User login
How to interpret surveys in medical research: A practical approach
Surveys are common in medical research. Although survey research may be subject to inherent self-report bias, surveys have a great impact on policies and practices in medicine, often forming the basis for recommendations or new guidelines.1,2 To interpret and use survey research results, clinicians should be familiar with key elements involved in the creation and validation of surveys.
The purpose of this article is to provide readers with a basic framework for evaluating surveys to allow them to be more informed as consumers of survey research.
IMPORTANT TOOLS IN MEDICAL RESEARCH
Surveys are important tools for answering questions on topics that are difficult to assess using other methods.3 They allow us to gather data systematically from subjects by asking questions, in order to make inferences about a larger population.3,4 Clinicians use surveys to explore the opinions, beliefs, and perceptions of a group, or to investigate physician practice patterns and adherence to clinical guidelines. They may also use surveys to better understand why patients are not engaging in recommended behavioral or lifestyle changes.
Survey methods include interviews (in person, by phone) and questionnaires (paper-and-pencil, e-mailed, online).4
A well-constructed, validated survey can provide powerful data that may influence clinical practice, guide future research development, or drive the development and provision of needed programs and services. Surveys have the potential to transform the ways in which we think about and practice medicine.
READER BEWARE
While survey research in health care appears to have grown exponentially, the quality of reported survey research has not necessarily increased over time.
For consumers of survey research, the adage “reader beware” is apt. Although a considerable number of studies have examined the effects of survey methodology on the validity, reliability, and generalizability of the results,4 medical journals differ in their requirements for reporting survey methods.
In an analysis of 117 articles, Bennett et al3 found that more than 80% did not fully describe the survey development process or pretesting methods. They also found limited guidance and lack of consensus about the best way to report survey research. Of 95 surveys requiring scoring, 66% did not report scoring practices.
Duffett et al5 noted that of 127 critical care medicine surveys, only 36% had been pretested or pilot-tested, and half of all surveys reviewed did not include participant demographics or included only minimal information.
Because journal reporting practices differ, physicians may be unaware of the steps involved in survey construction and validation. Knowledge of these steps is helpful not only in constructing surveys but also in assessing published articles that used survey research.
LIMITATIONS OF SURVEY RESEARCH
Indirect measures of attitudes and behaviors
Surveys that rely on participants’ self-reports of behaviors, attitudes, beliefs, or actions are indirect measures and are susceptible to self-report and social-desirability biases. Participants may overestimate their own expertise or knowledge in self-report surveys. They may wish to reduce embarrassment6 or answer in ways that would make them “look better,”7 resulting in social-desirability bias. These issues need to be mentioned in the limitations section in papers reporting survey research.
Questions and response choices
The data derived from surveys are only as good as the questions that are asked.8 Stone9 noted that questions should be intelligible, unambiguous, and unbiased. If respondents do not comprehend questions as researchers intended, if questionnaire response choices are inadequate, or if questions trigger unintended emotional responses,10–14 researchers may unwittingly introduce error, which will affect the validity of results. Even seemingly objective questions, such as those related to clinical algorithm use, practice patterns, or equipment available to hospital staff, may be interpreted differently by different respondents.
In their eagerness to launch a survey, clinician researchers may not realize that it must be carefully constructed. A focus on question development and validation is critical, as the questions determine the quality of the data derived from the survey.8 Even the position of the question or answer in the survey can affect how participants respond,15 as they may be guided to a response choice by preceding questions.16
WHAT DO YOU NEED TO KNOW ABOUT ASSESSING SURVEY RESEARCH?

What follows are questions and a basic framework that can be used to evaluate published survey research. Recommendations are based on the work of survey scientists,4,7,10,14,15,17,18 survey researchers in medicine and the social sciences, and national standards for test and questionnaire construction and validation (Table 1).4,19,20
Who created the survey? How did they do it?
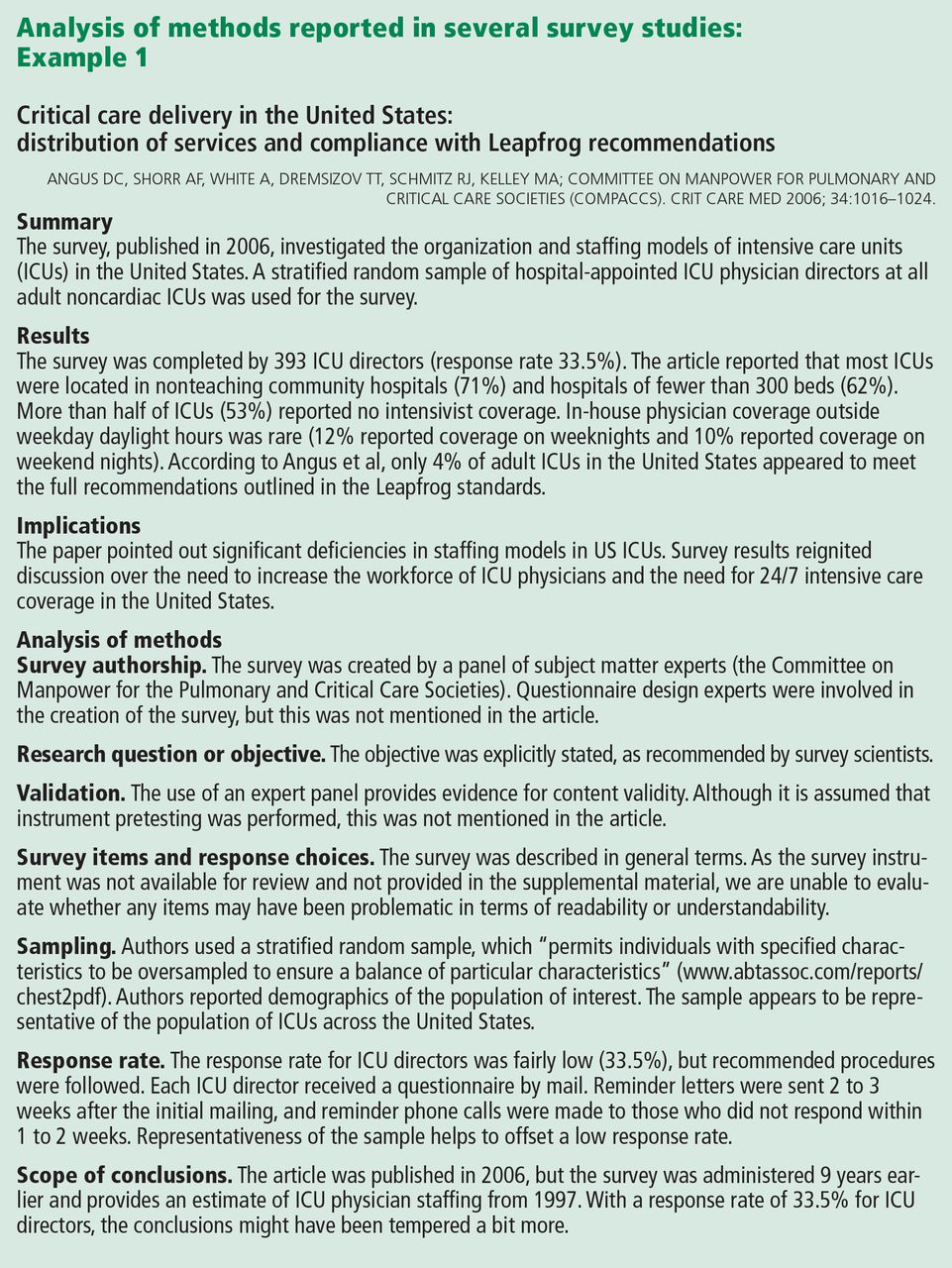
How the survey was created should be sufficiently described to allow readers to judge the adequacy of instrument development.3–5 It is generally recommended that feedback from multiple sources be solicited during survey creation. Both questionnaire-design experts and subject-matter experts are considered critical in the process.4
What question was the survey designed to answer?
Is the objective of the study articulated in the paper? 3,20 To judge survey research, readers need to know if the survey appears to adequately address the research question or questions and the objectives of the study in terms of methods used.4
Was evidence on validity gathered?
Instrument pretesting and field testing are considered best practices by the American Association for Public Opinion Research, a professional organization for US survey scientists.4
Pretesting can include cognitive interviewing, the use of questionnaire appraisal tools, and hybrid methods, all of which are aimed at addressing validity issues.21 Pretesting with a group of participants similar to the target population allows for assessment of item ambiguity, instrument ease of use, adequacy of response categories (response choices), and time to completion.4,12
Cognitive interviewing is designed to explore respondents’ comprehension of questions, response processes, and decision processes governing how they answer questions.4,7,10,11 In cognitive interviewing, respondents are generally interviewed one on one. Techniques vary, but typically include “think alouds” (in which a respondent is asked to verbalize thoughts while responding to questions) and “verbal probing” (in which the respondent answers a question, then is asked follow-up questions as the interviewer probes for information related to the response choice or question itself).7 These techniques can provide evidence that researchers are actually measuring what they set out to measure and not an unrelated construct.4,19
Field testing of a survey under realistic conditions can help to uncover problems in administration, such as issues in standardization of key procedures, and to ensure that the survey was administered as the researchers intended.21,22 Field testing is vital before phone or in-person interviews to ensure standardization of any critical procedures. Pilot testing in a sample similar to the intended population allows for further refinement, with deletion of problem items, before the survey is launched.15
Because even “objective” questions can be somewhat subjective, all research surveys should go through some type of pretesting.4,21 Based on the results of pretesting and field testing, surveys should then be revised before launch.4,21 If an article on a self-report survey makes no mention of survey validation steps, readers may well question the validity of the results.
Are the survey questions and response choices understandable?
Is the meaning of each question unambiguous? Is the reading level appropriate for the sample population (a critical consideration in patient surveys)? Do any of the items actually ask two different questions?13 An example would be: “Was the representative courteous and prompt?” as it is possible to be courteous, but not prompt, and vice versa. If so, respondents may be confused or frustrated in attempting to answer it. If a rating scale is used throughout the questionnaire, are the anchors appropriate? For example, a question may be written in such a way that respondents want to answer “yes/no” or “agree/disagree,” but the scale used may include response options such as “poor,” “marginal,” “good,” and “excellent.” Items with Likert-response formats are commonly used in self-report surveys and allow participants to respond to a statement by choosing from a range of responses (eg, strongly disagree to strongly agree), often spaced horizontally under a line.
It is recommended that surveys also include options for answers beyond the response choices provided,20 such as comment boxes or fill-in-the-blank items. Surveys with a closed-response format may constrain the quality of data collected because investigators may not foresee all possible answers. Surveys need to be available for review either within the article itself, in an appendix, or as supplementary material that is available elsewhere.
Does the sample appear to be appropriate?

Articles that report the results of surveys should describe the target population, the sample design, and, in a demographic table, respondents and nonrespondents. To judge appropriateness, several questions can be asked regarding sampling:
Target population. Is the population of interest (ie, the target population) described, including regional demographics, if applicable? The relationship between the sample and the target population is important, as a nonrepresentative sample may result in misleading conclusions about the population of interest.
Sampling frame. Who had an opportunity to participate in the survey? At its simplest, the sampling frame establishes who (or what, in the case of institutions) should be included within the sample. This is typically a list of elements (Groves et al4) that acts to “frame” or define the sample to be selected. Where the target population may be all academic internal medicine physicians in the United States, the sampling frame may be all male and female US physicians who are members of particular internal medicine professional organizations, identified by their directory email addresses.
Sample design. How was the sample actually selected?4 For example, did investigators use a convenience sample of colleagues at other institutions or use a stratified random sample, ensuring adequate representation of respondents with certain characteristics?
Description of respondents. How is the sample of respondents described? Are demographic features reported, including statistics on regional or national representativeness?5 Does the sample of survey respondents appear to be representative of the researcher’s population of interest (ie, the target population)?3,23 If not, is this adequately described in the limitations section? Although outcomes will not be available on nonrespondents, demographic and baseline data often are available and should be reported. Are there systematic differences between respondents and nonrespondents?
Was the response rate adequate?

Was the response rate adequate, given the number of participants initially recruited? If the response rate was not adequate, did the researchers discuss this limitation?
Maximum response rate, defined as the total number of surveys returned divided by the total number of surveys sent,18 may be difficult to calculate with electronic or Web-based survey platforms. When the maximum response rate cannot be calculated, this issue needs to be addressed in the article’s limitations section.
The number of surveys has increased across fields over the past few decades, but survey response rates in general have decreased.17,21,24,25 In fields outside of clinical medicine, response rates in the 40% range are common.17 In the 1990s, the mean response rate for surveys published in medical journals (mailed surveys) was approximately 60%.26 A 2001 review of physician questionnaire studies found a similar average response rate (61%), with a 52% response rate for large-sample surveys.27 In 2002, Field et al28 examined the impact of incentives in physician survey studies and found response rates ranging from 8.5% to 80%.
Importantly, electronically delivered surveys (e-mail, Web-based) often have lower response rates than mailed surveys.24,29 Nominal financial incentives have been associated with enhanced response rates.28
A relatively low response rate does not necessarily mean you cannot trust the data. Survey scientists note that the representativeness of the sample may be more critical than response rate alone.17 Studies with small sample sizes may be more representative—and findings more valid—than those with large samples, if large samples are nonrepresentative when considering the target population.17
Do the conclusions go beyond the data?
Are the inferences overreaching, in view of the survey design? In studies with low response rates and nonrepresentative samples, researchers must be careful in interpreting the results. If the results cannot be generalized beyond the research sample, is this clear from the limitations, discussion, and conclusion sections?
In this review, we have summarized the findings of three published surveys1,2,30 and commented on how they appear to meet—or don’t quite meet—recommendations for survey development, validation, and use. The papers chosen were deemed strong examples in particular categories, such as description of survey authorship,1 instrument validation,30 sampling methodology,2 and response rate.1
It should be noted that even when surveys are conducted with the utmost rigor, survey reporting may leave out critical details. Survey methodology may not be adequately described for a variety of reasons, including researchers’ training in survey design and methodology; a lack of universally accepted journal-reporting guidelines3; and even journals’ space limitations. At times, journals may excise descriptions of survey development and validation, deeming these sections superfluous. Limitations sections can be critical to interpreting the results of survey research and evaluating the scope of conclusions.
- Jha AK, DesRoches CM, Campbell EG, et al. Use of electronic health records in US hospitals. N Engl J Med 2009; 360:1628–1638.
- Angus DC, Shorr AF, White A, Dremsizov TT, Schmitz RJ, Kelley MA; Committee on Manpower for Pulmonary and Critical Care Societies (COMPACCS). Critical care delivery in the United States: distribution of services and compliance with Leapfrog recommendations. Crit Care Med 2006; 34:1016–1024.
- Bennett C, Khangura S, Brehaut JC, et al. Reporting guidelines for survey research: an analysis of published guidance and reporting practices. PLoS Med 2010; 8:e1001069.
- Groves RM, Fowler FJ, Couper MP, Lepkowski JM, Singer E, Tourangeau R. Survey Methodology. 2nd ed. Hoboken, NJ: John Wiley and Sons, Inc; 2009.
- Duffett M, Burns KE, Adhikari NK, et al. Quality of reporting of surveys in critical care journals: a methodologic review. Crit Care Med 2012; 40:441–449.
- Mattell MS, Jacoby J. Is there an optimal number of alternatives for Likert-scale items? Effects of testing time and scale properties. J Appl Psychol 1972; 56:506–509.
- Willis GB. Cognitive Interviewing. A “How To” Guide. Research Triangle Institute. Presented at the meeting of the American Statistical Association; 1999. http://fog.its.uiowa.edu/~c07b209/interview.pdf. Accessed June 3, 2013.
- Schwarz N. Self-reports. How the questions shape the answers. Amer Psychol 1999; 54:93–105.
- Stone DH. Design a questionnaire. BMJ 1993; 307:1264–1266.
- Willis GB, Royston P, Bercini D. The use of verbal report methods in the development and testing of survey questionnaires. Appl Cogn Psychol 1991; 5:251–267.
- Desimone LM, LeFloch KC. Are we asking the right questions? Using cognitive interviews to improve surveys in education research. Educ Eval Policy Anal 2004; 26:1–22.
- Presser S, Couper MP, Lessler JT, et al. Methods for testing and evaluating survey questions. Public Opin Q 2004; 68:109–130.
- Rogers G. Accreditation Board for Engineering and Technology (ABET), Inc. Sample Protocol for Pilot Testing Survey Items. www.abet.org/WorkArea/DownloadAsset.aspx?id=1299. Accessed January 22, 2013.
- Schwarz N, Oyserman D. Asking questions about behavior: cognition, communication, and questionnaire construction. Am J Eval 2001; 22:127–160.
- Bradburn N, Sudman S, Wansink B. Asking Questions. The Definitive Guide to Questionnaire Design—For Market Research, Political Polls, and Social and Health Questionnaires. San Francisco, CA: Jossey-Bass; 2004.
- Stone AA, Broderick JE, Schwartz JE, Schwarz N. Context effects in survey ratings of health, symptoms, and satisfaction. Med Care 2008; 46:662–667.
- Cook C, Heath F, Thompson RL. A meta-analysis of response rates in Web or internet-based surveys. Educ Psychol Meas 2000; 60:821–836.
- Kaplowitz MD, Hadlock TD, Levine R. A comparison of Web and mail survey response rates. Public Opin Q 2004; 68:94–101.
- American Educational Research Association. Standards for Educational and Psychological Testing/American Educational Research Association, American Psychological Association, National Council on Measurement in Education. Washington, DC: American Educational Research Association; 1999.
- Burns KE, Duffett M, Kho ME, et al; ACCADEMY Group. A guide for the design and conduct of self-administered surveys of clinicians. CMAJ 2008; 179:245–252.
- American Association for Public Opinion Research (AAPOR). http://www.aapor.org/Home.htm. Accessed June 3, 2013.
- National Center for Education Statistics. Planning and Design of Surveys. http://nces.ed.gov/statprog/2002/std2_1.asp. Accessed January 22, 2013.
- Bordens KS, Abbott BB. Research Design and Methods. A Process Approach. 6th ed. New York, NY: McGraw-Hill; 2004.
- Sheehan K. Email survey response rates: a review. JCMC 2001. http://jcmc.indiana.edu/vol6/issue2/sheehan.html. Accessed January 22, 2013.
- Baruch Y, Holtom BC. Survey response rate levels and trends in organizational research. Hum Relat 2008; 61:1139–1160.
- Asch DA, Jedrziewski MK, Christakis NA. Response rates to mail surveys published in medical journals. J Clin Epidemiol 1997; 50:1129–1136.
- Cummings SM, Savitz LA, Konrad TR. Reported response rates to mailed physician questionnaires. Health Services Res 2001; 35:1347–1355.
- Field TS, Cadoret CA, Brown ML, et al. Surveying physicians. Do components of the “Total Design Approach” to optimizing survey response rates apply to physicians? Med Care 2002; 40:596–606.
- Converse PD, Wolfe EW, Huang X, Oswald FL. Response rates for mixed-mode surveys using mail and e-mail/Web. Am J Eval 2008; 29:99–107.
- Hirshberg E, Lacroix J, Sward K, Willson D, Morris AH. Blood glucose control in critically ill adults and children: a survey on stated practice. Chest 2008; 133:1328–1335.
Surveys are common in medical research. Although survey research may be subject to inherent self-report bias, surveys have a great impact on policies and practices in medicine, often forming the basis for recommendations or new guidelines.1,2 To interpret and use survey research results, clinicians should be familiar with key elements involved in the creation and validation of surveys.
The purpose of this article is to provide readers with a basic framework for evaluating surveys to allow them to be more informed as consumers of survey research.
IMPORTANT TOOLS IN MEDICAL RESEARCH
Surveys are important tools for answering questions on topics that are difficult to assess using other methods.3 They allow us to gather data systematically from subjects by asking questions, in order to make inferences about a larger population.3,4 Clinicians use surveys to explore the opinions, beliefs, and perceptions of a group, or to investigate physician practice patterns and adherence to clinical guidelines. They may also use surveys to better understand why patients are not engaging in recommended behavioral or lifestyle changes.
Survey methods include interviews (in person, by phone) and questionnaires (paper-and-pencil, e-mailed, online).4
A well-constructed, validated survey can provide powerful data that may influence clinical practice, guide future research development, or drive the development and provision of needed programs and services. Surveys have the potential to transform the ways in which we think about and practice medicine.
READER BEWARE
While survey research in health care appears to have grown exponentially, the quality of reported survey research has not necessarily increased over time.
For consumers of survey research, the adage “reader beware” is apt. Although a considerable number of studies have examined the effects of survey methodology on the validity, reliability, and generalizability of the results,4 medical journals differ in their requirements for reporting survey methods.
In an analysis of 117 articles, Bennett et al3 found that more than 80% did not fully describe the survey development process or pretesting methods. They also found limited guidance and lack of consensus about the best way to report survey research. Of 95 surveys requiring scoring, 66% did not report scoring practices.
Duffett et al5 noted that of 127 critical care medicine surveys, only 36% had been pretested or pilot-tested, and half of all surveys reviewed did not include participant demographics or included only minimal information.
Because journal reporting practices differ, physicians may be unaware of the steps involved in survey construction and validation. Knowledge of these steps is helpful not only in constructing surveys but also in assessing published articles that used survey research.
LIMITATIONS OF SURVEY RESEARCH
Indirect measures of attitudes and behaviors
Surveys that rely on participants’ self-reports of behaviors, attitudes, beliefs, or actions are indirect measures and are susceptible to self-report and social-desirability biases. Participants may overestimate their own expertise or knowledge in self-report surveys. They may wish to reduce embarrassment6 or answer in ways that would make them “look better,”7 resulting in social-desirability bias. These issues need to be mentioned in the limitations section in papers reporting survey research.
Questions and response choices
The data derived from surveys are only as good as the questions that are asked.8 Stone9 noted that questions should be intelligible, unambiguous, and unbiased. If respondents do not comprehend questions as researchers intended, if questionnaire response choices are inadequate, or if questions trigger unintended emotional responses,10–14 researchers may unwittingly introduce error, which will affect the validity of results. Even seemingly objective questions, such as those related to clinical algorithm use, practice patterns, or equipment available to hospital staff, may be interpreted differently by different respondents.
In their eagerness to launch a survey, clinician researchers may not realize that it must be carefully constructed. A focus on question development and validation is critical, as the questions determine the quality of the data derived from the survey.8 Even the position of the question or answer in the survey can affect how participants respond,15 as they may be guided to a response choice by preceding questions.16
WHAT DO YOU NEED TO KNOW ABOUT ASSESSING SURVEY RESEARCH?
What follows are questions and a basic framework that can be used to evaluate published survey research. Recommendations are based on the work of survey scientists,4,7,10,14,15,17,18 survey researchers in medicine and the social sciences, and national standards for test and questionnaire construction and validation (Table 1).4,19,20
Who created the survey? How did they do it?
How the survey was created should be sufficiently described to allow readers to judge the adequacy of instrument development.3–5 It is generally recommended that feedback from multiple sources be solicited during survey creation. Both questionnaire-design experts and subject-matter experts are considered critical in the process.4
What question was the survey designed to answer?
Is the objective of the study articulated in the paper? 3,20 To judge survey research, readers need to know if the survey appears to adequately address the research question or questions and the objectives of the study in terms of methods used.4
Was evidence on validity gathered?
Instrument pretesting and field testing are considered best practices by the American Association for Public Opinion Research, a professional organization for US survey scientists.4
Pretesting can include cognitive interviewing, the use of questionnaire appraisal tools, and hybrid methods, all of which are aimed at addressing validity issues.21 Pretesting with a group of participants similar to the target population allows for assessment of item ambiguity, instrument ease of use, adequacy of response categories (response choices), and time to completion.4,12
Cognitive interviewing is designed to explore respondents’ comprehension of questions, response processes, and decision processes governing how they answer questions.4,7,10,11 In cognitive interviewing, respondents are generally interviewed one on one. Techniques vary, but typically include “think alouds” (in which a respondent is asked to verbalize thoughts while responding to questions) and “verbal probing” (in which the respondent answers a question, then is asked follow-up questions as the interviewer probes for information related to the response choice or question itself).7 These techniques can provide evidence that researchers are actually measuring what they set out to measure and not an unrelated construct.4,19
Field testing of a survey under realistic conditions can help to uncover problems in administration, such as issues in standardization of key procedures, and to ensure that the survey was administered as the researchers intended.21,22 Field testing is vital before phone or in-person interviews to ensure standardization of any critical procedures. Pilot testing in a sample similar to the intended population allows for further refinement, with deletion of problem items, before the survey is launched.15
Because even “objective” questions can be somewhat subjective, all research surveys should go through some type of pretesting.4,21 Based on the results of pretesting and field testing, surveys should then be revised before launch.4,21 If an article on a self-report survey makes no mention of survey validation steps, readers may well question the validity of the results.
Are the survey questions and response choices understandable?
Is the meaning of each question unambiguous? Is the reading level appropriate for the sample population (a critical consideration in patient surveys)? Do any of the items actually ask two different questions?13 An example would be: “Was the representative courteous and prompt?” as it is possible to be courteous, but not prompt, and vice versa. If so, respondents may be confused or frustrated in attempting to answer it. If a rating scale is used throughout the questionnaire, are the anchors appropriate? For example, a question may be written in such a way that respondents want to answer “yes/no” or “agree/disagree,” but the scale used may include response options such as “poor,” “marginal,” “good,” and “excellent.” Items with Likert-response formats are commonly used in self-report surveys and allow participants to respond to a statement by choosing from a range of responses (eg, strongly disagree to strongly agree), often spaced horizontally under a line.
It is recommended that surveys also include options for answers beyond the response choices provided,20 such as comment boxes or fill-in-the-blank items. Surveys with a closed-response format may constrain the quality of data collected because investigators may not foresee all possible answers. Surveys need to be available for review either within the article itself, in an appendix, or as supplementary material that is available elsewhere.
Does the sample appear to be appropriate?
Articles that report the results of surveys should describe the target population, the sample design, and, in a demographic table, respondents and nonrespondents. To judge appropriateness, several questions can be asked regarding sampling:
Target population. Is the population of interest (ie, the target population) described, including regional demographics, if applicable? The relationship between the sample and the target population is important, as a nonrepresentative sample may result in misleading conclusions about the population of interest.
Sampling frame. Who had an opportunity to participate in the survey? At its simplest, the sampling frame establishes who (or what, in the case of institutions) should be included within the sample. This is typically a list of elements (Groves et al4) that acts to “frame” or define the sample to be selected. Where the target population may be all academic internal medicine physicians in the United States, the sampling frame may be all male and female US physicians who are members of particular internal medicine professional organizations, identified by their directory email addresses.
Sample design. How was the sample actually selected?4 For example, did investigators use a convenience sample of colleagues at other institutions or use a stratified random sample, ensuring adequate representation of respondents with certain characteristics?
Description of respondents. How is the sample of respondents described? Are demographic features reported, including statistics on regional or national representativeness?5 Does the sample of survey respondents appear to be representative of the researcher’s population of interest (ie, the target population)?3,23 If not, is this adequately described in the limitations section? Although outcomes will not be available on nonrespondents, demographic and baseline data often are available and should be reported. Are there systematic differences between respondents and nonrespondents?
Was the response rate adequate?
Was the response rate adequate, given the number of participants initially recruited? If the response rate was not adequate, did the researchers discuss this limitation?
Maximum response rate, defined as the total number of surveys returned divided by the total number of surveys sent,18 may be difficult to calculate with electronic or Web-based survey platforms. When the maximum response rate cannot be calculated, this issue needs to be addressed in the article’s limitations section.
The number of surveys has increased across fields over the past few decades, but survey response rates in general have decreased.17,21,24,25 In fields outside of clinical medicine, response rates in the 40% range are common.17 In the 1990s, the mean response rate for surveys published in medical journals (mailed surveys) was approximately 60%.26 A 2001 review of physician questionnaire studies found a similar average response rate (61%), with a 52% response rate for large-sample surveys.27 In 2002, Field et al28 examined the impact of incentives in physician survey studies and found response rates ranging from 8.5% to 80%.
Importantly, electronically delivered surveys (e-mail, Web-based) often have lower response rates than mailed surveys.24,29 Nominal financial incentives have been associated with enhanced response rates.28
A relatively low response rate does not necessarily mean you cannot trust the data. Survey scientists note that the representativeness of the sample may be more critical than response rate alone.17 Studies with small sample sizes may be more representative—and findings more valid—than those with large samples, if large samples are nonrepresentative when considering the target population.17
Do the conclusions go beyond the data?
Are the inferences overreaching, in view of the survey design? In studies with low response rates and nonrepresentative samples, researchers must be careful in interpreting the results. If the results cannot be generalized beyond the research sample, is this clear from the limitations, discussion, and conclusion sections?
In this review, we have summarized the findings of three published surveys1,2,30 and commented on how they appear to meet—or don’t quite meet—recommendations for survey development, validation, and use. The papers chosen were deemed strong examples in particular categories, such as description of survey authorship,1 instrument validation,30 sampling methodology,2 and response rate.1
It should be noted that even when surveys are conducted with the utmost rigor, survey reporting may leave out critical details. Survey methodology may not be adequately described for a variety of reasons, including researchers’ training in survey design and methodology; a lack of universally accepted journal-reporting guidelines3; and even journals’ space limitations. At times, journals may excise descriptions of survey development and validation, deeming these sections superfluous. Limitations sections can be critical to interpreting the results of survey research and evaluating the scope of conclusions.
Surveys are common in medical research. Although survey research may be subject to inherent self-report bias, surveys have a great impact on policies and practices in medicine, often forming the basis for recommendations or new guidelines.1,2 To interpret and use survey research results, clinicians should be familiar with key elements involved in the creation and validation of surveys.
The purpose of this article is to provide readers with a basic framework for evaluating surveys to allow them to be more informed as consumers of survey research.
IMPORTANT TOOLS IN MEDICAL RESEARCH
Surveys are important tools for answering questions on topics that are difficult to assess using other methods.3 They allow us to gather data systematically from subjects by asking questions, in order to make inferences about a larger population.3,4 Clinicians use surveys to explore the opinions, beliefs, and perceptions of a group, or to investigate physician practice patterns and adherence to clinical guidelines. They may also use surveys to better understand why patients are not engaging in recommended behavioral or lifestyle changes.
Survey methods include interviews (in person, by phone) and questionnaires (paper-and-pencil, e-mailed, online).4
A well-constructed, validated survey can provide powerful data that may influence clinical practice, guide future research development, or drive the development and provision of needed programs and services. Surveys have the potential to transform the ways in which we think about and practice medicine.
READER BEWARE
While survey research in health care appears to have grown exponentially, the quality of reported survey research has not necessarily increased over time.
For consumers of survey research, the adage “reader beware” is apt. Although a considerable number of studies have examined the effects of survey methodology on the validity, reliability, and generalizability of the results,4 medical journals differ in their requirements for reporting survey methods.
In an analysis of 117 articles, Bennett et al3 found that more than 80% did not fully describe the survey development process or pretesting methods. They also found limited guidance and lack of consensus about the best way to report survey research. Of 95 surveys requiring scoring, 66% did not report scoring practices.
Duffett et al5 noted that of 127 critical care medicine surveys, only 36% had been pretested or pilot-tested, and half of all surveys reviewed did not include participant demographics or included only minimal information.
Because journal reporting practices differ, physicians may be unaware of the steps involved in survey construction and validation. Knowledge of these steps is helpful not only in constructing surveys but also in assessing published articles that used survey research.
LIMITATIONS OF SURVEY RESEARCH
Indirect measures of attitudes and behaviors
Surveys that rely on participants’ self-reports of behaviors, attitudes, beliefs, or actions are indirect measures and are susceptible to self-report and social-desirability biases. Participants may overestimate their own expertise or knowledge in self-report surveys. They may wish to reduce embarrassment6 or answer in ways that would make them “look better,”7 resulting in social-desirability bias. These issues need to be mentioned in the limitations section in papers reporting survey research.
Questions and response choices
The data derived from surveys are only as good as the questions that are asked.8 Stone9 noted that questions should be intelligible, unambiguous, and unbiased. If respondents do not comprehend questions as researchers intended, if questionnaire response choices are inadequate, or if questions trigger unintended emotional responses,10–14 researchers may unwittingly introduce error, which will affect the validity of results. Even seemingly objective questions, such as those related to clinical algorithm use, practice patterns, or equipment available to hospital staff, may be interpreted differently by different respondents.
In their eagerness to launch a survey, clinician researchers may not realize that it must be carefully constructed. A focus on question development and validation is critical, as the questions determine the quality of the data derived from the survey.8 Even the position of the question or answer in the survey can affect how participants respond,15 as they may be guided to a response choice by preceding questions.16
WHAT DO YOU NEED TO KNOW ABOUT ASSESSING SURVEY RESEARCH?
What follows are questions and a basic framework that can be used to evaluate published survey research. Recommendations are based on the work of survey scientists,4,7,10,14,15,17,18 survey researchers in medicine and the social sciences, and national standards for test and questionnaire construction and validation (Table 1).4,19,20
Who created the survey? How did they do it?
How the survey was created should be sufficiently described to allow readers to judge the adequacy of instrument development.3–5 It is generally recommended that feedback from multiple sources be solicited during survey creation. Both questionnaire-design experts and subject-matter experts are considered critical in the process.4
What question was the survey designed to answer?
Is the objective of the study articulated in the paper? 3,20 To judge survey research, readers need to know if the survey appears to adequately address the research question or questions and the objectives of the study in terms of methods used.4
Was evidence on validity gathered?
Instrument pretesting and field testing are considered best practices by the American Association for Public Opinion Research, a professional organization for US survey scientists.4
Pretesting can include cognitive interviewing, the use of questionnaire appraisal tools, and hybrid methods, all of which are aimed at addressing validity issues.21 Pretesting with a group of participants similar to the target population allows for assessment of item ambiguity, instrument ease of use, adequacy of response categories (response choices), and time to completion.4,12
Cognitive interviewing is designed to explore respondents’ comprehension of questions, response processes, and decision processes governing how they answer questions.4,7,10,11 In cognitive interviewing, respondents are generally interviewed one on one. Techniques vary, but typically include “think alouds” (in which a respondent is asked to verbalize thoughts while responding to questions) and “verbal probing” (in which the respondent answers a question, then is asked follow-up questions as the interviewer probes for information related to the response choice or question itself).7 These techniques can provide evidence that researchers are actually measuring what they set out to measure and not an unrelated construct.4,19
Field testing of a survey under realistic conditions can help to uncover problems in administration, such as issues in standardization of key procedures, and to ensure that the survey was administered as the researchers intended.21,22 Field testing is vital before phone or in-person interviews to ensure standardization of any critical procedures. Pilot testing in a sample similar to the intended population allows for further refinement, with deletion of problem items, before the survey is launched.15
Because even “objective” questions can be somewhat subjective, all research surveys should go through some type of pretesting.4,21 Based on the results of pretesting and field testing, surveys should then be revised before launch.4,21 If an article on a self-report survey makes no mention of survey validation steps, readers may well question the validity of the results.
Are the survey questions and response choices understandable?
Is the meaning of each question unambiguous? Is the reading level appropriate for the sample population (a critical consideration in patient surveys)? Do any of the items actually ask two different questions?13 An example would be: “Was the representative courteous and prompt?” as it is possible to be courteous, but not prompt, and vice versa. If so, respondents may be confused or frustrated in attempting to answer it. If a rating scale is used throughout the questionnaire, are the anchors appropriate? For example, a question may be written in such a way that respondents want to answer “yes/no” or “agree/disagree,” but the scale used may include response options such as “poor,” “marginal,” “good,” and “excellent.” Items with Likert-response formats are commonly used in self-report surveys and allow participants to respond to a statement by choosing from a range of responses (eg, strongly disagree to strongly agree), often spaced horizontally under a line.
It is recommended that surveys also include options for answers beyond the response choices provided,20 such as comment boxes or fill-in-the-blank items. Surveys with a closed-response format may constrain the quality of data collected because investigators may not foresee all possible answers. Surveys need to be available for review either within the article itself, in an appendix, or as supplementary material that is available elsewhere.
Does the sample appear to be appropriate?
Articles that report the results of surveys should describe the target population, the sample design, and, in a demographic table, respondents and nonrespondents. To judge appropriateness, several questions can be asked regarding sampling:
Target population. Is the population of interest (ie, the target population) described, including regional demographics, if applicable? The relationship between the sample and the target population is important, as a nonrepresentative sample may result in misleading conclusions about the population of interest.
Sampling frame. Who had an opportunity to participate in the survey? At its simplest, the sampling frame establishes who (or what, in the case of institutions) should be included within the sample. This is typically a list of elements (Groves et al4) that acts to “frame” or define the sample to be selected. Where the target population may be all academic internal medicine physicians in the United States, the sampling frame may be all male and female US physicians who are members of particular internal medicine professional organizations, identified by their directory email addresses.
Sample design. How was the sample actually selected?4 For example, did investigators use a convenience sample of colleagues at other institutions or use a stratified random sample, ensuring adequate representation of respondents with certain characteristics?
Description of respondents. How is the sample of respondents described? Are demographic features reported, including statistics on regional or national representativeness?5 Does the sample of survey respondents appear to be representative of the researcher’s population of interest (ie, the target population)?3,23 If not, is this adequately described in the limitations section? Although outcomes will not be available on nonrespondents, demographic and baseline data often are available and should be reported. Are there systematic differences between respondents and nonrespondents?
Was the response rate adequate?
Was the response rate adequate, given the number of participants initially recruited? If the response rate was not adequate, did the researchers discuss this limitation?
Maximum response rate, defined as the total number of surveys returned divided by the total number of surveys sent,18 may be difficult to calculate with electronic or Web-based survey platforms. When the maximum response rate cannot be calculated, this issue needs to be addressed in the article’s limitations section.
The number of surveys has increased across fields over the past few decades, but survey response rates in general have decreased.17,21,24,25 In fields outside of clinical medicine, response rates in the 40% range are common.17 In the 1990s, the mean response rate for surveys published in medical journals (mailed surveys) was approximately 60%.26 A 2001 review of physician questionnaire studies found a similar average response rate (61%), with a 52% response rate for large-sample surveys.27 In 2002, Field et al28 examined the impact of incentives in physician survey studies and found response rates ranging from 8.5% to 80%.
Importantly, electronically delivered surveys (e-mail, Web-based) often have lower response rates than mailed surveys.24,29 Nominal financial incentives have been associated with enhanced response rates.28
A relatively low response rate does not necessarily mean you cannot trust the data. Survey scientists note that the representativeness of the sample may be more critical than response rate alone.17 Studies with small sample sizes may be more representative—and findings more valid—than those with large samples, if large samples are nonrepresentative when considering the target population.17
Do the conclusions go beyond the data?
Are the inferences overreaching, in view of the survey design? In studies with low response rates and nonrepresentative samples, researchers must be careful in interpreting the results. If the results cannot be generalized beyond the research sample, is this clear from the limitations, discussion, and conclusion sections?
In this review, we have summarized the findings of three published surveys1,2,30 and commented on how they appear to meet—or don’t quite meet—recommendations for survey development, validation, and use. The papers chosen were deemed strong examples in particular categories, such as description of survey authorship,1 instrument validation,30 sampling methodology,2 and response rate.1
It should be noted that even when surveys are conducted with the utmost rigor, survey reporting may leave out critical details. Survey methodology may not be adequately described for a variety of reasons, including researchers’ training in survey design and methodology; a lack of universally accepted journal-reporting guidelines3; and even journals’ space limitations. At times, journals may excise descriptions of survey development and validation, deeming these sections superfluous. Limitations sections can be critical to interpreting the results of survey research and evaluating the scope of conclusions.
- Jha AK, DesRoches CM, Campbell EG, et al. Use of electronic health records in US hospitals. N Engl J Med 2009; 360:1628–1638.
- Angus DC, Shorr AF, White A, Dremsizov TT, Schmitz RJ, Kelley MA; Committee on Manpower for Pulmonary and Critical Care Societies (COMPACCS). Critical care delivery in the United States: distribution of services and compliance with Leapfrog recommendations. Crit Care Med 2006; 34:1016–1024.
- Bennett C, Khangura S, Brehaut JC, et al. Reporting guidelines for survey research: an analysis of published guidance and reporting practices. PLoS Med 2010; 8:e1001069.
- Groves RM, Fowler FJ, Couper MP, Lepkowski JM, Singer E, Tourangeau R. Survey Methodology. 2nd ed. Hoboken, NJ: John Wiley and Sons, Inc; 2009.
- Duffett M, Burns KE, Adhikari NK, et al. Quality of reporting of surveys in critical care journals: a methodologic review. Crit Care Med 2012; 40:441–449.
- Mattell MS, Jacoby J. Is there an optimal number of alternatives for Likert-scale items? Effects of testing time and scale properties. J Appl Psychol 1972; 56:506–509.
- Willis GB. Cognitive Interviewing. A “How To” Guide. Research Triangle Institute. Presented at the meeting of the American Statistical Association; 1999. http://fog.its.uiowa.edu/~c07b209/interview.pdf. Accessed June 3, 2013.
- Schwarz N. Self-reports. How the questions shape the answers. Amer Psychol 1999; 54:93–105.
- Stone DH. Design a questionnaire. BMJ 1993; 307:1264–1266.
- Willis GB, Royston P, Bercini D. The use of verbal report methods in the development and testing of survey questionnaires. Appl Cogn Psychol 1991; 5:251–267.
- Desimone LM, LeFloch KC. Are we asking the right questions? Using cognitive interviews to improve surveys in education research. Educ Eval Policy Anal 2004; 26:1–22.
- Presser S, Couper MP, Lessler JT, et al. Methods for testing and evaluating survey questions. Public Opin Q 2004; 68:109–130.
- Rogers G. Accreditation Board for Engineering and Technology (ABET), Inc. Sample Protocol for Pilot Testing Survey Items. www.abet.org/WorkArea/DownloadAsset.aspx?id=1299. Accessed January 22, 2013.
- Schwarz N, Oyserman D. Asking questions about behavior: cognition, communication, and questionnaire construction. Am J Eval 2001; 22:127–160.
- Bradburn N, Sudman S, Wansink B. Asking Questions. The Definitive Guide to Questionnaire Design—For Market Research, Political Polls, and Social and Health Questionnaires. San Francisco, CA: Jossey-Bass; 2004.
- Stone AA, Broderick JE, Schwartz JE, Schwarz N. Context effects in survey ratings of health, symptoms, and satisfaction. Med Care 2008; 46:662–667.
- Cook C, Heath F, Thompson RL. A meta-analysis of response rates in Web or internet-based surveys. Educ Psychol Meas 2000; 60:821–836.
- Kaplowitz MD, Hadlock TD, Levine R. A comparison of Web and mail survey response rates. Public Opin Q 2004; 68:94–101.
- American Educational Research Association. Standards for Educational and Psychological Testing/American Educational Research Association, American Psychological Association, National Council on Measurement in Education. Washington, DC: American Educational Research Association; 1999.
- Burns KE, Duffett M, Kho ME, et al; ACCADEMY Group. A guide for the design and conduct of self-administered surveys of clinicians. CMAJ 2008; 179:245–252.
- American Association for Public Opinion Research (AAPOR). http://www.aapor.org/Home.htm. Accessed June 3, 2013.
- National Center for Education Statistics. Planning and Design of Surveys. http://nces.ed.gov/statprog/2002/std2_1.asp. Accessed January 22, 2013.
- Bordens KS, Abbott BB. Research Design and Methods. A Process Approach. 6th ed. New York, NY: McGraw-Hill; 2004.
- Sheehan K. Email survey response rates: a review. JCMC 2001. http://jcmc.indiana.edu/vol6/issue2/sheehan.html. Accessed January 22, 2013.
- Baruch Y, Holtom BC. Survey response rate levels and trends in organizational research. Hum Relat 2008; 61:1139–1160.
- Asch DA, Jedrziewski MK, Christakis NA. Response rates to mail surveys published in medical journals. J Clin Epidemiol 1997; 50:1129–1136.
- Cummings SM, Savitz LA, Konrad TR. Reported response rates to mailed physician questionnaires. Health Services Res 2001; 35:1347–1355.
- Field TS, Cadoret CA, Brown ML, et al. Surveying physicians. Do components of the “Total Design Approach” to optimizing survey response rates apply to physicians? Med Care 2002; 40:596–606.
- Converse PD, Wolfe EW, Huang X, Oswald FL. Response rates for mixed-mode surveys using mail and e-mail/Web. Am J Eval 2008; 29:99–107.
- Hirshberg E, Lacroix J, Sward K, Willson D, Morris AH. Blood glucose control in critically ill adults and children: a survey on stated practice. Chest 2008; 133:1328–1335.
- Jha AK, DesRoches CM, Campbell EG, et al. Use of electronic health records in US hospitals. N Engl J Med 2009; 360:1628–1638.
- Angus DC, Shorr AF, White A, Dremsizov TT, Schmitz RJ, Kelley MA; Committee on Manpower for Pulmonary and Critical Care Societies (COMPACCS). Critical care delivery in the United States: distribution of services and compliance with Leapfrog recommendations. Crit Care Med 2006; 34:1016–1024.
- Bennett C, Khangura S, Brehaut JC, et al. Reporting guidelines for survey research: an analysis of published guidance and reporting practices. PLoS Med 2010; 8:e1001069.
- Groves RM, Fowler FJ, Couper MP, Lepkowski JM, Singer E, Tourangeau R. Survey Methodology. 2nd ed. Hoboken, NJ: John Wiley and Sons, Inc; 2009.
- Duffett M, Burns KE, Adhikari NK, et al. Quality of reporting of surveys in critical care journals: a methodologic review. Crit Care Med 2012; 40:441–449.
- Mattell MS, Jacoby J. Is there an optimal number of alternatives for Likert-scale items? Effects of testing time and scale properties. J Appl Psychol 1972; 56:506–509.
- Willis GB. Cognitive Interviewing. A “How To” Guide. Research Triangle Institute. Presented at the meeting of the American Statistical Association; 1999. http://fog.its.uiowa.edu/~c07b209/interview.pdf. Accessed June 3, 2013.
- Schwarz N. Self-reports. How the questions shape the answers. Amer Psychol 1999; 54:93–105.
- Stone DH. Design a questionnaire. BMJ 1993; 307:1264–1266.
- Willis GB, Royston P, Bercini D. The use of verbal report methods in the development and testing of survey questionnaires. Appl Cogn Psychol 1991; 5:251–267.
- Desimone LM, LeFloch KC. Are we asking the right questions? Using cognitive interviews to improve surveys in education research. Educ Eval Policy Anal 2004; 26:1–22.
- Presser S, Couper MP, Lessler JT, et al. Methods for testing and evaluating survey questions. Public Opin Q 2004; 68:109–130.
- Rogers G. Accreditation Board for Engineering and Technology (ABET), Inc. Sample Protocol for Pilot Testing Survey Items. www.abet.org/WorkArea/DownloadAsset.aspx?id=1299. Accessed January 22, 2013.
- Schwarz N, Oyserman D. Asking questions about behavior: cognition, communication, and questionnaire construction. Am J Eval 2001; 22:127–160.
- Bradburn N, Sudman S, Wansink B. Asking Questions. The Definitive Guide to Questionnaire Design—For Market Research, Political Polls, and Social and Health Questionnaires. San Francisco, CA: Jossey-Bass; 2004.
- Stone AA, Broderick JE, Schwartz JE, Schwarz N. Context effects in survey ratings of health, symptoms, and satisfaction. Med Care 2008; 46:662–667.
- Cook C, Heath F, Thompson RL. A meta-analysis of response rates in Web or internet-based surveys. Educ Psychol Meas 2000; 60:821–836.
- Kaplowitz MD, Hadlock TD, Levine R. A comparison of Web and mail survey response rates. Public Opin Q 2004; 68:94–101.
- American Educational Research Association. Standards for Educational and Psychological Testing/American Educational Research Association, American Psychological Association, National Council on Measurement in Education. Washington, DC: American Educational Research Association; 1999.
- Burns KE, Duffett M, Kho ME, et al; ACCADEMY Group. A guide for the design and conduct of self-administered surveys of clinicians. CMAJ 2008; 179:245–252.
- American Association for Public Opinion Research (AAPOR). http://www.aapor.org/Home.htm. Accessed June 3, 2013.
- National Center for Education Statistics. Planning and Design of Surveys. http://nces.ed.gov/statprog/2002/std2_1.asp. Accessed January 22, 2013.
- Bordens KS, Abbott BB. Research Design and Methods. A Process Approach. 6th ed. New York, NY: McGraw-Hill; 2004.
- Sheehan K. Email survey response rates: a review. JCMC 2001. http://jcmc.indiana.edu/vol6/issue2/sheehan.html. Accessed January 22, 2013.
- Baruch Y, Holtom BC. Survey response rate levels and trends in organizational research. Hum Relat 2008; 61:1139–1160.
- Asch DA, Jedrziewski MK, Christakis NA. Response rates to mail surveys published in medical journals. J Clin Epidemiol 1997; 50:1129–1136.
- Cummings SM, Savitz LA, Konrad TR. Reported response rates to mailed physician questionnaires. Health Services Res 2001; 35:1347–1355.
- Field TS, Cadoret CA, Brown ML, et al. Surveying physicians. Do components of the “Total Design Approach” to optimizing survey response rates apply to physicians? Med Care 2002; 40:596–606.
- Converse PD, Wolfe EW, Huang X, Oswald FL. Response rates for mixed-mode surveys using mail and e-mail/Web. Am J Eval 2008; 29:99–107.
- Hirshberg E, Lacroix J, Sward K, Willson D, Morris AH. Blood glucose control in critically ill adults and children: a survey on stated practice. Chest 2008; 133:1328–1335.
KEY POINTS
- Most survey reports do not adequately describe their methods.
- Surveys that rely on participants’ self-reports of behaviors, attitudes, beliefs, or actions are indirect measures and are susceptible to self-report and social-desirability biases.
- Informed readers need to consider a survey’s authorship, objective, validation, items, response choices, sampling representativeness, response rate, generalizability, and scope of the conclusions.
Paget disease of bone: Diagnosis and drug therapy
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.

Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).

