User login
Implementing the Exercise Guidelines for Cancer Survivors
Kathleen Y. Wolin, ScD, Anna L. Schwartz, PhD, Charles E. Matthews, PhD, FACSM, Kerry S. Courneya, PhD, Kathryn H. Schmitz, PhD

Abstract
In 2009, the American College of Sports Medicine convened an expert roundtable to issue guidelines on exercise for cancer survivors. This multidisciplinary group evaluated the strength of the evidence for the safety and benefits of exercise as a therapeutic intervention for survivors. The panel concluded that exercise is safe and offers myriad benefits for survivors including improvements in physical function, strength, fatigue, quality of life, and possibly recurrence and survival. Recommendations for situations in which deviations from the US Physical Activity Guidelines for Americans are appropriate were provided. Here, we outline a process for implementing the guidelines in clinical practice and provide recommendations for how the oncology care provider can interface with the exercise and physical therapy community.
*For a PDF of the full article and accompanying commentary by Nicole Stout, click on the links to the left of this introduction.
Kathleen Y. Wolin, ScD, Anna L. Schwartz, PhD, Charles E. Matthews, PhD, FACSM, Kerry S. Courneya, PhD, Kathryn H. Schmitz, PhD
Abstract
In 2009, the American College of Sports Medicine convened an expert roundtable to issue guidelines on exercise for cancer survivors. This multidisciplinary group evaluated the strength of the evidence for the safety and benefits of exercise as a therapeutic intervention for survivors. The panel concluded that exercise is safe and offers myriad benefits for survivors including improvements in physical function, strength, fatigue, quality of life, and possibly recurrence and survival. Recommendations for situations in which deviations from the US Physical Activity Guidelines for Americans are appropriate were provided. Here, we outline a process for implementing the guidelines in clinical practice and provide recommendations for how the oncology care provider can interface with the exercise and physical therapy community.
*For a PDF of the full article and accompanying commentary by Nicole Stout, click on the links to the left of this introduction.
Kathleen Y. Wolin, ScD, Anna L. Schwartz, PhD, Charles E. Matthews, PhD, FACSM, Kerry S. Courneya, PhD, Kathryn H. Schmitz, PhD
Abstract
In 2009, the American College of Sports Medicine convened an expert roundtable to issue guidelines on exercise for cancer survivors. This multidisciplinary group evaluated the strength of the evidence for the safety and benefits of exercise as a therapeutic intervention for survivors. The panel concluded that exercise is safe and offers myriad benefits for survivors including improvements in physical function, strength, fatigue, quality of life, and possibly recurrence and survival. Recommendations for situations in which deviations from the US Physical Activity Guidelines for Americans are appropriate were provided. Here, we outline a process for implementing the guidelines in clinical practice and provide recommendations for how the oncology care provider can interface with the exercise and physical therapy community.
*For a PDF of the full article and accompanying commentary by Nicole Stout, click on the links to the left of this introduction.
Using the ankle-brachial index to diagnose peripheral artery disease and assess cardiovascular risk
In this article, we seek to convince you to measure the ankle-brachial index for any patient you suspect may have peripheral artery disease. This would include patients who are elderly, who smoke, or who have diabetes, regardless of whether or not they have symptoms. The ankle-brachial index is a simple test that involves taking the blood pressure in all four limbs using a hand-held Doppler device and then dividing the leg systolic pressure by the arm systolic pressure.
This simple test is both sensitive and specific for peripheral artery disease. It also gives a good assessment of cardiovascular risk. The downside: you or a member of your staff spends a few minutes doing it. Also, for patients without leg symptoms or abnormal findings on physical examination, you may not be paid for doing it.
Despite these limitations, the ankle-brachial index is a powerful clinical tool that deserves to be performed more often in primary care.
PERIPHERAL ARTERY DISEASE IS COMMON AND SERIOUS
Peripheral artery disease is important to detect, as it is common, it has serious consequences, and effective treatments are available. However, many patients with the disease do not have typical symptoms.
Peripheral artery disease affects up to 29% of people over age 70, depending on the population sampled.1,2 Its classic symptom is intermittent claudication, ie, leg pain with walking that improves with rest. However, most patients do not have intermittent claudication; they have atypical leg symptoms or no symptoms at all.2,3 While the risk factors for peripheral artery disease are similar to those for coronary artery disease, the factors most strongly associated with peripheral artery disease are older age, tobacco smoking, and diabetes mellitus.4 Blacks are twice as likely to have it compared with whites, even after adjusting for other cardiovascular risk factors.5
Untreated peripheral artery disease may have serious consequences, such as amputation, impaired functional capacity, poor quality of life, and depression.3,6,7 In addition, it is a strong marker of atherosclerotic burden and cardiovascular risk and has been recognized as a coronary risk equivalent. Patients with peripheral artery disease are at higher risk of death, myocardial infarction, stroke, and hospitalization, with event rates as high as 21% per year.8
Fortunately, simple therapies have been shown to prevent adverse cardiovascular events in peripheral artery disease, including antiplatelet drugs, statins, and angiotensin-converting enzyme inhibitors.9–11
THE ANKLE-BRACHIAL INDEX IS SENSITIVE AND SPECIFIC
The evaluation for possible peripheral artery disease should begin with the medical history, a cardiovascular review of systems, and a focused physical examination in which one should:
- Measure the blood pressure in both arms to assess for occult subclavian stenosis
- Auscultate for bruits over the carotid, abdominal, and femoral arteries
- Palpate the pulses in the lower extremities and the abdominal aorta
- Inspect the bare legs and feet for thinning of the skin, hair loss, thickening of the nails (which are nonspecific signs), and ulceration.
However, the physical examination has limited sensitivity and specificity for diagnosing peripheral artery disease. In general, the most reliable finding is the absence of a palpable posterior tibial artery pulse, which has been reported to have a specificity of 71% and a sensitivity of 91% for peripheral artery disease.12
The ankle-brachial index is the first-line test for both screening for peripheral artery disease and for diagnosing it. It is inexpensive and noninvasive to obtain and has a high sensitivity (79% to 95%) and specificity (95% to 96%) compared with angiography as the gold standard.13–18 It can be measured easily in the office, and every practitioner who cares for patients at risk of cardiovascular disease can be trained to measure it competently.
HOW TO MEASURE THE ANKLE-BRACHIAL INDEX
The ankle-brachial index is the ratio of the systolic pressure in the ankle to the systolic pressure in the arm. In healthy people, this ratio is typically greater than 1.0 or 1.1.
You can measure the ankle-brachial index in the office with a blood pressure cuff, sphygmomanometer, and handheld Doppler device. Alternatively, it can be measured in a noninvasive vascular laboratory as part of a more detailed examination that allows for assessment of blood pressures and waveforms (Doppler or pulse-volume recordings) at multiple segments along the limb. These more detailed vascular studies are generally reserved for patients with confirmed peripheral artery disease to locate the level and extent of blockage or for patients in whom lower-extremity revascularization is contemplated.
The use of a stethoscope to measure blood pressures for the ankle-brachial index has been studied in a few small series,19,20 but is thought to be less accurate than Doppler, especially in the setting of significant arterial occlusive disease. Because of this, it is recommended and assumed that a Doppler device be used to measure all blood pressures for the ankle-brachial index.
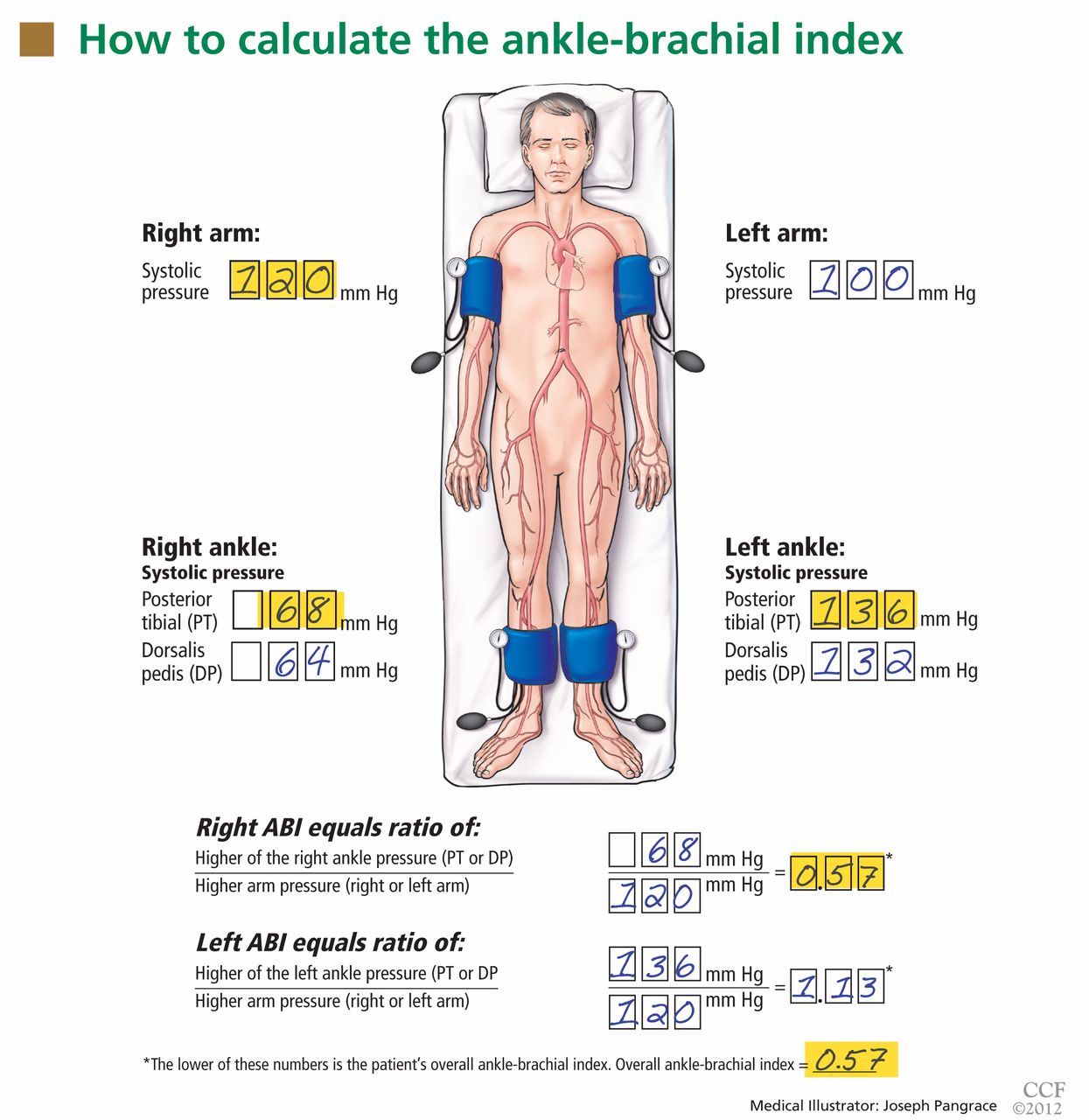
After the patient has been resting quietly for 5 to 10 minutes in the supine position, the systolic blood pressure is measured in both arms and in both ankles in the dorsalis pedis and posterior tibial arteries (Figure 1). The blood pressure cuff is placed about 1 inch above the antecubital fossa for the brachial pressure and about 2 inches above the medial malleolus for the ankle pressures. A clear arterial pulse signal should be heard using the Doppler probe before inflating the blood pressure cuff. The cuff is then inflated to at least 20 mm Hg above the point where the arterial Doppler sounds disappear and then slowly deflated until the Doppler sounds reappear. The blood pressure at which the Doppler signal of the arterial pulse reappears is the systolic pressure for that vessel.
The ankle-brachial index is calculated by dividing the higher of the two ankle systolic blood pressures in each leg by the higher of the two brachial systolic blood pressures. The higher of the two brachial pressures is used as the denominator to account for the possibility of subclavian artery stenosis, which can decrease the blood pressure in the upper extremity. The ankle-brachial index is calculated for each leg, and the lower value is the patient’s overall ankle-brachial index. An abnormal value in either leg indicates peripheral artery disease.
While other ways of calculating the ankle-brachial index have been proposed, such as averaging the two pressures at each ankle or reporting the lower of the two ankle pressures, these methods are not standard for use in clinical practice.
Similarly, the use of oscillometric blood pressure devices has been proposed, which would eliminate the need for a Doppler device and personnel trained in its use. However, results of validation studies of oscillometric measurement of the ankle-brachial index have been inconsistent, likely because the devices were designed for measuring blood pressure in nonobstructed arms, not the legs, and especially not diseased legs.21–25 In general, oscillometric devices tend to overestimate ankle pressure, giving a falsely high ankle-brachial index in patients with moderate to severe peripheral artery disease.21 Their utility in screening for peripheral artery disease has not been evaluated in broad, population-based studies. Efforts to develop and validate new oscillometric devices for diagnosing peripheral artery disease are ongoing.
INTERPRETING THE ANKLE-BRACHIAL INDEX
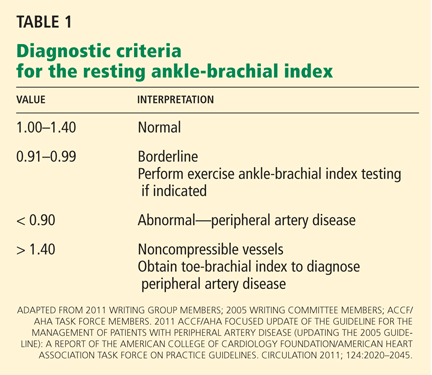
Diagnostic criteria for the ankle-brachial index were standardized in 2011 (Table 1).26 Most healthy adults have a value greater than 1.0. A value of less than 0.91 is consistent with significant peripheral artery disease, and a value lower than 0.40 at rest generally indicates severe disease. A value between 0.91 and 0.99 is borderline abnormal and does not rule out peripheral artery disease. A value greater than 1.40 reflects noncompressibility of the leg arteries and is not diagnostic (see below).
The ankle-brachial index after exercise. In patients strongly suspected of having peripheral artery disease but who have a normal ankle-brachial index at rest, and especially if the resting value is borderline (ie, 0.91–0.99), the measurement should be repeated after exercise, the better to detect “mild” peripheral artery disease.15 With exercise, increased flow across a fixed stenosis leads to a significant fall in ankle pressure and a lower ankle-brachial index. In one study,27 the ankle-brachial index fell below 0.9 after exercise in 31% of outpatients with symptoms who had initially tested normal.
The exercise is optimally done on a motorized treadmill set at an incline. A number of exercise protocols are in use; at our institution, we use a fixed workload protocol. The ankle-brachial index and ankle pulse-volume recordings are recorded on both sides at rest, after which the patient generally walks for 5 minutes at a 12% grade at 2.0 mph or until symptoms force the patient to stop. The advantage of treadmill testing is the ability to assess functional capacity by measuring the time to the onset of pain and the total walking time.
Alternatively, active pedal plantar flexion maneuvers (heel raises) or corridor walking to the point at which limiting symptoms occur can be done if a treadmill is not available, though this is not the favored approach and does not qualify as formal exercise testing for reimbursement purposes. The patient is asked to do heel raises as high and as fast as possible for 30 seconds or until limiting pain symptoms occur. Results with this maneuver have been shown to correlate well with those of treadmill exercise testing.28
Immediately after any exercise maneuver, arm and ankle pressures are remeasured and bilateral ankle-brachial indices are recalculated. A fall in ankle pressure or the ankle-brachial index after exercise (generally, a fall of more than 20%) supports the diagnosis of peripheral artery disease. If the patient develops leg symptoms during exercise while his or her ankle-brachial index falls significantly, this also supports the vasculogenic nature of the leg symptoms.
An ankle-brachial index greater than 1.40 means that the pedal arteries are stiff and cannot be compressed by the blood pressure cuff. This is considered abnormal, though not necessarily diagnostic of peripheral artery disease. Noncompressible leg arteries are common among patients with long-standing diabetes mellitus or end-stage renal disease, and also can be found in obese patients.
Because toe arteries are usually compressible even when the pedal arteries are not, a toe-brachial index can be obtained to confirm the diagnosis of peripheral artery disease in these cases. This is calculated by measuring the blood pressure in the great toe using a small digital blood pressure cuff and a Doppler probe or a plethysmographic flow sensor. The toe-brachial index is calculated by dividing the toe blood pressure by the higher of the two brachial artery pressures; a value of 0.7 or less generally indicates peripheral artery disease.
WHAT SHOULD BE DONE WITH AN ABNORMAL RESULT?
An abnormal ankle-brachial index establishes the diagnosis of peripheral artery disease, and in many cases no additional diagnostic testing is necessary.
Care of patients with peripheral artery disease has three elements:
- Cardiovascular risk factor assessment and reduction to prevent myocardial infarction, stroke, and death
- Assessment and treatment of leg symptoms to improve function and quality of life
- Foot care to prevent ulcers and amputation.
Risk factor reduction. Because they have a markedly greater risk of cardiovascular disease and death, all patients with peripheral artery disease should undergo aggressive cardiovascular risk factor modification,26,29 including:
- Antiplatelet therapy in the form of aspirin 75–325 mg daily or clopidogrel 75 mg daily as an alternative to aspirin
- Counseling and therapy for immediate smoking cessation if the patient smokes
- Treatment of hypertension to Seventh Joint National Committee goals30
- Treatment of lipids to Adult Treatment Panel III goals31 (generally to a goal low-density lipoprotein cholesterol of less than 100 mg/dL, and less than 70 mg/dL if possible)
- Treatment of diabetes to a goal hemoglobin A1c of less than 7% (in the absence of contraindications).32
Exercise and anticlaudication medication. Patients with an abnormal ankle-brachial index and intermittent claudication may benefit from a supervised exercise program, a trial of drug therapy for claudication, or both. All patients with peripheral artery disease, regardless of symptoms, should be advised to incorporate aerobic exercise (ideally, walking) into their daily routine.
Cilostazol (Pletal), a phosphodiesterase inhibitor, has been given a class IA recommendation in the American College of Cardiology/American Heart Association guidelines for the treatment of intermittent claudication. The dose is generally 100 mg by mouth twice daily.29
Revascularization. Patients with an abnormal ankle-brachial index and lifestyle-limiting claudication that has failed to improve with medical therapy or a course of supervised exercise training should be referred to a vascular specialist for evaluation for revascularization (endovascular therapy or surgical bypass). 29 Endovascular therapy is particularly attractive for patients with claudication and evidence of aortoiliac disease (suspected in patients with gluteal or thigh claudication, diminution of the femoral pulse, or a bruit over the femoral artery on examination and confirmed by noninvasive vascular laboratory testing).
Patients who have ischemic pain at rest, gangrene, or a nonhealing lower-extremity wound that has been present for at least 2 weeks should be referred for revascularization on an urgent basis, given the risk of impending limb loss associated with critical limb ischemia.
A detailed review of the medical, endovascular, and surgical management of peripheral artery disease can be found in a supplement to the Cleveland Clinic Journal of Medicine published in 200633 and in comprehensive multi-society guidelines.26,29
THE ANKLE-BRACHIAL INDEX AS A MARKER OF RISK
Low values: Peripheral artery disease
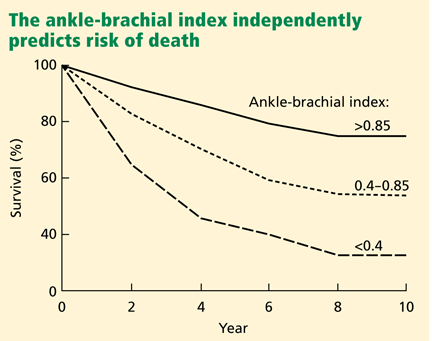
Peripheral artery disease, as diagnosed by a low ankle-brachial index, confers an excess risk of death from all causes in a graded fashion: ie, the more severe the disease, the lower the survival rate (Figure 2).34 Because peripheral artery disease is a sign of systemic atherosclerosis and one-third to one-half of patients with peripheral artery disease have evidence of cerebrovascular or coronary artery disease,35–37 peripheral artery disease also confers a higher risk of cardiovascular death.
The Edinburgh Artery Study,38 a prospective cohort study of 1,592 randomly selected patients age 55 to 74 years, demonstrated the relationship between a low ankle-brachial index and an increased risk of cardiovascular death. Over 5 years of follow-up, compared with patients with a normal ankle-brachial index, the relative risk of cardiovascular death in symptomatic patients with a value of 0.9 or lower was 2.67 (95% confidence interval [CI] 1.34–5.29). The relative risk in patients with asymptomatic disease was between 1.74 (95% CI 1.09–2.76) and 2.08 (95% CI 1.13–3.83), depending on the level of ankle-brachial index decrement and ankle blood pressure response to hyperemia.
(Reactive hyperemia is an alternative to exercise testing. It is performed by inflating a blood pressure cuff at the thigh above the systolic pressure for 3 to 5 minutes or until the patient can no longer tolerate the inflation. Blood pressures at the ankle are remeasured after cuff release.)
Several other epidemiologic studies have established the association between low ankle-brachial index and the risk of cardiovascular death.
Heald et al39 performed a meta-analysis of 44,590 patients in 11 epidemiologic studies and found that, after adjustment for age, sex, conventional cardiovascular risk factors, and prevalent cardiovascular disease, an ankle-brachial index lower than 0.9 conferred a higher risk of:
- All-cause mortality (pooled risk ratio [RR] 1.60, 95% CI 1.32–1.95)
- Cardiovascular mortality (pooled RR 1.96, 95% CI 1.46–2.64)
- Coronary heart disease (pooled RR 1.45, 95% CI 1.08–1.93)
- Stroke (pooled RR 1.35, 95% CI 1.10–1.65).
Fowkes et al,40 in a meta-analysis of 16 population cohort studies including 48,294 patients over 480,325 person-years of follow-up, found that a low ankle-brachial index predicted cardiovascular events and death even after adjusting for the Framingham risk score, Hazard ratios for cardiovascular death were:
- 2.92 (95% CI 2.31–3.70) in men
- 2.97 (95% CI 2.02–4.35) in women.
Hazard ratios for death from any cause were:
- 2.34 (95% CI 1.97–2.78) in men
- 2.35 (95% CI 1.76–3.13) in women.
Adding the ankle-brachial index to the Framingham risk score resulted in reclassification of risk category in approximately 19% of men and 36% of women.40
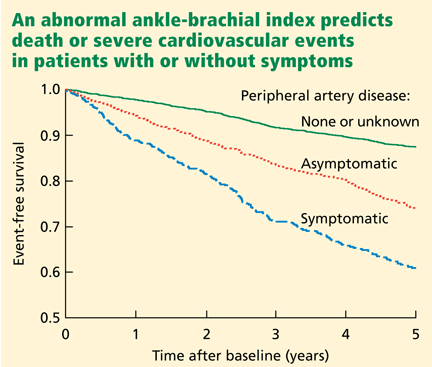
The German Epidemiological Trial on Ankle Brachial Index (getABI) screened 6,880 patients 65 years of age and found an abnormal ankle-brachial index in 20.9% of them.41 In more than 5 years of follow-up, a value of less than 0.90 was associated with a higher rate of cardiovascular events and death from any cause in patients with both symptomatic and asymptomatic peripheral artery disease (Figure 3).41
In addition, the lower the ankle-brachial index, the greater the rate of death or severe cardiovascular events. An index between 0.7 and 0.9 was associated with a statistically significant twofold increase (adjusted hazard ratio 2.03), and a value lower than 0.5 was associated with a nearly fivefold increase (hazard ratio 4.65) in the risk of events compared with the group of patients with normal values.41
Abnormal results after exercise
Exercise testing may increase the sensitivity of the ankle-brachial index to detect peripheral artery disease in patients with normal resting values and especially in patients with borderline values. As such, abnormal exercise values have also been associated with an increased risk of death due to any cause and of cardiovascular death.
In a prospective cohort study of 3,209 patients with suspected or known peripheral artery disease referred to a vascular surgery clinic in the Netherlands, patients with lower postexercise values had a higher rate of overall and cardiac death (hazard ratio per 10% lower value 1.16 [95% CI 1.13–1.18] and 1.10 [95% CI 1.09–1.13], respectively).42
Sheikh et al43 reported similar findings in patients with normal resting ankle-brachial indices at Cleveland Clinic. In this study, an abnormal postexercise ankle-brachial index (defined as < 0.85) was associated with a hazard ratio of 1.67 for all-cause mortality compared with a normal postexercise value among individuals with no history of cardiovascular events.
High values: Noncompressible vessels
While the relationship between low values and increased mortality and cardiovascular risk is well accepted, there have been conflicting reports regarding high values (> 1.4) and adverse outcomes.44,45
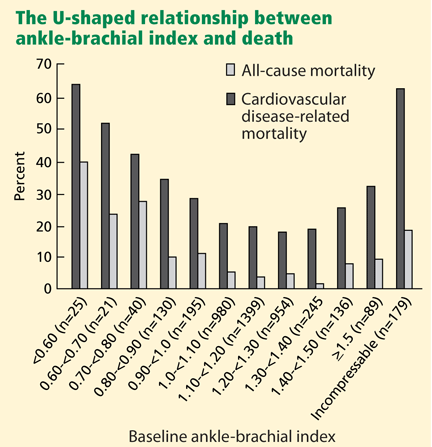
The Strong Heart Study44 was a population-based study in 4,393 Native Americans followed for more than 8 years for the rate of all-cause and cardiovascular mortality. Most (n = 3,773) of the cohort had a normal ankle-brachial index (≥ 0.9 and ≤ 1.4); 4.9% (n = 216) had a low value (< 0.9); and 9.2% (n = 404) had a high value (> 1.4 or noncompressible). Relative risk ratios for all-cause mortality were 1.69 (95% CI 1.34–2.14) for low values and 1.77 (95% CI 1.48–2.13) for high values compared with those with normal values. Low and high ankle-brachial indices also conferred a risk of cardiovascular death, with relative risk ratios of 2.52 (95% CI 1.74–3.64) and 2.09 (95% CI 1.49–2.94), respectively. There was a U-shaped relationship between the ankle-brachial index and the mortality rate (Figure 4).44
The Atherosclerosis Risk in Communities (ARIC) study45 had different findings. In 14,777 participants followed for a mean of 12.2 years, the cardiovascular disease event rates of patients whose ankle-brachial index-was categorized as high (> 1.3, > 1.4, or > 1.5) were similar to those of patients with a normal value (between 0.9 and 1.3).
Differences in event rates between the two studies may be due to a higher prevalence of values greater than 1.4 in the Strong Heart Study cohort as well as to a higher prevalence of concomitant risk factors (diabetes, older age, hypertension, lipid abnormality) in the high ankle-brachial index group in the Strong Heart Study compared with the ARIC study.
DIFFERING RECOMMENDATIONS
The ankle-brachial index can be used to screen for asymptomatic peripheral artery disease. It can also be used to confirm the diagnosis in patients with symptoms such as intermittent claudication, ischemic pain at rest, or lower extremity ulcers or in patients with signs such as abnormal pulses, bruits, or lower-extremity skin changes. It is also used to reassess the severity of known peripheral artery disease and as a part of a routine surveillance program to assess the patency of bypass grafts and endovascular stents after revascularization procedures.
The complication of peripheral artery disease that patients dread the most is limb loss, but of greater clinical consequence are the alarming rates of cardiovascular events and death in these patients. Epidemiologic studies have shown that fewer than 5% of patients age 55 or older with claudication or asymptomatic peripheral artery disease experience major amputation over a 5-year follow-up period, but 20% of these patients will have a stroke or myocardial infarction and 15% to 30% will die. Of those who die, 75% die of a coronary or cerebrovascular cause.36 Because of the markedly increased risk of death or cardiovascular morbidity in patients with peripheral artery disease, many have advocated screening patients at high risk using the ankle-brachial index. However, there have been conflicting recommendations from national societies and agencies.29,46–48
The United States Preventive Services Task Force (USPSTF) updated its 1996 recommendations on screening for peripheral artery disease in 2005 and recommended against routinely screening for it, giving the practice a “D” recommendation (not recommended). Specifically, it stated that it found “fair evidence that screening asymptomatic adults with the ankle brachial index could lead to some small degree of harm, including false-positive results and unnecessary work-ups,”46 and concluded that “for asymptomatic adults, harms of routine screening for [peripheral artery disease] exceed benefits.”46
This negative recommendation was intensely debated among vascular specialty groups, and a rebuttal was published in 2006.49 The major area of contention was the task force’s assumption that decreased disease-specific morbidity (especially limb loss) is the most important outcome to be prevented by screening for asymptomatic peripheral artery disease, rather than adverse cardiovascular events. The USPSTF has announced plans for an update on screening for peripheral artery disease, anticipated for 2013.50
The American College of Cardiology/American Heart Association task force in 2005 recommended that a history of walking impairment, intermittent claudication, ischemic rest pain, or nonhealing wounds be solicited as part of a standard review of systems for adults age 70 and older or adults age 50 and older who have risk factors for atherosclerosis (class IC recommendation—based only on a consensus opinion of experts, case studies, or standard of care).29 In contrast to the USPSTF recommendations, the joint guidelines further recommended that patients with asymptomatic lower-extremity peripheral artery disease be identified by physical examination, ankle-brachial index, or both, to prevent myocardial infarction, stroke, or death (class IC).29 Patients at risk for lower-extremity peripheral artery disease for whom ankle-brachial index measurement is recommended include those with exertional leg symptoms, those with nonhealing ulcers, those age 70 and older, and those age 50 and older who have a history of moking or diabetes.
The American Diabetes Association and the second Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II) issued similar recommendations.48
In 2011, the American College of Cardiology/American Heart Association task force issued a focused update to its 2005 guidelines, broadening the recommendation for testing to include patients age 65 and older on the basis of the getABI study, as well as maintaining the recommendation for testing for those age 50 and older with a history of smoking or diabetes (class IB recommendation).26,41
The task force’s Guideline for the Assessment of Cardiovascular Risk in Asymptomatic Adults says that measuring the ankle-brachial index is reasonable for cardiovascular risk assessment in asymptomatic adults at intermediate risk (class IIA—conflicting evidence or divergence of opinion, from multiple randomized clinical trials).51 Also recommended as risk stratification tools for this patient population are measurement of carotid intima-media thickness and measurement of coronary artery calcium (both class IIA recommendations).
Unlike these tests, however, the ankle-brachial index does not require highly trained technical and medical personnel to perform and interpret. In addition, there is no risk of radiation exposure as is the case in coronary calcium measurement. It is a simpler, lower-cost, and more widely available tool for cardiovascular risk assessment.
LIMITATIONS TO ANKLE-BRACHIAL SCREENING IN PRACTICE
Although this test is relatively simple and noninvasive, it is not widely performed in primary care and cardiovascular medicine. In a study by Mohler and colleagues,52 the most common barriers to its use among primary care providers were the time required to perform it, lack of reimbursement for it, and limited staff availability. Currently, third-party payers do not generally reimburse for an ankle-brachial index examination performed to screen a patient who is asymptomatic but at risk for peripheral artery disease. Unfortunately, this has limited the widespread adoption of a program to detect peripheral artery disease in patients at risk.
Despite these limitations, the ankle-brachial index is an invaluable tool to both screen for peripheral artery disease in the appropriate at-risk patient populations and to diagnose it in patients who present with lower extremity symptoms. There are few diagnostic tests available today that provide such a high degree of diagnostic accuracy with as much prognostic information as the ankle-brachial index and with such little expense and risk to the patient.
- Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 2004; 110:738–743.
- Hirsch AT, Criqui MH, Treat-Jacobson D, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 2001; 286:1317–1324.
- McDermott MM, Greenland P, Liu K, et al. Leg symptoms in peripheral arterial disease: associated clinical characteristics and functional impairment. JAMA 2001; 286:1599–1606.
- McDermott MM. The magnitude of the problem of peripheral arterial disease: epidemiology and clinical significance. Cleve Clin J Med 2006; 73(suppl 4):S2–S7.
- Criqui MH, Vargas V, Denenberg JO, et al. Ethnicity and peripheral arterial disease: the San Diego Population Study. Circulation 2005; 112:2703–2707.
- Regensteiner JG, Hiatt WR, Coll JR, et al The impact of peripheral arterial disease on health-related quality of life in the Peripheral Arterial Disease Awareness, Risk, and Treatment: New Resources for Survival (PARTNERS) Program. Vasc Med 2008; 13:15–24.
- Arseven A, Guralnik JM, O’Brien E, Liu K, McDermott MM. Peripheral arterial disease and depressed mood in older men and women. Vasc Med 2001; 6:229–234.
- Steg PG, Bhatt DL, Wilson PW, et al; REACH Registry Investigators. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Gornik HL, Creager MA. Contemporary management of peripheral arterial disease: I. cardiovascular risk-factor modification. Cleve Clin J Med 2006; 73(suppl 4):S30–S37.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Heart Protection Study Collaborative Group. Randomized trial of the effects of cholesterol-lowering with simvastatin on peripheral vascular and other major vascular outcomes in 20,536 people with peripheral arterial disease and other high-risk conditions. J Vasc Surg 2007; 45:645–654.
- Criqui MH, Fronek A, Klauber MR, Barrett-Connor E, Gabriel S. The sensitivity, specificity, and predictive value of traditional clinical evaluation of peripheral arterial disease: results from noninvasive testing in a defined population. Circulation 1985; 71:516–522.
- Carter SA. Clinical measurement of systolic pressures in limbs with arterial occlusive disease. JAMA 1969; 207:1869–1874.
- Ouriel K, Zarins CK. Doppler ankle pressure: an evaluation of three methods of expression. Arch Surg 1982; 117:1297–1300.
- Yao ST, Hobbs JT, Irvine WT. Ankle systolic pressure measurements in arterial disease affecting the lower extremities. Br J Surg 1969; 56:676–679.
- Carter SA. Indirect systolic pressures and pulse waves in arterial occlusive diseases of the lower extremities. Circulation 1968; 37:624–637.
- Belch JJ, Topol EJ, Agnelli G, et al; Prevention of Atherothrombotic Disease Network. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med 2003; 163:884–892.
- Lijmer JG, Hunink MG, van den Dungen JJ, Loonstra J, Smit AJ. ROC analysis of noninvasive tests for peripheral arterial disease. Ultrasound Med Biol 1996; 22:391–398.
- Carmo GA, Mandil A, Nascimento BR, et al. Can we measure the ankle-brachial index using only a stethoscope? A pilot study. Fam Pract 2009; 26:22–26.
- Chesbro SB, Asongwed ET, Brown J, John EB. Reliability of Doppler and stethoscope methods of determining systolic blood pressures: considerations for calculating an ankle-brachial index. J Natl Med Assoc 2011; 103:863–869.
- Jönsson B, Lindberg LG, Skau T, Thulesius O. Is oscillometric ankle pressure reliable in leg vascular disease? Clin Physiol 2001; 21:155–163.
- Ramanathan A, Conaghan PJ, Jenkinson AD, Bishop CR. Comparison of ankle-brachial pressure index measurements using an automated oscillometric device with the standard Doppler ultrasound technique. ANZ J Surg 2003; 73:105–108.
- Beckman JA, Higgins CO, Gerhard-Herman M. Automated oscillometric determination of the ankle-brachial index provides accuracy necessary for office practice. Hypertension 2006; 47:35–38.
- Mehlsen J, Wiinberg N, Bruce C. Oscillometric blood pressure measurement: a simple method in screening for peripheral arterial disease. Clin Physiol Funct Imaging 2008; 28:426–429.
