User login
Omega-3 fatty acids for psychiatric illness
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.

Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Epidemiologic data suggest that people who consume diets rich in omega-3 fatty acids (FAs)—long-chain polyunsaturated FAs such as eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)—have a decreased risk of major depressive disorder (MDD), postpartum depression, and bipolar disorder (BD).1-5 Omega-3 FA concentration may impact serotonin and dopamine transmission via effects on cell membrane fluidity.6 Therefore, decreased intake may increase the risk of several psychiatric disorders. As the average Western diet has changed over the last 2 centuries, omega-3 FA consumption has decreased.7 Omega-3 FAs cannot be synthesized by the body and must come from exogenous sources, such as fish and nuts. For a discussion of different types of dietary fats, see Box 1.8
Should we advise our patients to increase their omega-3 FA consumption? The American Psychiatric Association (APA) and the American Heart Association (AHA) recommend omega-3 FA consumption for the general population and in some cases, supplementation for specific disorders (Box 2).9-12 New data has been published since Current Psychiatry last reviewed the evidence for using omega-3 FAs for psychiatric conditions in 2004.8 This article looks at the latest evidence on the use of omega-3 FAs to treat mood disorders, schizophrenia, dementia, and other psychiatric conditions.
Dietary fat is saturated or unsaturated. Unsaturated fats are further categorized as monounsaturated or polyunsaturated (PUFA). PUFAs contain a hydrocarbon chain with ≥2 double bonds.8 The position of this double bond relative to the methyl end carbon—or “omega” carbon—groups the PUFAs into 2 categories:8
- omega-6 fatty acids, including arachidonic acid (AA) and linoleic acid (LA)
- omega-3 fatty acids, including eicosapentaenoic acid (EPA), docosahexaenoic acid (DHA), and alpha-linolenic acid (ALA). ALA is a metabolic precursor to EPA and DHA.
PUFAs—in particular AA and DHA—are thought to contribute to cell membrane fluidity, modulation of neurotransmitters, and signal transduction pathways. As precursors to eicosanoids and cytokines, PUFAs may affect anti-inflammatory response systems.
Consumption of omega-3 fatty acids (FAs) reduces risk for arrhythmia, thrombosis, and atherosclerotic plaque, according to American Heart Association (AHA) guidelines. Omega-3 FA intake also may improve endothelial function, slightly lower blood pressure, and reduce inflammatory response. Replacing dietary saturated fat with polyunsaturated fat reduces coronary heart disease risk by 19%.9 The AHA recommends that all adults eat fish, particularly oily fish such as salmon or tuna, ≥2 times per week. Patients with documented coronary heart disease should consume 1 g/d eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) combined10 either via oily fish or omega-3 FA capsules. Side effects of omega-3 FA supplements are minor and include mild gastrointestinal discomfort, mostly burping or an unpleasant aftertaste; no cases of bleeding have been reported.11
For patients with hypertriglyceridemia, 2 to 4 g/d may be useful. Because of a theoretical risk of bleeding, doses >3 g/d should be supervised by a physician.
Because psychiatric illnesses and cardiovascular disease may be comorbid, the Omega-3 FA Subcommittee of the American Psychiatric Association supports the AHA’s guidelines regarding fish consumption, and further recommends that patients with mood, impulse control, or psychotic disorders consume ≥1 g/d of combined EPA and DHA.12
Limitations of the data
Reviewing the literature on omega-3 FAs to treat psychiatric disorders is hampered by several difficulties:13
- studies may evaluate the use of EPA alone, EPA combined with DHA, or DHA alone
- the doses of EPA and DHA and ratio of EPA to DHA of the supplements used in clinical trials varies greatly
- patients’ dietary consumption of omega-3 FAs is difficult to control
- DSM diagnostic criteria, as well as severity of illness, differ within studies.
In addition, studies may use omega-3 FAs as monotherapy or as adjuncts. All of these factors lead to difficulty interpreting the literature, as well as trouble in extracting data for meta-analysis.
Omega-3 FAs for mood disorders
MDD and other depressive diagnoses. Several meta-analyses examining the use of omega-3 FAs for treating depressive disorders have had equivocal findings. Variability in results might be partially explained by differences in the severity of baseline depression among diverse study populations, diagnostic variation, differing omega-3 supplementation protocols, or other issues.13 In addition, publication bias also may affect results.
In a 2011 literature review and meta-analysis of omega-3 FAs as monotherapy or an adjunct to antidepressants to treat MDD, Bloch and Hannestad6 concluded that omega-3 FAs offer a small but nonsignificant benefit in treating MDD. This review suggested that omega-3 FAs may be more effective in patients with more severe depression. The effects of varying levels of EPA vs DHA were not examined.
In a systematic review and meta-analysis, Appleton et al14 concluded that omega-3 FA supplements have little beneficial effect on depressed mood in individuals who do not have a depressive illness diagnosis (eg, MDD). However, this study did not consider the differential effects of EPA vs DHA on treatment response. Patients diagnosed with a depressive illness received greater benefits from omega-3 FA supplementation, although the patients in this study were heterogeneous. Similar to Bloch and Hannestad, Appleton et al14 found that omega-3 FA supplementation may be most beneficial for depressed patients with more severe symptoms, but is unlikely to help those with mild-to-moderate symptoms or individuals without symptoms who aim to prevent depression.
A meta-analysis by Martins15 looked at EPA vs DHA to treat depressive illness and found that only supplements that were mostly or completely EPA effectively treated depressive symptoms. Martins also found that severity of illness is key for positive treatment outcomes; there was a significant relationship between higher baseline depression levels and efficacy.15 Martins noted that omega-3 FA therapy was more effective as a treatment than a preventive strategy, and that adding omega-3 FAs to antidepressants was more efficacious than omega-3 FAs alone.15
A meta-analysis of clinical trials of omega-3 FAs for depressive illness suggested EPA should be ≥60% of total EPA + DHA.16
BD. A recent meta-analysis of 6 randomized controlled trials (RCTs) found that adding omega-3 supplements to mood stabilizers in patients with BD was associated with a statistically significant reduction of depressive symptoms, but was not effective for treating mania.17 The authors suggested patients with BD—especially those with comorbid cardiovascular or metabolic conditions— increase their dietary consumption of foods containing omega-3 FAs (Table)18 and, if necessary, take a supplement of 1 to 1.5 g/d of mixed EPA and DHA, with a higher ratio of EPA.19 See Box 3 for a box on how to read omega-3 supplement labels.
In a small RCT of 51 children and adolescents (age 6 to 17) with symptomatic bipolar I or bipolar II disorder, supplementation with flax oil (alpha-linolenic acid, a polyunsaturated omega-3 FA that is a precursor to EPA and DHA) did not affect symptoms as measured by several rating scales.20
Perinatal and postpartum depression. Omega-3 FAs are considered a safe treatment for depressive disorders during pregnancy because they provide neurodevelopmental benefits for neonates and have few contraindications during pregnancy.21 RCTs of omega-3 FA monotherapy for perinatal depression have been small (≤51 patients) and produced mixed findings.21 A pilot study (N = 16) of patients with postpartum depression found a significant decrease in depressive symptoms with EPA treatment.22 More research is needed before omega-3 FA supplementation can be recommended during pregnancy.
Table
Foods with healthy fats: From best to worst
| Polyunsaturated fats | Omega-3 | Fish-based: oily fish, including salmon, tuna, mackerel, lake trout, herring, and sardines Plant-based: tofu and other forms of soybeans; walnuts and flaxseed and their oils, and canola oil |
| Omega-6 | Only available in plant-based form: corn, soy, and safflower oil | |
| Monosaturated fats | Olive and peanut oil | |
| Saturated fats | Red meats, high-fat dairy, and partially hydrogenated oils | |
| Source: Reference 18 | ||
Because nutritional supplements vary, advise patients to look at the supplement facts on the back of a bottle of omega-3 fatty acids. The American Psychiatric Association recommends patients take a total eicosapentaenoic acid (EPA) + docosahexaenoic acid (DHA) of 1 g/d; EPA should be ≥60% of total EPA + DHA.
This image is an example of a label that would meet the appropriate criteria. Total EPA + DHA = 1,490 mg and EPA is 60% of this combined total.
Source: Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584
Schizophrenia
In a Cochrane review of 8 studies of patients with schizophrenia, adjunctive treatment with omega-3 FAs led to >25% reduction in the Positive and Negative Syndrome Scale, but this improvement was not statistically significant.23 Omega-3 FAs did not decrease tardive dyskinesia symptoms as measured by the Abnormal Involuntary Movement Scale. The authors stated that results were inconclusive, and use of omega-3 FAs in patients with schizophrenia remains experimental. In a separate meta-analysis that included 335 patients with schizophrenia, EPA augmentation had no beneficial effect on psychotic symptoms.24
In a double-blind RCT of 81 adolescents and young adults (age 13 to 25) at ultra-high risk of psychotic illness, 5% of patients who received 1.2 g/d of omega-3 FAs developed a psychotic disorder compared with 28% of patients receiving placebo.25 The authors concluded that supplementation with omega-3 FAs may be a safe and effective strategy for young patients with subthreshold psychotic symptoms.
Dementia
Studies evaluating the relationship between omega-3 FAs and dementia risk have revealed mixed findings.26,27 In a pilot study of 10 geriatric patients with moderately severe dementia related to thrombotic cerebrovascular disorder, DHA supplementation led to improved Hamilton Depression Rating Scale and Mini-Mental State Examination (MMSE) scores compared with controls.28 In another study, administering EPA to 64 patients with Alzheimer’s disease significantly improved MMSE scores, with maximum improvement at 3 months, but this benefit dissipated after 6 months of treatment.29 In a study of 22 patients with various types of dementia, Suzuki et al30 found that DHA supplementation improved scores on a Japanese dementia scale. These studies show promise, but more evidence is necessary before recommendations can be made.
Other psychiatric disorders
Omega-3 FAs as monotherapy or an adjunct to psychostimulants does not seem to improve symptoms in children who meet DSM-IV-TR criteria for attention-deficit/hyperactivity disorder (ADHD).31-33 Studies of omega-3 FAs as treatment for anxiety and personality disorders are limited. To date, omega-3 FAs as adjunctive treatment in obsessive-compulsive disorder (OCD) and monotherapy in borderline personality disorder have not shown efficacy.34,35
Using omega-3 FAs in practice
Based on new data and several recent meta-analyses, clinical recommendations have emerged. Sarris et al17 suggested patients with BD increase dietary intake of omega-3 FAs or take a supplement with 1 to 1.5 g/d of mixed EPA and DHA (with a higher ratio of EPA). In MDD, the type of omega-3 FA supplementation seems to be important; EPA seems to be the primary component for efficacy.15,19 Additionally, the more severe the depression, the more likely symptoms will respond to omega-3 FAs.6,14,15 Omega-3 FAs are not effective at preventing depression14,15 and evidence is equivocal for treating perinatal depression.21 Omega-3 FA supplementation has not shown efficacy for patients with schizophrenia,23,24 although it may prevent transition to psychosis in adolescents and young adults at ultra-high risk for a psychotic disorder.25 Data examining omega-3 FA supplementation in postpartum depression22 and dementia28,29 are limited but show promise. Omega-3 FAs appear to lack efficacy in ADHD,31-33 OCD,34 and borderline personality disorder.35
Related Resources
- National Center for Complementary and Alternative Medicine. Omega-3 fatty acids. http://nccam.nih.gov/health/omega3.
- National Institutes of Health. Office of Dietary Supplements. Working group report: Omega-3 fatty acids and cardiovascular disease. http://ods.od.nih.gov/Health_Information/omega_3_fatty_acids.aspx.
Disclosure
Dr. Morreale reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
1. Hibbeln JR. Fish consumption and major depression. Lancet. 1998;351(9110):1213.-
2. Tanskanen A, Hibbeln JR, Tuomilehto J, et al. Fish consumption and depressive symptoms in the general population in Finland. Psychiatr Serv. 2001;52(4):529-531.
3. Silvers KM, Scott KM. Fish consumption and self-reported physical and mental health status. Public Health Nutr. 2002;5(3):427-431.
4. Timonen M, Horrobin DF, Jokelaienen J, et al. Fish consumption and depression: the northern Finland 1966 birth cohort study. J Affect Disord. 2004;82(3):447-452.
5. Freeman MP, Rapaport MH. Omega-3 fatty acids and depression: from cellular mechanisms to clinical care. J Clin Psychiatry. 2011;72(2):258-259.
6. Bloch MH, Hannestad J. Omega-3 fatty acids for the treatment of depression: systematic review and meta-analysis [published online ahead of print September 20 2011]. Mol Psychiatry. doi: 10.1038/mp.2011.100.
7. Parker G, Gibson NA, Brotchie H, et al. Omega-3 fatty acids and mood disorders. Am J Psychiatry. 2006;163(6):969-978.
8. Martinez JM, Marangell LB. Omega-3 fatty acids: do ‘fish oils’ have a therapeutic role in psychiatry? Current Psychiatry. 2004;3(1):25-52.
9. Mozaffarian D, Micha R, Wallace S. Effects of coronary heart disease of increasing polyunsaturated fat in place of saturated fat: a systematic review and meta-analysis of randomized controlled trials. PLoS Med. 2010;7(3):e1000252.-
10. Kris-Etherton PM, Harris WS, Appel LJ. AHA Nutrition Committee. American Heart Association. Omega-3 fatty acids and cardiovascular disease: new recommendations from the American Heart Association. Arterioscler Thromb Vasc Biol. 2003;23(2):151-152.
11. Freeman MP, Fava M, Lake J, et al. Complementary and alternative medicine in major depressive disorder: the American Psychiatric Association Task Force report. J Clin Psychiatry. 2010;71(6):669-681.
12. Freeman MP, Hibbeln J, Wisner KL, et al. Omega-3 fatty acids: evidence basis for treatment and future research in psychiatry. J Clin Psychiatry. 2006;67(12):1954-1967.
13. Mischoulon D. The impact of omega-3 fatty acids on depressive disorders and suicidality: can we reconcile 2 studies with seemingly contradictory results? J Clin Psychiatry. 2011;72(12):1574-1576.
14. Appleton KM, Rogers PJ, Andrew RN. Updated systematic review and meta-analysis of the effects of n-3 long-chain polyunsaturated fatty acids on depressed mood. Am J Clin Nutr. 2010;91(31):757-770.
15. Martins JG. EPA but not DHA appears to be responsible for the efficacy of omega-3 long chain polyunsaturated fatty acid supplementation in depression: evidence from a meta-analysis of randomized controlled trials. J Am Coll Nutr. 2009;28(5):525-542.
16. Young G, Conquer J. Omega-3 fatty acids and neuropsychiatric disorders. Reprod Nutr Dev. 2005;45(1):1-28.
17. Sarris J, Mischoulon D, Schweitzer I. Omega-3 for bipolar disorder: meta-analyses of use in mania and bipolar depression. J Clin Psychiatry. 2012;73(1):81-86.
18. Sacks F. Ask the expert: omega-3 fatty acids. The Nutrition Source.http://www.hsph.harvard.edu/nutritionsource/questions/omega-3/index.html. Accessed July 23 2012.
19. Sublette ME, Ellis SP, Geant AL, et al. Meta-analysis of the effects of eicosapentaenoic acid (EPA) in clinical trials in depression. J Clin Psychiatry. 2011;72(12):1577-1584.
20. Gracious BL, Chirieac MC, Costescu S, et al. Randomized, placebo-controlled trial of flax oil in pediatric bipolar disorder. Bipolar Disord. 2010;12(2):142-154.
21. Freeman MP. Omega-3 fatty acids in major depressive disorder. J Clin Psychiatry. 2009;70(suppl 5):7-11.
22. Freeman MP, Hibbeln JR, Wisner KL, et al. Randomized dose-ranging pilot trial of omega-3 fatty acids for postpartum depression. Acta Psychiatr Scand. 2006;113(1):31-35.
23. Joy CB, Mumby-Croft R, Joy LA. Polyunsaturated fatty acid supplementation for schizophrenia. Cochrane Database Syst Rev. 2006;(3):CD001257.-
24. Fusar-Poli P, Berger G. Eicosapentaenoic acid interventions in schizophrenia: meta-analysis of randomized placebo-controlled studies. J Clin Psychopharmacol. 2012;32(2):179-185.
25. Amminger GP, Schäfer MR, Papageorgiou K, et al. Long-chain omega-3 fatty acids for indicated prevention of psychotic disorders: a randomized, placebo-controlled trial. Arch Gen Psychiatry. 2010;67(2):146-154.
26. Morris MC, Evans DA, Bienias JL, et al. Consumption of fish and n-3 fatty acids and risk of incident Alzheimer disease. Arch Neurol. 2003;60(7):940-946.
27. Engelhart MJ, Geerlings MI, Ruitenberg A, et al. Diet and risk of dementia: does fat matter? The Rotterdam Study. Neurology. 2002;59(12):1915-1921.
28. Terano T, Fujishiro S, Ban T, et al. Docosahexaenoic acid supplementation improves the moderately severe dementia from thrombotic cerebrovascular diseases. Lipids. 1999;34 suppl:S345-S346.
29. Otsuka M. Analysis of dietary factors in Alzheimer’s disease: clinical use of nutritional intervention for prevention and treatment of dementia [in Japanese]. Nihon Ronen Igakkai Zasshi. 2000;37(12):970-973.
30. Suzuki H, Morikawa Y, Takahashi H. Effect of DHA oil supplementation in intelligence and visual acuity in the elderly. World Rev Nutr Diet. 2001;88:68-71.
31. Joshi K, Lad S, Kale M, et al. Supplementation with flax oil and vitamin C improves the outcome of attention deficit hyperactivity disorder (ADHD). Prostaglandins Leukot Essent Fatty Acids. 2006;74(1):17-21.
32. Voigt RG, Llorente AM, Jensen CL, et al. A randomized, double-blind, placebo-controlled trial of docosahexaenoic acid supplementation in children with attention-deficit/hyperactivity disorder. J Pediatr. 2001;139(2):189-196.
33. Hirayama S, Hamazaki T, Terasawa K. Effect of docosahexaenoic acid-containing food administration on symptoms of attention-deficit/hyperactivity disorder - a placebo-controlled double-blind study. Eur J Clin Nutr. 2004;58(3):467-473.
34. Fux M, Benjamin J, Nemets B. A placebo-controlled cross-over trial of adjunctive EPA in OCD. J Psychiatr Res. 2004;38(3):323-325.
35. Zanarini MC, Frankenburg FR. Omega-3 Fatty acid treatment of women with borderline personality disorder: a double-blind placebo-controlled pilot study. Am J Psychiatry. 2003;160(1):167-169.
Intravenous iron in chemotherapy and cancer-related anemia
Recent guidance from the Centers for Medicare and Medicaid Services restricting erythropoiesis-stimulating agents (ESAs) in chemotherapy and cancer-related anemias has resulted in an increase in transfusions. Nine studies, without published contradictory evidence, have shown optimization of the response to ESAs by intravenous (IV) iron when the iron was added to the treatment of chemotherapy-induced anemia. The synergy observed, although greater in iron deficiency, was independent of pretreatment iron parameters. Three studies demonstrated decreased transfusions when IV iron is administered without ESAs. Discordant recommendations regarding IV iron currently exist among the American Society of Hematology/American Society of Clinical Oncology guidelines, the National Comprehensive Cancer Network, and the European Society of Medical Oncology. This discordance is at least partly the result of misconceptions about the clinical nature and incidence of adverse effects with IV iron. Other reasons for this discordance are presented in this review. Based on thousands of studied patients, we conclude that IV iron is safe and probably safer than most physicians realize. Education is needed relating to the interpretation of minor, subclinical infusion reactions that resolve without therapy. IV iron without ESAs may be an effective treatment for chemotherapy-induced anemia and warrants further study. We present evidence supporting the conclusion that baseline serum hepcidin levels may predict responses to IV iron, and we examine the published evidence supporting the conclusion that IV iron should be a standard addition to the management of chemotherapy and cancer-related anemia.
Click on the PDF icon at the top of this introduction to read the full article.
Recent guidance from the Centers for Medicare and Medicaid Services restricting erythropoiesis-stimulating agents (ESAs) in chemotherapy and cancer-related anemias has resulted in an increase in transfusions. Nine studies, without published contradictory evidence, have shown optimization of the response to ESAs by intravenous (IV) iron when the iron was added to the treatment of chemotherapy-induced anemia. The synergy observed, although greater in iron deficiency, was independent of pretreatment iron parameters. Three studies demonstrated decreased transfusions when IV iron is administered without ESAs. Discordant recommendations regarding IV iron currently exist among the American Society of Hematology/American Society of Clinical Oncology guidelines, the National Comprehensive Cancer Network, and the European Society of Medical Oncology. This discordance is at least partly the result of misconceptions about the clinical nature and incidence of adverse effects with IV iron. Other reasons for this discordance are presented in this review. Based on thousands of studied patients, we conclude that IV iron is safe and probably safer than most physicians realize. Education is needed relating to the interpretation of minor, subclinical infusion reactions that resolve without therapy. IV iron without ESAs may be an effective treatment for chemotherapy-induced anemia and warrants further study. We present evidence supporting the conclusion that baseline serum hepcidin levels may predict responses to IV iron, and we examine the published evidence supporting the conclusion that IV iron should be a standard addition to the management of chemotherapy and cancer-related anemia.
Click on the PDF icon at the top of this introduction to read the full article.
Recent guidance from the Centers for Medicare and Medicaid Services restricting erythropoiesis-stimulating agents (ESAs) in chemotherapy and cancer-related anemias has resulted in an increase in transfusions. Nine studies, without published contradictory evidence, have shown optimization of the response to ESAs by intravenous (IV) iron when the iron was added to the treatment of chemotherapy-induced anemia. The synergy observed, although greater in iron deficiency, was independent of pretreatment iron parameters. Three studies demonstrated decreased transfusions when IV iron is administered without ESAs. Discordant recommendations regarding IV iron currently exist among the American Society of Hematology/American Society of Clinical Oncology guidelines, the National Comprehensive Cancer Network, and the European Society of Medical Oncology. This discordance is at least partly the result of misconceptions about the clinical nature and incidence of adverse effects with IV iron. Other reasons for this discordance are presented in this review. Based on thousands of studied patients, we conclude that IV iron is safe and probably safer than most physicians realize. Education is needed relating to the interpretation of minor, subclinical infusion reactions that resolve without therapy. IV iron without ESAs may be an effective treatment for chemotherapy-induced anemia and warrants further study. We present evidence supporting the conclusion that baseline serum hepcidin levels may predict responses to IV iron, and we examine the published evidence supporting the conclusion that IV iron should be a standard addition to the management of chemotherapy and cancer-related anemia.
Click on the PDF icon at the top of this introduction to read the full article.
