User login
Brain stimulation can improve prognosis following a stroke and other neurological diseases
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
HAMBURG, GERMANY – Around 86 billion nerve cells in our brain work together in complex dynamic networks to control almost every sensorimotor and cognitive process. However, the way in which the information is processed in the different regions of the brain is still unclear. There are already some promising approaches to specifically influence the dynamics of neuronal networks to treat neurological and psychiatric diseases.
One of the main topics at the Congress for Clinical Neuroscience of the German Society for Clinical Neurophysiology and Functional Neuroimaging (DGKN), recently held in Hamburg, Germany, was the dynamics of cerebral networks in sensorimotor and cognitive processes, as well as disruptions to network dynamics in neurological and psychiatric diseases.
“We will be unable to develop innovative therapies for widespread neurological and psychiatric diseases until we understand neuronal functions on every level of complexity,” Andreas K. Engel, PhD, director of the Institute for Neurophysiology and Pathophysiology at the University Hospital of Hamburg-Eppendorf, president of the DGKN, and congress president, said during an online press conference.
Characterizing states of consciousness
For more than 30 years, it has been known that neuronal signals in the brain are dynamically coupled. Despite intensive research, the functional significance of this coupling on information processing is still largely unknown.
Neuroimaging methods such as electroencephalography (EEG), magnetoencephalography (MEG), structural and functional magnetic resonance imaging (MRI), and electrophysiological examinations were used. Model calculations of the data suggest that dynamic couplings of signals in the cortex play a crucial role in memory performance, thinking processes, and developing perception, among other things.
It has already been shown that the network dynamics of neuronal signals could possibly characterize states of consciousness. Neuronal signals and coupling patterns differ significantly between healthy individuals in a waking state and those who are asleep, under general anesthetic, or in a vegetative state. In Dr. Engel’s view, it may be possible in the future for machine learning algorithms to be used to classify states of consciousness.
Changes in brain activity as a biomarker?
The differences in the dynamics of neuronal signals between healthy individuals and patients with psychiatric diseases such as schizophrenia appear much more important for clinical practice. “The characteristic changes in brain activity in the primary auditory cortex could be considered a potential biomarker and used to predict the clinical course of psychiatric diseases, such as psychoses,” reported Dr. Engel.
The gamma-band activity in the auditory cortex could be a potential marker for schizophrenia. According to MEG examinations, the values are decreased both in people at increased risk of psychosis and experiencing first symptoms compared with controls.
Activation or inhibition of cerebral networks as new therapeutic approaches
New therapeutic approaches based on the activation or inhibition of cerebral networks are currently areas of intensive research. Close interdisciplinary collaboration between basic science researchers and clinicians is necessary, stressed Dr. Engel. The use of noninvasive brain stimulation is already within reach for the neurorehabilitation of stroke patients. “I am optimistic that in a few years brain stimulation will be established as an integral element of stroke therapy,” said Christian Grefkes-Hermann, MD, PhD, director of the department of neurology at University Hospital of Frankfurt and first vice president of the DGKN.
Despite great advances in acute stroke therapy, many patients must endure permanent deficits in their everyday life, he said. According to Dr. Grefkes-Hermann, rehabilitation procedures often have a dissatisfactory effect, and results greatly vary. He hopes that in the future it may be possible to personalize therapy by using network patterns, thereby improving results.
“The most important factor for functional recovery after a stroke is neuronal reorganization,” said Dr. Grefkes-Hermann. With the new methods of neurorehabilitation, network-connectivity disruptions, which are associated with motor function deficits, are first visualized using functional MRI (fMRI).
The imaging or the EEG makes visible the area of the brain that may benefit most from neurostimulation. Subsequently, nerve cells in this region may be precisely stimulated with TMS. Because the healthy hemisphere of the brain is usually overactive after a stroke, there are simultaneous attempts to inhibit the contralesional motor cortex.
Initial results are hopeful. In the initial period after a stroke, TMS can be used in some patients to correct pathological connectivities and thereby improve motor deficits, reported Dr. Grefkes-Hermann. The fMRI pattern can also be used to predict recovery and intervention effects on an individual basis. A phase 3 trial is currently underway of 150 patients who have had a stroke and aims to study the efficacy of the new procedure.
Combined TMS and EEG
With the combination of TMS and the simultaneous measurement of EEG activity, a further development of fMRI connectivity analyses is currently being tested. Dr. Grefkes-Hermann believes that this procedure, which is more cost-effective, has higher temporal resolution, can be used directly at the bedside, and has more potential for personalized therapy planning in clinical practice.
The TMS-EEG procedure also makes it possible to predict the risk of post-stroke delirium, which affects around 30% of stroke patients and greatly worsens the outcome, underlined Ulf Ziemann, MD, medical director of the department of neurology at Tübingen (Germany) University Hospital. In a study of 33 patients with acute stroke, the onset of post-stroke delirium could be predicted with a high degree of accuracy by using the TMS-EEG procedure no later than 48 hours after the event.
Other promising, noninvasive methods for neuron activation mentioned by Dr. Ziemann include transcranial focused ultrasound stimulation (tFUS) with low intensity, which is being studied for chronic pain, dementia, epilepsy, traumatic brain injury, and depression, as well as transcranial pulse stimulation (TPS), which is also based on ultrasound. In a pilot study of 35 patients with Alzheimer’s disease, use of TPS within 3 months had positive effects on cognition. However, the study was not controlled and therefore further assessments are needed.
Custom deep brain stimulation
For deep brain stimulation (DBS), an established therapy for Parkinson’s disease and other movement disorders, the aim is individualized, symptom-related network stimulation, reported Andrea Kühn, MD, head of the movement disorders and neuromodulation section in the department of neurology at Charité University Hospital Berlin.
At the panregional collaborative research center ReTune, which has been supported for 4 years now by €10 million from the German Research Foundation (DFG), imaging and computer-assisted programming algorithms are being developed for DBS. They will greatly simplify the time-consuming standard procedure for the best possible setting of the stimulation parameters, which requires a hospital stay of several days.
A randomized crossover study of 35 patients with Parkinson’s disease proved the equivalence of the fast, algorithm-assisted DBS for the control of motor symptoms compared with standard procedures.
The new methods have the potential to considerably improve the outcome of patients with neurological and psychiatric diseases, according to scientists. However, the positive data must still be validated in further studies.
This article was translated from Medscape’s German edition. A version of this article appeared on Medscape.com.
Antidepressants benefit some patients with osteoarthritis pain
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
DENVER – Using antidepressants to treat osteoarthritis pain can benefit some individuals but appears to have a clinically unimportant reduction in pain when looking at all patients who have tried them, according to a study presented at the OARSI 2023 World Congress. The review was also published in the Cochrane Database of Systematic Reviews in October 2022.
In terms of implications for clinical practice, the findings “seem to suggest there is a subgroup that is more likely to respond to antidepressants,” Anita Wluka, PhD, MBBS, a professor in the School of Public Health and Preventive Medicine at Monash University in Melbourne, told attendees. The findings also raise an important research question: “How can we identify the patient phenotype likely to benefit so we can [minimize the] risk of those adverse events and effects?”
Osteoarthritis pain is heterogeneous, and an estimated 30% of the pain is neuropathic-like, likely including central and peripheral sensitization, Dr. Wluka said. Given that antidepressants affect multiple sites along these pathways, multiple organizations have issued a conditional recommendation for duloxetine in their osteoarthritis guidelines, including OARSI, the European Alliance of Associations for Rheumatology, and the American College of Rheumatology.
The Cochrane Collaboration therefore conducted a systematic review and meta-analysis of research on the benefits and harms of using antidepressants to treat symptomatic knee and hip osteoarthritis. The review included studies through January 2021 whose participants had knee and/or hip osteoarthritis and which compared antidepressant therapy with placebo or another intervention for at least 6 weeks. The authors looked at seven outcomes: overall pain on a 0-10 scale, clinical response (at least a 50% reduction in 24‐hour mean pain), physical function using the Western Ontario and McMaster Universities Arthritis Index (WOMAC), quality of life using the EQ-5D, the proportion of participants withdrawing because of adverse events, the proportion who experienced any adverse events, and the proportion who experienced serious adverse events.
The researchers considered a change on the pain scale of 0.5-1 points to be “slight to small,” a difference above 1 up to 2 to be “moderate,” and a difference greater than 2 points to be “large.” In assessing quality of life function on a scale of 0-100, a slight to small difference was 5-10, a moderate difference was 11-20, and a large difference was above 20.