It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications

Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies

Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
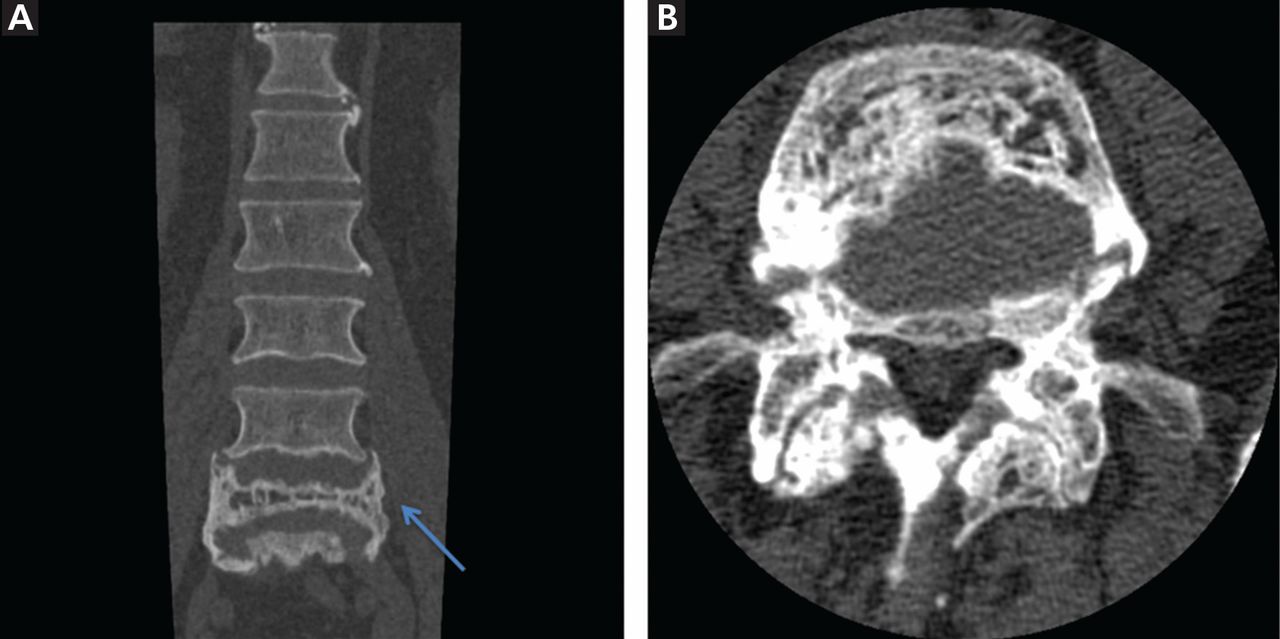
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
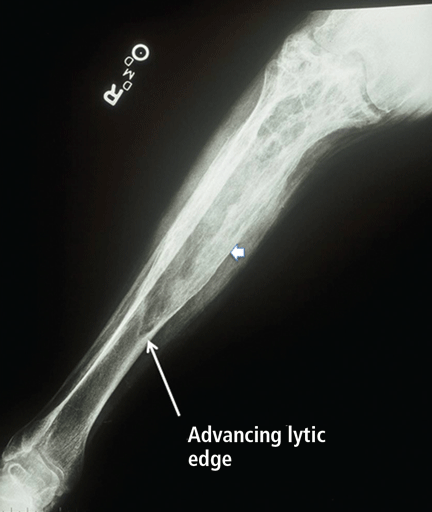
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed

If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
Paget disease of bone is a focal disorder of the aging skeleton that can be asymptomatic or can present with pain, bowing deformities, fractures, or nonspecific rheumatic complaints. Physicians often discover it in asymptomatic patients when serum alkaline phosphatase levels are elevated or as an incidental finding on radiography. Despite evidence of germline mutations and polymorphisms that predispose to Paget disease, the environmental determinants that permit disease expression in older people remain unknown.
A STRIKING GEOGRAPHIC DISTRIBUTION
Researchers have been studying the determinants and distribution of Paget disease ever since Sir James Paget first described it in 1877.1
Paget disease has a predilection for the axial skeleton, particularly the lumbosacral spine and pelvis, as well as the skull, femur, and tibia.2 Knowing this, investigators have used screening plain films of the abdomen (kidney-ureter-bladder views) to estimate its prevalence in different populations, as these images capture the lumbosacral spine, pelvis, and proximal femurs. Other means of assessing prevalence have included autopsy series, questionnaires, and screens for biochemical markers of bone turnover, such as elevated serum alkaline phosphatase from bone.3–6
Using these methods, Paget disease has been estimated to occur in 1% to 3% of people over age 55, and in as many as 8% of people over age 80 in certain countries.7
This disease has a striking geographic distribution, being frequent in Europe, Canada, the United States, Australia, New Zealand, and cities of South America, but rare in Scandinavia and Japan. It seems to be equally rare in other countries of the Far East and in India, Russia, and Africa, although its prevalence in these areas has not been thoroughly investigated.8
That it is an ancient disease has been corroborated by excavations in churchyards in Great Britain.9,10 It may be familial or sporadic, but its expression is delayed until late middle age in most persons, and it does not occur in children. For reasons unclear, the prevalence seems to be decreasing in many countries.11–13
GENETICS IS NOT THE WHOLE STORY
The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition, environmental factor, or both.
Mutations in SQSTM1
In 2002, scientists investigating a cohort of French Canadian families found a mutation in the SQSTM1 gene that was present in almost 50% of people with familial Paget disease and in 16% of those with sporadic Paget disease.14 Hocking and his colleagues in the United Kingdom subsequently found the same mutation in 19% of cases of familial Paget disease and in 9% of sporadic cases.15
Further, investigators noted that the mutation was often present on a conserved haplotype, consistent with a stable genetic change occurring in the affected population.16 This observation of a “founder effect” dovetailed with the epidemiology of Paget disease,17 but only with this SQSTM1 mutation.
Throughout Europe, Australia, and the United States, comparable rates of the SQSTM1 mutation were reported in or around the ubiquitin-associated domain. Several specific mutations exist, the most common one being P392L, ie, a prolineto-leucine substitution at amino acid 392. Scientists have tried to correlate severity of disease with genotype, but the findings have been inconsistent.18–21
Investigations into the mechanism of disease have pointed to the role of p62, the product of SQSTM1, in signaling osteoclast activation via nuclear factor kappa B. Since this initial discovery, polymorphisms in the genes affecting osteoclast maturation, activation, and fusion pathways have been shown to predispose to Paget disease. Examples:
- TNFRSF11A, which codes for receptor activator of nuclear factor kappa B, or RANK
- TNFRSF11B, which codes for osteoprotegerin, or OPG
- CSF1, which codes for macrophage colony-stimulating factor 1, and
- OPTN, which codes for optineurin, a member of the nuclear factor kappa B-modulating protein family.
Clinicians interested in these details can read an excellent review of the pathogenesis of Paget disease.22
Other possible factors
Although there is good evidence that measles and canine distemper virus can infect osteoclasts and modify their phenotype, there is no good evidence that these infections by themselves cause Paget disease.23–25 It is, however, tempting to think of these RNA paramyxoviruses as precipitating factors; conceivably, an infectious agent might seed the ends of long bones, accounting for the fixed distribution of Paget disease and its late expression.
Epidemiologic studies from around the world have failed to identify conclusively any environmental exposure that predisposes to Paget disease, although a rural setting, trauma, infection, and milk ingestion have all been proposed.26–28 It is also possible that as bone ages and the marrow becomes less cellular and more fatty, these changes may permit the disease to develop.
The greatest risk factor for Paget disease is perhaps aging, followed by ancestry and a known family history of it. That genetics is not the whole story is evident by reports of people with SQSTM1 mutations who show no clinical evidence of Paget disease in their old age, and patients with Paget disease who have no SQSTM1 mutation.20,29
CLINICAL PRESENTATION
Most patients with Paget disease have no symptoms and come to medical attention because of an elevated serum alkaline phosphatase level or characteristic findings on radiographs ordered for other indications.11 Paget disease is the second most common disorder of aging bone after osteoporosis. Yet unlike osteoporosis, which presents as a systemic fragility of bone, the clinical manifestations of Paget disease depend on which bones are affected and how enlarged or misshapen they have become.
Common complications
As a consequence of this abnormal bone remodeling and overgrowth, many patients present with bone pain. Bone deformity, headache, and hearing loss may also occur (Figure 1), as well as fractures and nerve compression syndromes (eg, spinal stenosis, sciatica, cauda equina syndrome).
It is important to remember that “pagetic” bone may not be the source of pain, and that functional impairment caused by degenerative changes at affected sites is common (Figure 2).30,31
In a study from the New England Registry for Paget’s Disease,32 most patients knew fairly well which bones were affected and what complications resulted from this when deformity, fracture, or total joint replacement had occurred.32 Although Paget disease did affect their quality of life as measured by physical functioning on the Short Form-12 assessment, these impairments did not seem to affect their outlook, which was as good as or better than that in other people their age.
Metabolic complications
Metabolic complications of Paget disease are rare today but can occur in an elderly patient who has active, polyostotic (multibone) disease.33 The accelerated rate of bone remodeling and the increased vascularity of pagetic bone have been reported to lead to high-output heart failure. In theory, treatment should ease this by diminishing blood flow to pagetic bone and restoring bone turnover to more normal levels.34
Hypercalcemia can occur when patients with Paget disease are immobilized for any reason, and there is probably a higher incidence of renal stones in patients with Paget disease.35,36
Malignant complications
Osteosarcoma rarely arises in pagetic bone. Yet Paget disease may account for a significant number of cases of this cancer in the elderly.37 In these cases, osteosarcoma is presumed to be driven by a second genetic mutation, has a genetic signature distinct from that in osteosarcomas occurring in youth, and is quite resistant to treatment.38 In Scandinavia and Japan, where Paget disease is rare, the second peak of osteosarcoma that occurs with aging seems muted as well.39,40 These cancers present with pain, soft-tissue swelling, and variable elevations in serum alkaline phosphatase. Investigations to date suggest that pagetic lesions and osteosarcomas arising in pagetic bone are probably both driven to some extent by stromal cells overexpressing RANK ligand and may not represent defects intrinsic to the osteoclast.41
Giant-cell tumors of bone are also rare but can arise in pagetic bone. A cluster of cases was reported in Avellino and other towns of southern Italy.42 Again, the lesions occur in older individuals and in different sites than those seen in the benign giant-cell tumors recorded in patients without Paget disease.
Metastases from lymphomas, prostate cancer, and breast cancer certainly occur in bone, but rarely in pagetic sites.43 A recent case study noted that patients with prostate cancer who also had Paget disease had a later onset of metastasis to bone than patients without coincident Paget disease.44
A THOUGHTFUL ASSESSMENT
Evaluating a patient with Paget disease requires a thoughtful assessment of its musculoskeletal consequences in an aging skeleton. Pain in Paget disease is often multifactorial. In the elderly, end-stage degenerative disease of the spine, hip, and knees, mechanical instability, compression fractures of the spine, and neuropathies may compound the clinical picture. Therefore, a thorough evaluation is required to plan effective therapy.
Alkaline phosphatase and other markers
A screening serum alkaline phosphatase level is usually sufficient to measure bone turnover. Produced by osteoblasts, alkaline phosphatase is a marker of bone formation, but an imperfect one. Often it is elevated in active Paget disease—but not always.45 Many patients have normal serum alkaline phosphatase levels, particularly if they have monostotic (single-bone) disease. It is unclear why, in a disorder marked by accelerated bone remodeling, the biochemical markers are inconsistent measures of bone turnover.
Research into biochemical markers of Paget disease has had two aims: to identify the single best marker for baseline assessment of pagetic bone activity and to find out whether this measurement responds to therapy.46,47 Measures of bone formation such as bone-specific alkaline phosphatase, osteocalcin, and the procollagen type I peptides, and measures of bone resorption including the pyridinolines, hydroxyproline, and cross-linked collagens, have been analyzed as markers of bone remodeling and show no real advantage over the serum alkaline phosphatase level as reflections of bone turnover. As alkaline phosphatase measurement is inexpensive, available, and reliable, it should be used preferentially, with gamma-glutamyl transpeptidate or 5′ nucleotidase confirming the source as either liver or bone. Readers are directed to a recent review in which the utility of these markers is explored in more detail.48
Imaging studies
Bone scans can give us an idea of the extent, location, and general activity of the disease (Figure 3). Uptake is avid in affected bones, beginning in the subchondral region and spreading throughout the bone. Bone scans can be particularly useful in defining sites of active disease when the serum alkaline phosphatase level is normal.
Plain radiography of the affected bones outlines the anatomy of the problem and gives some insight into the cause of pain (Figure 3).
Computed tomography or magnetic resonance imaging may prove useful in cases of spinal stenosis, cauda equina syndrome, compression fractures, or suspected malignancy (Figure 4), but these studies are expensive and generally are not needed.
Radiographic features. Paget disease is presumed to be a disease of the osteoclast, and the earliest lesion is described as lytic. In my own experience, it is unusual to see a purely lytic lesion, although sometimes the disease presents in the skull in this way—osteoporosis circumscripta—or in the femur or tibia with an advancing edge of pure osteolysis.
More often, one sees evidence of both resorption by osteoclasts and formation by osteoblasts, reflecting the coupling of these two processes in this disease. Radiographic findings on plain films are usually definitive, showing enlargement of the affected bone, deformity, coarsened trabeculae, and thickened cortices with tunneling (Figure 5).49 In weightbearing bones, pseudofractures may stud the convex surface. These incongruities of bone may persist for years, heralding fracture only when there is focal pain (Figure 6).50
Biopsy is infrequently needed
If these diagnostic findings are not present, then biopsy is indicated. In the United Sates and Canada, where Paget disease is fairly common, biopsy is infrequently needed and is usually reserved for situations in which the differential diagnosis includes cancer, as when the cortex cannot be clearly visualized, the lesions are atypical in pattern or location, or there is a single sclerotic vertebral body on imaging.51
The other indication for biopsy is a “new” pagetic lesion. For reasons unknown, the pattern of skeletal involvement in Paget disease tends to be stable throughout the patient’s lifetime. This is another reason why a baseline bone scan is useful.
TREATMENT WITH BISPHOSPHONATES
Treatment of Paget disease today relies for the most part on the new generation of nitrogen-containing bisphosphonates. As a class, these are antiresorptive agents that inhibit osteoclasts; in this way they slow bone remodeling and enhance the deposition of normal lamellar bone. Their clinical efficacy in Paget disease, coupled with the observation that the earliest lesion in Paget disease is lytic, underscores the principle that Paget disease is a disorder of the osteoclast.
Oral bisphosphonates
Etidronate, approved in 1977, was the first bisphosphonate licensed to treat Paget disease, and it remains available for this indication in the United States. Used in 6-month regimens, it lowers the serum alkaline phosphatase level in some patients, but it has a narrow therapeutic margin. Drug-induced osteomalacia and worsening lytic lesions and fractures in weight-bearing bones are some of the complications.52 When the nitrogen-containing bisphosphonates were developed, they proved to be more potent antiresorptive agents that pose less risk of mineralization defects at prescribed doses.
Alendronate, approved in 1995, is an oral nitrogen-containing bisphosphonate that is effective in treating Paget disease.53 Alendronate is now available in the United States only through special programs (eg, the CVS ProCare Program); the paperwork required to secure this drug is onerous, so the drug is used infrequently. Studies in Paget disease showed that it normalizes the serum alkaline phosphatase level, improves the radiographic appearance, and eases pain in many patients.54 The dosage is 40 mg daily for 6 months.
Risedronate, approved in 1998, is another oral nitrogen-containing bisphosphonate and is comparable to alendronate in efficacy.55 The dosage is 30 mg daily for 2 months.
Tiludronate is another oral bisphosphonate with a different mechanism of action from the nitrogen-containing bisphosphonates.56 It is safe, often effective, but less potent than the newer agents.
The oral bisphosphonates are well tolerated, with few side effects other than gastrointestinal distress. As a class, they are poorly absorbed and so must be taken fasting with a full glass of water on rising, after which the patient should remain upright without food or drink for 30 to 60 minutes. This is a nuisance for elderly patients already on multiple medications and thus makes intravenous agents appealing.
Intravenous bisphosphonates
Pamidronate was approved in 1994. It is quite effective in many patients with Paget disease. There is no consensus around the world on dosing, with regimens ranging from 30 mg to 90 mg or more intravenously in divided doses given over 2 to 4 hours from once a day to once a week. In the United States, 30 mg is given over 4 hours on 3 consecutive days. Resistance to pamidronate has been described; the mechanism is unknown.
Zoledronic acid is a nitrogen-containing bisphosphonate. It is given as a single infusion over 15 minutes, and re-treatment may not be necessary for years. A randomized clinical trial in 2005 demonstrated the efficacy of zoledronic acid 5 mg by infusion compared with oral risedronate in the treatment of Paget disease.57 In observational extension studies lasting as long as 6.5 years, zoledronic acid has been shown to be superior to risedronate in terms of the proportion of patients experiencing a sustained clinical remission.58
While there are many bisphosphonates on the market, an infusion of 5 mg of zoledronic acid seems optimal in most patients who do not have a contraindication or an aversion to intravenous therapy. It tends to normalize the serum alkaline phosphatase level quickly and to leave more patients in sustained biochemical remission than do older bisphosphonates, as noted above. It also tends to be more effective in normalizing the serum alkaline phosphatase level when a patient has used other bisphosphonates in the past or has become resistant to them.
Bisphosphonates reduce bone turnover but do not correct deformities
In randomized clinical trials, bisphosphonates have been shown to restore bone remodeling to more normal levels, to ease pain from pagetic bone, to lower the serum alkaline phosphatase level, and to heal radiographic lesions, but these drugs have not been proven to prevent progression of deformity or to restore the structural integrity of bone (Figure 6).
The Paget’s Disease: Randomized Trial of Intensive Versus Symptomatic Management (PRISM), in 1,324 people with Paget disease in the United Kingdom, showed no difference in the incidence of fracture, orthopedic surgery, quality of life, or hearing thresholds over 2 to 5 years in patients treated with bisphosphonates vs those treated symptomatically, despite a significant difference in serum alkaline phosphatase in the two groups (P < .001).59
In the observational extension study of zoledronic acid described above,58 three of four fractures occurred in the group treated with zoledronic acid, echoing the findings of the PRISM study.
Adverse effects of bisphosphonates
The more potent the bisphosphonate is as an antiresorptive agent, the more it suppresses normal bone remodeling, which can lead to osteonecrosis of the jaw and to atypical femoral fractures.60,61 These complications are unusual in patients with Paget disease because the treatment is intermittent. Sometimes a single dose of zoledronic acid or one course of risedronate or alendronate will last for years.
All the nitrogen-containing bisphosphonates, particularly zoledronic acid, may provoke flulike symptoms of fever, arthralgias, and bone pain. This effect is self-limited, resolves in days, and does not tend to recur. Bone pain may be more sustained, but this also passes, and within weeks the antiresorptive process has abated and pagetic bone pain will ease. Atrial fibrillation is not an anticipated complication of treatment with a bisphosphonate.62 The risk of esophageal cancer is not confirmed at this time.63 Other rare complications of the bisphosphonates include iritis, acute renal failure, and allergy.
Bisphosphonates are not approved for use in patients with creatinine clearance less than 30 mL/min, or in pregnancy.
Other treatments
Calcitonin, an older agent, can still be useful in easing the pain of Paget disease, healing bone lesions, and reducing the metabolic activity of pagetic bone in patients who cannot receive bisphosphonates. It is given by injection in doses of 50 to 100 IU daily or every other day. Although unlikely to effect a sustained clinical remission, calcitonin remains a safe, well-tolerated, and well-studied medication in Paget disease and is approved for this indication.64,65
Denosumab has not been formally studied in Paget disease, but a recent case report indicated it was effective.66
A conservative strategy
Guidelines for treating Paget disease have been written at various times in many countries, including Italy (2007),67 the United Kingdom (2004),68 Japan (2006),69 and Canada (2007).70 Recommendations differ, in part because it is hard to ascertain whether long-term outcomes are improved by treatment, and in part because the prevalence of Paget disease is decreasing and its severity is lessening.11,12 Some guidelines are outdated, since they do not include the newer bisphosphonates.
If the natural history of untreated Paget disease involves the gradual evolution over more than 20 years of bowing deformities in the lower limbs, rigidity and overgrowth of the spine, and softening and enlargement of the skull, as described by Sir James Paget, then treatment should be initiated in hopes that it will modify the outcome. We have no lens to better focus this question on the effect of treatment on the natural history of the disease. We have the PRISM study, designed before zoledronic acid was approved and only 2 to 5 years in duration. And we have the epidemiologic data demonstrating that most patients have no symptoms during their lifetime.
We see the crippling bone disease described by Sir James Paget so infrequently today in the United States that we forget the profound morbidity that may attend the skeletal changes of Paget disease that were common in the early 20th century. Once the bones of the skull are overgrown, the limbs are bowed, and the degenerative joint disease is present, no medication can reverse these changes. Then, the integrity of the bone is lost, and the vulnerability to fracture, early osteoarthritis, nerve compression syndromes, and hearing loss persist. Understanding these consequences prompts the recommendation of early treatment in patients with Paget disease, in hopes of mitigating disease progression.
Patients with active Paget disease, documented either by an elevated serum alkaline phosphatase or by a bone scan, should be treated with a bisphosphonate if the disease is found in sites where remodeling of bone may lead to complications. Such sites include the skull, spine, and long bones of the lower extremity. Paget disease of bone in the pelvis tends to give little trouble (Figure 2) unless it is proximal to a joint, when pain and early arthritis may result. Treatment is safe and, I think, prudent to undertake in any person over age 55 with active disease. To prevent hypocalcemia during treatment, all patients should be repleted with vitamin D and maintained on calcium 1,200 mg daily through diet or supplements with meals.
Throughout the evaluation and treatment, it is important to remember that pain may not emanate from pagetic bone. If medication for Paget disease proves ineffective in the first few months, analgesics, bracing, walking aids, and operative management71 are adjunctive therapies to improve the functional status of these patients.
It is a remarkable clinical observation that treatment of Paget disease may rapidly reverse neurologic syndromes, resolve the erythema or warmth overlying active pagetic bone, and diminish the risk of bleeding with surgery. This response to therapy suggests that there is prompt inhibition and apoptosis of the osteoclasts, accompanied by diminished vascularity of bone. Whatever the mechanism, it is worth treating patients who have spinal stenosis, arthritis, and nerve compression syndromes with calcitonin or bisphosphonates before surgical intervention, whenever possible.34,72
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
- Paget J. On a form of chronic inflammation of bones (osteitis deformans). Med Chir Trans 1877; 60:37–64.9.
- Guyer PB, Chamberlain AT, Ackery DM, Rolfe EB. The anatomic distribution of osteitis deformans. Clin Orthop Relat Res 1981; 156:141–144.
- Tiegs RD, Lohse CM, Wollan PC, Melton LJ. Long-term trends in the incidence of Paget’s disease of bone. Bone 2000; 27:423–427.
- Altman RD, Bloch DA, Hochberg MC, Murphy WA. Prevalence of pelvic Paget’s disease of bone in the United States. J Bone Miner Res 2000; 15:461–465.
- Barker DJ. The epidemiology of Paget’s disease of bone. Br Med Bull 1984; 40:396–400.
- Detheridge FM, Guyer PB, Barker DJ. European distribution of Paget’s disease of bone. Br Med J (Clin Res Ed) 1982; 285:1005–1008.
- van Staa TP, Selby P, Leufkens HG, Lyles K, Sprafka JM, Cooper C. Incidence and natural history of Paget’s disease of bone in England and Wales. J Bone Miner Res 2002; 17:465–471.
- Barker DJ. The epidemiology of Paget’s disease. Metab Bone Dis Relat Res 1981; 3:231–233.
- Rogers J, Jeffrey DR, Watt I. Paget’s disease in an archeological population. J Bone Miner Res 2002; 17:1127–1134.
- Aaron JE, Rogers J, Kanis JA. Paleohistology of Paget’s disease in two medieval skeletons. Am J Phys Anthropol 1992; 89:325–331.
- Poór G, Donáth J, Fornet B, Cooper C. Epidemiology of Paget’s disease in Europe: the prevalence is decreasing. J Bone Miner Res 2006; 21:1545–1549.
- Cundy HR, Gamble G, Wattie D, Rutland M, Cundy T. Paget’s disease of bone in New Zealand: continued decline in disease severity. Calcif Tissue Int 2004; 75:358–364.
- Doyle T, Gunn J, Anderson G, Gill M, Cundy T. Paget’s disease in New Zealand: evidence for declining prevalence. Bone 2002; 31:616–619.
- Laurin N, Brown JP, Morissette J, Raymond V. Recurrent mutation of the gene encoding sequestosome 1 (SQSTM1/p62) in Paget disease of bone. Am J Hum Genet 2002; 70:1582–1588.
- Hocking LJ, Lucas GJ, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet 2002; 11:2735–2739.
- Lucas GJ, Hocking LJ, Daroszewska A, et al. Ubiquitin-associated domain mutations of SQSTM1 in Paget’s disease of bone: evidence for a founder effect in patients of British descent. J Bone Miner Res 2005; 20:227–231.
- Mays S. Archaeological skeletons support a northwest European origin for Paget’s disease of bone. J Bone Miner Res 2010; 25:1839–1841.
- Bolland MJ, Tong PC, Naot D, et al. Delayed development of Paget’s disease in offspring inheriting SQSTM1 mutations. J Bone Miner Res 2007; 22:411–415.
- Rea SL, Walsh JP, Ward L, et al. A novel mutation (K378X) in the sequestosome 1 gene associated with increased NF-kappaB signaling and Paget’s disease of bone with a severe phenotype. J Bone Miner Res 2006; 21:1136–1145.
- Morissette J, Laurin N, Brown JP. Sequestosome 1: mutation frequencies, haplotypes, and phenotypes in familial Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P38–P44.
- Eekhoff EW, Karperien M, Houtsma D, et al. Familial Paget’s disease in The Netherlands: occurrence, identification of new mutations in the sequestosome 1 gene, and their clinical associations. Arthritis Rheum 2004; 50:1650–1654.
- Ralston SH, Layfield R. Pathogenesis of Paget disease of bone. Calcif Tissue Int 2012; 91:97–113.
- Kurihara N, Hiruma Y, Yamana K, et al. Contributions of the measles virus nucleocapsid gene and the SQSTM1/p62(P392L) mutation to Paget’s disease. Cell Metab 2011; 13:23–34.
- Kurihara N, Zhou H, Reddy SV, et al. Expression of measles virus nucleocapsid protein in osteoclasts induces Paget’s disease-like bone lesions in mice. J Bone Miner Res 2006; 21:446–455.
- Reddy SV, Singer FR, Roodman GD. Bone marrow mononuclear cells from patients with Paget’s disease contain measles virus nucleocapsid messenger ribonucleic acid that has mutations in a specific region of the sequence. J Clin Endocrinol Metab 1995; 80:2108–2111.
- Gennari L, Merlotti D, Martini G, Nuti R. Paget’s disease of bone in Italy. J Bone Miner Res 2006; 21(suppl 2):P14–P21.
- Seton M, Choi HK, Hansen MF, Sebaldt RJ, Cooper C. Analysis of environmental factors in familial versus sporadic Paget’s disease of bone—the New England Registry for Paget’s Disease of Bone. J Bone Miner Res 2003; 18:1519–1524.
- Siris ES. Extensive personal experience: Paget’s disease of bone. J Clin Endocrinol Metab 1995; 80:335–338.
- Lucas GJ, Daroszewska A, Ralston SH. Contribution of genetic factors to the pathogenesis of Paget’s disease of bone and related disorders. J Bone Miner Res 2006; 21(suppl 2):P31–P37.
- Seton M. Diagnosis, complications and treatment of Paget’s disease of bone. Aging Health 2009; 5:497–508.
- Siris E, Roodman GD. Paget’s Disease of Bone. 7th ed. Washington, DC: American Society for Bone and Mineral Research; 2008.
- Seton M, Moses AM, Bode RK, Schwartz C. Paget’s disease of bone: the skeletal distribution, complications and quality of life as perceived by patients. Bone 2011; 48:281–285.
- Seton M. Paget’s disease of bone. In:Hochberg MC, Silman AJ, Smolen JS, Weinblatt ME, Weisman MH, editors. Rheumatology. 5th ed. Philadelphia, PA: Mosby Elsevier; 2010:2021–2028.
- Douglas DL, Duckworth T, Kanis JA, Jefferson AA, Martin TJ, Russell RG. Spinal cord dysfunction in Paget’s disease of bone. Has medical treatment a vascular basis? J Bone Joint Surg Br 1981; 63B:495–503.
- Siris ES. Epidemiological aspects of Paget’s disease: family history and relationship to other medical conditions. Semin Arthritis Rheum 1994; 23:222–225.
- Kanis JA, Evanson JM, Russell RG. Paget’s disease of bone: diagnosis and management. Metab Bone Dis Relat Res 1981; 3:219–230.
- Mangham DC, Davie MW, Grimer RJ. Sarcoma arising in Paget’s disease of bone: declining incidence and increasing age at presentation. Bone 2009; 44:431–436.
- Hansen MF, Seton M, Merchant A. Osteosarcoma in Paget’s disease of bone. J Bone Miner Res 2006; 21(suppl 2):P58–P63.
- Price CH. The incidence of osteogenic sarcoma in South-West England and its relationship to Paget’s disease of bone. J Bone Joint Surg Br 1962; 44-B:366–376.
- Ishikawa Y, Tsukuma H, Miller RW. Low rates of Paget’s disease of bone and osteosarcoma in elderly Japanese. Lancet 1996; 347:1559.
- Sun SG, Lau YS, Itonaga I, Sabokbar A, Athanasou NA. Bone stromal cells in pagetic bone and Paget’s sarcoma express RANKL and support human osteoclast formation. J Pathol 2006; 209:114–120.
- Rendina D, Gennari L, De Filippo G, et al. Evidence for increased clinical severity of familial and sporadic Paget’s disease of bone in Campania, southern Italy. J Bone Miner Res 2006; 21:1828–1835.
- Fenton P, Resnick D. Metastases to bone affected by Paget’s disease. A report of three cases. Int Orthop 1991; 15:397–399.
- Tu SM, Som A, Tu B, Logothetis CJ, Lee MH, Yeung SC. Effect of Paget’s disease of bone (osteitis deformans) on the progression of prostate cancer bone metastasis. Br J Cancer 2012; 107:646–651.
- Eekhoff ME, van der Klift M, Kroon HM, et al. Paget’s disease of bone in The Netherlands: a population-based radiological and biochemical survey—the Rotterdam Study. J Bone Miner Res 2004; 19:566–570.
- Reid IR, Davidson JS, Wattie D, et al. Comparative responses of bone turnover markers to bisphosphonate therapy in Paget’s disease of bone. Bone 2004; 35:224–230.
- Alvarez L, Guañabens N, Peris P, et al. Usefulness of biochemical markers of bone turnover in assessing response to the treatment of Paget’s disease. Bone 2001; 29:447–452.
- Cundy T, Reid IR. Paget’s disease of bone. Clin Biochem 2012; 45:43–48.
- Cortis K, Micallef K, Mizzi A. Imaging Paget’s disease of bone—from head to toe. Clin Radiol 2011; 66:662–672.
- Redden JF, Dixon J, Vennart W, Hosking DJ. Management of fissure fractures in Paget’s disease. Int Orthop 1981; 5:103–106.
- Case records of the Massachusetts General Hospital. Weekly clinicopathological exercises. Case 25-1993. A 67-year-old man with osteolytic lesions of T11 and T12. N Engl J Med 1993; 328:1836–1841.
- Evans RA, Dunstan CR, Hills E, Wong SY. Pathologic fracture due to severe osteomalacia following low-dose diphosphonate treatment of Paget’s disease of bone. Aust N Z J Med 1983; 13:277–279.
- Siris E, Weinstein RS, Altman R, et al. Comparative study of alendronate versus etidronate for the treatment of Paget’s disease of bone. J Clin Endocrinol Metab 1996; 81:961–967.
- Reid IR, Siris E. Alendronate in the treatment of Paget’s disease of bone. Int J Clin Pract Suppl 1999; 101:62–66.
- Miller PD, Brown JP, Siris ES, Hoseyni MS, Axelrod DW, Bekker PJ. A randomized, double-blind comparison of risedronate and etidronate in the treatment of Paget’s disease of bone. Paget’s Risedronate/Etidronate Study Group. Am J Med 1999; 106:513–520.
- Peris P, Alvarez L, Vidal S, Martínez MA, Monegal A, Guañabens N. Treatment with tiludronate has a similar effect to risedronate on Paget’s disease activity assessed by bone markers and bone scintigraphy. Clin Exp Rheumatol 2007; 25:206–210.
- Reid IR, Miller P, Lyles K, et al. Comparison of a single infusion of zoledronic acid with risedronate for Paget’s disease. N Engl J Med 2005; 353:898–908.
- Reid IR, Lyles K, Su G, et al. A single infusion of zoledronic acid produces sustained remissions in Paget disease: data to 6.5 years. J Bone Miner Res 2011; 26:2261–2270.
- Langston AL, Campbell MK, Fraser WD, MacLennan GS, Selby PL, Ralston SH; PRISM Trial Group. Randomized trial of intensive bisphosphonate treatment versus symptomatic management in Paget’s disease of bone. J Bone Miner Res 2010; 25:20–31.
- Abrahamsen B, Einhorn TA. Beyond a reasonable doubt? Bisphosphonates and atypical femur fractures. Bone 2012; 50:1196–1200.
- Seton M, Krane SM. Use of zoledronic acid in the treatment of Paget’s disease. Ther Clin Risk Manag 2007; 3:913–918.
- Sørensen HT, Christensen S, Mehnert F, et al. Use of bisphosphonates among women and risk of atrial fibrillation and flutter: Population based case-control study. BMJ 2008; 336:813–816.
- Dixon WG, Solomon DH. Bisphosphonates and esophageal cancer—a pathway through the confusion. Nat Rev Rheumatol 2011; 7:369–372.
- Singer FR, Krane SM. Paget’s disease of bone. In:Avioli LV, Krane SM, editors. Metabolic Bone Disease and Clinically Related Disorders. 2nd ed. Philadelphia, PA: W.B. Saunders Company; 1990:546–615.
- Kanis JA, Horn DB, Scott RD, Strong JA. Treatment of Paget’s disease of bone with synthetic salmon calcitonin. Br Med J 1974; 3:727–731.
- Schwarz P, Rasmussen AQ, Kvist TM, Andersen UB, Jørgensen NR. Paget’s disease of the bone after treatment with denosumab: a case report. Bone 2012; 50:1023–1025.
- Adami S, Bartolozzi P, Brandi ML, et al; Societa Italiana di Ortopedia e Traumatologia. [Italian guidelines for the diagnosis and treatment of Paget’s disease of bone.] Reumatismo 2007; 59:153–168. (Article in Italian.)
- Scarsbrok A, Brown M, Wilson D. UK guidelines on management of Paget’s disease of bone. Rheumatology (Oxford) 2004; 43:399–400.
- Takata S, Hashimoto J, Nakatsuka K, et a.l Guidelines for diagnosis and management of Paget’s disease of bone in Japan. J Bone Miner Metab 2006; 24:359–367.
- Josse RG, Hanley DA, Kendler D, Ste Marie L-G, Adachi JD, Brown J. Diagnosis and treatment of Paget’s disease of bone. Clin Invest Med 2007; 30:E210–E223.
- Kaplan FS. Paget’s disease of bone: orthopedic complications. Semin Arthritis Rheum 1994; 23:250–252.
- Kanis JA, Gray RE. Long-term follow-up observations on treatment in Paget’s disease of bone. Clin Orthop Relat Res 1987; 217:99–125.
KEY POINTS
- The variable prevalence of Paget disease in different geographic regions and its sometimes-familial expression suggest a genetic predisposition or an environmental factor, or both.
- Because Paget disease tends to occur in an aging skeleton, “pagetic” bone may not always be the source of pain. Rather, the pain may be from secondary degenerative changes of the spine or joints or from compression fractures.
- An elevated serum alkaline phosphatase level may signal Paget disease, but many patients have a normal serum alkaline phosphatase.
- Plain radiography of the affected bones outlines the anatomy of the problem and provides insight into the cause of pain.
- Treatment of Paget disease relies primarily on the new generation of nitrogen-containing bisphosphonates.

Practical management of bleeding due to the anticoagulants dabigatran, rivaroxaban, and apixaban
In the past several years, three new oral anticoagulants—dabigatran etexilate (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis)—have been approved for use in the United States. These long-awaited agents are appealing because they are easy to use, do not require laboratory monitoring, and have demonstrated equivalence, or in some cases, superiority to warfarin in preventing stroke or systemic embolism in at-risk populations.1–4 However, unlike warfarin, they have no specific reversal agents. How then should one manage spontaneous bleeding problems and those due to drug overdose, and how can we quickly reverse anticoagulation if emergency surgery is needed?
For these reasons, physicians and patients have been wary of these agents. However, with a systematic approach based on an understanding of the properties of these drugs, the appropriate use and interpretation of coagulation tests, and awareness of available therapeutic strategies, physicians can more confidently provide care for patients who require urgent reversal of anticoagulant effects.
Here, we review the available literature and suggest practical strategies for management based on an understanding of the pharmacokinetic and pharmacodynamic effects of these drugs and our current knowledge of the coagulation tests.
NEED FOR ANTICOAGULANTS
Anticoagulants are important in preventing systemic embolization in patients with atrial fibrillation and preventing pulmonary embolism in patients with venous thromboembolism.
And the numbers are staggering. The estimated prevalence of atrial fibrillation in the United States was 3.03 million in 2005 and is projected to increase to 7.56 million by 2050.5 Ischemic stroke is the most serious complication of atrial fibrillation, which accounts for 23.5% of strokes in patients ages 80 through 89 according to Framingham data.6 Venous thromboembolism accounts for 900,000 incident or recurrent fatal and nonfatal events in the United States yearly.7
HOW THE NEW AGENTS BLOCK COAGULATION
Thrombin (factor IIa), a serine protease, is central to the process of clot formation during hemostasis. It activates factors V, VIII, and XI (thus generating more thrombin), catalyzes the conversion of fibrinogen to fibrin, and stimulates platelet aggregation. Its role in the final steps of the coagulation cascade has made it a target for new direct thrombin inhibitors such as dabigatran.
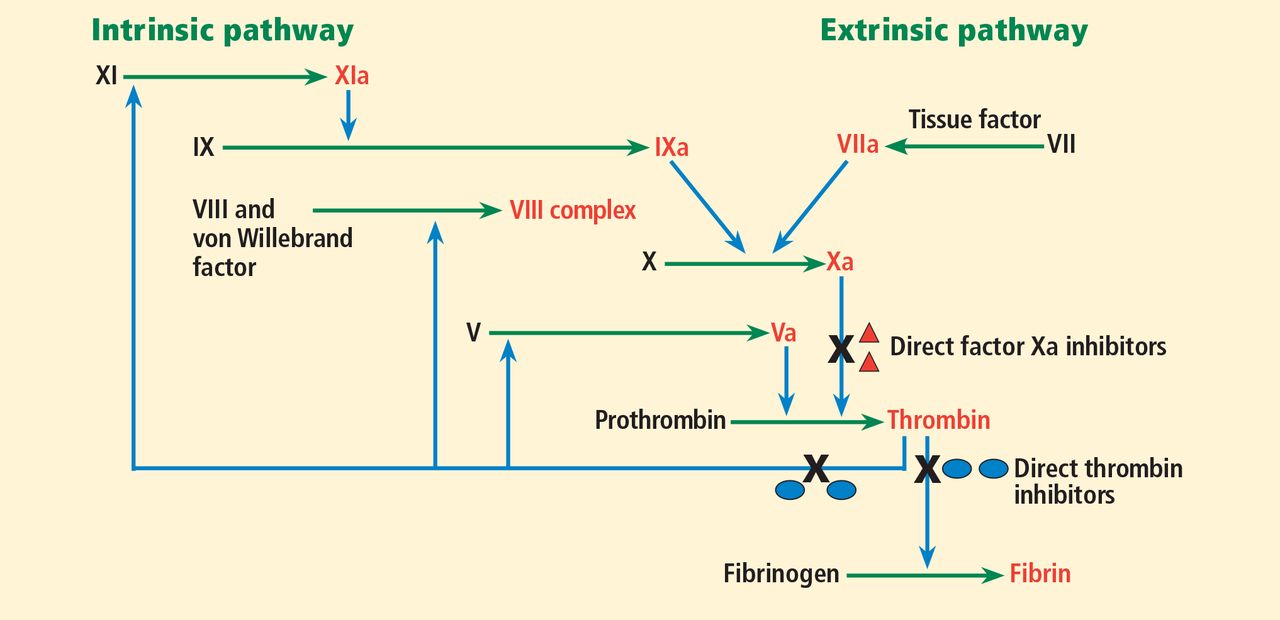
Factor Xa is a serine protease that plays a central role in the coagulation cascade. It is a desirable target for anticoagulation because it is the convergence point for the extrinsic and the intrinsic coagulation pathways. It converts prothrombin to thrombin. Rivaroxaban and apixaban are direct factor Xa inhibitors (Figure 1).
Dabigatran, a direct thrombin inhibitor
Dabigatran etexilate is a synthetic, orally available prodrug that is rapidly absorbed and converted by esterases to its active form, dabigatran, a potent direct inhibitor of both free thrombin and clot-bound thrombin.8

Plasma levels of dabigatran peak within 2 hours of administration, and its half-life is 14 to 17 hours.9 Dabigatran is eliminated mainly via the kidneys, with more that 80% of the drug excreted unchanged in the urine (Table 1).
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is a potent, selective, direct factor Xa inhibitor.
Plasma levels of rivaroxaban peak 2 to 3 hours after administration, and it is cleared with a terminal half-life of 7 to 11 hours.10,11
Rivaroxaban is eliminated by the kidneys and in the feces. The kidneys eliminate one-third of the active drug unchanged and another one-third as inactive metabolites. The remaining one-third is metabolized by the liver and then excreted in the feces. Rivaroxaban has a predictable and dose-dependent pharmacodynamic and pharmacokinetic profile that is not affected by age, sex, or body weight (Table 1).12
Apixaban, an oral factor Xa inhibitor
Apixaban is a selective, direct oral factor Xa inhibitor.
Plasma levels of apixaban peak about 3 hours after administration, and its terminal half-life is 8 to 14 hours.13 Apixaban is eliminated by oxidative metabolism, by the kidney, and in the feces. It has predictable pharmacodynamic and pharmacokinetic profiles and has the least renal dependence of the three agents (Table 1).
THE NEW ORAL ANTICOAGULANTS AND BLOOD COAGULATION ASSAYS
Assessment of the anticoagulant activity of the new oral anticoagulants is not necessary in routine clinical practice, but it may be useful in planning intervention in patients with major bleeding, those with drug overdose, or those who need emergency surgery.
The activated partial thromboplastin time
The activated partial thromboplastin time (aPTT) is a measure of the activity of the intrinsic pathway of the coagulation cascade.
Dabigatran. There is a curvilinear relationship between the aPTT and the plasma concentration of dabigatran and other direct thrombin inhibitors, although the aPTT prolongation appears to vary with different reagents and coagulometers.9,14,15 However, Stangier et al9 found a linear relationship between the aPTT and the square root of the dabigatran plasma concentration.
Rivaroxaban prolongs the aPTT in a dose-dependent manner, but there is no standard for calibration of this assay. Hence, the aPTT is not recommended for monitoring rivaroxaban in clinical practice.
Apixaban may also prolong the aPTT, but there are limited data on its reactivity with different reagents.
The prothrombin time and international normalized ratio
The prothrombin time and international normalized ratio (INR) are measures of the extrinsic pathway of the coagulation cascade.
Dabigatran. The INR has a linear response to the dabigatran concentration, but it is insensitive.9 Hence, it is not suitable for monitoring the anticoagulant effects of direct thrombin inhibitors.
Rivaroxaban. The prothrombin time correlates strongly with the plasma concentration of rivaroxaban in healthy trial participants11 and in patients undergoing total hip arthroplasty or total knee arthroplasty.16 Samama et al17 noted that, unlike with vitamin K antagonists, the INR cannot be used to monitor patients on rivaroxaban because the prothrombin time results varied with different reagents. They used a standard calibration curve to express the prothrombin time results in plasma concentrations of rivaroxaban rather than in seconds or the INR.
Apixaban increases the INR in a dose-dependent manner.18 Its effect on different reagents remains unknown.
The thrombin time
The thrombin time reflects the activity of thrombin in the plasma. The amount of thrombin and the concentration of thrombin inhibitors in the plasma sample determine the time to clot formation.
Dabigatran. The thrombin time displays a linear dose-response to dabigatran, but only over the range of therapeutic concentrations. At a dabigatran concentration greater than 600 ng/mL, the test often exceeds the maximum measurement time of coagulometers.9 Hence, this test is too sensitive for emergency monitoring, especially in cases of drug overdose. However, it is well suited for determining if any dabigatran is present.
Rivaroxaban and apixaban have no effect on the thrombin time.
The Hemoclot direct thrombin inhibitor assay and dabigatran
The Hemoclot direct thrombin inhibitor assay (Hyphen BioMed, France) is a sensitive diluted thrombin time assay that can be used for quantitative measurement of dabigatran activity in plasma. This test is based on inhibition of a constant amount of highly purified human alpha-thrombin by adding it to diluted test plasma (1:8 to 1:20) mixed with normal pooled human plasma.19,20
Stangier et al19 found that the Hemoclot assay was suitable for calculating a wide range of dabigatran concentrations up to 4,000 nmol/L (1,886 ng/mL). Although this finding has not been confirmed in larger studies, this test may provide a rapid and accurate assessment of dabigatran’s anticoagulant activity in cases of emergency surgery or overdose.
The ecarin clotting time and dabigatran
The ecarin clotting time is a measure of the activity of direct thrombin inhibitors, but not the factor Xa inhibitors.
Ecarin is a highly purified metalloprotease isolated from the venom of a snake, Echis carinatus, and it generates meizothrombin from prothrombin.21 Meizothrombin facilitates clot formation by converting fibrinogen to fibrin and, like thrombin, it can be inactivated by direct thrombin inhibitors, thereby prolonging the clotting time.
The limitations of the ecarin clotting time include dependence on the plasma levels of fibrinogen and prothrombin.
The ecarin chromogenic assay and dabigatran
The ecarin chromogenic assay is an improvement on the principle of the ecarin clotting time that can be used to measure the activity of direct thrombin inhibitors.22 In this test, ecarin is added to a plasma sample to generate meizothrombin, and the amidolytic activity of meizothrombin towards a chromogenic substrate is then determined.
Results of the ecarin chromogenic assay are not influenced by the levels of fibrinogen or prothrombin. Another advantage is that this assay can be used in automated and manual analyzers, thus enabling its use at the bedside. However, to our knowledge, it is not being regularly used to monitor direct thrombin inhibitors in the clinical setting, and there is no standard calibration of the ecarin clotting time method.
Assays of factor Xa activity
A variety of assays to monitor the anticoagulant activity of factor Xa inhibitors have been proposed.23–25 All measure inhibition of the activity of factor Xa using methods similar to those used in monitoring heparin levels. All require calibrators with a known concentration of the Xa inhibitor; many are easily adapted for laboratories currently providing measurement of factor Xa inhibition from heparin.23 These assays have been suggested as a better indicator of plasma concentration of factor Xa inhibitor drugs than the prothrombin time.25
CONTROLLING BLEEDING IN PATIENTS ON THE NEW ORAL ANTICOAGULANTS
Bleeding is an anticipated adverse event in patients taking anticoagulants. It is associated with significant morbidity and risk of death.26,27
Many physicians still have limited experience with using the new oral anticoagulants and managing the attendant bleeding risks. Hence, we recommend that every health institution have a treatment policy or algorithm to guide all clinical staff in the management of such emergencies.
Prevention of bleeding
Management of bleeding from these agents should begin with preventing bleeding in the first place.
The physician should adhere to the recommended dosages of these medications. Studies have shown that the plasma concentration of these drugs and the risk of bleeding increase with increasing dosage.1,28,29
In addition, these medications should be used for the shortest time for which anticoagulation is required, especially when used for preventing deep vein thrombosis. Prolonged use increases the risk of bleeding.30,31
Most patients who need anticoagulation have comorbidities such as heart failure, renal failure, diabetes mellitus, and hypertension. Although the kidneys play a major role in the excretion of dabigatran and, to some extent, rivaroxaban and apixaban, patients with severe renal impairment were excluded from the major trials of all three drugs.1–3 Hence, to avoid excessive drug accumulation and bleeding, these medications should not be used in such patients pending further studies. Further, patients taking these medications should be closely followed to detect new clinical situations, such as acute renal failure, that will necessitate their discontinuation or dose adjustment.
If surgery is needed
If a patient taking a new oral anticoagulant needs to undergo elective surgery, it is important to temporarily discontinue the drug, assess the risk of bleeding, and test for renal impairment.
Renal impairment is particularly relevant in the case of dabigatran, since more than 80% of the unchanged drug is cleared by the kidneys. Decreasing the dose, prolonging the dosing interval, or both have been suggested as means to reduce the risk of bleeding in patients with renal impairment who are taking dabigatran.32,33 Patients with normal renal function undergoing low-risk surgery should discontinue dabigatran at least 24 hours before the surgery. If the creatinine clearance is 31 to 50 mL/min, inclusively, the last dose should be at least 48 hours before the procedure for low-risk surgery, and 4 days before a procedure that poses a high risk of bleeding.32–34 Some experts have given the same recommendations for rivaroxaban and apixaban (Table 2).34
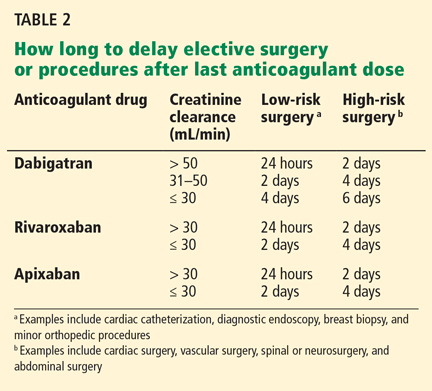
The aPTT and prothrombin time are readily available tests, but they cannot determine the residual anticoagulant effects of dabigatran, rivaroxaban, or apixaban. However, in many (but not all) cases, a normal aPTT suggests that the hemostatic function is not impaired by dabigatran, and a normal prothrombin time or an absence of anti-factor Xa activity would similarly exclude hemostatic dysfunction caused by rivaroxaban or apixaban. These tests are potentially useful as adjuncts before surgical procedures that require complete hemostasis.
Furthermore, a normal thrombin time rules out the presence of a significant amount of dabigatran. Therefore, a normal thrombin time might be particularly useful in a patient undergoing a high-risk intervention such as epidural cannulation or neurosurgery and who is normally receiving dabigatran.
Managing overdose and bleeding complications