Autoinflammatory syndromes: Fever is not always a sign of infection
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
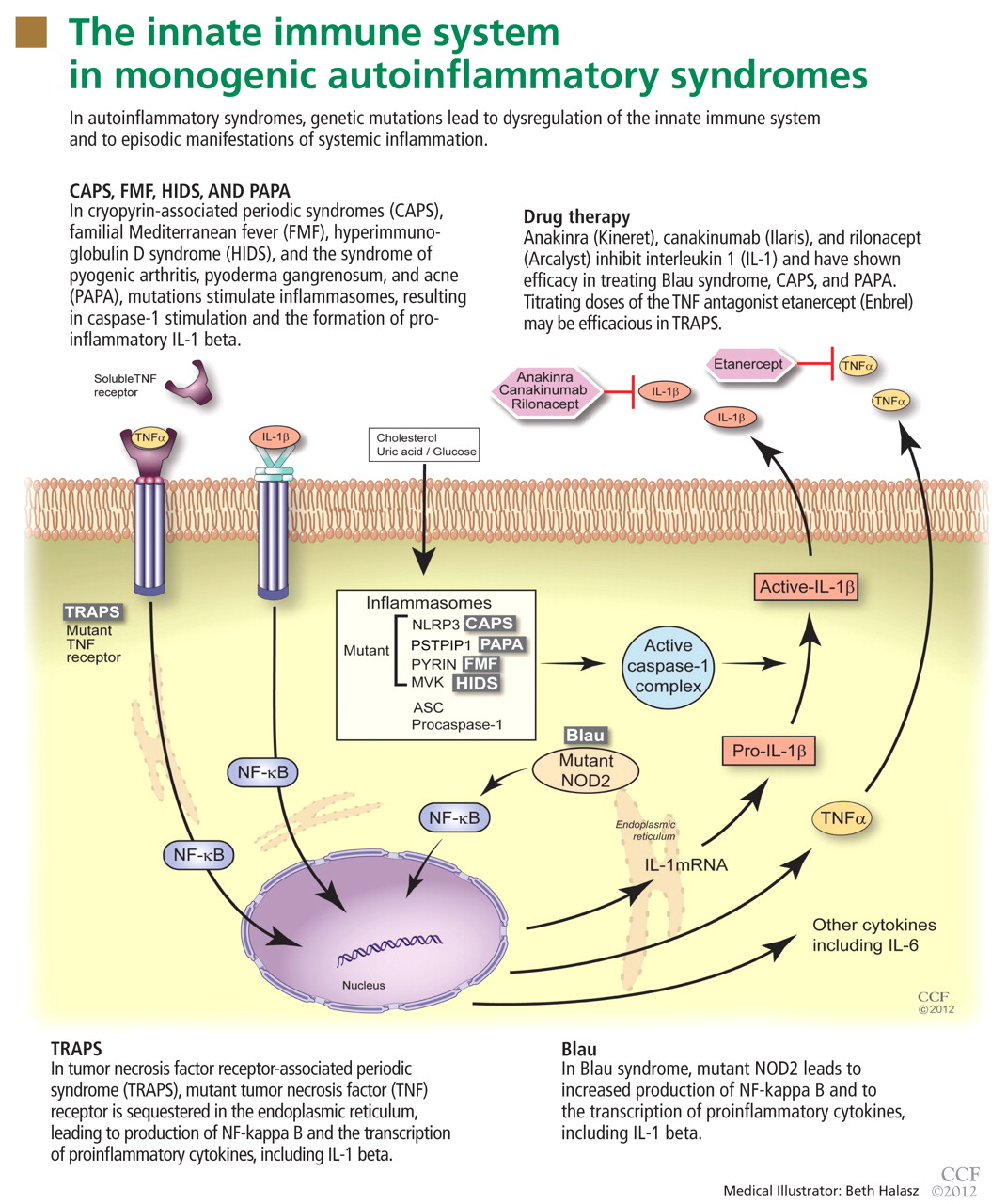
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
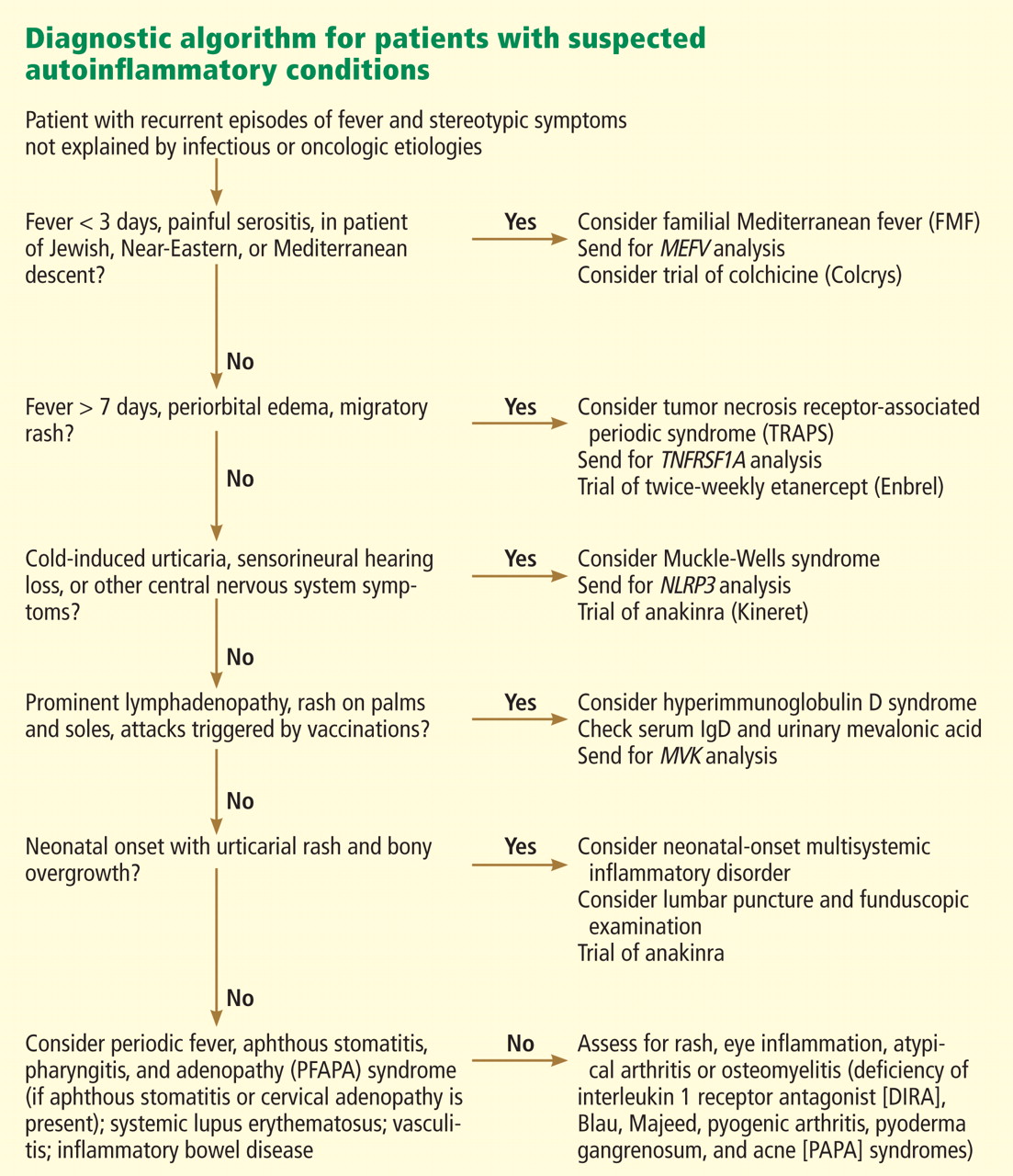
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
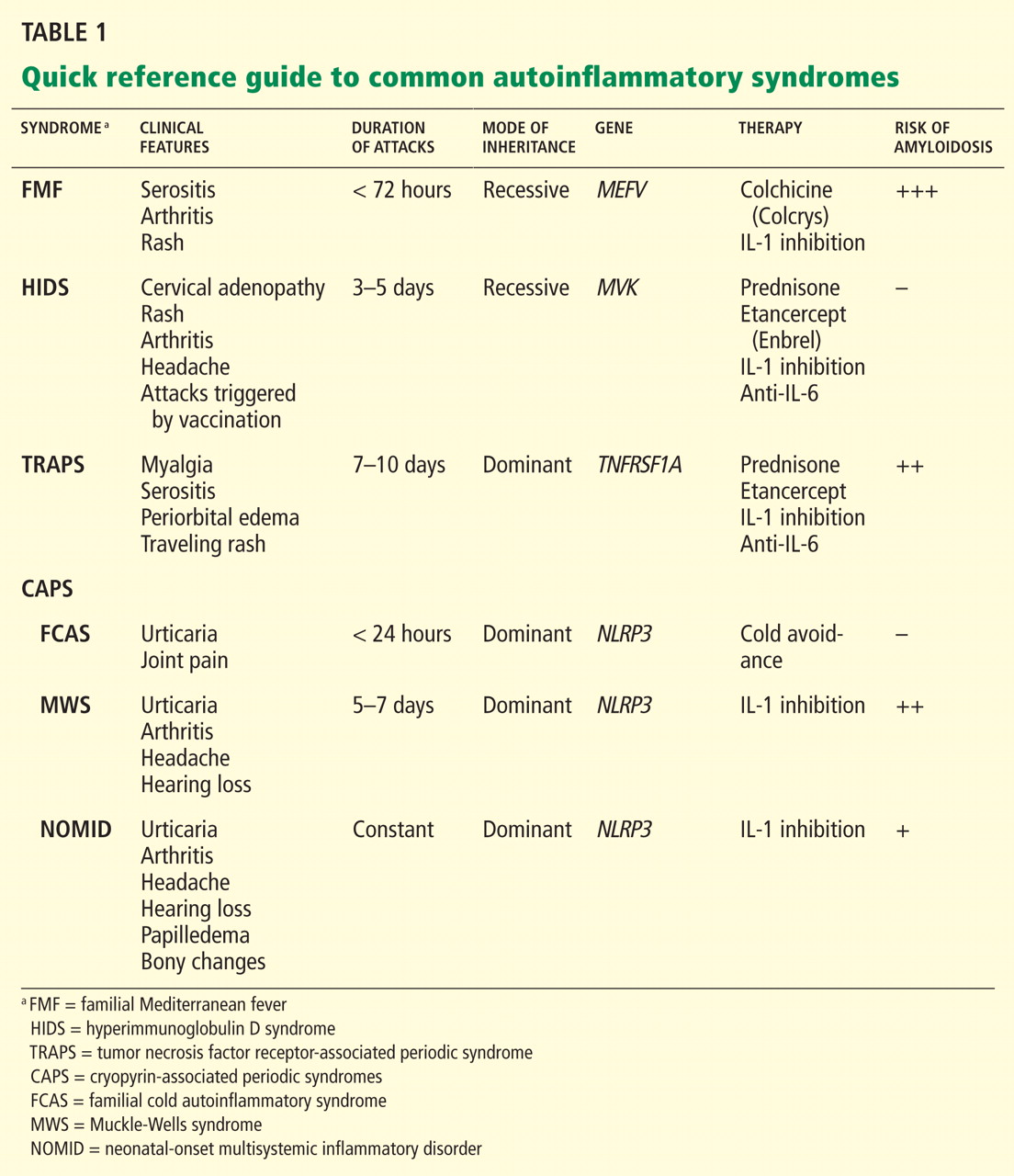
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74

Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74
Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
A 22-year-old man of Turkish ancestry presents to your office for an urgent visit. One day before the visit, he abruptly developed a fever with temperatures as high as 104°F (40°C), abdominal pain, joint pain, and a red rash on the lower right leg. He has no cough, nasal congestion, rhinorrhea, ear or eye pain, oral ulcers, vomiting, or diarrhea. After reviewing his chart, it becomes apparent that he has experienced similar intermittent, random, and self-limited episodes over the last 4 years.
On examination, he is febrile with diffuse abdominal tenderness and guarding. Bowel sounds are normal, and there is no rebound. The left knee is slightly swollen and limited in range of motion, and there is a large, non-palpable, blanching, erythematous lesion over the anterior lower leg.
While pondering diagnostic possibilities, you remember reading about autoinflammatory syndromes that result in recurrent episodes of fever and multisystemic inflammatory symptoms but cannot recall the evaluation and therapeutic options for these conditions.
These syndromes pose diagnostic challenges for physicians. Although these conditions are uncommon and may mimic malignancy or infection, they should be considered in patients who have recurrent febrile illness. While the autoinflammatory syndrome of familial Mediterranean fever (FMF), the diagnosis in the case above, is well known, our growing understanding of genetics and the immune system has unearthed a growing number of autoinflammatory syndromes.
A GENETICALLY DIVERSE BUT CLINICALLY SIMILAR GROUP OF CONDITIONS
The autoinflammatory syndromes are a group of genetically diverse but clinically similar conditions characterized by recurrent attacks of fever, rash, serositis, lymphadenopathy, and musculoskeletal involvement. This category of diseases is rapidly expanding and was built on the discovery of the genetics behind FMF, hyperimmunoglobulin D syndrome (HIDS), tumor necrosis factor receptor-associated periodic syndrome (TRAPS), and the cryopyrin-associated periodic syndromes (CAPS). More recent additions to the list include Blau syndrome and the syndrome of pyogenic arthritis, pyoderma gangrenosum, and acne (PAPA).
In autoinflammatory syndromes, genetic mutations lead to dysregulation of the innate immune system and to episodic manifestations of systemic inflammation. Many patients have first- or second-degree relatives with similar symptoms, reflecting the genetic abnormalities underlying this class of conditions. Unlike patients with other rheumatic diseases, patients with autoinflammatory diseases do not have autoreactive T lymphocytes, and they typically lack pathogenic autoantibodies.
The characterization of genetic autoinflammatory syndromes shows the importance of a well-regulated innate immune system and sheds light on the role of the innate immune system in common medical conditions such as gout and type 2 diabetes (see below).
THE INNATE IMMUNE SYSTEM : OUR FIRST LINE OF DEFENSE
The innate immune system is the first line of immune defense. It is evolutionarily conserved. Unlike the adaptive immune response, the innate immune response is not antigen-specific, and its activation does not produce a memory response. Generally speaking, it is composed of certain white blood cells (neutrophils, dendritic cells, macrophages, natural killer cells), proinflammatory signaling proteins (cytokines), and the complement system. Interleukin 1 (IL-1), interleukin 6 (IL-6), and tumor necrosis factor (TNF) alpha are the critical and most potent proinflammatory cytokines of the innate immune system.
To date, nearly all mutations that have been linked to the autoinflammatory syndromes disrupt regulation of inflammatory signaling within the innate immune system. This disruption generates a proinflammatory state, often leading to a final common pathway ending with activation of the inflammasome.
The inflammasome is a complex of distinct proteins which, when brought together, serve to convert inactive prointerleukin 1 beta to the active proinflammatory cytokine IL-1 beta.1 Formation of the inflammasome can be mediated by multiple different signals including microbial products, endogenously produced substances such as cholesterol and uric acid, or by proinflammatory cytokines and chemokines (Figure 1).
FAMILIAL MEDITERRANEAN FEVER
FMF is the most common and well characterized autoinflammatory syndrome. Described in 1949, its etiology was not understood until the genetic mutation that causes it was discovered in 1997.2–4
The Mediterranean fever gene MEFV encodes pyrin, a protein with an important role in controlling IL-1 production. Mutations in MEFV affect pyrin-mediated regulation, and IL-1 production increases.
Classically, FMF is described as autosomal recessive, although many patients have only one abnormal allele.5 Possibly, the abnormal allele confers an evolutionary advantage in resisting an endemic pathogen, an idea reflected in the carrier frequencies of different MEFV mutations in certain Mediterranean and Middle Eastern ethnic populations (Sephardic Jews, Turks, Arabs, Armenians).6,7 Also, carriage of mutations in MEFV in patients with Crohn disease has been associated with a higher risk of extraintestinal manifestations and colonic stricture,8 and their carriage in patients with multiple sclerosis has been associated with a rapid progression of that disease.9
Brief episodes of fever and serositis
Although FMF usually presents at ages 5 to 15, about 20% of patients with FMF suffer their first inflammatory attack after age 20 years.
Attacks are characterized by brief episodes of fever with temperatures higher than 102°F (38.9°C), lasting less than 72 hours, accompanied by intense serositis. Abdominal serositis may be severe enough to mimic appendicitis and lead to exploratory surgery.
About 70% of patients experience arthritis (predominantly in the legs), and 40% develop erysipeloid erythema, an intensely erythematous, warm, tender, and plaque-like lesion on the lower extremities. Biopsy of involved skin shows a diffuse, primarily neutrophilic, inflammatory cell infiltrate.
Laboratory examination reveals marked elevation of acute-phase reactants, which may normalize between episodes. The diagnosis can be made using a combination of clinical suspicion, sequencing of the MEFV gene, and a positive response to a trial of colchicine (Colcrys).
Without treatment, repetitive attacks of inflammation may result in amyloidosis of the kidneys or liver. The risk of amyloidosis is related to several discrete risk factors, such as country of residence, MEFV genotype, and serum amyloid A genotype.10–12 Patients should be monitored for physical manifestations of amyloidosis at least annually.
FMF patients have also been described who develop vasculitides such as Henoch-Schönlein purpura, polyarteritis nodosa, or Behçet disease.
Colchicine is the mainstay of FMF treatment
Colchicine has been the mainstay of therapy for patients with FMF for almost 40 years.13–15 Its benefits in FMF are clear: symptoms cease in nearly 70% of patients treated with colchicine, and an additional 25% have a reduction in the severity and frequency of attacks.
Only 5% to 10% of patients have no response to colchicine; this may be partially due to individual dose limitations imposed by common drug-associated gastrointestinal side effects.16–18 For these patients, newer biologic drugs that inhibit IL-1 activity, such as anakinra (Kineret) and rilonacept (Arcalyst), offer great promise.
Typically, FMF attacks become less frequent and less severe with age. However, the overall prognosis in FMF is related mainly to the individual’s genotype and the associated risk of amyloidosis.19
HYPERIMMUNOGLOBULIN D SYNDROME
HIDS is another autosomal recessive autoinflammatory syndrome.20
The genetic defect underlying HIDS lies within the mevalonate kinase gene MVK.21 Mevalonate kinase, an enzyme, plays an important role in the cholesterol biosynthesis pathway, following the initial step by 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. Mutations are primarily missense mutations in highly conserved areas of protein that result in decreased MVK activity (1% to 5% of normal).22,23 Decreased production of geranylgeranyl pyrophosphate resulting from disruption in the HMG-CoA reductase pathway subsequently leads to increased release of IL-1 beta from peripheral blood mononuclear cells and triggers inflammatory symptoms.24
Attacks of HIDS begin early in life
HIDS attacks begin early in life, with more than 70% of patients suffering their first attack before age 2, but adult-onset disease has been reported. Patients may report that routine childhood vaccinations triggered attacks, a historical finding unique to HIDS.
Attacks typically last 4 days; a longer duration can help the clinician differentiate HIDS from FMF.
More than 90% of patients have cervical lymphadenopathy, and 80% have an erythematous rash characteristically located on the palms and soles. About 70% of patients have headache, arthritis, and abdominal pain.
During attacks, laboratory examination reveals elevated acute inflammatory reactants. As the name implies, serum levels of immunoglobulin D (IgD) are elevated. However, this finding is not specific to HIDS and may also be found in patients with Still disease or FMF or in those who smoke cigarettes. Serum IgD levels fluctuate throughout life, and the sensitivity of commercially available IgD test kits is variable.
Assessment of mevalonic acid levels in the urine during febrile attacks offers a more sensitive, specific, and reliable diagnostic test for HIDS.25 While genetic sequencing is the gold standard of diagnostic testing, close to 30% of patients meeting clinical criteria for HIDS have no definable mutation.26
Treatment of HIDS can be challenging
Oral corticosteroids are effective in HIDS, but their long-term side effects are undesirable. Patients rarely respond to colchicine, differentiating them from FMF patients.
Etanercept (Enbrel), a fusion protein composed of the soluble TNF receptor and the Fc portion of the human IgG1 protein, has been efficacious in some patients.27,28 IL-1 inhibitors have also been used with increasing efficacy in the treatment of HIDS attacks.29,30
Although the frequency of attacks decreases with age, long-term follow-up of 28 Dutch HIDS patients found that their quality of life was still lower than that in country-matched controls.31
TUMOR NECROSIS FACTOR RECEPTOR-ASSOCIATED PERIODIC SYNDROME
In 1982, a large multiplex family from Scotland and Ireland was described who had a newly recognized syndrome termed familial Hibernian fever, characterized by recurrent fever, rash, and abdominal pain.32 In 1998, the genetics of this autosomal dominant condition were characterized,33–35 and it is now known by the acronym TRAPS.
TRAPS has a variable presentation owing to a variety of mutations in the gene encoding the cell surface receptor for TNF (TNFRSF1A). TNFRSF1A mutations affecting conserved cysteine residues important for protein folding correspond to severe disease phenotypes.
The R92Q mutation has an allele frequency of up to 4% of the population. It has no impact on the structure and function of the TNF receptor protein and is associated with a heterogeneous disease course. In contrast, the P46L mutation has an allele frequency of 1% of the population and typically is associated with a milder disease course characterized by older age of onset, shorter episodes, and a low frequency of amyloidosis.36–39
The R92Q and T61I mutations, which have low penetrance, have been increasingly reported in adult patients with the autoimmune diseases systemic lupus erythematosus, rheumatoid arthritis, and multiple sclerosis.40–42 Their influence is believed to contribute to proinflammatory responses but not to provide additional genetic susceptibility as provided by human leukocyte antigen (HLA) genotypes for susceptibility for these autoimmune diseases.
TRAPS attacks last longer than FMF and HIDS attacks
TRAPS attacks last 7 days or more, differentiating TRAPS from FMF and HIDS. Patients may present from infancy into adulthood, but more typically present in the toddler period.
Most patients experience intense myalgia as well as abdominal and pleuritic chest pain. A single-center series in 2002 described close to half of patients diagnosed with TRAPS as having had an intra-abdominal surgical procedure; in 10% necrotic bowel was identified, yet the biopsy typically revealed only a serosal mononuclear infiltrate.43
Like FMF and HIDS, TRAPS can cause an erythematous rash. The rash usually appears on an extremity, is centrifugal, and travels proximal-to-distal in concert with symptoms of myalgia. Deep tissue biopsy often demonstrates an intense, neutrophilic fasciitis sparing the underlying musculature. Painful conjunctivitis with periorbital edema also may occur.
Laboratory values reflecting widespread systemic inflammation and elevated acute-phase reactants are encountered during attacks and in some cases may persist between episodes.
Genetic testing can be used to confirm the diagnosis. The probability of finding a mutation in TNFRSF1A depends highly on whether the patient has affected relatives. In a series of 28 patients with recurrent inflammatory syndromes and TNFRSF1A mutations, 9 (32%) had a family history of recurrent inflammatory syndromes, while in 176 patients with sporadic, nonfamilial “TRAPS-like” symptoms, TNFRSF1A mutations were uncommon.37,38
Etanercept is effective for TRAPS
Systemic corticosteroids may be effective for treating TRAPS, but ever-increasing doses are often required.
Etanercept’s ability to bind both soluble and bound TNF explains its relative efficacy in treating TRAPS even though other TNF inhibitors have proven ineffective.44,45 With etanercept, the prognosis of TRAPS patients is typically good. Etanercept has even been effective in treating cases of renal amyloidosis from long-standing TRAPS, although it has not been shown to facilitate regression of renal amyloid mass.46,47 However, responses to treatment with etanercept may wane with time, and resistant cases have been reported.
IL-1 blockade with anakinra has been shown to be effective in the short term and long term in small case series, providing a reasonable alternative for patients who are difficult to manage.
CRYOPYRIN-ASSOCIATED PERIODIC SYNDROMES
- Perhaps the most clinically diverse hereditary autoinflammatory syndromes are the cryopyrin-associated periodic syndromes (CAPS). There are three overlapping phenotypes: Familial cold autoinflammatory syndrome (FCAS)
- Muckle-Wells syndrome (MWS)
- Neonatal-onset multisystemic inflammatory disorder (NOMID).
Mutations in NLRP3
CAPS symptoms stem from mutations within the NLRP3 gene (NOD-like receptor family, pyrin domain), which encodes the protein, cyropyrin.48NLRP3 mutations result in an abnormal cryopyrin structure, abnormal inflammasome activity, and increased IL-1 beta production.49,50
There is poor genotype-phenotype association in CAPS; the same NLRP3 point mutation can result in variable features, typically of either FCAS and MWS or MWS and NOMID overlapping phenotypes, supporting the hypothesis that modifier genes play a role in phenotypic expression.