- Aboyans V, Lacroix P, Doucet S, Preux PM, Criqui MH, Laskar M. Diagnosis of peripheral arterial disease in general practice: can the ankle-brachial index be measured either by pulse palpation or an automatic blood pressure device? Int J Clin Pract 2008; 62:1001–1007.
- 2011 Writing Group Members. 2011 ACCF/AHA Focused Update of the Guideline for the Management of patients with peripheral artery disease (updating the 2005 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011; 124:2020–2045.
- Stein R, Hriljac I, Halperin JL, Gustavson SM, Teodorescu V, Olin JW. Limitation of the resting ankle-brachial index in symptomatic patients with peripheral arterial disease. Vasc Med 2006; 11:29–33.
- Amirhamzeh MM, Chant HJ, Rees JL, Hands LJ, Powell RJ, Campbell WB. A comparative study of treadmill tests and heel raising exercise for peripheral arterial disease. Eur J Vasc Endovasc Surg 1997; 13:301–305.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Skyler JS, Bergenstal R, Bonow RO, et al; American Diabetes Association; American College of Cardiology Foundation; American Heart Association. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol 2009; 53:298–304.
- Almahameed A, Bartholomew JR, editors. Peripheral arterial disease: recognition and contemporary management. Cleve Clin J Med 2006; 73(suppl 4):S1–S51.
- McKenna M, Wolfson S, Kuller L. The ratio of ankle and arm arterial pressure as an independent predictor of mortality. Atherosclerosis 1991; 87:119–128.
- Valentine RJ, Grayburn PA, Eichhorn EJ, Myers SI, Clagett GP. Coronary artery disease is highly prevalent among patients with premature peripheral vascular disease. J Vasc Surg 1994; 19:668–674.
- Weitz JI, Byrne J, Clagett GP, et al. Diagnosis and treatment of chronic arterial insufficiency of the lower extremities: a critical review. Circulation 1996; 94:3026–3049.
- Hertzer NR, Beven EG, Young JR, et al. Coronary artery disease in peripheral vascular patients. A classification of 1000 coronary angiograms and results of surgical management. Ann Surg 1984; 199:223–233.
- Leng GC, Lee AJ, Fowkes FG, et al. Incidence, natural history and cardiovascular events in symptomatic and asymptomatic peripheral arterial disease in the general population. Int J Epidemiol 1996; 25:1172–1181.
- Heald CL, Fowkes FG, Murray GD, Price JF; Ankle Brachial Index Collaboration. Risk of mortality and cardiovascular disease associated with the ankle-brachial index: Systematic review. Atherosclerosis 2006; 189:61–69.
- Ankle Brachial Index; Fowkes FG, Murray GD, Butcher I, et al. Collaboration Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA 2008; 300:197–208.
- Diehm C, Allenberg JR, Pittrow D, et al; German Epidemiological Trial on Ankle Brachial Index Study Group. Mortality and vascular morbidity in older adults with asymptomatic versus symptomatic peripheral artery disease. Circulation 2009; 120:2053–2061.
- Feringa HH, Bax JJ, van Waning VH, et al. The long-term prognostic value of the resting and postexercise ankle-brachial index. Arch Intern Med 2006; 166:529–535.
- Sheikh MA, Bhatt DL, Li J, Lin S, Bartholomew JR. Usefulness of postexercise ankle-brachial index to predict all-cause mortality. Am J Cardiol 2011; 107:778–782.
- Resnick HE, Lindsay RS, McDermott MM, et al. Relationship of high and low ankle brachial index to all-cause and cardiovascular disease mortality: the Strong Heart Study. Circulation 2004; 109:733–739.
- Wattanakit K, Folsom AR, Duprez DA, Weatherley BD, Hirsch AT. Clinical significance of a high ankle-brachial index: insights from the Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis 2007; 190:459–464.
- US Preventive Services Task Force. Screening for peripheral arterial disease: Recommendation statement. http://www.ahrq.gov/clinic/uspstf05/pad/padrs.pdf. Accessed July 25, 2012.
- Mayfield JA, Reiber GE, Sanders LJ, Janisse D, Pogach LM; American Diabetes Association. Preventive foot care in diabetes. Diabetes Care 2004; 27(suppl 1):S63–S64.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl S):S5–S67.
- Beckman JA, Jaff MR, Creager MA. The United States preventive services task force recommendation statement on screening for peripheral arterial disease: more harm than benefit? Circulation 2006; 114:861–866.
- US Preventive Services Task Force. Screening for peripheral artery disease draft research plan. http://www.uspreventiveservicestaskforce.org/uspstf12/pad/padslides.htm. Accessed July 26, 2012.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2010; 122:e584–e636.
- Mohler ER, Treat-Jacobson D, Reilly MP, et al. Utility and barriers to performance of the ankle-brachial index in primary care practice. Vasc Med 2004; 9:253–260.
In this article, we seek to convince you to measure the ankle-brachial index for any patient you suspect may have peripheral artery disease. This would include patients who are elderly, who smoke, or who have diabetes, regardless of whether or not they have symptoms. The ankle-brachial index is a simple test that involves taking the blood pressure in all four limbs using a hand-held Doppler device and then dividing the leg systolic pressure by the arm systolic pressure.
This simple test is both sensitive and specific for peripheral artery disease. It also gives a good assessment of cardiovascular risk. The downside: you or a member of your staff spends a few minutes doing it. Also, for patients without leg symptoms or abnormal findings on physical examination, you may not be paid for doing it.
Despite these limitations, the ankle-brachial index is a powerful clinical tool that deserves to be performed more often in primary care.
PERIPHERAL ARTERY DISEASE IS COMMON AND SERIOUS
Peripheral artery disease is important to detect, as it is common, it has serious consequences, and effective treatments are available. However, many patients with the disease do not have typical symptoms.
Peripheral artery disease affects up to 29% of people over age 70, depending on the population sampled.1,2 Its classic symptom is intermittent claudication, ie, leg pain with walking that improves with rest. However, most patients do not have intermittent claudication; they have atypical leg symptoms or no symptoms at all.2,3 While the risk factors for peripheral artery disease are similar to those for coronary artery disease, the factors most strongly associated with peripheral artery disease are older age, tobacco smoking, and diabetes mellitus.4 Blacks are twice as likely to have it compared with whites, even after adjusting for other cardiovascular risk factors.5
Untreated peripheral artery disease may have serious consequences, such as amputation, impaired functional capacity, poor quality of life, and depression.3,6,7 In addition, it is a strong marker of atherosclerotic burden and cardiovascular risk and has been recognized as a coronary risk equivalent. Patients with peripheral artery disease are at higher risk of death, myocardial infarction, stroke, and hospitalization, with event rates as high as 21% per year.8
Fortunately, simple therapies have been shown to prevent adverse cardiovascular events in peripheral artery disease, including antiplatelet drugs, statins, and angiotensin-converting enzyme inhibitors.9–11
THE ANKLE-BRACHIAL INDEX IS SENSITIVE AND SPECIFIC
The evaluation for possible peripheral artery disease should begin with the medical history, a cardiovascular review of systems, and a focused physical examination in which one should:
- Measure the blood pressure in both arms to assess for occult subclavian stenosis
- Auscultate for bruits over the carotid, abdominal, and femoral arteries
- Palpate the pulses in the lower extremities and the abdominal aorta
- Inspect the bare legs and feet for thinning of the skin, hair loss, thickening of the nails (which are nonspecific signs), and ulceration.
However, the physical examination has limited sensitivity and specificity for diagnosing peripheral artery disease. In general, the most reliable finding is the absence of a palpable posterior tibial artery pulse, which has been reported to have a specificity of 71% and a sensitivity of 91% for peripheral artery disease.12
The ankle-brachial index is the first-line test for both screening for peripheral artery disease and for diagnosing it. It is inexpensive and noninvasive to obtain and has a high sensitivity (79% to 95%) and specificity (95% to 96%) compared with angiography as the gold standard.13–18 It can be measured easily in the office, and every practitioner who cares for patients at risk of cardiovascular disease can be trained to measure it competently.
HOW TO MEASURE THE ANKLE-BRACHIAL INDEX
The ankle-brachial index is the ratio of the systolic pressure in the ankle to the systolic pressure in the arm. In healthy people, this ratio is typically greater than 1.0 or 1.1.
You can measure the ankle-brachial index in the office with a blood pressure cuff, sphygmomanometer, and handheld Doppler device. Alternatively, it can be measured in a noninvasive vascular laboratory as part of a more detailed examination that allows for assessment of blood pressures and waveforms (Doppler or pulse-volume recordings) at multiple segments along the limb. These more detailed vascular studies are generally reserved for patients with confirmed peripheral artery disease to locate the level and extent of blockage or for patients in whom lower-extremity revascularization is contemplated.
The use of a stethoscope to measure blood pressures for the ankle-brachial index has been studied in a few small series,19,20 but is thought to be less accurate than Doppler, especially in the setting of significant arterial occlusive disease. Because of this, it is recommended and assumed that a Doppler device be used to measure all blood pressures for the ankle-brachial index.
After the patient has been resting quietly for 5 to 10 minutes in the supine position, the systolic blood pressure is measured in both arms and in both ankles in the dorsalis pedis and posterior tibial arteries (Figure 1). The blood pressure cuff is placed about 1 inch above the antecubital fossa for the brachial pressure and about 2 inches above the medial malleolus for the ankle pressures. A clear arterial pulse signal should be heard using the Doppler probe before inflating the blood pressure cuff. The cuff is then inflated to at least 20 mm Hg above the point where the arterial Doppler sounds disappear and then slowly deflated until the Doppler sounds reappear. The blood pressure at which the Doppler signal of the arterial pulse reappears is the systolic pressure for that vessel.
The ankle-brachial index is calculated by dividing the higher of the two ankle systolic blood pressures in each leg by the higher of the two brachial systolic blood pressures. The higher of the two brachial pressures is used as the denominator to account for the possibility of subclavian artery stenosis, which can decrease the blood pressure in the upper extremity. The ankle-brachial index is calculated for each leg, and the lower value is the patient’s overall ankle-brachial index. An abnormal value in either leg indicates peripheral artery disease.
While other ways of calculating the ankle-brachial index have been proposed, such as averaging the two pressures at each ankle or reporting the lower of the two ankle pressures, these methods are not standard for use in clinical practice.
Similarly, the use of oscillometric blood pressure devices has been proposed, which would eliminate the need for a Doppler device and personnel trained in its use. However, results of validation studies of oscillometric measurement of the ankle-brachial index have been inconsistent, likely because the devices were designed for measuring blood pressure in nonobstructed arms, not the legs, and especially not diseased legs.21–25 In general, oscillometric devices tend to overestimate ankle pressure, giving a falsely high ankle-brachial index in patients with moderate to severe peripheral artery disease.21 Their utility in screening for peripheral artery disease has not been evaluated in broad, population-based studies. Efforts to develop and validate new oscillometric devices for diagnosing peripheral artery disease are ongoing.
INTERPRETING THE ANKLE-BRACHIAL INDEX
Diagnostic criteria for the ankle-brachial index were standardized in 2011 (Table 1).26 Most healthy adults have a value greater than 1.0. A value of less than 0.91 is consistent with significant peripheral artery disease, and a value lower than 0.40 at rest generally indicates severe disease. A value between 0.91 and 0.99 is borderline abnormal and does not rule out peripheral artery disease. A value greater than 1.40 reflects noncompressibility of the leg arteries and is not diagnostic (see below).
The ankle-brachial index after exercise. In patients strongly suspected of having peripheral artery disease but who have a normal ankle-brachial index at rest, and especially if the resting value is borderline (ie, 0.91–0.99), the measurement should be repeated after exercise, the better to detect “mild” peripheral artery disease.15 With exercise, increased flow across a fixed stenosis leads to a significant fall in ankle pressure and a lower ankle-brachial index. In one study,27 the ankle-brachial index fell below 0.9 after exercise in 31% of outpatients with symptoms who had initially tested normal.
The exercise is optimally done on a motorized treadmill set at an incline. A number of exercise protocols are in use; at our institution, we use a fixed workload protocol. The ankle-brachial index and ankle pulse-volume recordings are recorded on both sides at rest, after which the patient generally walks for 5 minutes at a 12% grade at 2.0 mph or until symptoms force the patient to stop. The advantage of treadmill testing is the ability to assess functional capacity by measuring the time to the onset of pain and the total walking time.
Alternatively, active pedal plantar flexion maneuvers (heel raises) or corridor walking to the point at which limiting symptoms occur can be done if a treadmill is not available, though this is not the favored approach and does not qualify as formal exercise testing for reimbursement purposes. The patient is asked to do heel raises as high and as fast as possible for 30 seconds or until limiting pain symptoms occur. Results with this maneuver have been shown to correlate well with those of treadmill exercise testing.28
Immediately after any exercise maneuver, arm and ankle pressures are remeasured and bilateral ankle-brachial indices are recalculated. A fall in ankle pressure or the ankle-brachial index after exercise (generally, a fall of more than 20%) supports the diagnosis of peripheral artery disease. If the patient develops leg symptoms during exercise while his or her ankle-brachial index falls significantly, this also supports the vasculogenic nature of the leg symptoms.
An ankle-brachial index greater than 1.40 means that the pedal arteries are stiff and cannot be compressed by the blood pressure cuff. This is considered abnormal, though not necessarily diagnostic of peripheral artery disease. Noncompressible leg arteries are common among patients with long-standing diabetes mellitus or end-stage renal disease, and also can be found in obese patients.
Because toe arteries are usually compressible even when the pedal arteries are not, a toe-brachial index can be obtained to confirm the diagnosis of peripheral artery disease in these cases. This is calculated by measuring the blood pressure in the great toe using a small digital blood pressure cuff and a Doppler probe or a plethysmographic flow sensor. The toe-brachial index is calculated by dividing the toe blood pressure by the higher of the two brachial artery pressures; a value of 0.7 or less generally indicates peripheral artery disease.
WHAT SHOULD BE DONE WITH AN ABNORMAL RESULT?
An abnormal ankle-brachial index establishes the diagnosis of peripheral artery disease, and in many cases no additional diagnostic testing is necessary.
Care of patients with peripheral artery disease has three elements:
- Cardiovascular risk factor assessment and reduction to prevent myocardial infarction, stroke, and death
- Assessment and treatment of leg symptoms to improve function and quality of life
- Foot care to prevent ulcers and amputation.
Risk factor reduction. Because they have a markedly greater risk of cardiovascular disease and death, all patients with peripheral artery disease should undergo aggressive cardiovascular risk factor modification,26,29 including:
- Antiplatelet therapy in the form of aspirin 75–325 mg daily or clopidogrel 75 mg daily as an alternative to aspirin
- Counseling and therapy for immediate smoking cessation if the patient smokes
- Treatment of hypertension to Seventh Joint National Committee goals30
- Treatment of lipids to Adult Treatment Panel III goals31 (generally to a goal low-density lipoprotein cholesterol of less than 100 mg/dL, and less than 70 mg/dL if possible)
- Treatment of diabetes to a goal hemoglobin A1c of less than 7% (in the absence of contraindications).32
Exercise and anticlaudication medication. Patients with an abnormal ankle-brachial index and intermittent claudication may benefit from a supervised exercise program, a trial of drug therapy for claudication, or both. All patients with peripheral artery disease, regardless of symptoms, should be advised to incorporate aerobic exercise (ideally, walking) into their daily routine.
Cilostazol (Pletal), a phosphodiesterase inhibitor, has been given a class IA recommendation in the American College of Cardiology/American Heart Association guidelines for the treatment of intermittent claudication. The dose is generally 100 mg by mouth twice daily.29
Revascularization. Patients with an abnormal ankle-brachial index and lifestyle-limiting claudication that has failed to improve with medical therapy or a course of supervised exercise training should be referred to a vascular specialist for evaluation for revascularization (endovascular therapy or surgical bypass). 29 Endovascular therapy is particularly attractive for patients with claudication and evidence of aortoiliac disease (suspected in patients with gluteal or thigh claudication, diminution of the femoral pulse, or a bruit over the femoral artery on examination and confirmed by noninvasive vascular laboratory testing).
Patients who have ischemic pain at rest, gangrene, or a nonhealing lower-extremity wound that has been present for at least 2 weeks should be referred for revascularization on an urgent basis, given the risk of impending limb loss associated with critical limb ischemia.
A detailed review of the medical, endovascular, and surgical management of peripheral artery disease can be found in a supplement to the Cleveland Clinic Journal of Medicine published in 200633 and in comprehensive multi-society guidelines.26,29
THE ANKLE-BRACHIAL INDEX AS A MARKER OF RISK
Low values: Peripheral artery disease
Peripheral artery disease, as diagnosed by a low ankle-brachial index, confers an excess risk of death from all causes in a graded fashion: ie, the more severe the disease, the lower the survival rate (Figure 2).34 Because peripheral artery disease is a sign of systemic atherosclerosis and one-third to one-half of patients with peripheral artery disease have evidence of cerebrovascular or coronary artery disease,35–37 peripheral artery disease also confers a higher risk of cardiovascular death.
The Edinburgh Artery Study,38 a prospective cohort study of 1,592 randomly selected patients age 55 to 74 years, demonstrated the relationship between a low ankle-brachial index and an increased risk of cardiovascular death. Over 5 years of follow-up, compared with patients with a normal ankle-brachial index, the relative risk of cardiovascular death in symptomatic patients with a value of 0.9 or lower was 2.67 (95% confidence interval [CI] 1.34–5.29). The relative risk in patients with asymptomatic disease was between 1.74 (95% CI 1.09–2.76) and 2.08 (95% CI 1.13–3.83), depending on the level of ankle-brachial index decrement and ankle blood pressure response to hyperemia.
(Reactive hyperemia is an alternative to exercise testing. It is performed by inflating a blood pressure cuff at the thigh above the systolic pressure for 3 to 5 minutes or until the patient can no longer tolerate the inflation. Blood pressures at the ankle are remeasured after cuff release.)
Several other epidemiologic studies have established the association between low ankle-brachial index and the risk of cardiovascular death.
Heald et al39 performed a meta-analysis of 44,590 patients in 11 epidemiologic studies and found that, after adjustment for age, sex, conventional cardiovascular risk factors, and prevalent cardiovascular disease, an ankle-brachial index lower than 0.9 conferred a higher risk of:
- All-cause mortality (pooled risk ratio [RR] 1.60, 95% CI 1.32–1.95)
- Cardiovascular mortality (pooled RR 1.96, 95% CI 1.46–2.64)
- Coronary heart disease (pooled RR 1.45, 95% CI 1.08–1.93)
- Stroke (pooled RR 1.35, 95% CI 1.10–1.65).
Fowkes et al,40 in a meta-analysis of 16 population cohort studies including 48,294 patients over 480,325 person-years of follow-up, found that a low ankle-brachial index predicted cardiovascular events and death even after adjusting for the Framingham risk score, Hazard ratios for cardiovascular death were:
- 2.92 (95% CI 2.31–3.70) in men
- 2.97 (95% CI 2.02–4.35) in women.
Hazard ratios for death from any cause were:
- 2.34 (95% CI 1.97–2.78) in men
- 2.35 (95% CI 1.76–3.13) in women.
Adding the ankle-brachial index to the Framingham risk score resulted in reclassification of risk category in approximately 19% of men and 36% of women.40
The German Epidemiological Trial on Ankle Brachial Index (getABI) screened 6,880 patients 65 years of age and found an abnormal ankle-brachial index in 20.9% of them.41 In more than 5 years of follow-up, a value of less than 0.90 was associated with a higher rate of cardiovascular events and death from any cause in patients with both symptomatic and asymptomatic peripheral artery disease (Figure 3).41
In addition, the lower the ankle-brachial index, the greater the rate of death or severe cardiovascular events. An index between 0.7 and 0.9 was associated with a statistically significant twofold increase (adjusted hazard ratio 2.03), and a value lower than 0.5 was associated with a nearly fivefold increase (hazard ratio 4.65) in the risk of events compared with the group of patients with normal values.41
Abnormal results after exercise
Exercise testing may increase the sensitivity of the ankle-brachial index to detect peripheral artery disease in patients with normal resting values and especially in patients with borderline values. As such, abnormal exercise values have also been associated with an increased risk of death due to any cause and of cardiovascular death.
In a prospective cohort study of 3,209 patients with suspected or known peripheral artery disease referred to a vascular surgery clinic in the Netherlands, patients with lower postexercise values had a higher rate of overall and cardiac death (hazard ratio per 10% lower value 1.16 [95% CI 1.13–1.18] and 1.10 [95% CI 1.09–1.13], respectively).42
Sheikh et al43 reported similar findings in patients with normal resting ankle-brachial indices at Cleveland Clinic. In this study, an abnormal postexercise ankle-brachial index (defined as < 0.85) was associated with a hazard ratio of 1.67 for all-cause mortality compared with a normal postexercise value among individuals with no history of cardiovascular events.
High values: Noncompressible vessels
While the relationship between low values and increased mortality and cardiovascular risk is well accepted, there have been conflicting reports regarding high values (> 1.4) and adverse outcomes.44,45
The Strong Heart Study44 was a population-based study in 4,393 Native Americans followed for more than 8 years for the rate of all-cause and cardiovascular mortality. Most (n = 3,773) of the cohort had a normal ankle-brachial index (≥ 0.9 and ≤ 1.4); 4.9% (n = 216) had a low value (< 0.9); and 9.2% (n = 404) had a high value (> 1.4 or noncompressible). Relative risk ratios for all-cause mortality were 1.69 (95% CI 1.34–2.14) for low values and 1.77 (95% CI 1.48–2.13) for high values compared with those with normal values. Low and high ankle-brachial indices also conferred a risk of cardiovascular death, with relative risk ratios of 2.52 (95% CI 1.74–3.64) and 2.09 (95% CI 1.49–2.94), respectively. There was a U-shaped relationship between the ankle-brachial index and the mortality rate (Figure 4).44
The Atherosclerosis Risk in Communities (ARIC) study45 had different findings. In 14,777 participants followed for a mean of 12.2 years, the cardiovascular disease event rates of patients whose ankle-brachial index-was categorized as high (> 1.3, > 1.4, or > 1.5) were similar to those of patients with a normal value (between 0.9 and 1.3).
Differences in event rates between the two studies may be due to a higher prevalence of values greater than 1.4 in the Strong Heart Study cohort as well as to a higher prevalence of concomitant risk factors (diabetes, older age, hypertension, lipid abnormality) in the high ankle-brachial index group in the Strong Heart Study compared with the ARIC study.
DIFFERING RECOMMENDATIONS
The ankle-brachial index can be used to screen for asymptomatic peripheral artery disease. It can also be used to confirm the diagnosis in patients with symptoms such as intermittent claudication, ischemic pain at rest, or lower extremity ulcers or in patients with signs such as abnormal pulses, bruits, or lower-extremity skin changes. It is also used to reassess the severity of known peripheral artery disease and as a part of a routine surveillance program to assess the patency of bypass grafts and endovascular stents after revascularization procedures.
The complication of peripheral artery disease that patients dread the most is limb loss, but of greater clinical consequence are the alarming rates of cardiovascular events and death in these patients. Epidemiologic studies have shown that fewer than 5% of patients age 55 or older with claudication or asymptomatic peripheral artery disease experience major amputation over a 5-year follow-up period, but 20% of these patients will have a stroke or myocardial infarction and 15% to 30% will die. Of those who die, 75% die of a coronary or cerebrovascular cause.36 Because of the markedly increased risk of death or cardiovascular morbidity in patients with peripheral artery disease, many have advocated screening patients at high risk using the ankle-brachial index. However, there have been conflicting recommendations from national societies and agencies.29,46–48
The United States Preventive Services Task Force (USPSTF) updated its 1996 recommendations on screening for peripheral artery disease in 2005 and recommended against routinely screening for it, giving the practice a “D” recommendation (not recommended). Specifically, it stated that it found “fair evidence that screening asymptomatic adults with the ankle brachial index could lead to some small degree of harm, including false-positive results and unnecessary work-ups,”46 and concluded that “for asymptomatic adults, harms of routine screening for [peripheral artery disease] exceed benefits.”46
This negative recommendation was intensely debated among vascular specialty groups, and a rebuttal was published in 2006.49 The major area of contention was the task force’s assumption that decreased disease-specific morbidity (especially limb loss) is the most important outcome to be prevented by screening for asymptomatic peripheral artery disease, rather than adverse cardiovascular events. The USPSTF has announced plans for an update on screening for peripheral artery disease, anticipated for 2013.50
The American College of Cardiology/American Heart Association task force in 2005 recommended that a history of walking impairment, intermittent claudication, ischemic rest pain, or nonhealing wounds be solicited as part of a standard review of systems for adults age 70 and older or adults age 50 and older who have risk factors for atherosclerosis (class IC recommendation—based only on a consensus opinion of experts, case studies, or standard of care).29 In contrast to the USPSTF recommendations, the joint guidelines further recommended that patients with asymptomatic lower-extremity peripheral artery disease be identified by physical examination, ankle-brachial index, or both, to prevent myocardial infarction, stroke, or death (class IC).29 Patients at risk for lower-extremity peripheral artery disease for whom ankle-brachial index measurement is recommended include those with exertional leg symptoms, those with nonhealing ulcers, those age 70 and older, and those age 50 and older who have a history of moking or diabetes.
The American Diabetes Association and the second Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II) issued similar recommendations.48
In 2011, the American College of Cardiology/American Heart Association task force issued a focused update to its 2005 guidelines, broadening the recommendation for testing to include patients age 65 and older on the basis of the getABI study, as well as maintaining the recommendation for testing for those age 50 and older with a history of smoking or diabetes (class IB recommendation).26,41
The task force’s Guideline for the Assessment of Cardiovascular Risk in Asymptomatic Adults says that measuring the ankle-brachial index is reasonable for cardiovascular risk assessment in asymptomatic adults at intermediate risk (class IIA—conflicting evidence or divergence of opinion, from multiple randomized clinical trials).51 Also recommended as risk stratification tools for this patient population are measurement of carotid intima-media thickness and measurement of coronary artery calcium (both class IIA recommendations).
Unlike these tests, however, the ankle-brachial index does not require highly trained technical and medical personnel to perform and interpret. In addition, there is no risk of radiation exposure as is the case in coronary calcium measurement. It is a simpler, lower-cost, and more widely available tool for cardiovascular risk assessment.
LIMITATIONS TO ANKLE-BRACHIAL SCREENING IN PRACTICE
Although this test is relatively simple and noninvasive, it is not widely performed in primary care and cardiovascular medicine. In a study by Mohler and colleagues,52 the most common barriers to its use among primary care providers were the time required to perform it, lack of reimbursement for it, and limited staff availability. Currently, third-party payers do not generally reimburse for an ankle-brachial index examination performed to screen a patient who is asymptomatic but at risk for peripheral artery disease. Unfortunately, this has limited the widespread adoption of a program to detect peripheral artery disease in patients at risk.
Despite these limitations, the ankle-brachial index is an invaluable tool to both screen for peripheral artery disease in the appropriate at-risk patient populations and to diagnose it in patients who present with lower extremity symptoms. There are few diagnostic tests available today that provide such a high degree of diagnostic accuracy with as much prognostic information as the ankle-brachial index and with such little expense and risk to the patient.
In this article, we seek to convince you to measure the ankle-brachial index for any patient you suspect may have peripheral artery disease. This would include patients who are elderly, who smoke, or who have diabetes, regardless of whether or not they have symptoms. The ankle-brachial index is a simple test that involves taking the blood pressure in all four limbs using a hand-held Doppler device and then dividing the leg systolic pressure by the arm systolic pressure.
This simple test is both sensitive and specific for peripheral artery disease. It also gives a good assessment of cardiovascular risk. The downside: you or a member of your staff spends a few minutes doing it. Also, for patients without leg symptoms or abnormal findings on physical examination, you may not be paid for doing it.
Despite these limitations, the ankle-brachial index is a powerful clinical tool that deserves to be performed more often in primary care.
PERIPHERAL ARTERY DISEASE IS COMMON AND SERIOUS
Peripheral artery disease is important to detect, as it is common, it has serious consequences, and effective treatments are available. However, many patients with the disease do not have typical symptoms.
Peripheral artery disease affects up to 29% of people over age 70, depending on the population sampled.1,2 Its classic symptom is intermittent claudication, ie, leg pain with walking that improves with rest. However, most patients do not have intermittent claudication; they have atypical leg symptoms or no symptoms at all.2,3 While the risk factors for peripheral artery disease are similar to those for coronary artery disease, the factors most strongly associated with peripheral artery disease are older age, tobacco smoking, and diabetes mellitus.4 Blacks are twice as likely to have it compared with whites, even after adjusting for other cardiovascular risk factors.5
Untreated peripheral artery disease may have serious consequences, such as amputation, impaired functional capacity, poor quality of life, and depression.3,6,7 In addition, it is a strong marker of atherosclerotic burden and cardiovascular risk and has been recognized as a coronary risk equivalent. Patients with peripheral artery disease are at higher risk of death, myocardial infarction, stroke, and hospitalization, with event rates as high as 21% per year.8
Fortunately, simple therapies have been shown to prevent adverse cardiovascular events in peripheral artery disease, including antiplatelet drugs, statins, and angiotensin-converting enzyme inhibitors.9–11
THE ANKLE-BRACHIAL INDEX IS SENSITIVE AND SPECIFIC
The evaluation for possible peripheral artery disease should begin with the medical history, a cardiovascular review of systems, and a focused physical examination in which one should:
- Measure the blood pressure in both arms to assess for occult subclavian stenosis
- Auscultate for bruits over the carotid, abdominal, and femoral arteries
- Palpate the pulses in the lower extremities and the abdominal aorta
- Inspect the bare legs and feet for thinning of the skin, hair loss, thickening of the nails (which are nonspecific signs), and ulceration.
However, the physical examination has limited sensitivity and specificity for diagnosing peripheral artery disease. In general, the most reliable finding is the absence of a palpable posterior tibial artery pulse, which has been reported to have a specificity of 71% and a sensitivity of 91% for peripheral artery disease.12
The ankle-brachial index is the first-line test for both screening for peripheral artery disease and for diagnosing it. It is inexpensive and noninvasive to obtain and has a high sensitivity (79% to 95%) and specificity (95% to 96%) compared with angiography as the gold standard.13–18 It can be measured easily in the office, and every practitioner who cares for patients at risk of cardiovascular disease can be trained to measure it competently.
HOW TO MEASURE THE ANKLE-BRACHIAL INDEX
The ankle-brachial index is the ratio of the systolic pressure in the ankle to the systolic pressure in the arm. In healthy people, this ratio is typically greater than 1.0 or 1.1.
You can measure the ankle-brachial index in the office with a blood pressure cuff, sphygmomanometer, and handheld Doppler device. Alternatively, it can be measured in a noninvasive vascular laboratory as part of a more detailed examination that allows for assessment of blood pressures and waveforms (Doppler or pulse-volume recordings) at multiple segments along the limb. These more detailed vascular studies are generally reserved for patients with confirmed peripheral artery disease to locate the level and extent of blockage or for patients in whom lower-extremity revascularization is contemplated.
The use of a stethoscope to measure blood pressures for the ankle-brachial index has been studied in a few small series,19,20 but is thought to be less accurate than Doppler, especially in the setting of significant arterial occlusive disease. Because of this, it is recommended and assumed that a Doppler device be used to measure all blood pressures for the ankle-brachial index.
After the patient has been resting quietly for 5 to 10 minutes in the supine position, the systolic blood pressure is measured in both arms and in both ankles in the dorsalis pedis and posterior tibial arteries (Figure 1). The blood pressure cuff is placed about 1 inch above the antecubital fossa for the brachial pressure and about 2 inches above the medial malleolus for the ankle pressures. A clear arterial pulse signal should be heard using the Doppler probe before inflating the blood pressure cuff. The cuff is then inflated to at least 20 mm Hg above the point where the arterial Doppler sounds disappear and then slowly deflated until the Doppler sounds reappear. The blood pressure at which the Doppler signal of the arterial pulse reappears is the systolic pressure for that vessel.
The ankle-brachial index is calculated by dividing the higher of the two ankle systolic blood pressures in each leg by the higher of the two brachial systolic blood pressures. The higher of the two brachial pressures is used as the denominator to account for the possibility of subclavian artery stenosis, which can decrease the blood pressure in the upper extremity. The ankle-brachial index is calculated for each leg, and the lower value is the patient’s overall ankle-brachial index. An abnormal value in either leg indicates peripheral artery disease.
While other ways of calculating the ankle-brachial index have been proposed, such as averaging the two pressures at each ankle or reporting the lower of the two ankle pressures, these methods are not standard for use in clinical practice.
Similarly, the use of oscillometric blood pressure devices has been proposed, which would eliminate the need for a Doppler device and personnel trained in its use. However, results of validation studies of oscillometric measurement of the ankle-brachial index have been inconsistent, likely because the devices were designed for measuring blood pressure in nonobstructed arms, not the legs, and especially not diseased legs.21–25 In general, oscillometric devices tend to overestimate ankle pressure, giving a falsely high ankle-brachial index in patients with moderate to severe peripheral artery disease.21 Their utility in screening for peripheral artery disease has not been evaluated in broad, population-based studies. Efforts to develop and validate new oscillometric devices for diagnosing peripheral artery disease are ongoing.
INTERPRETING THE ANKLE-BRACHIAL INDEX
Diagnostic criteria for the ankle-brachial index were standardized in 2011 (Table 1).26 Most healthy adults have a value greater than 1.0. A value of less than 0.91 is consistent with significant peripheral artery disease, and a value lower than 0.40 at rest generally indicates severe disease. A value between 0.91 and 0.99 is borderline abnormal and does not rule out peripheral artery disease. A value greater than 1.40 reflects noncompressibility of the leg arteries and is not diagnostic (see below).
The ankle-brachial index after exercise. In patients strongly suspected of having peripheral artery disease but who have a normal ankle-brachial index at rest, and especially if the resting value is borderline (ie, 0.91–0.99), the measurement should be repeated after exercise, the better to detect “mild” peripheral artery disease.15 With exercise, increased flow across a fixed stenosis leads to a significant fall in ankle pressure and a lower ankle-brachial index. In one study,27 the ankle-brachial index fell below 0.9 after exercise in 31% of outpatients with symptoms who had initially tested normal.
The exercise is optimally done on a motorized treadmill set at an incline. A number of exercise protocols are in use; at our institution, we use a fixed workload protocol. The ankle-brachial index and ankle pulse-volume recordings are recorded on both sides at rest, after which the patient generally walks for 5 minutes at a 12% grade at 2.0 mph or until symptoms force the patient to stop. The advantage of treadmill testing is the ability to assess functional capacity by measuring the time to the onset of pain and the total walking time.
Alternatively, active pedal plantar flexion maneuvers (heel raises) or corridor walking to the point at which limiting symptoms occur can be done if a treadmill is not available, though this is not the favored approach and does not qualify as formal exercise testing for reimbursement purposes. The patient is asked to do heel raises as high and as fast as possible for 30 seconds or until limiting pain symptoms occur. Results with this maneuver have been shown to correlate well with those of treadmill exercise testing.28
Immediately after any exercise maneuver, arm and ankle pressures are remeasured and bilateral ankle-brachial indices are recalculated. A fall in ankle pressure or the ankle-brachial index after exercise (generally, a fall of more than 20%) supports the diagnosis of peripheral artery disease. If the patient develops leg symptoms during exercise while his or her ankle-brachial index falls significantly, this also supports the vasculogenic nature of the leg symptoms.