Assessing the severity of bleeding is the key to managing bleeding complications (Table 3).
Minor bleeding such as epistaxis and ecchymosis can be managed symptomatically (eg, with nasal packing), perhaps with short-term withdrawal of the anticoagulant. Moderate bleeding such as upper or lower gastrointestinal bleeding can be managed by withdrawal of the anticoagulant, clinical monitoring, blood transfusion if needed, and treatment directed at the etiology.
Major and life-threatening bleeding (eg, intracerebral hemorrhage) requires aggressive treatment in the intensive care unit, withdrawal of the anticoagulant, mechanical compression of the bleeding site if accessible, fluid replacement and blood transfusion as appropriate, and interventional procedures. Nonspecific reversal agents might be considered in patients with major or life-threatening bleeding.
The half-life of dabigatran after multiple doses is approximately 14 to 17 hours and is not dose-dependent.9 Hence, if there is no active bleeding after a dabigatran overdose, stopping the drug may be sufficient. Since the pharmacodynamic effect of dabigatran declines in parallel to its plasma concentration, urgent but not emergency surgery may need to be delayed for only about 12 hours from the last dose of dabigatran.
The 2011 American College of Cardiology Foundation/American Heart Association guidelines recommend that patients with severe hemorrhage resulting from dabigatran should receive supportive therapy, including transfusion of fresh-frozen plasma, transfusion of packed red blood cells, or surgical intervention if appropriate.35 However, transfusion of fresh-frozen plasma is debatable because there is no evidence to support its use in this situation. While fresh-frozen plasma may be useful in cases of coagulation factor depletion, it does not effectively reverse inhibition of coagulation factors.36
Off-label use of nonspecific hemostatic agents
To date, no specific agent has been demonstrated to reverse excessive bleeding in patients taking the new oral anticoagulants. However, in view of their procoagulant capabilities, nonspecific hemostatic agents have been suggested for use in reversal of major bleeding resulting from these drugs.37–39 Examples are:
Recombinant factor VIIa (NovoSeven) initiates thrombin generation by activating factor X.
Four-factor prothrombin complex concentrate (Beriplex, recently approved in the United States) contains relatively large amounts of four nonactive vitamin K-dependent procoagulant factors (factors II, VII, IX, and X) that stimulate thrombin formation.
Three-factor prothrombin complex concentrate (Bebulin VH and Profilnine SD) contains low amounts of nonactive factor VII relative to factors II, IX, and X. In some centers a four-factor equivalent is produced by transfusion of a three-factor product with the addition of small amounts of recombinant factor VIIa or fresh-frozen plasma to replace the missing factor VII.40
Activated prothrombin complex concentrate (FEIBA NF) contains activated factor VII and factors II, IX, and X, mainly in nonactivated form.36 Therefore, it combines the effect of both recombinant factor VIIa and four-factor prothrombin complex concentrate.37
Studies of nonspecific hemostatic agents
In a study of rats infused with high doses of dabigatran, van Ryn et al38 observed that activated prothrombin complex concentrate at a dose of 50 or 100 U/kg and recombinant factor VIIa at a dose of 0.1 or 0.5 mg/kg reduced the rat-tail bleeding time in a dose-dependent manner but not the blood loss, compared with controls, even with a higher dose of recombinant factor VIIa (1 mg/kg). Recombinant factor VIIa also reversed the prolonged aPTT induced by dabigatran, whereas activated prothrombin complex concentrate did not. They suggested that recombinant factor VIIa and activated prothrombin complex concentrate may be potential antidotes for dabigatran-induced severe bleeding in humans.
In an ex vivo study of healthy people who took a single dose of dabigatran 150 mg or rivaroxaban 20 mg, Marlu et al37 found that activated prothrombin complex concentrate and four-factor prothrombin complex concentrate could be reasonable antidotes to these drugs.
Dabigatran-associated bleeding after cardiac surgery in humans has been successfully managed with hemodialysis and recombinant factor VIIa, although the efficacy of the latter cannot be individually assessed in the study.41
In a randomized placebo-controlled trial aimed at reversing rivaroxaban and dabigatran in healthy participants, Eerenberg et al39 showed that four-factor prothrombin complex concentrate at a dose of 50 IU/kg reversed prolongation of the prothrombin time and decreased the endogenous thrombin potential in those who received rivaroxaban, but it failed to reverse the aPTT, the endogenous thrombin potential, and thrombin time in those who received dabigatran.
However, Marlu et al reported that four-factor prothrombin complex concentrate at three doses (12.5 U/kg, 25 U/kg, and 50 U/kg)—or better still, activated prothrombin complex concentrate (40–80 U/kg)—could be a useful antidote to dabigatran.37
It is important to note that the healthy participants in the Eerenberg et al study39 took dabigatran 150 mg twice daily and rivaroxaban 20 mg daily for 2.5 days, whereas those in the Marlu et al study37 took the same dose of each medication, but only once.
The three-factor prothrombin complex concentrate products have been shown to be less effective than four-factor ones in reversing supratherapeutic INRs in patients with warfarin overdose, but whether this will be true with the new oral anticoagulants remains unknown. Furthermore, the four-factor concentrates effectively reversed warfarin-induced coagulopathy and bleeding in patients,42 but to our knowledge, the same is yet to be demonstrated in bleeding related to the newer agents.
Other measures
Gastric lavage or the administration of activated charcoal (or in some cases both) may reduce drug absorption if done within 2 or 3 hours of drug ingestion (Table 1). Because it is lipophilic, more than 99.9% of dabigatran etexilate was adsorbed by activated charcoal from water prepared to simulate gastric fluid in an in vitro experiment by van Ryn et al.43 This has not been tested in patients, and no similar study has been done for rivaroxaban or apixaban. However, use of charcoal in cases of recent ingestion, particularly with intentional overdose of these agents, seems reasonable.
Hemodialysis may reverse the anticoagulant effects of dabigatran overdose or severe bleeding because only about 35% of dabigatran is bound to plasma proteins (Table 1). In a single-center study, 50 mg of dabigatran etexilate was given orally to six patients with end-stage renal disease before dialysis, and the mean fraction of the drug removed by the dialyzer was 62% at 2 hours and 68% at 4 hours.32 This study suggests that hemodialysis may be useful to accelerate the removal of the drug in cases of life-threatening bleeding.
Rivaroxaban and apixaban are not dialyzable: the plasma protein binding of rivaroxaban is 95% and that of apixaban is 87%.
FUTURE DIRECTIONS
Because the new oral anticoagulants, unlike warfarin, have a wide therapeutic window, routine anticoagulant monitoring is not needed and might be misleading. However, there are times when monitoring might be useful; at such times, a validated, widely available, easily understood test would be good to have—but we don’t have it—at least not yet.
Therapeutic ranges for the aPTT have been established empirically for heparin in various indications.44 Additional study is needed to determine if an appropriate aPTT range can be determined for the new oral anticoagulants, particularly dabigatran.
Similarly, as with low-molecular-weight heparins, anti-factor Xa activity monitoring may become a more available validated means of testing for exposure to rivaroxaban and apixaban. More promising, using concepts derived from the development of the INR for warfarin monitoring,45 Tripodi et al46 have derived normalized INR-like assays to report rivaroxaban levels. A standardized schema for reporting results is being developed.46 Studies are required to determine if and how this assay may be useful. Initial trials in this regard are encouraging.47
Finally, the thrombotic risk associated with the use of nonspecific prohemostatic agents is unknown.37,48 Additional studies are required to standardize their dosages, frequency of administration, and duration of action, as well as to quantify their complications in bleeding patients.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke 1991; 22:983–988.
- Heit JA, Cohen AT, Anderson FA; on behalf of the VTE Impact Assessment Group. Estimated annual number of incident and recurrent, non-fatal and fatal venous thromboembolism (VTE) events in the US. Blood (ASH Annual Meeting Abstracts) 2005; 106:abstract 910.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in healthy subjects. Int J Clin Pharmacol Ther 2007; 45:335–344.
- Weitz JI, Eikelboom JW, Samama MM; American College of Chest Physicians. New antithrombotic drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e120S–e151S.
- Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos 2009; 37:74–81.
- Cullberg M, Eriksson UG, Larsson M, Karlsson MO. Population modelling of the effect of inogatran, at thrombin inhibitor, on ex vivo coagulation time (APTT) in healthy subjects and patients with coronary artery disease. Br J Clin Pharmacol 2001; 51:71–79.
- Carlsson SC, Mattsson C, Eriksson UG, et al. A review of the effects of the oral direct thrombin inhibitor ximelagatran on coagulation assays. Thromb Res 2005; 115:9–18.
- Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in patients undergoing major orthopaedic surgery. Clin Pharmacokinet 2008; 47:203–216.
- Samama MM, Martinoli JL, LeFlem L, et al. Assessment of laboratory assays to measure rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2010; 103:815–825.
- Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 2008; 6:820–829.
- Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis 2012; 23:138–143.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Nowak G. The ecarin clotting time, a universal method to quantify direct thrombin inhibitors. Pathophysiol Haemost Thromb 2003–2004; 33:173–183.
- Lange U, Nowak G, Bucha E. Ecarin chromogenic assay—a new method for quantitative determination of direct thrombin inhibitors like hirudin. Pathophysiol Haemost Thromb 2003–2004; 33:184–191.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Anti-Factor Xa Chromogenic Assay Field Trial Laboratories. Evaluation of the anti-factor Xa chromogenic assay for the measurement of rivaroxaban plasma concentrations using calibrators and controls. Thromb Haemost 2012; 107:379–387.
- Miyares MA, Davis K. Newer oral anticoagulants: a review of laboratory monitoring options and reversal agents in the hemorrhagic patient. Am J Health Syst Pharm 2012; 69:1473–1484.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct factor Xa inhibitor. J Thromb Haemost 2005; 3:514–521.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- US Food and Drug Administration (FDA). Medication Guide: Pradaxa (dabigatran etexilate mesylate) capsules. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM231720.pdf. Accessed June 5, 2013.
- Schulman S, Crowther MA. How I treat with anticoagulants in 2012: new and old anticoagulants, and when and how to switch. Blood 2012; 119:3016–3023.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/ AHA/ HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol 2011; 57:1330–1337.
- Crowther MA, Warkentin TE. Managing bleeding in anticoagulated patients with a focus on novel therapeutic agents. J Thromb Haemost 2009; 7(suppl 1):107–110.
- Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost 2012; 108:217–224.
- van Ryn J, Ruehl D, Priepke H, Hauel N, Wienen W. Reversibility of the anticoagulant effect of high doses of the direct thrombin inhibitor dabigatran, by recombinant factor VIIa or activated prothrombin complex concentrate. 13th Congress of the European Hematology Association, June 12–15, 2008. Hematologica 2008; 93( s1):148Abs.0370.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Holland L, Warkentin TE, Refaai M, Crowther MA, Johnston MA, Sarode R. Suboptimal effect of a three-factor prothrombin complex concentrate (Profilnine-SD) in correcting supratherapeutic international normalized ratio due to warfarin overdose. Transfusion 2009; 49:1171–1177.
- Warkentin TE, Margetts P, Connolly SJ, Lamy A, Ricci C, Eikelboom JW. Recombinant factor VIIa (rFVIIa) and hemodialysis to manage massive dabigatran-associated postcardiac surgery bleeding. Blood 2012; 119:2172–2174.
- Song MM, Warne CP, Crowther MA. Prothrombin complex concentrate (PCC, Octaplex) in patients requiring immediate reversal of vitamin K antagonist anticoagulation. Thromb Res 2012; 129:526–529.
- van Ryn J, Sieger P, Kink-Eiband M, Gansser D, Clemens A. Adsorption of dabigatran etexilate in water or dabigatran in pooled human plasma by activated charcoal in vitro. 51st ASH Annual Meeting and Exposition. Abstract no. 1065. http://ash.confex.com/ash/2009/webprogram/Paper21383.html. Accessed June 5, 2013.
- Hirsh J. Heparin. N Engl J Med 1991; 324:1565–1574.
- van den Besselaar AMHP, Poller L, Tripodi A. Guidelines for thromboplastins and plasmas used to control for oral anticoagulant therapy. WHO Technical Report Series 1999; 889:64–93.
- Tripodi A, Chantarangkul V, Guinet C, Samama MM. The international normalized ratio calibrated for rivaroxaban has the potential to normalize prothrombin time results for rivaroxaban-treated patients: Results of an in vitro study. J Thromb Haemost 2011; 9:226–228.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Prothrombin Time Field Trial Laboratories. Evaluation of the prothrombin time for measuring rivaroxaban plasma concentrations using calibrators and controls: results of a multicenter field trial. Clin Appl Thromb Hemost 2012; 18:150–158.
- Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse events. Haemophilia 2002; 8:83–90.
In the past several years, three new oral anticoagulants—dabigatran etexilate (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis)—have been approved for use in the United States. These long-awaited agents are appealing because they are easy to use, do not require laboratory monitoring, and have demonstrated equivalence, or in some cases, superiority to warfarin in preventing stroke or systemic embolism in at-risk populations.1–4 However, unlike warfarin, they have no specific reversal agents. How then should one manage spontaneous bleeding problems and those due to drug overdose, and how can we quickly reverse anticoagulation if emergency surgery is needed?
For these reasons, physicians and patients have been wary of these agents. However, with a systematic approach based on an understanding of the properties of these drugs, the appropriate use and interpretation of coagulation tests, and awareness of available therapeutic strategies, physicians can more confidently provide care for patients who require urgent reversal of anticoagulant effects.
Here, we review the available literature and suggest practical strategies for management based on an understanding of the pharmacokinetic and pharmacodynamic effects of these drugs and our current knowledge of the coagulation tests.
NEED FOR ANTICOAGULANTS
Anticoagulants are important in preventing systemic embolization in patients with atrial fibrillation and preventing pulmonary embolism in patients with venous thromboembolism.
And the numbers are staggering. The estimated prevalence of atrial fibrillation in the United States was 3.03 million in 2005 and is projected to increase to 7.56 million by 2050.5 Ischemic stroke is the most serious complication of atrial fibrillation, which accounts for 23.5% of strokes in patients ages 80 through 89 according to Framingham data.6 Venous thromboembolism accounts for 900,000 incident or recurrent fatal and nonfatal events in the United States yearly.7
HOW THE NEW AGENTS BLOCK COAGULATION
Thrombin (factor IIa), a serine protease, is central to the process of clot formation during hemostasis. It activates factors V, VIII, and XI (thus generating more thrombin), catalyzes the conversion of fibrinogen to fibrin, and stimulates platelet aggregation. Its role in the final steps of the coagulation cascade has made it a target for new direct thrombin inhibitors such as dabigatran.
Factor Xa is a serine protease that plays a central role in the coagulation cascade. It is a desirable target for anticoagulation because it is the convergence point for the extrinsic and the intrinsic coagulation pathways. It converts prothrombin to thrombin. Rivaroxaban and apixaban are direct factor Xa inhibitors (Figure 1).
Dabigatran, a direct thrombin inhibitor
Dabigatran etexilate is a synthetic, orally available prodrug that is rapidly absorbed and converted by esterases to its active form, dabigatran, a potent direct inhibitor of both free thrombin and clot-bound thrombin.8
Plasma levels of dabigatran peak within 2 hours of administration, and its half-life is 14 to 17 hours.9 Dabigatran is eliminated mainly via the kidneys, with more that 80% of the drug excreted unchanged in the urine (Table 1).
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is a potent, selective, direct factor Xa inhibitor.
Plasma levels of rivaroxaban peak 2 to 3 hours after administration, and it is cleared with a terminal half-life of 7 to 11 hours.10,11
Rivaroxaban is eliminated by the kidneys and in the feces. The kidneys eliminate one-third of the active drug unchanged and another one-third as inactive metabolites. The remaining one-third is metabolized by the liver and then excreted in the feces. Rivaroxaban has a predictable and dose-dependent pharmacodynamic and pharmacokinetic profile that is not affected by age, sex, or body weight (Table 1).12
Apixaban, an oral factor Xa inhibitor
Apixaban is a selective, direct oral factor Xa inhibitor.
Plasma levels of apixaban peak about 3 hours after administration, and its terminal half-life is 8 to 14 hours.13 Apixaban is eliminated by oxidative metabolism, by the kidney, and in the feces. It has predictable pharmacodynamic and pharmacokinetic profiles and has the least renal dependence of the three agents (Table 1).
THE NEW ORAL ANTICOAGULANTS AND BLOOD COAGULATION ASSAYS
Assessment of the anticoagulant activity of the new oral anticoagulants is not necessary in routine clinical practice, but it may be useful in planning intervention in patients with major bleeding, those with drug overdose, or those who need emergency surgery.
The activated partial thromboplastin time
The activated partial thromboplastin time (aPTT) is a measure of the activity of the intrinsic pathway of the coagulation cascade.
Dabigatran. There is a curvilinear relationship between the aPTT and the plasma concentration of dabigatran and other direct thrombin inhibitors, although the aPTT prolongation appears to vary with different reagents and coagulometers.9,14,15 However, Stangier et al9 found a linear relationship between the aPTT and the square root of the dabigatran plasma concentration.
Rivaroxaban prolongs the aPTT in a dose-dependent manner, but there is no standard for calibration of this assay. Hence, the aPTT is not recommended for monitoring rivaroxaban in clinical practice.
Apixaban may also prolong the aPTT, but there are limited data on its reactivity with different reagents.
The prothrombin time and international normalized ratio
The prothrombin time and international normalized ratio (INR) are measures of the extrinsic pathway of the coagulation cascade.
Dabigatran. The INR has a linear response to the dabigatran concentration, but it is insensitive.9 Hence, it is not suitable for monitoring the anticoagulant effects of direct thrombin inhibitors.
Rivaroxaban. The prothrombin time correlates strongly with the plasma concentration of rivaroxaban in healthy trial participants11 and in patients undergoing total hip arthroplasty or total knee arthroplasty.16 Samama et al17 noted that, unlike with vitamin K antagonists, the INR cannot be used to monitor patients on rivaroxaban because the prothrombin time results varied with different reagents. They used a standard calibration curve to express the prothrombin time results in plasma concentrations of rivaroxaban rather than in seconds or the INR.
Apixaban increases the INR in a dose-dependent manner.18 Its effect on different reagents remains unknown.
The thrombin time
The thrombin time reflects the activity of thrombin in the plasma. The amount of thrombin and the concentration of thrombin inhibitors in the plasma sample determine the time to clot formation.
Dabigatran. The thrombin time displays a linear dose-response to dabigatran, but only over the range of therapeutic concentrations. At a dabigatran concentration greater than 600 ng/mL, the test often exceeds the maximum measurement time of coagulometers.9 Hence, this test is too sensitive for emergency monitoring, especially in cases of drug overdose. However, it is well suited for determining if any dabigatran is present.
Rivaroxaban and apixaban have no effect on the thrombin time.
The Hemoclot direct thrombin inhibitor assay and dabigatran
The Hemoclot direct thrombin inhibitor assay (Hyphen BioMed, France) is a sensitive diluted thrombin time assay that can be used for quantitative measurement of dabigatran activity in plasma. This test is based on inhibition of a constant amount of highly purified human alpha-thrombin by adding it to diluted test plasma (1:8 to 1:20) mixed with normal pooled human plasma.19,20
Stangier et al19 found that the Hemoclot assay was suitable for calculating a wide range of dabigatran concentrations up to 4,000 nmol/L (1,886 ng/mL). Although this finding has not been confirmed in larger studies, this test may provide a rapid and accurate assessment of dabigatran’s anticoagulant activity in cases of emergency surgery or overdose.
The ecarin clotting time and dabigatran
The ecarin clotting time is a measure of the activity of direct thrombin inhibitors, but not the factor Xa inhibitors.
Ecarin is a highly purified metalloprotease isolated from the venom of a snake, Echis carinatus, and it generates meizothrombin from prothrombin.21 Meizothrombin facilitates clot formation by converting fibrinogen to fibrin and, like thrombin, it can be inactivated by direct thrombin inhibitors, thereby prolonging the clotting time.
The limitations of the ecarin clotting time include dependence on the plasma levels of fibrinogen and prothrombin.
The ecarin chromogenic assay and dabigatran
The ecarin chromogenic assay is an improvement on the principle of the ecarin clotting time that can be used to measure the activity of direct thrombin inhibitors.22 In this test, ecarin is added to a plasma sample to generate meizothrombin, and the amidolytic activity of meizothrombin towards a chromogenic substrate is then determined.
Results of the ecarin chromogenic assay are not influenced by the levels of fibrinogen or prothrombin. Another advantage is that this assay can be used in automated and manual analyzers, thus enabling its use at the bedside. However, to our knowledge, it is not being regularly used to monitor direct thrombin inhibitors in the clinical setting, and there is no standard calibration of the ecarin clotting time method.
Assays of factor Xa activity
A variety of assays to monitor the anticoagulant activity of factor Xa inhibitors have been proposed.23–25 All measure inhibition of the activity of factor Xa using methods similar to those used in monitoring heparin levels. All require calibrators with a known concentration of the Xa inhibitor; many are easily adapted for laboratories currently providing measurement of factor Xa inhibition from heparin.23 These assays have been suggested as a better indicator of plasma concentration of factor Xa inhibitor drugs than the prothrombin time.25
CONTROLLING BLEEDING IN PATIENTS ON THE NEW ORAL ANTICOAGULANTS
Bleeding is an anticipated adverse event in patients taking anticoagulants. It is associated with significant morbidity and risk of death.26,27
Many physicians still have limited experience with using the new oral anticoagulants and managing the attendant bleeding risks. Hence, we recommend that every health institution have a treatment policy or algorithm to guide all clinical staff in the management of such emergencies.
Prevention of bleeding
Management of bleeding from these agents should begin with preventing bleeding in the first place.
The physician should adhere to the recommended dosages of these medications. Studies have shown that the plasma concentration of these drugs and the risk of bleeding increase with increasing dosage.1,28,29
In addition, these medications should be used for the shortest time for which anticoagulation is required, especially when used for preventing deep vein thrombosis. Prolonged use increases the risk of bleeding.30,31
Most patients who need anticoagulation have comorbidities such as heart failure, renal failure, diabetes mellitus, and hypertension. Although the kidneys play a major role in the excretion of dabigatran and, to some extent, rivaroxaban and apixaban, patients with severe renal impairment were excluded from the major trials of all three drugs.1–3 Hence, to avoid excessive drug accumulation and bleeding, these medications should not be used in such patients pending further studies. Further, patients taking these medications should be closely followed to detect new clinical situations, such as acute renal failure, that will necessitate their discontinuation or dose adjustment.
If surgery is needed
If a patient taking a new oral anticoagulant needs to undergo elective surgery, it is important to temporarily discontinue the drug, assess the risk of bleeding, and test for renal impairment.
Renal impairment is particularly relevant in the case of dabigatran, since more than 80% of the unchanged drug is cleared by the kidneys. Decreasing the dose, prolonging the dosing interval, or both have been suggested as means to reduce the risk of bleeding in patients with renal impairment who are taking dabigatran.32,33 Patients with normal renal function undergoing low-risk surgery should discontinue dabigatran at least 24 hours before the surgery. If the creatinine clearance is 31 to 50 mL/min, inclusively, the last dose should be at least 48 hours before the procedure for low-risk surgery, and 4 days before a procedure that poses a high risk of bleeding.32–34 Some experts have given the same recommendations for rivaroxaban and apixaban (Table 2).34
The aPTT and prothrombin time are readily available tests, but they cannot determine the residual anticoagulant effects of dabigatran, rivaroxaban, or apixaban. However, in many (but not all) cases, a normal aPTT suggests that the hemostatic function is not impaired by dabigatran, and a normal prothrombin time or an absence of anti-factor Xa activity would similarly exclude hemostatic dysfunction caused by rivaroxaban or apixaban. These tests are potentially useful as adjuncts before surgical procedures that require complete hemostasis.
Furthermore, a normal thrombin time rules out the presence of a significant amount of dabigatran. Therefore, a normal thrombin time might be particularly useful in a patient undergoing a high-risk intervention such as epidural cannulation or neurosurgery and who is normally receiving dabigatran.
Managing overdose and bleeding complications
Assessing the severity of bleeding is the key to managing bleeding complications (Table 3).
Minor bleeding such as epistaxis and ecchymosis can be managed symptomatically (eg, with nasal packing), perhaps with short-term withdrawal of the anticoagulant. Moderate bleeding such as upper or lower gastrointestinal bleeding can be managed by withdrawal of the anticoagulant, clinical monitoring, blood transfusion if needed, and treatment directed at the etiology.
Major and life-threatening bleeding (eg, intracerebral hemorrhage) requires aggressive treatment in the intensive care unit, withdrawal of the anticoagulant, mechanical compression of the bleeding site if accessible, fluid replacement and blood transfusion as appropriate, and interventional procedures. Nonspecific reversal agents might be considered in patients with major or life-threatening bleeding.
The half-life of dabigatran after multiple doses is approximately 14 to 17 hours and is not dose-dependent.9 Hence, if there is no active bleeding after a dabigatran overdose, stopping the drug may be sufficient. Since the pharmacodynamic effect of dabigatran declines in parallel to its plasma concentration, urgent but not emergency surgery may need to be delayed for only about 12 hours from the last dose of dabigatran.
The 2011 American College of Cardiology Foundation/American Heart Association guidelines recommend that patients with severe hemorrhage resulting from dabigatran should receive supportive therapy, including transfusion of fresh-frozen plasma, transfusion of packed red blood cells, or surgical intervention if appropriate.35 However, transfusion of fresh-frozen plasma is debatable because there is no evidence to support its use in this situation. While fresh-frozen plasma may be useful in cases of coagulation factor depletion, it does not effectively reverse inhibition of coagulation factors.36
Off-label use of nonspecific hemostatic agents
To date, no specific agent has been demonstrated to reverse excessive bleeding in patients taking the new oral anticoagulants. However, in view of their procoagulant capabilities, nonspecific hemostatic agents have been suggested for use in reversal of major bleeding resulting from these drugs.37–39 Examples are:
Recombinant factor VIIa (NovoSeven) initiates thrombin generation by activating factor X.
Four-factor prothrombin complex concentrate (Beriplex, recently approved in the United States) contains relatively large amounts of four nonactive vitamin K-dependent procoagulant factors (factors II, VII, IX, and X) that stimulate thrombin formation.
Three-factor prothrombin complex concentrate (Bebulin VH and Profilnine SD) contains low amounts of nonactive factor VII relative to factors II, IX, and X. In some centers a four-factor equivalent is produced by transfusion of a three-factor product with the addition of small amounts of recombinant factor VIIa or fresh-frozen plasma to replace the missing factor VII.40
Activated prothrombin complex concentrate (FEIBA NF) contains activated factor VII and factors II, IX, and X, mainly in nonactivated form.36 Therefore, it combines the effect of both recombinant factor VIIa and four-factor prothrombin complex concentrate.37
Studies of nonspecific hemostatic agents
In a study of rats infused with high doses of dabigatran, van Ryn et al38 observed that activated prothrombin complex concentrate at a dose of 50 or 100 U/kg and recombinant factor VIIa at a dose of 0.1 or 0.5 mg/kg reduced the rat-tail bleeding time in a dose-dependent manner but not the blood loss, compared with controls, even with a higher dose of recombinant factor VIIa (1 mg/kg). Recombinant factor VIIa also reversed the prolonged aPTT induced by dabigatran, whereas activated prothrombin complex concentrate did not. They suggested that recombinant factor VIIa and activated prothrombin complex concentrate may be potential antidotes for dabigatran-induced severe bleeding in humans.
In an ex vivo study of healthy people who took a single dose of dabigatran 150 mg or rivaroxaban 20 mg, Marlu et al37 found that activated prothrombin complex concentrate and four-factor prothrombin complex concentrate could be reasonable antidotes to these drugs.
Dabigatran-associated bleeding after cardiac surgery in humans has been successfully managed with hemodialysis and recombinant factor VIIa, although the efficacy of the latter cannot be individually assessed in the study.41
In a randomized placebo-controlled trial aimed at reversing rivaroxaban and dabigatran in healthy participants, Eerenberg et al39 showed that four-factor prothrombin complex concentrate at a dose of 50 IU/kg reversed prolongation of the prothrombin time and decreased the endogenous thrombin potential in those who received rivaroxaban, but it failed to reverse the aPTT, the endogenous thrombin potential, and thrombin time in those who received dabigatran.
However, Marlu et al reported that four-factor prothrombin complex concentrate at three doses (12.5 U/kg, 25 U/kg, and 50 U/kg)—or better still, activated prothrombin complex concentrate (40–80 U/kg)—could be a useful antidote to dabigatran.37
It is important to note that the healthy participants in the Eerenberg et al study39 took dabigatran 150 mg twice daily and rivaroxaban 20 mg daily for 2.5 days, whereas those in the Marlu et al study37 took the same dose of each medication, but only once.
The three-factor prothrombin complex concentrate products have been shown to be less effective than four-factor ones in reversing supratherapeutic INRs in patients with warfarin overdose, but whether this will be true with the new oral anticoagulants remains unknown. Furthermore, the four-factor concentrates effectively reversed warfarin-induced coagulopathy and bleeding in patients,42 but to our knowledge, the same is yet to be demonstrated in bleeding related to the newer agents.
Other measures
Gastric lavage or the administration of activated charcoal (or in some cases both) may reduce drug absorption if done within 2 or 3 hours of drug ingestion (Table 1). Because it is lipophilic, more than 99.9% of dabigatran etexilate was adsorbed by activated charcoal from water prepared to simulate gastric fluid in an in vitro experiment by van Ryn et al.43 This has not been tested in patients, and no similar study has been done for rivaroxaban or apixaban. However, use of charcoal in cases of recent ingestion, particularly with intentional overdose of these agents, seems reasonable.
Hemodialysis may reverse the anticoagulant effects of dabigatran overdose or severe bleeding because only about 35% of dabigatran is bound to plasma proteins (Table 1). In a single-center study, 50 mg of dabigatran etexilate was given orally to six patients with end-stage renal disease before dialysis, and the mean fraction of the drug removed by the dialyzer was 62% at 2 hours and 68% at 4 hours.32 This study suggests that hemodialysis may be useful to accelerate the removal of the drug in cases of life-threatening bleeding.
Rivaroxaban and apixaban are not dialyzable: the plasma protein binding of rivaroxaban is 95% and that of apixaban is 87%.
FUTURE DIRECTIONS
Because the new oral anticoagulants, unlike warfarin, have a wide therapeutic window, routine anticoagulant monitoring is not needed and might be misleading. However, there are times when monitoring might be useful; at such times, a validated, widely available, easily understood test would be good to have—but we don’t have it—at least not yet.
Therapeutic ranges for the aPTT have been established empirically for heparin in various indications.44 Additional study is needed to determine if an appropriate aPTT range can be determined for the new oral anticoagulants, particularly dabigatran.
Similarly, as with low-molecular-weight heparins, anti-factor Xa activity monitoring may become a more available validated means of testing for exposure to rivaroxaban and apixaban. More promising, using concepts derived from the development of the INR for warfarin monitoring,45 Tripodi et al46 have derived normalized INR-like assays to report rivaroxaban levels. A standardized schema for reporting results is being developed.46 Studies are required to determine if and how this assay may be useful. Initial trials in this regard are encouraging.47
Finally, the thrombotic risk associated with the use of nonspecific prohemostatic agents is unknown.37,48 Additional studies are required to standardize their dosages, frequency of administration, and duration of action, as well as to quantify their complications in bleeding patients.
In the past several years, three new oral anticoagulants—dabigatran etexilate (Pradaxa), rivaroxaban (Xarelto), and apixaban (Eliquis)—have been approved for use in the United States. These long-awaited agents are appealing because they are easy to use, do not require laboratory monitoring, and have demonstrated equivalence, or in some cases, superiority to warfarin in preventing stroke or systemic embolism in at-risk populations.1–4 However, unlike warfarin, they have no specific reversal agents. How then should one manage spontaneous bleeding problems and those due to drug overdose, and how can we quickly reverse anticoagulation if emergency surgery is needed?
For these reasons, physicians and patients have been wary of these agents. However, with a systematic approach based on an understanding of the properties of these drugs, the appropriate use and interpretation of coagulation tests, and awareness of available therapeutic strategies, physicians can more confidently provide care for patients who require urgent reversal of anticoagulant effects.
Here, we review the available literature and suggest practical strategies for management based on an understanding of the pharmacokinetic and pharmacodynamic effects of these drugs and our current knowledge of the coagulation tests.
NEED FOR ANTICOAGULANTS
Anticoagulants are important in preventing systemic embolization in patients with atrial fibrillation and preventing pulmonary embolism in patients with venous thromboembolism.
And the numbers are staggering. The estimated prevalence of atrial fibrillation in the United States was 3.03 million in 2005 and is projected to increase to 7.56 million by 2050.5 Ischemic stroke is the most serious complication of atrial fibrillation, which accounts for 23.5% of strokes in patients ages 80 through 89 according to Framingham data.6 Venous thromboembolism accounts for 900,000 incident or recurrent fatal and nonfatal events in the United States yearly.7
HOW THE NEW AGENTS BLOCK COAGULATION
Thrombin (factor IIa), a serine protease, is central to the process of clot formation during hemostasis. It activates factors V, VIII, and XI (thus generating more thrombin), catalyzes the conversion of fibrinogen to fibrin, and stimulates platelet aggregation. Its role in the final steps of the coagulation cascade has made it a target for new direct thrombin inhibitors such as dabigatran.
Factor Xa is a serine protease that plays a central role in the coagulation cascade. It is a desirable target for anticoagulation because it is the convergence point for the extrinsic and the intrinsic coagulation pathways. It converts prothrombin to thrombin. Rivaroxaban and apixaban are direct factor Xa inhibitors (Figure 1).
Dabigatran, a direct thrombin inhibitor
Dabigatran etexilate is a synthetic, orally available prodrug that is rapidly absorbed and converted by esterases to its active form, dabigatran, a potent direct inhibitor of both free thrombin and clot-bound thrombin.8
Plasma levels of dabigatran peak within 2 hours of administration, and its half-life is 14 to 17 hours.9 Dabigatran is eliminated mainly via the kidneys, with more that 80% of the drug excreted unchanged in the urine (Table 1).
Rivaroxaban, a factor Xa inhibitor
Rivaroxaban is a potent, selective, direct factor Xa inhibitor.
Plasma levels of rivaroxaban peak 2 to 3 hours after administration, and it is cleared with a terminal half-life of 7 to 11 hours.10,11
Rivaroxaban is eliminated by the kidneys and in the feces. The kidneys eliminate one-third of the active drug unchanged and another one-third as inactive metabolites. The remaining one-third is metabolized by the liver and then excreted in the feces. Rivaroxaban has a predictable and dose-dependent pharmacodynamic and pharmacokinetic profile that is not affected by age, sex, or body weight (Table 1).12
Apixaban, an oral factor Xa inhibitor
Apixaban is a selective, direct oral factor Xa inhibitor.
Plasma levels of apixaban peak about 3 hours after administration, and its terminal half-life is 8 to 14 hours.13 Apixaban is eliminated by oxidative metabolism, by the kidney, and in the feces. It has predictable pharmacodynamic and pharmacokinetic profiles and has the least renal dependence of the three agents (Table 1).
THE NEW ORAL ANTICOAGULANTS AND BLOOD COAGULATION ASSAYS
Assessment of the anticoagulant activity of the new oral anticoagulants is not necessary in routine clinical practice, but it may be useful in planning intervention in patients with major bleeding, those with drug overdose, or those who need emergency surgery.
The activated partial thromboplastin time
The activated partial thromboplastin time (aPTT) is a measure of the activity of the intrinsic pathway of the coagulation cascade.
Dabigatran. There is a curvilinear relationship between the aPTT and the plasma concentration of dabigatran and other direct thrombin inhibitors, although the aPTT prolongation appears to vary with different reagents and coagulometers.9,14,15 However, Stangier et al9 found a linear relationship between the aPTT and the square root of the dabigatran plasma concentration.
Rivaroxaban prolongs the aPTT in a dose-dependent manner, but there is no standard for calibration of this assay. Hence, the aPTT is not recommended for monitoring rivaroxaban in clinical practice.
Apixaban may also prolong the aPTT, but there are limited data on its reactivity with different reagents.
The prothrombin time and international normalized ratio
The prothrombin time and international normalized ratio (INR) are measures of the extrinsic pathway of the coagulation cascade.
Dabigatran. The INR has a linear response to the dabigatran concentration, but it is insensitive.9 Hence, it is not suitable for monitoring the anticoagulant effects of direct thrombin inhibitors.
Rivaroxaban. The prothrombin time correlates strongly with the plasma concentration of rivaroxaban in healthy trial participants11 and in patients undergoing total hip arthroplasty or total knee arthroplasty.16 Samama et al17 noted that, unlike with vitamin K antagonists, the INR cannot be used to monitor patients on rivaroxaban because the prothrombin time results varied with different reagents. They used a standard calibration curve to express the prothrombin time results in plasma concentrations of rivaroxaban rather than in seconds or the INR.
Apixaban increases the INR in a dose-dependent manner.18 Its effect on different reagents remains unknown.
The thrombin time
The thrombin time reflects the activity of thrombin in the plasma. The amount of thrombin and the concentration of thrombin inhibitors in the plasma sample determine the time to clot formation.
Dabigatran. The thrombin time displays a linear dose-response to dabigatran, but only over the range of therapeutic concentrations. At a dabigatran concentration greater than 600 ng/mL, the test often exceeds the maximum measurement time of coagulometers.9 Hence, this test is too sensitive for emergency monitoring, especially in cases of drug overdose. However, it is well suited for determining if any dabigatran is present.
Rivaroxaban and apixaban have no effect on the thrombin time.
The Hemoclot direct thrombin inhibitor assay and dabigatran
The Hemoclot direct thrombin inhibitor assay (Hyphen BioMed, France) is a sensitive diluted thrombin time assay that can be used for quantitative measurement of dabigatran activity in plasma. This test is based on inhibition of a constant amount of highly purified human alpha-thrombin by adding it to diluted test plasma (1:8 to 1:20) mixed with normal pooled human plasma.19,20
Stangier et al19 found that the Hemoclot assay was suitable for calculating a wide range of dabigatran concentrations up to 4,000 nmol/L (1,886 ng/mL). Although this finding has not been confirmed in larger studies, this test may provide a rapid and accurate assessment of dabigatran’s anticoagulant activity in cases of emergency surgery or overdose.
The ecarin clotting time and dabigatran
The ecarin clotting time is a measure of the activity of direct thrombin inhibitors, but not the factor Xa inhibitors.
Ecarin is a highly purified metalloprotease isolated from the venom of a snake, Echis carinatus, and it generates meizothrombin from prothrombin.21 Meizothrombin facilitates clot formation by converting fibrinogen to fibrin and, like thrombin, it can be inactivated by direct thrombin inhibitors, thereby prolonging the clotting time.
The limitations of the ecarin clotting time include dependence on the plasma levels of fibrinogen and prothrombin.
The ecarin chromogenic assay and dabigatran
The ecarin chromogenic assay is an improvement on the principle of the ecarin clotting time that can be used to measure the activity of direct thrombin inhibitors.22 In this test, ecarin is added to a plasma sample to generate meizothrombin, and the amidolytic activity of meizothrombin towards a chromogenic substrate is then determined.
Results of the ecarin chromogenic assay are not influenced by the levels of fibrinogen or prothrombin. Another advantage is that this assay can be used in automated and manual analyzers, thus enabling its use at the bedside. However, to our knowledge, it is not being regularly used to monitor direct thrombin inhibitors in the clinical setting, and there is no standard calibration of the ecarin clotting time method.
Assays of factor Xa activity
A variety of assays to monitor the anticoagulant activity of factor Xa inhibitors have been proposed.23–25 All measure inhibition of the activity of factor Xa using methods similar to those used in monitoring heparin levels. All require calibrators with a known concentration of the Xa inhibitor; many are easily adapted for laboratories currently providing measurement of factor Xa inhibition from heparin.23 These assays have been suggested as a better indicator of plasma concentration of factor Xa inhibitor drugs than the prothrombin time.25
CONTROLLING BLEEDING IN PATIENTS ON THE NEW ORAL ANTICOAGULANTS
Bleeding is an anticipated adverse event in patients taking anticoagulants. It is associated with significant morbidity and risk of death.26,27
Many physicians still have limited experience with using the new oral anticoagulants and managing the attendant bleeding risks. Hence, we recommend that every health institution have a treatment policy or algorithm to guide all clinical staff in the management of such emergencies.
Prevention of bleeding
Management of bleeding from these agents should begin with preventing bleeding in the first place.
The physician should adhere to the recommended dosages of these medications. Studies have shown that the plasma concentration of these drugs and the risk of bleeding increase with increasing dosage.1,28,29
In addition, these medications should be used for the shortest time for which anticoagulation is required, especially when used for preventing deep vein thrombosis. Prolonged use increases the risk of bleeding.30,31
Most patients who need anticoagulation have comorbidities such as heart failure, renal failure, diabetes mellitus, and hypertension. Although the kidneys play a major role in the excretion of dabigatran and, to some extent, rivaroxaban and apixaban, patients with severe renal impairment were excluded from the major trials of all three drugs.1–3 Hence, to avoid excessive drug accumulation and bleeding, these medications should not be used in such patients pending further studies. Further, patients taking these medications should be closely followed to detect new clinical situations, such as acute renal failure, that will necessitate their discontinuation or dose adjustment.
If surgery is needed
If a patient taking a new oral anticoagulant needs to undergo elective surgery, it is important to temporarily discontinue the drug, assess the risk of bleeding, and test for renal impairment.
Renal impairment is particularly relevant in the case of dabigatran, since more than 80% of the unchanged drug is cleared by the kidneys. Decreasing the dose, prolonging the dosing interval, or both have been suggested as means to reduce the risk of bleeding in patients with renal impairment who are taking dabigatran.32,33 Patients with normal renal function undergoing low-risk surgery should discontinue dabigatran at least 24 hours before the surgery. If the creatinine clearance is 31 to 50 mL/min, inclusively, the last dose should be at least 48 hours before the procedure for low-risk surgery, and 4 days before a procedure that poses a high risk of bleeding.32–34 Some experts have given the same recommendations for rivaroxaban and apixaban (Table 2).34
The aPTT and prothrombin time are readily available tests, but they cannot determine the residual anticoagulant effects of dabigatran, rivaroxaban, or apixaban. However, in many (but not all) cases, a normal aPTT suggests that the hemostatic function is not impaired by dabigatran, and a normal prothrombin time or an absence of anti-factor Xa activity would similarly exclude hemostatic dysfunction caused by rivaroxaban or apixaban. These tests are potentially useful as adjuncts before surgical procedures that require complete hemostasis.
Furthermore, a normal thrombin time rules out the presence of a significant amount of dabigatran. Therefore, a normal thrombin time might be particularly useful in a patient undergoing a high-risk intervention such as epidural cannulation or neurosurgery and who is normally receiving dabigatran.
Managing overdose and bleeding complications
Assessing the severity of bleeding is the key to managing bleeding complications (Table 3).
Minor bleeding such as epistaxis and ecchymosis can be managed symptomatically (eg, with nasal packing), perhaps with short-term withdrawal of the anticoagulant. Moderate bleeding such as upper or lower gastrointestinal bleeding can be managed by withdrawal of the anticoagulant, clinical monitoring, blood transfusion if needed, and treatment directed at the etiology.
Major and life-threatening bleeding (eg, intracerebral hemorrhage) requires aggressive treatment in the intensive care unit, withdrawal of the anticoagulant, mechanical compression of the bleeding site if accessible, fluid replacement and blood transfusion as appropriate, and interventional procedures. Nonspecific reversal agents might be considered in patients with major or life-threatening bleeding.
The half-life of dabigatran after multiple doses is approximately 14 to 17 hours and is not dose-dependent.9 Hence, if there is no active bleeding after a dabigatran overdose, stopping the drug may be sufficient. Since the pharmacodynamic effect of dabigatran declines in parallel to its plasma concentration, urgent but not emergency surgery may need to be delayed for only about 12 hours from the last dose of dabigatran.
The 2011 American College of Cardiology Foundation/American Heart Association guidelines recommend that patients with severe hemorrhage resulting from dabigatran should receive supportive therapy, including transfusion of fresh-frozen plasma, transfusion of packed red blood cells, or surgical intervention if appropriate.35 However, transfusion of fresh-frozen plasma is debatable because there is no evidence to support its use in this situation. While fresh-frozen plasma may be useful in cases of coagulation factor depletion, it does not effectively reverse inhibition of coagulation factors.36
Off-label use of nonspecific hemostatic agents
To date, no specific agent has been demonstrated to reverse excessive bleeding in patients taking the new oral anticoagulants. However, in view of their procoagulant capabilities, nonspecific hemostatic agents have been suggested for use in reversal of major bleeding resulting from these drugs.37–39 Examples are:
Recombinant factor VIIa (NovoSeven) initiates thrombin generation by activating factor X.
Four-factor prothrombin complex concentrate (Beriplex, recently approved in the United States) contains relatively large amounts of four nonactive vitamin K-dependent procoagulant factors (factors II, VII, IX, and X) that stimulate thrombin formation.
Three-factor prothrombin complex concentrate (Bebulin VH and Profilnine SD) contains low amounts of nonactive factor VII relative to factors II, IX, and X. In some centers a four-factor equivalent is produced by transfusion of a three-factor product with the addition of small amounts of recombinant factor VIIa or fresh-frozen plasma to replace the missing factor VII.40
Activated prothrombin complex concentrate (FEIBA NF) contains activated factor VII and factors II, IX, and X, mainly in nonactivated form.36 Therefore, it combines the effect of both recombinant factor VIIa and four-factor prothrombin complex concentrate.37
Studies of nonspecific hemostatic agents
In a study of rats infused with high doses of dabigatran, van Ryn et al38 observed that activated prothrombin complex concentrate at a dose of 50 or 100 U/kg and recombinant factor VIIa at a dose of 0.1 or 0.5 mg/kg reduced the rat-tail bleeding time in a dose-dependent manner but not the blood loss, compared with controls, even with a higher dose of recombinant factor VIIa (1 mg/kg). Recombinant factor VIIa also reversed the prolonged aPTT induced by dabigatran, whereas activated prothrombin complex concentrate did not. They suggested that recombinant factor VIIa and activated prothrombin complex concentrate may be potential antidotes for dabigatran-induced severe bleeding in humans.
In an ex vivo study of healthy people who took a single dose of dabigatran 150 mg or rivaroxaban 20 mg, Marlu et al37 found that activated prothrombin complex concentrate and four-factor prothrombin complex concentrate could be reasonable antidotes to these drugs.
Dabigatran-associated bleeding after cardiac surgery in humans has been successfully managed with hemodialysis and recombinant factor VIIa, although the efficacy of the latter cannot be individually assessed in the study.41
In a randomized placebo-controlled trial aimed at reversing rivaroxaban and dabigatran in healthy participants, Eerenberg et al39 showed that four-factor prothrombin complex concentrate at a dose of 50 IU/kg reversed prolongation of the prothrombin time and decreased the endogenous thrombin potential in those who received rivaroxaban, but it failed to reverse the aPTT, the endogenous thrombin potential, and thrombin time in those who received dabigatran.
However, Marlu et al reported that four-factor prothrombin complex concentrate at three doses (12.5 U/kg, 25 U/kg, and 50 U/kg)—or better still, activated prothrombin complex concentrate (40–80 U/kg)—could be a useful antidote to dabigatran.37
It is important to note that the healthy participants in the Eerenberg et al study39 took dabigatran 150 mg twice daily and rivaroxaban 20 mg daily for 2.5 days, whereas those in the Marlu et al study37 took the same dose of each medication, but only once.
The three-factor prothrombin complex concentrate products have been shown to be less effective than four-factor ones in reversing supratherapeutic INRs in patients with warfarin overdose, but whether this will be true with the new oral anticoagulants remains unknown. Furthermore, the four-factor concentrates effectively reversed warfarin-induced coagulopathy and bleeding in patients,42 but to our knowledge, the same is yet to be demonstrated in bleeding related to the newer agents.
Other measures
Gastric lavage or the administration of activated charcoal (or in some cases both) may reduce drug absorption if done within 2 or 3 hours of drug ingestion (Table 1). Because it is lipophilic, more than 99.9% of dabigatran etexilate was adsorbed by activated charcoal from water prepared to simulate gastric fluid in an in vitro experiment by van Ryn et al.43 This has not been tested in patients, and no similar study has been done for rivaroxaban or apixaban. However, use of charcoal in cases of recent ingestion, particularly with intentional overdose of these agents, seems reasonable.
Hemodialysis may reverse the anticoagulant effects of dabigatran overdose or severe bleeding because only about 35% of dabigatran is bound to plasma proteins (Table 1). In a single-center study, 50 mg of dabigatran etexilate was given orally to six patients with end-stage renal disease before dialysis, and the mean fraction of the drug removed by the dialyzer was 62% at 2 hours and 68% at 4 hours.32 This study suggests that hemodialysis may be useful to accelerate the removal of the drug in cases of life-threatening bleeding.
Rivaroxaban and apixaban are not dialyzable: the plasma protein binding of rivaroxaban is 95% and that of apixaban is 87%.
FUTURE DIRECTIONS
Because the new oral anticoagulants, unlike warfarin, have a wide therapeutic window, routine anticoagulant monitoring is not needed and might be misleading. However, there are times when monitoring might be useful; at such times, a validated, widely available, easily understood test would be good to have—but we don’t have it—at least not yet.
Therapeutic ranges for the aPTT have been established empirically for heparin in various indications.44 Additional study is needed to determine if an appropriate aPTT range can be determined for the new oral anticoagulants, particularly dabigatran.
Similarly, as with low-molecular-weight heparins, anti-factor Xa activity monitoring may become a more available validated means of testing for exposure to rivaroxaban and apixaban. More promising, using concepts derived from the development of the INR for warfarin monitoring,45 Tripodi et al46 have derived normalized INR-like assays to report rivaroxaban levels. A standardized schema for reporting results is being developed.46 Studies are required to determine if and how this assay may be useful. Initial trials in this regard are encouraging.47
Finally, the thrombotic risk associated with the use of nonspecific prohemostatic agents is unknown.37,48 Additional studies are required to standardize their dosages, frequency of administration, and duration of action, as well as to quantify their complications in bleeding patients.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke 1991; 22:983–988.
- Heit JA, Cohen AT, Anderson FA; on behalf of the VTE Impact Assessment Group. Estimated annual number of incident and recurrent, non-fatal and fatal venous thromboembolism (VTE) events in the US. Blood (ASH Annual Meeting Abstracts) 2005; 106:abstract 910.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in healthy subjects. Int J Clin Pharmacol Ther 2007; 45:335–344.
- Weitz JI, Eikelboom JW, Samama MM; American College of Chest Physicians. New antithrombotic drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e120S–e151S.
- Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos 2009; 37:74–81.
- Cullberg M, Eriksson UG, Larsson M, Karlsson MO. Population modelling of the effect of inogatran, at thrombin inhibitor, on ex vivo coagulation time (APTT) in healthy subjects and patients with coronary artery disease. Br J Clin Pharmacol 2001; 51:71–79.
- Carlsson SC, Mattsson C, Eriksson UG, et al. A review of the effects of the oral direct thrombin inhibitor ximelagatran on coagulation assays. Thromb Res 2005; 115:9–18.
- Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in patients undergoing major orthopaedic surgery. Clin Pharmacokinet 2008; 47:203–216.
- Samama MM, Martinoli JL, LeFlem L, et al. Assessment of laboratory assays to measure rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2010; 103:815–825.
- Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 2008; 6:820–829.
- Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis 2012; 23:138–143.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Nowak G. The ecarin clotting time, a universal method to quantify direct thrombin inhibitors. Pathophysiol Haemost Thromb 2003–2004; 33:173–183.
- Lange U, Nowak G, Bucha E. Ecarin chromogenic assay—a new method for quantitative determination of direct thrombin inhibitors like hirudin. Pathophysiol Haemost Thromb 2003–2004; 33:184–191.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Anti-Factor Xa Chromogenic Assay Field Trial Laboratories. Evaluation of the anti-factor Xa chromogenic assay for the measurement of rivaroxaban plasma concentrations using calibrators and controls. Thromb Haemost 2012; 107:379–387.
- Miyares MA, Davis K. Newer oral anticoagulants: a review of laboratory monitoring options and reversal agents in the hemorrhagic patient. Am J Health Syst Pharm 2012; 69:1473–1484.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct factor Xa inhibitor. J Thromb Haemost 2005; 3:514–521.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- US Food and Drug Administration (FDA). Medication Guide: Pradaxa (dabigatran etexilate mesylate) capsules. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM231720.pdf. Accessed June 5, 2013.
- Schulman S, Crowther MA. How I treat with anticoagulants in 2012: new and old anticoagulants, and when and how to switch. Blood 2012; 119:3016–3023.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/ AHA/ HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol 2011; 57:1330–1337.
- Crowther MA, Warkentin TE. Managing bleeding in anticoagulated patients with a focus on novel therapeutic agents. J Thromb Haemost 2009; 7(suppl 1):107–110.
- Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost 2012; 108:217–224.
- van Ryn J, Ruehl D, Priepke H, Hauel N, Wienen W. Reversibility of the anticoagulant effect of high doses of the direct thrombin inhibitor dabigatran, by recombinant factor VIIa or activated prothrombin complex concentrate. 13th Congress of the European Hematology Association, June 12–15, 2008. Hematologica 2008; 93( s1):148Abs.0370.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Holland L, Warkentin TE, Refaai M, Crowther MA, Johnston MA, Sarode R. Suboptimal effect of a three-factor prothrombin complex concentrate (Profilnine-SD) in correcting supratherapeutic international normalized ratio due to warfarin overdose. Transfusion 2009; 49:1171–1177.
- Warkentin TE, Margetts P, Connolly SJ, Lamy A, Ricci C, Eikelboom JW. Recombinant factor VIIa (rFVIIa) and hemodialysis to manage massive dabigatran-associated postcardiac surgery bleeding. Blood 2012; 119:2172–2174.
- Song MM, Warne CP, Crowther MA. Prothrombin complex concentrate (PCC, Octaplex) in patients requiring immediate reversal of vitamin K antagonist anticoagulation. Thromb Res 2012; 129:526–529.
- van Ryn J, Sieger P, Kink-Eiband M, Gansser D, Clemens A. Adsorption of dabigatran etexilate in water or dabigatran in pooled human plasma by activated charcoal in vitro. 51st ASH Annual Meeting and Exposition. Abstract no. 1065. http://ash.confex.com/ash/2009/webprogram/Paper21383.html. Accessed June 5, 2013.
- Hirsh J. Heparin. N Engl J Med 1991; 324:1565–1574.
- van den Besselaar AMHP, Poller L, Tripodi A. Guidelines for thromboplastins and plasmas used to control for oral anticoagulant therapy. WHO Technical Report Series 1999; 889:64–93.
- Tripodi A, Chantarangkul V, Guinet C, Samama MM. The international normalized ratio calibrated for rivaroxaban has the potential to normalize prothrombin time results for rivaroxaban-treated patients: Results of an in vitro study. J Thromb Haemost 2011; 9:226–228.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Prothrombin Time Field Trial Laboratories. Evaluation of the prothrombin time for measuring rivaroxaban plasma concentrations using calibrators and controls: results of a multicenter field trial. Clin Appl Thromb Hemost 2012; 18:150–158.
- Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse events. Haemophilia 2002; 8:83–90.
- Granger CB, Alexander JH, McMurray JJ, et al; ARISTOTLE Committees and Investigators. Apixaban versus warfarin in patients with atrial fibrillation. N Engl J Med 2011; 365:981–992.
- Connolly SJ, Ezekowitz MD, Yusuf S, et al; RE-LY Steering Committee and Investigators. Dabigatran versus warfarin in patients with atrial fibrillation. N Engl J Med 2009; 361:1139–1151.
- Patel MR, Mahaffey KW, Garg J, et al; ROCKET AF Investigators. Rivaroxaban versus warfarin in nonvalvular atrial fibrillation. N Engl J Med 2011; 365:883–891.
- Schulman S, Kearon C, Kakkar AK, et al; RE-COVER Study Group. Dabigatran versus warfarin in the treatment of acute venous thromboembolism. N Engl J Med 2009; 361:2342–2352.
- Naccarelli GV, Varker H, Lin J, Schulman KL. Increasing prevalence of atrial fibrillation and flutter in the United States. Am J Cardiol 2009; 104:1534–1539.
- Wolf PA, Abbott RD, Kannel WB. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke 1991; 22:983–988.
- Heit JA, Cohen AT, Anderson FA; on behalf of the VTE Impact Assessment Group. Estimated annual number of incident and recurrent, non-fatal and fatal venous thromboembolism (VTE) events in the US. Blood (ASH Annual Meeting Abstracts) 2005; 106:abstract 910.
- Stangier J, Clemens A. Pharmacology, pharmacokinetics, and pharmacodynamics of dabigatran etexilate, an oral direct thrombin inhibitor. Clin Appl Thromb Hemost 2009; 15(suppl 1):9S–16S.
- Stangier J, Rathgen K, Stähle H, Gansser D, Roth W. The pharmacokinetics, pharmacodynamics and tolerability of dabigatran etexilate, a new oral direct thrombin inhibitor, in healthy male subjects. Br J Clin Pharmacol 2007; 64:292–303.
- Kubitza D, Becka M, Wensing G, Voith B, Zuehlsdorf M. Safety, pharmacodynamics, and pharmacokinetics of BAY 59-7939—an oral, direct factor Xa inhibitor—after multiple dosing in healthy male subjects. Eur J Clin Pharmacol 2005; 61:873–880.
- Mueck W, Becka M, Kubitza D, Voith B, Zuehlsdorf M. Population model of the pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in healthy subjects. Int J Clin Pharmacol Ther 2007; 45:335–344.
- Weitz JI, Eikelboom JW, Samama MM; American College of Chest Physicians. New antithrombotic drugs: Antithrombotic Therapy and Prevention of Thrombosis, 9th ed: American College of Chest Physicians Evidence-Based Clinical Practice Guidelines. Chest 2012; 141(suppl 2):e120S–e151S.
- Raghavan N, Frost CE, Yu Z, et al. Apixaban metabolism and pharmacokinetics after oral administration to humans. Drug Metab Dispos 2009; 37:74–81.
- Cullberg M, Eriksson UG, Larsson M, Karlsson MO. Population modelling of the effect of inogatran, at thrombin inhibitor, on ex vivo coagulation time (APTT) in healthy subjects and patients with coronary artery disease. Br J Clin Pharmacol 2001; 51:71–79.
- Carlsson SC, Mattsson C, Eriksson UG, et al. A review of the effects of the oral direct thrombin inhibitor ximelagatran on coagulation assays. Thromb Res 2005; 115:9–18.
- Mueck W, Eriksson BI, Bauer KA, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban—an oral, direct factor Xa inhibitor—in patients undergoing major orthopaedic surgery. Clin Pharmacokinet 2008; 47:203–216.
- Samama MM, Martinoli JL, LeFlem L, et al. Assessment of laboratory assays to measure rivaroxaban—an oral, direct factor Xa inhibitor. Thromb Haemost 2010; 103:815–825.
- Wong PC, Crain EJ, Xin B, et al. Apixaban, an oral, direct and highly selective factor Xa inhibitor: in vitro, antithrombotic and antihemostatic studies. J Thromb Haemost 2008; 6:820–829.
- Stangier J, Feuring M. Using the HEMOCLOT direct thrombin inhibitor assay to determine plasma concentrations of dabigatran. Blood Coagul Fibrinolysis 2012; 23:138–143.
- van Ryn J, Stangier J, Haertter S, et al. Dabigatran etexilate—a novel, reversible, oral direct thrombin inhibitor: interpretation of coagulation assays and reversal of anticoagulant activity. Thromb Haemost 2010; 103:1116–1127.
- Nowak G. The ecarin clotting time, a universal method to quantify direct thrombin inhibitors. Pathophysiol Haemost Thromb 2003–2004; 33:173–183.
- Lange U, Nowak G, Bucha E. Ecarin chromogenic assay—a new method for quantitative determination of direct thrombin inhibitors like hirudin. Pathophysiol Haemost Thromb 2003–2004; 33:184–191.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Anti-Factor Xa Chromogenic Assay Field Trial Laboratories. Evaluation of the anti-factor Xa chromogenic assay for the measurement of rivaroxaban plasma concentrations using calibrators and controls. Thromb Haemost 2012; 107:379–387.
- Miyares MA, Davis K. Newer oral anticoagulants: a review of laboratory monitoring options and reversal agents in the hemorrhagic patient. Am J Health Syst Pharm 2012; 69:1473–1484.
- Barrett YC, Wang Z, Frost C, Shenker A. Clinical laboratory measurement of direct factor Xa inhibitors: anti-Xa assay is preferable to prothrombin time assay. Thromb Haemost 2010; 104:1263–1271.
- Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation 2006; 114:774–782.
- Manoukian SV, Feit F, Mehran R, et al. Impact of major bleeding on 30-day mortality and clinical outcomes in patients with acute coronary syndromes: an analysis from the ACUITY Trial. J Am Coll Cardiol 2007; 49:1362–1368.
- Perzborn E, Strassburger J, Wilmen A, et al. In vitro and in vivo studies of the novel antithrombotic agent BAY 59-7939—an oral, direct factor Xa inhibitor. J Thromb Haemost 2005; 3:514–521.
- Eriksson BI, Dahl OE, Rosencher N, et al; RE-NOVATE Study Group. Dabigatran etexilate versus enoxaparin for prevention of venous thromboembolism after total hip replacement: a randomised, double-blind, non-inferiority trial. Lancet 2007; 370:949–956.
- Eriksson BI, Borris LC, Friedman RJ, et al; RECORD1 Study Group. Rivaroxaban versus enoxaparin for thromboprophylaxis after hip arthroplasty. N Engl J Med 2008; 358:2765–2775.
- Lassen MR, Ageno W, Borris LC, et al; RECORD3 Investigators. Rivaroxaban versus enoxaparin for thromboprophylaxis after total knee arthroplasty. N Engl J Med 2008; 358:2776–2786.
- Stangier J, Rathgen K, Stähle H, Mazur D. Influence of renal impairment on the pharmacokinetics and pharmacodynamics of oral dabigatran etexilate: an open-label, parallel-group, single-centre study. Clin Pharmacokinet 2010; 49:259–268.
- US Food and Drug Administration (FDA). Medication Guide: Pradaxa (dabigatran etexilate mesylate) capsules. http://www.fda.gov/downloads/Drugs/DrugSafety/UCM231720.pdf. Accessed June 5, 2013.
- Schulman S, Crowther MA. How I treat with anticoagulants in 2012: new and old anticoagulants, and when and how to switch. Blood 2012; 119:3016–3023.
- Wann LS, Curtis AB, Ellenbogen KA, et al. 2011 ACCF/ AHA/ HRS focused update on the management of patients with atrial fibrillation (update on dabigatran): a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. J Am Coll Cardiol 2011; 57:1330–1337.
- Crowther MA, Warkentin TE. Managing bleeding in anticoagulated patients with a focus on novel therapeutic agents. J Thromb Haemost 2009; 7(suppl 1):107–110.
- Marlu R, Hodaj E, Paris A, Albaladejo P, Cracowski JL, Pernod G. Effect of non-specific reversal agents on anticoagulant activity of dabigatran and rivaroxaban: a randomised crossover ex vivo study in healthy volunteers. Thromb Haemost 2012; 108:217–224.
- van Ryn J, Ruehl D, Priepke H, Hauel N, Wienen W. Reversibility of the anticoagulant effect of high doses of the direct thrombin inhibitor dabigatran, by recombinant factor VIIa or activated prothrombin complex concentrate. 13th Congress of the European Hematology Association, June 12–15, 2008. Hematologica 2008; 93( s1):148Abs.0370.
- Eerenberg ES, Kamphuisen PW, Sijpkens MK, Meijers JC, Buller HR, Levi M. Reversal of rivaroxaban and dabigatran by prothrombin complex concentrate: a randomized, placebo-controlled, crossover study in healthy subjects. Circulation 2011; 124:1573–1579.
- Holland L, Warkentin TE, Refaai M, Crowther MA, Johnston MA, Sarode R. Suboptimal effect of a three-factor prothrombin complex concentrate (Profilnine-SD) in correcting supratherapeutic international normalized ratio due to warfarin overdose. Transfusion 2009; 49:1171–1177.
- Warkentin TE, Margetts P, Connolly SJ, Lamy A, Ricci C, Eikelboom JW. Recombinant factor VIIa (rFVIIa) and hemodialysis to manage massive dabigatran-associated postcardiac surgery bleeding. Blood 2012; 119:2172–2174.
- Song MM, Warne CP, Crowther MA. Prothrombin complex concentrate (PCC, Octaplex) in patients requiring immediate reversal of vitamin K antagonist anticoagulation. Thromb Res 2012; 129:526–529.
- van Ryn J, Sieger P, Kink-Eiband M, Gansser D, Clemens A. Adsorption of dabigatran etexilate in water or dabigatran in pooled human plasma by activated charcoal in vitro. 51st ASH Annual Meeting and Exposition. Abstract no. 1065. http://ash.confex.com/ash/2009/webprogram/Paper21383.html. Accessed June 5, 2013.
- Hirsh J. Heparin. N Engl J Med 1991; 324:1565–1574.
- van den Besselaar AMHP, Poller L, Tripodi A. Guidelines for thromboplastins and plasmas used to control for oral anticoagulant therapy. WHO Technical Report Series 1999; 889:64–93.
- Tripodi A, Chantarangkul V, Guinet C, Samama MM. The international normalized ratio calibrated for rivaroxaban has the potential to normalize prothrombin time results for rivaroxaban-treated patients: Results of an in vitro study. J Thromb Haemost 2011; 9:226–228.
- Samama MM, Contant G, Spiro TE, et al; Rivaroxaban Prothrombin Time Field Trial Laboratories. Evaluation of the prothrombin time for measuring rivaroxaban plasma concentrations using calibrators and controls: results of a multicenter field trial. Clin Appl Thromb Hemost 2012; 18:150–158.
- Ehrlich HJ, Henzl MJ, Gomperts ED. Safety of factor VIII inhibitor bypass activity (FEIBA): 10-year compilation of thrombotic adverse events. Haemophilia 2002; 8:83–90.
KEY POINTS
- Thromboprophylaxis with anticoagulants is an important aspect of managing patients at risk of systemic or pulmonary embolization.
- Dabigatran is a direct inhibitor of thrombin (factor IIa); rivaroxaban and apixaban inhibit factor Xa.
- Monitoring of coagulation function is not routinely necessary with the new drugs but may be useful in emergencies.
- Nonspecific hemostatic agents that have been suggested for off-label use in reversing excessive bleeding in patients taking the new oral anticoagulants include recombinant factor VIIa, three-factor and four-factor prothrombin complex concentrate, and activated prothrombin complex concentrate.
Four pillars of a successful practice: 3. Obtain and maintain physician referrals
READ THE REST OF THE SERIES
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 4: Motivate your staff (August 2013)
Discussions of medical marketing often begin with the three As: availability, affability, and affordability. But most physicians already think of themselves as available, likeable, and offering appropriately priced services.
How do you differentiate yourself from the competition?
Fancy stationery; a slick, three-color brochure; a catchy logo; and a Web site will not do the trick. In fact, these are the last things you need.
One of the biggest misconceptions about marketing is that, to do it well, you must spend lots of money on peripherals. In truth, there are many other actions that are far more effective and essential to marketing than merely polishing your public relations image. The most essential element of your marketing plan is to make your practice user-friendly.
Nowhere is this need greater than when it comes to working with colleagues who are capable of referring patients to you—or are already doing so. In this article, I describe 10 strategies you can use to enhance your relationships with referring physicians.
1. WRITE AN EFFECTIVE REFERRAL LETTER
To obtain referrals from your colleagues, you need to ensure that your name crosses their mind and desk as frequently as possible—and in a positive fashion.
If you interview referring physicians, you will find that prompt communication is one of the most important reasons they refer a patient to a particular provider. According to the Annals of Family Medicine, more than 50% of physicians state that effective communication is the reason they select a doctor for referral (TABLE).1
| How primary care physicians select a doctor for referral | |
| Medical skill of the specialist | 87.5% |
| Access to the practice and acceptance of insurance | 59.0% |
| Previous experience with the specialist | 59.2% |
| Quality of communication | 52.5% |
| Board certification of the specialist | 33.9% |
| Medical school, residency | <1% |
Source: Kinchen et al1
Keep your referral letter short
The traditional referral letter is far too long, often 2 or 3 pages. It usually arrives 10 to 14 days after the patient was seen and is very expensive, costing a practice $12–$15 for each letter sent. The goal of an effective referral letter: Get it there before the patient returns to the primary care provider.
The key ingredients of an effective referral letter are:
diagnosis
medications you have prescribed for the patient
your treatment plan.
The referring doctor is not interested in the nuances of your history or physical exam. They just want the three ingredients listed above.
For example, let’s say that Dr. Bill Smith refers Jane Doe, who has an overactive bladder and cystocele. Her urinalysis is negative, so you prescribe an anticholinergic agent and schedule a follow-up visit in 1 month to check symptoms and to conduct a urodynamic study if she has not improved. Your letter to Dr. Smith would read as follows:
Now the letter can be faxed to the referring doctor, often before the patient leaves the office. That way you can be certain that the letter arrives before the patient calls the physician with questions or concerns.
This is the best way to keep the referring physician informed and to function as the captain of the patient’s health-care ship.
EHRs can smooth the referral process
Most electronic health records (EHRs) have the capability to fax the entire note to the referring physician. However, if you were to ask a referring physician if she would like to read your entire note, the answer would probably be “No.” Most EHRs will allow you to select fields that contain the diagnosis, medications prescribed, and the treatment plan. A sample of this kind of letter appears in the FIGURE.