Inheritance patterns in CAPS are autosomal dominant, but spontaneous mutations are also common. In fact, approximately two-thirds of patients with mutation-negative NOMID have somatic NLRP3 mutations, indicating that somatic NLRP3 mosaicism contributes to the clinical syndrome.51
Clinical features of the CAPS
The hallmarks of the CAPS include recurrent fevers, urticarial rash, and central nervous system inflammation. Characteristically, CAPS patients present in the neonatal period through early childhood, but adult-onset cases, which may have less typical features, have been reported.
Patients with FCAS develop brief episodes (< 24 hours) of fever, joint pain, and urticarial rash when exposed to sudden drops in ambient temperature.
Patients with MWS have more frequent, prolonged attacks, which may or may not be related to changes in ambient temperature. They also develop fever and urticarial rash and may develop arthritis and headaches from aseptic meningitis.
Patients with NOMID often present with fever and persistent urticarial rash shortly after birth and suffer from chronic aseptic meningitis, which can lead to papilledema and optic nerve atrophy. Frontal bossing of the skull and overgrowth of the epiphyseal regions of long bones with accompanying growth delay are also characteristic of NOMID.
IL-1 antagonists offer relief from CAPS
Many patients with FCAS do not require treatment and may move to a warmer climate to avoid rapid swings in ambient temperature. Otherwise, control of IL-1 beta activity is essential to the successful treatment of CAPS. Patients with MWS and NOMID require treatment with IL-1 antagonists, and the biologic drugs anakinra, rilonacept, and canakinumab (Ilaris) offer the possibility of symptomatic relief and long-term control of the disease.52–54
Prognosis depends on the phenotype
The overall prognosis for patients with CAPS largely depends on phenotype.
Patients with FCAS generally have progressive improvement in attack frequency and severity over time and are at minimal risk of amyloidosis.
Patients with MWS have a relatively good prognosis when treated with IL-1 antagonists, making them at low risk of amyloidosis and sensorineural hearing loss.
However, patients with NOMID are at high risk of sensorineural hearing loss, growth delay, and amyloidosis unless the condition is recognized and treated early in its course. Mortality rates historically are as high as 20% in untreated patients with NOMID.55
OTHER AUTOINFLAMMATORY SYNDROMES
More recently, other autoinflammatory syndromes of known genetic etiology have been described.
NLRP12-associated autoinflammatory disorders
A subset of patients with clinical manifestations attributable to CAPS but without mutations at the NLRP3 locus have mutations in another NLRP family member expressed in peripheral blood mononuclear cells on the NLRP12 gene. They are therefore labeled as having an NLRP12-associated autoinflammatory disorder.56,57
Deficiency of interleukin 1 receptor antagonist
IL-1 receptor antagonist is a naturally occurring antagonist of IL-1 alpha and IL-1 beta. In patients with deficiency of IL-1 receptor antagonist (DIRA), the action of these potent proinflammatory proteins is unopposed, leading to severe pustular rash and osteitis.58,59
Pyogenic arthritis, pyoderma gangrenosum, and acne syndrome
Patients with PAPA syndrome also have increased IL-1 production, in this case due to a mutation in the cytoplasmic adapter protein proline-serine-threonine phosphatase-interacting protein (PSTPIP1) gene, leading to the development of the symptoms included in the PAPA acronym.60
Majeed syndrome
Majeed syndrome is caused by a mutation in the LPIN2 gene, resulting in the early onset of chronic recurrent multifocal osteomyelitis, neutrophilic dermatosis, and dyserythropoietic anemia.61
Blau syndrome
Some patients with Blau syndrome (granulomatosis, arthritis, and uveitis) have NOD2/CARD15 gene mutations.62 Cases of DIRA, PAPA, and Blau syndrome have been reported that responded favorably to treatment with IL-1 antagonists.
Periodic fever, aphthous stomatitis, pharyngitis, and adenopathy syndrome
Although symptoms of the periodic fever, aphthous stomatitis, pharyngitis, and adenopathy (PFAPA) syndrome typically begin in childhood, adult-onset cases have been reported.63
Patients with PFAPA syndrome develop predictable, stereotypic febrile attacks that last on average 5 days and occur approximately every 4 weeks. Between attacks, patients are healthy; during attacks, they may experience oral ulceration (aphthous stomatitis), exudative or nonexudative pharyngitis, and enlarged and tender cervical lymph nodes. Up to 60% of PFAPA patients also experience abdominal pain.
No single genetic mutation has been identified, although it has been shown that 45% of PFAPA patients have a parent or sibling with recurrent fever and 12% have a parent or sibling with a PFAPA-like phenotype, suggesting that the disease has a genetic basis.64 Recent studies have demonstrated that T-cell–regulated complement activation and IL-1 production are altered in PFAPA patients, thus supporting the hypothesis that PFAPA is an autoinflammatory syndrome.65
Treatment. In view of the syndrome’s self-limited nature, treatment is reserved for patients with a severe presentation or for patients whose condition is especially burdensome.
The fever’s height may partially respond to nonsteroidal anti-inflammatory drugs, but these drugs have little effect on the duration or frequency of fever.
One or two doses of prednisone (1 mg/kg) within 6 hours of fever onset is effective in aborting the febrile episode in 90% of patients; however, up to 50% of patients may experience an increased frequency of attacks after treatment with systemic corticosteroids.66,67
Additional options include daily colchicine, which may lengthen the time between attacks, and cimetidine (Tagamet), which has been shown to prevent PFAPA attacks in approximately one-third of patients.67–69
The prognosis of PFAPA is quite favorable, and without intervention 40% of patients experience a significant reduction in the severity and frequency of fever attacks within 5 years of diagnosis. To date, there have been no reports of amyloidosis or hearing loss in PFAPA patients.
DIAGNOSTIC EVALUATION OF SUSPECTED AUTOINFLAMMATORY DISEASE
The autoinflammatory syndromes pose a true diagnostic challenge for physicians. Tremendous advances have been made in molecular and genetic testing. Nevertheless, the history and careful physical examination can lead the astute clinician to the proper diagnosis when evaluating a patient with a suspected autoinflammatory syndrome.
Critical elements in the history include age at the onset of attacks, duration of attacks, associated symptoms (serositis, adenopathy, myalgias, arthralgias, arthritis, ocular symptoms, central nervous system symptoms, rash), family members with similar symptoms, and ethnic background.
Internists should remember that autoinflammatory syndromes are part of the differential diagnosis in adult patients with a recurrent febrile illness. A vigorous search for malignancy and infection (especially tuberculosis) should be conducted in all patients. However, the repetitive, stereotypic nature of autoinflammatory syndromes differentiates them from typical confounders.
The utility of acute-phase reactants in the diagnostic evaluation is limited, as many conditions result in abnormal values. However, serial monitoring of inflammatory markers such as the erythrocyte sedimentation rate and C-reactive protein level in patients with a formally diagnosed autoinflammatory syndrome can be useful in tracking disease activity, identifying flares, and monitoring the efficacy of therapy.
In cases of suspected HIDS, assessment of IgD levels is not recommended, since IgD can be elevated in a number of autoinflammatory and rheumatologic conditions. Instead, preference should be given to testing mevalonic acid levels in the urine in patients with HIDS or suspected HIDS.
Patients with central nervous system symptoms should undergo a thorough examination, including a formal ophthalmologic evaluation, imaging, and possibly lumbar puncture to assess intracranial pressure and inflammatory changes in the cerebrospinal fluid.
Dermatologic manifestations should be examined firsthand and assessed as needed with magnetic resonance imaging to elucidate fascial inflammation or with full-thickness biopsy.
Gross bony abnormalities should be evaluated with plain radiography.
Audiologic testing may be indicated in the diagnostic evaluation of patients with recurrent fever.
Renal or hepatic biopsy may be indicated in the evaluation for amyloidosis; amyloid deposition has been reported in patients with TRAPS and clinical FMF not presenting with fever.70,71
Genetic testing is commercially available for patients with suspected hereditary autoinflammatory syndromes. However, clinicians should be cautioned that up to 30% of patients with phenotypic manifestations characteristic of a given autoinflammatory syndrome have normal results on genetic testing. In addition, the results of genetic testing may take several months to return and may cost patients and families up to several thousand dollars, as some insurers refuse to cover this procedure. Genetic testing may ultimately be indicated for proper counseling of reproductive risk.
Responses to short courses of medications such as colchicine, prednisone, and IL-1 receptor antagonists also represent diagnostic tools.
Figure 2 provides a proposed diagnostic algorithm for patients with suspected recurrent fever syndromes. Table 1 summarizes clinical and genetic features of the common autoinflammatory syndromes.
NEW INSIGHT INTO MORE COMMON CONDITIONS
Advances in the understanding of the autoinflammatory syndromes have provided new insight into the role of the innate immune system in other, more common conditions.72 Indeed, abnormal regulation of the innate inflammatory pathway has been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.73,74
Table 2 presents examples of the innate immune system’s involvement in the pathogenesis of several common chronic conditions.
Further study of autoinflammatory syndromes will add to our understanding of the innate immune system. These advances will lead to continued improvement in the care we give patients, both for the classic autoinflammatory syndromes and for other, more common, genetically complex conditions.
Our 22-year-old patient’s fever, abdominal pain (presumed peritonitis), erysipelas-like skin lesion, and arthritis are typical of FMF. Therefore, genetic testing was performed, which revealed a single MEFV gene mutation (M694V). Colchicine has been efficacious in preventing flares of his disease.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10:417–426.
- Siegal S. Benign paroxysmal peritonitis. Gastroenterology 1949; 12:234–247.
- International FMF Consortium. Ancient missense mutations in a new member of the RoRet gene family are likely to cause familial Mediterranean fever. Cell 1997; 90:797–807.
- French FMF Consortium. A candidate gene for familial Mediterranean fever. Nat Genet 1997; 17:25–31.
- Marek-Yagel D, Berkun Y, Padeh S, et al. Clinical disease among patients heterozygous for familial Mediterranean fever. Arthritis Rheum 2009; 60:1862–1866.
- Cattan D. Familial Mediterranean fever: is low mortality from tuberculosis a specific advantage for MEFV mutations carriers? Mortality from tuberculosis among Muslims, Jewish, French, Italian and Maltese patients in Tunis (Tunisia) in the first half of the 20th century. Clin Exp Rheumatol 2003; 21(suppl 30):S53–S54.
- Masters SL, Simon A, Aksentijevich I, Kastner DL. Horror autoinflammaticus: the molecular pathophysiology of autoinflammatory disease. Annu Rev Immunol 2009; 27:621–668.
- Fidder H, Chowers Y, Ackerman Z, et al. The familial Mediterranean fever (MEVF) gene as a modifier of Crohn’s disease. Am J Gastroenterol 2005; 100:338–343.
- Shinar Y, Livneh A, Villa Y, et al. Common mutations in the familial Mediterranean fever gene associate with rapid progression to disability in non-Ashkenazi Jewish multiple sclerosis patients. Genes Immun 2003; 4:197–203.
- Medlej-Hashim M, Delague V, Chouery E, et al. Amyloidosis in familial Mediterranean fever patients: correlation with MEFV genotype and SAA1 and MICA polymorphisms effects. BMC Med Genet 2004; 5:4.
- Mimouni A, Magal N, Stoffman N, et al. Familial Mediterranean fever: effects of genotype and ethnicity on inflammatory attacks and amyloidosis. Pediatrics 2000; 105:E70.
- Touitou I, Sarkisian T, Medlej-Hashim M, et al; International Study Group for Phenotype-Genotype Correlation in Familial Mediterranean Fever. Country as the primary risk factor for renal amyloidosis in familial Mediterranean fever. Arthritis Rheum 2007; 56:1706–1712.
- Goldfinger SE. Colchicine for familial Mediterranean fever. N Engl J Med 1972; 287:1302.
- Wolff SM, Dinarello CA, Dale DC, Goldfinger SE, Alling DW. Colchicine therapy of familial Mediterranean fever. Trans Assoc Am Physicians 1974; 87:186–194.
- Dinarello CA, Wolff SM, Goldfinger SE, Dale DC, Alling DW. Colchicine therapy for familial mediterranean fever. A double-blind trial. N Engl J Med 1974; 291:934–937.
- Putterman C, Ben-Chetrit E, Caraco Y, Levy M. Colchicine intoxication: clinical pharmacology, risk factors, features, and management. Semin Arthritis Rheum 1991; 21:143–155.
- Lidar M, Scherrmann JM, Shinar Y, et al. Colchicine nonresponsiveness in familial Mediterranean fever: clinical, genetic, pharmacokinetic, and socioeconomic characterization. Semin Arthritis Rheum 2004; 33:273–282.
- Ben-Chetrit E, Ozdogan H. Non-response to colchicine in FMF—definition, causes and suggested solutions. Clin Exp Rheumatol 2008; 26(suppl 50):S49–S51.
- Ben-Chetrit E, Touitou I. Familial Mediterranean fever in the world. Arthritis Rheum 2009; 61:1447–1453.
- van der Meer JW, Vossen JM, Radl J, et al. Hyperimmunoglobulinaemia D and periodic fever: a new syndrome. Lancet 1984; 1:1087–1090.
- Drenth JP, Cuisset L, Grateau G, et al. Mutations in the gene encoding mevalonate kinase cause hyper-IgD and periodic fever syndrome. International Hyper-IgD Study Group. Nat Genet 1999; 22:178–181.
- Houten SM, Frenkel J, Kuis W, Wanders RJ, Poll-The BT, Waterham HR. Molecular basis of classical mevalonic aciduria and the hyperimmunoglobulinaemia D and periodic fever syndrome: high frequency of 3 mutations in the mevalonate kinase gene. J Inherit Metab Dis 2000; 23:367–370.
- Poll-The BT, Frenkel J, Houten SM, et al. Mevalonic aciduria in 12 unrelated patients with hyperimmunoglobulinaemia D and periodic fever syndrome. J Inherit Metab Dis 2000; 23:363–366.
- Mandey SH, Kuijk LM, Frenkel J, Waterham HR. A role for geranylgeranylation in interleukin-1beta secretion. Arthritis Rheum 2006; 54:3690–3695.
- van der Hilst JC, Frenkel J. Hyperimmunoglobulin D syndrome in childhood. Curr Rheumatol Rep 2010; 12:101–107.
- Simon A, Cuisset L, Vincent MF, et al. Molecular analysis of the mevalonate kinase gene in a cohort of patients with the hyper-igd and periodic fever syndrome: its application as a diagnostic tool. Ann Intern Med 2001; 135:338–343.
- Takada K, Aksentijevich I, Mahadevan V, Dean JA, Kelley RI, Kastner DL. Favorable preliminary experience with etanercept in two patients with the hyperimmunoglobulinemia D and periodic fever syndrome. Arthritis Rheum 2003; 48:2645–2651.
- Korppi M, Van Gijn ME, Antila K. Hyperimmunoglobulinemia D and periodic fever syndrome in children. Review on therapy with biological drugs and case report. Acta Paediatr 2011; 100:21–25.
- Rigante D, Ansuini V, Bertoni B, et al. Treatment with anakinra in the hyperimmunoglobulinemia D/periodic fever syndrome. Rheumatol Int 2006; 27:97–100.
- Bodar EJ, Kuijk LM, Drenth JP, van der Meer JW, Simon A, Frenkel J. On-demand anakinra treatment is effective in mevalonate kinase deficiency. Ann Rheum Dis 2011; 70:2155–2158.
- van der Hilst JC, Bodar EJ, Barron KS, et al; International HIDS Study Group. Long-term follow-up, clinical features, and quality of life in a series of 103 patients with hyperimmunoglobulinemia D syndrome. Medicine (Baltimore) 2008; 87:301–310.
- Williamson LM, Hull D, Mehta R, Reeves WG, Robinson BH, Toghill PJ. Familial Hibernian fever. Q J Med 1982; 51:469–480.
- Mulley J, Saar K, Hewitt G, et al. Gene localization for an autosomal dominant familial periodic fever to 12p13. Am J Hum Genet 1998; 62:884–889.
- McDermott MF, Ogunkolade BW, McDermott EM, et al. Linkage of familial Hibernian fever to chromosome 12p13. Am J Hum Genet 1998; 62:1446–1451.
- McDermott MF, Aksentijevich I, Galon J, et al. Germline mutations in the extracellular domains of the 55 kDa TNF receptor, TNFR1, define a family of dominantly inherited autoinflammatory syndromes. Cell 1999; 97:133–144.
- Aksentijevich I, Galon J, Soares M, et al. The tumor-necrosis-factor receptor-associated periodic syndrome: new mutations in TNFRSF1A, ancestral origins, genotype-phenotype studies, and evidence for further genetic heterogeneity of periodic fevers. Am J Hum Genet 2001; 69:301–314.
- Dodé C, André M, Bienvenu T, et al; French Heraditary Recurrent Inflammatory Disorder Study Group. The enlarging clinical, genetic, and population spectrum of tumor necrosis factor receptor-associated periodic syndrome. Arthritis Rheum 2002; 46:2181–2188.
- Aganna E, Hammond L, Hawkins PN, et al. Heterogeneity among patients with tumor necrosis factor receptor-associated periodic syndrome phenotypes. Arthritis Rheum 2003; 48:2632–2644.
- Ravet N, Rouaghe S, Dodé C, et al. Clinical significance of P46L and R92Q substitutions in the tumour necrosis factor superfamily 1A gene. Ann Rheum Dis 2006; 65:1158–1162.
- Dieudé P, Goossens M, Cornélis F, Michou L, Bardin T, Tchernitchko DO; European Consortium on Rheumatoid Arthritis Families. The TNFRSF1A R92Q mutation is frequent in rheumatoid arthritis but shows no evidence for association or linkage with the disease. Ann Rheum Dis 2007; 66:1113–1115.
- Ida H, Kawasaki E, Miyashita T, et al. A novel mutation (T61I) in the gene encoding tumour necrosis factor receptor superfamily 1A (TNFRSF1A) in a Japanese patient with tumour necrosis factor receptor-associated periodic syndrome (TRAPS) associated with systemic lupus erythematosus. Rheumatology (Oxford) 2004; 43:1292–1299.
- Kümpfel T, Hoffmann LA, Pellkofer H, et al. Multiple sclerosis and the TNFRSF1A R92Q mutation: clinical characteristics of 21 cases. Neurology 2008; 71:1812–1820.
- Hull KM, Drewe E, Aksentijevich I, et al. The TNF receptor-associated periodic syndrome (TRAPS): emerging concepts of an autoinflammatory disorder. Medicine (Baltimore) 2002; 81:349–368.
- Drewe E, McDermott EM, Powell PT, Isaacs JD, Powell RJ. Prospective study of anti-tumour necrosis factor receptor superfamily 1B fusion protein, and case study of anti-tumour necrosis factor receptor superfamily 1A fusion protein, in tumour necrosis factor receptor associated periodic syndrome (TRAPS): clinical and laboratory findings in a series of seven patients. Rheumatology (Oxford) 2003; 42:235–239.
- Bulua AC, Mogul DB, Aksentijevich I, et al. Efficacy of etanercept in the tumor necrosis factor receptor–associated periodic syndrome: a prospective, open-label, dose-escalation study. Arthritis Rheum 2012; 64:908–913.
- Drewe E, McDermott EM, Powell RJ. Treatment of the nephrotic syndrome with etanercept in patients with the tumor necrosis factor receptor-associated periodic syndrome. N Engl J Med 2000; 343:1044–1045.
- Simsek I, Kaya A, Erdem H, Pay S, Yenicesu M, Dinc A. No regression of renal amyloid mass despite remission of nephrotic syndrome in a patient with TRAPS following etanercept therapy. J Nephrol 2010; 23:119–123.
- Hoffman HM, Mueller JL, Broide DH, Wanderer AA, Kolodner RD. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat Genet 2001; 29:301–315.
- Aganna E, Martinon F, Hawkins PN, et al. Association of mutations in the NALP3/CIAS1/PYPAF1 gene with a broad phenotype including recurrent fever, cold sensitivity, sensorineural deafness, and AA amyloidosis. Arthritis Rheum 2002; 46:2445–2452.
- Aksentijevich I, Nowak M, Mallah M, et al. De novo CIAS1 mutations, cytokine activation, and evidence for genetic heterogeneity in patients with neonatal-onset multisystem inflammatory disease (NOMID): a new member of the expanding family of pyrin-associated autoinflammatory diseases. Arthritis Rheum 2002; 46:3340–3348.
- Tanaka N, Izawa K, Saito MK, et al. High incidence of NLRP3 somatic mosaicism in patients with chronic infantile neurologic, cutaneous, articular syndrome: results of an International Multicenter Collaborative Study. Arthritis Rheum 2011; 63:3625–3632.
- Hoffman HM, Throne ML, Amar NJ, et al. Efficacy and safety of rilonacept (interleukin-1 Trap) in patients with cryopyrin-associated periodic syndromes: results from two sequential placebo-controlled studies. Arthritis Rheum 2008; 58:2443–2652.
- Lachmann HJ, Kone-Paut I, Kuemmerle-Deschner JB, et al. Use of canakinumab in the cryopyrin-associated periodic syndrome. N Engl J Med 2009; 360:2416–2425.
- Kuemmerle-Deschner JB, Tyrrell PN, Koetter I, et al. Efficacy and safety of anakinra therapy in pediatric and adult patients with the autoinflammatory Muckle-Wells syndrome. Arthritis Rheum 2011; 63:840–849.
- Prieur AM, Griscelli C, Lampert F, et al. A chronic, infantile, neurological, cutaneous and articular (CINCA) syndrome. A specific entity analysed in 30 patients. Scand J Rheumatol Suppl 1987; 66:57–68.
- Jéru I, Duquesnoy P, Fernandes-Alnemri T, et al. Mutations in NALP12 cause hereditary periodic fever syndromes. Proc Natl Acad Sci U S A 2008; 105:1614–1619.
- Borghini S, Tassi S, Chiesa S, et al. Clinical presentation and pathogenesis of cold-induced autoinflammatory disease in a family with recurrence of an NLRP12 mutation. Arthritis Rheum 2011; 63:830–839.
- Aksentijevich I, Masters SL, Ferguson PJ, et al. An autoinflammatory disease with deficiency of the interleukin-1-receptor antagonist. N Engl J Med 2009; 360:2426–2437.
- Reddy S, Jia S, Geoffrey R, et al. An autoinflammatory disease due to homozygous deletion of the IL1RN locus. N Engl J Med 2009; 360:2438–2444.
- Wise CA, Gillum JD, Seidman CE, et al. Mutations in CD2BP1 disrupt binding to PTP PEST and are responsible for PAPA syndrome, an autoinflammatory disorder. Hum Mol Genet 2002; 11:961–969.
- Ferguson PJ, Chen S, Tayeh MK, et al. Homozygous mutations in LPIN2 are responsible for the syndrome of chronic recurrent multifocal osteomyelitis and congenital dyserythropoietic anaemia (Majeed syndrome). J Med Genet 2005; 42:551–557.
- Miceli-Richard C, Lesage S, Rybojad M, et al. CARD15 mutations in Blau syndrome. Nat Genet 2001; 29:19–20.
- Padeh S, Stoffman N, Berkun Y. Periodic fever accompanied by aphthous stomatitis, pharyngitis and cervical adenitis syndrome (PFAPA syndrome) in adults. Isr Med Assoc J 2008; 10:358–360.
- Cochard M, Clet J, Le L, et al. PFAPA syndrome is not a sporadic disease. Rheumatology (Oxford) 2010; 49:1984–1987.
