikhan A, Tintle S, et al. Cutaneous porphyrias part I: epidemiology, pathogenesis, presentation, diagnosis, and histopathology. Int J Dermatol. 2013;52:1464-1480.
- Michaels BD, Del Rosso JQ, Mobini N, et al. Erythropoietic protoporphyria: a case report and literature review. J Clin Aesthet Dermatol. 2010;3:44-48.
- Calonje E, Brenn T, Lazar A, et al, eds. McKee's Pathology of the Skin. 4th ed. China: Elsevier Saunders; 2012.
- Patterson JW. Weedon's Skin Pathology. 4th ed. China: Elsevier Limited; 2016.
- MacDonald DM, Black MM, Ramnarain N. Immunofluorescence studies in primary localized cutaneous amyloidosis. Br J Dermatol. 1977;96:635-641.
- Ortiz-Romero PL, Ballestin-Carcavilla C, Lopez-Estebaranz JL, et al. Clinicopathologic and immunohistochemical studies on lichen amyloidosis and macular amyloidosis. Arch Dermatol. 1994;130:1559-1560.
- Raso DS, Greene WB, Maize JC, et al. Caterpillar bodies of porphyria cutanea tarda ultrastructurally represent a unique arrangement of colloid and basement membrane bodies. Am J Dermatopathol. 1996;18:24-29.
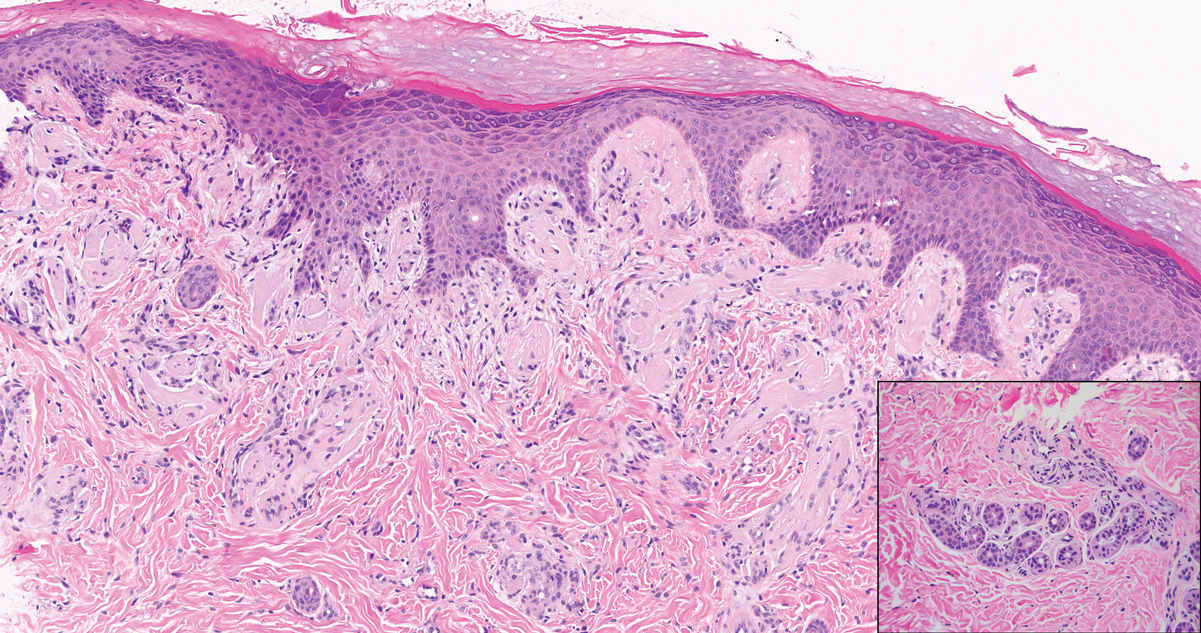
A 9-year-old girl presented with unexplained burning pain on the face, hands, and feet of 3 years' duration. Physical examination showed depressed shiny scars and crusted erosions on the dorsal aspect of the nose, arms, hands, and fingers. A 3-mm punch biopsy specimen was obtained from the right hand.
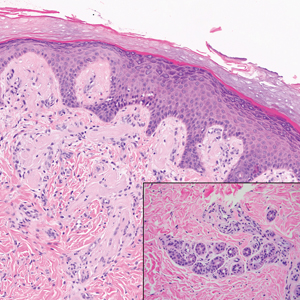
Woody Erythematous Induration on the Posterior Neck
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8

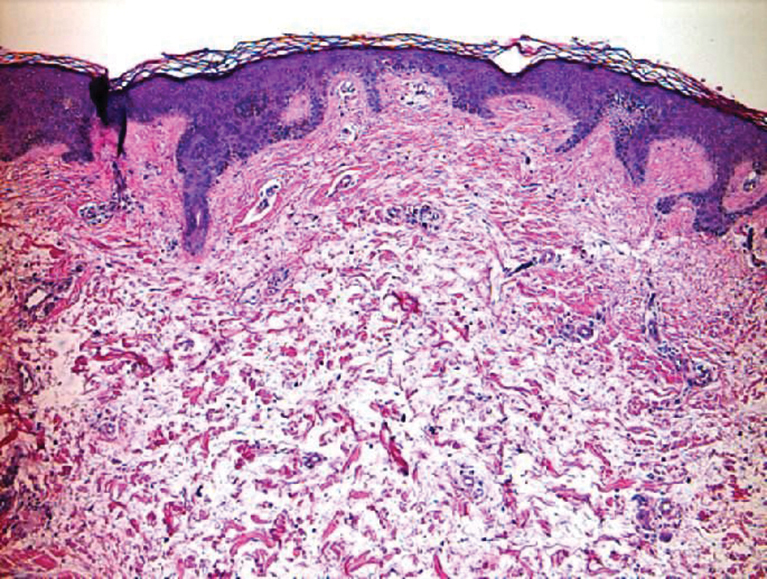
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
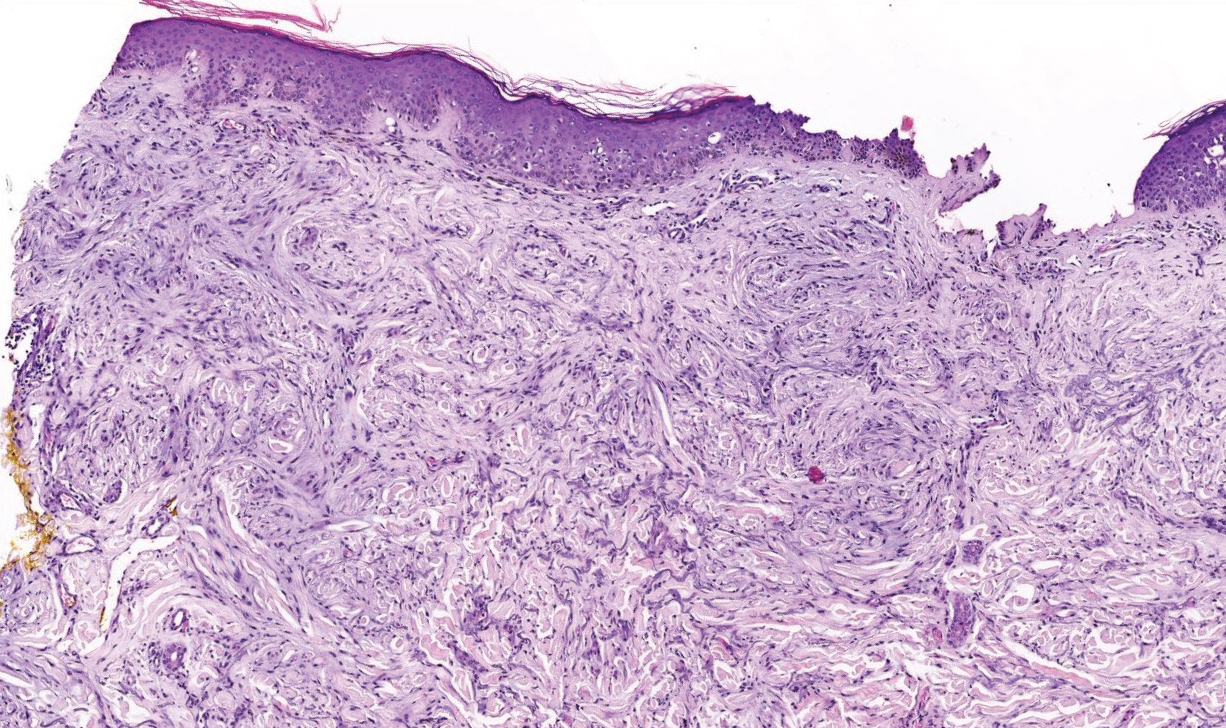
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
The Diagnosis: Scleredema Diabeticorum
Histologically, scleredema is characterized by mucin deposition between collagen bundles in the deep dermis. Clinically, it is characterized by a progressive indurated plaque with associated stiffness of the involved area. It most commonly presents on the posterior aspect of the neck, though it can extend to involve the shoulders and upper torso.1 Scleredema is divided into 3 subtypes based on clinical associations. Type 1 often is preceded by an infection, most commonly group A Streptococcus. This type occurs acutely and often resolves completely over a few months.2 Type 2, which has progressive onset, is associated with monoclonal gammopathy.3 Type 3 is the most common type and is associated with diabetes mellitus. A study of 484 patients with type 2 diabetes mellitus demonstrated a prevalence of 2.5%.4 Although the exact pathogenesis has not been defined, it is hypothesized that irreversible glycosylation of collagen and alterations in collagenase activity may lead to accumulation of collagen and mucin in the dermis.5 Similar to type 2, type 3 scleredema appears subtly, progresses slowly, and tends to be chronic.1,6 Scleredema is characterized by marked dermal thickening and enlarged collagen bundles separated by mucin deposition (Figure 1). Fibroblast proliferation is characteristically absent.1
Clinically, tumid lupus erythematosus presents with erythematous edematous plaques on sun-exposed areas.7 Pretibial myxedema (PM) classically is associated with Graves disease; however, it can present in association with other types of thyroid dysfunction. Classically, PM presents on the pretibial regions as well-demarcated erythematous or hyperpigmented plaques.8 Similar to scleredema, histologic examination of tumid lupus erythematosus and PM reveals mucin deposition. Tumid lupus erythematosus also may demonstrate periadnexal and perivascular lymphocytic inflammation (Figure 2).7 The collagen bundles present in PM often are thin in comparison to scleredema (Figure 3).8
Scleroderma also presents with skin induration, erythema, and stiffening. However, unlike scleredema, scleroderma commonly involves the fingers, toes, and face. It presents with symptoms of Raynaud phenomenon, painful digital nonpitting edema, perioral skin tightening, mucocutaneous telangiectasia, and calcinosis cutis. Scleroderma also can involve organs such as the lungs, heart, kidneys, and gastrointestinal tract.9 Histologically, scleroderma is characterized by a compact dermis with closely packed collagen bundles. Other features of scleroderma can include perivascular mononuclear inflammatory cell infiltration, progressive atrophy of intradermal and perieccrine fat, and fibrosis (Figure 4).10
Scleromyxedema, also called papular mucinosis, is primary dermal mucinosis that often presents with waxy, dome-shaped papules that may coalesce into plaques. Similar to scleredema, scleromyxedema shows increased mucin deposition. However, scleromyxedema commonly is associated with fibroblast proliferation, which is characteristically absent in scleredema (Figure 5).11
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
- Beers WH, Ince A, Moore TL. Scleredema adultorum of Buschke: a case report and review of the literature. Semin Arthritis Rheum. 2006;35:355-359.