An ankle-brachial index greater than 1.40 means that the pedal arteries are stiff and cannot be compressed by the blood pressure cuff. This is considered abnormal, though not necessarily diagnostic of peripheral artery disease. Noncompressible leg arteries are common among patients with long-standing diabetes mellitus or end-stage renal disease, and also can be found in obese patients.
Because toe arteries are usually compressible even when the pedal arteries are not, a toe-brachial index can be obtained to confirm the diagnosis of peripheral artery disease in these cases. This is calculated by measuring the blood pressure in the great toe using a small digital blood pressure cuff and a Doppler probe or a plethysmographic flow sensor. The toe-brachial index is calculated by dividing the toe blood pressure by the higher of the two brachial artery pressures; a value of 0.7 or less generally indicates peripheral artery disease.
WHAT SHOULD BE DONE WITH AN ABNORMAL RESULT?
An abnormal ankle-brachial index establishes the diagnosis of peripheral artery disease, and in many cases no additional diagnostic testing is necessary.
Care of patients with peripheral artery disease has three elements:
- Cardiovascular risk factor assessment and reduction to prevent myocardial infarction, stroke, and death
- Assessment and treatment of leg symptoms to improve function and quality of life
- Foot care to prevent ulcers and amputation.
Risk factor reduction. Because they have a markedly greater risk of cardiovascular disease and death, all patients with peripheral artery disease should undergo aggressive cardiovascular risk factor modification,26,29 including:
- Antiplatelet therapy in the form of aspirin 75–325 mg daily or clopidogrel 75 mg daily as an alternative to aspirin
- Counseling and therapy for immediate smoking cessation if the patient smokes
- Treatment of hypertension to Seventh Joint National Committee goals30
- Treatment of lipids to Adult Treatment Panel III goals31 (generally to a goal low-density lipoprotein cholesterol of less than 100 mg/dL, and less than 70 mg/dL if possible)
- Treatment of diabetes to a goal hemoglobin A1c of less than 7% (in the absence of contraindications).32
Exercise and anticlaudication medication. Patients with an abnormal ankle-brachial index and intermittent claudication may benefit from a supervised exercise program, a trial of drug therapy for claudication, or both. All patients with peripheral artery disease, regardless of symptoms, should be advised to incorporate aerobic exercise (ideally, walking) into their daily routine.
Cilostazol (Pletal), a phosphodiesterase inhibitor, has been given a class IA recommendation in the American College of Cardiology/American Heart Association guidelines for the treatment of intermittent claudication. The dose is generally 100 mg by mouth twice daily.29
Revascularization. Patients with an abnormal ankle-brachial index and lifestyle-limiting claudication that has failed to improve with medical therapy or a course of supervised exercise training should be referred to a vascular specialist for evaluation for revascularization (endovascular therapy or surgical bypass). 29 Endovascular therapy is particularly attractive for patients with claudication and evidence of aortoiliac disease (suspected in patients with gluteal or thigh claudication, diminution of the femoral pulse, or a bruit over the femoral artery on examination and confirmed by noninvasive vascular laboratory testing).
Patients who have ischemic pain at rest, gangrene, or a nonhealing lower-extremity wound that has been present for at least 2 weeks should be referred for revascularization on an urgent basis, given the risk of impending limb loss associated with critical limb ischemia.
A detailed review of the medical, endovascular, and surgical management of peripheral artery disease can be found in a supplement to the Cleveland Clinic Journal of Medicine published in 200633 and in comprehensive multi-society guidelines.26,29
THE ANKLE-BRACHIAL INDEX AS A MARKER OF RISK
Low values: Peripheral artery disease
Peripheral artery disease, as diagnosed by a low ankle-brachial index, confers an excess risk of death from all causes in a graded fashion: ie, the more severe the disease, the lower the survival rate (Figure 2).34 Because peripheral artery disease is a sign of systemic atherosclerosis and one-third to one-half of patients with peripheral artery disease have evidence of cerebrovascular or coronary artery disease,35–37 peripheral artery disease also confers a higher risk of cardiovascular death.
The Edinburgh Artery Study,38 a prospective cohort study of 1,592 randomly selected patients age 55 to 74 years, demonstrated the relationship between a low ankle-brachial index and an increased risk of cardiovascular death. Over 5 years of follow-up, compared with patients with a normal ankle-brachial index, the relative risk of cardiovascular death in symptomatic patients with a value of 0.9 or lower was 2.67 (95% confidence interval [CI] 1.34–5.29). The relative risk in patients with asymptomatic disease was between 1.74 (95% CI 1.09–2.76) and 2.08 (95% CI 1.13–3.83), depending on the level of ankle-brachial index decrement and ankle blood pressure response to hyperemia.
(Reactive hyperemia is an alternative to exercise testing. It is performed by inflating a blood pressure cuff at the thigh above the systolic pressure for 3 to 5 minutes or until the patient can no longer tolerate the inflation. Blood pressures at the ankle are remeasured after cuff release.)
Several other epidemiologic studies have established the association between low ankle-brachial index and the risk of cardiovascular death.
Heald et al39 performed a meta-analysis of 44,590 patients in 11 epidemiologic studies and found that, after adjustment for age, sex, conventional cardiovascular risk factors, and prevalent cardiovascular disease, an ankle-brachial index lower than 0.9 conferred a higher risk of:
- All-cause mortality (pooled risk ratio [RR] 1.60, 95% CI 1.32–1.95)
- Cardiovascular mortality (pooled RR 1.96, 95% CI 1.46–2.64)
- Coronary heart disease (pooled RR 1.45, 95% CI 1.08–1.93)
- Stroke (pooled RR 1.35, 95% CI 1.10–1.65).
Fowkes et al,40 in a meta-analysis of 16 population cohort studies including 48,294 patients over 480,325 person-years of follow-up, found that a low ankle-brachial index predicted cardiovascular events and death even after adjusting for the Framingham risk score, Hazard ratios for cardiovascular death were:
- 2.92 (95% CI 2.31–3.70) in men
- 2.97 (95% CI 2.02–4.35) in women.
Hazard ratios for death from any cause were:
- 2.34 (95% CI 1.97–2.78) in men
- 2.35 (95% CI 1.76–3.13) in women.
Adding the ankle-brachial index to the Framingham risk score resulted in reclassification of risk category in approximately 19% of men and 36% of women.40
The German Epidemiological Trial on Ankle Brachial Index (getABI) screened 6,880 patients 65 years of age and found an abnormal ankle-brachial index in 20.9% of them.41 In more than 5 years of follow-up, a value of less than 0.90 was associated with a higher rate of cardiovascular events and death from any cause in patients with both symptomatic and asymptomatic peripheral artery disease (Figure 3).41
In addition, the lower the ankle-brachial index, the greater the rate of death or severe cardiovascular events. An index between 0.7 and 0.9 was associated with a statistically significant twofold increase (adjusted hazard ratio 2.03), and a value lower than 0.5 was associated with a nearly fivefold increase (hazard ratio 4.65) in the risk of events compared with the group of patients with normal values.41
Abnormal results after exercise
Exercise testing may increase the sensitivity of the ankle-brachial index to detect peripheral artery disease in patients with normal resting values and especially in patients with borderline values. As such, abnormal exercise values have also been associated with an increased risk of death due to any cause and of cardiovascular death.
In a prospective cohort study of 3,209 patients with suspected or known peripheral artery disease referred to a vascular surgery clinic in the Netherlands, patients with lower postexercise values had a higher rate of overall and cardiac death (hazard ratio per 10% lower value 1.16 [95% CI 1.13–1.18] and 1.10 [95% CI 1.09–1.13], respectively).42
Sheikh et al43 reported similar findings in patients with normal resting ankle-brachial indices at Cleveland Clinic. In this study, an abnormal postexercise ankle-brachial index (defined as < 0.85) was associated with a hazard ratio of 1.67 for all-cause mortality compared with a normal postexercise value among individuals with no history of cardiovascular events.
High values: Noncompressible vessels
While the relationship between low values and increased mortality and cardiovascular risk is well accepted, there have been conflicting reports regarding high values (> 1.4) and adverse outcomes.44,45
The Strong Heart Study44 was a population-based study in 4,393 Native Americans followed for more than 8 years for the rate of all-cause and cardiovascular mortality. Most (n = 3,773) of the cohort had a normal ankle-brachial index (≥ 0.9 and ≤ 1.4); 4.9% (n = 216) had a low value (< 0.9); and 9.2% (n = 404) had a high value (> 1.4 or noncompressible). Relative risk ratios for all-cause mortality were 1.69 (95% CI 1.34–2.14) for low values and 1.77 (95% CI 1.48–2.13) for high values compared with those with normal values. Low and high ankle-brachial indices also conferred a risk of cardiovascular death, with relative risk ratios of 2.52 (95% CI 1.74–3.64) and 2.09 (95% CI 1.49–2.94), respectively. There was a U-shaped relationship between the ankle-brachial index and the mortality rate (Figure 4).44
The Atherosclerosis Risk in Communities (ARIC) study45 had different findings. In 14,777 participants followed for a mean of 12.2 years, the cardiovascular disease event rates of patients whose ankle-brachial index-was categorized as high (> 1.3, > 1.4, or > 1.5) were similar to those of patients with a normal value (between 0.9 and 1.3).
Differences in event rates between the two studies may be due to a higher prevalence of values greater than 1.4 in the Strong Heart Study cohort as well as to a higher prevalence of concomitant risk factors (diabetes, older age, hypertension, lipid abnormality) in the high ankle-brachial index group in the Strong Heart Study compared with the ARIC study.
DIFFERING RECOMMENDATIONS
The ankle-brachial index can be used to screen for asymptomatic peripheral artery disease. It can also be used to confirm the diagnosis in patients with symptoms such as intermittent claudication, ischemic pain at rest, or lower extremity ulcers or in patients with signs such as abnormal pulses, bruits, or lower-extremity skin changes. It is also used to reassess the severity of known peripheral artery disease and as a part of a routine surveillance program to assess the patency of bypass grafts and endovascular stents after revascularization procedures.
The complication of peripheral artery disease that patients dread the most is limb loss, but of greater clinical consequence are the alarming rates of cardiovascular events and death in these patients. Epidemiologic studies have shown that fewer than 5% of patients age 55 or older with claudication or asymptomatic peripheral artery disease experience major amputation over a 5-year follow-up period, but 20% of these patients will have a stroke or myocardial infarction and 15% to 30% will die. Of those who die, 75% die of a coronary or cerebrovascular cause.36 Because of the markedly increased risk of death or cardiovascular morbidity in patients with peripheral artery disease, many have advocated screening patients at high risk using the ankle-brachial index. However, there have been conflicting recommendations from national societies and agencies.29,46–48
The United States Preventive Services Task Force (USPSTF) updated its 1996 recommendations on screening for peripheral artery disease in 2005 and recommended against routinely screening for it, giving the practice a “D” recommendation (not recommended). Specifically, it stated that it found “fair evidence that screening asymptomatic adults with the ankle brachial index could lead to some small degree of harm, including false-positive results and unnecessary work-ups,”46 and concluded that “for asymptomatic adults, harms of routine screening for [peripheral artery disease] exceed benefits.”46
This negative recommendation was intensely debated among vascular specialty groups, and a rebuttal was published in 2006.49 The major area of contention was the task force’s assumption that decreased disease-specific morbidity (especially limb loss) is the most important outcome to be prevented by screening for asymptomatic peripheral artery disease, rather than adverse cardiovascular events. The USPSTF has announced plans for an update on screening for peripheral artery disease, anticipated for 2013.50
The American College of Cardiology/American Heart Association task force in 2005 recommended that a history of walking impairment, intermittent claudication, ischemic rest pain, or nonhealing wounds be solicited as part of a standard review of systems for adults age 70 and older or adults age 50 and older who have risk factors for atherosclerosis (class IC recommendation—based only on a consensus opinion of experts, case studies, or standard of care).29 In contrast to the USPSTF recommendations, the joint guidelines further recommended that patients with asymptomatic lower-extremity peripheral artery disease be identified by physical examination, ankle-brachial index, or both, to prevent myocardial infarction, stroke, or death (class IC).29 Patients at risk for lower-extremity peripheral artery disease for whom ankle-brachial index measurement is recommended include those with exertional leg symptoms, those with nonhealing ulcers, those age 70 and older, and those age 50 and older who have a history of moking or diabetes.
The American Diabetes Association and the second Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II) issued similar recommendations.48
In 2011, the American College of Cardiology/American Heart Association task force issued a focused update to its 2005 guidelines, broadening the recommendation for testing to include patients age 65 and older on the basis of the getABI study, as well as maintaining the recommendation for testing for those age 50 and older with a history of smoking or diabetes (class IB recommendation).26,41
The task force’s Guideline for the Assessment of Cardiovascular Risk in Asymptomatic Adults says that measuring the ankle-brachial index is reasonable for cardiovascular risk assessment in asymptomatic adults at intermediate risk (class IIA—conflicting evidence or divergence of opinion, from multiple randomized clinical trials).51 Also recommended as risk stratification tools for this patient population are measurement of carotid intima-media thickness and measurement of coronary artery calcium (both class IIA recommendations).
Unlike these tests, however, the ankle-brachial index does not require highly trained technical and medical personnel to perform and interpret. In addition, there is no risk of radiation exposure as is the case in coronary calcium measurement. It is a simpler, lower-cost, and more widely available tool for cardiovascular risk assessment.
LIMITATIONS TO ANKLE-BRACHIAL SCREENING IN PRACTICE
Although this test is relatively simple and noninvasive, it is not widely performed in primary care and cardiovascular medicine. In a study by Mohler and colleagues,52 the most common barriers to its use among primary care providers were the time required to perform it, lack of reimbursement for it, and limited staff availability. Currently, third-party payers do not generally reimburse for an ankle-brachial index examination performed to screen a patient who is asymptomatic but at risk for peripheral artery disease. Unfortunately, this has limited the widespread adoption of a program to detect peripheral artery disease in patients at risk.
Despite these limitations, the ankle-brachial index is an invaluable tool to both screen for peripheral artery disease in the appropriate at-risk patient populations and to diagnose it in patients who present with lower extremity symptoms. There are few diagnostic tests available today that provide such a high degree of diagnostic accuracy with as much prognostic information as the ankle-brachial index and with such little expense and risk to the patient.
- Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 2004; 110:738–743.
- Hirsch AT, Criqui MH, Treat-Jacobson D, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 2001; 286:1317–1324.
- McDermott MM, Greenland P, Liu K, et al. Leg symptoms in peripheral arterial disease: associated clinical characteristics and functional impairment. JAMA 2001; 286:1599–1606.
- McDermott MM. The magnitude of the problem of peripheral arterial disease: epidemiology and clinical significance. Cleve Clin J Med 2006; 73(suppl 4):S2–S7.
- Criqui MH, Vargas V, Denenberg JO, et al. Ethnicity and peripheral arterial disease: the San Diego Population Study. Circulation 2005; 112:2703–2707.
- Regensteiner JG, Hiatt WR, Coll JR, et al The impact of peripheral arterial disease on health-related quality of life in the Peripheral Arterial Disease Awareness, Risk, and Treatment: New Resources for Survival (PARTNERS) Program. Vasc Med 2008; 13:15–24.
- Arseven A, Guralnik JM, O’Brien E, Liu K, McDermott MM. Peripheral arterial disease and depressed mood in older men and women. Vasc Med 2001; 6:229–234.
- Steg PG, Bhatt DL, Wilson PW, et al; REACH Registry Investigators. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Gornik HL, Creager MA. Contemporary management of peripheral arterial disease: I. cardiovascular risk-factor modification. Cleve Clin J Med 2006; 73(suppl 4):S30–S37.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Heart Protection Study Collaborative Group. Randomized trial of the effects of cholesterol-lowering with simvastatin on peripheral vascular and other major vascular outcomes in 20,536 people with peripheral arterial disease and other high-risk conditions. J Vasc Surg 2007; 45:645–654.
- Criqui MH, Fronek A, Klauber MR, Barrett-Connor E, Gabriel S. The sensitivity, specificity, and predictive value of traditional clinical evaluation of peripheral arterial disease: results from noninvasive testing in a defined population. Circulation 1985; 71:516–522.
- Carter SA. Clinical measurement of systolic pressures in limbs with arterial occlusive disease. JAMA 1969; 207:1869–1874.
- Ouriel K, Zarins CK. Doppler ankle pressure: an evaluation of three methods of expression. Arch Surg 1982; 117:1297–1300.
- Yao ST, Hobbs JT, Irvine WT. Ankle systolic pressure measurements in arterial disease affecting the lower extremities. Br J Surg 1969; 56:676–679.
- Carter SA. Indirect systolic pressures and pulse waves in arterial occlusive diseases of the lower extremities. Circulation 1968; 37:624–637.
- Belch JJ, Topol EJ, Agnelli G, et al; Prevention of Atherothrombotic Disease Network. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med 2003; 163:884–892.
- Lijmer JG, Hunink MG, van den Dungen JJ, Loonstra J, Smit AJ. ROC analysis of noninvasive tests for peripheral arterial disease. Ultrasound Med Biol 1996; 22:391–398.
- Carmo GA, Mandil A, Nascimento BR, et al. Can we measure the ankle-brachial index using only a stethoscope? A pilot study. Fam Pract 2009; 26:22–26.
- Chesbro SB, Asongwed ET, Brown J, John EB. Reliability of Doppler and stethoscope methods of determining systolic blood pressures: considerations for calculating an ankle-brachial index. J Natl Med Assoc 2011; 103:863–869.
- Jönsson B, Lindberg LG, Skau T, Thulesius O. Is oscillometric ankle pressure reliable in leg vascular disease? Clin Physiol 2001; 21:155–163.
- Ramanathan A, Conaghan PJ, Jenkinson AD, Bishop CR. Comparison of ankle-brachial pressure index measurements using an automated oscillometric device with the standard Doppler ultrasound technique. ANZ J Surg 2003; 73:105–108.
- Beckman JA, Higgins CO, Gerhard-Herman M. Automated oscillometric determination of the ankle-brachial index provides accuracy necessary for office practice. Hypertension 2006; 47:35–38.
- Mehlsen J, Wiinberg N, Bruce C. Oscillometric blood pressure measurement: a simple method in screening for peripheral arterial disease. Clin Physiol Funct Imaging 2008; 28:426–429.
- Aboyans V, Lacroix P, Doucet S, Preux PM, Criqui MH, Laskar M. Diagnosis of peripheral arterial disease in general practice: can the ankle-brachial index be measured either by pulse palpation or an automatic blood pressure device? Int J Clin Pract 2008; 62:1001–1007.
- 2011 Writing Group Members. 2011 ACCF/AHA Focused Update of the Guideline for the Management of patients with peripheral artery disease (updating the 2005 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011; 124:2020–2045.
- Stein R, Hriljac I, Halperin JL, Gustavson SM, Teodorescu V, Olin JW. Limitation of the resting ankle-brachial index in symptomatic patients with peripheral arterial disease. Vasc Med 2006; 11:29–33.
- Amirhamzeh MM, Chant HJ, Rees JL, Hands LJ, Powell RJ, Campbell WB. A comparative study of treadmill tests and heel raising exercise for peripheral arterial disease. Eur J Vasc Endovasc Surg 1997; 13:301–305.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Skyler JS, Bergenstal R, Bonow RO, et al; American Diabetes Association; American College of Cardiology Foundation; American Heart Association. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol 2009; 53:298–304.
- Almahameed A, Bartholomew JR, editors. Peripheral arterial disease: recognition and contemporary management. Cleve Clin J Med 2006; 73(suppl 4):S1–S51.
- McKenna M, Wolfson S, Kuller L. The ratio of ankle and arm arterial pressure as an independent predictor of mortality. Atherosclerosis 1991; 87:119–128.
- Valentine RJ, Grayburn PA, Eichhorn EJ, Myers SI, Clagett GP. Coronary artery disease is highly prevalent among patients with premature peripheral vascular disease. J Vasc Surg 1994; 19:668–674.
- Weitz JI, Byrne J, Clagett GP, et al. Diagnosis and treatment of chronic arterial insufficiency of the lower extremities: a critical review. Circulation 1996; 94:3026–3049.
- Hertzer NR, Beven EG, Young JR, et al. Coronary artery disease in peripheral vascular patients. A classification of 1000 coronary angiograms and results of surgical management. Ann Surg 1984; 199:223–233.
- Leng GC, Lee AJ, Fowkes FG, et al. Incidence, natural history and cardiovascular events in symptomatic and asymptomatic peripheral arterial disease in the general population. Int J Epidemiol 1996; 25:1172–1181.
- Heald CL, Fowkes FG, Murray GD, Price JF; Ankle Brachial Index Collaboration. Risk of mortality and cardiovascular disease associated with the ankle-brachial index: Systematic review. Atherosclerosis 2006; 189:61–69.
- Ankle Brachial Index; Fowkes FG, Murray GD, Butcher I, et al. Collaboration Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA 2008; 300:197–208.
- Diehm C, Allenberg JR, Pittrow D, et al; German Epidemiological Trial on Ankle Brachial Index Study Group. Mortality and vascular morbidity in older adults with asymptomatic versus symptomatic peripheral artery disease. Circulation 2009; 120:2053–2061.
- Feringa HH, Bax JJ, van Waning VH, et al. The long-term prognostic value of the resting and postexercise ankle-brachial index. Arch Intern Med 2006; 166:529–535.
- Sheikh MA, Bhatt DL, Li J, Lin S, Bartholomew JR. Usefulness of postexercise ankle-brachial index to predict all-cause mortality. Am J Cardiol 2011; 107:778–782.
- Resnick HE, Lindsay RS, McDermott MM, et al. Relationship of high and low ankle brachial index to all-cause and cardiovascular disease mortality: the Strong Heart Study. Circulation 2004; 109:733–739.
- Wattanakit K, Folsom AR, Duprez DA, Weatherley BD, Hirsch AT. Clinical significance of a high ankle-brachial index: insights from the Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis 2007; 190:459–464.
- US Preventive Services Task Force. Screening for peripheral arterial disease: Recommendation statement. http://www.ahrq.gov/clinic/uspstf05/pad/padrs.pdf. Accessed July 25, 2012.
- Mayfield JA, Reiber GE, Sanders LJ, Janisse D, Pogach LM; American Diabetes Association. Preventive foot care in diabetes. Diabetes Care 2004; 27(suppl 1):S63–S64.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl S):S5–S67.
- Beckman JA, Jaff MR, Creager MA. The United States preventive services task force recommendation statement on screening for peripheral arterial disease: more harm than benefit? Circulation 2006; 114:861–866.
- US Preventive Services Task Force. Screening for peripheral artery disease draft research plan. http://www.uspreventiveservicestaskforce.org/uspstf12/pad/padslides.htm. Accessed July 26, 2012.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2010; 122:e584–e636.
- Mohler ER, Treat-Jacobson D, Reilly MP, et al. Utility and barriers to performance of the ankle-brachial index in primary care practice. Vasc Med 2004; 9:253–260.
- Selvin E, Erlinger TP. Prevalence of and risk factors for peripheral arterial disease in the United States: results from the National Health and Nutrition Examination Survey, 1999–2000. Circulation 2004; 110:738–743.
- Hirsch AT, Criqui MH, Treat-Jacobson D, et al. Peripheral arterial disease detection, awareness, and treatment in primary care. JAMA 2001; 286:1317–1324.
- McDermott MM, Greenland P, Liu K, et al. Leg symptoms in peripheral arterial disease: associated clinical characteristics and functional impairment. JAMA 2001; 286:1599–1606.
- McDermott MM. The magnitude of the problem of peripheral arterial disease: epidemiology and clinical significance. Cleve Clin J Med 2006; 73(suppl 4):S2–S7.
- Criqui MH, Vargas V, Denenberg JO, et al. Ethnicity and peripheral arterial disease: the San Diego Population Study. Circulation 2005; 112:2703–2707.
- Regensteiner JG, Hiatt WR, Coll JR, et al The impact of peripheral arterial disease on health-related quality of life in the Peripheral Arterial Disease Awareness, Risk, and Treatment: New Resources for Survival (PARTNERS) Program. Vasc Med 2008; 13:15–24.
- Arseven A, Guralnik JM, O’Brien E, Liu K, McDermott MM. Peripheral arterial disease and depressed mood in older men and women. Vasc Med 2001; 6:229–234.
- Steg PG, Bhatt DL, Wilson PW, et al; REACH Registry Investigators. One-year cardiovascular event rates in outpatients with atherothrombosis. JAMA 2007; 297:1197–1206.
- Gornik HL, Creager MA. Contemporary management of peripheral arterial disease: I. cardiovascular risk-factor modification. Cleve Clin J Med 2006; 73(suppl 4):S30–S37.
- Antithrombotic Trialists’ Collaboration. Collaborative meta-analysis of randomised trials of antiplatelet therapy for prevention of death, myocardial infarction, and stroke in high risk patients. BMJ 2002; 324:71–86.
- Heart Protection Study Collaborative Group. Randomized trial of the effects of cholesterol-lowering with simvastatin on peripheral vascular and other major vascular outcomes in 20,536 people with peripheral arterial disease and other high-risk conditions. J Vasc Surg 2007; 45:645–654.
- Criqui MH, Fronek A, Klauber MR, Barrett-Connor E, Gabriel S. The sensitivity, specificity, and predictive value of traditional clinical evaluation of peripheral arterial disease: results from noninvasive testing in a defined population. Circulation 1985; 71:516–522.
- Carter SA. Clinical measurement of systolic pressures in limbs with arterial occlusive disease. JAMA 1969; 207:1869–1874.
- Ouriel K, Zarins CK. Doppler ankle pressure: an evaluation of three methods of expression. Arch Surg 1982; 117:1297–1300.
- Yao ST, Hobbs JT, Irvine WT. Ankle systolic pressure measurements in arterial disease affecting the lower extremities. Br J Surg 1969; 56:676–679.
- Carter SA. Indirect systolic pressures and pulse waves in arterial occlusive diseases of the lower extremities. Circulation 1968; 37:624–637.
- Belch JJ, Topol EJ, Agnelli G, et al; Prevention of Atherothrombotic Disease Network. Critical issues in peripheral arterial disease detection and management: a call to action. Arch Intern Med 2003; 163:884–892.
- Lijmer JG, Hunink MG, van den Dungen JJ, Loonstra J, Smit AJ. ROC analysis of noninvasive tests for peripheral arterial disease. Ultrasound Med Biol 1996; 22:391–398.
- Carmo GA, Mandil A, Nascimento BR, et al. Can we measure the ankle-brachial index using only a stethoscope? A pilot study. Fam Pract 2009; 26:22–26.
- Chesbro SB, Asongwed ET, Brown J, John EB. Reliability of Doppler and stethoscope methods of determining systolic blood pressures: considerations for calculating an ankle-brachial index. J Natl Med Assoc 2011; 103:863–869.
- Jönsson B, Lindberg LG, Skau T, Thulesius O. Is oscillometric ankle pressure reliable in leg vascular disease? Clin Physiol 2001; 21:155–163.
- Ramanathan A, Conaghan PJ, Jenkinson AD, Bishop CR. Comparison of ankle-brachial pressure index measurements using an automated oscillometric device with the standard Doppler ultrasound technique. ANZ J Surg 2003; 73:105–108.
- Beckman JA, Higgins CO, Gerhard-Herman M. Automated oscillometric determination of the ankle-brachial index provides accuracy necessary for office practice. Hypertension 2006; 47:35–38.
- Mehlsen J, Wiinberg N, Bruce C. Oscillometric blood pressure measurement: a simple method in screening for peripheral arterial disease. Clin Physiol Funct Imaging 2008; 28:426–429.
- Aboyans V, Lacroix P, Doucet S, Preux PM, Criqui MH, Laskar M. Diagnosis of peripheral arterial disease in general practice: can the ankle-brachial index be measured either by pulse palpation or an automatic blood pressure device? Int J Clin Pract 2008; 62:1001–1007.
- 2011 Writing Group Members. 2011 ACCF/AHA Focused Update of the Guideline for the Management of patients with peripheral artery disease (updating the 2005 guideline): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation 2011; 124:2020–2045.
- Stein R, Hriljac I, Halperin JL, Gustavson SM, Teodorescu V, Olin JW. Limitation of the resting ankle-brachial index in symptomatic patients with peripheral arterial disease. Vasc Med 2006; 11:29–33.
- Amirhamzeh MM, Chant HJ, Rees JL, Hands LJ, Powell RJ, Campbell WB. A comparative study of treadmill tests and heel raising exercise for peripheral arterial disease. Eur J Vasc Endovasc Surg 1997; 13:301–305.
- Hirsch AT, Haskal ZJ, Hertzer NR, et al; American Association for Vascular Surgery; Society for Vascular Surgery; Society for Cardiovascular Angiography and Interventions; Society for Vascular Medicine and Biology; Society of Interventional Radiology; ACC/AHA Task Force on Practice Guidelines Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease; American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; Vascular Disease Foundation. ACC/AHA 2005 practice guidelines for the management of patients with peripheral arterial disease (lower extremity, renal, mesenteric, and abdominal aortic): a collaborative report from the American Association for Vascular Surgery/Society for Vascular Surgery, Society for Cardiovascular Angiography and Interventions, Society for Vascular Medicine and Biology, Society of Interventional Radiology, and the ACC/AHA Task Force on Practice Guidelines (Writing Committee to Develop Guidelines for the Management of Patients With Peripheral Arterial Disease): endorsed by the American Association of Cardiovascular and Pulmonary Rehabilitation; National Heart, Lung, and Blood Institute; Society for Vascular Nursing; TransAtlantic Inter-Society Consensus; and Vascular Disease Foundation. Circulation 2006; 113:e463–e654.
- Chobanian AV, Bakris GL, Black HR, et al; Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure; National Heart, Lung, and Blood Institute; National High Blood Pressure Education Program Coordinating Committee. Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure. Hypertension 2003; 42:1206–1252.
- National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III). Third Report of the National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (Adult Treatment Panel III) final report. Circulation 2002; 106:3143–3421.
- Skyler JS, Bergenstal R, Bonow RO, et al; American Diabetes Association; American College of Cardiology Foundation; American Heart Association. Intensive glycemic control and the prevention of cardiovascular events: implications of the ACCORD, ADVANCE, and VA Diabetes Trials: a position statement of the American Diabetes Association and a Scientific Statement of the American College of Cardiology Foundation and the American Heart Association. J Am Coll Cardiol 2009; 53:298–304.
- Almahameed A, Bartholomew JR, editors. Peripheral arterial disease: recognition and contemporary management. Cleve Clin J Med 2006; 73(suppl 4):S1–S51.
- McKenna M, Wolfson S, Kuller L. The ratio of ankle and arm arterial pressure as an independent predictor of mortality. Atherosclerosis 1991; 87:119–128.
- Valentine RJ, Grayburn PA, Eichhorn EJ, Myers SI, Clagett GP. Coronary artery disease is highly prevalent among patients with premature peripheral vascular disease. J Vasc Surg 1994; 19:668–674.
- Weitz JI, Byrne J, Clagett GP, et al. Diagnosis and treatment of chronic arterial insufficiency of the lower extremities: a critical review. Circulation 1996; 94:3026–3049.
- Hertzer NR, Beven EG, Young JR, et al. Coronary artery disease in peripheral vascular patients. A classification of 1000 coronary angiograms and results of surgical management. Ann Surg 1984; 199:223–233.
- Leng GC, Lee AJ, Fowkes FG, et al. Incidence, natural history and cardiovascular events in symptomatic and asymptomatic peripheral arterial disease in the general population. Int J Epidemiol 1996; 25:1172–1181.
- Heald CL, Fowkes FG, Murray GD, Price JF; Ankle Brachial Index Collaboration. Risk of mortality and cardiovascular disease associated with the ankle-brachial index: Systematic review. Atherosclerosis 2006; 189:61–69.
- Ankle Brachial Index; Fowkes FG, Murray GD, Butcher I, et al. Collaboration Ankle brachial index combined with Framingham Risk Score to predict cardiovascular events and mortality: a meta-analysis. JAMA 2008; 300:197–208.
- Diehm C, Allenberg JR, Pittrow D, et al; German Epidemiological Trial on Ankle Brachial Index Study Group. Mortality and vascular morbidity in older adults with asymptomatic versus symptomatic peripheral artery disease. Circulation 2009; 120:2053–2061.
- Feringa HH, Bax JJ, van Waning VH, et al. The long-term prognostic value of the resting and postexercise ankle-brachial index. Arch Intern Med 2006; 166:529–535.
- Sheikh MA, Bhatt DL, Li J, Lin S, Bartholomew JR. Usefulness of postexercise ankle-brachial index to predict all-cause mortality. Am J Cardiol 2011; 107:778–782.
- Resnick HE, Lindsay RS, McDermott MM, et al. Relationship of high and low ankle brachial index to all-cause and cardiovascular disease mortality: the Strong Heart Study. Circulation 2004; 109:733–739.
- Wattanakit K, Folsom AR, Duprez DA, Weatherley BD, Hirsch AT. Clinical significance of a high ankle-brachial index: insights from the Atherosclerosis Risk in Communities (ARIC) Study. Atherosclerosis 2007; 190:459–464.
- US Preventive Services Task Force. Screening for peripheral arterial disease: Recommendation statement. http://www.ahrq.gov/clinic/uspstf05/pad/padrs.pdf. Accessed July 25, 2012.
- Mayfield JA, Reiber GE, Sanders LJ, Janisse D, Pogach LM; American Diabetes Association. Preventive foot care in diabetes. Diabetes Care 2004; 27(suppl 1):S63–S64.
- Norgren L, Hiatt WR, Dormandy JA, Nehler MR, Harris KA, Fowkes FG; TASC II Working Group. Inter-Society Consensus for the Management of Peripheral Arterial Disease (TASC II). J Vasc Surg 2007; 45(suppl S):S5–S67.
- Beckman JA, Jaff MR, Creager MA. The United States preventive services task force recommendation statement on screening for peripheral arterial disease: more harm than benefit? Circulation 2006; 114:861–866.
- US Preventive Services Task Force. Screening for peripheral artery disease draft research plan. http://www.uspreventiveservicestaskforce.org/uspstf12/pad/padslides.htm. Accessed July 26, 2012.
- Greenland P, Alpert JS, Beller GA, et al; American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. 2010 ACCF/AHA guideline for assessment of cardiovascular risk in asymptomatic adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines. Circulation 2010; 122:e584–e636.
- Mohler ER, Treat-Jacobson D, Reilly MP, et al. Utility and barriers to performance of the ankle-brachial index in primary care practice. Vasc Med 2004; 9:253–260.
KEY POINTS
- The ankle-brachial index is the systolic pressure in the ankle (either the dorsalis pedis or the posterior tibial artery, whichever has the higher pressure) divided by the systolic pressure in the arm (either the left or right, whichever is higher). The lower of the two values obtained (left and right) is the patient’s overall ankle-brachial index.
- Most healthy adults have a value greater than 1.0. A value of less than 0.91 indicates significant peripheral artery disease, and a value lower than 0.40 at rest generally indicates severe disease. Values higher than 1.4 indicate stiffened, noncompressible arteries.
- Measuring the ankle-brachial index after exercise can uncover peripheral artery disease in patients with a normal resting ankle-brachial index.