- Stojanov S, Lapidus S, Chitkara P, et al. Periodic fever, aphthous stomatitis, pharyngitis, and adenitis (PFAPA) is a disorder of innate immunity and Th1 activation responsive to IL-1 blockade. Proc Natl Acad Sci U S A 2011; 108:7148–7153.
- Thomas KT, Feder HM, Lawton AR, Edwards KM. Periodic fever syndrome in children. J Pediatr 1999; 135:15–21.
- Feder HM. Cimetidine treatment for periodic fever associated with aphthous stomatitis, pharyngitis and cervical adenitis. Pediatr Infect Dis J 1992; 11:318–321.
- Tasher D, Stein M, Dalal I, Somekh E. Colchicine prophylaxis for frequent periodic fever, aphthous stomatitis, pharyngitis and adenitis episodes. Acta Paediatr 2008; 97:1090–1092.
- Pillet P, Ansoborlo S, Carrère A, Perel Y, Guillard JM. [(P)FAPA syndrome: value of cimetidine]. In French. Arch Pediatr 2000; 7:54–57.
- Kallinich T, Haffner D, Rudolph B, et al. ”Periodic fever” without fever: two cases of non-febrile TRAPS with mutations in the TNFRSF1A gene presenting with episodes of inflammation or monosymptomatic amyloidosis. Ann Rheum Dis 2006; 65:958–960.
- Sohar E, Gafni J, Pras M, Heller H. Familial Mediterranean fever. A survey of 470 cases and review of the literature. Am J Med 1967; 43:227–253.
- Goldbach-Mansky R, Kastner DL. Autoinflammation: the prominent role of IL-1 in monogenic autoinflammatory diseases and implications for common illnesses. J Allergy Clin Immunol 2009; 124:1141–1149.
- Martinon F, Pétrilli V, Mayor A, Tardivel A, Tschopp J. Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 2006; 440:237–241.
- Masters SL, Dunne A, Subramanian SL, et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1ß in type 2 diabetes. Nat Immunol 2010; 11:897–904.
KEY POINTS
- In many of the autoinflammatory syndromes, genetic abnormalities and consequent disordered regulation of the innate immune system lead to overactivity of proinflammatory cytokines and subsequent inflammatory symptoms.
- Early recognition and treatment with immunoregulatory agents may improve quality of life and reduce the risk of disease sequelae.
- Abnormal regulation of the innate inflammatory pathway has also been implicated in the pathogenesis of conditions as phenotypically diverse as gout, type 2 diabetes, atherosclerosis, and epilepsy.
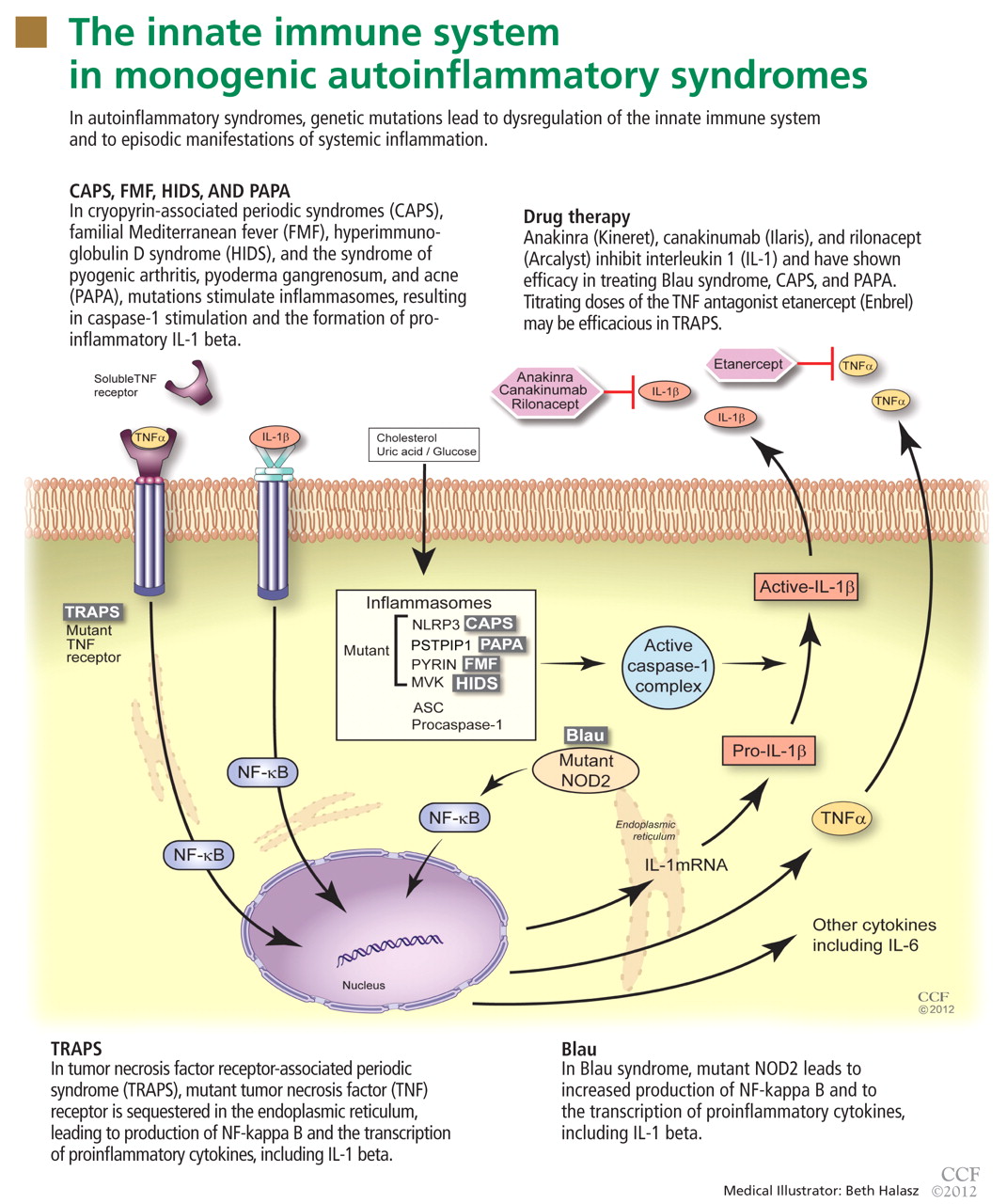
Distinguishing cellulitis from its mimics
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
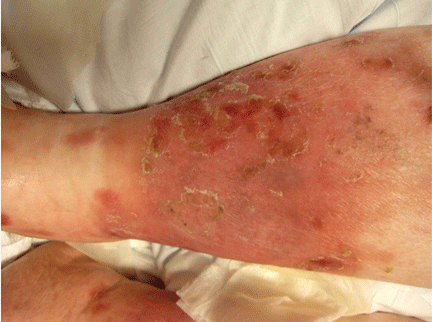
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
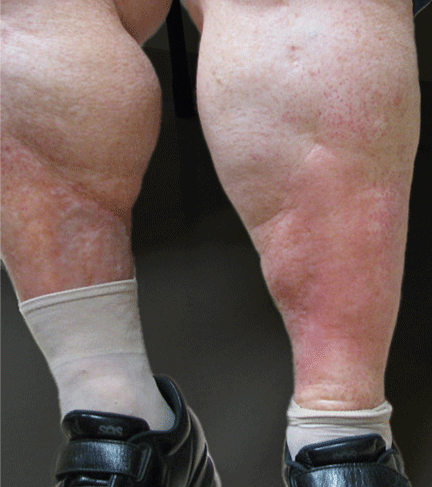
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
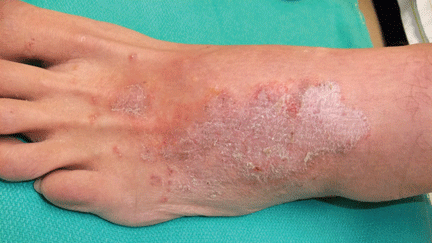
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
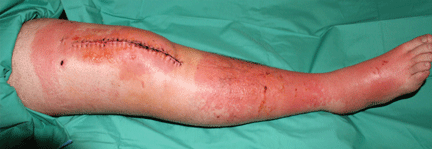
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
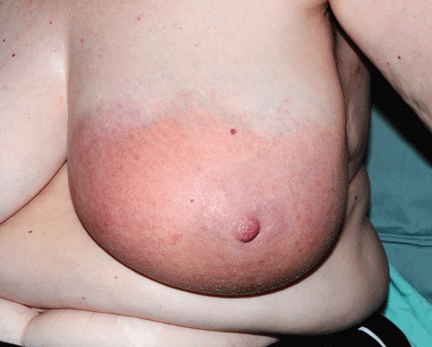
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
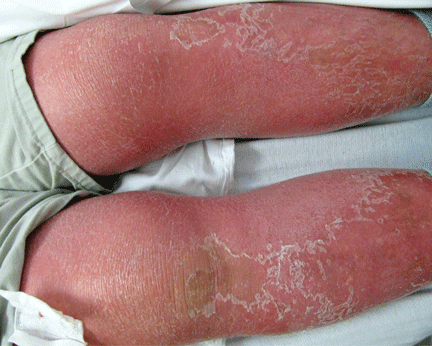
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
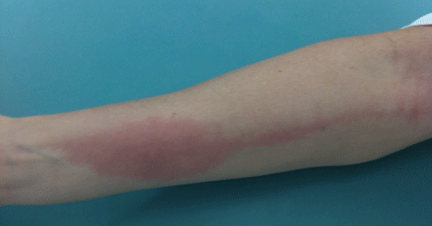
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
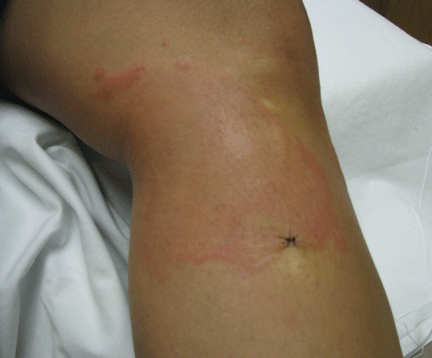
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
More than 10% of patients labeled as having cellulitis do not have cellulitis.1 This is unfortunate, as it leads to excessive and incorrect use of antibiotics and to delays in appropriate therapy.2 However, it is not surprising, given the number of conditions that bear a striking similarity to cellulitis. A familiarity with the features of true cellulitis and with the handful of conditions that can bear a striking similarity to it is the way out of this potential diagnostic quagmire.
WHAT CELLULITIS IS—AND IS NOT
The key characteristics of cellulitis are redness, warmth, tenderness, and swelling of the skin. A history of trauma and pain in the affected area and evidence of leukocytosis3 suggest cellulitis. A symmetric or diffusely scattered pattern indicates a condition other than cellulitis, which is overwhelmingly unilateral, with smooth, indistinct borders4,5 Other factors pointing to cellulitis are underlying immunosuppression, a more rapid progression, previous episodes, systemic symptoms (eg, fever, leukocytosis), new medications, new travel or outdoor exposure, and comorbidities such as diabetes and peripheral vascular disease. A long-standing, slowly progressive course and a history of unsuccessful treatment with antibiotics are strong indicators of a condition other than cellulitis.
Consultation with a dermatologist is recommended to narrow the differential diagnosis. The dermatologist can determine if biopsy is necessary, as many dermatoses that mimic cellulitis can be diagnosed by visual recognition alone.
STASIS DERMATITIS
The most common mimic of cellulitis is stasis dermatitis (Figure 1).2 Patients can present with ill-defined, bilateral, pitting edema of the lower extremities, typically with erythema, hyperpigmentation, serous drainage, and superficial desquamation.3,6,7
The inciting factor is chronic venous insufficiency, leading to interstitial edema, extravasation of red blood cells, and decreased tissue oxygenation. This process causes micro-vascular changes and microthrombi that up-regulate transforming growth factor beta and fibroblastic growth factor.7 If the process is allowed to continue, stasis dermatitis may progress to lipodermatosclerosis.
Tip: Stasis dermatitis is generally bilateral, the process will have been ongoing for years, there is often pitting edema, and the legs should be nontender.
LIPODERMATOSCLEROSIS
Lipodermatosclerosis is a sclerosing panniculitis classically described as an “inverted champagne bottle” or “inverted bowling pin” appearance of the leg, ie, the diameter of the leg is sharply narrowed directly below the calf (Figure 2).
There is an acute and a chronic phase. The acute phase is characterized by inflammation and erythema, and the chronic phase is characterized by fibrosis.8 The acute phase presents with severe lower-extremity pain above the medial malleolus, erythema, edema, and warmth; there is no sharp demarcation between affected and unaffected skin.9,10 This phase can be difficult to distinguish from cellulitis, so the history plays a key role. Known venous insufficiency, cutaneous changes of stasis dermatitis, and the absence of systemic symptoms all point to lipodermatosclerosis.
The chronic phase is characterized by unilateral or bilateral, indurated, sclerotic plaques with a “bound-down” appearance (ie, they appear as if tethered—or bound—to the subcutaneous tissue) affecting the skin from below the knee to the ankle; there is a sharp demarcation between affected and unaffected skin.9–11 The skin is often bronze or brown secondary to hemosiderin deposits. There can be prominent varicosities and scattered ulcerations depending on the course of the disease.
This condition is thought to be the result of long-standing chronic venous insufficiency.7,8,9,11 It is proposed that venous incompetence leads to extravasation of interstitial fluid and red blood cells, decreased diffusion of oxygen to the tissues, and eventual tissue and endothelial damage. As the endothelium is damaged, microthrombi formation and infarction ensue, stimulating fibroblasts to form granulation tissue.
Tip: The history helps to distinguish acute lipodermatosclerosis from cellulitis. Chroniclipodermatoslcerosis will have been ongoing for years, the legs should be nontender, the skin will be bound-down, and the diameter of the leg will sharply decrease from knee to ankle.
CONTACT DERMATITIS
Allergic and irritant forms of contact dermatitis are often mistaken for cellulitis. Irritant contact dermatitis (Figure 3) presents with erythematous patches and plaques with well-defined borders, often in a geometric distribution where the skin was exposed to an irritant.12 Allergic contact dermatitis is a delayed hypersensitivity dermatitis that can be secondary to something ingested, applied to the skin, or airborne (Figure 4). It presents as erythematous macules, papules, and plaques that may have serous drainage or vesiculation. Lesions of allergic contact dermatitis are usually confined to the site of contact with the allergen, but they can infrequently be found at distant sites, in which case it is considered systemic contact dermatitis.3,5 Depending on the severity of the allergy, patients may complain of intense pain and pruritus.3
Additionally, chronic, nonhealing leg ulcers may have a confounding allergic contact dermatitis.7 Although patients may believe they are helping the ulcer heal by applying topical antibiotics or other lubricants, they may in fact be impeding the healing process. Always inquire as to what the patient is applying if he or she has leg ulceration with surrounding edema and erythema that has not resolved with conventional treatments.13,14
Tip: The key to distinguishing contact dermatitis from cellulitis is the history. For example, ask about recent changes in medications, soaps, and laundry detergents, new hobbies, or recent surgeries. The involved site is often confined to the area where the allergen contacted the skin, except in cases of exposure to an airborne allergen.
LYMPHEDEMA
Lymphedema is characterized by localized edema of an affected extremity, with induration, erythema, and secondary cutaneous changes such as hyperkeratosis, dyspigmentation, and wart-like architecture (Figure 5).
Primary lymphedema appears in the setting of congenital abnormalities, whereas secondary lymphedema results from an interruption of a previously functioning lymphatic system (eg, after radical mastectomy).
Patients often present with unilateral nonpitting edema and erythema in the absence of systemic symptoms.12 Many patients presenting with lower-extremity lymphedema are overweight or obese, as the weight they carry causes obstruction of the inguinal lymphatics.6
The pathophysiology is not clearly delineated but is thought to be a consequence of decreased oxygenation of tissue secondary to extravasated lymph. As the oxygen is compromised, macrophages and fibroblasts are recruited, resulting in fibrosis.6
Patients with lymphedema are more susceptible to superficial and deep skin infections, as the natural defense system in the epidermis and papillary dermis is compromised by impaired lymphatic drainage.15
To differentiate uncomplicated lymphedema from a secondary cutaneous infection, the clinician should take into account the presence or absence of warmth, pain, increased erythema, and systemic symptoms (Figure 6).
Tip: Primary lymphedema will most likely present in childhood with no inciting factors and will require a full workup. Obtaining a history should make secondary lymphedema a relatively straightforward diagnosis: Has the patient undergone lymph node dissection? Has the patient had an injury in the affected leg? Lymphedema is overwhelmingly unilateral and nonpitting, and is often seen in overweight people (if no precipitating factor is present).
EOSINOPHILIC CELLULITIS
Eosinophilic cellulitis, or Wells syndrome, was first described in 1971 as a granulomatous dermatitis.16 It is a recurrent hypersensitivity reaction to a drug, to a vaccine, or to an insect bite, or to a viral or fungal infection that presents on the extremities as localized erythema, edema, and induration with sharp borders and a green or gray hue (Figure 7).17–19 The lesions commonly progress to firm, indurated plaques that resemble morphea. The plaques may take weeks or years to resolve, but they do so without scarring.12,17,20,21
As patients tend to have recurrent bouts of eosinophilic cellulitis, they may have lesions in different stages of healing. Patients tend to report itching and burning that precedes the onset of plaques.22 The complete blood count typically shows a transient hypereosinophilia.12,16,17,23–25
Tip: This diagnosis often requires biopsy for confirmation, but helpful clues are a history of recurrent episodes, the color of the lesions, and peripheral eosinophilia.
PAPULAR URTICARIA
Papular urticaria is a dermal hypersensitivity reaction to an insect bite, most commonly from a flea or mosquito.26 Patients are often children, as their immune system may be hypersensitive. But children often develop tolerance before puberty.27
The presentation may vary, from numerous urticarial papules near the site of a bite, to generalized, large, indurated, erythematous plaques reminiscent of cellulitis (Figure 8).5,26 The lesions usually develop within hours of a bite and persist for an average of 1 to 2 weeks.28 The areas typically affected are the head and neck or the upper or lower extremities; the palms, soles, and trunk are usually spared.27
Patients most often complain of intense itching.12 The pathogenesis is proposed to be mediated by the immune complex, and tissue biopsy study shows increased eosinophils. The eosinophils stimulate mast cells, causing release of histamine, leading to increased vascular permeability, edema, and erythema.28,29
Tip: Biopsy may be necessary to confirm the diagnosis, though often the history may be sufficient. The patient may or may not recall a bite, so probe into recent activities such as outdoor sports or contact with a new pet. The papules and plaques are generally very pruritic but not painful.
DERMATOLOGY CONSULT
If the clinical presentation and history do not correlate, or if the skin condition has been treated with antibiotics yet has failed to respond, the possibility of other cutaneous dermatoses should be entertained. A dermatology consult can help determine the diagnosis, the need for further evaluation, and the best treatment course.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
- Hepburn MJ, Dooley DP, Ellis MW. Alternative diagnoses that often mimic cellulitis. Am Fam Physician 2003; 67:2471.
- David CV, Chira S, Eells SJ, et al. Diagnostic accuracy in patients admitted to hospitals with cellulitis. Dermatol Online J 2011; 17:1.
- Bailey E, Kroshinsky D. Cellulitis: diagnosis and management. Dermatol Ther 2011; 24:229–239.
- Stevens DL, Bisno AL, Chambers HF, et al; Infectious Diseases Society of America. Practice guidelines for the diagnosis and management of skin and soft-tissue infections. Clin Infect Dis 2005; 41:1373–1406.
- Lio PA. The many faces of cellulitis. Arch Dis Child Educ Pract Ed 2009; 94:50–54.
- Yosipovitch G, DeVore A, Dawn A. Obesity and the skin: skin physiology and skin manifestations of obesity. J Am Acad Dermatol 2007; 56:901–916.
- Farage MA, Miller KW, Berardesca E, Maibach HI. Clinical implications of aging skin: cutaneous disorders in the elderly. Am J Clin Dermatol 2009; 10:73–86.
- Kirsner RS, Pardes JB, Eaglstein WH, Falanga V. The clinical spectrum of lipodermatosclerosis. J Am Acad Dermatol 1993; 28:623–627.
- Miteva M, Romanelli P, Kirsner RS. Lipodermatosclerosis. Dermatol Ther 2010; 23:375–388.
- Barron GS, Jacob SE, Kirsner RS. Dermatologic complications of chronic venous disease: medical management and beyond. Ann Vasc Surg 2007; 21:652–662.
- Bruce AJ, Bennett DD, Lohse CM, Rooke TW, Davis MD. Lipodermatosclerosis: review of cases evaluated at Mayo Clinic. J Am Acad Dermatol 2002; 46:187–192.
- Falagas ME, Vergidis PI. Narrative review: diseases that masquerade as infectious cellulitis. Ann Intern Med 2005; 142:47–55.
- Wilson CL, Cameron J, Powell SM, Cherry G, Ryan TJ. High incidence of contact dermatitis in leg-ulcer patients—implications for management. Clin Exp Dermatol 1991; 16:250–253.
- Wolf R. The lanolin paradox. Dermatology 1996; 192:198–202.
- Keeley VL. Lymphoedema and cellulitis: chicken or egg? Br J Dermatol 2008; 158:1175–1176.
- Wells GC. Recurrent granulomatous dermatitis with eosinophilia. Trans St Johns Hosp Dermatol Soc 1971; 57:46–56.
- Ferreli C, Pinna AL, Atzori L, Aste N. Eosinophilic cellulitis (Well’s syndrome): a new case description. J Eur Acad Dermatol Venereol 1999; 13:41–45.
- Ladoyanni E, Vlachou C, Thushara R, Snead D. A patient with Wells’ syndrome. Clin Exp Dermatol 2010; 35:e3–e4.
- Moon HS, Park K, Lee JH, Son SJ. Eosinophilic cellulitis in an infant. Int J Dermatol 2010; 49:592–593.
- Walker P, Long D, James C, Marshman G. Exaggerated insect bite reaction exacerbated by a pyogenic infection in a patient with chronic lymphocytic leukaemia. Australas J Dermatol 2007; 48:165–169.
- Laliwala NM, Kulshrestha R, Singh R, Balasubramaniam P. A case of eosinophilic cellulitis of the hand mimicking bacterial cellulitis. J Hand Surg Eur Vol 2009; 34:410–411.
- Chung CL, Cusack CA. Wells syndrome: an enigmatic and therapeutically challenging disease. J Drugs Dermatol 2006; 5:908–911.
- Melski JW. Wells’ syndrome, insect bites, and eosinophils. Dermatol Clin 1990; 8:287–293.
- Spigel GT, Winkelmann RK. Wells’ syndrome. Recurrent granulomatous dermatitis with eosinophilia. Arch Dermatol 1979; 115:611–613.
- Clark DP, Anderson PC. Eosinophilic cellulitis caused by arthropod bites. Int J Dermatol 1988; 27:411–412.
- Howard R, Frieden IJ. Papular urticaria in children. Pediatr Dermatol 1996; 13:246–249.
- Hernandez RG, Cohen BA. Insect bite-induced hypersensitivity and the SCRATCH principles: a new approach to papular urticaria. Pediatrics 2006; 118:e189–e196.
- Heng MC, Kloss SG, Haberfelde GC. Pathogenesis of papular urticaria. J Am Acad Dermatol 1984; 10:1030–1034.
- Kossard S, Hamann I, Wilkinson B. Defining urticarial dermatitis: a subset of dermal hypersensitivity reaction pattern. Arch Dermatol 2006; 142:29–34.
KEY POINTS
- Cellulitis is rarely bilateral.
- Patients with cellulitis often have systemic symptoms, such as fever and leukocytosis.
- A chronic course points to a diagnosis other than cellulitis.