Of the 18 articles the researchers identified for qualitative synthesis, 9 met the criteria for qualitative synthesis in the meta-analysis, including 7 studies only on the knee and 2 that included the knees and hips. All nine studies compared antidepressants with placebo, with or without NSAIDs. Most focused on serotonin and norepinephrine reuptake inhibitors (SNRIs) – six studies on duloxetine and one on milnacipran – while one included fluvoxamine and one included nortriptyline.
The trials included a combined 2,122 participants who were predominantly female with an average age range of 54-66. Trials ranged from 8 to 16 weeks. Five of the trials carried risk of attrition and reporting bias, and only one trial had low risk of bias across all domains.
In five trials with SNRIs and one trial with tricyclics (nortriptyline) totaling 1,904 participants, 45% of those receiving antidepressants had a clinical response, compared with 29% of patients who received placebo (risk ratio, 1.55; 95% CI, 1.31-1.92). This absolute improvement in pain occurred in 16% more participants taking antidepressants, giving a number needed to treat (NNT) of 6. Average improvement in WOMAC physical function was 10.5 points with placebo and 16.2 points with antidepressants, indicating a “small, clinically unimportant response,” the researchers concluded.
Withdrawals because of adverse events included 11% of the antidepressant group and 5% of the placebo group (RR, 2.15; 95% CI, 1.56-2.87), putting the NNT for a harmful outcome at 17.
For all nine trials together, however, the mean reduction in pain from antidepressants was 2.3 points, compared with 1.7 points with placebo, a statistically significant but ”clinically unimportant improvement,” the researchers concluded. Adverse events occurred in 64% of the antidepressant group, compared with 49% of the placebo group (RR, 1.27; 95% CI, 1.15-1.41), which put the NNT for a harmful outcome at 7. No significant difference in serious adverse events occurred between the groups.
The analysis was limited by the low number of trials, most of which were sponsored by industry and most of which used duloxetine. Further, few of the studies enrolled patients with osteoarthritis of the hip, none assessed medium- or long-term effects, and none stratified the participants for different types of pain (neuropathic-like or central or peripheral pain sensitization).
“My general impression is that there was a statistically significant difference found in favor of duloxetine and the antidepressants,” David J. Hunter, MBBS, PhD, MSc, of the University of Sydney, said after the presentation. “There is a real risk of harm, which I think is important to take into consideration, but at least for me as a clinician and in advising other clinicians, it’s one tool in our armamentarium. I think it’s really important to allow patients to make an informed decision about the potential benefit, the real risk of harm, and the fact that it is quite useful in some patients, and I use it in my clinical practice.”
Jeffrey N. Katz, MD, MS, of Brigham and Women’s Hospital in Boston, said he uses antidepressants in the same way in his practice and that other types of medications, such as TNF inhibitors, also carry risk of harm that may exceed that of antidepressants.
“I’ve had lots of people start duloxetine, and if they stop it, it’s usually because they just don’t tolerate it very well,” Dr. Katz said.
“We don’t want to throw too many things away,” Dr. Hunter added. “Our patients don’t necessarily have a lot of choices here from a pharmacologic perspective, so I think it’s one of those options that I want to keep in my tool kit, and that’s not necessarily going to change.”
The research did not involve outside funding, and Dr. Wluka reported having no industry disclosures. Disclosure information was unavailable for Dr. Katz and Dr. Hunter. The Congress was sponsored by the Osteoarthritis Research Society International.
FROM OARSI 2023
JAK inhibitor ivarmacitinib shows efficacy for atopic dermatitis in a pivotal trial
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.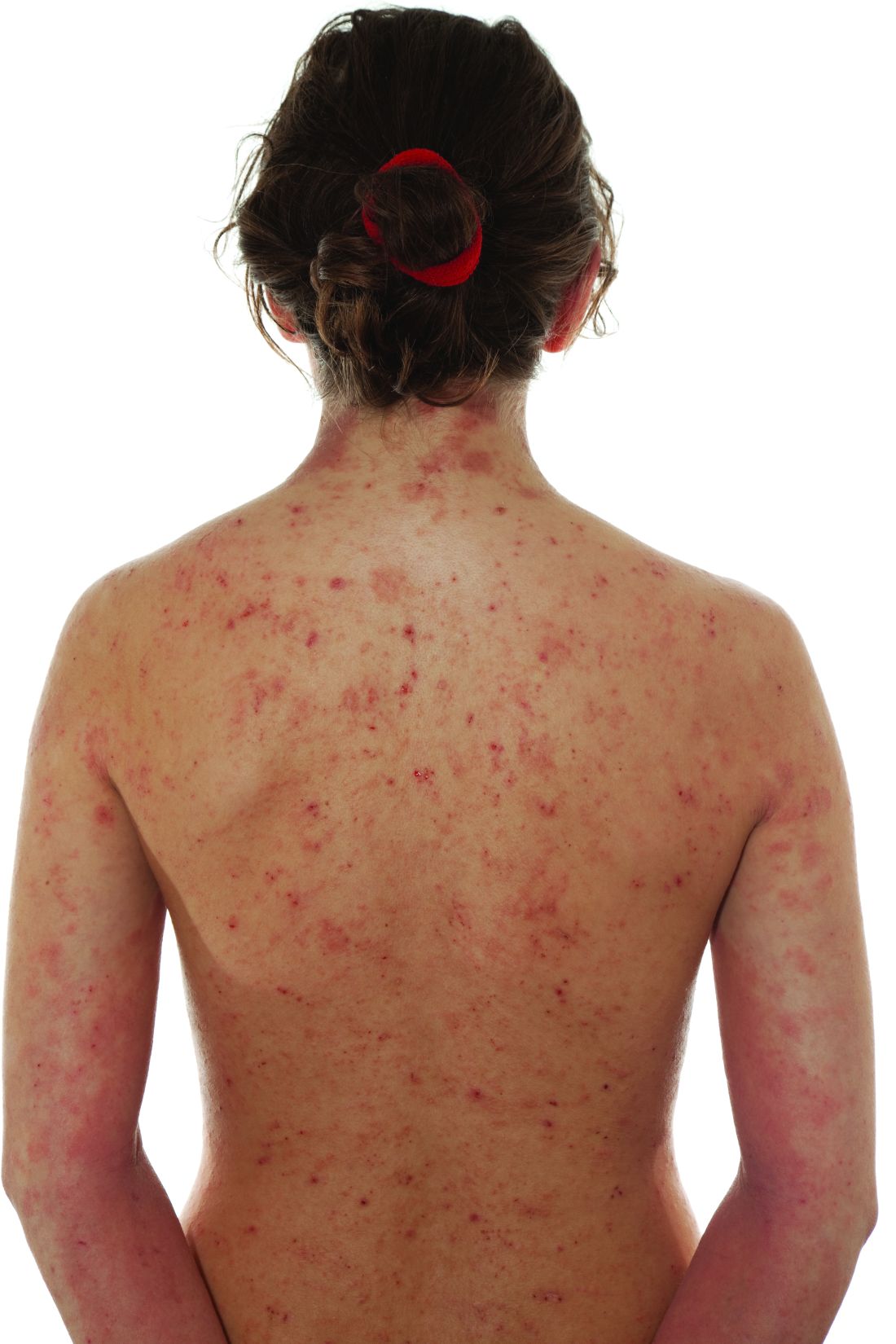
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
NEW ORLEANS – The presented as a late-breaker at the annual meeting of the American Academy of Dermatology.
Two doses were studied in the placebo-controlled trial and both demonstrated “a favorable benefit-to-risk profile in patients with moderate to severe AD,” reported Yan Zhao, MD, a clinician and researcher in the department of dermatology, Peking University People’s Hospital, Beijing.
In the study, called QUARTZ3, 336 patients aged 12 and older at 51 sites in China and Canada were randomized to 4 mg once-daily ivarmacitinib, 8 mg once-daily QD ivarmacitinib, or placebo. The mean age of the population was 32 years and approximately one-third were female.
The mean duration of AD for participants was 10 years. The mean baseline Eczema Area and Severity Index (EASI) score was near 30. On the Investigator Global Assessment (IGA) tool, approximately 40% had a score of 4, which is the highest score on the scale and indicates severe disease. The remaining patients had an IGA score of 3.
The co-primary endpoints were change in IGA and EASI scores at 16 weeks, and both improved rapidly, showing statistical significance relative to placebo by 4 weeks with no plateauing effect at the end of the 16-week trial. By week 16, the proportion of patients with an EASI score of 75, signifying a 75% improvement, was 66%, 54%, and 22% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups (P < .001 versus placebo for both doses of active therapy), respectively.