- Cron RQ, Swetter SM. Scleredema revisited. a poststreptococcal complication. Clin Pediatr (Phila). 1994;33:606-610.
- Kövary PM, Vakilzadeh F, Macher E, et al. Monoclonal gammopathy in scleredema. observations in three cases. Arch Dermatol. 1981;117:536-539.
- Cole GW, Headley J, Skowsky R. Scleredema diabeticorum: a common and distinct cutaneous manifestation of diabetes mellitus. Diabetes Care. 1983;6:189-192.
- Namas R, Ashraf A. Scleredema of Buschke. Eur J Rheumatol. 2016;3:191-192.
- Knobler R, Moinzadeh P, Hunzelmann N, et al. European Dermatology Forum S1-guideline on the diagnosis and treatment of sclerosing diseases of the skin, part 2: scleromyxedema, scleredema and nephrogenic systemic fibrosis. J Eur Acad Dermatol Venereol. 2017;31:1581-1594.
- Kuhn A, Richter-Hintz D, Oslislo C, et al. Lupus erythematosus tumidus--a neglected subset of cutaneous lupus erythematosus: report of 40 cases. Arch Dermatol. 2000;136:1033-1041.
- Fatourechi V. Pretibial myxedema: pathophysiology and treatment options. Am J Clin Dermatol. 2005;6:295-309.
- van den Hoogen F, Khanna D, Fransen J, et al. 2013 Classification Criteria for Systemic Sclerosis: An American College of Rheumatology/European League Against Rheumatism Collaborative Initiative. 2013;65:2737-2747.
- Ferreli C, Gasparini G, Parodi A, et al. Cutaneous manifestations of scleroderma and scleroderma-like disorders: a comprehensive review. Clin Rev Allergy Immunol. 2017;53:306-336.
- Rongioletti F, Merlo G, Cinotti E, et al. Scleromyxedema: a multicenter study of characteristics, comorbidities, course, and therapy in 30 patients. J Am Acad Dermatol. 2013;69:66-72.
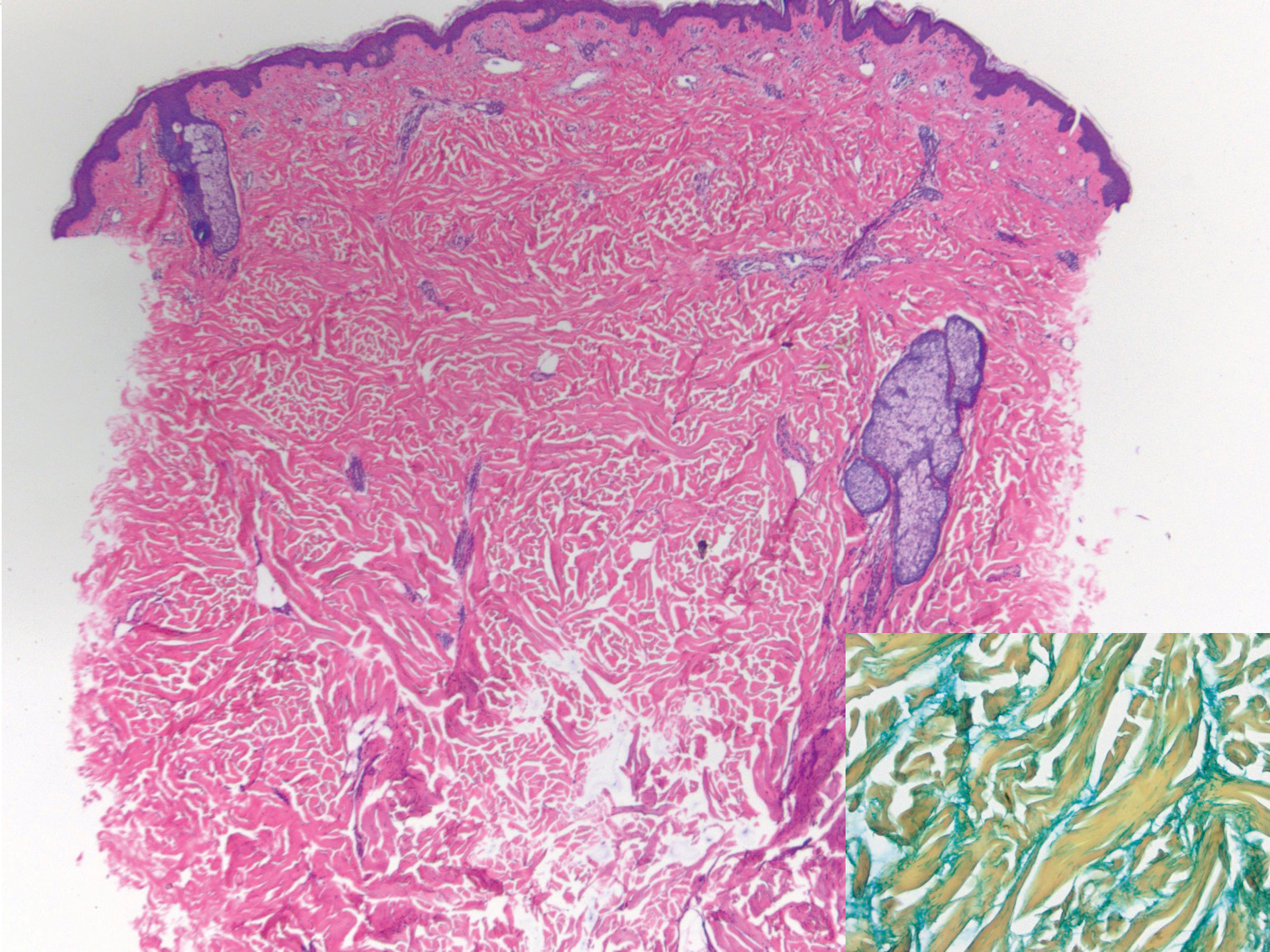
A 39-year-old white woman with a medical history of type 1 diabetes mellitus and rheumatoid arthritis presented to the dermatology clinic with pain and thickened skin on the posterior neck of 4 weeks’ duration. The patient noted stiffness in the neck and shoulders but denied any pain, pruritus, fever, chills, night sweats, fatigue, cough, dyspnea, dysphagia, weight loss, or change in appetite. Physical examination revealed a woody indurated plaque with slight erythema that was present diffusely on the posterior neck and upper back. The patient reported that a recent complete blood cell count and complete metabolic panel performed by her primary care physician were within reference range. Hemoglobin A1C was 8.6% of total hemoglobin (reference range, 4%–7%). A punch biopsy was performed.