- Plaques with a “bound-down” appearance or dark pigmentation point to a chronic disease rather than cellulitis.
- Stasis dermatitis is the most common mimic of cellulitis.
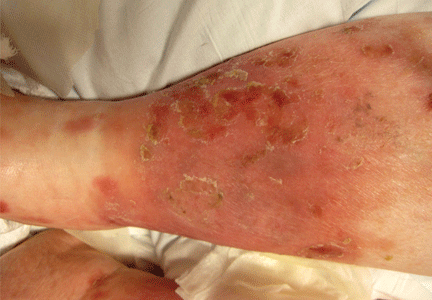
Prolonged delivery: child with CP awarded $70M … and more

AFTER MORE THAN 4 HOURS of second-stage labor followed by prolonged pushing and crowning, the baby was born depressed. Later, the child was found to have cerebral palsy.
PARENTS’ CLAIM The ObGyn was negligent in failing to perform an episiotomy, not attempting vacuum extraction, and not using forceps to assist delivery. Although fetal heart-rate monitoring results deteriorated, the ObGyn did not assess contractions for 30 minutes at one point. Hospital staff members were unable to adequately intubate or ventilate the newborn. The hospital staff disposed of the baby’s cord blood. Records were altered.
The parents’ counsel proposed that the defendants’ insurance company refused all settlement efforts prior to trial because the case venue was known to be conservative regarding jury verdicts.
DEFENDANTS’ DEFENSE The hospital and the ObGyn were not negligent; the mother and baby received proper care. Hospital staff acted appropriately.
VERDICT During the trial, the hospital settled for an undisclosed amount. An additional $2 million was offered on behalf of the ObGyn later in the trial, but the parents refused settlement at that time.
A California jury returned a $74.525 million verdict against the ObGyn. The child was awarded $70.725 million for medical expenses, lost earnings, and damages. The parents were awarded $3.8 million for emotional distress.
Was ectopic pregnancy missed?
A WOMAN IN SEVERE ABDOMINAL PAIN saw her internist. CT scans revealed a right ovarian cyst. When pain continued, she saw her ObGyn 3 weeks later, and her bowel was full of hard stool. Ultrasonography (US) showed a multicystic right ovary and a thin endometrial stripe. She was taking birth control pills and her husband had a vasectomy. She was told her abdominal pain was from constipation and ovarian cysts. A week later, she had laparoscopic surgery to remove an ectopic pregnancy.
PATIENT’S CLAIM The ObGyn did not perform a pregnancy test, and did not diagnose an ectopic pregnancy in a timely manner. An earlier diagnosis would have allowed medical rather than surgical resolution.
PHYSICIAN’S DEFENSE It was too early to determine if the pregnancy was intrauterine or ectopic. An earlier diagnosis would have resulted in laparoscopic surgery rather than medical treatment, as the medication (methotrexate) can cause increased pain.
VERDICT An Illinois defense verdict was returned.
Foreshortened vagina inhibits intercourse
A 65-YEAR-OLD WOMAN underwent anterior and posterior colphorrhaphy to repair a cystocele and rectocele, sacrospinous ligament fixation for vaginal prolapse, and a TVT mid-urethral suspension procedure to correct stress urinary incontinence. During two follow-up visits, the gynecologist determined that she was healing normally.
Within the next few weeks, the patient came to believe that her vagina had been sewn shut. She did not return to her gynecologist, but sought treatment with another physician 6 months later. It was determined that she had a stenotic and foreshortened vagina.
PATIENT’S CLAIM Too much vaginal tissue was removed during surgery.
PHYSICIAN’S DEFENSE The stenotic and foreshortened vagina was an unexpected result of the healing process after surgery.
VERDICT An Illinois defense verdict was returned.
Hydrocephalus in utero not seen until too late
A WOMAN HAD PRENATAL TREATMENT at a federally funded health clinic. A certified nurse midwife (CNM) ordered second-trimester US, with normal results. During the third trimester, the mother switched to a private ObGyn who ordered testing. US indicated the fetus was hydrocephalic. The child was born with cognitive disabilities and will need lifelong care.
PARENTS’ CLAIM The CNM ordered US too early in the pregnancy to be of diagnostic value; no further testing was undertaken. When hydrocephalus was seen, an abortion was not legally available because of fetal age.
DEFENDANT’S DEFENSE Even if US had been performed later in the second trimester, the defect would not have shown.
VERDICT A $4 million New Jersey settlement was reached.

WHEN SHOULDER DYSTOCIA WAS ENCOUNTERED, the ObGyn used standard maneuvers to deliver the child. The baby suffered a severe brachial plexus injury with rupture of C7 nerve and avulsions at C8 and T1.
Nerve-graft surgery at 6 months and tendon transfer surgery at 2 years resulted in recovery of good shoulder and elbow function, but the child has inadequate use of his wrist and hand. Additional surgeries are planned.
PARENTS’ CLAIM The ObGyn did not inform the mother that she was at risk for shoulder dystocia, nor did he discuss cesarean delivery. The mother’s risk factors included short stature, gestational diabetes, excessive weight gain during pregnancy, and two previous deliveries that involved vacuum assistance and a broken clavicle. The ObGyn applied excessive traction to the fetal head during delivery.
PHYSICIAN’S DEFENSE The mother’s risk factors were not severe enough to consider the chance of shoulder dystocia. The baby’s injuries were due to the normal forces of labor. Traction placed on the baby’s head during delivery was gentle and appropriate.
VERDICT A $5.5 million Iowa verdict was returned.
Faulty testing: baby has Down syndrome
AT 13 WEEKS’ GESTATION, a 34-year-old woman underwent chorionic villus sampling (CVS) at a maternal-fetal medicine center. Results showed a normal chromosomal profile. Later, two sonograms indicated possible Down syndrome. The parents were assured that the baby did not have a genetic disorder; amniocentesis was never suggested.
A week before the baby’s birth, the parents were told the child has Down syndrome.
PARENTS’ CLAIM Maternal tissue, not fetal tissue, had been removed and tested during CVS. The parents would have aborted the fetus had they known she had Down syndrome.
DEFENDANTS’ DEFENSE CVS was properly administered.
VERDICT A $3 million Missouri verdict was returned against the center where the testing was performed.
Why did the uterus seem to be growing?
A 52-YEAR-OLD WOMAN’S UTERUS was larger than normal in February 2007. By November 2008, her uterus was the size of a 14-week gestation. In September 2009, she complained of abdominal discomfort. Her uterus was larger than at the previous visit. The gynecologist suggested a hysterectomy, but nothing was scheduled.
In November 2009, she reported increasing pelvic pressure; her uterus was the size of an 18-week gestation. US and MRI showed large masses on both ovaries although the uterus had no masses or fibroids within it. A gynecologic oncologist performed abdominal hysterectomy with bilateral salpingo-oophorectomy and bilateral peri-aortic lymph node dissection. Pathology returned a diagnosis of ovarian cancer. The patient underwent chemotherapy.
PATIENT’S CLAIM The gynecologist was negligent in not ordering testing in 2007 when the larger-than-normal uterus was first detected, or in subsequent visits through September 2009. A more timely reaction would have given her an opportunity to treat the cancer at an earlier stage.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT A $650,000 Maryland settlement was reached.
Erb’s palsy after shoulder dystocia
DURING VAGINAL DELIVERY, the ObGyn encountered shoulder dystocia. The child suffered a brachial plexus injury and has Erb’s palsy. There was some improvement after two operations, but she still has muscle weakness, arm-length discrepancy, and limited range of motion.
PARENTS’ CLAIM The ObGyn applied excessive downward traction on the baby’s head when her left shoulder could not pass under the pubic bone.
PHYSICIAN’S DEFENSE The injury was caused by uterine contractions and maternal pushing. Proper maneuvers and gentle pressure were used.
VERDICT A $1.34 million New Jersey verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
AFTER MORE THAN 4 HOURS of second-stage labor followed by prolonged pushing and crowning, the baby was born depressed. Later, the child was found to have cerebral palsy.
PARENTS’ CLAIM The ObGyn was negligent in failing to perform an episiotomy, not attempting vacuum extraction, and not using forceps to assist delivery. Although fetal heart-rate monitoring results deteriorated, the ObGyn did not assess contractions for 30 minutes at one point. Hospital staff members were unable to adequately intubate or ventilate the newborn. The hospital staff disposed of the baby’s cord blood. Records were altered.
The parents’ counsel proposed that the defendants’ insurance company refused all settlement efforts prior to trial because the case venue was known to be conservative regarding jury verdicts.
DEFENDANTS’ DEFENSE The hospital and the ObGyn were not negligent; the mother and baby received proper care. Hospital staff acted appropriately.
VERDICT During the trial, the hospital settled for an undisclosed amount. An additional $2 million was offered on behalf of the ObGyn later in the trial, but the parents refused settlement at that time.
A California jury returned a $74.525 million verdict against the ObGyn. The child was awarded $70.725 million for medical expenses, lost earnings, and damages. The parents were awarded $3.8 million for emotional distress.
Was ectopic pregnancy missed?
A WOMAN IN SEVERE ABDOMINAL PAIN saw her internist. CT scans revealed a right ovarian cyst. When pain continued, she saw her ObGyn 3 weeks later, and her bowel was full of hard stool. Ultrasonography (US) showed a multicystic right ovary and a thin endometrial stripe. She was taking birth control pills and her husband had a vasectomy. She was told her abdominal pain was from constipation and ovarian cysts. A week later, she had laparoscopic surgery to remove an ectopic pregnancy.
PATIENT’S CLAIM The ObGyn did not perform a pregnancy test, and did not diagnose an ectopic pregnancy in a timely manner. An earlier diagnosis would have allowed medical rather than surgical resolution.
PHYSICIAN’S DEFENSE It was too early to determine if the pregnancy was intrauterine or ectopic. An earlier diagnosis would have resulted in laparoscopic surgery rather than medical treatment, as the medication (methotrexate) can cause increased pain.
VERDICT An Illinois defense verdict was returned.
Foreshortened vagina inhibits intercourse
A 65-YEAR-OLD WOMAN underwent anterior and posterior colphorrhaphy to repair a cystocele and rectocele, sacrospinous ligament fixation for vaginal prolapse, and a TVT mid-urethral suspension procedure to correct stress urinary incontinence. During two follow-up visits, the gynecologist determined that she was healing normally.
Within the next few weeks, the patient came to believe that her vagina had been sewn shut. She did not return to her gynecologist, but sought treatment with another physician 6 months later. It was determined that she had a stenotic and foreshortened vagina.
PATIENT’S CLAIM Too much vaginal tissue was removed during surgery.
PHYSICIAN’S DEFENSE The stenotic and foreshortened vagina was an unexpected result of the healing process after surgery.
VERDICT An Illinois defense verdict was returned.
Hydrocephalus in utero not seen until too late
A WOMAN HAD PRENATAL TREATMENT at a federally funded health clinic. A certified nurse midwife (CNM) ordered second-trimester US, with normal results. During the third trimester, the mother switched to a private ObGyn who ordered testing. US indicated the fetus was hydrocephalic. The child was born with cognitive disabilities and will need lifelong care.
PARENTS’ CLAIM The CNM ordered US too early in the pregnancy to be of diagnostic value; no further testing was undertaken. When hydrocephalus was seen, an abortion was not legally available because of fetal age.
DEFENDANT’S DEFENSE Even if US had been performed later in the second trimester, the defect would not have shown.
VERDICT A $4 million New Jersey settlement was reached.
WHEN SHOULDER DYSTOCIA WAS ENCOUNTERED, the ObGyn used standard maneuvers to deliver the child. The baby suffered a severe brachial plexus injury with rupture of C7 nerve and avulsions at C8 and T1.
Nerve-graft surgery at 6 months and tendon transfer surgery at 2 years resulted in recovery of good shoulder and elbow function, but the child has inadequate use of his wrist and hand. Additional surgeries are planned.
PARENTS’ CLAIM The ObGyn did not inform the mother that she was at risk for shoulder dystocia, nor did he discuss cesarean delivery. The mother’s risk factors included short stature, gestational diabetes, excessive weight gain during pregnancy, and two previous deliveries that involved vacuum assistance and a broken clavicle. The ObGyn applied excessive traction to the fetal head during delivery.
PHYSICIAN’S DEFENSE The mother’s risk factors were not severe enough to consider the chance of shoulder dystocia. The baby’s injuries were due to the normal forces of labor. Traction placed on the baby’s head during delivery was gentle and appropriate.
VERDICT A $5.5 million Iowa verdict was returned.
Faulty testing: baby has Down syndrome
AT 13 WEEKS’ GESTATION, a 34-year-old woman underwent chorionic villus sampling (CVS) at a maternal-fetal medicine center. Results showed a normal chromosomal profile. Later, two sonograms indicated possible Down syndrome. The parents were assured that the baby did not have a genetic disorder; amniocentesis was never suggested.
A week before the baby’s birth, the parents were told the child has Down syndrome.
PARENTS’ CLAIM Maternal tissue, not fetal tissue, had been removed and tested during CVS. The parents would have aborted the fetus had they known she had Down syndrome.
DEFENDANTS’ DEFENSE CVS was properly administered.
VERDICT A $3 million Missouri verdict was returned against the center where the testing was performed.
Why did the uterus seem to be growing?
A 52-YEAR-OLD WOMAN’S UTERUS was larger than normal in February 2007. By November 2008, her uterus was the size of a 14-week gestation. In September 2009, she complained of abdominal discomfort. Her uterus was larger than at the previous visit. The gynecologist suggested a hysterectomy, but nothing was scheduled.
In November 2009, she reported increasing pelvic pressure; her uterus was the size of an 18-week gestation. US and MRI showed large masses on both ovaries although the uterus had no masses or fibroids within it. A gynecologic oncologist performed abdominal hysterectomy with bilateral salpingo-oophorectomy and bilateral peri-aortic lymph node dissection. Pathology returned a diagnosis of ovarian cancer. The patient underwent chemotherapy.
PATIENT’S CLAIM The gynecologist was negligent in not ordering testing in 2007 when the larger-than-normal uterus was first detected, or in subsequent visits through September 2009. A more timely reaction would have given her an opportunity to treat the cancer at an earlier stage.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT A $650,000 Maryland settlement was reached.
Erb’s palsy after shoulder dystocia
DURING VAGINAL DELIVERY, the ObGyn encountered shoulder dystocia. The child suffered a brachial plexus injury and has Erb’s palsy. There was some improvement after two operations, but she still has muscle weakness, arm-length discrepancy, and limited range of motion.
PARENTS’ CLAIM The ObGyn applied excessive downward traction on the baby’s head when her left shoulder could not pass under the pubic bone.
PHYSICIAN’S DEFENSE The injury was caused by uterine contractions and maternal pushing. Proper maneuvers and gentle pressure were used.
VERDICT A $1.34 million New Jersey verdict was returned.
AFTER MORE THAN 4 HOURS of second-stage labor followed by prolonged pushing and crowning, the baby was born depressed. Later, the child was found to have cerebral palsy.
PARENTS’ CLAIM The ObGyn was negligent in failing to perform an episiotomy, not attempting vacuum extraction, and not using forceps to assist delivery. Although fetal heart-rate monitoring results deteriorated, the ObGyn did not assess contractions for 30 minutes at one point. Hospital staff members were unable to adequately intubate or ventilate the newborn. The hospital staff disposed of the baby’s cord blood. Records were altered.
The parents’ counsel proposed that the defendants’ insurance company refused all settlement efforts prior to trial because the case venue was known to be conservative regarding jury verdicts.
DEFENDANTS’ DEFENSE The hospital and the ObGyn were not negligent; the mother and baby received proper care. Hospital staff acted appropriately.
VERDICT During the trial, the hospital settled for an undisclosed amount. An additional $2 million was offered on behalf of the ObGyn later in the trial, but the parents refused settlement at that time.
A California jury returned a $74.525 million verdict against the ObGyn. The child was awarded $70.725 million for medical expenses, lost earnings, and damages. The parents were awarded $3.8 million for emotional distress.
Was ectopic pregnancy missed?
A WOMAN IN SEVERE ABDOMINAL PAIN saw her internist. CT scans revealed a right ovarian cyst. When pain continued, she saw her ObGyn 3 weeks later, and her bowel was full of hard stool. Ultrasonography (US) showed a multicystic right ovary and a thin endometrial stripe. She was taking birth control pills and her husband had a vasectomy. She was told her abdominal pain was from constipation and ovarian cysts. A week later, she had laparoscopic surgery to remove an ectopic pregnancy.
PATIENT’S CLAIM The ObGyn did not perform a pregnancy test, and did not diagnose an ectopic pregnancy in a timely manner. An earlier diagnosis would have allowed medical rather than surgical resolution.
PHYSICIAN’S DEFENSE It was too early to determine if the pregnancy was intrauterine or ectopic. An earlier diagnosis would have resulted in laparoscopic surgery rather than medical treatment, as the medication (methotrexate) can cause increased pain.
VERDICT An Illinois defense verdict was returned.
Foreshortened vagina inhibits intercourse
A 65-YEAR-OLD WOMAN underwent anterior and posterior colphorrhaphy to repair a cystocele and rectocele, sacrospinous ligament fixation for vaginal prolapse, and a TVT mid-urethral suspension procedure to correct stress urinary incontinence. During two follow-up visits, the gynecologist determined that she was healing normally.
Within the next few weeks, the patient came to believe that her vagina had been sewn shut. She did not return to her gynecologist, but sought treatment with another physician 6 months later. It was determined that she had a stenotic and foreshortened vagina.
PATIENT’S CLAIM Too much vaginal tissue was removed during surgery.
PHYSICIAN’S DEFENSE The stenotic and foreshortened vagina was an unexpected result of the healing process after surgery.
VERDICT An Illinois defense verdict was returned.
Hydrocephalus in utero not seen until too late
A WOMAN HAD PRENATAL TREATMENT at a federally funded health clinic. A certified nurse midwife (CNM) ordered second-trimester US, with normal results. During the third trimester, the mother switched to a private ObGyn who ordered testing. US indicated the fetus was hydrocephalic. The child was born with cognitive disabilities and will need lifelong care.
PARENTS’ CLAIM The CNM ordered US too early in the pregnancy to be of diagnostic value; no further testing was undertaken. When hydrocephalus was seen, an abortion was not legally available because of fetal age.
DEFENDANT’S DEFENSE Even if US had been performed later in the second trimester, the defect would not have shown.
VERDICT A $4 million New Jersey settlement was reached.
WHEN SHOULDER DYSTOCIA WAS ENCOUNTERED, the ObGyn used standard maneuvers to deliver the child. The baby suffered a severe brachial plexus injury with rupture of C7 nerve and avulsions at C8 and T1.
Nerve-graft surgery at 6 months and tendon transfer surgery at 2 years resulted in recovery of good shoulder and elbow function, but the child has inadequate use of his wrist and hand. Additional surgeries are planned.
PARENTS’ CLAIM The ObGyn did not inform the mother that she was at risk for shoulder dystocia, nor did he discuss cesarean delivery. The mother’s risk factors included short stature, gestational diabetes, excessive weight gain during pregnancy, and two previous deliveries that involved vacuum assistance and a broken clavicle. The ObGyn applied excessive traction to the fetal head during delivery.
PHYSICIAN’S DEFENSE The mother’s risk factors were not severe enough to consider the chance of shoulder dystocia. The baby’s injuries were due to the normal forces of labor. Traction placed on the baby’s head during delivery was gentle and appropriate.
VERDICT A $5.5 million Iowa verdict was returned.
Faulty testing: baby has Down syndrome
AT 13 WEEKS’ GESTATION, a 34-year-old woman underwent chorionic villus sampling (CVS) at a maternal-fetal medicine center. Results showed a normal chromosomal profile. Later, two sonograms indicated possible Down syndrome. The parents were assured that the baby did not have a genetic disorder; amniocentesis was never suggested.
A week before the baby’s birth, the parents were told the child has Down syndrome.
PARENTS’ CLAIM Maternal tissue, not fetal tissue, had been removed and tested during CVS. The parents would have aborted the fetus had they known she had Down syndrome.
DEFENDANTS’ DEFENSE CVS was properly administered.
VERDICT A $3 million Missouri verdict was returned against the center where the testing was performed.
Why did the uterus seem to be growing?
A 52-YEAR-OLD WOMAN’S UTERUS was larger than normal in February 2007. By November 2008, her uterus was the size of a 14-week gestation. In September 2009, she complained of abdominal discomfort. Her uterus was larger than at the previous visit. The gynecologist suggested a hysterectomy, but nothing was scheduled.
In November 2009, she reported increasing pelvic pressure; her uterus was the size of an 18-week gestation. US and MRI showed large masses on both ovaries although the uterus had no masses or fibroids within it. A gynecologic oncologist performed abdominal hysterectomy with bilateral salpingo-oophorectomy and bilateral peri-aortic lymph node dissection. Pathology returned a diagnosis of ovarian cancer. The patient underwent chemotherapy.
PATIENT’S CLAIM The gynecologist was negligent in not ordering testing in 2007 when the larger-than-normal uterus was first detected, or in subsequent visits through September 2009. A more timely reaction would have given her an opportunity to treat the cancer at an earlier stage.
PHYSICIAN’S DEFENSE The case was settled before trial.
VERDICT A $650,000 Maryland settlement was reached.
Erb’s palsy after shoulder dystocia
DURING VAGINAL DELIVERY, the ObGyn encountered shoulder dystocia. The child suffered a brachial plexus injury and has Erb’s palsy. There was some improvement after two operations, but she still has muscle weakness, arm-length discrepancy, and limited range of motion.
PARENTS’ CLAIM The ObGyn applied excessive downward traction on the baby’s head when her left shoulder could not pass under the pubic bone.
PHYSICIAN’S DEFENSE The injury was caused by uterine contractions and maternal pushing. Proper maneuvers and gentle pressure were used.
VERDICT A $1.34 million New Jersey verdict was returned.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
These cases were selected by the editors of OBG Management from Medical Malpractice Verdicts, Settlements & Experts, with permission of the editor, Lewis Laska (www.verdictslaska.com). The information available to the editors about the cases presented here is sometimes incomplete. Moreover, the cases may or may not have merit. Nevertheless, these cases represent the types of clinical situations that typically result in litigation and are meant to illustrate nationwide variation in jury verdicts and awards.
We want to hear from you! Tell us what you think.
Treating ‘depression’ in patients with schizophrenia
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
Discuss this article at www.facebook.com/CurrentPsychiatry
Approximately 25% of schizophrenia patients experience course-related depression.1-4 Depression in patients with schizophrenia is linked to reduced social and vocational functioning, increased likelihood of psychotic relapse and rehospitalization, and other problems.2-4 Depression in patients with schizophrenia also has been linked to undesirable life events, especially “exit events” such as losing people in their lives, as well as suicidal ideation, suicide attempts, and completed suicides. Overall, it has been noted that approximately 10% of patients with schizophrenia commit suicide.5 Depressed schizophrenia patients are at particularly high risk for suicide the first few months after diagnosis and after hospital discharge.
Confirm the diagnosis
The best approach to treating depressive symptoms in schizophrenia patients is to formulate a thorough differential diagnosis (Table 1).
Table 1
Differential diagnosis of ‘depression’ in schizophrenia
| Organic factors |
| Antipsychotic-induced dysphoria |
| Akinesia |
| Akathisia |
| Negative symptoms |
| Acute disappointment reactions |
| Chronic disappointment reactions |
| Prodrome of psychotic relapse |
| Depression |
Antipsychotic-induced dysphoria. Blockade of dopamine receptors is an important feature of all antipsychotics; however, dopamine neurotransmission also is involved in the brain’s “pleasure” pathways. Individuals who take antipsychotics may experience reduced joy from once-pleasurable activities. Results of studies on the link between depression and antipsychotics have been mixed.2,4 Although some researchers have found depressed mood common among patients receiving antipsychotics, others have failed to show differences between patients treated with antipsychotics and those randomized to placebo.