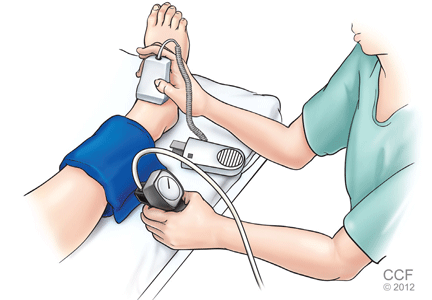
The role of aldosterone receptor antagonists in the management of heart failure: An update
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23

Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
Over the past 30 years, the focus of treating heart failure has shifted from managing symptoms to prolonging lives. When the neurohormonal hypothesis (ie, the concept that neurohormonal dysregulation and not merely hemodynamic changes are responsible for the onset and progression of heart failure) was introduced, it brought a dramatic change that included new classes of drugs that interfere with the renin-angiotensin-aldosterone system, ie, angiotensin-converting enzyme (ACE) inhibitors, angiotensin II receptor blockers (ARBs), and, most recently, aldosterone receptor antagonists (ARAs) (Figure 1).
Evidence supporting the use of the ARAs spironolactone (Aldactone) and eplerenone (Inspra) in heart failure has been growing, as has evidence of their usefulness in treating diabetes and chronic renal disease. Still, these drugs must be used cautiously, as they can cause hyperkalemia.
This paper will review the clinical use of ARAs in symptomatic systolic heart failure, their side effects, the findings and implications of recent trials, and controversies in this area, notably whether there is any evidence favoring the use of one drug over another.
ALDOSTERONE IN HEART FAILURE
Aldosterone, a hormone secreted by the zona glomerulosa of the adrenal gland, was first isolated by Simpson and Tait more than half a century ago.1 Later, it was found to promote reabsorption of sodium and excretion of potassium in the kidneys and hence was categorized as a mineralocorticoid hormone.
Release of aldosterone is stimulated by decreased renal perfusion via angiotensin II, hyperkalemia, and possibly adrenocorticotropic hormone.2 Aldosterone exerts its effects by binding to mineralocorticoid receptors in renal epithelial cells.
Aldosterone has several deleterious effects on the failing heart, primarily sodium and fluid retention, but also endothelial dysfunction, left ventricular hypertrophy, and myocardial fibrosis.2,3 Plasma aldosterone levels can be markedly elevated in patients with heart failure, likely due to activation of the renin-angiotensin-aldosterone system. Elevated aldosterone and angiotensin II levels have been associated with higher mortality rates.4
ALDOSTERONE ‘ESCAPE’ BLUNTS THE EFFECT OF ACE INHIBITORS AND ARBs
ACE inhibitors and ARBs have become standards of care for patients with systolic heart failure, and for many years, it was believed that these drugs suppressed aldosterone levels sufficiently. But elevated aldosterone levels have been noted in up to 38% of patients on chronic ACE inhibitor therapy.5 In one study, patients on dual blockade, ie, on both an ACE inhibitor and an ARB, had significantly lower aldosterone levels at 17 weeks of therapy, but not at 43 weeks.6 This phenomenon is known as “aldosterone escape.”
Several mechanisms might explain this phenomenon. Angiotensin II, a potent inducer of aldosterone, is “reactivated” during long-term ACE inhibitor therapy. Interestingly, patients progress toward aldosterone escape regardless of whether the ACE inhibitor dose is low or high.7 There is evidence that some aldosterone is produced by endothelial cells and vascular smooth muscle in the heart and blood vessels,8 but ACE inhibitors and ARBs suppress only the aldosterone secreted by the adrenal glands.
Regardless of the mechanism, aldosterone escape can blunt the effects of ACE inhibitors and ARBs, reducing their favorable effects on the risk of death in heart failure patients. This is the rationale for also using ARAs.
ARAs IN HEART FAILURE
Aldosterone acts by regulating gene expression after binding to mineralocorticoid receptors. These receptors are found not only in epithelial tissue in the kidneys and glands, but also in nonepithelial tissues such as cardiomyocytes, vessel walls, and the hippocampus of the brain.9 The nonepithelial effects were first demonstrated 2 decades ago by Brilla et al,10 who noted that chronically elevated aldosterone levels in rats promoted cardiac fibroblast growth, collagen accumulation, and, hence, ventricular remodeling.
The hypertensive effect of aldosterone may also be mediated through mineralocorticoid receptors in the brain. Gomez-Sanchez et al11 found that infusing aldosterone into the cerebral ventricles caused significant hypertension. A selective mineralocorticoid antagonist inhibited this effect when infused into the cerebral ventricles but not when given systemically.
In 1959, Cella and Kagawa created spironolactone, a nonselective ARA, by combining elements of progesterone for its antimineralocorticoid effect and elements of digitoxin for its cardiotonic effect.12 Although spironolactone is very effective in treating hypertension and heart failure, its use is limited by progestational and antiandrogenic side effects. This led, in 1987, to the invention by de Gasparo et al of a newer molecule, a selective ARA now called eplerenone.13 Although eplerenone may be somewhat less potent than spironolactone in blocking mineralocorticoid receptors, no significant difference in efficacy has been noted in randomized clinical trials, and its antiandrogenic action is negligible.12
Although these drugs target aldosterone receptors, newer drugs may target different aspects of mineralocorticoid activities, and thus the term “mineralocorticoid receptor antagonist” has been proposed.
TRIALS OF ARAs IN HEART FAILURE
An online data supplement that accompanies this paper at provides a detailed comparison of the three major trials of ARAs in patients with heart failure.
The Randomized Aldactone Evaluation Study (RALES)
The first major clinical trial of an ARA was the Randomized Aldactone Evaluation Study (RALES),14 a randomized, double-blind, controlled comparison of spironolactone and placebo.
The 1,663 patients in the trial all had severe heart failure (New York Heart Association class [NYHA] III and ambulatory class IV symptoms) and a left ventricular ejection fraction of 35% or less. Most were on an ACE inhibitor, a loop diuretic, and digoxin, but only 10% of patients in both groups were on a beta-blocker. Patients with chronic renal failure (serum creatinine > 2.5 mg/dL) or hyperkalemia (potassium > 5.0 mmol/L) were excluded.
RALES was halted early when an interim analysis at a mean follow-up of 24 months showed that significantly fewer patients were dying in the spironolactone group; their all-cause mortality rate was 30% lower (relative risk [RR] 0.70, 95% confidence interval [CI] 0.60–0.82, P < .001), and their cardiac mortality rate was 31% lower (RR 0.69, 95% CI 0.58–0.82, P < .001). This was concordant with a lower risk of both sudden cardiac death and death from progressive heart failure. The risk of hospitalization for cardiac causes was also 30% lower for patients in the spironolactone group, who also experienced significant symptom improvement.
Gynecomastia and breast pain occurred in about 10% of patients in the spironolactone group, and adverse effects leading to study drug discontinuation occurred in 2%.14
The Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS)
The next landmark trial of an ARA was the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS).15 A total of 6,632 patients were randomized to receive eplerenone or placebo in this multicenter, double-blind trial. To be enrolled, patients had to have acute myocardial infarction, a left ventricular ejection fraction of 40% or less, and either clinical signs of heart failure 3 to 14 days after the infarction or a history of diabetes mellitus. Patients were excluded if they had chronic kidney disease (defined as a serum creatinine > 2.5 mg/dL or an estimated glomerular filtration rate < 30 mL/min/1.73 m2) or hyperkalemia (a serum potassium > 5.0 mmol/L). All the patients received optimal medical therapy and reperfusion therapy, if warranted.
This event-driven trial was stopped when 1,012 deaths had occurred. During a mean follow-up of 16 months, there was a 15% lower rate of all-cause mortality in the eplerenone group (RR 0.85, 95% CI 0.75–0.96, P = .008) and a 13% lower rate of cardiovascular mortality (RR 0.83, 95% CI 0.72–0.94, P = .005). The reduction in the cardiovascular mortality rate was attributed to a 21% reduction in the rate of sudden cardiac deaths. The rate of heart failure hospitalization was also lower in the eplerenone group.
Serious hyperkalemia occurred significantly more frequently in the eplerenone group (5.5% vs 3.9%, P = .002), but similar rates of gynecomastia were observed. The incidence of hyperkalemia was higher in patients with a creatinine clearance less than 50 mL/min.
Further analyses revealed a 31% lower rate of all-cause mortality (95% CI 0.54–0.89, P = .004) and a 32% lower rate of cardiovascular mortality (95% CI 0.53–0.88, P = .003) at 30 days after randomization in the eplerenone group.16 Importantly, 25% of all deaths in the EPHESUS study during the 16-month follow-up period occurred in the first 30 days after randomization. The Kaplan-Meier survival curves showed separation as early as 5 days after randomization. Hence, the 30-day mortality results from EPHESUS further indicated that starting eplerenone early may be particularly beneficial.
The Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF)
After RALES and EPHESUS, a gap remained in our knowledge, ie, how to use ARAs in patients with mild heart failure, who account for most cases. This led to the EMPHASIS-HF (Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure) trial, which expanded the indications for ARAs to patients with chronic systolic heart failure with mild symptoms.17
In this double-blind trial, 2,737 patients with NYHA class II heart failure with a left ventricular ejection fraction of 35% or less were randomized to receive oral eplerenone 25 mg or placebo once daily. All patients were already on a beta-blocker; they were also all on an ACE inhibitor, an ARB, or both at the recommended or maximal tolerated dose. Patients with a glomerular filtration rate between 30 and 49 mL/min were started on alternate-day dosing, and those with glomerular filtration rates below 30 mL/min were excluded.
To ensure that the event rate was high enough to give this trial sufficient power:
- Only patients age 55 years or older were included
- Patients with a left ventricular ejection fraction greater than 30% were enrolled only if the QRS duration was greater than 130 ms (only 3.5% of patients in both groups were enrolled based on this criterion)
- Patients either had to have been hospitalized for cardiovascular reasons in the 6 months before randomization or had to have elevated natriuretic peptides (B-type natriuretic peptide [BNP] level > 250 pg/mL or N-terminal pro-BNP > 500 pg/mL in men and > 750 pg/mL in women).
The study was stopped early at a median follow-up of 21 months after an interim analysis showed a significantly lower rate of the primary composite end point (death from a cardiovascular cause or hospitalization for heart failure) in the eplerenone group: 18.3% vs 25.9% (hazard ratio [HR] 0.63, 95% CI 0.54– 0.74, P < .001). The rates of all-cause mortality were 12.5% vs 15.5% (HR 0.76, 95% CI 0.62–0.93, P = .008), and the rates of cardiovascular mortality were 10.8% vs 13.5% (HR 0.76, 95% CI 0.61–0.94, P = .01). Kaplan-Meier curves for all-cause mortality showed significant separation only after 1 year, which was not the case in EPHESUS and RALES. But the curves for hospitalization separated within a few weeks after randomization.
The incidence of hyperkalemia (serum potassium level > 5.5 mmol/L) was significantly higher in the eplerenone group (11.8% vs 7.2%, P < .001), but there was no statistically significant difference between groups when potassium levels above 6 mmol/L were considered (2.5% vs 1.9%, P = .29). This is despite one-third of patients having an estimated glomerular filtration rate less than 60 mL/min/1.73 m2. Breast symptoms were very rare, occurring in 1% or fewer patients in both groups. The discontinuation rate of the study drug was similar in both groups.
HOW DO ARAs PREVENT DEATH?
Multiple studies show that spironolactone and eplerenone lower blood pressure in a dose-related manner.18 These drugs reduce fluid volume and pulmonary congestion, which could have been the primary mechanism for the reduction in heart failure hospitalizations in the EMPHASIS-HF trial. But other mechanisms might explain the reduction in cardiovascular mortality rates in the trials summarized above.
Transcardiac extraction of aldosterone was increased in a study of patients with heart failure. 19 The transcardiac gradient of plasma aldosterone correlated with levels of procollagen III N-terminal propeptide, a biochemical marker of myocardial fibrosis. This suggests that aldosterone could be a stimulant of myocardial fibrosis. Spironolactone inhibited the transcardiac extraction of aldosterone in the same study.19
In another study,20 spironolactone significantly suppressed elevation of procollagen III N-terminal propeptide after myocardial infarction. It was also demonstrated that spironolactone prevented left ventricular remodeling after infarction, even in patients receiving an ACE inhibitor. Similar results, ie, decreased left ventricular myocardial fibrosis and remodeling, were noted in another trial in which eplerenone was added to an ARB.21
Myocardial fibrosis is a known substrate for ventricular arrhythmias. In a randomized study in 35 patients, spironolactone decreased the incidence of ventricular arrhythmias.22 This finding correlates with the decreased incidence of sudden cardiac death in the RALES and EPHESUS trials.
ADVERSE EFFECTS OF ARAs
Hyperkalemia, hyperkalemia, hyperkalemia
Potassium excretion is physiologically regulated by the serum aldosterone concentration and by the delivery of sodium to the distal nephron. Aldosterone increases potassium excretion. As a result of decreased renal perfusion that occurs with heart failure, sodium is intensely reabsorbed in the proximal tubule, and very little sodium reaches the distal nephron. When aldosterone receptors are blocked by ARAs, the risk of hyperkalemia increases.23
Other electrolyte abnormalities associated with ARAs are hyponatremia and hyperchloremic metabolic acidosis (Table 1). There could be a reversible decline in the glomerular filtration rate as well.24 Of note, most patients with chronic systolic heart failure in the RALES and EMPHASIS-HF trials were already receiving a diuretic; thus, the adverse effect profile of ARAs in otherwise euvolemic (or even hypovolemic) patients is not well appreciated.
Failure to closely monitor electrolyte levels increases the risk of hyperkalemia and renal failure, so there is a need for regular follow-up visits for patients taking an ARA.25 This was made clear when a population-based analysis from Canada compared the rates of hyperkalemia-related hospitalization and death before and after the RALES trial was published. The prescription rate for spironolactone increased threefold, but the rate of hyperkalemia-related hospitalization increased fourfold and the rate of death increased sixfold.26
Although caution is recommended when starting a patient on an ARA, a recent trial conducted in 167 cardiology practices noted that ARAs were the most underused drugs for heart failure. In this study, an ARA was prescribed to only 35% of eligible patients. The prescription rate was not significantly higher even in dedicated heart failure clinics.27 Possible reasons suggested by the authors were drug side effects, the need for closer monitoring of laboratory values, and a lack of knowledge.
A population-based analysis from the United Kingdom found a significant increase over time in spironolactone prescriptions after the release of the RALES trial results, but there was no increase in the rate of serious hyperkalemia (serum potassium > 6 mmol/L) or hyperkalemia-related hospitalization.28 The authors suggested that careful monitoring could prevent hyperkalemia-related complications. They also observed that 75% of patients who had spironolactone-associated hyperkalemia were over 65 years old. Hence, we recommend closer monitoring when starting an elderly patient on an ARA.
Breast, gastrointestinal symptoms
The nonselective ARA spironolactone is associated with antiandrogenic side effects. In a smaller study in patients with resistant hypertension, Nishizaka et al noted that low-dose spironolactone (up to 50 mg/day) was associated with breast tenderness in about 10%.29 Breast symptoms with spironolactone are dose-related, and the incidence can be as high as 50% when the drug is used in dosages of 150 mg/day or higher.30
In one population-based case-control study, spironolactone was associated with a 2.7 times higher risk of gastrointestinal side effects (bleeding or ulcer).31
ARAs IN HEART FAILURE WITH PRESERVED EJECTION FRACTION
The concept of diastolic heart failure or “heart failure with preserved ejection fraction” has been growing. A significant proportion of patients with a diagnosis of heart failure have preserved left ventricular ejection fraction (≥ 50%) and diastolic dysfunction.
Despite multiple trials, no treatment has been shown to lower the mortality rate in heart failure with preserved ejection fraction.32,33 A recently published randomized controlled trial in 44 patients with this condition showed reduction in serum biochemical markers of collagen turnover and improvement in diastolic function with ARAs, but there was no difference in exercise capacity.34 A larger double-blind randomized control trial, Aldosterone Receptor Blockade in Diastolic Heart Failure (Aldo-DHF), is under way to evaluate the effects of ARAs on exercise capacity and diastolic function in patients with heart failure with preserved ejection fraction.35
In January 2012, the Trial of Aldosterone Antagonist Therapy in Adults With Preserved Ejection Fraction Congestive Heart Failure (TOPCAT) completed enrollment of 3,445 patients to study the effect of ARAs in reducing the composite end point of cardiovascular mortality, aborted cardiac arrest, and heart failure hospitalization. Long-term follow-up of this event-driven study is currently under way.
ARAs IN DIABETES MELLITUS AND CHRONIC KIDNEY DISEASE
Under physiologic conditions, the serum aldosterone level is regulated by volume status through the renin-angiotensin system. But in patients with chronic kidney disease, the serum aldosterone level could be elevated without renin-angiotensin system stimulation.36
High aldosterone levels were associated with proteinuria and glomerulosclerosis in rats.37 In a study in 83 patients, aldosterone receptor blockade was shown to decrease proteinuria and possibly to retard the progression of chronic kidney disease. In this trial, baseline serum aldosterone levels correlated with proteinuria.38 Animal studies suggest that adipocyte-derived factors may stimulate aldosterone, which may be relevant in patients who have both chronic kidney disease and metabolic syndrome.39
The impact of ARAs in patients with diabetes mellitus is often overlooked. In EPHESUS, diabetes mellitus was an inclusion criterion even in the absence of heart failure signs and symptoms in the postinfarction setting of impaired left ventricular ejection fraction.15
In patients with diabetic nephropathy, there is growing evidence that ARAs can decrease proteinuria, even if the serum aldosterone level is normal. For example, in a study in 20 patients with diabetic nephropathy, spironolactone reduced proteinuria by 32%. This reduction was independent of serum aldosterone levels.40
In diabetic rats, hyperglycemia was noted to cause podocyte injury through mineralocorticoid receptor-mediated production of reactive oxygen species, independently of serum aldosterone levels. Spironolactone decreased the production of reactive oxygen species, thereby potentially reducing proteinuria.41
RECOMMENDATIONS ARE BEING REVISED
The most recent joint guidelines of the American Heart Association and the American College of Cardiology for the management of heart failure42 were published in 2009, which was before the EMPHASIS-HF results. An update is expected soon. In the 2009 version, ARAs received a class I recommendation for patients with moderately severe to severe symptoms, decreased ejection fraction, normal renal function, and normal potassium levels. The guidelines also said that the risks of ARAs may outweigh their benefits if regular monitoring is not possible.
The recommended starting dosage is 12.5 mg/day of spironolactone or 25 mg/day of eplerenone; the dose can be doubled, if tolerated.
Close monitoring is recommended, ie, measuring serum potassium and renal function 3 and 7 days after starting therapy and then monthly for the first 3 months. Closer monitoring is needed if an ACE inhibitor or an ARB is added later. In elderly patients, the glomerular filtration rate is preferred over the serum creatinine level, and ARA therapy is not advisable if the glomerular filtration rate is less than 30 mL/min/1.73 m2.
Avoid concomitant use of the following:
- Potassium supplements (unless persistent hypokalemia is present)
- Nonsteroidal anti-inflammatory drugs
- An ACE inhibitor and an ARB in combination
- A high dose of an ACE inhibitor or ARB.
Conditions that can lead to dehydration (eg, diarrhea, excessive use of diuretics) or acute illness should warrant reduction (or even withholding) of ARAs. When to discontinue ARA therapy is not well described, nor is the safety of starting ARAs in the hospital. However, it is clear that many patients who are potentially eligible for ARAs are not prescribed them.43
The guidelines are currently being revised, and will likely incorporate the new data from EMPHASIS-HF to extend to a broader population. The benefits of ARAs can be met only if the risks are minimized.
WHICH ARA IS BETTER?
The pharmacologic differences between the two ARAs have been described earlier, and guidelines have advocated evidence-based use of ARAs for their respective indications. There have been no large-scale, head-to-head comparisons of spironolactone and eplerenone in the heart failure population, and in clinical practice the drugs are prescribed interchangeably in most patients.
A double-blind randomized controlled trial in 141 patients with hypertension and primary hyperaldosteronism found that spironolactone lowered diastolic blood pressure more, but it also caused antiandrogenic effects more often.44
There is some evidence to suggest that eplerenone has a better metabolic profile than spironolactone. The data came from a small randomized controlled trial in 107 stable outpatients with mild heart failure.45 Patients who were prescribed spironolactone had a higher cortisol level and hemoglobin A1c level 4 months after starting treatment. This effect was not seen in patients who were on eplerenone. However, these findings need to be confirmed in larger trials.
While the differences between the two drugs remain to be determined, the most important differences in clinical practice are selectivity for receptors (and hence their antiandrogenic side effects) and price. Even though it is available as a generic drug, eplerenone still costs at least three times more than spironolactone for the same dosage and indication.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
- Simpson SA, Tait JF, Bush IE. Secretion of a salt-retaining hormone by the mammalian adrenal cortex. Lancet 1952; 2:226–228.
- Struthers AD, MacDonald TM. Review of aldosterone- and angiotensin II-induced target organ damage and prevention. Cardiovasc Res 2004; 61:663–670.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the “aldosterone receptor blockade in diastolic heart failure” trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Swedberg K, Eneroth P, Kjekshus J, Wilhelmsen L. Hormones regulating cardiovascular function in patients with severe congestive heart failure and their relation to mortality. CONSENSUS Trial Study Group. Circulation 1990; 82:1730–1736.
- MacFadyen RJ, Lee AF, Morton JJ, Pringle SD, Struthers AD. How often are angiotensin II and aldosterone concentrations raised during chronic ACE inhibitor treatment in cardiac failure? Heart 1999; 82:57–61.
- McKelvie RS, Yusuf S, Pericak D, et al. Comparison of candesartan, enalapril, and their combination in congestive heart failure: randomized evaluation of strategies for left ventricular dysfunction (RESOLVD) pilot study. The RESOLVD Pilot Study Investigators. Circulation 1999; 100:1056–1064.
- Tang WH, Vagelos RH, Yee YG, et al. Neurohormonal and clinical responses to high- versus low-dose enalapril therapy in chronic heart failure. J Am Coll Cardiol 2002; 39:70–78.
- Weber KT. Aldosterone in congestive heart failure. N Engl J Med 2001; 345:1689–1697.
- Funder JW. The role of aldosterone and mineralocorticoid receptors in cardiovascular disease. Am J Cardiovasc Drugs 2007; 7:151–157.
- Brilla CG, Pick R, Tan LB, Janicki JS, Weber KT. Remodeling of the rat right and left ventricles in experimental hypertension. Circ Res 1990; 67:1355–1364.
- Gomez-Sanchez EP, Fort C, Thwaites D. Central mineralocorticoid receptor antagonism blocks hypertension in Dahl S/JR rats. Am J Physiol 1992; 262:E96–E99.
- Garthwaite SM, McMahon EG. The evolution of aldosterone antagonists. Mol Cell Endocrinol 2004; 217:27–31.
- de Gasparo M, Joss U, Ramjoué HP, et al. Three new epoxy-spirolactone derivatives: characterization in vivo and in vitro. J Pharmacol Exp Ther 1987; 240:650–656.
- Pitt B, Zannad F, Remme WJ, et al. The effect of spironolactone on morbidity and mortality in patients with severe heart failure. Randomized Aldactone Evaluation Study Investigators. N Engl J Med 1999; 341:709–717.
- Pitt B, Remme W, Zannad F, et al; Eplerenone Post-Acute Myocardial Infarction Heart Failure Efficacy and Survival Study Investigators. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. N Engl J Med 2003; 348:1309–1321.
- Pitt B, White H, Nicolau J, et al; EPHESUS Investigators. Eplerenone reduces mortality 30 days after randomization following acute myocardial infarction in patients with left ventricular systolic dysfunction and heart failure. J Am Coll Cardiol 2005; 46:425–431.
- Zannad F, McMurray JJ, Krum H, et al; EMPHASIS-HF Study Group. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med 2011; 364:11–21.
- Weinberger MH, Roniker B, Krause SL, Weiss RJ. Eplerenone, a selective aldosterone blocker, in mild-to-moderate hypertension. Am J Hypertens 2002; 15:709–716.
- Tsutamoto T, Wada A, Maeda K, et al. Spironolactone inhibits the transcardiac extraction of aldosterone in patients with congestive heart failure. J Am Coll Cardiol 2000; 36:838–844.
- Hayashi M, Tsutamoto T, Wada A, et al. Immediate administration of mineralocorticoid receptor antagonist spironolactone prevents postinfarct left ventricular remodeling associated with suppression of a marker of myocardial collagen synthesis in patients with first anterior acute myocardial infarction. Circulation 2003; 107:2559–2565.
- Fraccarollo D, Galuppo P, Schmidt I, Ertl G, Bauersachs J. Additive amelioration of left ventricular remodeling and molecular alterations by combined aldosterone and angiotensin receptor blockade after myocardial infarction. Cardiovasc Res 2005; 67:97–105.
- Ramires FJ, Mansur A, Coelho O, et al. Effect of spironolactone on ventricular arrhythmias in congestive heart failure secondary to idiopathic dilated or to ischemic cardiomyopathy. Am J Cardiol 2000; 85:1207–1211.
- Palmer BF. Managing hyperkalemia caused by inhibitors of the reninangiotensin-aldosterone system. N Engl J Med 2004; 351:585–592.
- Sica DA. The risks and benefits of therapy with aldosterone receptor antagonist therapy. Curr Drug Saf 2007; 2:71–77.
- Shah KB, Rao K, Sawyer R, Gottlieb SS. The adequacy of laboratory monitoring in patients treated with spironolactone for congestive heart failure. J Am Coll Cardiol 2005; 46:845–849.
- Juurlink DN, Mamdani MM, Lee DS, et al. Rates of hyperkalemia after publication of the Randomized Aldactone Evaluation Study. N Engl J Med 2004; 351:543–551.
- Albert NM, Fonarow GC, Yancy CW, et al. Influence of dedicated heart failure clinics on delivery of recommended therapies in outpatient cardiology practices: findings from the Registry to Improve the Use of Evidence-Based Heart Failure Therapies in the Outpatient Setting (IMPROVE HF). Am Heart J 2010; 159:238–244.
- Wei L, Struthers AD, Fahey T, Watson AD, Macdonald TM. Spironolactone use and renal toxicity: population based longitudinal analysis. BMJ 2010; 340:c1768.
- Nishizaka MK, Zaman MA, Calhoun DA. Efficacy of low-dose spironolactone in subjects with resistant hypertension. Am J Hypertens 2003; 16:925–930.
- Jeunemaitre X, Chatellier G, Kreft-Jais C, et al. Efficacy and tolerance of spironolactone in essential hypertension. Am J Cardiol 1987; 60:820–825.
- Verhamme K, Mosis G, Dieleman J, Stricker B, Sturkenboom M. Spironolactone and risk of upper gastrointestinal events: population based case-control study. BMJ 2006; 333:330.
- Massie BM, Carson PE, McMurray JJ, et al; I-PRESERVE Investigators. Irbesartan in patients with heart failure and preserved ejection fraction. N Engl J Med 2008; 359:2456–2467.
- Yusuf S, Pfeffer MA, Swedberg K, et al; CHARM Investigators and Committees. Effects of candesartan in patients with chronic heart failure and preserved left-ventricular ejection fraction: the CHARM-Preserved Trial. Lancet 2003; 362:777–781.
- Deswal A, Richardson P, Bozkurt B, Mann DL. Results of the Randomized Aldosterone Antagonism in Heart Failure With Preserved Ejection Fraction Trial (RAAM-PEF). J Card Fail 2011; 17:634–642.
- Edelmann F, Schmidt AG, Gelbrich G, et al. Rationale and design of the ‘aldosterone receptor blockade in diastolic heart failure’ trial: a double-blind, randomized, placebo-controlled, parallel group study to determine the effects of spironolactone on exercise capacity and diastolic function in patients with symptomatic diastolic heart failure (Aldo-DHF). Eur J Heart Fail 2010; 12:874–882.
- Hené RJ, Boer P, Koomans HA, Mees EJ. Plasma aldosterone concentrations in chronic renal disease. Kidney Int 1982; 21:98–101.
- Greene EL, Kren S, Hostetter TH. Role of aldosterone in the remnant kidney model in the rat. J Clin Invest 1996; 98:1063–1068.
- Bianchi S, Bigazzi R, Campese VM. Long-term effects of spironolactone on proteinuria and kidney function in patients with chronic kidney disease. Kidney Int 2006; 70:2116–2123.
- Nagase M, Yoshida S, Shibata S, et al. Enhanced aldosterone signaling in the early nephropathy of rats with metabolic syndrome: possible contribution of fat-derived factors. J Am Soc Nephrol 2006; 17:3438–3446.
- Schjoedt KJ, Rossing K, Juhl TR, et al. Beneficial impact of spironolactone on nephrotic range albuminuria in diabetic nephropathy. Kidney Int 2006; 70:536–542.
- Toyonaga J, Tsuruya K, Ikeda H, et al. Spironolactone inhibits hyperglycemia-induced podocyte injury by attenuating ROS production. Nephrol Dial Transplant 2011; 26:2475–2484.
- Hunt SA, Abraham WT, Chin MH, et al. 2009 focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults: a report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines: developed in collaboration with the International Society for Heart and Lung Transplantation. Circulation 2009; 119:e391–e479.
- Albert NM, Yancy CW, Liang L, et al. Use of aldosterone antagonists in heart failure. JAMA 2009; 302:1658–1665.
- Parthasarathy HK, Ménard J, White WB, et al. A double-blind, randomized study comparing the antihypertensive effect of eplerenone and spironolactone in patients with hypertension and evidence of primary aldosteronism. J Hypertens 2011; 29:980–990.
- Yamaji M, Tsutamoto T, Kawahara C, et al. Effect of eplerenone versus spironolactone on cortisol and hemoglobin A1(c) levels in patients with chronic heart failure. Am Heart J 2010; 160:915–921.
KEY POINTS
- Although caution is advised in starting ARAs, these drugs are commonly underused in heart failure.
- Aldosterone “escape” can blunt the effects of angiotensin-converting enzyme inhibitors and angiotensin receptor blockers. This is the rationale for also using ARAs.
- The major trials of ARAs in heart failure to date have been the Randomized Aldactone Evaluation Study (RALES), the Eplerenone Post-acute Myocardial Infarction Heart Failure Efficacy and Survival Study (EPHESUS), and the Eplerenone in Mild Patients Hospitalization and Survival Study in Heart Failure (EMPHASIS-HF).
- Close monitoring is essential when starting an ARA, as severe hyperkalemia and renal insufficiency can occur.
Immune thrombocytopenia in adults: An update
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14

A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7

Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14
A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7
Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
Immune thrombocytopenia (ITP), formerly known as idiopathic thrombocytopenic purpura, is an autoimmune disorder characterized by a low platelet count and increased risk of mucocutaneous bleeding. During the last decade its management has changed, with the advent of new medications and with increased awareness of treatment side effects. This article will focus on the pathophysiology, diagnosis, and management of ITP in adults.
A SLIGHT FEMALE PREDOMINANCE UNTIL AGE 65
The estimated age-adjusted prevalence of ITP in the United States is 9.5 to 23.6 cases per 100,000.1 In a recent study in the United Kingdom, the incidence was 4.4 per 100,000 patient-years among women and 3.4 among men.2 A slight female predominance was seen until age 65; thereafter, the incidence rates in men and women were about equal.
INCREASED PLATELET DESTRUCTION AND DECREASED PRODUCTION
ITP is a complex immune process in which cellular and humoral immunity are involved in the destruction of platelets3 as well as impaired platelet production. Several theories have emerged in the last decade to explain this autoimmune process.
Autoantibodies form against platelets
The triggering event for antibody initiation in ITP is unknown.3 Autoantibodies (mostly immunoglobulin G [IgG] but sometimes IgM and IgA) are produced against the platelet membrane glycoprotein GPIIb-IIIa. The antibody-coated platelets are rapidly cleared by the reticuloendothelial system in the spleen and liver, in a process mediated by Fc-receptor expression on macrophages and dendritic cells. Autoantibodies may also affect platelet production by inhibiting megakaryocyte maturation and inducing apoptosis.4,5
Patients with ITP also have CD4+ T cells that are autoreactive to GPIIb-IIIa and that stimulate B-cell clones to produce antiplatelet antibodies. Although autoreactive T cells are present in healthy individuals, they appear to be activated in patients with ITP by exposure to fragments of GPIIb-IIIa rather than native GPIIb-IIIa proteins.6 Activated macrophages internalize antibody-coated platelets and degrade GPIIb-IIIa and other glycoproteins to form “cryptic” epitopes that are expressed on the macrophage surface as novel peptides that induce further proliferation of CD4+ T-cell clones. Epitope spread thereby sustains a continuous loop that amplifies the production of GPIIb-IIIa antibodies.7
Defective T-regulatory cells appear to be critical to the pathogenesis of ITP by breaking self-tolerance, allowing the autoimmune process to progress.8 This, together with several other immune mechanisms such as molecular mimicry, abnormal cytokine profile, and B-cell abnormalities, may lead to enhanced platelet clearance.9
In addition to destroying platelets, antibodies may impair platelet production.10 Good evidence for platelets being underproduced in patients with ITP is that treating with thrombopoietin agonists results in increased platelet counts.
A DIAGNOSIS OF EXCLUSION
ITP is defined as isolated thrombocytopenia with no clinically apparent associated conditions or other causes of thrombocytopenia.11 No diagnostic criteria currently exist, and the diagnosis is established only after excluding other causes of thrombocytopenia.
A recent report12 from an international working group established a platelet count threshold of less than 100 × 109/L for diagnosing ITP, down from the previous threshold of 150 × 109/L. The panel also recommended using the term “immune” rather than “idiopathic” thrombocytopenia, emphasizing the role of underlying immune mechanisms. The term “purpura” was removed, because many patients have no or minimal signs of bleeding at the time of diagnosis.12
The 2011 American Society of Hematology’s evidenced-based guidelines for the treatment of ITP present the most recent authoritative diagnostic and therapeutic recommendations.13
ITP is considered to be primary if it occurs in isolation, and secondary if it is associated with an underlying disorder. It is further classified according to its duration since diagnosis: newly diagnosed (< 3 months), persistent (3−12 months), and chronic (> 12 months).
In adults, ITP tends to be chronic, presenting with a more indolent course than in childhood, and unlike childhood ITP, infrequently following a viral infection.
Clinical features associated with ITP are related to thrombocytopenia: petechiae (pinpoint microvascular hemorrhages that do not blanch with pressure), purpura (appearing like large bruises), epistaxis (nosebleeds), menorrhagia, gum bleeding, and other types of mucocutaneous bleeding. Other common clinical features include fatigue, impaired quality of life, and treatment-related side effects (eg, infection).14
A low platelet count may be the sole initial manifestation. The patient’s history, physical examination, blood counts, and findings on blood smear are essential to rule out other diagnoses. Few diagnostic tests are useful in the initial evaluation (Table 1). Abnormalities in the blood count or blood smear may be further investigated with bone marrow biopsy but is not required if the patient has typical features of ITP, regardless of age.
Because there are no specific criteria for diagnosing ITP, other causes of thrombocytopenia must be excluded. The differential diagnosis can be further classified as ITP due to other underlying disease (ie, secondary ITP) vs nonautoimmune causes that are frequently encountered in clinical practice.