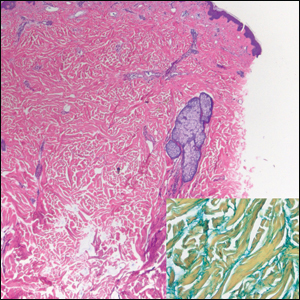
Granular Parakeratosis
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
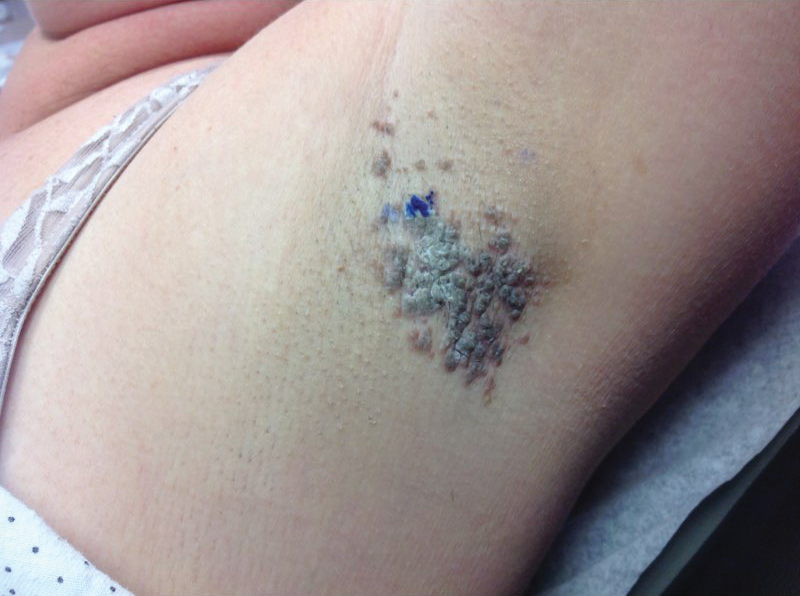
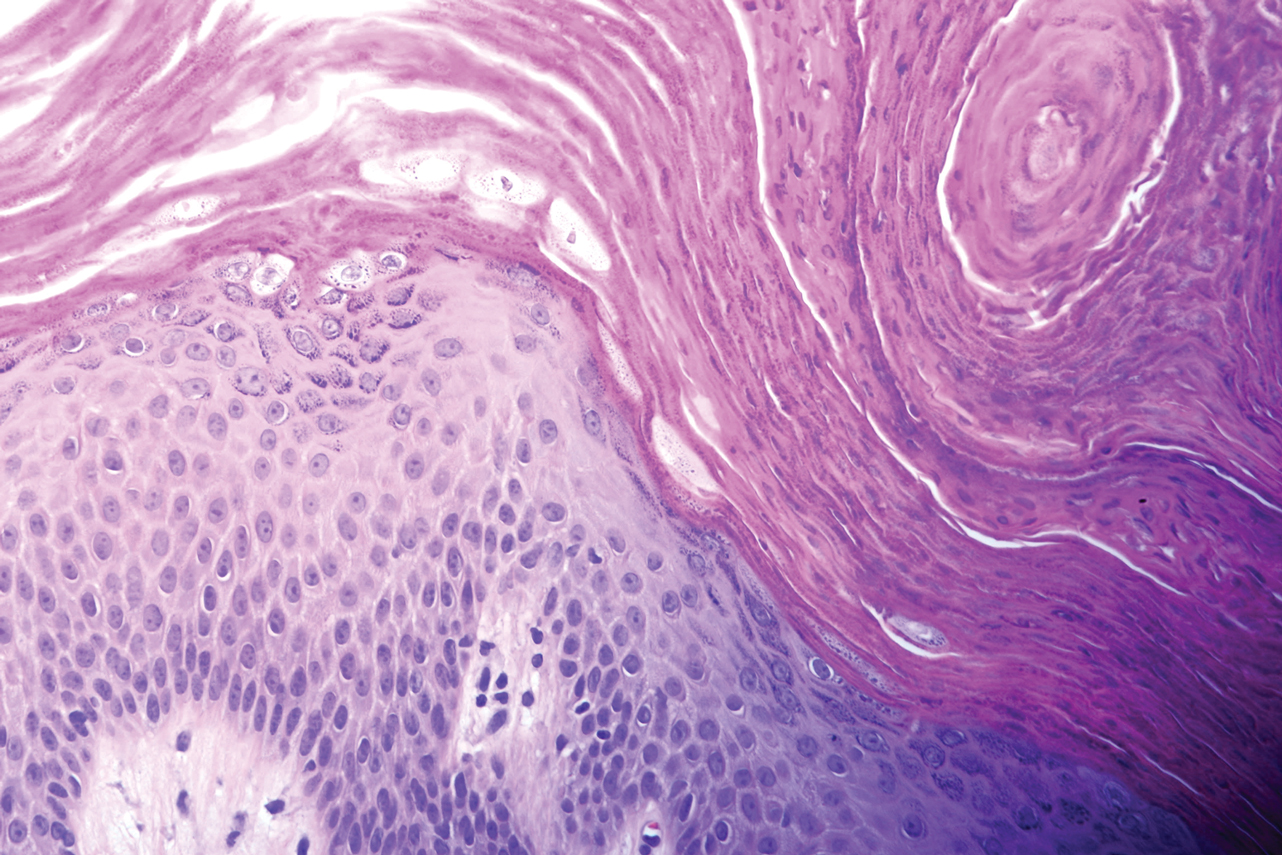
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
To the Editor:
A 46-year-old overweight woman presented with a rash in the axillae of 2 months’ duration. She did not report any additional symptoms such as pruritus or pain. She reported changing her deodorant recently from Secret Original to Secret Clinical Strength (both Procter & Gamble). Her medical history was remarkable for asthma and gastroesophageal reflux disease. Clinical examination revealed erythematous-brown, stuccolike, hyperkeratotic papules coalescing into plaques in recently shaved axillae, affecting the left axilla more than the right axilla (Figure 1). The clinical differential diagnosis included granular parakeratosis, intertrigo, Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, acanthosis nigricans, seborrheic keratoses, and irritant or allergic contact dermatitis. A punch biopsy revealed a marked compact parakeratotic horn with retention of keratohyalin granules (Figure 2). The subjacent epidermis showed some acanthosis and spongiosis with mild chronic inflammation of the dermal rim. Based on histopathology, granular parakeratosis was diagnosed.
At a subsequent visit 2 weeks later, we prescribed glycolic acid lotion 10% applied to the axillae twice daily, plus tretinoin gel 0.05% applied to the axillae each evening. She reported clearing after 1 week of therapy. She also had changed her deodorant from Secret Clinical Strength back to the usual Secret Original. The patient discontinued topical treatment after clearing of the lesions. Three weeks later, clinical examination revealed postinflammatory hyperpigmentation in the axillae, and the prior lesions had resolved (Figure 3).
Granular parakeratosis is an unusual condition most commonly presenting in middle-aged women in the axillae, with a clinical presentation of erythematous to brownish hyperkeratotic papules coalescing into plaques. Although few cases have been reported, granular parakeratosis likely is more common than has been reported. There have been reports involving the scalp, cheeks, abdomen, thighs, and other intertriginous areas including inguinal folds and the submammary region.1-4 There also is an infantile form related to diapers and zinc oxide paste.5 Although uncommon, granular parakeratosis can occur as a single papule or plaque and is termed granular parakeratotic acanthoma.6 Lesions may persist for months, spontaneously resolve and recur, and occasionally evolve into fissures and erosions due to irritation. Pruritus is a common concern. Histology of granular parakeratosis reveals hyperkeratosis with eosinophilic staining, compact parakeratosis with retention of basophilic keratohyalin granules, and vascular proliferation and ectasia.5
The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.5,7 Cases indicate a link to obesity. Hypotheses as to the etiology include the disruption of cornification. Normally, filaggrin maintains the keratohyaline granules in the stratum corneum during cornification. Therefore, the retention of keratohyaline granules in granular parakeratosis may be due to a defect in processing profilaggrin to filaggrin, which has been proposed based on ultrastructural and immunohistochemical studies.8
The differential diagnosis includes granular parakeratosis, intertrigo (caused by seborrheic dermatitis, candidiasis, inverse psoriasis, or erythrasma), Hailey-Hailey disease, Darier disease, pemphigus vegetans, confluent and reticulated papillomatosis, and irritant or allergic contact dermatitis. The papules may resemble seborrheic keratoses, while the plaques can be mistaken for acanthosis nigricans.
Therapeutic success has been reported with topical corticosteroids, vitamin D analogues, topical or oral retinoids, ammonium lactate, calcineurin inhibitors, topical or oral antifungals, cryotherapy, and botulinum toxin injections.3,9-11 In addition, parakeratosis has decreased in biopsies from psoriatic patients after acitretin, methotrexate, and phototherapy, which may be alternative treatments for unusually difficult or recalcitrant cases of granular parakeratosis. To minimize side effects and resolve the papules quickly, we combined 2 synergistic agents—glycolic acid and tretinoin—each with different mechanisms of action, and we observed excellent clinical response.
Granular parakeratosis is possibly related to a combination of topical products that potentiate irritation, rubbing, and occlusion of sweat. Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products. Our patient’s change in deodorant may have been the inciting factor for the disease. Withdrawal of the Secret Clinical Strength deodorant prompted clearing, though topical retinoid and glycolic acid acted as facilitating therapies for timely results. A thorough history, as highlighted by this case, may help pinpoint etiologic factors. By identifying a seemingly innocuous change in hygienic routine, we were able to minimize the need for ongoing therapy.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
- Graham R. Intertriginous granular parakeratosis: a case report and review of the literature. J Am Acad Dermatol. 2011;64:AB45-AB45.
- Compton AK, Jackson JM. Isotretinoin as a treatment for axillary granular parakeratosis. Cutis. 2007;80:55-56.
- Channual J, Fife DJ, Wu JJ. Axillary granular parakeratosis. Cutis. 2013;92;61, 65-66.
- Streams S, Gottwald L, Zaher A, et al. Granular parakeratosis of the scalp: a case report. J Am Acad Dermatol. 2007;56:AB81-AB81.
- James WD, Berger T, Elston D. Andrews’ Diseases of the Skin. 12th ed. Philadelphia, PA: Elsevier, Inc; 2015.
- Resnik KS, Kantor GR, DiLeonardo M. Granular parakeratotic acanthoma. Am J Dermatopathol. 2005;27:393-396.
- Naylor E, Wartman D, Telang G, et al. Granular parakeratosis secondary to postsurgical occlusion. J Am Acad Dermatol. 2008;58:AB126.
- Bolognia JL, Jorizzo JL, Schaffer JV. Dermatology. 3rd ed. Philadelphia, PA: Elsevier, Inc; 2012.
- Baum B, Skopit S. Granular parakeratosis treatment with tacrolimus 0.1% ointment: a case presentation and discussion. J Am Osteo Coll Dermatol. 2013;26:40-41.
- Brown SK, Heilman ER. Granular parakeratosis: resolution with topical tretinoin. J Am Acad Dermatol. 2002;47:S279-S280.
- Webster CG, Resnik KS, Webster GF. Axillary granular parakeratosis: response to isotretinoin. J Am Acad Dermatol. 1997;37:789790.
Practice Points
- Granular parakeratosis most commonly presents in middle-aged women in the axillae.
- The cause is unknown but possibly related to irritation from rubbing, occlusion, sweating, or deodorants.
- Multiple treatment modalities likely contribute to clearing, the most important being removal of any triggering topical products.