Akinesia, a parkinsonian side effect of antipsychotics, can be blatant or subtle. The blatant form involves large muscle groups; these patients present with diminished arm swing, stooped posture, and parkinsonian gait. Easily spotted, such patients are unlikely to be considered depressed.
The more subtle form of akinesia is easier to confuse with depression. It can affect small muscle groups, such as in the face or vocal cords. Lack of responsiveness of facial expression is easily confused with blunted affect, low mood, lack of interest, or emotional unresponsiveness. Subtle akinesia also can impair a patient’s ability to initiate or sustain motor behavior. Many activities, from striking up a conversation to changing television channels, involve initiating and sustaining motor behavior, which these patients’ basal ganglia are underequipped to do. Life becomes boring and patients criticize themselves for “being lazy.” Patients with akinesia also are prone to dysphoria.6,7 When the lack of spontaneous motor behavior found in subtle akinesia is combined with diminished experience of pleasure due to antipsychotic blockade of dopamine, a patient may feel that “nothing is worth the effort.”
Akathisia is another movement disorder of the basal ganglia that can be triggered by antipsychotics. Whereas a patient with akinesia experiences having a “broken starter motor,” the akathisia patient experiences “a starter motor that won’t turn off.” Akathisia can be blatant or subtle. A patient with blatant akathisia has difficulty remaining seated and often paces. In subtle akathisia, increased motor activity is less dramatic, and patients may simply wander or talk excessively. Akathisia also has a dysphoric component that, when the movement is interpreted as restlessness or agitation, may look like depression.8
Negative symptoms. Primary negative symptoms in schizophrenia have several features in common with depression, which can create diagnostic challenges.9 These include anhedonia, social withdrawal, lack of initiative, lowered energy, diminished expectations and/or self confidence, and reduced speech or activity. The main feature that distinguishes the primary negative symptom syndrome from depression is prominent blue mood, which is present in depression but not in negative symptoms. Cognitive features—such as guilt, pessimism, and suicidal thoughts—are common in depression, but usually are absent in negative symptoms.
4 While the acute disappointment reaction is ongoing, the emotional burden may be substantial. With bereavement or grief reactions the loss is clear; however, be vigilant for situations where the patient’s loss may be idiosyncratic or symbolic.
Chronic disappointment reactions, also known as the demoralization syndrome, involve long-term convictions of defeat, despair, incompetence, and loss of control.10 These reactions can be devastating and prolonged. These reactions are important to identify because they may be ameliorated by rehabilitative interventions or other psychosocial supports.
Prodrome of psychotic relapse. Longitudinal observations of patients with schizophrenia have found depressive symptoms may occur during the early stages of psychotic decompensation.4,11,12 These symptoms include dysphoria, anxiety, agitation, sleep and/or appetite disturbances, impaired concentration, hopelessness, helplessness, feelings of loss of control or alienation, and social withdrawal. These features usually last a few days to a couple of weeks before they are overtaken by psychotic phenomena.
Treatment: A suggested approach
Based on my clinical experience in managing newly emergent “depression” episodes in patients correctly diagnosed with schizophrenia, I suggest the following approach:
First, assess the patient for medical disorders that could present with depressive features. Collaborate with the patient’s primary care physician to determine which medications the patient is taking and whether there have been any recent changes in these agents or their doses, including adherence issues, potential substance use or abuse, and changes between brand name and generic agents. Thoroughly evaluate the patient’s psychiatric status, including symptoms, suicidal risk, and changes in life circumstances. A patient who is at high risk of suicide may require hospitalization. Also assess for the presence of extrapyramidal side effects.
Do not change your patient’s medication regimen at this early stage, but provide him or her structure and support, and schedule an early appointment for the next visit (eg, 1 week later). A planned telephone call before the appointment may be helpful as well. If the “depression” is an acute disappointment reaction, it may run its course and resolve. However, if your patient’s depressive symptoms are a prodrome of psychotic relapse, the quick follow-up contact will improve the chances of preventing a psychotic episode by increasing the antipsychotic dosage or making other reasonable changes in pharmacotherapy.
If at the follow-up visit the patient’s psychotic symptoms have not progressed but depressive symptoms persist, evaluate for the possibility of parkinsonian symptoms, which may be subtle and difficult to rule out. If your patient is restless or tends to be physically active, a trial of a benzodiazepine can be added to treat akathisia. If the patient is underactive, consider a trial of an anticholinergic antiparkinsonian agent, such as benztropine, for akinesia. Dosages of benztropine can be raised in a stepwise manner up to 6 mg/d if there are no side effects, such as constipation, dry mouth, blurry vision, or memory impairment. Advantages of treating extrapyramidal side effects first include:
- response to antiparkinsonian medications occurs rapidly—if your patient shows no response within a week, future response at this dose is unlikely
- the presence of anticholinergic side effects is a biologic marker indicating that the treatment dose is adequate
- the clinician has more time to get to know the patient and his or her condition before committing to lowering, raising, or changing the antipsychotic dosage.
Table 2
Antidepressant effects of antipsychotics in schizophrenia patients
| Study | Design | Results |
|---|---|---|
| Marder et al, 199714 | In 2 double-blind trials, 513 patients with chronic schizophrenia received risperidone (2, 6, 10, or 16 mg/d), haloperidol (20 mg/d), or placebo for 8 weeks | Patients receiving risperidone showed greater reductions in anxiety and depression symptoms as measured by PANSS scores than patients receiving haloperidol or placebo |
| Tollefson et al, 199815 | In a prospective, blinded trial, 1,996 patients with schizophrenia received olanzapine (5 to 20 mg/d) or haloperidol (5 to 20 mg/d) | Among patients with depressive signs and symptoms, those who received olanzapine showed better improvement in MADRS scores than patients receiving haloperidol |
| Emsley et al, 200316 | Patients with schizophrenia (N = 269) who had not responded to 4 weeks of fluphenazine (20 mg/d) were randomized to receive quetiapine (600 mg/d) or haloperidol (20 mg/d) for 8 weeks | Quetiapine produced greater reduction on PANSS depression scores than haloperidol |
| Mauri et al, 200817 | In a retrospective study, 222 patients in the reexacerbation phase of schizophrenia received fluphenazine, haloperidol decanoate, haloperidol, clozapine, olanzapine, quetiapine, risperidone, or L-sulpiride monotherapy | All antipsychotics led to improvements in depressive symptoms as measured by the BPRS scale, but improvements were statistically significant only with fluphenazine, haloperidol, olanzapine, risperidone, and L-sulpiride |
| BPRS: Brief Psychiatric Rating Scale; MADRS: Montgomery-Åsberg Depression Rating Scale; PANSS: Positive and Negative Syndrome Scale | ||
Antidepressants. If depressive symptoms persist after lowering or changing the antipsychotic, consider a trial of an adjunctive antidepressant. Titrate antidepressants to the recommended dose over 1 month, and continue antiparkinsonian medications. See patients frequently, and ensure that they receive psychosocial support.
No randomized trials have compared the efficacy of antidepressants for treating patients with schizophrenia; therefore, it is unclear if there is a preferred agent. Newer antidepressants often are used in depressed patients with schizophrenia because they are less likely to cause anticholinergic side effects. However, anticholinergic activity may be desirable, eg, for patients with akinesia. Caution is required when combining a selective serotonin reuptake inhibitor with clozapine because metabolism interactions could lead to toxic clozapine levels in some patients.19
If your patient’s depressive symptoms improve after adding an antidepressant, continue that agent along with the antipsychotic and any antiparkinsonian medications. Only 1 study has evaluated maintenance adjunctive antidepressant therapy for depressed patients with schizophrenia who initially responded to antidepressants. It found that imipramine appeared to protect patients from depressive relapse, and patients who received maintenance adjunctive imipramine were less likely to experience worsening psychotic symptoms.20
Depressed schizophrenia patients are most likely to improve if they receive optimal psychosocial intervention,21 which consists of nonspecific support and, when indicated, psychosocial rehabilitation services. Change, even positive change, can be stressful, and patients with schizophrenia need every advantage they can get to be successful in moving their lives in a positive direction.
- Rybakowski JK, Vansteelandt K, Szafranski T, et al. Treatment of depression in first episode of schizophrenia: Results from EUFEST [published online ahead of print May 22, 2012]. Eur Neuropsychopharmacol. doi:10.1016/j.euroneuro.2012.04.001.
- Addington D, Addington J. Calgary Depression Scale for Schizophrenia. www.ucalgary.ca/cdss.
- Benztropine • Cogentin
- Clozapine • Clozaril
- Fluphenazine • Permitil, Prolixin
- Haloperidol • Haldol
- Imipramine • Tofranil
- Olanzapine • Zyprexa
- Quetiapine • Seroquel
- Risperidone • Risperdal
Dr. Siris reports no financial relationships with any company whose products are mentioned in this article or with manufacturers of competing products.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
1. Möller HJ. Drug treatment of depressive symptoms in schizophrenia. Clin Schizophr Relat Psychoses. 2008;1(4):328-340.
2. Buckley PF, Miller BJ, Lehrer DS, et al. Psychiatric comorbidities and schizophrenia. Schizophr Bull. 2009;35(2):383-402.
3. Hausmann A, Fleischhacker WW. Differential diagnosis of depressed mood in patients with schizophrenia: a diagnostic algorithm based on a review. Acta Psychiatr Scand. 2002;106(2):83-96.
4. Siris SG, Bench C. Depression and schizophrenia. In: Schizophrenia. Hirsch SR Weinberger DR, eds. Malden, MA: Blackwell Publishing Company; 2003:142-167.
5. Hawton K, Sutton L, Haw C, et al. Schizophrenia and suicide: systematic review of risk factors. Br J Psychiatry. 2005;187:9-20.
6. Rifkin A, Quitkin F, Klein DF. Akinesia: a poorly recognized drug-induced extrapyramidal behavioral disorder. Arch Gen Psychiatry. 1975;32(5):672-674.
7. Van Putten T, May RP. “Akinetic depression” in schizophrenia. Arch Gen Psychiatry. 1978;35(9):1101-1107.
8. Van Putten T. The many faces of akathisia. Compr Psychiatry. 1975;16(1):43-47.
9. Bermanzohn PC, Siris SG. Akinesia: a syndrome common to parkinsonism retarded depression, and negative symptoms of schizophrenia. Compr Psychiatry. 1992;33(4):221-232.
10. Clarke DM, Kissame DW. Demoralization: its phenomenology and importance. Aust N Z J Psychiatry. 2002;36(6):733-742.
11. Herz MI, Melville C. Relapse in schizophrenia. Am J Psychiatry. 1980;137(7):801-805.
12. Rosen JL, Miller TJ, D’Andrea JT, et al. Comorbid diagnoses in patients meeting criteria for the schizophrenia prodrome. Schizophr Res. 2006;85(1-3):124-131.
13. Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62(6):593-602.
14. Marder SR, Davis JM, Chouinard G. The effects of risperidone on the five dimensions of schizophrenia derived by factor analysis: combined results of the North American trials. J Clin Psychiatry. 1997;58(12):538-546.
15. Tollefson GD, Sanger TM, Lu Y, et al. Depressive signs and symptoms in schizophrenia: a prospective blinded trial of olanzapine and haloperidol. Arch Gen Psychiatry. 1998;55(3):250-258.
16. Emsley RA, Buckley P, Jones AM, et al. Differential effect of quetiapine on depressive symptoms in patients with partially responsive schizophrenia. J Psychopharmacol. 2003;17(2):210-215.
17. Mauri MC, Moliterno D, Rossattini M, et al. Depression in schizophrenia: comparison of first- and second-generation antipsychotic drugs. Schizophr Res. 2008;99(1-3):7-12.
18. Siris SG. Depression in schizophrenia: perspective in the era of “atypical” antipsychotic agents. Am J Psychiatry. 2000;157(9):1379-1389.
19. Centorrino F, Baldessarini RJ, Frankenburg FR, et al. Serum levels of clozapine and norclozapine in patients treated with selective serotonin reuptake inhibitors. Am J Psychiatry. 1996;153(6):820-822.
20. Siris SG, Bermanzohn PC, Mason SE, et al. Maintenance imipramine therapy for secondary depression in schizophrenia. A controlled trial. Arch Gen Psychiatry. 1994;51(2):109-115.
21. Bustillo J, Lauriello J, Horan W, et al. The psychosocial treatment of schizophrenia: an update. Am J Psychiatry. 2001;158(2):163-175.
Recognizing and treating complicated grief
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
Nearly 2.5 million persons die each year in the United States.1 For the bereaved, these deaths may be among the most painful and disruptive events they will experience. In this article, we evaluate the growing body of research on complicated grief (CG)—which also has been called prolonged grief, chronic grief, traumatic grief, and pathological grief—with an emphasis on how to identify CG and distinguish it from other adaptive and maladaptive reactions to the loss of a loved one. In addition, we review empirical evidence on treating CG, including psychotherapy, pharmacotherapy, and combined treatment approaches.
The bereavement-specific syndrome we refer to as CG currently is being reviewed for possible inclusion in DSM-5 as an official diagnosis. At press time, proposals for DSM-5 included a bereavement-related adjustment disorder within the new Trauma- and Stressor-Related Disorders category, as well as a provisional diagnosis of CG entitled Persistent Complex Bereavement-Related Disorder, which, upon acceptance, would be listed in Section III.2
What is ‘normal’ grief?
Grief is highly variable across individuals and time and may range from an absence of distress to severe and persistent pain and anguish. There’s no simple definition of “normal grief.” However, as clinicians, it’s necessary to understand the range of usual reactions. We recommend 2 considerations when evaluating grief reactions.
First, be aware that grief encompasses a range of cognitions, emotions, and behaviors. It may range from a relative lack of painful thoughts and emotions to intense and disruptive sadness, loneliness, anger, guilt, intrusive thoughts, difficulty concentrating, preoccupation with loss, social withdrawal, and a sense of being overwhelmed by the loss and its consequences. In the months after a loss, bereaved individuals may look for the deceased in a crowd, speak to them, or even experience auditory or visual hallucinations of the deceased. Nonetheless, positive feelings such as relief, peace, and happiness also are common following a loss.3 Moreover, laughter and smiling when discussing a lost loved one predicts reductions in grief symptoms over time.4 Overall, grief research suggests that, far from proceeding along standard and uniform stages,5 grief is complex and comprises a broad spectrum of thoughts, feelings, and behaviors that vary within and among individuals.
Second, note that in the absence of complications, grief progresses. For those who experience elevated levels of distress, the pain and disruption of loss initially may feel overwhelming but will subside in intensity over time for most individuals.5 This is not to say that an individual will never again feel sadness or longing for the deceased; elements of grief are likely to remain. Although the trajectory of grief symptoms varies among individuals and may progress in fits and starts, over time grief becomes more intermittent, less interfering, and is balanced with a sense of interest and purpose in life.
What is CG?
As research on grief experiences has grown, there’s increasing recognition that a minority of bereaved individuals experience more extreme grief symptoms that cause substantial, persistent distress and impairment despite the passage of many months or years. Shear et al6 proposed a set of CG diagnostic criteria (Table) in which a cluster of symptoms of intense and persistent separation distress are defined as core symptoms. Similar to other psychiatric diagnoses, the symptoms must be associated with significant distress or impairment.
Table
Proposed diagnostic criteria for complicated grief
| Symptom domain | Criteria |
|---|---|
| Separation distress | The patient has ≥1 of the following 4 symptoms: 1) Persistent, intense yearning or longing for the deceased 2) Frequent feelings of intense loneliness or emptiness 3) Recurrent negative thoughts about life without the deceased or recurrent urge to join the deceased 4) Preoccupying thoughts about the deceased that impair daily functioning |
| Thoughts | The patient has ≥2 of the following 8 symptoms: 1) Rumination about circumstances of the death 2) Frequent disbelief or inability to accept the death |
| Feelings | 3) Persistent feeling of being shocked, stunned, or emotionally numb since the death 4) Recurrent feelings of anger or bitterness regarding the death 5) Difficulty trusting or caring about others since the loss 6) Experiencing pain or other somatic symptoms the deceased person had, hearing the voice of the deceased, or seeing the deceased person 7) Intense emotional reactions to memories of the deceased |
| Behaviors | 8) Excessive avoidance or excessive preoccupation with places, people, and things related to the deceased or death |
| Source: Adapted from reference 6 | |
Assessing CG symptoms
Among those with persistent elevated distress, a CG diagnosis must be considered in the context of the individual’s social and cultural environment, time since the loss, and duration of symptoms. The hallmark symptom of CG is separation distress with a focus of cognitive, behavioral, and emotional symptoms on the loss and its consequences. CG is associated with substantial distress, functional impairment, and an increased risk for suicide. See the Box for a case study.
Many individuals with CG remain undiagnosed and untreated for years despite high levels of distress and impairment and high risk for negative consequences such as suicide.7 Accordingly, there’s a need for greater CG screening. Clinically useful tools for assessing CG include a brief, 5-item dimensional screening assessment6 and the patient-rated Inventory of Complicated Grief.8
Distinguishing complicated and uncomplicated grief. Exhibiting CG symptoms in the first several months after a loss does not mean an individual has or will develop CG. Most bereaved adults report painful thoughts and emotions in the weeks and months following the loss, including distressed yearning, waves of intense grief, persistent and intrusive thoughts, images related to the death, somatic distress, and a feeling of being disconnected from others. For most individuals, the intensity of this response diminishes within 6 to 18 months after the loved one’s death.5 Although the optimal length of time to wait before establishing a diagnosis remains debatable, the earliest CG should be diagnosed is 6 months after a loss.
It’s common for grief to occasionally rise in intensity for days or weeks. This surge may occur many months or years after the loss, even in people who exhibited relatively little distress or impairment. In particular, anniversaries, holidays, or periods of stress may trigger increased grief intensity. However, these surges typically subside naturally within a short time. Accordingly, CG should be diagnosed only when symptoms persist for >1 month.
CG vs other post-loss disorders. CG, major depressive disorder (MDD), and posttraumatic stress disorder (PTSD) often are comorbid in bereaved adults. Simon et al9 found 72% of CG patients in a treatment- seeking sample reported a lifetime history of MDD and 53% reported a lifetime history of PTSD. However, CG can be distinguished from these disorders. In the same study, 25% of CG patients had no other axis I diagnosis.9 After accounting for comorbid disorders, researchers associated CG severity with work and social impairment. These findings provide clear evidence for the incremental validity of CG—ie, a CG diagnosis gives clinicians additional information that predicts impairment above and beyond other disorders. However, future research needs to further examine CG and its overlap and differentiation from MDD and PTSD.
Distinguishing CG and MDD. Intense yearning or preoccupation with the deceased is a common symptom of CG but not MDD. In addition, CG symptoms possess intentionality. For example, emotional distress such as sadness and anger are prominent features of both CG and MDD. However, in CG, these symptoms are specific to the loss or circumstances of the loss, whereas in MDD they generally are more nebulous and generalized. Similarly, CG entails proximity seeking related to the deceased, and avoidance of reminders of the deceased, whereas MDD includes a more general social withdrawal and anhedonia.
Distinguishing CG and PTSD. CG and loss-related PTSD are distinguished by the predominant emotions and focus of concern associated with each disorder. The predominant emotion in PTSD is fear, whereas in CG it is sadness and longing. In PTSD, intrusive thoughts and memories associated with the trauma generally are associated with the event itself and produce an ongoing sense of threat.10 Avoidance in PTSD is intended to reduce this threat feeling. By contrast, in CG, intrusive memories focus on the deceased or the circumstances of the death, and avoidance is aimed at preventing painful reminders of the loss or its permanence. Importantly, both syndromes may be present.
Mr. C, age 67, presents to a local emergency department (ED) with his daughter. His daughter reports that he has not been himself since his wife died in a car accident 2 years ago. He continues to live in the house he shared with his wife, despite not needing the extra space and being unable to maintain it. Although Mr. C and his daughter used to talk about her mother a great deal, she says she now tries to avoid the subject because it upsets him. More recently she became concerned when Mr. C began to tell her that his life was meaningless without his wife. He said he frequently thinks about taking his own life to end his pain and loneliness.
Mr. C tells the ED psychiatrist he feels an intense wave of grief and loneliness every morning when he realizes his wife is not with him. He often stays in bed for hours, longing for her and thinking about their time together. At times, he thinks he hears her voice downstairs but when he searches for her, she is not there. Mr. C has been unable to go through his wife’s belongings, and feels nothing should be moved in their home. He will look at her photos, yet avoids other reminders of her (eg, partaking in their favorite hobbies, going to their favorite restaurants). He feels bitter and angry about his wife’s death, and becomes agitated when describing the car accident that took her life. Mr. C feels guilty for not being with his wife when she died. He assures the psychiatrist that he loves his children, but says he feels increasingly distant from them and doesn’t understand how they can move on after their mother’s death.
Mr. C reports symptoms consistent with a diagnosis of complicated grief. Further assessment is appropriate to determine if his symptoms are severe enough to warrant treatment.
Treating CG
When is treatment indicated? For years, bereavement theorists emphasized the need to work through emotions and memories related to the deceased with particular focus on negative material. However, evidence suggests that universal application of treatment to all bereaved individuals is unhelpful. In a recent meta-analysis, Neimeyer et al11 found that the outcomes of grief therapy applied indiscriminately to all bereaved adults or all members of high-risk populations—such as parents whose child experienced a violent death—were no better than would be expected by the passage of time. In contrast, grief therapy applied only to those who develop elevated and persistent distress (eg, CG) led to greater and more enduring improvement in post-loss distress than was observed in control conditions.
These results suggest that most grieving individuals who do not meet criteria for CG (or other psychiatric disorders) will not require intervention. Those who do seek treatment for grief-related distress in the acute grief period should be assessed for bereavement-related depression, anxiety, and suicidality, and treated or referred to professional or community-based resources for support or counseling as clinically indicated.
Evidence for psychotherapy. For those who meet CG criteria, psychotherapy targeting the specific symptoms of CG is helpful. The evidence is strongest for CG treatment (CGT), a 16-session, manualized psychotherapy developed by M. Katherine Shear, MD.12 CGT is based on an attachment model and cognitive-behavioral therapy (CBT) principles, and is informed by the dual-process theory proposed by Stroebe et al.13 According to this theory, natural healing following loss comprises 2 processes:
- a loss-oriented process in which the patient comes to terms with the loss, and
- a restoration-oriented process in which the patient reinvigorates a sense of purpose and meaning in life without the deceased.
CGT focuses on both processes. To address the former, it includes clinician-guided exercises in which the patient revisits the time of the death and planned activities in which the patient reengages with people, places, or thoughts that remind him or her of the deceased. CGT aims to allow the patient to gain an increased tolerance of the distressing thoughts and emotions associated with the loss so that these thoughts can be processed and the finality of the death and its circumstances can be accepted.
The restoration process is addressed by having patients generate and discuss personal goals and aspirations for the near and distant future, as well as scheduling pleasurable and rewarding events. This is accomplished by having patients imagine what they would want for themselves if their grief was less intense and planning concrete steps to take toward these goals. The restoration-oriented process is addressed concurrent with the loss-oriented process to encourage the oscillation between processes thought to be characteristic of a natural healing process following the loss of a loved one.
Other psychotherapy approaches (eg, support groups) may have a role for some individuals, and future research may suggest alternative approaches to CGT. To date, CGT is the most targeted evidence-based psychotherapy with randomized controlled data supporting its use in CG.