2. MAKE AN EFFORT TO PERSONALLY MEET EVERY PHYSICIAN WHO REFERS A PATIENT
Not only that, but try to meet all new physicians in your area. It is important to coddle your existing sources of referrals, but don’t forget to reach out to new physicians to let them know about your areas of interest or expertise.
3. REFER YOUR NEW PATIENTS TO REFERRING PHYSICIANS
Don’t refer to the same colleagues time after time. If a doctor starts sending new patients your way, it’s in your best interest to “reverse-refer” when a patient needs a primary care doctor, endocrinologist, or cardiologist.
You can be sure these referring doctors will appreciate your recommendations.
Related Article Complex atypical endometrial hyperplasia: When to refer
4. CREATE A LUNCH-AND-LEARN PROGRAM
You want other offices and medical staffs to get to know your staff and to be familiar with what you do. There’s no better way than to create a lunch-and-learn program in your office and extend an invitation to other offices in the area. At the program, have all of the staff members introduce themselves. Provide a tour of your office and give a 3- to 5-minute lecture on areas of your gynecologic interest and expertise.
5. ACKNOWLEDGE THE ACCOMPLISHMENTS OF REFERRING PHYSICIANS AND THEIR FAMILIES
If you see that one of your referring physicians has received an honor or award, send him a congratulatory note. If her children have been recognized for academic or athletic achievement, acknowledge this accomplishment with a note. You can be sure it will be one of the only acknowledgments they receive and will be deeply appreciated.
6. SHARE INFORMATION WITH A NO-MEETING JOURNAL CLUB
It’s very difficult to keep up with the medical literature. It’s challenging enough to keep up with the literature in your own specialty, let alone articles appearing in other specialty publications. One of the nicest gestures you can make is to copy any article that may be of interest to your colleagues and send it to them. Include a sticky note indicating where you would like them to look so that they don’t have to read the entire article.
7. SHARE NONMEDICAL INFORMATION, TOO
Your colleagues will appreciate it when you share nonmedical information to let them know you are thinking of them even when you are not discussing patient care. For example, one of my colleagues collects fine pens. When I saw an article about a very expensive pen made with diamonds, I sent the story to my friend, suggesting that he tell his wife what was on his wish list.
8. KEEP THE REFERRING DOCTOR IN THE MEDICAL LOOP
If you are caring for a patient and plan to discharge her from the hospital, make sure that you or someone in your office contacts the referring doctor to inform him that the patient is being discharged so he doesn’t make unnecessary rounds. Other times to notify the referring doctor:
upon admission of her patient to the hospital
after surgery or a procedure
when you receive a significant laboratory or pathology report.
9. BE USER-FRIENDLY
If you perform gynecologic surgery on a referred patient, be sure to dictate a discharge summary. If the patient is to be discharged with gynecologic medications, give the patient their names in writing. Another convenience for the patient: Arrange your follow-up appointment on the same day she is to return to see the referring physician.
10. DON’T FORGET NONPHYSICIAN REFERRAL SOURCES
Nurses, pharmacists, pharmaceutical representatives, social workers, lawyers, beauticians, and manicurists—all of these professionals are likely to refer patients to you if you keep them in the loop.
11. BOTTOM LINE
You can build a practice by word of mouth by doing a great job of caring for patients, hoping that they will tell others about their positive experience. However, there are other opportunities to enhance your practice—notably, by nurturing your relationship with referring physicians. Try a few of these ideas and you will certainly see your referrals increase significantly.

Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
Neil H. Baum, MD
Dr. Baum practices urology in New Orleans, Louisiana. He is Associate Clinical Professor of Urology at Tulane Medical School and Louisiana State University Medical School, both in New Orleans. He is also on the medical staff at Touro Infirmary in New Orleans, and East Jefferson General Hospital in Metairie, Louisiana. And he is the author of Marketing Your Clinical Practice: Ethically, Effectively, Economically (4th edition, 2009; Jones & Bartlett).
The author reports no financial relationships relevant to this article.
READ THE REST OF THE SERIES
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 4: Motivate your staff (August 2013)
Discussions of medical marketing often begin with the three As: availability, affability, and affordability. But most physicians already think of themselves as available, likeable, and offering appropriately priced services.
How do you differentiate yourself from the competition?
Fancy stationery; a slick, three-color brochure; a catchy logo; and a Web site will not do the trick. In fact, these are the last things you need.
One of the biggest misconceptions about marketing is that, to do it well, you must spend lots of money on peripherals. In truth, there are many other actions that are far more effective and essential to marketing than merely polishing your public relations image. The most essential element of your marketing plan is to make your practice user-friendly.
Nowhere is this need greater than when it comes to working with colleagues who are capable of referring patients to you—or are already doing so. In this article, I describe 10 strategies you can use to enhance your relationships with referring physicians.
1. WRITE AN EFFECTIVE REFERRAL LETTER
To obtain referrals from your colleagues, you need to ensure that your name crosses their mind and desk as frequently as possible—and in a positive fashion.
If you interview referring physicians, you will find that prompt communication is one of the most important reasons they refer a patient to a particular provider. According to the Annals of Family Medicine, more than 50% of physicians state that effective communication is the reason they select a doctor for referral (TABLE).1
| How primary care physicians select a doctor for referral | |
| Medical skill of the specialist | 87.5% |
| Access to the practice and acceptance of insurance | 59.0% |
| Previous experience with the specialist | 59.2% |
| Quality of communication | 52.5% |
| Board certification of the specialist | 33.9% |
| Medical school, residency | <1% |
Source: Kinchen et al1
Keep your referral letter short
The traditional referral letter is far too long, often 2 or 3 pages. It usually arrives 10 to 14 days after the patient was seen and is very expensive, costing a practice $12–$15 for each letter sent. The goal of an effective referral letter: Get it there before the patient returns to the primary care provider.
The key ingredients of an effective referral letter are:
diagnosis
medications you have prescribed for the patient
your treatment plan.
The referring doctor is not interested in the nuances of your history or physical exam. They just want the three ingredients listed above.
For example, let’s say that Dr. Bill Smith refers Jane Doe, who has an overactive bladder and cystocele. Her urinalysis is negative, so you prescribe an anticholinergic agent and schedule a follow-up visit in 1 month to check symptoms and to conduct a urodynamic study if she has not improved. Your letter to Dr. Smith would read as follows:
Now the letter can be faxed to the referring doctor, often before the patient leaves the office. That way you can be certain that the letter arrives before the patient calls the physician with questions or concerns.
This is the best way to keep the referring physician informed and to function as the captain of the patient’s health-care ship.
EHRs can smooth the referral process
Most electronic health records (EHRs) have the capability to fax the entire note to the referring physician. However, if you were to ask a referring physician if she would like to read your entire note, the answer would probably be “No.” Most EHRs will allow you to select fields that contain the diagnosis, medications prescribed, and the treatment plan. A sample of this kind of letter appears in the FIGURE.
2. MAKE AN EFFORT TO PERSONALLY MEET EVERY PHYSICIAN WHO REFERS A PATIENT
Not only that, but try to meet all new physicians in your area. It is important to coddle your existing sources of referrals, but don’t forget to reach out to new physicians to let them know about your areas of interest or expertise.
3. REFER YOUR NEW PATIENTS TO REFERRING PHYSICIANS
Don’t refer to the same colleagues time after time. If a doctor starts sending new patients your way, it’s in your best interest to “reverse-refer” when a patient needs a primary care doctor, endocrinologist, or cardiologist.
You can be sure these referring doctors will appreciate your recommendations.
Related Article Complex atypical endometrial hyperplasia: When to refer
4. CREATE A LUNCH-AND-LEARN PROGRAM
You want other offices and medical staffs to get to know your staff and to be familiar with what you do. There’s no better way than to create a lunch-and-learn program in your office and extend an invitation to other offices in the area. At the program, have all of the staff members introduce themselves. Provide a tour of your office and give a 3- to 5-minute lecture on areas of your gynecologic interest and expertise.
5. ACKNOWLEDGE THE ACCOMPLISHMENTS OF REFERRING PHYSICIANS AND THEIR FAMILIES
If you see that one of your referring physicians has received an honor or award, send him a congratulatory note. If her children have been recognized for academic or athletic achievement, acknowledge this accomplishment with a note. You can be sure it will be one of the only acknowledgments they receive and will be deeply appreciated.
6. SHARE INFORMATION WITH A NO-MEETING JOURNAL CLUB
It’s very difficult to keep up with the medical literature. It’s challenging enough to keep up with the literature in your own specialty, let alone articles appearing in other specialty publications. One of the nicest gestures you can make is to copy any article that may be of interest to your colleagues and send it to them. Include a sticky note indicating where you would like them to look so that they don’t have to read the entire article.
7. SHARE NONMEDICAL INFORMATION, TOO
Your colleagues will appreciate it when you share nonmedical information to let them know you are thinking of them even when you are not discussing patient care. For example, one of my colleagues collects fine pens. When I saw an article about a very expensive pen made with diamonds, I sent the story to my friend, suggesting that he tell his wife what was on his wish list.
8. KEEP THE REFERRING DOCTOR IN THE MEDICAL LOOP
If you are caring for a patient and plan to discharge her from the hospital, make sure that you or someone in your office contacts the referring doctor to inform him that the patient is being discharged so he doesn’t make unnecessary rounds. Other times to notify the referring doctor:
upon admission of her patient to the hospital
after surgery or a procedure
when you receive a significant laboratory or pathology report.
9. BE USER-FRIENDLY
If you perform gynecologic surgery on a referred patient, be sure to dictate a discharge summary. If the patient is to be discharged with gynecologic medications, give the patient their names in writing. Another convenience for the patient: Arrange your follow-up appointment on the same day she is to return to see the referring physician.
10. DON’T FORGET NONPHYSICIAN REFERRAL SOURCES
Nurses, pharmacists, pharmaceutical representatives, social workers, lawyers, beauticians, and manicurists—all of these professionals are likely to refer patients to you if you keep them in the loop.
11. BOTTOM LINE
You can build a practice by word of mouth by doing a great job of caring for patients, hoping that they will tell others about their positive experience. However, there are other opportunities to enhance your practice—notably, by nurturing your relationship with referring physicians. Try a few of these ideas and you will certainly see your referrals increase significantly.
READ THE REST OF THE SERIES
Pillar 1: Keep your current patients happy (March 2013)
Dr. Baum describes his number one strategy to retain patients (Audiocast, March 2013)
Pillar 2: Attract new patients (May 2013)
Pillar 4: Motivate your staff (August 2013)
Discussions of medical marketing often begin with the three As: availability, affability, and affordability. But most physicians already think of themselves as available, likeable, and offering appropriately priced services.
How do you differentiate yourself from the competition?
Fancy stationery; a slick, three-color brochure; a catchy logo; and a Web site will not do the trick. In fact, these are the last things you need.
One of the biggest misconceptions about marketing is that, to do it well, you must spend lots of money on peripherals. In truth, there are many other actions that are far more effective and essential to marketing than merely polishing your public relations image. The most essential element of your marketing plan is to make your practice user-friendly.
Nowhere is this need greater than when it comes to working with colleagues who are capable of referring patients to you—or are already doing so. In this article, I describe 10 strategies you can use to enhance your relationships with referring physicians.
1. WRITE AN EFFECTIVE REFERRAL LETTER
To obtain referrals from your colleagues, you need to ensure that your name crosses their mind and desk as frequently as possible—and in a positive fashion.
If you interview referring physicians, you will find that prompt communication is one of the most important reasons they refer a patient to a particular provider. According to the Annals of Family Medicine, more than 50% of physicians state that effective communication is the reason they select a doctor for referral (TABLE).1
| How primary care physicians select a doctor for referral | |
| Medical skill of the specialist | 87.5% |
| Access to the practice and acceptance of insurance | 59.0% |
| Previous experience with the specialist | 59.2% |
| Quality of communication | 52.5% |
| Board certification of the specialist | 33.9% |
| Medical school, residency | <1% |
Source: Kinchen et al1
Keep your referral letter short
The traditional referral letter is far too long, often 2 or 3 pages. It usually arrives 10 to 14 days after the patient was seen and is very expensive, costing a practice $12–$15 for each letter sent. The goal of an effective referral letter: Get it there before the patient returns to the primary care provider.
The key ingredients of an effective referral letter are:
diagnosis
medications you have prescribed for the patient
your treatment plan.
The referring doctor is not interested in the nuances of your history or physical exam. They just want the three ingredients listed above.
For example, let’s say that Dr. Bill Smith refers Jane Doe, who has an overactive bladder and cystocele. Her urinalysis is negative, so you prescribe an anticholinergic agent and schedule a follow-up visit in 1 month to check symptoms and to conduct a urodynamic study if she has not improved. Your letter to Dr. Smith would read as follows:
Now the letter can be faxed to the referring doctor, often before the patient leaves the office. That way you can be certain that the letter arrives before the patient calls the physician with questions or concerns.
This is the best way to keep the referring physician informed and to function as the captain of the patient’s health-care ship.
EHRs can smooth the referral process
Most electronic health records (EHRs) have the capability to fax the entire note to the referring physician. However, if you were to ask a referring physician if she would like to read your entire note, the answer would probably be “No.” Most EHRs will allow you to select fields that contain the diagnosis, medications prescribed, and the treatment plan. A sample of this kind of letter appears in the FIGURE.
2. MAKE AN EFFORT TO PERSONALLY MEET EVERY PHYSICIAN WHO REFERS A PATIENT
Not only that, but try to meet all new physicians in your area. It is important to coddle your existing sources of referrals, but don’t forget to reach out to new physicians to let them know about your areas of interest or expertise.
3. REFER YOUR NEW PATIENTS TO REFERRING PHYSICIANS
Don’t refer to the same colleagues time after time. If a doctor starts sending new patients your way, it’s in your best interest to “reverse-refer” when a patient needs a primary care doctor, endocrinologist, or cardiologist.
You can be sure these referring doctors will appreciate your recommendations.
Related Article Complex atypical endometrial hyperplasia: When to refer
4. CREATE A LUNCH-AND-LEARN PROGRAM
You want other offices and medical staffs to get to know your staff and to be familiar with what you do. There’s no better way than to create a lunch-and-learn program in your office and extend an invitation to other offices in the area. At the program, have all of the staff members introduce themselves. Provide a tour of your office and give a 3- to 5-minute lecture on areas of your gynecologic interest and expertise.
5. ACKNOWLEDGE THE ACCOMPLISHMENTS OF REFERRING PHYSICIANS AND THEIR FAMILIES
If you see that one of your referring physicians has received an honor or award, send him a congratulatory note. If her children have been recognized for academic or athletic achievement, acknowledge this accomplishment with a note. You can be sure it will be one of the only acknowledgments they receive and will be deeply appreciated.
6. SHARE INFORMATION WITH A NO-MEETING JOURNAL CLUB
It’s very difficult to keep up with the medical literature. It’s challenging enough to keep up with the literature in your own specialty, let alone articles appearing in other specialty publications. One of the nicest gestures you can make is to copy any article that may be of interest to your colleagues and send it to them. Include a sticky note indicating where you would like them to look so that they don’t have to read the entire article.
7. SHARE NONMEDICAL INFORMATION, TOO
Your colleagues will appreciate it when you share nonmedical information to let them know you are thinking of them even when you are not discussing patient care. For example, one of my colleagues collects fine pens. When I saw an article about a very expensive pen made with diamonds, I sent the story to my friend, suggesting that he tell his wife what was on his wish list.
8. KEEP THE REFERRING DOCTOR IN THE MEDICAL LOOP
If you are caring for a patient and plan to discharge her from the hospital, make sure that you or someone in your office contacts the referring doctor to inform him that the patient is being discharged so he doesn’t make unnecessary rounds. Other times to notify the referring doctor:
upon admission of her patient to the hospital
after surgery or a procedure
when you receive a significant laboratory or pathology report.
9. BE USER-FRIENDLY
If you perform gynecologic surgery on a referred patient, be sure to dictate a discharge summary. If the patient is to be discharged with gynecologic medications, give the patient their names in writing. Another convenience for the patient: Arrange your follow-up appointment on the same day she is to return to see the referring physician.
10. DON’T FORGET NONPHYSICIAN REFERRAL SOURCES
Nurses, pharmacists, pharmaceutical representatives, social workers, lawyers, beauticians, and manicurists—all of these professionals are likely to refer patients to you if you keep them in the loop.
11. BOTTOM LINE
You can build a practice by word of mouth by doing a great job of caring for patients, hoping that they will tell others about their positive experience. However, there are other opportunities to enhance your practice—notably, by nurturing your relationship with referring physicians. Try a few of these ideas and you will certainly see your referrals increase significantly.

Preventing prescription drug abuse: Make it LAST
Medications that psychiatrists routinely prescribe—such as benzodiazepines for anxiety and psychostimulants for attention-deficit/hyperactivity disorder—often are diverted and abused. In 2011, 6.1 million Americans age ≥12 abused prescription drugs.1
The mnemonic LAST can bring to mind 4 clinical “red flags” that can assist you in determining whether prescription abuse or diversion is occurring. Incorporating these 4 warning signs in your clinical assessment and medication reviews will make it easier for you to detect when medications are not being taken as prescribed.
Lost or stolen prescriptions. Patients who want to obtain a new or replacement prescription may claim that their medication was lost or stolen. Although this can occur, the prescriber should be suspicious if this becomes a recurrent situation. Some clinicians require patients to produce a filed police report for stolen medications before they will consider writing a new prescription.
Alternating medications/providers. Patients may obtain similar medications from multiple providers. Prescription Drug Monitoring Programs (PDMPs), which are databases that allow physicians to track where patients are getting their prescriptions, may help prevent this. According to the Alliance of States with Prescription Monitoring Programs, as of January 2010, 48 states had instituted PDMPs or passed legislation to implement them.2
Specific medication. Patients may have an allergy or respond better to a particular drug; however, be cautious when a patient refuses to consider an alternate medication or claims he or she has taken a specific medication without a prescription and it was the only thing that worked for them.
Time between prescriptions. Patients may get a prescription for a medication, then shortly after their visit claim the medication doesn’t work and request a second prescription for a similar medication. One way to address this is to require the patient to return the unused portion of the first medication before writing a new prescription. A patient also may complain that they have to come to your office too frequently and ask for multiple refills of medication, which would decrease your ability to monitor his or her response to treatment.
A patient who meets ≥1 of the above criteria could be a higher risk for prescription drug abuse or diversion. Documenting these findings and talking with the patient could help justify the need to switch to a medication with a lower abuse potential or possibly referral to a drug treatment program.
In a 2009 survey, 56% of teens stated that prescription medications were easier to obtain than illicit drugs.3 Medications such as benzodiazepines and stimulants can be beneficial to patients, but because of their abuse potential, they may be underprescribed. Be vigilant when prescribing these medications, and monitor patients carefully to ensure that they are taking all medications as directed.
Disclosure
Dr. Wiley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Substance Abuse and Mental Health Services Administration. Results from the 2011 National Survey on Drug Use and Health: Summary of findings. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. http://www.samhsa.gov/data/NSDUH/2k11Results/NSDUHresults2011.htm. Accessed May 2, 2013.
2. Alliance of States with Prescription Monitoring Programs. http://www.pmpalliance.org/content/about-alliance. Accessed May 1, 2013.
3. Partnership for a Drug-Free America. 2009 parents and teens attitude tracking study report. New York, NY: Partnership for a Drug-Free America; 2010. http://www.drugfree.org/wp-content/uploads/2011/04/FULL-REPORT-PATS-2009-3-2-10.pdf. Accessed May 2, 2013.
Medications that psychiatrists routinely prescribe—such as benzodiazepines for anxiety and psychostimulants for attention-deficit/hyperactivity disorder—often are diverted and abused. In 2011, 6.1 million Americans age ≥12 abused prescription drugs.1
The mnemonic LAST can bring to mind 4 clinical “red flags” that can assist you in determining whether prescription abuse or diversion is occurring. Incorporating these 4 warning signs in your clinical assessment and medication reviews will make it easier for you to detect when medications are not being taken as prescribed.
Lost or stolen prescriptions. Patients who want to obtain a new or replacement prescription may claim that their medication was lost or stolen. Although this can occur, the prescriber should be suspicious if this becomes a recurrent situation. Some clinicians require patients to produce a filed police report for stolen medications before they will consider writing a new prescription.
Alternating medications/providers. Patients may obtain similar medications from multiple providers. Prescription Drug Monitoring Programs (PDMPs), which are databases that allow physicians to track where patients are getting their prescriptions, may help prevent this. According to the Alliance of States with Prescription Monitoring Programs, as of January 2010, 48 states had instituted PDMPs or passed legislation to implement them.2
Specific medication. Patients may have an allergy or respond better to a particular drug; however, be cautious when a patient refuses to consider an alternate medication or claims he or she has taken a specific medication without a prescription and it was the only thing that worked for them.
Time between prescriptions. Patients may get a prescription for a medication, then shortly after their visit claim the medication doesn’t work and request a second prescription for a similar medication. One way to address this is to require the patient to return the unused portion of the first medication before writing a new prescription. A patient also may complain that they have to come to your office too frequently and ask for multiple refills of medication, which would decrease your ability to monitor his or her response to treatment.
A patient who meets ≥1 of the above criteria could be a higher risk for prescription drug abuse or diversion. Documenting these findings and talking with the patient could help justify the need to switch to a medication with a lower abuse potential or possibly referral to a drug treatment program.
In a 2009 survey, 56% of teens stated that prescription medications were easier to obtain than illicit drugs.3 Medications such as benzodiazepines and stimulants can be beneficial to patients, but because of their abuse potential, they may be underprescribed. Be vigilant when prescribing these medications, and monitor patients carefully to ensure that they are taking all medications as directed.
Disclosure
Dr. Wiley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Substance Abuse and Mental Health Services Administration. Results from the 2011 National Survey on Drug Use and Health: Summary of findings. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. http://www.samhsa.gov/data/NSDUH/2k11Results/NSDUHresults2011.htm. Accessed May 2, 2013.
2. Alliance of States with Prescription Monitoring Programs. http://www.pmpalliance.org/content/about-alliance. Accessed May 1, 2013.
3. Partnership for a Drug-Free America. 2009 parents and teens attitude tracking study report. New York, NY: Partnership for a Drug-Free America; 2010. http://www.drugfree.org/wp-content/uploads/2011/04/FULL-REPORT-PATS-2009-3-2-10.pdf. Accessed May 2, 2013.
Medications that psychiatrists routinely prescribe—such as benzodiazepines for anxiety and psychostimulants for attention-deficit/hyperactivity disorder—often are diverted and abused. In 2011, 6.1 million Americans age ≥12 abused prescription drugs.1
The mnemonic LAST can bring to mind 4 clinical “red flags” that can assist you in determining whether prescription abuse or diversion is occurring. Incorporating these 4 warning signs in your clinical assessment and medication reviews will make it easier for you to detect when medications are not being taken as prescribed.
Lost or stolen prescriptions. Patients who want to obtain a new or replacement prescription may claim that their medication was lost or stolen. Although this can occur, the prescriber should be suspicious if this becomes a recurrent situation. Some clinicians require patients to produce a filed police report for stolen medications before they will consider writing a new prescription.
Alternating medications/providers. Patients may obtain similar medications from multiple providers. Prescription Drug Monitoring Programs (PDMPs), which are databases that allow physicians to track where patients are getting their prescriptions, may help prevent this. According to the Alliance of States with Prescription Monitoring Programs, as of January 2010, 48 states had instituted PDMPs or passed legislation to implement them.2
Specific medication. Patients may have an allergy or respond better to a particular drug; however, be cautious when a patient refuses to consider an alternate medication or claims he or she has taken a specific medication without a prescription and it was the only thing that worked for them.
Time between prescriptions. Patients may get a prescription for a medication, then shortly after their visit claim the medication doesn’t work and request a second prescription for a similar medication. One way to address this is to require the patient to return the unused portion of the first medication before writing a new prescription. A patient also may complain that they have to come to your office too frequently and ask for multiple refills of medication, which would decrease your ability to monitor his or her response to treatment.
A patient who meets ≥1 of the above criteria could be a higher risk for prescription drug abuse or diversion. Documenting these findings and talking with the patient could help justify the need to switch to a medication with a lower abuse potential or possibly referral to a drug treatment program.
In a 2009 survey, 56% of teens stated that prescription medications were easier to obtain than illicit drugs.3 Medications such as benzodiazepines and stimulants can be beneficial to patients, but because of their abuse potential, they may be underprescribed. Be vigilant when prescribing these medications, and monitor patients carefully to ensure that they are taking all medications as directed.
Disclosure
Dr. Wiley reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Substance Abuse and Mental Health Services Administration. Results from the 2011 National Survey on Drug Use and Health: Summary of findings. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2012. http://www.samhsa.gov/data/NSDUH/2k11Results/NSDUHresults2011.htm. Accessed May 2, 2013.
2. Alliance of States with Prescription Monitoring Programs. http://www.pmpalliance.org/content/about-alliance. Accessed May 1, 2013.
3. Partnership for a Drug-Free America. 2009 parents and teens attitude tracking study report. New York, NY: Partnership for a Drug-Free America; 2010. http://www.drugfree.org/wp-content/uploads/2011/04/FULL-REPORT-PATS-2009-3-2-10.pdf. Accessed May 2, 2013.
Ending a physician/patient relationship: 8 tips for writing a termination letter
For many valid reasons, a physician-patient relationship may need to end before treatment is completed. When terminating a clinical relationship, send a letter to the patient, even if the patient initiated the termination. Here are 8 tips for writing and sending a termination letter:
1. Don’t send a form letter. Start with a standard letter but personalize it for each patient. Address the patient by name and, if possible, allude to specifics of the patient’s situation.
2. Wish the patient well, but avoid hyperbole, such as “It truly has been an honor and a privilege to participate in your treatment.” Also, be unambiguous in stating that the treatment relationship is terminated.
3. Don’t mention confidential information. Because someone other than the patient may open the letter, do not include confidential information.
4. Provide appropriate notice. Specify a date after which you can no longer provide care. A reasonable period is 30 days from the date of the letter, but if you expect the patient will need more time to find an appropriate clinician, a longer period may be necessary.1,2 Occasionally, a patient’s care may need to be terminated immediately because of a serious problem such as actual or threatened violence. Even in these cases, communicate and document how the patient can obtain emergency psychiatric care.
5. State the reason for termination. Although you are not required legally to do so, briefly state the reason for terminating the relationship, although you should never use emotional or harshly critical language. Use nonjudgmental language and avoid referring to your “policy,” which can imply unthinking application of rigid rules.
6. Recommend continued treatment. Make a clear recommendation that the patient continue treatment elsewhere. Provide a list of mental health professionals with whom the patient could continue treatment or offer to provide referrals. Offer to send a copy of your records to the patient’s new clinician. Consider enclosing a blank copy of the release form you use so that the patient can mail it to you to request his or her records.
7. Sign the letter yourself. Don’t have a staff member sign the letter or use a stamp.
8. Send the letter by certified mail. Request a return receipt and put a copy of the letter, along with the certified mail form, in the patient’s chart. When the return receipt is received, put it in the chart. If a certified letter is returned to you, put the undelivered letter and envelope in the chart, then send a copy of the letter through regular mail and document that you did so.
If the patient requests an appointment after the notice period is over, including saying that he or she did not receive the letter, you are not legally obligated to resume his or her care.2
1. The Psychiatrist’s Program. Termination of the psychiatrist-patient relationship dos and donts. http://www.psychprogram.com/risk-management/tip-termination.html. Accessed February 1, 2013.
2. Willis DR, Zerr A. Terminating a patient: is it time to part ways? Fam Pract Manag. 2005;12(8):34-38.
For many valid reasons, a physician-patient relationship may need to end before treatment is completed. When terminating a clinical relationship, send a letter to the patient, even if the patient initiated the termination. Here are 8 tips for writing and sending a termination letter:
1. Don’t send a form letter. Start with a standard letter but personalize it for each patient. Address the patient by name and, if possible, allude to specifics of the patient’s situation.
2. Wish the patient well, but avoid hyperbole, such as “It truly has been an honor and a privilege to participate in your treatment.” Also, be unambiguous in stating that the treatment relationship is terminated.
3. Don’t mention confidential information. Because someone other than the patient may open the letter, do not include confidential information.
4. Provide appropriate notice. Specify a date after which you can no longer provide care. A reasonable period is 30 days from the date of the letter, but if you expect the patient will need more time to find an appropriate clinician, a longer period may be necessary.1,2 Occasionally, a patient’s care may need to be terminated immediately because of a serious problem such as actual or threatened violence. Even in these cases, communicate and document how the patient can obtain emergency psychiatric care.
5. State the reason for termination. Although you are not required legally to do so, briefly state the reason for terminating the relationship, although you should never use emotional or harshly critical language. Use nonjudgmental language and avoid referring to your “policy,” which can imply unthinking application of rigid rules.
6. Recommend continued treatment. Make a clear recommendation that the patient continue treatment elsewhere. Provide a list of mental health professionals with whom the patient could continue treatment or offer to provide referrals. Offer to send a copy of your records to the patient’s new clinician. Consider enclosing a blank copy of the release form you use so that the patient can mail it to you to request his or her records.
7. Sign the letter yourself. Don’t have a staff member sign the letter or use a stamp.
8. Send the letter by certified mail. Request a return receipt and put a copy of the letter, along with the certified mail form, in the patient’s chart. When the return receipt is received, put it in the chart. If a certified letter is returned to you, put the undelivered letter and envelope in the chart, then send a copy of the letter through regular mail and document that you did so.
If the patient requests an appointment after the notice period is over, including saying that he or she did not receive the letter, you are not legally obligated to resume his or her care.2
For many valid reasons, a physician-patient relationship may need to end before treatment is completed. When terminating a clinical relationship, send a letter to the patient, even if the patient initiated the termination. Here are 8 tips for writing and sending a termination letter:
1. Don’t send a form letter. Start with a standard letter but personalize it for each patient. Address the patient by name and, if possible, allude to specifics of the patient’s situation.
2. Wish the patient well, but avoid hyperbole, such as “It truly has been an honor and a privilege to participate in your treatment.” Also, be unambiguous in stating that the treatment relationship is terminated.
3. Don’t mention confidential information. Because someone other than the patient may open the letter, do not include confidential information.
4. Provide appropriate notice. Specify a date after which you can no longer provide care. A reasonable period is 30 days from the date of the letter, but if you expect the patient will need more time to find an appropriate clinician, a longer period may be necessary.1,2 Occasionally, a patient’s care may need to be terminated immediately because of a serious problem such as actual or threatened violence. Even in these cases, communicate and document how the patient can obtain emergency psychiatric care.
5. State the reason for termination. Although you are not required legally to do so, briefly state the reason for terminating the relationship, although you should never use emotional or harshly critical language. Use nonjudgmental language and avoid referring to your “policy,” which can imply unthinking application of rigid rules.
6. Recommend continued treatment. Make a clear recommendation that the patient continue treatment elsewhere. Provide a list of mental health professionals with whom the patient could continue treatment or offer to provide referrals. Offer to send a copy of your records to the patient’s new clinician. Consider enclosing a blank copy of the release form you use so that the patient can mail it to you to request his or her records.
7. Sign the letter yourself. Don’t have a staff member sign the letter or use a stamp.
8. Send the letter by certified mail. Request a return receipt and put a copy of the letter, along with the certified mail form, in the patient’s chart. When the return receipt is received, put it in the chart. If a certified letter is returned to you, put the undelivered letter and envelope in the chart, then send a copy of the letter through regular mail and document that you did so.
If the patient requests an appointment after the notice period is over, including saying that he or she did not receive the letter, you are not legally obligated to resume his or her care.2
1. The Psychiatrist’s Program. Termination of the psychiatrist-patient relationship dos and donts. http://www.psychprogram.com/risk-management/tip-termination.html. Accessed February 1, 2013.
2. Willis DR, Zerr A. Terminating a patient: is it time to part ways? Fam Pract Manag. 2005;12(8):34-38.
1. The Psychiatrist’s Program. Termination of the psychiatrist-patient relationship dos and donts. http://www.psychprogram.com/risk-management/tip-termination.html. Accessed February 1, 2013.
2. Willis DR, Zerr A. Terminating a patient: is it time to part ways? Fam Pract Manag. 2005;12(8):34-38.
Depression and inflammation: Examining the link
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
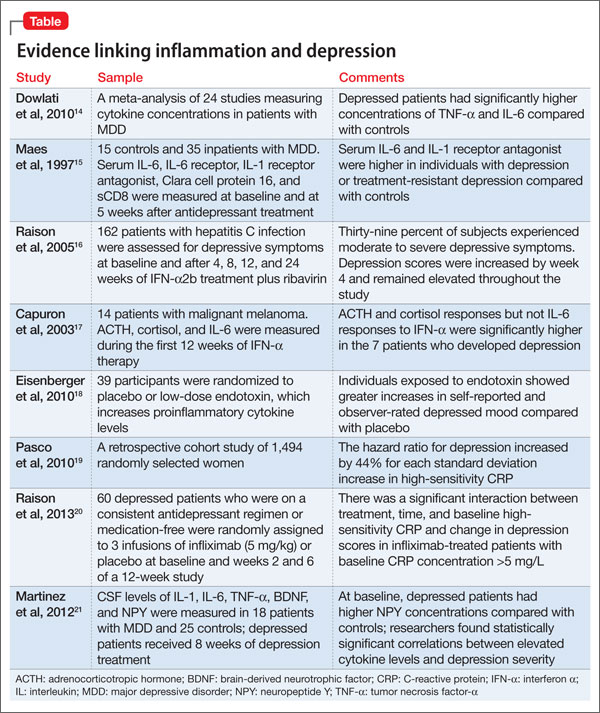
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).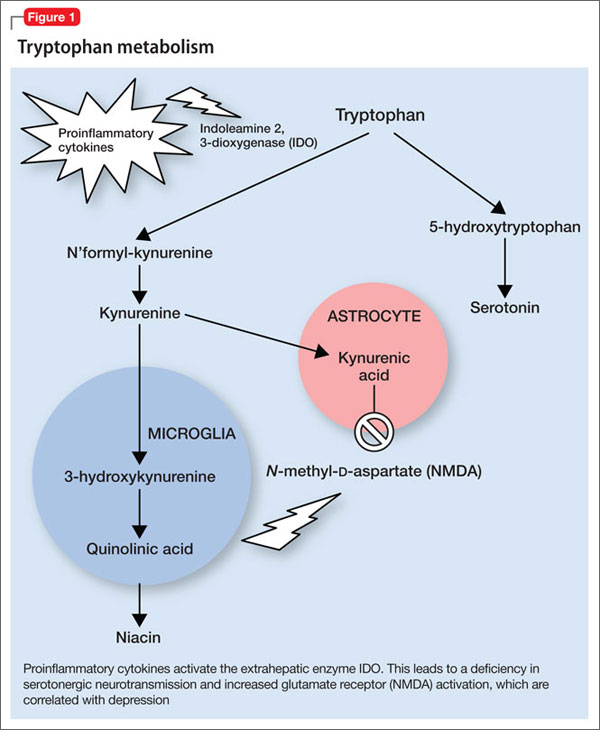
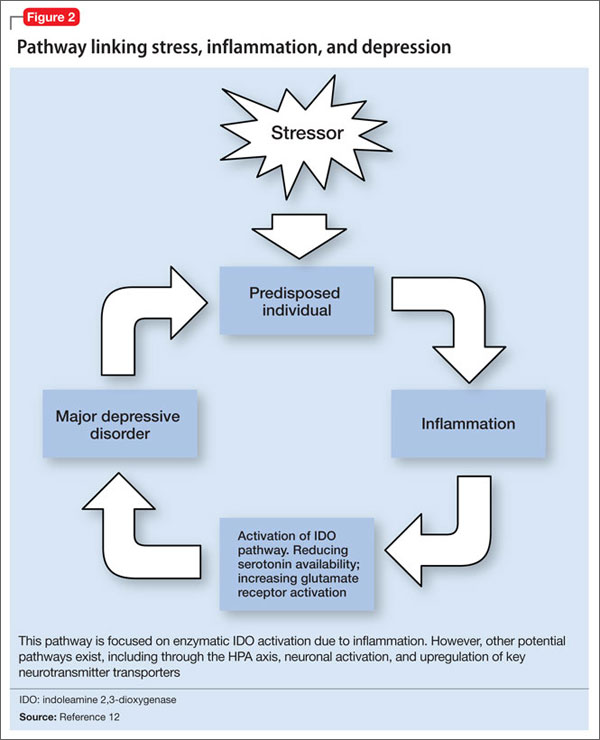
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.
Sneezing, coughing, and a sore throat are hallmark symptoms of a common cold, but what keeps you in bed are the accompanying fatigue, inattentiveness, loss of appetite, change in sleep pattern, heightened perception of pain, and apathetic withdrawal. This “sickness behavior” is induced by inflammatory markers released in response to illness.1,2 These symptoms are similar to the constellation of symptoms that define depression. Within the inflammatory response to illness, we see the shadow of depression, but the precise relationship remains murky.
Is depression part of a normal somatic inflammatory response run amok? Some researchers have argued that “sickness behavior” is adaptive, forcing the body into a constricted pattern in order to funnel energy into healing.1,3 If depression and inflammation are related, depression pushes past these adaptive roots and is less a forced pause than a debilitating withdrawal. Perhaps depression, or a subtype, is a sign of inflammation along with heat, pain, redness, and swelling. In some instances, depression may be a sign of an underlying inflammatory process.4
In our progression toward understanding depression’s pathophysiology, we see factors that point to a relationship between depression and inflammation:
• depression frequently is comorbid with many inflammatory illnesses
• increased inflammatory biomarkers are associated with major depressive disorder (MDD)
• exposure to immunomodulating agents may increase the risk of developing depression
• stress can activate proinflammatory pathways
• antidepressants can decrease inflammatory response
• inhibition of inflammatory pathways can improve mood.
Exploring these factors and a possible pathway linking inflammation and neurobiologic changes found in depression allows us to look closer at the possible integration of the inflammatory process and depressive symptoms.
Illness and depression rates
Individuals with inflammatory illnesses—autoimmune diseases, cardiovascular disease, diabetes, and cancer—often struggle with depression. Nearly 1 in 5 persons with cardiovascular disease experiences MDD.5 A diabetes diagnosis doubles the odds of having depression.6 Up to 70% of patients with autoimmune diseases, such as rheumatoid arthritis or systemic lupus erythematosus, experience depression.7,8 In a large-scale longitudinal study, having a prior autoimmune disease increased the risk of depression by 45% and history of hospitalization with infection increased a patient’s risk by 62%; the risk more than doubled in individuals with both.9 Several studies show that 15% to 25% of cancer patients experience depression,10 compared with 9% in the general population.11
Role of inflammatory markers
During an inflammatory episode the body releases cytokines, which are small, cell-signaling protein molecules. These inflammatory markers launch signaling cascades that incite the immune system into action. Type 1 cytokines (interferon-ã, tumor necrosis factor-á [TNF-á], interleukin [IL]-1) enhance cellular immune responses, and type 2 cytokines (IL-6, IL-10, IL-13) engage antibody responses. These cytokines also induce acute phase proteins, such as C-reactive protein (CRP), which can activate the immune system. Significantly higher levels of inflammatory markers are associated with a range of depressive symptoms, which grants insight into disease severity and treatment response.3,12,13
Multiple studies have explored the link between depression and inflammatory markers (Table).14-21 Peripheral inflammatory markers such as IL-6, IL-1â, CRP, and TNF-á are elevated in inflammatory diseases and in otherwise healthy individuals with MDD.12 In a meta-analysis of 24 studies measuring cytokines in depressed patients, Dowlati et al14 found individuals with MDD had significantly higher concentrations of TNF-á and IL-6 compared with controls. Increased peripheral inflammatory markers were found among antidepressant nonresponders more often than those who responded to treatment.15,22
Cytokines and depression risk
Administering immunomodulating agents has been shown to increase the risk of developing depression. Injecting animals with IL-1â or TNF-á causes sickness behavior in a dose- and time-related manner.1 As these inflammatory signaling proteins increase, sickness behaviors become more pronounced.
In humans, a natural model arises in the use of the cytokine interferon-á (INF-á) for treating hepatitis C, multiple sclerosis, malignant melanoma, and some blood cancers. Patients receiving INF-á have higher rates of depression than those not administered interferon.16 Patients receiving chronic immunotherapy treatment show long-term changes in monoamine neurotransmitters and along the HPA axis; these changes mimic those seen in depressed individuals.17,23 Acutely administered immunotherapeutic agents, such as the typhoid vaccine, have led to depressive symptoms with brain changes similar to those seen in MDD.18 Low levels of IL-6 and CRP independently predicted development of depression over several years.19
Immunotherapy-induced depression looks similar to any other major depressive episode through our current diagnostic framework and at the molecular and anatomical level.
Stress and inflammation
Depression can develop in the absence of inflammatory illness. Knowing that depressive symptoms may be associated with increased peripheral inflammatory markers, what induces the inflammatory process in some persons who are depressed but medically healthy? One theory is that psychological stress can activate inflammation.
Acute and chronic stress is associated with increased availability of proinflammatory cytokines and decreases in anti-inflammatory cytokines.3,24 One theory looks to glucocorticoid response to stress as an explanation. Miller et al25 found glucocorticoid sensitivity decreased among depressed women after exposure to a mock job interview stressor and increased among nondepressed controls. Because glucocorticoids normally stop the inflammatory cascade, this finding suggests depressed individuals may not be able to control inflammation during stress.26 At the level of genetic expression, there is increased transcription of proinflammatory genes in response to stress as a result of increased activation of nuclear factor kappa B.3,27
Shared pathways
If there is a relationship between inflammation and depression, what is the possible shared pathway?
There are 4 pathways by which cytokines effect changes in the CNS:12
• cytokines can activate primary afferent neurons (eg, vagal nerve)
• cytokines, released by macrophage-like cells in response to pathogens, diffuse through the brain’s circumventricular organs
• cytokine transporters saturate the blood-brain barrier
• cytokine IL-1 activates receptors on perivascular macrophages and endothelial cells of brain venules, causing local release of prostaglandin E2.
Through these pathways, cytokines initiate a cascade of reactions that lower serotonin levels and boost glutamatergic actions, possibly contributing to development of depressive symptoms. Depression correlates with a deficiency in serotonergic neurotransmission and increased glutamate receptor N-methyl-d-aspartate (NMDA) activation.28
Proinflammatory cytokines activate theextrahepatic enzyme indoleamine 2,3-dioxygenase (IDO), which degrades tryptophan, a precursor to serotonin (Figure 1). Tryptophan is channeled increasingly toward production of kynurenine via IDO degradation, competing with the serotonin pathway. Within the microglia, which are preferentially activated over astrocytes during inflammatory states, kynurenine is metabolized into quinolinic acid, which is an agonist of glutamatergic NMDA receptors.28 Therefore, there is a serotonergic deficiency and glutamatergic overdrive in proinflammatory states that paves the way toward a likely depressive syndrome (Figure 2).
Antidepressants’ effects
The symptoms of cytokine-induced depression are no different from MDD with unknown etiology29 and both are effectively treated with antidepressants. Even sickness behavior can be improved with antidepressant treatment.30
Antidepressants not only decrease immunotherapy-induced depressive symptoms but have been shown to decrease inflammatory response and lower proinflammatory factors (IL-2, IL-6, TNF-á, and INF-ã).31-33 Electroconvulsive therapy has been shown to normalize elevated TNF-á levels.34
Enhancing depression treatment
Researchers are investigating whether treatment with anti-inflammatory agents can ease depressive symptoms. In animal studies, normal behavioral reactions to a stressor—similar to sickness behavior and overlapping with several features of depression—were reduced with administration of cytokine antagonists or anti-inflammatory cytokines directly into the brain.35 However, there have been few successful trials in humans. Both anti-inflammatory agents such as cyclooxygenase-2 (COX-2) inhibitors, acetylsalicylic acid (aspirin), and TNF receptor antagonists can enhance depression treatments. Persoons et al36 found that Crohn’s disease patients who had higher pretreatment CRP levels and MDD had greater remission of depressive symptoms after treatment with the TNF-á antagonist infliximab. In studies, depression within the context of other autoimmune disorders or any condition with increased inflammation has responded to treatment with TNF-á antagonists.37,38 COX-2 inhibitors added to a standard antidepressant regimen improved depressive symptoms in medically healthy individuals during an acute depressive episode.39 Aspirin has shown some benefits as an adjuvant agent in persons who have failed selective serotonin reuptake inhibitor monotherapy.40,41
These anti-inflammatory agents have shown benefits in treating depression in some persons, but not in all. The key difference between those subsets of patients is elusive, mired in the complex interactions of the many systems that contribute to the symptoms we label as depression.
Future clinical applications
The association between depression and inflammation raises the possibility of a tantalizing line of future theories and treatment options. However, when considered individually, these pieces are limited in defining the precise relationship - a task nearly impossible for such a diffuse symptom as inflammation and such a complex disease as depression.
It is evident that inflammation and depression form a strong relationship to each other in individuals, which suggests the possibility of an inflammatory subtype of depression. At least within that limited group, there is the possibility of successful intervention and treatment of depression by directly treating inflammation with anti-inflammatory agents.
Perhaps once the relationship between depression and inflammation is further defined and a high-risk population identified—maybe even by genotype—depressive symptoms might be used to flag a provider’s attention to a possible disease process and serve as a new tool for identifying dangerous inflammatory activity at an early stage. Managing stress and depression may become the next tool to prevent inflammatory diseases.
Given our current knowledge, clinicians treating patients with inflammatory conditions should be aware of the increased risk of depression and ensure that depression screening is routinely completed and treatment is initiated or referrals made as needed. Ensuring appropriate depression treatment may help improve patients’ quality of life and ease the inflammatory response itself.
For psychiatrists seeing patients with an inflammatory condition, brief explanations of the known links between depression and inflammation can provide patients—particularly those ambivalent about seeking mental health care—support for engaging in treatment and adhering to medication. Describing the links between inflammation and depression also can help encourage regular exercise and healthy diets rich in fruits, vegetables, and omega-3 fatty acids. In cases of treatment-resistant depression, particularly in those with known high inflammatory factors, it may be worthwhile to consider anti-inflammatory agents, such as infliximab, as an adjuvant treatment.
The relationship between inflammation and depression is rapidly unfolding, but the full intricacies have not yet described. However, this beginning awareness of the interplay among stress, inflammation, and depression can broaden our approach to care and treatment.
Bottom Line
Depression and inflammation are linked in many ways, although neither appears to be wholly necessary or sufficient for the other. Most likely there exists a particular subset of patients for whom inflammation will precipitate and perpetuate depression.
Related Resources
- The Emory University Mind-Body Program. www.
psychiatry.emory.edu/PROGRAMS/mindbody/index.html. - Gabriel B. The evolutionary advantage of depression. The Atlantic. October 2, 2012. www.theatlantic.com/health/archive/2012/10/the-evolutionary-advantage-of-depression/263124.
Drug Brand Names
Infliximab • Remicade Ribavirin • Rebetol, Virazole
Interferon-α • Intron
Disclosure
Dr. Almond reports no financial relationship with any company whose products are mentioned in this article or with manufacturers of competing products.
References
1. Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9(1):46-56.
2. Hart BL. Biological basis of the behavior of sick animals. Neurosci Biobehav Rev. 1988;12(2):123-137.
3. Raison CL, Capuron L, Miller AH. Cytokines sing the blues: inflammation and the pathogenesis of depression. Trends Immunol. 2006;27(1):24-31.
4. Lamers F, Vogelzangs N, Merikangas KR, et al. Evidence for a differential role of HPA-axis function, inflammation and metabolic syndrome in melancholic versus atypical depression [published online October 23, 2012]. Mol Psychiatry. doi: 10.1038/mp.2012.144.
5. Hoen P, Kupper N, de Jonge P. Depression and cardiovascular disease progression: epidemiology, mechanisms and treatment. In: Hjemdahl P, Rosengren A, Steptoe A, eds. Stress and cardiovascular disease. London, United Kingdom: Springer; 2012:211-233.
6. Anderson RJ, Freedland KE, Clouse RE, et al. The prevalence of comorbid depression in adults with diabetes: a meta-analysis. Diabetes Care. 2001;24(6):1069-1078.
7. Bachen EA, Chesney MA, Criswell LA. Prevalence of mood and anxiety disorders in women with systemic lupus erythematosus. Arthritis Rheum. 2009;61(6):822-829.
8. Dickens C, McGowan L, Clark-Carter D, et al. Depression in rheumatoid arthritis: a systematic review of the literature with meta-analysis. Psychosom Med. 2002;64(1):52-60.
9. Benros ME, Waltoft BL, Nordentoft M, et al. Autoimmunity and infections as risk factors for depression and other severe mental illnesses. Neurology, Psychiatry and Brain Research. 2012;18(2):40-41.
10. National Cancer Institute. Depression (PDQ). http://www.cancer.gov/cancertopics/pdq/supportivecare/depression/HealthProfessional/page1. Updated January 9, 2013. Accessed April 23, 2013.
11. Centers for Disease Control and Prevention. Current depression among adults—United States, 2006 and 2008. Morb Mortal Wkly Rep. 2010;59(38):1229-1235.
12. Krishnadas R, Cavanagh J. Depression: an inflammatory illness? J Neurol Neurosurg Psychiatry. 2012;83(5):495-502.
13. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732-741.
14. Dowlati Y, Herrmann N, Swardfager W, et al. A meta-analysis of cytokines in major depression. Biol Psychiatry. 2010;67(5):446-457.
15. Maes M, Bosmans E, De Jongh R, et al. Increased serum IL-6 and IL-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine. 1997;9(11):853-858.
16. Raison CL, Borisov AS, Broadwell SD, et al. Depression during pegylated interferon-alpha plus ribavirin therapy: prevalence and prediction. J Clin Psychiatry. 2005;66(1):41-48.
17. Capuron L, Raison CL, Musselman DL, et al. Association of exaggerated HPA axis response to the initial injection of interferon-alpha with development of depression during interferon-alpha therapy. Am J Psychiatry. 2003;160(7):1342-1345.
18. Eisenberger NI, Berkman ET, Inagaki TK, et al. Inflammation-induced anhedonia: endotoxin reduces ventral striatum responses to reward. Biol Psychiatry. 2010;68(8):748-754.
19. Pasco JA, Nicholson GC, Williams LJ, et al. Association of high-sensitivity C-reactive protein with de novo major depression. Br J Psychiatry. 2010;197(5):372-377.
20. Raison CL, Rutherford RE, Woolwine BJ, et al. A randomized controlled trial of the tumor necrosis factor antagonist infliximab for treatment-resistant depression: the role of baseline inflammatory biomarkers. JAMA Psychiatry. 2013;70(1):31-41.
21. Martinez JM, Garakani A, Yehuda R, et al. Proinflammatory and “resiliency” proteins in the CSF of patients with major depression. Depress Anxiety. 2012;29(1):32-38.
22. Lanquillon S, Krieg JC, Bening-Abu-Shach U, et al. Cytokine production and treatment response in major depressive disorder. Neuropsychopharmacology. 2000;22(4):370-379.
23. Raison CL, Miller AH. Is depression an inflammatory disorder? Curr Psychiatry Rep. 2011;13(6):467-775.
24. Maes M, Song C, Lin A, et al. The effects of psychological stress on humans: increased production of pro-inflammatory cytokines and Th1-like response in stress-induced anxiety. Cytokine. 1998;10(4):313-318.
25. Miller GE, Rohleder N, Stetler C, et al. Clinical depression and regulation of the inflammatory response during acute stress. Psychosom Med. 2005;67(5):679-687.
26. Raison CL, Miller AH. When not enough is too much: the role of insufficient glucocorticoid signaling in the pathophysiology of stress-related disorders. Am J Psychiatry. 2003;160(9):1554-1565.
27. Tak PP, Firestein GS. NF-êB: a key role in inflammatory diseases. J Clin Invest. 2001;107(1):7-12.
28. Müller N, Schwarz MJ. The immune-mediated alteration of serotonin and glutamate: towards an integrated view of depression. Mol Psychiatry. 2007;12(11):988-1000.
29. Capuron L, Fornwalt FB, Knight BT, et al. Does cytokine-induced depression differ from idiopathic major depression in medically healthy individuals? J Affect Disord. 2009;119(1-3):181-185.
30. Yirmiya R, Pollak Y, Morag M, et al. Illness, cytokines, and depression. Ann N Y Acad Sci. 2000;917(1):478-487.
31. Maes M. The immunoregulatory effects of antidepressants. Hum Psychopharmacol. 2001;16(1):95-103.
32. Szuster-Ciesielska A, Tustanowska-Stachura A, Słotwin`ska M, et al. In vitro immunoregulatory effects of antidepressants in healthy volunteers. Pol J Pharmacol. 2003;55(3):353-362.
33. Maes M, Berk M, Goehler L, et al. Depression and sickness behavior are Janus-faced responses to shared inflammatory pathways. BMC Med. 2012;10(1):66.
34. Hestad KA, Tønseth S, Støen CD, et al. Raised plasma levels of tumor necrosis factor [alpha] in patients with depression: normalization during electroconvulsive therapy. J ECT. 2003;19(4):183-188.
35. Maier SF, Watkins LR. Intracerebroventricular interleukin-1 receptor antagonist blocks the enhancement of fear conditioning and interference with escape produced by inescapable shock. Brain Res. 1995;695(2):279-282.
36. Persoons P, Vermeire S, Demyttenaere K, et al. The impact of major depressive disorder on the short- and long-term outcome of Crohn’s disease treatment with infliximab. Aliment Pharmacol Ther. 2005;22(2):101-110.
37. Mathias SD, Colwell HH, Miller DP, et al. Health-related quality of life and functional status of patients with rheumatoid arthritis randomly assigned to receive etanercept or placebo. Clin Ther. 2000;22(1):128-139.
38. Raison C, Rutherford RE, Woolwine B, et al. The tumor necrosis factor-alpha antagonist infliximab reduces depressive symptoms in patients with treatment resistant depression and high inflammation. Brain, Behavior, and Immunity. 2012;26(suppl 1):S49.
39. Müller N, Schwarz MJ, Dehning S, et al. The cyclooxygenase-2 inhibitor celecoxib has therapeutic effects in major depression: results of a double-blind, randomized, placebo controlled, add-on pilot study to reboxetine. Mol Psychiatry. 2006;11(7):680-684.
40. Mendlewicz J, Kriwin P, Oswald P, et al. Shortened onset of action of antidepressants in major depression using acetylsalicylic acid augmentation: a pilot open-label study. Int Clin Psychopharmacol. 2006;21(4):227-231.
41. Brunello N, Alboni S, Capone G, et al. Acetylsalicylic acid accelerates the antidepressant effect of fluoxetine in the chronic escape deficit model of depression. Int Clin Psychopharmacol. 2006;21(4):219-225.