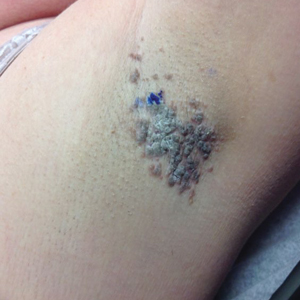
Skin patterns of COVID-19 vary widely
according to Christine Ko, MD.

“Things are very fluid,” Dr. Ko, professor of dermatology and pathology at Yale University, New Haven, Conn., said during the virtual annual meeting of the American Academy of Dermatology. “New studies are coming out daily. Due to the need for rapid dissemination, a lot of the studies are case reports, but there are some nice case series. Another caveat for the literature is that a lot of these cases were not necessarily confirmed with testing for SARS-CoV-2, but some were.”
Dr. Ko framed her remarks largely on a case collection survey of images and clinical data from 375 patients in Spain with suspected or confirmed COVID-19 that was published online April 29, 2020, in the British Journal of Dermatology (doi: 10.1111/bjd.19163). Cutaneous manifestations included early vesicular eruptions mainly on the trunk or limbs (9%), maculopapular (47%) to urticarial lesions (19%) mainly on the trunk, and acral areas of erythema sometimes with vesicles or erosion (perniosis-like) (19%) that seemed to be a later manifestation of COVID-19. Retiform purpura or necrosis (6%) was most concerning in terms of skin disease, with an associated with a mortality of 10%.
On histology, the early vesicular eruptions are typically marked by dyskeratotic keratinocytes, Dr. Ko said, while urticarial lesions are characterized by a mixed dermal infiltrate; maculopapular lesions were a broad category. “There are some case reports that show spongiotic dermatitis or parakeratosis with a lymphocytic infiltrate,” she said. “A caveat to keep in mind is that, although these patients may definitely have COVID-19 and be confirmed to have it by testing, hypersensitivity reactions may be due to the multiple medications they’re on.”
Patients can develop a spectrum of lesions that are suggestive of vascular damage or occlusion, Dr. Ko continued. Livedoid lesions may remain static and not eventuate into necrosis or purpura but will self-resolve. Purpuric lesions and acral gangrene have been described, and these lesions correspond to vascular occlusion on biopsy.
A later manifestation are the so-called “COVID toes” with a superficial and deep lymphocytic infiltrate, as published June 1, 2020, in JAAD Case Reports: (doi: 10.1016/j.jdcr.2020.04.011).
“There are patients in the literature that have slightly different pathology, with lymphocytic inflammation as well as occlusion of vessels,” Dr. Ko said. A paper published June 20, 2020, in the British Journal of Dermatology used immunohistochemical staining against the SARS-CoV-2 spike protein, and biopsies of “COVID toes” had positive staining of endothelial cells, supporting the notion that “COVID toes” are a direct manifestation of viral infection (doi: 10.1111/bjd.19327).
“There’s a lot that we still don’t know, and some patterns are going to be outliers,” Dr. Ko concluded. “[As for] determining which skin manifestations are directly from coronavirus infection within the skin, more study is needed and likely time will tell.” She reported having no financial disclosures relevant to her talk.
according to Christine Ko, MD.
“Things are very fluid,” Dr. Ko, professor of dermatology and pathology at Yale University, New Haven, Conn., said during the virtual annual meeting of the American Academy of Dermatology. “New studies are coming out daily. Due to the need for rapid dissemination, a lot of the studies are case reports, but there are some nice case series. Another caveat for the literature is that a lot of these cases were not necessarily confirmed with testing for SARS-CoV-2, but some were.”
Dr. Ko framed her remarks largely on a case collection survey of images and clinical data from 375 patients in Spain with suspected or confirmed COVID-19 that was published online April 29, 2020, in the British Journal of Dermatology (doi: 10.1111/bjd.19163). Cutaneous manifestations included early vesicular eruptions mainly on the trunk or limbs (9%), maculopapular (47%) to urticarial lesions (19%) mainly on the trunk, and acral areas of erythema sometimes with vesicles or erosion (perniosis-like) (19%) that seemed to be a later manifestation of COVID-19. Retiform purpura or necrosis (6%) was most concerning in terms of skin disease, with an associated with a mortality of 10%.
On histology, the early vesicular eruptions are typically marked by dyskeratotic keratinocytes, Dr. Ko said, while urticarial lesions are characterized by a mixed dermal infiltrate; maculopapular lesions were a broad category. “There are some case reports that show spongiotic dermatitis or parakeratosis with a lymphocytic infiltrate,” she said. “A caveat to keep in mind is that, although these patients may definitely have COVID-19 and be confirmed to have it by testing, hypersensitivity reactions may be due to the multiple medications they’re on.”
Patients can develop a spectrum of lesions that are suggestive of vascular damage or occlusion, Dr. Ko continued. Livedoid lesions may remain static and not eventuate into necrosis or purpura but will self-resolve. Purpuric lesions and acral gangrene have been described, and these lesions correspond to vascular occlusion on biopsy.
A later manifestation are the so-called “COVID toes” with a superficial and deep lymphocytic infiltrate, as published June 1, 2020, in JAAD Case Reports: (doi: 10.1016/j.jdcr.2020.04.011).
“There are patients in the literature that have slightly different pathology, with lymphocytic inflammation as well as occlusion of vessels,” Dr. Ko said. A paper published June 20, 2020, in the British Journal of Dermatology used immunohistochemical staining against the SARS-CoV-2 spike protein, and biopsies of “COVID toes” had positive staining of endothelial cells, supporting the notion that “COVID toes” are a direct manifestation of viral infection (doi: 10.1111/bjd.19327).
“There’s a lot that we still don’t know, and some patterns are going to be outliers,” Dr. Ko concluded. “[As for] determining which skin manifestations are directly from coronavirus infection within the skin, more study is needed and likely time will tell.” She reported having no financial disclosures relevant to her talk.
according to Christine Ko, MD.
“Things are very fluid,” Dr. Ko, professor of dermatology and pathology at Yale University, New Haven, Conn., said during the virtual annual meeting of the American Academy of Dermatology. “New studies are coming out daily. Due to the need for rapid dissemination, a lot of the studies are case reports, but there are some nice case series. Another caveat for the literature is that a lot of these cases were not necessarily confirmed with testing for SARS-CoV-2, but some were.”
Dr. Ko framed her remarks largely on a case collection survey of images and clinical data from 375 patients in Spain with suspected or confirmed COVID-19 that was published online April 29, 2020, in the British Journal of Dermatology (doi: 10.1111/bjd.19163). Cutaneous manifestations included early vesicular eruptions mainly on the trunk or limbs (9%), maculopapular (47%) to urticarial lesions (19%) mainly on the trunk, and acral areas of erythema sometimes with vesicles or erosion (perniosis-like) (19%) that seemed to be a later manifestation of COVID-19. Retiform purpura or necrosis (6%) was most concerning in terms of skin disease, with an associated with a mortality of 10%.
On histology, the early vesicular eruptions are typically marked by dyskeratotic keratinocytes, Dr. Ko said, while urticarial lesions are characterized by a mixed dermal infiltrate; maculopapular lesions were a broad category. “There are some case reports that show spongiotic dermatitis or parakeratosis with a lymphocytic infiltrate,” she said. “A caveat to keep in mind is that, although these patients may definitely have COVID-19 and be confirmed to have it by testing, hypersensitivity reactions may be due to the multiple medications they’re on.”
Patients can develop a spectrum of lesions that are suggestive of vascular damage or occlusion, Dr. Ko continued. Livedoid lesions may remain static and not eventuate into necrosis or purpura but will self-resolve. Purpuric lesions and acral gangrene have been described, and these lesions correspond to vascular occlusion on biopsy.
A later manifestation are the so-called “COVID toes” with a superficial and deep lymphocytic infiltrate, as published June 1, 2020, in JAAD Case Reports: (doi: 10.1016/j.jdcr.2020.04.011).
“There are patients in the literature that have slightly different pathology, with lymphocytic inflammation as well as occlusion of vessels,” Dr. Ko said. A paper published June 20, 2020, in the British Journal of Dermatology used immunohistochemical staining against the SARS-CoV-2 spike protein, and biopsies of “COVID toes” had positive staining of endothelial cells, supporting the notion that “COVID toes” are a direct manifestation of viral infection (doi: 10.1111/bjd.19327).
“There’s a lot that we still don’t know, and some patterns are going to be outliers,” Dr. Ko concluded. “[As for] determining which skin manifestations are directly from coronavirus infection within the skin, more study is needed and likely time will tell.” She reported having no financial disclosures relevant to her talk.