Pharmacotherapy for CG. Early research suggested that antidepressants—in particular tricyclics—may effectively reduce depressive symptoms in bereavement-related depression; their effect on CG symptoms, however, may not be as strong.14 Research on pharmacologic treatment that targets CG symptoms is developing. Because of the overlap between CG, PTSD, and MDD, researchers have hypothesized that antidepressants may be effective. Two open-label studies reported that the selective serotonin reuptake inhibitor (SSRI) escitalopram may be effective for CG.15,16 Although a post-hoc comparison of paroxetine and nortriptyline17 showed significant reduction in CG and depressive symptoms with both agents, effects could not be separated from concomitant psychotherapy. Furthermore, an examination of naturalistic data on combining antidepressants with CGT suggested that antidepressants may improve outcomes for individuals receiving CGT.18 A multicenter, randomized controlled trial funded by the National Institute of Mental Health is examining the potential efficacy of citalopram, an SSRI, alone or in combination with CGT.19
The efficacy of benzodiazepines, which commonly are prescribed for bereaved individuals, has not been assessed in CG. However, recent research suggests they may not be useful for medically managing recent grief20 and that their use in the aftermath of a loss may lead to long-term dependence in geriatric patients.21
Related Resources
- Center for Anxiety and Traumatic Stress Disorders. Massachusetts General Hospital. www.bostongrief.com.
- Zisook S, Shear K. Grief and bereavement: what psychiatrists need to know. World Psychiatry. 2009;8(2):67-74.
- Bonanno G. The other side of sadness: what the new science of bereavement tells us about loss. New York, NY: Basic Books; 2009.
Drug Brand Names
- Citalopram • Celexa
- Nortriptyline • Aventyl, Pamelor
- Escitalopram • Lexapro
- Paroxetine • Paxil
Disclosures
Dr. Simon receives grant or research support from the American Cancer Society, the American Foundation for Suicide Prevention, the Department of Defense, Forest Laboratories, and the National Institute of Mental Health.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
1. Kochanek KD, Xu J, Murphy SL, et al. U.S. Department of Health and Human Services. Deaths: preliminary data for 2009. http://www.cdc.gov/nchs/data/nvsr/nvsr59/nvsr59_04.pdf. Published March 16 2011. Accessed June 19, 2012.
2. American Psychiatric Association. Trauma- and stressor-related disorders. http://www.dsm5.org/ProposedRevision/Pages/TraumaandStressorRelatedDisorders.aspx. Accessed June 19 2012.
3. Bonanno GA, Kaltman S. Toward an integrative perspective on bereavement. Psychol Bull. 1999;125(6):760-776.
4. Bonanno GA, Keltner D. Facial expressions of emotion and the course of conjugal bereavement. J Abnorm Psychol. 1997;106(1):126-137.
5. Bonanno GA, Wortman CB, Lehman DR, et al. Resilience to loss and chronic grief: a prospective study from preloss to 18-months postloss. J Pers Soc Psychol. 2002;83(5):1150-1164.
6. Shear MK, Simon N, Wall M, et al. Complicated grief and related bereavement issues for DSM-5. Depress Anxiety. 2011;28(2):103-117.
7. Boelen PA, Prigerson HG. The influence of symptoms of prolonged grief disorder depression, and anxiety on quality of life among bereaved adults: a prospective study. Eur Arch Psychiatry Clin Neurosci. 2007;257(8):444-452.
8. Prigerson HG, Maciejewski PK, Reynolds CF, 3rd, et al. Inventory of Complicated Grief: a scale to measure maladaptive symptoms of loss. Psychiatry Res. 1995;59 (1-2):65-79.
9. Simon NM, Shear KM, Thompson EH, et al. The prevalence and correlates of psychiatric comorbidity in individuals with complicated grief. Compr Psychiatry. 2007;48(5):395-399.
10. Brewin CR, Holmes EA. Psychological theories of posttraumatic stress disorder. Clin Psychol Rev. 2003;23(3):339-376.
11. Neimeyer RA, Currier JM. Grief therapy: evidence of efficacy and emerging directions. Curr Dir Psychol Sci. 2009;18(6):352-356.
12. Shear K, Frank E, Houck PR, et al. Treatment of complicated grief: a randomized controlled trial. JAMA. 2005;293(21):2601-2608.
13. Stroebe M, Schut H. The dual process model of coping with bereavement: rationale and description. Death Stud. 1999;23(3):197-224.
14. Reynolds CF, 3rd, Miller MD, Pasternak RE, et al. Treatment of bereavement-related major depressive episodes in later life: a controlled study of acute and continuation treatment with nortriptyline and interpersonal psychotherapy. Am J Psychiatry. 1999;156(2):202-208.
15. Simon NM, Thompson EH, Pollack MH, et al. Complicated grief: a case series using escitalopram. Am J Psychiatry. 2007;164(11):1760-1761.
16. Hensley PL, Slonimski CK, Uhlenhuth EH, et al. Escitalopram: an open-label study of bereavement-related depression and grief. J Affect Disord. 2009;113(1-2):142-149.
17. Zygmont M, Prigerson HG, Houck PR, et al. A post hoc comparison of paroxetine and nortriptyline for symptoms of traumatic grief. J Clin Psychiatry. 1998;59(5):241-245.
18. Simon NM, Shear MK, Fagiolini A, et al. Impact of concurrent naturalistic pharmacotherapy on psychotherapy of complicated grief. Psychiatry Res. 2008;159(1-2):31-36.
19. U.S. National Institutes of Health. A study of medication with or without psychotherapy for complicated grief (HEAL). http://clinicaltrials.gov/ct2/show/NCT01179568. Published June 24, 2012. Accessed June 25, 2012.
20. Warner J, Metcalfe C, King M. Evaluating the use of benzodiazepines following recent bereavement. Br J Psychiatry. 2001;178(1):36-41.
21. Cook JM, Biyanova T, Marshall R. Medicating grief with benzodiazepines: physician and patient perspectives. Arch Intern Med. 2007;167(18):2006-2007.
When to treat subthreshold hypomanic episodes
According to DSM-IV-TR, the minimal duration of a hypomanic episode is 4 days.1 Should we treat patients for hypomanic symptoms that last <4 days? Could antidepressants’ high failure rate2 be because many depressed patients have untreated “subthreshold hypomanic episodes”? Aripiprazole, quetiapine, and lithium all have been shown to alleviate depression when added to an antidepressant.3-5 Is it possible that these medications are treating subthreshold hypomanic episodes rather than depression?
The literature does not answer these questions. To further confuse matters, a subthreshold hypomanic episode may not be a discrete episode. In such episodes, hypomanic symptoms may overlap at some point and the duration of each symptom may vary.
When I administer the Mood Disorder Questionnaire,6,7 I ask patients about 13 hypomanic symptoms. Patient responses to questions about 7 of these symptoms—increased energy, irritability, talking, and activity, feeling “hyper,” racing thoughts, and decreased need for sleep—can help demonstrate the variability of symptom duration. For example, a patient may complain of increased energy and irritability for 3 days, increased activity and feeling “hyper” for 2 days, increased talking and a decreased need to sleep for 1 day, and racing thoughts every day.
Alternative criteria
Considering this variation, I often use the following criteria when considering whether to treat subthreshold hypomanic symptoms:
- ≥4 symptoms must last ≥2 consecutive days
- ≥3 symptoms must overlap at some point, and
- ≥2 of the symptoms must be increased energy, increased activity, or racing thoughts.
However, some patients have hypomanic symptoms that do not meet these relaxed criteria but require treatment.8 I also need to know when these episodes started, how frequently they occur, and how much of a problem they cause in the patient’s life. I often treat subthreshold hypomanic episodes with an antipsychotic or a mood stabilizer. As with all patients I see, I consider the patient’s reliability, substance abuse history, and mental status during the interview.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Pigott HE, Leventhal AM, Alter GS, et al. Efficacy and effectiveness of antidepressants: current status of research. Psychother Psychosom. 2010;79(5):267-279.
3. Nelson JC, Pikalov A, Berman RM. Augmentation treatment in major depressive disorder: focus on aripiprazole. Neuropsychiatr Dis Treat. 2008;4(5):937-948.
4. Daly EJ, Trivedi MH. A review of quetiapine in combination with antidepressant therapy in patients with depression. Neuropsychiatr Dis Treat. 2007;3(6):855-867.
5. Price LH, Carpenter LL, Tyrka AR. Lithium augmentation for refractory depression: a critical reappraisal. Prim Psychiatry. 2008;15(11):35-42.
6. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
7. The Mood Disorder Questionnaire. http://www.drpaddison.com/mood.pdf. Accessed June 20 2012.
8. Angst J, Azorin JM, Bowden CL, et al. Prevalence and characteristics of undiagnosed bipolar disorders in patients with a major depressive episode: the BRIDGE study. Arch Gen Psychiatry. 2011;68(8):791-798.
According to DSM-IV-TR, the minimal duration of a hypomanic episode is 4 days.1 Should we treat patients for hypomanic symptoms that last <4 days? Could antidepressants’ high failure rate2 be because many depressed patients have untreated “subthreshold hypomanic episodes”? Aripiprazole, quetiapine, and lithium all have been shown to alleviate depression when added to an antidepressant.3-5 Is it possible that these medications are treating subthreshold hypomanic episodes rather than depression?
The literature does not answer these questions. To further confuse matters, a subthreshold hypomanic episode may not be a discrete episode. In such episodes, hypomanic symptoms may overlap at some point and the duration of each symptom may vary.
When I administer the Mood Disorder Questionnaire,6,7 I ask patients about 13 hypomanic symptoms. Patient responses to questions about 7 of these symptoms—increased energy, irritability, talking, and activity, feeling “hyper,” racing thoughts, and decreased need for sleep—can help demonstrate the variability of symptom duration. For example, a patient may complain of increased energy and irritability for 3 days, increased activity and feeling “hyper” for 2 days, increased talking and a decreased need to sleep for 1 day, and racing thoughts every day.
Alternative criteria
Considering this variation, I often use the following criteria when considering whether to treat subthreshold hypomanic symptoms:
- ≥4 symptoms must last ≥2 consecutive days
- ≥3 symptoms must overlap at some point, and
- ≥2 of the symptoms must be increased energy, increased activity, or racing thoughts.
However, some patients have hypomanic symptoms that do not meet these relaxed criteria but require treatment.8 I also need to know when these episodes started, how frequently they occur, and how much of a problem they cause in the patient’s life. I often treat subthreshold hypomanic episodes with an antipsychotic or a mood stabilizer. As with all patients I see, I consider the patient’s reliability, substance abuse history, and mental status during the interview.
According to DSM-IV-TR, the minimal duration of a hypomanic episode is 4 days.1 Should we treat patients for hypomanic symptoms that last <4 days? Could antidepressants’ high failure rate2 be because many depressed patients have untreated “subthreshold hypomanic episodes”? Aripiprazole, quetiapine, and lithium all have been shown to alleviate depression when added to an antidepressant.3-5 Is it possible that these medications are treating subthreshold hypomanic episodes rather than depression?
The literature does not answer these questions. To further confuse matters, a subthreshold hypomanic episode may not be a discrete episode. In such episodes, hypomanic symptoms may overlap at some point and the duration of each symptom may vary.
When I administer the Mood Disorder Questionnaire,6,7 I ask patients about 13 hypomanic symptoms. Patient responses to questions about 7 of these symptoms—increased energy, irritability, talking, and activity, feeling “hyper,” racing thoughts, and decreased need for sleep—can help demonstrate the variability of symptom duration. For example, a patient may complain of increased energy and irritability for 3 days, increased activity and feeling “hyper” for 2 days, increased talking and a decreased need to sleep for 1 day, and racing thoughts every day.
Alternative criteria
Considering this variation, I often use the following criteria when considering whether to treat subthreshold hypomanic symptoms:
- ≥4 symptoms must last ≥2 consecutive days
- ≥3 symptoms must overlap at some point, and
- ≥2 of the symptoms must be increased energy, increased activity, or racing thoughts.
However, some patients have hypomanic symptoms that do not meet these relaxed criteria but require treatment.8 I also need to know when these episodes started, how frequently they occur, and how much of a problem they cause in the patient’s life. I often treat subthreshold hypomanic episodes with an antipsychotic or a mood stabilizer. As with all patients I see, I consider the patient’s reliability, substance abuse history, and mental status during the interview.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Pigott HE, Leventhal AM, Alter GS, et al. Efficacy and effectiveness of antidepressants: current status of research. Psychother Psychosom. 2010;79(5):267-279.
3. Nelson JC, Pikalov A, Berman RM. Augmentation treatment in major depressive disorder: focus on aripiprazole. Neuropsychiatr Dis Treat. 2008;4(5):937-948.
4. Daly EJ, Trivedi MH. A review of quetiapine in combination with antidepressant therapy in patients with depression. Neuropsychiatr Dis Treat. 2007;3(6):855-867.
5. Price LH, Carpenter LL, Tyrka AR. Lithium augmentation for refractory depression: a critical reappraisal. Prim Psychiatry. 2008;15(11):35-42.
6. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
7. The Mood Disorder Questionnaire. http://www.drpaddison.com/mood.pdf. Accessed June 20 2012.
8. Angst J, Azorin JM, Bowden CL, et al. Prevalence and characteristics of undiagnosed bipolar disorders in patients with a major depressive episode: the BRIDGE study. Arch Gen Psychiatry. 2011;68(8):791-798.
1. Diagnostic and statistical manual of mental disorders, 4th ed, text rev. Washington DC: American Psychiatric Association; 2000.
2. Pigott HE, Leventhal AM, Alter GS, et al. Efficacy and effectiveness of antidepressants: current status of research. Psychother Psychosom. 2010;79(5):267-279.
3. Nelson JC, Pikalov A, Berman RM. Augmentation treatment in major depressive disorder: focus on aripiprazole. Neuropsychiatr Dis Treat. 2008;4(5):937-948.
4. Daly EJ, Trivedi MH. A review of quetiapine in combination with antidepressant therapy in patients with depression. Neuropsychiatr Dis Treat. 2007;3(6):855-867.
5. Price LH, Carpenter LL, Tyrka AR. Lithium augmentation for refractory depression: a critical reappraisal. Prim Psychiatry. 2008;15(11):35-42.
6. Hirschfeld RM, Williams JB, Spitzer RL, et al. Development and validation of a screening instrument for bipolar spectrum disorder: the Mood Disorder Questionnaire. Am J Psychiatry. 2000;157(11):1873-1875.
7. The Mood Disorder Questionnaire. http://www.drpaddison.com/mood.pdf. Accessed June 20 2012.
8. Angst J, Azorin JM, Bowden CL, et al. Prevalence and characteristics of undiagnosed bipolar disorders in patients with a major depressive episode: the BRIDGE study. Arch Gen Psychiatry. 2011;68(8):791-798.
Distinguishing between adult ADHD and mild cognitive impairment
There is considerable overlap between symptoms of adult attention-deficit/hyperactivity disorder (ADHD) and mild cognitive impairment (MCI), including problems with sustained attention or concentration, anterograde memory, and executive functioning. Differentiating these clinical syndromes based on symptomatic presentation alone can be difficult, but considering the following factors can help you make a more informed diagnosis:
Neurodevelopmental disorder history. DSM-IV-TR stipulates onset for some ADHD symptoms by age 7, although a DSM-5 Work Group is considering symptom onset as late as age 12.1 Initial onset or a dramatic worsening of longstanding ADHD symptoms in middle-age or older adults is atypical for this neurodevelopmental disorder.
Detailed self-diagnosed symptoms. Patients with ADHD usually can give a satisfactory history of their symptoms. Patients with MCI often are less able to provide a useful history because they have prominent difficulties with anterograde memory, which may be associated with emerging anosognosia.
Educational learning difficulties. Patients with ADHD frequently have comorbid learning difficulties and substance abuse disorders, which are uncommon in MCI.
Rating scales. When in doubt, use rating scales to assess for ADHD.2 Ask your patient to complete the rating scale based on how he or she remembers behaving in elementary through middle school, most of their adult life after age 20, and since symptom onset. Obtain collateral ratings from a reliable informant based on his or her knowledge of the patient’s long-term behavioral functioning.
Worsening symptoms. The typical ADHD patient will have a “positive” screen for symptoms, but will report fewer and less severe symptoms from childhood or adolescence through young adulthood and into middle and older age. Suspect MCI when your patient or an informant reports a clear worsening of symptoms in recent months or years despite a lack of evidence of a significant intervening psychiatric disorder.
Psychopharmacotherapy. Patients with MCI usually do not benefit from medications for ADHD. Patients with ADHD often report improvement in at least some of their symptoms with psychopharmacologic treatment.
When your patient’s history, rating scale assessment, and medication trials do not allow you to make a confident differential diagnosis, consider referring him or her for psychological or neuropsychological testing.
There can be overlap in psychometric test findings of middle-age and older adults with a history of ADHD and those who may have MCI. Still, MCI patients’ cognitive difficulties usually are more concerning and dramatic, including problems with spontaneous recall as well as “recognition memory.”
When findings from psychometric testing are equivocal because of possible co-occurrence, retesting in 12 to 18 months usually will help you make a reliable differential diagnosis. Specifically, progression of cognitive dysfunction—including evidence of worsening anterograde memory—is common in MCI but not in ADHD.
Current symptoms of major depressive disorder may further “muddy the waters.” However, parameters such as response to adequate medication trials, progression of cognitive dysfunction, and worsening of test-based cognitive or neuropsychological deficits over time can be useful in reaching a satisfactory differential diagnosis.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
There is considerable overlap between symptoms of adult attention-deficit/hyperactivity disorder (ADHD) and mild cognitive impairment (MCI), including problems with sustained attention or concentration, anterograde memory, and executive functioning. Differentiating these clinical syndromes based on symptomatic presentation alone can be difficult, but considering the following factors can help you make a more informed diagnosis:
Neurodevelopmental disorder history. DSM-IV-TR stipulates onset for some ADHD symptoms by age 7, although a DSM-5 Work Group is considering symptom onset as late as age 12.1 Initial onset or a dramatic worsening of longstanding ADHD symptoms in middle-age or older adults is atypical for this neurodevelopmental disorder.
Detailed self-diagnosed symptoms. Patients with ADHD usually can give a satisfactory history of their symptoms. Patients with MCI often are less able to provide a useful history because they have prominent difficulties with anterograde memory, which may be associated with emerging anosognosia.
Educational learning difficulties. Patients with ADHD frequently have comorbid learning difficulties and substance abuse disorders, which are uncommon in MCI.
Rating scales. When in doubt, use rating scales to assess for ADHD.2 Ask your patient to complete the rating scale based on how he or she remembers behaving in elementary through middle school, most of their adult life after age 20, and since symptom onset. Obtain collateral ratings from a reliable informant based on his or her knowledge of the patient’s long-term behavioral functioning.
Worsening symptoms. The typical ADHD patient will have a “positive” screen for symptoms, but will report fewer and less severe symptoms from childhood or adolescence through young adulthood and into middle and older age. Suspect MCI when your patient or an informant reports a clear worsening of symptoms in recent months or years despite a lack of evidence of a significant intervening psychiatric disorder.
Psychopharmacotherapy. Patients with MCI usually do not benefit from medications for ADHD. Patients with ADHD often report improvement in at least some of their symptoms with psychopharmacologic treatment.
When your patient’s history, rating scale assessment, and medication trials do not allow you to make a confident differential diagnosis, consider referring him or her for psychological or neuropsychological testing.
There can be overlap in psychometric test findings of middle-age and older adults with a history of ADHD and those who may have MCI. Still, MCI patients’ cognitive difficulties usually are more concerning and dramatic, including problems with spontaneous recall as well as “recognition memory.”
When findings from psychometric testing are equivocal because of possible co-occurrence, retesting in 12 to 18 months usually will help you make a reliable differential diagnosis. Specifically, progression of cognitive dysfunction—including evidence of worsening anterograde memory—is common in MCI but not in ADHD.
Current symptoms of major depressive disorder may further “muddy the waters.” However, parameters such as response to adequate medication trials, progression of cognitive dysfunction, and worsening of test-based cognitive or neuropsychological deficits over time can be useful in reaching a satisfactory differential diagnosis.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
There is considerable overlap between symptoms of adult attention-deficit/hyperactivity disorder (ADHD) and mild cognitive impairment (MCI), including problems with sustained attention or concentration, anterograde memory, and executive functioning. Differentiating these clinical syndromes based on symptomatic presentation alone can be difficult, but considering the following factors can help you make a more informed diagnosis:
Neurodevelopmental disorder history. DSM-IV-TR stipulates onset for some ADHD symptoms by age 7, although a DSM-5 Work Group is considering symptom onset as late as age 12.1 Initial onset or a dramatic worsening of longstanding ADHD symptoms in middle-age or older adults is atypical for this neurodevelopmental disorder.
Detailed self-diagnosed symptoms. Patients with ADHD usually can give a satisfactory history of their symptoms. Patients with MCI often are less able to provide a useful history because they have prominent difficulties with anterograde memory, which may be associated with emerging anosognosia.
Educational learning difficulties. Patients with ADHD frequently have comorbid learning difficulties and substance abuse disorders, which are uncommon in MCI.
Rating scales. When in doubt, use rating scales to assess for ADHD.2 Ask your patient to complete the rating scale based on how he or she remembers behaving in elementary through middle school, most of their adult life after age 20, and since symptom onset. Obtain collateral ratings from a reliable informant based on his or her knowledge of the patient’s long-term behavioral functioning.
Worsening symptoms. The typical ADHD patient will have a “positive” screen for symptoms, but will report fewer and less severe symptoms from childhood or adolescence through young adulthood and into middle and older age. Suspect MCI when your patient or an informant reports a clear worsening of symptoms in recent months or years despite a lack of evidence of a significant intervening psychiatric disorder.
Psychopharmacotherapy. Patients with MCI usually do not benefit from medications for ADHD. Patients with ADHD often report improvement in at least some of their symptoms with psychopharmacologic treatment.
When your patient’s history, rating scale assessment, and medication trials do not allow you to make a confident differential diagnosis, consider referring him or her for psychological or neuropsychological testing.
There can be overlap in psychometric test findings of middle-age and older adults with a history of ADHD and those who may have MCI. Still, MCI patients’ cognitive difficulties usually are more concerning and dramatic, including problems with spontaneous recall as well as “recognition memory.”
When findings from psychometric testing are equivocal because of possible co-occurrence, retesting in 12 to 18 months usually will help you make a reliable differential diagnosis. Specifically, progression of cognitive dysfunction—including evidence of worsening anterograde memory—is common in MCI but not in ADHD.
Current symptoms of major depressive disorder may further “muddy the waters.” However, parameters such as response to adequate medication trials, progression of cognitive dysfunction, and worsening of test-based cognitive or neuropsychological deficits over time can be useful in reaching a satisfactory differential diagnosis.
Disclosure
Dr. Pollak reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Prescription opioid use disorder: A complex clinical challenge
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
Discuss this article at www.facebook.com/CurrentPsychiatry
You’ve been treating Mr. H, a 54-year-old factory worker and tobacco user, for depression that developed after a work-related back injury and subsequent disability. His depression has had a fair response to an antidepressant. He also has been maintained on chronic opioids (morphine and oxycodone/acetaminophen) for 18 months by his primary care physician (PCP). At the end of your appointment, he asks you for a refill of the opioids because he “ran out” early because of increased night pain and resultant insomnia and “stress.” He clarifies he has asked for early refills before from his PCP, but lately he has been denied. Because you “seem to listen to me more,” he asks for your help. How should you manage Mr. H?
Opioids are among the most commonly misused prescription drugs in the United States.1 In 2008, poisoning was the leading cause of death from injury in the United States; roughly 90% of poisonings resulted from drug exposure, and >40% of these drug poisonings were from prescription opioids.2 The Centers for Disease Control and Prevention estimates that the number of emergency department (ED) visits for nonmedical use of opioids increased 111% between 2004 and 2008, from 144,600 to 305,900 visits.3 The highest number of visits were for use of oxycodone, hydrocodone, and methadone.3
Increased prescribing of opioids and overdose deaths attributable to prescribed opioids have raised concern among physicians about how to effectively treat pain as well as prevent, recognize, and manage aberrant medication-taking behaviors (AMTBs). Psychiatrists are well-positioned to screen and manage their own patients for prescription opioid use disorder (POUD) or collaborate with opioid prescribers to accomplish the same.