Chest Tube Management
A pneumothorax is a collection of air in the space outside the lungs that is trapped within the thorax. This abnormality can occur spontaneously or as the result of trauma. Traumatic pneumothoraces include those resulting from medical interventions such as a transthoracic and transbronchial needle biopsy, central line placement, and positive‐pressure mechanical ventilation. This group is most accurately described as iatrogenic pneumothorax (IP).[1]
IP can be an expected complication of many routine thoracic procedures, but it can also occur accidentally during procedures near the lung or thoracic cavity. Some IPs may be asymptomatic and go undiagnosed, or their diagnosis may be delayed.[2] The majority of iatrogenic pneumothoraces will resolve without complications, and patients will not require medical attention. A small percentage can, however, expand and have the potential to develop into a tension pneumothorax causing severe respiratory distress and mediastinal shift.[3, 4]
The incidence of IP ranges from 0.11% with mechanical ventilation to 2.68% with thoracentesis, according to an analysis of 7.5 million uniform hospital discharge abstracts from 2000.5 A 2010 systematic review of 24 studies that included 6605 patients suggested a 6.0% incidence of pneumothorax following thoracentesis.[3] The highest risk of IP is seen with computed tomography (CT)‐guided lung biopsy, with 1 series of 1098 biopsies showing a 42% incidence; chest tube evacuation was required in 12% of these cases.[4] A Veterans Administration study of patient safety indicators from 2001 to 2004 found that risk‐adjusted rates of IP were increasing over time.[6] It is unclear whether this increase is due to increasing numbers of interventional procedures or to better rates of detection. IP poses a considerable cost to the medical system, with safety studies finding that patients with IP will stay in the hospital approximately 4 days longer and incur an additional $17,000 in charges.[7]
In addition to this financial burden, the lack of consistency in training and guidelines for management of pneumothorax is thought to add to chest tube‐related complications.[8] In 2001, the American College of Chest Physicians (ACCP) published guidelines for the management of spontaneous pneumothorax that do not specifically address IP.[9] In 2010, the British Thoracic Society (BTS) updated their guidelines and included a brief statement on IP that described a higher incidence for it than for spontaneous pneumothorax and noted its relative ease of management.[10] Despite the lack of specific guidelines dedicated to IPs, common clinical practice is to manage iatrogenic defects in a manner similar to that for spontaneous ones. However, studies have shown that the management of pneumothorax remains diverse and that the adherence to these published guidelines is suboptimal.[10, 11] The BTS guidelines favor needle aspiration as the first‐line treatment,[10] whereas the ACCP recommends drainage with catheters over aspiration.[9]
The possibility of this complication, along with the rising rate of invasive interventions being performed, has led to expanded surveillance criteria for IP. Surveillance imaging, clinical observation, or a combination of the 2 may be required, depending on the institution, the risk of the procedure, and the preference of the treating clinician. The algorithms presented here were designed in alignment with both major society guidelines and with the intention of simplifying the treatment regimen for the ease of adoption by hospitalists.
ETIOLOGY AND RISK FACTORS
The etiology and risk factors for IP are multiple, with the most common being interventional‐based procedures. In 535 Veterans Administration patients, the most common precursor procedures were transthoracic needle biopsy (24%), subclavian vein catheterization (22%), thoracentesis (20%), transbronchial biopsy (10%), pleural biopsy (8%), and positive pressure ventilation.[12] IP can also be a rare complication of pacemaker manipulations,[5] and less commonly, bronchoscopy.[13] Patient factors that increase the risk of pneumothorax in the setting of an intervention include age, chronic obstructive lung disease, primary lung cancer, malignant and parapneumonic pleural effusions, empyema, and chronic corticosteroid use.[4] As might be expected, patients with structural lung disease (eg, emphysema with bullae) and poor healing ability (eg, corticosteroid dependent), tend to have IPs more often and to require more complicated interventions for resolution.[14, 15] In some studies, operator experience seems to be inversely related to the rate of IP, and the use of ultrasound is correlated with lower rates of this complication.[1, 3]
PATIENT PRESENTATIONS AND DIAGNOSIS
Clinical signs and symptoms of a significant pneumothorax vary in severity but most often include dyspnea, tachypnea, chest pain, and pleurisy (see Box 1). Post procedure signs or symptoms require further evaluation with imaging, usually a plain chest radiograph. CT can be useful for further evaluation. Small anterior pneumothoraces may be difficult to detect without lateral radiographic imaging or computed tomogram. Ultrasound is being used more frequently at the bedside to make this diagnosis, and various studies of trauma patients have found that it has good sensitivity and specificity.[16, 17] These results have been validated by a recent meta‐analysis comparing ultrasound to chest radiographs for the detection of pneumothorax among trauma, critically ill, and postprocedural patients.[18] This study demonstrated superior sensitivity and similar specificity for ultrasound versus chest radiographs for detection of pneumothorax. More ominous signs, such as tachycardia or hypotension, can be indicative of tension pneumothorax, which requires emergent evacuation.
MANAGEMENT
Once the diagnosis of pneumothorax has been established, treatment options should be guided by defect size and clinical assessment following a defined treatment algorithm (Figure 1). As emphasized by the BTS and ACCP guidelines, we advocate considering the use of symptoms along with defect size to determine the best management course.
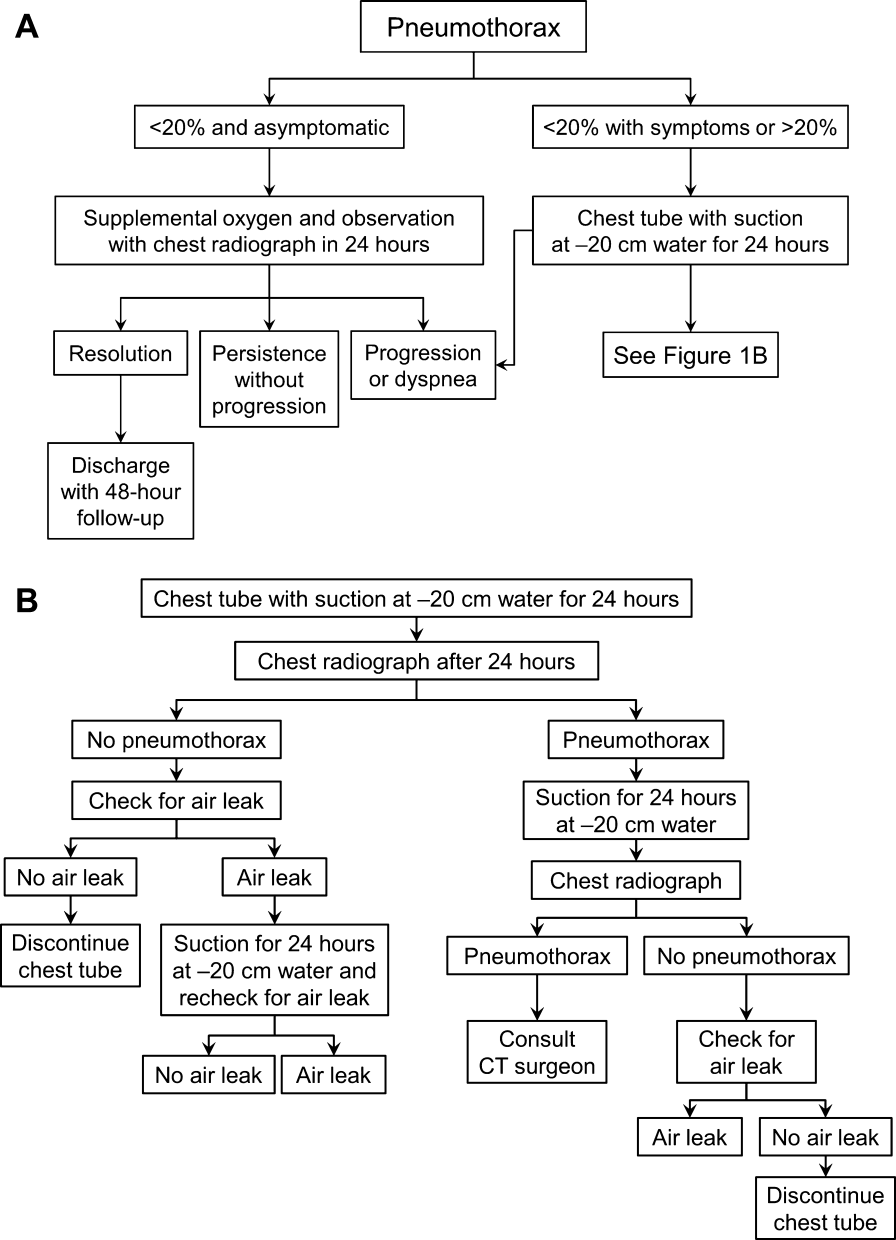
Observation
Defects that involve<20% of the hemithorax in a patient who is clinically asymptomatic and hemodynamically stable can be safely managed by oxygen supplementation and hospital observation. Repeat imaging can be obtained after 12 to 24 hours of defect detection or with symptom change. Patients who display resolution may be discharged home.
Patients who show persistence without progression but are asymptomatic may also be discharged safely, with follow‐up imaging and clinical evaluation 48 hours later.[9, 10] This was demonstrated by Kelly and colleagues,[19] who described the outcomes of 154 patients in a retrospective cohort study. Of the 91 patients treated with outpatient observation, 82 resolved without additional interventions. A recent review article by the same author cites conservative management of small pneumothoraces as being widely accepted.[20] If reimaging shows progression of defect or if the patient becomes more symptomatic, the pneumothorax should be evacuated by 1 of the methods described below.
Aspiration
Aspiration is defined by the ACCP Delphi consensus statement as the removal of pleural air via needle or cannula followed by immediate removal of needle or cannula.[9] This option mandates careful patient selection. It should be considered for small pneumothoraces that cause only mild dyspnea in patients who have no known parenchymal disease. These patients should be observed overnight in the hospital and reimaged 24 hours after aspiration of the pneumothorax. Several authors have reported success with aspiration alone. Yamagami et al.[21] noted the efficacy of manual aspiration immediately after CT‐guided biopsy, with a success rate exceeding 90%. They also noted that evacuated volumes >543 mL correlated with the need for further intervention with a chest tube. This technique is advocated for small pneumothoraces that are recognized shortly after the procedure.
Similarly, Delius and colleagues[22] managed 131 pneumothoraces with aspiration as an alternative to chest tube placement. Of these, 79 were iatrogenic. Aspiration achieved a 75% success rate for all IPs. Small defects defined as <20% of volume had an even higher resolution rate of 87%. Similar findings were demonstrated by Talbot‐Stern et al.[23] in their prospective study of 76 pneumothoraces. Among those that were iatrogenic, 82% resolved after simple aspiration. Faruqi et al.[24] also showed that aspiration is a viable option for IPs. Of the 57 patients with pneumothorax included in their study, 35 were treated with aspiration alone. Iatrogenesis was the culprit in 12 of the 35 manually aspirated cases. Aspiration achieved a success rate of 91.7% in IP. A recent Cochrane database systematic review compared simple aspiration with intercostal tube drainage for primary spontaneous pneumothorax.[25] The authors reported no difference between these methods in terms of success rate, early failure rate, duration of hospital stay, 1‐year success rate, or number of patients who required pleurodesis at 1‐year follow‐up.
Because the algorithms presented in this article were specifically designed for the use by hospitalists, we intentionally omitted aspiration from the decision trees. Most hospitalists would not be expected to evacuate IPs. However, knowledge regarding this option and appropriate follow‐up are valuable to internists, because many interventionalists admit patients to the hospital service for overnight observation. An asymptomatic postaspiration patient, who on subsequent imaging demonstrates resolution or persistence without progression of pneumothorax, may be discharged with 48‐hour follow‐up.
Placement of Catheter or Chest Tube Drainage
Most patients with a clinically significant pneumothorax will require evacuation of the air. Pneumothoraces larger than 20% or that produce symptoms warrant chest tube management and inpatient observation (Figure 1B). Traditionally, large tubes with 20 cm of water on continuous suction are used and have been studied the most widely. Several authors have shown that smaller tubes can effectively drain a pneumothorax.[26, 27, 28, 29] Small‐bore catheters (8F14F), which can be inserted percutaneously, have been shown to provide effective lung re‐expansion with minimal morbidity[8] and may be better tolerated by patients with uncomplicated pneumothoraces (Figure 2). Terzi and colleagues[30] have shown that smaller tubes cause less discomfort to patients at rest, with cough, and at the time of tube removal.

At most US institutions, catheters and chest tubes are connected to all‐purpose drainage systems. Although commercially available through a variety of manufacturers, they share similar design principles because they replicate the 3‐bottle system described in detail elsewhere in the literature.[31] We have limited our discussion to 3 pleural evacuation systems because it is our intention to familiarize hospitalists with the units that they are most likely to encounter. The first 2 systems have been studied and described by Baumann and colleagues[32] as being commonplace and reasonably reliable. These include the Oasis (Atrium Medical Corp., Hudson, NH) (Figure 3A) and the Pleur‐evac (Teleflex Inc., Limerick, PA). The third unit is the Thopaz digital thoracic drainage system (Medela Inc., McHenry, IL) (Figure 3B). The Thopaz is unique in its inclusion of a suction source and digital capability. Although it utilizes the same principles of all pleural evacuation devices, its setup and information output require that one be familiar with its digital format.