SECONDARY ITP
The differential diagnosis of thrombocytopenia due to known underlying immune disease includes the following:
Drug-induced ITP
Recurrent episodes of acute thrombocytopenia not explained by other causes should trigger consideration of drug-induced thrombocytopenia. 11 Patients should be questioned about drug use, especially of sulfonamides, antiepileptics, and quinine. Thrombocytopenia usually occurs 5 to 7 days after beginning the inciting drug for the first time and more quickly when the drug is given intermittently. Heparin is the most common cause of drug-related thrombocytopenia among hospitalized patients; the mechanism is unique and involves formation of a heparin-PF4 immune complex.
Human immunodeficiency virus infection
Approximately 40% of patients with human immunodeficiency virus (HIV) infection develop thrombocytopenia at some time.15 HIV infection can initially manifest as isolated thrombocytopenia and is sometimes clinically indistinguishable from chronic ITP, making it an important consideration in a newly diagnosed case of thrombocytopenia.
The mechanism of thrombocytopenia in early HIV is similar to that in primary ITP: as the disease progresses, low platelet counts can result from ineffective hematopoiesis due to megakaryocyte infection and marrow infiltration.16
Hepatitis C virus infection
Hepatitis C virus (HCV) infection can also cause immune thrombocytopenia. A recent study demonstrated the potential of the HCV core envelope protein 1 to induce antiplatelet antibodies (to platelet surface integrin GPIIIa49-66) by molecular mimicry.17 Other causes of thrombocytopenia in HCV infection may be related to chronic liver disease, such as portal hypertension-related hypersplenism, as well as decreased thrombopoietin production.18 Antiviral treatment with pegylated interferon may also cause mild thrombocytopenia.19
Helicobacter pylori
The association between H pylori infection and ITP remains uncertain. Eradication of infection appears to completely correct ITP in some places where the prevalence of H pylori is high (eg, Italy and Japan) but not in the United States and Canada, where the prevalence is low.20 The different response may be due not only to the differences in prevalence, but to different H pylori genotypes: most H pylori strains in Japan express CagA, whereas the frequency of CagA-positive strains is much lower in western countries.20
In areas where eradication therapy may be useful, the presence of H pylori infection should be determined by either a urea breath test or stool antigen testing.
Lymphoproliferative disorders
Secondary forms of ITP can occur in association with chronic lymphocytic leukemia, non-Hodgkin lymphoma, and Hodgkin lymphoma. These diagnoses should especially be considered in patients presenting with thrombocytopenia accompanied by systemic illness. ITP occurs in at least 2% of patients with chronic lymphocytic leukemia and is usually difficult to distinguish from thrombocytopenia secondary to marrow infiltration or from fludarabine (Fludora) therapy.21
It is especially important to determine if a lymphoproliferative disorder is present because it changes the treatment of ITP. Treatment of ITP complicating chronic lymphocytic leukemia is challenging and includes corticosteroids and steroid-sparing agents such as cyclosporine (Gengraf, Neoral, Sandimmune), rituximab (Rituxan), and intravenous immunoglobulin.22
Systemic lupus erythematosus and other autoimmune diseases
Thrombocytopenia is a frequent clinical manifestation of systemic lupus erythematosus, occurring in 7% to 30% of patients,23 and is an independent risk factor for death.24 Lupus should be suspected in patients with ITP who have multiorgan involvement and other clinical and laboratory abnormalities. A small percentage of patients with ITP (about 2%−5%) develop lupus after several years.21
Thrombocytopenia can also result from other autoimmune disorders such as antiphospholipid antibody syndrome25 and autoimmune thyroid diseases as well as immunodeficient states such as IgA deficiency and common variable immunodeficiency with low IgG levels.
NONAUTOIMMUNE THROMBOCYTOPENIA
Thrombocytopenia can also be caused by a number of nonautoimmune conditions.
Pseudothrombocytopenia
Pseudothrombocytopenia can occur if ex-vivo agglutination of platelets is induced by antiplatelet antibodies to EDTA, a standard blood anticoagulant. Automated counters cannot differentiate the agglutinated platelet clumps from individual cells such as red cells. This can frequently be overcome by running the counts in a citrate or ACD reagent tube. A peripheral blood smear can demonstrate whether platelet clumps are present.
Thrombotic thrombocytopenic purpura
Thrombotic thrombocytopenic purpura presents with thrombocytopenia, purpura, and anemia. Associated clinical abnormalities (fever, neurologic symptoms, and renal failure) and the presence of fragmented red cells on blood smear help to distinguish it from ITP. Plasma exchange is the treatment of choice.
Gestational thrombocytopenia
Five percent of pregnant women develop mild thrombocytopenia (platelet counts typically > 70 × 109/L) near the end of gestation.26 It requires no treatment and resolves after delivery. The fetus’ platelet count remains unaffected.
Gestational thrombocytopenia should be differentiated from the severe thrombocytopenia of preeclampsia and HELLP syndrome (hemolysis, elevated liver enzymes, and low platelet count), which requires immediate attention.
Myelodysplastic syndrome
Myelodysplastic syndrome is common among elderly patients and should be considered in cases of unexplained cytopenia and abnormalities in the peripheral blood smear suggestive of dysplastic cytologic features. It can be diagnosed by bone marrow biopsy. Thrombocytopenia occurs in about 40% to 65% of cases of myelodysplastic syndrome.27
MANAGE ITP TO KEEP PLATELET COUNT ABOVE 30 × 109/L
ITP does not necessarily require treatment, and the initial challenge is to determine whether treatment or observation is indicated. Treatment is based on two major factors: the platelet count and degree of bleeding. The goals of management are to achieve a safe platelet count to prevent serious bleeding while minimizing treatment-related toxicity.7
Adults with platelet counts of less than 30 × 109/L are usually treated. In multiple large cohort studies, patients with platelet counts above that level have been safely observed without treatment.11,28
Table 2 outlines a comprehensive approach to therapy.
INITIAL TREATMENT: STEROIDS AND IMMUNOGLOBULINS
Oral corticosteroids are the initial agents of choice
Oral prednisone 1 mg/kg/day in tapering doses for 4 to 6 weeks is the most common initial regimen. Other regimens, such as high-dose dexamethasone (Decadron) (40 mg daily for 4 days per month) for several cycles, have been reported to be more effective29 but have not been studied in head-to-head trials with oral prednisone.
Due to their effectiveness, low cost, and convenience of use, corticosteroids have been the backbone of initial treatment in ITP. However, in most patients the platelet count decreases once the dose is tapered or stopped; remission is sustained in only 10% to 30% of cases.30 Continuation of corticosteroids is limited by long-term complications such as opportunistic infections, osteoporosis, and emotional lability.31
Intravenous immunoglobulin and anti-D immunoglobulin are alternatives
Intravenous immunoglobulin is recommended for patients who have not responded to corticosteroids and is often used in pregnancy. It is thought to act by blocking Fc receptors in the reticuloendothelial system. Intravenous immunoglobulin rapidly increases platelet counts in 65% to 80% of patients,32 but the effect is transient and the drug requires frequent administration. It is usually well tolerated, although about 5% of patients experience headache, chills, myalgias, arthralgias, and back pain. Rare, serious complications include thrombotic events, anaphylaxis (in IgA-deficient patients), and renal failure.
Anti-D immunoglobulin, a pooled IgG product, is derived from the plasma of Rh(D)-negative donors and can be given only to patients who are Rh(D)-positive. Response rates as high as 70% have been reported, with platelet effects lasting for more than 21 days.33 Studies have shown better results at a high dose (75 μg/kg) than with the approved dose of 50 μg/kg.34
Anti-D immunoglobulin can also be given intermittently whenever the platelet count falls below a specific level (ie, 30 × 109/L). This allows some patients to avoid splenectomy and may even trigger long-term remission.32
Common side effects of anti-D immunoglobulin include fever and chills; these can be prevented by premedication with acetaminophen or corticosteroids. Rare but fatal cases of intravascular hemolysis, renal failure, and disseminated intravascular coagulation have been reported, precluding its use for ITP in some countries, including those of the European Union.
Emergency treatment: Combination therapy
Evidence-based guidelines are limited for treating patients with active bleeding or who are at high risk of bleeding. For uncontrolled bleeding, a combination of first-line therapies is recommended, using prednisone and intravenous immunoglobulin.35 Other options include high-dose methylprednisolone and platelet transfusions, alone or in combination with intravenous immunoglobulin.36
SECOND-LINE TREATMENTS
Splenectomy produces complete remission in most patients
Patients who relapse and have a platelet count of less than 20 × 109/L are traditionally considered for splenectomy. More than two-thirds of patients respond with no need for further treatment.37
Although splenectomy has the highest rate of durable platelet response, the risks associated with surgery are an important concern. Even with a laparoscopic splenectomy, complications occur in 10% of patients and death in 0.2%. Long-term risks include the rare occurrence of sepsis with an estimated mortality rate of 0.73 per 1,000 patient-years, and possible increased risk of thrombosis.38,39
Adherence to recommended vaccination protocols and early administration of antibiotics for systemic febrile illness reduce the risk of sepsis.40 Patients are advised to receive immunization against encapsulated bacteria with pneumococcal, Haemophilus influenzae type b, and meningococcal vaccines. These vaccines should be given at least 2 weeks before elective splenectomy.41
Treatment of patients refractory to splenectomy is challenging and requires further immunosuppressive therapy, which is associated with an increased risk of infections and infection-related deaths.42
Rituximab in addition to or possibly instead of splenectomy
Rituximab (Rituxan) is a chimeric anti-CD20 monoclonal antibody that targets B cells. Although initially approved for treatment of lymphomas, rituximab has gained popularity in treating ITP due to its safety profile and ability to deplete CD20+ B cells responsible for antiplatelet antibody production by Fc-mediated cell lysis.
In the largest systematic review of published reports of rituximab use in ITP (19 studies, 313 patients), Arnold and colleagues43 reported an overall platelet response (defined as platelet count > 50 × 109/L) in 62.5% (95% confidence interval [CI] 52.6%−72.5%) of patients. The median duration of response was 10.5 months (range 3–20), and median follow-up was 9.5 months (range 2–25). Nearly all patients had received corticosteroid treatment and half of them had undergone splenectomy.
Rituximab has also been investigated as an alternative to splenectomy. In a prospective, single-arm, phase 2 trial, 60 patients with chronic ITP (platelet counts < 30 × 109/L) for whom one or more previous treatments had failed received rituximab infusions and were followed for up to 2 years. A good response (defined as a platelet count ≥ 50 × 109/L, with at least a doubling from baseline) was obtained in 24 (40%) of 60 patients (95% CI 28%–52%) at 1 year and 33.3% at 2 years. The authors concluded that rituximab could be used as a presplenectomy therapeutic option, particularly in patients with chronic ITP who are at increased surgical risk or who are reluctant to undergo surgery.44 Based on these results, rituximab may spare some patients from splenectomy, or at least delay it. However, it has never been tested in randomized controlled trials to establish its role as a splenectomy-sparing agent in ITP.
Side effects include infusion reactions, which are usually mild but in rare cases can be severe. Recently, progressive multifocal leukoencephalopathy has been recognized as a complication of rituximab treatment in patients with lymphoproliferative and autoimmune disorders.45 Although this complication is rare in patients with ITP, careful monitoring is required until additional long-term safety data are available.
Thrombopoietic receptor agonists require continuous treatment
In the early 1990s, recombinant thrombopoietin was tested in clinical studies. These were halted when antibodies developed to recombinant thrombopoietin that cross-reacted with endogenous thrombopoietin, resulting in severe thrombocytopenia.46
This led to the development of nonimmunogenic thrombopoietin receptor agonists that mimic the effect of thrombopoietin and stimulate the production of platelets. In 2008, the US Food and Drug Administration approved two drugs of this class for treating ITP: romiplostim (Nplate) and eltrombopag (Promacta). They are mainly used to treat patients with chronic ITP who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy.
Although well tolerated and effective in increasing platelet counts, these agents share common drawbacks. They do not modify the course of the disease, they are used only to sustain the platelet count, they require repeated administration, and they must be given for about 7 days to achieve an adequate platelet response, so they cannot be used in emergencies. Long-term adverse effects include bone marrow fibrosis and thrombosis.
Romiplostim is a synthetic peptide capable of binding to the thrombopoietin receptor c-Mpl. It has no sequence homology with endogenous thrombopoietin,47 so does not induce cross-reacting antibodies. It has a half-life of 120 to 160 hours and is usually given subcutaneously 1 to 10 μg/kg weekly.
Phase III clinical trials have shown the effectiveness of romiplostim in attaining a durable platelet response (platelet count > 50 × 109/L) in splenectomized and nonsplenectomized populations. It is well tolerated, and only two uncommon serious adverse effects have been reported: bone marrow reticulin formation and thromboembolism.48
A long-term open-label extension study of 142 patients treated with romiplostim for up to 156 weeks showed that 124 (87%) achieved a platelet count of more than 50 × 109/L at some point, and 84% of patients were able to reduce or discontinue concurrent medications for ITP.49
Kuter et al,50 in a randomized controlled trial, confirmed the efficacy of romiplostim in attaining durable increased platelet counts. Patients treated with romiplostim at a mean weekly dose of 3.9 μg/kg ± 2.1 μg/kg demonstrated a higher rate of platelet response, lower incidence of treatment failure, and improved quality of life vs patients treated with standard care.
Eltrombopag is a nonpeptide thrombopoietin agonist that binds to the transmembrane domain of the thrombopoietin receptor and stimulates the proliferation and differentiation of megakaryocytes in bone marrow. It is given orally in doses of 25 to 75 mg daily.
Eltrombopag has been shown to be effective in increasing platelet counts in chronic ITP.51 In a phase III trial conducted by Cheng and colleagues, 197 patients were randomized to eltrombopag or placebo.52 Patients treated with eltrombopag were eight times more likely to achieve platelet counts of more than 50 × 109/L during the 6-month treatment period (odds ratio 8.2, 95% CI 4.32–15.38, P < .001) vs placebo. Patients treated with eltrombopag had fewer bleeding episodes and were more likely to reduce or discontinue the dose of concurrent ITP medications. The only significant side effect seen was a rise in aminotransferases (seen in 7% of eltrombopag recipients vs 2% with placebo).52
Additional thrombopoietin agonists under investigation include ARK-501, totrombopag, and LGD-4665. MDX-33, a monoclonal antibody against the Fc-receptor, is also being studied; it acts by preventing opsonization of autoantibody-coated platelets.53
THIRD-LINE TREATMENTS FOR REFRACTORY CASES
Patients with ITP that is resistant to standard therapies have an increased risk of death, disease, and treatment-related complications.28,42
Combination chemotherapy
Immunosuppressants such as azathioprine (Imuran), cyclosporine (Neoral, Sandimmune), cyclophosphamide (Cytoxan), and mycophenolate (CellCept) were used in the past in single-agent regimens with some efficacy, but their use was limited due to drug-related toxicity and a low safety profile.3 However, there is increasing evidence for a role of combination chemotherapy to treat chronic refractory ITP to achieve greater efficacy and fewer adverse effects.54
Arnold and colleagues55 reported that combined azathioprine, mycophenolate, and cyclosporine achieved an overall response (platelet count > 30 × 109/L and doubling of the baseline) in 14 (73.7%) of 19 patients with chronic refractory ITP, lasting a median of 24 months.
Hematopoietic stem cell transplantation
Hematopoietic stem cell transplantation has provided remission in a limited number of patients. However, it is associated with fatal toxicities such as graft-vs-host disease and septicemia, and therefore it is reserved for severe refractory ITP with bleeding complications unresponsive to other therapies.56,57
THERAPY FOR SECONDARY ITP DEPENDS ON THE CAUSE
Treatments for secondary ITP vary depending on the cause of thrombocytopenia and are often more complex than therapy for primary disease. Optimal management involves treating the underlying condition (eg, chronic lymphocytic leukemia or systemic lupus erythematosus).
Drug-induced thrombocytopenia requires prompt recognition and withdrawal of the inciting agent.
Treating ITP due to HCV infection primarily involves antiviral agents to suppress viral replication. If treating ITP is required, then intravenous immunoglobulin is preferable to glucocorticoids because of the risk of increasing viral load with the latter.58 Eltrombopag may effectively increase platelet counts, allowing patients to receive interferon therapy for HCV.59 However, a recent study was halted due to increased incidence of portal vein thrombosis, raising concerns about the safety of eltrombopag for patients with chronic liver disease.60
Secondary ITP due to HIV infection should always be treated first with antivirals targeting HIV unless thrombocytopenia-related bleeding complications warrant treatment. If treatment for ITP is necessary, it should include corticosteroids, intravenous immunoglobulin, or anti-D immunoglobulin as first-line therapy.
Eradication therapy for H pylori is recommended for patients who are positive for the organism based on urea breath testing, stool antigen testing, or endoscopic biopsies.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
- Feudjo-Tepie MA, Robinson NJ, Bennett D. Prevalence of diagnosed chronic immune thrombocytopenic purpura in the US: analysis of a large US claim database: a rebuttal. J Thromb Haemost 2008; 6:711–712.
- Abrahamson PE, Hall SA, Feudjo-Tepie M, Mitrani-Gold FS, Logie J. The incidence of idiopathic thrombocytopenic purpura among adults: a population-based study and literature review. Eur J Haematol 2009; 83:83–89.
- Gernsheimer T. Chronic idiopathic thrombocytopenic purpura: mechanisms of pathogenesis. Oncologist 2009; 14:12–21.
- McMillan R, Wang L, Tomer A, Nichol J, Pistillo J. Suppression of in vitro megakaryocyte production by antiplatelet auto-antibodies from adult patients with chronic ITP. Blood 2004; 103:1364–1369.
- Houwerzijl EJ, Blom NR, van der Want JJ, et al. Ultrastructural study shows morphologic features of apoptosis and para-apoptosis in megakaryocytes from patients with idiopathic thrombocytopenic purpura. Blood 2004; 103:500–506.
- Kuwana M, Kaburaki J, Kitasato H, et al. Immunodominant epitopes on glycoprotein IIb-IIIa recognized by autoreactive T cells in patients with immune thrombocytopenic purpura. Blood 2001; 98:130–139.
- Cines DB, Blanchette VS. Immune thrombocytopenic purpura. N Engl J Med 2002; 346:995–1008.
- Littman DR, Rudensky AY. Th17 and regulatory T cells in mediating and restraining inflammation. Cell 2010; 140:845–858.
- Semple JW, Provan D, Garvey MB, Freedman J. Recent progress in understanding the pathogenesis of immune thrombocytopenia. Curr Opin Hematol 2010; 17:590–595.
- Ballem PJ, Segal GM, Stratton JR, Gernsheimer T, Adamson JW, Slichter SJ. Mechanisms of thrombocytopenia in chronic autoimmune thrombocytopenic purpura. Evidence of both impaired platelet production and increased platelet clearance. J Clin Invest 1987; 80:33–40.
- George JN. Definition, diagnosis and treatment of immune thrombocytopenic purpura. Haematologica 2009; 94:759–762.
- Rodeghiero F, Stasi R, Gernsheimer T, et al. Standardization of terminology, definitions and outcome criteria in immune thrombocytopenic purpura of adults and children: report from an international working group. Blood 2009; 113:2386–2393.
- Neunert C, Lim W, Crowther M, Cohen A, Solberg L, Crowther MA; American Society of Hematology. The American Society of Hematology 2011 evidence-based practice guideline for immune thrombocytopenia. Blood 2011; 117:4190–4207.
- Newton JL, Reese JA, Watson SI, et al. Fatigue in adult patients with primary immune thrombocytopenia. Eur J Haematol 2011; 86:420–429.
- Stasi R, Willis F, Shannon MS, Gordon-Smith EC. Infectious causes of chronic immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1275–1297.
- Moses A, Nelson J, Bagby GC. The influence of human immunodeficiency virus-1 on hematopoiesis. Blood 1998; 91:1479–1495.
- Zhang W, Nardi MA, Borkowsky W, Li Z, Karpatkin S. Role of molecular mimicry of hepatitis C virus protein with platelet GPIIIa in hepatitis C-related immunologic thrombocytopenia. Blood 2009; 113:4086–4093.
- Peck-Radosavljevic M. Thrombocytopenia in liver disease. Can J Gastroenterol 2000; 14(suppl D):60D–66D.
- Roomer R, Hansen BE, Janssen HL, de Knegt RJ. Thrombocytopenia and the risk of bleeding during treatment with peginterferon alfa and ribavirin for chronic hepatitis C. J Hepatol 2010; 53:455–459.
- Stasi R, Sarpatwari A, Segal JB, et al. Effects of eradication of Helicobacter pylori infection in patients with immune thrombocytopenic purpura: a systematic review. Blood 2009; 113:1231–1240.
- Cines DB, Bussel JB, Liebman HA, Luning Prak ET. The ITP syndrome: pathogenic and clinical diversity. Blood 2009; 113:6511–6521.
- Zent CS, Kay NE. Autoimmune complications in chronic lymphocytic leukaemia (CLL). Best Pract Res Clin Haematol 2010; 23:47–59.
- Hepburn AL, Narat S, Mason JC. The management of peripheral blood cytopenias in systemic lupus erythematosus. Rheumatology (Oxford) 2010; 49:2243–2254.
- Mok CC, Lee KW, Ho CT, Lau CS, Wong RW. A prospective study of survival and prognostic indicators of systemic lupus erythematosus in a southern Chinese population. Rheumatology (Oxford) 2000; 39:399–406.
- Cervera R, Piette JC, Font J, et al; Euro-Phospholipid Project Group. Antiphospholipid syndrome: clinical and immunologic manifestations and patterns of disease expression in a cohort of 1,000 patients. Arthritis Rheum 2002; 46:1019–1027.
- Burrows RF, Kelton JG. Fetal thrombocytopenia and its relation to maternal thrombocytopenia. N Engl J Med 1993; 329:1463–1466.
- Kantarjian H, Giles F, List A, et al. The incidence and impact of thrombocytopenia in myelodysplastic syndromes. Cancer 2007; 109:1705–1714.
- Portielje JE, Westendorp RG, Kluin-Nelemans HC, Brand A. Morbidity and mortality in adults with idiopathic thrombocytopenic purpura. Blood 2001; 97:2549–2554.
- Cheng Y, Wong RS, Soo YO, et al. Initial treatment of immune thrombocytopenic purpura with high-dose dexamethasone. N Engl J Med 2003; 349:831–836.
- Bromberg ME. Immune thrombocytopenic purpura—the changing therapeutic landscape. N Engl J Med 2006; 355:1643–1645.
- Guidry JA, George JN, Vesely SK, Kennison SM, Terrell DR. Corticosteroid side-effects and risk for bleeding in immune thrombocytopenic purpura: patient and hematologist perspectives. Eur J Haematol 2009; 83:175–182.
- Cooper N. Intravenous immunoglobulin and anti-RhD therapy in the management of immune thrombocytopenia. Hematol Oncol Clin North Am 2009; 23:1317–1327.
- Scaradavou A, Woo B, Woloski BM, et al. Intravenous anti-D treatment of immune thrombocytopenic purpura: experience in 272 patients. Blood 1997; 89:2689–2700.
- Newman GC, Novoa MV, Fodero EM, Lesser ML, Woloski BM, Bussel JB. A dose of 75 microg/kg/d of i.v. anti-D increases the platelet count more rapidly and for a longer period of time than 50 microg/kg/d in adults with immune thrombocytopenic purpura. Br J Haematol 2001; 112:1076–1078.
- Provan D, Stasi R, Newland AC, et al. International consensus report on the investigation and management of primary immune thrombocytopenia. Blood 2010; 115:168–186.
- Spahr JE, Rodgers GM. Treatment of immune-mediated thrombocytopenia purpura with concurrent intravenous immunoglobulin and platelet transfusion: a retrospective review of 40 patients. Am J Hematol 2008; 83:122–125.
- Kojouri K, Vesely SK, Terrell DR, George JN. Splenectomy for adult patients with idiopathic thrombocytopenic purpura: a systematic review to assess long-term platelet count responses, prediction of response, and surgical complications. Blood 2004; 104:2623–2634.
- Schilling RF. Estimating the risk for sepsis after splenectomy in hereditary spherocytosis. Ann Intern Med 1995; 122:187–188.
- Crary SE, Buchanan GR. Vascular complications after splenectomy for hematologic disorders. Blood 2009; 114:2861–2868.
- Davies JM, Barnes R, Milligan D; British Committee for Standards in Haematology. Update of guidelines for the prevention and treatment of infection in patients with an absent or dysfunctional spleen. Clin Med 2002; 2:440–443.
- Centers for Disease Control and Prevention (CDC). Recommended adult immunization schedule—United States, 2011. MMWR Morb Mortal Wkly Rep 2011; 60:1–4.
- McMillan R, Durette C. Long-term outcomes in adults with chronic ITP after splenectomy failure. Blood 2004; 104:956–960.
- Arnold DM, Dentali F, Crowther MA, et al. Systematic review: efficacy and safety of rituximab for adults with idiopathic thrombocytopenic purpura. Ann Intern Med 2007; 146:25–33.
- Godeau B, Porcher R, Fain O, et al. Rituximab efficacy and safety in adult splenectomy candidates with chronic immune thrombocytopenic purpura: results of a prospective multicenter phase 2 study. Blood 2008; 112:999–1004.
- Carson KR, Evens AM, Richey EA, et al. Progressive multifocal leukoencephalopathy after rituximab therapy in HIV-negative patients: a report of 57 cases from the Research on Adverse Drug Events and Reports project. Blood 2009; 113:4834–4840.
- Li J, Yang C, Xia Y, et al. Thrombocytopenia caused by the development of antibodies to thrombopoietin. Blood 2001; 98:3241–3248.
- Kuter DJ. New thrombopoietic growth factors. Blood 2007; 109:4607–4616.
- Kuter DJ, Bussel JB, Lyons RM, et al. Efficacy of romiplostim in patients with chronic immune thrombocytopenic purpura: a double-blind randomised controlled trial. Lancet 2008; 371:395–403.
- Bussel JB, Kuter DJ, Pullarkat V, Lyons RM, Guo M, Nichol JL. Safety and efficacy of long-term treatment with romiplostim in thrombocytopenic patients with chronic ITP. Blood 2009; 113:2161–2171.
- Kuter DJ, Rummel M, Boccia R, et al. Romiplostim or standard of care in patients with immune thrombocytopenia. N Engl J Med 2010; 363:1889–1899.
- Bussel JB, Provan D, Shamsi T, et al. Effect of eltrombopag on platelet counts and bleeding during treatment of chronic idiopathic thrombocytopenic purpura: a randomised, double-blind, placebo-controlled trial. Lancet 2009; 373:641–648.
- Cheng G, Saleh MN, Marcher C, et al. Eltrombopag for management of chronic immune thrombocytopenia (RAISE): a 6-month, randomised, phase 3 study. Lancet 2011; 377:393–402.
- Arnold DM, Nazi I, Kelton JG. New treatments for idiopathic thrombocytopenic purpura: rethinking old hypotheses. Expert Opin Investig Drugs 2009; 18:805–819.
- Boruchov DM, Gururangan S, Driscoll MC, Bussel JB. Multiagent induction and maintenance therapy for patients with refractory immune thrombocytopenic purpura (ITP). Blood 2007; 110:3526–3531.
- Arnold DM, Nazi I, Santos A, et al. Combination immunosuppressant therapy for patients with chronic refractory immune thrombocytopenic purpura. Blood 2010; 115:29–31.
- Passweg JR, Rabusin M. Hematopoetic stem cell transplantation for immune thrombocytopenia and other refractory autoimmune cytopenias. Autoimmunity 2008; 41:660–665.
- Huhn RD, Fogarty PF, Nakamura R, et al. High-dose cyclophosphamide with autologous lymphocyte-depleted peripheral blood stem cell (PBSC) support for treatment of refractory chronic autoimmune thrombocytopenia. Blood 2003; 101:71–77.
- Magrin S, Craxi A, Fabiano C, et al. Hepatitis C viremia in chronic liver disease: relationship to interferon-alpha or corticosteroid treatment. Hepatology 1994; 19:273–279.
- McHutchison JG, Dusheiko G, Shiffman ML, et al; TPL102357 Study Group. Eltrombopag for thrombocytopenia in patients with cirrhosis associated with hepatitis C. N Engl J Med 2007; 357:2227–2236.
- US Department of Health & Human Services. Promacta (eltrombopag): Portal Venous System Thromboses in Study of Patients With Chronic Liver Disease http://www.fda.gov/Safety/MedWatch/SafetyInformation/SafetyAlertsforHumanMedicalProducts/ucm211796.htm. Accessed June 27, 2012.
KEY POINTS
- Secondary ITP can be drug-induced or be a manifestation of human immunodeficiency virus (HIV), hepatitis C virus (HCV), a lymphoproliferative disorder, or systemic lupus erythematosus.
- Nonautoimmune conditions should also be considered, including pseudothrombocytopenia (a laboratory artifact induced by EDTA), thrombotic thrombocytopenic purpura, thrombocytopenia in pregnancy, and myelodysplastic syndrome (common in the elderly).
- Treatment is indicated to keep the platelet count above 30 × 109/L or to control bleeding.
- Initial treatment usually begins with glucocorticoids, with the duration limited by side effects.
- Patients for whom glucocorticoids fail generally require splenectomy, rituximab, or thrombopoietin receptor agonists.
Implementation of ICD-10 codes delayed 1 year
Change has come again to ICD-9 diagnostic codes
Melanie Witt, RN, CPC, COBGC, MA (Reimbursement Advisor, November 2011)
Health and Human Services (HHS) Secretary Kathleen G. Sebelius recently announced that the date for compliance with International Classification of Diseases, 10th Edition, diagnosis and procedure codes, or ICD-10, has been changed from October 1, 2013, to October 1, 2014.
Secretary Sebelius noted that the date change has been made in reaction to many providers’ concerns about the administrative burdens they will face in years ahead due to implementation of electronic health records and the Affordable Care Act.1
“ICD-10 codes provide more robust and specific data that will help improve patient care and enable the exchange of our health-care data with that of the rest of the world that has long been using ICD-10. Entities covered under the Health Insurance Portability and Accountability Act of 1996 (HIPAA) will be required to use the ICD-10 diagnostic and procedure codes,” said Secretary Sebelius.”1
The final rule was issued by the Centers for Medicare & Medicaid Services, and published in the Federal Register on September 5, 2012. The rule states that, “The change in the compliance date is intended to give covered health-care providers and other covered entities more time to prepare and fully test their systems to ensure a smooth and coordinated transition by all covered entities.”2
Adoption will “increase standardization within HIPAA standard transactions and provide a platform for other regulatory and industry initiatives. Their adoption will allow for a higher level of automation for health-care provider offices, particularly for provider processing of billing and insurance related tasks, eligibility responses from health plans, and remittance advice that describes health-care claim payments.”2
Decision based on costs versus savings
HHS is aware that health-care plans, hospitals, and physician practices are currently in the process of implementing the ICD-10 due to the anticipated 2013 compliance date. It is estimated that the 1-year delay may cost the entire health-care industry $1 billion to $6.6 billion. However, HHS also anticipates substantial savings ($3.6 billion to $8 billion) by avoiding costs that could incur if a significant number of providers are unprepared for the transition to ICD-10. Possible consequences of implementing ICD-10 in 2013 are: 1) health-care providers and plans could have to manually process claims for claims to be paid and 2) small health-care providers might have to arrange for loans or lines of credit to continue to provide health-care services because of delayed payments. The decision to delay administration of ICD-10 was based on these factors.3
The American Health Information Management Association (AHIMA) will continue to assist the industry through the implementation process. AHIMA CEO Lynne Thomas Gordon, MBA, RHIA, FACHE, CAE, wrote on August 24, 2012, “ICD-10-CM/PCS implementation is inevitable, but today’s news gives the health-care community the certainty and clarity it needs to move forward with implementation, testing, and training. We realize that a few are still apprehensive about the implementation process, and that is why AHIMA remains committed to assisting the health-care community with its transition to a new code set that will lead to improved patient care and reduced costs.”3
We want to hear from you! Tell us what you think.
Change has come again to ICD-9 diagnostic codes
Melanie Witt, RN, CPC, COBGC, MA (Reimbursement Advisor, November 2011)
Health and Human Services (HHS) Secretary Kathleen G. Sebelius recently announced that the date for compliance with International Classification of Diseases, 10th Edition, diagnosis and procedure codes, or ICD-10, has been changed from October 1, 2013, to October 1, 2014.
Secretary Sebelius noted that the date change has been made in reaction to many providers’ concerns about the administrative burdens they will face in years ahead due to implementation of electronic health records and the Affordable Care Act.1
“ICD-10 codes provide more robust and specific data that will help improve patient care and enable the exchange of our health-care data with that of the rest of the world that has long been using ICD-10. Entities covered under the Health Insurance Portability and Accountability Act of 1996 (HIPAA) will be required to use the ICD-10 diagnostic and procedure codes,” said Secretary Sebelius.”1
The final rule was issued by the Centers for Medicare & Medicaid Services, and published in the Federal Register on September 5, 2012. The rule states that, “The change in the compliance date is intended to give covered health-care providers and other covered entities more time to prepare and fully test their systems to ensure a smooth and coordinated transition by all covered entities.”2
Adoption will “increase standardization within HIPAA standard transactions and provide a platform for other regulatory and industry initiatives. Their adoption will allow for a higher level of automation for health-care provider offices, particularly for provider processing of billing and insurance related tasks, eligibility responses from health plans, and remittance advice that describes health-care claim payments.”2
Decision based on costs versus savings
HHS is aware that health-care plans, hospitals, and physician practices are currently in the process of implementing the ICD-10 due to the anticipated 2013 compliance date. It is estimated that the 1-year delay may cost the entire health-care industry $1 billion to $6.6 billion. However, HHS also anticipates substantial savings ($3.6 billion to $8 billion) by avoiding costs that could incur if a significant number of providers are unprepared for the transition to ICD-10. Possible consequences of implementing ICD-10 in 2013 are: 1) health-care providers and plans could have to manually process claims for claims to be paid and 2) small health-care providers might have to arrange for loans or lines of credit to continue to provide health-care services because of delayed payments. The decision to delay administration of ICD-10 was based on these factors.3
The American Health Information Management Association (AHIMA) will continue to assist the industry through the implementation process. AHIMA CEO Lynne Thomas Gordon, MBA, RHIA, FACHE, CAE, wrote on August 24, 2012, “ICD-10-CM/PCS implementation is inevitable, but today’s news gives the health-care community the certainty and clarity it needs to move forward with implementation, testing, and training. We realize that a few are still apprehensive about the implementation process, and that is why AHIMA remains committed to assisting the health-care community with its transition to a new code set that will lead to improved patient care and reduced costs.”3
We want to hear from you! Tell us what you think.
Change has come again to ICD-9 diagnostic codes
Melanie Witt, RN, CPC, COBGC, MA (Reimbursement Advisor, November 2011)
Health and Human Services (HHS) Secretary Kathleen G. Sebelius recently announced that the date for compliance with International Classification of Diseases, 10th Edition, diagnosis and procedure codes, or ICD-10, has been changed from October 1, 2013, to October 1, 2014.