Clarifying terminology
Terminology used to describe POUD and related conditions often is poorly defined or loosely applied. Because emotions often enter discussions between patients and physicians about problems related to opioid therapy, nonstigmatizing and more objective terminology is needed, and clinicians are working toward standardizing this. Relevant terms are defined in Table 1.4
The DSM-5 Substance Use Disorders Work Group has proposed using the term opioid use disorder (OUD) to replace the term opioid dependence.5 The hope is that removing the word “dependence” from the diagnostic term will reduce confusion between “dependence” due to expected physical dependence (tolerance, withdrawal) on medically prescribed opioids vs true addiction (currently defined as “opioid dependence” in DSM-IV-TR). This Work Group also has proposed combining opioid abuse and opioid dependence criteria into a single diagnosis of OUD, and adding “craving” to the criteria. For the complete proposed criteria, see www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460.These changes are still under review. In this article, we use the term POUD.
Table 1
Terminology related to prescription opioid use disorder
| Term | Definition |
|---|---|
| Chronic paina | Pain that extends beyond the expected period for healing (6 months), initiated by tissue damage, but perpetrated by the interaction of physiologic, affective, and environmental factors |
| Chronic nonmalignant paina | Chronic pain associated with diverse diagnoses and syndromes that are not terminal but affect the patient’s function |
| Appropriate usea | Taking a prescription as prescribed, and only for the condition indicated |
| Misusea | Taking a prescription for a reason or at a dose or frequency other than for which it was prescribed; this may or may not reflect POUD |
| Drug-seeking behaviors | Patient behaviors directed toward obtaining controlled substances, driven not by amelioration of the condition for which the medication was indicated but rather by other maladaptive gains; this may or may not reflect POUD |
| Chemical coping | Taking a controlled substance medication to relieve psychological problems (eg, to relieve low mood, anxiety, insomnia) and for reasons other than the purpose for which it was prescribed; this may or may not reflect POUD |
| Aberrant medication-taking behaviorsa | Taking a controlled substance medication in a manner that is not prescribed; causes for this may include:
|
| Pseudoaddiction | An iatrogenic syndrome of “addiction-like” behaviors in which the patient seeks opioids to relieve pain—such as seeking different doctors, self-adjusting the opioid dose, early refills of opioids, etc.—rather than to achieve pleasure or other nonpain-related effect. At times mistaken for true addiction, these behaviors tend to resolve and function improves once analgesia is better addressed |
| a These terms and definitions are adapted from reference 4. The remaining terms and definitions were developed by the authors POUD: prescription opioid use disorder | |
POUD and chronic pain
The incidence of POUD during opioid therapy for pain is unknown.6 Some researchers have suggested it may be as low as 0.2%,7 while others estimate that rates of POUD in patients with chronic pain may be similar to those in the general population: 3% to 16%.8 When applying the proposed DSM-5 criteria to patients receiving long-term opioid therapy for noncancer pain, the lifetime prevalence of POUD may be as high as 35%.9
Prescribers may be contributing to POUD. Roughly 76% of opioids used for nonmedical purposes were prescribed to someone else, 20% were prescribed to the user, and 4% came from other sources.1 Strategies to reduce POUD risk may be underused. In a retrospective cohort study of 1,612 patient electronic medical records from 8 primary care clinics that managed patients with long-term opioids for chronic noncancer pain (average prescribing duration of 2 years duration, ≥3 monthly prescriptions in 6 months), researchers evaluated how often prescribers used 3 risk reduction practices:
- urine drug tests
- regular office visits (≥1 every 6 months and within 30 days of changing opioid treatment)
- restricted early refills (≤1 opioid refill more than a week early).10
Risk factors for opioid misuse included age 1 early refill. Researchers found that even for high-risk patients, these strategies were used infrequently. Less than one-quarter of patients with ≥3 risk factors ever had a drug test, and those at increased risk were more likely to receive >1 early refill but no more likely to have more frequent visits. Issues such as patient entitlement, lack of physician education, and time constraints may explain why these strategies are not used more often.11
No one procedure or set of variables is sufficient to identify chronic pain patients who may be at risk for POUD. However, a history of drug or alcohol use disorders may be a significant risk factor.12,13
Few tools have been developed to help identify those at risk of AMTBs or POUD, and all have limitations.4,14 Recommended self-report measures include the Current Opioid Misuse Measure and the Opioid Risk Tool.15 A review of studies in which these kinds of tools were developed revealed limited evidence for their use; most studies had methodological shortcomings, did not use standardized AMTB criteria, and provided little assessment of whether these tools changed clinician behaviors or improved patient outcomes.16
Evaluating AMTBs
Although diagnosing POUD in pain patients receiving chronic opioids can be challenging, assessing for AMTBs typically is helpful. Once AMTBs are identified, they can be examined to determine what drives their expression (Table 14 and Table 217). However, often it is easier to identify AMTBs than to interpret their origins; as much as 30% to 50% of patients who complain of chronic pain may have primary substance dependence to sedatives, opioids, or both.11
Table 2
Aberrant medication-taking behaviors and POUD risk
| Behaviors more suggestive of POUD |
|---|
| Deterioration in function (work, social) |
| Illegal activities (selling medication, forging prescriptions, buying from non-medical sources) |
| Altering the route of administration (snorting, injecting) |
| Multiple episodes of ‘lost’ or ‘stolen’ prescriptions |
| Resistance to change therapy despite negative outcomes |
| Refusal to comply with toxicology testing |
| Concurrent, active abuse of alcohol, illegal drugs |
| Use of multiple physicians or pharmacies to obtain the prescription |
| Behaviors less suggestive of POUD |
| Complaints for more medication |
| Medication hoarding |
| Requesting specific pain medications |
| Openly acquiring similar medications from other providers |
| Occasional unsanctioned dose escalation |
| Nonadherence to other recommendations for pain therapy |
| POUD: prescription opioid use disorder Source: Reference 17 |
Although AMTBs are common among chronic nonmalignant pain patients,18,19 how often AMTBs reflect underlying POUD is uncertain.7 It is critical to interpret AMTBs with a balance of caution and care: “react therapeutically, not punitively.”20 Categorizing a patient’s AMTB as more or less likely to support a POUD diagnosis can be helpful, but is not conclusive (Table 2).17 Clinical correlation often is required. No single AMTB alone is indicative of POUD. When evaluating AMTBs, the treating provider should use a nonjudgmental stance, and consider obtaining collateral data from people who can provide differing perspectives of the patient’s behaviors, such as other clinicians, significant others, family, etc. (a release of information from the patient may be required). Another source of collateral data is prescription monitoring databases. These databases typically are state-based and provide electronic access to prescription information, allowing you to search for patterns—ie, use of multiple prescribers or pharmacies, undisclosed prescriptions, etc. Interest in establishing a single, federal database has been increasing, but striking a balance between carefully monitoring for AMTBs and protecting privacy remains unresolved.
DSM-IV-TR diagnostic criteria for opioid dependence21 can be challenging to interpret in patients who are prescribed opioids for pain (Table 3
).6 To clarify interpretation, the Liaison Committee on Pain and Addiction of the American Society of Addiction Medicine (ASAM) has provided an outline of possible indicators of addiction in pain patients (Table 4).6 This was a consensus statement from the American Pain Society, the American Academy of Pain Medicine, and ASAM.
Assessment is primarily clinical and requires an awareness of appropriate terminology, an index of clinical suspicion, and expertise teasing apart pain, addiction, and pseudoaddiction. In our experience, it is helpful to ask a chronic pain patient whom you suspect might have POUD, “Have you ever used your prescribed opioids for reasons other than improving function or reducing pain, such as for getting a ‘high,’ managing stress, escaping from problems, etc.?” An affirmative response suggests an underlying problem with use of prescribed opioids, indicating a need for more careful questioning to determine if AMTBs or POUD coexist with chronic pain.
Drug testing can help determine if a patient is taking opioids that are not prescribed—as well as illicit drugs or alcohol—and confirm the presence of those that are prescribed. Toxicology screening should include opioids typically screened for (eg, morphine, codeine, heroin) and those for which additional tests may be required (eg, semi-synthetics such as oxycodone and hydrocodone and synthetics such as fentanyl).
Table 3
Identifying addiction in pain patients: Limitations of DSM-IV-TR
| DSM-IV-TR substance dependence criteria | Challenges in using criterion to diagnose prescription opioid use disorder |
|---|---|
| Tolerance | Expected with prolonged opioid compliance |
| Physical dependence, withdrawal | Expected with prolonged opioid compliance |
| Use of larger amounts or longer than initially intended | Emergence of pain may demand increased dose or prolonged use |
| Multiple failed attempts to cut down or control | Emergence of pain may deter dose reduction or cessation |
| Time spent finding, using, or recovering | Difficulty finding adequate pain treatment may increase time spent pursuing analgesics. However, time spent recovering from overuse may suggest addiction |
| Given up or reduced important activities | Valid criteria—engaging in activities is expected to increase, not decline, with effective pain treatment |
| Continued use despite knowledge of negative consequences | Valid criteria—no harm is anticipated from analgesic opioid use for pain (see Table 4) |
| Source: Adapted from reference 6 | |
Table 4
Possible indicators of addiction in pain patients
| ASAM-APS-AAPM behavioral criteria | Examples of specific behaviors in opioid therapy for pain |
|---|---|
| Impaired control over opioid use | Patient requests early refills, frequently reports loss or theft of medication. Withdrawal noted at follow-up appointments despite having an adequate quantity of medication prescribed |
| Continued use despite harm from opioids | Patient exhibits declining function, opioid intoxication, persistent oversedation from opioids |
| Preoccupation with opioids | Patient ignores non-opioid interventions for pain, makes recurrent requests for opioid dose escalation (or complains of increasing pain) despite absence of disease progression or despite opioid dose increase by provider |
| AAPM: American Academy of Pain Medicine; APS: American Pain Society; ASAM: American Society of Addiction Medicine Source: Adapted from reference 6 | |
Helping POUD patients
Goals of treatment include establishing a therapeutic alliance, educating patients about POUD, reducing relapse risk, and optimizing overall health (including pain and physical function). The ASAM Patient Placement Criteria22 provide guidance regarding level-of-care decisions. Treatment ideally includes a combination of education about POUD and its relationship to chronic pain, pharmacotherapy, psychotherapy—such as motivational enhancement therapy, 12-step facilitation therapy, cognitive-behavioral therapy, and relapse prevention—and referral to self-help groups such as Narcotics Anonymous or Pills Anonymous. Importantly, if pain is genuine, it requires treatment.
Pharmacotherapy. Methadone is recommended as the standard of care for OUD by the National Institutes of Health. Methadone is a full opioid agonist that decreases illicit opioid use, mortality, and related problems and requires highly structured treatment approaches under federal and state regulation. POUD patients may have higher rates of methadone maintenance treatment retention than heroin-dependent patients.23 Published trials of buprenorphine for OUD have shown good treatment retention and reduction in illicit drug use and adverse events.24 Buprenorphine also decreases mortality among OUD patients.
The first large-scale, randomized clinical trial of buprenorphine specifically for POUD included 653 treatment-seeking outpatients.25 This study was designed to approximate clinical practice and included buprenorphine/naloxone, recommended abstinence, and self-help; one-half of participants received intensive addiction counseling. POUD patients were most likely to reduce prescription opioid misuse during buprenorphine/naloxone treatment. If tapered off buprenorphine/naloxone, even after 12 weeks of treatment, the likelihood of an unsuccessful outcome was high. Moreover, opioid dependence counseling did not seem to afford any difference in outcomes. However, despite clinical effectiveness, over the last decade only 19% of patients admitted primarily for OUD treatment (other than heroin) were planned to be offered buprenorphine or methadone.26
A Cochrane review of oral naltrexone for OUD found that the drug was no better than placebo but concluded that available evidence does not allow an adequate evaluation.27 Opioid antagonists may be of value to patients who do not want to take agonists or partial agonists. Extended-release naltrexone also is available to treat OUD.
See the Box below that details steps the FDA and others have taken to prevent POUD and Table 5 for precautions to incorporate when prescribing opioids long-term.
The FDA has moved toward a risk evaluation and mitigation strategy (REMS) for opioids prescribed for pain that requires clinicians to receive training and certification in prescribing opioids for pain as well as identifying and reducing the risk for prescription opioid use disorder (POUD).a In 2011, the Obama administration developed an action plan to better address prescription drug abuse that required several federal agencies to develop programs and policies to address this growing problem; this plan was updated for 2012 (the complete National Drug Control Strategy 2012 is available at www.whitehouse.gov/sites/default/files/ondcp/2012_ndcs.pdf). The American Society of Addiction Medicine has issued a public policy statement that supports the federal approach and outlines other means to reduce POUD.b
Some pain specialists recommend requiring patients to sign an Opioid Pain Management Agreement that includes an “exit strategy” before the first opioid prescription is written. These agreements incorporate elements of “universal precautions” to take when prescribing opioids long term.c,d Although not well-studied, prescribing agreements may help educate patients and providers on how to interact in the management of pain with opioids in a way that is objective and empathic, and may reduce POUD risk.
References
- U.S. Department of Health and Human Services. U.S. Food and Drug Administration. Opioid drugs and risk evaluation and mitigation strategies (REMS). http://www.fda.gov/drugs/drugsafety/informationbydrugclass/ucm163647.htm. Updated April 5, 2012. Accessed June 28, 2012.
- American Society of Addiction Medicine. Measures to counteract prescription drug diversion, misuse and addiction. http://www.asam.org/advocacy/find-a-policy-statement/view-policy-statement/public-policy-statements/2012/01/26/measures-to-counteract-prescription-drug-diversion-misuse-and-addiction. Published January 25, 2012. Accessed June 20, 2012.
- Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112.
- Gourlay DL, Heit HA. Universal precautions revisited: managing the inherited pain patient. Pain Med. 2009; 10(suppl 2):S115-S123.
Table 5
Universal precautions with chronic opioid management
| Goals of therapy: partial pain relief and improvement in physical, emotional, and/or social functioning |
| Requirement for a single prescribing provider or treatment team |
| Limitation on dose and number of prescribed medications |
| Prohibition of changing dosage without discussion with the provider first |
| Monitoring patient adherence; discuss the use of ‘pill counts’ |
| Prohibition of use with alcohol, other sedating medications, or illegal drugs without discussion with the provider |
| Agreement not to drive or operate heavy machinery until abatement of medication-related drowsiness |
| Responsibility to keep medication safe and secure |
| Prohibition of selling, lending, sharing, or giving medication to others |
| Limitations on refills—only by appointment, in person, and no extra refills for running out early |
| Compliance with all components of overall treatment plan (including consultations and referrals) |
| Biological testing to screen for drugs of abuse or alcohol as well as to confirm the presence of prescribed opioids |
| Adverse effects and safety issues, such as the risk of physical dependence and addiction behaviors |
| The option of sharing information with family members and other providers, as necessary, with the patient’s consent |
| Need for periodic reevaluation of treatment |
| Reasons for stopping opioid therapy |
| Consequences of nonadherence with the treatment agreement |
| Source: Gourlay DL, Heit HA, Almahrezi A. Universal precautions in pain medicine: a rational approach to the treatment of chronic pain. Pain Med. 2005;6(2):107-112. |
CASE CONTINUED: A closer evaluation
After expressing your appreciation for Mr. H’s kind words and empathy for his chronic pain, you redirect him to his PCP. You ask him to sign a release of information so you and his other clinicians can coordinate his care. When discussing Mr. H with his PCP, you learn the patient has made limited requests for early refills and dose escalation primarily in relation to inadequate pain control and function, has genuine pain pathology, and is greatly distressed over his inability to work. No other AMTBs are present, and a check of the state prescribing database reveals that Mr. H did receive a small quantity of opioids from an ED on 1 occasion.
You and Mr. H’s PCP agree this is “pseudo-addiction” but want to watch Mr. H more closely and look for ways to coordinate his care. The PCP agrees to implement a prescribing agreement, start drug testing (including for the prescribed opioids), and reassess maximizing Mr. H’s function and pain management while you address his combined pain, depression, insomnia, and tobacco use.
Related Resources
- Ries RK, Fiellin D, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009.
- Department of Veterans Affairs. Department of Defense. VA/DoD clinical practice guideline for management of opioid therapy for chronic pain. Appendix C: sample opioid pain care agreement. http://www.healthquality.va.gov/COT_312_Full-er.pdf. Published May 2010. Accessed June 21, 2012.
- Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic non-malignant pain. J Addiction Med. 2007;1(1):2-10.
- Weaver M, Heit HA, Savage S, et al. Clinical case discussion: chronic pain management. J Addiction Med. 2007;1(1):11-14.
Drug Brand Names
- Buprenorphine • Subutex
- Buprenorphine/naloxone • Suboxone
- Codeine • Tylenol with codeine, others
- Fentanyl • Duragesic, Actiq
- Hydrocodone • Lortab, Vicodin, others
- Methadone • Dolophine, Methadose
- Morphine • Roxanol
- Naltrexone extended-release • Vivitrol
- Oxycodone • OxyContin, Roxicodone
Disclosures
The authors report no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
Dr. Frankowski’s time toward this project was provided by the American Board of Addiction Medicine-accredited Cincinnati VA Addiction Medicine Research Fellowship, affiliated with the CeTREAD, Department of Psychiatry and Behavioral Neuroscience, University of Cincinnati, Cincinnati, OH.
The statements in this publication do not necessarily reflect the views or opinions of the Department of Veterans Affairs.
Acknowledgement
The authors thank Catherine Constance and Sandra Mason at the Cincinnati VA Medical Center for their administrative assistance.
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-
1. U.S. Department of Health and Human Services. Substance Abuse and Mental Health Services Administration. Office of Applied Studies. Results from the 2009 national survey on drug use and health: volume I. http://www.samhsa.gov/data/NSDUH/2k9NSDUH/2k9Results.htm. Accessed June 20, 2012.
2. Warner M, Chen LH, Makuc DM, et al. Drug poisoning deaths in the United States, 1980-2008. http://www.cdc.gov/nchs/data/databriefs/db81.htm. Published December 2011. Accessed June 20, 2012.
3. Centers for Disease Control and Prevention (CDC). Emergency department visits involving nonmedical use of selected prescription drugs - United States 2004-2008. MMWR Morb Mortal Wkly Rep. 2010;59(23):705-709.
4. Weaver M, Schnoll S. Addiction issues in prescribing opioids for chronic nonmalignant pain. J Addict Med. 2007;1(1):2-10.
5. American Psychiatric Association. R 19 opioid use disorder. http://www.dsm5.org/ProposedRevisions/Pages/proposedrevision.aspx?rid=460. Updated April 30 2012. Accessed June 20, 2012.
6. Savage SR, Horvath R. Opioid therapy of pain. In: Ries RK Fiellin DA, Miller SC, et al, eds. Principles of addiction medicine. 4th ed. Hagerstown, MD: Lippincott Williams & Wilkins; 2009:1329-1351.
7. Fishbain DA, Cole B, Lewis J, et al. What percentage of chronic nonmalignant pain patients exposed to chronic opioid analgesic therapy develop abuse/addiction and/or aberrant drug-related behaviors? A structured evidence-based review. Pain Med. 2008;9(4):444-459.
8. Gourlay DL, Heit HA. Pain and addiction: managing risk through comprehensive care. J Addict Dis. 2008;27(3):23-30.
9. Boscarino JA, Rukstalis MR, Hoffman SN, et al. Prevalence of prescription opioid-use disorder among chronic pain patients: comparison of the DSM-5 vs. DSM-4 diagnostic criteria. J Addict Dis. 2011;30(3):185-194.
10. Starrels JL, Becker WC, Weiner MG, et al. Low use of opioid risk reduction strategies in primary care even for high risk patients with chronic pain. J Gen Intern Med. 2011;26(9):958-964.
11. Miller NS. Failure of enforcement controlled substance laws in health policy for prescribing opiate medications: a painful assessment of morbidity and mortality. Am J Ther. 2006;13(6):527-533.
12. Turk DC, Swanson KS, Gatchel RJ. Predicting opioid misuse by chronic pain patients: a systematic review and literature synthesis. Clin J Pain. 2008;24(6):497-508.
13. Miller NS, Greenfeld A. Patient characteristics and risks factors for development of dependence on hydrocodone and oxycodone. Am J Ther. 2004;11(1):26-32.
14. Butler SF, Budman SH, Fernandez KC, et al. Cross-validation of a Screener to Predict Opioid Misuse in Chronic Pain Patients (SOAPP-R). J Addict Med. 2009;3(2):66-73.
15. Passik SD, Kirsh KL, Casper D. Addiction-related assessment tools and pain management: instruments for screening treatment planning, and monitoring compliance. Pain Med. 2008;9(suppl 2):S145-S166.
16. Chou R, Fanciullo GJ, Fine PG, et al. Opioids for chronic noncancer pain: prediction and identification of aberrant drug-related behaviors: a review of the evidence for an American Pain Society and American Academy of Pain Medicine clinical practice guideline. J Pain. 2009;10(2):131-146.
17. Alford DP, Liebschutz J, Jackson A, et al. Prescription drug abuse: an introduction. http://www.drugabuse.gov/sites/default/files/prescription-drug-abuse-alt.pdf. Published November 8, 2009. Accessed June 20, 2012.
18. Passik SD, Kirsh KL, Whitcomb L, et al. Monitoring outcomes during long-term opioid therapy for noncancer pain: results with the Pain Assessment and Documentation Tool. J Opioid Manag. 2005;1(5):257-266.
19. Webster LR, Webster RM. Predicting aberrant behaviors in opioid-treated patients: preliminary validation of the Opioid Risk Tool. Pain Med. 2005;6(6):432-442.
20. Passik SD. Pain management misstatements: ceiling effects red and yellow flags. Pain Med. 2006;7(1):76-77.
21. Diagnostic and statistical manual of mental disorders 4th ed text rev. Washington DC: American Psychiatric Association; 2000.
22. Mee-Lee D, Shulman GD, Fishman MJ, et al. eds. ASAM patient placement criteria for the treatment of substance-related disorders. 2nd ed. Chevy Chase, MD: American Society of Addiction Medicine, Inc.; 2001.
23. Banta-Green CJ, Maynard C, Koepsell TD, et al. Retention in methadone maintenance drug treatment for prescription-type opioid primary users compared to heroin users. Addiction. 2009;104(5):775-783.
24. Moore BA, Fiellin DA, Barry DT, et al. Primary care office-based buprenorphine treatment: comparison of heroin and prescription opioid dependent patients. J Gen Intern Med. 2007;22(4):527-530.
25. Weiss RD, Potter JS, Fiellin DA, et al. Adjunctive counseling during brief and extended buprenorphine-naloxone treatment for prescription opioid dependence: a 2-phase randomized controlled trial. Arch Gen Psychiatry. 2011;68(12):1238-1246.
26. U.S. Department of Health and Human Services (HHS). Substance Abuse and Mental Health Services Administration (SAMHSA). Office of Applied Studies. Treatment Episode Data Set (TEDS). 1998 - 2008. National Admissions to Substance Abuse Treatment Services, DASIS Series: S-50, HHS Publication No. (SMA) 09-4471. Rockville, MD; 2010.
27. Minozzi S, Amato L, Vecchi S, et al. Oral naltrexone maintenance treatment for opioid dependence. Cochrane Database Syst Rev. 2011;(4):CD001333.-