The pattern of the IGA response was similar. By week 16, the proportion of patients achieving an IGA score of 0 (clear) or 1 (almost clear) was 42%, 36%, and 9% for the 8-mg dose of ivarmacitinib, 4-mg dose of ivarmacitinib, and placebo groups, respectively. The advantage of either dose over placebo was highly significant (P < .001) at 8, 12, and 16 weeks.
For the WI-NRS (Worst Itch – Numeric Rating Scale), the advantage of the 8-mg dose relative to placebo was significant (P < .001) at the 1-week evaluation. By 2 weeks, the 4-mg dose had gained the same degree of statistical significance relative to placebo. After week 4, when the maximum proportion of patients with a WI-NRS score ≤ 4 was reached (50%, 35%, and 10% in the 8-mg, 4-mg, and placebo groups), and the relative advantage of active treatment persisted until the end of the 16-week study.
Two scales were used to evaluate change in quality of life. On the DLQI (Dermatology Life Quality Index) and POEM (Patient-Oriented Eczema Measure), improvements were again rapid and sustained. By week 4, improvement with the 8-mg dose was about fourfold greater (P < .001) than improvement with placebo for DLQI and about sixfold greater (P < .001) for POEM. For the 4-mg dose, the relative differences were approximately threefold and fourfold greater, and both were significant (P <.001).
There was no further gain in these quality-of-life scales from week 4 to week 16, but the advantages relative to placebo were generally sustained, Dr. Zhao reported.
Ivarmacitinib was safe and well-tolerated, according to Dr. Zhao. The proportion of patients with a treatment-emergent adverse event that led to drug discontinuation was numerically higher (5.4%) in the placebo group than in the 8-mg (3.6%) or 4-mg group (2.7%). Rates of infection in the three groups were similar, and there were no major adverse cardiovascular events (MACE) or thromboembolism observed in any group.
Ivarmacitinib, which has about a 10-fold greater selectivity for JAK1 than JAK2 and a more than 70-fold greater selectivity for JAK1 than JAK3, is being tested for rheumatoid arthritis, inflammatory bowel disease, and alopecia areata in addition to AD, Dr. Zhao said. She also reported that an application for new drug approval has been submitted in China. Efforts to pursue regulatory approval elsewhere are anticipated.
Currently, there are three JAK inhibitors licensed for the treatment of AD in the United States. Upadacitinib (Rinvoq) and abrocitinib (Cibinqo) are also once-daily oral JAK1-selective inhibitors. Regulatory approval for AD by the Food and Drug Administration was granted to both in early 2022 and both now have an indication for moderate to severe disease in patients ages 12 years and older.
In September 2021, the first U.S. approval of a drug in this class for AD was granted for a topical formulation of ruxolitinib (Opzelura), which has selectivity for both JAK1 and JAK2. The indication is for mild to moderate AD in patients aged 12 years and older.
In the phase 3 clinical trial that led to approval of abrocitinib for AD, the comparator groups included placebo and active treatment with 300 mg dupilumab administered subcutaneously every other week. The higher of two doses of abrocitinib (100 mg) was numerically superior to dupilumab in terms of EASI 75 response at week 12 and was statistically superior for relief of itch at week 2.
Relative to the first-generation JAK inhibitor tofacitinib (Xeljanz), both of the approved oral JAK inhibitors for AD, abrocitinib and upadacitinib, have greater JAK1-selectivity. However, selectivity for all JAK inhibitors is relative rather than absolute, according to a recent review article on oral JAK inhibitors for AD. Efficacy and safety are likely determined by relative inhibition of each of the four JAK enzymes (JAK1, JAK2, JAK3, and TYK2). Although JAK1 appears to be an important target for AD treatment, the clinical significance of the degree of selectivity among oral JAK inhibitors is not yet clear.
In an interview, the senior author of that review article, Emma Guttman-Yassky, MD, PhD, emphasized this point. She said there is no evidence and no basis on which to speculate that any one drug in this class is better than another for AD. Dr. Guttman-Yassky is a professor and system chair of dermatology and immunology at the Icahn School of Medicine at Mount Sinai, New York.
“The efficacy [of ivarmacitinib] seems, in general, to be in line with other JAK inhibitors,” said Dr. Guttman-Yassky, who attended the late-breaker session during which these data were presented. Although she acknowledged that rapid control of pruritus is important clinically, she said the speed of itch relief as reported in the phase 3 ivarmacitinib trial does not distinguish it from other oral drugs in the class.
Shawn Kwatra, MD, director of the Johns Hopkins Itch Center, Johns Hopkins University, Baltimore, agreed.
“The rapid effects on itch of ivarmacitinib are consistent with those observed by the already approved JAK1-selective inhibitors abrocitinib and upadacitinib,” he said in an interview.
This suggests that head-to-head trials will be needed to draw any conclusions about the relative efficacy and safety of existing and emerging oral JAK inhibitors for AD.
Dr. Zhao has reported a financial relationship with Reistone Biopharma, which is developing ivarmacitinib and provided funding for the trial. Dr. Guttman-Yassky has reported financial relationships with more than 20 pharmaceutical companies, including companies that make JAK inhibitors. Dr. Kwatra has reported financial relationships with AbbVie, Aslan, Arcutis Biotherapeutics, Castle Biosciences, Celldex, Galderma, Genzada, Incyte, Johnson & Johnson, Leo Pharma, Novartis, Pfizer, Regeneron, and Sanofi.
A version of this article first appeared on Medscape.com.
AT AAD 2023
Melatonin: A new way to reduce self-harm?
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
. However, at least one expert has some concerns about the strength of the evidence.
The results suggest improving sleep hygiene in this population may reduce self-injury, study investigator Sarah E. Bergen, PhD, associate professor, department of medical epidemiology and biostatistics, Karolinska Institute, Stockholm, said in an interview.
In addition, she noted, for “pediatric patients who are experiencing sleep problems, melatonin is a safe and effective way to help them.”
Dr. Bergen believes clinicians should recommend melatonin to all teens because “there’s little harm that could come from it and possibly a lot of benefit.”
The findings were published online in the Journal of Child Psychology and Psychiatry.
Few treatments available
Research shows sleep disorders like insomnia are common in youth, particularly among those with psychiatric disorders. Sleep disorders can significantly affect daytime functioning, cognition, emotional regulation, and behavior, and can be a risk factor for unintentional injuries such as falls and vehicular accidents, as well as for intentional self-harm.
The lifetime prevalence of self-harm in youth is estimated to be 17%, but this varies across study designs. There are few treatments for self-harm in youth, although psychosocial treatments appear promising.
Melatonin is a naturally occurring hormone secreted primarily by the pineal gland in response to darkness. It helps promote and maintain the normal sleep-wake cycle and is involved in other biological functions.
In Sweden, melatonin is the most commonly prescribed drug for sleep disturbances in children and adolescents. Prior to 2020, during the course of the study, it was only available by prescription.
The study, which used linked national databases, included 25,575 children and adolescents, 58.2% of them male, who initiated a melatonin treatment between the ages of 6 and 18 years.
Researchers estimated the risks of self-harm, including poisoning (57%) and cutting (34%). The fact that poisoning was more common than cutting was somewhat surprising, said Dr. Bergen. “I would have thought the opposite would be true; that cutting was more prevalent.”
The study examined the risk of self-harm in individual participants by comparing the last unmedicated month with the 12 months after initiating melatonin treatment. In this way, they accounted for potential confounders such as genetics, sleep disorder severity, and psychiatric disorders.
The median age at first melatonin prescription was 13 years for males and 15 years for females.
While there were no statistically significant changes in relative risk for body injuries, falls, and transport accidents, the relative risk for self-injury was statistically significantly lower during the months following melatonin treatment initiation.
The incidence rate ratio in the month following treatment was 0.58 (95% confidence interval, 0.46-0.73) for self-harm and 0.59 (95% CI, 0.45-0.78) for poisoning.
Higher risks in females
The relative risk of self-harm was higher in females than males. This, said Dr. Bergen, is possibly because self-harm is more common in adolescence than in childhood. Female study participants were older than their male counterparts.
Melatonin may help male teens, too, she said. “It’s just that the problem is not that great in males to begin with, so a decrease is not very dramatic after melatonin initiation.”
About 87.2% of participants treated with melatonin were diagnosed with at least one psychiatric disorder. Attention-deficit hyperactivity disorder, the most common comorbidity, was diagnosed in more than 50% of new melatonin users. This isn’t surprising, because sleep disturbances are associated with this psychiatric condition and are frequent side effects of ADHD medications.