FROM AAD 20
Purpuric Bullae on the Lower Extremities
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
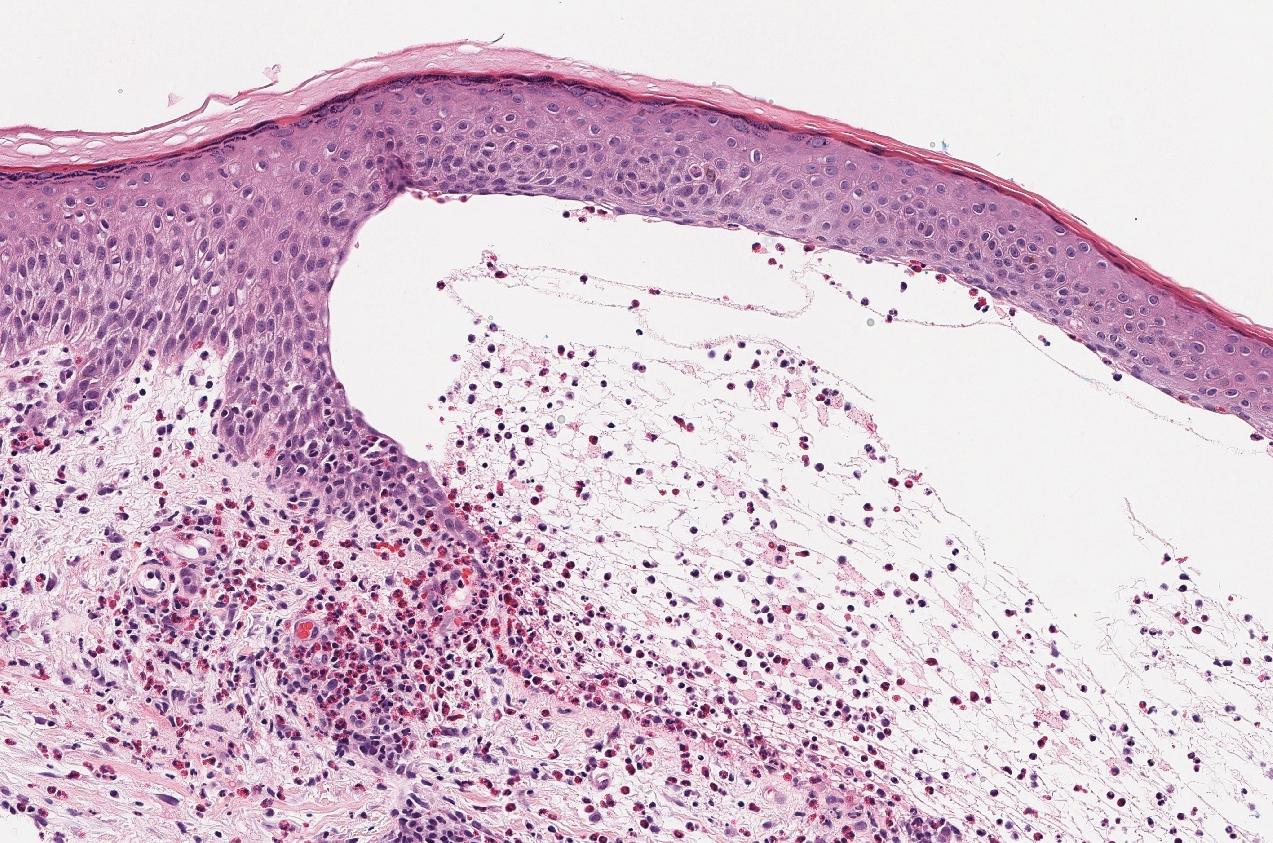
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
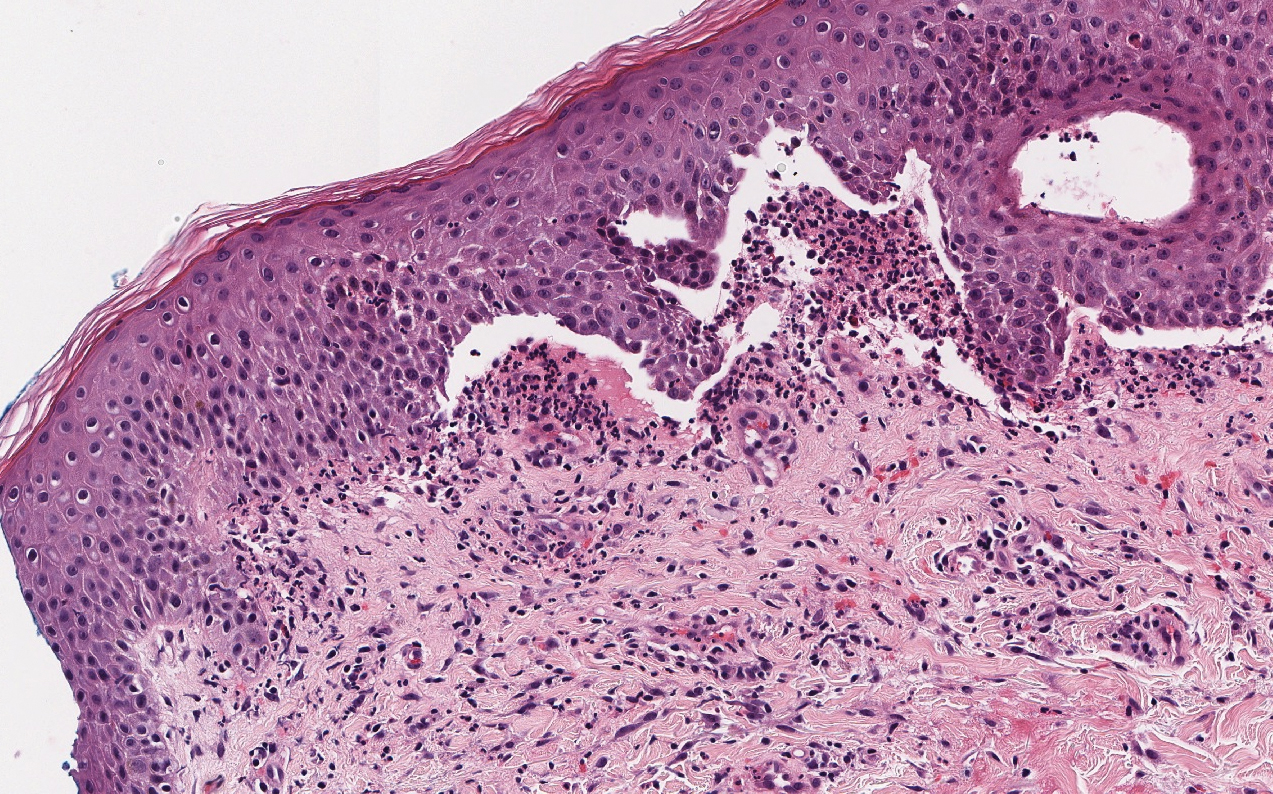
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.

Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
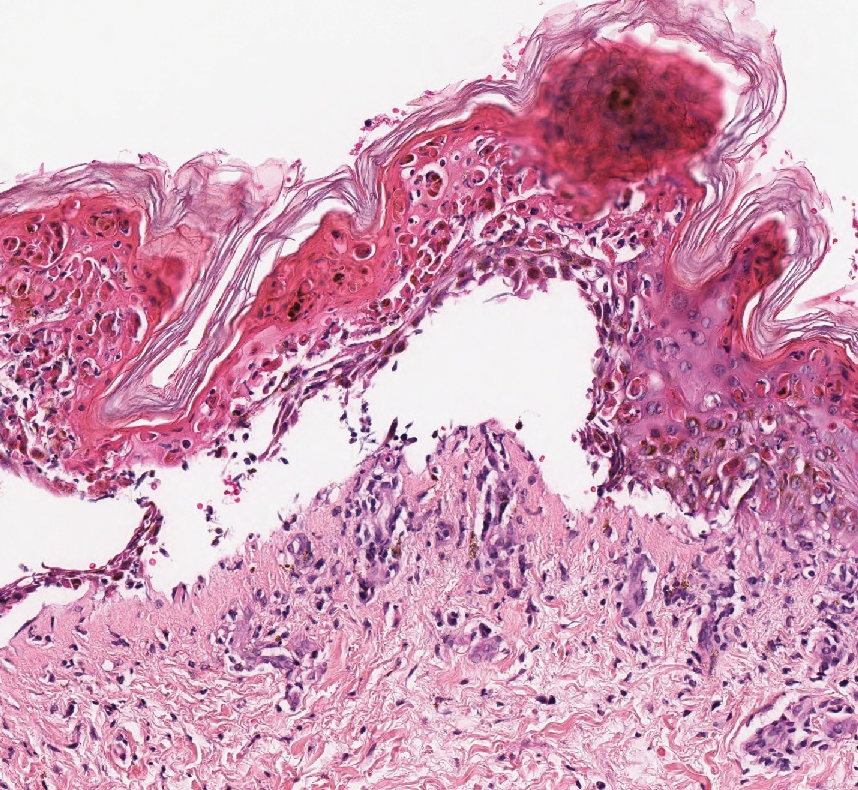
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
The Diagnosis: Bullous Leukocytoclastic Vasculitis
Histopathology with hematoxylin and eosin (H&E) stain showed a perivascular neutrophilic infiltrate, karyorrhexis, red blood cell extravasation, and fibrin deposition in the vessel wall (quiz images). Direct immunofluorescence (DIF) showed fibrin surrounding the vasculature, consistent with vasculitis. The clinical and histopathological evaluation supported the diagnosis of bullous leukocytoclastic vasculitis (LCV). The patient had a full LCV workup including antinuclear antibody, rheumatoid factor, hepatitis B and hepatitis C screening, erythrocyte sedimentation rate, C-reactive protein, and C3/C4/total complement level, which were all within reference range. The patient denied that she had taken any medications prior to the onset of the rash. She was started on a 12-day prednisone taper starting at 60 mg, and the rash resolved in 1 week.
Although the incidence of LCV is estimated to be 30 cases per million individuals per year,1 bullous LCV is a rarer entity with only a few cases reported in the literature.2,3 As in our patient's case, up to 50% of LCV cases are idiopathic or the etiology cannot be determined despite laboratory workup and medication review. Other cases can be secondary to medication, infection, collagen vascular disease, or malignancy.3 Despite the exact pathogenesis of bullous LCV being unknown,4 it likely is related to a type III hypersensitivity reaction with immune complex deposition in postcapillary venules leading to endothelial injury, activation of the complement cascade, and development of intraepidermal or subepidermal blister formation depending on location of inflammation and edema.2 Clinically, an intraepidermal split would be more flaccid, similar to pemphigus vulgaris, while a subepidermal split, as in our patient, would be taut bullae. The subepidermal split more commonly is seen in bullous LCV.2
Leukocytoclastic vasculitis on H&E staining characteristically has a perivascular inflammatory infiltrate, neutrophilic fragments called leukocytoclasis, and blood extravasation.3 Extravasated blood presents clinically as petechiae. In this case, the petechiae helped distinguish this entity from the differential diagnosis. Furthermore, DIF would be helpful in distinguishing bullous diseases such as bullous pemphigoid (BP) and pemphigus vulgaris from LCV.2 Direct immunofluorescence in bullous LCV would have fibrinogen surrounding the vasculature without C3 and IgG deposition (intraepidermal or subepidermal).