Suction Versus Water Seal
The chest tube should be placed initially to a suction pressure level of 20 cm of water for 24 hours to maximize lung expansion and evacuate all extrapulmonary air. Suction pressure is set on the Pleur‐evac and Atrium drainage systems by a manual dial that reads to a water pressure of 0 to 40 cm. The default setting from the manufacturer is 20 cm of water. This level of suction is present only when the drainage system is connected to a wall or a portable suction device. The only confirmation of suction presence in the Atrium system is the deployment of the orange bellows (located under the dial) to the level of the arrow tip (Figure 3A). The Pleur‐evac system has a red stripe along the circular edge of the dial that appears at the set level of suction when negative pressure is being applied. It is important to be aware that when patients are disconnected from the wall or the portable suction apparatus, they are on water seal or gravity. These terms are synonymous with no suction. On the Thopaz, a digital menu directs operation, and levels of suction can be selected from water seal (no suction) up to 40 cm of water. We recommend using suction to 20 cm of water given the scarce evidence supporting higher levels of negative pressure. Some clinicians prefer placing patients on water seal for some time before moving toward tube discontinuance, but this is a matter of preference, and no substantial evidence exists to show that any 1 method is superior.[8, 33]
Assessing for Air Leak
If there is improvement or resolution of the pneumothorax after 24 hours, the presence of an air leak should be assessed; if no leak is present, the chest tube can be safely removed. In the context of chest tubes, the term air leak refers to residual air between the lung and the chest wall. It is possible to see resolution of a pneumothorax on chest radiographs and still have an air leak. This situation is created by a perfect balance between the pleural air evacuation by the catheter and the flow of air exiting from the lung puncture. This would result in reaccumulation of the pneumothorax if the chest tube is removed prematurely. It should also be kept in mind that chest radiographs may miss a small pneumothorax given their relatively low sensitivity.[18] Therefore, the absence of an air leak needs to be documented before the chest tube is discontinued. Depending on the type of drainage system (Atrium, Pleur‐evac, or Thopaz), this assessment can be done in several ways. All systems can be assessed for air leak by clamping the actual chest tube for 2 to 4 hours and then repeating the chest radiograph. Clamping a chest tube simulates the condition of not having a chest tube. Chest tubes should never be clamped without supervision and only with the knowledge of nursing personnel. The onset of chest pain or dyspnea in a patient with a clamped tube mandates immediate removal of the clamp and a return to suction. A repeat chest radiograph showing reaccumulation or expansion of the pneumothorax after clamping indicates that the air leak has not resolved and the chest tube must remain in place and returned to suction. Simpler and more time‐efficient methods of detecting air leaks are available with both cardiothoracic drainage systems.
For the Atrium and Pleur‐evac models, there is a graded panel through which one can visualize air leaks being funneled through water (Figure 4A). Having the patient cough several times or perform a Valsalva maneuver should release any air trapped within the chest into this chamber, where bubbles can be visualized as they travel through the water. The presence of bubbles indicates the presence of residual air in the chest, pointing to a possible leak. In contrast, the Thopaz system offers a graphical display of the air flowing into the system that can be reviewed over the 24‐hour period. When the graph line reaches a 0 flatline graph, no airflow is being detected and no air leakage is suspected (Figure 4B). If no air leaks are detected, the chest tube may be discontinued. Those patients with a failed air leak test should have their chest tubes continued under suction for another 24 hours, with the above tests then repeated. The same holds true for those patients with persistent pneumothorax at 24 hours.
Removal of chest tubes is a simple process that requires the tube to be pulled out of the patient without allowing air to enter the site where the tube was present and where it entered the thorax. Most interventionalists will discontinue the tubes that they have placed. Some small catheters have an internal string that has to be released so the catheter will straighten and pull out easily. Knowledge of the type of catheter or tube that was placed is critical before removal to prevent complications and patient discomfort. Standard chest tubes are straight, smooth plastic and pull out easily but require rapid occlusion of the larger puncture site in the chest wall with an occlusive dressing that often includes petroleum or water‐soluble gel. Some physicians will leave a suture tie when placing the chest tube so that it can be tied down to occlude the site instead of using a dressing.
Consultation of the Cardiothoracic Surgeon or Interventional Pulmonologist
We recommend the involvement of cardiothoracic surgery or interventional pulmonology for patients with nonresolving pneumothorax lasting longer than 48 hours because additional procedures may be necessary. One of the rare but serious complications of a persistent pneumothorax is the formation of a bronchopleural fistula. This communication between the bronchial tree and the pleural space can lead to significant morbidity and mortality. The treatment of a bronchopleural fistula includes medical and surgical options that are beyond the scope of this article but require the expertise of a cardiothoracic surgeon or interventional pulmonologist.[34] Most patients who will not require additional procedures will heal within 48 hours.[11, 35] Decisions regarding more invasive treatment measures can then be made as necessary.[26]
PRACTICAL TIPS
Hospitalists caring for patients with chest tubes are often asked to troubleshoot at the bedside. Scenarios that may be encountered include nonfunctioning tubes, catheter migration, and tube discomfort. Ensuring patency of the tube entails visualizing the tube from the point of entry into the chest wall to the collection chamber and inspecting for kinks or debris clogging the tube. Smaller catheters can be easily kinked during patient positioning and can become clogged. Respiratory variation, which is the movement of the column of fluid in the collection chamber or in the tubing with inspiration and expiration, suggests that the chest tube is patent. This should be part of the daily examination in a patient with a chest tube, and it should also be the first step in assessing sudden dyspnea, hypoxia, pain, or hemodynamic instability. Clogged tubes should be referred to the interventionalists or other physicians who placed them. Chest tubes are typically sutured at the site of entry and securely bandaged to avoid migration but occasionally can be dislodged. This should prompt placement of another tube by an experienced operator. Last, chest tubes can be uncomfortable for patients who may require systemic analgesics. Additionally, tube positioning may ease some of the discomfort. Chest tubes are commonly placed along the midaxillary line and the posterior thorax, leading to discomfort in the recumbent position. Directing the tube anteriorly helps ease some of the discomfort. This can be done using all‐purpose sponges to build a barrier between the skin and the chest tube as it is directed anteriorly. Additional sponges are placed above the tube for extra protection. The gentle curve accomplished by padding the underside of the tube also keeps the tube patent by avoiding sharp kinks as the catheter exits the thorax.
FUTURE TRENDS
Future trends in the management of IP may include shorter duration of tube management (14 hours of suction with air leak evaluation and removal) and the use of even smaller catheters. Outpatient management of IP with small pigtail catheters or small‐caliber tubes in addition to 1‐way valves is also being investigated. The benefits of these practices may include greater patient comfort and lower cost. These approaches will need larger‐scale replication and careful patient selection before they become standard practice.[28, 36]
Ultrasound can be used to assess the presence and size of pneumothoraces that are difficult to visualize by standard chest radiographs. Several studies have established ultrasonography as an effective method of diagnosing pneumothorax and have shown it to have superior sensitivity compared with chest radiography.[1, 18] In the future, the use of ultrasound will likely be more widespread given its performance, portability, ease of use, and relatively low cost.
SUMMARY
IP is a known and costly complication of many medical procedures. The aforementioned algorithms help simplify the management of chest tubes for hospitalists caring for patients with this common complication. This stepwise approach may not only help curtail added expenses related to IPs by decreasing the length of inpatient stays but may also improve patient satisfaction.
Acknowledgments
Disclosure: Mayo does not endorse the products mentioned in this article. The authors report no conflicts of interest.
Box
Clinical Signs and Symptoms of Pneumothorax
Dyspnea
Pleuritic chest pain
Tachypnea
Hypoxia
Decreased breath sounds on affected side
Hyper‐resonant percussion on affected side
Subcutaneous emphysema
- , . Management of pneumothorax. Semin Respir Crit Care Med. 2010;31(6):769–780.
- , . Pneumothorax. Respirology. 2004;9(2):157–164.
- , , , . Pneumothorax following thoracentesis: a systematic review and meta‐analysis. Arch Intern Med. 2010;170(4):332–339.
- , , , et al. Incidence of and risk factors for pneumothorax and chest tube placement after CT fluoroscopy‐guided percutaneous lung biopsy: retrospective analysis of the procedures conducted over a 9‐year period. AJR Am J Roentgenol. 2010;194(3):809–814.
- , , . Accidental iatrogenic pneumothorax in hospitalized patients. Med Care. 2006;44(2):182–186.
- , , , et al. Tracking rates of patient safety indicators over time: lessons from the Veterans Administration. Med Care. 2006;44(9):850–861.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA. 2003;290(14):1868–1874.
- , , , , . Preliminary report of a prospective, randomized trial of underwater seal for spontaneous and iatrogenic pneumothorax. J Am Coll Surg. 2007;204(1):84–90.
- , , , et al.; AACP Pneumothorax Consensus Group. Management of spontaneous pneumothorax: an American College of Chest Physicians Delphi consensus statement. Chest. 2001;119(2):590–602.
- , , ; BTS Pleural Disease Guideline Group. Management of spontaneous pneumothorax: British Thoracic Society Pleural Disease Guideline 2010. Thorax. 2010;65(suppl 2):ii18–ii31.
- , . The clinician's perspective on pneumothorax management. Chest. 1997;112(3):822–828.
- , , , . Iatrogenic pneumothorax: etiology and morbidity: results of a Department of Veterans Affairs Cooperative Study. Respiration. 1992;59(4):215–220.
- , , , , , . Severe complications of bronchoscopy. Respiration. 2008;76(4):429–433.
- , , , et al. Incidence and risk factors of delayed pneumothorax after transthoracic needle biopsy of the lung. Chest. 2004;126(5):1516–1521.
- , , , , , . Factors associated with pneumothorax and pneumothorax requiring treatment after percutaneous lung biopsy in 443 consecutive patients. J Vasc Interv Radiol. 2004;15(5):479–483.
- , . Ultrasound detection of pneumothorax compared with chest X‐ray and computed tomography scan. Am Surg. 2011;77(4):480–484.
- , . Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med. 2010;17(1):11–17.
- , , , , . Diagnosis of pneumothorax by radiography and ultrasonography: a meta‐analysis. Chest. 2011;140(4):859–866.
- , , . Outcomes of emergency department patients treated for primary spontaneous pneumothorax. Chest. 2008;134(5):1033–1036.
- . Review of management of primary spontaneous pneumothorax: is the best evidence clearer 15 years on? Emerg Med Australas. 2007;19(4):303–308.
- , , , , , . Efficacy of manual aspiration immediately after complicated pneumothorax in CT‐guided lung biopsy. J Vasc Interv Radiol. 2005;16(4):477–483.
- , , , , , . Catheter aspiration for simple pneumothorax: experience with 114 patients. Arch Surg. 1998;124(7):833–836.
- , , , , . Catheter aspiration for simple pneumothorax. J Emerg Med. 1986;4(6):437–442.
- , , , . Role of simple needle aspiration in the management of pneumothorax. Indian J Chest Dis Allied Sci. 2004;46(3):183–190.
- , , . Simple aspiration versus intercostal tube drainage for primary spontaneous pneumothorax in adults. Cochrane Database Syst Rev. 2007;(1):CD004479.
- , , , . Evaluation of conventional chest tube therapy for iatrogenic pneumothorax. Chest. 1993;104(6):1770–1772.
- , , , . Outpatient treatment of iatrogenic pneumothorax after needle biopsy. Radiology. 1997;205(1):249–252.
- , , , , , . Outpatient management of postbiopsy pneumothorax with small‐caliber chest tubes: factors affecting the need for prolonged drainage and additional interventions. Cardiovasc Intervent Radiol. 2008;31(2):342–348.
- , . Management of primary and secondary pneumothorax using a small‐bore thoracic catheter. Interact Cardiovasc Thorac Surg. 2010;11(2):146–149.
- , , , et al. The use of flexible spiral drains after non‐cardiac thoracic surgery: a clinical study. Eur J Cardiothorac Surg. 2005;27(1):134–137.
- . Pleural Diseases. 5th ed. Philadelphia: PA: Lippincott Williams 2007.
- , , , . Comparison of function of commercially available pleural drainage units and catheters. Chest. 2003;123(6):1878–1886.
- , . Catheter drainage of spontaneous pneumothorax: suction or no suction, early or late removal? Thorax. 1982;37(1):46–48.
- , . Bronchopleural fistulas: an overview of the problem with special focus on endoscopic management. Chest. 2005;128(6):3955–3965.
- , , , . Timing of invasive procedures in therapy for primary and secondary spontaneous pneumothorax. Arch Surg. 1991;126(6):764–766.
- , , , , . A treatment algorithm for pneumothoraces complicating central venous catheter insertion. Am J Surg. 2000;180(6):523–526.
A pneumothorax is a collection of air in the space outside the lungs that is trapped within the thorax. This abnormality can occur spontaneously or as the result of trauma. Traumatic pneumothoraces include those resulting from medical interventions such as a transthoracic and transbronchial needle biopsy, central line placement, and positive‐pressure mechanical ventilation. This group is most accurately described as iatrogenic pneumothorax (IP).[1]
IP can be an expected complication of many routine thoracic procedures, but it can also occur accidentally during procedures near the lung or thoracic cavity. Some IPs may be asymptomatic and go undiagnosed, or their diagnosis may be delayed.[2] The majority of iatrogenic pneumothoraces will resolve without complications, and patients will not require medical attention. A small percentage can, however, expand and have the potential to develop into a tension pneumothorax causing severe respiratory distress and mediastinal shift.[3, 4]
The incidence of IP ranges from 0.11% with mechanical ventilation to 2.68% with thoracentesis, according to an analysis of 7.5 million uniform hospital discharge abstracts from 2000.5 A 2010 systematic review of 24 studies that included 6605 patients suggested a 6.0% incidence of pneumothorax following thoracentesis.[3] The highest risk of IP is seen with computed tomography (CT)‐guided lung biopsy, with 1 series of 1098 biopsies showing a 42% incidence; chest tube evacuation was required in 12% of these cases.[4] A Veterans Administration study of patient safety indicators from 2001 to 2004 found that risk‐adjusted rates of IP were increasing over time.[6] It is unclear whether this increase is due to increasing numbers of interventional procedures or to better rates of detection. IP poses a considerable cost to the medical system, with safety studies finding that patients with IP will stay in the hospital approximately 4 days longer and incur an additional $17,000 in charges.[7]
In addition to this financial burden, the lack of consistency in training and guidelines for management of pneumothorax is thought to add to chest tube‐related complications.[8] In 2001, the American College of Chest Physicians (ACCP) published guidelines for the management of spontaneous pneumothorax that do not specifically address IP.[9] In 2010, the British Thoracic Society (BTS) updated their guidelines and included a brief statement on IP that described a higher incidence for it than for spontaneous pneumothorax and noted its relative ease of management.[10] Despite the lack of specific guidelines dedicated to IPs, common clinical practice is to manage iatrogenic defects in a manner similar to that for spontaneous ones. However, studies have shown that the management of pneumothorax remains diverse and that the adherence to these published guidelines is suboptimal.[10, 11] The BTS guidelines favor needle aspiration as the first‐line treatment,[10] whereas the ACCP recommends drainage with catheters over aspiration.[9]
The possibility of this complication, along with the rising rate of invasive interventions being performed, has led to expanded surveillance criteria for IP. Surveillance imaging, clinical observation, or a combination of the 2 may be required, depending on the institution, the risk of the procedure, and the preference of the treating clinician. The algorithms presented here were designed in alignment with both major society guidelines and with the intention of simplifying the treatment regimen for the ease of adoption by hospitalists.
ETIOLOGY AND RISK FACTORS
The etiology and risk factors for IP are multiple, with the most common being interventional‐based procedures. In 535 Veterans Administration patients, the most common precursor procedures were transthoracic needle biopsy (24%), subclavian vein catheterization (22%), thoracentesis (20%), transbronchial biopsy (10%), pleural biopsy (8%), and positive pressure ventilation.[12] IP can also be a rare complication of pacemaker manipulations,[5] and less commonly, bronchoscopy.[13] Patient factors that increase the risk of pneumothorax in the setting of an intervention include age, chronic obstructive lung disease, primary lung cancer, malignant and parapneumonic pleural effusions, empyema, and chronic corticosteroid use.[4] As might be expected, patients with structural lung disease (eg, emphysema with bullae) and poor healing ability (eg, corticosteroid dependent), tend to have IPs more often and to require more complicated interventions for resolution.[14, 15] In some studies, operator experience seems to be inversely related to the rate of IP, and the use of ultrasound is correlated with lower rates of this complication.[1, 3]
PATIENT PRESENTATIONS AND DIAGNOSIS
Clinical signs and symptoms of a significant pneumothorax vary in severity but most often include dyspnea, tachypnea, chest pain, and pleurisy (see Box 1). Post procedure signs or symptoms require further evaluation with imaging, usually a plain chest radiograph. CT can be useful for further evaluation. Small anterior pneumothoraces may be difficult to detect without lateral radiographic imaging or computed tomogram. Ultrasound is being used more frequently at the bedside to make this diagnosis, and various studies of trauma patients have found that it has good sensitivity and specificity.[16, 17] These results have been validated by a recent meta‐analysis comparing ultrasound to chest radiographs for the detection of pneumothorax among trauma, critically ill, and postprocedural patients.[18] This study demonstrated superior sensitivity and similar specificity for ultrasound versus chest radiographs for detection of pneumothorax. More ominous signs, such as tachycardia or hypotension, can be indicative of tension pneumothorax, which requires emergent evacuation.
MANAGEMENT
Once the diagnosis of pneumothorax has been established, treatment options should be guided by defect size and clinical assessment following a defined treatment algorithm (Figure 1). As emphasized by the BTS and ACCP guidelines, we advocate considering the use of symptoms along with defect size to determine the best management course.
Observation
Defects that involve<20% of the hemithorax in a patient who is clinically asymptomatic and hemodynamically stable can be safely managed by oxygen supplementation and hospital observation. Repeat imaging can be obtained after 12 to 24 hours of defect detection or with symptom change. Patients who display resolution may be discharged home.
Patients who show persistence without progression but are asymptomatic may also be discharged safely, with follow‐up imaging and clinical evaluation 48 hours later.[9, 10] This was demonstrated by Kelly and colleagues,[19] who described the outcomes of 154 patients in a retrospective cohort study. Of the 91 patients treated with outpatient observation, 82 resolved without additional interventions. A recent review article by the same author cites conservative management of small pneumothoraces as being widely accepted.[20] If reimaging shows progression of defect or if the patient becomes more symptomatic, the pneumothorax should be evacuated by 1 of the methods described below.
Aspiration
Aspiration is defined by the ACCP Delphi consensus statement as the removal of pleural air via needle or cannula followed by immediate removal of needle or cannula.[9] This option mandates careful patient selection. It should be considered for small pneumothoraces that cause only mild dyspnea in patients who have no known parenchymal disease. These patients should be observed overnight in the hospital and reimaged 24 hours after aspiration of the pneumothorax. Several authors have reported success with aspiration alone. Yamagami et al.[21] noted the efficacy of manual aspiration immediately after CT‐guided biopsy, with a success rate exceeding 90%. They also noted that evacuated volumes >543 mL correlated with the need for further intervention with a chest tube. This technique is advocated for small pneumothoraces that are recognized shortly after the procedure.
Similarly, Delius and colleagues[22] managed 131 pneumothoraces with aspiration as an alternative to chest tube placement. Of these, 79 were iatrogenic. Aspiration achieved a 75% success rate for all IPs. Small defects defined as <20% of volume had an even higher resolution rate of 87%. Similar findings were demonstrated by Talbot‐Stern et al.[23] in their prospective study of 76 pneumothoraces. Among those that were iatrogenic, 82% resolved after simple aspiration. Faruqi et al.[24] also showed that aspiration is a viable option for IPs. Of the 57 patients with pneumothorax included in their study, 35 were treated with aspiration alone. Iatrogenesis was the culprit in 12 of the 35 manually aspirated cases. Aspiration achieved a success rate of 91.7% in IP. A recent Cochrane database systematic review compared simple aspiration with intercostal tube drainage for primary spontaneous pneumothorax.[25] The authors reported no difference between these methods in terms of success rate, early failure rate, duration of hospital stay, 1‐year success rate, or number of patients who required pleurodesis at 1‐year follow‐up.
Because the algorithms presented in this article were specifically designed for the use by hospitalists, we intentionally omitted aspiration from the decision trees. Most hospitalists would not be expected to evacuate IPs. However, knowledge regarding this option and appropriate follow‐up are valuable to internists, because many interventionalists admit patients to the hospital service for overnight observation. An asymptomatic postaspiration patient, who on subsequent imaging demonstrates resolution or persistence without progression of pneumothorax, may be discharged with 48‐hour follow‐up.
Placement of Catheter or Chest Tube Drainage
Most patients with a clinically significant pneumothorax will require evacuation of the air. Pneumothoraces larger than 20% or that produce symptoms warrant chest tube management and inpatient observation (Figure 1B). Traditionally, large tubes with 20 cm of water on continuous suction are used and have been studied the most widely. Several authors have shown that smaller tubes can effectively drain a pneumothorax.[26, 27, 28, 29] Small‐bore catheters (8F14F), which can be inserted percutaneously, have been shown to provide effective lung re‐expansion with minimal morbidity[8] and may be better tolerated by patients with uncomplicated pneumothoraces (Figure 2). Terzi and colleagues[30] have shown that smaller tubes cause less discomfort to patients at rest, with cough, and at the time of tube removal.
At most US institutions, catheters and chest tubes are connected to all‐purpose drainage systems. Although commercially available through a variety of manufacturers, they share similar design principles because they replicate the 3‐bottle system described in detail elsewhere in the literature.[31] We have limited our discussion to 3 pleural evacuation systems because it is our intention to familiarize hospitalists with the units that they are most likely to encounter. The first 2 systems have been studied and described by Baumann and colleagues[32] as being commonplace and reasonably reliable. These include the Oasis (Atrium Medical Corp., Hudson, NH) (Figure 3A) and the Pleur‐evac (Teleflex Inc., Limerick, PA). The third unit is the Thopaz digital thoracic drainage system (Medela Inc., McHenry, IL) (Figure 3B). The Thopaz is unique in its inclusion of a suction source and digital capability. Although it utilizes the same principles of all pleural evacuation devices, its setup and information output require that one be familiar with its digital format.
Suction Versus Water Seal
The chest tube should be placed initially to a suction pressure level of 20 cm of water for 24 hours to maximize lung expansion and evacuate all extrapulmonary air. Suction pressure is set on the Pleur‐evac and Atrium drainage systems by a manual dial that reads to a water pressure of 0 to 40 cm. The default setting from the manufacturer is 20 cm of water. This level of suction is present only when the drainage system is connected to a wall or a portable suction device. The only confirmation of suction presence in the Atrium system is the deployment of the orange bellows (located under the dial) to the level of the arrow tip (Figure 3A). The Pleur‐evac system has a red stripe along the circular edge of the dial that appears at the set level of suction when negative pressure is being applied. It is important to be aware that when patients are disconnected from the wall or the portable suction apparatus, they are on water seal or gravity. These terms are synonymous with no suction. On the Thopaz, a digital menu directs operation, and levels of suction can be selected from water seal (no suction) up to 40 cm of water. We recommend using suction to 20 cm of water given the scarce evidence supporting higher levels of negative pressure. Some clinicians prefer placing patients on water seal for some time before moving toward tube discontinuance, but this is a matter of preference, and no substantial evidence exists to show that any 1 method is superior.[8, 33]
Assessing for Air Leak
If there is improvement or resolution of the pneumothorax after 24 hours, the presence of an air leak should be assessed; if no leak is present, the chest tube can be safely removed. In the context of chest tubes, the term air leak refers to residual air between the lung and the chest wall. It is possible to see resolution of a pneumothorax on chest radiographs and still have an air leak. This situation is created by a perfect balance between the pleural air evacuation by the catheter and the flow of air exiting from the lung puncture. This would result in reaccumulation of the pneumothorax if the chest tube is removed prematurely. It should also be kept in mind that chest radiographs may miss a small pneumothorax given their relatively low sensitivity.[18] Therefore, the absence of an air leak needs to be documented before the chest tube is discontinued. Depending on the type of drainage system (Atrium, Pleur‐evac, or Thopaz), this assessment can be done in several ways. All systems can be assessed for air leak by clamping the actual chest tube for 2 to 4 hours and then repeating the chest radiograph. Clamping a chest tube simulates the condition of not having a chest tube. Chest tubes should never be clamped without supervision and only with the knowledge of nursing personnel. The onset of chest pain or dyspnea in a patient with a clamped tube mandates immediate removal of the clamp and a return to suction. A repeat chest radiograph showing reaccumulation or expansion of the pneumothorax after clamping indicates that the air leak has not resolved and the chest tube must remain in place and returned to suction. Simpler and more time‐efficient methods of detecting air leaks are available with both cardiothoracic drainage systems.
For the Atrium and Pleur‐evac models, there is a graded panel through which one can visualize air leaks being funneled through water (Figure 4A). Having the patient cough several times or perform a Valsalva maneuver should release any air trapped within the chest into this chamber, where bubbles can be visualized as they travel through the water. The presence of bubbles indicates the presence of residual air in the chest, pointing to a possible leak. In contrast, the Thopaz system offers a graphical display of the air flowing into the system that can be reviewed over the 24‐hour period. When the graph line reaches a 0 flatline graph, no airflow is being detected and no air leakage is suspected (Figure 4B). If no air leaks are detected, the chest tube may be discontinued. Those patients with a failed air leak test should have their chest tubes continued under suction for another 24 hours, with the above tests then repeated. The same holds true for those patients with persistent pneumothorax at 24 hours.
Removal of chest tubes is a simple process that requires the tube to be pulled out of the patient without allowing air to enter the site where the tube was present and where it entered the thorax. Most interventionalists will discontinue the tubes that they have placed. Some small catheters have an internal string that has to be released so the catheter will straighten and pull out easily. Knowledge of the type of catheter or tube that was placed is critical before removal to prevent complications and patient discomfort. Standard chest tubes are straight, smooth plastic and pull out easily but require rapid occlusion of the larger puncture site in the chest wall with an occlusive dressing that often includes petroleum or water‐soluble gel. Some physicians will leave a suture tie when placing the chest tube so that it can be tied down to occlude the site instead of using a dressing.
Consultation of the Cardiothoracic Surgeon or Interventional Pulmonologist
We recommend the involvement of cardiothoracic surgery or interventional pulmonology for patients with nonresolving pneumothorax lasting longer than 48 hours because additional procedures may be necessary. One of the rare but serious complications of a persistent pneumothorax is the formation of a bronchopleural fistula. This communication between the bronchial tree and the pleural space can lead to significant morbidity and mortality. The treatment of a bronchopleural fistula includes medical and surgical options that are beyond the scope of this article but require the expertise of a cardiothoracic surgeon or interventional pulmonologist.[34] Most patients who will not require additional procedures will heal within 48 hours.[11, 35] Decisions regarding more invasive treatment measures can then be made as necessary.[26]
PRACTICAL TIPS
Hospitalists caring for patients with chest tubes are often asked to troubleshoot at the bedside. Scenarios that may be encountered include nonfunctioning tubes, catheter migration, and tube discomfort. Ensuring patency of the tube entails visualizing the tube from the point of entry into the chest wall to the collection chamber and inspecting for kinks or debris clogging the tube. Smaller catheters can be easily kinked during patient positioning and can become clogged. Respiratory variation, which is the movement of the column of fluid in the collection chamber or in the tubing with inspiration and expiration, suggests that the chest tube is patent. This should be part of the daily examination in a patient with a chest tube, and it should also be the first step in assessing sudden dyspnea, hypoxia, pain, or hemodynamic instability. Clogged tubes should be referred to the interventionalists or other physicians who placed them. Chest tubes are typically sutured at the site of entry and securely bandaged to avoid migration but occasionally can be dislodged. This should prompt placement of another tube by an experienced operator. Last, chest tubes can be uncomfortable for patients who may require systemic analgesics. Additionally, tube positioning may ease some of the discomfort. Chest tubes are commonly placed along the midaxillary line and the posterior thorax, leading to discomfort in the recumbent position. Directing the tube anteriorly helps ease some of the discomfort. This can be done using all‐purpose sponges to build a barrier between the skin and the chest tube as it is directed anteriorly. Additional sponges are placed above the tube for extra protection. The gentle curve accomplished by padding the underside of the tube also keeps the tube patent by avoiding sharp kinks as the catheter exits the thorax.
FUTURE TRENDS
Future trends in the management of IP may include shorter duration of tube management (14 hours of suction with air leak evaluation and removal) and the use of even smaller catheters. Outpatient management of IP with small pigtail catheters or small‐caliber tubes in addition to 1‐way valves is also being investigated. The benefits of these practices may include greater patient comfort and lower cost. These approaches will need larger‐scale replication and careful patient selection before they become standard practice.[28, 36]
Ultrasound can be used to assess the presence and size of pneumothoraces that are difficult to visualize by standard chest radiographs. Several studies have established ultrasonography as an effective method of diagnosing pneumothorax and have shown it to have superior sensitivity compared with chest radiography.[1, 18] In the future, the use of ultrasound will likely be more widespread given its performance, portability, ease of use, and relatively low cost.
SUMMARY
IP is a known and costly complication of many medical procedures. The aforementioned algorithms help simplify the management of chest tubes for hospitalists caring for patients with this common complication. This stepwise approach may not only help curtail added expenses related to IPs by decreasing the length of inpatient stays but may also improve patient satisfaction.
Acknowledgments
Disclosure: Mayo does not endorse the products mentioned in this article. The authors report no conflicts of interest.
Box
Clinical Signs and Symptoms of Pneumothorax
Dyspnea
Pleuritic chest pain
Tachypnea
Hypoxia
Decreased breath sounds on affected side
Hyper‐resonant percussion on affected side
Subcutaneous emphysema
A pneumothorax is a collection of air in the space outside the lungs that is trapped within the thorax. This abnormality can occur spontaneously or as the result of trauma. Traumatic pneumothoraces include those resulting from medical interventions such as a transthoracic and transbronchial needle biopsy, central line placement, and positive‐pressure mechanical ventilation. This group is most accurately described as iatrogenic pneumothorax (IP).[1]
IP can be an expected complication of many routine thoracic procedures, but it can also occur accidentally during procedures near the lung or thoracic cavity. Some IPs may be asymptomatic and go undiagnosed, or their diagnosis may be delayed.[2] The majority of iatrogenic pneumothoraces will resolve without complications, and patients will not require medical attention. A small percentage can, however, expand and have the potential to develop into a tension pneumothorax causing severe respiratory distress and mediastinal shift.[3, 4]
The incidence of IP ranges from 0.11% with mechanical ventilation to 2.68% with thoracentesis, according to an analysis of 7.5 million uniform hospital discharge abstracts from 2000.5 A 2010 systematic review of 24 studies that included 6605 patients suggested a 6.0% incidence of pneumothorax following thoracentesis.[3] The highest risk of IP is seen with computed tomography (CT)‐guided lung biopsy, with 1 series of 1098 biopsies showing a 42% incidence; chest tube evacuation was required in 12% of these cases.[4] A Veterans Administration study of patient safety indicators from 2001 to 2004 found that risk‐adjusted rates of IP were increasing over time.[6] It is unclear whether this increase is due to increasing numbers of interventional procedures or to better rates of detection. IP poses a considerable cost to the medical system, with safety studies finding that patients with IP will stay in the hospital approximately 4 days longer and incur an additional $17,000 in charges.[7]
In addition to this financial burden, the lack of consistency in training and guidelines for management of pneumothorax is thought to add to chest tube‐related complications.[8] In 2001, the American College of Chest Physicians (ACCP) published guidelines for the management of spontaneous pneumothorax that do not specifically address IP.[9] In 2010, the British Thoracic Society (BTS) updated their guidelines and included a brief statement on IP that described a higher incidence for it than for spontaneous pneumothorax and noted its relative ease of management.[10] Despite the lack of specific guidelines dedicated to IPs, common clinical practice is to manage iatrogenic defects in a manner similar to that for spontaneous ones. However, studies have shown that the management of pneumothorax remains diverse and that the adherence to these published guidelines is suboptimal.[10, 11] The BTS guidelines favor needle aspiration as the first‐line treatment,[10] whereas the ACCP recommends drainage with catheters over aspiration.[9]
The possibility of this complication, along with the rising rate of invasive interventions being performed, has led to expanded surveillance criteria for IP. Surveillance imaging, clinical observation, or a combination of the 2 may be required, depending on the institution, the risk of the procedure, and the preference of the treating clinician. The algorithms presented here were designed in alignment with both major society guidelines and with the intention of simplifying the treatment regimen for the ease of adoption by hospitalists.
ETIOLOGY AND RISK FACTORS
The etiology and risk factors for IP are multiple, with the most common being interventional‐based procedures. In 535 Veterans Administration patients, the most common precursor procedures were transthoracic needle biopsy (24%), subclavian vein catheterization (22%), thoracentesis (20%), transbronchial biopsy (10%), pleural biopsy (8%), and positive pressure ventilation.[12] IP can also be a rare complication of pacemaker manipulations,[5] and less commonly, bronchoscopy.[13] Patient factors that increase the risk of pneumothorax in the setting of an intervention include age, chronic obstructive lung disease, primary lung cancer, malignant and parapneumonic pleural effusions, empyema, and chronic corticosteroid use.[4] As might be expected, patients with structural lung disease (eg, emphysema with bullae) and poor healing ability (eg, corticosteroid dependent), tend to have IPs more often and to require more complicated interventions for resolution.[14, 15] In some studies, operator experience seems to be inversely related to the rate of IP, and the use of ultrasound is correlated with lower rates of this complication.[1, 3]
PATIENT PRESENTATIONS AND DIAGNOSIS
Clinical signs and symptoms of a significant pneumothorax vary in severity but most often include dyspnea, tachypnea, chest pain, and pleurisy (see Box 1). Post procedure signs or symptoms require further evaluation with imaging, usually a plain chest radiograph. CT can be useful for further evaluation. Small anterior pneumothoraces may be difficult to detect without lateral radiographic imaging or computed tomogram. Ultrasound is being used more frequently at the bedside to make this diagnosis, and various studies of trauma patients have found that it has good sensitivity and specificity.[16, 17] These results have been validated by a recent meta‐analysis comparing ultrasound to chest radiographs for the detection of pneumothorax among trauma, critically ill, and postprocedural patients.[18] This study demonstrated superior sensitivity and similar specificity for ultrasound versus chest radiographs for detection of pneumothorax. More ominous signs, such as tachycardia or hypotension, can be indicative of tension pneumothorax, which requires emergent evacuation.
MANAGEMENT
Once the diagnosis of pneumothorax has been established, treatment options should be guided by defect size and clinical assessment following a defined treatment algorithm (Figure 1). As emphasized by the BTS and ACCP guidelines, we advocate considering the use of symptoms along with defect size to determine the best management course.
Observation
Defects that involve<20% of the hemithorax in a patient who is clinically asymptomatic and hemodynamically stable can be safely managed by oxygen supplementation and hospital observation. Repeat imaging can be obtained after 12 to 24 hours of defect detection or with symptom change. Patients who display resolution may be discharged home.
Patients who show persistence without progression but are asymptomatic may also be discharged safely, with follow‐up imaging and clinical evaluation 48 hours later.[9, 10] This was demonstrated by Kelly and colleagues,[19] who described the outcomes of 154 patients in a retrospective cohort study. Of the 91 patients treated with outpatient observation, 82 resolved without additional interventions. A recent review article by the same author cites conservative management of small pneumothoraces as being widely accepted.[20] If reimaging shows progression of defect or if the patient becomes more symptomatic, the pneumothorax should be evacuated by 1 of the methods described below.
Aspiration
Aspiration is defined by the ACCP Delphi consensus statement as the removal of pleural air via needle or cannula followed by immediate removal of needle or cannula.[9] This option mandates careful patient selection. It should be considered for small pneumothoraces that cause only mild dyspnea in patients who have no known parenchymal disease. These patients should be observed overnight in the hospital and reimaged 24 hours after aspiration of the pneumothorax. Several authors have reported success with aspiration alone. Yamagami et al.[21] noted the efficacy of manual aspiration immediately after CT‐guided biopsy, with a success rate exceeding 90%. They also noted that evacuated volumes >543 mL correlated with the need for further intervention with a chest tube. This technique is advocated for small pneumothoraces that are recognized shortly after the procedure.
Similarly, Delius and colleagues[22] managed 131 pneumothoraces with aspiration as an alternative to chest tube placement. Of these, 79 were iatrogenic. Aspiration achieved a 75% success rate for all IPs. Small defects defined as <20% of volume had an even higher resolution rate of 87%. Similar findings were demonstrated by Talbot‐Stern et al.[23] in their prospective study of 76 pneumothoraces. Among those that were iatrogenic, 82% resolved after simple aspiration. Faruqi et al.[24] also showed that aspiration is a viable option for IPs. Of the 57 patients with pneumothorax included in their study, 35 were treated with aspiration alone. Iatrogenesis was the culprit in 12 of the 35 manually aspirated cases. Aspiration achieved a success rate of 91.7% in IP. A recent Cochrane database systematic review compared simple aspiration with intercostal tube drainage for primary spontaneous pneumothorax.[25] The authors reported no difference between these methods in terms of success rate, early failure rate, duration of hospital stay, 1‐year success rate, or number of patients who required pleurodesis at 1‐year follow‐up.
Because the algorithms presented in this article were specifically designed for the use by hospitalists, we intentionally omitted aspiration from the decision trees. Most hospitalists would not be expected to evacuate IPs. However, knowledge regarding this option and appropriate follow‐up are valuable to internists, because many interventionalists admit patients to the hospital service for overnight observation. An asymptomatic postaspiration patient, who on subsequent imaging demonstrates resolution or persistence without progression of pneumothorax, may be discharged with 48‐hour follow‐up.
Placement of Catheter or Chest Tube Drainage
Most patients with a clinically significant pneumothorax will require evacuation of the air. Pneumothoraces larger than 20% or that produce symptoms warrant chest tube management and inpatient observation (Figure 1B). Traditionally, large tubes with 20 cm of water on continuous suction are used and have been studied the most widely. Several authors have shown that smaller tubes can effectively drain a pneumothorax.[26, 27, 28, 29] Small‐bore catheters (8F14F), which can be inserted percutaneously, have been shown to provide effective lung re‐expansion with minimal morbidity[8] and may be better tolerated by patients with uncomplicated pneumothoraces (Figure 2). Terzi and colleagues[30] have shown that smaller tubes cause less discomfort to patients at rest, with cough, and at the time of tube removal.
At most US institutions, catheters and chest tubes are connected to all‐purpose drainage systems. Although commercially available through a variety of manufacturers, they share similar design principles because they replicate the 3‐bottle system described in detail elsewhere in the literature.[31] We have limited our discussion to 3 pleural evacuation systems because it is our intention to familiarize hospitalists with the units that they are most likely to encounter. The first 2 systems have been studied and described by Baumann and colleagues[32] as being commonplace and reasonably reliable. These include the Oasis (Atrium Medical Corp., Hudson, NH) (Figure 3A) and the Pleur‐evac (Teleflex Inc., Limerick, PA). The third unit is the Thopaz digital thoracic drainage system (Medela Inc., McHenry, IL) (Figure 3B). The Thopaz is unique in its inclusion of a suction source and digital capability. Although it utilizes the same principles of all pleural evacuation devices, its setup and information output require that one be familiar with its digital format.
Suction Versus Water Seal
The chest tube should be placed initially to a suction pressure level of 20 cm of water for 24 hours to maximize lung expansion and evacuate all extrapulmonary air. Suction pressure is set on the Pleur‐evac and Atrium drainage systems by a manual dial that reads to a water pressure of 0 to 40 cm. The default setting from the manufacturer is 20 cm of water. This level of suction is present only when the drainage system is connected to a wall or a portable suction device. The only confirmation of suction presence in the Atrium system is the deployment of the orange bellows (located under the dial) to the level of the arrow tip (Figure 3A). The Pleur‐evac system has a red stripe along the circular edge of the dial that appears at the set level of suction when negative pressure is being applied. It is important to be aware that when patients are disconnected from the wall or the portable suction apparatus, they are on water seal or gravity. These terms are synonymous with no suction. On the Thopaz, a digital menu directs operation, and levels of suction can be selected from water seal (no suction) up to 40 cm of water. We recommend using suction to 20 cm of water given the scarce evidence supporting higher levels of negative pressure. Some clinicians prefer placing patients on water seal for some time before moving toward tube discontinuance, but this is a matter of preference, and no substantial evidence exists to show that any 1 method is superior.[8, 33]
Assessing for Air Leak
If there is improvement or resolution of the pneumothorax after 24 hours, the presence of an air leak should be assessed; if no leak is present, the chest tube can be safely removed. In the context of chest tubes, the term air leak refers to residual air between the lung and the chest wall. It is possible to see resolution of a pneumothorax on chest radiographs and still have an air leak. This situation is created by a perfect balance between the pleural air evacuation by the catheter and the flow of air exiting from the lung puncture. This would result in reaccumulation of the pneumothorax if the chest tube is removed prematurely. It should also be kept in mind that chest radiographs may miss a small pneumothorax given their relatively low sensitivity.[18] Therefore, the absence of an air leak needs to be documented before the chest tube is discontinued. Depending on the type of drainage system (Atrium, Pleur‐evac, or Thopaz), this assessment can be done in several ways. All systems can be assessed for air leak by clamping the actual chest tube for 2 to 4 hours and then repeating the chest radiograph. Clamping a chest tube simulates the condition of not having a chest tube. Chest tubes should never be clamped without supervision and only with the knowledge of nursing personnel. The onset of chest pain or dyspnea in a patient with a clamped tube mandates immediate removal of the clamp and a return to suction. A repeat chest radiograph showing reaccumulation or expansion of the pneumothorax after clamping indicates that the air leak has not resolved and the chest tube must remain in place and returned to suction. Simpler and more time‐efficient methods of detecting air leaks are available with both cardiothoracic drainage systems.
For the Atrium and Pleur‐evac models, there is a graded panel through which one can visualize air leaks being funneled through water (Figure 4A). Having the patient cough several times or perform a Valsalva maneuver should release any air trapped within the chest into this chamber, where bubbles can be visualized as they travel through the water. The presence of bubbles indicates the presence of residual air in the chest, pointing to a possible leak. In contrast, the Thopaz system offers a graphical display of the air flowing into the system that can be reviewed over the 24‐hour period. When the graph line reaches a 0 flatline graph, no airflow is being detected and no air leakage is suspected (Figure 4B). If no air leaks are detected, the chest tube may be discontinued. Those patients with a failed air leak test should have their chest tubes continued under suction for another 24 hours, with the above tests then repeated. The same holds true for those patients with persistent pneumothorax at 24 hours.
Removal of chest tubes is a simple process that requires the tube to be pulled out of the patient without allowing air to enter the site where the tube was present and where it entered the thorax. Most interventionalists will discontinue the tubes that they have placed. Some small catheters have an internal string that has to be released so the catheter will straighten and pull out easily. Knowledge of the type of catheter or tube that was placed is critical before removal to prevent complications and patient discomfort. Standard chest tubes are straight, smooth plastic and pull out easily but require rapid occlusion of the larger puncture site in the chest wall with an occlusive dressing that often includes petroleum or water‐soluble gel. Some physicians will leave a suture tie when placing the chest tube so that it can be tied down to occlude the site instead of using a dressing.
Consultation of the Cardiothoracic Surgeon or Interventional Pulmonologist
We recommend the involvement of cardiothoracic surgery or interventional pulmonology for patients with nonresolving pneumothorax lasting longer than 48 hours because additional procedures may be necessary. One of the rare but serious complications of a persistent pneumothorax is the formation of a bronchopleural fistula. This communication between the bronchial tree and the pleural space can lead to significant morbidity and mortality. The treatment of a bronchopleural fistula includes medical and surgical options that are beyond the scope of this article but require the expertise of a cardiothoracic surgeon or interventional pulmonologist.[34] Most patients who will not require additional procedures will heal within 48 hours.[11, 35] Decisions regarding more invasive treatment measures can then be made as necessary.[26]
PRACTICAL TIPS
Hospitalists caring for patients with chest tubes are often asked to troubleshoot at the bedside. Scenarios that may be encountered include nonfunctioning tubes, catheter migration, and tube discomfort. Ensuring patency of the tube entails visualizing the tube from the point of entry into the chest wall to the collection chamber and inspecting for kinks or debris clogging the tube. Smaller catheters can be easily kinked during patient positioning and can become clogged. Respiratory variation, which is the movement of the column of fluid in the collection chamber or in the tubing with inspiration and expiration, suggests that the chest tube is patent. This should be part of the daily examination in a patient with a chest tube, and it should also be the first step in assessing sudden dyspnea, hypoxia, pain, or hemodynamic instability. Clogged tubes should be referred to the interventionalists or other physicians who placed them. Chest tubes are typically sutured at the site of entry and securely bandaged to avoid migration but occasionally can be dislodged. This should prompt placement of another tube by an experienced operator. Last, chest tubes can be uncomfortable for patients who may require systemic analgesics. Additionally, tube positioning may ease some of the discomfort. Chest tubes are commonly placed along the midaxillary line and the posterior thorax, leading to discomfort in the recumbent position. Directing the tube anteriorly helps ease some of the discomfort. This can be done using all‐purpose sponges to build a barrier between the skin and the chest tube as it is directed anteriorly. Additional sponges are placed above the tube for extra protection. The gentle curve accomplished by padding the underside of the tube also keeps the tube patent by avoiding sharp kinks as the catheter exits the thorax.
FUTURE TRENDS
Future trends in the management of IP may include shorter duration of tube management (14 hours of suction with air leak evaluation and removal) and the use of even smaller catheters. Outpatient management of IP with small pigtail catheters or small‐caliber tubes in addition to 1‐way valves is also being investigated. The benefits of these practices may include greater patient comfort and lower cost. These approaches will need larger‐scale replication and careful patient selection before they become standard practice.[28, 36]
Ultrasound can be used to assess the presence and size of pneumothoraces that are difficult to visualize by standard chest radiographs. Several studies have established ultrasonography as an effective method of diagnosing pneumothorax and have shown it to have superior sensitivity compared with chest radiography.[1, 18] In the future, the use of ultrasound will likely be more widespread given its performance, portability, ease of use, and relatively low cost.
SUMMARY
IP is a known and costly complication of many medical procedures. The aforementioned algorithms help simplify the management of chest tubes for hospitalists caring for patients with this common complication. This stepwise approach may not only help curtail added expenses related to IPs by decreasing the length of inpatient stays but may also improve patient satisfaction.
Acknowledgments
Disclosure: Mayo does not endorse the products mentioned in this article. The authors report no conflicts of interest.
Box
Clinical Signs and Symptoms of Pneumothorax
Dyspnea
Pleuritic chest pain
Tachypnea
Hypoxia
Decreased breath sounds on affected side
Hyper‐resonant percussion on affected side
Subcutaneous emphysema
- , . Management of pneumothorax. Semin Respir Crit Care Med. 2010;31(6):769–780.
- , . Pneumothorax. Respirology. 2004;9(2):157–164.
- , , , . Pneumothorax following thoracentesis: a systematic review and meta‐analysis. Arch Intern Med. 2010;170(4):332–339.
- , , , et al. Incidence of and risk factors for pneumothorax and chest tube placement after CT fluoroscopy‐guided percutaneous lung biopsy: retrospective analysis of the procedures conducted over a 9‐year period. AJR Am J Roentgenol. 2010;194(3):809–814.
- , , . Accidental iatrogenic pneumothorax in hospitalized patients. Med Care. 2006;44(2):182–186.
- , , , et al. Tracking rates of patient safety indicators over time: lessons from the Veterans Administration. Med Care. 2006;44(9):850–861.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA. 2003;290(14):1868–1874.
- , , , , . Preliminary report of a prospective, randomized trial of underwater seal for spontaneous and iatrogenic pneumothorax. J Am Coll Surg. 2007;204(1):84–90.
- , , , et al.; AACP Pneumothorax Consensus Group. Management of spontaneous pneumothorax: an American College of Chest Physicians Delphi consensus statement. Chest. 2001;119(2):590–602.
- , , ; BTS Pleural Disease Guideline Group. Management of spontaneous pneumothorax: British Thoracic Society Pleural Disease Guideline 2010. Thorax. 2010;65(suppl 2):ii18–ii31.
- , . The clinician's perspective on pneumothorax management. Chest. 1997;112(3):822–828.
- , , , . Iatrogenic pneumothorax: etiology and morbidity: results of a Department of Veterans Affairs Cooperative Study. Respiration. 1992;59(4):215–220.
- , , , , , . Severe complications of bronchoscopy. Respiration. 2008;76(4):429–433.
- , , , et al. Incidence and risk factors of delayed pneumothorax after transthoracic needle biopsy of the lung. Chest. 2004;126(5):1516–1521.
- , , , , , . Factors associated with pneumothorax and pneumothorax requiring treatment after percutaneous lung biopsy in 443 consecutive patients. J Vasc Interv Radiol. 2004;15(5):479–483.
- , . Ultrasound detection of pneumothorax compared with chest X‐ray and computed tomography scan. Am Surg. 2011;77(4):480–484.
- , . Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med. 2010;17(1):11–17.
- , , , , . Diagnosis of pneumothorax by radiography and ultrasonography: a meta‐analysis. Chest. 2011;140(4):859–866.
- , , . Outcomes of emergency department patients treated for primary spontaneous pneumothorax. Chest. 2008;134(5):1033–1036.
- . Review of management of primary spontaneous pneumothorax: is the best evidence clearer 15 years on? Emerg Med Australas. 2007;19(4):303–308.
- , , , , , . Efficacy of manual aspiration immediately after complicated pneumothorax in CT‐guided lung biopsy. J Vasc Interv Radiol. 2005;16(4):477–483.
- , , , , , . Catheter aspiration for simple pneumothorax: experience with 114 patients. Arch Surg. 1998;124(7):833–836.
- , , , , . Catheter aspiration for simple pneumothorax. J Emerg Med. 1986;4(6):437–442.
- , , , . Role of simple needle aspiration in the management of pneumothorax. Indian J Chest Dis Allied Sci. 2004;46(3):183–190.
- , , . Simple aspiration versus intercostal tube drainage for primary spontaneous pneumothorax in adults. Cochrane Database Syst Rev. 2007;(1):CD004479.
- , , , . Evaluation of conventional chest tube therapy for iatrogenic pneumothorax. Chest. 1993;104(6):1770–1772.
- , , , . Outpatient treatment of iatrogenic pneumothorax after needle biopsy. Radiology. 1997;205(1):249–252.
- , , , , , . Outpatient management of postbiopsy pneumothorax with small‐caliber chest tubes: factors affecting the need for prolonged drainage and additional interventions. Cardiovasc Intervent Radiol. 2008;31(2):342–348.
- , . Management of primary and secondary pneumothorax using a small‐bore thoracic catheter. Interact Cardiovasc Thorac Surg. 2010;11(2):146–149.
- , , , et al. The use of flexible spiral drains after non‐cardiac thoracic surgery: a clinical study. Eur J Cardiothorac Surg. 2005;27(1):134–137.
- . Pleural Diseases. 5th ed. Philadelphia: PA: Lippincott Williams 2007.
- , , , . Comparison of function of commercially available pleural drainage units and catheters. Chest. 2003;123(6):1878–1886.
- , . Catheter drainage of spontaneous pneumothorax: suction or no suction, early or late removal? Thorax. 1982;37(1):46–48.
- , . Bronchopleural fistulas: an overview of the problem with special focus on endoscopic management. Chest. 2005;128(6):3955–3965.
- , , , . Timing of invasive procedures in therapy for primary and secondary spontaneous pneumothorax. Arch Surg. 1991;126(6):764–766.
- , , , , . A treatment algorithm for pneumothoraces complicating central venous catheter insertion. Am J Surg. 2000;180(6):523–526.
- , . Management of pneumothorax. Semin Respir Crit Care Med. 2010;31(6):769–780.
- , . Pneumothorax. Respirology. 2004;9(2):157–164.
- , , , . Pneumothorax following thoracentesis: a systematic review and meta‐analysis. Arch Intern Med. 2010;170(4):332–339.
- , , , et al. Incidence of and risk factors for pneumothorax and chest tube placement after CT fluoroscopy‐guided percutaneous lung biopsy: retrospective analysis of the procedures conducted over a 9‐year period. AJR Am J Roentgenol. 2010;194(3):809–814.
- , , . Accidental iatrogenic pneumothorax in hospitalized patients. Med Care. 2006;44(2):182–186.
- , , , et al. Tracking rates of patient safety indicators over time: lessons from the Veterans Administration. Med Care. 2006;44(9):850–861.
- , . Excess length of stay, charges, and mortality attributable to medical injuries during hospitalization. JAMA. 2003;290(14):1868–1874.
- , , , , . Preliminary report of a prospective, randomized trial of underwater seal for spontaneous and iatrogenic pneumothorax. J Am Coll Surg. 2007;204(1):84–90.
- , , , et al.; AACP Pneumothorax Consensus Group. Management of spontaneous pneumothorax: an American College of Chest Physicians Delphi consensus statement. Chest. 2001;119(2):590–602.
- , , ; BTS Pleural Disease Guideline Group. Management of spontaneous pneumothorax: British Thoracic Society Pleural Disease Guideline 2010. Thorax. 2010;65(suppl 2):ii18–ii31.
- , . The clinician's perspective on pneumothorax management. Chest. 1997;112(3):822–828.
- , , , . Iatrogenic pneumothorax: etiology and morbidity: results of a Department of Veterans Affairs Cooperative Study. Respiration. 1992;59(4):215–220.
- , , , , , . Severe complications of bronchoscopy. Respiration. 2008;76(4):429–433.
- , , , et al. Incidence and risk factors of delayed pneumothorax after transthoracic needle biopsy of the lung. Chest. 2004;126(5):1516–1521.
- , , , , , . Factors associated with pneumothorax and pneumothorax requiring treatment after percutaneous lung biopsy in 443 consecutive patients. J Vasc Interv Radiol. 2004;15(5):479–483.
- , . Ultrasound detection of pneumothorax compared with chest X‐ray and computed tomography scan. Am Surg. 2011;77(4):480–484.
- , . Sensitivity of bedside ultrasound and supine anteroposterior chest radiographs for the identification of pneumothorax after blunt trauma. Acad Emerg Med. 2010;17(1):11–17.
- , , , , . Diagnosis of pneumothorax by radiography and ultrasonography: a meta‐analysis. Chest. 2011;140(4):859–866.
- , , . Outcomes of emergency department patients treated for primary spontaneous pneumothorax. Chest. 2008;134(5):1033–1036.
- . Review of management of primary spontaneous pneumothorax: is the best evidence clearer 15 years on? Emerg Med Australas. 2007;19(4):303–308.
- , , , , , . Efficacy of manual aspiration immediately after complicated pneumothorax in CT‐guided lung biopsy. J Vasc Interv Radiol. 2005;16(4):477–483.
- , , , , , . Catheter aspiration for simple pneumothorax: experience with 114 patients. Arch Surg. 1998;124(7):833–836.
- , , , , . Catheter aspiration for simple pneumothorax. J Emerg Med. 1986;4(6):437–442.
- , , , . Role of simple needle aspiration in the management of pneumothorax. Indian J Chest Dis Allied Sci. 2004;46(3):183–190.
- , , . Simple aspiration versus intercostal tube drainage for primary spontaneous pneumothorax in adults. Cochrane Database Syst Rev. 2007;(1):CD004479.
- , , , . Evaluation of conventional chest tube therapy for iatrogenic pneumothorax. Chest. 1993;104(6):1770–1772.
- , , , . Outpatient treatment of iatrogenic pneumothorax after needle biopsy. Radiology. 1997;205(1):249–252.
- , , , , , . Outpatient management of postbiopsy pneumothorax with small‐caliber chest tubes: factors affecting the need for prolonged drainage and additional interventions. Cardiovasc Intervent Radiol. 2008;31(2):342–348.
- , . Management of primary and secondary pneumothorax using a small‐bore thoracic catheter. Interact Cardiovasc Thorac Surg. 2010;11(2):146–149.
- , , , et al. The use of flexible spiral drains after non‐cardiac thoracic surgery: a clinical study. Eur J Cardiothorac Surg. 2005;27(1):134–137.
- . Pleural Diseases. 5th ed. Philadelphia: PA: Lippincott Williams 2007.
- , , , . Comparison of function of commercially available pleural drainage units and catheters. Chest. 2003;123(6):1878–1886.
- , . Catheter drainage of spontaneous pneumothorax: suction or no suction, early or late removal? Thorax. 1982;37(1):46–48.
- , . Bronchopleural fistulas: an overview of the problem with special focus on endoscopic management. Chest. 2005;128(6):3955–3965.
- , , , . Timing of invasive procedures in therapy for primary and secondary spontaneous pneumothorax. Arch Surg. 1991;126(6):764–766.
- , , , , . A treatment algorithm for pneumothoraces complicating central venous catheter insertion. Am J Surg. 2000;180(6):523–526.
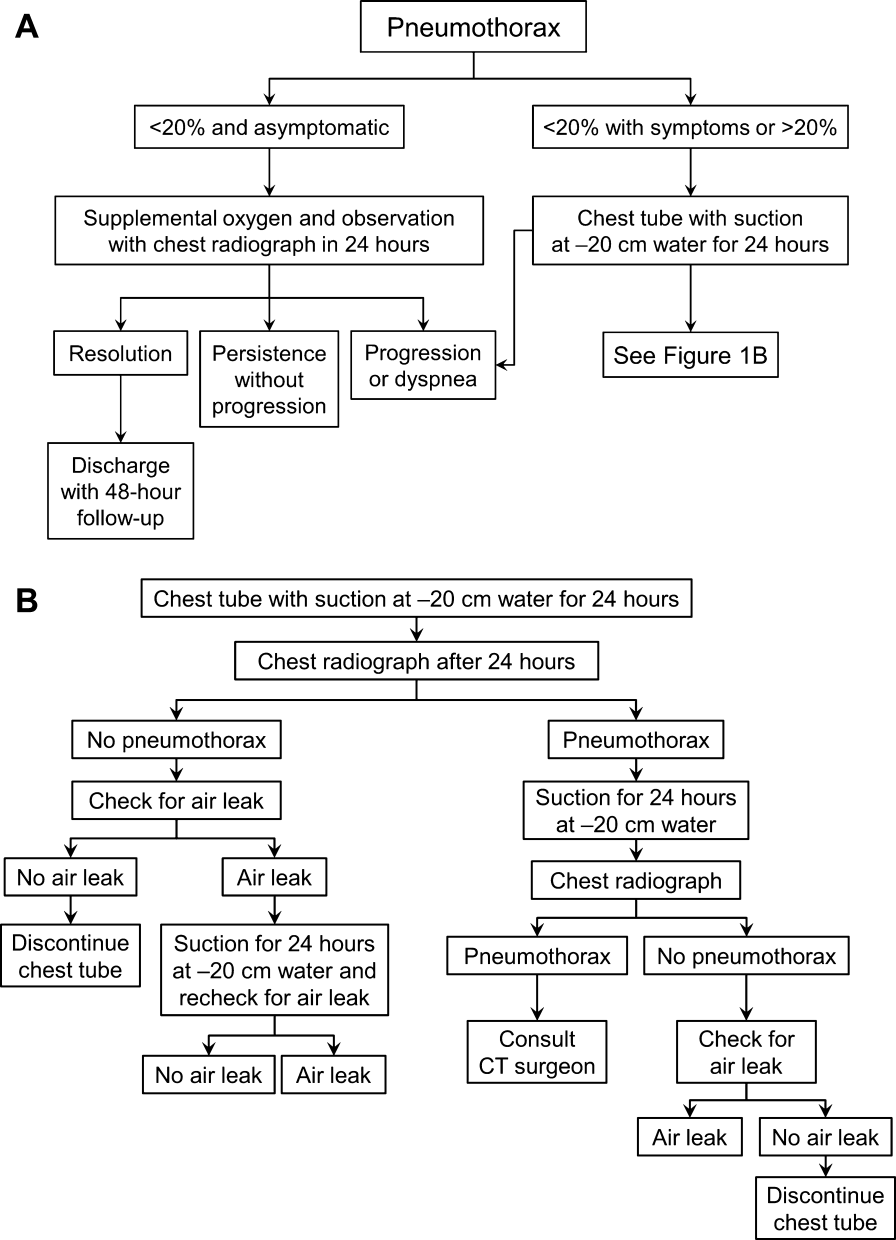
Hospitalist Teaching Rounds for FUTURE
The implementation of resident duty hour restrictions has created a clinical learning environment on the wards quite different from any previous era. The Accreditation Council for Graduate Medical Education issued its first set of regulations limiting consecutive hours worked for residents in 2003, and further restricted hours in 2011.[1] These restrictions have had many implications across several aspects of patient care, education, and clinical training, particularly for hospitalists who spend the majority of their time in this setting and are heavily involved in undergraduate and graduate clinical education in academic medical centers.[2, 3]
As learning environments have been shifting, so has the composition of learners. The Millennial Generation (or Generation Y), defined as those born approximately between 1980 and 2000, represents those young clinicians currently filling the halls of medical schools and ranks of residency and fellowship programs.[4] Interestingly, the current system of restricted work hours is the only system under which the Millennial Generation has ever trained.
As this new generation represents the bulk of current trainees, hospitalist faculty must consider how their teaching styles can be adapted to accommodate these learners. For teaching hospitalists, an approach that considers the learning environment as affected by duty hours, as well as the preferences of Millennial learners, is necessary to educate the next generation of trainees. This article aimed to introduce potential strategies for hospitalists to better align teaching on the wards with the preferences of Millennial learners under the constraints of residency duty hours.
THE NEWEST GENERATION OF LEARNERS
The Millennial Generation has been well described.[4, 5, 6, 7, 8, 9, 10] Broadly speaking, this generation is thought to have been raised by attentive and involved parents, influencing relationships with educators and mentors; they respect authority but do not hesitate to question the relevance of assignments or decisions. Millennials prefer structured learning environments that focus heavily on interaction and experiential learning, and they value design and appearance in how material is presented.[7] Millennials also seek clear expectations and immediate feedback on their performance, and though they have sometimes been criticized for a strong sense of entitlement, they have a strong desire for collaboration and group‐based activity.[5, 6]
One of the most notable and defining characteristics of the Millennial Generation is an affinity for technology and innovation.[7, 8, 9] Web‐based learning tools that are interactive and engaging, such as blogs, podcasts, or streaming videos are familiar and favored methods of learning. Millennials are skilled at finding information and providing answers and data, but may need help with synthesis and application.[5] They take pride in their ability to multitask, but can be prone to doing so inappropriately, particularly with technology that is readily available.[11]
Few studies have explored characteristics of the Millennial Generation specific to medical trainees. One study examined personality characteristics of Millennial medical students compared to Generation X students (those born from 19651980) at a single institution. Millennial students scored higher on warmth, reasoning, emotional stability, rule consciousness, social boldness, sensitivity, apprehension, openness to change, and perfectionism compared to Generation X students. They scored lower on measures for self‐reliance.[12] Additionally, when motives for behavior were studied, Millennial medical students scored higher on needs for affiliation and achievement, and lower on needs for power.[13]
DUTY HOURS: A GENERATION APART
As noted previously, the Millennial Generation is the first to train exclusively in the era of duty hours restrictions. The oldest members of this generation, those born in 1981, were entering medical school at the time of the first duty hours restrictions in 2003, and thus have always been educated, trained, and practiced in an environment in which work hours were an essential part of residency training.
Though duty hours have been an omnipresent part of training for the Millennial Generation, the clinical learning environment that they have known continues to evolve and change. Time for teaching, in particular, has been especially strained by work hour limits, and this has been noted by both attending physicians and trainees with each iteration of work hours limits. Attendings in one study estimated that time spent teaching on general medicine wards was reduced by about 20% following the 2003 limits, and over 40% of residents in a national survey reported that the 2011 limits had worsened the quality of education.[14, 15]
GENERATIONAL STRATEGIES FOR SUCCESS FOR HOSPITALIST TEACHING ATTENDINGS
The time limitations imposed by duty hours restrictions have compelled teaching rounds to become more patient‐care centered and often less learner‐centered, as providing patient care becomes the prime obligation for this limited time period. Millennial learners are accustomed to being the center of attention in educational environments, and changing the focus from education to patient care in the wards setting may be an abrupt transition for some learners.[6] However, hospitalists can help restructure teaching opportunities on the clinical wards by using teaching methods of the highest value to Millennial learners to promote learning under the conditions of duty hours limitations.
An approach using these methods was developed by reviewing recent literature as well as educational innovations that have been presented at scholarly meetings (eg, Sal Khan's presentation at the 2012 Association of American Medical Colleges meeting).[16] The authors discussed potential teaching techniques that were thought to be feasible to implement in the context of the current learning environment, with consideration of learning theories that would be most effective for the target group of learners (eg, adult learning theory).[17] A mnemonic was created to consolidate strategies thought to best represent these techniques. FUTURE is a group of teaching strategies that can be used by hospitalists to improve teaching rounds by Flipping the Wards, Using Documentation to Teach, Technology‐Enabled Teaching, Using Guerilla Teaching Tactics, Rainy Day Teaching, and Embedding Teaching Moments into Rounds.
Flipping the Wards
Millennial learners prefer novel methods of delivery that are interactive and technology based.[7, 8, 9] Lectures and slide‐based presentations frequently do not feature the degree of interactive engagement that they seek, and methods such as case‐based presentations and simulation may be more suitable. The Khan Academy is a not‐for‐profit organization that has been proposed as a model for future directions for medical education.[18] The academy's global classroom houses over 4000 videos and interactive modules to allow students to progress through topics on their own time.[19] Teaching rounds can be similarly flipped such that discussion and group work take place during rounds, whereas lectures, modules, and reading are reserved for individual study.[18]
As time pressures shift the focus of rounds exclusively toward discussion of patient‐care tasks, finding time for teaching outside of rounds can be emphasized to inspire self‐directed learning. When residents need time to tend to immediate patient‐care issues, hospitalist attendings could take the time to search for articles to send to team members. Rather than distributing paper copies that may be lost, cloud‐based data management systems such as Dropbox (Dropbox, San Francisco, CA) or Google Drive (Google Inc., Mountain View, CA) can be used to disseminate articles, which can be pulled up in real time on mobile devices during rounds and later deposited in shared folders accessible to all team members.[20, 21] The advantage of this approach is that it does not require all learners to be present on rounds, which may not be possible with duty hours.
Using Documentation to Teach
Trainees report that one of the most desirable attributes of clinical teachers is when they delineate their clinical reasoning and thought process.[22] Similarly, Millennial learners specifically desire to understand the rationale behind their teachers' actions.[6] Documentation in the medical chart or electronic health record (EHR) can be used to enhance teaching and role‐model clinical reasoning in a transparent and readily available fashion.
Billing requirements necessitate daily attending documentation in the form of an attestation. Hospitalist attendings can use attestations to model thought process and clinical synthesis in the daily assessment of a patient. For example, an attestation one‐liner can be used to concisely summarize the patient's course or highlight the most pressing issue of the day, rather than simply serve as a placeholder for billing or agree with above in reference to housestaff documentation. This practice can demonstrate to residents how to write a short snapshot of a patient's care in addition to improving communication.
Additionally, the EHR can be a useful platform to guide feedback for residents on their clinical performance. Millennial learners prefer specific, immediate feedback, and trainee documentation can serve as a template to show examples of good documentation and clinical reasoning as well as areas needing improvement.[5] These tangible examples of clinical performance are specific and understandable for trainees to guide their self‐learning and improvement.
Technology‐Enabled Teaching
Using technology wisely on the wards can improve efficiency while also taking advantage of teaching methods familiar to Millennial learners. Technology can be used in a positive manner to keep the focus on the patient and enhance teaching when time is limited on rounds. Smartphones and tablets have become an omnipresent part of the clinical environment.[23] Rather than distracting from rounds, these tools can be used to answer clinical questions in real time, thus directly linking the question to the patient's care.
The EHR is a powerful technological resource that is readily available to enhance teaching during a busy ward schedule. Clinical information is electronically accessible at all hours for both trainees and attendings, rather than only at prespecified times on daily rounds, and the Millennial Generation is accustomed to receiving and sharing information in this fashion.[24] Technology platforms that enable simultaneous sharing of information among multiple members of a team can also be used to assist in sharing clinical information in this manner. Health Insurance Portability and Accountability Act‐compliant group text‐messaging applications for smartphones and tablets such as GroupMD (GroupMD, San Francisco, CA) allow members of a team to connect through 1 portal.[25] These discussions can foster communication, inspire clinical questions, and model the practice of timely response to new information.
Using Guerilla Teaching Tactics