After ADHD, anxiety and depression were the next most common psychiatric disorders among study subjects. The analysis found risks for self-harm and poisoning were largely driven by patients suffering from one or both of these disorders, particularly among females.
The IRR in the month following melatonin treatment initiation was 0.46 (95% CI, 0.27-0.76] among adolescent females with psychiatric disorders, after excluding antidepressant users.
Melatonin may reduce the risk of self-harm by treating sleep problems related to psychiatric comorbidities, especially anxiety and depression. It could also decrease pain sensitivity experienced by adolescents who self-harm.
Other factors could play a role in treating sleep problems and/or preventing self-harm in these patients. For example, increased clinician awareness and monitoring, behavioral interventions, a placebo effect, and concurrent use of other medications.
When researchers ran an analysis that excluded individuals taking an antidepressant, “surprisingly, there wasn’t much difference,” said Dr. Bergen. “We thought antidepressants might be causing some of the effect we observed, but when we removed antidepressant users, we saw a very similar pattern of intentional self-harm rates following melatonin use, which suggests melatonin is causal, but we can’t prove that.”
Other sleep medications such as sedatives could also affect self-harm rates by improving sleep. However, these are not typically prescribed to children because of their side effects and overdose potential, said Dr. Bergen.
“Melatonin is extremely safe and side effects are rare; it’s impossible to overdose, and people really can’t hurt themselves with it.”
More research needed
Adrian Jacques Ambrose, MD, medical director, Columbia University Irving Medical Center, and assistant professor of psychiatry, Columbia University, New York, pointed out some evidence in the study is relatively weak.
“When the authors separated out the on- and off-melatonin groups, it looks like there wasn’t a statistically significant difference [in IRRs] between the two groups – for example, in any injury, self-harm, or poisoning – and this weakens their argument that melatonin is associated with self-harm and poisoning.”
Given the current youth mental health crisis, more research “would absolutely be indicated” to better explore possible additional variables, said Dr. Ambrose.
“For example, some additional follow-up studies may add on covariates in conjunction with melatonin usage, such as the number of medical appointments, the presence of psychotherapeutic interventions, dosage of melatonin, or even the sleepiness scale, to evaluate whether the symptoms of sleep disturbances are more directly correlated with the self-harm behaviors.”
The study was supported by the European Union’s Horizon 2020 Research and Innovation Programme. Dr. Bergen and Dr. Ambrose report no relevant financial relationships.
A version of this article first appeared on Medscape.com.
FROM THE JOURNAL OF CHILD PSYCHOLOGY AND PSYCHIATRY
Dupilumab moves forward as possible COPD treatment
of more than 900 adults with uncontrolled chronic obstructive pulmonary disease.
In the study, known as the BOREAS trial, dupilumab met its primary and secondary endpoints, with a significant reduction compared with placebo in exacerbations for adults with chronic obstructive pulmonary disease (COPD) that was uncontrolled despite use of the maximal standard-of-care inhaled therapy (triple therapy), according to a press release from manufacturers Regeneron and Sanofi.
Dupilumab, which inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways, is currently approved in multiple countries for certain patients with conditions including atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, or prurigo nodularis in different age groups. The drug is not an immunosuppressant, and would be the first biologic approved for COPD, according to the manufacturers.
In the BOREAS trial, 468 adults with COPD who were current or former smokers aged 40-80 years were randomized to dupilumab and 471 to placebo; both groups continued to receive maximal standard of care.
Over 52 weeks, patients in the dupilumab group experienced a 30% reduction in moderate to severe COPD exacerbations compared with placebo (P = .0005).
In addition, patients treated with dupilumab met the key secondary endpoints of significant improvement in lung function from baseline to 12 weeks compared with placebo (160 mL vs. 77 mL, P < .0001); this difference persisted at 52 weeks (P = .0003).
Dupilumab also met endpoints for improvement in patient-reported health-related quality of life based on the St. George’s Respiratory Questionnaire (SGRQ) and reduction in the severity of respiratory symptoms of COPD based on the Evaluation Respiratory Symptoms: COPD (E-RS: COPD) Scale, according to the companies’ statement.
The results represent a previously unreported magnitude of improvement for COPD patients treated with a biologic, principal investigator George D. Yancopoulos, MD, said in the statement. “These results also validate the role type 2 inflammation plays in driving COPD in these patients, advancing the scientific community’s understanding of the underlying biology of this disease,” he added.
The safety results in the BOREAS trial were generally consistent with the known safety profile of Dupixent in its approved indications. Overall adverse event rates were similar for dupilumab and placebo patients (77% and 76%, respectively) and the overall safety profiles were consistent with the currently approved dupilumab indications, according to the manufacturers.
The adverse events that were more common in dupilumab patients compared with placebo patients were headache (8.1% vs. 6.8%), diarrhea (5.3% vs. 3.6%), and back pain (5.1% vs. 3.4%).
Adverse events leading to deaths were similar between the groups (1.7% in placebo patients and 1.5% in dupilumab patients).
Complete safety and efficacy results from the BOREAS trial are scheduled to be presented in a future scientific forum, and a second phase 3 trial of dupilumab for COPD, known as NOTUS, is ongoing, with data expected in 2024, according to the manufacturers.
The Boreas trial was sponsored by Sanofi and Regeneron Pharmaceuticals.
of more than 900 adults with uncontrolled chronic obstructive pulmonary disease.
In the study, known as the BOREAS trial, dupilumab met its primary and secondary endpoints, with a significant reduction compared with placebo in exacerbations for adults with chronic obstructive pulmonary disease (COPD) that was uncontrolled despite use of the maximal standard-of-care inhaled therapy (triple therapy), according to a press release from manufacturers Regeneron and Sanofi.
Dupilumab, which inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways, is currently approved in multiple countries for certain patients with conditions including atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, or prurigo nodularis in different age groups. The drug is not an immunosuppressant, and would be the first biologic approved for COPD, according to the manufacturers.
In the BOREAS trial, 468 adults with COPD who were current or former smokers aged 40-80 years were randomized to dupilumab and 471 to placebo; both groups continued to receive maximal standard of care.
Over 52 weeks, patients in the dupilumab group experienced a 30% reduction in moderate to severe COPD exacerbations compared with placebo (P = .0005).
In addition, patients treated with dupilumab met the key secondary endpoints of significant improvement in lung function from baseline to 12 weeks compared with placebo (160 mL vs. 77 mL, P < .0001); this difference persisted at 52 weeks (P = .0003).
Dupilumab also met endpoints for improvement in patient-reported health-related quality of life based on the St. George’s Respiratory Questionnaire (SGRQ) and reduction in the severity of respiratory symptoms of COPD based on the Evaluation Respiratory Symptoms: COPD (E-RS: COPD) Scale, according to the companies’ statement.
The results represent a previously unreported magnitude of improvement for COPD patients treated with a biologic, principal investigator George D. Yancopoulos, MD, said in the statement. “These results also validate the role type 2 inflammation plays in driving COPD in these patients, advancing the scientific community’s understanding of the underlying biology of this disease,” he added.
The safety results in the BOREAS trial were generally consistent with the known safety profile of Dupixent in its approved indications. Overall adverse event rates were similar for dupilumab and placebo patients (77% and 76%, respectively) and the overall safety profiles were consistent with the currently approved dupilumab indications, according to the manufacturers.
The adverse events that were more common in dupilumab patients compared with placebo patients were headache (8.1% vs. 6.8%), diarrhea (5.3% vs. 3.6%), and back pain (5.1% vs. 3.4%).
Adverse events leading to deaths were similar between the groups (1.7% in placebo patients and 1.5% in dupilumab patients).
Complete safety and efficacy results from the BOREAS trial are scheduled to be presented in a future scientific forum, and a second phase 3 trial of dupilumab for COPD, known as NOTUS, is ongoing, with data expected in 2024, according to the manufacturers.
The Boreas trial was sponsored by Sanofi and Regeneron Pharmaceuticals.
of more than 900 adults with uncontrolled chronic obstructive pulmonary disease.
In the study, known as the BOREAS trial, dupilumab met its primary and secondary endpoints, with a significant reduction compared with placebo in exacerbations for adults with chronic obstructive pulmonary disease (COPD) that was uncontrolled despite use of the maximal standard-of-care inhaled therapy (triple therapy), according to a press release from manufacturers Regeneron and Sanofi.