Secretary Sebelius noted that the date change has been made in reaction to many providers’ concerns about the administrative burdens they will face in years ahead due to implementation of electronic health records and the Affordable Care Act.1
“ICD-10 codes provide more robust and specific data that will help improve patient care and enable the exchange of our health-care data with that of the rest of the world that has long been using ICD-10. Entities covered under the Health Insurance Portability and Accountability Act of 1996 (HIPAA) will be required to use the ICD-10 diagnostic and procedure codes,” said Secretary Sebelius.”1
The final rule was issued by the Centers for Medicare & Medicaid Services, and published in the Federal Register on September 5, 2012. The rule states that, “The change in the compliance date is intended to give covered health-care providers and other covered entities more time to prepare and fully test their systems to ensure a smooth and coordinated transition by all covered entities.”2
Adoption will “increase standardization within HIPAA standard transactions and provide a platform for other regulatory and industry initiatives. Their adoption will allow for a higher level of automation for health-care provider offices, particularly for provider processing of billing and insurance related tasks, eligibility responses from health plans, and remittance advice that describes health-care claim payments.”2
Decision based on costs versus savings
HHS is aware that health-care plans, hospitals, and physician practices are currently in the process of implementing the ICD-10 due to the anticipated 2013 compliance date. It is estimated that the 1-year delay may cost the entire health-care industry $1 billion to $6.6 billion. However, HHS also anticipates substantial savings ($3.6 billion to $8 billion) by avoiding costs that could incur if a significant number of providers are unprepared for the transition to ICD-10. Possible consequences of implementing ICD-10 in 2013 are: 1) health-care providers and plans could have to manually process claims for claims to be paid and 2) small health-care providers might have to arrange for loans or lines of credit to continue to provide health-care services because of delayed payments. The decision to delay administration of ICD-10 was based on these factors.3
The American Health Information Management Association (AHIMA) will continue to assist the industry through the implementation process. AHIMA CEO Lynne Thomas Gordon, MBA, RHIA, FACHE, CAE, wrote on August 24, 2012, “ICD-10-CM/PCS implementation is inevitable, but today’s news gives the health-care community the certainty and clarity it needs to move forward with implementation, testing, and training. We realize that a few are still apprehensive about the implementation process, and that is why AHIMA remains committed to assisting the health-care community with its transition to a new code set that will lead to improved patient care and reduced costs.”3
We want to hear from you! Tell us what you think.
Faulty equipment blamed for improper diagnosis: $78M verdict … and more
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
AT 36 WEEKS’ GESTATION, a woman went to the emergency department (ED) with abdominal pain. After ultrasonography (US), a nurse told her the fetus had died in utero, but the mother continued to feel fetal movement. The ED physician requested a second US, but it took 75 minutes for a radiology technician to arrive. This US showed a beating fetal heart with placental abruption. After cesarean delivery, the child was found to have cerebral palsy.
PATIENT’S CLAIM The first US was performed by an inexperienced technician using outdated equipment and the wrong transducer. An experienced technician with newer equipment should have been immediately available. The ED physician did not react when fetal distress was first identified.
DEFENDANTS’ DEFENSE The ED physician was told that the baby had died. Perhaps the child’s heart had started again by the time the second US was performed and a heartbeat found. The hospital denied negligence.
VERDICT A Pennsylvania jury found the ED physician not negligent; the hospital was 100% at fault. A $78.5 million verdict included $1.5 million in emotional distress to the mother, $10 million in pain and suffering for the child, $2million in lost future earnings, and the rest in future medical expenses.
Ligated ureter found after hysterectomy
A 50-YEAR-OLD WOMAN underwent laparoscopically assisted vaginal hysterectomy. She went to the ED with pain 6 days later. Imaging studies indicated a ligated ureter; a nephrostomy tube was placed. She required a nephrostomy bag for 4 months and underwent two repair operations.
PATIENT’S CLAIM The patient’s ureter was ligated and/or constricted during surgery. The gynecologist was negligent in failing to recognize and repair the injury during surgery.
PHYSICIAN’S DEFENSE The ureter was not ligated during surgery; therefore it could not have been discovered. In addition, injury to a ureter is a known risk of the procedure.
VERDICT An Arizona defense verdict was returned.
Myomectomy after cesarean; mother dies
IMMEDIATELY AFTER A WOMAN with preeclampsia had a cesarean delivery, she underwent a myomectomy. The day before discharge, her abdominal incision opened and a clear liquid drained. The day after discharge, she went to the ED with intense abdominal pain. Necrotizing fasciitis was found and debridement surgery performed. She was transferred to another hospital but died of sepsis several days later.
ESTATE’S CLAIM The infection occurred because the myomectomy was performed immediately following cesarean delivery. Prophylactic antibiotics were not prescribed before surgery. The mother was not fully informed as to the risks of concurrent operations. The ObGyns failed to recognize the infection before discharging the patient.
PHYSICIANS’ DEFENSE The patient was fully informed of the risks of surgery; it was reasonable to perform myomectomy immediately following cesarean delivery. There were no signs or symptoms of infection before discharge. Cesarean incisions open about 30% of the time—not a cause for concern. Prophylactic antibiotics for cesarean procedures are not standard of care. The patient’s infection was caused by a rapidly spreading, rare bacterium.
VERDICT A Michigan defense verdict was returned.
TOLAC to cesarean: baby has cerebral palsy
A WOMAN WANTED A TRIAL OF LABOR after a previous cesarean delivery (TOLAC). During labor, fetal distress was noted, and a cesarean delivery was performed. Uterine rupture had occurred. The baby has spastic cerebral palsy with significantly impaired neuromotor and cognitive abilities.
PARENTS’ CLAIM The hospital staff and physicians overlooked earlier fetal distress. A timelier delivery would have prevented the child’s injuries.
DEFENDANTs’ DEFENSE The hospital reached a confidential settlement. The ObGyns claimed fetal tracings were not suggestive of uterine rupture; they met the standard of care.
VERDICT A Texas defense verdict was returned.
WHEN GESTATIONAL DIABETES WAS DIAGNOSED at 33 weeks’ gestation, a family practitioner (FP) referred the mother to an ObGyn practice. Two ObGyns performed amniocentesis to check fetal lung maturity. After the procedure, fetal distress was noted, and the ObGyns instructed the FP to induce labor.
The baby suffered brain damage, had seizures, and has cerebral palsy. She was born without kidney function. By age 10, she had 2 kidney transplant operations and functions at a pre-kindergarten level.
PATIENT’S CLAIM The mother was not fully informed of the risks of and alternatives to amniocentesis. Although complications arose before amniocentesis, the test proceeded. The ObGyns were negligent in not performing cesarean delivery when fetal distress was detected.
DEFENDANTS’ DEFENSE The FP and hospital settled prior to trial. The ObGyns claimed that their care was an appropriate alternative to the actions the patient claimed should have been taken.
VERDICT Costs for the child’s care had reached $1.4 million before trial. A $9 million Virginia verdict was returned that included $7 million for the child and $2 million for the mother, but the settlement was reduced by the state cap.
Did HT cause breast cancer?
A 52-YEAR-OLD WOMAN was prescribed conjugated estrogens/medroxyprogesterone acetate (Prempro, Wyeth, Inc.) for hormone therapy by her gynecologist.
After taking the drug for 5 years, the patient developed invasive breast cancer. She underwent a lumpectomy, chemotherapy, and three reconstructive surgeries.
PATIENT’S CLAIM The manufacturer failed to warn of a woman’s risk of developing breast cancer while taking the product.
DEFENDANT’S DEFENSE Prempro alone does not cause cancer. The drug is just one of many contributing factors that may or may not increase the risk of developing breast cancer.
VERDICT A $3.75 million Connecticut verdict was returned for the patient plus $250,000 to her husband for loss of consortium.
Failure to diagnose preeclampsia—twice
AT 28 WEEKS’ GESTATION, a woman with a history of hypertension went to an ED with headache, nausea, vomiting, cramping, and ringing in her ears. After waiting 4 hours before being seen, her BP was 150/108 mm Hg and normal fetal heart tones were heard. The ED physician diagnosed otitis media and discharged her.
Later that evening, the patient returned to the ED with similar symptoms. A urine specimen showed significant proteinuria and the fetal heart rate was 158 bpm. A second ED physician diagnosed a urinary tract infection, prescribed antibiotics and pain medication, and sent her home.
A few hours later, she returned to the ED by ambulance suffering from eclamptic seizures. Her BP was 174/121 mm Hg, and no fetal heart tones were heard. She delivered a stillborn child by cesarean delivery.
PATIENT’S CLAIM The ED physicians were negligent in failing to diagnose preeclampsia at the first two visits.
PHYSICIANS’ DEFENSE The first ED physician settled for $45,000 while serving a prison sentence after conviction on two sex-abuse felonies related to his treatment of female patients at the same ED. The second ED physician denied negligence.
VERDICT A $50,000 Alabama verdict was returned for compensatory damages for the mother and $600,000 punitive damages for the stillborn child.
Inflated Foley catheter injures mother
DURING A LONG AND DIFFICULT LABOR, an ObGyn used forceps to complete delivery of a 31-year-old woman’s first child. The baby was healthy, but the mother has suffered urinary incontinence since delivery. Despite several repair operations, the condition remains.
PATIENT’S CLAIM The ObGyn was negligent in failing to remove a fully inflated Foley catheter before beginning delivery, leading to a urethral sphincter injury.
PHYSICIAN’S DEFENSE The decision regarding removal of the catheter was a matter of hospital policy. The ObGyn blamed improper catheter placement on the nurses.
VERDICT A Kentucky defense verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Women’s health under the Affordable Care Act: What is covered?
Lay midwives and the ObGyn: Is collaboration risky?
(May 2012)
How state budget crises are putting the squeeze on Medicaid (and you)
(February 2012)
Is private ObGyn practice on its way out?
(October 2011)
14 questions (and answers) about health reform and you
with Janelle Yates, Senior Editor (August 2010)
For the first half of 2012, the big question was: Will anything be covered under the Affordable Care Act (ACA)? After considering constitutional challenges to the Act that had the potential to invalidate the entire law, the US Supreme Court ruled, on June 28, that the ACA met constitutional muster in National Federation of Independent Business v. Sebelius (2012).
Now that the Court has upheld the ACA, let’s review the major women’s health services included under the law. This Web version incorporates 10 more women's health provisions from the ACA, from smoking cessation to young women’s breast cancer, that were not in the print version.

Preventive services guaranteed without copays
A major component of the health reform law went into effect August 1, 2012; it requires most health plans to cover women’s preventive services without requiring enrollees to pay a copay or deductibles. This provision reflects Congress’ understanding that women have a longer life expectancy and bear a greater burden of chronic disease, disability, and reproductive and gender-specific conditions. In addition, women often have a different response to treatment than men do.
The federal Department of Health and Human Services (HHS) estimates that Americans use preventive services at only about half of the recommended rate. By 2013, as many as 73 million individuals will benefit from preventive care offered under the law.
The American Congress of Obstetricians and Gynecologists (ACOG) worked with the Institute of Medicine (IOM)—which was charged with advising HHS—to encourage the inclusion of women’s preventive services specified in ACOG guidelines to ensure women’s health and well-being. As ACOG Executive Vice President Hal C. Lawrence, MD, told the IOM in January 2011:
- The College’s clinical guidelines…offer an excellent resource…and encompass the entire field of women’s preventive care. Our guidance is based on the best available evidence and is developed by committees with expertise reflecting the breadth of women’s health care and subject to a rigorous conflict of interest policy.
Dr. Lawrence further urged the IOM “to recommend coverage of the following services and products without cost-sharing”:
- well-woman visits
- preconception care
- family planning counseling and services
- HIV screening (for women at average risk)
- screening for intimate partner violence
- testing for human papillomavirus (HPV) as part of cervical cancer screening.
ACOG’s recommendations were approved by the IOM and, subsequently, by HHS. As a result, all private health plans that began on or after September 30, 2010, are required to cover these services at no out-of-pocket cost to patients (TABLE).
Women’s preventive services guaranteed under ACA*
| Service | Frequency | HHS guidelines for health insurance coverage |
|---|---|---|
| Well-woman visit | Annual for adult women, although HHS recognizes that several visits may be needed to obtain all necessary recommended preventive services, depending on a woman’s health status, health needs, and other risk factors** | The visit should focus on preventive services that are appropriate for the patient’s age and development, including preconception and prenatal care. This visit should, where appropriate, include other preventive services listed in this set of guidelines, as well as others referenced in section 2713 |
| Screening for gestational diabetes | Between 24 and 28 weeks of gestation and at the first prenatal visit for pregnant women identified to be at high risk for diabetes | |
| Testing for human papillomavirus (HPV) | At age 30 and older, no more frequently than every 3 years | High-risk HPV DNA testing in women who have normal cervical cytology |
| Counseling about sexually transmitted infection (STI) | Annual | All sexually active women |
| Counseling about and screening for HIV | Annual | All sexually active women |
| Counseling about and provision of contraception† | As prescribed | All FDA-approved contraceptive methods and sterilization procedures. Counseling for all women with reproductive capacity |
| Breastfeeding support, supplies, and counseling | In conjunction with each birth | Comprehensive lactation support and counseling by a trained provider during pregnancy or postpartum (or both), as well as costs for renting breastfeeding equipment |
| Screening for and counseling about interpersonal and domestic violence | Annual | |
| HHS = Health and Human Services * HHS guidelines are effective August 1, 2011. Nongrandfathered plans and insurers are required to provide coverage without cost-sharing consistent with HHS guidelines in the first plan year (in the individual market, policy year) that begins on or after August 1, 2012. ** The July 2011 Institute of Medicine report titled “Clinical preventive services for women: closing the gap” lists recommendations on individual preventive services that may be obtained during a well-woman preventive service visit. † Group health plans sponsored by certain religious employers, and group health insurance coverage in connection with such plans, are exempt from the requirement to cover contraceptive services. SOURCE: Adapted from Healthcare.gov. Affordable Care Act Expands Prevention Coverage for Women’s Health and Well-Being. http://www.hrsa.gov/womensguidelines/. Accessed August 8, 2012. | ||
Contraceptive mandate triggers a firestorm
On February 10, 2012, under pressure from religious groups, the Obama Administration offered a religious exemption to the contraception mandate for certain employers and group health plans. Under this “accommodation,” certain religious employers are exempt from the requirement to cover contraceptive services in their group health plans. An employer qualifies for this exemption if it:
- has the inculcation of religious values as one of its purposes
- primarily employs individuals who share its religious tenets
- primarily serves individuals who share its religious tenets, and
- qualifies for nonprofit status under
Internal Revenue Service (IRS) rules. At the same time that the Obama Administration wanted to accommodate employers’ religious beliefs, it also wanted to ensure that every woman would have access to free preventive care, including contraceptive services, regardless of where she works. And so while the Administration requires insurers to offer group health plan coverage without contraceptive coverage to religious-affiliated organizations, it also requires insurers to provide contraceptive coverage directly to individuals covered under the organization’s group health plan with no cost sharing.
This contraceptive mandate—even with the accommodation—has set off a firestorm on Capitol Hill that will eventually be settled in the courts.
Medicaid expansion falls short of original goal
In National Federation of Independent Business v. Sebelius, the plaintiffs asked the Supreme Court to rule on the federal government’s authority to require states to expand their Medicaid programs. Medicaid costs are typically shared by the federal and state governments. Under the ACA, state Medicaid programs were required to cover nearly all individuals who have incomes below 133% of the federal poverty level—$30,656 for a family of four in 2012—paid entirely by the federal government from 2014 through 2016. After that, the federal share gradually declines to, and then stays at, 90%. States that did not expand their Medicaid programs risked losing all federal Medicaid funding.
The Court ruled that the federal government can expand Medicaid but can’t penalize states that don’t accept the expansion mandate—effectively turning the mandate into a state option. States will receive the additional federal funds if they expand coverage, but states that don’t expand will not be penalized by losing existing federal funds for other parts of the program.
Since the ruling, a number of governors have announced that they will not expand their Medicaid programs, including governors of Florida and Louisiana. Those two states alone are home to 20% of all individuals intended to be covered under the Medicaid expansion.
This part of the ACA is particularly important to women because it strikes, for the first time, the requirement that a low-income woman must be pregnant to receive Medicaid coverage.
The figure below shows the dramatic potential improvement in coverage for women if all states fully implement the Medicaid expansion. Time, court decisions, elections, and state budget fights will determine how much of this change is realized for women’s health.
Percentage of insured women will increase under ACA
Percentage of women aged 19 to 64 years who were uninsured in 2009–2012 and under the Affordable Care Act when fully implemented.
SOURCE: Commonwealth Fund. Analysis of the March 2011 and 2010 Current Population Surveys by N. Tilipman and B. Sampat of Columbia University.
Women gain direct access to ObGyns
The ACA guarantees women in all states and all plans direct access to their ObGyns. Before the ACA, women in nine states lacked this guarantee, and women in 16 other states had only limited direct access. Now, a woman can go directly to her ObGyn without having to get a referral from her primary care physician or insurer.
Direct access is especially important because the ACA establishes new delivery systems, such as medical homes and accountable care organizations, designed to capture patients to maximize savings. An ObGyn does not have to be the patient’s primary care provider, and the patient’s access to her ObGyn cannot be limited to a certain number of visits or types of services.
ACA encourages states to cover family-planning services
Under the ACA, states have an easier time covering family-planning services, up to the same eligibility levels as pregnant women. Family planning is still an optional service that a state can choose to extend to women who have incomes above the Medicaid income eligibility level but, before the ACA was enacted, states had to apply to HHS for permission to waive the federal rules, often a very cumbersome process.
Prior to the ACA, 27 states had family planning waivers to provide services to nonpregnant women who had incomes above the Medicaid eligibility level—most at or near 200% of poverty. Now, states can provide family planning services to this population without federal approval.
Don’t miss Dr. Robert L. Barbieri’s October article titled “Gynecologic care across a woman’s life”
Insurance reforms end lifetime limits on coverage
Insurance reforms are important to us and our patients. The better the private health insurance system works—allowing us to provide our best possible care to patients and making sure they can see us when they need our care—the less our nation relies on the public safety net.
Beginning in 2010, the ACA eliminated all lifetime limits on how much insurance companies would cover when beneficiaries get sick; it also bans insurance companies from dropping people from coverage when they get sick. So if your patient has private health insurance and has faithfully paid her premiums and hasn’t committed fraud, her insurer cannot drop her or impose a limit on her coverage once she claims benefits.
This may be especially important for patients who need the most care, such as those who have cancer or another long-term, expensive, and unforeseen diagnosis. Because of this provision, you will not have to worry about your patient losing coverage in the middle of a long course of treatment.
The insurance practice of charging women more than men for equivalent policies ended on January 1, 2011, making insurance more affordable for our patients. Insurers in the individual and small group markets are allowed to vary premiums only for age, geographic location, family size, and tobacco use, not for gender—another important aspect of the law.
2014 is a key year in health reform
Exchanges begin
In 2014, under the ACA, state health insurance exchanges become reality.
An exchange is a marketplace where people can shop for health insurance; private health insurers can market their insurance products in state and multistate exchanges if they comply with new federal insurance reforms established in the ACA and offer the minimum benefits packages established by each state. Exchanges are intended to offer patients a choice of health insurance plans that are affordable, comprehensive, and easy to compare. Low-income individuals will be able to purchase private insurance in the exchanges with the federal premium subsidies or tax credits.
Insurers wanting to market their policies in an exchange may not deny coverage for preexisting conditions, including pregnancy, domestic violence, and previous cesarean delivery. They can’t deny coverage on the basis of an individual’s medical history, health status, genetic information, or disability. And they can’t impose waiting periods longer than 90 days before coverage takes effect, including 9-month waiting periods before maternity coverage.
Essential benefits are established
The ACA sets a minimum standard of health-care coverage that must be included in nearly every private insurance policy. The intent is that every person in the United States, regardless of where they live, who employs them, and what their income is, should have access to the same basic care.
Effective January 1, 2014, all insurance plans, except plans that existed before the ACA was enacted on March 23, 2010, must offer an “essential health benefits” (EHB) package, which must include:
- ambulatory patient services
- emergency services
- hospitalization
- maternity and newborn care
- mental health and substance use disorder services
- prescription drugs
- rehabilitative and habilitative services and devices
- laboratory services
- women’s preventive and wellness services and chronic disease management
- pediatric services, including oral and vision care.
Last December, HHS surprised many by giving states flexibility to design their own EHB packages, as long as the packages included each service on the list.
To choose its EHB package, a state must select a “benchmark” plan from the top- selling plans in four markets: federal and state public employee plans, commercial HMO plans, and small business plans. If a state doesn’t select a benchmark plan, the EHB defaults to the largest small-group market plan in the state. Each state must also choose an EHB package for its Medicaid program using the same 10 benefit categories.
State EHB plans must follow ACA requirements on annual and lifetime dollar limits but may impose limits on the scope and duration of coverage.
As for state-mandated benefits, if a state selects an EHB package that does not include a benefit already mandated by the state, the state must fund coverage for that service on its own—a decision HHS has promised to revisit in 2016.
Abortion decisions reside with the states
ACA requirements regarding abortion coverage 1) take effect in 2014 and 2) apply only to private health insurance plans marketed in the state exchanges that 3) cover abortions beyond those eligible for Medicaid coverage now, which are those that involve cases of rape or incest or that are necessary to save the life of the mother. Medicaid coverage for these categories of abortion is allowed under the Hyde Amendment.
Each insurer marketing a health plan in an exchange can determine whether or not its plan will cover abortion and, if it does, whether coverage will be limited to or go beyond those allowed under the Hyde Amendment. No federal tax or premium subsidies may be used to pay for abortions beyond those permitted by the Hyde Amendment.
The Secretary of HHS must ensure that at least one plan in each state exchange covers abortion, and that at least one plan either covers no abortions or limits abortions to those allowed under the Hyde Amendment. Insurers who offer abortion coverage beyond Hyde have to comply with a number of administrative requirements.
Congress was clear that the ultimate decisions about abortion should be made at the state rather than the federal level, and it gave states the ultimate trump card: Any state can pass legislation that prohibits any plan from offering abortion coverage of any kind within that state’s exchange. Any state can prohibit insurers offering plans within that state’s exchange from including any abortion coverage.
10 additional health provisions under the ACA
1. Creation of women's medical homes
The law points the way for creation of medical homes for women in the Medicare and Medicaid programs. The bill establishes an Innovation Center within the Centers for Medicare and Medicaid Services that has broad authority to evaluate, test, and adopt systems that foster patient-centered care, improve quality, and contain costs under Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP)—and this includes patient-centered medical homes that address women’s unique health needs. ObGyn practices are eligible to participate and to receive additional reimbursement if they do.
2. Smoking-cessation counseling in pregnancy
The framers of the ACA recognized the large negative impact that smoking has on health, especially during pregnancy. Studies suggest that the intervention of a physician—most notably, counseling of the patient to quit smoking—has strong potential to modify this behavior. The new law provides reimbursement for this intervention. There are no copays or deductibles for patients, and smoking-cessation services can include diagnostic, therapeutic, and counseling modalities in addition to prescription of pharmacotherapy.
Before this bill became law, only 24 state Medicaid programs paid ObGyns or other physicians for smoking-cessation counseling of pregnant patients, and five states provided no coverage at all. Now, all pregnant Medicaid patients can get this counseling, and you’ll be paid for this important service.
3. Payments to nonphysician providers in freestanding birth centers
Before the ACA became law, Medicaid was authorized to pay hospitals and other facilities operated by and under the supervision of a physician; no payments were authorized for services of an ambulatory center operated by other health professionals. The ACA authorizes Medicaid payments to state-recognized freestanding birth centers not operated by or under the supervision of a physician. A state that doesn’t currently license birth centers must pass legislation and license these centers before the centers can receive these payments.
Medicaid will also reimburse providers who practice in state-recognized freestanding birth centers, as long as the individuals are practicing within their state’s scope of practice laws and regulations. Because the type of provider is not specified but instead left up to each state’s scope of practice laws and regulations, this provision could allow for separate provider payments for physicians, certified nurse midwives, certified professional midwives, and doulas.
4. Immigrant coverage
Legal immigrants are bound by the individual coverage mandate and must purchase health insurance. These individuals are eligible for income-related premium credits and subsidies for insurance purchased through an exchange. Legal immigrants who are barred from Medicaid during their first 5 years in the United States (by earlier law) are eligible for premium credits only.
Undocumented immigrants are not eligible for Medicaid, premium credits, or subsidies and are barred from purchasing insurance in the exchange, even with their own money.
5. Postpartum depression
Health reform will help bring perinatal and postpartum depression out of the shadows by providing federal funds for research, patient education, and clinical treatment. For example, the federal Department of Health and Human Services (HHS) will:
- conduct research into the causes of, and treatments for, postpartum conditions
- create a national public awareness campaign to increase awareness and knowledge of postpartum depression and psychosis
- provide grants to study the benefits of screening for postpartum depression and psychosis
- establish grants to deliver or enhance outpatient, inpatient, and home-based health and support services, including case management and comprehensive treatment services for individuals with, or at risk for, postpartum conditions.
The National Institute of Mental Health is encouraged to conduct a 10-year longitudinal study on the mental health consequences of pregnancy. This study is intended to focus on perinatal depression.
Community health centers will be eligible for grants in 2012 (as they were in 2011) to the tune of $3 million for inpatient and outpatient counseling and services.
And a federal public awareness campaign will educate the public through radio and television ads.
These endeavors point to the need for ObGyns to familiarize themselves with postpartum depression—if they aren’t already well versed in the subject—because patients are likely to become more aware of this issue and look to their ObGyns for answers.
6. Maternal home visits
Congress established a new Maternal, Infant, and Early Childhood Home Visiting program to improve maternal and fetal health in underserved areas of our country. This program will provide funds to states, tribes, and territories to develop and implement evidence-based home-visitation models to reduce infant and maternal mortality and its causes by producing improvements in:
- prenatal, maternal, and newborn health
- child health and development
- parenting skills
- school readiness
- juvenile delinquency
- family economic self-sufficiency.
These programs will have to demonstrate effectiveness and improved outcomes. HHS recently requested suggestions on ways of demonstrating the effectiveness of home-visiting program models for pregnant women, expectant fathers, and caregivers of children from birth through entry into kindergarten.
The law appropriates $350 million to this program in 2012 and $400 million in both 2013 and 2014.
7. Assistance for pregnant students
A new Pregnancy Assistance Fund—$25 million annually over 10 years (fiscal years 2010–2019)—requires the Secretary of HHS (in collaboration with the Secretary of Education) to establish a state grant program to help pregnant and parenting teens and young women. The aim of this program is to help teens who become pregnant and who choose to bring their pregnancies to term or keep their babies, or both, to stay in school. Grants will go to institutions of higher education, high schools and community service centers, as well as state attorneys general.
Institutions that receive grants must work with providers to meet specific practical needs of pregnant or parenting students:
- housing
- childcare
- parenting education
- postpartum counseling
- assistance in finding and accessing needed services
- referrals for prenatal care and delivery, infant or foster care, or adoption.
Funds to attorneys general will be used to combat domestic violence among pregnant teens.
8. Young women's breast cancer
A new program is intended to help educate young women about the importance of breast health and screening, in two ways:
- The National Institutes of Health (NIH) will conduct research to develop and test screening measures for prevention and early detection of breast cancer in women 15 to 44 years old.
- The US Department of HHS will create a national awareness campaign, with $9 million in funding each year from 2010 to 2014, to encourage young women to talk with their doctors about breast cancer and early detection.
ObGyns can expect to see more interest and questions about breast health among young women and their mothers. It pays to be prepared with good information for these important conversations.
9. Personal responsibility education
From 2010 through 2014, each state will receive funds for personal responsibility education programs aimed at reducing pregnancy in youths. Funds are $75 million for each fiscal year, allocated to each state depending on the size of its youth population but not intended to be less than $250,000 per state.
Educational programs eligible for federal funds must include both abstinence and contraception information for prevention of teenage pregnancy and sexually transmitted infections, including HIV/AIDS, as well as three or more adulthood-preparation subjects.
10. Community-based support of Patient-Centered Medical Homes
Federal funding is available to states for the development of community-based health teams to support medical homes run by primary care practices. These teams may include specialists, nurses, pharmacists, nutritionists, dieticians, social workers, behavioral and mental health providers, and physician assistants. Primary care practices in this program function as medical homes and are responsible for addressing a patient’s personal health-care needs. The team links the medical home to community support services for its patients.
Eligible ObGyn practices can qualify as primary care practices, and ObGyns are eligible to serve as specialist members of the community-based health team.
ACA is a mixed bag for ObGyns
Women have much to gain from the provisions of the ACA. It’s also true that many parts of the law are terrible for practicing ObGyns, including the Independent Payment Advisory Board (IPAB) and the absence of meaningful medical liability reform. For more on these issues, see “Is private ObGyn practice on its way out?” which appears in the October 2011 issue of OBG Management (available in the archive at obgmanagement.com). ACOG is committed to working with Congress to repeal or remedy those aspects of the law.
A recent study reveals that almost one-third of physicians are no longer accepting Medicaid patients
If all states expanded Medicaid to cover people with incomes at or below 138% of the federal poverty level in 2014, as the Affordable Care Act (ACA) proposes, 23 million people would become eligible for the program.1
That statistic prompts important questions:
- Would the health-care workforce be able to meet the demand of caring for all these new patients?
- Would it be willing?
A recent analysis of data from 4,326 office-based physicians suggests that the answer to both questions is “No”: Almost one-third of these providers were already declining to accept new Medicaid patients in 2011.2
Although 96% of physicians in the analysis accepted new patients in 2011, the percentage of physicians accepting new patients covered by Medicaid was lower (69%), as was the percentage accepting new self-paying patients (91.7%), patients covered by Medicare (83%), and patients with private insurance (82%).2
Physicians who were in solo practice were 23.5% less likely to accept new Medicaid patients, compared with those who practiced in an office with 10 or more other physicians.2
The data from this study come from the 2011 National Ambulatory Medical Care Survey Electronic Medical Records Supplement, a survey conducted by the Centers for Disease Control and Prevention National Center for Health Statistics. The survey included questions exploring whether physicians were accepting new patients.2
Earlier studies have found that the low reimbursement levels for care delivered through Medicaid has deterred many physicians from accepting patients.3
The view in the ObGyn specialty
The findings of this analysis were not broken down by specialty—only by primary care versus non–primary care. To get an idea of conditions in the ObGyn specialty, OBG Management surveyed the members of its Virtual Board of Editors (VBE). Of the 117 members contacted, 61 responded—a response rate of 52.1%. Roughly three-quarters (75.4%) reported that they currently treat patients covered by Medicaid, but only 60.7% are accepting new patients covered by Medicaid. Twenty-one percent of respondents reported that they have not and will not accept patients covered by Medicaid.
When asked to comment on their level of satisfaction with Medicaid, the most common response among VBE members was dissatisfaction due to “insufficient reimbursement.”
“I am not satisfied with Medicaid,” commented one VBE member. “The reimbursement is terrible….I have certainly thought of stopping care for Medicaid patients and, if Congress ever allows the big cuts to reimbursement that are threatened every year, I think I would stop.”
Another VBE member reported extreme dissatisfaction with Medicaid because of “lousy” reimbursement. He also pointed to “all the paperwork and crazy regulations that require inordinate time and additional personnel just to handle….and then [the claim] gets denied for reasons beyond reason.” He added that physicians who do accept Medicaid “are on the fast track to sainthood.”
Other reasons for refusing to accept patients with Medicaid (or, if Medicaid was accepted, for high levels of aggravation with the program):
- payment rejections
- too many different categories of coverage “that patients are completely uninformed about”
- difficulty finding a specialist who will manage high-risk patients covered by Medicaid
- red tape
- the complex health problems that Medicaid patients tend to have, compared with patients who have other types of coverage.
One VBE member summed up his feelings in one word: “Phooey.”
Several VBE members suggested that health reform should focus on the Medicaid program.
“These plans are just sucking up the state’s money and paying docs peanuts and their administrators big bucks!” wrote Mary Vanko, MD, of Munster, Indiana.
“I’m tired of how much Medicaid is being abused by people,” commented another VBE member. “People using other people’s cards, people with regular insurance getting Medicaid to cover their copays. The whole system needs reform!”
Some physicians were satisfied with Medicaid
Among the respondents were several who reported being satisfied with the program, including one who called the experience “good” and another who reported being “shielded from the reimbursement issues.”
“I have no problems with Medicaid,” wrote another.
—Janelle Yates, Senior Editor
References
1. Kenney GM, Dubay L, Zuckerman S, Huntress M. Making the Medicaid Expansion an ACA Option: How Many Low-Income Americans Could Remain Uninsured? Washington, DC: Urban Institute Health Policy Center; June 29, 2012. http://www.urban.org/UploadedPDF/412606-Making-the-Medicaid-Expansion-an-ACA-Option.pdf. Accessed August 18, 2012.
2. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Affairs. 2012;31(8):1673–1679.
3. Centers for Disease Control and Prevention. QuickStats: percentage of office-based physicians accepting new patients, by types of payment accepted—United States, 1999–2000 and 2008–2009. MMWR Morb Mortal Wkly Rep. 2011;60(27):928.
We want to hear from you! Tell us what you think.

Lucia DiVenere, MA
Ms. DiVenere is Senior Director of Government Affairs at the American Congress of Obstetricians and Gynecologists.
Ms. DiVenere reports no financial relationships relevant to this article.
Lucia DiVenere, MA
Ms. DiVenere is Senior Director of Government Affairs at the American Congress of Obstetricians and Gynecologists.
Ms. DiVenere reports no financial relationships relevant to this article.
Lucia DiVenere, MA
Ms. DiVenere is Senior Director of Government Affairs at the American Congress of Obstetricians and Gynecologists.
Ms. DiVenere reports no financial relationships relevant to this article.
Lay midwives and the ObGyn: Is collaboration risky?
(May 2012)
How state budget crises are putting the squeeze on Medicaid (and you)
(February 2012)
Is private ObGyn practice on its way out?
(October 2011)
14 questions (and answers) about health reform and you
with Janelle Yates, Senior Editor (August 2010)
For the first half of 2012, the big question was: Will anything be covered under the Affordable Care Act (ACA)? After considering constitutional challenges to the Act that had the potential to invalidate the entire law, the US Supreme Court ruled, on June 28, that the ACA met constitutional muster in National Federation of Independent Business v. Sebelius (2012).
Now that the Court has upheld the ACA, let’s review the major women’s health services included under the law. This Web version incorporates 10 more women's health provisions from the ACA, from smoking cessation to young women’s breast cancer, that were not in the print version.
Preventive services guaranteed without copays
A major component of the health reform law went into effect August 1, 2012; it requires most health plans to cover women’s preventive services without requiring enrollees to pay a copay or deductibles. This provision reflects Congress’ understanding that women have a longer life expectancy and bear a greater burden of chronic disease, disability, and reproductive and gender-specific conditions. In addition, women often have a different response to treatment than men do.
The federal Department of Health and Human Services (HHS) estimates that Americans use preventive services at only about half of the recommended rate. By 2013, as many as 73 million individuals will benefit from preventive care offered under the law.
The American Congress of Obstetricians and Gynecologists (ACOG) worked with the Institute of Medicine (IOM)—which was charged with advising HHS—to encourage the inclusion of women’s preventive services specified in ACOG guidelines to ensure women’s health and well-being. As ACOG Executive Vice President Hal C. Lawrence, MD, told the IOM in January 2011:
- The College’s clinical guidelines…offer an excellent resource…and encompass the entire field of women’s preventive care. Our guidance is based on the best available evidence and is developed by committees with expertise reflecting the breadth of women’s health care and subject to a rigorous conflict of interest policy.