Though time may be limited by work hours, there are opportunities embedded into clinical practice to create teaching moments. The principle of guerilla marketing uses unconventional marketing tactics in everyday locales to aggressively promote a product.[26] Similarly, guerilla teaching might be employed on rounds to make teaching points about common patient care issues that occur at nearly every room, such as Foley catheters after seeing one at the beside or hand hygiene after leaving a room. These types of topics are familiar to trainees as well as hospitalist attendings and fulfill the relevance that Millennial learners seek by easily applying them to the patient at hand.
Memory triggers or checklists are another way to systematically introduce guerilla teaching on commonplace topics. The IBCD checklist, for example, has been successfully implemented at our institution to promote adherence to 4 quality measures.[27] IBCD, which stands for immunizations, bedsores, catheters, and deep vein thrombosis prophylaxis, is easily and quickly tacked on as a checklist item at the end of the problem list during a presentation. Similar checklists can serve as teaching points on quality and safety in inpatient care, as well as reminders to consider these issues for every patient.
Rainy Day Teaching
Hospitalist teaching attendings recognize that duty hours have shifted the preferred time for teaching away from busy admission periods such as postcall rounds.[28] The limited time spent reviewing new admissions is now often focused on patient care issues, with much of the discussion eliminated. However, hospitalist attendings can be proactive and save certain teaching moments for rainy day teaching, anticipating topics to introduce during lower census times. Additionally, attending access to the EHRs allows attendings to preview cases the residents have admitted during a call period and may facilitate planning teaching topics for future opportunities.[23]
Though teaching is an essential part of the hospitalist teaching attending role, the Millennial Generation's affinity for teamwork makes it possible to utilize additional team members as teachers for the group. This type of distribution of responsibility, or outsourcing of teaching, can be done in the form of a teaching or float resident. These individuals can be directed to search the literature to answer clinical questions the team may have during rounds and report back, which may influence decision making and patient care as well as provide education.[29]
Embedding Teaching Moments Into Rounds
Dr. Francis W. Peabody may have been addressing students many generations removed from Millennial learners when he implored them to remember that the secret of the care of the patient is in caring for the patient, but his maxim still rings true today.[30] This advice provides an important insight on how the focus can be kept on the patient by emphasizing physical examination and history‐taking skills, which engages learners in hands‐on activity and grounds that education in a patient‐based experience.[31] The Stanford 25 represents a successful project that refocuses the doctorpatient encounter on the bedside.[32] Using a Web‐based platform, this initiative instructs on 25 physical examination maneuvers, utilizing teaching methods that are familiar to Millennial learners and are patient focused.
In addition to emphasizing bedside teaching, smaller moments can be used during rounds to establish an expectation for learning. Hospitalist attendings can create a routine with daily teaching moments, such as an electrocardiogram or a daily Medical Knowledge Self‐Assessment Program question, a source of internal medicine board preparation material published by the American College of Physicians.[33] These are opportunities to inject a quick educational moment that is easily relatable to the patients on the team's service. Using teaching moments that are routine, accessible, and relevant to patient care can help shape Millennial learners' expectations that teaching be a daily occurrence interwoven within clinical care provided during rounds.
There are several limitations to our work. These strategies do not represent a systematic review, and there is little evidence to support that our approach is more effective than conventional teaching methods. Though we address hospitalists specifically, these strategies are likely suitable for all inpatient educators as they have not been well studied in specific groups. With the paucity of literature regarding learning preferences of Millennial medical trainees, it is difficult to know what methods may truly be most desirable in the wards setting, as many of the needs and learning styles considered in our approach are borrowed from other more traditional learning environments. It is unclear how adoptable our strategies may be for educators from other generations; these faculty may have different approaches to teaching. Further research is necessary to identify areas for faculty development in learning new techniques as well as compare the efficacy of our approach to conventional methods with respect to standardized educational outcomes such as In‐Training Exam performance, as well as patient outcomes.
ACCEPTING THE CHALLENGE
The landscape of clinical teaching has shifted considerably in recent years, in both the makeup of learners for whom educators are responsible for teaching as well as the challenges in teaching under the duty hours restrictions. Though rounds are more focused on patient care than in the past, it is possible to work within the current structure to promote successful learning with an approach that considers the preferences of today's learners.
A hospitalist's natural habitat, the busy inpatient wards, is a clinical learning environment with rich potential for innovation and excellence in teaching. The challenges in practicing hospital medicine closely parallel the challenges in teaching under the constraints of duty hours restrictions; both require a creative approach to problem solving and an affinity for teamwork. The hospitalist community is well suited to not only meet these challenges but become leaders in embracing how to teach effectively on today's wards. Maximizing interaction, embracing technology, and encouraging group‐based learning may represent the keys to a successful approach to teaching the Millennial Generation in a post‐duty hours world.
- , , ; ACGME Duty Hour Task Force. The new recommendations on duty hours from the ACGME Task Force. N Engl J Med. 2010;363(2):e3.
- , . The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514–517.
- , , , et al. Hospital medicine in the internal medicine clerkship: results from a national survey. J Hosp Med. 2012;7(7):557–561.
- , . Millennials Rising: The Next Great Generation. New York, NY: Random House/Vintage Books; 2000.
- Eckleberry‐Hunt J, Tucciarone J. The challenges and opportunities of teaching “Generation Y.” J Grad Med Educ.2011;3(4):458–461.
- . Generational changes and their impact in the classroom: teaching Generation Me. Med Educ. 2009;43(5):398–405.
- , , . Twelve tips for facilitating Millennials' learning. Med Teach. 2012;34(4):274–278.
- Pew Research Center. Millennials: a portrait of generation next. Available at: http://pewsocialtrends.org/files/2010/10/millennials‐confident‐connected‐open‐to‐change.pdf. Accessed February 28, 2013.
- , , , et al. Generational influences in academic emergency medicine: teaching and learning, mentoring, and technology (part I). Acad Emerg Med. 2011;18(2):190–199.
- , , , et al. Generational influences in academic emergency medicine: structure, function, and culture (part II). Acad Emerg Med. 2011;18(2):200–207.
- , , , . Smartphone use during inpatient attending rounds: prevalence, patterns, and potential for distraction. J Hosp Med. 2012;8:595–599.
- , , , et al. Comparing millennial and generation X medical students at one medical school. Acad Med. 2006;81(6):571–576.
- , , , . Differences in motives between Millennial and Generation X students. Med Educ. 2010;44(6):570–576.
- , . Effect of ACGME duty hours on attending physician teaching and satisfaction. Arch Intern Med. 2008;168(11):1226–1227.
- , , . Residents' response to duty‐hours regulations—a follow‐up national survey. N Engl J Med. 2012; 366(24):e35.
- . Innovation arc: new approaches. Presented at: Association of American Colleges of Medicine National Meeting; November 2012; San Francisco, CA.
- , . Learner‐centered approaches in medical education. BMJ. 1999;318:1280–1283.
- , . Lecture halls without lectures—a proposal for medical education. N Engl J Med. 2012;366(18):1657–1659.
- The Khan Academy. Available at: https://www.khanacademy.org/. Accessed March 4, 2013.
- Dropbox. Dropbox Inc. Available at: https://www.dropbox.com/. Accessed April 19, 2013.
- Google Drive. Google Inc. Available at: https://drive.google.com/. Accessed April 19, 2013.
- , , , et al. What makes a good clinical teacher in medicine? A review of the literature. Acad Med. 2008;83(5):452–466.
- . Smartphones in clinical practice, medical education, and research. Arch Intern Med. 2011;171(14):1294–1296.
- , , , et al. Attending use of the electronic health record (EHR) and implications for housestaff supervision. Presented at: Midwest Society of General Internal Medicine Regional Meeting; September 2012; Chicago, IL.
- GroupMD. GroupMD Inc. Available at http://group.md. Accessed April 19, 2013.
- . Guerilla Marketing: Secrets for Making Big Profits From Your Small Business. Boston, MA: Houghton Mifflin; 1984.
- , , , et al. IBCD: development and testing of a checklist to improve quality of care for hospitalized general medical patients. Jt Comm J Qual Patient Saf. 2013;39(4):147–156.
- , . Ice cream rounds. Acad Med. 2013;88(1):66.
- , , , et al. The impact of evidence on physicians' inpatient treatment decisions. J Gen Intern Med. 2004; 19(5 pt 1):402–409.
- . Landmark article March 19, 1927: the care of the patient. By Francis W. Peabody. JAMA. 1984;252(6):813–818.
- , , , et al. The art of bedside rounds: a multi‐center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412–420.
- Stanford University School of Medicine. Stanford Medicine 25. Available at: http://stanfordmedicine25.stanford.edu/. Accessed February 28, 2013.
- Medical Knowledge Self‐Assessment Program 16. The American College of Physicians. Available at: https://mksap.acponline.org. Accessed April 19, 2013.
The implementation of resident duty hour restrictions has created a clinical learning environment on the wards quite different from any previous era. The Accreditation Council for Graduate Medical Education issued its first set of regulations limiting consecutive hours worked for residents in 2003, and further restricted hours in 2011.[1] These restrictions have had many implications across several aspects of patient care, education, and clinical training, particularly for hospitalists who spend the majority of their time in this setting and are heavily involved in undergraduate and graduate clinical education in academic medical centers.[2, 3]
As learning environments have been shifting, so has the composition of learners. The Millennial Generation (or Generation Y), defined as those born approximately between 1980 and 2000, represents those young clinicians currently filling the halls of medical schools and ranks of residency and fellowship programs.[4] Interestingly, the current system of restricted work hours is the only system under which the Millennial Generation has ever trained.
As this new generation represents the bulk of current trainees, hospitalist faculty must consider how their teaching styles can be adapted to accommodate these learners. For teaching hospitalists, an approach that considers the learning environment as affected by duty hours, as well as the preferences of Millennial learners, is necessary to educate the next generation of trainees. This article aimed to introduce potential strategies for hospitalists to better align teaching on the wards with the preferences of Millennial learners under the constraints of residency duty hours.
THE NEWEST GENERATION OF LEARNERS
The Millennial Generation has been well described.[4, 5, 6, 7, 8, 9, 10] Broadly speaking, this generation is thought to have been raised by attentive and involved parents, influencing relationships with educators and mentors; they respect authority but do not hesitate to question the relevance of assignments or decisions. Millennials prefer structured learning environments that focus heavily on interaction and experiential learning, and they value design and appearance in how material is presented.[7] Millennials also seek clear expectations and immediate feedback on their performance, and though they have sometimes been criticized for a strong sense of entitlement, they have a strong desire for collaboration and group‐based activity.[5, 6]
One of the most notable and defining characteristics of the Millennial Generation is an affinity for technology and innovation.[7, 8, 9] Web‐based learning tools that are interactive and engaging, such as blogs, podcasts, or streaming videos are familiar and favored methods of learning. Millennials are skilled at finding information and providing answers and data, but may need help with synthesis and application.[5] They take pride in their ability to multitask, but can be prone to doing so inappropriately, particularly with technology that is readily available.[11]
Few studies have explored characteristics of the Millennial Generation specific to medical trainees. One study examined personality characteristics of Millennial medical students compared to Generation X students (those born from 19651980) at a single institution. Millennial students scored higher on warmth, reasoning, emotional stability, rule consciousness, social boldness, sensitivity, apprehension, openness to change, and perfectionism compared to Generation X students. They scored lower on measures for self‐reliance.[12] Additionally, when motives for behavior were studied, Millennial medical students scored higher on needs for affiliation and achievement, and lower on needs for power.[13]
DUTY HOURS: A GENERATION APART
As noted previously, the Millennial Generation is the first to train exclusively in the era of duty hours restrictions. The oldest members of this generation, those born in 1981, were entering medical school at the time of the first duty hours restrictions in 2003, and thus have always been educated, trained, and practiced in an environment in which work hours were an essential part of residency training.
Though duty hours have been an omnipresent part of training for the Millennial Generation, the clinical learning environment that they have known continues to evolve and change. Time for teaching, in particular, has been especially strained by work hour limits, and this has been noted by both attending physicians and trainees with each iteration of work hours limits. Attendings in one study estimated that time spent teaching on general medicine wards was reduced by about 20% following the 2003 limits, and over 40% of residents in a national survey reported that the 2011 limits had worsened the quality of education.[14, 15]
GENERATIONAL STRATEGIES FOR SUCCESS FOR HOSPITALIST TEACHING ATTENDINGS
The time limitations imposed by duty hours restrictions have compelled teaching rounds to become more patient‐care centered and often less learner‐centered, as providing patient care becomes the prime obligation for this limited time period. Millennial learners are accustomed to being the center of attention in educational environments, and changing the focus from education to patient care in the wards setting may be an abrupt transition for some learners.[6] However, hospitalists can help restructure teaching opportunities on the clinical wards by using teaching methods of the highest value to Millennial learners to promote learning under the conditions of duty hours limitations.
An approach using these methods was developed by reviewing recent literature as well as educational innovations that have been presented at scholarly meetings (eg, Sal Khan's presentation at the 2012 Association of American Medical Colleges meeting).[16] The authors discussed potential teaching techniques that were thought to be feasible to implement in the context of the current learning environment, with consideration of learning theories that would be most effective for the target group of learners (eg, adult learning theory).[17] A mnemonic was created to consolidate strategies thought to best represent these techniques. FUTURE is a group of teaching strategies that can be used by hospitalists to improve teaching rounds by Flipping the Wards, Using Documentation to Teach, Technology‐Enabled Teaching, Using Guerilla Teaching Tactics, Rainy Day Teaching, and Embedding Teaching Moments into Rounds.
Flipping the Wards
Millennial learners prefer novel methods of delivery that are interactive and technology based.[7, 8, 9] Lectures and slide‐based presentations frequently do not feature the degree of interactive engagement that they seek, and methods such as case‐based presentations and simulation may be more suitable. The Khan Academy is a not‐for‐profit organization that has been proposed as a model for future directions for medical education.[18] The academy's global classroom houses over 4000 videos and interactive modules to allow students to progress through topics on their own time.[19] Teaching rounds can be similarly flipped such that discussion and group work take place during rounds, whereas lectures, modules, and reading are reserved for individual study.[18]
As time pressures shift the focus of rounds exclusively toward discussion of patient‐care tasks, finding time for teaching outside of rounds can be emphasized to inspire self‐directed learning. When residents need time to tend to immediate patient‐care issues, hospitalist attendings could take the time to search for articles to send to team members. Rather than distributing paper copies that may be lost, cloud‐based data management systems such as Dropbox (Dropbox, San Francisco, CA) or Google Drive (Google Inc., Mountain View, CA) can be used to disseminate articles, which can be pulled up in real time on mobile devices during rounds and later deposited in shared folders accessible to all team members.[20, 21] The advantage of this approach is that it does not require all learners to be present on rounds, which may not be possible with duty hours.
Using Documentation to Teach
Trainees report that one of the most desirable attributes of clinical teachers is when they delineate their clinical reasoning and thought process.[22] Similarly, Millennial learners specifically desire to understand the rationale behind their teachers' actions.[6] Documentation in the medical chart or electronic health record (EHR) can be used to enhance teaching and role‐model clinical reasoning in a transparent and readily available fashion.
Billing requirements necessitate daily attending documentation in the form of an attestation. Hospitalist attendings can use attestations to model thought process and clinical synthesis in the daily assessment of a patient. For example, an attestation one‐liner can be used to concisely summarize the patient's course or highlight the most pressing issue of the day, rather than simply serve as a placeholder for billing or agree with above in reference to housestaff documentation. This practice can demonstrate to residents how to write a short snapshot of a patient's care in addition to improving communication.
Additionally, the EHR can be a useful platform to guide feedback for residents on their clinical performance. Millennial learners prefer specific, immediate feedback, and trainee documentation can serve as a template to show examples of good documentation and clinical reasoning as well as areas needing improvement.[5] These tangible examples of clinical performance are specific and understandable for trainees to guide their self‐learning and improvement.
Technology‐Enabled Teaching
Using technology wisely on the wards can improve efficiency while also taking advantage of teaching methods familiar to Millennial learners. Technology can be used in a positive manner to keep the focus on the patient and enhance teaching when time is limited on rounds. Smartphones and tablets have become an omnipresent part of the clinical environment.[23] Rather than distracting from rounds, these tools can be used to answer clinical questions in real time, thus directly linking the question to the patient's care.
The EHR is a powerful technological resource that is readily available to enhance teaching during a busy ward schedule. Clinical information is electronically accessible at all hours for both trainees and attendings, rather than only at prespecified times on daily rounds, and the Millennial Generation is accustomed to receiving and sharing information in this fashion.[24] Technology platforms that enable simultaneous sharing of information among multiple members of a team can also be used to assist in sharing clinical information in this manner. Health Insurance Portability and Accountability Act‐compliant group text‐messaging applications for smartphones and tablets such as GroupMD (GroupMD, San Francisco, CA) allow members of a team to connect through 1 portal.[25] These discussions can foster communication, inspire clinical questions, and model the practice of timely response to new information.
Using Guerilla Teaching Tactics
Though time may be limited by work hours, there are opportunities embedded into clinical practice to create teaching moments. The principle of guerilla marketing uses unconventional marketing tactics in everyday locales to aggressively promote a product.[26] Similarly, guerilla teaching might be employed on rounds to make teaching points about common patient care issues that occur at nearly every room, such as Foley catheters after seeing one at the beside or hand hygiene after leaving a room. These types of topics are familiar to trainees as well as hospitalist attendings and fulfill the relevance that Millennial learners seek by easily applying them to the patient at hand.
Memory triggers or checklists are another way to systematically introduce guerilla teaching on commonplace topics. The IBCD checklist, for example, has been successfully implemented at our institution to promote adherence to 4 quality measures.[27] IBCD, which stands for immunizations, bedsores, catheters, and deep vein thrombosis prophylaxis, is easily and quickly tacked on as a checklist item at the end of the problem list during a presentation. Similar checklists can serve as teaching points on quality and safety in inpatient care, as well as reminders to consider these issues for every patient.
Rainy Day Teaching
Hospitalist teaching attendings recognize that duty hours have shifted the preferred time for teaching away from busy admission periods such as postcall rounds.[28] The limited time spent reviewing new admissions is now often focused on patient care issues, with much of the discussion eliminated. However, hospitalist attendings can be proactive and save certain teaching moments for rainy day teaching, anticipating topics to introduce during lower census times. Additionally, attending access to the EHRs allows attendings to preview cases the residents have admitted during a call period and may facilitate planning teaching topics for future opportunities.[23]
Though teaching is an essential part of the hospitalist teaching attending role, the Millennial Generation's affinity for teamwork makes it possible to utilize additional team members as teachers for the group. This type of distribution of responsibility, or outsourcing of teaching, can be done in the form of a teaching or float resident. These individuals can be directed to search the literature to answer clinical questions the team may have during rounds and report back, which may influence decision making and patient care as well as provide education.[29]
Embedding Teaching Moments Into Rounds
Dr. Francis W. Peabody may have been addressing students many generations removed from Millennial learners when he implored them to remember that the secret of the care of the patient is in caring for the patient, but his maxim still rings true today.[30] This advice provides an important insight on how the focus can be kept on the patient by emphasizing physical examination and history‐taking skills, which engages learners in hands‐on activity and grounds that education in a patient‐based experience.[31] The Stanford 25 represents a successful project that refocuses the doctorpatient encounter on the bedside.[32] Using a Web‐based platform, this initiative instructs on 25 physical examination maneuvers, utilizing teaching methods that are familiar to Millennial learners and are patient focused.
In addition to emphasizing bedside teaching, smaller moments can be used during rounds to establish an expectation for learning. Hospitalist attendings can create a routine with daily teaching moments, such as an electrocardiogram or a daily Medical Knowledge Self‐Assessment Program question, a source of internal medicine board preparation material published by the American College of Physicians.[33] These are opportunities to inject a quick educational moment that is easily relatable to the patients on the team's service. Using teaching moments that are routine, accessible, and relevant to patient care can help shape Millennial learners' expectations that teaching be a daily occurrence interwoven within clinical care provided during rounds.
There are several limitations to our work. These strategies do not represent a systematic review, and there is little evidence to support that our approach is more effective than conventional teaching methods. Though we address hospitalists specifically, these strategies are likely suitable for all inpatient educators as they have not been well studied in specific groups. With the paucity of literature regarding learning preferences of Millennial medical trainees, it is difficult to know what methods may truly be most desirable in the wards setting, as many of the needs and learning styles considered in our approach are borrowed from other more traditional learning environments. It is unclear how adoptable our strategies may be for educators from other generations; these faculty may have different approaches to teaching. Further research is necessary to identify areas for faculty development in learning new techniques as well as compare the efficacy of our approach to conventional methods with respect to standardized educational outcomes such as In‐Training Exam performance, as well as patient outcomes.
ACCEPTING THE CHALLENGE
The landscape of clinical teaching has shifted considerably in recent years, in both the makeup of learners for whom educators are responsible for teaching as well as the challenges in teaching under the duty hours restrictions. Though rounds are more focused on patient care than in the past, it is possible to work within the current structure to promote successful learning with an approach that considers the preferences of today's learners.
A hospitalist's natural habitat, the busy inpatient wards, is a clinical learning environment with rich potential for innovation and excellence in teaching. The challenges in practicing hospital medicine closely parallel the challenges in teaching under the constraints of duty hours restrictions; both require a creative approach to problem solving and an affinity for teamwork. The hospitalist community is well suited to not only meet these challenges but become leaders in embracing how to teach effectively on today's wards. Maximizing interaction, embracing technology, and encouraging group‐based learning may represent the keys to a successful approach to teaching the Millennial Generation in a post‐duty hours world.
The implementation of resident duty hour restrictions has created a clinical learning environment on the wards quite different from any previous era. The Accreditation Council for Graduate Medical Education issued its first set of regulations limiting consecutive hours worked for residents in 2003, and further restricted hours in 2011.[1] These restrictions have had many implications across several aspects of patient care, education, and clinical training, particularly for hospitalists who spend the majority of their time in this setting and are heavily involved in undergraduate and graduate clinical education in academic medical centers.[2, 3]
As learning environments have been shifting, so has the composition of learners. The Millennial Generation (or Generation Y), defined as those born approximately between 1980 and 2000, represents those young clinicians currently filling the halls of medical schools and ranks of residency and fellowship programs.[4] Interestingly, the current system of restricted work hours is the only system under which the Millennial Generation has ever trained.
As this new generation represents the bulk of current trainees, hospitalist faculty must consider how their teaching styles can be adapted to accommodate these learners. For teaching hospitalists, an approach that considers the learning environment as affected by duty hours, as well as the preferences of Millennial learners, is necessary to educate the next generation of trainees. This article aimed to introduce potential strategies for hospitalists to better align teaching on the wards with the preferences of Millennial learners under the constraints of residency duty hours.
THE NEWEST GENERATION OF LEARNERS
The Millennial Generation has been well described.[4, 5, 6, 7, 8, 9, 10] Broadly speaking, this generation is thought to have been raised by attentive and involved parents, influencing relationships with educators and mentors; they respect authority but do not hesitate to question the relevance of assignments or decisions. Millennials prefer structured learning environments that focus heavily on interaction and experiential learning, and they value design and appearance in how material is presented.[7] Millennials also seek clear expectations and immediate feedback on their performance, and though they have sometimes been criticized for a strong sense of entitlement, they have a strong desire for collaboration and group‐based activity.[5, 6]
One of the most notable and defining characteristics of the Millennial Generation is an affinity for technology and innovation.[7, 8, 9] Web‐based learning tools that are interactive and engaging, such as blogs, podcasts, or streaming videos are familiar and favored methods of learning. Millennials are skilled at finding information and providing answers and data, but may need help with synthesis and application.[5] They take pride in their ability to multitask, but can be prone to doing so inappropriately, particularly with technology that is readily available.[11]
Few studies have explored characteristics of the Millennial Generation specific to medical trainees. One study examined personality characteristics of Millennial medical students compared to Generation X students (those born from 19651980) at a single institution. Millennial students scored higher on warmth, reasoning, emotional stability, rule consciousness, social boldness, sensitivity, apprehension, openness to change, and perfectionism compared to Generation X students. They scored lower on measures for self‐reliance.[12] Additionally, when motives for behavior were studied, Millennial medical students scored higher on needs for affiliation and achievement, and lower on needs for power.[13]
DUTY HOURS: A GENERATION APART
As noted previously, the Millennial Generation is the first to train exclusively in the era of duty hours restrictions. The oldest members of this generation, those born in 1981, were entering medical school at the time of the first duty hours restrictions in 2003, and thus have always been educated, trained, and practiced in an environment in which work hours were an essential part of residency training.
Though duty hours have been an omnipresent part of training for the Millennial Generation, the clinical learning environment that they have known continues to evolve and change. Time for teaching, in particular, has been especially strained by work hour limits, and this has been noted by both attending physicians and trainees with each iteration of work hours limits. Attendings in one study estimated that time spent teaching on general medicine wards was reduced by about 20% following the 2003 limits, and over 40% of residents in a national survey reported that the 2011 limits had worsened the quality of education.[14, 15]
GENERATIONAL STRATEGIES FOR SUCCESS FOR HOSPITALIST TEACHING ATTENDINGS
The time limitations imposed by duty hours restrictions have compelled teaching rounds to become more patient‐care centered and often less learner‐centered, as providing patient care becomes the prime obligation for this limited time period. Millennial learners are accustomed to being the center of attention in educational environments, and changing the focus from education to patient care in the wards setting may be an abrupt transition for some learners.[6] However, hospitalists can help restructure teaching opportunities on the clinical wards by using teaching methods of the highest value to Millennial learners to promote learning under the conditions of duty hours limitations.
An approach using these methods was developed by reviewing recent literature as well as educational innovations that have been presented at scholarly meetings (eg, Sal Khan's presentation at the 2012 Association of American Medical Colleges meeting).[16] The authors discussed potential teaching techniques that were thought to be feasible to implement in the context of the current learning environment, with consideration of learning theories that would be most effective for the target group of learners (eg, adult learning theory).[17] A mnemonic was created to consolidate strategies thought to best represent these techniques. FUTURE is a group of teaching strategies that can be used by hospitalists to improve teaching rounds by Flipping the Wards, Using Documentation to Teach, Technology‐Enabled Teaching, Using Guerilla Teaching Tactics, Rainy Day Teaching, and Embedding Teaching Moments into Rounds.
Flipping the Wards
Millennial learners prefer novel methods of delivery that are interactive and technology based.[7, 8, 9] Lectures and slide‐based presentations frequently do not feature the degree of interactive engagement that they seek, and methods such as case‐based presentations and simulation may be more suitable. The Khan Academy is a not‐for‐profit organization that has been proposed as a model for future directions for medical education.[18] The academy's global classroom houses over 4000 videos and interactive modules to allow students to progress through topics on their own time.[19] Teaching rounds can be similarly flipped such that discussion and group work take place during rounds, whereas lectures, modules, and reading are reserved for individual study.[18]
As time pressures shift the focus of rounds exclusively toward discussion of patient‐care tasks, finding time for teaching outside of rounds can be emphasized to inspire self‐directed learning. When residents need time to tend to immediate patient‐care issues, hospitalist attendings could take the time to search for articles to send to team members. Rather than distributing paper copies that may be lost, cloud‐based data management systems such as Dropbox (Dropbox, San Francisco, CA) or Google Drive (Google Inc., Mountain View, CA) can be used to disseminate articles, which can be pulled up in real time on mobile devices during rounds and later deposited in shared folders accessible to all team members.[20, 21] The advantage of this approach is that it does not require all learners to be present on rounds, which may not be possible with duty hours.
Using Documentation to Teach
Trainees report that one of the most desirable attributes of clinical teachers is when they delineate their clinical reasoning and thought process.[22] Similarly, Millennial learners specifically desire to understand the rationale behind their teachers' actions.[6] Documentation in the medical chart or electronic health record (EHR) can be used to enhance teaching and role‐model clinical reasoning in a transparent and readily available fashion.
Billing requirements necessitate daily attending documentation in the form of an attestation. Hospitalist attendings can use attestations to model thought process and clinical synthesis in the daily assessment of a patient. For example, an attestation one‐liner can be used to concisely summarize the patient's course or highlight the most pressing issue of the day, rather than simply serve as a placeholder for billing or agree with above in reference to housestaff documentation. This practice can demonstrate to residents how to write a short snapshot of a patient's care in addition to improving communication.
Additionally, the EHR can be a useful platform to guide feedback for residents on their clinical performance. Millennial learners prefer specific, immediate feedback, and trainee documentation can serve as a template to show examples of good documentation and clinical reasoning as well as areas needing improvement.[5] These tangible examples of clinical performance are specific and understandable for trainees to guide their self‐learning and improvement.
Technology‐Enabled Teaching
Using technology wisely on the wards can improve efficiency while also taking advantage of teaching methods familiar to Millennial learners. Technology can be used in a positive manner to keep the focus on the patient and enhance teaching when time is limited on rounds. Smartphones and tablets have become an omnipresent part of the clinical environment.[23] Rather than distracting from rounds, these tools can be used to answer clinical questions in real time, thus directly linking the question to the patient's care.
The EHR is a powerful technological resource that is readily available to enhance teaching during a busy ward schedule. Clinical information is electronically accessible at all hours for both trainees and attendings, rather than only at prespecified times on daily rounds, and the Millennial Generation is accustomed to receiving and sharing information in this fashion.[24] Technology platforms that enable simultaneous sharing of information among multiple members of a team can also be used to assist in sharing clinical information in this manner. Health Insurance Portability and Accountability Act‐compliant group text‐messaging applications for smartphones and tablets such as GroupMD (GroupMD, San Francisco, CA) allow members of a team to connect through 1 portal.[25] These discussions can foster communication, inspire clinical questions, and model the practice of timely response to new information.
Using Guerilla Teaching Tactics
Though time may be limited by work hours, there are opportunities embedded into clinical practice to create teaching moments. The principle of guerilla marketing uses unconventional marketing tactics in everyday locales to aggressively promote a product.[26] Similarly, guerilla teaching might be employed on rounds to make teaching points about common patient care issues that occur at nearly every room, such as Foley catheters after seeing one at the beside or hand hygiene after leaving a room. These types of topics are familiar to trainees as well as hospitalist attendings and fulfill the relevance that Millennial learners seek by easily applying them to the patient at hand.
Memory triggers or checklists are another way to systematically introduce guerilla teaching on commonplace topics. The IBCD checklist, for example, has been successfully implemented at our institution to promote adherence to 4 quality measures.[27] IBCD, which stands for immunizations, bedsores, catheters, and deep vein thrombosis prophylaxis, is easily and quickly tacked on as a checklist item at the end of the problem list during a presentation. Similar checklists can serve as teaching points on quality and safety in inpatient care, as well as reminders to consider these issues for every patient.
Rainy Day Teaching
Hospitalist teaching attendings recognize that duty hours have shifted the preferred time for teaching away from busy admission periods such as postcall rounds.[28] The limited time spent reviewing new admissions is now often focused on patient care issues, with much of the discussion eliminated. However, hospitalist attendings can be proactive and save certain teaching moments for rainy day teaching, anticipating topics to introduce during lower census times. Additionally, attending access to the EHRs allows attendings to preview cases the residents have admitted during a call period and may facilitate planning teaching topics for future opportunities.[23]
Though teaching is an essential part of the hospitalist teaching attending role, the Millennial Generation's affinity for teamwork makes it possible to utilize additional team members as teachers for the group. This type of distribution of responsibility, or outsourcing of teaching, can be done in the form of a teaching or float resident. These individuals can be directed to search the literature to answer clinical questions the team may have during rounds and report back, which may influence decision making and patient care as well as provide education.[29]
Embedding Teaching Moments Into Rounds
Dr. Francis W. Peabody may have been addressing students many generations removed from Millennial learners when he implored them to remember that the secret of the care of the patient is in caring for the patient, but his maxim still rings true today.[30] This advice provides an important insight on how the focus can be kept on the patient by emphasizing physical examination and history‐taking skills, which engages learners in hands‐on activity and grounds that education in a patient‐based experience.[31] The Stanford 25 represents a successful project that refocuses the doctorpatient encounter on the bedside.[32] Using a Web‐based platform, this initiative instructs on 25 physical examination maneuvers, utilizing teaching methods that are familiar to Millennial learners and are patient focused.
In addition to emphasizing bedside teaching, smaller moments can be used during rounds to establish an expectation for learning. Hospitalist attendings can create a routine with daily teaching moments, such as an electrocardiogram or a daily Medical Knowledge Self‐Assessment Program question, a source of internal medicine board preparation material published by the American College of Physicians.[33] These are opportunities to inject a quick educational moment that is easily relatable to the patients on the team's service. Using teaching moments that are routine, accessible, and relevant to patient care can help shape Millennial learners' expectations that teaching be a daily occurrence interwoven within clinical care provided during rounds.
There are several limitations to our work. These strategies do not represent a systematic review, and there is little evidence to support that our approach is more effective than conventional teaching methods. Though we address hospitalists specifically, these strategies are likely suitable for all inpatient educators as they have not been well studied in specific groups. With the paucity of literature regarding learning preferences of Millennial medical trainees, it is difficult to know what methods may truly be most desirable in the wards setting, as many of the needs and learning styles considered in our approach are borrowed from other more traditional learning environments. It is unclear how adoptable our strategies may be for educators from other generations; these faculty may have different approaches to teaching. Further research is necessary to identify areas for faculty development in learning new techniques as well as compare the efficacy of our approach to conventional methods with respect to standardized educational outcomes such as In‐Training Exam performance, as well as patient outcomes.
ACCEPTING THE CHALLENGE
The landscape of clinical teaching has shifted considerably in recent years, in both the makeup of learners for whom educators are responsible for teaching as well as the challenges in teaching under the duty hours restrictions. Though rounds are more focused on patient care than in the past, it is possible to work within the current structure to promote successful learning with an approach that considers the preferences of today's learners.
A hospitalist's natural habitat, the busy inpatient wards, is a clinical learning environment with rich potential for innovation and excellence in teaching. The challenges in practicing hospital medicine closely parallel the challenges in teaching under the constraints of duty hours restrictions; both require a creative approach to problem solving and an affinity for teamwork. The hospitalist community is well suited to not only meet these challenges but become leaders in embracing how to teach effectively on today's wards. Maximizing interaction, embracing technology, and encouraging group‐based learning may represent the keys to a successful approach to teaching the Millennial Generation in a post‐duty hours world.
- , , ; ACGME Duty Hour Task Force. The new recommendations on duty hours from the ACGME Task Force. N Engl J Med. 2010;363(2):e3.
- , . The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514–517.
- , , , et al. Hospital medicine in the internal medicine clerkship: results from a national survey. J Hosp Med. 2012;7(7):557–561.
- , . Millennials Rising: The Next Great Generation. New York, NY: Random House/Vintage Books; 2000.
- Eckleberry‐Hunt J, Tucciarone J. The challenges and opportunities of teaching “Generation Y.” J Grad Med Educ.2011;3(4):458–461.
- . Generational changes and their impact in the classroom: teaching Generation Me. Med Educ. 2009;43(5):398–405.
- , , . Twelve tips for facilitating Millennials' learning. Med Teach. 2012;34(4):274–278.
- Pew Research Center. Millennials: a portrait of generation next. Available at: http://pewsocialtrends.org/files/2010/10/millennials‐confident‐connected‐open‐to‐change.pdf. Accessed February 28, 2013.
- , , , et al. Generational influences in academic emergency medicine: teaching and learning, mentoring, and technology (part I). Acad Emerg Med. 2011;18(2):190–199.
- , , , et al. Generational influences in academic emergency medicine: structure, function, and culture (part II). Acad Emerg Med. 2011;18(2):200–207.
- , , , . Smartphone use during inpatient attending rounds: prevalence, patterns, and potential for distraction. J Hosp Med. 2012;8:595–599.
- , , , et al. Comparing millennial and generation X medical students at one medical school. Acad Med. 2006;81(6):571–576.
- , , , . Differences in motives between Millennial and Generation X students. Med Educ. 2010;44(6):570–576.
- , . Effect of ACGME duty hours on attending physician teaching and satisfaction. Arch Intern Med. 2008;168(11):1226–1227.
- , , . Residents' response to duty‐hours regulations—a follow‐up national survey. N Engl J Med. 2012; 366(24):e35.
- . Innovation arc: new approaches. Presented at: Association of American Colleges of Medicine National Meeting; November 2012; San Francisco, CA.
- , . Learner‐centered approaches in medical education. BMJ. 1999;318:1280–1283.
- , . Lecture halls without lectures—a proposal for medical education. N Engl J Med. 2012;366(18):1657–1659.
- The Khan Academy. Available at: https://www.khanacademy.org/. Accessed March 4, 2013.
- Dropbox. Dropbox Inc. Available at: https://www.dropbox.com/. Accessed April 19, 2013.
- Google Drive. Google Inc. Available at: https://drive.google.com/. Accessed April 19, 2013.
- , , , et al. What makes a good clinical teacher in medicine? A review of the literature. Acad Med. 2008;83(5):452–466.
- . Smartphones in clinical practice, medical education, and research. Arch Intern Med. 2011;171(14):1294–1296.
- , , , et al. Attending use of the electronic health record (EHR) and implications for housestaff supervision. Presented at: Midwest Society of General Internal Medicine Regional Meeting; September 2012; Chicago, IL.
- GroupMD. GroupMD Inc. Available at http://group.md. Accessed April 19, 2013.
- . Guerilla Marketing: Secrets for Making Big Profits From Your Small Business. Boston, MA: Houghton Mifflin; 1984.
- , , , et al. IBCD: development and testing of a checklist to improve quality of care for hospitalized general medical patients. Jt Comm J Qual Patient Saf. 2013;39(4):147–156.
- , . Ice cream rounds. Acad Med. 2013;88(1):66.
- , , , et al. The impact of evidence on physicians' inpatient treatment decisions. J Gen Intern Med. 2004; 19(5 pt 1):402–409.
- . Landmark article March 19, 1927: the care of the patient. By Francis W. Peabody. JAMA. 1984;252(6):813–818.
- , , , et al. The art of bedside rounds: a multi‐center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412–420.
- Stanford University School of Medicine. Stanford Medicine 25. Available at: http://stanfordmedicine25.stanford.edu/. Accessed February 28, 2013.
- Medical Knowledge Self‐Assessment Program 16. The American College of Physicians. Available at: https://mksap.acponline.org. Accessed April 19, 2013.
- , , ; ACGME Duty Hour Task Force. The new recommendations on duty hours from the ACGME Task Force. N Engl J Med. 2010;363(2):e3.
- , . The emerging role of “hospitalists” in the American health care system. N Engl J Med. 1996;335(7):514–517.
- , , , et al. Hospital medicine in the internal medicine clerkship: results from a national survey. J Hosp Med. 2012;7(7):557–561.
- , . Millennials Rising: The Next Great Generation. New York, NY: Random House/Vintage Books; 2000.
- Eckleberry‐Hunt J, Tucciarone J. The challenges and opportunities of teaching “Generation Y.” J Grad Med Educ.2011;3(4):458–461.
- . Generational changes and their impact in the classroom: teaching Generation Me. Med Educ. 2009;43(5):398–405.
- , , . Twelve tips for facilitating Millennials' learning. Med Teach. 2012;34(4):274–278.
- Pew Research Center. Millennials: a portrait of generation next. Available at: http://pewsocialtrends.org/files/2010/10/millennials‐confident‐connected‐open‐to‐change.pdf. Accessed February 28, 2013.
- , , , et al. Generational influences in academic emergency medicine: teaching and learning, mentoring, and technology (part I). Acad Emerg Med. 2011;18(2):190–199.
- , , , et al. Generational influences in academic emergency medicine: structure, function, and culture (part II). Acad Emerg Med. 2011;18(2):200–207.
- , , , . Smartphone use during inpatient attending rounds: prevalence, patterns, and potential for distraction. J Hosp Med. 2012;8:595–599.
- , , , et al. Comparing millennial and generation X medical students at one medical school. Acad Med. 2006;81(6):571–576.
- , , , . Differences in motives between Millennial and Generation X students. Med Educ. 2010;44(6):570–576.
- , . Effect of ACGME duty hours on attending physician teaching and satisfaction. Arch Intern Med. 2008;168(11):1226–1227.
- , , . Residents' response to duty‐hours regulations—a follow‐up national survey. N Engl J Med. 2012; 366(24):e35.
- . Innovation arc: new approaches. Presented at: Association of American Colleges of Medicine National Meeting; November 2012; San Francisco, CA.
- , . Learner‐centered approaches in medical education. BMJ. 1999;318:1280–1283.
- , . Lecture halls without lectures—a proposal for medical education. N Engl J Med. 2012;366(18):1657–1659.
- The Khan Academy. Available at: https://www.khanacademy.org/. Accessed March 4, 2013.
- Dropbox. Dropbox Inc. Available at: https://www.dropbox.com/. Accessed April 19, 2013.
- Google Drive. Google Inc. Available at: https://drive.google.com/. Accessed April 19, 2013.
- , , , et al. What makes a good clinical teacher in medicine? A review of the literature. Acad Med. 2008;83(5):452–466.
- . Smartphones in clinical practice, medical education, and research. Arch Intern Med. 2011;171(14):1294–1296.
- , , , et al. Attending use of the electronic health record (EHR) and implications for housestaff supervision. Presented at: Midwest Society of General Internal Medicine Regional Meeting; September 2012; Chicago, IL.
- GroupMD. GroupMD Inc. Available at http://group.md. Accessed April 19, 2013.
- . Guerilla Marketing: Secrets for Making Big Profits From Your Small Business. Boston, MA: Houghton Mifflin; 1984.
- , , , et al. IBCD: development and testing of a checklist to improve quality of care for hospitalized general medical patients. Jt Comm J Qual Patient Saf. 2013;39(4):147–156.
- , . Ice cream rounds. Acad Med. 2013;88(1):66.
- , , , et al. The impact of evidence on physicians' inpatient treatment decisions. J Gen Intern Med. 2004; 19(5 pt 1):402–409.
- . Landmark article March 19, 1927: the care of the patient. By Francis W. Peabody. JAMA. 1984;252(6):813–818.
- , , , et al. The art of bedside rounds: a multi‐center qualitative study of strategies used by experienced bedside teachers. J Gen Intern Med. 2013;28(3):412–420.
- Stanford University School of Medicine. Stanford Medicine 25. Available at: http://stanfordmedicine25.stanford.edu/. Accessed February 28, 2013.
- Medical Knowledge Self‐Assessment Program 16. The American College of Physicians. Available at: https://mksap.acponline.org. Accessed April 19, 2013.
Review of VTE Prophylaxis Strategies
Venous thromboembolism (VTE), including deep venous thrombosis (DVT) and pulmonary embolism (PE), is estimated to affect 900,000 Americans each year and is a cause of significant morbidity and mortality with associated high healthcare costs.[1] Accordingly, the comparative effectiveness and safety of interventions for the prevention and treatment of VTE are among the national priorities for comparative effectiveness research.[2] Whereas we have evidence‐based guidelines for the prophylaxis of VTE in the general population, there are no guidelines informing the care of select patient populations. Select populations are those patients in whom there is decisional uncertainty about the optimal choice, timing, and dose of VTE prophylaxis. Not only do these patients have an increased risk of DVT and PE, but most are also at high risk of bleeding, the most important complication of VTE prophylaxis.[3, 4, 5, 6]
The objectives of this systematic review were to define the comparative effectiveness and safety of pharmacologic and mechanical strategies for VTE prevention in some of these select medical populations including obese patients, patients on concomitant antiplatelet therapy, patients with renal insufficiency, patients who are underweight, and patients with coagulopathy due to liver disease.
METHODS
The methods for this comparative effectiveness review (CER) follow the guidelines suggested in the Agency for Healthcare Research and Quality (AHRQ) Methods Guide for Effectiveness and Comparative Effectiveness Reviews.[7] The protocol was publically posted.[8]
Search Strategy
We searched MEDLINE, EMBASE, and SCOPUS through August 2011, CINAHL, International Pharmaceutical Abstracts,
Study Selection
We reviewed titles followed by abstracts to identify randomized controlled trials (RCTs) or observational studies with comparison groups reporting on the effectiveness or safety of VTE prevention in our populations. Two investigators independently reviewed abstracts, and we excluded the abstracts if both investigators agreed that the article met 1 or more of the exclusion criteria. We included only English‐language articles that evaluated the effectiveness of pharmacological or mechanical interventions that have been approved for clinical use in the United States. To be eligible, the studies must have addressed relevant key questions in the population of our interest. We resolved disagreements by consensus. We used DistillerSR (Evidence Partners Inc., Ottawa, Ontario, Canada), a Web‐based database management program to manage the review process. Two investigators assessed the risk of bias in each study independently, using the Downs and Black instrument for observational studies and trials.[10]
Data Synthesis
For each select population, we created detailed evidence tables containing the information abstracted from the eligible studies. After synthesizing the evidence, we graded the quantity, quality, and consistency of the best available evidence for each select population by adapting an evidence‐grading scheme recommended in the Methods Guide for Conducting Comparative Effectiveness Reviews.[7]
RESULTS
We identified 30,902 unique citations and included 9 studies (Figure 1). There were 5 RCTs with relevant subgroups and 4 observational studies (Table 1). Two studies reported on the risk of bleeding in patients given pharmacologic prophylaxis while they are concomitantly taking nonsteroidal anti‐inflammatory drugs (NSAIDS) or antiplatelet agents/aspirin, 1 RCT and 1 prospective observational study reported on obese patients, and 5 studies described outcomes of patients with renal insufficiency (see Supporting Information, Table 1, in the online version of this article). No study tested prophylaxis in underweight patients or those with liver disease.
| Study | Arm, n | Total VTE (DVT and PE) | Bleeding | Other Outcomes | |
|---|---|---|---|---|---|
| |||||
| Obese patients | |||||
| Kucher et al., 2005[11] | Arm 1 (dalteparin), 558 | 2.8% (95% CI: 1.34.3) | 0% | Mortality at 21 days: 4.6% | |
| Arm 2 (placebo), 560 | 4.3% (95% CI: 2.56.2) | 0.7% | Mortality at 21 days: 2.7% | ||
| Freeman et al., [12] | Arm 1 (fixed‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 19 % | |
| Arm 2 (lower‐dose enoxaparin), 9 | NR | NR | Peak anti‐factor Xa level 32 % | ||
| Arm 3 (higher‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 86 % | ||
| Patients on antiplatelet agents | |||||
| Eriksson et al., 2012[14] | Arm 1 (rivaroxaban), 563 | NR | 20 (3.6%), rate ratio for use vs nonuse: 1.32 (95% CI: 0.85‐2.05) | NR | |
| Arm 2 (enoxaparin/placebo), 526 | NR | 17 (3.2%), rate ratio for use vs nonuse: 1.40 (95% CI: 0.87‐2.25) | NR | ||
| Friedman et al., 2012[15] | Arm 2 (150 mg dabigatran, no ASA), 1149 | NR | 11 (1.0%)a | NR | |
| Arm 5 (150 mg dabigatran+ASA), 128 | NR | 2 (1.6%)a | NR | ||
| Arm 3 (enoxaparin, no ASA), 1167 | NR | 14 (1.2%)a | NR | ||
| Arm 6 (enoxaparin+ASA), 132 | NR | 4 (3.0%) | NR | ||
| 150 mg dabigatran compared with enoxaparinNo concomitant ASA therapy | NR | RR: 0.82 (95% CI: 0.37‐1.84) | NR | ||
| 150 mg dabigatran compared with enoxaparinWith concomitant ASA therapy | NR | RR: 0.55 (95% CI: 0.11‐2.78) | NR | ||
| Patients with renal insufficiency | |||||
| Bauersachs et al., 2011[16] | Arm 2 (GFR <30), 92 | Total DVT: 11.11%; Total PE: 0% | Major bleeding: 4/92 (4.35%), minor bleeding: 9/92 (9.78%) | Mortality: 5.81% | |
| Mah et al., 2007[17] | Arm 2 (tinzaparin), 27 | NR | Major bleeding: 2/27 (7.4%), minor bleeding: 3/27 (11.1%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.05 | |
| Arm 3 (enoxaparin), 28 | NR | Major bleeding: 1/28 (3.6%), minor bleeding: 3/28 (10.7%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.22 | ||
| Dahl et al., 2012[18] | Arm 1 (enoxaparin), 332 | Major VTE: 8 (9.0%) | Major bleeding: 6 (4.7%) | Infections and infestations: 25 (7.5%), Wound infection: 4 (1.2%) | |
| Arm 2 (dabigatran), 300 | Major VTE: 3 (4.3%) | Major bleeding: 0 (0%) | Infections and infestations: 21 (7.0%), Wound Infection: 3 (1.0%) | ||
| Shorr et al., 2012[19] | Arm 1 (enoxaparin, CrCL 60 mL/min), 353 | Total VTE: 17/275 (6.2%) | Major bleeding: 0/351 (0%) | NR | |
| Arm 2 (desirudin, CrCL 60 mL/min), 353 | Total VTE: 13/284 (4.3%) | Major bleeding: 2/349 (0.27%) | NR | ||
| Arm 3 (enoxaparin, CrCL 4559 mL/min), 369 | Total VTE: 18/282 (6.2%) | Major bleeding: 1/365 (0.27%) | NR | ||
| Arm 4 (desirudin, CrCL 4559 mL/min), 395 | Total VTE: 17/303 (5.6%) | Major bleeding: 1/393 (0.25%) | NR | ||
| Arm 5 (enoxaparin, CrCL <45 mL/min), 298 | Total VTE: 24/216 (11.1%) | Major bleeding: 1/294 (0.34%) | NR | ||
| Arm 6 (desirudin, CrCL <45 mL/min), 279 | Total VTE: 7/205 (3.4%) | Major bleeding: 5/275 (1.82%) | NR | ||
| Elsaid et al., 2012[20] | Arm 1 (enoxaparin, CrCL 60 mL/min), 17 | NR | Major bleeding: 2 (11.8%) | NR | |
| Arm 2 (enoxaparin, CrCL 3059 mL/min), 86 | NR | Major bleeding: 9 (10.5%) | NR | ||
| Arm 3 (enoxaparin, CrCL 30 mL/min), 53 | NR | Major bleeding: 10 (18.9%) | NR | ||
| Arm 4 (UFH, CrCL 60 mL/min), 19 | NR | Major bleeding: 2 (10.5%) | NR | ||
| Arm 5 (UFH, CrCL 3059 mL/min), 99 | NR | Major bleeding: 3 (3%) | NR | ||
| Arm 6 (UFH, CrCL 30 mL/min), 49 | NR | Major bleeding: 2 (4.1%) | NR | ||
Obese Patients
We found 1 subgroup analysis of an RCT (total 3706 patients, 2563 nonobese and 1118 obese patients) that reported on the comparative effectiveness and safety of fixed low‐dose dalteparin 5000 IU/day compared to placebo among 1118 hospitalized medically ill patients with body mass indices (BMI) greater than 30 kg/m2.11 Neither group received additional concurrent prophylactic therapies. The 3 most prevalent medical diagnoses prompting hospitalization were congestive heart failure, respiratory failure, and infectious diseases. Compression ultrasound was performed in all patients by day 21 of hospitalization. The primary end point was the composite of VTE, fatal PE, and sudden death, and secondary end points included DVT, bleeding, and thrombocytopenia by day 21 (Table 1). In obese patients, the primary end point occurred in 2.8% (95% confidence interval [CI]: 1.34.3) of the dalteparin group and in 4.3% (95% CI: 2.56.2) of the placebo group (relative risk [RR]: 0.64; 95% CI: 0.32‐1.28). In nonobese patients, the primary end point occurred in 2.8% (95% CI: 1.8‐3.8) and 5.2% (95% CI: 3.9‐6.6) of the dalteparin and placebo groups, respectively (RR: 0.53; 95% CI: 0.34‐0.82). When weight was modeled as a continuous variable, no statistically significant interaction between weight and dalteparin efficacy was observed (P=0.97). The authors calculated the RR in predefined BMI subgroups and found that dalteparin was effective in reducing VTE in patients with BMIs up to 40, with RRs of <1.0 for all (approximate range, 0.20.8). However, a fixed dose of dalteparin 5000 IU/day was not better than placebo for individuals with BMI >40 kg/m2. There was no significant difference in mortality or major hemorrhage by day 21 between treatment and placebo groups.
Freeman and colleagues prospectively assigned 31 medically ill patients with extreme obesity (BMI >40 kg/m2) to 1 of 3 dosing regimens of enoxaparin: a fixed dose of 40 mg daily enoxaparin (control group, n=11), enoxaparin at 0.4 mg/kg (n=9), or enoxaparin at 0.5 mg/kg (n=11).[12] The average BMI of the entire cohort was 62.1 kg/m2 (range, 40.582.4). All patients had anti‐factor Xa levels drawn on the day of enrollment and daily for 3 days (Table 2). The relationship between anti‐factor Xa levels and clinical efficacy of low‐molecular weight heparin (LMWH) in VTE prophylaxis is still unclear; however, an anti‐factor Xa level of 0.2 to 0.5 IU/mL, measured 4 hours after the fourth dose of LMWH, is the target level recommended for VTE prophylaxis.[13] Patients who received weight‐based enoxaparin at 0.5mg/kg achieved target anti‐factor Xa level 86% of the time compared to 32% of the time in those receiving 0.4 mg/kg and 19% of the time for those in the fixed‐dose group (P<0.001). No clinical outcomes were reported in this study.
| Intervention | Outcome | Risk of Bias | Evidence Statement and Magnitude of Effect |
|---|---|---|---|
| |||
| Patients on antiplatelet agents | |||
| Rivaroxaban vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic rivaroxaban or enoxaparin in patients concomitantly treated with antiplatelet agents; 3.6% vs 3.25% |
| Dabigatran vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic dabigatran or enoxaparin in patients concomitantly treated with aspirin; 1.6% vs 3.0% |
| Obese patients | |||
| Dalteparin vs placebo | VTE | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing total VTE in obese patients; 2.8% vs 4.3%, RR: 0.64, 95% CI: 0.32‐1.28 |
| Dalteparin vs placebo | Mortality | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing mortality in obese patients; 9.9% vs 8.6%, P=0.36 |
| Dalteparin vs placebo | Major bleeding | Moderate | Insufficient evidence for safety of dalteparin vs placebo in reducing major bleeding in obese patients; 0% vs 0.7%, P>0.99 |
| Enoxaparin 40 mg daily vs 0.4 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.4 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 32%, P=NR |
| Enoxaparin 40 mg daily vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 86%, P<0.001 |
| Enoxaparin 0.4 mg/kg vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 0.4 mg/kg versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 32% vs 86%, P=NR |
| Patients with renal insufficiency | |||
| Tinzaparin vs enoxaparin | VTE | High | Insufficient evidence about superiority of either drug for preventing VTE in patients with renal insufficiency, 0/27 vs 0/28* |
| Tinzaparin vs enoxaparin | Bleeding | High | Insufficient evidence about safety of either drug in patients with renal insufficiency; 5/27 vs 4/28, P=0.67 |
| Dabigatran vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of dabigatran in reducing VTE in severe renal compromise patients vs enoxaparin; 4.3% vs 9%, OR: 0.48, 95% CI: 0.13‐1.73, P=0.271 |
| Dabigatran vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of dabigatran vs enoxaparin in patients with renal impairment; 0 vs 4.7%, P=0.039 |
| Desirudin vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of desirudin vs enoxaparin in reducing VTE in patients with renal impairment; 4.9% vs 7.6%, P=0.019 |
| Desirudin vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of desirudin vs enoxaparin in patients with renal impairment; 0.8% vs 0.2%, P=0.109 |
| Enoxaparin vs UFH | Bleeding | High | Insufficient evidence for increased risk of bleeding with enoxaparin vs unfractionated heparin in patients with all levels of renal impairment, 13.5% vs 4.2%, RR: 3.2, 95% CI: 1.47.3; and for the subgroup of patients with creatinine clearance <30 mL/min; 18.9% vs 4.1%, RR: 4.68, 95% CI: 1.120.6 |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | VTE | Moderate | Insufficient evidence regarding differential benefit of unfractionated heparin by renal function; 2.6% of patients had a VTE event |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | Bleeding | Moderate | Insufficient evidence for differential harm from unfractionated heparin by renal function; 13 events in 92 patients |
Patients on Antiplatelet Drugs
We did not find studies that directly looked at the comparative effectiveness of VTE prophylaxis in patients who were on antiplatelet drugs including aspirin. However, there were 2 studies that looked at the risk of bleeding in patients who received VTE pharmacologic prophylaxis while concurrently taking antiplatelet agents including aspirin. Both studies used pooled data from large phase III trials.
The study by Eriksson et al. used data from the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) trial where over 12,000 patients undergoing elective total knee or hip replacement were randomized to receive VTE prophylaxis with oral rivaroxaban or subcutaneous enoxaparin.[14] Nine percent of participants in each arm (563 in rivaroxaban and 526 in enoxaparin/placebo) were concomitantly using antiplatelet agents or aspirin at least once during the at risk period, defined as starting at day 1 of surgery up to 2 days after the last intake of the study drug. The only end point evaluated was bleeding, and the authors found no statistically significant bleeding difference among the 2 arms (Table 1). Any bleeding event in the rivaroxaban with antiplatelets or aspirin arm was found in 20 (3.6%) patients, whereas in those on enoxaparin/placebo with antiplatelets or aspirin arm it was 17 (3.2%). The relative rate of bleeding among users versus nonusers of antiplatelet drugs or aspirin was 1.32 (95% CI: 0.85‐2.05) in the rivaroxaban group and 1.40 (95% CI: 0.87‐2.25) in the enoxaparin arm (Table 1).
Friedman et al. used pooled data from the RE‐MODEL, RENOVATE, and REMOBILIZE trials, where patients who were undergoing hip or knee arthroplasty were randomized to 220 mg of dabigatran once daily, 150 mg of dabigatran once daily (we focused on this lower dosage as this is the only available dose used in the US), 40 mg of enoxaparin once daily, or 30 mg of enoxaparin twice a day.[15] Of the 8135 patients, 4.7% were on concomitant aspirin. The baseline characteristics of those on aspirin were similar to the other enrollees. The primary outcome was major bleeding events requiring transfusion, symptomatic internal bleeding, or bleeding requiring surgery. Among patients receiving 150 mg of dabigatran, bleeding events with and without concomitant aspirin occurred in 1.6% and 1.0%, respectively (odds ratio [OR]: 1.64; 95% CI: 0.36‐7.49; P=0.523). The percentages of participants with bleeding who received enoxaparin, with and without aspirin, were 3.0% and 1.2%, respectively (OR: 2.57; 95% CI: 0.83‐7.94; P=0.101). The RR of bleeding on dabigatran compared to enoxaparin with and without aspirin therapy was 0.55 (95% CI: 0.11‐2.78) and 0.82 (95% CI: 0.37‐1.84), respectively (Table 1).
Patients With Renal Insufficiency
We found 5 studies that evaluated the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE in patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, or patients receiving dialysis. Four studies were RCTs,[16, 17, 18, 19] and 1 used a cohort design assessing separate cohorts before and after a quality improvement intervention.[20] Bauersachs and colleagues conducted an RCT comparing unfractionated heparin at 5000 IU, 3 times daily to certoparin, which is not approved in the United States and is not further discussed here.[16] The rate of DVT among patients treated with unfractionated heparin in patients with a glomerular filtration rate >30 mL/min was marginally lower than those with severe renal dysfunction (10.3 vs 11.1%) (Table 1).
Patients with severe renal dysfunction who received 5000 IU of unfractionated heparin 3 times a day were at increased risk of all bleeds (RR: 3.4; 95% CI: 2.05.9), major bleeds (RR: 7.3; 95% CI: 3.316), and minor bleeds (RR: 2.6; 95% CI: 1.4‐4.9) compared to patients treated with unfractionated heparin without severe renal dysfunction.[16]
A randomized trial by Mah and colleagues compared drug accumulation and anti‐Xa activity in elderly patients with renal dysfunction (defined as a glomerular filtration rate of 20 to 50 mL/min) who received either tinzaparin at 4500 IU once daily or enoxaparin at 4000 IU once daily.[17] Enoxaparin accumulated to a greater extent from day 1 to day 8 than did tinzaparin; the ratio of maximum concentration on day 8 compared to day 1 was 1.22 for enoxaparin and 1.05 for tinzaparin (P=0.016). No VTE events were reported in patients who received tinzaparin or enoxaparin. There was no statistical difference in the incidence of bleeding events between patients receiving tinzaparin (5, including 2 major events) and enoxaparin (4, including 3 major events, P=0.67) (Table 1).
The trial by Dahl and colleagues randomly assigned patients who were over 75 years of age and/or who had moderate renal dysfunction (defined as creatinine clearance between 30 and 49 mL/min) to receive enoxaparin 40 mg daily or dabigatran 150 mg daily.[18] There was no significant difference in the rate of major VTE events between patients receiving dabigatran (4.3%) and enoxaparin (9%) (OR: 0.48; 95% CI: 0.13‐1.73; P=0.271) (Table 1). The rate of major bleeding was significantly higher among patients randomly assigned to receive enoxaparin (4.7%) versus dabigatran (0%) (P=0.039).[18]
Shorr and colleagues published a post hoc subgroup analysis of a multicenter trial in which orthopedic patients were randomly assigned to receive desirudin 15 mg twice daily or enoxaparin 40 mg once daily.[19] Evaluable patients (1565 of the 2079 patients randomized in the trial) receiving desirudin experienced a significantly lower rate of major VTE compared with patients receiving enoxaparin (4.9% vs 7.6%, P=0.019). This relationship was particularly pronounced for evaluable patients whose creatinine clearance was between 30 and 44 mL/min. In evaluable patients with this degree of renal dysfunction, 11% of patients taking enoxaparin compared to 3.4% of those taking desirudin had a major VTE (OR: 3.52; 95% CI: 1.48‐8.4; P=0.004). There was no significant difference in the rates of major bleeding among a subset of patients assessed for safety outcomes (2078 of the 2079 patients randomized in the trial) who received desirudin (0.8%) or enoxaparin (0.2%) (Table 1).
Elsaid and Collins assessed VTE and bleeding events associated with the use of unfractionated heparin 5000U either 2 or 3 times daily and enoxaparin 30 mg once or twice daily across patients stratified by renal function (creatinine clearance <30, 3059, and 60 mL/min). The investigators made assessments before and after a quality improvement intervention that was designed to eliminate the use of enoxaparin in patients whose creatinine clearance was <30 mL/min. No VTE events were reported. Patients receiving enoxaparin were significantly more likely to experience a major bleeding episode compared with patients receiving unfractionated heparin (overall rates for all levels of renal function: 13.5% vs 4. 2%; RR: 3.2; 95% CI: 1.47.3) (Table 2). This association was largely driven by the subgroup of patients with a creatinine clearance <30 mL/min. For this subgroup with severe renal insufficiency, patients receiving enoxaparin were significantly more likely to have a bleed compared with patients receiving unfractionated heparin (18.9% vs 4.1%; RR: 4.68; 95% CI: 1.120.6) (Tables 1 and 2). There was no difference in the bleeding rates for patients whose creatinine clearances were >60 mL/min.[20]
Strength of Evidence
Obese Patients
Overall, we found that the strength of evidence was insufficient regarding the composite end point of DVT, PE, and sudden death, and the outcomes of mortality and bleeding (Table 2). This was based on a paucity of available data, and a moderate risk of bias in the reviewed studies. Additionally, 92% of the enrolled patients in the studies were white, limiting the generalizability of the results to other ethnic groups.
Patients on Antiplatelets
The strength of evidence was insufficient in the studies reviewed here to conclude that there is no difference in rates of bleeding in patients who are concomitantly taking antiplatelet drugs while getting VTE prophylaxis with rivaroxaban, dabigatran, or enoxaparin. We based this rating because of the imprecision of results and unknown consistencies across multiple studies.
Patients With Renal Insufficiency
One RCT had a high risk of bias for our key question because data from only 1 study arm were useful for our review.[16] The other RCTs were judged to have a moderate risk of bias. The analyses led by Dahl and Shorr[18, 19] were based on post hoc (ie, not prespecified) analysis of data from RCTs. Additionally, outcomes in the Shorr et al. trial were reported for evaluable subpopulations of the cohort that was initially randomized in the clinical trial.
We rated the strength of evidence as insufficient to know the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE during hospitalization of patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, and patients receiving dialysis. We based this rating on the risk of bias associated with published studies and a lack of consistent evidence regarding associations that were reported. Similarly, we rated the strength of evidence as insufficient that 5000 U of unfractionated heparin 3 times daily increases the risk of major and minor bleeding events in patients with severely compromised renal function compared to this dose in patients without severely compromised renal function. We based this rating on a high risk of bias of included studies and inconsistent evidence. Likewise, we rated the strength of evidence as insufficient that enoxaparin significantly increases the risk of major bleeding compared with unfractionated heparin in patients with severe renal insufficiency. We based this rating on a high risk of bias and inconsistent published evidence.
We similarly found insufficient evidence to guide treatment decisions for patients with renal insufficiency. Our findings are consistent with other recent reviews. The American College of Chest Physicians (ACCP) practice guidelines[21] make dosing recommendations for the therapeutic use of enoxaparin. However, their assessment is that the data are insufficient to make direct recommendations about prophylaxis. Their assessment of the indirect evidence regarding bioaccumulation and increased anti‐factor Xa levels are consistent with ours. The ACCP guidelines also suggest that decreased clearance of enoxaparin has been associated with increased risk of bleeding events for patients with severe renal insufficiency. However, the cited study[20] compares patients with and without severe renal dysfunction who received the same therapy. Therefore, it is not possible to determine the additional risk conveyed by enoxaparin therapy, that is, above the baseline increased risk of bleeding among patients with renal insufficiency, particularly those receiving an alternate pharmacologic VTE prevention strategy, such as unfractionated heparin.
DISCUSSION