Dupilumab, which inhibits the signaling of the interleukin-4 (IL-4) and interleukin-13 (IL-13) pathways, is currently approved in multiple countries for certain patients with conditions including atopic dermatitis, asthma, chronic rhinosinusitis with nasal polyps, eosinophilic esophagitis, or prurigo nodularis in different age groups. The drug is not an immunosuppressant, and would be the first biologic approved for COPD, according to the manufacturers.
In the BOREAS trial, 468 adults with COPD who were current or former smokers aged 40-80 years were randomized to dupilumab and 471 to placebo; both groups continued to receive maximal standard of care.
Over 52 weeks, patients in the dupilumab group experienced a 30% reduction in moderate to severe COPD exacerbations compared with placebo (P = .0005).
In addition, patients treated with dupilumab met the key secondary endpoints of significant improvement in lung function from baseline to 12 weeks compared with placebo (160 mL vs. 77 mL, P < .0001); this difference persisted at 52 weeks (P = .0003).
Dupilumab also met endpoints for improvement in patient-reported health-related quality of life based on the St. George’s Respiratory Questionnaire (SGRQ) and reduction in the severity of respiratory symptoms of COPD based on the Evaluation Respiratory Symptoms: COPD (E-RS: COPD) Scale, according to the companies’ statement.
The results represent a previously unreported magnitude of improvement for COPD patients treated with a biologic, principal investigator George D. Yancopoulos, MD, said in the statement. “These results also validate the role type 2 inflammation plays in driving COPD in these patients, advancing the scientific community’s understanding of the underlying biology of this disease,” he added.
The safety results in the BOREAS trial were generally consistent with the known safety profile of Dupixent in its approved indications. Overall adverse event rates were similar for dupilumab and placebo patients (77% and 76%, respectively) and the overall safety profiles were consistent with the currently approved dupilumab indications, according to the manufacturers.
The adverse events that were more common in dupilumab patients compared with placebo patients were headache (8.1% vs. 6.8%), diarrhea (5.3% vs. 3.6%), and back pain (5.1% vs. 3.4%).
Adverse events leading to deaths were similar between the groups (1.7% in placebo patients and 1.5% in dupilumab patients).
Complete safety and efficacy results from the BOREAS trial are scheduled to be presented in a future scientific forum, and a second phase 3 trial of dupilumab for COPD, known as NOTUS, is ongoing, with data expected in 2024, according to the manufacturers.
The Boreas trial was sponsored by Sanofi and Regeneron Pharmaceuticals.
Forceps may help moms with obesity avoid cesareans
Among patients who undergo forceps-assisted vaginal delivery, obesity does not appear to be associated with increased risk for complications such as injuries to the anal sphincter or the need for their babies to be admitted to the neonatal intensive care unit, researchers have found.
But obesity does appear to increase the chances that when physicians attempt operative vaginal delivery with either forceps or a vacuum, patients will wind up undergoing cesarean delivery, another study found.
Taken together, the new data may help inform physicians’ decisions about when to consider operative vaginal delivery as an alternative to emergency cesarean births.
A prospective study showed that failed operative vaginal delivery – that is, a cesarean delivery after an attempted operative vaginal delivery – occurred for 10.1% of patients with obesity and 4.2% of those without obesity.
Researchers presented the findings at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We want to really try to reduce the rate of C-sections and primary cesarean deliveries. One of the ways to do that is to attempt operative vaginal delivery,” said Marissa Platner, MD, assistant professor of maternal-fetal medicine at Emory University School of Medicine, Atlanta, who was not involved in the new research.
Data on how obesity influences risks with operative vaginal delivery have been limited and mixed, the researchers said.
To examine how often attempted operative vaginal delivery fails in patients with obesity, Jennifer Grasch, MD, a maternal-fetal medicine fellow at the Ohio State University Wexner Medical Center, Columbus, and her colleagues conducted a secondary analysis of data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be, which included more than 10,000 participants.
“We know that cesarean sections among people with obesity are associated with increased complications, such as higher rates of infection and wound complications, than for people with lower BMI,” Dr. Grasch said. “Operative vaginal delivery can be an alternative to cesarean delivery in some situations, so we were interested in whether attempted operative vaginal delivery was also associated with higher rates of complications in individuals with obesity than those without obesity.”
The researchers focused on 791 patients with an attempted operative vaginal delivery. About 40% had a BMI of 30 or greater. Clinicians used a vacuum in approximately 60% of the attempts.
After an attempted vacuum-assisted delivery, neonatal morbidity was more common for infants whose mothers had obesity than for those whose mothers did not (32.7% vs. 22.3%; adjusted odds ratio, 1.61 [1.07-2.43]). Neonatal morbidity did not differ by obesity status following forceps-attempted delivery. Other adverse outcomes, including measures of maternal morbidity, did not significantly differ by obesity status, according to the researchers.
Choice may come down to experience
Several factors influence whether a clinician chooses forceps- or vacuum-assisted delivery or cesarean delivery, “but one of the most important is experience,” Dr. Grasch said. “Complication rates with both forms of operative vaginal delivery are low, yet there has been a trend toward lower rates of both in the last few decades.”
Elizabeth Cochrane, MD, a maternal-fetal medicine fellow at Mount Sinai Hospital, New York, and her colleagues investigated the relationship between obesity and adverse outcomes among patients with forceps-assisted vaginal deliveries.
The researchers analyzed data from 897 patients who underwent a forceps-assisted vaginal delivery between 2017 and 2021; 29% had a BMI of 30 or greater.
Injuries to the anal sphincter – which can lead to fecal incontinence – occurred in 18.7% of patients without obesity and in 17.7% of those with obesity. Admission to the neonatal intensive care unit occurred in 11.5% of patients without obesity and in 12.3% of patients with obesity. The differences were not statistically significant.
The bottom line: For forceps-assisted vaginal delivery, “obesity does not appear to be associated with increased rates” of adverse outcomes for mothers or newborns, the researchers concluded.
Reassuring data
The study by Dr. Cochrane’s group “provides helpful information for providers to be reassured when they are performing forceps deliveries” for patients with obesity, Dr. Platner said.
Rates of obesity have risen in the United States, and physicians often wonder whether a patient with obesity could be a candidate for forceps-assisted delivery, Dr. Cochrane said. In 2019, 29% of women had obesity before becoming pregnant.
“It all really comes down to how comfortable the provider is in that skill set and also the overall clinical scenario,” she said. “Sometimes an operative delivery with forceps or a vacuum can be the fastest way to deliver a baby when there is acute concern for maternal decompensation or fetal decompensation.”
The alternative is an emergency cesarean delivery. Given that those operations can be riskier and more difficult for patients with higher BMIs, a forceps-assisted delivery may be “an interesting alternative to emergency caesarean sections, as long as it is in an appropriate clinical setting with providers who feel very confident and comfortable using those devices,” Dr. Cochrane said.
A version of this article first appeared on Medscape.com.
Among patients who undergo forceps-assisted vaginal delivery, obesity does not appear to be associated with increased risk for complications such as injuries to the anal sphincter or the need for their babies to be admitted to the neonatal intensive care unit, researchers have found.
But obesity does appear to increase the chances that when physicians attempt operative vaginal delivery with either forceps or a vacuum, patients will wind up undergoing cesarean delivery, another study found.
Taken together, the new data may help inform physicians’ decisions about when to consider operative vaginal delivery as an alternative to emergency cesarean births.
A prospective study showed that failed operative vaginal delivery – that is, a cesarean delivery after an attempted operative vaginal delivery – occurred for 10.1% of patients with obesity and 4.2% of those without obesity.
Researchers presented the findings at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We want to really try to reduce the rate of C-sections and primary cesarean deliveries. One of the ways to do that is to attempt operative vaginal delivery,” said Marissa Platner, MD, assistant professor of maternal-fetal medicine at Emory University School of Medicine, Atlanta, who was not involved in the new research.
Data on how obesity influences risks with operative vaginal delivery have been limited and mixed, the researchers said.
To examine how often attempted operative vaginal delivery fails in patients with obesity, Jennifer Grasch, MD, a maternal-fetal medicine fellow at the Ohio State University Wexner Medical Center, Columbus, and her colleagues conducted a secondary analysis of data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be, which included more than 10,000 participants.
“We know that cesarean sections among people with obesity are associated with increased complications, such as higher rates of infection and wound complications, than for people with lower BMI,” Dr. Grasch said. “Operative vaginal delivery can be an alternative to cesarean delivery in some situations, so we were interested in whether attempted operative vaginal delivery was also associated with higher rates of complications in individuals with obesity than those without obesity.”