Mild cases of LCV often resolve with supportive measures including elevation of the legs, ice packs applied to the affected area, and removal of the inciting drug or event.4 In the few cases reported in the literature, bullous LCV presented more diffusely than classic LCV with bullous lesions on the forearms and the lower extremities. Oral steroids are efficacious for extensive bullous LCV.4
The differential diagnosis of bullous LCV includes bullous diseases with subepidermal split including BP and linear IgA bullous dermatosis (LABD). Bullous pemphigoid is an autoimmune subepidermal blistering disease typically affecting patients older than 60 years.5 The pathogenesis of BP is related to development of autoantibodies directed against hemidesmosome components, bullous pemphigoid antigen (BPAG) 1 or BPAG2.5 Bullous pemphigoid presents clinically as widespread, generally pruritic, erythematous, urticarial plaques with bullae. Histologically, BP characteristically has a subepidermal split with superficial dermal edema and eosinophils at the dermoepidermal junction (Figure 1). Direct immunofluorescence confirms the diagnosis with IgG and C3 deposition in an n-serrated pattern at the dermoepidermal junction.6 Bullous pemphigoid can be distinguished from bullous LCV by the older age of presentation, DIF findings, and the absence of purpura.
Linear IgA bullous dermatosis represents a rare subepidermal vesiculobullous disease occurring in patients in their 60s.7 Clinically, this entity presents as tense bullae often located on the periphery of an urticarial plaque, classically called the "string of pearls sign." Histologically, LABD also presents with subepidermal split; however, neutrophils are the predominant cell type vs eosinophils in BP (Figure 2).7 Direct immunofluorescence is specific with a linear deposition of IgA at the dermoepidermal junction. Linear IgA bullous dermatosis most commonly is induced by vancomycin. Unlike bullous LCV, the bullae of LABD have an annular peripheral pattern on an erythematous base and lack purpura.
Stasis dermatitis is inflammation of the dermis due to venous insufficiency that often is present in the bilateral lower extremities. The disorder affects approximately 7% of adults older than 50 years, but it also can occur in younger patients.8 The pathophysiology of stasis dermatitis is caused by edema, which leads to extracellular fluid, plasma proteins, macrophages, and erythrocytes passing into the interstitial space. Patients with stasis dermatitis present with scaly erythematous papules and plaques or edematous blisters on the lower extremities. Diagnosis usually can be made clinically; however, a skin biopsy also can be helpful. Hematoxylin and eosin shows a pauci-inflammatory subepidermal bulla with fibrin (Figure 3).8 The overlying epidermis is intact. The dermis has cannon ball angiomatosis, red blood cell extravasation, and fibrosis typical of stasis dermatitis. Stasis dermatitis with bullae is cell poor and lacks the perivascular inflammatory infiltrate and neutrophilic fragments that often are present in LCV, making the 2 entities distinguishable.
Stevens-Johnson syndrome/toxic epidermal necrolysis (SJS/TEN) lies on a spectrum of severe cutaneous drug reactions involving the skin and mucous membranes. Cutaneous involvement typically begins on the trunk and face and later can involve the palms and soles.9 Similar drugs have been implicated in bullous LCV and SJS/TEN, including nonsteroidal anti-inflammatory drugs and antibiotics. Histologically, SJS/TEN has full-thickness epidermal necrolysis, vacuolar interface, and keratinocyte apoptosis (Figure 4).9 The clinical presentation of sloughing of skin with positive Nikolsky sign, oral involvement, and H&E and DIF findings can help differentiate this entity from bullous LCV.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
- Einhorn J, Levis JT. Dermatologic diagnosis: leukocytoclastic vasculitis. Perm J. 2015;19:77-78.
- Davidson KA, Ringpfeil F, Lee JB. Ibuprofen-induced bullous leukocytoclastic vasculitis. Cutis. 2001;67:303-307.
- Lazic T, Fonder M, Robinson-Bostom L, et al. Orlistat-induced bullous leukocytoclastic vasculitis. Cutis. 2013;91:148-149.
- Mericliler M, Shnawa A, Al-Qaysi D, et al. Oxacillin-induced leukocytoclastic vasculitis. IDCases. 2019;17:E00539.
- Bernard P, Antonicelli F. Bullous pemphigoid: a review of its diagnosis, associations and treatment. Am J Clin Dermatol. 2017;18:513-528.
- High WA. Blistering disorders. In: Elston DM, Ferringer T, Ko C, et al, eds. Dermatopathology. 3rd ed. Philadelphia, PA: Elsevier; 2019:161-171.
- Visentainer L, Massuda JY, Cintra ML, et al. Vancomycin-induced linear IgA bullous dermatosis (LABD)--an atypical presentation. Clin Case Rep. 2019;7:1091-1093.
- Hyman DA, Cohen PR. Stasis dermatitis as a complication of recurrent levofloxacin-associated bilateral leg edema. Dermatol Online J. 2013;19:20399.
- Harr T, French LE. Toxic epidermal necrolysis and Stevens-Johnson syndrome. Orphanet J Rare Dis. 2010;5:39.
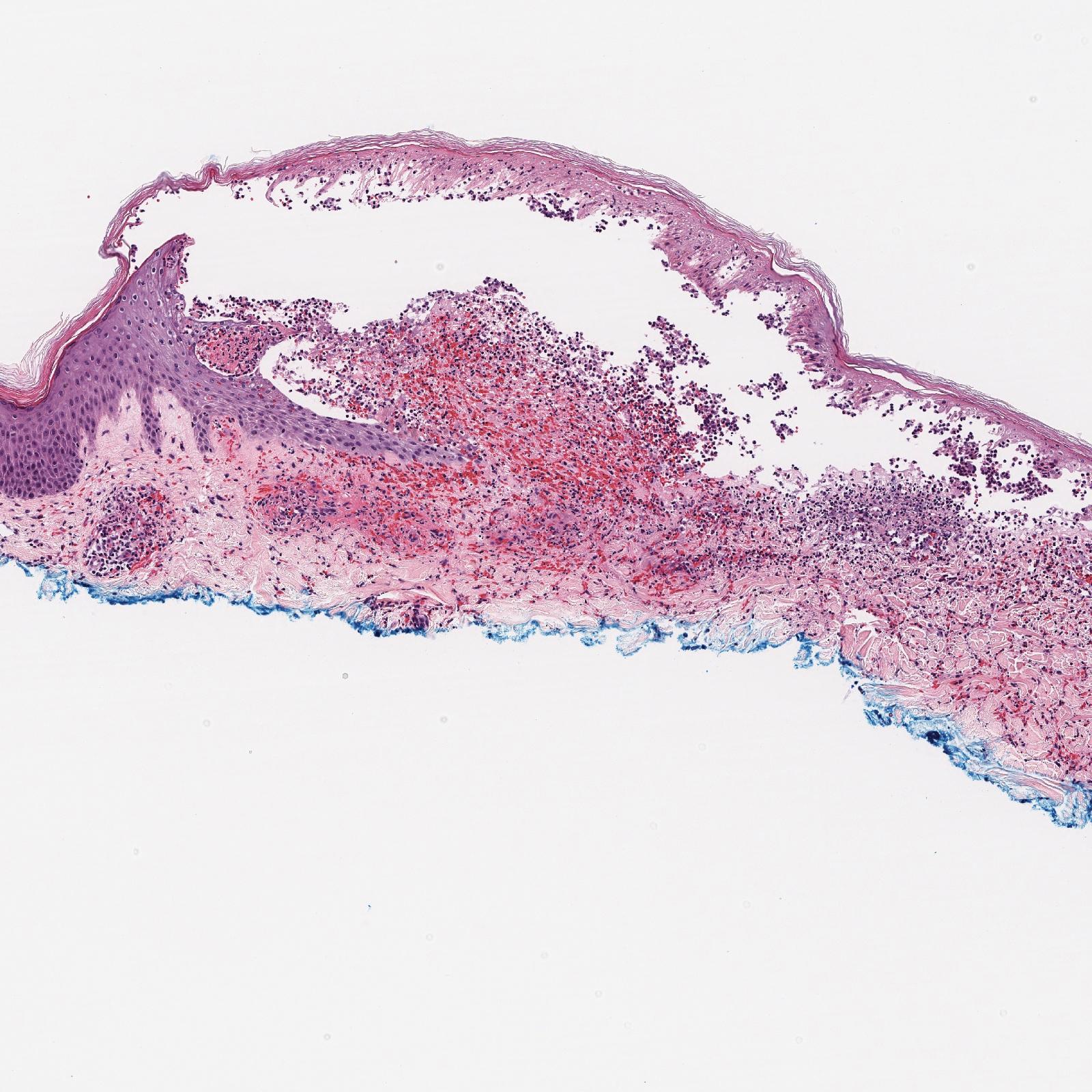
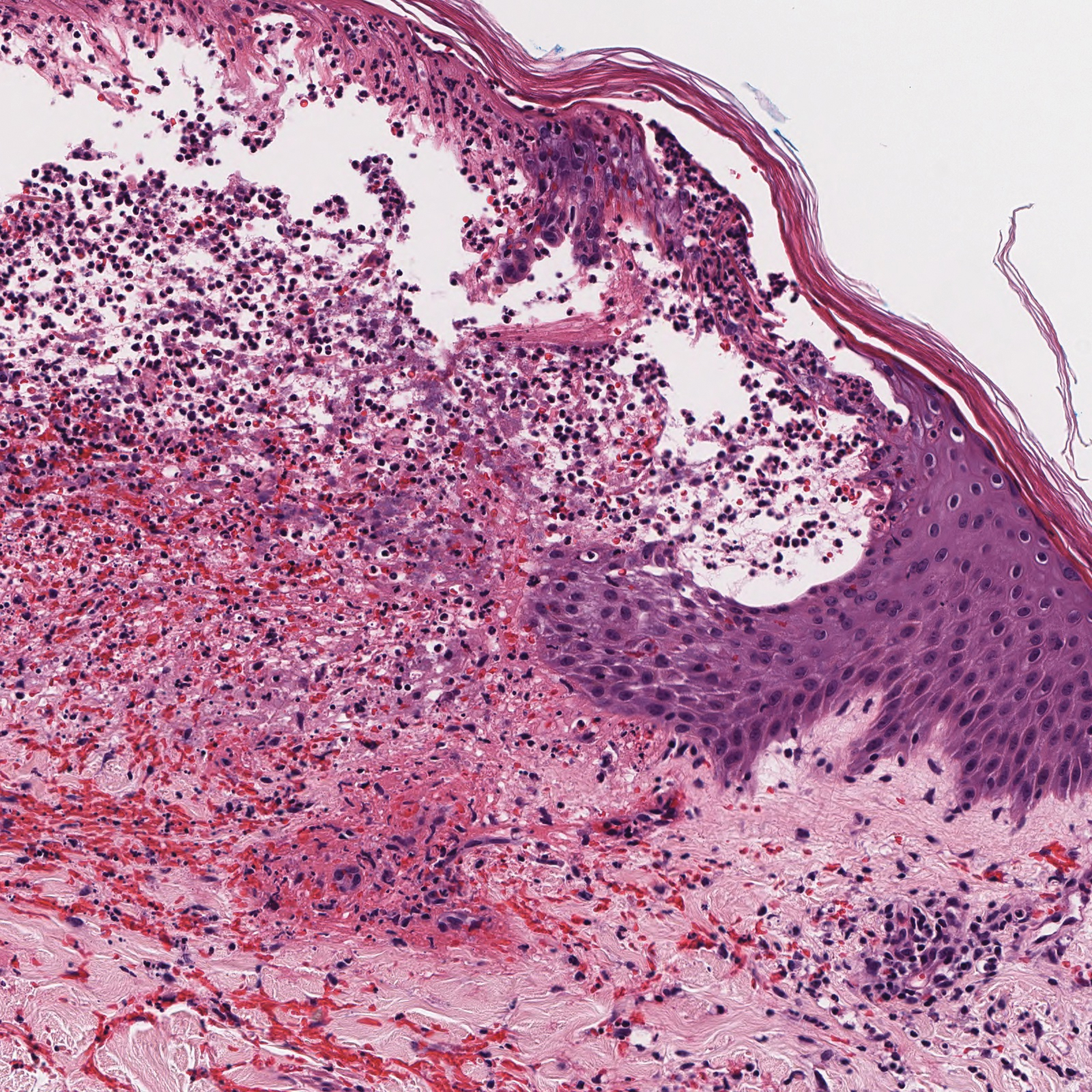