We found that the evidence was very limited about prevention of VTE in these select and yet prevalent patient populations. Despite the fact that there is an increasing number of obese patients and patients who are on antiplatelet therapies, most clinical practice guidelines do not address the care of these populations, which may be entirely appropriate given the state of the evidence.
The ACCP practice guidelines[21] suggest using a higher dose of enoxaparin for the prevention of VTE in obese patients. The subgroup analysis by Kucher et al.[11] showed effect attenuation of dalteparin when given at a fixed dose of 5000 IU/mL to patients with a BMI of >40 kg/m2. The Freeman study[12] showed that extremely obese patients (average BMI >62.1 kg/m2) who are given a fixed dose of enoxaparin achieved target anti‐factor Xa levels significantly less often than those who received a higher dose of enoxaparin. The 2 separate findings, although not conclusive, lend some credence to the current ACCP guidelines.[21]
The studies we reviewed on VTE prophylaxis in patients who are concomitantly on antiplatelets including aspirin reported no major increased risk of bleeding; however, in the Friedman et al. study,[15] 3.0% of patients who were put on enoxaparin while still on aspirin had a bleeding event compared to 1.2% of those on enoxaparin alone. This difference is not statistically significant but is a trend possibly worth noting, especially when one looks at the lower RR of bleeding at 0.55 compared to 0.82 when dabigatran is compared with enoxaparin with and without concomitant aspirin therapy, respectively (Table 1). The highest dose of aspirin used in either of the studies was 160 mg/day, and neither study addressed other potent antiplatelets such as clopidogrel or ticlopidine separately, which limits the generalizability of the finding to all antiplatelets. Current ACCP guidelines do not recommend aspirin as a sole option for the prevention of VTE in orthopedic surgery patients.[22] Concerns remain among clinicians that antiplatelets, including aspirin, on their own are unlikely to be fully effective to thwart venous thrombotic processes for most patients, and yet the risk of bleeding is not fully known when these agents are combined with other anticoagulants for VTE prophylaxis.
Our review has several limitations, including the possibility that we may have missed some observational studies, as the identification of relevant observational studies in electronic searches is more challenging than that of RCTs. The few studies made it impossible to quantitatively pool results. These results, however, have important implications, namely that additional research on the comparative effectiveness and safety of pharmacologic and mechanical strategies to prevent VTE is needed for the optimal care of these patient subgroups. This might be achieved with trials dedicated to enrolling these patients or prespecified subgroup analyses within larger trials. Observational data may be appropriate as long as attention is paid to confounding.
APPENDIX
MEDLINE Search Strategy
((pulmonary embolism[mh] OR PE[tiab] OR Pulmonary embolism[tiab] OR thromboembolism[mh] OR thromboembolism[tiab] OR thromboembolisms[tiab] OR Thrombosis[mh] OR thrombosis[tiab] OR DVT[tiab] OR VTE[tiab] OR clot[tiab]) AND (Anticoagulants[mh] OR Anticoagulants[tiab] OR Anticoagulant[tiab] OR thrombin inhibitors[tiab] OR Aspirin[mh] or aspirin[tiab] OR aspirins[tiab] or clopidogrel[nm] OR clopidogrel[tiab] OR Plavix[tiab] or ticlopidine[mh] or ticlopidine[tiab]OR ticlid[tiab] OR prasugrel[nm]Or prasugrel[tiab]OR effient[tiab]OR ticagrelor[NM] OR ticagrelor[tiab]OR Brilinta[tiab] OR cilostazol[NM] OR cilostazol[tiab]OR pletal[tiab] OR warfarin[mh]OR warfarin[tiab]OR coumadin[tiab] OR coumadine[tiab] OR Dipyridamole[mh]OR dipyridamole[tiab]OR persantine[tiab] OR dicoumarol[MH] OR dicoumarol[tiab] OR dicumarol[tiab] OR Dextran sulfate[mh] OR dextran sulfate[tiab] ORthrombin inhibitors[tiab] OR thrombin inhibitor[tiab] OR heparin[mh] OR Heparin[tiab] OR Heparins[tiab] OR LMWH[tiab] OR LDUH[tiab] OR Enoxaparin[mh] OR Enoxaparin[tiab] OR Lovenox[tiab] OR Dalteparin[tiab] OR Fragmin[tiab] OR Tinzaparin[tiab] OR innohep[tiab] OR Nadroparin[tiab] OR Fondaparinux[nm] OR Fondaparinux[tiab] OR Arixtra[tiab] OR Idraparinux[nm] OR Idraparinux[tiab] OR Rivaroxaban[nm] OR Rivaroxaban[tiab] OR novastan[tiab] OR Desirudin[nm] OR Desirudin[tiab] OR Iprivask[tiab]OR direct thrombin inhibitor[tiab] OR Argatroban[nm] OR Argatroban[tiab] OR Acova[tiab] OR Bivalirudin[nm] OR Bivalirudin[tiab] OR Angiomax[tiab] OR Lepirudin[nm] OR Lepirudin[tiab] OR Refludan[tiab] OR Dabigatran[nm] OR Dabigatran[tiab] OR Pradaxa[tiab] OR factor xa[mh] OR factor Xa[tiab] OR vena cava filters[mh] OR filters[tiab] OR filter[tiab] OR compression stockings[mh] OR intermittent pneumatic compression devices[mh] OR compression [tiab] OR Venous foot pump[tiab])) AND(prevent*[tiab] OR prophyla*[tiab] OR prevention and control[subheading]) NOT (animals[mh] NOT humans[mh]) NOT (editorial[pt] OR comment[pt]) NOT ((infant[mh] OR infant[tiab] OR child[mh] OR child[tiab] OR children[tiab] OR adolescent[mh] OR adolescent[tiab] OR teen‐age[tiab] OR pediatric[tiab] OR perinatal[tiab]) NOT (adult[tiab] OR adults[tiab] OR adult[mh])) NOT (mechanical valve[tiab] OR heart valve[tiab] OR atrial fibrillation[mh] OR atrial fibrillation[tiab] OR thrombophilia[mh] OR thrombophilia[tiab] OR pregnancy[mh])
- , , . Estimated annual number of incident and recurrent, non‐fatal and fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106:910.
- Institute of Medicine. Institute of Medicine. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: National Academies Press; 2009.
- Lovenox (enoxaparin sodium injection for subcutaneous and intravenous use: prescribing information). Bridgewater, NJ: SanofiAventis; 2011. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed October 17, 2012.
- Innohep (tinzaparin sodium injection). Ballerup, Denmark: LEO Pharmaceutical Products; 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed October 17, 2012.
- Leizorovicz A. Tinzaparin compared to unfractionated heparin for initial treatment of deep vein thrombosis in very elderly patients with renal insufficiency‐ the IRIS trial. [50th ASH Annual Meeting and Exposition abstract 434]. Blood. 2008;11:112.
- Fragmin (dalteparin sodium injection). New York, NY: Pfizer Inc.; 2007. Available at: http://www.pfizer.com/files/products/uspi_fragmin.pdf. Accessed October 17, 2012.
- Methods guide for effectiveness and comparative effectiveness reviews. Rockville, MD: Agency for Healthcare Research and Quality; August 2011. AHRQ publication No. 10 (11)‐EHC063‐EF. Available at: http://www.effectivehealthcare.ahrq.gov. Accessed October 17, 2012.
- Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/928/VTE‐Special‐Populations_Protocol_20120112.pdf. Accessed April 17, 2012.
- , , , et al. Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Evidence Report/Technology Assessment (AHRQ). Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/1501/venous‐thromboembolism‐special‐populations‐report‐130529.pdf. 2013.
- , . The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non‐randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377–384.
- , , , et al. Efficacy and safety of fixed low‐dose dalteparin in preventing venous thromboembolism among obese or elderly hospitalized patients: a subgroup analysis of the PREVENT trial. Arch Intern Med. 2005;165(3):341–345.
- , , , . Prospective comparison of three enoxaparin dosing regimens to achieve target anti‐factor Xa levels in hospitalized, medically ill patients with extreme obesity. Am J Hematol. 2012;87(7):740–743.
- , , . Effect of prophylactic dalteparin on anti‐factor xa levels in morbidly obese patients after bariatric surgery. Obes Surg. 2010;20(4):487–491.
- , , , , . Concomitant use of medication with antiplatelet effects in patients receiving either rivaroxaban or enoxaparin after total hip or knee arthroplasty. Thromb Res. 2012;130(2):147–151.
- , , , , , . Dabigatran etexilate and concomitant use of non‐steroidal anti‐inflammatory drugs or acetylsalicylic acid in patients undergoing total hip and total knee arthroplasty: No increased risk of bleeding. Thromb Haemost. 2012;108(1):183–190.
- , , , et al. CERTIFY: prophylaxis of venous thromboembolism in patients with severe renal insufficiency. Thromb Haemost. 2011;105(6):981–988.
- , , , et al. Tinzaparin and enoxaparin given at prophylactic dose for eight days in medical elderly patients with impaired renal function: a comparative pharmacokinetic study. Thromb Haemost. 2007;97(4):581–586.
- , , , , , . Thromboprophylaxis in patients older than 75 years or with moderate renal impairment undergoing knee or hip replacement surgery [published correction appears in Int Orthop. 2012;36(5):1113]. Int Orthop. 2012;36(4):741–748.
- , , , . Impact of stage 3B chronic kidney disease on thrombosis and bleeding outcomes after orthopedic surgery in patients treated with desirudin or enoxaparin: insights from a randomized trial. J Thromb Haemost. 2012;10(8):1515–1520.
- , . Initiative to improve thromboprophylactic enoxaparin exposure in hospitalized patients with renal impairment. Am J Health Syst Pharm. 2012;69(5):390–396.
- , , , , ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):7S–47S.
- , . Aspirin for the prophylaxis of venous thromboembolic events in orthopedic surgery patients: a comparison of the AAOS and ACCP guidelines with review of the evidence. Ann Pharmacother. 2013;47(1):63–74.
Venous thromboembolism (VTE), including deep venous thrombosis (DVT) and pulmonary embolism (PE), is estimated to affect 900,000 Americans each year and is a cause of significant morbidity and mortality with associated high healthcare costs.[1] Accordingly, the comparative effectiveness and safety of interventions for the prevention and treatment of VTE are among the national priorities for comparative effectiveness research.[2] Whereas we have evidence‐based guidelines for the prophylaxis of VTE in the general population, there are no guidelines informing the care of select patient populations. Select populations are those patients in whom there is decisional uncertainty about the optimal choice, timing, and dose of VTE prophylaxis. Not only do these patients have an increased risk of DVT and PE, but most are also at high risk of bleeding, the most important complication of VTE prophylaxis.[3, 4, 5, 6]
The objectives of this systematic review were to define the comparative effectiveness and safety of pharmacologic and mechanical strategies for VTE prevention in some of these select medical populations including obese patients, patients on concomitant antiplatelet therapy, patients with renal insufficiency, patients who are underweight, and patients with coagulopathy due to liver disease.
METHODS
The methods for this comparative effectiveness review (CER) follow the guidelines suggested in the Agency for Healthcare Research and Quality (AHRQ) Methods Guide for Effectiveness and Comparative Effectiveness Reviews.[7] The protocol was publically posted.[8]
Search Strategy
We searched MEDLINE, EMBASE, and SCOPUS through August 2011, CINAHL, International Pharmaceutical Abstracts,
Study Selection
We reviewed titles followed by abstracts to identify randomized controlled trials (RCTs) or observational studies with comparison groups reporting on the effectiveness or safety of VTE prevention in our populations. Two investigators independently reviewed abstracts, and we excluded the abstracts if both investigators agreed that the article met 1 or more of the exclusion criteria. We included only English‐language articles that evaluated the effectiveness of pharmacological or mechanical interventions that have been approved for clinical use in the United States. To be eligible, the studies must have addressed relevant key questions in the population of our interest. We resolved disagreements by consensus. We used DistillerSR (Evidence Partners Inc., Ottawa, Ontario, Canada), a Web‐based database management program to manage the review process. Two investigators assessed the risk of bias in each study independently, using the Downs and Black instrument for observational studies and trials.[10]
Data Synthesis
For each select population, we created detailed evidence tables containing the information abstracted from the eligible studies. After synthesizing the evidence, we graded the quantity, quality, and consistency of the best available evidence for each select population by adapting an evidence‐grading scheme recommended in the Methods Guide for Conducting Comparative Effectiveness Reviews.[7]
RESULTS
We identified 30,902 unique citations and included 9 studies (Figure 1). There were 5 RCTs with relevant subgroups and 4 observational studies (Table 1). Two studies reported on the risk of bleeding in patients given pharmacologic prophylaxis while they are concomitantly taking nonsteroidal anti‐inflammatory drugs (NSAIDS) or antiplatelet agents/aspirin, 1 RCT and 1 prospective observational study reported on obese patients, and 5 studies described outcomes of patients with renal insufficiency (see Supporting Information, Table 1, in the online version of this article). No study tested prophylaxis in underweight patients or those with liver disease.
| Study | Arm, n | Total VTE (DVT and PE) | Bleeding | Other Outcomes | |
|---|---|---|---|---|---|
| |||||
| Obese patients | |||||
| Kucher et al., 2005[11] | Arm 1 (dalteparin), 558 | 2.8% (95% CI: 1.34.3) | 0% | Mortality at 21 days: 4.6% | |
| Arm 2 (placebo), 560 | 4.3% (95% CI: 2.56.2) | 0.7% | Mortality at 21 days: 2.7% | ||
| Freeman et al., [12] | Arm 1 (fixed‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 19 % | |
| Arm 2 (lower‐dose enoxaparin), 9 | NR | NR | Peak anti‐factor Xa level 32 % | ||
| Arm 3 (higher‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 86 % | ||
| Patients on antiplatelet agents | |||||
| Eriksson et al., 2012[14] | Arm 1 (rivaroxaban), 563 | NR | 20 (3.6%), rate ratio for use vs nonuse: 1.32 (95% CI: 0.85‐2.05) | NR | |
| Arm 2 (enoxaparin/placebo), 526 | NR | 17 (3.2%), rate ratio for use vs nonuse: 1.40 (95% CI: 0.87‐2.25) | NR | ||
| Friedman et al., 2012[15] | Arm 2 (150 mg dabigatran, no ASA), 1149 | NR | 11 (1.0%)a | NR | |
| Arm 5 (150 mg dabigatran+ASA), 128 | NR | 2 (1.6%)a | NR | ||
| Arm 3 (enoxaparin, no ASA), 1167 | NR | 14 (1.2%)a | NR | ||
| Arm 6 (enoxaparin+ASA), 132 | NR | 4 (3.0%) | NR | ||
| 150 mg dabigatran compared with enoxaparinNo concomitant ASA therapy | NR | RR: 0.82 (95% CI: 0.37‐1.84) | NR | ||
| 150 mg dabigatran compared with enoxaparinWith concomitant ASA therapy | NR | RR: 0.55 (95% CI: 0.11‐2.78) | NR | ||
| Patients with renal insufficiency | |||||
| Bauersachs et al., 2011[16] | Arm 2 (GFR <30), 92 | Total DVT: 11.11%; Total PE: 0% | Major bleeding: 4/92 (4.35%), minor bleeding: 9/92 (9.78%) | Mortality: 5.81% | |
| Mah et al., 2007[17] | Arm 2 (tinzaparin), 27 | NR | Major bleeding: 2/27 (7.4%), minor bleeding: 3/27 (11.1%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.05 | |
| Arm 3 (enoxaparin), 28 | NR | Major bleeding: 1/28 (3.6%), minor bleeding: 3/28 (10.7%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.22 | ||
| Dahl et al., 2012[18] | Arm 1 (enoxaparin), 332 | Major VTE: 8 (9.0%) | Major bleeding: 6 (4.7%) | Infections and infestations: 25 (7.5%), Wound infection: 4 (1.2%) | |
| Arm 2 (dabigatran), 300 | Major VTE: 3 (4.3%) | Major bleeding: 0 (0%) | Infections and infestations: 21 (7.0%), Wound Infection: 3 (1.0%) | ||
| Shorr et al., 2012[19] | Arm 1 (enoxaparin, CrCL 60 mL/min), 353 | Total VTE: 17/275 (6.2%) | Major bleeding: 0/351 (0%) | NR | |
| Arm 2 (desirudin, CrCL 60 mL/min), 353 | Total VTE: 13/284 (4.3%) | Major bleeding: 2/349 (0.27%) | NR | ||
| Arm 3 (enoxaparin, CrCL 4559 mL/min), 369 | Total VTE: 18/282 (6.2%) | Major bleeding: 1/365 (0.27%) | NR | ||
| Arm 4 (desirudin, CrCL 4559 mL/min), 395 | Total VTE: 17/303 (5.6%) | Major bleeding: 1/393 (0.25%) | NR | ||
| Arm 5 (enoxaparin, CrCL <45 mL/min), 298 | Total VTE: 24/216 (11.1%) | Major bleeding: 1/294 (0.34%) | NR | ||
| Arm 6 (desirudin, CrCL <45 mL/min), 279 | Total VTE: 7/205 (3.4%) | Major bleeding: 5/275 (1.82%) | NR | ||
| Elsaid et al., 2012[20] | Arm 1 (enoxaparin, CrCL 60 mL/min), 17 | NR | Major bleeding: 2 (11.8%) | NR | |
| Arm 2 (enoxaparin, CrCL 3059 mL/min), 86 | NR | Major bleeding: 9 (10.5%) | NR | ||
| Arm 3 (enoxaparin, CrCL 30 mL/min), 53 | NR | Major bleeding: 10 (18.9%) | NR | ||
| Arm 4 (UFH, CrCL 60 mL/min), 19 | NR | Major bleeding: 2 (10.5%) | NR | ||
| Arm 5 (UFH, CrCL 3059 mL/min), 99 | NR | Major bleeding: 3 (3%) | NR | ||
| Arm 6 (UFH, CrCL 30 mL/min), 49 | NR | Major bleeding: 2 (4.1%) | NR | ||
Obese Patients
We found 1 subgroup analysis of an RCT (total 3706 patients, 2563 nonobese and 1118 obese patients) that reported on the comparative effectiveness and safety of fixed low‐dose dalteparin 5000 IU/day compared to placebo among 1118 hospitalized medically ill patients with body mass indices (BMI) greater than 30 kg/m2.11 Neither group received additional concurrent prophylactic therapies. The 3 most prevalent medical diagnoses prompting hospitalization were congestive heart failure, respiratory failure, and infectious diseases. Compression ultrasound was performed in all patients by day 21 of hospitalization. The primary end point was the composite of VTE, fatal PE, and sudden death, and secondary end points included DVT, bleeding, and thrombocytopenia by day 21 (Table 1). In obese patients, the primary end point occurred in 2.8% (95% confidence interval [CI]: 1.34.3) of the dalteparin group and in 4.3% (95% CI: 2.56.2) of the placebo group (relative risk [RR]: 0.64; 95% CI: 0.32‐1.28). In nonobese patients, the primary end point occurred in 2.8% (95% CI: 1.8‐3.8) and 5.2% (95% CI: 3.9‐6.6) of the dalteparin and placebo groups, respectively (RR: 0.53; 95% CI: 0.34‐0.82). When weight was modeled as a continuous variable, no statistically significant interaction between weight and dalteparin efficacy was observed (P=0.97). The authors calculated the RR in predefined BMI subgroups and found that dalteparin was effective in reducing VTE in patients with BMIs up to 40, with RRs of <1.0 for all (approximate range, 0.20.8). However, a fixed dose of dalteparin 5000 IU/day was not better than placebo for individuals with BMI >40 kg/m2. There was no significant difference in mortality or major hemorrhage by day 21 between treatment and placebo groups.
Freeman and colleagues prospectively assigned 31 medically ill patients with extreme obesity (BMI >40 kg/m2) to 1 of 3 dosing regimens of enoxaparin: a fixed dose of 40 mg daily enoxaparin (control group, n=11), enoxaparin at 0.4 mg/kg (n=9), or enoxaparin at 0.5 mg/kg (n=11).[12] The average BMI of the entire cohort was 62.1 kg/m2 (range, 40.582.4). All patients had anti‐factor Xa levels drawn on the day of enrollment and daily for 3 days (Table 2). The relationship between anti‐factor Xa levels and clinical efficacy of low‐molecular weight heparin (LMWH) in VTE prophylaxis is still unclear; however, an anti‐factor Xa level of 0.2 to 0.5 IU/mL, measured 4 hours after the fourth dose of LMWH, is the target level recommended for VTE prophylaxis.[13] Patients who received weight‐based enoxaparin at 0.5mg/kg achieved target anti‐factor Xa level 86% of the time compared to 32% of the time in those receiving 0.4 mg/kg and 19% of the time for those in the fixed‐dose group (P<0.001). No clinical outcomes were reported in this study.
| Intervention | Outcome | Risk of Bias | Evidence Statement and Magnitude of Effect |
|---|---|---|---|
| |||
| Patients on antiplatelet agents | |||
| Rivaroxaban vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic rivaroxaban or enoxaparin in patients concomitantly treated with antiplatelet agents; 3.6% vs 3.25% |
| Dabigatran vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic dabigatran or enoxaparin in patients concomitantly treated with aspirin; 1.6% vs 3.0% |
| Obese patients | |||
| Dalteparin vs placebo | VTE | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing total VTE in obese patients; 2.8% vs 4.3%, RR: 0.64, 95% CI: 0.32‐1.28 |
| Dalteparin vs placebo | Mortality | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing mortality in obese patients; 9.9% vs 8.6%, P=0.36 |
| Dalteparin vs placebo | Major bleeding | Moderate | Insufficient evidence for safety of dalteparin vs placebo in reducing major bleeding in obese patients; 0% vs 0.7%, P>0.99 |
| Enoxaparin 40 mg daily vs 0.4 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.4 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 32%, P=NR |
| Enoxaparin 40 mg daily vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 86%, P<0.001 |
| Enoxaparin 0.4 mg/kg vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 0.4 mg/kg versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 32% vs 86%, P=NR |
| Patients with renal insufficiency | |||
| Tinzaparin vs enoxaparin | VTE | High | Insufficient evidence about superiority of either drug for preventing VTE in patients with renal insufficiency, 0/27 vs 0/28* |
| Tinzaparin vs enoxaparin | Bleeding | High | Insufficient evidence about safety of either drug in patients with renal insufficiency; 5/27 vs 4/28, P=0.67 |
| Dabigatran vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of dabigatran in reducing VTE in severe renal compromise patients vs enoxaparin; 4.3% vs 9%, OR: 0.48, 95% CI: 0.13‐1.73, P=0.271 |
| Dabigatran vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of dabigatran vs enoxaparin in patients with renal impairment; 0 vs 4.7%, P=0.039 |
| Desirudin vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of desirudin vs enoxaparin in reducing VTE in patients with renal impairment; 4.9% vs 7.6%, P=0.019 |
| Desirudin vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of desirudin vs enoxaparin in patients with renal impairment; 0.8% vs 0.2%, P=0.109 |
| Enoxaparin vs UFH | Bleeding | High | Insufficient evidence for increased risk of bleeding with enoxaparin vs unfractionated heparin in patients with all levels of renal impairment, 13.5% vs 4.2%, RR: 3.2, 95% CI: 1.47.3; and for the subgroup of patients with creatinine clearance <30 mL/min; 18.9% vs 4.1%, RR: 4.68, 95% CI: 1.120.6 |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | VTE | Moderate | Insufficient evidence regarding differential benefit of unfractionated heparin by renal function; 2.6% of patients had a VTE event |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | Bleeding | Moderate | Insufficient evidence for differential harm from unfractionated heparin by renal function; 13 events in 92 patients |
Patients on Antiplatelet Drugs
We did not find studies that directly looked at the comparative effectiveness of VTE prophylaxis in patients who were on antiplatelet drugs including aspirin. However, there were 2 studies that looked at the risk of bleeding in patients who received VTE pharmacologic prophylaxis while concurrently taking antiplatelet agents including aspirin. Both studies used pooled data from large phase III trials.
The study by Eriksson et al. used data from the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) trial where over 12,000 patients undergoing elective total knee or hip replacement were randomized to receive VTE prophylaxis with oral rivaroxaban or subcutaneous enoxaparin.[14] Nine percent of participants in each arm (563 in rivaroxaban and 526 in enoxaparin/placebo) were concomitantly using antiplatelet agents or aspirin at least once during the at risk period, defined as starting at day 1 of surgery up to 2 days after the last intake of the study drug. The only end point evaluated was bleeding, and the authors found no statistically significant bleeding difference among the 2 arms (Table 1). Any bleeding event in the rivaroxaban with antiplatelets or aspirin arm was found in 20 (3.6%) patients, whereas in those on enoxaparin/placebo with antiplatelets or aspirin arm it was 17 (3.2%). The relative rate of bleeding among users versus nonusers of antiplatelet drugs or aspirin was 1.32 (95% CI: 0.85‐2.05) in the rivaroxaban group and 1.40 (95% CI: 0.87‐2.25) in the enoxaparin arm (Table 1).
Friedman et al. used pooled data from the RE‐MODEL, RENOVATE, and REMOBILIZE trials, where patients who were undergoing hip or knee arthroplasty were randomized to 220 mg of dabigatran once daily, 150 mg of dabigatran once daily (we focused on this lower dosage as this is the only available dose used in the US), 40 mg of enoxaparin once daily, or 30 mg of enoxaparin twice a day.[15] Of the 8135 patients, 4.7% were on concomitant aspirin. The baseline characteristics of those on aspirin were similar to the other enrollees. The primary outcome was major bleeding events requiring transfusion, symptomatic internal bleeding, or bleeding requiring surgery. Among patients receiving 150 mg of dabigatran, bleeding events with and without concomitant aspirin occurred in 1.6% and 1.0%, respectively (odds ratio [OR]: 1.64; 95% CI: 0.36‐7.49; P=0.523). The percentages of participants with bleeding who received enoxaparin, with and without aspirin, were 3.0% and 1.2%, respectively (OR: 2.57; 95% CI: 0.83‐7.94; P=0.101). The RR of bleeding on dabigatran compared to enoxaparin with and without aspirin therapy was 0.55 (95% CI: 0.11‐2.78) and 0.82 (95% CI: 0.37‐1.84), respectively (Table 1).
Patients With Renal Insufficiency
We found 5 studies that evaluated the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE in patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, or patients receiving dialysis. Four studies were RCTs,[16, 17, 18, 19] and 1 used a cohort design assessing separate cohorts before and after a quality improvement intervention.[20] Bauersachs and colleagues conducted an RCT comparing unfractionated heparin at 5000 IU, 3 times daily to certoparin, which is not approved in the United States and is not further discussed here.[16] The rate of DVT among patients treated with unfractionated heparin in patients with a glomerular filtration rate >30 mL/min was marginally lower than those with severe renal dysfunction (10.3 vs 11.1%) (Table 1).
Patients with severe renal dysfunction who received 5000 IU of unfractionated heparin 3 times a day were at increased risk of all bleeds (RR: 3.4; 95% CI: 2.05.9), major bleeds (RR: 7.3; 95% CI: 3.316), and minor bleeds (RR: 2.6; 95% CI: 1.4‐4.9) compared to patients treated with unfractionated heparin without severe renal dysfunction.[16]
A randomized trial by Mah and colleagues compared drug accumulation and anti‐Xa activity in elderly patients with renal dysfunction (defined as a glomerular filtration rate of 20 to 50 mL/min) who received either tinzaparin at 4500 IU once daily or enoxaparin at 4000 IU once daily.[17] Enoxaparin accumulated to a greater extent from day 1 to day 8 than did tinzaparin; the ratio of maximum concentration on day 8 compared to day 1 was 1.22 for enoxaparin and 1.05 for tinzaparin (P=0.016). No VTE events were reported in patients who received tinzaparin or enoxaparin. There was no statistical difference in the incidence of bleeding events between patients receiving tinzaparin (5, including 2 major events) and enoxaparin (4, including 3 major events, P=0.67) (Table 1).
The trial by Dahl and colleagues randomly assigned patients who were over 75 years of age and/or who had moderate renal dysfunction (defined as creatinine clearance between 30 and 49 mL/min) to receive enoxaparin 40 mg daily or dabigatran 150 mg daily.[18] There was no significant difference in the rate of major VTE events between patients receiving dabigatran (4.3%) and enoxaparin (9%) (OR: 0.48; 95% CI: 0.13‐1.73; P=0.271) (Table 1). The rate of major bleeding was significantly higher among patients randomly assigned to receive enoxaparin (4.7%) versus dabigatran (0%) (P=0.039).[18]
Shorr and colleagues published a post hoc subgroup analysis of a multicenter trial in which orthopedic patients were randomly assigned to receive desirudin 15 mg twice daily or enoxaparin 40 mg once daily.[19] Evaluable patients (1565 of the 2079 patients randomized in the trial) receiving desirudin experienced a significantly lower rate of major VTE compared with patients receiving enoxaparin (4.9% vs 7.6%, P=0.019). This relationship was particularly pronounced for evaluable patients whose creatinine clearance was between 30 and 44 mL/min. In evaluable patients with this degree of renal dysfunction, 11% of patients taking enoxaparin compared to 3.4% of those taking desirudin had a major VTE (OR: 3.52; 95% CI: 1.48‐8.4; P=0.004). There was no significant difference in the rates of major bleeding among a subset of patients assessed for safety outcomes (2078 of the 2079 patients randomized in the trial) who received desirudin (0.8%) or enoxaparin (0.2%) (Table 1).
Elsaid and Collins assessed VTE and bleeding events associated with the use of unfractionated heparin 5000U either 2 or 3 times daily and enoxaparin 30 mg once or twice daily across patients stratified by renal function (creatinine clearance <30, 3059, and 60 mL/min). The investigators made assessments before and after a quality improvement intervention that was designed to eliminate the use of enoxaparin in patients whose creatinine clearance was <30 mL/min. No VTE events were reported. Patients receiving enoxaparin were significantly more likely to experience a major bleeding episode compared with patients receiving unfractionated heparin (overall rates for all levels of renal function: 13.5% vs 4. 2%; RR: 3.2; 95% CI: 1.47.3) (Table 2). This association was largely driven by the subgroup of patients with a creatinine clearance <30 mL/min. For this subgroup with severe renal insufficiency, patients receiving enoxaparin were significantly more likely to have a bleed compared with patients receiving unfractionated heparin (18.9% vs 4.1%; RR: 4.68; 95% CI: 1.120.6) (Tables 1 and 2). There was no difference in the bleeding rates for patients whose creatinine clearances were >60 mL/min.[20]
Strength of Evidence
Obese Patients
Overall, we found that the strength of evidence was insufficient regarding the composite end point of DVT, PE, and sudden death, and the outcomes of mortality and bleeding (Table 2). This was based on a paucity of available data, and a moderate risk of bias in the reviewed studies. Additionally, 92% of the enrolled patients in the studies were white, limiting the generalizability of the results to other ethnic groups.
Patients on Antiplatelets
The strength of evidence was insufficient in the studies reviewed here to conclude that there is no difference in rates of bleeding in patients who are concomitantly taking antiplatelet drugs while getting VTE prophylaxis with rivaroxaban, dabigatran, or enoxaparin. We based this rating because of the imprecision of results and unknown consistencies across multiple studies.
Patients With Renal Insufficiency
One RCT had a high risk of bias for our key question because data from only 1 study arm were useful for our review.[16] The other RCTs were judged to have a moderate risk of bias. The analyses led by Dahl and Shorr[18, 19] were based on post hoc (ie, not prespecified) analysis of data from RCTs. Additionally, outcomes in the Shorr et al. trial were reported for evaluable subpopulations of the cohort that was initially randomized in the clinical trial.
We rated the strength of evidence as insufficient to know the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE during hospitalization of patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, and patients receiving dialysis. We based this rating on the risk of bias associated with published studies and a lack of consistent evidence regarding associations that were reported. Similarly, we rated the strength of evidence as insufficient that 5000 U of unfractionated heparin 3 times daily increases the risk of major and minor bleeding events in patients with severely compromised renal function compared to this dose in patients without severely compromised renal function. We based this rating on a high risk of bias of included studies and inconsistent evidence. Likewise, we rated the strength of evidence as insufficient that enoxaparin significantly increases the risk of major bleeding compared with unfractionated heparin in patients with severe renal insufficiency. We based this rating on a high risk of bias and inconsistent published evidence.
We similarly found insufficient evidence to guide treatment decisions for patients with renal insufficiency. Our findings are consistent with other recent reviews. The American College of Chest Physicians (ACCP) practice guidelines[21] make dosing recommendations for the therapeutic use of enoxaparin. However, their assessment is that the data are insufficient to make direct recommendations about prophylaxis. Their assessment of the indirect evidence regarding bioaccumulation and increased anti‐factor Xa levels are consistent with ours. The ACCP guidelines also suggest that decreased clearance of enoxaparin has been associated with increased risk of bleeding events for patients with severe renal insufficiency. However, the cited study[20] compares patients with and without severe renal dysfunction who received the same therapy. Therefore, it is not possible to determine the additional risk conveyed by enoxaparin therapy, that is, above the baseline increased risk of bleeding among patients with renal insufficiency, particularly those receiving an alternate pharmacologic VTE prevention strategy, such as unfractionated heparin.
DISCUSSION
We found that the evidence was very limited about prevention of VTE in these select and yet prevalent patient populations. Despite the fact that there is an increasing number of obese patients and patients who are on antiplatelet therapies, most clinical practice guidelines do not address the care of these populations, which may be entirely appropriate given the state of the evidence.
The ACCP practice guidelines[21] suggest using a higher dose of enoxaparin for the prevention of VTE in obese patients. The subgroup analysis by Kucher et al.[11] showed effect attenuation of dalteparin when given at a fixed dose of 5000 IU/mL to patients with a BMI of >40 kg/m2. The Freeman study[12] showed that extremely obese patients (average BMI >62.1 kg/m2) who are given a fixed dose of enoxaparin achieved target anti‐factor Xa levels significantly less often than those who received a higher dose of enoxaparin. The 2 separate findings, although not conclusive, lend some credence to the current ACCP guidelines.[21]
The studies we reviewed on VTE prophylaxis in patients who are concomitantly on antiplatelets including aspirin reported no major increased risk of bleeding; however, in the Friedman et al. study,[15] 3.0% of patients who were put on enoxaparin while still on aspirin had a bleeding event compared to 1.2% of those on enoxaparin alone. This difference is not statistically significant but is a trend possibly worth noting, especially when one looks at the lower RR of bleeding at 0.55 compared to 0.82 when dabigatran is compared with enoxaparin with and without concomitant aspirin therapy, respectively (Table 1). The highest dose of aspirin used in either of the studies was 160 mg/day, and neither study addressed other potent antiplatelets such as clopidogrel or ticlopidine separately, which limits the generalizability of the finding to all antiplatelets. Current ACCP guidelines do not recommend aspirin as a sole option for the prevention of VTE in orthopedic surgery patients.[22] Concerns remain among clinicians that antiplatelets, including aspirin, on their own are unlikely to be fully effective to thwart venous thrombotic processes for most patients, and yet the risk of bleeding is not fully known when these agents are combined with other anticoagulants for VTE prophylaxis.
Our review has several limitations, including the possibility that we may have missed some observational studies, as the identification of relevant observational studies in electronic searches is more challenging than that of RCTs. The few studies made it impossible to quantitatively pool results. These results, however, have important implications, namely that additional research on the comparative effectiveness and safety of pharmacologic and mechanical strategies to prevent VTE is needed for the optimal care of these patient subgroups. This might be achieved with trials dedicated to enrolling these patients or prespecified subgroup analyses within larger trials. Observational data may be appropriate as long as attention is paid to confounding.
APPENDIX
MEDLINE Search Strategy
((pulmonary embolism[mh] OR PE[tiab] OR Pulmonary embolism[tiab] OR thromboembolism[mh] OR thromboembolism[tiab] OR thromboembolisms[tiab] OR Thrombosis[mh] OR thrombosis[tiab] OR DVT[tiab] OR VTE[tiab] OR clot[tiab]) AND (Anticoagulants[mh] OR Anticoagulants[tiab] OR Anticoagulant[tiab] OR thrombin inhibitors[tiab] OR Aspirin[mh] or aspirin[tiab] OR aspirins[tiab] or clopidogrel[nm] OR clopidogrel[tiab] OR Plavix[tiab] or ticlopidine[mh] or ticlopidine[tiab]OR ticlid[tiab] OR prasugrel[nm]Or prasugrel[tiab]OR effient[tiab]OR ticagrelor[NM] OR ticagrelor[tiab]OR Brilinta[tiab] OR cilostazol[NM] OR cilostazol[tiab]OR pletal[tiab] OR warfarin[mh]OR warfarin[tiab]OR coumadin[tiab] OR coumadine[tiab] OR Dipyridamole[mh]OR dipyridamole[tiab]OR persantine[tiab] OR dicoumarol[MH] OR dicoumarol[tiab] OR dicumarol[tiab] OR Dextran sulfate[mh] OR dextran sulfate[tiab] ORthrombin inhibitors[tiab] OR thrombin inhibitor[tiab] OR heparin[mh] OR Heparin[tiab] OR Heparins[tiab] OR LMWH[tiab] OR LDUH[tiab] OR Enoxaparin[mh] OR Enoxaparin[tiab] OR Lovenox[tiab] OR Dalteparin[tiab] OR Fragmin[tiab] OR Tinzaparin[tiab] OR innohep[tiab] OR Nadroparin[tiab] OR Fondaparinux[nm] OR Fondaparinux[tiab] OR Arixtra[tiab] OR Idraparinux[nm] OR Idraparinux[tiab] OR Rivaroxaban[nm] OR Rivaroxaban[tiab] OR novastan[tiab] OR Desirudin[nm] OR Desirudin[tiab] OR Iprivask[tiab]OR direct thrombin inhibitor[tiab] OR Argatroban[nm] OR Argatroban[tiab] OR Acova[tiab] OR Bivalirudin[nm] OR Bivalirudin[tiab] OR Angiomax[tiab] OR Lepirudin[nm] OR Lepirudin[tiab] OR Refludan[tiab] OR Dabigatran[nm] OR Dabigatran[tiab] OR Pradaxa[tiab] OR factor xa[mh] OR factor Xa[tiab] OR vena cava filters[mh] OR filters[tiab] OR filter[tiab] OR compression stockings[mh] OR intermittent pneumatic compression devices[mh] OR compression [tiab] OR Venous foot pump[tiab])) AND(prevent*[tiab] OR prophyla*[tiab] OR prevention and control[subheading]) NOT (animals[mh] NOT humans[mh]) NOT (editorial[pt] OR comment[pt]) NOT ((infant[mh] OR infant[tiab] OR child[mh] OR child[tiab] OR children[tiab] OR adolescent[mh] OR adolescent[tiab] OR teen‐age[tiab] OR pediatric[tiab] OR perinatal[tiab]) NOT (adult[tiab] OR adults[tiab] OR adult[mh])) NOT (mechanical valve[tiab] OR heart valve[tiab] OR atrial fibrillation[mh] OR atrial fibrillation[tiab] OR thrombophilia[mh] OR thrombophilia[tiab] OR pregnancy[mh])
Venous thromboembolism (VTE), including deep venous thrombosis (DVT) and pulmonary embolism (PE), is estimated to affect 900,000 Americans each year and is a cause of significant morbidity and mortality with associated high healthcare costs.[1] Accordingly, the comparative effectiveness and safety of interventions for the prevention and treatment of VTE are among the national priorities for comparative effectiveness research.[2] Whereas we have evidence‐based guidelines for the prophylaxis of VTE in the general population, there are no guidelines informing the care of select patient populations. Select populations are those patients in whom there is decisional uncertainty about the optimal choice, timing, and dose of VTE prophylaxis. Not only do these patients have an increased risk of DVT and PE, but most are also at high risk of bleeding, the most important complication of VTE prophylaxis.[3, 4, 5, 6]
The objectives of this systematic review were to define the comparative effectiveness and safety of pharmacologic and mechanical strategies for VTE prevention in some of these select medical populations including obese patients, patients on concomitant antiplatelet therapy, patients with renal insufficiency, patients who are underweight, and patients with coagulopathy due to liver disease.
METHODS
The methods for this comparative effectiveness review (CER) follow the guidelines suggested in the Agency for Healthcare Research and Quality (AHRQ) Methods Guide for Effectiveness and Comparative Effectiveness Reviews.[7] The protocol was publically posted.[8]
Search Strategy
We searched MEDLINE, EMBASE, and SCOPUS through August 2011, CINAHL, International Pharmaceutical Abstracts,
Study Selection
We reviewed titles followed by abstracts to identify randomized controlled trials (RCTs) or observational studies with comparison groups reporting on the effectiveness or safety of VTE prevention in our populations. Two investigators independently reviewed abstracts, and we excluded the abstracts if both investigators agreed that the article met 1 or more of the exclusion criteria. We included only English‐language articles that evaluated the effectiveness of pharmacological or mechanical interventions that have been approved for clinical use in the United States. To be eligible, the studies must have addressed relevant key questions in the population of our interest. We resolved disagreements by consensus. We used DistillerSR (Evidence Partners Inc., Ottawa, Ontario, Canada), a Web‐based database management program to manage the review process. Two investigators assessed the risk of bias in each study independently, using the Downs and Black instrument for observational studies and trials.[10]
Data Synthesis
For each select population, we created detailed evidence tables containing the information abstracted from the eligible studies. After synthesizing the evidence, we graded the quantity, quality, and consistency of the best available evidence for each select population by adapting an evidence‐grading scheme recommended in the Methods Guide for Conducting Comparative Effectiveness Reviews.[7]
RESULTS
We identified 30,902 unique citations and included 9 studies (Figure 1). There were 5 RCTs with relevant subgroups and 4 observational studies (Table 1). Two studies reported on the risk of bleeding in patients given pharmacologic prophylaxis while they are concomitantly taking nonsteroidal anti‐inflammatory drugs (NSAIDS) or antiplatelet agents/aspirin, 1 RCT and 1 prospective observational study reported on obese patients, and 5 studies described outcomes of patients with renal insufficiency (see Supporting Information, Table 1, in the online version of this article). No study tested prophylaxis in underweight patients or those with liver disease.
| Study | Arm, n | Total VTE (DVT and PE) | Bleeding | Other Outcomes | |
|---|---|---|---|---|---|
| |||||
| Obese patients | |||||
| Kucher et al., 2005[11] | Arm 1 (dalteparin), 558 | 2.8% (95% CI: 1.34.3) | 0% | Mortality at 21 days: 4.6% | |
| Arm 2 (placebo), 560 | 4.3% (95% CI: 2.56.2) | 0.7% | Mortality at 21 days: 2.7% | ||
| Freeman et al., [12] | Arm 1 (fixed‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 19 % | |
| Arm 2 (lower‐dose enoxaparin), 9 | NR | NR | Peak anti‐factor Xa level 32 % | ||
| Arm 3 (higher‐dose enoxaparin), 11 | NR | NR | Peak anti‐factor Xa level 86 % | ||
| Patients on antiplatelet agents | |||||
| Eriksson et al., 2012[14] | Arm 1 (rivaroxaban), 563 | NR | 20 (3.6%), rate ratio for use vs nonuse: 1.32 (95% CI: 0.85‐2.05) | NR | |
| Arm 2 (enoxaparin/placebo), 526 | NR | 17 (3.2%), rate ratio for use vs nonuse: 1.40 (95% CI: 0.87‐2.25) | NR | ||
| Friedman et al., 2012[15] | Arm 2 (150 mg dabigatran, no ASA), 1149 | NR | 11 (1.0%)a | NR | |
| Arm 5 (150 mg dabigatran+ASA), 128 | NR | 2 (1.6%)a | NR | ||
| Arm 3 (enoxaparin, no ASA), 1167 | NR | 14 (1.2%)a | NR | ||
| Arm 6 (enoxaparin+ASA), 132 | NR | 4 (3.0%) | NR | ||
| 150 mg dabigatran compared with enoxaparinNo concomitant ASA therapy | NR | RR: 0.82 (95% CI: 0.37‐1.84) | NR | ||
| 150 mg dabigatran compared with enoxaparinWith concomitant ASA therapy | NR | RR: 0.55 (95% CI: 0.11‐2.78) | NR | ||
| Patients with renal insufficiency | |||||
| Bauersachs et al., 2011[16] | Arm 2 (GFR <30), 92 | Total DVT: 11.11%; Total PE: 0% | Major bleeding: 4/92 (4.35%), minor bleeding: 9/92 (9.78%) | Mortality: 5.81% | |
| Mah et al., 2007[17] | Arm 2 (tinzaparin), 27 | NR | Major bleeding: 2/27 (7.4%), minor bleeding: 3/27 (11.1%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.05 | |
| Arm 3 (enoxaparin), 28 | NR | Major bleeding: 1/28 (3.6%), minor bleeding: 3/28 (10.7%) | Factor Xa level: AF: CmaxD8/Cmax D1=1.22 | ||
| Dahl et al., 2012[18] | Arm 1 (enoxaparin), 332 | Major VTE: 8 (9.0%) | Major bleeding: 6 (4.7%) | Infections and infestations: 25 (7.5%), Wound infection: 4 (1.2%) | |
| Arm 2 (dabigatran), 300 | Major VTE: 3 (4.3%) | Major bleeding: 0 (0%) | Infections and infestations: 21 (7.0%), Wound Infection: 3 (1.0%) | ||
| Shorr et al., 2012[19] | Arm 1 (enoxaparin, CrCL 60 mL/min), 353 | Total VTE: 17/275 (6.2%) | Major bleeding: 0/351 (0%) | NR | |
| Arm 2 (desirudin, CrCL 60 mL/min), 353 | Total VTE: 13/284 (4.3%) | Major bleeding: 2/349 (0.27%) | NR | ||
| Arm 3 (enoxaparin, CrCL 4559 mL/min), 369 | Total VTE: 18/282 (6.2%) | Major bleeding: 1/365 (0.27%) | NR | ||
| Arm 4 (desirudin, CrCL 4559 mL/min), 395 | Total VTE: 17/303 (5.6%) | Major bleeding: 1/393 (0.25%) | NR | ||
| Arm 5 (enoxaparin, CrCL <45 mL/min), 298 | Total VTE: 24/216 (11.1%) | Major bleeding: 1/294 (0.34%) | NR | ||
| Arm 6 (desirudin, CrCL <45 mL/min), 279 | Total VTE: 7/205 (3.4%) | Major bleeding: 5/275 (1.82%) | NR | ||
| Elsaid et al., 2012[20] | Arm 1 (enoxaparin, CrCL 60 mL/min), 17 | NR | Major bleeding: 2 (11.8%) | NR | |
| Arm 2 (enoxaparin, CrCL 3059 mL/min), 86 | NR | Major bleeding: 9 (10.5%) | NR | ||
| Arm 3 (enoxaparin, CrCL 30 mL/min), 53 | NR | Major bleeding: 10 (18.9%) | NR | ||
| Arm 4 (UFH, CrCL 60 mL/min), 19 | NR | Major bleeding: 2 (10.5%) | NR | ||
| Arm 5 (UFH, CrCL 3059 mL/min), 99 | NR | Major bleeding: 3 (3%) | NR | ||
| Arm 6 (UFH, CrCL 30 mL/min), 49 | NR | Major bleeding: 2 (4.1%) | NR | ||
Obese Patients
We found 1 subgroup analysis of an RCT (total 3706 patients, 2563 nonobese and 1118 obese patients) that reported on the comparative effectiveness and safety of fixed low‐dose dalteparin 5000 IU/day compared to placebo among 1118 hospitalized medically ill patients with body mass indices (BMI) greater than 30 kg/m2.11 Neither group received additional concurrent prophylactic therapies. The 3 most prevalent medical diagnoses prompting hospitalization were congestive heart failure, respiratory failure, and infectious diseases. Compression ultrasound was performed in all patients by day 21 of hospitalization. The primary end point was the composite of VTE, fatal PE, and sudden death, and secondary end points included DVT, bleeding, and thrombocytopenia by day 21 (Table 1). In obese patients, the primary end point occurred in 2.8% (95% confidence interval [CI]: 1.34.3) of the dalteparin group and in 4.3% (95% CI: 2.56.2) of the placebo group (relative risk [RR]: 0.64; 95% CI: 0.32‐1.28). In nonobese patients, the primary end point occurred in 2.8% (95% CI: 1.8‐3.8) and 5.2% (95% CI: 3.9‐6.6) of the dalteparin and placebo groups, respectively (RR: 0.53; 95% CI: 0.34‐0.82). When weight was modeled as a continuous variable, no statistically significant interaction between weight and dalteparin efficacy was observed (P=0.97). The authors calculated the RR in predefined BMI subgroups and found that dalteparin was effective in reducing VTE in patients with BMIs up to 40, with RRs of <1.0 for all (approximate range, 0.20.8). However, a fixed dose of dalteparin 5000 IU/day was not better than placebo for individuals with BMI >40 kg/m2. There was no significant difference in mortality or major hemorrhage by day 21 between treatment and placebo groups.
Freeman and colleagues prospectively assigned 31 medically ill patients with extreme obesity (BMI >40 kg/m2) to 1 of 3 dosing regimens of enoxaparin: a fixed dose of 40 mg daily enoxaparin (control group, n=11), enoxaparin at 0.4 mg/kg (n=9), or enoxaparin at 0.5 mg/kg (n=11).[12] The average BMI of the entire cohort was 62.1 kg/m2 (range, 40.582.4). All patients had anti‐factor Xa levels drawn on the day of enrollment and daily for 3 days (Table 2). The relationship between anti‐factor Xa levels and clinical efficacy of low‐molecular weight heparin (LMWH) in VTE prophylaxis is still unclear; however, an anti‐factor Xa level of 0.2 to 0.5 IU/mL, measured 4 hours after the fourth dose of LMWH, is the target level recommended for VTE prophylaxis.[13] Patients who received weight‐based enoxaparin at 0.5mg/kg achieved target anti‐factor Xa level 86% of the time compared to 32% of the time in those receiving 0.4 mg/kg and 19% of the time for those in the fixed‐dose group (P<0.001). No clinical outcomes were reported in this study.
| Intervention | Outcome | Risk of Bias | Evidence Statement and Magnitude of Effect |
|---|---|---|---|
| |||
| Patients on antiplatelet agents | |||
| Rivaroxaban vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic rivaroxaban or enoxaparin in patients concomitantly treated with antiplatelet agents; 3.6% vs 3.25% |
| Dabigatran vs enoxaparin | Major bleeding | Low | Insufficient to support no difference in rates of major bleeding with prophylactic dabigatran or enoxaparin in patients concomitantly treated with aspirin; 1.6% vs 3.0% |
| Obese patients | |||
| Dalteparin vs placebo | VTE | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing total VTE in obese patients; 2.8% vs 4.3%, RR: 0.64, 95% CI: 0.32‐1.28 |
| Dalteparin vs placebo | Mortality | Moderate | Insufficient evidence for effectiveness of dalteparin vs placebo in reducing mortality in obese patients; 9.9% vs 8.6%, P=0.36 |
| Dalteparin vs placebo | Major bleeding | Moderate | Insufficient evidence for safety of dalteparin vs placebo in reducing major bleeding in obese patients; 0% vs 0.7%, P>0.99 |
| Enoxaparin 40 mg daily vs 0.4 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.4 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 32%, P=NR |
| Enoxaparin 40 mg daily vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 40 mg daily versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 19% vs 86%, P<0.001 |
| Enoxaparin 0.4 mg/kg vs 0.5 mg/kg | Percentage of patients achieving target anti‐factor Xa level | Moderate | Insufficient evidence for effectiveness of enoxaparin 0.4 mg/kg versus 0.5 mg/kg in achieving peak anti‐factor Xa level in obese patients; 32% vs 86%, P=NR |
| Patients with renal insufficiency | |||
| Tinzaparin vs enoxaparin | VTE | High | Insufficient evidence about superiority of either drug for preventing VTE in patients with renal insufficiency, 0/27 vs 0/28* |
| Tinzaparin vs enoxaparin | Bleeding | High | Insufficient evidence about safety of either drug in patients with renal insufficiency; 5/27 vs 4/28, P=0.67 |
| Dabigatran vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of dabigatran in reducing VTE in severe renal compromise patients vs enoxaparin; 4.3% vs 9%, OR: 0.48, 95% CI: 0.13‐1.73, P=0.271 |
| Dabigatran vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of dabigatran vs enoxaparin in patients with renal impairment; 0 vs 4.7%, P=0.039 |
| Desirudin vs enoxaparin | VTE | Moderate | Insufficient evidence for effectiveness of desirudin vs enoxaparin in reducing VTE in patients with renal impairment; 4.9% vs 7.6%, P=0.019 |
| Desirudin vs enoxaparin | Bleeding | Moderate | Insufficient evidence for safety of desirudin vs enoxaparin in patients with renal impairment; 0.8% vs 0.2%, P=0.109 |
| Enoxaparin vs UFH | Bleeding | High | Insufficient evidence for increased risk of bleeding with enoxaparin vs unfractionated heparin in patients with all levels of renal impairment, 13.5% vs 4.2%, RR: 3.2, 95% CI: 1.47.3; and for the subgroup of patients with creatinine clearance <30 mL/min; 18.9% vs 4.1%, RR: 4.68, 95% CI: 1.120.6 |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | VTE | Moderate | Insufficient evidence regarding differential benefit of unfractionated heparin by renal function; 2.6% of patients had a VTE event |
| UFH in severe renal compromise vs all other renal status (undifferentiated) | Bleeding | Moderate | Insufficient evidence for differential harm from unfractionated heparin by renal function; 13 events in 92 patients |
Patients on Antiplatelet Drugs
We did not find studies that directly looked at the comparative effectiveness of VTE prophylaxis in patients who were on antiplatelet drugs including aspirin. However, there were 2 studies that looked at the risk of bleeding in patients who received VTE pharmacologic prophylaxis while concurrently taking antiplatelet agents including aspirin. Both studies used pooled data from large phase III trials.
The study by Eriksson et al. used data from the RECORD (Regulation of Coagulation in Orthopedic Surgery to Prevent Deep Venous Thrombosis and Pulmonary Embolism) trial where over 12,000 patients undergoing elective total knee or hip replacement were randomized to receive VTE prophylaxis with oral rivaroxaban or subcutaneous enoxaparin.[14] Nine percent of participants in each arm (563 in rivaroxaban and 526 in enoxaparin/placebo) were concomitantly using antiplatelet agents or aspirin at least once during the at risk period, defined as starting at day 1 of surgery up to 2 days after the last intake of the study drug. The only end point evaluated was bleeding, and the authors found no statistically significant bleeding difference among the 2 arms (Table 1). Any bleeding event in the rivaroxaban with antiplatelets or aspirin arm was found in 20 (3.6%) patients, whereas in those on enoxaparin/placebo with antiplatelets or aspirin arm it was 17 (3.2%). The relative rate of bleeding among users versus nonusers of antiplatelet drugs or aspirin was 1.32 (95% CI: 0.85‐2.05) in the rivaroxaban group and 1.40 (95% CI: 0.87‐2.25) in the enoxaparin arm (Table 1).
Friedman et al. used pooled data from the RE‐MODEL, RENOVATE, and REMOBILIZE trials, where patients who were undergoing hip or knee arthroplasty were randomized to 220 mg of dabigatran once daily, 150 mg of dabigatran once daily (we focused on this lower dosage as this is the only available dose used in the US), 40 mg of enoxaparin once daily, or 30 mg of enoxaparin twice a day.[15] Of the 8135 patients, 4.7% were on concomitant aspirin. The baseline characteristics of those on aspirin were similar to the other enrollees. The primary outcome was major bleeding events requiring transfusion, symptomatic internal bleeding, or bleeding requiring surgery. Among patients receiving 150 mg of dabigatran, bleeding events with and without concomitant aspirin occurred in 1.6% and 1.0%, respectively (odds ratio [OR]: 1.64; 95% CI: 0.36‐7.49; P=0.523). The percentages of participants with bleeding who received enoxaparin, with and without aspirin, were 3.0% and 1.2%, respectively (OR: 2.57; 95% CI: 0.83‐7.94; P=0.101). The RR of bleeding on dabigatran compared to enoxaparin with and without aspirin therapy was 0.55 (95% CI: 0.11‐2.78) and 0.82 (95% CI: 0.37‐1.84), respectively (Table 1).
Patients With Renal Insufficiency
We found 5 studies that evaluated the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE in patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, or patients receiving dialysis. Four studies were RCTs,[16, 17, 18, 19] and 1 used a cohort design assessing separate cohorts before and after a quality improvement intervention.[20] Bauersachs and colleagues conducted an RCT comparing unfractionated heparin at 5000 IU, 3 times daily to certoparin, which is not approved in the United States and is not further discussed here.[16] The rate of DVT among patients treated with unfractionated heparin in patients with a glomerular filtration rate >30 mL/min was marginally lower than those with severe renal dysfunction (10.3 vs 11.1%) (Table 1).
Patients with severe renal dysfunction who received 5000 IU of unfractionated heparin 3 times a day were at increased risk of all bleeds (RR: 3.4; 95% CI: 2.05.9), major bleeds (RR: 7.3; 95% CI: 3.316), and minor bleeds (RR: 2.6; 95% CI: 1.4‐4.9) compared to patients treated with unfractionated heparin without severe renal dysfunction.[16]
A randomized trial by Mah and colleagues compared drug accumulation and anti‐Xa activity in elderly patients with renal dysfunction (defined as a glomerular filtration rate of 20 to 50 mL/min) who received either tinzaparin at 4500 IU once daily or enoxaparin at 4000 IU once daily.[17] Enoxaparin accumulated to a greater extent from day 1 to day 8 than did tinzaparin; the ratio of maximum concentration on day 8 compared to day 1 was 1.22 for enoxaparin and 1.05 for tinzaparin (P=0.016). No VTE events were reported in patients who received tinzaparin or enoxaparin. There was no statistical difference in the incidence of bleeding events between patients receiving tinzaparin (5, including 2 major events) and enoxaparin (4, including 3 major events, P=0.67) (Table 1).
The trial by Dahl and colleagues randomly assigned patients who were over 75 years of age and/or who had moderate renal dysfunction (defined as creatinine clearance between 30 and 49 mL/min) to receive enoxaparin 40 mg daily or dabigatran 150 mg daily.[18] There was no significant difference in the rate of major VTE events between patients receiving dabigatran (4.3%) and enoxaparin (9%) (OR: 0.48; 95% CI: 0.13‐1.73; P=0.271) (Table 1). The rate of major bleeding was significantly higher among patients randomly assigned to receive enoxaparin (4.7%) versus dabigatran (0%) (P=0.039).[18]
Shorr and colleagues published a post hoc subgroup analysis of a multicenter trial in which orthopedic patients were randomly assigned to receive desirudin 15 mg twice daily or enoxaparin 40 mg once daily.[19] Evaluable patients (1565 of the 2079 patients randomized in the trial) receiving desirudin experienced a significantly lower rate of major VTE compared with patients receiving enoxaparin (4.9% vs 7.6%, P=0.019). This relationship was particularly pronounced for evaluable patients whose creatinine clearance was between 30 and 44 mL/min. In evaluable patients with this degree of renal dysfunction, 11% of patients taking enoxaparin compared to 3.4% of those taking desirudin had a major VTE (OR: 3.52; 95% CI: 1.48‐8.4; P=0.004). There was no significant difference in the rates of major bleeding among a subset of patients assessed for safety outcomes (2078 of the 2079 patients randomized in the trial) who received desirudin (0.8%) or enoxaparin (0.2%) (Table 1).
Elsaid and Collins assessed VTE and bleeding events associated with the use of unfractionated heparin 5000U either 2 or 3 times daily and enoxaparin 30 mg once or twice daily across patients stratified by renal function (creatinine clearance <30, 3059, and 60 mL/min). The investigators made assessments before and after a quality improvement intervention that was designed to eliminate the use of enoxaparin in patients whose creatinine clearance was <30 mL/min. No VTE events were reported. Patients receiving enoxaparin were significantly more likely to experience a major bleeding episode compared with patients receiving unfractionated heparin (overall rates for all levels of renal function: 13.5% vs 4. 2%; RR: 3.2; 95% CI: 1.47.3) (Table 2). This association was largely driven by the subgroup of patients with a creatinine clearance <30 mL/min. For this subgroup with severe renal insufficiency, patients receiving enoxaparin were significantly more likely to have a bleed compared with patients receiving unfractionated heparin (18.9% vs 4.1%; RR: 4.68; 95% CI: 1.120.6) (Tables 1 and 2). There was no difference in the bleeding rates for patients whose creatinine clearances were >60 mL/min.[20]
Strength of Evidence
Obese Patients
Overall, we found that the strength of evidence was insufficient regarding the composite end point of DVT, PE, and sudden death, and the outcomes of mortality and bleeding (Table 2). This was based on a paucity of available data, and a moderate risk of bias in the reviewed studies. Additionally, 92% of the enrolled patients in the studies were white, limiting the generalizability of the results to other ethnic groups.
Patients on Antiplatelets
The strength of evidence was insufficient in the studies reviewed here to conclude that there is no difference in rates of bleeding in patients who are concomitantly taking antiplatelet drugs while getting VTE prophylaxis with rivaroxaban, dabigatran, or enoxaparin. We based this rating because of the imprecision of results and unknown consistencies across multiple studies.
Patients With Renal Insufficiency
One RCT had a high risk of bias for our key question because data from only 1 study arm were useful for our review.[16] The other RCTs were judged to have a moderate risk of bias. The analyses led by Dahl and Shorr[18, 19] were based on post hoc (ie, not prespecified) analysis of data from RCTs. Additionally, outcomes in the Shorr et al. trial were reported for evaluable subpopulations of the cohort that was initially randomized in the clinical trial.
We rated the strength of evidence as insufficient to know the comparative effectiveness and safety of pharmacologic prophylaxis for prevention of VTE during hospitalization of patients with acute kidney injury, moderate renal insufficiency, severe renal insufficiency not undergoing dialysis, and patients receiving dialysis. We based this rating on the risk of bias associated with published studies and a lack of consistent evidence regarding associations that were reported. Similarly, we rated the strength of evidence as insufficient that 5000 U of unfractionated heparin 3 times daily increases the risk of major and minor bleeding events in patients with severely compromised renal function compared to this dose in patients without severely compromised renal function. We based this rating on a high risk of bias of included studies and inconsistent evidence. Likewise, we rated the strength of evidence as insufficient that enoxaparin significantly increases the risk of major bleeding compared with unfractionated heparin in patients with severe renal insufficiency. We based this rating on a high risk of bias and inconsistent published evidence.
We similarly found insufficient evidence to guide treatment decisions for patients with renal insufficiency. Our findings are consistent with other recent reviews. The American College of Chest Physicians (ACCP) practice guidelines[21] make dosing recommendations for the therapeutic use of enoxaparin. However, their assessment is that the data are insufficient to make direct recommendations about prophylaxis. Their assessment of the indirect evidence regarding bioaccumulation and increased anti‐factor Xa levels are consistent with ours. The ACCP guidelines also suggest that decreased clearance of enoxaparin has been associated with increased risk of bleeding events for patients with severe renal insufficiency. However, the cited study[20] compares patients with and without severe renal dysfunction who received the same therapy. Therefore, it is not possible to determine the additional risk conveyed by enoxaparin therapy, that is, above the baseline increased risk of bleeding among patients with renal insufficiency, particularly those receiving an alternate pharmacologic VTE prevention strategy, such as unfractionated heparin.
DISCUSSION
We found that the evidence was very limited about prevention of VTE in these select and yet prevalent patient populations. Despite the fact that there is an increasing number of obese patients and patients who are on antiplatelet therapies, most clinical practice guidelines do not address the care of these populations, which may be entirely appropriate given the state of the evidence.
The ACCP practice guidelines[21] suggest using a higher dose of enoxaparin for the prevention of VTE in obese patients. The subgroup analysis by Kucher et al.[11] showed effect attenuation of dalteparin when given at a fixed dose of 5000 IU/mL to patients with a BMI of >40 kg/m2. The Freeman study[12] showed that extremely obese patients (average BMI >62.1 kg/m2) who are given a fixed dose of enoxaparin achieved target anti‐factor Xa levels significantly less often than those who received a higher dose of enoxaparin. The 2 separate findings, although not conclusive, lend some credence to the current ACCP guidelines.[21]
The studies we reviewed on VTE prophylaxis in patients who are concomitantly on antiplatelets including aspirin reported no major increased risk of bleeding; however, in the Friedman et al. study,[15] 3.0% of patients who were put on enoxaparin while still on aspirin had a bleeding event compared to 1.2% of those on enoxaparin alone. This difference is not statistically significant but is a trend possibly worth noting, especially when one looks at the lower RR of bleeding at 0.55 compared to 0.82 when dabigatran is compared with enoxaparin with and without concomitant aspirin therapy, respectively (Table 1). The highest dose of aspirin used in either of the studies was 160 mg/day, and neither study addressed other potent antiplatelets such as clopidogrel or ticlopidine separately, which limits the generalizability of the finding to all antiplatelets. Current ACCP guidelines do not recommend aspirin as a sole option for the prevention of VTE in orthopedic surgery patients.[22] Concerns remain among clinicians that antiplatelets, including aspirin, on their own are unlikely to be fully effective to thwart venous thrombotic processes for most patients, and yet the risk of bleeding is not fully known when these agents are combined with other anticoagulants for VTE prophylaxis.
Our review has several limitations, including the possibility that we may have missed some observational studies, as the identification of relevant observational studies in electronic searches is more challenging than that of RCTs. The few studies made it impossible to quantitatively pool results. These results, however, have important implications, namely that additional research on the comparative effectiveness and safety of pharmacologic and mechanical strategies to prevent VTE is needed for the optimal care of these patient subgroups. This might be achieved with trials dedicated to enrolling these patients or prespecified subgroup analyses within larger trials. Observational data may be appropriate as long as attention is paid to confounding.
APPENDIX
MEDLINE Search Strategy
((pulmonary embolism[mh] OR PE[tiab] OR Pulmonary embolism[tiab] OR thromboembolism[mh] OR thromboembolism[tiab] OR thromboembolisms[tiab] OR Thrombosis[mh] OR thrombosis[tiab] OR DVT[tiab] OR VTE[tiab] OR clot[tiab]) AND (Anticoagulants[mh] OR Anticoagulants[tiab] OR Anticoagulant[tiab] OR thrombin inhibitors[tiab] OR Aspirin[mh] or aspirin[tiab] OR aspirins[tiab] or clopidogrel[nm] OR clopidogrel[tiab] OR Plavix[tiab] or ticlopidine[mh] or ticlopidine[tiab]OR ticlid[tiab] OR prasugrel[nm]Or prasugrel[tiab]OR effient[tiab]OR ticagrelor[NM] OR ticagrelor[tiab]OR Brilinta[tiab] OR cilostazol[NM] OR cilostazol[tiab]OR pletal[tiab] OR warfarin[mh]OR warfarin[tiab]OR coumadin[tiab] OR coumadine[tiab] OR Dipyridamole[mh]OR dipyridamole[tiab]OR persantine[tiab] OR dicoumarol[MH] OR dicoumarol[tiab] OR dicumarol[tiab] OR Dextran sulfate[mh] OR dextran sulfate[tiab] ORthrombin inhibitors[tiab] OR thrombin inhibitor[tiab] OR heparin[mh] OR Heparin[tiab] OR Heparins[tiab] OR LMWH[tiab] OR LDUH[tiab] OR Enoxaparin[mh] OR Enoxaparin[tiab] OR Lovenox[tiab] OR Dalteparin[tiab] OR Fragmin[tiab] OR Tinzaparin[tiab] OR innohep[tiab] OR Nadroparin[tiab] OR Fondaparinux[nm] OR Fondaparinux[tiab] OR Arixtra[tiab] OR Idraparinux[nm] OR Idraparinux[tiab] OR Rivaroxaban[nm] OR Rivaroxaban[tiab] OR novastan[tiab] OR Desirudin[nm] OR Desirudin[tiab] OR Iprivask[tiab]OR direct thrombin inhibitor[tiab] OR Argatroban[nm] OR Argatroban[tiab] OR Acova[tiab] OR Bivalirudin[nm] OR Bivalirudin[tiab] OR Angiomax[tiab] OR Lepirudin[nm] OR Lepirudin[tiab] OR Refludan[tiab] OR Dabigatran[nm] OR Dabigatran[tiab] OR Pradaxa[tiab] OR factor xa[mh] OR factor Xa[tiab] OR vena cava filters[mh] OR filters[tiab] OR filter[tiab] OR compression stockings[mh] OR intermittent pneumatic compression devices[mh] OR compression [tiab] OR Venous foot pump[tiab])) AND(prevent*[tiab] OR prophyla*[tiab] OR prevention and control[subheading]) NOT (animals[mh] NOT humans[mh]) NOT (editorial[pt] OR comment[pt]) NOT ((infant[mh] OR infant[tiab] OR child[mh] OR child[tiab] OR children[tiab] OR adolescent[mh] OR adolescent[tiab] OR teen‐age[tiab] OR pediatric[tiab] OR perinatal[tiab]) NOT (adult[tiab] OR adults[tiab] OR adult[mh])) NOT (mechanical valve[tiab] OR heart valve[tiab] OR atrial fibrillation[mh] OR atrial fibrillation[tiab] OR thrombophilia[mh] OR thrombophilia[tiab] OR pregnancy[mh])
- , , . Estimated annual number of incident and recurrent, non‐fatal and fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106:910.
- Institute of Medicine. Institute of Medicine. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: National Academies Press; 2009.
- Lovenox (enoxaparin sodium injection for subcutaneous and intravenous use: prescribing information). Bridgewater, NJ: SanofiAventis; 2011. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed October 17, 2012.
- Innohep (tinzaparin sodium injection). Ballerup, Denmark: LEO Pharmaceutical Products; 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed October 17, 2012.
- Leizorovicz A. Tinzaparin compared to unfractionated heparin for initial treatment of deep vein thrombosis in very elderly patients with renal insufficiency‐ the IRIS trial. [50th ASH Annual Meeting and Exposition abstract 434]. Blood. 2008;11:112.
- Fragmin (dalteparin sodium injection). New York, NY: Pfizer Inc.; 2007. Available at: http://www.pfizer.com/files/products/uspi_fragmin.pdf. Accessed October 17, 2012.
- Methods guide for effectiveness and comparative effectiveness reviews. Rockville, MD: Agency for Healthcare Research and Quality; August 2011. AHRQ publication No. 10 (11)‐EHC063‐EF. Available at: http://www.effectivehealthcare.ahrq.gov. Accessed October 17, 2012.
- Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/928/VTE‐Special‐Populations_Protocol_20120112.pdf. Accessed April 17, 2012.
- , , , et al. Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Evidence Report/Technology Assessment (AHRQ). Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/1501/venous‐thromboembolism‐special‐populations‐report‐130529.pdf. 2013.
- , . The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non‐randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377–384.
- , , , et al. Efficacy and safety of fixed low‐dose dalteparin in preventing venous thromboembolism among obese or elderly hospitalized patients: a subgroup analysis of the PREVENT trial. Arch Intern Med. 2005;165(3):341–345.
- , , , . Prospective comparison of three enoxaparin dosing regimens to achieve target anti‐factor Xa levels in hospitalized, medically ill patients with extreme obesity. Am J Hematol. 2012;87(7):740–743.
- , , . Effect of prophylactic dalteparin on anti‐factor xa levels in morbidly obese patients after bariatric surgery. Obes Surg. 2010;20(4):487–491.
- , , , , . Concomitant use of medication with antiplatelet effects in patients receiving either rivaroxaban or enoxaparin after total hip or knee arthroplasty. Thromb Res. 2012;130(2):147–151.
- , , , , , . Dabigatran etexilate and concomitant use of non‐steroidal anti‐inflammatory drugs or acetylsalicylic acid in patients undergoing total hip and total knee arthroplasty: No increased risk of bleeding. Thromb Haemost. 2012;108(1):183–190.
- , , , et al. CERTIFY: prophylaxis of venous thromboembolism in patients with severe renal insufficiency. Thromb Haemost. 2011;105(6):981–988.
- , , , et al. Tinzaparin and enoxaparin given at prophylactic dose for eight days in medical elderly patients with impaired renal function: a comparative pharmacokinetic study. Thromb Haemost. 2007;97(4):581–586.
- , , , , , . Thromboprophylaxis in patients older than 75 years or with moderate renal impairment undergoing knee or hip replacement surgery [published correction appears in Int Orthop. 2012;36(5):1113]. Int Orthop. 2012;36(4):741–748.
- , , , . Impact of stage 3B chronic kidney disease on thrombosis and bleeding outcomes after orthopedic surgery in patients treated with desirudin or enoxaparin: insights from a randomized trial. J Thromb Haemost. 2012;10(8):1515–1520.
- , . Initiative to improve thromboprophylactic enoxaparin exposure in hospitalized patients with renal impairment. Am J Health Syst Pharm. 2012;69(5):390–396.
- , , , , ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):7S–47S.
- , . Aspirin for the prophylaxis of venous thromboembolic events in orthopedic surgery patients: a comparison of the AAOS and ACCP guidelines with review of the evidence. Ann Pharmacother. 2013;47(1):63–74.
- , , . Estimated annual number of incident and recurrent, non‐fatal and fatal venous thromboembolism (VTE) events in the US. Blood. 2005;106:910.
- Institute of Medicine. Institute of Medicine. Initial National Priorities for Comparative Effectiveness Research. Washington, DC: National Academies Press; 2009.
- Lovenox (enoxaparin sodium injection for subcutaneous and intravenous use: prescribing information). Bridgewater, NJ: SanofiAventis; 2011. Available at: http://products.sanofi.us/lovenox/lovenox.html. Accessed October 17, 2012.
- Innohep (tinzaparin sodium injection). Ballerup, Denmark: LEO Pharmaceutical Products; 2008. Available at: http://www.accessdata.fda.gov/drugsatfda_docs/label/2008/020484s011lbl.pdf. Accessed October 17, 2012.
- Leizorovicz A. Tinzaparin compared to unfractionated heparin for initial treatment of deep vein thrombosis in very elderly patients with renal insufficiency‐ the IRIS trial. [50th ASH Annual Meeting and Exposition abstract 434]. Blood. 2008;11:112.
- Fragmin (dalteparin sodium injection). New York, NY: Pfizer Inc.; 2007. Available at: http://www.pfizer.com/files/products/uspi_fragmin.pdf. Accessed October 17, 2012.
- Methods guide for effectiveness and comparative effectiveness reviews. Rockville, MD: Agency for Healthcare Research and Quality; August 2011. AHRQ publication No. 10 (11)‐EHC063‐EF. Available at: http://www.effectivehealthcare.ahrq.gov. Accessed October 17, 2012.
- Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/928/VTE‐Special‐Populations_Protocol_20120112.pdf. Accessed April 17, 2012.
- , , , et al. Comparative effectiveness of pharmacologic and mechanical prophylaxis of venous thromboembolism among special populations. Evidence Report/Technology Assessment (AHRQ). Available at: http://effectivehealthcare.ahrq.gov/ehc/products/341/1501/venous‐thromboembolism‐special‐populations‐report‐130529.pdf. 2013.
- , . The feasibility of creating a checklist for the assessment of the methodological quality both of randomised and non‐randomised studies of health care interventions. J Epidemiol Community Health. 1998;52(6):377–384.
- , , , et al. Efficacy and safety of fixed low‐dose dalteparin in preventing venous thromboembolism among obese or elderly hospitalized patients: a subgroup analysis of the PREVENT trial. Arch Intern Med. 2005;165(3):341–345.
- , , , . Prospective comparison of three enoxaparin dosing regimens to achieve target anti‐factor Xa levels in hospitalized, medically ill patients with extreme obesity. Am J Hematol. 2012;87(7):740–743.
- , , . Effect of prophylactic dalteparin on anti‐factor xa levels in morbidly obese patients after bariatric surgery. Obes Surg. 2010;20(4):487–491.
- , , , , . Concomitant use of medication with antiplatelet effects in patients receiving either rivaroxaban or enoxaparin after total hip or knee arthroplasty. Thromb Res. 2012;130(2):147–151.
- , , , , , . Dabigatran etexilate and concomitant use of non‐steroidal anti‐inflammatory drugs or acetylsalicylic acid in patients undergoing total hip and total knee arthroplasty: No increased risk of bleeding. Thromb Haemost. 2012;108(1):183–190.
- , , , et al. CERTIFY: prophylaxis of venous thromboembolism in patients with severe renal insufficiency. Thromb Haemost. 2011;105(6):981–988.
- , , , et al. Tinzaparin and enoxaparin given at prophylactic dose for eight days in medical elderly patients with impaired renal function: a comparative pharmacokinetic study. Thromb Haemost. 2007;97(4):581–586.
- , , , , , . Thromboprophylaxis in patients older than 75 years or with moderate renal impairment undergoing knee or hip replacement surgery [published correction appears in Int Orthop. 2012;36(5):1113]. Int Orthop. 2012;36(4):741–748.
- , , , . Impact of stage 3B chronic kidney disease on thrombosis and bleeding outcomes after orthopedic surgery in patients treated with desirudin or enoxaparin: insights from a randomized trial. J Thromb Haemost. 2012;10(8):1515–1520.
- , . Initiative to improve thromboprophylactic enoxaparin exposure in hospitalized patients with renal impairment. Am J Health Syst Pharm. 2012;69(5):390–396.
- , , , , ; American College of Chest Physicians Antithrombotic Therapy and Prevention of Thrombosis Panel. Executive summary: antithrombotic therapy and prevention of thrombosis, 9th ed: American College of Chest Physicians evidence‐based clinical practice guidelines. Chest. 2012;141(2 suppl):7S–47S.
- , . Aspirin for the prophylaxis of venous thromboembolic events in orthopedic surgery patients: a comparison of the AAOS and ACCP guidelines with review of the evidence. Ann Pharmacother. 2013;47(1):63–74.