The researchers focused on 791 patients with an attempted operative vaginal delivery. About 40% had a BMI of 30 or greater. Clinicians used a vacuum in approximately 60% of the attempts.
After an attempted vacuum-assisted delivery, neonatal morbidity was more common for infants whose mothers had obesity than for those whose mothers did not (32.7% vs. 22.3%; adjusted odds ratio, 1.61 [1.07-2.43]). Neonatal morbidity did not differ by obesity status following forceps-attempted delivery. Other adverse outcomes, including measures of maternal morbidity, did not significantly differ by obesity status, according to the researchers.
Choice may come down to experience
Several factors influence whether a clinician chooses forceps- or vacuum-assisted delivery or cesarean delivery, “but one of the most important is experience,” Dr. Grasch said. “Complication rates with both forms of operative vaginal delivery are low, yet there has been a trend toward lower rates of both in the last few decades.”
Elizabeth Cochrane, MD, a maternal-fetal medicine fellow at Mount Sinai Hospital, New York, and her colleagues investigated the relationship between obesity and adverse outcomes among patients with forceps-assisted vaginal deliveries.
The researchers analyzed data from 897 patients who underwent a forceps-assisted vaginal delivery between 2017 and 2021; 29% had a BMI of 30 or greater.
Injuries to the anal sphincter – which can lead to fecal incontinence – occurred in 18.7% of patients without obesity and in 17.7% of those with obesity. Admission to the neonatal intensive care unit occurred in 11.5% of patients without obesity and in 12.3% of patients with obesity. The differences were not statistically significant.
The bottom line: For forceps-assisted vaginal delivery, “obesity does not appear to be associated with increased rates” of adverse outcomes for mothers or newborns, the researchers concluded.
Reassuring data
The study by Dr. Cochrane’s group “provides helpful information for providers to be reassured when they are performing forceps deliveries” for patients with obesity, Dr. Platner said.
Rates of obesity have risen in the United States, and physicians often wonder whether a patient with obesity could be a candidate for forceps-assisted delivery, Dr. Cochrane said. In 2019, 29% of women had obesity before becoming pregnant.
“It all really comes down to how comfortable the provider is in that skill set and also the overall clinical scenario,” she said. “Sometimes an operative delivery with forceps or a vacuum can be the fastest way to deliver a baby when there is acute concern for maternal decompensation or fetal decompensation.”
The alternative is an emergency cesarean delivery. Given that those operations can be riskier and more difficult for patients with higher BMIs, a forceps-assisted delivery may be “an interesting alternative to emergency caesarean sections, as long as it is in an appropriate clinical setting with providers who feel very confident and comfortable using those devices,” Dr. Cochrane said.
A version of this article first appeared on Medscape.com.
Among patients who undergo forceps-assisted vaginal delivery, obesity does not appear to be associated with increased risk for complications such as injuries to the anal sphincter or the need for their babies to be admitted to the neonatal intensive care unit, researchers have found.
But obesity does appear to increase the chances that when physicians attempt operative vaginal delivery with either forceps or a vacuum, patients will wind up undergoing cesarean delivery, another study found.
Taken together, the new data may help inform physicians’ decisions about when to consider operative vaginal delivery as an alternative to emergency cesarean births.
A prospective study showed that failed operative vaginal delivery – that is, a cesarean delivery after an attempted operative vaginal delivery – occurred for 10.1% of patients with obesity and 4.2% of those without obesity.
Researchers presented the findings at the meeting sponsored by the Society for Maternal-Fetal Medicine.
“We want to really try to reduce the rate of C-sections and primary cesarean deliveries. One of the ways to do that is to attempt operative vaginal delivery,” said Marissa Platner, MD, assistant professor of maternal-fetal medicine at Emory University School of Medicine, Atlanta, who was not involved in the new research.
Data on how obesity influences risks with operative vaginal delivery have been limited and mixed, the researchers said.
To examine how often attempted operative vaginal delivery fails in patients with obesity, Jennifer Grasch, MD, a maternal-fetal medicine fellow at the Ohio State University Wexner Medical Center, Columbus, and her colleagues conducted a secondary analysis of data from the Nulliparous Pregnancy Outcomes Study: Monitoring Mothers-to-Be, which included more than 10,000 participants.
“We know that cesarean sections among people with obesity are associated with increased complications, such as higher rates of infection and wound complications, than for people with lower BMI,” Dr. Grasch said. “Operative vaginal delivery can be an alternative to cesarean delivery in some situations, so we were interested in whether attempted operative vaginal delivery was also associated with higher rates of complications in individuals with obesity than those without obesity.”
The researchers focused on 791 patients with an attempted operative vaginal delivery. About 40% had a BMI of 30 or greater. Clinicians used a vacuum in approximately 60% of the attempts.
After an attempted vacuum-assisted delivery, neonatal morbidity was more common for infants whose mothers had obesity than for those whose mothers did not (32.7% vs. 22.3%; adjusted odds ratio, 1.61 [1.07-2.43]). Neonatal morbidity did not differ by obesity status following forceps-attempted delivery. Other adverse outcomes, including measures of maternal morbidity, did not significantly differ by obesity status, according to the researchers.
Choice may come down to experience
Several factors influence whether a clinician chooses forceps- or vacuum-assisted delivery or cesarean delivery, “but one of the most important is experience,” Dr. Grasch said. “Complication rates with both forms of operative vaginal delivery are low, yet there has been a trend toward lower rates of both in the last few decades.”
Elizabeth Cochrane, MD, a maternal-fetal medicine fellow at Mount Sinai Hospital, New York, and her colleagues investigated the relationship between obesity and adverse outcomes among patients with forceps-assisted vaginal deliveries.
The researchers analyzed data from 897 patients who underwent a forceps-assisted vaginal delivery between 2017 and 2021; 29% had a BMI of 30 or greater.
Injuries to the anal sphincter – which can lead to fecal incontinence – occurred in 18.7% of patients without obesity and in 17.7% of those with obesity. Admission to the neonatal intensive care unit occurred in 11.5% of patients without obesity and in 12.3% of patients with obesity. The differences were not statistically significant.
The bottom line: For forceps-assisted vaginal delivery, “obesity does not appear to be associated with increased rates” of adverse outcomes for mothers or newborns, the researchers concluded.
Reassuring data
The study by Dr. Cochrane’s group “provides helpful information for providers to be reassured when they are performing forceps deliveries” for patients with obesity, Dr. Platner said.
Rates of obesity have risen in the United States, and physicians often wonder whether a patient with obesity could be a candidate for forceps-assisted delivery, Dr. Cochrane said. In 2019, 29% of women had obesity before becoming pregnant.
“It all really comes down to how comfortable the provider is in that skill set and also the overall clinical scenario,” she said. “Sometimes an operative delivery with forceps or a vacuum can be the fastest way to deliver a baby when there is acute concern for maternal decompensation or fetal decompensation.”
The alternative is an emergency cesarean delivery. Given that those operations can be riskier and more difficult for patients with higher BMIs, a forceps-assisted delivery may be “an interesting alternative to emergency caesarean sections, as long as it is in an appropriate clinical setting with providers who feel very confident and comfortable using those devices,” Dr. Cochrane said.
A version of this article first appeared on Medscape.com.
FROM SMFM 2023
Topical delgocitinib shows promise for chronic hand eczema, pivotal trial shows
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.

“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
NEW ORLEANS – , compared with those who received vehicle cream, results from a pivotal phase 3 trial showed.
“Chronic hand eczema is the most frequent chronic inflammatory disorder affecting the hands,” Robert Bissonnette, MD, a dermatologist who is founder and CEO of Innovaderm Research, said at the annual meeting of the American Academy of Dermatology, where the study was presented during a late-breaking research session. “It’s associated with pain, pruritus, and has a huge impact on quality of life,” and results with current topical treatments are often unsatisfactory, he noted.
Delgocitinib is an investigational topical pan-JAK inhibitor that inhibits activation of the JAK-STAT pathway and targets key mediators of chronic hand eczema. In a phase 2b dose-ranging trial, twice-daily treatment with delgocitinib cream demonstrated significantly greater efficacy, compared with the cream vehicle, and was well tolerated in adults with mild to severe chronic hand eczema.
For the phase 3 study, known as DELTA 1, researchers randomized 487 adults with moderate to severe chronic hand eczema to receive twice-daily applications of delgocitinib cream 20 mg/g or cream vehicle for 16 weeks. After week 16, patients had the option to enter a long-term extension trial, which is currently ongoing. DELTA 1 was limited to adults with a diagnosis of chronic hand eczema defined as hand eczema that had persisted for more than 3 months or had returned more than twice within the past 12 months; an Investigator’s Global Assessment for chronic hand eczema (IGA-CHE) score of 3 (moderate) or 4 (severe); a weekly average Hand Eczema Symptom Diary (HESD) itch score of 4 or more points, and a medical history of inadequate response to topical corticosteroids within the past 12 months or for whom treatment with topical corticosteroids was not medically advisable.