Dr. Lawrence further urged the IOM “to recommend coverage of the following services and products without cost-sharing”:
- well-woman visits
- preconception care
- family planning counseling and services
- HIV screening (for women at average risk)
- screening for intimate partner violence
- testing for human papillomavirus (HPV) as part of cervical cancer screening.
ACOG’s recommendations were approved by the IOM and, subsequently, by HHS. As a result, all private health plans that began on or after September 30, 2010, are required to cover these services at no out-of-pocket cost to patients (TABLE).
Women’s preventive services guaranteed under ACA*
| Service | Frequency | HHS guidelines for health insurance coverage |
|---|---|---|
| Well-woman visit | Annual for adult women, although HHS recognizes that several visits may be needed to obtain all necessary recommended preventive services, depending on a woman’s health status, health needs, and other risk factors** | The visit should focus on preventive services that are appropriate for the patient’s age and development, including preconception and prenatal care. This visit should, where appropriate, include other preventive services listed in this set of guidelines, as well as others referenced in section 2713 |
| Screening for gestational diabetes | Between 24 and 28 weeks of gestation and at the first prenatal visit for pregnant women identified to be at high risk for diabetes | |
| Testing for human papillomavirus (HPV) | At age 30 and older, no more frequently than every 3 years | High-risk HPV DNA testing in women who have normal cervical cytology |
| Counseling about sexually transmitted infection (STI) | Annual | All sexually active women |
| Counseling about and screening for HIV | Annual | All sexually active women |
| Counseling about and provision of contraception† | As prescribed | All FDA-approved contraceptive methods and sterilization procedures. Counseling for all women with reproductive capacity |
| Breastfeeding support, supplies, and counseling | In conjunction with each birth | Comprehensive lactation support and counseling by a trained provider during pregnancy or postpartum (or both), as well as costs for renting breastfeeding equipment |
| Screening for and counseling about interpersonal and domestic violence | Annual | |
| HHS = Health and Human Services * HHS guidelines are effective August 1, 2011. Nongrandfathered plans and insurers are required to provide coverage without cost-sharing consistent with HHS guidelines in the first plan year (in the individual market, policy year) that begins on or after August 1, 2012. ** The July 2011 Institute of Medicine report titled “Clinical preventive services for women: closing the gap” lists recommendations on individual preventive services that may be obtained during a well-woman preventive service visit. † Group health plans sponsored by certain religious employers, and group health insurance coverage in connection with such plans, are exempt from the requirement to cover contraceptive services. SOURCE: Adapted from Healthcare.gov. Affordable Care Act Expands Prevention Coverage for Women’s Health and Well-Being. http://www.hrsa.gov/womensguidelines/. Accessed August 8, 2012. | ||
Contraceptive mandate triggers a firestorm
On February 10, 2012, under pressure from religious groups, the Obama Administration offered a religious exemption to the contraception mandate for certain employers and group health plans. Under this “accommodation,” certain religious employers are exempt from the requirement to cover contraceptive services in their group health plans. An employer qualifies for this exemption if it:
- has the inculcation of religious values as one of its purposes
- primarily employs individuals who share its religious tenets
- primarily serves individuals who share its religious tenets, and
- qualifies for nonprofit status under
Internal Revenue Service (IRS) rules. At the same time that the Obama Administration wanted to accommodate employers’ religious beliefs, it also wanted to ensure that every woman would have access to free preventive care, including contraceptive services, regardless of where she works. And so while the Administration requires insurers to offer group health plan coverage without contraceptive coverage to religious-affiliated organizations, it also requires insurers to provide contraceptive coverage directly to individuals covered under the organization’s group health plan with no cost sharing.
This contraceptive mandate—even with the accommodation—has set off a firestorm on Capitol Hill that will eventually be settled in the courts.
Medicaid expansion falls short of original goal
In National Federation of Independent Business v. Sebelius, the plaintiffs asked the Supreme Court to rule on the federal government’s authority to require states to expand their Medicaid programs. Medicaid costs are typically shared by the federal and state governments. Under the ACA, state Medicaid programs were required to cover nearly all individuals who have incomes below 133% of the federal poverty level—$30,656 for a family of four in 2012—paid entirely by the federal government from 2014 through 2016. After that, the federal share gradually declines to, and then stays at, 90%. States that did not expand their Medicaid programs risked losing all federal Medicaid funding.
The Court ruled that the federal government can expand Medicaid but can’t penalize states that don’t accept the expansion mandate—effectively turning the mandate into a state option. States will receive the additional federal funds if they expand coverage, but states that don’t expand will not be penalized by losing existing federal funds for other parts of the program.
Since the ruling, a number of governors have announced that they will not expand their Medicaid programs, including governors of Florida and Louisiana. Those two states alone are home to 20% of all individuals intended to be covered under the Medicaid expansion.
This part of the ACA is particularly important to women because it strikes, for the first time, the requirement that a low-income woman must be pregnant to receive Medicaid coverage.
The figure below shows the dramatic potential improvement in coverage for women if all states fully implement the Medicaid expansion. Time, court decisions, elections, and state budget fights will determine how much of this change is realized for women’s health.
Percentage of insured women will increase under ACA
Percentage of women aged 19 to 64 years who were uninsured in 2009–2012 and under the Affordable Care Act when fully implemented.
SOURCE: Commonwealth Fund. Analysis of the March 2011 and 2010 Current Population Surveys by N. Tilipman and B. Sampat of Columbia University.
Women gain direct access to ObGyns
The ACA guarantees women in all states and all plans direct access to their ObGyns. Before the ACA, women in nine states lacked this guarantee, and women in 16 other states had only limited direct access. Now, a woman can go directly to her ObGyn without having to get a referral from her primary care physician or insurer.
Direct access is especially important because the ACA establishes new delivery systems, such as medical homes and accountable care organizations, designed to capture patients to maximize savings. An ObGyn does not have to be the patient’s primary care provider, and the patient’s access to her ObGyn cannot be limited to a certain number of visits or types of services.
ACA encourages states to cover family-planning services
Under the ACA, states have an easier time covering family-planning services, up to the same eligibility levels as pregnant women. Family planning is still an optional service that a state can choose to extend to women who have incomes above the Medicaid income eligibility level but, before the ACA was enacted, states had to apply to HHS for permission to waive the federal rules, often a very cumbersome process.
Prior to the ACA, 27 states had family planning waivers to provide services to nonpregnant women who had incomes above the Medicaid eligibility level—most at or near 200% of poverty. Now, states can provide family planning services to this population without federal approval.
Don’t miss Dr. Robert L. Barbieri’s October article titled “Gynecologic care across a woman’s life”
Insurance reforms end lifetime limits on coverage
Insurance reforms are important to us and our patients. The better the private health insurance system works—allowing us to provide our best possible care to patients and making sure they can see us when they need our care—the less our nation relies on the public safety net.
Beginning in 2010, the ACA eliminated all lifetime limits on how much insurance companies would cover when beneficiaries get sick; it also bans insurance companies from dropping people from coverage when they get sick. So if your patient has private health insurance and has faithfully paid her premiums and hasn’t committed fraud, her insurer cannot drop her or impose a limit on her coverage once she claims benefits.
This may be especially important for patients who need the most care, such as those who have cancer or another long-term, expensive, and unforeseen diagnosis. Because of this provision, you will not have to worry about your patient losing coverage in the middle of a long course of treatment.
The insurance practice of charging women more than men for equivalent policies ended on January 1, 2011, making insurance more affordable for our patients. Insurers in the individual and small group markets are allowed to vary premiums only for age, geographic location, family size, and tobacco use, not for gender—another important aspect of the law.
2014 is a key year in health reform
Exchanges begin
In 2014, under the ACA, state health insurance exchanges become reality.
An exchange is a marketplace where people can shop for health insurance; private health insurers can market their insurance products in state and multistate exchanges if they comply with new federal insurance reforms established in the ACA and offer the minimum benefits packages established by each state. Exchanges are intended to offer patients a choice of health insurance plans that are affordable, comprehensive, and easy to compare. Low-income individuals will be able to purchase private insurance in the exchanges with the federal premium subsidies or tax credits.
Insurers wanting to market their policies in an exchange may not deny coverage for preexisting conditions, including pregnancy, domestic violence, and previous cesarean delivery. They can’t deny coverage on the basis of an individual’s medical history, health status, genetic information, or disability. And they can’t impose waiting periods longer than 90 days before coverage takes effect, including 9-month waiting periods before maternity coverage.
Essential benefits are established
The ACA sets a minimum standard of health-care coverage that must be included in nearly every private insurance policy. The intent is that every person in the United States, regardless of where they live, who employs them, and what their income is, should have access to the same basic care.
Effective January 1, 2014, all insurance plans, except plans that existed before the ACA was enacted on March 23, 2010, must offer an “essential health benefits” (EHB) package, which must include:
- ambulatory patient services
- emergency services
- hospitalization
- maternity and newborn care
- mental health and substance use disorder services
- prescription drugs
- rehabilitative and habilitative services and devices
- laboratory services
- women’s preventive and wellness services and chronic disease management
- pediatric services, including oral and vision care.
Last December, HHS surprised many by giving states flexibility to design their own EHB packages, as long as the packages included each service on the list.
To choose its EHB package, a state must select a “benchmark” plan from the top- selling plans in four markets: federal and state public employee plans, commercial HMO plans, and small business plans. If a state doesn’t select a benchmark plan, the EHB defaults to the largest small-group market plan in the state. Each state must also choose an EHB package for its Medicaid program using the same 10 benefit categories.
State EHB plans must follow ACA requirements on annual and lifetime dollar limits but may impose limits on the scope and duration of coverage.
As for state-mandated benefits, if a state selects an EHB package that does not include a benefit already mandated by the state, the state must fund coverage for that service on its own—a decision HHS has promised to revisit in 2016.
Abortion decisions reside with the states
ACA requirements regarding abortion coverage 1) take effect in 2014 and 2) apply only to private health insurance plans marketed in the state exchanges that 3) cover abortions beyond those eligible for Medicaid coverage now, which are those that involve cases of rape or incest or that are necessary to save the life of the mother. Medicaid coverage for these categories of abortion is allowed under the Hyde Amendment.
Each insurer marketing a health plan in an exchange can determine whether or not its plan will cover abortion and, if it does, whether coverage will be limited to or go beyond those allowed under the Hyde Amendment. No federal tax or premium subsidies may be used to pay for abortions beyond those permitted by the Hyde Amendment.
The Secretary of HHS must ensure that at least one plan in each state exchange covers abortion, and that at least one plan either covers no abortions or limits abortions to those allowed under the Hyde Amendment. Insurers who offer abortion coverage beyond Hyde have to comply with a number of administrative requirements.
Congress was clear that the ultimate decisions about abortion should be made at the state rather than the federal level, and it gave states the ultimate trump card: Any state can pass legislation that prohibits any plan from offering abortion coverage of any kind within that state’s exchange. Any state can prohibit insurers offering plans within that state’s exchange from including any abortion coverage.
10 additional health provisions under the ACA
1. Creation of women's medical homes
The law points the way for creation of medical homes for women in the Medicare and Medicaid programs. The bill establishes an Innovation Center within the Centers for Medicare and Medicaid Services that has broad authority to evaluate, test, and adopt systems that foster patient-centered care, improve quality, and contain costs under Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP)—and this includes patient-centered medical homes that address women’s unique health needs. ObGyn practices are eligible to participate and to receive additional reimbursement if they do.
2. Smoking-cessation counseling in pregnancy
The framers of the ACA recognized the large negative impact that smoking has on health, especially during pregnancy. Studies suggest that the intervention of a physician—most notably, counseling of the patient to quit smoking—has strong potential to modify this behavior. The new law provides reimbursement for this intervention. There are no copays or deductibles for patients, and smoking-cessation services can include diagnostic, therapeutic, and counseling modalities in addition to prescription of pharmacotherapy.
Before this bill became law, only 24 state Medicaid programs paid ObGyns or other physicians for smoking-cessation counseling of pregnant patients, and five states provided no coverage at all. Now, all pregnant Medicaid patients can get this counseling, and you’ll be paid for this important service.
3. Payments to nonphysician providers in freestanding birth centers
Before the ACA became law, Medicaid was authorized to pay hospitals and other facilities operated by and under the supervision of a physician; no payments were authorized for services of an ambulatory center operated by other health professionals. The ACA authorizes Medicaid payments to state-recognized freestanding birth centers not operated by or under the supervision of a physician. A state that doesn’t currently license birth centers must pass legislation and license these centers before the centers can receive these payments.
Medicaid will also reimburse providers who practice in state-recognized freestanding birth centers, as long as the individuals are practicing within their state’s scope of practice laws and regulations. Because the type of provider is not specified but instead left up to each state’s scope of practice laws and regulations, this provision could allow for separate provider payments for physicians, certified nurse midwives, certified professional midwives, and doulas.
4. Immigrant coverage
Legal immigrants are bound by the individual coverage mandate and must purchase health insurance. These individuals are eligible for income-related premium credits and subsidies for insurance purchased through an exchange. Legal immigrants who are barred from Medicaid during their first 5 years in the United States (by earlier law) are eligible for premium credits only.
Undocumented immigrants are not eligible for Medicaid, premium credits, or subsidies and are barred from purchasing insurance in the exchange, even with their own money.
5. Postpartum depression
Health reform will help bring perinatal and postpartum depression out of the shadows by providing federal funds for research, patient education, and clinical treatment. For example, the federal Department of Health and Human Services (HHS) will:
- conduct research into the causes of, and treatments for, postpartum conditions
- create a national public awareness campaign to increase awareness and knowledge of postpartum depression and psychosis
- provide grants to study the benefits of screening for postpartum depression and psychosis
- establish grants to deliver or enhance outpatient, inpatient, and home-based health and support services, including case management and comprehensive treatment services for individuals with, or at risk for, postpartum conditions.
The National Institute of Mental Health is encouraged to conduct a 10-year longitudinal study on the mental health consequences of pregnancy. This study is intended to focus on perinatal depression.
Community health centers will be eligible for grants in 2012 (as they were in 2011) to the tune of $3 million for inpatient and outpatient counseling and services.
And a federal public awareness campaign will educate the public through radio and television ads.
These endeavors point to the need for ObGyns to familiarize themselves with postpartum depression—if they aren’t already well versed in the subject—because patients are likely to become more aware of this issue and look to their ObGyns for answers.
6. Maternal home visits
Congress established a new Maternal, Infant, and Early Childhood Home Visiting program to improve maternal and fetal health in underserved areas of our country. This program will provide funds to states, tribes, and territories to develop and implement evidence-based home-visitation models to reduce infant and maternal mortality and its causes by producing improvements in:
- prenatal, maternal, and newborn health
- child health and development
- parenting skills
- school readiness
- juvenile delinquency
- family economic self-sufficiency.
These programs will have to demonstrate effectiveness and improved outcomes. HHS recently requested suggestions on ways of demonstrating the effectiveness of home-visiting program models for pregnant women, expectant fathers, and caregivers of children from birth through entry into kindergarten.
The law appropriates $350 million to this program in 2012 and $400 million in both 2013 and 2014.
7. Assistance for pregnant students
A new Pregnancy Assistance Fund—$25 million annually over 10 years (fiscal years 2010–2019)—requires the Secretary of HHS (in collaboration with the Secretary of Education) to establish a state grant program to help pregnant and parenting teens and young women. The aim of this program is to help teens who become pregnant and who choose to bring their pregnancies to term or keep their babies, or both, to stay in school. Grants will go to institutions of higher education, high schools and community service centers, as well as state attorneys general.
Institutions that receive grants must work with providers to meet specific practical needs of pregnant or parenting students:
- housing
- childcare
- parenting education
- postpartum counseling
- assistance in finding and accessing needed services
- referrals for prenatal care and delivery, infant or foster care, or adoption.
Funds to attorneys general will be used to combat domestic violence among pregnant teens.
8. Young women's breast cancer
A new program is intended to help educate young women about the importance of breast health and screening, in two ways:
- The National Institutes of Health (NIH) will conduct research to develop and test screening measures for prevention and early detection of breast cancer in women 15 to 44 years old.
- The US Department of HHS will create a national awareness campaign, with $9 million in funding each year from 2010 to 2014, to encourage young women to talk with their doctors about breast cancer and early detection.
ObGyns can expect to see more interest and questions about breast health among young women and their mothers. It pays to be prepared with good information for these important conversations.
9. Personal responsibility education
From 2010 through 2014, each state will receive funds for personal responsibility education programs aimed at reducing pregnancy in youths. Funds are $75 million for each fiscal year, allocated to each state depending on the size of its youth population but not intended to be less than $250,000 per state.
Educational programs eligible for federal funds must include both abstinence and contraception information for prevention of teenage pregnancy and sexually transmitted infections, including HIV/AIDS, as well as three or more adulthood-preparation subjects.
10. Community-based support of Patient-Centered Medical Homes
Federal funding is available to states for the development of community-based health teams to support medical homes run by primary care practices. These teams may include specialists, nurses, pharmacists, nutritionists, dieticians, social workers, behavioral and mental health providers, and physician assistants. Primary care practices in this program function as medical homes and are responsible for addressing a patient’s personal health-care needs. The team links the medical home to community support services for its patients.
Eligible ObGyn practices can qualify as primary care practices, and ObGyns are eligible to serve as specialist members of the community-based health team.
ACA is a mixed bag for ObGyns
Women have much to gain from the provisions of the ACA. It’s also true that many parts of the law are terrible for practicing ObGyns, including the Independent Payment Advisory Board (IPAB) and the absence of meaningful medical liability reform. For more on these issues, see “Is private ObGyn practice on its way out?” which appears in the October 2011 issue of OBG Management (available in the archive at obgmanagement.com). ACOG is committed to working with Congress to repeal or remedy those aspects of the law.
A recent study reveals that almost one-third of physicians are no longer accepting Medicaid patients
If all states expanded Medicaid to cover people with incomes at or below 138% of the federal poverty level in 2014, as the Affordable Care Act (ACA) proposes, 23 million people would become eligible for the program.1
That statistic prompts important questions:
- Would the health-care workforce be able to meet the demand of caring for all these new patients?
- Would it be willing?
A recent analysis of data from 4,326 office-based physicians suggests that the answer to both questions is “No”: Almost one-third of these providers were already declining to accept new Medicaid patients in 2011.2
Although 96% of physicians in the analysis accepted new patients in 2011, the percentage of physicians accepting new patients covered by Medicaid was lower (69%), as was the percentage accepting new self-paying patients (91.7%), patients covered by Medicare (83%), and patients with private insurance (82%).2
Physicians who were in solo practice were 23.5% less likely to accept new Medicaid patients, compared with those who practiced in an office with 10 or more other physicians.2
The data from this study come from the 2011 National Ambulatory Medical Care Survey Electronic Medical Records Supplement, a survey conducted by the Centers for Disease Control and Prevention National Center for Health Statistics. The survey included questions exploring whether physicians were accepting new patients.2
Earlier studies have found that the low reimbursement levels for care delivered through Medicaid has deterred many physicians from accepting patients.3
The view in the ObGyn specialty
The findings of this analysis were not broken down by specialty—only by primary care versus non–primary care. To get an idea of conditions in the ObGyn specialty, OBG Management surveyed the members of its Virtual Board of Editors (VBE). Of the 117 members contacted, 61 responded—a response rate of 52.1%. Roughly three-quarters (75.4%) reported that they currently treat patients covered by Medicaid, but only 60.7% are accepting new patients covered by Medicaid. Twenty-one percent of respondents reported that they have not and will not accept patients covered by Medicaid.
When asked to comment on their level of satisfaction with Medicaid, the most common response among VBE members was dissatisfaction due to “insufficient reimbursement.”
“I am not satisfied with Medicaid,” commented one VBE member. “The reimbursement is terrible….I have certainly thought of stopping care for Medicaid patients and, if Congress ever allows the big cuts to reimbursement that are threatened every year, I think I would stop.”
Another VBE member reported extreme dissatisfaction with Medicaid because of “lousy” reimbursement. He also pointed to “all the paperwork and crazy regulations that require inordinate time and additional personnel just to handle….and then [the claim] gets denied for reasons beyond reason.” He added that physicians who do accept Medicaid “are on the fast track to sainthood.”
Other reasons for refusing to accept patients with Medicaid (or, if Medicaid was accepted, for high levels of aggravation with the program):
- payment rejections
- too many different categories of coverage “that patients are completely uninformed about”
- difficulty finding a specialist who will manage high-risk patients covered by Medicaid
- red tape
- the complex health problems that Medicaid patients tend to have, compared with patients who have other types of coverage.
One VBE member summed up his feelings in one word: “Phooey.”
Several VBE members suggested that health reform should focus on the Medicaid program.
“These plans are just sucking up the state’s money and paying docs peanuts and their administrators big bucks!” wrote Mary Vanko, MD, of Munster, Indiana.
“I’m tired of how much Medicaid is being abused by people,” commented another VBE member. “People using other people’s cards, people with regular insurance getting Medicaid to cover their copays. The whole system needs reform!”
Some physicians were satisfied with Medicaid
Among the respondents were several who reported being satisfied with the program, including one who called the experience “good” and another who reported being “shielded from the reimbursement issues.”
“I have no problems with Medicaid,” wrote another.
—Janelle Yates, Senior Editor
References
1. Kenney GM, Dubay L, Zuckerman S, Huntress M. Making the Medicaid Expansion an ACA Option: How Many Low-Income Americans Could Remain Uninsured? Washington, DC: Urban Institute Health Policy Center; June 29, 2012. http://www.urban.org/UploadedPDF/412606-Making-the-Medicaid-Expansion-an-ACA-Option.pdf. Accessed August 18, 2012.
2. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Affairs. 2012;31(8):1673–1679.
3. Centers for Disease Control and Prevention. QuickStats: percentage of office-based physicians accepting new patients, by types of payment accepted—United States, 1999–2000 and 2008–2009. MMWR Morb Mortal Wkly Rep. 2011;60(27):928.
We want to hear from you! Tell us what you think.
Lay midwives and the ObGyn: Is collaboration risky?
(May 2012)
How state budget crises are putting the squeeze on Medicaid (and you)
(February 2012)
Is private ObGyn practice on its way out?
(October 2011)
14 questions (and answers) about health reform and you
with Janelle Yates, Senior Editor (August 2010)
For the first half of 2012, the big question was: Will anything be covered under the Affordable Care Act (ACA)? After considering constitutional challenges to the Act that had the potential to invalidate the entire law, the US Supreme Court ruled, on June 28, that the ACA met constitutional muster in National Federation of Independent Business v. Sebelius (2012).
Now that the Court has upheld the ACA, let’s review the major women’s health services included under the law. This Web version incorporates 10 more women's health provisions from the ACA, from smoking cessation to young women’s breast cancer, that were not in the print version.
Preventive services guaranteed without copays
A major component of the health reform law went into effect August 1, 2012; it requires most health plans to cover women’s preventive services without requiring enrollees to pay a copay or deductibles. This provision reflects Congress’ understanding that women have a longer life expectancy and bear a greater burden of chronic disease, disability, and reproductive and gender-specific conditions. In addition, women often have a different response to treatment than men do.
The federal Department of Health and Human Services (HHS) estimates that Americans use preventive services at only about half of the recommended rate. By 2013, as many as 73 million individuals will benefit from preventive care offered under the law.
The American Congress of Obstetricians and Gynecologists (ACOG) worked with the Institute of Medicine (IOM)—which was charged with advising HHS—to encourage the inclusion of women’s preventive services specified in ACOG guidelines to ensure women’s health and well-being. As ACOG Executive Vice President Hal C. Lawrence, MD, told the IOM in January 2011:
- The College’s clinical guidelines…offer an excellent resource…and encompass the entire field of women’s preventive care. Our guidance is based on the best available evidence and is developed by committees with expertise reflecting the breadth of women’s health care and subject to a rigorous conflict of interest policy.
Dr. Lawrence further urged the IOM “to recommend coverage of the following services and products without cost-sharing”:
- well-woman visits
- preconception care
- family planning counseling and services
- HIV screening (for women at average risk)
- screening for intimate partner violence
- testing for human papillomavirus (HPV) as part of cervical cancer screening.
ACOG’s recommendations were approved by the IOM and, subsequently, by HHS. As a result, all private health plans that began on or after September 30, 2010, are required to cover these services at no out-of-pocket cost to patients (TABLE).
Women’s preventive services guaranteed under ACA*
| Service | Frequency | HHS guidelines for health insurance coverage |
|---|---|---|
| Well-woman visit | Annual for adult women, although HHS recognizes that several visits may be needed to obtain all necessary recommended preventive services, depending on a woman’s health status, health needs, and other risk factors** | The visit should focus on preventive services that are appropriate for the patient’s age and development, including preconception and prenatal care. This visit should, where appropriate, include other preventive services listed in this set of guidelines, as well as others referenced in section 2713 |
| Screening for gestational diabetes | Between 24 and 28 weeks of gestation and at the first prenatal visit for pregnant women identified to be at high risk for diabetes | |
| Testing for human papillomavirus (HPV) | At age 30 and older, no more frequently than every 3 years | High-risk HPV DNA testing in women who have normal cervical cytology |
| Counseling about sexually transmitted infection (STI) | Annual | All sexually active women |
| Counseling about and screening for HIV | Annual | All sexually active women |
| Counseling about and provision of contraception† | As prescribed | All FDA-approved contraceptive methods and sterilization procedures. Counseling for all women with reproductive capacity |
| Breastfeeding support, supplies, and counseling | In conjunction with each birth | Comprehensive lactation support and counseling by a trained provider during pregnancy or postpartum (or both), as well as costs for renting breastfeeding equipment |
| Screening for and counseling about interpersonal and domestic violence | Annual | |
| HHS = Health and Human Services * HHS guidelines are effective August 1, 2011. Nongrandfathered plans and insurers are required to provide coverage without cost-sharing consistent with HHS guidelines in the first plan year (in the individual market, policy year) that begins on or after August 1, 2012. ** The July 2011 Institute of Medicine report titled “Clinical preventive services for women: closing the gap” lists recommendations on individual preventive services that may be obtained during a well-woman preventive service visit. † Group health plans sponsored by certain religious employers, and group health insurance coverage in connection with such plans, are exempt from the requirement to cover contraceptive services. SOURCE: Adapted from Healthcare.gov. Affordable Care Act Expands Prevention Coverage for Women’s Health and Well-Being. http://www.hrsa.gov/womensguidelines/. Accessed August 8, 2012. | ||
Contraceptive mandate triggers a firestorm
On February 10, 2012, under pressure from religious groups, the Obama Administration offered a religious exemption to the contraception mandate for certain employers and group health plans. Under this “accommodation,” certain religious employers are exempt from the requirement to cover contraceptive services in their group health plans. An employer qualifies for this exemption if it:
- has the inculcation of religious values as one of its purposes
- primarily employs individuals who share its religious tenets
- primarily serves individuals who share its religious tenets, and
- qualifies for nonprofit status under
Internal Revenue Service (IRS) rules. At the same time that the Obama Administration wanted to accommodate employers’ religious beliefs, it also wanted to ensure that every woman would have access to free preventive care, including contraceptive services, regardless of where she works. And so while the Administration requires insurers to offer group health plan coverage without contraceptive coverage to religious-affiliated organizations, it also requires insurers to provide contraceptive coverage directly to individuals covered under the organization’s group health plan with no cost sharing.
This contraceptive mandate—even with the accommodation—has set off a firestorm on Capitol Hill that will eventually be settled in the courts.
Medicaid expansion falls short of original goal
In National Federation of Independent Business v. Sebelius, the plaintiffs asked the Supreme Court to rule on the federal government’s authority to require states to expand their Medicaid programs. Medicaid costs are typically shared by the federal and state governments. Under the ACA, state Medicaid programs were required to cover nearly all individuals who have incomes below 133% of the federal poverty level—$30,656 for a family of four in 2012—paid entirely by the federal government from 2014 through 2016. After that, the federal share gradually declines to, and then stays at, 90%. States that did not expand their Medicaid programs risked losing all federal Medicaid funding.
The Court ruled that the federal government can expand Medicaid but can’t penalize states that don’t accept the expansion mandate—effectively turning the mandate into a state option. States will receive the additional federal funds if they expand coverage, but states that don’t expand will not be penalized by losing existing federal funds for other parts of the program.
Since the ruling, a number of governors have announced that they will not expand their Medicaid programs, including governors of Florida and Louisiana. Those two states alone are home to 20% of all individuals intended to be covered under the Medicaid expansion.
This part of the ACA is particularly important to women because it strikes, for the first time, the requirement that a low-income woman must be pregnant to receive Medicaid coverage.
The figure below shows the dramatic potential improvement in coverage for women if all states fully implement the Medicaid expansion. Time, court decisions, elections, and state budget fights will determine how much of this change is realized for women’s health.
Percentage of insured women will increase under ACA
Percentage of women aged 19 to 64 years who were uninsured in 2009–2012 and under the Affordable Care Act when fully implemented.
SOURCE: Commonwealth Fund. Analysis of the March 2011 and 2010 Current Population Surveys by N. Tilipman and B. Sampat of Columbia University.
Women gain direct access to ObGyns
The ACA guarantees women in all states and all plans direct access to their ObGyns. Before the ACA, women in nine states lacked this guarantee, and women in 16 other states had only limited direct access. Now, a woman can go directly to her ObGyn without having to get a referral from her primary care physician or insurer.
Direct access is especially important because the ACA establishes new delivery systems, such as medical homes and accountable care organizations, designed to capture patients to maximize savings. An ObGyn does not have to be the patient’s primary care provider, and the patient’s access to her ObGyn cannot be limited to a certain number of visits or types of services.
ACA encourages states to cover family-planning services
Under the ACA, states have an easier time covering family-planning services, up to the same eligibility levels as pregnant women. Family planning is still an optional service that a state can choose to extend to women who have incomes above the Medicaid income eligibility level but, before the ACA was enacted, states had to apply to HHS for permission to waive the federal rules, often a very cumbersome process.
Prior to the ACA, 27 states had family planning waivers to provide services to nonpregnant women who had incomes above the Medicaid eligibility level—most at or near 200% of poverty. Now, states can provide family planning services to this population without federal approval.
Don’t miss Dr. Robert L. Barbieri’s October article titled “Gynecologic care across a woman’s life”
Insurance reforms end lifetime limits on coverage
Insurance reforms are important to us and our patients. The better the private health insurance system works—allowing us to provide our best possible care to patients and making sure they can see us when they need our care—the less our nation relies on the public safety net.
Beginning in 2010, the ACA eliminated all lifetime limits on how much insurance companies would cover when beneficiaries get sick; it also bans insurance companies from dropping people from coverage when they get sick. So if your patient has private health insurance and has faithfully paid her premiums and hasn’t committed fraud, her insurer cannot drop her or impose a limit on her coverage once she claims benefits.
This may be especially important for patients who need the most care, such as those who have cancer or another long-term, expensive, and unforeseen diagnosis. Because of this provision, you will not have to worry about your patient losing coverage in the middle of a long course of treatment.
The insurance practice of charging women more than men for equivalent policies ended on January 1, 2011, making insurance more affordable for our patients. Insurers in the individual and small group markets are allowed to vary premiums only for age, geographic location, family size, and tobacco use, not for gender—another important aspect of the law.
2014 is a key year in health reform
Exchanges begin
In 2014, under the ACA, state health insurance exchanges become reality.
An exchange is a marketplace where people can shop for health insurance; private health insurers can market their insurance products in state and multistate exchanges if they comply with new federal insurance reforms established in the ACA and offer the minimum benefits packages established by each state. Exchanges are intended to offer patients a choice of health insurance plans that are affordable, comprehensive, and easy to compare. Low-income individuals will be able to purchase private insurance in the exchanges with the federal premium subsidies or tax credits.
Insurers wanting to market their policies in an exchange may not deny coverage for preexisting conditions, including pregnancy, domestic violence, and previous cesarean delivery. They can’t deny coverage on the basis of an individual’s medical history, health status, genetic information, or disability. And they can’t impose waiting periods longer than 90 days before coverage takes effect, including 9-month waiting periods before maternity coverage.
Essential benefits are established
The ACA sets a minimum standard of health-care coverage that must be included in nearly every private insurance policy. The intent is that every person in the United States, regardless of where they live, who employs them, and what their income is, should have access to the same basic care.
Effective January 1, 2014, all insurance plans, except plans that existed before the ACA was enacted on March 23, 2010, must offer an “essential health benefits” (EHB) package, which must include:
- ambulatory patient services
- emergency services
- hospitalization
- maternity and newborn care
- mental health and substance use disorder services
- prescription drugs
- rehabilitative and habilitative services and devices
- laboratory services
- women’s preventive and wellness services and chronic disease management
- pediatric services, including oral and vision care.
Last December, HHS surprised many by giving states flexibility to design their own EHB packages, as long as the packages included each service on the list.
To choose its EHB package, a state must select a “benchmark” plan from the top- selling plans in four markets: federal and state public employee plans, commercial HMO plans, and small business plans. If a state doesn’t select a benchmark plan, the EHB defaults to the largest small-group market plan in the state. Each state must also choose an EHB package for its Medicaid program using the same 10 benefit categories.
State EHB plans must follow ACA requirements on annual and lifetime dollar limits but may impose limits on the scope and duration of coverage.
As for state-mandated benefits, if a state selects an EHB package that does not include a benefit already mandated by the state, the state must fund coverage for that service on its own—a decision HHS has promised to revisit in 2016.
Abortion decisions reside with the states
ACA requirements regarding abortion coverage 1) take effect in 2014 and 2) apply only to private health insurance plans marketed in the state exchanges that 3) cover abortions beyond those eligible for Medicaid coverage now, which are those that involve cases of rape or incest or that are necessary to save the life of the mother. Medicaid coverage for these categories of abortion is allowed under the Hyde Amendment.
Each insurer marketing a health plan in an exchange can determine whether or not its plan will cover abortion and, if it does, whether coverage will be limited to or go beyond those allowed under the Hyde Amendment. No federal tax or premium subsidies may be used to pay for abortions beyond those permitted by the Hyde Amendment.
The Secretary of HHS must ensure that at least one plan in each state exchange covers abortion, and that at least one plan either covers no abortions or limits abortions to those allowed under the Hyde Amendment. Insurers who offer abortion coverage beyond Hyde have to comply with a number of administrative requirements.
Congress was clear that the ultimate decisions about abortion should be made at the state rather than the federal level, and it gave states the ultimate trump card: Any state can pass legislation that prohibits any plan from offering abortion coverage of any kind within that state’s exchange. Any state can prohibit insurers offering plans within that state’s exchange from including any abortion coverage.
10 additional health provisions under the ACA
1. Creation of women's medical homes
The law points the way for creation of medical homes for women in the Medicare and Medicaid programs. The bill establishes an Innovation Center within the Centers for Medicare and Medicaid Services that has broad authority to evaluate, test, and adopt systems that foster patient-centered care, improve quality, and contain costs under Medicare, Medicaid, and the Children’s Health Insurance Program (CHIP)—and this includes patient-centered medical homes that address women’s unique health needs. ObGyn practices are eligible to participate and to receive additional reimbursement if they do.
2. Smoking-cessation counseling in pregnancy
The framers of the ACA recognized the large negative impact that smoking has on health, especially during pregnancy. Studies suggest that the intervention of a physician—most notably, counseling of the patient to quit smoking—has strong potential to modify this behavior. The new law provides reimbursement for this intervention. There are no copays or deductibles for patients, and smoking-cessation services can include diagnostic, therapeutic, and counseling modalities in addition to prescription of pharmacotherapy.