A 30-year-old woman with a medical history of uncontrolled type 2 diabetes mellitus and morbid obesity presented to the dermatology clinic with a painful blistering rash on the lower extremities with scattered red-purple papules of 1 week's duration. The rash began on the left dorsal foot. Physical examination showed nonblanching, 2- to 4-mm, violaceous papules with numerous vesiculopustular bullae on the lower extremities from the dorsal feet to the proximal knee. A shave biopsy with hematoxylin and eosin stain and a punch biopsy for direct immunofluorescence were performed.
Vulvar Syringoma
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.

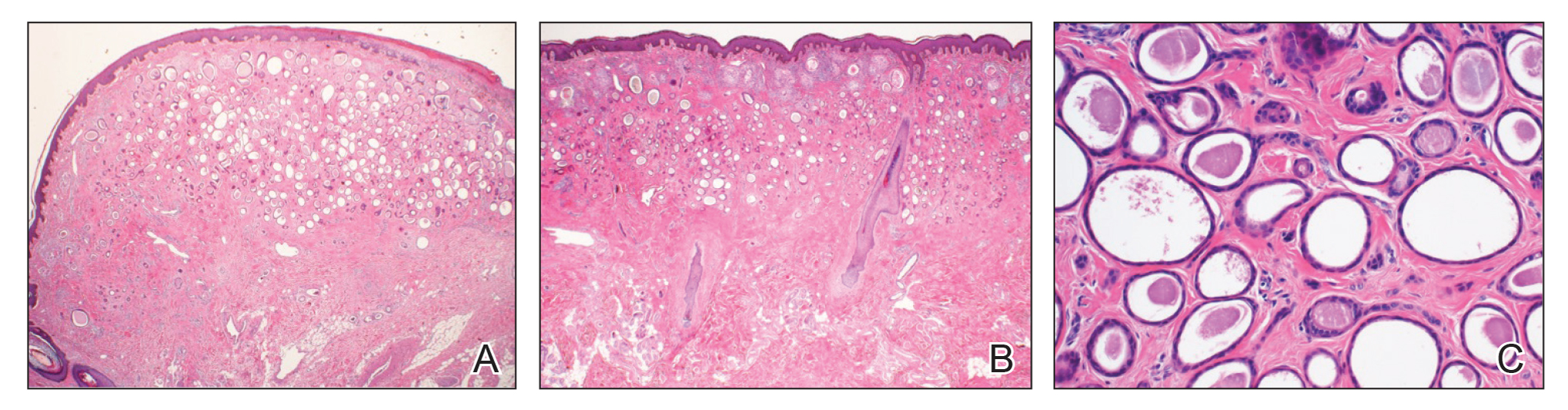
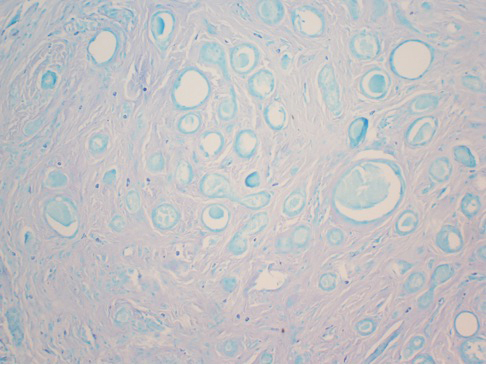
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
To the Editor:
Syringomas are common benign tumors of the eccrine sweat glands that usually manifest clinically as multiple flesh-colored papules. They are most commonly seen on the face, neck, and chest of adolescent girls. Syringomas may appear at any site of the body but are rare in the vulva. We present a case of a 51-year-old woman who was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of a tumor carrying a differential diagnosis of vulvar syringoma vs microcystic adnexal carcinoma (MAC).
A 51-year-old woman presented to dermatology (G.G.) and was referred to the Division of Gynecologic Oncology at the University of Alabama at Birmingham for further management of possible vulvar syringoma vs MAC. The patient previously had been evaluated at an outside community practice due to dyspareunia, vulvar discomfort, and vulvar irregularities of 1 month’s duration. At that time, a small biopsy was performed, and the histologic differential diagnosis included syringoma vs an adnexal carcinoma. Consequently, she was referred to gynecologic oncology for further management.
Pelvic examination revealed multilobular nodular areas overlying the clitoral hood that extended down to the labia majora. The nodular processes did not involve the clitoris, labia minora, or perineum. A mobile isolated lymph node measuring 2.0×1.0 cm in the right inguinal area also was noted. The patient’s clinical history was notable for right breast carcinoma treated with a right mastectomy with axillary lymph node dissection that showed metastatic disease. She also underwent adjuvant chemotherapy with paclitaxel and doxorubicin for breast carcinoma.
After discussing the diagnostic differential and treatment options, the patient elected to undergo a bilateral partial radical vulvectomy with reconstruction and resection of the right inguinal lymph node. Gross examination of the vulvectomy specimen showed multiple flesh-colored papules (Figure 1). Histologic examination revealed a neoplasm with sweat gland differentiation that was broad and poorly circumscribed but confined to the dermis (Figures 2A and 2B). The neoplasm was composed of epithelial cells that formed ductlike structures, lined by 2 layers of cuboidal epithelium within a fibrous stroma (Figure 2C). A toluidine blue special stain was performed and demonstrated an increased amount of mast cells in the tissue (Figure 3). Immunohistochemical stains for gross cystic disease fluid protein, estrogen receptor (ER), and progesterone receptor (PR) were negative in the tumor cells. The lack of cytologic atypia, perineural invasion, and deep infiltration into the subcutis favored a syringoma. One month later, the case was presented at the Tumor Board Conference at the University of Alabama at Birmingham where a final diagnosis of vulvar syringoma was agreed upon and discussed with the patient. At that time, no recurrence was evident and follow-up was recommended.
Syringomas are benign tumors of the sweat glands that are fairly common and appear to have a predilection for women. Although most of the literature classifies them as eccrine neoplasms, the term syringoma can be used to describe neoplasms of either apocrine or eccrine lineage.1 To rule out an apocrine lineage of the tumor in our patient, we performed immunohistochemistry for gross cystic disease fluid protein, a marker of apocrine differentiation. This stain highlighted normal apocrine glands that were not involved in the tumor proliferation.
Syringomas may occur at any site on the body but are prone to occur on the periorbital area, especially the eyelids.1 Some of the atypical locations for a syringoma include the anterior neck, chest, abdomen, genitals, axillae, groin, and buttocks.2 Vulvar syringomas were first reported by Carneiro3 in 1971 as usually affecting adolescent girls and middle-aged women. There have been approximately 40 reported cases affecting women aged 8 to 78 years.4,5 Vulvar syringomas classically appear as firm or soft, flesh-colored to transparent, papular lesions. The 2 other clinical variants are miliumlike, whitish, cystic papules as well as lichenoid papules.6 Pérez-Bustillo et al5 reported a case of the lichenoid papule variant on the labia majora of a 78-year-old woman who presented with intermittent vulvar pruritus of 4 years’ duration. Due to this patient’s 9-year history of urinary incontinence, the lesions had been misdiagnosed as irritant dermatitis and associated lichen simplex chronicus (LSC). This case is a reminder to consider vulvar syringoma in patients with LSC who respond poorly to oral antihistamines and topical steroids.5 Rarely, multiple clinical variants may coexist. In a case reported by Dereli et al,7 a 19-year-old woman presented with concurrent classical and miliumlike forms of vulvar syringoma.