The IGA-CHE scale used in the trial was new, “where, in order to be almost clear, the only sign that could be present on the skin was barely perceptible erythema,” Dr. Bissonnette said. He noted that he has used many IGA scales over the more than 25 years he has been involved with clinical trials, and “this was the first that used a scale with a bar so high.” Key secondary endpoints include a 75% and 90% improvement in Hand Eczema Severity Index (HECSI) from baseline at week 16 and a 4-point or greater improvement in the Dermatology Life Quality Index (DLQI) from baseline at week 16.
The median age of patients was 44 years, 88% were White, 4% were Asian, 1% were Black, and the remainder were from other racial groups. One-third of patients (33%) had severe hand eczema based on their IGA-CHE score, the median HECSI was 65 (in line with severe disease), and the median DLQI was 12. As for previous chronic hand eczema treatments, 19% had undergone phototherapy, 14% had tried oral retinoids, and 12% had tried oral corticosteroids.
In the study, a greater proportion of delgocitinib-treated patients achieved the primary endpoint of IGA-CHE 0/1, compared with the cream vehicle group at week 4 (15.4% vs. 4.9%; P < .001); week 8 (22.8% vs. 10.5%; P = .001), and week 16 (19.7% vs. 9.9%; P = .006). “As early as week 2, there is a separation between cream and vehicle,” Dr. Bissonnette said. When reviewing the results and the patients in the trial, he said that, in his personal opinion, “I don’t think this is uniquely representative of the efficacy of the drug,” because of the IGA scale that was used, which set such a high bar for efficacy.
As for secondary endpoints, a greater proportion of delgocitinib-treated patients than those in the vehicle group achieved a HESCI-75 (49.2% vs. 23.5%), a HECSI-90 (29.5% vs. 12.3%), and a 4-point or greater improvement on the DLQI (74.4% vs 50%; P < .001 for all endpoints).
Delgocitinib had a similar safety profile as the vehicle over 16 weeks, with no difference between the delgocitinib and vehicle arms in the proportion of patients who had adverse events (45.2% vs. 50.6%, respectively) and serious adverse events (1.8% vs. 1.9%). The most common adverse events (defined as 5% or greater in any treatment group) during the study were COVID-19 infections and nasopharyngitis; rates were comparable in the two arms.
Raj Chovatiya, MD, PhD, a dermatologist who directs the Center for Eczema and Itch at Northwestern University, Chicago, who was asked to comment on the study, said that chronic hand eczema can be functionally limiting for many patients. “Given its focal symptoms but multifaceted immunopathogenesis, topical JAK inhibition represents a rational strategy for targeted treatment,” Dr. Chovatiya told this news organization. He was not an investigator in the trial.
“In the phase 3 DELTA 1 study, topical delgocitinib cream was superior to vehicle control with nearly one out of five patients achieving clear or almost clear skin, with no difference in total adverse events between groups. While both comparative and long-term data would be helpful to better assess how delgocitinib cream stacks up against common topical anti-inflammatories and how it may be used for a chronic condition that typically requires ongoing treatment, these findings move us closer to a potential first-in-class approved therapy for chronic hand eczema.”
Dr. Bissonnette disclosed that he served as a consultant and investigator for the developer of delgocitinib, LEO Pharma, on this study. He has also received grants and research funding from many other pharmaceutical companies. Dr. Chovatiya disclosed that he is a consultant to, a speaker for, and/or a member of the advisory board for several pharmaceutical companies, including LEO Pharma.
AT AAD 2023
FDA approves new formulation of Hyrimoz adalimumab biosimilar
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
The Food and Drug Administration has approved a citrate-free, 100 mg/mL formulation of the biosimilar adalimumab-adaz (Hyrimoz), according to a statement from manufacturer Sandoz.
Hyrimoz, a tumor necrosis factor (TNF) blocker that is biosimilar to its reference product Humira, was approved by the FDA in 2018 at a concentration of 50 mg/mL for rheumatoid arthritis, juvenile idiopathic arthritis, psoriatic arthritis, ankylosing spondylitis, Crohn’s disease, ulcerative colitis, and plaque psoriasis. The high-concentration formula is indicated for these same conditions.
Sandoz said that it intends to launch the citrate-free formulation in the United States on July 1. It will be one of up to nine other adalimumab biosimilars that are expected to launch in July. On January 31, Amjevita (adalimumab-atto) became the first adalimumab biosimilar to launch in the United States.
The current label for Hyrimoz contains a black box warning emphasizing certain risks, notably the increased risk for serious infections, such as tuberculosis or sepsis, and an increased risk of malignancy, particularly lymphomas.
Adverse effects associated with Hyrimoz with an incidence greater than 10% include upper respiratory infections and sinusitis, injection-site reactions, headache, and rash.
The approval for the high-concentration formulation was based on data from a phase 1 pharmacokinetics bridging study that compared Hyrimoz 50 mg/mL and citrate-free Hyrimoz 100 mg/mL.
“This study met all of the primary objectives, demonstrating comparable pharmacokinetics and showing similar safety and immunogenicity of the Hyrimoz 50 mg/mL and Hyrimoz [100 mg/mL],” according to Sandoz, a division of Novartis.
The approval for Hyrimoz 50 mg/mL in 2018 was based on preclinical and clinical research comparing Hyrimoz and Humira. In a phase 3 trial published in the British Journal of Dermatology, which included adults with clinically stable but active moderate to severe chronic plaque psoriasis, Hyrimoz and Humira showed a similar percentage of patients met the primary endpoint of a 75% reduction or more in Psoriasis Area and Severity Index (PASI 75) score at 16 weeks, compared with baseline (66.8% and 65%, respectively).
A version of this article originally appeared on Medscape.com.
Anifrolumab shows promise in refractory discoid lupus erythematosus
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.

Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.
Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
a small retrospective study reports.
DLE, the most common form of chronic cutaneous lupus erythematosus, can permanently scar and disfigure patients, and traditional treatments such as antimalarials, steroid-sparing immunosuppressive agents, thalidomide, retinoids, and lenalidomide don’t consistently improve refractory DLE, the authors noted.
“All patients demonstrated significant improvement in symptomatology and disease activity within 2 months of initiating anifrolumab,” lead study author Katharina Shaw, MD, of the department of dermatology of Brigham and Women’s Hospital and Harvard Medical School, both in Boston, and colleagues wrote in a research letter published in JAMA Dermatology. “These early results highlight the potential for anifrolumab to be a viable therapeutic option for patients with DLE, particularly those with severe or recalcitrant disease.”
The Food and Drug Administration approved anifrolumab (Saphnelo), a human monoclonal antibody targeting type 1 interferon receptor subunit 1, in 2021 for adults with moderate to severe systemic lupus erythematosus, but it has not been approved for the treatment of DLE.
Dr. Shaw and colleagues queried the medical records from Brigham and Women’s Hospital and Massachusetts General Hospital, Boston, to find all cases of DLE based on biopsy, expert opinion, or both from January 2000 to October 2022.
The researchers identified eight female patients who had received anifrolumab for at least 8 weeks. The women were aged between 19 and 75 years (median, 42.5 years), and all had DLE recalcitrant to standard therapies and had been treated with hydroxychloroquine and between 1 and 10 other drugs, most commonly methotrexate and mycophenolate mofetil (MMF).
The authors looked for improvements in patient-reported symptoms and Cutaneous Lupus Erythematosus Disease Area and Severity Index scores, including CLASI A (activity) score 0-70, and CLASI-D (damage) score 0-56.
All patients showed significantly improved symptoms and disease activity within 2 months of their first infusion of the treatment. The mean decrease and mean percentage decrease in CLASI-A scores were 17.1 and 65.1%, respectively. The mean decrease and mean percentage decrease in CLASI-D scores were 0.5 and 2.9%, respectively.
The rapid clinical improvements with anifrolumab, compared with improvements with traditional medications, were striking, the authors wrote. “Given the risk for permanent scarring, dyspigmentation, and alopecia with poorly controlled DLE, the importance of rapidly mitigating disease activity cannot be overemphasized.”
They acknowledged that the results are limited by the study’s small sample size and retrospective design, and they recommend larger related prospective studies.