Before this bill became law, only 24 state Medicaid programs paid ObGyns or other physicians for smoking-cessation counseling of pregnant patients, and five states provided no coverage at all. Now, all pregnant Medicaid patients can get this counseling, and you’ll be paid for this important service.
3. Payments to nonphysician providers in freestanding birth centers
Before the ACA became law, Medicaid was authorized to pay hospitals and other facilities operated by and under the supervision of a physician; no payments were authorized for services of an ambulatory center operated by other health professionals. The ACA authorizes Medicaid payments to state-recognized freestanding birth centers not operated by or under the supervision of a physician. A state that doesn’t currently license birth centers must pass legislation and license these centers before the centers can receive these payments.
Medicaid will also reimburse providers who practice in state-recognized freestanding birth centers, as long as the individuals are practicing within their state’s scope of practice laws and regulations. Because the type of provider is not specified but instead left up to each state’s scope of practice laws and regulations, this provision could allow for separate provider payments for physicians, certified nurse midwives, certified professional midwives, and doulas.
4. Immigrant coverage
Legal immigrants are bound by the individual coverage mandate and must purchase health insurance. These individuals are eligible for income-related premium credits and subsidies for insurance purchased through an exchange. Legal immigrants who are barred from Medicaid during their first 5 years in the United States (by earlier law) are eligible for premium credits only.
Undocumented immigrants are not eligible for Medicaid, premium credits, or subsidies and are barred from purchasing insurance in the exchange, even with their own money.
5. Postpartum depression
Health reform will help bring perinatal and postpartum depression out of the shadows by providing federal funds for research, patient education, and clinical treatment. For example, the federal Department of Health and Human Services (HHS) will:
- conduct research into the causes of, and treatments for, postpartum conditions
- create a national public awareness campaign to increase awareness and knowledge of postpartum depression and psychosis
- provide grants to study the benefits of screening for postpartum depression and psychosis
- establish grants to deliver or enhance outpatient, inpatient, and home-based health and support services, including case management and comprehensive treatment services for individuals with, or at risk for, postpartum conditions.
The National Institute of Mental Health is encouraged to conduct a 10-year longitudinal study on the mental health consequences of pregnancy. This study is intended to focus on perinatal depression.
Community health centers will be eligible for grants in 2012 (as they were in 2011) to the tune of $3 million for inpatient and outpatient counseling and services.
And a federal public awareness campaign will educate the public through radio and television ads.
These endeavors point to the need for ObGyns to familiarize themselves with postpartum depression—if they aren’t already well versed in the subject—because patients are likely to become more aware of this issue and look to their ObGyns for answers.
6. Maternal home visits
Congress established a new Maternal, Infant, and Early Childhood Home Visiting program to improve maternal and fetal health in underserved areas of our country. This program will provide funds to states, tribes, and territories to develop and implement evidence-based home-visitation models to reduce infant and maternal mortality and its causes by producing improvements in:
- prenatal, maternal, and newborn health
- child health and development
- parenting skills
- school readiness
- juvenile delinquency
- family economic self-sufficiency.
These programs will have to demonstrate effectiveness and improved outcomes. HHS recently requested suggestions on ways of demonstrating the effectiveness of home-visiting program models for pregnant women, expectant fathers, and caregivers of children from birth through entry into kindergarten.
The law appropriates $350 million to this program in 2012 and $400 million in both 2013 and 2014.
7. Assistance for pregnant students
A new Pregnancy Assistance Fund—$25 million annually over 10 years (fiscal years 2010–2019)—requires the Secretary of HHS (in collaboration with the Secretary of Education) to establish a state grant program to help pregnant and parenting teens and young women. The aim of this program is to help teens who become pregnant and who choose to bring their pregnancies to term or keep their babies, or both, to stay in school. Grants will go to institutions of higher education, high schools and community service centers, as well as state attorneys general.
Institutions that receive grants must work with providers to meet specific practical needs of pregnant or parenting students:
- housing
- childcare
- parenting education
- postpartum counseling
- assistance in finding and accessing needed services
- referrals for prenatal care and delivery, infant or foster care, or adoption.
Funds to attorneys general will be used to combat domestic violence among pregnant teens.
8. Young women's breast cancer
A new program is intended to help educate young women about the importance of breast health and screening, in two ways:
- The National Institutes of Health (NIH) will conduct research to develop and test screening measures for prevention and early detection of breast cancer in women 15 to 44 years old.
- The US Department of HHS will create a national awareness campaign, with $9 million in funding each year from 2010 to 2014, to encourage young women to talk with their doctors about breast cancer and early detection.
ObGyns can expect to see more interest and questions about breast health among young women and their mothers. It pays to be prepared with good information for these important conversations.
9. Personal responsibility education
From 2010 through 2014, each state will receive funds for personal responsibility education programs aimed at reducing pregnancy in youths. Funds are $75 million for each fiscal year, allocated to each state depending on the size of its youth population but not intended to be less than $250,000 per state.
Educational programs eligible for federal funds must include both abstinence and contraception information for prevention of teenage pregnancy and sexually transmitted infections, including HIV/AIDS, as well as three or more adulthood-preparation subjects.
10. Community-based support of Patient-Centered Medical Homes
Federal funding is available to states for the development of community-based health teams to support medical homes run by primary care practices. These teams may include specialists, nurses, pharmacists, nutritionists, dieticians, social workers, behavioral and mental health providers, and physician assistants. Primary care practices in this program function as medical homes and are responsible for addressing a patient’s personal health-care needs. The team links the medical home to community support services for its patients.
Eligible ObGyn practices can qualify as primary care practices, and ObGyns are eligible to serve as specialist members of the community-based health team.
ACA is a mixed bag for ObGyns
Women have much to gain from the provisions of the ACA. It’s also true that many parts of the law are terrible for practicing ObGyns, including the Independent Payment Advisory Board (IPAB) and the absence of meaningful medical liability reform. For more on these issues, see “Is private ObGyn practice on its way out?” which appears in the October 2011 issue of OBG Management (available in the archive at obgmanagement.com). ACOG is committed to working with Congress to repeal or remedy those aspects of the law.
A recent study reveals that almost one-third of physicians are no longer accepting Medicaid patients
If all states expanded Medicaid to cover people with incomes at or below 138% of the federal poverty level in 2014, as the Affordable Care Act (ACA) proposes, 23 million people would become eligible for the program.1
That statistic prompts important questions:
- Would the health-care workforce be able to meet the demand of caring for all these new patients?
- Would it be willing?
A recent analysis of data from 4,326 office-based physicians suggests that the answer to both questions is “No”: Almost one-third of these providers were already declining to accept new Medicaid patients in 2011.2
Although 96% of physicians in the analysis accepted new patients in 2011, the percentage of physicians accepting new patients covered by Medicaid was lower (69%), as was the percentage accepting new self-paying patients (91.7%), patients covered by Medicare (83%), and patients with private insurance (82%).2
Physicians who were in solo practice were 23.5% less likely to accept new Medicaid patients, compared with those who practiced in an office with 10 or more other physicians.2
The data from this study come from the 2011 National Ambulatory Medical Care Survey Electronic Medical Records Supplement, a survey conducted by the Centers for Disease Control and Prevention National Center for Health Statistics. The survey included questions exploring whether physicians were accepting new patients.2
Earlier studies have found that the low reimbursement levels for care delivered through Medicaid has deterred many physicians from accepting patients.3
The view in the ObGyn specialty
The findings of this analysis were not broken down by specialty—only by primary care versus non–primary care. To get an idea of conditions in the ObGyn specialty, OBG Management surveyed the members of its Virtual Board of Editors (VBE). Of the 117 members contacted, 61 responded—a response rate of 52.1%. Roughly three-quarters (75.4%) reported that they currently treat patients covered by Medicaid, but only 60.7% are accepting new patients covered by Medicaid. Twenty-one percent of respondents reported that they have not and will not accept patients covered by Medicaid.
When asked to comment on their level of satisfaction with Medicaid, the most common response among VBE members was dissatisfaction due to “insufficient reimbursement.”
“I am not satisfied with Medicaid,” commented one VBE member. “The reimbursement is terrible….I have certainly thought of stopping care for Medicaid patients and, if Congress ever allows the big cuts to reimbursement that are threatened every year, I think I would stop.”
Another VBE member reported extreme dissatisfaction with Medicaid because of “lousy” reimbursement. He also pointed to “all the paperwork and crazy regulations that require inordinate time and additional personnel just to handle….and then [the claim] gets denied for reasons beyond reason.” He added that physicians who do accept Medicaid “are on the fast track to sainthood.”
Other reasons for refusing to accept patients with Medicaid (or, if Medicaid was accepted, for high levels of aggravation with the program):
- payment rejections
- too many different categories of coverage “that patients are completely uninformed about”
- difficulty finding a specialist who will manage high-risk patients covered by Medicaid
- red tape
- the complex health problems that Medicaid patients tend to have, compared with patients who have other types of coverage.
One VBE member summed up his feelings in one word: “Phooey.”
Several VBE members suggested that health reform should focus on the Medicaid program.
“These plans are just sucking up the state’s money and paying docs peanuts and their administrators big bucks!” wrote Mary Vanko, MD, of Munster, Indiana.
“I’m tired of how much Medicaid is being abused by people,” commented another VBE member. “People using other people’s cards, people with regular insurance getting Medicaid to cover their copays. The whole system needs reform!”
Some physicians were satisfied with Medicaid
Among the respondents were several who reported being satisfied with the program, including one who called the experience “good” and another who reported being “shielded from the reimbursement issues.”
“I have no problems with Medicaid,” wrote another.
—Janelle Yates, Senior Editor
References
1. Kenney GM, Dubay L, Zuckerman S, Huntress M. Making the Medicaid Expansion an ACA Option: How Many Low-Income Americans Could Remain Uninsured? Washington, DC: Urban Institute Health Policy Center; June 29, 2012. http://www.urban.org/UploadedPDF/412606-Making-the-Medicaid-Expansion-an-ACA-Option.pdf. Accessed August 18, 2012.
2. Decker SL. In 2011 nearly one-third of physicians said they would not accept new Medicaid patients, but rising fees may help. Health Affairs. 2012;31(8):1673–1679.
3. Centers for Disease Control and Prevention. QuickStats: percentage of office-based physicians accepting new patients, by types of payment accepted—United States, 1999–2000 and 2008–2009. MMWR Morb Mortal Wkly Rep. 2011;60(27):928.
We want to hear from you! Tell us what you think.
An evidence-based approach to treating pediatric anxiety disorders
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders

GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
Anxiety disorders are remarkably common among pediatric patients1,2 and are associated with significant morbidity3 and increased risk of suicidality in adolescents.4,5 Effective diagnosis and treatment of pediatric anxiety disorders are critical for reducing psychosocial morbidity,3,6 suicidality, and the risk of secondary mood disorders.7
This article summarizes open-label studies and randomized controlled trials (RCTs) of selective serotonin reuptake inhibitors (SSRIs), selective serotonin-norepinephrine reuptake inhibitors, atypical anxiolytics, and benzodiazepines in children and adolescents with generalized anxiety disorder (GAD), social phobia, separation anxiety disorder, and panic disorder. Although we focus on psychopharmacologic treatments, the best outcomes generally are observed with multimodal treatments that combine psychotherapy and pharmacotherapy.
Generalized anxiety disorder
Researchers have evaluated SSRIs, benzodiazepines, and buspirone in pediatric patients with GAD. In a double-blind, placebo-controlled trial of 22 patients age 5 to 17, sertraline, 50 mg/d, was associated with improvement in Hamilton Anxiety Rating Scale (HAM-A), Clinical Global Impression-Severity (CGI-S), and Clinical Global Impression-Improvement (CGI-I) scores over 9 weeks.8 The Child-Adolescent Anxiety Multimodal Study compared cognitive-behavioral therapy (CBT) to sertraline or sertraline plus CBT in 488 patients age 7 to 17, 78% of whom had GAD.9 Sertraline monotherapy was superior to placebo and not statistically different from CBT, while combination treatment was superior to both monotherapy conditions in improving CGI score. In both trials, sertraline was well tolerated.
One study evaluated fluoxetine, 5 to 40 mg/d, or CBT in 14 youths with GAD; both treatments improved symptoms.10 In a study of 320 GAD patients age 6 to 17, venlafaxine extended-release (XR) initiated at 37.5 mg/d was associated with improved HAM-A scores.11 In general, venlafaxine was well tolerated; adverse effects included increased blood pressure, asthenia, pain, anorexia, somnolence, weight loss, and possibly treatment-emergent suicidal ideation.
Two RCTs of buspirone, 15 to 60 mg/d, that evaluated 559 children and adolescents age 6 to 17 with GAD did not observe significant differences between buspirone and placebo.12 By contrast, 2 open-label studies of youths with anxiety suggested improvement associated with buspirone.12 Treatment-emergent adverse events included nausea, stomachache, and headache.
Clinical trials of benzodiazepines in anxious children and adolescents have yielded mixed results. A 4-week, open-label trial of alprazolam, 0.5 mg to 1.5 mg/d, in 12 adolescents with overanxious disorder—the DSM-III forerunner of GAD—found improvements in anxiety, depression, psychomotor excitation, and hyperactivity, but patients experienced sedation, activation, headache, and nausea.13 However, a double-blind RCT in 30 youths age 8 to 16 found no statistically significant difference between alprazolam and placebo.14 Alprazolam generally was well tolerated; fatigue and dry mouth were reported, but no withdrawal symptoms. Additionally, benzodiazepine use may be associated with tolerance and—in young children—disinhibition.
Social phobia
Researchers have evaluated paroxetine, citalopram, fluoxetine, and venlafaxine for treating social phobia in pediatric patients. In an RCT, 78% of paroxetine-treated patients with social phobia responded compared with 38% for placebo over 16 weeks. Adverse events—including withdrawal symptoms—were twice as likely in patients who received paroxetine. Additionally, 4 paroxetine patients exhibited suicidal ideation vs 0 patients who received placebo.15
In an RCT of 293 children and adolescents age 8 to 17 with social phobia, venlafaxine XR was initiated at 37.5 mg/d and titrated to 112.5 mg/d, 150 mg/d, or 225 mg/d, depending on body weight.16 The venlafaxine group experienced significantly improved anxiety symptoms and the medication generally was well tolerated, although 3 venlafaxine-treated patients developed suicidal ideation compared with 0 in the placebo group.
An RCT compared Social Effectiveness Therapy for Children (SET-C) and fluoxetine, 10 to 40 mg/d, for 139 patients age 7 to 17 with social phobia.17 SET-C is a CBT for children and adolescents that focuses on increasing interpersonal skills and becoming more comfortable in social situations; it involves psychoeducation, social skills training, and exposure exercises. At endpoint, 53% of patients in the SET-C group no longer met diagnostic criteria for social phobia. Fluoxetine was well tolerated; no severe adverse events were reported.
In an open-label study of sertraline (mean dose = 123 mg/d) for 14 young persons with social phobia, 36% of patients responded and 29% partially responded at 8 weeks.18 Adverse events generally were mild and included nausea, diarrhea, and headache. In a 12-week study, 12 pediatric patients with social phobia received citalopram, 10 to 40 mg/d, and eight 15-minute counseling sessions. At endpoint, clinicians rated 83% of patients as much improved or very much improved. The medication generally was well tolerated.19
Separation anxiety disorder
In a 4-week, double-blind crossover pilot study, researchers randomly assigned 15 children age 7 to 13 with separation anxiety disorder to clonazepam, up to 2 mg/d, or placebo.20 There was no significant difference in CGI-I score between clonazepam and placebo. Side effects—including drowsiness, irritability and “oppositional behavior”—were more frequent in patients treated with clonazepam.
Panic disorder
Only 2 open-label studies of SSRIs have been conducted in pediatric patients with panic disorder. The first evaluated the effectiveness and tolerability of fluoxetine, sertraline, or paroxetine over 6 months in 12 patients; 67% no longer met criteria for panic disorder at endpoint.21 In this study, benzodiazepines—including clonazepam and lorazepam—were used in 67% of patients at the start of SSRI treatment. The authors suggested this strategy may be clinically useful for patients with panic disorder.
In the second study, Fairbanks et al22 examined the use of fluoxetine for 6 to 9 weeks in 16 outpatients with mixed anxiety disorders who did not respond to psychotherapy. Patients age ≤12 were given 5 to 40 mg/d and those age ≥13 received 5 to 80 mg/d. Fluoxetine was associated with clinically significant improvement in 3 of the 5 patients who had panic disorder. Although overall fluoxetine was well tolerated, drowsiness, dyssomnia, decreased appetite, nausea, and abdominal pain were the most common side effects. Fluoxetine was not associated with suicidal ideation.
Mixed anxiety disorders
Most trials of pediatric anxiety have evaluated patients with “mixed anxiety disorders” because GAD, social phobia, and separation anxiety disorder are highly comorbid and share diagnostic features (Figure 1).9 An RCT of fluvoxamine, up to 300 mg/d, in 128 pediatric patients with ≥1 anxiety disorders found significant differences in CGI-I and endpoint Pediatric Anxiety Rating Scale (PARS) scores.23 Fluvoxamine was well tolerated but associated with increased motor activity and abdominal discomfort compared with placebo.
Two open-label trials of pediatric patients with mixed anxiety disorders suggested fluoxetine may be beneficial. Fairbanks et al22 documented clinical improvement in 10 of 10 patients with separation anxiety disorder, 8 of 10 with social phobia, 4 of 6 with specific phobia, 3 of 5 with panic disorder, and 1 of 7 with GAD. Birmaher et al24 evaluated 21 pediatric patients with overanxious disorder, social phobia, or separation anxiety who had not responded to psychotherapy and were not depressed; all patients received flexibly-dosed fluoxetine for up to 10 months. Fluoxetine was well tolerated and 81% of patients improved.
Finally, in a 12-week RCT of 74 patients age 7 to 17 with GAD, separation anxiety disorder, and/or social phobia, fluoxetine, 10 to 20 mg/d, was associated with improved scores on the Screen for Anxiety Related Emotional Disorders, PARS, CGI-I, CGI-S, and Children’s Global Assessment Scale.25 A follow-up open-label trial suggested that maintenance treatment is associated with sustained improvement.26
Figure 1: The pediatric anxiety disorders triad: Comorbidity is common
In the Child-Adolescent Multimodal Treatment Study, GAD was the most common disorder; however, GAD, SAD, and SoP were highly comorbid
GAD: generalized anxiety disorder; SAD: separation anxiety disorder; SoP: social phobia
Source: Reference 9
Anxiety disorders with ADHD
Anxiety disorders often are comorbid with attention-deficit/hyperactivity disorder (ADHD). An RCT of patients age 8 to 17 with ADHD and comorbid anxiety found that atomoxetine was associated with improved PARS scores and ADHD symptoms.27 The target dose was 1.2 mg/kg/d. Atomoxetine was well-tolerated; decreased appetite was the only significant adverse event in the treatment group vs placebo.
Multimodal treatment
Although this article reviews evidence for psychopharmacologic treatments, psychotherapeutic treatment of young patients with anxiety disorders has seen significant advances.28 Most psychotherapy studies have evaluated the efficacy of CBT,29-31 although there is evidence for psychodynamic therapy and interpersonal therapy.32 The American Academy of Child & Adolescent Psychiatry recommends a multimodal treatment approach because combination treatment appears to be more effective than monotherapy.8,28,33 Also, clinicians who treat pediatric patients who have an anxiety disorder should evaluate the family’s role on anxiety symptoms and may consider family therapy.
Treatment considerations
Evidence supports the efficacy of sertraline, citalopram, paroxetine, fluvoxamine, fluoxetine, and venlafaxine for treating children and adolescents with anxiety disorders (Figure 2).8,9,11,15,16,23,25 Some practitioners suggest using differing dosing strategies for pediatric anxiety disorders compared with those used to treat adults (Table).34 When considering SSRIs for children and adolescents, keep in mind the “black-box” warning regarding suicidality in these patients. Carefully monitor patients for treatment-emergent suicidality and routinely reassess for the presence and severity of suicidal ideation and suicide risk.
Figure 2: Number needed to treat for SSRIs and SNRIs in pediatric anxiety disorders
GAD: generalized anxiety disorder; RUPP: Research Unit on Pediatric Psychopharmacology; SAD: separation anxiety disorder; SNRI: serotonin-norepinephrine reuptake inhibitor; SoP: social phobia; SSRI: selective serotonin reuptake inhibitorTable
Practical dosing of SSRIs and SNRIs in pediatric patients with anxietya
| Medication | Initial child dose (age <12; mg/d) | Initial adolescent dose (age 12 to 17; mg/d) | Target dose (mg/d) |
|---|---|---|---|
| Citalopram | 5 to 10 | 10 | 20 to 40 |
| Escitalopram | 2.5 to 5 | 5 to 10 | 10 to 20 |
| Fluoxetineb | 10 | 20 | 20 to 40 (children), 40 to 60 (adolescents) |
| Paroxetineb | 5 to 10 | 10 | 20 |
| Sertralinec | 10 to 12.5 | 25 | 150 |
| Venlafaxine | 37.5 | 37.5 | 150 |
| aGeneralized anxiety disorder, social phobia, and separation anxiety disorder bMay consider cytochrome P450 genotyping for 2D6, which may suggest an alternate dosing strategy cSertraline is available in a liquid formulation (20 mg/mL) SNRI: serotonin-norepinephrine reuptake inhibitor; SSRI: selective serotonin reuptake inhibitor Source: Adapted from reference 34 | |||
Related Resources
- Connolly SD, Bernstein GA; Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
- Anxiety and Depression Association of America. www.adaa.org.
- American Academy of Child & Adolescent Psychiatry. www.aacap.org.
Drug Brand Names
- Alprazolam • Xanax
- Atomoxetine • Strattera
- Buspirone • BuSpar
- Citalopram • Celexa
- Clonazepam • Klonopin
- Fluoxetine • Prozac
- Fluvoxamine • Luvox, Luvox CR
- Lorazepam • Ativan
- Paroxetine • Paxil, Paxil CR
- Sertraline • Zoloft
- Venlafaxine • Effexor, Effexor XR
Disclosures
Dr. Strawn has received research support from the American Academy of Child & Adolescent Psychiatry, Eli Lilly and Company, and Shire, and is an employee of the University of Cincinnati, Cincinnati, OH.
Dr. McReynolds was employed by Eli Lilly and Company from 1997 to 2005.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
1. Beesdo K, Knappe S, Pine DS. Anxiety and anxiety disorders in children and adolescents: developmental issues and implications for DSM-V. Psychiatr Clin North Am. 2009;32(3):483-524.
2. Beesdo K, Pine DS, Lieb R, et al. Incidence and risk patterns of anxiety and depressive disorders and categorization of generalized anxiety disorder. Arch Gen Psychiatry. 2010;67(1):47-57.
3. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first grade children: prediction to anxious symptoms and adaptive functioning in fifth grade. J Child Psychol Psychiatry. 1995;36(3):427-437.
4. Foley DL, Goldston DB, Costello EJ, et al. Proximal psychiatric risk factors for suicidality in youth: the Great Smoky Mountains Study. Arch Gen Psychiatry. 2006;63(9):1017-1024.
5. Jacobson CM, Muehlenkamp JJ, Miller AL, et al. Psychiatric impairment among adolescents engaging in different types of deliberate self-harm. J Clin Child Adolesc Psychol. 2008;37(2):363-375.
6. Ialongo N, Edelsohn G, Werthamer-Larsson L, et al. The significance of self-reported anxious symptoms in first-grade children. J Abnorm Child Psychol. 1994;22(4):441-455.
7. Pine DS, Cohen P, Gurley D, et al. The risk for early-adulthood anxiety and depressive disorders in adolescents with anxiety and depressive disorders. Arch Gen Psychiatry. 1998;55(1):56-64.
8. Rynn MA, Siqueland L, Rickels K. Placebo-controlled trial of sertraline in the treatment of children with generalized anxiety disorders. Am J Psychiatry. 2001;158(12):2008-2014.
9. Walkup JT, Albano AM, Piacentini J, et al. Cognitive behavioral therapy, sertraline, or a combination in childhood anxiety. N Engl J Med. 2008;359(26):2753-2766.
10. Maslowsky J, Mogg K, Bradley BP, et al. A preliminary investigation of neural correlates of treatment in adolescents with generalized anxiety disorder. J Child Adolesc Psychopharmacol. 2010;20(2):105-111.
11. Rynn MA, Riddle MA, Yeung PP, et al. Efficacy and safety of extended-release venlafaxine in the treatment of generalized anxiety disorder in children and adolescents: two placebo-controlled trials. Am J Psychiatry. 2007;164(2):290-300.
12. BuSpar [package insert] Princeton NJ: Bristol-Myers Squibb; 2010.
13. Simeon JG, Ferguson HB. Alprazolam effects in children with anxiety disorders. Can J Psychiatry. 1987;32(7):570-574.
14. Simeon JG, Ferguson HB, Knott V, et al. Clinical, cognitive, and neurophysiological effects of alprazolam in children and adolescents with overanxious and avoidant disorders. J Am Acad Child Adolesc Psychiatry. 1992;31(1):29-33.
15. Wagner KD, Berard R, Stein MB, et al. A multicenter, randomized, double-blind, placebo-controlled trial of paroxetine in children and adolescents with social anxiety disorder. Arch Gen Psychiatry. 2004;61(11):1153-1162.
16. March JS, Entusah AR, Rynn M, et al. A randomized controlled trial of venlafaxine ER versus placebo in pediatric social anxiety disorder. Biol Psychiatry. 2007;62(10):1149-1154.
17. Beidel DC, Turner SM, Sallee FR, et al. SET-C versus fluoxetine in the treatment of childhood social phobia. J Am Acad Child Adolesc Psychiatry. 2007;46(12):1622-1632.
18. Compton SN, Grant PJ, Chrisman AK, et al. Sertraline in children and adolescents with social anxiety disorder: an open trial. J Am Acad Child Adolesc Psychiatry. 2001;40(5):564-571.
19. Chavira DA, Stein MB. Combined psychoeducation and treatment with selective serotonin reuptake inhibitors for youth with generalized social anxiety disorder. J Child Adolesc Psychopharmacol. 2002;12(1):47-54.
20. Graae F, Milner J, Rizzotto L, et al. Clonazepam in childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(3):372-376.
21. Renaud J, Birmaher B, Wassick SC, et al. Use of selective serotonin reuptake inhibitors for the treatment of childhood panic disorder: a pilot study. J Child Adolesc Psychopharmacol. 1999;9(2):73-83.
22. Fairbanks JM, Pine DS, Tancer NK, et al. Open fluoxetine treatment of mixed anxiety disorders in children and adolescents. J Child Adolesc Psychopharmacol. 1997;7(1):17-29.
23. The Research Unit on Pediatric Psychopharmacology Anxiety Study Group. Fluvoxamine for the treatment of anxiety disorders in children and adolescents. N Engl J Med. 2001;344(17):1279-1285.
24. Birmaher B, Waterman GS, Ryan N, et al. Fluoxetine for childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 1994;33(7):993-999.
25. Birmaher B, Axelson DA, Monk K, et al. Fluoxetine for the treatment of childhood anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2003;42(4):415-423.
26. Clark DB, Birmaher B, Axelson D, et al. Fluoxetine for the treatment of childhood anxiety disorders: open-label, long-term extension to a controlled trial. J Am Acad Child Adolesc Psychiatry. 2005;44(12):1263-1270.
27. Geller D, Donnelly C, Lopez F, et al. Atomoxetine treatment for pediatric patients with attention-deficit/hyperactivity disorder with comorbid anxiety disorder. J Am Acad Child Adolesc Psychiatry. 2007;46(9):1119-1127.
28. Connolly SD, Bernstein GA. Work Group on Quality Issues. Practice parameter for the assessment and treatment of children and adolescents with anxiety disorders. J Am Acad Child Adolesc Psychiatry. 2007;46(2):267-283.
29. Kendall PC. Treating anxiety disorders in children: results of a randomized clinical trial. J Consult Clin Psychol. 1994;62(1):100-110.
30. Kendall PC, Flannery-Schroeder E, Panichelli-Mindel SM, et al. Therapy for youths with anxiety disorders: a second randomized clinical trial. J Consult Clin Psychol. 1997;65(3):366-380.
31. Reynolds S, Wilson C, Austin J, et al. Effects of psychotherapy for anxiety in children and adolescents: a meta-analytic review. Clin Psychol Rev. 2012;32(4):251-262.
32. Strawn JR, Wehry AM, DelBello MP, et al. Establishing the neurobiologic basis of treatment in children and adolescents with generalized anxiety disorder. Depress Anxiety. 2012;29(4):328-339.
33. Ginsburg GS, Kendall PC, Sakolsky D, et al. Remission after acute treatment in children and adolescents with anxiety disorders: findings from the CAMS. J Consult Clin Psychol. 2011;79(6):806-813.
34. Findling RL, Kowatch RA. How (not) to dose antidepressants and antipsychotics for children. Current Psychiatry. 2007;6(6):79-83.
Reducing CYP450 drug interactions caused by antidepressants
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
Most psychiatrists are aware that some antidepressants can cause clinically significant drug interactions, especially through the cytochrome P450 (CYP450) hepatic enzyme system. Antidepressants’ potential for drug interactions is especially important for patients who take >1 other medication, including cardiovascular agents.1
Unfortunately, drug interactions can be difficult to remember and are commonly missed. One strategy to help remember a list of antidepressants with a relatively low potential for CYP450 drug interactions is to use the mnemonic Various Medicines Definitely Commingle Very Easily (VMDCVE) to recall venlafaxine, mirtazapine, desvenlafaxine,2 citalopram, vilazodone,3 and escitalopram. The order in which these medications are listed does not indicate a preference for any of the 6 antidepressants. Bupropion and duloxetine are not included in this list because they are moderately potent inhibitors of the 2D6 isoenzyme.4,5
A few caveats
There are some important caveats in using this mnemonic:
- None of these antidepressants is completely devoid of effects on the CYP450 system. However, compared with the antidepressants included in this mnemonic, fluoxetine, paroxetine, fluvoxamine, duloxetine, bupropion, and nefazodone are more likely to have clinically significant effects on CYP450.4,5
- Although sertraline has a lower potential for CYP450-mediated drug interactions at low doses, it is not included in this mnemonic because it may have greater effects on 2D6 inhibition in some patients, especially at higher doses, such as ≥150 mg/d.5 Also, sertraline may significantly increase lamotrigine levels through a different mechanism: inhibition of uridine 5’-diphosphate glucuronosyltransferase 1A4.4
- Antidepressants also may be the substrates for CYP450 drug interactions caused by other medications.
- This mnemonic refers only to CYP450-mediated drug interactions. Antidepressants included in this mnemonic may have a high potential for drug interactions mediated by displacement from carrier proteins— eg, with digoxin or warfarin.
- Pharmacodynamic drug interactions also are possible—eg, serotonin syndrome as a result of combining a selective serotonin reuptake inhibitor with another serotonergic medication.
To remain vigilant for drug-drug interactions, routinely use a drug interaction software, in addition to this mnemonic.
Disclosure
Dr. Mago receives grant/research, support from Bristol-Myers Squibb, Eli Lilly and Company, and NARSAD.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
1. Williams S, Wynn G, Cozza K, et al. Cardiovascular medications. Psychosomatics. 2007;48(6):537-547.
2. Nichols AI, Tourian KA, Tse SY, et al. Desvenlafaxine for major depressive disorder: incremental clinical benefits from a second-generation serotonin-norepinephrine reuptake inhibitor. Expert Opin Drug Metab Toxicol. 2010;6(12):1565-1574.
3. Laughren TP, Gobburu J, Temple RJ, et al. Vilazodone: clinical basis for the US Food and Drug Administration’s approval of a new antidepressant. J Clin Psychiatry. 2011;72(9):1166-1173.
4. Sandson NB, Armstrong SC, Cozza KL. An overview of psychotropic drug-drug interactions. Psychosomatics. 2005;46(5):464-494.
5. Spina E, Santoro V, D’Arrigo C. Clinically relevant pharmacokinetic drug interactions with second-generation antidepressants: an update. Clin Ther. 2008;30(7):1206-1227.
Sleep terrors in adults: How to help control this potentially dangerous condition
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Sleep terrors (STs)—also known as night terrors—are characterized by sudden arousal accompanied by a piercing scream or cry in the first few hours after falling asleep. These parasomnias arise out of slow-wave sleep (stages 3 and 4 of nonrapid eye movement [non-REM] sleep) and affect approximately 5% of adults.1 The condition is twice as common in men than women, and usually affects children but may not develop until adulthood.1
During STs, a patient may act scared, afraid, agitated, anxious, or panicky without being fully aware of his or her surroundings. The episode may last 30 seconds to 5 minutes; most patients don’t remember the event the next morning. STs may leave individuals feeling exhausted and perplexed the next day. Verbalization during the episode is incoherent and a patient’s perception of the environment seems altered. Tachycardia, tachypnea, sweating, flushed skin, or mydriasis are prominent. When ST patients walk, they may do so violently and can cause harm to themselves or others.
The differential diagnosis of STs includes posttraumatic stress disorder; nocturnal seizures characterized by excessive motor activity and organic CNS lesions; REM sleep behavior disorder; sleep choking syndrome; and nocturnal panic attacks. Patients with STs report high rates of stressful events—eg, divorce or bereavement—in the previous year. They are more likely to have a history of mood and anxiety disorders and high levels of depression, anxiety, and obsessive-compulsive and phobic traits. One study found patients with STs were 4.3 times more likely to have had a car accident in the past year.2
Evaluating and treating STs
Rule out comorbid conditions such as obstructive sleep apnea and periodic limb movement disorder. Encourage your patient to improve his or her sleep hygiene by maintaining a regular sleep/wake cycle, exercising, and limiting caffeine and alcohol and exposure to bright light before bedtime.
Self-help techniques. To avoid injury, encourage your patient to remove dangerous objects from their sleeping area. Suggest locking the doors to the room or home, and putting medications in a secure place. Patients also may consider keeping their mattress close to the floor to limit the risk of injury.
Pharmacotherapy and psychotherapy. Along with counseling and support, your patient may benefit from cognitive-behavioral therapy, relaxation therapy, or hypnosis.3 Anticipatory arousal therapy may help by interrupting the altered underlying electrophysiology of partial arousal.
If your patient is concerned about physical injury during STs, consider prescribing clonazepam, temazepam, or diazepam.4 Trazodone and selective serotonin reuptake inhibitors such as paroxetine5 also have been used to treat STs.
Disclosure
Dr. Jain reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.
1. Crisp AH. The sleepwalking/night terrors syndrome in adults. Postgrad Med J. 1996;72(852):599-604.
2. Oudiette D, Leu S, Pottier M, et al. Dreamlike mentations during sleepwalking and sleep terrors in adults. Sleep. 2009;32(12):1621-1627.
3. Lowe P, Humphreys C, Williams SJ. Night terrors: women’s experiences of (not) sleeping where there is domestic violence. Violence Against Women. 2007;13(6):549-561.
4. Schenck CH, Mahowald MW. Long-term nightly benzodiazepine treatment of injurious parasomnias and other disorders of disrupted nocturnal sleep in 170 adults. Am J Med. 1996;100(3):333-337.
5. Lillywhite AR, Wilson SJ, Nutt DJ. Successful treatment of night terrors and somnambulism with paroxetine. Br J Psychiatry. 1994;164(4):551-554.