Vulvar syringomas usually present as multiple lesions involving both sides of the labia majora; however, Blasdale and McLelland8 reported a single isolated syringoma of the vulva on the anterior right labia minora that measured 1.0×0.5 cm, leading the lesion to be described as a giant syringoma.
Vulvar syringomas usually are asymptomatic and noticed during routine gynecologic examination. Therefore, it is believed that they likely are underdiagnosed.5 When symptomatic, they commonly present with constant9 or intermittent5 pruritus, which may intensify during menstruation, pregnancy, and summertime.6,10-12 Gerdsen et al10 documented a 27-year-old woman who presented with a 2-year history of pruritic vulvar skin lesions that became exacerbated during menstruation, which raised the possibility of cyclical hormonal changes being responsible for periodic exacerbation of vulvar pruritus during menstruation. In addition, patients may experience an increase in size and number of the lesions during pregnancy. Bal et al11 reported a 24-year-old primigravida with vulvar papular lesions that intensified during pregnancy. She had experienced intermittent vulvar pruritus for 12 years but had no change in symptoms during menstruation.11 Few studies have attempted to evaluate the presence of ER and PR in the syringomas. A study of 9 nonvulvar syringomas by Wallace and Smoller13 showed ER positivity in 1 case and PR positivity in 8 cases, lending support to the hormonal theory; however, in another case series of 15 vulvar syringomas, Huang et al6 failed to show ER and PR expression by immunohistochemical staining. A case report published 3 years earlier documented the first case of PR positivity on a vulvar syringoma.14 Our patient also was negative for ER and PR, which suggested that hormonal status is important in some but not all syringomas.
Patients with vulgar syringomas also might have coexisting extragenital syringomas in the neck,4 eyelids,6,7,10 and periorbital area,6 and thorough examination of the body is essential. If an extragenital syringoma is diagnosed, a vulvar syringoma should be considered, especially when the patient presents with unexplained genital symptoms. Although no proven hereditary transmission pattern has been established, family history of syringomas has been established in several cases.15 In a case series reported by Huang et al,6 4 of 18 patients reported a family history of periorbital syringomas. In our case, the patient did not report a family history of syringomas.
The differential diagnosis of vulvar lesions with pruritus is broad and includes Fox-Fordyce disease, lichen planus, LSC, epidermal cysts, senile angiomas, dystrophic calcinosis, xanthomas, steatocytomas, soft fibromas, condyloma acuminatum, and candidiasis. Vulvar syringomas might have a nonspecific appearance, and histologic examination is essential to confirm the diagnosis and rule out any malignant process such as MAC, vulvar intraepithelial neoplasia, extramammary Paget disease, or other glandular neoplasms of the vulva.
Microcystic adnexal carcinoma was first reported in 1982 by Goldstein et al16 as a locally aggressive neoplasm that can be confused with benign adnexal neoplasms, particularly desmoplastic trichoepithelioma, trichoadenoma, and syringoma. Microcystic adnexal carcinomas present as slow-growing, flesh-colored papules that may resemble syringomas and appear in similar body sites. Histologic examination is essential to differentiate between these two entities. Syringomas are tumors confined to the dermis and are composed of multiple small ducts lined by 2 layers of cuboidal epithelium within a dense fibrous stroma. Unlike syringomas, MACs usually infiltrate diffusely into the dermis and subcutis and may extend into the underlying muscle. Although bland cytologic features predominate, perineural invasion frequently is present in MACs. A potential pitfall of misdiagnosis can be caused by a superficial biopsy that may reveal benign histologic appearance, particularly in the upper level of the tumor where it may be confused with a syringoma or a benign follicular neoplasm.17
The initial biopsy performed on our patient was possibly not deep enough to render an unequivocal diagnosis and therefore bilateral partial radical vulvectomy was considered. After surgery, histologic examination of the resection specimen revealed a poorly circumscribed tumor confined to the dermis. The tumor was broad and the lack of deep infiltration into the subcutis and perineural invasion favored a syringoma (Figures 2A and 2B). These findings were consistent with case reports that documented syringomas as being more wide than deep on microscopic examination, whereas the opposite pertained to MAC.18 Cases of plaque-type syringomas that initially were misdiagnosed as MACs also have been reported.19 Because misdiagnosis may affect the treatment plan and potentially result in unnecessary surgery, caution should be taken when differentiating between these two entities. When a definitive diagnosis cannot be rendered on a superficial biopsy, a recommendation should be made for a deeper biopsy sampling the subcutis.
For the majority of the patients with vulvar syringomas, treatment is seldom required due to their asymptomatic nature; however, patients who present with symptoms usually report pruritus of variable intensities and patterns. A standardized treatment does not exist for vulvar syringomas, and oral or topical treatment might be used as an initial approach. Commonly prescribed medications with variable results include topical corticosteroids, oral antihistamines, and topical retinoids. In a case reported by Iwao et al,20 vulvar syringomas were successfully treated with tranilast, which has anti-inflammatory and immunomodulatory effects. This medication could have a possible dual action—inhibiting the release of chemical mediators from the mast cells and inhibiting the release of IL-1β from the eccrine duct, which could suppress the proliferation of stromal connective tissue. Our case was stained with toluidine blue and showed an increased number of mast cells in the tissue (Figure 3). Patients who are unresponsive to tranilast or have extensive disease resulting in cosmetic disfigurement might benefit from more invasive treatment methods including a variety of lasers, cryotherapy, electrosurgery, and excision. Excisions should include the entire tumor to avoid recurrence. In a case reported by Garman and Metry,21 the lesions were surgically excised using small 2- to 3-mm punches; however, several weeks later the lesions recurred. Our patient presented with a 1-month evolution of dyspareunia, vulvar discomfort, and vulvar irregularities that were probably not treated with oral or topical medications before being referred for surgery.
We report a case of a vulvar syringoma that presented diagnostic challenges in the initial biopsy, which prevented the exclusion of an MAC. After partial radical vulvectomy, histologic examination was more definitive, showing lack of deep infiltration into the subcutis or perineural invasion that are commonly seen in MAC. This case is an example of a notable pitfall in the diagnosis of vulvar syringoma on a limited biopsy leading to overtreatment. Raising awareness of this entity is the only modality to prevent misdiagnosis. We encourage reporting of further cases of syringomas, particularly those with atypical locations or patterns that may cause diagnostic problems.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
- Bolognia JL, Jorizzo JL, Rapini RP. Dermatology. 2nd ed. Spain: Mosby Elsevier; 2008.
- Weedon D. Skin Pathology. 3rd ed. China: Churchill Livingstone Elsevier; 2010.
- Carneiro SJ, Gardner HL, Knox JM. Syringoma of the vulva. Arch Dermatol. 1971;103:494-496.
- Trager JD, Silvers J, Reed JA, et al. Neck and vulvar papules in an 8-year-old girl. Arch Dermatol. 1999;135:203, 206.
- Pérez-Bustillo A, Ruiz-González I, Delgado S, et al. Vulvar syringoma: a rare cause of vulvar pruritus. Actas Dermo-Sifiliográficas. 2008;99:580-581.
- Huang YH, Chuang YH, Kuo TT, et al. Vulvar syringoma: a clinicopathologic and immunohistologic study of 18 patients and results of treatment. J Am Acad Dermatol. 2003;48:735-739.
- Dereli T, Turk BG, Kazandi AC. Syringomas of the vulva. Int J Gynaecol Obstet. 2007;99:65-66.
- Blasdale C, McLelland J. Solitary giant vulval syringoma. Br J Dermatol. 1999;141:374-375.
- Kavala M, Can B, Zindanci I, et al. Vulvar pruritus caused by syringoma of the vulva. Int J Dermatol. 2008;47:831-832.
- Gerdsen R, Wenzel J, Uerlich M, et al. Periodic genital pruritus caused by syringoma of the vulva. Acta Obstet Gynecol Scand. 2002;81:369-370.
- Bal N, Aslan E, Kayaselcuk F, et al. Vulvar syringoma aggravated by pregnancy. Pathol Oncol Res. 2003;9:196-197.
- Turan C, Ugur M, Kutluay L, et al. Vulvar syringoma exacerbated during pregnancy. Eur J Obstet Gynecol Reprod Biol. 1996;64:141-142.
- Wallace ML, Smoller BR. Progesterone receptor positivity supports hormonal control of syringomas. J Cutan Pathol. 1995;22:442-445.
- Yorganci A, Kale A, Dunder I, et al. Vulvar syringoma showing progesterone receptor positivity. BJOG. 2000;107:292-294.
- Draznin M. Hereditary syringomas: a case report. Dermatol Online J. 2004;10:19.
- Goldstein DJ, Barr RJ, Santa Cruz DJ. Microcystic adnexal carcinoma: a distinct clinicopathologic entity. Cancer. 1982;50:566-572.
- Hamsch C, Hartschuh W. Microcystic adnexal carcinoma - aggressive infiltrative tumor often with innocent clinical appearance. J Dtsch Dermatol Ges. 2010;8:275-278.
- Henner MS, Shapiro PE, Ritter JH, et al. Solitary syringoma. report of five cases and clinicopathologic comparison with microcystic adnexal carcinoma of the skin. Am J Dermatopathol. 1995;17:465-470.
- Suwattee P, McClelland MC, Huiras EE, et al. Plaque-type syringoma: two cases misdiagnosed as microcystic adnexal carcinoma. J Cutan Pathol. 2008;35:570-574.
- Iwao F, Onozuka T, Kawashima T. Vulval syringoma successfully treated with tranilast. Br J Dermatol. 2005;153:1228-1230.
- Garman M, Metry D. Vulvar syringomas in a 9-year-old child with review of the literature. Pediatr Dermatol. 2006;23:369-372.
Practice Points
- Ensure adequate depth of biopsy to assist in the histologic diagnosis of syringoma vs microcystic adnexal carcinoma.
- Vulvar syringomas also may contribute to notable pruritus and ultimately be the underlying etiology for secondary skin changes leading to a lichen simplex chronicus–like phenotype.