Asked to comment on the results, Kaveh Ardalan, MD, MS, assistant professor of pediatrics in the division of pediatric rheumatology at Duke University, Durham, N.C., said that finding new DLE therapeutics is important because of the huge impact of uncontrolled DLE on patients’ quality of life, body image, and social roles.
Dr. Ardalan noted that he sees DLE in his pediatric patients, “either as an isolated finding or in the context of systemic lupus erythematosus. Anifrolumab is not approved by the FDA to treat DLE or children.
“Randomized controlled trials, including the TULIP-1 and TULIP-2 studies of anifrolumab in systemic lupus, have indicated that lupus skin manifestations can improve in patients who receive anifrolumab,” said Dr. Ardalan, who was not involved in the study. “And we know that type I interferons are major drivers of cutaneous disease activity in patients with lupus, so targeting that mechanism with anifrolumab makes biological sense.”
The authors’ use of the validated CLASI classification system to quantify disease activity and damage over time, and their determination of the length of time for the drug to take effect are strengths of the study, he added.
Funding information was not provided. Two authors reported financial relationships with Pfizer, which does not manufacture anifrolumab. Dr. Ardalan reported no conflicts of interest with the study.
FROM JAMA DERMATOLOGY
New JAK inhibitor study data confirm benefit in alopecia areata
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
from clinical trials of two drugs presented at a late-breaker research session at the annual meeting of the American Academy of Dermatology.
Based on phase 3 studies that document robust hair growth in about one third of patients, deuruxolitinib (CTP-543), an inhibitor of the JAK1 and JAK2 enzymes, has the potential to become the second JAK inhibitor available for the treatment of alopecia areata. If approved, it will join baricitinib (Olumiant), which received U.S. approval almost 1 year ago.
In his talk on THRIVE-AA2, a phase 3 trial of the investigational medicine deuruxolitinib, the principal investigator, Brett A. King, MD, PhD, displayed several before-and-after photos and said, “The photos tell the whole story. This is why there is so much excitement about these drugs.”
THRIVE-AA2 was the second of two phase 3 studies of deuruxolitinib. King was a principal investigator for both pivotal trials, called THRIVE-AA1 and THRIVE AA-2. He characterized the results of the two THRIVE trials as “comparable.”
Dr. King also was a principal investigator for the trials with baricitinib, called BRAVE-AA1 and BRAVE AA-2, which were published last year in the New England Journal of Medicine. The trials for both drugs had similar designs and endpoints.
Deuruxolitinib and the THRIVE studies
In the THRIVE-AA2 trial, 517 adult patients were enrolled with moderate to severe alopecia areata, defined as a SALT (Severity of Alopecia Tool) score of ≥ 50%, which signifies a hair loss of at least 50%. Like THRIVE-AA1, patients participated at treatment centers in North America and Europe. About two-thirds were female. The mean age was 39 years. The majority of patients had complete or near complete hair loss at baseline.
“Many of these patients are the ones we have historically characterized as having alopecia totalis or universalis,” Dr. King said.
Participating patients were randomly assigned to 8 mg deuruxolitinib twice daily, 12 mg deuruxolitinib twice daily, or placebo. The primary endpoint was a SALT score of ≤ 20% at week 24.
At 24 weeks, almost no patients in the placebo group (1%) vs. 33% and 38% in the 8 mg and 12 mg twice-daily groups, respectively, met the primary endpoint. Each active treatment group was highly significant vs. placebo.
Of the responders, the majority achieved complete or near complete hair growth as defined by a SALT score of ≤ 10%, Dr. King reported.
Based on a graph that showed a relatively steep climb over the entire 24-week study period, deuruxolitinib “had a really fast onset of action,” Dr. King said. By week 8, which was the time of the first assessment, both doses of deuruxolitinib were superior to placebo.
The majority of patients had complete or significant loss of eyebrows and eye lashes at baseline, but more than two-thirds of these patients had regrowth by week 24, Dr. King said. Again, no significant regrowth was observed in the placebo arm.
On the Satisfaction of Hair Patient Reported Outcomes (SPRO), more than half of patients on both doses reported being satisfied or very satisfied with the improvement when evaluated at 24 weeks.
“The patient satisfaction overshot what one would expect by looking at the SALT scores, but a lot of subjects were at the precipice of the primary endpoint, sitting on SALT scores of 21, 25, or 30,” Dr. King said.
High participation in extension trial
More than 90% of the patients assigned to deuruxolitinib completed the trial and have entered an open-label extension (OLE). Dr. King credited the substantial rates of hair growth and the low rate of significant adverse events for the high rate of transition to OLE. Those who experienced the response were motivated to maintain it.
“This is a devastating disease. Patients want to get better,” Dr. King said.
There were no serious treatment-emergent adverse events associated with deuruxolitinib, including no thromboembolic events or other off-target events that have been reported previously with other JAK inhibitors in other disease states, such as rheumatoid arthritis. Although some adverse events, such as nasopharyngitis, were observed more often in those taking deuruxolitinib than placebo, there were “very few” discontinuations because of an adverse event, he said.
The data of THRIVE-AA2 are wholly compatible with the previously reported 706-patient THRIVE-AA1, according to Dr. King. In THRIVE-AA1, the primary endpoint of SALT ≤ 20% was reached by 29.6%, 41.5%, and 0.8% of the 8 mg, 12 mg, and placebo groups, respectively. Patient satisfaction scores, safety, and tolerability were also similar, according to Dr. King.
The experience with deuruxolitinib in the THRIVE-AA phase 3 program is similar to the experience with baricitinib in the BRAVE-AA trials. Although they cannot be compared directly because of potential differences between study populations, the 4-mg dose of baricitinib also achieved SALT score ≤ 20 in about 35% of patients, he said. The proportion was lower in the 2-mg group but was also superior to the placebo group.
“JAK inhibitors are changing the paradigm of alopecia areata,” Dr. King said. Responding to a question about payers reluctant to reimburse therapies for a “cosmetic” condition, Dr. King added that the effective treatments are “changing the landscape of how we think about this disease.” Dr. King believes these kinds of data show that “we are literally transforming lives forever.”
Baricitinib and the BRAVE studies
When baricitinib received regulatory approval for alopecia areata last year, it was not just the first JAK inhibitor approved for this disease, but the first systemic therapy of any kind, according to Maryanne Senna, MD, an assistant professor of dermatology at Harvard Medical School, Boston, and the director of the Lahey Hair Loss Center of Excellence, Burlington, Mass. Dr. Senna was a clinical investigator of BRAVE-AA1, as well as of THRIVE-AA2.
Providing an update on the BRAVE-AA program, Dr. Senna reported 104-week data that appear to support the idea of a life-changing benefit from JAK inhibitor therapy. This is because the effects appear durable.
In the data she presented at the AAD, responders and mixed responders at 52 weeks were followed to 104 weeks. Mixed responders were defined as those without a SALT response of ≤ 20 at week 52 but who had achieved this degree of hair regrowth at some earlier point.
Of the responders, 90% maintained their response at 104 weeks. In addition, many of the mixed responders and patients with a partial response but who never achieved a SALT score ≤ 20% gained additional hair growth, including complete or near complete hair growth, when maintained on treatment over the 2 years of follow-up.
“The follow-up suggests that, if you keep patients on treatment, you can get many of them to a meaningful response,” she said.
Meanwhile, “there have been no new safety signals,” Dr. Senna said. She based this statement not only of the 104-week data but on follow-up of up to 3.6 years among patients who have remained on treatment after participating in previous studies.
According to Dr. Senna, the off-target events that have been reported previously in other diseases with other JAK inhibitors, such as major adverse cardiovascular events and thromboembolic events, have not so far been observed in the BRAVE-AA phase 3 program.
Baricitinib, much like all but one of the JAK inhibitors with dermatologic indications, carries a black box warning that lists multiple risks for drugs in this class, based on a rheumatoid arthritis study.
The Food and Drug Administration has granted deuruxolitinib Breakthrough Therapy designation for the treatment of adult patients with moderate to severe alopecia areata and Fast Track designation for the treatment of alopecia areata, according to its manufacturer Concert Pharmaceuticals.
Dr. King reports financial relationships with more than 15 pharmaceutical companies, including Concert Pharmaceuticals, which provided the funding for the THRIVE-AA trial program, and for Eli Lilly, which provided funding for the BRAVE-AA trial program. Dr. Senna reports financial relationships with Arena pharmaceuticals, Follica, and both Concert Pharmaceuticals and Eli Lilly.
A version of this article originally appeared on Medscape.com.
AT AAD 2023